- Tổng quan
- Nội dung
- Tiêu chuẩn liên quan
- Lược đồ
- Tải về
Tiêu chuẩn TCVN I-1:2017 Bộ tiêu chuẩn về phương pháp kiểm nghiệm thuốc
| Số hiệu: | TCVN I-1:2017 | Loại văn bản: | Tiêu chuẩn Việt Nam |
| Cơ quan ban hành: | Bộ Khoa học và Công nghệ | Lĩnh vực: | Y tế-Sức khỏe , Thực phẩm-Dược phẩm |
|
Ngày ban hành:
Ngày ban hành là ngày, tháng, năm văn bản được thông qua hoặc ký ban hành.
|
2017 |
Hiệu lực:
|
Đã biết
|
| Người ký: | Đang cập nhật |
Tình trạng hiệu lực:
Cho biết trạng thái hiệu lực của văn bản đang tra cứu: Chưa áp dụng, Còn hiệu lực, Hết hiệu lực, Hết hiệu lực 1 phần; Đã sửa đổi, Đính chính hay Không còn phù hợp,...
|
Đã biết
|
TÓM TẮT TIÊU CHUẨN VIỆT NAM TCVN I-1:2017
Nội dung tóm tắt đang được cập nhật, Quý khách vui lòng quay lại sau!
Tải tiêu chuẩn Việt Nam TCVN I-1:2017
 Tiêu chuẩn Việt Nam TCVN I-1:2017 PDF (Bản có dấu đỏ)
Tiêu chuẩn Việt Nam TCVN I-1:2017 PDF (Bản có dấu đỏ) Tiêu chuẩn Việt Nam TCVN I-1:2017 DOC (Bản Word)
Tiêu chuẩn Việt Nam TCVN I-1:2017 DOC (Bản Word)TIÊU CHUẨN QUỐC GIA
TCVN 1-1:2017
BỘ TIÊU CHUẨN QUỐC GIA VỀ THUỐC - PHẦN 1: PHƯƠNG PHÁP KIỂM NGHIỆM THUỐC (GỒM 201 TIÊU CHUẨN)
Set of national standards for medicines -
Part 1: General methods for quality control of medicines
Mục lục
Lời nói đầu
Lời giới thiệu
1 Phạm vi áp dụng
2 Quy định chung
3 Kí hiệu và các chữ viết tắt
Phụ lục 1
1.1 Cao thuốc
1.2 Cồn thuốc
1.3 Dung dịch thuốc
1.4 Sirô thuốc
1.5 Hỗn dịch thuốc
1.6 Nhũ tương thuốc
1.7 Thuốc bột
1.8 Thuốc cốm
1.9 Thuốc dán thấm qua da và cao dán
1.10 Thuốc đặt
1.11 Thuốc hoàn
1.12 Thuốc mềm dùng trên da và niêm mạc
1.13 Thuốc nang
1.14 Thuốc nhỏ mắt
1.15 Thuốc nhỏ mũi và thuốc xịt mũi dạng lỏng
1.16 Thuốc nhỏ tai và thuốc xịt vào tai
1.17 Thuốc hít
1.18 Thuốc khí dung
1.19 Thuốc tiêm, thuốc tiêm truyền
1.20 Thuốc viên nén
1.21 Thuốc bọt y tế
1.22 Rượu thuốc
1.23 Thuốc thang
1.24 Chè thuốc
Phụ lục 2
2.1 Các thuốc thử chung
2.1.1 Hóa chất và thuốc thử
2.1.2 Các chất chỉ thị
2.2 Các dung dịch chuẩn độ
2.3 Các dung dịch đệm
2.4 Các dung dịch mẫu
2.5 Các chất đối chiếu
Phụ lục 3
3.1 Cân và xác định khối lượng
3.2 Nhiệt kế
3.3 Dụng cụ đo thể tích
3.4 Phễu lọc thủy tinh xốp
3.5 Cỡ bột và rây
3.6 Rửa dụng cụ thủy tinh
Phụ lục 4
4.1 Phương pháp quang phổ hấp thụ tử ngoại và khả kiến
4.2 Phương pháp quang phổ hồng ngoại
4.3 Phương pháp quang phổ huỳnh quang
4.4 Phương pháp quang phổ nguyên tử phát xạ và hấp thụ
Phụ lục 5
5 Các kĩ thuật tách sắc ký
5.1 Phương pháp sắc ký giấy
5.2 Phương pháp sắc ký khí
5.3 Phương pháp sắc ký lỏng
5.4 Phương pháp sắc ký lớp mỏng
5.5 Phương pháp sắc ký rây phân tử
5.6 Phương pháp điện di
5.7 Phương pháp điện di mao quản
Phụ lục 6
6.1 Xác định chỉ số khúc xạ
6.2 Xác định chỉ số pH
6.3 Xác định độ nhớt của chất lỏng
6.4 Xác định góc quay cực và góc quay cực riêng
6.5 Xác định khối lượng riêng và tỷ trọng
6.6 Xác định nhiệt độ đông đặc
6.7 Xác định nhiệt độ nóng chảy, khoảng nóng chảy và điểm nhỏ giọt
6.8 Xác định nhiệt độ sôi và khoảng chưng cất
6.9 Xác định độ thẩm thấu
6.10 Xác định điện dẫn suất
6.11 Xác định khối lượng riêng của chất rắn
Phụ lục 7
7.1 Xác định chỉ số acetyl
7.2 Xác định chỉ số acid
7.3 Xác định chỉ số ester
7.4 Xác định chỉ số hydroxyl
7.5 Xác định chỉ số iod
7.6 Xác định chỉ số peroxyd
7.7 Xác định chỉ số xà phòng hóa
7.8 Xác định chất không bị xà phòng hóa
7.9 Xác định lưu huỳnh dioxyd
7.10 Xác định các chất oxy hóa
7.11 Xác định carbon hữu cơ toàn phần trong nước dùng cho ngành dược
Phụ lục 8
8.1 Các phản ứng định tính
8.2 Định tính các penicilin
8.3 Phản ứng màu của các penicilin và cephalosporin
Phụ lục 9
9.1 Ống nghiệm dùng trong các phép thử so sánh
9.2 Xác định độ trong của dung dịch
9.3 Xác định màu sắc của dung dịch
9.4 Xác định giới hạn các tạp chất
9.4.1 Amoni
9.4.2 Arsen
9.4.3 Calci
9.4.4 Chì trong đường
9.4.5 Clorid
9.4.6 Fluorid
9.4.7 Kali
9.4.8 Kim loại nặng
9.4.9 Nhôm
9.4.10 Nickel trong polyol
9.4.11 Kim loại nặng trong dược liệu và trong dầu béo
9.4.12 Phosphat
9.4.13 Sắt
9.4.14 Sulfat
9.4.15 Magnesi
9.4.16 Magnesi và kim loại kiềm thổ
9.5 Xác định giới hạn carbon monoxyd trong khí y tế
9.6 Xác định mất khối lượng do làm khô
9.7 Xác định tro không tan trong acid
9.8 Xác định tro toàn phần
9.9 Xác định tro sulfat
9.10 Xác định tro tan trong nước
Phụ lục 10
10.1 Phương pháp chuẩn độ ampe
10.2 Phương pháp chuẩn độ đo điện thế
10.3 Định lượng nước
10.4 Phương pháp chuẩn độ bằng nitrit
10.5 Phương pháp chuẩn độ complexon
10.6 Phương pháp chuẩn độ trong môi trường khan
10.7 Định lượng các kháng sinh họ penicilin bằng phương pháp đo iod
10.8 Định lượng các steroid bằng tetrazolium
10.9 Định lượng nitrogen trong hợp chất hữu cơ
10.10 Định lượng vitamin A
10.11 Phương pháp phân tích acid amin
10.12 Xác định hàm lượng ethanol
10.13 Xác định hàm lượng methanol và propan-2-ol
10.14 Xác định dung môi tồn dư
10.14.1 Quy định đối với tạp chất là dung môi tồn dư
10.15 Xác định ethylen oxyd và dioxan tồn dư
10.16 Định lượng N,N-dimethylanilin
10.17 Định lượng acid 2-ethylhexanoic
10.18 Xác định acid acetic trong peptid tổng hợp
10.19 Đốt trong oxygen
Phụ lục 11
11.1 Giới hạn cho phép về thể tích của các dạng thuốc lỏng
11.2 Phép thử đồng đều hàm lượng
11.3 Phép thử đồng đều khối lượng
11.4 Phép thử độ hòa tan của dạng thuốc rắn phân liều
11.5 Phép thử độ rã của thuốc đạn và thuốc trứng
11.6 Phép thử độ rã của viên nén và nang
11.7 Phép thử độ rã của viên bao tan trong ruột
11.8 Xác định giới hạn tiểu phân
Phụ lục 12
12.1 Lấy mẫu dược liệu
12.2 Những quy định chung về kiểm tra chất lượng dược liệu
12.3 Phép thử xác định chiết kiệt alcaloid
12.4 Định lượng aldehyd trong tinh dầu
12.5 Định lượng cineol trong tinh dầu
12.6 Định lượng taninoid trong dược liệu
12.7 Định lượng tinh dầu trong dược liệu
12.8 Các phép thử của tinh dầu
12.9 Dầu béo
12.10 Xác định các chất chiết được trong dược liệu
12.11 Xác định tạp chất lẫn trong dược liệu
12.12 Xác định tỷ lệ vụn nát của dược liệu
12.13 Xác định hàm lượng nước bằng phương pháp cất với dung môi
12.15 Cặn khô của các chất chiết được trong dược liệu
12.16 Mất khối lượng do làm khô của các chất chiết được trong dược liệu
12.17 Dư lượng hóa chất bảo vệ thực vật
12.18 Định tính dược liệu và các chế phẩm bằng kính hiển vi
12.19 Xác định chỉ số trương nở
12.20 Phương pháp chế biến đông dược
Phụ lục 13
13.1 Phép thử histamin
13.2 Phép thử nội độc tố vi khuẩn
13.3 Phép thử các chất hạ áp
13.4 Phép thử chất gây sốt
13.5 Thử độc tính bất thường
13.6 Thử giới hạn nhiễm khuẩn
13.7 Thử vô khuẩn
13.8 Xác định hiệu quả kháng khuẩn của chất bảo quản
13.9 Xác định hoạt lực thuốc kháng sinh bằng phương pháp thử vi sinh vật
13.10 Phân tích thống kê các kết quả định lượng sinh học
Phụ lục 14
Hướng dẫn đánh giá sinh khả dụng và tương đương sinh học invivo thuốc generic
Phụ lục 15
15.1 Xác định độ sống của vắc xin BCG
15.2 Xác định độ chân không của vắc xin BCG
15.3 Xác định độ phân tán của vắc xin BCG
15.4 Xác định tính an toàn của vắc xin DTwP hấp phụ
15.5 Xác định đậm độ vi khuẩn ho gà
15.6 Xác định hàm lượng Tween 20 trong vắc xin và sinh phẩm
15.7 Kiểm tra vô trùng vắc xin/sinh phẩm
15.8 Môi trường dùng để phát hiện vi khuẩn hiếu khí, kỵ khí và nấm
15.9 Kiểm tra độc tính đặc hiệu (an toàn đặc hiệu) trong vắc xin BCG đông khô
15.10 Thử nghiệm nhận dạng huyết thanh miễn dịch
15.11 Xác định an toàn chung của vắc xin và sinh phẩm
15.12 Xác định chất gây sốt trong vắc xin và sinh phẩm
15.14 Phương pháp lấy mẫu và lưu mẫu
15.15 Xác định hiệu giá huyết thanh kháng độc tố bạch hầu
15.16 Xác định hiệu giá huyết thanh kháng độc tố uốn ván
15.17 Xác định hiệu giá huyết thanh kháng dại
15.18 Xác định hàm lượng nitơ toàn phần của vắc xin và sinh phẩm bằng thuốc thử Nessler
15.19 Thử nghiệm nhận dạng thành phần Bạch hầu-uốn ván-ho gà trong vắc xin DTwP hấp phụ
15.20 Xác định độc tố thần kinh tồn dư trong vắc xin bại liệt uống
15.21 Xác định công hiệu của vắc xin bại liệt uống
15.22 Xác định công hiệu thành phần uốn ván trong vắc xin hấp phụ chứa giải độc tố uốn ván
15.23 Xác định công hiệu thành phần bạch hầu trong vắc xin hấp phụ chứa giải độc tố bạch hầu
12.24 Xác định công hiệu thành phần ho gà toàn tế bào trong vắc xin phối hợp, hấp phụ
15.25 Xác định hàm lượng formaldehyd tồn dư trong vắc xin và sinh phẩm
15.26 Xác định hàm lượng natri clorid với sự có mặt của protein bằng phương pháp định lượng gián tiếp (phương pháp Charpentier Volhard)
15.27 Xác định hàm lượng nhôm (Al+++) trong vắc xin và sinh phẩm
15.28 Xác định hàm lượng phenol trong vắc xin và sinh phẩm
15.29 Xác định hàm lượng thimerosal trong vắc xin và sinh phẩm
15.31 Xác định hiệu lực vắc xin dại theo phương pháp NIH
15.32 Xác định hàm lượng nitơ protein của vắc xin và sinh phẩm bằng thuốc thử Nessler
15.33 Xác định pH của các vắc xin và sinh phẩm
15.34 Xác định hàm lượng protein toàn phần trong vắc xin và sinh phẩm
15.35 Xác định độ ẩm tồn dư trong vắc xin đông khô
15.36 Phát hiện mycoplasma bằng phương pháp nuôi cấy
15.37 Xác định hàm lượng Vi polysarcarid của vắc xin thương hàn Vi polysacarid
15.38 Xác định hàm lượng polysacarid trong vắc xin và sinh phẩm
15.39 Xác định độ tinh khiết kháng nguyên HbsAg
15.40 Xác định hàm lượng lipid trong vắc xin và sinh phẩm
15.41 Xác định hàm lượng cesi trong vắc xin và sinh phẩm
Phụ lục 16
16.1 Các phương pháp tiệt khuẩn
16.2 Chỉ thị sinh học dùng cho tiệt khuẩn
Phụ lục 17. Đồ đựng cấp 1 cho các chế phẩm dược
17.1 Đồ đựng bằng thủy tinh dùng cho chế phẩm dược
17.2 Đồ đựng bằng kim loại cho thuốc mỡ tra mắt
17.3 Đồ đựng và nút bằng chất dẻo
17.3.1 Đồ đựng bằng chất dẻo dùng cho những chế phẩm không phải thuốc tiêm
17.3.2 Đồ đựng bằng chất dẻo dùng cho chế phẩm thuốc tiêm
17.3.3 Đồ đựng bằng chất dẻo dùng cho chế phẩm nhỏ mắt
17.4 Dụng cụ tiêm truyền đã tiệt khuẩn (Bộ đây truyền dịch)
17.5 Nút cao su dùng cho chai đựng thuốc tiêm và thuốc tiêm truyền
17.6 Bơm tiêm vô khuẩn bằng chất dẻo sử dụng một lần
17.7 Silicon
Phụ lục 18. Bảng nguyên tử lượng các nguyên tố
Phổ Hồng ngoại đối chiếu
Lời nói đầu
Bộ tiêu chuẩn quốc gia về thuốc TCVN I:2017 thay thế bộ TCVN I:2009. Bộ TCVN I:2017 gồm 5 phần:
TCVN I-1:2017 - Phần 1: Phương pháp kiểm nghiệm thuốc và các chuyên mục;
TCVN I-2:2017 - Phần 2: Nguyên liệu hóa dược;
TCVN I-3:2017 - Phần 3: Thành phẩm hóa dược;
TCVN I-4:2017 - Phần 4: Dược liệu và thuốc từ dược liệu;
TCVN I-5:2017 - Phần 5: Vắc xin và sinh phẩm y tế.
Bộ tiêu chuẩn quốc gia về thuốc TCVN I:2017 do Hội đồng Dược điển Việt Nam biên soạn, Bộ Y tế đề nghị, Tổng cục Tiêu chuẩn Đo lường Chất lượng thẩm định, Bộ Khoa học và Công nghệ công bổ.
Lời giới thiệu
Tiêu chuẩn quốc gia về thuốc là văn bản kỹ thuật về tiêu chuẩn hóa và kiểm nghiệm chất lượng thuốc. Bộ tiêu chuẩn quốc gia về thuốc TCVN I:2017 có 1158 tiêu chuẩn, bao gồm:
Phần 1: 201 tiêu chuẩn về phương pháp kiểm nghiệm thuốc và chuyên mục;
Phần 2: 362 tiêu chuẩn về nguyên liệu hóa dược;
Phần 3: 257 tiêu chuẩn về thành phẩm hóa dược;
Phần 4: 315 tiêu chuẩn về dược liệu và thuốc từ dược liệu;
Phần 5: 23 tiêu chuẩn về vắc xin và sinh phẩm y tế.
Danh pháp, thuật ngữ trong Bộ tiêu chuẩn quốc gia về thuốc được viết theo quy định của Hội đồng Dược điển Việt Nam, Bộ Y tế. Các thuật ngữ dược phẩm được viết dựa trên nguyên tắc việt hóa tên chung quốc tế Latin (DCI Latin) một cách hợp lý nhằm giữ các ký tự cho sát với thuật ngữ quốc tế. Tên hợp chất hữu cơ được viết theo danh pháp do Hiệp hội quốc tế hóa học thuần túy và ứng dụng (I.U.P.A.C) qui định. Trong một số trường hợp cá biệt, các thuật ngữ tiếng Việt đã quen dùng đối với một số nguyên tố, hóa chất hay tên dược liệu vẫn tiếp tục sử dụng.
BỘ TIÊU CHUẨN QUỐC GIA VỀ THUỐC - PHẦN 1: PHƯƠNG PHÁP KIỂM NGHIỆM THUỐC
Set of national standards for medicines - Part 1: General methods for quality control of medicines
1 Phạm vi áp dụng
Bộ tiêu chuẩn này quy định các phương pháp kiểm nghiệm thuốc; áp dụng để kiểm tra, đánh giá chất lượng thuốc thành phẩm, nguyên liệu làm thuốc, vắc xin và sinh phẩm y tế, dược liệu và thuốc từ dược liệu.
2 QUY ĐỊNH CHUNG
1. Tên chính của các chuyên luận là tên Việt Nam, sau tên Việt Nam là tên Latin và những tên Việt Nam thông dụng khác nếu có.
Đối với dược liệu: Có thể dùng tên quy ước của dược liệu hoặc dùng tên cây kèm theo bộ phận dùng làm thuốc để làm tên chuyên luận, những từ chỉ bộ phận dùng làm thuốc để trong dấu ngoặc đơn, ví dụ: (Lá), (Quả), (Thân rễ).... Tên quy ước của dược liệu là tên của vị thuốc đã được dùng trong y học cồ truyền, ví dụ: Phù bình, Bạch giới tử...
Mỗi chuyên luận của Dược điển Việt Nam V (chuyên luận riêng hay phụ lục) là một tiêu chuẩn về chất lượng thuốc hoặc phương pháp kiểm nghiệm thuốc của Việt Nam.
2. Nguyên tử lượng các nguyên tố trong Dược điển Việt Nam V là các giá trị đã được thừa nhận ghi trong Phụ lục 18. Bảng nguyên tử lượng các nguyên tố.
3. Các đơn vị đo lường dùng trong Dược điển Việt Nam V đều tuân theo Luật Đo lường ban hành 11/11/2011 và Nghị định của Chính phủ số 86/2012/NĐ-CP ngày 19/10/2012.
4. Các đơn vị đo lường được viết tắt như sau:
| Mét: m | Giờ: h |
| Decimét: dm | Phút: min |
| Centimét: cm | Giây: s |
| Milimét: mm | KilôPascan: kPa |
| Micrômét: μm | Pascan: Pa |
| Nanômét: nm | Pascan giây: Pa∙s |
| Lít: I hoặc L | Ampe: A |
| Mililít: ml hoặc mL | Miliampe: mA |
| Micrôlít: μl hoặc μL | Vôn: V |
| Kilôgam: kg | Milivôn: mV |
| Gam: g | Mol: mol |
| Miligam: mg | Mol trên lít: mol/l, Mol/L, M |
| Centigam: cg | Becơren: Bq |
| Micrôgam: μg | Đơn vị quốc tế: IU hoặc đvqt |
| Nanôgam: ng | Độ Celsius: °C |
|
| Phần trăm: % |
5. Nếu không có chỉ dẫn khác, mọi nhiệt độ được ghi trong Dược điển Việt Nam V đều được biểu thị bằng độ bách phân Celsius, ký hiệu là “°C”.
6. Nhiệt độ tiêu chuẩn được quy định là 20 °C, nhiệt độ bình thường của phòng thí nghiệm (nhiệt độ phòng) được quy định là 20 °C đến 30 °C. Nếu không có chỉ dẫn gì khác, tất cả thử nghiệm đối với thuốc phải thực hiện ở nhiệt độ phòng (20 °c đến 30 °C) và những nhận xét kết quả phải thực hiện ngay sau khi thao tác. Tuy nhiên khi đánh giá kết quả một thử nghiệm bị ảnh hưởng bởi nhiệt độ thì phải thực hiện ở điều kiện nhiệt độ tiêu chuẩn (20 °C).
Trong thử nghiệm “Mất khối lượng do làm khô”, nếu chỉ quy định tiến hành ở một nhiệt độ nào đó, thì giới hạn cho phép về nhiệt độ được hiểu là: Nhiệt độ quy định ± 2 °C (ví dụ: 100 °C nghĩa là 100 °C ± 2 °C).
7. Nhiệt độ nước: Nước cách thủy là nước có nhiệt độ 98 °C đến 100 °C, trừ khi có chỉ dẫn khác; cụm từ “trong cách thủy” có nghĩa là dụng cụ ngâm trong nước đun sôi, “trên cách thủy” có nghĩa là dụng cụ chỉ tiếp xúc với hơi nước đun sôi.
Nước nóng: 70 °C đến 80 °C.
Nước ấm: 40 °C đến 50 °C.
Nước lạnh: 2 °C đến 10 °C.
Nước đá: 0 °C
8. Nhiệt độ nơi bảo quản:
Lạnh sâu: Dưới -10 °C.
Lạnh: 2 °C đến 10 °C.
Mát: 10 °C đến 20 °C.
Nhiệt độ phòng: 20 °C đến 30 °C (là nhiệt độ phổ biến ở nơi làm việc).
Nhiệt độ phòng có điều nhiệt: 20 °C đến 25 °C (là nhiệt độ được duy trì bằng máy điều hòa nhiệt độ). Nóng: 35 °C đến 40 °C.
Rất nóng: Trên 40 °C.
9. Áp suất được biểu thị bằng số đơn vị kilôPascan (kPa).
1 kPa = 7,5006 Torr.
1 Torr là áp suất dưới cột thủy ngân 1 mm.
Nếu ghi “chân không” mà không có chỉ dẫn gì khác thì có nghĩa là áp suất không quá 2,0 kPa (15 mm thủy ngân).
10. Khái niệm “cân chính xác” là cân tới 0,1 mg, 0,01 mg hoặc 0,001 mg tùy theo độ nhạy của loại cân phân tích dùng để cân sao cho sai số của phép cân không quá 0,1%.
Khối lượng cân được có độ chính xác phù hợp với độ lặp lại xác định. Độ lặp lại đó tương ứng với +5 hoặc -5 đơn vị sau chữ số có nghĩa cuối cùng đã cho; ví dụ: Lượng cân 0,25 g nghĩa là lượng cân đó nằm trong khoảng 0,245 g đến 0,255 g.
Khái niệm “cân” nghĩa là phép cân được thực hiện với sai số dưới 1 %.
Khái niệm “cân khoảng” là cân để lấy một lượng không quá ± 10 % lượng chỉ định trong Dược điển. Khái niệm “sấy đến khối lượng không đổi” và “nung đến khối lượng không đổi” nghĩa là hai lần cân liên tiếp không khác nhau quá 0,5 mg. Lần cân thứ hai tiến hành sau một thời gian sấy hoặc nung thêm (thường 1 h là thích hợp) tùy theo tính chất và lượng cân.
Khái niệm “đã cân trước” (đối với chén nung, bình, vại...) nghĩa là dụng cụ được xử lý đến khối lượng không đổi. Nếu trong chuyên luận có quy định phải cân một cặn hay một tủa (sấy khô, nung, đun bốc hơi) trong những dụng cụ thì có nghĩa là những dụng cụ này được sấy hoặc nung đến khối lượng không đổi.
Khái niệm “cặn không đáng kể” hay “cặn không thể cân được” là cặn không nặng quá 0,5 mg.
11. Khi đo thể tích, nếu chữ số sau dấu thập phân là 0 hoặc tận cùng bằng 0 thì thể tích đó phải đong, đo chính xác (ví dụ 10,0 ml hoặc 0,50 ml). Khái niệm “đong, đo chính xác” để lấy một thể tích dung dịch hay chất lỏng là phải đong đo bằng pipet chính xác, bình định mức hay buret chuẩn. Còn “đong, đo” được hiểu là dùng ống đong hoặc những phương tiện khác thích hợp để đo thể tích. Những thể tích cỡ micrôlít được đo bằng micropipet hay micrôsyringe (bơm chất lỏng siêu vi).
Để đếm giọt, dùng ống đếm giọt chuẩn, 20 giọt nước tinh khiết của ống này ở 20 °C có khối lượng từ 0,90 g đến 1,10 g.
12. “Nước” có nghĩa là nước tinh khiết. Nước tinh khiết được sử dụng trong tất cả các thử nghiệm thuốc nhưng không dùng với những chế phẩm tiêm.
13. Một dung dịch, nếu không ghi rõ dung môi sử dụng thì được hiểu là dung dịch trong nước tinh khiết hay nước cất.
14. Khái niệm “ngay” và “ngay lập tức” dùng trong thử nghiệm thuốc có nghĩa là thao tác này phải thực hiện trong vòng 30 s sau thao tác trước đó.
15. Thử định tính là phép thử cần thiết để nhận biết một chất thuốc hay những thành phần chính của thuốc dựa trên tính chất vật lý hay hóa học đặc trưng. Nhận biết một dược liệu hay thuốc từ dược liệu dựa trên mô tả, đặc điểm vi phẫu, bột và các phép thử vật lý hóa học đặc trưng.
Phổ hồng ngoại là phương pháp chuẩn xác để định tính, vì mỗi một chất thuốc chỉ cho một vùng “điểm chỉ” của phổ, không trùng lặp với phổ của những chất khác. Những đặc tính của phổ hồng ngoại có thể được dùng như là phép thử hàng đầu để định tính. Thường thì phép thử phổ hồng ngoại tự nó đã đủ tin cậy và không cần thêm phép thử nào khác. Tuy nhiên, khi một sản phẩm là một muối thì cần thiết thử thêm “ion đặc hiệu”. Những phép thử định tính tiếp theo trong mỗi chuyên luận là để khẳng định lại về định tính của phổ hồng ngoại đã làm trước. Trong trường hợp cần thiết, cần tiến hành xác định thêm điểm chảy của thuốc; khi điểm chảy không có sự lặp lại đúng như nhiệt độ đã quy định thì có thể dùng một giá trị xấp xỉ, giới hạn sai số được quy định trong chuyên luận riêng.
16. Thử tinh khiết là tập hợp các phép thử nhằm phát hiện những tạp chất nhiễm vào thuốc. Để kiểm tra độ tinh khiết, phải thử xác định sự có mặt các tạp chất, số lượng và giới hạn của chúng cũng như những yêu cầu khác tùy theo mỗi chuyên luận. Những tạp chất được coi là đối tượng phải thử là những chất thường được đưa vào trong quá trình sản xuất hoặc xuất hiện trong quá trình bảo quản và những tạp chất bất thường như những kim loại nặng, arsen...Dược điển chỉ quy định kiểm tra những tạp chất thường gặp. Điều này có nghĩa là Dược điển không chấp nhận những tạp chất khác có trong thuốc mặc dù không được ghi trong Dược điển. Mỗi khi có sự thay thế chất này bằng chất khác hoặc cho thêm một chất mới trong thực hiện quy trình thì phải có thêm những phép thử tương ứng. Nồng độ của tạp chất được biểu thị bằng phần triệu của khối lượng, hoặc khi giới hạn vượt quá 500 phần triệu thì được biểu thị bằng phần trăm (%). Những giá trị này chỉ là khoảng giá trị phù hợp với những yêu cầu đã được xác định dựa trên sự thích hợp với những thử nghiệm đã cho.
17. Trong chuyên luận kháng sinh, tại mục định lượng có ghi đồng thời hai phương pháp là phương pháp vi sinh vật và phương pháp hóa học hay hóa lý; mỗi phương pháp đều có thể được sử dụng. Phương pháp định lượng vi sinh vật được coi như phương pháp được khuyến cáo áp dụng, nếu dùng phương pháp khác để định lượng thì phương pháp đó không được kém tin cậy hơn phương pháp vi sinh vật. Nếu kết quả của hai phương pháp quá chênh lệch thì lấy kết quả theo phương pháp vi sinh vật để kết luận (trừ khi có chỉ dẫn khác).
18. Phương pháp thử quy định trong Dược điển Việt Nam có thể được thay thế bằng những phương pháp khác có độ đúng và độ chính xác cao hơn. Tuy nhiên, khi có sự chênh lệch, nghi ngờ, thì chỉ kết quả thu được từ phương pháp ghi trong Dược điển là có giá trị để đưa ra kết luận cuối cùng.
19. Nếu trong chuyên luận có ghi việc xác định phải tiến hành so sánh với một chất chuẩn hay chất đối chiếu (ĐC) thì phải dùng các chất này theo quy định tại Phụ lục 2.5.
20. Khi thử nghiệm hay định lượng (trừ chỉ dẫn khác) nếu quy định mẫu thử phải so sánh với mẫu kiểm tra trắng thì phải chuẩn bị mẫu này như mẫu thử nhưng không cho chất cần thử hay cần định lượng và phải tiến hành song song cùng điều kiện như mẫu thử. Cũng tương tự như vậy khi thử nghiệm được yêu cầu tiến hành song song với mẫu đối chiếu (chứng).
21. Các kết quả định lượng được tính đến một số lẻ thập phân cần thiết nhiều hơn yêu cầu một chữ số rồi làm tròn lên hay xuống như sau:
Nếu con số cuối cùng đã tính được là 5 đến 9 thì con số đứng trước nó được tăng thêm 1.
Nếu con số cuối cùng đã tính được là dưới 5 thì con số đứng trước nó không thay đổi.
Các phép tính khác, thí dụ chuẩn hóa các dung dịch chuẩn độ cũng tiến hành tương tự.
Thí dụ: 8,2758 làm tròn số là 8,276.
1,2634 làm tròn số là 1,263.
22. Hàm lượng tiêu chuẩn: Hàm lượng tiêu chuẩn của một chất quy định trong một chuyên luận được thể hiện tính theo công thức hóa học có thể có giới hạn trên 100 % của chất đó, giới hạn trên này áp dụng với kết quả định lượng tính theo hàm lượng tương đương của công thức hóa học mà chất đó quy định. Ví dụ: Ghi chứa không ít hơn 98,5 % và không nhiều hơn 102,0 % của C12H22CaO14.H2O nghĩa là kết quả định lượng không được ít hơn 98,5 % và không nhiều hơn 102,0 % tính theo hàm lượng tương đương của C12H22CaO14.H2O.
Nếu trong chuyên luận riêng không ghi giới hạn trên thì có nghĩa là giới hạn trên không quá 101,0 %.
23. Trong các chuyên luận, ở mục mô tả hoặc tính chất, thuật ngữ “trắng” có nghĩa là trắng hoặc gần như trắng; “không màu” có nghĩa là không có màu hoặc gần như không màu; “không mùi” có nghĩa là không có mùi hoặc thực tế không có mùi. Trừ khi có các chỉ dẫn khác, cách thử màu sắc hoặc mùi được tiến hành như sau:
a) Màu:
Chất rắn: Lấy 1 g chất thử cho lên trên một tờ giấy trắng hoặc mặt kính đồng hồ không màu đặt lên trên tờ giấy trắng rồi quan sát.
Chất lỏng: Cho chất thử vào trong một ống nghiệm không màu, đường kính bên trong 15 mm, đặt trước một nền trắng cách ống 30 mm, nhìn ngang ống dưới ánh sáng ban ngày.
b) Mùi:
Chất rắn: Trên một mặt kính đồng hồ, đường kính từ 6 cm đến 8 cm, lấy từ 0,5 g đến 2,0 g chất thử trải thành lớp mỏng, sau 15 min, xác định mùi bằng cảm quan.
Chất lỏng: Lấy 2 ml chất thử cho vào mặt kính đồng hồ như trên rồi xác định mùi bằng cảm quan.
24. Trong cách biểu thị nồng độ dung dịch, nếu không có chỉ dẫn gì khác, nồng độ được biểu thị là phần trăm (%) khối lượng trên thể tích (kl/tt), tính theo số gam chất hòa tan trong 100 ml dung dịch, ký hiệu là % (kl/tt) hoặc %.
Các trường hợp khác biểu thị bằng các ký hiệu và được hiểu như sau:
% (kl/kl): Số gam chất hòa tan trong 100 g dung dịch.
% (tt/tt): Số mililít chất hòa tan trong 100 ml dung dịch.
% (tt/kl): Số mililít chất hòa tan trong 100 g dung dịch.
25. Ethanol không có chỉ dẫn gì khác thì có nghĩa là ethanol tuyệt đối.
Khái niệm “alcol” không có chỉ dẫn gì có nghĩa là alcol chứa khoảng 96 % (tt/tt) ethanol (C2H6O). Dung dịch ethanol trong nước ở những nồng độ khác được chỉ bằng từ ethanol kèm theo tỷ lệ phần trăm (tt/tt) hoặc (kl/kl) ethanol (C2H6O) trong dung dịch đó, ví dụ: Ethanol 70 %.
26. Ether có nghĩa là ether ethylic.
Các ether dầu hỏa: Khi viết kèm giới hạn về nhiệt độ thì giới hạn này được hiểu là khoảng sôi. Ví dụ: Ether dầu hỏa (30 °C đến 40 °C) nghĩa là ether dầu hỏa có khoảng sôi từ 30 °C đến 40 °C.
27. Hỗn hợp của các chất lỏng được ghi theo ký hiệu 10 : 1, hoặc 50 : 9 : 1, v.v... có nghĩa là hỗn hợp các chất đó thứ tự theo thể tích. Thí dụ: cloroform - methanol - amoniac (50 : 9 : 1) có nghĩa là lấy lần lượt 50 ml cloroform trộn đều với 9 ml methanol và 1 ml amoniac thành một hỗn hợp. Trong chuyên luận, tên các chất lỏng trong hỗn hợp được in nghiêng và không có chữ (TT) đi kèm sau.
28. Trong chuyên luận, tên các hóa chất, thuốc thử, dung dịch thử, chất chỉ thị, dung dịch chuẩn độ, dung dịch mẫu, dung dịch đệm được trình bày bằng chữ nghiêng và có chữ (TT) nếu có trong Phụ lục 2.1. Các thuốc thử chung; Phụ lục 2.3. Các dung dịch đệm và Phụ lục 2.4. Các dung dịch mẫu hoặc có chữ (CĐ) nếu có trong Phụ lục 2.2. Các dung dịch chuẩn độ.
29. Các định nghĩa về độ tan như sau:
“Tan” có nghĩa là chất thử (đã được tán nhỏ thành bột nếu là chất rắn) hòa tan được trong dung môi tạo thành một dung dịch trong, đồng nhất, không còn những phần tử của chất thử. Xác định độ tan bằng cách cho lượng dung môi vào chất thử để ở nhiệt độ (25 ± 2) °C trong 30 min, cứ cách 5 min lại lắc 30 s.
Độ tan được biểu thị như sau:
| Độ tan | Số ml dung môi hòa tan 1 g chất thử |
| Rất tan Dễ tan Tan Hơi tan Khó tan Rất khó tan Thực tế không tan | Dưới 1 Từ 1 đến 10 Trên 10 đến 30 Trên 30 đến 100 Trên 100 đến 1 000 Trên 1 000 đến 10 000 Trên 10 000 |
30. Độ acid hay độ kiềm của dung dịch, nếu không có chỉ dẫn gì khác, được xác định bằng giấy quỳ xanh hay đỏ. Muốn xác định những tính chất này chính xác hơn thì phải đo pH bằng pH kế.
31. Bình hút ẩm: Cụm từ “trong bình hút ẩm” có nghĩa là dùng một bình kín có kích thước thích hợp, bên trong chứa silica gel hoặc một chất làm khô khác để giữ cho không khí trong bình có độ ẩm thấp.
Bình hút ẩm chân không: Là bình hút ẩm chứa một chất làm khô thích hợp và có áp suất không quá 2,0 kPa (15 mm thủy ngân), nếu không có chỉ dẫn khác.
32. Danh pháp thực vật, động vật của cây con làm thuốc gồm tên chi và tên loài, họ thực vật hay động vật.
33. Dược liệu được ghi trong Dược điển là bộ phận dùng làm thuốc có lưu hành trên thị trường, không lẫn tạp chất. Trong Dược điển, việc thu thập xử lý dược liệu tại chỗ chỉ bao hàm riêng bộ phận dùng.
34. Quy cách đặc điểm dược liệu được mô tả dựa trên dược liệu khô nói chung. Chất lượng tiêu chuẩn của dược liệu tươi cũng được quy định nếu đem dùng tươi.
35. Phần mô tả dược liệu chỉ đề cập đến bộ phận dùng làm thuốc, không mô tả toàn bộ con vật hay toàn bộ cây cung cấp dược liệu đó.
36. Việc làm khô dược liệu tại chỗ, hay làm khô trong quá trình thu hái dược liệu được hiểu như sau:
a. Thuật ngữ “làm khô”, có nghĩa là có thể sấy, nướng khô, phơi dưới ánh sáng mặt trời hoặc phơi trong bóng râm (phơi âm can).
b. Thuật ngữ “làm khô ở nhiệt độ thấp” có nghĩa là phơi, sấy khô ở nhiệt độ không quá 60 °C, được dùng cho dược liệu không chịu được nhiệt độ cao.
c. Thuật ngữ “phơi âm can” có nghĩa là phơi trong bóng râm ngoài không khí, được dùng cho dược liệu không sấy nướng, không phơi nắng được.
d. Trong một số trường hợp, dược liệu phải phơi khô trong thời gian ngắn, thuật ngữ được dùng là “phơi nhanh dưới nắng to”, “phơi đúng thời gian”.
e. Thuật ngữ “dược liệu khô kiệt” có nghĩa là dược liệu được điều chỉnh khối lượng bằng cách trừ đi khối lượng nước được xác định theo phương pháp sấy hoặc cất với dung môi ghi trong chuyên luận riêng.
37. Đặc điểm vi phẫu (thường là đặc điểm mặt cắt ngang dược liệu), soi bột dược liệu và những vi đặc điểm khác của một dược liệu là những đặc điểm của vật mẫu quan sát dưới kính hiển vi.
38. Các dược liệu, dược chất, tá dược và chất phụ gia dùng trong một chế phẩm phải tuân theo những quy định của Dược điển. Những điều Dược điển không quy định thì phải tuân theo quy định hiện hành của Bộ Y tế.
Tá dược, chất phụ gia đem dùng không được phương hại tới tính an toàn và tính hiệu quả của thuốc. Cần chú ý tránh làm cản trở phương pháp phân tích đã quy định trong chuyên luận Dược điển.
39. Dược liệu dùng sản xuất thuốc thành phẩm (thuốc từ dược liệu) phải đạt tiêu chuẩn Dược điển Việt Nam. Nếu có yêu cầu dùng dược liệu đã bào chế thì quy trình bào chế phải được tiến hành theo phương pháp quy định trong chuyên luận Dược điển. Trừ khi có những chỉ dẫn khác.
40. Khối lượng quy định cho mỗi thành phần trong công thức của một chế phẩm thuốc từ dược liệu được tính theo khối lượng dược liệu sạch, đã tán thành bột hoặc đã được xử lý.
41. Dược liệu sử dụng theo đường uống thường dùng dưới dạng thuốc sắc, trừ khi có những chỉ dẫn khác.
42. Liều lượng quy định dùng trong dược điển là liều dùng thông thường đối với người trưởng thành. Việc sửa đổi, điều chỉnh liều lượng tùy thuộc vào tình trạng bệnh tật của từng người bệnh cụ thể. Liều tối đa của một thuốc là liều cao nhất mà người trưởng thành có thể chịu được và không được dùng quá liều; trừ khi có những chỉ dẫn khác.
43. Nước dùng trong bào chế thuốc từ dược liệu là nước sạch, nước uống đạt theo tiêu chuẩn vệ sinh y tế.
44. Rượu dùng trong bào chế thuốc từ dược liệu là ethanol 40 % đến 45 %.
45. Những yêu cầu cơ bản về bảo quản thuốc được ghi ở mục “Bảo quản”. Đồ đựng là phương tiện để bảo quản thuốc, yêu cầu của đồ đựng cũng bao gồm cả những phần hợp thành như là nút hay nắp. Các đồ đựng phải kín, không được làm ảnh hưởng đến chất lượng thuốc đựng bên trong, không cho môi trường bên ngoài tác động ảnh hưởng đến chất lượng và phải đạt theo quy định của Dược điển Việt Nam.
Về nhiệt độ nơi bảo quản phải thực hiện đúng yêu cầu quy định.
“Tránh ánh sáng” có nghĩa là chất đựng được để trong chai lọ thủy tinh màu hổ phách, hoặc chai lọ thủy tinh màu, chai lọ thủy tinh bọc bằng giấy đen hoặc bất kỳ đồ đựng nào không bị ảnh hưởng bởi ánh sáng.
“Đậy kín” có nghĩa là đồ đựng có thể tránh cho các chất đựng bên trong không bị bụi bẩn và các chất lạ bên ngoài nhiễm vào.
“Hàn kín” có nghĩa là đồ đựng phải kín hơi và có thể bảo vệ chất đựng trong đó chống được hơi ẩm và các vi khuẩn.
46. Nhãn thuốc phải theo đúng quy định hiện hành.
3 KÝ HIỆU CÁC CHỮ VIẾT TẮT
| DĐVN V: | Dược điển Việt Nam lần xuất bản thứ năm. |
| P.t.l: | Phân tử lượng |
| TT: | Thuốc thử. |
| TT1, TT2, TT3: | Thuốc thử 1, thuốc thử 2, thuốc thử 3... |
| ĐC: | Chất đối chiếu. |
| CĐ: | Chuẩn độ (Dung dịch). |
| BCG: | Bacillus Calmette - Guérin. |
| Lf/ mg PN: | Đơn vị lên bông trong 1 mg nitrogen protein. |
| Lf/ mg N: | Đơn vị lên bông trong 1 mg nitrogen toàn phần. |
| Kf: | Thời gian xuất hiện hiện tượng lên bông (tính theo phút) khi được theo dõi trong phản ứng lên bông. |
| Lf: | Lượng độc tố hoặc giải độc tố khi trộn với 1 IU kháng độc tố sẽ xuất hiện lên bông trong thời gian ngắn nhất. |
| L+: | Lượng độc tố tối thiểu khi kết hợp với 1 IU kháng độc tố có thể giết chết một con vật có trọng lượng xác định trong bốn ngày (Liều L+ phụ thuộc vào từng loại động vật thí nghiệm). |
| L+/ 10: | Lượng độc tố tối thiểu khi kết hợp với 0,1 IU kháng độc tố có thể giết chết một vật thí nghiệm có trọng lượng xác định trong bốn ngày. |
| Lr: | Lượng độc tố tối thiểu khi kết hợp với một lượng kháng độc tố cố định (thường là 0,002 IU kháng độc tố) trong thể tích 0,2 ml sẽ gây phản ứng da tại chỗ có thể nhìn thấy được (chỉ đối với bạch cầu). |
| LD50: | Lượng độc tố giết 50 % của một nhóm động vật trong vòng 4 ngày (LD50 khác nhau tùy theo từng loại động vật thí nghiệm) |
| MLD: | Liều gây chết nhỏ nhất, là lượng độc tố có thể giết chết các súc vật trong vòng 4 ngày (MLD khác nhau tùy theo loại động vật thí nghiệm). Nói chung, MLD đã dược thay thế bằng LD50. |
| ED50: | Liều vắc xin bảo vệ được 50 % động vật đã được gây miễn dịch chống lại một liều thử thách của vi khuẩn độc hoặc độc tố. |
| ABV: | Đơn vị kết hợp kháng độc tố (Antitoxin Binding Value), là giá trị xác định lượng độc tố thêm vào giải độc tố thành một hỗn hợp (xác định kiểm tra trên động vật thí nghiệm). |
| CPE: | Tác động gây bệnh tế bào (Cytopathic effect). |
| MEM: | Môi trường MEM (Minimum Essential Medium). |
| FCS: | Huyết thanh bào thai bê (Foetal Calf Sera). |
| CCID50: | Lượng virus gây nhiễm 50% tế bào (Cell culture infective dose). |
| NMSL: | Nước muối sinh lý. |
| EU: | Đơn vị nội độc tố (Endotoxin Unit). |
| Nước BET: | Nước để thử nội độc tố. |
| Me: | - CH3 (methyl) |
| Et: | - CH2CH3 (ethyl) |
| Pri: | - CH(CH3)2 (iso-propyl) |
| Prn: | - CH2CH2CH3 (n-propyl) |
| Bui: | - CH2CH(CH3)2 (iso-butyl) |
| Bus: | - CH (CH3)CH2CH3 (sec-butyl) |
| Bun: | - CH2CH2CH2CH3 (n-butyl) |
| But: | - C(CH3)3 (tert-butyl) |
| Ph: | - C6H5 (phenyl) |
| Ac: | - COCH3 (acetyl) |
PHỤ LỤC 1
(Quy định)
1.1 CAO THUỐC
Extracta
Định nghĩa
Cao thuốc là chế phẩm được chế bằng cách cô hoặc sấy đến thể chất quy định các dịch chiết thu được từ dược liệu thực vật hay động vật với các dung môi thích hợp.
Các dược liệu trước khi chiết xuất được xử lý sơ bộ (rửa sạch, phơi khô hoặc sấy khô và chia nhỏ đến kích thước thích hợp). Đối với một số dược liệu đặc biệt có chứa men làm phân hủy hoạt chất, cần phải diệt men trước khi đưa vào sử dụng bằng cách dùng hơi cồn sôi, hơi nước sôi hoặc bằng phương pháp thích hợp khác.
Cao thuốc được chia làm 3 loại:
Cao lỏng: Là chất lỏng hơi sánh, có mùi vị đặc trưng của dược liệu sử dụng trong đó cồn và nước đóng vai trò dung môi chính (hay chất bảo quản hay cả hai). Nếu không có chỉ dẫn khác, quy ước 1 ml cao lỏng tương ứng với 1 g dược liệu dùng để điều chế cao thuốc.
Cao đặc: Là khối đặc quánh. Hàm lượng dung môi sử dụng còn lại trong cao không quá 20 %.
Cao khô: Là khối hoặc bột khô, đồng nhất nhưng rất dễ hút ẩm. Cao khô không được có độ ẩm lớn hơn 5 %.
Phương pháp điều chế
Quá trình điều chế cao thuốc thường có 2 giai đoạn:
Giai đoạn I
Chiết xuất dược liệu bằng các dung môi thích hợp. Tùy theo bản chất của dược liệu, dung môi, tiêu chuẩn chất lượng của thành phẩm cũng như điều kiện, quy mô sản xuất và trang thiết bị, có thể sử dụng các phương pháp chiết xuất: Ngâm, hầm, hãm, sắc, ngâm nhỏ giọt, chiết xuất ngược dòng, chiết xuất bằng thiết bị siêu âm, chiết xuất bằng phương pháp sử dụng điện trường và các phương pháp khác. Phương pháp ngâm nhỏ giọt thường được sử dụng. Khi đó, dược liệu thô đã được chia nhỏ đến kích thước phù hợp, được làm ẩm với một lượng dung môi vừa đủ rồi đậy kín để yên trong khoảng 2 h đến 4 h. Sau đó, chuyển khối dược liệu vào bình ngấm kiệt, thêm lượng dung môi vừa đủ đến khi ngập hoàn toàn khối dược liệu. Thời gian ngâm lạnh và tốc độ chảy trong quá trình chiết có thể thay đổi theo khối lượng và bản chất của dược liệu thô đem chiết.
Giai đoạn II
Cao lỏng: Sau khi thu được dịch chiết, tiến hành lọc và cô dịch chiết bằng các phương pháp khác nhau để thu được cao lỏng có tỷ lệ theo như quy ước (1 ml cao lỏng tương ứng với 1 g dược liệu). Trong trường hợp điều chế cao lỏng bằng phương pháp ngâm nhỏ giọt, tốc độ chảy của dịch chiết có thể chậm, vừa hay nhanh. Nếu chiết xuất 1000 g dược liệu thì:
Ở tốc độ chậm: Không quá 1 ml dịch chiết trong 1 min.
Ở tốc độ vừa: 1 ml đến 3 ml dịch chiết trong 1 min.
Ở tốc độ nhanh: 3 ml đến 5 ml dịch chiết trong 1 min.
Để riêng phần dịch chiết đầu đậm đặc bằng 4/5 lượng dược liệu đem chiết. Sau đó cô các phần dịch chiết tiếp theo trên cách thủy hoặc cô dưới áp suất giảm ở nhiệt độ không quá 60 °C cho đến khi loại hết dung môi. Hòa tan cặn thu được vào trong dịch chiết đầu đậm đặc và nếu cần, thêm dung môi vào để thu được cao lỏng đạt tỷ lệ quy định. Cao lỏng có khuynh hướng bị lắng cặn vì vậy để cao lỏng ở chỗ mát trong thời gian ít nhất 3 ngày, rồi lọc.
Cao đặc và cao khô: Dịch chiết được cô đặc đến khi dung môi dùng để chiết xuất còn lại không quá 20 % được cao đặc. Trong trường hợp điều chế cao khô, tiếp tục sấy khô để độ ẩm còn lại không quá 5 %. Để đạt đến thể chất quy định, quá trình cô đặc và sấy khô dịch chiết thường được tiến hành trong các thiết bị cô dưới áp suất giảm ở nhiệt độ không quá 60 °C. Nếu không có các thiết bị cô đặc và sấy dưới áp suất giảm thì được phép cô cách thủy (không được cô trực tiếp trên lửa) và sấy ở nhiệt độ không quá 80 °C.
Trường hợp muốn thu được cao thuốc có tỷ lệ tạp chất thấp, phải tiến hành loại tạp chất bằng các phương pháp thích hợp tùy thuộc vào bản chất của dược liệu, dung môi và phương pháp chiết xuất.
Có thể cho thêm chất bảo quản hoặc các chất trơ để làm chất mang hay để cải thiện các tính chất vật lý. Đối với cao khô có thể sử dụng các bột trơ thích hợp để điều chỉnh nồng độ hoạt chất đến tỷ lệ quy định.
Yêu cầu chất lượng
Đạt các yêu cầu theo quy định trong chuyên luận riêng và đạt các yêu cầu chung sau đây:
Độ tan: Cao lỏng phải tan hoàn toàn trong dung môi đã sử dụng để điều chế cao.
Độ trong, mùi vị, độ đồng nhất và màu sắc: Cao thuốc phải đúng màu sắc đã mô tả trong chuyên luận riêng, có mùi và vị đặc trưng của dược liệu sử dụng. Ngoài ra, cao lỏng còn phải đồng nhất, không có váng mốc, không có cặn bã dược liệu và vật lạ.
Cách tiến hành: Lấy riêng phần phía trên của chai thuốc chỉ để lại khoảng 10 ml đến 15 ml. Chuyển phần còn lại trong chai vào một bát sứ men trắng, nghiêng bát cho chúng chảy trên thành bát tạo thành một lớp dễ quan sát. Quan sát dưới ánh sáng tự nhiên, thuốc phải đạt các yêu cầu quy định. Nếu không đạt, phải thử lại lần hai với chai thuốc khác, nếu không đạt, coi như lô thuốc không đạt chỉ tiêu này.
Mất khối lượng do làm khô (nếu không có chỉ dẫn khác);
Cao đặc không quá 20 %.
Cao khô không quá 5 %.
Hàm lượng cồn: Đạt từ 90 % đến 110 % lượng ethanol ghi trên nhãn (áp dụng cho cao lỏng và cao đặc).
Kim loại nặng (Nếu không có chỉ dẫn khác).
Không được quá 20 phần triệu (Phụ lục 9.4.8)
Cách tiến hành: Lấy 1,0 g chế phẩm tiến hành thử theo phương pháp 3, Phụ lục 9.4.8. Dùng 2 ml dung dịch chì mẫu 10 phần triệu (TT) để chuẩn bị mẫu đối chiếu.
Dung môi tồn dư: Nếu điều chế với dung môi không phải là cồn, nước hay hỗn hợp cồn - nước, dư lượng dung môi sử dụng phải đáp ứng yêu cầu quy định trong Phụ lục 10.14 Xác định dung môi tồn dư.
Dư lượng hóa chất bảo vệ thực vật: Đáp ứng yêu cầu quy định trong Phụ lục 12.17 Dư lượng hóa chất bảo vệ thực vật.
Giới hạn nhiễm khuẩn: Đáp ứng yêu cầu quy định trong Phụ lục 13.6 Thử giới hạn nhiễm khuẩn.
Bảo quản
Cao thuốc được đựng trong bao bì kín, để nơi khô, mát, tránh ánh sáng, nhiệt độ ít thay đổi.
Nhãn: Ghi tên bộ phận dùng của cây thuốc, tên dung môi (nếu không phải là dung môi cồn, nước hay hỗn hợp cồn - nước), hàm lượng (%) của hoạt chất hoặc của hợp chất nhận dạng được quy định theo từng chuyên luận riêng, tên và nồng độ của chất bảo quản thêm vào. Khi hoạt chất chưa biết, tỷ lệ giữa dược liệu và sản phẩm cuối cùng phải được nêu rõ. Đối với cao đặc và cao khô, loại và số lượng tá dược thêm vào cũng được nêu ra và tỷ lệ phần trăm của cao tự nhiên cũng phải được ghi rõ.
1.2 CỒN THUỐC
Tincturae
Định nghĩa
Cồn thuốc là những chế phẩm lỏng, được điều chế bằng cách chiết dược liệu thực vật, động vật hoặc hòa tan cao thuốc, dược chất theo tỷ lệ quy định với ethanol ở các nồng độ khác nhau.
Cồn thuốc được điều chế từ một nguyên liệu gọi là cồn thuốc đơn.
Cồn thuốc được điều chế từ nhiều nguyên liệu khác nhau gọi là cồn thuốc kép.
Cách biểu thị hoạt tính:
Các cồn thuốc có nguồn gốc từ thực vật có chứa các thành phần có hoạt tính mạnh, biểu thị hoạt tính theo 10 g dược liệu trong mỗi 100 ml cồn thuốc.
Phần lớn những cồn thuốc từ dược liệu khác biểu thị theo 20 g dược liệu trong mỗi 100 ml cồn thuốc. Các cồn thuốc khác nhau không nhất thiết phải pha loãng để đạt cùng một tỷ lệ giữa dược liệu ban đầu và cồn thuốc. Tỷ lệ này sẽ tùy thuộc vào các yêu cầu được mô tả trong các thử nghiệm xác định hàm lượng của hoạt chất hay của nhóm hoạt chất trong các chuyên luận riêng. Trong khi điều chế, cồn thuốc được định lượng theo những thử nghiệm xác định hàm lượng này. Sử dụng các giá trị thu được từ kết quả định lượng, điều chỉnh nồng độ cuối cùng của cồn thuốc bằng cách cho thêm dung môi hoặc làm bay hơi một phần dung môi.
Phương pháp điều chế
Cồn thuốc có thể được điều chế theo ba phương pháp: Ngâm, ngâm nhỏ giọt và hòa tan.
Phương pháp ngâm
Cho dược liệu đã chia nhỏ vào một dụng cụ thích hợp và thêm khoảng 3/4 lượng ethanol sử dụng. Đậy kín, để ở nhiệt độ thường, ngâm từ 3 ngày đến 10 ngày, thỉnh thoảng khuấy trộn. Sau đó gạn lọc thu dịch chiết. Rửa bã và ép bã bằng lượng ethanol còn lại. Gộp dịch chiết, dịch ép và bổ sung ethanol để thu được lượng dịch chiết quy định, để yên từ 1 ngày đến 3 ngày, gạn lọc lấy dịch trong.
Phương pháp ngâm nhỏ giọt
Dùng bình ngâm nhỏ giọt có thể tích phù hợp với khối lượng dược liệu đem chiết. Cho dược liệu đã chia nhỏ vào một dụng cụ thích hợp, trộn với ethanol vừa đủ ẩm. Đậy nắp kín, để yên 2 h đến 4 h ở nhiệt độ phòng. Cho dược liệu đã làm ẩm vào bình ngâm nhỏ giọt đến khoảng 3/4 thể tích của bình, đặt trên mặt dược liệu những vật liệu thích hợp để tránh xáo trộn khi đổ dung môi vào. Mở khóa bình, đổ từ từ ethanol lên khối dược liệu cho đến khi có vài giọt dịch chiết chảy ra, đóng khóa bình lại và tiếp tục thêm ethanol cho đến khi ngập hoàn toàn khối dược liệu. Để ngâm trong khoảng 24 h hoặc tùy theo mỗi chuyên luận, sau đó rút dịch chiết.
Nếu trong chuyên luận không yêu cầu phải định lượng hoạt chất hoặc nhóm hoạt chất, tiến hành rút dịch chiết với tốc độ nhỏ giọt phù hợp (xem tốc độ nhỏ giọt ở mục Cao lỏng, chuyên luận Cao thuốc - Phụ lục 1.1). Thêm ethanol vào và tiếp tục rút dịch chiết đến khi thu được lượng dịch chiết quy định. Trộn đều và để yên trong 2 ngày đến 3 ngày, gạn lọc lấy dịch trong.
Nếu có yêu cầu phải định lượng hoạt chất hoặc nhóm hoạt chất gộp các dịch chiết lại, trộn, rồi định lượng theo hướng dẫn trong chuyên luận. Pha loãng phần dịch chiết còn lại với một lượng dung môi theo tính toán từ thử nghiệm xác định hàm lượng để thu được cồn thuốc theo yêu cầu.
Phương pháp hòa tan
Hòa tan cao thuốc, dược chất hoặc tinh dầu vào ethanol có nồng độ quy định. Để lắng sau đó lọc để loại tủa.
Yêu cầu chất lượng
Trừ trường hợp đặc biệt được nêu trong chuyên luận riêng, yêu cầu chung đối với cồn thuốc như sau: Tỷ trọng, tạp chất, định tính, hàm lượng hoạt chất, hàm lượng ethanol: Đáp ứng yêu cầu quy định trong chuyên luận riêng.
Giới hạn methanol (Nếu không có chỉ dẫn khác):
Không quá 0,05 % (tt/tt) (Phụ lục 10.13).
Cặn sau khi bay hơi: Giới hạn quy định theo chuyên luận riêng.
Cách tiến hành: Lấy chính xác 5,0 ml hoặc 5,000 g cồn thuốc cho vào một cốc có đường kính 5 cm đến 7 cm và cao 2 cm đến 3 cm đã cân bì trước, làm bay hơi đến khô trên cách thủy và sấy khô ở 100 °C đến 105 °C trong 3 h, để nguội trong bình hút ẩm có chứa diphosphor pentoxyd và cân. Tính % khối lượng hay số g cặn trong 1 lít chế phẩm.
Bảo quản
Cồn thuốc được đựng trong bao bì kín, để nơi thoáng mát và tránh ánh sáng.
Nhãn phải nêu tên của bộ phận dùng của cây, tên dung môi hoặc hỗn hợp dung môi được sử dụng, nồng độ các thành phần quan trọng và tỷ lệ giữa dược liệu thô ban đầu so với cồn thuốc.
1.3 DUNG DỊCH THUỐC
Solutiones
Định nghĩa
Dung dịch thuốc là những chế phẩm lỏng trong suốt chứa một hoặc nhiều dược chất hòa tan trong một dung môi hay hỗn hợp dung môi thích hợp.
Tính chất: Do các phân tử trong dung dịch phân tán đồng nhất, nên các dung dịch thuốc đảm bảo sự phân liều đồng nhất khi sử dụng và độ chính xác cao khi pha loãng hoặc khi trộn các dung dịch với nhau.
Các dược chất trong dung dịch thường ít ổn định về mặt hóa học so với dạng rắn.
Các dung dịch thuốc thường cần bao bì lớn và có dung tích lớn hơn so với dạng thuốc rắn.
Phân loại: có thể phân loại dung dịch thuốc theo hai cách sau đây
Phân loại theo đường sử dụng như:
• Dung dịch thuốc uống: Các dạng thuốc dùng được uống, bao gồm cả sirô thuốc (quy định tại Phụ lục 1.4).
• Dung dịch thuốc dùng tại chỗ: Thuốc dùng ngoài da, thuốc nhỏ mắt (quy định tại Phụ lục 1.14), thuốc nhỏ mũi (quy định tại Phụ lục 1.15), thuốc nhỏ tai (quy định tại Phụ lục 1.16).
• Dung dịch thuốc tiêm được quy định riêng trong chuyên luận Thuốc tiêm, thuốc tiêm truyền (Phụ lục 1.19).
• Dung dịch thuốc khí dung quy định tại Phụ lục 1.18; dung dịch thuốc hít quy định tại Phụ lục 1.17.
• Phân loại theo hệ dung môi và chất tan, ví dụ: Rượu thuốc (quy định tại Phụ lục 1.22), cồn thuốc (quy định tại Phụ lục 1.2), cồn ngọt, nước thơm.
Phương pháp điều chế
Dung dịch thuốc thường được điều chế bằng cách hòa tan dược chất vào trong dung môi.
Với một số dung dịch thuốc uống có thể điều chế bằng cách pha loãng dung dịch đậm đặc hoặc hòa tan bột hoặc cốm thuốc, thuốc viên vào dung môi thích hợp.
Dung môi dùng để pha chế dung dịch thuốc được lựa chọn dựa trên tính chất của dược chất và đường dùng đồng thời phải mang lại tính chất cảm quan phù hợp với yêu cầu của chế phẩm.
Có thể cho thêm các chất bảo quản chống kháng vi khuẩn, nấm mốc, chất chống oxy hóa và các tá dược khác như chất làm tăng độ tan, chất làm ngọt hay tạo mùi vị, tạo màu...
Yêu cầu chất lượng
Tính chất: Khi quan sát bằng mắt thường, dung dịch phải trong, có thể có màu hoặc không màu.
Yêu cầu về pH, định tính, định lượng, sai số thể tích và các yêu cầu kỹ thuật khác: Phải đạt yêu cầu kỹ thuật chung của từng loại thuốc và theo chuyên luận riêng.
Bảo quản
Các dung dịch, đặc biệt là các dung dịch chứa dung môi dễ bay hơi, phải bảo quản trong bao bì kín, để nơi mát. Cần xem xét để sử dụng các bao bì tránh ánh sáng khi sự biến đổi hóa học do ánh sáng có thể ảnh hưởng đến độ ổn định của thuốc.
1.4 SIRÔ THUỐC
Sirupi
Định nghĩa
Sirô thuốc là chế phẩm thuốc lỏng hay hỗn dịch dùng đường uống, có vị ngọt, chứa nồng độ cao đường trắng (sucrose) hay chất tạo ngọt khác và dược chất hoặc các dịch chiết từ dược liệu.
Sirô đơn là dung dịch đường trắng gần bão hòa trong nước tinh khiết.
Sirô cũng bao gồm những chế phẩm được hòa tan hay tạo thành hỗn dịch bằng nước ngay trước khi sử dụng tùy theo tính chất của dược chất (sirô khô).
Phương pháp điều chế
Chuẩn bị:
Dung dịch thuốc: Dược chất được hòa tan trong dung môi thích hợp.
Dịch chiết dược liệu: Các dược liệu được chiết xuất, lọc, làm đậm đặc theo những phương pháp thích hợp.
Sirô đơn: Hòa tan đường trắng vào nước tinh khiết bằng phương pháp hòa tan nóng hay hòa tan nguội.
Lọc. Nồng độ đường trắng là 64 % (khối lượng/khối lượng).
Điều chế sirô thuốc:
Tùy theo tính chất của dược chất, sirô được điều chế bằng cách hòa tan, nhũ hóa hay trộn đều dược chất hay dung dịch thuốc, dịch chiết dược liệu vào trong dung dịch của đường trắng hay của các chất tạo ngọt khác, hoặc trong sirô đơn. Ngoài ra có thể điều chế siro bằng cách hòa tan đường vào dung dịch dược chất. Lọc đối với sirô dạng dung dịch nếu cần thiết.
Sirô có thể được điều chế từ dạng bột hay cốm khô được hòa tan hay tạo thành hỗn dịch bằng nước ngay trước khi sử dụng tùy theo tính chất của dược chất.
Có thể cho thêm chất phụ gia như: chất bảo quản, chất làm thơm, chất ổn định... với nồng độ thích hợp (ví dụ: ethanol, glycerin, poly-alcohol). số lượng và chủng loại các chất phụ gia phải đáp ứng các yêu cầu của cơ quan quản lý, các chất này không được làm ảnh hưởng đến độ ổn định và việc kiểm tra chất lượng đối với chế phẩm.
Điều chế sirô trong môi trường sản xuất có cấp độ sạch theo quy định.
Yêu cầu chất lượng
Tính chất: Trừ các quy định khác, sirô phải trong (nếu là dạng dung dịch), không được lẫn tạp chất, không có mùi lạ, bọt khí hoặc có sự biến chất khác trong quá trình bảo quản.
Nồng độ đường: Không được ít hơn 45 % nếu dùng đường trắng làm chất tạo ngọt.
Thể tích: Đáp ứng yêu cầu của Phụ lục 11.1.
Giới hạn nhiễm khuẩn: Đáp ứng yêu cầu của Phụ lục 13.6.
Yêu cầu về pH, tỷ trọng, định tính, định lượng và các yêu cầu kỹ thuật khác được quy định trong chuyên luận riêng.
Bột hoặc cốm để pha sirô phải đáp ứng yêu cầu chung của dạng Thuốc bột (Phụ lục 1.7) hoặc Thuốc cốm (Phụ lục 1.8). Sau khi hòa tan hay tạo thành hỗn dịch, chế phẩm thu được phải đáp ứng các yêu cầu đối với sirô.
Sirô là hỗn dịch phải đáp ứng yêu cầu chung của Hỗn dịch thuốc (Phụ lục 1.5).
Bảo quản
Đựng trong chai lọ khô sạch, đậy nút kín, để nơi mát.
1.5 HỖN DỊCH THUỐC
Suspensiones
Định nghĩa
Hỗn dịch là dạng thuốc lỏng để uống, tiêm hoặc dùng ngoài, chứa ít nhất một dược chất rắn không hòa tan được phân tán đều dưới dạng tiểu phân mịn hoặc cực mịn trong chất dẫn là nước hoặc dầu. Hỗn dịch có thể lắng xuống đáy và khi lắc phải phân tán đều thành dạng huyền phù ổn định trong một khoảng thời gian đủ để lấy ra liều đúng theo quy định.
Hỗn dịch có thể chứa chất hoạt động bề mặt, chất tăng độ nhớt nhằm duy trì trạng thái phân tán đều và ngăn cản hiện tượng các chất lắng xuống bị đóng bánh và trở lên rắn chắc. Hỗn dịch uống có thể chứa chất bảo quản kháng khuẩn, chất chống oxy hóa và các tá dược thích hợp khác như chất phân tán, chất tạo hương, chất tạo màu, chất làm ngọt, chất ổn định. Các chất trong thành phần bào chế của hỗn dịch phải đạt tiêu chuẩn Dược điển hoặc tuân thủ các quy định hiện hành của cơ quan có thẩm quyền.
Tùy theo hình thức cảm quan, hỗn dịch được chia làm hai loại:
Dạng hỗn dịch có thể sử dụng ngay: Là chất lỏng đục hay thể lỏng có một lớp cặn ở đáy chai, khi lắc nhẹ cặn này phải phân tán đều trở lại trong chất dẫn.
Dạng bột hoặc cốm để pha hỗn dịch: Trước khi sử dụng, chuyển thành hỗn dịch bằng cách lắc với một lượng chất dẫn thích hợp.
Thuốc tiêm hỗn dịch không được tiêm tĩnh mạch và không được tiêm ống sống.
Phương pháp điều chế
Có hai phương pháp điều chế hỗn dịch:
Phương pháp phân tán: Hỗn dịch được điều chế bằng cách thêm các chất tạo hỗn dịch, các tá dược thích hợp khác và nước hoặc dầu vào dược chất rắn đã được nghiền mịn và làm thành hỗn dịch đồng nhất bằng phương pháp thích hợp.
Phương pháp ngưng kết: Tạo ra dược chất rắn kết tủa trong chất dẫn ngay khi pha chế bằng cách làm thay đổi dung môi hay bằng phản ứng trao đổi ion.
Hỗn dịch dùng để tiêm hoặc nhỏ mắt phải pha chế trong điều kiện vô khuẩn và được thêm chất sát khuẩn thích hợp sau khi pha chế.
Hỗn dịch uống có thể chứa trong đồ đựng đơn liều hoặc đa liều. Đối với hỗn dịch đựng trong đồ đựng đơn liều, phải chứng tỏ có thể lấy ra lượng thuốc theo quy định từ đồ đựng. Đối với hỗn dịch chứa trong đồ đựng đa liều, phải có dụng cụ phù hợp để lấy ra thể tích đúng theo liều như đã quy định. Dụng cụ đo thể tích có thể là thìa, cốc đong 5 ml hoặc hơn có chia vạch hoặc bơm tiêm với thể tích khác nhau.
Yêu cầu chất lượng
Yêu cầu chung: Hỗn dịch khi để yên thì dược chất rắn phân tán có thể tách riêng nhưng phải trở lại trạng thái phân tán đồng nhất trong chất dẫn khi lắc nhẹ trong 1 min đến 2 min và giữ nguyên trạng thái đó trong vài phút.
Yêu cầu về cảm quan, pH, định tính, đinh lượng, sai số thể tích và các yêu cầu kỹ thuật khác: Phải đạt yêu cầu kỹ thuật chung của từng loại thuốc và theo quy định trong chuyên luận riêng.
Hỗn dịch dùng tiêm hoặc nhỏ mắt: Phải đáp ứng yêu cầu về Thử vô khuẩn (Phụ lục 13.7) và yêu cầu về kích thước tiểu phân cũng như các quy định theo chuyên luận chung. Hỗn dịch nhỏ mắt không được phân phối và sử dụng nếu có dấu hiệu đóng bánh hoặc kết khối.
Bột hoặc cốm để pha hỗn dịch: Phải đáp ứng yêu cầu chung của Thuốc bột (Phụ lục 1.7) hoặc Thuốc cốm (Phụ lục 1.8).
Độ hòa tan: Yêu cầu được chỉ ra trong chuyên luận riêng. Phương pháp thử được ghi trong chuyên luận Phép thử độ hòa tan (Phụ lục 11.4).
Bảo quản và nhãn
Đóng hỗn dịch vào chai, lọ hoặc đồ đựng kín có dung tích lớn hơn thể tích thuốc.
Nhãn có ghi “Lắc trước khi dùng”.
Bảo quản ở nơi khô, mát.
1.6 NHŨ TƯƠNG THUỐC
Emulsiones
Định nghĩa
Nhũ tương thuốc là dạng thuốc lỏng hoặc mềm để uống, tiêm hoặc dùng ngoài, được điều chế bằng cách sử dụng các chất nhũ hóa để trộn đều hai chất lỏng không đồng tan được gọi theo quy ước là:
Dầu (bao gồm các dầu, mỡ, sáp, tinh dầu, chất nhựa và những dược chất không tan trong nước) và Nước (bao gồm nước cất, nước thơm, nước sắc, nước hãm hoặc các dung dịch nước của các dược chất, v.v...).
Trong nhũ tương thuốc, một trong hai chất lỏng là pha phân tán hoặc pha nội, ở dạng tiểu phân có đường kính từ 0,1 μm trở lên, phân tán đều trong chất lỏng kia gọi là môi trường phân tán hoặc pha ngoại.
Khi dầu là pha phân tán và nước là môi trường phân tán thì nhũ tương là kiểu dầu trong nước, có ký hiệu là D/N;
Khi nước là pha phân tán và dầu là môi trường phân tán thì nhũ tương là kiểu nước trong dầu có ký hiệu là N/D.
Chất nhũ hóa quyết định kiểu nhũ tương và giúp ổn định chúng do ngăn cản sự kết tụ các giọt nhỏ thành giọt lớn, dẫn đến sự tách lớp. Chất nhũ hóa hòa tan trong nước sẽ tạo ra kiểu nhũ tương D/N; chất nhũ hóa hòa tan trong dầu, mỡ, sáp sẽ tạo ra kiểu nhũ tương N/D. Các chất nhũ hóa là chất hoạt động bề mặt, khi có lực phân tán, sẽ tập trung lên bề mặt tiếp xúc giữa hai pha tạo ra hàng rào ngăn cản không cho các giọt kết tụ lại, mặt khác làm giảm sức căng liên bề mặt giữa hai pha, nhờ vậy giúp sự nhũ hóa được dễ dàng. Các chất cao phân tử thân nước thiên nhiên, bán tổng hợp hay tổng hợp có thể được sử dụng phối hợp với chất nhũ hóa hoạt động bề mặt trong các nhũ tương kiểu D/N do chúng tích tụ lên bề mặt tiếp xúc và cũng làm tăng độ nhớt của pha nước, như vậy làm giảm sự kết hợp của các giọt. Sự kết hợp này thường sẽ dẫn đến hiện tượng: Sự nổi kem do các giọt dầu lớn nổi lên (trong nhũ tương D/N) hoặc sự lắng xuống đáy của các giọt nước lớn (trong nhũ tương N/D).
Phương pháp điều chế
Tùy theo điều kiện trang thiết bị, nhũ tương có thể được điều chế bằng cách:
Điều chế nhũ tương đậm đặc với chất nhũ hóa, dược chất lỏng và lượng vừa đủ pha ngoại, sau đó pha loãng nhũ tương đậm đặc với pha ngoại.
Hòa tan chất nhũ hóa vào pha ngoại, sau đó cho dược chất lỏng vào từ từ vừa dùng lực gây phân tán mạnh để nhũ hóa.
Cần cho thêm các chất bảo quản thích hợp vào nhũ tương do pha nước là môi trường thuận lợi cho sự phát triển của vi khuẩn và nấm mốc. Hiệu lực của các chất bảo quản trong thành phẩm phải được kiểm tra.
Yêu cầu chất lượng
Tính chất: Khi quan sát bằng mắt thường, nhũ tương đặc phải mịn và đồng nhất giống như kem; còn nhũ tương lỏng phải đục trắng và đồng nhất giống như sữa.
Nhũ tương được coi như đã bị hỏng khi hai pha lỏng đã tách riêng nhau và bằng cách khuấy lắc cũng không thể khôi phục lại trạng thái phân tán đồng nhất nữa.
Yêu cầu về pH, định tính, định lượng, sai số thể tích và các yêu cầu kỹ thuật khác: Phải đạt quy định theo chuyên luận riêng.
Nhũ tương dùng để tiêm: Phải đáp ứng yêu cầu về Thử vô khuẩn (Phụ lục 13.7).
Bảo quản
Ở nơi mát, nhiệt độ ít thay đổi.
Đóng nhũ tương thuốc vào chai lọ có dung tích hơi lớn hơn thể tích thuốc một chút và trên nhãn có ghi dòng chữ: Lắc trước khi dùng.
1.7 THUỐC BỘT
Pulveres
Định nghĩa
Thuốc bột là dạng thuốc rắn, gồm các hạt nhỏ, khô tơi, có độ mịn xác định, có chứa một hay nhiều loại dược chất. Ngoài dược chất, thuốc bột còn có thể thêm các tá dược như tá dược độn, tá dược hút, tá dược màu, tá dược điều hương, vị...
Thuốc bột có thể dùng để uống, để pha tiêm hay để dùng ngoài.
Các yêu cầu chất lượng chung
Tính chất
Quan sát màu sắc bằng mắt thường, dưới ánh sáng tự nhiên, với một lượng bột vừa đủ, được phân tán đều trên một tờ giấy trắng mịn. Bột phải khô tơi, không bị ẩm, vón, màu sắc đồng nhất.
Độ ẩm
Xác định độ ẩm thuốc bột theo phương pháp Xác định mất khối lượng do làm khô (Phụ lục 9.6), hoặc Định lượng nước bằng thuốc thử Karl - Fischer (Phụ lục 10.3), tùy theo chỉ dẫn trong chuyên luận riêng.
Thuốc bột không được chứa hàm lượng nước quá 9,0 %, trừ các chỉ dẫn khác.
Độ mịn
Nếu không có chỉ dẫn khác, độ mịn của thuốc bột được xác định qua phép thử Cỡ bột và rây (Phụ lục 3.5). Thuốc bột phải đạt độ mịn quy định trong chuyên luận.
Độ đồng đều hàm lượng (Phụ lục 11.2)
Trừ khi có chỉ dẫn khác, phép thử này áp dụng cho thuốc bột để uống, để tiêm, được trình bày trong các đơn vị đóng gói 1 liều, trong đó có các dược chất có hàm lượng dưới 2 mg hoặc dưới 2 % (kl/kl) so với khối lượng bột đóng gói trong 1 liều.
Phép thử đồng đều hàm lượng được tiến hành sau phép thử định lượng và hàm lượng dược chất đã đạt trong giới hạn quy định.
Độ đồng đều khối lượng (Phụ lục 11.3)
Những thuốc bột không quy định thử độ đồng đều hàm lượng thì phải thử độ đồng đều khối lượng.
Nếu thuổc bột chứa nhiều hoạt chất, thì chỉ khi tất cả các dược chất đã được thử độ đồng đều hàm lượng mới không thử độ đồng đều khối lượng.
Định tính
Theo chuyên luận riêng.
Định lượng
Theo chuyên luận riêng.
Giới hạn nhiễm khuẩn
Đáp ứng yêu cầu Thử giới hạn nhiễm khuẩn (Phụ lục 13.6).
Ghi nhãn
Theo quy định hiện hành.
Đối với thuốc bột trong một đơn vị đóng gói 1 liều phải ghi tên và hàm lượng dược chất.
Thuốc bột đóng gói nhiều liều phải ghi tên, lượng dược chất trên tổng khối lượng.
Trên nhãn phải ghi tên và lượng chất bảo quản chống vi khuẩn, hạn dùng, điều kiện bảo quản.
Bảo quản
Thuốc bột phải được bảo quản trong đồ đựng kín. Để nơi khô mát.
Thuốc bột để uống
Thuốc bột để uống có thể dùng nuốt trực tiếp hoặc được sử dụng sau khi đã hòa tan hay phân tán trong nước hoặc chất lỏng thích hợp. Thuốc bột để uống phải đáp ứng các yêu cầu chất lượng chung của thuốc bột.
Thuốc bột sủi bọt để uống thường chứa tá dược sủi bọt, gồm các acid hữu cơ và muối carbonat hoặc hydrocarbonat, phản ứng khi có nước để giải phóng khí carbon dioxyd. Thuốc bột sủi bọt để uống phải đáp ứng các yêu cầu chung của thuốc bột. Ngoài ra thuốc bột sủi bọt để uống phải đạt yêu cầu về Độ tan như sau:
Độ tan: Cho một lượng bột tương ứng với một liều vào một cốc thủy tinh chứa 200 ml nước ở 15 °C đến 25 °C, xuất hiện nhiều bọt khí bay ra. Khi hết bọt khí, thuốc phải tan hoàn toàn. Thử như vậy với 6 liều đơn. Mẫu thử đạt yêu cầu nếu mỗi liều thử đều tan trong vòng 5 min, trừ khi có chỉ dẫn riêng.
Thuốc bột dùng ngoài
Thuốc bột dùng ngoài thường đóng gói nhiều liều, có thể dùng để đắp, rắc trực tiếp lên da, vết thương hoặc được hòa tan, phân tán trong dung môi thích hợp để nhỏ mắt, rửa hoặc thụt.
Thuốc bột dùng ngoài phải đáp ứng các yêu cầu chung của thuốc bột, ngoài ra phải đạt các chỉ tiêu riêng sau:
Thử vô khuẩn (Phụ lục 13.7)
Thuốc bột để đắp, dùng cho vết thương rộng hoặc trên da bị tổn thương nặng, thuốc bột dùng cho mắt phải vô khuẩn.
Độ mịn
Thuốc bột dùng để đắp hoặc rắc phải là bột mịn hoặc rất mịn (Phụ lục 3.5).
Thuốc bột để pha tiêm
Thuốc bột pha tiêm phải đáp ứng các yêu cầu chung của thuốc bột và yêu cầu chất lượng đối với thuốc tiêm, thuốc tiêm truyền dạng bột (Phụ lục 1.19).
1.8 THUỐC CỐM
Granulae
Định nghĩa
Thuốc cốm hay thuốc hạt là dạng thuốc rắn có dạng hạt nhỏ xốp hay sợi ngắn xốp, thường dùng để uống với một ít nước hay một chất lỏng thích hợp, hoặc pha thành dung dịch, hỗn dịch hay sirô. Thuốc cốm chứa một hoặc nhiều dược chất, ngoài ra có thêm các tá dược như tá dược độn, tá dược dính, tá dược điều hương vị, tá dược màu...
Các yêu cầu chất lượng chung
Tính chất
Thuốc cốm phải khô, đồng đều về kích thước hạt, không có hiện tượng hút ẩm, không bị mềm và biến màu.
Độ ẩm
Xác định nước trong các thuốc cốm nói chung theo phương pháp Xác định mất khối lượng do làm khô (Phụ lục 9.6), trong các thuốc cốm chứa tinh dầu theo phương pháp cất với dung môi (Phụ lục 12.13). Các thuốc cốm có độ ẩm không quá 5,0 %, trừ các chỉ dẫn khác.
Độ đồng đều khối lượng (Phụ lục 11.3)
Thuốc cốm không quy định thử độ đồng đều về hàm lượng thì phải thử độ đồng đều khối lượng.
Độ đồng đều hàm lượng (Phụ lục 11.2)
Trừ khi có chỉ dẫn khác, phép thử này áp dụng cho các thuốc cốm đóng gói một liều, có chứa một hoặc nhiều dược chất, phải thử đồng đều hàm lượng với các dược chất có hàm lượng dưới 2 mg hoặc dưới 2 % (kl/kl) so với khối lượng cốm trong 1 liều.
Định tính, định lượng và các yêu cầu kỹ thuật khác
Theo chuyên luận riêng.
Bảo quản
Thuốc cốm phải được bảo quản trong các đồ đựng kín, đóng từng liều hoặc nhiều liều, có nhãn đúng quy định. Để nơi khô mát.
Cốm sủi bọt
Cốm sủi bọt thường chứa tá dược sủi bọt gồm các acid hữu cơ và muối carbonat hoặc hydrocarbonat, phản ứng khi có nước để giải phóng khí carbon dioxyd. Cốm được hòa tan hoặc phân tán trong nước trước khi dùng.
Cốm sủi bọt phải đáp ứng các yêu cầu chất lượng chung của thuốc cốm, ngoài ra phải đạt yêu cầu về độ rã như sau:
Độ rã
Cho một lượng cốm đóng gói trong một đơn vị phân liều vào cốc chứa 200 ml nước ở 15 °C đến 25 °C, phải có nhiều bọt khí bay ra. Cốm được coi là rã hết nếu hòa tan hoặc phân tán hết trong nước. Thử với 6 liều, chế phẩm đạt yêu cầu phép thử nếu mỗi liều rã trong vòng 5 min, trừ khi có các chỉ dẫn khác trong chuyên luận riêng.
1.9 THUỐC DÁN THẤM QUA DA VÀ CAO DÁN
I. THUỐC DÁN THẤM QUA DA
Định nghĩa
Thuốc dán thấm qua da (transdermal system, transdermal patch) là những chế phẩm bán rắn, giải phóng dược chất có kiểm soát, được dán trên vùng da nguyên vẹn nhằm đưa dược chất thấm qua da vào hệ tuần hoàn để gây tác dụng tại chỗ hoặc toàn thân. Trong đó, hệ trị liệu qua da (transdermal therapeutic system) thường được thiết kế để giải phóng dược chất ở tốc độ hằng định nhằm đạt được nồng độ trong máu ở trạng thái cân bằng và duy trì cho đến lúc bóc khỏi da
Cấu tạo chung
Thuốc dán thấm qua da thường có 4 lớp: lớp nền, lớp chứa dược chất, lớp nền dính và màng bảo vệ. Lớp nền là tấm mỏng dùng để trải lớp chứa thuốc, cấu tạo từ các vật liệu không thấm như màng polymer hoặc lá kim loại. Lớp này không tương kị với lớp chứa dược chất, ngoài vai trò mang thuốc, còn có tác dụng bảo vệ dược chất tránh tác động của ngoại môi.
Lớp chứa dược chất: có cấu tạo dưới dạng hệ cốt (matrix) hoặc bình chứa (recevoir). Ở hệ cốt, dược chất được hòa tan hoặc phân tán đồng nhất dưới dạng tiểu phân trong cốt polymer (như Eudragit, polyvinyl alcohol, polyvinyl pyrolidon, chitosan, HPMC,...); khi dùng, dược chất được giải phóng bằng cách khuếch tán qua cốt polymer và qua nền dính. Với hệ bình chứa, dược chất được hòa tan hoặc phân tán trong chất lỏng có độ nhớt cao (silicon, PEG lỏng,...) hoặc trong môi trường bột nhão, gel,...Khi dùng, dược chất được khuếch tán qua một màng kiểm soát giải phóng gắn với lớp chứa dược chất và khuếch tán tiếp qua nền dính.
Lớp nền dính: có tác dụng làm cho hệ bắt dính da, giữ thuốc tại nơi dùng, gồm những polymer nhạy cảm với áp suất (polyacrylat, polyisobutilen, polysiloxan,...), có khả năng bắt dính da, không độc và không gây kích ứng hay dị ứng với da. Trong một số thuốc dán thấm qua da, lớp chứa dược chất có thể đồng thời đóng vai trò là lớp nền dính.
Màng bảo vệ: Thường là màng mỏng polyethylen phủ lên mặt ngoài của lớp nền dính để bảo vệ thuốc dán trong quá trình bảo quản và được bóc bỏ trước khi dán. Khi bóc bỏ, màng không được làm hỏng cấu trúc của lớp nền dính
Ngoài dược chất, hệ trị liệu qua da còn chứa các chất phụ như chất hóa dẻo, chất ổn định, chất tăng độ tan, chất tăng thấm, chất bảo quản,...
Kỹ thuật bào chế
Chủ yếu sử dụng phương pháp tráng phim và đổ khuôn với các bước cơ bản như sau:
1. Chế tạo lớp chứa dược chất bằng cách đun chảy polymer tạo cốt hoặc hòa tan polymer trong dung môi hữu cơ. Thêm dược chất và các thành phần khác, khuấy trộn đồng nhất, kiểm nghiệm bán thành phẩm.
2. Trải hỗn hợp cốt lên lớp nền trong máy trải hoặc đổ dung dịch thuốc vào khuôn rồi bốc hơi dung môi, kiểm soát bề dày lớp chứa dược chất.
3. Sấy lớp chứa dược chất hoặc làm khô ở nhiệt độ phòng.
4. Gắn màng kiểm soát (nếu có).
5. Tráng tiếp lớp nền dính lên lớp chứa dược chất (nếu có).
6. Phủ màng bảo vệ lên nền dính.
7. Đóng gói sản phẩm.
Yêu cầu chất lượng
Thuốc thấm qua da phải đạt các yêu cầu quy định trong chuyên luận riêng và các yêu cầu sau đây:
Tính chất
Chế phẩm phải đồng nhất, có độ bắt dính thích hợp (dễ dính và dễ bóc), không gây kích ứng trên da.
Độ đồng đều hàm lượng
Nếu như không có chỉ dẫn khác trong chuyên luận riêng, tiến hành trên 10 đơn vị riêng rẽ được lấy bất kỳ, kết quả được đánh giá như sau:
Chế phẩm đem kiểm tra đạt yêu cầu phép thử nếu hàm lượng trung bình của 10 đơn vị không nằm ngoài giới hạn từ 90 % đến 110 % so với hàm lượng ghi trên nhãn và không có đơn vị nào có hàm lượng nằm ngoài giới hạn từ 75 % đến 125 % so với hàm lượng trung bình.
Độ đồng đều khối lượng lớp chứa dược chất
Nếu phép thử độ đồng đều hàm lượng đã được tiến hành với tất cả các dược chất có trong thuốc dán qua da thì không cần phải thử độ đồng đều khối lượng.
Tiến hành trên 20 đơn vị bất kỳ, xác định khối lượng lớp chứa dược chất của từng đơn vị và tính khối lượng trung bình của lớp chứa dược chất. Cho phép không quá 2 đơn vị có khối lượng lệch ra ngoài 5 % so với khối lượng trung bình và không có đơn vị nào có khối lượng lệch ra ngoài 10 % so với khối lượng trung bình.
Tiến hành: Cân từng đơn vị đã được loại bỏ lớp bảo vệ, ta được khối lượng m1, dùng hỗn hợp dung môi hữu cơ phù hợp để rửa hết lớp polymer chứa dược chất, làm khô, rồi cân lại khối lượng của lớp, ta được khối lượng m2. Khối lượng lớp chứa dược chất là hiệu số của m1 và m2.
Độ đồng đều diện tích
Tiến hành trên 20 đơn vị bất kỳ. Đo diện tích của từng đơn vị (tùy hình dạng của thuốc dán là hình tròn hay hình vuông mà có phương pháp đo và tính toán phù hợp), tính diện tích trung bình. Cho phép không quá 2 đơn vị có diện tích lệch ra ngoài 5 % so với diện tích trung bình và không có đơn vị nào có diện tích lệch ra ngoài 10 % so với diện tích trung bình.
Tiến hành: bóc lớp màng bảo vệ của từng đơn vị, đo và tính diện tích của lớp chứa dược chất.
Định tính, định lượng
Theo quy định trong chuyên luận riêng.
Giải phóng dược chất
Phương pháp thử nghiệm thích hợp được yêu cầu theo từng chuyên luận riêng để chứng minh sự giải phóng của dược chất là phù hợp. Thiết bị kiểu giỏ quay, kiểu cánh khuấy hoặc kiểu dòng chảy có thể được sử dụng tùy theo thành phần, kích thước và hình dạng của mẫu thử.
Sự giải phóng dược chất qua màng cũng được sử dụng. Màng có thể là màng celulose hoặc màng silicon và phải không ảnh hưởng đến động học của giải phóng dược chất từ miếng thuốc dán. Màng có thể được xử lý một cách phù hợp trước khi thử nghiệm, được lưu giữ trong môi trường thích hợp để sử dụng cho thử nghiệm trong 24 h. Đặt bề mặt giải phóng dược chất của miếng thuốc dán lên màng, tránh sự hình thành bọt khí.
Điều kiện thử nghiệm và các yêu cầu tiến hành theo chuyên luận riêng.
Thử tính kích ứng
Tiến hành theo quy định hiện hành về thử tính kích ứng trên da áp dụng cho các sản phẩm dùng trong y tế và mỹ phẩm.
Bảo quản
Nếu không có quy định khác, chế phẩm được đựng trong bao bì kín, bảo quản nhiệt độ phòng, tránh ánh sáng.
Nhãn
Nhãn ghi rõ nơi dán thuốc, tổng hàm lượng hoạt chất có trong một đơn vị, liều giải phóng cho mỗi đơn vị thời gian và diện tích bề mặt phóng thích và theo đúng quy định hiện hành.
II. CAO DÁN
Định nghĩa
Cao dán (medicated plasters) là dạng thuốc quy ước có thể chất mềm dẻo, có khả năng bắt dính da, chứa một hoặc nhiều dược chất hòa tan hay phân tán trong nền dính được trải đều thành lớp mỏng trên một lớp vải hoặc vật liệu thích hợp. Cao dán thường được dán trên vùng da lành hoặc da tổn thương (viêm, sưng,...) để gây tác dụng tại chỗ.
Cấu tạo chung
Cấu tạo tương tự cấu tạo chung của thuốc dán thấm qua da.
Kỹ thuật bào chế
Ngoại trừ các trường hợp đặc biệt, cao dán thường được điều chế bởi phương pháp trộn đều hợp chất cao phân tử trong tự nhiên hoặc tổng hợp có thể tan hoặc không tan trong nước với các dược chất, rồi trải đều trên một miếng vải hoặc phim với một hình dạng thích hợp.
Các chất cao phân tử thường gặp: chất béo, dầu béo, muối của acid béo, sáp, nhựa, chất dẻo, lanolin tinh chế, cao su hoặc một hỗn hợp đồng nhất của các chất trên.
Yêu cầu chất lượng
Cao dán phải đạt các yêu cầu quy định trong chuyên luận riêng và các yêu cầu chung sau đây:
Tính chất
Phải đồng nhất, có độ bắt dính thích hợp (dễ dính và dễ bóc), không gây kích ứng trên da.
Định tính, định lượng
Theo quy định trong chuyên luận riêng.
Giải phóng dược chất
Phương pháp thử nghiệm thích hợp được yêu cầu theo từng chuyên luận riêng để chứng minh sự giải phóng của hoạt chất là phù hợp. Thiết bị kiểu giỏ quay, kiểu cánh khuấy hoặc kiểu dòng chảy cỏ thể được sử dụng tùy theo thành phần, kích thước và hình dạng của miếng thuốc dán.
Sự giải phóng dược chất qua màng cũng được sử dụng. Màng có thể là màng celulose hoặc màng silicon và phải không ảnh hưởng đến động học của giải phóng hoạt chất từ miếng thuốc dán. Màng có thể được xử lý một cách phù hợp trước khi thử nghiệm, được lưu giữ trong môi trường thích hợp để sử dụng cho thử nghiệm trong 24 h. Đặt bề mặt giải phóng dược chất của miếng thuốc dán lên màng, tránh sự hình thành bọt khí.
Điều kiện thử nghiệm và các yêu cầu tiến hành theo chuyên luận riêng.
Thử tính kích ứng
Tiến hành theo quy định hiện hành về thử tính kích ứng trên da áp dụng cho các sản phẩm dùng trong y tế và mỹ phẩm.
Bảo quản
Nếu không có quy định khác, chế phẩm được đựng trong bao bì kín, bảo quản nhiệt độ phòng, tránh ánh sáng.
Nhãn
Theo quy định hiện hành.
1.10 THUỐC ĐẶT
Suppositoria
Định nghĩa
Thuốc đặt là dạng thuốc rắn, chứa một hoặc nhiều dược chất, dùng để đặt vào các hốc tự nhiên của cơ thể. Thuốc có thể có tác dụng tại chỗ hoặc toàn thân.
Khi đặt vào vị trí trên cơ thể, thuốc đặt thường chảy ra, mềm ở thân nhiệt hoặc hòa tan dần trong niêm dịch để giải phóng dược chất.
Tá dược dùng cho thuốc đặt bao gồm: Bơ cacao và chế phẩm của bơ cacao, hỗn hợp gelatin - glycerin - nước, dầu thực vật hydrogen hóa, glycerid bán tổng hợp, hỗn hợp polyethylen glycol khối lượng phân tử khác nhau (PEG 400, PEG 1500, PEG 1540, PEG 3000, PEG 4000), este của acid béo với PEG.
Phân loại
Thuốc đặt trực tràng: Thường có hình dạng giống như đầu viên đạn (hình nón cụt) hoặc hình thủy lôi, khối lượng khoảng 1 g đến 3 g.
Thuốc đặt âm đạo: Thường có hình trái xoan, khối lượng khoảng 3 g đến 10 g.
Thuốc đặt niệu đạo: Đường kính 1 mm đến 4 mm, chiều dài 6 cm đến 20 cm, khối lượng khoảng 0,5 g đến 4,0 g.
Yêu cầu chất lượng
Độ rã
Đạt yêu cầu Phép thử độ rã của thuốc đạn và thuốc trứng (Phụ lục 11.5).
Độ đồng đều khối lượng
Đạt yêu cầu Phép thử độ đồng đều khối lượng (Phụ lục 11.3).
Độ đồng đều hàm lượng (Phụ lục 11.2)
Nếu không có chỉ dẫn khác, thuốc đặt đơn liều có hàm lượng hoạt chất dưới 2 mg hoặc dưới 2 % (kl/kl) phải thử độ đồng đều hàm lượng. Đối với chế phẩm có từ 2 dược chất trở lên, chỉ áp dụng yêu cầu này với thành phần có hàm lượng nhỏ như quy định ở trên. Thuốc đặt đã thử độ đồng đều về hàm lượng với tất cả các dược chất có trong thành phần thì không phải thử độ đồng đều khối lượng.
Các yêu cầu kỹ thuật khác
Thử theo quy định trong chuyên luận riêng.
Bảo quản: Trong bao bì kín, thích hợp (hộp, màng nhôm, polymer), để nơi mát (nhiệt độ dưới 30 °C).
1.11 THUỐC HOÀN
Pilula
Định nghĩa
Thuốc hoàn là dạng thuốc rắn, hình cầu, được bào chế từ bột hoặc cao dược liệu với các loại tá dược thích hợp, thường dùng để uống.
Phân loại
Trong Y học cổ truyền, tùy theo tá dược dính sử dụng mà người ta chia ra các loại hoàn như sau:
Thủy hoàn: Là hoàn được điều chế với tá dược dính là nước, rượu, dấm, dịch chiết dược liệu bằng phương pháp bồi viên và thường là hoàn nhỏ (khối lượng viên dưới 0,5 g).
Hồ hoàn: Là hoàn dùng hồ tinh bột làm tá dược dính, điều chế bằng phương pháp chia viên hay bồi viên, thường là hoàn nhỏ.
Mật hoàn: Là hoàn bào chế với tá dược dính là mật ong. Mật được luyện thành châu, trộn với bột thuốc khi còn nóng và bào chế hoàn bằng phương pháp chia viên. Hoàn mật thường gọi là “tễ”, khối lượng có thể đến 12 gam, có thể chất nhuận dẻo.
Lạp hoàn: Lạp hoàn được điều chế với sáp ong bằng cách đun chảy và vê viên ở nhiệt độ gần nhiệt độ đông rắn của sáp, thường có khối lượng từ 0,3 g đến 0,5 g.
Phương pháp điều chế
Thuốc hoàn được điều chế bằng 2 phương pháp: Chia viên và bồi viên.
Phương pháp chia viên: Áp dụng khi dùng các tá dược dính có độ nhớt cao như mật, hồ, sáp. Bột thuốc được trộn với tá dược dính ở nhiệt độ thích hợp thành khối bánh viên đồng nhất rồi chia viên bằng bàn hay máy chia viên.
Phương pháp bồi viên: Áp dụng cho các tá dược có độ dính thấp như nước, dịch chiết dược liệu, hồ loãng, sirô hay mật ong pha loãng. Tá dược dính lỏng và bột thuốc được bồi dần từng lớp lên nhân đã gây sẵn kết hợp với sấy cho đến khi viên đạt kích thước yêu cầu.
Thuốc hoàn có thể được bao bằng các lớp áo khác nhau để bảo quản hay tăng giá trị thẩm mỹ, viên hoàn mềm thường được đóng trong vỏ sáp.
Yêu cầu chất lượng
Nếu không có quy định riêng trong chuyên luận, bột thuốc dùng bào chế thuốc hoàn phải là bột mịn hay rất mịn.
Mật ong dùng trong viên hoàn thường là loại mật luyện: Thêm khoảng 20 % nước vào mật, đun sôi vớt bỏ bọt rồi lọc qua gạc và cô nhỏ lửa cho đến khi giọt mật thành “châu” (không tan trong nước lạnh).
Tính chất
Hoàn phải tròn, đều, đồng nhất về hình dạng, màu sắc khi bảo quản, có mùi đặc trưng của dược liệu. Hoàn mềm phải nhuận, dẻo.
Hàm ẩm
Hoàn mật ong, hoàn chứa cao đặc: Không quá 15 %. Hoàn nước có kết hợp sirô, mật ong: Không quá 12 %. Hoàn nước và hoàn hồ: Không quá 9 % (hoàn sáp không xác định hàm ẩm).
Tiến hành theo phương pháp Xác định mất khối lượng do làm khô (Phụ lục 9.6) hoặc Xác định hàm lượng nước bằng phương pháp cất với dung môi (Phụ lục 12.13).
Độ rã
Chỉ áp dụng cho hoàn cứng: Viên rã trong vòng 1 h (riêng hoàn hồ trong vòng 2 h, hoàn sáp thử theo viên bao tan trong ruột).
Tiến hành theo Phép thử độ rã của viên nén và viên nang (Phụ lục 11.6).
Độ đồng đều khối lượng
Đối với hoàn uống theo số viên:
Cân 10 viên, xác định khối lượng từng viên. Sự chênh lệch khối lượng của từng viên so với khối lượng trung bình phải nằm trong giới hạn ở Bảng 1.11.1, trong đó, không được có quá 2 viên vượt giới hạn cho phép và không được có viên nào gấp đôi giới hạn cho phép.
Bảng 1.11.1
| Khối lượmg trung bình 1 viên | Giới hạn cho phép |
| Từ 0,05 g đến 1,5 g | ± 12 % |
| Trên 1,5 g đến 5 g | ± 10 % |
| Trên 5,0 g đến 9,0 g | ± 7 % |
| Trên 9,0 g | ± 5 % |
Đối với hoàn uống theo gam:
Cân 10 phần, mỗi phần 10 viên, xác định khối lượng trung bình chung. Sự chênh lệch khối lượng của từng phần so với khối lượng trung bình phải nằm trong giới hạn ở Bảng 1.11.2, trong đó, không được có quá 2 phần vượt giới hạn cho phép và không được có phần nào gấp đôi giới hạn cho phép.
Bảng 1.11.2
| Khối lượng trung bình mỗi phần | Giới hạn cho phép |
| Từ 0,05 g đến 0,1 g | ± 12 % |
| Trên 0,1 g đến 1,0 g | ± 10 % |
| Trên 1,0 g | ± 7 % |
Đối với đơn vị đóng gói đã chia liều:
Lấy 10 gói, cân từng gói. Sự chênh lệch khối lượng của từng gói so với khối lượng trên nhãn phải nằm trong giới hạn ở Bảng 1.11.3, trong đó, không được có quá 2 gói vượt giới hạn cho phép và không được có gói nào gấp đôi giới hạn đó.
Bảng 1.11.3
| Khối lượng trên nhãn | Giới hạn cho phép |
| Từ 0,5 g trở xuống | ± 12 % |
| Trên 0,5 g đến 1,0 g | ± 11 % |
| Trên 1,0 g đến 2,0 g | ± 10 % |
| Trên 2,0 g đến 3,0 g | ± 8 % |
| Trên 3,0 g đến 6,0 g | ± 6 % |
| Trên 6,0 g đến 9,0 g | ± 5 % |
| Trên 9,0 g | ± 4 % |
Định tính, định lượng
Đáp ứng theo quy định trong chuyên luận riêng.
Giới hạn nhiễm khuẩn
Thuốc hoàn phải đạt yêu cầu về giới hạn nhiễm khuẩn (Phụ lục 13.6).
1.12 THUỐC MỀM DÙNG TRÊN DA VÀ NIÊM MẠC
Praeparationes molles ad usum dermicum
Định nghĩa
Dạng thuốc có thể chất mềm, đồng nhất dùng để bôi lên da và niêm mạc nhằm gây tác dụng tại chỗ hoặc đưa dược chất thấm qua da và niêm mạc, làm trơn hoặc bảo vệ.
Thành phần của thuốc gồm một hay nhiều dược chất, được hòa tan hay phân tán đồng đều trong một hoặc hỗn hợp tá dược, thuộc hệ phân tán một pha hoặc nhiều pha.
Tá dược sử dụng có nguồn gốc thiên nhiên hoặc tổng hợp, thân dầu hay thân nước. Ngoài ra, trong thành phần tá dược còn có thêm chất bảo quản, chất chống oxy hóa, chất ổn định, chất nhũ hóa, chất làm thơm và các chất làm tăng tính thấm của dược chất.
Phân loại:
• Thuốc mỡ (ointments)
• Bột nhão (pastes)
• Kem (creams)
• Gel (gels)
Yêu cầu chất lượng chung
Độ đồng nhất
Cách thử: Lấy 4 đơn vị đóng gói, mỗi đơn vị khoảng 0,02 g đến 0,03 g, trải đều chế phẩm trên 4 phiến kính. Đậy mỗi phiến kính bằng một phiến kính thứ 2 và ép mạnh cho tới khi tạo thành một vết có đường kính khoảng 2 cm. Quan sát vết thu được bằng mắt thường (cách mắt khoảng 30 cm), ở 3 trong 4 tiêu bản không được nhận thấy các tiểu phân. Nếu có các tiểu phân nhìn thấy ở trong phần lớn số các vết thì phải làm lại với 8 đơn vị đóng gói. Trong số các tiêu bản này, các tiểu phân cho phép nhận thấy không được vượt quá 2 tiêu bản.
Độ đồng đều khối lượng
Đạt yêu cầu Phép thử độ đồng đều khối lượng (Phụ lục 11.3).
Độ nhiễm khuẩn
Đạt yêu cầu Phép thử giới hạn nhiễm khuẩn (Phụ lục 13.6).
Các yêu cầu kỹ thuật khác
Thử theo quy định trong chuyên luận riêng.
Bảo quản
Trong chai, lọ, tuýp kín chế tạo bằng vật liệu phù hợp (kim loại, polymer). Nếu chế phẩm vô khuẩn, cần bảo quản kín, vô khuẩn.
Thuốc mỡ
Định nghĩa
Tùy theo cách phối hợp và sử dụng tá dược, thuốc mỡ được chia ra 3 loại:
Thuốc mỡ thân dầu
Thuốc mỡ thân dầu có thể hút (hấp phụ) một lượng nhỏ nước hoặc dung môi phân cực. Tá dược điển hình gồm: Nhóm hydrocarbon no (vaselin, dầu parafin, parafin rắn), dầu, mỡ có nguồn gốc động vật, thực vật, glycerid bán tổng hợp, sáp và polyalkylsiloxan lỏng.
Thuốc mỡ thân nước
Thuốc mỡ thân nước có thể trộn lẫn với nước. Tá dược thường dùng là polyethylen glycol (macrogol, carbowax).
Thuốc mỡ nhũ hóa thân nước
Thuốc mỡ nhũ hóa thân nước có thể hút được một lượng lớn nước và chất lỏng phân cực để tạo thành nhũ tương nước-dầu (N/D) hoặc dầu-nước (D/N), tùy thuộc vào bản chất chất nhũ hóa có trong thành phần tá dược. Chất nhũ hóa cho nhũ tương N/D gồm: lanolin, este sorbitan (Span), monoglycerid và alcol béo. Chất nhũ hóa cho nhũ tương D/N gồm: Alcol béo sulfat, polysorbat (Tween), ether hoặc este của acid béo với polyethylen glycol.
Thuốc mỡ tra mắt
Thuốc mỡ tra mắt là những chế phẩm thuốc mỡ dùng cho mắt, chứa một hoặc nhiều dược chất hòa tan hoặc phân tán trong tá dược, được xếp vào nhóm các chế phẩm vô khuẩn. Tá dược và dược chất dùng cho thuốc mỡ tra mắt phải không bị phân hủy khi tiệt khuẩn bằng nhiệt.
Ngoài các yêu cầu của thuốc mỡ nói chung, thuốc mở tra mắt phải đạt các yêu cầu sau:
Thử vô khuẩn
Đạt yêu cầu Thử vô khuẩn (Phụ lục 13.7)
Các phần tử kim loại
Cách thử: Trừ trường hợp có chỉ dẫn riêng, lấy 10 tuýp thuốc, bóp hết thuốc chứa bên trong vào từng đĩa Petri riêng có đường kính 6 cm, đáy bằng, không có các vết xước và các phần tử lạ nhìn thấy được. Đậy các đĩa, đun nóng từ 80 °C đến 85 °C trong 2 h và để cho thuốc mỡ phân tán đồng đều. Làm nguội cho thuốc mỡ đông lại, lật ngược mỗi đĩa, đặt lên bản soi của kính hiển vi thích hợp. Chiếu sáng từ trên xuống bằng một đèn chiếu đặt ở góc 45° so với mặt phẳng của bản soi. Quan sát và đếm các phần tử kim loại sáng bóng, lớn hơn 50 μm ở bất kỳ kích thước nào. Không được có quá một tuýp trong 10 tuýp thuốc đem thử chứa nhiều hơn 8 phần tử và không được quá 50 phần tử tìm thấy trong 10 tuýp. Nếu chế phẩm không đạt ở lần thử thứ nhất, làm lại lần thử thứ hai với 20 tuýp thuốc khác. Mẫu thử được coi là đạt yêu cầu nếu không có quá 3 tuýp chứa quá 8 phần tử trong mỗi tuýp và tổng số không quá 150 phần tử trong 30 tuýp thử.
Giới hạn kích thước các phần tử
Trải một lượng nhỏ chế phẩm thành một lớp mỏng trên bản soi của kính hiển vi, phủ phiến kính lên trên và soi. Không được có phần tử nào của thuốc có kích thước lớn hơn 75 μm.
Bảo quản
Đựng trong lọ, tuýp có nắp hoặc nút kín. Để nơi khô mát, tránh ánh sáng.
Bao bì đựng thuốc mỡ tra mắt phải bền ở nhiệt độ tiệt khuẩn và không ảnh hưởng tới chất lượng của thuốc.
Bột nhão
Bột nhão bôi da là các chế phẩm nửa rắn, chứa một tỷ lệ lớn dược chất rắn phân tán trong tá dược.
Kem
Kem bôi da là những chế phẩm thuộc hệ phân tán nhiều pha, bao gồm: Pha dầu, pha nước và chất nhũ hóa.
Kem N/D: Pha nội thân nước, pha ngoại (pha liên tục) thân dầu. Trong thành phần có các chất nhũ hóa tạo nhũ tương N/D như: Lanolin, este sorbitan (Span), monoglycerid và alcol béo.
Kem D/N: Pha nội thân dầu, pha ngoại (pha liên tục) thân nước. Trong thành phần có các chất nhũ hóa tạo nhũ tương D/N như: Xà phòng kiềm hóa trị một (natri, kali), xà phòng amin (mono, di và triethanolamin), alcol béo sulfat, polysorbat (Tween), ether hoặc este của acid béo với polyethylen glycol.
Gel
Gel bôi da và niêm mạc là những chế phẩm thể chất mềm, sử dụng tá dược tạo gel thích hợp.
Gel thân dầu (oleogels): Trong thành phần sử dụng tá dược tạo gel, bao gồm dầu parafin phối hợp với tá dược thân dầu khác, có thêm keo silic, xà phòng nhôm hoặc xà phòng kẽm.
Gel thân nước (hydrogels): Thành phần bao gồm nước, glycerin, propylen glycol, có thêm các tá dược tạo gel như polysacarid (tinh bột, tinh bột biến tính, acid alginic và natri alginat), dẫn chất cellulose, polymer của acid acrylic (carbomer, carbomer copolymer, carbomer interpolymer, methyl acrylat) và các chất vô cơ (magnesi - nhôm silicat).
1.13 THUỐC NANG
Capsulae
Định nghĩa
Thuốc nang là dạng thuốc uống chứa một hay nhiều dược chất trong vỏ nang với nhiều hình dạng và kích thước khác nhau. Vỏ nang được làm chủ yếu từ gelatin hoặc polyme như HPMC... Ngoài ra trong vỏ nang còn chứa các tá dược khác như chất hóa dẻo, chất màu, chất bảo quản...
Thuốc chứa trong nang có thể là dạng rắn (bột, cốm, pellet...) hay lỏng, nửa rắn (hỗn dịch, nhũ tương, bột nhão...).
Yêu cầu chất lượng chung
Độ đồng đều hàm lượng (Phụ lục 11.2)
Nếu không có các chỉ dẫn khác, yêu cầu này áp dụng cho các thuốc nang có chứa một hoặc nhiều dược chất, trong đó có các dược chất có hàm lượng dưới 2 mg hoặc dưới 2 % (kl/kl) so với khối lượng thuốc trong 1 nang. Đối với nang có từ 2 dược chất trở lên, chỉ thử độ đồng đều hàm lượng với thành phần nào có hàm lượng nhỏ như quy định ở trên.
Độ đồng đều khối lượng (Phụ lục 11.3)
Nếu phép thử độ đồng đều hàm lượng đã được tiến hành với tất cả các dược chất có trong nang thì không phải thử độ đồng đều khối lượng.
Định tính, định lượng và các yêu cầu kỹ thuật khác
Theo quy định trong chuyên luận riêng.
Độ hòa tan (Phụ lục 11.4)
Các thuốc nang có yêu cầu thử độ hòa tan sẽ có quy định cụ thể trong chuyên luận riêng. Không yêu cầu thử độ rã đối với thuốc nang đã thử độ hòa tan.
Bảo quản
Bảo quản trong bao bì kín, nhiệt độ không quá 30 °C.
Thuốc nang cứng
Vỏ nang cứng gồm nắp nang và thân nang hình trụ lồng khít vào nhau bằng khớp trên vỏ nang hoặc được hàn kín sau khi đóng thuốc. Thuốc đóng trong nang thường ở dạng rắn (bột, hạt...). Quá trình chế tạo vỏ và đóng thuốc được thực hiện riêng.
Ngoài các yêu cầu chung của thuốc nang, thuốc nang cứng phải đạt các yêu cầu sau:
Độ rã (Phụ lục 11.6)
Nếu không có chỉ dẫn gì khác, dùng nước làm môi trường thử, thời gian rã phải trong vòng 30 min. Nếu thử trong môi trường nước không đạt, thay nước bằng dung dịch acid hydrocloric 0,1N (TT) hoặc dịch dạ dày giả (TT). Nếu nang nổi trên mặt nước, có thể dùng đĩa khi thử.
Thuốc nang mềm
Thuốc nang mềm là một khối mềm chứa dược chất và tá dược đóng trong vỏ kín với nhiều hình dạng và kích cỡ khác nhau, vỏ nang mềm làm bằng hỗn hợp gelatin, chất hóa dẻo, chất màu, chất bảo quản...
Thuốc đóng trong nang mềm thường ở dạng lỏng, dung dịch, hỗn dịch hay nhũ tương. Quá trình chế tạo vỏ và đóng thuốc được thực hiện đồng thời.
Thuốc nang mềm dùng để uống hoặc để đặt.
Ngoài các yêu cầu chung của thuốc nang, thuốc nang mềm phải đạt các yêu cầu sau:
Độ rã (Phụ lục 11.6)
Nếu không có chỉ dẫn gì khác, dùng nước làm môi trường thử, cho đĩa vào mỗi ống thử, thời gian rã phải trong vòng 30 min. Nếu thử trong môi trường nước không đạt, thay nước bằng dung dịch acid hydrocloric 0,1 N (TT) hoặc dịch dạ dày giả (TT).
Nếu dược chất có tương tác với đĩa, có thể thử không dùng đĩa.
Nếu nang không rã do dính vào đĩa, thử lại với 6 viên khác không dùng đĩa. Mẫu thử đạt yêu cầu nếu cả 6 viên đều rã.
Thuốc nang tan trong ruột
Thuốc nang tan trong ruột là các nang cứng hay nang mềm có vỏ nang hoặc hạt đóng nang rã trong dịch ruột. Đối với các thuốc nang đóng hạt được bao tan ở ruột phải tiến hành thử độ hòa tan để kiểm tra sự giải phóng dược chất theo Phụ lục 11.4 và theo chuyên luận riêng.
Ngoài các yêu cầu chung của thuốc nang, thuốc nang tan trong ruột phải đáp ứng được các yêu cầu:
Độ rã
Đối với thuốc nang có vỏ nang bền với dịch dạ dày phải thử độ rã (Phụ lục 11.6). Dùng dung dịch acid hydrocloric 0,1 N (TT) làm môi trường thử. Không dùng đĩa, nếu không có chỉ dẫn gì khác, vận hành thiết bị thử trong 2 h. Không có nang nào được rã hoặc nứt vỡ làm thuốc trong nang lọt ra ngoài.
Thay dung dịch acid bằng dung dịch đệm phosphat pH 6,8. Cho đĩa vào mỗi ống thử, vận hành thiết bị thử trong 60 min. Kiểm tra từng nang, cả 6 nang phải rã hết. Nếu nang không rã do dính vào đĩa, thử lại với 6 viên khác không dùng đĩa. Mẫu thử đạt yêu cầu nếu cả 6 viên đều rã.
Thuốc nang tác dụng kéo dài
Thuốc nang tác dụng kéo dài là thuốc nang cứng hay nang mềm, trong đó vỏ nang hay thuốc trong nang (hoặc cả 2) được bào chế để dược chất giải phóng kéo dài. Ngoài các yêu cầu chung của thuốc nang, thuốc nang tác dụng kéo dài phải thử độ hòa tan theo yêu cầu quy định trong chuyên luận riêng.
1.14 THUỐC NHỎ MẮT
Collyria
Định nghĩa
Thuốc nhỏ mắt là dung dịch nước, dung dịch dầu hoặc hỗn dịch vô khuẩn của một hay nhiều hoạt chất, dùng để nhỏ vào mắt. Chế phẩm cũng có thể được bào chế dưới dạng khô (bột, bột đông khô, viên nén) vô khuẩn, được hòa tan hoặc phân tán vào một chất lỏng vô khuẩn thích hợp khi dùng.
Yêu cầu chung
Thuốc nhỏ mắt phải được pha chế - sản xuất trong điều kiện vô khuẩn. Các dụng cụ, thiết bị và đồ đựng dùng trong pha chế - sản xuất phải sạch và vô khuẩn.
Dung môi để pha chế thuốc nhỏ mắt thường là nước tinh khiết hoặc các dung dịch nước thích hợp hoặc là dầu thực vật trung tính đạt tiêu chuẩn để pha thuốc tiêm.
Trong thành phần của thuốc nhỏ mắt có thể có thêm các tá dược, để điều chỉnh độ đẳng trương, độ nhớt, điều chỉnh hay ổn định pH của chế phẩm, tăng độ tan và độ ổn định của hoạt chất, nhưng không được ảnh hưởng xấu đến tác dụng của thuốc và không gây kích ứng đối với mắt ở nồng độ sử dụng trong chế phẩm.
Những chế phẩm thuốc nhỏ mắt nước đóng nhiều liều trong một đơn vị đóng gói phải cho thêm chất sát khuẩn với nồng độ thích hợp, trừ khi tự chế phẩm có đủ tính chất sát khuẩn. Chất sát khuẩn phải không tương kỵ với các thành phần khác có trong chế phẩm và phải duy trì được hiệu quả sát khuẩn trong thời gian sử dụng chế phẩm kể từ lần mở nắp đầu tiên.
Không được thêm chất sát khuẩn hoặc chất chống oxy hóa vào các thuốc nhỏ mắt dùng cho phẫu thuật ở mắt. Các thuốc nhỏ mắt này phải pha chế - sản xuất trong điều kiện vô khuẩn và đóng gói một liều.
Không được cho thêm chất màu vào thuốc nhỏ mắt chỉ với mục đích nhuộm màu chế phẩm.
Đồ đựng thuốc nhỏ mắt nên có đủ độ trong cần thiết để kiểm tra được bằng mắt độ trong của dung dịch hay độ đồng nhất của hỗn dịch nhỏ mắt chứa trong đó. Đồ đựng thuốc nhỏ mắt phải vô khuẩn và không có tương tác về mặt vật lý hay hóa học với thuốc. Đồ đựng thuốc nhỏ mắt phải đáp ứng các yêu cầu nêu trong Phụ lục 17.3.3. Đồ đựng thuốc nhỏ mắt chứa nhiều liều phải có bộ phận nhỏ giọt thích hợp, thể tích mỗi đơn vị đóng gói không nên vượt quá 10 ml.
Ghi nhãn: Nhãn thuốc nhỏ mắt phải ghi theo quy chế. Nhãn phải ghi tên chất sát khuẩn có trong chế phẩm. Đối với đơn vị đóng gói nhiều liều, trên nhãn phải ghi rõ thời hạn sử dụng tính từ khi thuốc được sử dụng lần đầu, sau thời gian đó thuốc còn lại phải bỏ đi, thường không quá 4 tuần, trừ khi có chỉ dẫn khác.
Yêu cầu chất lượng
Độ trong (Thử theo Phụ lục 11.8. Phần B)
Dung dịch thuốc nhỏ mắt phải trong suốt, không có các tiểu phân quan sát được bằng mắt thường
Hỗn dịch nhỏ mắt có thể lắng đọng khi để yên nhưng phải dễ dàng phân tán đồng nhất khi lắc và phải duy trì được sự phân tán đồng nhất đó trong khi nhỏ thuốc để sử dụng đúng liều.
Kích thước tiểu phân (Thử theo Phụ lục 11.8. Phần A)
Nếu không có chỉ dẫn khác, thuốc nhỏ mắt dạng hỗn dịch phải đạt yêu cầu của phép thử sau: Lắc mạnh và chuyển một lượng chế phẩm tương đương với khoảng 10 μg pha rắn vào buồng đếm hoặc lên một phiến kính thích hợp và quan sát dưới kính hiển vi có độ phóng đại thích hợp. Không được có quá 20 tiểu phân có kích thước lớn hơn 25 μm và không có quá 2 tiểu phân có kích thước lớn hơn 50 μm, không có tiểu phân nào có kích thước lớn hơn 90 μm.
Thử vô khuẩn
Đạt yêu cầu Thử vô khuẩn (Phụ lục 13.7).
Giới hạn cho phép về thể tích
+ 10 % (Phụ lục 11.1).
Các yêu cầu kỹ thuật khác
Thử theo quy định trong chuyên luận riêng.
Đối với dạng chế phẩm khô, dùng để pha thuốc nhỏ mắt trước khi dùng, sau khi pha phải đáp ứng các yêu cầu chất lượng của thuốc nhỏ mắt. Đối với chế phẩm đóng liều đơn phải đáp ứng các yêu cầu về phép thử độ đồng đều hàm lượng hoặc độ đồng đều khối lượng (Phụ lục 11.2 hoặc 11.3), trừ khi có chỉ dẫn khác.
Đối với các dung dịch dùng để rửa mắt, ngâm mắt hoặc để thấm vào băng mắt, nhất thiết phải là các dung dịch đẳng trương với dịch nước mắt và phải đáp ứng các yêu cầu chất lượng của thuốc nhỏ mắt. Thuốc rửa mắt dùng trong phẫu thuật hoặc trong điều trị sơ cứu về mắt, không được chứa chất sát khuẩn, phải pha chế vô khuẩn và đóng gói một liều. Thuốc rửa mắt đóng nhiều liều phải có chất sát khuẩn ở nồng độ thích hợp và không đóng gói quá thể tích 200 ml cho một đơn vị đóng gói nhỏ nhất. Nhãn thuốc theo quy định hiện hành và có ghi thời hạn sử dụng sau khi mở nắp.
1.15 THUỐC NHỎ MŨI VÀ THUỐC XỊT MŨI DẠNG LỎNG
Định nghĩa
Thuốc nhỏ mũi và thuốc xịt mũi dạng lỏng là các dung dịch, nhũ tương hay hỗn dịch dùng để nhỏ hoặc bơm xịt vào trong hốc mũi để gây tác dụng tại chỗ hay toàn thân.
Thuốc nhỏ mũi thường được đong vào lọ chứa đa liều có gắn bộ phận nhỏ giọt thích hợp. Thuốc xịt mũi dạng lỏng được đóng vào trong lọ chứa có gắn bộ phận phun hoặc đóng vào lọ dưới áp suất cao có hoặc không có van phân liều. Kích thước của các giọt thuốc khi phun ra phải cho phép thuốc bám được vào trong hốc mũi.
Phương pháp điều chế
Điều chế theo phương pháp thích hợp tùy theo thuốc là dung dịch, hỗn dịch hay nhũ tương.
Các chế phẩm có chất dẫn là nước thường phải đẳng trương và có thể cho thêm tá dược để điều chỉnh độ nhớt, điều chỉnh hay ổn định pH, để tăng tính tan của hoạt chất hay để ổn định chế phẩm. Trong sản xuất, đóng gói, tồn trữ và phân phối, phải có những biện pháp thích hợp để đảm bảo yêu cầu về giới hạn vi sinh vật. Đối với các chế phẩm vô khuẩn, phải điều chế với các nguyên liệu và phương pháp thích hợp để đảm bảo tính vô khuẩn, tránh ô nhiễm và ngăn cản sự phát triển của các vi sinh vật.
Thuốc nhỏ mũi đóng trong lọ chứa đa liều và thuốc xịt mũi có sử dụng chất dẫn là nước, phải cho thêm chất bảo quản ở nồng độ thích hợp trừ trường hợp bản thân chế phẩm có tính sát khuẩn.
Yêu cầu chất lượng
Đạt yêu cầu chất lượng theo các chuyên luận riêng.
Chế phẩm không được gây kích ứng và gây ảnh hưởng đến chức năng của màng nhày và hệ lông trong mũi.
Khi bảo quản, các chế phẩm dạng nhũ tương có thể có hiện tượng tách pha; dạng hỗn dịch có thể có hiện tượng lắng cặn xuống đáy lọ nhưng phải dễ dàng phân tán đều trở lại khi lắc để phân chia liều được chính xác.
Chế phẩm dạng hỗn dịch phải kiểm soát kích thước thích hợp của các tiểu phân tùy theo mục đích sử dụng.
Đồ đựng thuốc nhỏ mũi phải đáp ứng các yêu cầu nêu trong Phụ lục 17.1 hoặc 17.3.
Trừ khi có chỉ dẫn khác, thuốc nhỏ mũi được cung cấp dưới dạng đơn liều và các thuốc xịt mũi phân liều có tác động toàn thân, phải đáp ứng các yêu cầu của các thử nghiệm sau:
Độ đồng đều khối lượng
Thuốc nhỏ mũi đơn liều dạng dung dịch phải đáp ứng yêu cầu của thử nghiệm sau:
Cân riêng từng lượng thuốc được lấy cẩn thận ra từ 10 lọ và xác định khối lượng trung bình thuốc trong lọ. Không được có quá 2 đơn vị lệch hơn 10 % và không có đơn vị nào lệch hơn 20 % so với khối lượng trung bình của thuốc.
Thuốc xịt mũi phân liều dạng dung dịch phải đáp ứng yêu cầu của thử nghiệm sau:
Xịt bỏ liều đầu tiên, đợi ít nhất 5 s, xịt bỏ tiếp một lần nữa. Lặp lại quy trình này 3 lần nữa. Cân khối lượng của lọ thuốc, xịt bỏ một liều và cân khối lượng còn lại của lọ thuốc. Tính chênh lệch giữa hai khối lượng. Lặp lại quy trình như trên với 9 lọ thuốc xịt khác. Chế phẩm đạt yêu cầu của thử nghiệm nếu không có quá 2 giá trị lệch hơn 25 % và không có giá trị nào lệch hơn 35 % so với giá trị trung bình.
Độ đồng đều hàm lượng
Thuốc nhỏ mũi đơn liều dạng hỗn dịch hoặc nhũ tương phải đáp ứng thử nghiệm sau:
Lấy hết thuốc trong mỗi lọ ra và xác định hàm lượng riêng của thuốc trong từng lọ. Chế phẩm phải đáp ứng yêu cầu của Phụ lục 11.2, phương pháp 1.
Độ đồng đều phân liều
Thuốc xịt mũi phân liều dạng hỗn dịch hay nhũ tương phải đáp ứng theo thử nghiệm sau:
Dùng một dụng cụ có khả năng giữ lại số lượng thuốc vừa phun ra khỏi bộ phận phun. Lắc lọ thuốc trong 5 s và xịt bỏ liều đầu tiên, đợi ít nhất 5 s, lắc trong 5 s và xịt bỏ tiếp một lần nữa. Lặp lại quy trình này 3 lần nữa. Sau 2 s, xịt một liều thuốc vào trong dụng cụ hứng. Rửa dụng cụ hứng nhiều lần để thu hồi lại liều thuốc. Xác định hàm lượng của hoạt chất trong nước rửa. Lặp lại quy trình như trên với 9 lọ thuốc khác. Nếu không có chỉ dẫn khác, chế phẩm đáp ứng thử nghiệm nếu không có quá một liều vượt ngoài giới hạn 75 % đến 125 % và không có giá trị nào vượt ngoài giới hạn 65 % đến 135 % so với giá trị trung bình của 10 liều (lấy từ 10 lọ). Nếu có 2 hoặc 3 giá trị vượt qua giới hạn 75 % đến 125 % nhưng vẫn nằm trong giới hạn 65 % đến 135 %, lặp lại thử nghiệm với 20 lọ thuốc nữa. Chế phẩm đáp ứng thử nghiệm nếu không có quá 3 trong số 30 lọ vượt ra ngoài giới hạn 75 % đến 125 % và không có giá trị nào vượt ngoài giới hạn 65 % đến 135 % so với giá trị trung bình của 30 lọ.
Thể tích
+ 10 % thể tích ghi trên nhãn (Phụ lục 11.1).
Giới hạn nhiễm khuẩn
Đạt yêu cầu quy định trong Phụ lục 13.6.
Bảo quản
Đối với các thuốc vô khuẩn, bảo quản thuốc trong chai lọ vô khuẩn, kín, có niêm bảo đảm.
Các thuốc xịt mũi được đóng vào các lọ dưới áp suất cao phải được để nơi mát, xa nguồn nhiệt.
Nhãn thuốc theo quy định hiện hành và có ghi tên, nồng độ (hàm lượng) của chất bảo quản và thời hạn sử dụng sau khi mở nắp.
1.16 THUỐC NHỎ TAI VÀ THUỐC XỊT VÀO TAI
Định nghĩa
Thuốc nhỏ tai và thuốc xịt vào tai là các dung dịch, hỗn dịch hay nhũ tương của một hoặc nhiều dược chất trong các chất lỏng thích hợp (như nước, các glycol hoặc dầu béo) để đưa vào trong hốc tai nhưng không được gây ra áp lực có hại lên màng nhĩ. Thuốc cũng có thể dùng bôi vào hốc tai nhờ một que bông được tẩm thuốc.
Thuốc nhỏ tai thường được đóng gói trong các lọ chứa đa liều làm bằng thủy tinh hay chất dẻo thích hợp có gắn hoặc kèm theo một bộ phận thích hợp để nhỏ giọt. Các thuốc xịt vào tai thường được đóng gói trong các lọ chứa đa liều gắn theo một bộ phận để phun thuốc. Các bộ phận này được thiết kế sao cho tránh sự gây nhiễm.
Phương pháp điều chế
Điều chế theo phương pháp thích hợp tùy theo thuốc là dung dịch, hỗn dịch hay nhũ tương.
Có thể cho thêm các tá dược để điều chỉnh độ đẳng trương, độ nhớt, điều chỉnh hay ổn định pH, tăng tính tan của dược chất hoặc ổn định chế phẩm. Các tá dược này không được làm giảm tác dụng trị liệu của thuốc hay gây độc tính hoặc gây kích ứng tại chỗ ở nồng độ sử dụng.
Trừ khi có chỉ dẫn khác, các thuốc nhỏ tai và thuốc xịt có chất dẫn là nước khi được đóng gói trong lọ chứa đa liều phải được cho thêm vào các chất bảo quản ở nồng độ thích hợp trừ khi bản thân chế phẩm có tính sát khuẩn.
Thuốc dùng để điều trị tai bị tổn thương, đặc biệt khi màng nhĩ bị thủng hay để sử dụng trước khi phẫu thuật, phải vô khuẩn, không được chứa các chất bảo quản và được cung cấp trong lọ chứa đơn liều. Khi đó thuốc được điều chế với các nguyên liệu và phương pháp thích hợp để đảm bảo tính vô khuẩn, tránh ô nhiễm và tránh sự phát triển của các vi sinh vật.
Yêu cầu chất lượng
Đạt yêu cầu chất lượng theo các chuyên luận riêng.
Khi bảo quản, chế phẩm dạng nhũ tương có thể có hiện tượng tách pha nhưng phải dễ dàng phân tán đều trở lại khi lắc.
Chế phẩm dạng hỗn dịch khi bảo quản có thể có hiện tượng lắng cặn nhưng phải dễ dàng phân tán đều trở lại khi lắc và duy trì tình trạng ổn định này đủ để cho phép phân liều chính xác. Có biện pháp kiểm soát để đảm bảo kích thước tiểu phân phù hợp tùy theo mục đích sử dụng.
Đồ đựng thuốc nhỏ mũi phải đáp ứng các yêu cầu nêu trong Phụ lục 17.1 hoặc 17.3.
Độ đồng đều hàm lượng
Trừ khi có chỉ dẫn riêng, các chế phẩm đơn liều có hàm lượng dược chất dưới 2 mg hay chiếm dưới 2 % khối lượng của đơn vị phân liều và các thuốc xịt phân liều phải đáp ứng yêu cầu của Phụ lục 11.2 Phép thử độ đồng đều hàm lượng, phương pháp 1. Nếu thuốc có nhiều dược chất, yêu cầu này được áp dụng cho thành phần có các điều kiện như trên.
Độ đồng đều khối lượng
Các chế phẩm đơn liều phải đạt yêu cầu của Phụ lục 11.3 Phép thử độ đồng đều khối lượng. Nếu đã thử độ đồng đều hàm lượng của tất cả các dược chất thì không phải thử độ đồng đều khối lượng.
Giới hạn nhiễm khuẩn
Đạt yêu cầu quy định trong Phụ lục 13.6.
Nếu có yêu cầu vô khuẩn, thuốc phải đạt các yêu cầu của thử nghiệm về Thử vô khuẩn (Phụ lục 13.7) và tính chất này phải được ghi trên nhãn thuốc.
Thể tích
+ 10 % thể tích ghi trên nhãn (Phụ lục 11.1).
Bảo quản
Đối với các thuốc vô khuẩn, bảo quản thuốc trong chai lọ vô khuẩn, kín.
Thuốc xịt được đóng gói trong các lọ chứa đóng dưới áp suất cao được bảo quản nơi mát, xa nguồn nhiệt.
Nhãn thuốc theo quy định hiện hành và có ghi tên của chất bảo quản và thời hạn sử dụng sau khi mở nắp (thường không quá 4 tuần trừ khi có chỉ dẫn khác).
1.17 THUỐC HÍT
Inhalationis
Định nghĩa
Thuốc hít là dạng bào chế rắn hoặc lỏng, đóng gói kín, khi dùng thuốc sẽ bay hơi, thăng hoa trong không khí hoặc do khí đầy tạo ra những hạt thuốc mịn phân tán trong khí để hít vào đường hô hấp, có tác dụng tại chỗ hoặc toàn thân.
Thuốc hít chứa một hoặc nhiều hoạt chất hòa tan trong dung môi hoặc phân tán đều trong khối thuốc và các tá dược thích hợp như chất dẫn, chất sát trùng bảo quản, chất ổn định,... Các tá dược phải đảm bảo không ảnh hưởng tới chức năng hoặc gây tổn thương, kích ứng niêm mạc đường hô hấp.
Phân loại
Theo trạng thái: Thuốc hít có thể dưới dạng lỏng hoặc rắn.
Dạng lỏng như dung dịch tinh dầu dễ bay hơi hoặc dung dịch thuốc đóng trong bao bì chai, lọ được nạp khí để đẩy thuốc qua van kiểu khí dung hoặc dùng thuốc với dụng cụ thích hợp.
Dạng rắn là các bột để hít thường đóng dạng khí dung ở áp suất cao hoặc hít bởi các dụng cụ chuyên khoa mũi họng hoặc dạng khối rắn của hoạt chất dễ bay hơi, thăng hoa trong ống hít.
Theo dạng bào chế: Ở dạng bào chế hoàn chỉnh có thuốc khí dung để hít và ống hít.
Thuốc khí dung để hít: Dạng thuốc đóng gói dưới áp suất cao dùng qua đường hô hấp bằng cách hít vào miệng hoặc mũi. Hoạt chất trong thuốc có thể ở dạng dung dịch, hỗn dịch, hoặc nhũ tương. Nếu thuốc ở dạng hỗn dịch hoặc nhũ tương, cần lắc đều trước khi dùng. Thuốc có thể đóng gói với van phân liều hoạc không phân liều.
Tùy theo yêu cầu trị liệu mà hạt khí dung có độ mịn phù hợp. Riêng khí dung dùng để điều trị bệnh phổi có các hoạt chất kháng sinh, kháng viêm,... kích thước hạt phải nhỏ hơn 5 μm. Mặt khác, áp lực khí đẩy phải đảm bảo đưa thuốc tới được vị trí thuốc cần tác dụng, đồng thời phù hợp với đặc điểm sinh lý đường hô hấp và an toàn cho người sử dụng.
Thuốc ống hít: Dùng cho các hoạt chất dễ bay hơi, hay thăng hoa thành khí: Tinh dầu bạc hà, camphor, menthol, eucalytol, v.v... hoặc hoạt chất được hòa tan, phân tán đều trong tá dược dầu sáp, dung môi và trộn hoặc thấm vào các vật liệu thích hợp tạo thành khối xốp, đặt trong ống để hít.
Cấu tạo của ống hít gồm: Phần thân ống có đáy kín, chứa khối thuốc xốp và phía trên để rỗng chứa thuốc ở trạng thái khí. Phần đầu trên của ống có hình dạng thích hợp và đục lỗ để hít qua mũi. Nắp bảo vệ để thuốc không thoát ra khỏi ống khi bảo quản.
Bảo quản: cần bảo quản ống hít nơi khô, mát, tránh ánh sáng.
Các thuốc khác để xông hít:
Hộp chứa bột thuốc để hít: Hộp có ngăn chứa một liều thuốc ở dạng bột mịn và nối với một đầu để hít vào miệng. Hộp mang nhiều ngăn và có thể xoay các ngăn luân phiên cho mỗi lần sử dụng.
Nang thuốc để hít: Thuốc được cấp kèm theo một dụng cụ có bộ phận nghiền thuốc thành bột mịn để hít.
Dung dịch thuốc để xông hít: Thuốc được cấp để hít trên một máy hoặc dụng cụ phân tán thuốc thành thể hạt mịn trong khí để xông hít. Máy tạo hạt để hít từ dung dịch thuốc bằng rung động siêu âm hoặc kiểu máy nén khí qua đầu phun thuốc; máy xông hơi nước dùng nhiệt độ,... Ngoài ra, dụng cụ gây mê hồi sức cũng áp dụng kiểu trị liệu tương tự như thuốc hít.
Phương pháp sản xuất
Các dạng thuốc để xông hít nhất là thuốc hít theo đường hô hấp phải chọn lựa tá dược để phù hợp không gây kích ứng niêm mạc hô hấp, không gây nguy hiểm cho bệnh nhân. Các thuốc lỏng phải chọn pH trong khoảng 3,0 đến 8,5 vừa phù hợp sinh lý vừa giúp ổn định hoạt chất. Thuốc có chứa chất sát trùng bảo quản phải chứng tỏ hiệu lực ổn định theo quy định.
Đồ đựng của thuốc hít phải được lựa chọn theo quy định chung.
Tiêu chuẩn chất lượng
Đạt yêu cầu chất lượng theo các chuyên luận riêng.
Khí dung để hít đóng dưới áp suất khí đẩy phải đáp ứng các yêu cầu chung của thuốc khí dung, theo Phụ lục 1.18.
Các thuốc nếu bào chế để dùng kèm theo các y cụ để xông hít thuốc phải đáp ứng các quy định hiện hành về thuốc xông hít của ngành dược. Các y cụ để xông hít thuốc phải đáp ứng các quy định chung hiện hành về trang thiết bị y tế.
Nhãn thuốc
Nhãn thuốc theo quy định hiện hành.
Nhãn có các nội dung về liều lượng nếu ở dạng khí dung và các quy định khác theo từng chuyên luận riêng.
1.18 THUỐC KHÍ DUNG
Aerosolum
Thuốc khí dung là dạng bào chế mà trong quá trình sử dụng, hoạt chất được phân tán thành những hạt nhỏ trong không khí do thuốc được nén qua đầu phun bởi một luồng khí đẩy ở áp suất cao để tới vị trí tác dụng.
Thuốc khí dung có thể dùng ngoài da, tóc, mũi - họng, răng miệng hoặc tai,... và hay dùng để hít theo đường hô hấp có tác dụng tại chỗ hoặc tác dụng toàn thân.
Các dạng thuốc khí dung
Thuốc khí dung hoàn chỉnh: Dạng thuốc này là một hệ thống gồm: Thuốc, chai, lọ kín chứa thuốc có gắn đầu phun cùng với van và khí đẩy được nén ở áp suất thích hợp. Khi nhấn đầu phun, thuốc sẽ tự động đẩy ra khỏi đầu van. Van có thể là loại không phân liều, nhưng nếu cần thiết thuốc phải gắn van phân liều chính xác.
Thuốc khí dung khi đóng trong đồ đựng thường ở thể lỏng như: Dung dịch, hỗn dịch, nhũ tương. Đồ đựng thuốc có thể là chai, lọ hoặc bình được chế tạo đặc biệt để có thể gắn kết với các bộ phận gồm ống dẫn thuốc, van, đầu phun và nắp đậy. Đồ đựng thuốc được chế bằng vật liệu thích hợp như thủy tinh, nhựa, kim loại hoặc phối hợp. Khi đẩy có chức năng nén thuốc qua đầu phun và lượng khí nén đóng trong thuốc khí dung phải đảm bảo đủ để đẩy hết liều lượng thuốc theo chỉ định. Khí đẩy thường dùng là hỗn hợp các loại khí như khí trơ carbonic, nitơ hoặc khí hydrocarbon và dẫn chất halogen của hydrocarbon. Hai nhóm sau thường dùng ở dạng khí nén hóa lỏng.
Thuốc khí dung hoàn chỉnh được sản xuất công nghiệp, tuy nhiên, kiểu đóng thuốc dưới áp lực cao của khí đẩy còn gặp ở một số chế phẩm không phải là thuốc khí dung như thuốc bọt, thuốc lỏng để xoa da hoặc sirô thuốc và mỹ phẩm...
Thuốc khí dung kiểu piston: Thuốc này không nén sẵn khí đẩy như thuốc khí dung hoàn chỉnh, mà gắn van kiểu piston để người dùng tự bơm nén không khí để đẩy thuốc. Khi nhấn, piston hoạt động như van một chiều chỉ cho khí đi vào, sau vài lần ấn áp suất đạt tới mức nhất định, van mở cho thuốc phun ra. Tiếp tục nhấn piston, chu kỳ phun thuốc được lặp lại. Đây là một dạng thuốc khí dung chưa hoàn chỉnh.
Thuốc khí dung dùng quả bóp: Thuốc đựng trong đồ đựng riêng, khi dùng thuốc được cho vào một đầu phun có gắn quả bóp (bằng nhựa hoặc cao su). Khi bóp, không khí sẽ nén với áp lực đủ để đẩy thuốc ra khỏi đầu phun. Đầu phun có hình dạng khác nhau, tùy trường hợp cho thuốc qua miệng hoặc mũi... Đầu phun này là dụng cụ tạo khí dung cho cá nhân hay được dùng trong bệnh viện bằng cách nối chung với máy nén khí trị liệu khí dung.
Ngoài các dạng bào chế trên, còn có thể dùng dụng cụ, thiết bị khác như máy rung động siêu âm, máy dùng điện thế tạo ra thuốc khí dung để xông hít.
Kỹ thuật và điều kiện sản xuất
Kỹ thuật sản xuất thuốc khí dung phải đảm bảo cho hoạt chất ổn định trong thời hạn bảo quản. Các hoạt chất kém ổn định: Oxytetracyclin, hydrocortison, streptomycin, rifampicin,... phải sản xuất ở trạng thái khí dung khô, trong thành phần của thuốc không được chứa nước. Nếu hoạt chất ổn định với dung môi nước thì có thể bào chế thuốc ở thể dung dịch nhũ tương hoặc hỗn dịch.
Kích thước của các hạt mang hoạt chất trong thuốc khí dung phải đủ mịn và phân tán đều khi được đẩy ra khỏi đầu phun. Với khí dung trị bệnh phổi thường có hoạt chất kháng sinh, kháng viêm,... và kích thước hạt phải nhỏ hơn 5 μm. Mặt khác, áp lực khí đẩy của chế phẩm phải đảm bảo đưa thuốc tới được bề mặt vị trí thuốc cần tác dụng, đồng thời phù hợp với đặc điểm sinh lý đường hô hấp và an toàn cho người sử dụng.
Thuốc khí dung phải được sản xuất trong điều kiện đảm bảo chất lượng chung và phù hợp với đường sử dụng thuốc; phải kiểm tra giới hạn vi sinh vật (Phụ lục 13.6 Thử giới hạn nhiễm khuẩn). Thuốc sử dụng cho vết thương phải vô khuẩn (Phụ lục 13.7 Thử vô khuẩn). Chất liệu của đồ đựng thuốc phải lựa chọn đúng quy định.
Tiêu chuẩn chất lượng của thuốc thành phẩm
Đạt yêu cầu chất lượng theo các chuyên luận riêng.
Tiêu chuẩn áp dụng cho thuốc khí dung hoàn chỉnh gồm: Áp suất khí nén, cỡ hạt và phân bố cỡ hạt khi phun thuốc, khả năng phân liều của van, tốc độ phun. Đặc tính an toàn: Điểm bắt lửa của thuốc khi phun, khả năng chịu áp lực của bình, độ kín của bao bì.
Nếu dung môi dùng trong khí dung có chứa ethanol thì phải xây dựng tiêu chuẩn và phương pháp định lượng hàm lượng ethanol trong thuốc.
Thuốc thành phẩm phải đáp ứng các tiêu chuẩn chung về: Nồng độ, hàm lượng hoạt chất,...
Ghi nhãn theo quy định và có tên các chất khí đẩy, các chất phụ.
Đồ đựng
Đồ dựng thuốc khí dung cần có độ bền vững không ảnh hưởng tới hoạt chất và các thành phần của thuốc, đồng thời phải an toàn cho sản xuất và sử dụng. Phải chịu được áp lực cao của khí đẩy. Vật liệu có thể bằng thủy tinh, kim loại, nhựa hoặc phối hợp các vật liệu này.
Đồ đựng bằng thủy tinh: Nên dùng loại trung tính và có thể được bọc nhựa dẻo ở mặt ngoài.
Đồ đựng bằng kim loại: Phải dùng loại không rỉ như nhôm, thiếc và có thể tráng các lớp vecni bảo vệ bề mặt trong của bình hay chai lọ nhất là bình bằng nhôm.
Đồ đựng bằng nhựa dẻo: Như polyethylen PE, poly-propylen PP,...
Đồ đựng thuốc trước khi đưa vào quy trình sản xuất cần phải xử lý theo quy định. Riêng mặt trong của đồ đựng phải kiểm soát để giảm thiểu bụi, vi sinh vật theo yêu cầu và không chứa tạp chất bôi trơn khuôn đúc đồ đựng hoặc từ chất tẩy rửa.
Van và ống dẫn thuốc
Van giữ cho bình kín dưới áp suất cao và phun thuốc với lượng quy định. Có 2 loại van: Không phân liều và phân liều.
Van không phân liều: Van cho thuốc phun liên tục khi nhấn lên van. Thuốc sẽ phun những lượng phụ thuộc vào thời gian nhấn van, thường vài giây, song có thể kéo dài hơn nếu cần.
Van phân liều: Van chỉ phun những liều thuốc chính xác, không phun liên tục, mà theo từng liều. Liều thuốc thường tính theo thể tích thuốc lỏng tương đương với lượng hoạt chất đáp ứng yêu cầu trị liệu. Các van có thể được chế tạo để phun thuốc theo thế thẳng đứng hoặc dốc ngược bao bì để hít thuốc qua mũi hay miệng.
Ống nhúng: Là một ống nhỏ nhúng trong thuốc và nối với van. Ống nhúng có chiều dài phù hợp với chiều cao của bao bì và trạng thái tập hợp của thuốc. Đường kính trong của ống nhúng phải phù hợp với kích thước hạt và độ nhớt của thuốc, ống nhúng và thân van thường xuyên tiếp xúc với thuốc nên ngoài đặc tính bền hóa lý, không độc, còn phải có bề mặt trong nhẵn, trơn, không bị đọng thuốc, nghẹt thuốc trong sử dụng. Nhựa polyethylen PE, polypropylen PP và nylon hay được dùng.
Đầu phun, nút bấm và nắp bảo vệ
Đầu phun là một ống dẫn thuốc có lỗ nhỏ với cấu tạo đặc biệt, gắn liền với van để đưa thuốc ra khỏi van. Đầu phun thuốc có thể theo thế thẳng đứng hay nằm ngang và có hình dạng, kích cỡ phù hợp với cơ quan đưa thuốc tới như miệng, mũi, tai, để hít hoặc dùng ngoài da.
Nút bấm là bộ phận gắn liền trong hệ thống van - đầu phun, khi ấn hay di chuyển vị trí sẽ đẩy van về vị trí mở cho thuốc phát ra khỏi đầu phun. Đầu phun và nút bấm thường được chế tạo liền một khối để dễ sử dụng, nhưng đầu phun cũng có thể tháo rời, khi dùng mới lắp.
Nắp bảo vệ có chức năng bảo vệ giữ đầu phun khỏi biến dạng và luôn sạch trong sử dụng.
Độ bền và độ an toàn sinh học của đồ đựng
Đồ đựng thuốc cần có độ bền vững không ảnh hưởng tới hoạt chất, đặc biệt không giải phóng những thành phần độc hại khi thuốc tiếp xúc với da, niêm mạc đường hô hấp. Các bộ phận của đồ đựng tiếp xúc trực tiếp với thuốc như: Chai, lọ hoặc ống dẫn thuốc, van bằng nhựa, vòng đệm bằng cao su hoặc bao bì có lớp tráng bảo vệ bên trong cần phải chọn lựa kỹ và có thử nghiệm đánh giá độ bền và độ an toàn sinh học. Các chất nhả ra từ cao su lưu hóa hoặc nhựa như các chất đa nhân thơm, nitrosamin, chất chống oxy hóa, các monome dẫn chất từ nhựa dẻo.
Vật liệu chế tạo đồ đựng có bộ phận tiếp xúc trực tiếp với thuốc cần đạt giới hạn các chất trên thông qua những thử nghiệm hóa học thích hợp.
Van và ống dẫn thuốc bằng nhựa phải có đánh giá về an toàn sinh học, tương tự bao bì nhựa đựng thuốc tiêm (Phụ lục 17.3.2. Đồ đựng và nút bằng chất dẻo dùng cho chế phẩm thuốc tiêm).
Khí đẩy
Chức năng: Khí đẩy là thành phần đặc trưng trong thuốc khí dung, chúng có chức năng ép đẩy thuốc và tạo hệ phân tán mịn của thuốc qua đầu phun. Nhưng nhiều trường hợp khí đẩy còn tham gia với vai trò như là dung môi hòa tan hoạt chất, tham gia vào hệ phân tán trong bào chế để hình thành hệ nhũ tương trong khí dung bọt, như nhóm khí hóa lỏng. Khí đẩy có thể đóng sẵn nhưng cũng có thể nén vào khi dùng thuốc. Trong loại khí dung đóng sẵn khí đẩy, lượng khí đẩy được nén vào đồ đựng thuốc tương ứng với trạng thái áp suất dư, sao cho đảm bảo đẩy hết lượng thuốc đóng gói. Đa số thuốc khí dung đóng sẵn khí đẩy có áp suất ở khoảng 2 kg/cm2 đến 7 kg/cm2.
Các loại khí đẩy: Khí đẩy có thể ở trạng thái khí hoặc trạng thái lỏng (khí hóa lỏng). Khí đẩy dùng đơn giản nhất là không khí như trong trường hợp khí dung kiểu piston hoặc loại tương ứng, khi dùng khí mới được bơm vào theo chỉ dẫn. Với trường hợp khí đẩy đóng sẵn thường dùng hỗn hợp các loại khí như khí trơ, hidro carbon và dẫn chất. Nhóm khí trơ: Khí carbonic (CO2), nitơ (N2), nitơ oxyd (N2O). Nhóm khí hóa lỏng: Hydrocarbon và dẫn chất halogenocarbon thường là dẫn chất của butan, pentan. Khí được chọn phải an toàn, phù hợp với thành phần của thuốc và phối hợp ở tỷ lệ thích hợp để đạt được đặc tính về áp suất hơi nén.
Phương pháp sản xuất
Tùy vào kiểu thuốc khí dung hoàn chỉnh hay không hoặc là thiết bị, dụng cụ cung cấp khí dung mà có phương pháp chế tạo phù hợp. Sau đây chỉ nêu phương pháp sản xuất thuốc khí dung hoàn chỉnh chứa khí đẩy ở áp suất cao. Ở dạng thuốc này có thể tiến hành theo một trong hai quy trình: Quy trình tiến hành ở nhiệt độ thường, nén khí đẩy ở áp suất cao và quy trình tiến hành ở nhiệt độ lạnh.
Quy trình tiến hành ở nhiệt độ thường, nén khí đẩy ở áp suất cao: Áp dụng được cho cả hai trạng thái của khí đẩy: Dạng khí hoặc khí hóa lỏng. Các công đoạn của quy trình này gồm: Chuẩn bị nguyên phụ liệu và chuẩn bị bao bì để đựng thuốc. Pha chế thuốc và đóng thuốc vào bao bì sạch. Đặt van và phụ tùng, gắn chặt chúng với bao bì cho kín. Đóng khí đẩy đến áp suất ấn định. Dán nhãn. Nhập kho, bảo quản chế phẩm.
Quy trình tiến hành ở nhiệt độ lạnh: Dùng riêng cho khí hóa lỏng. Tiến hành pha chế thuốc ở nhiệt độ thích hợp nhưng công đoạn đóng khí đẩy phải tiến hành ở nhiệt độ thấp, thường dưới 0 °C. Các công đoạn của quy trình này gồm: Chuẩn bị nguyên phụ liệu và chuẩn bị bao bì để đựng thuốc. Pha chế thuốc và đóng thuốc vào bao bì sạch. Đóng khí đẩy với lượng thích hợp theo phương pháp cân. Đặt van và phụ tùng, gắn chặt chúng với bao bì cho kín. Dán nhãn. Nhập kho, bảo quản chế phẩm.
Quá trình pha chế, sản xuất thuốc khí dung cần phải phải kiểm soát các thông số như khối lượng thuốc, lượng khí đẩy và áp suất, khả năng hoạt động của van, độ kín của bao bì,...
Nhãn thuốc
Nhãn thuốc khí dung thành phẩm phải có nội dung phù hợp với quy định hiện hành. Ngoài ra, nhãn cần có những lưu ý, cảnh báo riêng biệt cần thiết cho người sử dụng ở từng loại thuốc khí dung, chẳng hạn:
Thuốc khí dung không được phun trực tiếp vào mắt hoặc niêm mạc.
Thuốc khí dung dùng riêng để hít, khi hít phải thận trọng, phải thực hiện theo những chỉ dẫn của đơn thuốc hoặc hướng dẫn của thầy thuốc. Không nên tự dùng thuốc khí dung để hít nếu không được chỉ định vì có thể ngạt thở hoặc nguy hiểm đến tính mạng.
Thuốc khí dung là chế phẩm đóng gói khí nén ở áp suất cao. Tuyệt đối không để những loại thuốc này gần lửa hoặc để ở nơi nhiệt độ cao từ 50 °C trở lên. Không đốt, không đè nén, không chọc vật nhọn vào thuốc.
Phải để thuốc khí dung xa tầm tay trẻ em.
Việc sản xuất các thuốc khí dung có sử dụng khí đẩy là các hydrocarbon hoặc dẫn chất halogenohydrocarbon, phải tuân thủ các quy định hiện hành.
1.19 THUỐC TIÊM, THUỐC TIÊM TRUYỀN
Injectiones, infusiones
Định nghĩa
Thuốc tiêm, thuốc tiêm truyền là những chế phẩm thuốc vô khuẩn dùng để tiêm hoặc tiêm truyền vào cơ thể.
Thuốc tiêm, thuốc tiêm truyền được phân thành 3 loại:
• Thuốc tiêm (dung dịch, hỗn dịch hay nhũ tương).
• Thuốc tiêm truyền (dung dịch nước hay nhũ tương dầu trong nước).
• Bột pha tiêm hoặc dung dịch đậm đặc để pha thuốc tiêm hay thuốc tiêm truyền.
A. Quy định chung
Yêu cầu về pha chế thuốc tiêm, thuốc tiêm truyền
Thuốc tiêm, thuốc tiêm truyền được pha chế - sản xuất bằng cách hòa tan, phân tán hoặc nhũ hóa dược chất và các tá dược vào một dung môi hoặc hỗn hợp dung môi hay chất dẫn thích hợp, trong điều kiện tuân thủ đầy đủ các yêu cầu về pha chế - sản xuất các chế phẩm thuốc vô khuẩn, để tránh nhiễm tạp và nhiễm vi sinh vật vào thuốc. Các thuốc tiêm, thuốc tiêm truyền phải được tiệt khuẩn theo phương pháp quy định (Phụ lục 16.1). Trường hợp vô khuẩn bằng cách lọc thì phải sử dụng dụng cụ, thiết bị, đồ đựng đã tiệt khuẩn và pha chế trong điều kiện tuyệt đối vô khuẩn.
Quá trình pha chế một mẻ thuốc tiêm, thuốc tiêm truyền từ khi pha thuốc, đóng thuốc hàn kín và tiệt khuẩn cần hoàn thành càng nhanh càng tốt, thường trong vòng 12 h. Nếu không, phải bảo quản thuốc trong điều kiện vô khuẩn.
Thành phần
Dược chất phải là loại nguyên liệu đáp ứng yêu cầu dùng để pha thuốc tiêm, thuốc tiêm truyền.
Dung môi thường dùng pha thuốc tiêm là nước cất để pha thuốc tiêm, dung môi hòa trộn với nước (ethanol, propylen glycol,...), dầu thực vật trung tính hoặc các dung môi thích hợp khác. Dung môi dùng trong thuốc tiêm phải đảm bảo an toàn và không ảnh hưởng đến hiệu lực điều trị ở thể tích tiêm.
Các tá dược: Tùy theo bản chất của dược chất, trong thành phần thuốc tiêm có thể thêm các chất để đẳng trương, điều chỉnh pH, tăng độ tan, độ ổn định của dược chất, chất gây thấm, nhũ hóa,... nhưng phải đảm bảo an toàn và không ảnh hưởng đến hiệu lực điều trị của thuốc ở nồng độ sử dụng trong chế phẩm.
Thuốc tiêm đóng nhiều liều trong một đơn vị đóng gói, thuốc tiêm không được tiệt khuẩn bằng nhiệt, sau khi đóng ống (lọ) phải cho thêm chất sát khuẩn với nồng độ thích hợp, trừ khi bản thân chế phẩm có đủ tính sát khuẩn cần thiết.
Không được cho chất sát khuẩn vào thuốc tiêm với liều trên 15 ml, trừ khi có chỉ dẫn khác.
Không được cho chất sát khuẩn vào thuốc tiêm để tiêm vào nội sọ, màng cứng, dịch não tủy hoặc các tổ chức ở mắt, trừ khi có chỉ dẫn riêng. Các chế phẩm này phải được đóng gói dưới dạng đơn liều. Không được cho chất màu vào thuốc tiêm với mục đích nhuộm màu chế phẩm.
Khi dược chất trong thuốc tiêm dễ bị oxy hóa có thể cho thêm các chất chống oxy hóa thích hợp và /hoặc đóng thuốc dưới dòng khí nitơ hoặc khí trơ thích hợp.
Đồ đựng
Đồ đựng thuốc tiêm, thuốc tiêm truyền (chai, lọ, ống, túi...) được làm từ các nguyên liệu, sao cho đồ đựng đủ trong để cho phép kiểm tra được bằng mắt thuốc chứa bên trong, trừ trường hợp đặc biệt. Đồ đựng không được tương tác về vật lý hay hóa học với thuốc chứa trong nó. Đồ đựng thuốc tiêm, thuốc tiêm truyền phải đạt các chỉ tiêu chất lượng quy định trong các Phụ lục 17.1, mục 17.3.2.
Nút chai lọ thuốc tiêm và thuốc tiêm truyền phải đạt các chỉ tiêu quy định trong Phụ lục 17.5.
Đồ đựng và nút chai lọ thuốc tiêm, thuốc tiêm truyền phải được xử lý theo những quy trình thích hợp, đảm bảo sạch và vô khuẩn mới được dùng để đóng thuốc tiêm, thuốc tiêm truyền.
Ghi nhãn
Nhãn thuốc tiêm theo quy định hiện hành.
Nhãn không che kín đồ đựng để có thể kiểm tra được thuốc ở bên trong.
Nhãn thuốc cần có tên chế phẩm, nồng độ dược chất hoặc lượng dược chất trong một thể tích xác định đối với thuốc tiêm lỏng, hoặc lượng dược chất đối với thuốc tiêm bột, tên và nồng độ chất bảo quản (nếu có).
Đối với bột pha tiêm và dung dịch đậm đặc để pha thuốc tiêm, thuốc tiêm truyền thì phải ghi rõ loại dung môi và lượng dung môi dùng để pha thuốc trước khi sử dụng, điều kiện bảo quản và thời gian sử dụng sau khi pha. Nếu có ống dung môi kèm theo thì trên ống phải có nhãn ghi rõ thành phần và thể tích dung môi.
B. Các loại thuốc tiêm, tiêm truyền
THUỐC TIÊM
Định nghĩa
Thuốc tiêm là các dung dịch, hỗn dịch hoặc nhũ tương vô khuẩn, để tiêm vào cơ thể bằng các đường tiêm khác nhau.
Đối với thuốc tiêm hỗn dịch, thông thường kích thước của phần lớn (trên 90 %) các tiểu phân dược chất phải dưới 15 μm, không quá 10 % số tiểu phân kích thước 15 μm đến 20 μm và hầu như không có tiểu phân kích thước 20 μm đến 50 μm.
Yêu cầu chất lượng
Cảm quan
Màu sắc: Không màu hoặc có màu của dược chất (theo chuyên luận riêng).
Trạng thái phân tán: Thuốc tiêm hỗn dịch có thể lắng cặn nhưng phải dễ dàng phân tán đồng nhất khi lắc và phải giữ được sự đồng nhất trong thời gian đủ để lấy đúng liều thuốc.
Thuốc tiêm nhũ tương phải không có bất kỳ biểu hiện nào của sự tách lớp.
Độ trong
Thuốc tiêm dạng dung dịch phải trong suốt và không có các tiểu phân không tan khi kiểm tra bằng mắt thường ở điều kiện quy định (Phụ lục 11.8, mục B) và phải đáp ứng các yêu cầu về số lượng và giới hạn kích thước các tiểu phân không quan sát được bằng mắt thường (Phụ lục 11.8, mục A).
Thể tích
Đáp ứng yêu cầu của Phụ lục 11.1.
Thử vô khuẩn
Phải vô khuẩn (Phụ lục 13.7).
Nội độc tố vi khuẩn
Phép thử nội độc tố vi khuẩn (Phụ lục 13.2) thực hiện trong những trường hợp có quy định trong chuyên luận riêng. Khi chuyên luận có quy định thử nội độc tố vi khuẩn thì không phải thử chất gây sốt, nếu không có chỉ dẫn khác.
Chất gây sốt
Không được có (Phụ lục 13.4).
Độ đồng đều hàm lượng
Áp dụng đối với thuốc tiêm hỗn dịch đóng liều đơn có hàm lượng dược chất nhỏ hơn 2 mg hoặc nhỏ hơn 2 % so với khối lượng thuốc thì phải đáp ứng yêu cầu về độ đồng đều hàm lượng (Phụ lục 11.2), trừ khi có chỉ dẫn riêng. Nếu chế phẩm có nhiều thành phần dược chất, yêu cầu này chỉ áp dụng cho thành phần dược chất nhỏ hơn 2 mg hoặc nhỏ hơn 2 % so với khối lượng thuốc. Yêu cầu này không áp dụng với thuốc tiêm chứa các vitamin và nguyên tố vi lượng.
Các yêu cầu kỹ thuật khác
Theo quy định trong chuyên luận riêng.
THUỐC TIÊM TRUYỀN
Định nghĩa
Thuốc tiêm truyền là dung dịch nước hoặc nhũ tương dầu trong nước vô khuẩn, không có chất gây sốt, không chứa chất sát khuẩn, thường đẳng trương với máu, dùng để tiêm truyền tĩnh mạch với thể tích lớn và tốc độ chậm.
Đối với thuốc tiêm truyền dạng nhũ tương, thông thường đường kính của phần lớn (80 %) các giọt phân tán phải nhỏ hơn 0,5 μm và không có giọt có đường kính lớn hơn 5 μm, trừ khi có chỉ dẫn riêng.
Yêu cầu chất lượng
Thuốc tiêm truyền phải đạt các yêu cầu chất lượng đối với thuốc tiêm và các yêu cầu sau đây:
Độ trong
Các dung dịch tiêm truyền phải đạt quy định về độ trong của thuốc tiêm khi kiểm tra bằng mắt thường (Phụ lục 11.8, mục B) và phải đáp ứng các yêu cầu về số lượng và giới hạn kích thước các tiểu phân không quan sát được bằng mắt thường (Phụ lục 11.8, mục A).
Các nhũ tương tiêm truyền không được có dấu hiệu của sự tách lớp.
Thể tích
Đáp ứng yêu cầu của Phụ lục 11.1.
Thử vô khuẩn
Phải vô khuẩn (Phụ lục 13.7).
Chất gây sốt
Không được có (Phụ lục 13.4). Chỉ không phải thử chất gây sốt nếu đã có quy định thử nội độc tố vi khuẩn, trừ những chỉ dẫn khác. Tiêm 10 ml cho mỗi kg thỏ nếu không có chỉ dẫn khác.
Các yêu cầu kỹ thuật khác
Theo quy định trong chuyên luận riêng.
BỘT PHA TIÊM ĐỂ PHA THUỐC TIÊM HAY THUỐC TIÊM TRUYỀN
Định nghĩa
Bột pha tiêm hay bột để pha thuốc tiêm (bao gồm cả các chế phẩm đông khô) để pha thuốc tiêm hay thuốc tiêm truyền là những chế phẩm vô khuẩn, phải pha với một thể tích quy định của một chất lỏng vô khuẩn thích hợp ngay trước khi dùng.
Yêu cầu chất lượng
Độ đồng đều khối lượng
Bột để pha thuốc tiêm hay thuốc tiêm truyền phải thử độ đồng đều khối lượng (Phụ lục 11.3). Yêu cầu này không áp dụng với các chế phẩm đã thử độ đồng đều về hàm lượng với tất cả các dược chất.
Độ đồng đều hàm lượng
Bột để pha thuốc tiêm hay thuốc tiêm truyền có hàm lượng dược chất nhỏ hơn 2 mg hoặc nhỏ hơn 2 % so với khối lượng thuốc hoặc có khối lượng thuốc bằng hay nhỏ hơn 40 mg thì phải đáp ứng yêu cầu độ đồng đều về hàm lượng (Phụ lục 11.2), trừ khi có chỉ dẫn riêng. Nếu chế phẩm có nhiều thành phần dược chất, yêu cầu này chỉ áp dụng cho thành phần dược chất nhỏ hơn 2 mg hoặc nhỏ hơn 2 % so với khối lượng thuốc. Yêu cầu này không áp dụng với thuốc tiêm chứa các vitamin và nguyên tố vi lượng.
Chất gây sốt - nội độc tố vi khuẩn
Sau khi pha, thuốc phải đáp ứng yêu cầu đối với thuốc tiêm hoặc thuốc tiêm truyền.
Các yêu cầu kỹ thuật khác
Theo quy định trong chuyên luận riêng.
Sau khi pha, thuốc phải đáp ứng các yêu cầu chất lượng đối với thuốc tiêm hoặc thuốc tiêm truyền.
DUNG DỊCH ĐẬM ĐẶC ĐỂ PHA THUỐC TIÊM HAY THUỐC TIÊM TRUYỀN
Dung dịch đậm đặc để pha thuốc tiêm hay thuốc tiêm truyền là những chế phẩm vô khuẩn, phải pha với một thể tích quy định của một chất lỏng vô khuẩn thích hợp ngay trước khi dùng.
Yêu cầu chất lượng
Chất gây sốt - nội độc tố vi khuẩn
Sau khi pha, thuốc phải đáp ứng yêu cầu đối với thuốc tiêm hoặc thuốc tiêm truyền.
Các yêu cầu kỹ thuật khác
Theo quy định trong chuyên luận riêng.
Sau khi pha, thuốc phải đáp ứng các yêu cầu chất lượng đối với thuốc tiêm hoặc thuốc tiêm truyền.
1.20 THUỐC VIÊN NÉN
Tabellae
Định nghĩa
Viên nén là dạng thuốc rắn, mỗi viên là một đơn vị phân liều, dùng để uống, nhai, ngậm, đặt hoặc hòa với nước để uống, để súc miệng, để rửa.... Viên nén chứa một hoặc nhiều dược chất, có thể thêm các tá dược độn, tá dược rã, tá dược dính, tá dược trơn, tá dược bao, tá dược màu... được nén thành khối hình trụ dẹt; thuôn (caplet) hoặc các hình dạng khác. Viên có thể được bao.
Yêu cầu kỹ thuật chung của thuốc viên nén
Tính chất
Viên rắn, mặt viên nhẵn hoặc lồi, trên mặt có thể có rãnh, chữ hoặc ký hiệu, cạnh và thành viên lành lặn. Viên không bị gãy vỡ, bở vụn trong quá trình bảo quản, phân phối và vận chuyển.
Độ rã
Nếu không có chỉ dẫn gì khác, viên nén phải đạt yêu cầu về độ rã quy định, được thử theo Phụ lục 11.6
Phép thử độ rã của viên nén và nang.
Viên nén và viên bao đã thử độ hòa tan với tất cả các dược chất có trong thành phần thì không phải thử độ rã.
Độ đồng đều khối lượng
Thử theo Phụ lục 11.3 Phép thử độ đồng đều khối lượng. Viên nén và viên bao đã thử độ đồng đều về hàm lượng với tất cả các dược chất có trong thành phần thì không phải thử độ đồng đều khối lượng.
Độ đồng đều hàm lượng (Phụ lục 11.2)
Nếu không có chỉ dẫn khác, viên nén có hàm lượng dược chất dưới 2 mg hoặc dưới 2 % (kl/kl) phải thử độ đồng đều hàm lượng. Đối với viên nén có từ 2 dược chất trở lên, chỉ áp dụng yêu cầu này với thành phần có hàm lượng nhỏ như quy định ở trên.
Độ hòa tan
Yêu cầu được chỉ ra trong chuyên luận riêng. Phương pháp thử được ghi trong chuyên luận Phép thử độ hòa tan của viên nén và nang ( Phụ lục 11.4).
Định lượng và các yêu cầu kỹ thuật khác
Thử theo quy định trong chuyên luận riêng.
Bảo quản - ghi nhãn
Thuốc viên nén phải đựng trong bao bì kín, chống ẩm và chống va chạm cơ học.
Ghi nhãn theo quy định. Nếu là viên bao cần phải ghi rõ: Bao đường, bao phim hay bao tan trong ruột.
Viên nén không bao
Viên nén không bao gồm các loại viên điều chế bằng cách nén các hạt nhỏ của một dược chất hoặc nhiều dược chất thành viên nén một lớp hoặc viên nén nhiều lớp. Các tá dược cho thêm vào viên không được làm thay đổi hoặc hạn chế việc giải phóng dược chất trong dịch tiêu hóa.
Độ rã
Viên nén không bao phải đáp ứng yêu cầu về độ rã quy định trong chuyên luận Phép thử độ rã của viên nén và nang (Phụ lục 11.6). Dùng nước làm môi trường thử, cho đĩa vào mỗi ống thử, thời gian rã không được quá 15 min, nếu không có chỉ dẫn khác.
Nếu viên không đáp ứng được yêu cầu do viên bị dính vào đĩa thì thử lại với 6 viên khác, nhưng không cho đĩa vào ống.
Viên nhai không phải thử độ rã.
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên sủi bọt
Viên sủi bọt là viên nén không bao, thường chứa tá dược sủi bọt gồm các acid hữu cơ và muối carbonat hoặc hydrocarbonat, phản ứng khi có nước giải phóng khí carbon dioxyd. Viên được hòa tan hoặc phân tán trong nước trước khi dùng.
Độ rã
Cho một viên vào cốc chứa 200 ml nước ở 15 °C đến 25 °C, phải có nhiều bọt khí bay ra. Viên được coi là rã hết nếu hòa tan hoặc phân tán hết trong nước, không còn các hạt kết vón. Thử với 6 viên, chế phẩm đạt yêu cầu phép thử nếu mỗi viên rã trong vòng 5 min, trừ khi có các chỉ dẫn khác trong chuyên luận riêng.
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên bao
Viên bao là viên nén được bao bằng một hay nhiều lớp của hỗn hợp các chất bao khác nhau như các chất nhựa tự nhiên hoặc tổng hợp, gôm, gelatin, chất bao không có hoạt tính và không tan, đường, chất hóa dẻo, chống dính, chất màu.... Tá dược bao thường được điều chế dưới dạng dung dịch hay hỗn dịch trong dung môi hay dẫn chất thích hợp. Sau khi bao, dung môi phải được loại bỏ khỏi viên. Khi viên nén có lớp bao là màng polymer rất mỏng thì gọi là viên nén bao phim.
Tính chất
Viên bao có bề mặt nhẵn, có thể có màu, được đánh bóng. Khi bẻ viên có thể quan sát thấy lớp bao.
Độ rã
Viên bao phải đáp ứng yêu cầu về độ rã quy định trong chuyên luận Phép thử độ rã của viên nén và nang (Phụ lục 11.6). Dùng nước làm môi trường thử, cho đĩa vào mỗi ống thử. Nếu không có chỉ dẫn khác, viên bao phim phải rã trong 30 min, viên bao khác phải rã trong 60 min.
Nếu có viên nào không rã thì thử lại với 6 viên khác, thay nước bằng dung dịch acid hydrocloric 0,1 N (TT).
Nếu phép thử không đạt yêu cầu do viên bị dính vào đĩa thì thử lại với 6 viên khác không dùng đĩa. Viên nén nhai có bao không phải thử độ rã.
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên ngậm
Viên ngậm thường là viên nén không bao được điều chế để dược chất giải phóng tại khoang miệng gây tác dụng tại chỗ hay hấp thu qua niêm mạc dưới lưỡi.
Độ rã
Viên ngậm phải rã trong vòng 4 h, thử theo chuyên luận Phép thử độ rã của viên nén và nang (Phụ lục 11.6). Nếu không có chỉ dẫn khác, dùng nước làm môi trường thử, cho đĩa vào mỗi ống thử.
Các yêu cầu kỹ thuật
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên nén tan trong nước
Viên nén tan trong nước là viên nén không bao hoặc viên nén bao phim, được hòa tan trong nước trước khi dùng. Dung dịch sau khi hòa tan phải trong suốt hoặc hơi đục nhẹ.
Độ rã
Viên nén phải rã trong vòng 3 min, thử theo chuyên luận Phép thử độ rã của viên nén và nang (Phụ lục 11.6). Dùng môi trường nước ở 15 °C đến 25 °C để thử, trừ khi có chỉ dẫn khác.
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên nén phân tán trong nước
Viên nén phân tán trong nước là viên nén không bao hoặc viên nén bao phim, được phân tán đồng đều trong nước tạo thành hỗn dịch đồng nhất trước khi dùng.
Độ rã
Theo yêu cầu của mục Viên nén tan trong nước.
Độ đồng đều phân tán
Cho 2 viên vào 100 ml nước, khuấy cho đến khi hoàn toàn phân tán. Độ phân tán đạt yêu cầu khi dung dịch phân tán chảy hết qua lỗ mắt rây có kích thước mắt rây 710 μm.
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên nén phân tán trong miệng
Viên nén phân tán trong miệng là viên nén không bao, khi đặt vào miệng phải phân tán nhanh trong nước bọt trước khi nuốt.
Độ rã
Viên nén phải rã trong 3 min, thử theo chuyên luận Phép thử độ rã của viên nén và viên nang (Phụ lục 11.6). Nếu không có chỉ dẫn khác, dùng nước làm môi trường thử, cho đĩa vào mỗi ống thử.
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên nén giải phóng biến đổi
Viên nén giải phóng biến đổi là viên nén không bao hay bao được bào chế với các tá dược đặc biệt hoặc bằng phương pháp đặc biệt hoặc bằng cả hai cách, để thay đổi tốc độ, vị trí hoặc thời gian giải phóng dược chất.
Viên nén giải phóng biến đổi bao gồm viên giải phóng kéo dài, viên giải phóng muộn và viên giải phóng theo nhịp.
Độ hòa tan
Phải thử độ hòa tan theo chuyên luận riêng để chứng minh sự giải phóng dược chất.
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
Viên nén tan trong ruột
Viên nén tan trong ruột là viên nén giải phóng muộn, kháng dịch dạ dày và giải phóng dược chất trong dịch ruột. Viên thường được bào chế từ cốm hoặc hạt được bao lớp bao kháng dịch dạ dày hoặc viên được bao lớp bao kháng dịch dạ dày.
Tính chất
Viên nén tan trong ruột phải đạt các tính chất chung của viên bao.
Độ hòa tan
Viên nén chứa cốm hoặc hạt đã được bao lớp bao kháng dịch dạ dày phải tiến hành thử độ hòa tan để kiểm tra sự giải phóng dược chất theo Phụ lục 11.4 Phép thử độ hòa tan của viên nén và nang và theo chuyên luận riêng.
Độ rã
Viên nén bao tan trong ruột nếu không thử độ hòa tan thì phải đáp ứng yêu cầu về độ rã quy định trong chuyên luận Phép thử độ rã của viên bao tan trong ruột (Phụ lục 11.7).
Các yêu cầu kỹ thuật khác
Theo yêu cầu kỹ thuật chung của thuốc viên nén và theo chuyên luận riêng.
1.21 THUỐC BỌT Y TẾ
Musci medicati
Thuốc bọt là dạng thuốc lỏng đóng trong bao bì kín với khí đẩy ở áp suất cao thích hợp, trong đó có một lượng khí phân tán đều trong môi trường lỏng tạo bọt, khi dùng bọt tự vỡ do khí thoát đi còn lại thuốc ở dạng mềm dễ bám dính.
Thuốc bọt y tế dùng để sát trùng da, niêm mạc, vết thương, trị bỏng,...hoặc vào việc thích hợp khác. Thuốc chứa một hoặc nhiều hoạt chất, thường gặp như menthol, tinh dầu tràm, clorocresol, hexetidin,...và các tá dược thích hợp như chất dẫn, chất sát trùng bảo quản, chất tạo nhũ, tạo bọt và chất ổn định. Các tá dược phải đảm bảo không ảnh hưởng tới chức năng hoặc gây tổn thương, kích ứng nơi dùng thuốc,…
Thuốc bọt được hình thành bởi sự phân tán khí trong môi trường lỏng chứa hoạt chất, tá dược và trạng thái bọt phải hình thành ổn định. Thuốc được đóng trong chai, lọ chứa với khí đẩy và có phụ tùng như van, nút bấm để đẩy thuốc ra khỏi bao bì khi sử dụng.
Phương pháp sản xuất
Thuốc bọt được bào chế dưới dạng nhũ tương, thường là nhũ tương Dầu trong Nước, trong đó hoạt chất được hòa tan vào một trong 2 pha tùy đặc tính thân nước hoặc thân dầu của chúng. Thuốc được đóng vào một bình chứa kiểu khí dung và đặt van, nút bấm phù hợp. Khí đẩy được nén vào bình với áp suất thích hợp, khí đẩy sẽ phân tán trong tương dầu của nhũ tương tạo ra bọt ở áp lực cao trong bình kín.
Khí đẩy thường dùng là khí hóa lỏng hydrocarbon và dẫn chất.
Tiêu chuẩn chất lượng
Đạt yêu cầu chất lượng theo các chuyên luận riêng.
Thuốc bọt y tế dạng đỏng khí đẩy dưới áp suất cao phải đáp ứng các yêu cầu chung của thuốc khí dung (Phụ lục 1.18).
Nếu thuốc bọt dùng trên vết thương, phải đạt yêu cầu vô khuẩn (Phụ lục 13.7).
Tiêu chuẩn riêng cho thuốc bọt gồm: Tỷ trọng của thuốc, khả năng tạo bọt.
Xác định tỷ trọng tương đối của thuốc bọt: Đặt chai (lọ) thuốc còn nguyên vẹn ở nhiệt độ 25 °C ít nhất trong 24 h. Lấy chai thuốc ra, cẩn thận không làm cho thuốc nóng lên, và gắn 1 ống cứng có đường kính trong 1 mm và chiều dài 70 mm đến 100 mm vào lỗ phun thuốc ở đầu bấm. Lắc chai để thuốc đồng nhất và bấm nút để bỏ lượng thuốc thoát ra ban đầu khoảng 5 ml đến 10 ml. Cân bì một đĩa đáy bằng có chiều cao khoảng 35 mm và dung tích khoảng 60 ml. Cho đầu ống chạm góc của đĩa và bấm cho thuốc chuyển vào đĩa, chuyển đầu ống xoay tròn trên khắp mặt đĩa cho bọt thuốc phủ đều và cao hơn mặt đĩa. Dùng một phiến mỏng gạt cho bọt bằng phẳng và vừa đúng thể tích của đĩa. Cân khối lượng thuốc (mt). Tiếp theo cho một thể tích nước cất tương tự. Cân khối lượng nước (mn).
Tỷ trọng tương đối của thuốc = mt / mn
Tiến hành thí nghiệm 3 lần, lấy kết quả trung bình.
Kết quả mỗi lần thử không được lệch quá 20 % so với giá trị trung bình.
Khả năng tạo bọt của thuốc: Chuẩn bị dụng cụ (xem hình) gồm: Một buret dung tích 50 ml, chia vạch đến khoảng 0,1 ml có đường kính trong 15 mm và khóa có lỗ thoát 4 mm. Gắn 1 ống dẫn bằng chất dẻo có đường kính trong 4 mm và chiều dài không quá 50 mm vào đầu ra của buret. Đặt chai (lọ) thuốc còn nguyên vẹn ở nhiệt độ 25 °C ít nhất trong 24 h. Lấy chai thuốc ra, cẩn thận không làm cho thuốc nóng lên. Lắc chai để thuốc đồng nhất và bấm nút để bỏ lượng thuốc thoát ra ban đầu khoảng 5 ml đến 10 ml. Gắn ống đã nối với buret vào lỗ phun thuốc ở đầu bấm. Bấm cho thuốc đi vào buret được 30 ml tính theo chiều cao của khối bọt cho một lần thử nghiệm. Khóa buret, đồng thời bấm đồng hồ và đọc thể tích khối bọt trên buret. Cứ mỗi 10 s đọc và ghi nhận thể tích tăng thêm đến khi khối bọt đạt thể tích cao nhất. Khi đọc, do bề mặt của khối bọt không phẳng, nên có thể đọc vạch cao nhất và vạch thấp nhất, cộng lại chia trung bình.
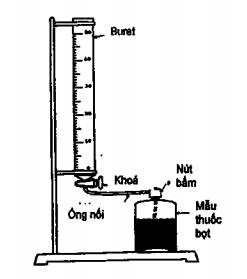
Hình 1.21 - Dụng cụ xác định khả năng tạo bọt của thuốc bọt y tế
Tiến hành thí nghiệm 3 lần.
Kết quả: Không có lần nào cần quá 5 min để đạt được thể tích cao nhất.
Nhãn thuốc
Thuốc bọt y tế phải dán nhãn theo quy định chung.
1.22 RƯỢU THUỐC
Alcoholaturae
Định nghĩa
Rượu thuốc là dạng thuốc lỏng dùng để uống hay đôi khi dùng ngoài, được điều chế bằng cách ngâm dược liệu (thảo mộc hoặc động vật) trong rượu trắng hoặc ethanol loãng trong một thời gian nhất định (tùy theo quy định của từng công thức) rồi gạn hoặc lọc lấy dịch trong.
Rượu thuốc phải đáp ứng các yêu cầu sau đây:
Dược liệu: Được chế biến và chuẩn bị theo yêu cầu của từng công thức. Các vị thuốc là thảo mộc được chia nhỏ đến kích thước thích hợp. Dược liệu động vật có thể dùng tươi hay sấy khô, tán bột.
Phương pháp điều chế: Rượu thuốc có thể điều chế theo phương pháp thích hợp (ngâm hoặc chiết). Khi điều chế rượu thuốc từ thang thuốc, do thành phần dược liệu đa dạng, bản chất dược liệu khác nhau nên có thể phải chiết riêng một số dược liệu để chiết xuất được tối đa hoạt chất.
Dung môi để ngâm thường dùng rượu trắng hoặc ethanol loãng. Tỷ lệ dung môi, hàm lượng ethanol, nhiệt độ ngâm, thời gian ngâm, tốc độ chiết theo yêu cầu quy định của từng công thức.
Yêu cầu chất lượng
Màu sắc: Theo yêu cầu quy định trong chuyên luận riêng.
Cách tiến hành: Lấy ở 2 chai rượu trong mỗi lô sản xuất, mỗi chai 5 ml, cho vào 2 ống nghiệm (thủy tinh không màu, đồng cỡ). Quan sát màu của hai ống ở ánh sáng thiên nhiên bằng cách nhìn ngang, màu sắc của hai ống phải như nhau và đúng như màu sắc đã quy định trong chuyên luận riêng.
Mùi vị: Theo yêu cầu quy định trong chuyên luận riêng. Rượu thuốc có thể có mùi thơm của dược liệu, có vị ngọt do thêm đường hoặc mật ong.
Độ trong và độ đồng nhất: Rượu thuốc phải trong, đồng nhất, không có cặn bã dược liệu và vật lạ.
Cách tiến hành: Quan sát toàn chai rượu, không được có váng mốc. Hút 5 ml rượu thuốc ở vị trí cách đáy chai khoảng 2 cm, cho vào ống nghiệm (thủy tinh không màu, dung tích 10 ml đến 20 ml), quan sát ở ánh sáng thiên nhiên bằng cách nhìn ngang. Thuốc phải trong và đồng nhất. Nếu không đạt yêu cầu, thử lại lần thứ hai với một chai thuốc khác. Lần này không đạt thì lô thuốc coi như không đạt tiêu chuẩn.
Hàm lượng ethanol: Theo yêu cầu quy định trong chuyên luận riêng. Xác định hàm lượng ethanol theo Phụ lục 10.12.
Tỷ trọng: Theo yêu cầu quy định trong chuyên luận. Xác định tỷ trọng theo Phụ lục 6.5.
Độ lắng cặn: Theo yêu cầu quy định trong từng chuyên luận.
Cách tiến hành: Quan sát toàn chai rượu (thể tích 500 ml, nếu không có quy định khác), nếu thấy có cặn thì để yên khoảng 48 h, sau đó mở nút và thận trọng dùng ống cao su hay ống nhựa làm xi phông, hút phần rượu ở phía trên, để còn lại 15 ml đến 20 ml (đối với rượu có thể tích cặn không quá 0,5 ml) hoặc 40 - 50 ml (đối với rượu có thể tích cặn trên 0,5 ml). Lắc cặn trong chai cho tan, rót hết sang ống đong 25 ml (chia độ 0,5 ml) hoặc 50 ml (chia độ 1 ml) có nút. Lấy phần rượu trong đã hút xiphông để tráng chai, đổ vào ống đong rồi thêm rượu thuốc vừa đủ 25 ml hoặc 50 ml. Để lắng 48 h, đọc kết quả trên vạch chia của ống đong. Mỗi loại rượu phải đạt được yêu cầu của tiêu chuẩn đề ra.
Sau khi đọc kết quả, nghiêng ống đong nhẹ để gạn lớp rượu ở trên, lấy lớp cặn ra bát sứ trắng để quan sát. Trong lớp cặn phải không được có bã dược liệu và vật lạ.
Cặn sau khi sấy khô: Theo yêu cầu quy định trong từng chuyên luận. Tiến hành theo một trong hai phương pháp sau đây:
Phương pháp 1: Áp dụng với các rượu thuốc có chứa đường hoặc mật ong.
Lấy chính xác 50 ml chế phẩm vào cốc miệng rộng, bốc hơi đến khô trên cách thủy, chiết bằng ethanol (TT) bằng cách thêm vào cặn lần lượt, 4 lần, mỗi lần 10 ml ethanol (TT), dùng đũa thủy tinh khuấy kỹ, lọc. Gộp các dịch lọc vào một chén sứ đã được xác định khối lượng, bay hơi trên cách thủy đến khô, sấy cặn ở 105 °C trong 3 h, để nguội trong bình hút ẩm 30 min, cân. Xác định khối lượng cặn thu được.
Phương pháp 2: Áp dụng với các rượu thuốc không chứa đường hoặc mật ong.
Lấy chính xác 50 ml chế phẩm vào một chén sứ đã được xác định khối lượng, bay hơi trên cách thủy đến khô, sấy cặn ở 105 °C trong 3 h, để nguội trong bình hút ẩm 30 min, cân. Xác định khối lượng cặn thu được.
Thể tích: Theo yêu cầu và cách thử ở Phụ lục 11.1.
Methanol (Nếu không có chỉ dẫn khác):
Không quá 0,05 % (tt/tt) (Phụ lục 10.13).
Giới hạn nhiễm khuẩn:
Rượu thuốc phải đạt yêu cầu về giới hạn nhiễm khuẩn (Phụ lục 13.6).
Định tính, định lượng và các chỉ tiêu khác: Theo yêu cầu quy định trong chuyên luận riêng.
Bảo quản
Trong đồ đựng thích hợp, đậy kín. Để ở nơi khô mát, tránh ánh sáng.
1.23 THUỐC THANG
Thuốc thang được cấu tạo từ các vị thuốc đã qua chế biến và phối hợp theo phương pháp của y học cổ truyền, bao gồm 2 dạng: Thuốc sắc và ngâm rượu. Với dạng sắc, thường được bào chế bằng cách đun sôi với nước sạch; dạng ngâm rượu, thường được ngâm với ethanol 30 % đến 40 %, trong thời gian thích hợp.
Đặc điểm của thuốc thang
Là dạng thuốc rất thông dụng, được dùng rộng rãi để phòng và điều trị nhiều chứng bệnh khác nhau, đối với các lứa tuổi, các mùa trong năm.
Dễ gia giảm theo triệu chứng của bệnh, do đó thường cho hiệu quả cao trong điều trị.
Được hấp thu nhanh qua đường tiêu hóa. Tuy vậy thuốc thang thường dùng cho từng người bệnh, bởi vậy, cũng gặp khó khăn nhất định trong bào chế.
Cấu tạo
Với thuốc thang dùng để uống thường được cấu tạo theo nguyên lý của y học cổ truyền; mỗi thang thuốc phải có đủ các thành phần: Quân,Thần, Tá, Sứ, với các công năng, chủ trị, cách dùng cụ thể của nó (cổ phương, tân phương).
Đôi khi, những phương thuốc gia truyền, hoặc các phương thuốc trong dân gian, cũng không hoàn toàn theo nguyên tắc đó. Nhưng phải đảm bảo tính an toàn và hiệu quả cao trong điều trị cho người bệnh.
Liều lượng của thuốc thang
Số vị trong thang, nhiều hay ít tùy theo từng thang, khối lượng của từng vị trong thang tùy theo tuổi, theo tình trạng của người bệnh (nếu là cổ phương thường được giữ nguyên số vị và khối lượng). Tuy nhiên, trong những điều kiện cụ thể có thể thay đổi cho phù hợp. Đối với các vị thuốc có tính độc, liều lượng phải thực hiện theo đúng với quy định hiện hành.
Yêu cầu chất lượng
Các vị thuốc trước khi làm thang phải đạt yêu cầu chất lượng quy định trong từng chuyên luận riêng.
Độ đồng đều khối lượng: Các vị thuốc trong thang phải đạt yêu cầu Phép thử độ đồng đều khối lượng của Thuốc hoàn (Phụ lục 1.11. Bảng 3).
Giới hạn nhiễm khuẩn: Thuốc thang phải đạt yêu cầu về giới hạn nhiễm khuẩn (Phụ lục 13.6).
Dụng cụ để sắc thuốc
Dụng cụ để sắc thuốc rất đa dạng, có thể dùng siêu đất, hoặc dụng cụ bằng thép không gỉ, không dùng các dụng cụ bằng gang, đồng, sắt. Hiện nay có nhiều loại dụng cụ sắc thuốc được đun nóng bằng điện, cần chú ý theo dõi để tránh ảnh hưởng đến chất lượng của thuốc.
Nước dùng để sắc thuốc
Nước dùng để sắc thuốc phải là nước sạch, dùng để uống được. Lượng nước đưa vào mỗi thang cần vừa đủ để hoạt chất có thể chiết xuất được. Tùy theo dung tích của dụng cụ sắc, mức nước thường cao hơn mặt thoáng của thuốc khoảng 2 cm.
Nhiệt độ và thời gian sắc thuốc
Nhiệt độ và thời gian sắc thuốc phụ thuộc vào tính chất của thang thuốc.
Nếu thang thuốc lấy khí là chính (chất bay hơi), trong thành phần chứa tinh dầu, và các chất bay hơi: Thuốc giải biểu, thuốc ôn trung..., lúc đầu thường dùng lửa to (vũ hỏa), để nhanh chóng nâng nhiệt độ của thuốc đến sôi, sau đó giảm độ lửa, duy trì ở 70 °C đến 80 °C trong thời gian 10 min đến 15 min. Nếu thang thuốc lấy vị là chính (thuốc bổ, thuốc thanh nhiệt...), có thể đun sôi 1 h đến 2 h. Tuy nhiên trên thực tế, trong một thang thuốc, thường bao gồm cả vị thuốc lấy khí và lấy vị, do đó trong quá trình sắc thuốc cần chú ý duy trì nhiệt độ và thời gian thích hợp.
Nhiên liệu dùng sắc thuốc
Nhiên liệu sắc thuốc rất đa dạng, có thể dùng củi, rơm, rạ..., song tránh dùng củi khi cháy cho chất độc hoặc có mùi khó chịu; có thể dùng than khi đã cháy hồng để tránh mùi khí than; hiện nay còn dùng điện, dùng khí đốt. Cần chú ý rằng, với nguồn nhiên liệu khác nhau thì nhiệt lượng cung cấp sẽ khác nhau, do đó thời gian sắc thuốc nên tính từ lúc sôi và cần theo dõi để tránh bị cháy.
Cách uống thuốc
Tùy theo tính chất của bệnh và tính chất của thang thuốc mà cách uống thuốc có khác nhau. Các bệnh thuộc thể hàn: cảm mạo phong hàn, trúng hàn...tính chất của thang thuốc thường ôn, nhiệt, cần uống khi còn nóng. Các bệnh thể nhiệt, tính chất của thang thuốc thường hàn, lương, thường uống nguội. Các thang thuốc mang tính chất tả (tả hạ, thanh nhiệt...), thường uống lúc đói. Tuy nhiên, nếu đói quá dễ cồn cào, buồn nôn, do đó cần ăn một chút ít để tránh cồn ruột.
Các thang thuốc mang tính chất bổ (bổ khí, huyết, âm, dương), thường uống sau khi ăn 1,5 h đến 2 h.
Kiêng kị khi dùng thuốc thang
Khi dùng thuốc thang, cũng kiêng kị theo nguyên tắc chung của y học cổ truyền. Nếu uống thuốc mang tính ôn nhiệt, cần kiêng các thức ăn sống lạnh: ốc, rau giền... Nếu uống thuốc mang tính hàn lương, cần kiêng các thức ăn cay nóng và kích thích: ớt, rượu, hạt tiêu... Một số vị thuốc kiêng đặc biệt: miết giáp kị rau giền, hành kị mật ong, kinh giới kị thịt gà... Ngoài ra cần chú ý đến một số thang thuốc có các vị có tính độc, phải kiêng cho phụ nữ có thai, hoặc sau khi đẻ, hoặc trẻ em dưới 15 tuổi: phụ tử chế, quế chi, cà độc dược...
Lưu ý khi dùng thuốc thang
Để phát huy được hiệu quả của thuốc thang, người ghi đơn cần chú ý:
Không ghi các vị thuốc mang tính chất tương kị.
Ghi các vị thuốc cần sắc sau, để người cân thuốc có thể để riêng.
Để có thể phối hợp hài hòa giữa các lần sắc thuốc: Lần 1, đun sôi (kể từ lúc sôi) 20 min đến 30 min; lần 2, đun sôi 40 min đến 1 h; lần 3, đun sôi 1h. Dịch thuốc sắc của mỗi lần, lấy khoảng 1 bát (100 ml). Có thể uống riêng từng lần; hoặc uống riêng nước lần 1, nước lần 2 và 3 trộn đều chia đôi, uống 2 lần nửa; hoặc trộn đều cả 3 lần sắc, rồi chia 3 lần uống.
1.24 CHÈ THUỐC
Định nghĩa
Là dạng thuốc rắn được bào chế từ hoa, lá hoặc bột thô dược liệu và các tá dược thích hợp dưới dạng gói hay bánh nhỏ. Khi dùng, đem hãm với nước sôi trong khoảng thời gian thích hợp để uống.
Phương pháp bào chế
Chè gói: Được bào chế chủ yếu từ các dược liệu là hoa lá mỏng manh. Dược liệu được sao nhỏ lửa hay sấy nhẹ (dưới 80 °C) đến khô rồi vò thành mảnh nhỏ, xát qua sàng (rây) có cỡ mắt rây 2 mm đến 5 mm. Các dược liệu có cấu tạo rắn chắc nếu có ở tỷ lệ thích hợp, thường được nghiền thành bột thô hoặc bào chế thành cao lỏng để phun vào chè trong khi sấy. Trộn đều dược liệu theo tỷ lệ trong công thức rồi đóng túi theo khối lượng quy định (nếu có cao lỏng thì phun vào chè, tiếp tục sấy khô rồi đóng gói). Túi đựng chè có thể là túi giấy thường, túi giấy lọc hay túi PE. Việc đóng túi có thể thực hiện bằng các loại máy đóng túi tự động. Túi chè cần được đóng kín, hạn chế sự hút ẩm trở lại trong quá trình bảo quản.
Chè bánh: Thường được bào chế từ các dược liệu có cấu tạo rắn chắc như thân, cành, rễ,... Dược liệu được chế biến, sấy khô rồi nghiền thành bột thô qua rây có số rây 1000 đến 2000. Thêm tá dược dính thích hợp tạo thành khối ẩm (hồ tinh bột, si rô, cao đặc, bột đường,...) rồi lèn hay nén thành bánh theo khuôn. Chè bánh sau khi tháo khỏi khuôn đưa sấy đến độ ẩm quy định rồi đóng gói thích hợp.
Tiêu chuẩn chất lượng
Phải đáp ứng các yêu cầu trong chuyên luận riêng và các quy định dưới đây:
Độ ẩm
Chè gói: Độ ẩm không được vượt quá 10 % (Phụ lục 9.6), nếu không có chỉ dẫn khác.
Chè bánh: Độ ẩm không được vượt quá 7 % (Phụ lục 9.6), nếu không có chỉ dẫn khác.
Độ đồng đều khối lượng
Cân riêng biệt 10 đơn vị đóng gói, xác định khối lượng trung bình. So sánh khối lượng từng đơn vị với khối lượng trung bình. Chênh lệch khối lượng phải nằm trong giới hạn sau:
| Khối lượng trung bình | Giới hạn chênh lệch cho phép (%) |
| Tới 2 g | 15 |
| Trên 2 g đến 5 g | 12 |
| Trên 5 g đến 10 g | 10 |
| Trên 10 g đến 20 g | 6 |
| Trên 20 g đến 40 g | 5 |
| Trên 40 g | 4 |
Giới hạn nhiễm khuẩn: Chè thuốc phải đạt yêu cầu về giới hạn nhiễm khuẩn (Phụ lục 13.6).
PHỤ LỤC 2
(Quy định)
2.1 CÁC THUỐC THỬ CHUNG
2.1.1 HÓA CHẤT VÀ THUỐC THỬ
Acetaldehyd
C2H4O = 44,1
Dùng loại tinh khiết hóa học.
Chất lỏng trong, không màu, dễ cháy. Hòa trộn với nước, ethanol, cloroform và ether.
Tỷ trọng ở 20 °C: Khoảng 0,788.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,332.
Điểm sôi: Khoảng 21 °C.
Aceton
Propan - 2 - on
C3H6O = 58,08
Dùng loại tinh khiết phân tích.
Chất lỏng dễ bay hơi, dễ bắt lửa.
Điểm sôi: Khoảng 56 °C.
Khối lượng riêng: Khoảng 0,79 g/ml.
Hàm lượng nước: Không được quá 0,3 % (kl/kl) (Phụ lục 10.3), dùng pyridin khan (TT) làm dung môi.
Acetonitril
Methyl cyanid
C2H3N = 41,05
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Tỷ trọng ở 20 °C: Khoảng 0,78.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,344.
Phần chưng cất được trong khoảng 80 °C đến 82 °C không được ít hơn 95 %.
Acetonitril dùng trong phương pháp quang phổ
Độ truyền quang: Không được nhỏ hơn 98 % trong khoảng bước sóng từ 255 đến 420 nm, dùng nước làm mẫu trắng.
Acetonitril dùng trong phương pháp sắc ký
Hàm lượng C2H3N không được ít hơn 99,8 %.
Độ truyền quang: Không được nhỏ hơn 98 % ở bước sóng 240 nm, dùng nước làm mẫu trắng.
Acetonitrile (TT1)
Hàm lượng C2H3N không được ít hơn 99,9 % và đạt yêu cầu sau:
Độ hấp thụ: Độ hấp thụ của chế phẩm ở bước sóng 200 nm (Phụ lục 4.1) không được lớn hơn 0,10; dùng nước làm mẫu trắng.
Acetaldehyd amoni trimer trihydrat
2,4,6-Trimethylhexahydro-1,3,5- triazin trihydrat
C6H15N3.3H2O = 183,3
Điểm chảy: Khoảng từ 95 °C đến 97 °C.
Acetylaceton
Pentan-2,4-dion
C5H8O2= 100,1
Dùng loại tinh khiết phân tích.
Chất lỏng dễ cháy, không màu hoặc màu vàng nhạt.
Chỉ số khúc xạ ở 20 °C: Khoảng từ 1,452 đến 1,453.
Điểm sôi: Khoảng từ 138 °C đến 140 °C.
Acetyl clorid
C2H3CIO = 78,50
Dùng loại tinh khiết phân tích.
Tỷ trọng ở 20 °C: Khoảng 1,10.
Phần chưng cất được trong khoảng 49 °C đến 53 °C không được ít hơn 95 %.
Acetyltyrosin ethyl ester
Ethyl N-acetyl-L-tyrosinat
C13H17NO4.H2O = 269,3
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng.
Góc quay cực riêng ở 20 °C: +21° đến +25°, dung dịch 1 % (kl/tt) trong ethanol (TT).
Giá trị A (1 %, 1 cm) ở bước sóng 278 nm: 60 đến 68, dung dịch trong ethanol (TT).
Acid acetic băng
Acid acetic kết tinh được.
CH3COOH = 60,1
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, mùi hăng cay.
Khối lượng riêng: Khoảng 1,05 g/ml.
Điểm đông đặc: Khoảng 16 °C.
Hàm lượng CH3COOH: Không được nhỏ hơn 98,0 % (kl/kl).
Dung dịch acid acetic xM
Pha loãng 57x ml (60x g) acid acetic băng (TT) với nước vừa đủ 1000 ml.
Acid acetic (Dung dịch acid acetic 30 % - Dung dịch acid acetic 5 M)
Pha loãng 30 g acid acetic băng (TT) với nước vừa đủ 100 ml.
Hàm lượng CH3COOH khoảng 29,0 % đến 31,0 %.
Acid acetic loãng (Dung dịch acid acetic 12 % - Dung dịch acid acetic 2 M)
Pha loãng 12 g acid acetic băng (TT) với nước vừa đủ 100 ml.
Hàm lượng CH3COOH khoảng 11,5 % đến 12,5 %.
Dung dịch acid acetic 6 %
Lấy 100 ml dung dịch acid acetic 30 % (TT), pha loãng với nước vừa đủ 500 ml.
Acid acetic khan
CH3COOH = 60,1
Acid acetic băng dùng trong chuẩn độ môi trường khan, hàm lượng CH3COOH không ít hơn 99,6 % (kl/kl).
Tỷ trọng ở 20 °C: Từ 1,052 đến 1,053.
Điểm sôi: 117 °C đến 119 °C.
Hàm lượng nước: Không được quá 0,4 % (kl/kl) (Phụ lục 10.3). Nếu hàm lượng nước lớn hơn 0,4 %, làm khan bằng cách cho thêm anhydrid acetic (TT) (7 ml cho mỗi gam nước).
Dung dịch acid peroxyacetic
Pha loãng 1 ml hydrogen peroxyd 100 tt (TT) thành 100 ml với acid acetic khan (TT). Lắc đều, để yên 12 h trước khi sử dụng.
Chỉ dùng trong vòng 24h.
Acid 4-aminobenzoic
C7H7NO2 = 137,1
Dùng loại tinh khiết hóa học.
Tinh thể màu trắng, chuyển sang màu vàng nhạt khi tiếp xúc với không khí và ánh sáng. Rất dễ tan trong nước sôi, ethanol, ether và acid acetic, tan rất ít trong nước.
Điểm chảy: Khoảng 188 °C.
Dung dịch acid 4-aminobenzoic
Hòa tan 1 g acid 4-aminobenzoic (TT) trong hỗn hợp gồm 18 ml acid acetic khan (TT), 20 ml nước và 1 ml acid phosphoric (TT). Trước khi dùng trộn 2 thể tích dung dịch trên với 3 thể tích aceton (TT).
Acid (4-aminobenzoyl)-L-glutamic
C12H14N2O5 = 266,2
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 173 °C.
Acid 3-aminomethylalirazin-N,N-diacetic
Acid aminomethylalirazindiacetic
C19H15NO8.2H2O = 421,4
Dùng loại tinh khiết phân tích.
Bột mịn màu nâu cam hay vàng nhạt.
Điểm chảy: Khoảng 185 °C.
Phải đạt yêu cầu sau:
Mất khối lượng do làm khô: Không quá 10,0 %, dùng 1 g.
Thuốc thử acid aminomethylalirazin-N,N-diacetic
Dung dịch I: Hòa tan 0,36 g ceri (III) nitrat (TT) trong nước vừa đủ 50 ml.
Dung dịch II: Phân tán 0,7 g acid 3-aminomethy-lalirazin-N,N-diacetic (TT) trong 50 ml nước. Hòa tan bằng cách thêm 0,25 ml amoniac (TT). Thêm 0,25 ml acid acetic băng (TT) và nước vừa đủ 100 ml.
Dung dịch III: Hòa tan 6 g natri acetat (TT) trong 50 ml nước, thêm 11,5 ml acid acetic băng (TT) và nước vừa đủ 100 ml.
Lấy 33 ml aceton (TT), thêm 6,8 ml dung dịch III, 1,0 ml dung dịch II, 1,0 ml dung dịch I và nước vừa đủ 50 ml. Chỉ dùng trong vòng 5 ngày.
Phải đạt yêu cầu sau:
Độ nhạy: Lấy 1,0 ml dung dịch fluorid chuẩn 10 phần triệu (TT), thêm 19,0 ml nước và 5,0 ml thuốc thử. Sau 20 min, dung dịch có màu xanh lam nhận thấy rõ.
Acid 4-aminomethylbenzoic
C8H9NO2= 151,2
Dùng loại tinh khiết hóa học.
Acid 8-aminonaphthalen-2-sulfonic
Acid 8-amino-2-naphthalensulfonic; Acid 1-naphthy-lamin-7-sulfonic
C10H9NO3S = 223,2
Dùng loại tinh khiết hóa học.
Dung dịch acid aminonaphthalensulfonic
Trộn 0,5 g acid 8-aminonaphthalen-2-sulfonic (TT), 30 ml acid acetic băng (TT) với 120 ml nước, đun nóng và khuấy cho đến khi hòa tan hoàn toàn. Để nguội, lọc.
Sử dụng dung dịch trong vòng 3 tuần.
Acid aspartic
Dùng loại tinh khiết hóa học.
Acid benzoic
C6H5COOH = 122,1
Dùng loại tinh khiết phân tích.
Acid boric
H3BO3 = 61,83
Dùng loại tinh khiết phân tích.
Dung dịch acid boric 5 %
Hòa tan 5 g acid boric (TT) trong nước nóng, thêm nước vừa đủ 100 ml.
Dung dịch acid boric 3%
Hòa tan 3 g acid boric (TT) trong nước nóng, thêm nước vừa đủ 100 ml.
Dung dịch acid boric
Hòa tan 5 g acid boric (TT) trong hỗn hợp 20 ml nước và 20 ml ethanol (TT). Thêm ethanol (TT) vừa đủ 250 ml.
Acid citric
C6H8O7.H2O = 210,1
Dùng loại tinh khiết phân tích.
Acid citric dùng trong phép thử giới hạn sắt phải thỏa mãn yêu cầu sau:
Hòa tan 0,5 g acid citric (TT) trong 10 ml nước, thêm 0,1 ml acid mercaptoacetic (TT), trộn đều, kiềm hóa bằng dung dịch amoniac 10 M(TT) và thêm nước vừa đủ 20 ml. Màu hồng không được xuất hiện.
Dung dịch acid citric 18%
Hòa tan 18 g acid citric (TT) với nước vừa đủ 100 ml.
Acid 2-clorobenzoic
C7H5CIO2 = 156,7
Tan trong nước, khó tan trong ethanol khan.
Điểm sôi: Khoảng 285 °C.
Điểm chảy: Khoảng 140 °C.
Acid cloroplatinic (IV)
Acid cloroplatinic, platinic clorid
H2PtCI6 + nước.
Chứa ít nhất 37,0 % (kl/kl) platin (p.t.l: 195,1).
Khối kết tinh hay tinh thể đỏ nâu. Rất tan trong nước, tan trong ethanol 96 %.
Định lượng: Nung 0,200 g chế phẩm đến khối lượng không đổi ở 900 °C ± 50 °C và cân khối lượng cặn (platin).
Bảo quản tránh ánh sáng.
Dung dịch acid cloroplatinic
Dung dịch acid cloroplatinic (IV) trong nước chứa 5.0 % H2PtCI6.6H2O
Acid cromic (dung dịch)
Hòa tan 84 g crom trioxyd (TT) trong 700 ml nước, vừa thêm vừa khuấy với 400 ml acid sulfuric (TT).
Acid cyclohexylenedinitrilotetra-acetic
Acid (±)-trans-1,2-diaminocyclohexan-N, N, N', N'-tetra-acetic
C14H22N2O8.H2O = 364,4
Dùng loại tinh khiết hóa học.
Bột kết tinh trắng.
Điểm chảy: Khoảng 204 °C.
Acid 3-cyclohexylpropionic
C9H16O2= 156,2
Dùng loại tinh khiết hóa học.
Tỷ trọng ở 20 °C: Khoảng 0,998.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,4648.
Điểm sôi: Khoảng 130 °C.
Acid ethylenedinitrilotetra-acetic
C10H16N2O8 = 292,2
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 250 °C.
Acid 2-ethylhexanoic
Acid 2-ethylhexoic
C8H16O2= 144,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Tỷ trọng ở 20 °C: Khoảng 0,91.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,425.
Phải đáp ứng phép thử sau:
Tạp chất liên quan: Tiến hành bằng phương pháp sắc ký khí (Phụ lục 5.2), dùng 1 μl dung dịch được chuẩn bị như sau: Trộn đều 0,2 g chế phẩm trong 5 ml nước, thêm 3 ml dung dịch acid hydrocloric 2 N (TT) và 5 ml hexan (TT), lắc trong 1 min, để yên cho tách lớp. Sử dụng lớp trên.
Sử dụng cách tiến hành như mô tả trong phép thử acid 2-ethylhexanoic, chuyên luận Amoxicilin natri. Tổng diện tích của các pic phụ không được lớn hơn 2,5 % diện tích của pic chính.
Acid fluofenamic
C14H10F3NO2 = 281,2
Dùng loại tinh khiết hóa học.
Tinh thể hình kim hay bột kết tinh màu vàng nhạt.
Điểm chảy: 132 °C đến 135 °C.
Acid formic
HCOOH = 46,03
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, mùi hăng cay và rất ăn da.
Khối lượng riêng: Khoảng 1,20 g/ml.
Hàm lượng HCOOH: Khoảng 90 % (kl/kl).
Acid formic khan
HCOOH = 46,03
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, mùi hăng cay và rất ăn da.
Tỷ trọng ở 20 °C: Khoảng 1,22.
Hàm lượng HCOOH: Không ít hơn 98,0 % (kl/kl).
Định lượng: Cân chính xác một bình nón đă chứa sẵn 10 ml nước, cho nhanh vào khoảng 1 ml acid formic (TT) rồi cân lại. Thêm 50 ml nước và chuẩn độ bằng dung dịch natri hydroxyd 1 N (CĐ), dùng 0,5 ml dung dịch phenolphtalein (TT) làm chỉ thị.
1 ml dung dịch natri hydroxyd 1 N (CĐ) tương đương với 46,03 mg HCOOH.
Acid hydrocloric (Acid hydrocloric đậm đặc)
HCI = 36,46
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, bốc khói.
Tỷ trọng ở 20 °C: Khoảng 1,18.
Hàm lượng HCI: 35 % đến 38 % (kl/kl), khoảng 11,5 M.
Bảo quản ở nhiệt độ không quá 30 °C, trong bao bì bằng polyethylen hoặc vật liệu không phản ứng với acid hydrocloric.
Dung dịch acid hydrocloric xM
Pha loãng 85x ml acid hydrocloric (TT) với nước vừa đủ 1000 ml.
Dung dịch acid hydrocloric 25 %
Pha loãng 61 ml acid hydrocloric (TT) với nước vừa đủ 100 ml.
Dung dịch acid hydrocloric 16%
Pha loãng 39 ml acid hydrocloric (TT) với nước vừa đủ 100 ml.
Dung dịch acid hydrocioric 10 %
Pha loãng 24 ml acid hydrocloric (TT) với nước vừa đủ 100 ml.
Dung dịch acid hydrocloric loãng
Pha loãng 17 ml acid hydrocloric (TT) với nước vừa đủ 100 ml.
Dung dịch acid hydrocloric 1 %
Pha loãng 2,4 ml acid hydrocloric (TT) với nước vừa đủ 100 ml.
Acid hydrocloric brom hóa
Dùng loại acid hydrocloric brom hóa có hàm lượng arsen thấp, hoặc điều chế bằng cách thêm 1 ml dung dịch brom (TT) vào 100 ml acid hydrocloric (TT).
Dung dịch acid hydrocloric thiếc hóa
Dùng loại acid hydrocloric thiếc hóa có hàm lượng arsen thấp, hoặc điều chế bằng cách thêm 1 ml dung dịch thiếc (II) clorid (TT) vào 100 ml acid hydrocloric (TT).
Dung dịch acid hydrocloric trong ethanol
Pha như các dung dịch acid hydrocloric khác, nhưng thay nước bằng ethanol 96 % (TT).
Nếu không ghi cụ thể nồng độ, dùng dung dịch sau: Pha loãng 5 ml dung dịch acid hydrocloric 1 M (TT) thành 500 ml với ethanol 96 % (TT).
Dung dịch acid hydrocloric trong methanol
Pha như các dung dịch acid hydrocloric khác, nhưng thay nước bằng methanol (TT).
Acid hydrocloric không có kim loại nặng
Phải đáp ứng các yêu cầu của acid hydrocloric (TT) và nồng độ tối đa của các kim loại nặng như sau: As: 0,005 phần triệu; Cd: 0,003 phần triệu; Cu: 0,003 phần triệu; Fe: 0,05 phần triệu; Hg: 0,005 phần triệu; Ni: 0,004 phần triệu; Pb: 0,001 phần triệu; Zn: 0,005 phần triệu.
Acid hydrofluoric
HF = 20,01
Dùng loại tinh khiết phân tích chứa không ít hơn 40 % (kl/kl) HF.
Chất lỏng không màu, mùi hăng, có tính ăn mòn, 1 ml acid có khối lượng khoảng 1,13 g.
Bảo quản trong lọ polyethylen.
Acid 2-hydroxyacetic
Acid glycollic
C2H4O3 = 76,05
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 80 °C.
Acid 4-hydroxybenzoic
Acid Parahydroxybenzoic
C7H6O3 = 138,1
Tinh thể khó tan trong nước, rất tan trong ethanol 96 %, tan trong aceton.
Điểm chảy: Khoảng từ 214 °C đến 215 °C.
Acid maleic
Acid (Z)-but-2-en-1,4-dioic
C4H4O4 = 116,1
Acid mercaptoacetic
Acid thioglycolic
HSCH2COOH = 92,12.
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, có mùi khó chịu.
Khối lượng riêng: Khoảng 1,33 g/ml.
Acid nitric (Acid nitric đậm đặc)
HN03 = 63,01
Dùng loại tinh khiết phân tích.
Chất lỏng bốc khói, ăn mòn, có nồng độ khoảng 16 M.
Khối lượng riêng: Khoảng 1,42 g/ml.
Hàm lượng HNO3: Khoảng 70 % (kl/kl).
Dung dịch acid nitric xM
Pha loãng 63x ml acid nitric (TT) với nước vừa đủ 1000 ml.
Dung dịch acid nitric 50 %
Pha loãng 80 g acid nitric (TT) với nước vừa đủ 100 ml.
Dung dịch acid nitric 32 %
Pha loãng 46 g acid nitric (TT) với nước vừa đủ 100 ml.
Dung dịch acid nitric 25 %
Pha loãng 40 g acid nitric (TT) với nước vừa đủ 100 ml.
Dung dịch acid nitric 16%
Pha loãng 23 g acid nitric (TT) với nước vừa đủ 100 ml.
Dung dịch acid nitric 12,5 % (Dung dịch acid nitric 2 M-Acid nitric loãng)
Pha loãng 20 g acid nitric (TT) với nước vừa đủ 100 ml.
Dung dịch acid nitric 10%
Pha loãng 15 g acid nitric (TT) với nước vừa đủ 100 ml.
Acid nitric bốc khói
HNO3 = 63,01
Dùng loại tinh khiết phân tích.
Chất lỏng màu vàng, bốc khói, ăn mòn.
Tỷ trọng ở 20 °C: Khoảng 1,5.
Hàm lượng HNO3: Khoảng 95 % (kl/kl).
Acid nitric không có chì
Phải đáp ứng phép thử sau:
Chì: không quá 0,1 phần triệu (Phụ lục 4.4). Đo độ hấp thụ ở 283,3 nm hoặc 217 nm, dùng đèn cathod rỗng chì, ngọn lửa acetylen - không khí. Dung dịch thử được chuẩn bị như sau: Thêm 0,1 g natri carbonat khan (TT) vào 100 g acid nitric (TT) và bốc hơi tới khô. Hòa tan cặn trong nước bằng cách đun nóng nhẹ và pha loãng thành 50 ml với nước.
Acid nitric không có cadmi và chì
Phải đáp ứng các phép thử sau:
Cadmi: Không được quá 0,1 phần triệu (Phụ lục 4.4). Đo độ hấp thụ ở 228,8 nm, dùng đèn cathod rỗng cadmi và ngọn lửa acetylen-không khí hay không khí - propan. Dung dịch thử được chuẩn bị như sau: Thêm 0,1 g natri carbonat khan (TT) vào 100 g acid nitric (TT) và bốc hơi tới khô. Hòa tan cặn trong nước bằng cách đun nóng nhẹ, pha loãng thành 50 ml với nước.
Chì: Không quá 0,1 phần triệu, xác định như acid nitric không có chì.
Acid nitric không có kim loại nặng
Phải đáp ứng các yêu cầu của acid nitric (TT) và nồng độ tối đa của các kim loại nặng như sau:
As: 0,005 phần triệu, Cd: 0,005 phần triệu, Cu: 0,001 phần triệu, Fe: 0,02 phần triệu, Hg: 0,002 phần triệu, Ni: 0,005 phần triệu, Pb: 0,001 phần triệu, Zn: 0,01 phần triệu.
Acid nitrilotriacetic
C9H9NO6 = 191,1
Bột kết tinh trắng hoặc gần như trắng. Thực tế không tan trong nước và trong hầu hết các dung môi hữu cơ.
Điểm chảy: Khoảng 246 °C, kèm phân hủy.
Acid oxalic
(C00H)2.2H2O = 126,07
Dùng loại tinh khiết hóa học.
Tinh thể màu trắng. Tan trong nước, dễ tan trong ethanol.
Dung dịch acid oxalic 10%
Hòa tan 10 g acid oxalic (TT) trong nước và thêm nước vừa đủ 100 ml.
Dung dịch acid oxalic 6,3 %
Hòa tan 6,3 g acid oxalic (TT) trong nước và thêm nước vừa đủ 100 ml.
Dung dịch acid oxalic 5 %
Hòa tan 5 g acid oxalic (TT) trong nước và thêm nước vừa đủ 100 ml.
Dung dịch acid oxalic 4 %
Hòa tan 4 g acid oxalic (TT) trong nước và thêm nước vừa đủ 100 ml.
Dung dịch acid oxalic trong acid sulfuric
Hòa tan 5 g acid oxalic (TT) trong hỗn hợp 50 ml nước và 50 ml acid sulfuric (TT) đã để nguội.
Acid percloric
HCIO4 = 100,46
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, hỗn hợp được với nước. Rất ăn mòn, có thể gây cháy nổ khi tiếp xúc với chất dễ oxy hóa.
Khối lượng riêng: Khoảng 1,7 g/ml.
Hàm lượng HClO4: 70,0 % đến 73,0 % (kl/kl).
Dung dịch acid percloric xM
Pha loãng 82x ml acid percloric (TT) với nước vừa đủ 1000 ml.
Dung dịch acid percloric
Pha loãng 8,5 ml acid percloric (TT) với nước vừa đủ 100 ml (khoảng 1 M).
Acid phenoldisulphonic, dung dịch
Dung dịch acid phenoldisulphonic
Chất lỏng trong, có thể có màu nâu nhạt trong quá trình bảo quản. Được điều chế bằng cách đun nóng 3,0 g phenol (TT) với 20 ml acid sulfuric đậm đặc (TT) trên cách thủy trong 6 h và chuyển dung dịch thu được vào bình nút mài. Hoặc bằng cách pha loãng dung dịch thương phẩm phenol 25 % với acid sulfuric (TT) thành dung dịch phenol 15% (kl/kl).
Dung dịch phải đạt phép thử sau:
Độ nhạy với nitrat: Cho bay hơi một lượng dung dịch chứa 0,1 mg kali nitrat (TT) đến khô trong chén sứ trên cách thủy. Thêm vào cặn đã nguội 1 ml thuốc thử và để yên 10 min. Thêm 10 ml nước, làm lạnh, thêm 10 ml dung dịch amoniac 5 M (TT) và pha loãng thành 25 ml với nước. Xuất hiện màu vàng, phân biệt rõ khi so với dung dịch được pha tương tự nhưng không có kali nitrat.
Acid phenylacetic
C8H8O2= 136,2
Dùng loại tinh khiết hóa học.
Điểm sôi: Khoảng 265 °C.
Điểm chảy: Khoảng 75 °C.
Acid phosphomolybdic
Acid dodecamolybdophosphoric
H3PO4.12MoO3.24H2O = 2258
Dùng loại tinh khiết phân tích.
Tinh thể mịn, màu vàng cam. Tan trong nước, ethanol và ether.
Dung dịch acid phosphomolybdic 10% trong ethanol
Hòa tan 10 g acid phosphomolybdic (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Dung dịch acid phosphomolybdic 5 % trong ethanol
Hòa tan 5 g acid phosphomolybdic (TT) trong ethanol 96 % (TT) và thêm ethanol 96 % (TT) vừa đủ 100 ml.
Acid phosphoric (Acid phosphoric đậm đặc)
Acid orthophosphoric
H3PO4 = 98,00
Dùng loại tinh khiết phân tích.
Chất lỏng ăn mòn.
Khối lượng riêng: Khoảng 1,75 g/ml.
Hàm lượng H3PO4: Không được nhỏ hơn 84 % (kl/kl).
Dung dịch acid phosphoric 25 %
Pha loãng 30 g acid phosphoric (TT) với nước vừa đủ 100 ml.
Dung dịch acid phosphoric 10%
Pha loãng 12 g acid phosphoric (TT) với nước vừa đủ 100 ml.
Dung dịch acid phosphoric 3 %
Pha loãng 3,5 g acid phosphoric (TT) với nước vừa đủ 100 ml.
Acid phosphotungstic, dung dịch
Dung dịch acid phosphotungstic
Hòa tan 10 g natri tungstat (TT) trong 75 ml nước và thêm 8 ml acid phosphoric (TT). Đun hồi lưu trong 3 h, để nguội, lọc và thêm nước vừa đủ 100 ml.
Acid picric
2,4,6- Trinitrophenol
C6H3O7N3 = 229,11
Dùng loại tinh khiết phân tích.
Tinh thể hình vẩy nhỏ hay lăng trụ, hay bột kết tinh màu vàng nhạt, bóng, không mùi, được làm ẩm đồng lượng với nước để đảm bảo an toàn, nổ khi đun nóng nhanh hoặc bị va đập.
Bảo quản ẩm với nước.
Dung dịch bão hòa acid picric
Cho dần 100 ml nước vào 12,3 g acid picric (TT), lắc liên tục, để yên 24 h, thỉnh thoảng lắc đều.
Bảo quản trong lọ thủy tinh màu nút mài, tránh ánh sáng.
Dung dịch acid picric 1 %
Hòa tan 1 g acid picric (TT) trong nước vừa đủ 100 ml.
Dung dịch acid picric
Thêm 0,25 ml dung dịch natri hydroxyd 10 M (TT) vào 100 ml dung dịch bão hòa acid picric (TT).
Dung dịch natri picrat kiềm
Trộn 20 ml dung dịch acid picric 1 % (TT) với 10 ml dung dịch natri hydroxyd 5 % (TT), thêm nước vừa đủ 100 ml.
Dung dịch chỉ dùng trong vòng 2 ngày.
Acid pteroic
Acid 4-{[(2-amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoic
C14H12N6O3= 312,3
Tinh thể, tan trong các dung dịch kiềm.
Acid salicylic
C7H6O3 = 138,1
Dùng loại tinh khiết phân tích.
Dung dịch acid salicylic 0,024 %
Hòa tan 0,24 g acid salicylic (TT) trong nước vừa đủ 1000 ml.
Pha trước khi dùng.
Dung dịch acid salicylic 0,01 %
Hòa tan 0,10 g acid salicylic (TT) trong nước vừa đủ 1000 ml.
Pha trước khi dùng.
Acid selenic
H2SeO3= 129,0
Dùng loại tinh khiết hóa học.
Bảo quản trong bao bì kín.
Acid silicowolframic
Acid silicotungstic
H4[Si(W3O10)4].xH2O = 2878,29 (khan)
Dùng loại tinh khiết hóa học.
Tinh thể màu trắng hay trắng ngà, dễ chảy nước. Rất dễ tan trong nước và ethanol.
Dung dịch acid silicowolframic 10 %
Hòa tan 10 g acid silicowolframic (TT) trong nước vừa đủ 100 ml.
Dung dịch acid silicowolframic 5 % (Thuốc thử Bertrand)
Hòa tan 5 g acid silicowolframic (TT) trong nước vừa đủ 100 ml.
Dung dịch acid silicowolframic 0,2 %
Hòa tan 0,2 g acid silicowolframic (TT) trong nước vừa đủ 100 ml.
Acid sulfamic
H3NO3S = 97,1
Dùng loại tinh khiết hóa học.
Bột kết tinh hay tinh thể màu trắng. Dễ tan trong nước, hơi tan trong aceton, ethanol 96 % và methanol, thực tế không tan trong ether.
Điểm chảy: Khoảng 205 °C kèm phân hủy.
Acid sulfanilic
C6H7NO3S = 173,2
Dùng loại tinh khiết phân tích.
Bột màu trắng hay gần như trắng. Rất dễ tan trong các dung dịch amoniac, natri hydroxyd và natri carbonat, tan trong nước nóng, tan rất ít trong nước.
Dung dịch acid sulfanilic 2,5%
Hòa tan 2,5 g acid sulfanilic (TT) trong nước vừa đủ 100 ml.
Dung dịch acid sulfanilic diazo hóa
Hòa tan 0,9 g acid sulfanilic (TT) trong 9 ml acid hydrocloric (TT) bằng cách đun nóng, pha loãng thành 100 ml với nước. Lấy 10 ml dung dịch thu được, làm lạnh trong nước đá và thêm 10 ml dung dịch natri nitrit 4,5 % (TT) đã được làm lạnh trước. Để trong nước đá 15 min, trước khi dùng thêm 20 ml dung dịch natri carbonat 10 % (TT).
Dung dịch acid sulfanilic diazo hóa (TT1)
Hòa tan bằng cách đun nóng 0,2 g acid sulfanilic (TT) trong 20 ml dung dịch acid hydrocloric 1 N (TT). Làm lạnh trong nước đá, thêm từng giọt 2,2 ml dung dịch natri nitrit 4 % (TT), lắc liên tục. Để yên trong nước đá 10 min, thêm 1 ml dung dịch acid sulfamic 5 %.
Acid sulfosalicylic
C6H3 (OH) (SO3H)COOH.2H2O = 254,22
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng. Rất dễ tan trong nước và ethanol, tan trong ether.
Điểm chảy: Khoảng 109 °C.
Dung dịch acid sulfosalicylic
Hòa tan 100 g acid sulfosalicylic (TT) trong nước vừa đủ 1000 ml.
Bảo quản trong lọ thủy tinh màu, tránh ánh sáng.
Acid sulfuric (Acid sulfuric đậm đặc)
H2SO4 = 98,08
Dùng loại tinh khiết phân tích.
Chất lỏng sánh, ăn mòn mạnh.
Khối lượng riêng: Khoảng 1,84 g/ml.
Hàm lượng H2SO4: Khoảng 96 % (kl/kl).
Dung dịch acid sulfuric xM
Cho cẩn thận 54x ml acid sulfuric (TT) vào đồng thể tích nước, thêm nước vừa đủ 1000 ml.
Dung dịch acid sulfuric 50 %
Cho từ từ 285 ml acid sulfuric (TT) vào 500 ml nước, lắc liên tục. Làm nguội, thêm nước vừa đủ 1000 ml.
Dung dịch acid sulfuric 38 %
Cho từ từ 22 ml acid sulfuric (TT) vào 60 ml nước, lắc liên tục. Làm nguội, thêm nước vừa đủ 100 ml.
Dung dịch acid sulfuric 20 %
Cho từ từ 11,5 ml acid sulfuric (TT) vào 80 ml nước, lắc liên tục. Làm nguội, thêm nước vừa đủ 100 ml.
Dung dịch acid sulfuric 10 % (Acid sulfuric loãng)
Cho từ từ 6 ml acid sulfuric (TT) vào 50 ml nước, lắc liên tục. Làm nguội, thêm nước vừa đủ 100 ml.
Dung dịch acid sulfuric 5 %
Pha loãng gấp đôi dung dịch acid sulfuric 10 % (TT) với nước.
Dung dịch acid sulfuric 2 %
Pha loãng gấp 5 lần dung dịch acid sulfuric 10% (TT) với nước.
Dung dịch acid sulfuric 1 %
Pha loãng gấp 10 lần dung dịch acid sulfuric 10% (TT) với nước.
Dung dịch acid sulfuric trong ethanol
Pha như các dung dịch acid sulfuric khác, nhưng thay nước bằng ethanol 96 % (TT).
Nếu không ghi rõ nồng độ, dùng dung dịch sau: cẩn thận và làm lạnh liên tục, khuấy 20 ml acid sulfuric (TT) với 60 ml ethanol 96 % (TT), để lạnh và pha loãng thành 100 ml với ethanol 96 % (TT).
Dung dịch chỉ pha khi dùng.
Dung dịch acid sulfuric trong methanol
Pha như các dung dịch acid sulfuric khác, nhưng thay nước bằng methanol (TT).
Acid sulfuric không có kim loại nặng
Phải đáp ứng các yêu cầu của acid sulfuric (TT) và nồng độ tối đa của các kim loại nặng như sau:
As: 0,005 phần triệu, Cd: 0,002 phần triệu, Cu: 0,001 phần triệu, Fe: 0,05 phần triệu, Hg: 0,005 phần triệu, Ni: 0,002 phần triệu, Pb: 0,001 phần triệu, Zn: 0,005 phần triệu.
Acid sulfuric không có nitrogen
H2SO4 = 98,08
Dùng loại tinh khiết phân tích.
Có chứa khoảng 96 % (kl/kl) H2SO4. Phải đáp ứng phép thử sau:
Nitrat: Lấy 5 ml nước, thêm cẩn thận vào 45 ml chế phẩm thử, để mát đến 40 °C và thêm 8 mg N,N’- diphenylbenzidin. Dung dịch có màu hồng nhạt hoặc màu xanh rất nhạt.
Acid tartric
C4H6O6 = 150,09
Dùng loại tinh khiết phân tích.
Tinh thể không màu hay bột kết tinh màu trắng. Dễ tan trong nước.
Dung dịch acid tartric 20 %
Hòa tan 20 g acid tartric (TT) trong nước vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Acid toluensulfonic
Acid 4-methyIbenzensulfonic; Acid toluen-p-sulfonic
C7H8O3S. H2O = 190,2
Dùng loại tinh khiết phân tích, chứa ít nhất 87,0 % C7H8O3S.
Bột kết tinh hay tinh thể màu trắng. Dễ tan trong nước, tan trong ethanol và ether.
Acid tricloroacetic
C2HCl3O2 = 163,4
Dùng loại tinh khiết phân tích.
Tinh thể không màu, dễ chảy nước, có mùi hăng cay. Rất dễ tan trong ether và ethanol, tan trong nước.
Dung dịch acid tricloroacetic
Hòa tan 40,0 g acid tricloroacetic (TT) trong nước và pha loãng thành 1000,0 ml với cùng dung môi. Chuẩn độ bằng dung dịch natri hydroxyd 0,1 M (CĐ) để xác định nồng độ dung dịch. Điều chỉnh nếu cần thiết để thu được dung dịch có nồng độ (4 ± 0,1) %.
Dung dịch acid tricloroacetic 10 % trong methanol
Hòa tan 10 g acid tricloroacetic (TT) trong methanol (TT) vừa đủ 100 ml.
Acid trifluoroacetic
C2HF3O2 = 114,0
Dùng loại phù hợp với trình tự protein có chứa không ít hơn 99 % C2HF3O2
Điểm sôi: Khoảng 72 °C.
Tỷ trọng ở 20 °C: Khoảng 1,53.
Bảo quản tránh ánh sáng.
Acid tropic
Acid (2RS)-3-hydroxy-2-phenylpropanoic
C9H10O3= 166,17
Adrenalon hydroclorid
1-(3,4-Dihydroxyphenyl)-2-(methylamino)ethanon hydroclorid;
3’, 4’-Dihydroxy-2-(methylamino)acetophenon hydroclorid
C9H12ClNO3 = 217,7
Dùng loại tinh khiết hóa học.
Alcol amylic
Alcol isoamylic; 3-methylbutan-1-ol
C5H12O = 88,15
Dùng loại tinh khiết phân tích.
Chất lỏng không màu.
Điểm sôi: Khoảng 130 °C.
Khối lượng riêng: Khoảng 0,81 g/ml.
Aldehyd dehydrogenase
Enzym thu được từ nấm men bánh mì để xúc tác quá trình oxy hóa acetaldehyd thành acid acetic với sự có mặt của nicotinamid adenin dinucleotid, muối kali và các thiol ở pH 8,0.
Dùng loại tinh khiết hóa học.
Dung dịch aldehyd dehydrogenase
Hòa tan một lượng aldehyd dehydrogenase (TT) tương đương với 70 đơn vị trong nước và pha loãng thành 10 ml với cùng dung môi. Dung dịch thu được ổn định trong 8 h ở 4 °C.
Alizarin S
Alizarin đỏ S
C14H7NaO7S. H2O = 360,3
Dùng loại tinh khiết hóa học.
Bột màu nâu vàng hay vàng cam. Dễ tan trong nước và ethanol 96 %.
Dung dịch alizarin S
Hòa tan 0,1 g alizarin S (TT) trong nước vừa đủ 100 ml.
Phải đáp ứng phép thử sau:
Độ nhạy với bari: Thêm 5 ml nước, 50 ml dung dịch đệm acetat pH 3,7 (TT) và 0,5 ml dung dịch alizarin S (TT) vào 5 ml dung dịch acid sulfuric 0,05 M (CĐ). Thêm từng giọt dung dịch bari perclorat 0,05 M (CĐ). Màu chuyển từ vàng sang đỏ cam.
Khoảng chuyển màu: pH 3,7 (vàng) đến pH 5,2 (tím).
Amaranth S
C20H11N2Na3O10S3= 604
Dùng loại tinh khiết hóa học.
Bột mịn màu nâu đậm hoặc màu nâu đỏ đậm.
Khi dùng để chuẩn độ iod và iodid bằng kali iodat, màu chuyển từ đỏ cam sang vàng.
Dung dịch amaranth S 0,2 %
Hòa tan 0,2 g amaranth S (TT) trong nước vừa đủ 100 ml.
2-Aminobutan-1-ol
Aminobutanol
C4H11NO = 89,14
Dùng loại tinh khiết hóa học.
Dạng dầu lỏng.
Điểm sôi: Khoảng 180 °C.
Khối lượng riêng ở 20 °C: Khoảng 0,94.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,453.
4-Aminophenol
C6H7NO = 109,1
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng hoặc có màu nhẹ.
Điểm chảy: Khoảng 186 °C, kèm theo phân hủy.
Hàm lượng C6H7NO: Không được ít hơn 95 %.
Bảo quản tránh ánh sáng.
3-Aminopropanol
3-Aminopropan-1-ol
C3H8NO = 75,1
Dùng loại tinh khiết hóa học.
Chất lỏng sánh, không màu, trong.
Tỷ trọng ở 20 °C: Khoảng 0,99.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,461.
Điểm chảy: Khoảng 11 °C.
Aminopyrazolon
4-Aminophenazon
C11H13N3O = 203,3
Dùng loại tinh khiết hóa học.
Bột hay tinh thể hình kim màu vàng nhạt.
Điểm chảy: Khoảng 108 °C.
Dung dịch aminophenazon (Dung dịch aminopy-razolon)
Dung dịch 4-aminophenazon 0,1 % trong dung dịch đệm borat pH 9,0.
Amoni acetat
CH3CO2NH4 = 77,08
Dùng loại tinh khiết phân tích.
Tinh thể không màu, rất dễ chảy nước. Rất dễ tan trong ether và ethanol, tan trong nước.
Dung dịch amoni acetat
Hòa tan 150 g amoni acetat (TT) trong nước, thêm 3 ml acid acetic băng (TT) và nước vừa đủ 1000 ml.
Chỉ dùng trong vòng 1 tuần sau khi pha.
Dung dịch amoni acetat 2 M (dung dịch amoni acetat 15,4 %)
Hòa tan 154 g amoni acetat (TT) trong nước vừa đủ 1000 ml.
Amoni carbonat
Amoni carbonat là hỗn hợp các tỷ lệ khác nhau của amoni hydrocarbonat (NH4HCO3 = 79,1) và amoni carbamat (NH2COONH4 = 78,1).
Dùng loại tinh khiết phân tích.
Bột màu trắng trong. Tan trong nước, phân hủy trong nước nóng, không tan trong ethanol và dung dịch amoniac đậm đặc.
Hàm lượng NH3 giải phóng ra không được ít hơn 30 % (kl/kl).
Bảo quản trong đồ đựng kín, ở nhiệt độ không quá 20 °C.
Dung dịch amoni carbonat 20 %
Hòa tan 20 g amoni carbonat (TT) trong nước vừa đủ 100 ml.
Dung dịch bão hòa amoni carbonat
Hòa tan amoni carbonat (TT) trong nước đến khi thu được dung dịch bão hòa, lọc.
Dung dịch amoni carbonat loãng
Hòa tan 5 g amoni carbonat (TT) trong hỗn hợp 7,5 ml dung dịch amoniac 5 M (TT) và 50 ml nước, pha loãng thành 100 ml với nước, lọc nếu cần.
Amoni ceri (IV) nitrat
Amoni and ceri nitrat, ceri amoni nitrat
(NH4)2[Ce(NO3)6] = 548,2
Dùng loại tinh khiết phân tích.
Amoni ceri sulfat
Xem Ceri amoni sulfat.
Amoni clorid
NH4CI = 53,49
Dùng loại tinh khiết phân tích.
Tinh thể trắng, không mùi, vị mát và hơi mặn. Tan trong nước, tan trong ethanol.
Dung dịch amoni clorid 10 %
Hòa tan 10 g amoni clorid (TT) trong nước vừa đủ 100 ml.
Amoni dihydrophosphat
(NH4)H2PO4 = 115,0
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước, không tan trong ethanol.
Amoni format
Ethyl nitro
C2H5NO2 = 63,1
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng từ 119 °C đến 121 °C.
Bảo quản trong bao bì kín.
Amoni hydrocarbonat
Amoni bicarbonat
NH4HCO3 = 79,06
Hàm lượng NH4HCO3: Không được ít hơn 99 %.
Dùng loại tinh khiết phân tích.
Amoni molybdat
(NH4)6Mo7O24. 4H2O = 1235,86
Dùng loại tinh khiết phân tích.
Tinh thể không màu hoặc màu lục vàng. Tan trong nước.
Dung dịch amoni molybdat
Hòa tan 15 g amoni molybdat (TT) trong 40 ml nước. Rót từ từ dung dịch thu được vào 130 ml acid nitric loãng (TT), khuấy liên tục. Để yên hỗn hợp trong 24 h, sau đó gạn lấy lớp trong.
Dung dịch không được vẩn đục khi đun tới 50 °C.
Trộn 5 ml dung dịch với 2 ml dung dịch natri hydro-phosphat 0,01 %, đun nhẹ. Tủa vàng phải xuất hiện rõ. Nếu kết tủa ít, phải pha dung dịch amoni molybdat mới.
Bảo quản dung dịch ở chỗ tối. Nếu trong quá trình bảo quản xuất hiện tủa, gạn lấy phần trong.
Dung dịch amoni molybdat 5 % trong acid sulfuric
Hòa tan 0,5 g amoni molybdat (TT) trong 10 ml acid sulfuric (TT).
Dung dịch chỉ pha khi dùng.
Dung dịch amoni molybdat - acid sulfuric
Hòa tan 10 g amoni molybdat (TT) trong nước vừa đủ 100 ml. Thêm từ từ dung dịch thu được vào 250 ml dung dịch acid sulfuric 10 M (TT) đã được làm lạnh.
Bảo quản trong chai nhựa, tránh ánh sáng.
Dung dịch sulphomolybdic
Hòa tan bằng cách đun nóng 2,5 g amoni molybdat (TT) trong 20 ml nước (dung dịch 1). Thêm từ từ 28 ml acid sulfuric (TT) vào 50 ml nước và làm lạnh (dung dịch 2). Trộn dung dịch 1 vào dung dịch 2, thêm nước vừa đủ 100 ml.
Bảo quản trong chai lọ polyethylen.
Amoni oxalat
(COONH4)2 H2O = 142,11
Dùng loại tinh khiết phân tích.
Tinh thể không màu. Tan trong nước.
Dung dịch amoni oxalat bão hòa
Hòa tan amoni oxalat (TT) trong nước đến khi thu được dung dịch bão hòa, lọc.
Dung dịch amoni oxalat 5 %
Hòa tan 5 g amoni oxalat (TT) trong nước vừa đủ 100 ml.
Dung dịch amoni oxalat 4 %
Hòa tan 4 g amoni oxalat (TT) trong nước vừa đủ 100 ml.
Dung dịch amoni oxalat 3,5%
Hòa tan 3,5 g amoni oxalat (TT) trong nước vừa đủ 100 ml.
Dung dịch amoni oxalat 2,5%
Hỏa tan 2,5 g amoni oxalat (TT) trong nước vừa đủ 100 ml.
Amoni phosphat
Diamoni hydrophosphat; Diamoni hydrogen ortho-phosphat
(NH4)2HPO4 = 132,1
Dùng loại tinh khiết phân tích.
Bột tinh thể hay tinh thể màu trắng. Tan trong nước, không tan trong ethanol.
Amoni persulfat
(NH4)2S2O8 = 228,20
Dùng loại tinh khiết phân tích.
Bột tinh thể màu trắng. Dễ tan trong nước.
Dung dịch amoni persulfat 20 %
Hòa tan 20 g amoni persulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch amoni persulfat 10 %
Hòa tan 10 g amoni persulfat (TT) trong nước vừa đủ 100 ml.
Amoni pyrolidin dithiocarbamat
Amoni tetramethylen dithiocarbamat
C5H12N2S2 = 164,3
Dùng loại tinh khiết hóa học.
Bột tinh thể màu trắng đến vàng nhạt. Hơi tan trong nước, rất khó tan trong ethanol.
Bảo quản trong lọ có chứa miếng amoni carbonat để ở trong túi vải mút-xơ-lin.
Dung dịch amoni pyrolidin dithiocarbamat
Hòa tan 1 g amoni pyrolidin dithiocarbamat (TT) với nước vừa đủ 100 ml. Ngay trước khi dùng, rửa 3 lần, mỗi lần với 25 ml 4-methylpentan-2-on (TT).
Amoni sulfamat
NH2SO3NH4 = 114,1
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng hay tinh thể không màu, hút ẩm. Dễ tan trong nước, khó tan trong ethanol.
Điểm chảy: Khoảng 130 °C.
Amoni sulfat
(NH4)2SO4 = 132,14
Dùng loại tinh khiết phân tích.
Tinh thể không màu hay bột cốm màu trắng. Rất dễ tan trong nước.
Amoni sulfocyanid
Amoni thiocyanat
NH4SCN = 76,12
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng. Rất dễ tan trong nước và ethanol, tan trong methanol và aceton, hầu như không tan trong cloroform và ethyl acetat.
Dung dịch amoni sulfocyanid (Dung dịch amoni sulfocyanid 1 M)
Hòa tan 7,6 g amoni sulfocyanid (TT) trong 50 ml nước và thêm nước vừa đủ 100 ml.
Dung dịch amoni cobalthiocyanat
Hòa tan 37,5 g cobalt (II) nitrat (TT) và 150 g amoni thiocyanat (TT) trong nước vừa đủ 1000 ml.
Pha trước khi dùng.
Amoni vanadat
Amonimeta vanadat
NH4VO3 = 116,98
Dùng loại tinh khiết phân tích.
Bột tinh thể màu trắng đến hơi vàng. Khó tan trong nước, tan trong dung dịch amoniac loãng.
Dung dịch amoni metavanadat (Dung dịch amoni vanadat)
Hòa tan 5 g amoni vanadat (TT) trong hỗn hợp gồm 10 ml dung dịch natri hydroxyd 5 N (TT) và 90 ml nước bằng cách đun nóng. Để nguội, lọc qua bông thủy tinh nếu cần.
Dung dịch amoni vanadat 1 % trong acid sulfuric
Hòa tan 0,1 g amoni vanadat (TT) trong 10 ml acid sulfuric (TT).
Amoniac (Amoniac đậm đặc)
Amoniac 13,5 M
NH3= 17,03
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, mùi mạnh đặc biệt.
Tỷ trọng ở 20 °C: Khoảng 0,91.
Hàm lượng NH3: Khoảng 25 % (kl/kl).
Dung dịch amoniac xM
Pha loãng 75x ml amoniac (TT) với nước thành 1000 ml.
Dung dịch amoniac loãng (Dung dịch amoniac 10 %)
Lấy 440 ml amoniac (TT) hòa vào nước và thêm nước vừa đủ 1000 ml.
Dung dịch amoniac 5 %
Lấy 500 ml dung dịch amoniac loãng (TT) hòa vào nước và thêm nước vừa đủ 1000 ml.
Dung dịch amoniac trong methanol
Pha như các dung dịch amoniac khác, nhưng thay nước bằng methanol (TT).
Dung dịch amoniac loãng (TT3)
Pha loãng 0,7 g amoniac đậm đặc (TT) thành 100 ml bằng nước. Dung dịch thu được không được chứa ít hơn 0,16 % (kl/tt) và không được lớn hơn 0,18 % (kl/tt) NH3.
Dung dịch kiềm - amoniac
Lấy 5 g natri hydroxyd (TT) thêm 2 ml amoniac đậm đặc (TT), thêm nước vừa đủ 100 ml.
Amoniac 18 M
NH3= 17,03
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, mùi mạnh đặc biệt.
Tỷ trọng ở 20 °C: Khoảng 0,88.
Hàm lượng NH3: Khoảng 35 % (kl/kl).
Anhydrid acetic
(CH3CO)2O = 102,09
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, mùi hắc. Dung dịch trong nước bị thủy phân nhanh và tỏa mùi acid acetic.
Điểm sôi: 136 - 142 °C.
Hàm lượng (CH3CO)2O: Không được ít hơn 97,0 %.
Anhydrid maleic
Furan-2,5-dione
C4H2O3 = 98,06
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 52 °C.
Cặn không tan trong toluen: Không quá 5 % (acid maleic).
Anhydrid pentafluoropropionic
C6F10O3 = 310,0
Anhydrid propionic
C6H10O3 = 130,1
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, mùi hăng. Tan trong ethanol và ether.
Khối lượng riêng: Khoảng 1,01 g/ml.
Điểm sôi: Khoảng 167 °c.
Thuốc thử anhydrid propionic
Hòa tan 1 g acid toluensulfonic (TT) trong 30 ml acid acetic băng (TT), thêm 5 ml anhydric propionic (TT), để yên ít nhất 15 min.
Sử dụng trong 24h.
Anilin
C6H5NH2 = 93,13
Dùng loại tinh khiết phân tích.
Chất lỏng sánh, không màu hay màu vàng nhạt.
Tỷ trọng ở 20 °C: Khoảng 1,02.
Điểm sôi: Khoảng 183 °C đến 186 °C.
Bảo quản tránh ánh sáng.
Dung dịch anilin 2,5%
Hòa tan 2,5 g anilin (TT) trong 80 ml nước và thêm nước vừa đủ 100 ml.
Dung dịch anilin trong cyclohexan
Hòa tan 0,9 g anilin (TT) trong cyclohexan (TT) vừa đủ 100 ml.
Anisaldehyd
4-Methoxybenzaldehyd
C8H8O2= 136,2
Dùng loại tinh khiết hóa học.
Chất lỏng sánh, không màu đến vàng nhạt, có mùi thơm.
Điểm sôi: Khoảng 248 °C.
Khối lượng riêng: Khoảng 1,125 g/ml.
Dung dịch anisaldehyd
Trộn đều theo thứ tự 0,5 ml anisaldehyd (TT), 10 ml acid acetic băng (TT), 85 ml methanol (TT) và 5 ml acid sulfuric (TT).
Dung dịch anisaldehyd trong ethanol
Thêm 90 ml ethanol 96 % (TT) vào 10 ml anisaldehyd (TT), trộn đều. Thêm 10 ml acid sulfuric (TT) và trộn đều.
Dung dịch anisaldehyd trong acid sulfuric
Trộn 10 ml acid sulfuric (TT) vào 5 ml anisaldehyd (TT).
Antimony triclorid
Stibi triclorid
SbCI3 = 228,1
Dùng loại tinh khiết phân tích.
Tinh thể hay vẩy không màu, bốc khói trong không khí ẩm. Tan trong ethanol, aceton, ether và benzen.
Dung dịch antimony triclorid
Rửa nhanh 30 g antimony triclorid (TT) 2 lần, mỗi lần với 15 ml cloroform không có ethanol (TT). Hòa tan ngay các tinh thể đã rửa trong 100 ml cloroform không có ethanol (TT) bằng cách làm nóng nhẹ.
Bảo quản với vài gam natri sulfat khan.
Thuốc thử antimony triclorid
Chuẩn bị 2 dung dịch gốc bằng cách sau:
Dung dịch A: Hòa tan 110 g antimony triclorid (TT) trong 400 ml ethylen clorid (TT), thêm 2 g nhôm oxyd khan (TT), trộn đều, lọc qua phễu xốp vào bình định mức 500 ml, pha loãng thành 500 ml với ethylen clorid (TT). Độ hấp thụ của dung dịch thu được (Phụ lục 4.1), đo trong cốc dày 2 cm, không được lớn hơn 0,07, dùng ethylen clorid (TT) làm mẫu trắng.
Dung dịch B: Trộn 100 ml acetyl clorid (TT) đã cất lại, không màu với 400 ml ethylen clorid (TT) và bảo quản ở chỗ lạnh.
Trộn đều 90 ml dung dịch A với 10 ml dung dịch B. Bảo quản trong chai thủy tinh màu nút mài. Dùng trong vòng 7 ngày. Nếu thuốc thử có màu thì không dùng.
Arginin
L-Arginin
C6H14N4O2= 174,2
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 235 °C, kèm phân hủy.
Arsen trioxyd
As2O3 = 197,84
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng. Khó tan trong nước, tan trong nước sôi.
Dung dịch natri arsenit 0,1 M
Hòa tan 4,946 g arsen trioxyd (TT) trong hỗn hợp gồm 20 ml dung dịch natri hydroxyd 10 M (TT) và 20 ml nước, pha loãng thành 400 ml với nước, thêm dung dịch acid hydrocloric 2 M (TT) đến khi trung tính với giấy quỳ. Hòa tan 2 g natri hydrocarbonat (TT) trong dung dịch này và thêm nước vừa đủ 500 ml.
Barbiton
Barbital; 5,5-diethylbarbituric acid
C8H12N2O3 = 184,2
Điểm chảy: Khoảng 190 °C.
Barbiton natri
Barbital natri; natri 5,5-diethylbarbiturat
C8H11N2NaO3 = 206,2
Dùng loại tinh khiết hóa học.
Bari clorid
BaCI2.2H2O = 244,28
Dùng loại tinh khiết phân tích.
Tinh thể không màu. Dễ tan trong nước.
Dung dịch bari clorid 5 %
Hòa tan 5 g bari clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch bari clorid 0,5 M
Hòa tan 12,2 g bari clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch bari clorid 0,25 M (6,1 %)
Hòa tan 6,1 g bari clorid (TT) trong nước vừa đủ 100 ml.
Bari hydroxyd
Ba(OH)2. 8H2O = 315,5
Dùng loại tinh khiết phân tích.
Tinh thể không màu. Tan trong nước.
Dung dịch bari hydroxyd
Hòa tan 4,73 g bari hydroxyd (TT) trong nước vừa đủ 100 ml.
Bạc nitrat
AgNO3 = 169,87
Dùng loại tinh khiết phân tích.
Tinh thể không màu hay màu trắng, chuyển thành màu xám đen khi để ngoài ánh sáng.
Dung dịch bạc nitrat 5 %
Hòa tan 5 g bạc nitrat (TT) trong nước vừa đủ 100 ml.
Bảo quản trong lọ thủy tinh màu, có nút mài.
Dung dịch bạc nitrat 4,25 %
Hòa tan 4,25 g bạc nitrat (TT) trong nước vừa đủ 100 ml.
Bảo quản trong chai lọ thủy tinh màu, có nút mài.
Dung dịch bạc nitrat 4 %
Hòa tan 4 g bạc nitrat (TT) trong nước vừa đủ 100 ml.
Bảo quản trong lọ thủy tinh màu, có nút mài.
Dung dịch bạc nitrat 2 %
Hòa tan 2 g bạc nitrat (TT) trong nước vừa đủ 100 ml.
Bảo quản trong lọ thủy tinh màu, có nút mài.
Bạc oxyd
Ag2O = 231,7
Dùng loại tinh khiết phân tích.
Bột màu đen nâu. Hầu như không tan trong nước và ethanol, dễ tan trong acid nitric loãng và amoniac. Bảo quản tránh ánh sáng.
Benzen
C6H6 = 78,11
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, dễ bắt lửa.
Điểm sôi: Khoảng 80 °C.
Benzil
Diphenylethandion
C14H10O2= 210,2
Dùng loại tinh khiết hóa học.
Điểm chảy: 95 °C.
Benzophenon
C13H10O = 182,2
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 48 °C.
1,4-Benzoquinon
Cyclohexa-2,5-dien-1,4-dion
C6H4O2 = 108,1
Dùng loại tinh khiết phân tích, chứa không ít hơn 98,0 % C6H4O2.
Bột kết tinh vàng, có mùi đặc trưng. Ít tan trong nước, tan trong ethanol và ether.
Benzoyl clorid
C7H5CIO = 140,6
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, gây chảy nước mắt, bốc khói trong không khí ẩm.
Tỷ trọng ở 20 °C: Khoảng 1,21.
Điểm sôi: Khoảng 197 °C.
Bismuth nitrat pentahydrat
Bi(NO3)3.5H2O = 485,1
Điểm chảy: Khoảng 30 °C.
Bismuth nitrat base
Bismuth oxynitrat; Bismuth subnitrat
4[BiNO3(OH2).BiO(OH)] = 1462
Muối base chứa khoảng 80 % Bi2O3.
Bột màu trắng, nặng, hơi hút ẩm. Tan trong acid hydrocloric, acid nitric, acid sulfuric loãng và acid acetic, hầu như không tan trong nước và ethanol.
Borneol
D-bomeol, endo-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ol
C10H18O = 154,3
Tinh thể không màu, dễ thăng hoa, thực tế không tan trong nước, dễ tan trong ethanol 96 % và ether dầu hỏa có nhiệt đội sôi từ 50 °C đến 70 °C.
Điểm chảy: Khoảng 208 °C.
Phải đạt yêu cầu sau:
Tính đồng nhất: Tiến hành phương pháp sắc lý lớp mỏng (Phụ lục 5.4). Bản mỏng silica gel G (TT), pha động cloroform (TT). Chấm lên bản mỏng 10 μl dung dịch chế phẩm 0,1 % trong toluen (TT). Triển khai sắc ký đến khi dung môi đi được 10 cm, để khô bản mỏng ngoài không khí và phun dung dịch anisaldehyd (TT) (dùng 10 ml dung dịch cho bản mỏng 200 mm x 200 mm) và sấy khô ở nhiệt độ 100 °C đến 105 °C trong 10 min. Sắc ký đo chỉ được có duy nhất một vết chính.
Boron triclorid
BCI3 = 117,2
Dùng loại tinh khiết hóa học.
Khí không màu, phản ứng rất mạnh với nước. Tồn tại ở dạng dung dịch trong các dung môi thích hợp (2-cloroethanol, dicloromethan, hexan, heptan, methanol).
Điểm sôi: Khoảng 12,6 °C.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,420.
Dung dịch boron triclorid trong methanol
Dung dịch 12 % boron triclorid (TT) trong methanol (TT).
Bảo quản tránh ánh sáng ở -20 °C, nắp kín.
Boron trifluorid
BF3 = 67,8
Dùng loại tinh khiết hóa học.
Khí không màu.
Dung dịch boron trifluorid (Dung dịch boron trif-luorid trong methanol)
Dung dịch chứa khoảng 14 % BF3 trong methanol (TT).
Brom
Br2 = 159,8
Dùng loại tinh khiết phân tích.
Chất lỏng nặng, bốc khói, màu đỏ nâu, ăn da mạnh.
Tỷ trọng ở 20 °C: Khoảng 3,1.
Nước brom (Dung dịch bão hòa brom)
Điều chế bằng cách thỉnh thoảng lắc 3 ml brom (TT) với 100 ml nước trong 24 h, rồi để yên cho tách lớp.
Bảo quản trong lọ thủy tinh màu nút mài, tránh ánh sáng và ở trên một lượng dư brom (TT).
Dung dịch brom
Hòa tan 30 g brom (TT) và 30 g kali bromid (TT) trong nước vừa đủ 100 ml.
Dung dịch brom trong acid acetic
Hòa tan 100 g kali acetat (TT) trong acid acetic băng (TT), thêm 4 ml brom (TT) và acid acetic băng (TT) vừa đủ 1000 ml.
Bromelain
Cô đặc các enzym thủy phân protein được lấy từ Ananas comosus Merr.
Bột màu vàng xám.
Phải đáp ứng phép thử sau:
Hoạt tính: 1 g chế phẩm làm giải phóng khoảng 1,2 g amino nitrogen từ một dung dịch gelatin chuẩn trong 20 min, ở nhiệt độ 45 °C và pH 4,5.
Butan-2-on
Methyl ethyl keton; Ethyl methyl keton
C4H8O = 72,11
Dùng loại tinh khiết sắc ký.
Chất lỏng không màu, dễ bắt lửa, có mùi đặc trưng.
Điểm sôi: Khoảng 79 °C.
Tỷ trọng ở 20 °C: Khoảng 0,81.
Butanol
n-Butanol, Butan-1-ol
CH3(CH2)2CH2OH = 74,12
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu.
Tỷ trọng ở 20 °C: Khoảng 0,81.
Điểm sôi: 116 đến 119 °C.
tert-Butanol
Cồn tert- butyl; 2-Methyl-2-propanol; 2-Methyl-propan-2-ol
C4H10O = 74,1
Sử dụng loại tinh khiết phân tích.
Chất rắn hay chất lỏng sánh không màu, mùi đặc trưng. Hòa trộn với ethanol và ether.
Điểm đông đặc: Khoảng 25 °C.
Điểm sôi: Khoảng 82 °C.
Khối lượng riêng ở 26 °C: Khoảng 0,78 g/ml.
Butyl Acetat
C6H12O2= 116,2
Chất lỏng không màu, trong, dễ chảy, khó tan trong nước, có thể trộn lẫn với ethanol 96 %.
Tỷ trọng ở 20 °C: Khoảng 0,88.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,395.
Phần chưng cất được trong khoảng 123 °C đến 126 °C không được ít hơn 95 %.
Butylamin
n-Butylamin
C4H11N = 73,1
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, có mùi amoniac.
Điểm sôi: Khoảng 78 °C.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,401.
Cất và dùng trong vòng 1 tháng.
Butyl hydroxytoluen
BHT; 2,6-Di-tert-butyl-p-cresol
C15H24O = 220,4
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng hay tinh thể không màu.
Điểm chảy: Khoảng 70 °C
Calcein
Bột dạng hạt màu trắng hoặc vàng nhạt, không mùi.
Không tan trong nước hoặc các dung môi tự nhiên khác; dễ tan trong dung dịch hydroxyd kiềm.
Hỗn hợp chỉ thị màu calcein
Trộn đều 0,1 g calcein (TT) và 10 g kali clorid (TT) bằng cách nghiền.
Calci carbonat
CaCO3 = 100,09
Dùng loại tinh khiết phân tích.
Calci clorid
CaCI2. 2H2O = 147,0
Dùng loại tinh khiết phân tích.
Dung dịch calci clorid 10 %
Hòa tan 10 g calci clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch calci clorid
Hòa tan 7,35 g calci cloiíd (TT) trong nước vừa đủ 100 ml.
Calci clorid khan
CaCI2 = 110,99
Dùng loại tinh khiết hóa học.
Cốm trắng, dễ chảy nước. Rất dễ tan trong nước và ethanol.
Hàm lượng CaCI2 không được ít hơn 98,0 %, tính theo chế phẩm đã sấy khô.
Mất khối lượng do làm khô: Không được quá 5,0 %, khi làm khô đến khối lượng không đổi ở 200 °C.
Calci clorid tetrahydrat
CaCI2. 4H2O = 183,1
Dùng loại tinh khiết hóa học, chứa không quá 0,05 phần triệu Fe.
Calci hydroxyd
Ca(OH)2 = 74,09
Dùng loại tinh khiết phân tích.
Bột màu trắng, tan gần như hoàn toàn trong 600 phần nước.
Dung dịch bão hòa calci hydroxyd
Lắc kỹ 10 g calci hydroxyd (TT) với 1000 ml nước và để yên đến khi dung dịch trong.
Calci lactat
Calci lactat pentahydrat
C6H10CaO6.5H2O = 218,2
Dùng loại tinh khiết phân tích.
Calci sulfat
CaSO4.1/2 H2O = 145,15
Bột màu trắng, khi trộn 1 phần với 1/2 phần nước, bột bị rắn lại thành khối cứng và xốp.
Dung dịch calci sulfat
Lắc 5 g calci sulfat (TT) với 100 ml nước trong 1 h và lọc.
Carbomer
Polymer liên kết ngang của acid acrylic, chứa 56 % đến 68 % nhóm acid carboxylic (COOH) sau khi sấy ở 80 °C trong 1 h.
Dùng loại tinh khiết hóa học.
Khối lượng phân tử tương đối trung bình khoảng 3 x 106.
Carbon disulfid
CS2 = 76,14
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, dễ bay hơi, dễ bắt lửa, mùi khó chịu.
Khối lượng riêng: Khoảng 1,26 g/ml.
Điểm sôi: Khoảng 46 °C.
Carbon tetraclorid
Tetracloromethan
CCI4 = 153,82
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, có mùi đặc trưng.
Tỷ trọng ở 20 °C: Khoảng 1,59.
Điểm sôi: Từ 76 °C đến 77 °C.
Carbophenothion
O,O-Diethyl S-[(4-clorophenyl)thio]methylphospho-rodithioat
C11H16CIO2PS3 = 342,9
Dùng loại tinh khiết thích hợp cho phân tích dư lượng thuốc trừ sâu. Có thể sử dụng loại nguyên liệu đối chiếu (10 ng/μl trong iso-octan).
Tỷ trọng ở 25 °C: Khoảng 1,27.
Celulose dùng cho sắc ký
Bột trắng mịn, đồng nhất, kích thước hạt trung bình nhỏ hơn 30 μm.
Chuẩn bị bản mỏng: Trộn 15 g chế phẩm với 100 ml nước và khuấy đều trong 60 s. Tráng lớp dày 0,1 mm lên bản sạch và để khô ngoài không khí.
Ceri amoni nitrat
Xem Amoni ceri (IV) nitrat
Ceri amoni sulfat
Ce(SO4)2.2(NH4)2SO4.2H2O = 632,6
Dùng loại tinh khiết hóa học.
Bột kết tinh hay tinh thể màu vàng cam. Tan chậm trong nước, tan nhanh hơn khi có mặt acid vô cơ.
Ceri nitrat
Ce(NO3)3.6H2O = 434,2
Dùng loại tinh khiết hóa học.
Tinh thể không màu hoặc màu vàng nhạt. Tan trong nước, ethanol và aceton.
Ceri sulfat
Ce(SO4)2.4H2O = 404,3
Dùng loại tinh khiết hóa học.
Tinh thể hoặc bột kết tinh màu vàng hay vàng cam. Rất ít tan trong nước lạnh, tan chậm trong các dung dịch acid vô cơ loãng lạnh, tan nhanh hơn trong các dung môi này khi đun nóng.
Dung dịch ceri sulfat 1 % trong acid sulfuric 10 %
Hòa tan 10 g ceri sulfat (TT) trong 850 ml dung dịch acid sulfuric 10 % (TT) bằng cách đun nóng nhẹ. Để nguội, thêm dung dịch acid sulfuric 10 % (TT) cho vừa đủ 1000 ml.
Cetrimid
C17H38BrN = 336,4
Dùng loại tinh khiết phân tích.
Bột trắng hoặc gần như trắng, dễ trơn chảy. Dễ tan trong nước và ethanol 96 %.
Hàm lượng C17H38BrN: Không được ít hơn 96,0 %, tính theo chế phẩm đã làm khô.
Chì acetat
Pb(CH3COO)2.3H2O = 379,35
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng. Dễ tan trong nước, tan trong ethanol.
Bông tẩm chì acetat
Ngâm bông hút nước trong hỗn hợp 10 thể tích dung dịch chì acetat 9,5% (TT) và 1 thể tích dung dịch acid acetic 2 M (TT). Loại bỏ dung dịch thừa bằng cách thấm bằng giấy lọc nhưng không làm khô kiệt khối bông. Để khô bông ở nhiệt độ phòng. Bảo quản trong bao bì kín.
Giấy tẩm chì acetat
Nhúng giấy lọc vào hỗn hợp gồm 10 thể tích dung dịch chì acetat 9,5% (TT) và 1 thể tích acid acetic 2 M (TT), để ráo rồi hong khô, tránh ánh sáng. Cắt thành từng băng dài khoảng 5 cm, rộng 6 mm.
Bảo quản trong lọ thủy tinh nút mài, tránh ánh sáng, tránh acid hay kiềm.
Dung dịch chì acetat 20 %
Hòa tan 20 g chì acetat (TT) trong nước, thêm acid acetic (TT) tới khi được dung dịch trong, thêm nước vừa đủ 100 ml.
Dung dịch chì acetat 9,5%
Hòa tan 9,5 g chì acetat (TT) trong nước không có carbon dioxyd (TT) vừa đủ 100 ml.
Chì nitrat
Pb(NO3)2 = 331,2
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng hay tinh thể không màu. Dễ tan trong nước.
Dung dịch chì nitrat
Hòa tan 3,3 g chì nitrat (TT) trong nước vừa đủ 100 ml.
Chromotrop II B
Chromotrop 2B
C16H9N3Na2O10S2 = 513,4
Dùng loại tinh khiết hóa học.
Bột màu nâu đỏ.
Dung dịch chromotrop II B
Hòa tan 0,005 g chromotrop II B (TT) trong 100 ml acid sulfuric (TT).
Clor
Cl2 = 70,91
Nước clor (Dung dịch bão hòa clor)
Điều chế bằng cách cho luồng khí clor chạy qua nước đến bão hòa.
Đóng đầy trong chai lọ thủy tinh màu nút mài, để chỗ mát, tránh ánh sáng.
Dung dịch không bảo quản được lâu.
Cloral hydrat
C2H3Cl3O2 = 165,40
Dùng loại tinh khiết hóa học.
Tinh thể không màu, mùi đặc biệt, dễ hút ẩm.
Điểm chảy: Khoảng 55 °C.
Dung dịch cloral hydrat
Hòa tan 50 g cloral hydrat (TT) trong hỗn hợp gồm 15 ml nước và 10 ml glycerin (TT).
Cloramin B
Muối natri của N-clorobenzen sulfonamid
C6H5ClNNaO2S = 213,6
Dùng loại tinh khiết hóa học.
Cloramin T
Muối natri của N-clorotoluen-p-sulfonamid
C7H7ClNNaO2S.3H2O = 281,7
Dùng loại tinh khiết hóa học.
Dung dịch cloramin T 2 %
Hòa tan 2 g cloramin T (TT) trong nước vừa đủ 100 ml.
Pha trước khi dùng.
Dung dịch cloramin T 0,02 %
Hòa tan 0,02 g cloramin T (TT) trong nước vừa đủ 100 ml.
Pha trước khi dùng.
4’-Cloroacetanilid
Cloroacetanilid
C8H8ClNO = 169,6
Hàm lượng C8H8ClNO: Không được ít hơn 95 %.
Bột kết tinh. Thực tế không tan trong nước, tan trong ethanol 96 %.
Điểm chảy: Khoảng 178 °C.
1- Clorobutan
Butyl clorid
C4H9Cl = 92,6
Dùng loại tinh khiết hóa học.
Chất lỏng trong, không màu, dễ bay hơi.
Điểm sôi: Khoảng 78 °C.
Khối lượng riêng: Khoảng 0,886 g/ml.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,402.
1-Cloro-2,4-dinitrobenzen
C6H3ClN2O4 = 202,6
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu vàng nhạt.
Điểm chảy: Khoảng 51 °C.
2-Cloro-4-nitroanilin
C6H5ClN2O2 = 172,6
Dùng loại tinh khiết phân tích.
Bột kết tinh màu vàng đến nâu.
Điểm chảy: Khoảng 107 °C.
Bảo quản tránh ánh sáng.
2-Cloro-N-(2,6-dimethylphenyl)acetamid
C10H12ClNO = 197,7
Cloroform
Tricloromethan
CHCl3 = 119,38
Dùng loại tinh khiết phân tích chứa 0,4 % đến 1,0 % (kl/kl) ethanol.
Chất lỏng trong, không màu, mùi đặc biệt.
Điểm sôi: Khoảng 60 °C.
Tỷ trọng ở 20 °C: 1,475 đến 1,481.
Cloroform không có ethanol
Rửa cloroform (TT) với nước nhiều lần, làm khan bằng natri sulfat khan (TT), rồi cất lại.
Xác định khối lượng riêng (Phụ lục 6.5), không được thấp hơn 1,49 g/ml.
Cloroform không có ethanol chỉ điều chế trước khi dùng.
Cloroform khan
Cho 100 g calci clorid khan (TT) vào 1000 ml cloroform (TT), lắc mạnh, để yên 24 h. Gạn chất lỏng vào bình khô có nút mài.
Cloroform dùng cho phổ hồng ngoại
Dùng loại tinh khiết quang phổ.
Cloroform được ổn định với amylen
Dùng loại tinh khiết phân tích, chứa không ít hơn 99,8 % CHCl3 được xác định bằng phương pháp sắc ký khí (Phụ lục 5.2) và phải đáp ứng các yêu cầu sau:
Nước: Không quá 0,05 %.
Cắn sau khi bay hơi: Không quá 0,001 %.
Độ truyền quang: Không ít hơn 50 % ở 255 nm, 80 % ở 260 nm và 98 % ở 300 nm, dùng nước làm mẫu trắng.
Cobalt acetat
(CH3CO2)2Co.4H2O = 249,1
Dùng loại tinh khiết hóa học.
Tinh thể màu đỏ tím. Tan trong nước, ethanol và acid loãng.
Dung dịch cobalt acetat 0,2% trong methanol
Hòa tan 0,2 g cobalt acetat (TT) trong methanol (TT) vừa đủ 100 ml.
Cobalt clorid
CoCl2.6H2O = 237,93
Dùng loại tinh khiết phân tích.
Bột kết tinh màu đỏ hay tinh thể đỏ đậm. Rất dễ tan trong nước, tan trong ethanol.
Dung dịch cobalt clorid 2 %
Hòa tan 2 g cobalt clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch cobalt clorid 1 % trong methanol
Hòa tan 1 g cobalt clorid (TT) trong methanol (TT) vừa đủ 100 ml.
Cobalt nitrat
Co(NO3)2.6H2O = 291,04
Dùng loại tinh khiết phân tích.
Tinh thể nhỏ màu đỏ ngọc. Rất dễ tan trong nước.
Dung dịch cobalt nitrat
Hòa tan 5 g cobalt nitrat (TT) trong nước vừa đủ 100 ml.
Dung dịch cobalt nitrat 10 %
Hòa tan 10 g cobalt nitrat (TT) trong nước vừa đủ 100 ml.
Copolymer styren - divinylbenzen
Dùng loại tinh khiết sắc ký khí.
Các hạt polymer liên kết ngang, cứng, xốp. Có nhiều loại với kích thước hạt khác nhau.
Cresol
o-Cresol
C7H8O = 108,1
Dùng loại tinh khiết hóa học.
Khối rắn tinh thể không màu hay có màu rất nhạt, hoặc chất lỏng chậm đông, có mùi phenol - hắc ín.
Điểm đông đặc: Không dưới 30,5 °C.
Tỷ trọng ở 20 °C: Khoảng 1,05.
Chỉ số khúc xạ ở 20 °C: 1,540 đến 1,550.
Điểm sôi: Khoảng 190 °C.
Bảo quản tránh ánh sáng, ẩm và oxygen. Cất lại trước khi dùng.
Crom (VI) oxyd
Crom trioxyd
CrO3= 100,0
Hình kim hay mảnh vụn hoặc chảy rữa, màu đỏ hơi nâu. Rất tan trong nước.
Bảo quản trong lọ thủy tinh kín.
Curcumin
C21H20O6 = 368,4
Dùng loại tinh khiết hóa học.
Bột kết tinh màu nâu vàng. Tan trong acid acetic băng, thực tế không tan trong nước và ether.
Điểm chảy: Khoảng 183 °C.
Cyanogen bromid (Dung dịch)
Thêm từng giọt dung dịch amoni thiocyanat 0,1 M (TT) vào nước brom (TT) trong điều kiện làm lạnh cho tới khi mất màu.
Pha ngay trước khi dùng.
Cyclohexan
C8H12 = 84,16
Dùng loại tinh khiết phân tích.
Chất lỏng không màu.
Điểm sôi: Khoảng 81 °C.
Tỷ trọng ở 20 °C: Khoảng 0,78.
Cyclohexan dùng cho phương pháp quang phổ
Độ truyền quang: Không được nhỏ hơn 45 % ở 220 nm; 70 % ở 235 nm; 90 % ở 240 nm và 98 % ở 250 nm, dùng nước làm mẫu trắng.
Cyclohexan (TT1)
Sử dụng loại cyclohexan dùng cho phương pháp quang phổ (TT) và đáp ứng phép thử sau:
Huỳnh quang: Dưới bức xạ ở bước sóng 365 nm, huỳnh quang đo được ở bước sóng 460 nm, trong cốc đo dày 1 cm, không được lớn hơn huỳnh quang của dung dịch chứa 0,002 μg quinin trong 1 ml dung dịch acid sulfuric 0,05 M (TT).
L-Cystein
C3H7NO2S = 121,2
Bột dễ tan trong nước, trong ethanol 96 % và trong acid acetic; thực tế không tan trong aceton.
Cytosin
C4H5N3O = 111,1
Hàm lượng không được dưới 95,0 %.
Dibenzosuberon
Dibenzo[a,d]cyclohepta-1,4-dien-3-on;10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-on
C15H12O = 208,3
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 34 °C.
Dibutylamine
Di-n-butylamine,N-Butylbutan-1-amine.
C8H19N = 129.3 (111-92-2)
Chất lỏng không màu.
Chỉ số khúc xạ ở 20 °C ( ): Khoảng 1.417; điểm sôi khoảng 159°.
): Khoảng 1.417; điểm sôi khoảng 159°.
Dibutylamoni phosphat dùng tạo cặp ion
Dung dịch không màu chứa 10 % đến 15 % (tt/tt) di-n-butylamin (TT) và 12 % đến 17 % (tt/tt) acid phosphoric (TT) trong nước, dùng tạo cặp ion trong sắc ký lỏng.
Dibutyl ether
C8H18O =130,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, dễ bắt lửa.
Điểm sôi: Khoảng 140 °C.
Tỷ trọng ở 20 °C: Khoảng 0,77.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,399.
Chú ý: Cẩn thận khi cất hoặc làm bay hơi dibutyl ether, vì có thể giải phóng peroxyd.
Di-n-butylamin
N-butylbutan-1-amin, dibutylamin
C8H19N = 129,3
Chất lỏng không màu.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,417.
Điểm sôi: Khoảng 159 °C.
Dibutyl phtalat
Di-n-butyl phtalat
C16H22O4 = 278,3
Dùng loại tinh khiết hóa học.
Chất lỏng không màu hay có màu rất nhạt.
Tỷ trọng ở 20 °C: 1,043 đến 1,048.
Chỉ số khúc xạ ở 20 °C: 1,490 đến 1,495.
Dịch dạ dày giả
Hòa tan 2,0 g natri clorid (TT) và 3,2 g pepsin (TT) trong nước, thêm 80 ml dung dịch acid hydrocloric 1 M (TT), thêm nước vừa đủ 1000 ml.
2,6-Diclorophenol-indophenol natri
C12H6Cl2NNaO2 = 290,08.
Là muối natri của 2,6 dicloro-N-(4-hydroxyphenyl)-1,4-benzoquinon monoimin.
Dùng loại tinh khiết phân tích.
Bột màu lục thẫm. Dung dịch nước có màu lam đậm, chuyển sang màu hồng khi acid hóa.
Dung dịch 2,6 diclorophenol - indophenol
Hòa tan 0,1 g 2,6 diclorophenol - indophenol natri (TT) với 100 ml nước ấm và lọc.
Dung dịch chỉ dùng trong vòng 3 ngày.
2,6- Dicloroquinon clorimid
C6H2Cl3NO = 210,45
Dùng loại tinh khiết hóa học.
Bột kết tinh màu vàng hay da cam. Không tan trong nước, tan trong ethanol và các dung dịch kiềm loãng.
Điểm chảy: Khoảng 66 °C.
Dung dịch 2,6-dicloroquinon clorimid
Hòa tan 0,02 g 2,6-dicloroquinon clorimid (TT) trong 50 ml n-butanol (TT) hay alcol isopropylic (TT).
Bảo quản trong lọ thủy tinh màu, ở chỗ mát.
Không dùng dung dịch khi đã có màu hồng.
2,5- Diethoxytetrahydrofuran
C8H16O3 = 160,2
Dùng loại tinh khiết hóa học, chứa hỗn hợp đồng phân cis và trans.
Chất lỏng không màu hay vàng nhạt.
Tỷ trọng ở 20 °C: Khoảng 0,98.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,418.
N,N-Diethylanilin
C10H15N = 149,2
Dùng loại tinh khiết hóa học.
Chất lỏng màu vàng nhạt, có mùi amoniac.
Điểm sôi: Khoảng 217 °C.
Khối lượng riêng: Khoảng 0,93 g/ml.
Diethylamin
(C2H5)2NH = 73,14
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, dễ bay hơi.
Điểm sôi: Khoảng 55 °C.
Tỷ trọng ở 20 °C: Khoảng 0,71.
1,3-Dihydroxynaphthalen
Naphthalen-1,3-diol; Naphthoresorcin
C10H8O2= 160,2
Dùng loại tinh khiết hóa học.
Bột kết tinh màu tím nâu. Dễ tan trong nước và ethanol.
Điểm chảy: Khoảng 125 °C.
2,7-Dihydroxynaphthalen
Naphthalen-2,7-diol
C10H8O2 = 160,2
Tinh thể hình kim, tan trong nước và trong ethanol 96 %.
Điểm chảy: Khoảng 190 °C.
Dung dịch 2,7-dihydroxynaphthalen
Hòa tan 10 mg 2,7-dihydroxynaphthalen (TT) trong 100 ml acid sulfuric (TT), để yên cho mất màu (dùng trong vòng 2 ngày sau khi pha).
Di-isopropyl ether
Isopropyl ether
C6H14O = 102,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, có mùi đặc trưng.
Điểm sôi: Từ 67 đến 69 °C.
Tỷ trọng ở 20 °C: Từ 0,723 đến 0,728.
Chú ý: Chỉ cất di-isopropyl ether khi đạt phép thử nghiệm sau:
Peroxyd: Cho 8 ml dung dịch hồ tinh bột có kali iodid (TT) vào ống nghiệm nút mài dung tích 12 ml, đường kính khoảng 1,5 cm, đổ đầy ống nghiệm với di-isopropyl ether cần thử. Đậy nút, lắc mạnh và để yên tránh ánh sáng trong 30 min. Không được có màu tạo thành.
Bảo quản tránh ánh sáng. Trên nhãn phải ghi tên và nồng độ chất bảo quản đưa vào.
Dikali hydrophosphat
K2HPO4 = 174,2
Dùng loại tinh khiết hóa học.
Bột kết tinh trắng, dễ hút ẩm. Rất dễ tan trong nước, ít tan trong ethanol.
Dimethylacetamid
C4H9NO = 87,12
Dùng loại tinh khiết hóa học, chứa không ít hơn 99,5 % C4H9NO.
Chất lỏng không màu.
Điểm sôi: Khoảng 165 °C.
Tỷ trọng ở 20 °C: Khoảng 0,94.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,437.
Dimethylanilin
N, N-dimethylanilin
C8H11N = 121,2
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, chuyển màu trong quá trình bảo quản. Thực tế không tan trong nước, dễ tan trong cồn và trong ether.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,568.
Không ít hơn 95 % chưng cất được ở 192 °C đến 194 °C.
26- Dimethylanilin
2,6-Xylidin
C8H11N = 121,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Tỷ trọng ở 20 °C: Khoảng 0,98.
p-Dimethylaminobenzaldehyd
4-Dimethylaminobenzaldehyd
(CH3)2NC6H4CHO = 149,19
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng hay vàng nhạt. Tan trong ethanol, aceton, cloroform, ether và acid acetic, tan ít trong nước.
Điểm chảy: Khoảng 74 °C.
Dung dịch p-dimethylaminobenzaldehyd 2 % trong ethanol.
Hòa tan 2 g p-dimethylaminobenzaldehyd (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Dung dịch p-dimethylaminobenzaldehyd (TT1)
Hòa tan 0,2 g p-dimethylaminobenzaldehyd (TT) trong 20 ml ethanol 96 % (TT), thêm 0,5 ml acid hydro-cloric (TT). Lắc dung dịch với than hoạt (TT), lọc. Dung dịch không được đậm hơn màu của dung dịch iod pha như sau: Lấy 10 ml dung dịch iod 0,1 N (CĐ), thêm 0,6 g kali iodid (TT) và nước vừa đủ 100 ml. Pha loãng 2,0 ml dung dịch thu được với nước thành 100 ml.
Pha ngay trước khi dùng.
Dung dịch p-dimethylaminobenzaldehyd (TT2)
Hòa tan 0,2 g p-dimethylaminobenzaldehyd (TT) trong hỗn hợp gồm 4,5 ml nước và 5,5 ml acid hydrocloric (TT). Pha ngay trước khi dùng.
Dung dịch p-dimethylaminobenzaldehyd (TT6)
Hòa tan 0,125 g p-dimethylaminobenzaldehyd (TT) trong hỗn hợp gồm 35 ml nước và 65 ml acid sulfuric (TT) đã được làm nguội, thêm 0,1 ml dung dịch sắt (III) clorid (TT) 5 % và lắc đều. Để yên 24 h trước khi dùng, tránh ánh sáng. Khi bảo quản dung dịch ở nhiệt độ phòng dùng được trong vòng 7 ngày, khi bảo quản trong tủ lạnh dùng được trong vài tháng.
4-Dimethylaminocinnamaldehyd
C11H13NO = 175,2
Dùng loại tinh khiết hóa học.
Bột hay tinh thể màu cam tới nâu cam.
Điểm chảy: Khoảng 138 °C.
Dung dịch 4-dimethylaminocinnamaldehyd
Hòa tan 2,0 g 4-dimethylaminocinnamadehyd (TT) trong hỗn hợp gồm 100 ml dung dịch acid hydrocloric 7 N (TT) và 100 ml ethanol (TT).
Bảo quản chỗ mát. Pha loãng gấp bốn với ethanol (TT) ngay trước khi dùng.
2,6-Dimethylanilin
2,6-Xylidin
C8H11N = 121,2
Chất lỏng không màu, hơi tan trong nước, tan trong ethanol 96 %.
Tỷ trọng ở 20 °C: Khoảng 0,98.
Dimethyl carbonat
C3H6O3 = 90,1
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, không mùi hoặc hơi có mùi khó chịu.
Điểm sôi: Khoảng 90 °C.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,368.
Tỉ trọng ở 17 °C: Khoảng 1,065.
1,1-Dimethylethyl methyl ether
Tert-butyl methyl ether
C5H12O = 88,1
Chất lỏng không màu, trong và dễ cháy.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,376.
Độ truyền quang: Không được quá 50 % ở bước sóng 240 nm, 80 % ở bước sóng 255 nm, 98 % ở bước sóng 280 nm, dùng nước làm mẫu trắng.
Dimethyldecylamin
C12H27N = 185,4
Dùng loại tinh khiết hóa học, chứa không ít hơn 98,0 % (kl/kl) C12H27N.
Điểm sôi: Khoảng 234 °C.
1,3-Dimethy-imidazolidinon
DMI
C5H10N2O= 114,2.
Dùng loại tinh khiết hóa học.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,472.
Điểm sôi: Khoảng 224 °C.
Dimethylformamid
HCON(CH3)2 = 73,10
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, mùi đặc biệt, hòa lẫn với nước và ethanol.
Điểm sôi: Khoảng 153 °C.
Tỷ trọng ở 20 °C: Khoảng 0,95.
Nước: Không được quá 0,1 % (Phụ lục 10.3).
N,N-Dimethyloctylamin
Octyldimethylamin
C10H23N = 157,3
Chất lỏng không màu.
Tỷ trọng ở 20 °C: Khoảng 0,765.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,424.
Điểm sôi: Khoảng 195 °C.
Dimethyl sulfoxid
C2H6OS = 78,1
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, không mùi hoặc hơi có mùi khó chịu.
Điểm sôi: Khoảng 189 °C.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,479.
Tỉ trọng ở 20 °C: Khoảng 1,10.
Phải đáp ứng yêu cầu sau:
Nước: Không quá 1,0 % (Phụ lục 10.3).
Dimethtyl sulfoxid dùng cho phương pháp quang phổ
Độ truyền quang: Không nhỏ hơn 10 % ở 262 nm, 35 % ở 270 nm, 70 % ở 290 nm và 98 % ở 340 nm và những bước sóng cao hơn, dùng nước làm mẫu trắng.
Dinatri hydrophosphat
Na2HPO4.12H2O = 358,17.
Dùng loại tinh khiết phân tích.
Tinh thể trong suốt, không màu. Tan trong nước, không tan trong ethanol.
Dung dịch dinatri hydrophosphat 12 %
Hòa tan 12 g dinatri hydrophosphat (TT) trong nước vừa đủ 100 ml.
Dung dịch dinatri hydrophosphat 4 %
Hòa tan 4 g dinatri hydrophosphat (TT) trong nước vừa đủ 100 ml.
Dinatri hydrophosphat khan
Na2HPO4 = 142,0
Dùng loại tinh khiết phân tích.
Dung dịch dinatri hydrophosphat 0,235 M
Hòa tan 33,36 g dinatri hydrophosphat khan (TT) trong 1000 ml nước, điều chỉnh pH tới 10,3 đến 10,5 với dung dịch natri hydroxyd 2 N (TT).
Dinitrobenzoyl clorid
3,5-Dinitrobenzoyl clorid
C7H3ClN2O5 = 230,6
Dùng loại tinh khiết hóa học.
Tinh thể hình kim màu vàng, phân hủy trong không khí ẩm.
Điểm chảy: Khoảng 68 °C.
2.4-Dinitrophenylhydrazin
C6H3(NO2)2NHNH2 = 198,14
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu cam đỏ. Ít tan trong ethanol, rất khó tan trong nước.
Điểm chảy: Khoảng 203 °C.
Dung dịch 2,4-dinitrophenylhydrazin trong acid hydrocloric
Thêm 4 ml acid hydrocloric (TT) vào 0,10 g 2,4-dini-trophenylhydrazin (TT), rồi thêm 20 ml nước nóng để hòa tan.
Dung dịch chỉ pha khi dùng.
Dung dịch dinitrophenylhydrazin-aceto-hydrocloric
Hòa tan 0,2 g 2,4-dinitrophenylhydrazin (TT) trong 20 ml methanol (TT) và thêm 80 ml hỗn hợp đồng thể tích acid acetic 30 % (TT) và dung dịch acid hydro-cloric 25 % (TT).
Dung dịch chỉ pha ngay trước khi dùng.
Dioctyl natri sulfosuccinat
Natri docusat
C20H37NaO7S = 444,6
Dùng loại tinh khiết hóa học, chứa 90 % C20H37NaO7S.
Các mảnh sáp màu trắng. Tan trong nước, methanol và aceton. Dễ thủy phân trong các dung dịch kiềm.
Dioxan
1,4- Dioxan
C4H8O2 = 88,11
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, có mùi ether.
Điểm đông đặc: 9 °C đến 11 °C.
Tỷ trọng ở 20 °C: Khoảng 1,03.
Nước: Không được quá 0,5 %.
Chú ý: Chỉ cất 1,4-dioxan khi đạt phép thử sau:
Peroxyd: Cho 8 ml dung dịch hồ tinh bột có kali iodid (TT) vào ống nghiệm nút mài dung tích 12 ml, đường kính 1,5 cm, đổ đầy ống nghiệm với dioxan cần thử. Đậy nút, lắc mạnh và để yên tránh ánh sáng trong 30 min. Không được có màu xuất hiện.
Dung dịch dioxan
Pha loãng 50 ml dung dịch gốc dioxan (TT) thành 100 ml với nước (0,5 mg/ml).
Dung dịch dioxan (TT1)
Pha loãng 10,0 ml dung dịch dioxan (TT) thành 50 ml với nước (0,1 mg/ml).
Dung dịch gốc dioxan
Hòa tan 1 g dioxan (TT) trong nước vừa đủ 100 ml. Pha loãng 5 ml dung dịch thu được thành 50 ml bằng nước (1,0 mg/ml).
Diphenylamin
C12H11N = 169,2
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng, có mùi đặc biệt. Ít tan trong nước, tan trong ethanol.
Điểm chảy: Khoảng 55 °C.
Dung dịch diphenylamin
Hòa tan 0,5 g diphenylamin (TT) trong hỗn hợp gồm 100 ml acid sulfuric đậm đặc (TT) và 20 ml nước.
Diphenylcarbazon
1.5- Diphenylcarbazon
C13H12N4O = 240,3
Dùng loại tinh khiết hóa học.
Bột kết tinh màu cam. Thực tế không tan trong nước, dễ tan trong ethanol.
Điểm chảy: Khoảng 157 °C cùng với phân hủy.
Thuốc thử diphenylcarbazon - thủy ngân
Hòa tan 0,1 g diphenylcarbazon (TT) trong ethanol (TT) vừa đủ 50 ml (dung dịch A).
Hòa tan 1 g thủy ngân (II) clorid (TT) trong ethanol (TT) vừa đủ 50 ml (dung dịch B).
Trộn đều đồng thể tích dung dịch A và dung dịch B.
Diphenylmethanol
Benzhydrol
C13H12O= 184,2
Tinh thể trắng hoặc gần như trắng.
Điểm chảy: Khoảng 66 °C.
2,2’-Dipyridylamin
N-(pyridin-2-yl)pyridin-2-amin
C10H9N3= 171,2
Điểm chảy: Khoảng 95 °C.
Dithizon
1,5-Diphenylthiocarbazon
C13H12N4S = 256,3
Dùng loại tinh khiết phân tích.
Bột màu xanh đen, nâu đen hay đen. Thực tế không tan trong nước, tan trong ethanol.
Dung dịch dithizon 0,025 % trong ethanol
Hòa tan 25 mg dithizon (TT) trong ethanol (TT) vừa đủ 100 ml.
Divanadi pentoxyd
Vanadi oxyd
V2O5 = 181,9
Dùng loại tinh khiết hóa học, chứa không ít hơn 98,5 % V2O5.
Bột tinh thể màu vàng cam hay tinh thể màu nâu đỏ. Khó tan trong nước, tan trong dung dịch acid và dung dịch kiềm.
Phải đáp ứng phép thử sau:
Độ nhạy với hydrogen peroxyd: Đun nóng 1 g divanadi pentoxyd (TT) với 10 ml acid sulfuric (TT) trong 30 min, để nguội, bổ sung acid sulfuric (TT) cho đủ 10 ml. Pha loãng cẩn thận 1 ml dung dịch trong thành 50 ml bằng nước. Lấy 0,5 ml dung dịch thu được, thêm 0,1 ml dung dịch hydrogen peroxyd 0,01 %. Xuất hiện màu cam, thấy rõ khi so sánh với mẫu trắng gồm 0,5 ml dung dịch trên và 0,1 ml nước. Thêm tiếp 0,4 ml dung dịch hydrogen peroxyd 0,01 %, màu chuyển sang vàng cam.
Dung dịch divanadi pentoxyd trong acid sulfuric
Hòa tan 0,2 g divanadi pentoxyd (TT) trong 4 ml acid sulfuric (TT) và thêm nước vừa đủ 100 ml.
Đỏ rutheni
H42Cl6N14O2Ru3.4H2O = 858
Dùng loại phẩm màu siêu mịn.
Bột màu đỏ nâu. Tan trong nước.
Dung dịch đỏ rutheni
Hòa tan 0,08 g đỏ rutheni (TT) trong dung dịch chì acetat 9,5 % (TT) vừa đủ 100 ml.
Đồng acetat
Cu (CH3COO)2.H2O = 199,65.
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu lục thẫm. Tan trong nước và ethanol, ít tan trong ether và glycerin.
Dung dịch đồng edetat
Lấy 2 ml dung dịch đồng acetat 2 %, thêm 2 ml dung dịch trilon B 0,1 M (CĐ) và pha loãng với nước thành 50 ml.
Dung dịch đồng acetat 5 %
Hòa tan 5 g đồng acetat (TT) trong nước đã acid hóa bằng vài mililit acid acetic loãng (TT) và thêm nước vừa đủ 100 ml.
Đồng phôi
Cu = 63,54
Dùng loại tinh khiết hóa học.
Đồng sulfat
CuSO4.5H2O = 249,69
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu xanh lam. Rất dễ tan trong nước, ít tan trong ethanol.
Dung dịch đồng sulfat 10 %
Hòa tan 10 g đồng sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch đồng sulfat 5 %
Hòa tan 5 g đồng sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch đồng sulfat 4 %
Hòa tan 4 g đồng sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch đồng sulfat 0,5 M (12,5 %)
Hòa tan 12,5 g đồng sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch đồng sulfat 0,01 M
Hòa tan 2,5 g đồng sulfat (TT) trong nước vừa đủ 1000 ml.
Dung dịch đồng - citrat
Hòa tan 2,5 g đồng sulfat (TT), 5 g acid citric (TT) và 14,4 g natri carbonat khan (TT) trong nước vừa đủ 100 ml.
Đồng sulfat khan
CuSO4= 159,6
Dùng loại tinh khiết phân tích, hoặc điều chế bằng cách sấy đồng sulfat (TT) ở 230 °C đến khối lượng không đổi.
Ethan-1,2-diol
Ethylen glycol
HOCH2-CH2OH = 62,07
Dùng loại tinh khiết phân tích.
Chất lỏng sánh, không màu.
Tỷ trọng ở 20 °C: 1,113 đến 1,115.
Chỉ số khúc xạ ở 20 °C: 1,430 đến 1,433.
Điểm sôi: Khoảng 196 °C.
Dung dịch ethan - 1,2 - diol 50 %
Hòa tan 50 g ethan-1,2-diol (TT) trong nước vừa đủ 100 ml.
Ethanol
Ethanol tuyệt đối
C2H5OH = 46,07
Dùng loại tinh khiết phân tích, chứa không ít hơn 99,5 % (tt/tt) C2H5OH.
Chất lỏng trong, không màu, dễ hút nước, có mùi đặc biệt.
Điểm sôi: 78 °C đến 79 °C.
Tỷ trọng ở 20 °C: 0,791 đến 0,794.
Ethanol 96 %
C2H5OH = 46,07
Dùng loại tinh khiết phân tích, chứa 95,1 % đến 96,9 % (tt/tt) C2H5OH.
Chất lỏng không màu.
Khối lượng riêng: Khoảng 0,81 g/ml.
Ethanol 96 % không có aldehyd
Trộn 1200 ml ethanol 96 % (TT) với 5 ml dung dịch bạc nitrat 40 % và 10 ml dung dịch kali hydroxyd 50 % đã làm lạnh. Lắc mạnh, để yên vài ngày và lọc, cất dịch lọc ngay trước khi sử dụng.
Ether
Ether ethylic; Diethylether
C4H10O = 74,12
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, trong suốt, dễ bắt lửa, dễ bay hơi, mùi đặc biệt.
Tỷ trọng ở 20 °C: 0,713 đến 0,715.
Điểm sôi: 34 °C đến 35 °C.
Chú ý: Chỉ cất ether khi đạt phép thử sau:
Peroxyd: Cho 8 ml dung dịch hồ tinh bột có kali iodid (TT) vào ống nghiệm nút mài dung tích 12 ml, đường kính khoảng 1,5 cm, đổ đầy ống nghiệm với ether cần thử. Đậy nút, lắc mạnh và để yên tránh ánh sáng trong 30 min. Không được có màu tạo thành.
Bảo quản tránh ánh sáng, ở nhiệt độ không quá 15 °C. Trên nhãn phải ghi tên và nồng độ chất bảo quản đưa vào.
Ether không có peroxyd
Dung dịch sắt (II) sulfat: Hòa tan 30 g sắt (II) sulfat (TT) trong hỗn hợp gồm 55 ml nước và 3 ml acid sulfuric (TT).
Lắc 1000 ml ether (TT) với 20 ml dung dịch sắt (II) sulfat. Tiếp tục lắc cho tới lúc không còn xuất hiện màu xanh lam, khi lắc lượng nhỏ mẫu thử với đồng thể tích dung dịch kali iodid 2 % (TT) và 0,1 ml dung dịch hồ tinh bột (TT).
Ether dầu hỏa
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, dễ bay hơi, rất dễ bắt lửa, chứa hỗn hợp các dãy hydrocarbon parafin bậc thấp, chia thành các phân đoạn sau:
Khoảng sôi 30 °C đến 40 °C; khối lượng riêng: Khoảng 0,63 g/ml.
Khoảng sôi 40 °C đến 60 °C; khối lượng riêng: Khoảng 0,64 g/ml.
Khoảng sôi 50 °C đến 70 °C; khối lượng riêng: Khoảng 0,66 g/ml.
Khoảng sôi 60 °C đến 80 °C; khối lượng riêng: Khoảng 0,67 g/ml.
Khoảng sôi 80 °C đến 100 °C; khối lượng riêng: Khoảng 0,70 g/ml.
Khoảng sôi 100 °C đến 120 °C; khối lượng riêng: Khoảng 0,72 g/ml.
Khoảng sôi 120 °C đến 160 °C; khối lượng riêng: Khoảng 0,75 g/ml.
p-Ethoxycrysoidin hydroclorid
4-p-Ethoxyphenylazo-m-phenylendiamin hydroclorid
C14H16N4O.HCl = 292,8
Dùng loại tinh khiết hóa học.
Bột màu đỏ. Tan trong nước và ethanol.
Dung dịch p-ethoxycrysoidin hydroclorid 0,1 % trong ethanol
Hòa tan 0,1 g p-ethoxycrysoidin hydroclorid (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Dung dịch phải đạt phép thử độ nhạy với brom: Thêm 0,05 ml dung dịch brom 0,1 N (TT) vào hỗn hợp 0,05 ml dung dịch p-ethoxycrysoidin hydroclorid 0,1 % trong ethanol (TT) và 5 ml dung dịch acid hydrocloric 2 M (TT). Màu chuyển từ đỏ sang vàng nhạt trong vòng 2 min.
Ethyl acetat
CH3COOC2H5 = 88,11 Dùng loại tinh khiết phân tích.
Chất lỏng không màu, mùi thơm hoa quả.
Tan trong nước, hòa lẫn với ethanol.
Tỷ trọng ở 20 ° C: 0,901 đến 0,904.
Điểm soi: 76 °C đến 78 °C.
Ethyl cyanoacetat
C5H7NO2 = 113,1
Dùng loại tinh khiết phân tích.
Chất lỏng không màu hoặc gần như không màu. Tan trong nước, hòa lẫn được với ethanol và ether.
Điểm sôi: 205 °C đến 209 °C kèm phân hủy.
Ethylen clorid
1,2- Dicloroethan
C2H4Cl2 = 98,96
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, có mùi giống mùi cloroform.
Điểm sôi: Khoảng 83 °C.
Tỷ trọng ở 20 °C: Khoảng 1,25.
Lượng cất được trong khoảng 82 °C đến 84 °C không ít hơn 95 %.
Ethylen diamin
1,2- Diaminoethan
C2H8N2 = 60,10
Dùng loại tinh khiết hóa học.
Điểm sôi: Khoảng 116 °C.
Khối lượng riêng: Khoảng 0,90 g/ml.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,457.
Ethylen oxyd
Oxiran
C2H4O = 44,05
Dùng loại tinh khiết hóa học.
Khí không màu.
Điểm hóa lỏng: Khoảng 12 °C.
Dung dịch gốc ethylen oxyd (Dung dịch đậm đặc ethylen oxyd)
Toàn bộ quá trình pha chế phải được tiến hành trong tủ hút. Người pha chế phải mang găng tay polyethylen và mặt nạ bảo vệ thích hợp. Bảo quản các dung dịch trong bình kín, ở nhiệt độ từ 4 °C đến 8°C. Tiến hành định lượng 3 lần.
Cho chậm luồng khí ethylen oxyd (TT) vào một ống nghiệm khô, sạch, đã được làm lạnh chứa hỗn hợp gồm 1 phần natri clorid (TT) và 3 phần nước đá nghiền, để tạo thành lớp đông đặc bám trên mặt trong thành ống nghiệm. Dùng bơm thủy tinh đã được làm lạnh ở -10 °C, tiêm khoảng 300 μl (ứng với khoảng 0,25 g) ethylen oxyd (TT) lỏng vào 50 ml macrogol 200 (TT1). Xác định lượng ethylen oxyd đã hấp thụ bằng cách cân trước và sau khi cho hấp thụ (Meo). Pha loãng thành 100 ml với macrogol 200 (TT1). Trộn kỹ trước khi dùng.
Xác định nồng độ chính xác của ethylen oxyd theo phương pháp sau:
Lấy 10 ml hỗn dịch magnesi clorid (TT) 50 % trong ethanol (TT) vào bình nón nút mài, thêm 20 ml dung dịch acid hydrocloric 0,1 N trong ethanol (TT), đậy nắp, lắc mạnh để thu được dung dịch bão hòa và để cân bằng qua đêm. Cân 5 g dung dịch cần định lượng vào bình đã chuẩn bị ở trên, để yên 30 min. Chuẩn độ bằng dung dịch kali hydroxyd 0,1 N trong ethanol (CĐ), xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2). Song song tiến hành một mẫu trắng, dùng macrogol 200 (TT1) thay thế cho dung dịch cần định lượng.
Nồng độ ethylen oxyd (mg/g) trong dung dịch được tính bằng công thức:
4,404(V0-V1)/m
Trong đó:
V0 và V1 là số ml dung dịch kali hydroxyd 0,1 N trong ethanol (CĐ) đã dùng cho mẫu trắng và mẫu thử; m là lượng mẫu thử đã cân để định lượng (g).
Dung dịch ethylen oxyd (TT1)
Cân chính xác một lượng dung dịch gốc ethylen oxyd (TT) đã được làm lạnh ứng với 2,5 mg ethylen oxyd vào một bình lạnh, pha loãng thành 50,0 g với macrogol 200 (TT1). Trộn kỹ, pha loãng 2,5 g dung dịch này thành
25 ml với macrogol 200 (TT1) (5 μg ethylen oxyd/g).
Dung dịch pha trước khi dùng.
Dung dịch ethylen oxyd (TT1)
Cân 1 g dung dịch gốc ethylen oxyd (TT) lạnh (tương ứng với 2,5 mg ethylen oxyd) vào bình lạnh có chứa 40 ml macrogol 200 (TT1) lạnh. Trộn, xác định khối lượng chính xác và pha loãng để thu được dung dịch chứa 50 μg ethylen oxyd trong 1 g dung dịch. Cân 10 g dung dịch vào bình chứa khoảng 30 ml nước, trộn và pha loãng thành 50 ml với nước (10 pg ethylen oxyd/ml).
Dung dịch pha chế ngay trước khi dùng.
Dung dịch ethylen oxyd (TT3)
Pha loãng 10 ml dung dịch ethylen oxyd (TT2) thành 50 ml với nước (2 μg ethylen oxyd/ml), Dung dịch pha ngay trước khi dùng.
Ethyl parahydroxybenzoat
Ethyl 4-hydroxybenzoat
C9H10O3 = 166,2
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 117 °C.
Ferroin
Dung dịch ferroin
Phức hợp tris(1,10-phenanthrolin) sắt(II) sulfat được chuẩn bị như sau: Hòa tan 0,7 g sắt (II) sulfat (TT) và 1,76 g phenanthrolin hydroclorid (TT) trong 70 ml nước và pha loãng thành 100 ml bằng nước.
Phải đáp ứng phép thử sau:
Độ nhạy của ceri (IV): Thêm 0,1 ml dung dịch trên và 0,15 ml dung dịch osmium tetroxyd (TT) vào 50 ml dung dịch acid sulfuric 1 M (TT). Thêm 0,1 ml dung dịch amoni ceri (IV) nitrat 0,1 M (TT). Màu chuyển từ màu đỏ sang xanh dương.
Fibrin đỏ Congo
Ngâm qua đêm 1,5 g fibrin trong 50 ml dung dịch đỏ congo 2 % (kl/tt) trong ethanol 90 %. Lọc, rửa fibrin với nước và bảo quản trong ether (TT).
1-Fluoro-2,4-dinitrobenzen
2,4-Dinitrofluorobenzen
C6H3FN2O4 = 186,1.
Dùng loại tinh khiết hóa học.
Chất lỏng, chất rắn hay tinh thể màu vàng nhạt với hơi làm cay mắt.
Điểm chảy: Khoảng 29 °C.
Khối lượng riêng: Khoảng 1,48 g/ml.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,569.
Formaldehyd
Formaldehyd; Dung dịch formol; Formalin
CH2O = 30,03
Dùng loại tinh khiết phân tích, chứa 34,0 % đến 37,0 % CH2O.
Chất lỏng trong, không màu hay gần như không màu, mùi hăng đặc biệt.
Khối lượng riêng: Khoảng 1,08 g/ml.
Bảo quản trong lọ kín, ở nhiệt độ 15 °C đến 25 °C.
Dung dịch formaldehyd trong acid sulfuric
Hòa tan 0,2 g formaldehyd (TT) trong 10 ml acid sulfuric (TT).
Formamid
CH3NO = 45,0.
Dùng loại tinh khiết phân tích.
Chất lỏng sánh, không màu. Hòa trộn được với nước và ethanol.
Khối lượng riêng: Khoảng 1,13 g/ml.
Fuchsin
Fuchsin base
Hỗn hợp rosanilin hydroclorid (C20H19N3.HCl = 337,9) và para-rosanilin hydroclorid (C19H17N3.HCl = 323,8). Bột màu đỏ đậm hay tinh thể xanh lục có ánh kim. Tan trong nước và ethanol.
Bảo quản tránh ánh sáng.
Dung dịch fuchsin đã khử màu
Hòa tan 0,1 g fuchsin base (TT) trong 60 ml nước, thêm 10 ml dung dịch natri sulfit khan 10 % (kl/tt). Thêm từ từ 2 ml acid hydrocloric (TT), lắc liên tục, pha loãng thành 100 ml với nước. Để yên tránh ánh sáng ít nhất 12 h. Lắc với 0,2 g đến 0,3 g than hoạt (TT) để khử màu và lọc ngay lập tức. Nếu dung dịch bị đục, lọc trước khi sử dụng. Trong quá trình bảo quản, nếu dung dịch có màu tím, khử màu bằng than hoạt (TT).
Dung dịch phải đạt phép thử độ nhạy với formaldehyd: Lấy 1,0 ml dung dịch, thêm 1,0 ml nước và 0,1 ml ethanol không có aldehyd (TT). Thêm 0,2 ml dung dịch fomnaldehyd 0,01 %, phải xuất hiện màu hồng nhạt trong vòng 5 min.
Bảo quản tránh ánh sáng.
Dung dịch fuchsin đã khử màu (TT1)
Hòa tan 0,1 g fuchsin base (TT) trong 100 ml nước, đun nóng đến 50 °C và để nguội, lắc liên tục. Để yên 48 h, lắc đều và lọc. Thêm 6 ml acid hydrocloric (TT) vào 4 ml dịch lọc, trộn đều và pha loãng thành 100 ml bằng nước. Để yên ít nhất 1 h để đảm bảo mất màu tối đa.
Gelatin
Dùng loại tinh khiết hóa học.
Gelatin thủy phân
Hòa tan 50 g gelatin (TT) trong 1000 ml nước. Hấp trong nồi hấp bão hòa hơi nước ở 121 °C trong 90 min và làm đông khô.
Thuốc thử gelatin - natri clorid
Hòa tan 1 g gelatin (TT) và 10 g natri clorid (TT) trong 100 ml nước bằng cách đun nóng trên cách thủy ở nhiệt độ dưới 60 °C. Dung dịch chỉ pha khi dùng.
L-γ-Glutamyl-L-cystein
C8H14N2O5S = 250,3
Dùng loại tinh khiết hóa học.
L-Glutathion đã oxy hóa
Bis(L-γ-glutamyl-L-cysteinylglycin) disulfid
C20H32N6O12S2 = 612,6
Dùng loại tinh khiết hóa học.
Glycerin
Propan-1,2,3 triol
C3H8O3 = 92,10
Dùng loại tinh khiết phân tích.
Chất lỏng sánh, không màu.
Khối lượng riêng: Khoảng 1,26 g/ml.
Dung dịch glycerin - acid acetic: xem Thuốc thử Smith.
Glycerin 85 %
Glycerin (TT) có chứa 12,0 % đến 16,0 % (kl/kl) nước.
Khối lượng riêng: 1,22 g/ml đến 1,24 g/ml.
Dung dịch glyoxal
Dùng loại tinh khiết hóa học chứa khoảng 40 % (kl/kl) glyoxal (C2H2O2).
Định lượng: Lấy 1 g vào bình nón nút mài, thêm 20 ml dung dịch hydroxylamin hydroclorid 7 % (TT) và 50 ml nước. Để yên 30 min và thêm 1 ml dung dịch hỗn hợp đỏ methyl (TT) và chuẩn độ bằng dung dịch natri hydroxyd 1 M (CĐ) đến khi màu chuyển sang xanh lục. Song song tiến hành một mẫu trắng. 1 ml dung dịch natri hydroxyd 1 M (CĐ) tương đương với 29,02 mg C2H2O2.
Guanidin hydroclorid
CH5N3.HCl = 95,5.
Dùng loại tinh khiết hóa học.
Bột tinh thể trắng. Dễ tan trong nước và ethanol.
Nhiệt độ nóng chảy: Khoảng178 °C đến 189 °C.
Haemoglobin
Dùng loại tinh khiết hóa học.
Hàm lượng sắt: Từ 0,2 % đến 0,3 %.
Hàm lượng nitrogen: Từ 15 % đến 16 %.
Mất khối lượng do làm khô: Không quá 2 %.
Tro sulfat: Không quá 1,5 %.
Dung dịch haemoglobin
Hòa tan 2 g haemoglobin (TT) trong 75 ml dung dịch acid hydrocloric 0,03 M (TT), điều chỉnh pH của dung dịch tới 1,5 đến 1,7 bằng dung dịch acid hydrocloric 1 M (TT), pha loãng thành 100 ml với dung dịch acid hydrocloric 0,03 M (TT) và thêm 25 mg thiomersal (TT).
Dung dịch pha trong ngày, bảo quản ở nhiệt độ từ 2 °C đến 8 °C, chỉnh pH đến 1,6 trước khi dùng.
Heli dùng cho sắc ký khí
He = 4,003
Dùng loại tinh khiết hóa học đóng ống dùng cho phòng thí nghiệm có chứa không ít hơn 99,5 % (tt/tt) He.
Heptan
n-Heptan
C7H16 = 100,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, dễ cháy.
Tỷ trọng ở 20 °C: 0,683 đến 0,686.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,387 đến 1,388.
Hexamethyldisilazan
C6H19NSi2= 161,4
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Điểm sôi: Khoảng 125 °C
Tỷ trọng ở 20 °C: Khoảng 0,78
Chỉ số khúc xạ ở 20 °C: Khoảng 1,408.
Bảo quản trong đồ chứa kín.
Hexan
C6H14 = 86,2
Chất lỏng không màu, dễ cháy.
Tỷ trọng ở 20 °C: Từ 0,659 đến 0,663.
Chỉ số khúc xạ ở 20 °C: Từ 1,375 đến 1,376.
Không ít hơn 95 % được cất ở 67 °C đến 69 °C.
Hexan dùng cho phương pháp quang phổ
Độ truyền quang: không ít hơn 97 % trong khoảng 260 nm đến 420 nm, dùng nước làm mẫu trắng.
n-Hexan
C6H14 = 86,2
Dùng loại tinh khiết phân tích, chứa không ít hơn 99 % đồng phân tinh khiết.
Chất lỏng không màu, dễ cháy.
Điểm sôi: Khoảng 68 °C.
Khối lượng riêng: Khoảng 0,66 g/ml.
Hexylamin
C6H15N = 101,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Điểm sôi: 127 °C đến 131 °C.
Tỷ trọng ở 20 °C: Khoảng 0,766.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,418.
Hydrazin sulfat
N2H4.H2SO4 = 130,1
Dùng loại tinh khiết phân tích.
Tinh thể không màu. Dễ tan trong nước nóng, ít tan trong nước và ethanol.
Hydridantin
2,2’-Dihydroxy-2,2’-bi-indan-1,1’,3,3’-tetraon dihydrat
C18H10O6.2H2O = 358,3.
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 258 °C.
Hydrogen
H2 = 2,02
Dùng loại tinh khiết hóa học đóng ống dùng cho phòng thí nghiệm, chứa không ít hơn 99,95 % (tt/tt) H2.
Hydrogen peroxyd
Nước oxy già
H2O2 =34,02
Dung dịch hydrogen peroxyd 200 tt
Dùng loại tinh khiết phân tích, chứa khoảng 60 % (kl/tt) H2O2.
Chất lỏng không màu.
Khối lượng riêng: Khoảng 1,18 g/ml.
Dung dịch hydrogen peroxyd 100 tt (Dung dịch hydrogen peroxyd đậm đặc)
Dùng loại tinh khiết phân tích, chứa khoảng 30 % (kl/tt) H2O2.
Chất lỏng không màu.
Khối lượng riêng: Khoảng 1,10 g/ml.
Dung dịch hydrogen peroxyd 20 tt
Dùng loại tinh khiết phân tích, chứa khoảng 6 % kl/tt H2O2 hay pha loãng dung dịch hydrogen peroxyd 100tt (TT) với 4 thể tích nước.
Chất lỏng không màu.
Khối lượng riêng: Khoảng 1,02 g/ml.
Dung dịch hydrogen peroxyd 10 tt (Dung dịch hydrogen peroxyd loãng)
Pha loãng dung dịch hydrogen peroxyd 20 tt (TT) với đồng thể thể tích nước.
Hydrogen sulfid
H2S =34,08
Dùng loại cho phòng thí nghiệm, hay điều chế bằng cách cho sắt sulfid tác dụng với acid hydrocloric (TT) đã pha loãng gấp đôi với nước và rửa khí thu được bằng cách cho chạy qua nước.
Khí độc, không màu, có mùi đặc trưng, khó chịu.
Hydrogen sulfid chứa không ít hơn 99,7 % (tt/tt) H2S.
Dung dịch hydrogen sulfid
Dung dịch bão hòa khí hydrogen sulfid trong nước mới điều chế.
Dung dịch này chứa khoảng 0,45 % (kl/tt) H2S ở 20 °C.
Bảo quản trong đồ đựng kín, tránh ánh sáng.
Dung dịch không bảo quản được lâu.
Hydroquinon
Quinol; Benzen-1,4-diol
C6H6O2= 110,1
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể không màu hay gần như không màu, bị sẫm màu khi tiếp xúc với không khí và ánh sáng. Tan trong nước, ethanol và ether.
Điểm chảy: Khoảng 173 °C.
Dung dịch hydroquinon 1 % trong ethanol
Hòa tan 1,0 g hydroquinon (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Hydroxylamin hydroclorid
NH2OH. HCl = 69,49
Dùng loại tinh khiết phân tích.
Bột kết tinh trắng. Rất dễ tan trong nước, tan trong ethanol.
Dung dịch hydroxylamin hydroclorid 20%
Hòa tan 20 g hydroxylamin hydroclorid (TT) trong nước vừa đủ 100 ml.
Dung dịch hydroxylamin hydroclorid 25 %
Hòa tan 25 g hydroxylamin hydroclorid (TT) trong nước vừa đủ 100 ml.
Dung dịch hydroxylamin hydroclorid 0,5 N
Hòa tan 6,95 g hydroxylamin hydroclorid (TT) trong nước vừa đủ 200 ml.
Dung dịch hydroxyiamin hydroclorid 7 %
Hòa tan 7 g hydroxylamin hydroclorid (TT) trong nước vừa đủ 100 ml.
Dung dịch hydroxylamin hydroclorid 0,5 %
Hòa tan 0,5 g hydroxylamin hydroclorid (TT) trong nước vừa đủ 100 ml.
Dung dịch hydroxylamin hydroclorid trong ethanol
Hòa tan 3,5 g hydroxylamin hydroclorid (TT) trong 95 ml ethanol 60 %, thêm 0,5 ml dung dịch methyl da cam 0,2% trong ethanol 60 % (TT) và thêm dung dịch kali hydroxyd 0,5 N trong ethanol 60 % (CĐ) đến khi xuất hiện màu vàng hoàn toàn, pha loãng thành 100 ml với ethanol 60 %.
Dung dịch hydroxylamin trong kiềm
Trước khi dùng, trộn đều đồng thể tích dung dịch hydroxylamin hydroclorid 13,9 % với dung dịch natri hydroxyd 15% (TT).
Dung dịch hydroxylamin hydroclorid
Hòa tan 3,5 g hydroxylamin hydroclorid (TT) trong ethanol 60 % vừa đủ 100 ml.
Hydroxyquinolin
8-Hydroxyquinolin; Quinolin-8-ol
C9H7NO = 145,2
Dùng loại tinh khiết phân tích.
Bột kết tinh trắng hay trắng ngà, có mùi phenol, dễ bị sẫm màu khi tiếp xúc với ánh sáng. Rất dễ tan trong ethanol, aceton, cloroform và acid vô cơ, hầu như không tan trong nước.
Điểm chảy: Khoảng 74 °C.
Dung dịch hydroxyquinolin 0,5% trong cloroform
Hòa tan 0,5 g hydroxyquinolin (TT) trong cloroform (TT) vừa đủ 100 ml. Chỉ pha trước khi dùng.
Imidazol
Glyoxalin
C3H4N2 = 68,1
Dùng loại tinh khiết hóa học.
Bột kết tinh trắng. Dễ tan trong nước.
Điểm chảy: Khoảng 90 °C.
Imidazol kết tinh lại
Lấy 25 g imidazol (TT), kết tinh lại 2 lần với 100 ml toluen (TT), làm lạnh trong nước đá, khuấy liên tục, rửa với ether (TT), làm khô trên silica gel khan, ở nhiệt độ phòng và áp suất 2 kPa.
Phải đáp ứng yêu cầu sau:
Độ hấp thụ ánh sáng: Độ hấp thụ của dung dịch 8 % (kl/tt) ở 325 nm không quá 0,10 (Phụ lục 4.1).
Thuốc thử imidazol thủy ngân
Hòa tan 8,25 g imidazol kết tinh lại (TT) trong 60 ml nước, thêm 10 ml dung dịch acid hydrocloric 5 M (TT). Dùng máy khuấy từ, khuấy và thêm từng giọt 10 ml dung dịch thủy ngân (II) clorid 0,27 %. Nếu dung dịch có tủa, pha lại dung dịch khác và nhỏ dung dịch thủy ngân (II) clorid chậm hơn. Điều chỉnh pH đến 6,8 ± 0,05 bằng dung dịch acid hydrocloric 5 M (TT) (khoảng 4 ml) và thêm nước vừa đủ 100 ml.
Iminodibenzyl
10,11-Dihydrodibenz[b,f]azepin
C14H13N = 195,3
Bột kết tinh vàng nhạt. Thực tế không tan trong nước, dễ tan trong aceton.
Điểm chảy: Khoảng 106 °C.
lod
l2 = 253,8
Dùng loại tinh khiết phân tích.
Tinh thể màu đen tía có ánh kim loại. Tan trong ethanol, ether và trong dung dịch kali iodid, tan rất ít trong nước.
Dung dịch iod-iodid (Thuốc thử Bouchardat)
Hòa tan 2 g iod (TT) và 4 g kali iodid (TT) trong 10 ml nước, lắc, để yên cho tan, rồi thêm nước vừa đủ 100 ml.
Dung dịch iod-iodid (TT1)
Hòa tan 500 mg iod (TT) và 1,5 g kali iodid (TT) trong nước và pha loãng thành 25 ml bằng cùng dung môi.
Dung dịch iod 1 % trong ethanol
Hòa tan 1 g iod (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Bảo quản tránh ánh sáng.
Dung dịch iod 5 % trong ethanol
Hòa tan 5 g iod (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Dung dịch iod trong ethanol
Dung dịch iod-iodid trong ethanol
Hòa tan 25 g iod (TT) và 25 g kali iodid (TT) trong 25 ml nước, thêm ethanol 90 % (TT) vừa đủ 1000 ml.
Dung dịch iod (TT1)
Dung dịch iod 0,05 M: Hòa tan 20 g kali iodid (TT) trong một lượng nước tối thiểu, thêm 13 g iod (TT), để yên để hòa tan rồi thêm nước vừa đủ 100 ml.
Thêm 0,6 g kali iodid (TT) vào 10,0 ml dung dịch iod 0,05 M thu được ở trên và pha loãng thành 100,0 ml bằng nước.
Chuẩn bị ngay trước khi dùng.
lod bromid
IBr = 206,8
Dùng loại tinh khiết phân tích.
Tinh thể màu đen nâu hay đen lam.
Điểm chảy: Khoảng 40 °C.
Điểm sôi: Khoảng 116 °C.
Bảo quản ở nơi lạnh, tránh ánh sáng.
Dung dịch iod bromid
Hòa tan 2 g iod bromid (TT) trong acid acetic băng (TT) vừa đủ 100 ml.
lod pentoxyd
lod pentoxyd kết tinh lại
l2O5 = 333,8.
Dùng loại tinh khiết hóa học được kết tinh lại theo phương pháp sau: Đun dung dịch bão hòa iod pentoxyd trong acid nitric (TT) trong 1 h, rồi để yên trong 24 h. Loại bỏ chất lỏng phía trên, làm khô các tinh thể thu được ban đầu bằng luồng không khí ở nhiệt độ phòng, sau sấy trên phospho pentoxyd (TT) ở áp suất không quá 5,2 mmHg (0,7 kPa).
Bột kết tinh màu trắng, dễ hút ẩm, chứa không ít hơn 99,5 % l2O5.
Định lượng: Hòa tan 0,1 g chế phẩm trong 50 ml nước, thêm 3,0 g kali iodid (TT) và 10 ml dung dịch acid hydrocloric 2 M (TT). Định lượng iod giải phóng bằng dung dịch natri thiosulfat 0,1 M (CĐ), dùng 1 ml dung dịch hồ tinh bột (TT) làm chỉ thị.
1 ml dung dịch natri thiosulfat 0,1 M tương đương với 2,782 mg l2O5.
Bảo quản trong bao bì kín, tránh ánh sáng.
lod triclorid
ICl3 = 233,3
Dùng loại tinh khiết phân tích.
Tinh thể màu cam đỏ.
Dung dịch iod monoclorid
Dung dịch A: Hòa tan 0,8 g iod triclorid (TT) trong 20 ml acid acetic băng (TT).
Dung dịch B: Hòa tan 0,9 g iod (TT) trong 30 ml dicloromethan (TT).
Trộn dung dịch A và dung dịch B, pha loãng với acid acetic băng (TT) vừa đủ 100 ml.
Bảo quản dung dịch trong bình nút kín, tránh ánh sáng, ở nhiệt độ không quá 15 °C.
Isopropanol
Propan-2-ol
C3H8O = 60,1
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, có mùi đặc biệt. Hòa tan trong nước và ethanol.
Tỷ trọng ở 20 °C: Khoảng 0,785.
Điểm sôi: 81 °C đến 83 °C.
Isopropanol (TT1)
Propan -2-ol (TT1)
Propan-2-ol thỏa mãn các yêu cầu sau:
Chỉ số khúc xạ: Khoảng 1,378.
Độ truyền quang: Không nhỏ hơn 25 % ở 210 nm, 55 % ở 220 nm, 75 % ở 230 nm, 95 % ở 250 nm và 98 % ở 260 nm, dùng nước làm mẫu trắng.
Nước: Không quá 0,05 % (kl/kl) (Phụ lục 10.3), dùng 10 g.
Kali acetat
C2H3O2K = 98,1
Dùng loại tinh khiết hóa học.
Bột hay tinh thể màu trắng, dễ hút ẩm. Dễ tan trong nước và ethanol.
Kali antimony tartrat
Kali antimony oxyde (+)-tartrat
KSbO.C4H4O6.½H2O = 333,9
Dùng loại tinh khiết phân tích.
Kali antimonat (V)
Kali pyroantimonat
KSbO3.3H2O = 262,9
Dùng loại tinh khiết phân tích.
Dung dịch kali antimonat (V)
Hòa tan 2 g kali antimonat (TT) trong 85 ml nước nóng, làm nguội nhanh, thêm 50 ml dung dịch kali hydroxyd 5 % (TT) và 1 ml dung dịch natri hydroxyd 8,5 % (TT). Để yên trong 24 h, lọc và pha loãng với nước thành 150 ml.
Kali biphtalat
Kali hydrophtalat
C8H5O4K = 204,23
Dùng loại tinh khiết phân tích.
Tinh thể trắng. Tan trong nước, ít tan trong ethanol.
Kali bromat
KBrO3 = 167,01
Dùng loại tinh khiết phân tích.
Bột cốm hay tinh thể màu trắng, tan trong nước, ít tan trong ethanol.
Kali bromid
KBr = 119,01
Dùng loại tinh khiết phân tích.
Kali bromid tinh khiết IR
Viên nén dày 2 mm, được làm từ kali bromid (TT) mới sấy khô ở 250 °C trong 1 h, phải cho đường nền phẳng trong khoảng từ 4000 cm-1 đến 620 cm'1, không được có cực đại với độ hấp thụ lớn hơn 0,02 so với đường nền, trừ cực đại của nước ở 3440 cm-1 và 1630 cm-1.
Dung dịch kali bromid 10 %
Hòa tan 10 g kali bromid (TT) trong nước vừa đủ 100 ml.
Dung dịch kali bromid 0,0015 %
Hòa tan 0,15 g kali bromid (TT) trong nước vừa đủ 100 ml. Lấy 1 ml dung dịch này pha loãng với nước thành 100 ml.
Dung dịch chỉ pha khi dùng.
Kali carbonat
K2CO3.1,5H2O= 165,2
Dùng loại tinh khiết hóa học.
Tinh thể hay cốm màu trắng. Tan trong nước, không tan trong ethanol.
Kali carbonat khan
K2CO3 = 138,2
Dùng loại tinh khiết phân tích.
Bột cốm màu trắng, dễ hút ẩm. Tan trong nước, không tan trong ethanol.
Dung dịch kali carbonat 15 %
Hòa tan 15 g kali carbonat khan (TT) trong 100 ml nước.
Kali clorat
Muối Berthollet
KClO3= 122,55
Dùng loại tinh khiết phân tích.
Bột, cốm hay tinh thể màu trắng. Rất dễ tan trong nước sôi, tan trong nước, hầu như không tan trong ethanol.
Kali clorid
KCl = 74,55
Dùng loại tinh khiết phân tích.
Dung dịch kali clorid 10%
Hòa tan 10 g kali clorid (TT) trong nước vừa đủ 100 ml.
Kali cromat
K2CrO4 = 194,20
Dùng loại tinh khiết phân tích.
Tinh thể màu vàng. Tan trong nước, không tan trong ethanol.
Dung dịch kali cromat 5 %
Hòa tan 5 g kali cromat (TT) trong nước vừa đủ 100 ml.
Kali cyanid
KCN = 65,12
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước, ít tan trong ethanol.
Dung dịch kali cyanid 10 %
Hòa tan 10 g kali cyanid (TT) trong nước vừa đủ 100 ml.
Kali dicromat
K2Cr2O7 = 294,20
Dùng loại tinh khiết phân tích.
Tinh thể màu da cam. Tan trong nước, không tan trong ethanol.
Dung dịch kali dicromat 5 %
Hòa tan 5 g kali dicromat (TT) trong nước vừa đủ 100 ml.
Dung dịch kali dicromat 10,6 %
Hòa tan 10,6 g kali dicromat (TT) trong nước vừa đủ 100 ml.
Kali dihydro citrat
C6H7KO7 = 230,2
Dùng loại tinh khiết hóa học.
Kali dihydrophosphat
KH2PO4 = 136,09
Dùng loại tinh khiết phân tích.
Tinh thể không màu. Tan trong nước, không tan trong ethanol.
Kali fericyanid
K3Fe(CN)6 = 329,26
Dùng loại tinh khiết phân tích.
Tinh thể màu đỏ. Dễ tan trong nước, không tan trong ethanol.
Dung dịch kali fericyanid 5 %
Hòa tan 5 g kali fericyanid (TT) trong nước vừa đủ 100 ml.
Dung dịch kali fericyanid 10 %
Hòa tan 10 g kali fericyanid (TT) trong nước vừa đủ 100 ml.
Dung dịch kali fericyanid 1 %
Hòa tan 1 g kali fericyanid (TT) trong nước vừa đủ 100 ml.
Pha trước khi dùng.
Dung dịch kali fericyanid loãng
Thêm 0,5 ml dung dịch sắt (III) clorid 5 % (TT) và 40 ml nước vào 10 ml dung dịch kali fericyanid 1 % (TT), lắc đều.
Pha trước khi dùng.
Kali ferocyanid
K4Fe(CN)6.3H2O = 422,41
Dùng loại tinh khiết phân tích.
Tinh thể trong, màu vàng. Dễ tan trong nước, không tan trong ethanol.
Dung dịch kali ferocyanid
Hòa tan 5,3 g kali ferocyanid (TT) trong nước vừa đủ 100 ml.
Dung dịch kali ferocyanid 10%
Hòa tan 10 g kali ferocyanid (TT) trong nước vừa đủ 100 ml.
Kali hydrocarbonat
Kali bicarbonat
KHCO3 = 100,1
Dùng loại tinh khiết phân tích.
Tinh thể không màu trong suốt. Dễ tan trong nước, thực tế không tan trong ethanol.
Dung dịch bão hòa kali hydrocarbonat trong methanol
Hòa tan 0,1 g kali hydrocarbonat (TT) trong 0,4 ml nước bằng cách đun nóng trên cách thủy. Thêm 25 ml methanol (TT) và khuấy, để trên cách thủy cho đến khi tan hoàn toàn.
Dung dịch chỉ pha khi dùng.
Kali hydrophtalat
C8H5KO4 = 204,2
Dùng loại tinh khiết phân tích.
Kali hydrosulfat
Kali bisulfat
KHSO4 = 136,17
Dùng loại tinh khiết phân tích.
Tinh thể không màu, trong suốt. Dễ tan trong nước, cho dung dịch có tính acid mạnh.
Kali hydroxyd
KOH = 56,11
Dùng loại tinh khiết phân tích, chứa không ít hơn 85,0 % lượng kiềm tính theo KOH và không quá 2,0 % K2CO3.
Hạt, que hay phiến màu trắng, dễ chảy nước.
Dung dịch kali hydroxyd 30 %
Hòa tan 300 g kali hydroxyd (TT) trong nước, để nguội, thêm nước vừa đủ 1000 ml. Để lắng và gạn lấy phần nước trong.
Dung dịch kali hydroxyd 5 %
Hòa tan 50 g kali hydroxyd (TT) trong nước vừa đủ 1000 ml.
Dung dịch kali hydroxyd 0,5 N
Hòa tan 34 g kali hydroxyd (TT) trong nước không có carbon dioxyd (TT) vừa đủ 1000 ml.
Dung dịch kali hydroxyd 3 %
Hòa tan 3 g kali hydroxyd (TT) trong nước không có carbon dioxyd (TT) vừa đủ 100 ml.
Dung dịch kali hydroxyd 1 % trong ethanol
Hòa tan 10 g kali hydroxyd (TT) trong ethanol 96 % (TT) vừa đủ 1000 ml.
Dung dịch kali hydroxyd 2 M trong ethanol
Hòa tan 12 g kali hydroxyd (TT) trong 10 ml nước, thêm ethanol 96 % (TT) vừa đủ 100 ml.
Dung dịch kali hydroxyd 1,5 M trong ethanol
Hòa tan 84 g kali hydroxyd (TT) trong ethanol 96 % (TT) vừa đủ 1000 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch kali hydroxyd 0,5 M trong ethanol
Hòa tan 28 g kali hydroxyd (TT) trong ethanol 96 % (TT) vừa đủ 1000 ml.
Dung dịch kali hydroxyd 0,1 M trong ethanol
Hòa tan 5,6 g kali hydroxyd (TT) trong ethanol 96 % (TT) vừa đủ 1000 ml.
Dung dịch kali hydroxyd trong ethanol
Hòa tan 3 g kali hydroxyd (TT) trong 5 ml nước và pha loãng thành 100 ml bằng ethanol không có aldehyd (TT). Gạn lấy dung dịch trong, dung dịch thu được gần như không màu.
Dung dịch kali hydroxyd trong ethanol (TT1)
Hòa tan 6,6 g kali hydroxyd (TT) trong 50 ml nước và pha loãng thành 1000 ml bằng ethanol (TT).
Dung dịch kali hydroxyd 0,1 M trong methanol
Hòa tan 5,6 g kali hydroxyd (TT) trong methanol (TT) vừa đủ 1000 ml.
Dung dịch kali hydroxyd 10 % trong methanol
Hòa tan 100 g kali hydroxyd (TT) trong methanol (TT) vừa đủ 1000 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch kali hydroxyd 3 % trong methanol
Hòa tan 3 g kali hydroxyd (TT) trong methanol (TT) vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Kali iodat
KIO3 = 214,0
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng. Tan trong nước và acid sulfuric loãng, không tan trong ethanol.
Dung dịch kali iodat 5 %
Hòa tan 5 g kali iodat (TT) trong nước vừa đủ 100 ml.
Kali iodid
KI = 166,01
Dùng loại tinh khiết phân tích.
Dung dịch bão hòa kali iodid
Dung dịch bão hòa kali iodid trong nước không có carbon dioxyd (TT), có chứa một vài tinh thể không tan.
Bảo quán tránh ánh sáng và gạn bỏ nếu không đáp ứng phép thử sau:
Thêm 0,1 ml dung dịch hồ tinh bột (TT) vào 0,5 ml thuốc thử trong 30 ml hỗn hợp gồm 3 thể tích dung dịch acid acetic 6 M (TT) và 2 thể tích cloroform (TT). Nếu xuất hiện màu xanh lam, màu phải mất khi cho thêm không quá 0,05 ml dung dịch natri thiosulfat 0,1 M (CĐ).
Dung dịch kali iodid 50%
Hòa tan 50 g kali iodid (TT) trong nước mới đun sôi để nguội vừa đủ 100 ml. Dung dịch không được có màu.
Dung dịch kali iodid
Hòa tan 16,6 g kali iodid (TT) trong nước mới đun sôi để nguội vừa đủ 100 ml. Dung dịch không được có màu.
Dung dịch kali iodid 10 % (Dung dịch kali iodid loãng)
Hòa tan 10 g kali iodid (TT) trong nước mới đun sôi để nguội vừa đủ 100 ml. Dung dịch không được có màu.
Dung dịch kali iodid 2 %
Hòa tan 2 g kali iodid (TT) trong nước mới đun sôi để nguội vừa đủ 100 ml. Dung dịch không được có màu.
Dung dịch kali iodid - hồ tinh bột
Hòa tan 0,75 g kali iodid (TT) trong 100 ml nước, đun sôi. Thêm 35 ml dung dịch chứa 0,5 g tinh bột (TT), khuấy liên tục. Đun sôi 2 min và để nguội.
Độ nhạy với iod: Lấy 15 ml thuốc thử, thêm 0,05 ml acid acetic băng (TT) và 0,3 ml dung dịch iod 0,0005 M, phải xuất hiện màu xanh lam.
Dung dịch kali tetraiodomercurat kiềm
Hòa tan 11 g kali iodid (TT) và 15 g thủy ngân (II) iodid (TT) trong nước vừa đủ 100 ml. Trước khi dùng, trộn đều đồng thể tích dung dịch trên với dung dịch natri hydroxyd 25 % (TT).
Kali periodat
Kali meta-periodat
KlO4 = 230,0
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng hay tinh thể không màu. Tan trong nước nóng, tan ít trong nước.
Dung dịch kali periodat 0,1 %
Hòa tan 1 g kali periodat (TT) trong nước vừa đủ 1000 ml.
Kail permanganat
KMnO4= 158,04
Dùng loại tinh khiết phân tích.
Dung dịch kali permanganat 5 %
Hòa tan 5 g kali penmanganat (TT) trong nước vừa đủ 100 ml.
Dung dịch kali permanganat trong acid phosphoric
Hòa tan 3 g kali permanganat (TT) trong hỗn hợp 15 ml acid phosphoric (TT) và 70 ml nước, thêm nước vừa đủ 100 ml.
Kali sulfat
Dikali sulfat
K2SO4 = 174,27
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước, không tan trong ethanol.
Dung dịch kali sulfat 0,001 M
Hòa tan 0,174 g kali sulfat (TT) trong nước vừa đủ 1000 ml.
Kali sulfocyanid
Kali thiocyanat
KSCN = 97,18
Dùng loại tinh khiết phân tích.
Tinh thể không màu, dễ chảy nước. Tan trong nước và ethanol.
Dung dịch kali sulfocyanld 10 %
Hòa tan 10 g kali suffocyanid (TT) trong nước vừa đủ 100 ml.
Kali tetraoxalat
Kali trihydro dioxalat
C4H3KO8.2H2O = 254,2
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng.
Kẽm
Zn = 65,37.
Dùng loại tinh khiết phân tích, chứa không được ít hơn 99,5 % Zn.
Hạt hay cốm màu trắng bạc. Tan trong acid loãng kèm giải phóng khí hydrogen.
Phải đáp ứng phép thử sau:
Arsen: Không quá 0,2 phần triệu (Phụ lục 9.4.2).
Lấy 5,0 g, thêm 15 ml acid hydrocloric (TT), 0,1 ml dung dịch thiếc (II) clorid AsT (TT) và 25 ml nước. Tiến hành theo phương pháp A.
Kẽm bột
Dùng loại tinh khiết phân tích, chứa không được ít hơn 90,0 % Zn.
Bột mịn, xanh xám nhạt. Tan trong acid loãng kèm giải phóng khí hydrogen.
Kẽm clorid
ZnCl2 = 136,3
Dùng loại tinh khiết hóa học.
Bột cốm màu trắng hay gần như trắng. Rất dễ tan trong nước, tan trong ethanol.
Dung dịch kẽm clorid-iod
Hòa tan 20 g kẽm clorid (TT) và 6,5 g kali iodid (TT) trong 10,5 ml nước. Thêm 0,5 g iod (TT) và lắc 15 min, lọc nếu cần. Bảo quản tránh ánh sáng.
Kẽm không có arsen
Ngâm một lượng kẽm (TT) trong dung dịch acid cloroplatinic 0,005 %. Để yên 10 min, rửa với nước, gạn kiệt và làm khô ngay lập tức.
Phải đáp ứng các phép thử sau:
Arsen (Phụ lục 9.4.2): Lấy 5,0 g, thêm 15 ml acid hydrocloric (TT), 0,1 ml dung dịch thiếc (II) clorid AsT (TT) và 25 ml nước. Tiến hành theo phương pháp A. Không được có vết màu trên giấy tẩm thủy ngân (II) bromid (TT).
Độ nhạy: Lặp lại phép thử arsen như mô tả ở trên, nhưng thêm 1 ml dung dịch arsen mẫu 1 phần triệu As (TT). Phải xuất hiện vết màu thấy rõ trên giấy tẩm thủy ngân (II) bromid (TT).
Kẽm oxyd
ZnO = 81,4
Dùng loại tinh khiết hóa học.
Kẽm sulfat
ZnSO4.7H2O = 287,5
Dùng loại tinh khiết phân tích.
Kieselguhr
Dùng loại tinh khiết hóa bằng acid.
Kieselguhr G
Kieselguhr (TT) được xử lý với acid hydrocloric và nung, kích thước hạt trung bình 10 mm đến 40 mm, chứa khoảng 15 % calci sulfat hemihydrat.
Phải đáp ứng phép thử sau:
Phân tách sắc ký: Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Chế từ kieselguhr trong dung dịch natri acetat 0,27 %.
Dung môi khai triển: Ethyl acetat - propan-2-ol - nước (65 : 23 :12).
Cách tiến hành: Chấm 5 ml dung dịch chứa 0,01 % (kl/tt) lactose, đường trắng, D-glucose và D-fructose trong pyridin (TT). Triển khai bản mỏng cho đến khi dung môi đi được 14 cm (thời gian triển khai khoảng 40 min). Lấy bản mỏng ra, để khô ngoài không khí và phun 10 ml dung dịch anisaldehyd (TT). Sấy 5 min đến 10 min ở 100 °C đến 105 °C. Trên sắc ký đồ thu được phải có 4 vết, tách nhau rõ ràng.
Lanthan nitrat
La(NO3)3.6H2O = 433,0
Dung loại tinh khiết sử dụng cho quang phổ hấp thụ nguyên tử.
Tinh thể màu trắng. Tan trong nước, ethanol va aceton.
Dung dịch lanthan nitrat 5 %
Hòa tan 5 g lanthan nitrat (TT) trong nước vừa đủ 100 ml.
Lanthan trioxyd
Lanthan oxyd
La2O3 = 325,8
Dùng loại tinh khiết hóa học có chứa không quá 5 phần triệu calci.
Dung dịch lanthan clorid
Thêm từ từ 100 ml acid hydrocloric (TT) vào 58,65 g lanthan trioxyd (TT) và đun nóng tới sôi. Để nguội và thêm nước vừa đủ 1000 ml.
Linalol
(RS)-3,7-Dimethylocta-1,6-dien-3-ol, linalool
C10H18O = 154,2
Hỗn hợp của 2 stereoisomer (licareol và coriandrol). Dạng lỏng và thực tế không tan trong nước.
Tỷ trọng ở 20 °C: Khoảng 0,860.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,462.
Điểm sôi: Khoảng 200 °C.
Linalol dùng trong sắc ký khí phải đạt yêu cầu: Hàm lượng không dưới 98,0 %. Được xác định bằng phương pháp sắc ký khí (Phụ lục 5.2).
Lithi
Li = 6,9
Dùng loại tinh khiết phân tích.
Kim loại mềm, bề mặt mới cắt có màu xám bạc. Phản ứng rất mạnh với nước. Trước khi sử dụng, rửa sạch dầu parafin bảo quản với toluen (TT).
Bảo quản trong ether dầu hoả nhẹ hay parafin lỏng.
Lithi clorid
LiCI = 42,4
Dùng loại tinh khiết phân tích.
Bảo quản trong đồ chứa kín.
Lithi sulfat
Li2SO4.H2O = 128,0
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng. Tan trong nước, không tan trong ethanol.
Lưu huỳnh (Lưu huỳnh kết tủa)
S = 32,06
Dùng loại tinh khiết hóa học.
Bột xốp, màu vàng xám nhạt hay vàng lục nhạt.
Dung dịch lưu huỳnh kết tủa 1 % trong carbon disulfid
Hòa tan 1 g lưu huỳnh (TT) trong carbon disulfid (TT) vừa đủ 100 ml.
Macrogol 200
Polyethylen glycol 200
Dùng loại tinh khiết hóa học.
Chất lỏng nhớt.
Tỷ trọng ở 20 °C: Khoảng 1,127.
Chỉ số khúc xạ: Khoảng 1,450.
Macrogol 200 (TT1)
Polyethylen glycol 200 (TT1)
Cho 500 ml polyethylen glycol 200 (TT) vào bình cầu dung tích 1000 ml. Cất quay ở 60 °C, với áp suất 1,5 đến 2,5 kPa, trong 6 h để loại các thành phần bay hơi.
Magnesi
Mg = 24,31
Dùng loại tinh khiết hóa học.
Bột màu xám hoặc vỏ bào màu trắng bạc.
Magnesi acetat
C4H6MgO4.4H2O = 214,5
Dùng loại tinh khiết phân tích.
Tinh thể không màu, dễ chảy nước. Dễ tan trong nước và ethanol.
Dung dịch magnesi uranyl acetat
Hòa tan bằng cách đun nóng trên cách thủy 3,2 g uranyl acetat (TT), 10 g magnesi acetat (TT), 2 ml acid acetic băng (TT) và khoảng 30 ml nước. Để nguội. Thêm 50 ml ethanol (TT) và nước vừa đủ 100 ml. Để yên 24 h, lọc.
Magnesi clorid
MgCl2.6H2O = 203,3
Dùng loại tinh khiết phân tích.
Magnesi nitrat
Mg(NO3)2.6H2O = 256,4
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng, dễ hút ẩm. Tan trong nước, ethanol và amoniac.
Dung dịch magnesi nitrat
Hòa tan 17,3 g magnesi nitrat (TT) trong 5 ml nước hơi ấm, thêm 80 ml ethanol 96 % (TT). Để nguội và pha loãng thành 100 ml với cùng dung môi.
Dung dịch magnesi nitrat (TT1)
Hòa tan 20 g magnesi nitrat (TT) trong nước trao đổi ion và pha loãng thành 100 ml với cùng dung môi. Ngay trước khi dùng, pha loãng 10 ml dung dịch thu được thành 100 ml bằng nước trao đổi ion. 5 μl dung dịch có chứa 0,06 mg Mg(NO3)2.
Magnesi oxyd
Magnesi oxyd nhẹ
MgO = 40,31
Dung loại tinh khiết hóa học.
Magnesi oxyd nặng
Dùng loại tinh khiết hóa học.
Magnesi sulfat
MgSO4.7H2O = 246,8
Dùng loại tinh khiết phân tích.
Dung dịch magnesi sulfat 25 %
Hòa tan 25 g magnesi sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch magnesi sulfat 12 %
Hòa tan 12 g magnesi sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch magnesi sulfat 5 %
Hòa tan 5 g magnesi sulfat (TT) trong nước vừa đủ 100 ml.
Mangan sulfat
MnSO4.H2O= 169,0
Dùng loại tinh khiết phân tích.
Tinh thể hay bột tinh thể màu hồng nhạt. Dễ tan trong nước, thực tế không tan trong ethanol 96 %.
Phải đáp ứng phép thử sau:
Mất khối lượng do nung: 10,0 % đến 12,0 %. Xác định trên 1,000 g ở 500 °C.
Giấy tẩm mangan bạc
Ngâm các dải giấy lọc trong dung dịch chứa 0,85 % mangan sulfat (TT) và 0,85 % bạc nitrat (TT), để vài min. Làm khô giấy đã tẩm trên phosphor pentoxyd (TT) tránh hơi acid và kiềm.
D-Mannose
Mannose
C6H12O6 = 180,2
Tinh thể nhỏ hay bột kết tinh trắng hoặc gần như trắng. Rất tan trong nước, khó tan trong ethanol khan.
Góc quay cực riêng: Từ +13,7° đến +14,7°. Dùng dung dịch chế phẩm 200 g/l trong nước chứa 0,05 % amoniac (TT) để đo.
Điểm chảy: Khoảng 132 °C, kèm theo phân hủy.
(-) Menthol
(-)-p-Menthan-3-ol
C10H20O = 156,3
Dùng loại tinh khiết hóa học.
Bột kết tinh trắng, mùi thơm bạc hà.
Điểm chảy: Khoảng 44 °C.
Năng suất quay cực ở 20 °C: Khoảng -50°, dùng dung dịch 5 % (kl/tt) trong ethanol (TT).
(±) Menthol
(±)-p- Menthan-3-ol
C10H20O = 156,3
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 34 °C.
Methanol
Alcol methylic
CH3OH = 32,04
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, dễ bắt lửa. Trộn lẫn với nước và ethanol.
Tỷ trọng ở 20 °C: 0,791 đến 0,793.
Điểm sôi: 64 °C đến 65 °C.
Methanol khan
Dùng cho phương pháp Karl Fischer và các phương pháp chuẩn độ môi trường khan.
Hàm lượng nước không được quá 0,03 % (kl/tt) (Phụ lục 10.3).
Có thể điều chế bằng phương pháp sau:
Xử lý 1000 ml methanol (TT) với 5 g magnesi (TT). Nếu cần, khơi mào phản ứng với 0,1 ml dung dịch thủy ngân (II) diclorid 5 % (TT). Khi bọt khí bay hết, cất và thu dịch cất vào bình khô, để tránh ẩm.
Methanol dùng cho phương pháp quang phổ
Methanol TT1
Phải có độ truyền quang tối thiểu là 20 % ở 210 nm, 50 % ở 220 nm, 75 % ở 230 nm, 95 % ở 250 nm và
98 % ở 260 nm, dùng nước làm mẫu trắng.
Methanol (TT2)
Hàm lượng methanol không nhỏ hơn 99,8 %.
Độ hấp thụ: Độ hấp thụ của chế phẩm ở 225 nm (Phụ lục 4.1) không được lớn hơn 0,17, dùng nước làm mẫu trắng.
Methanol không có aldehyd
Hòa tan 25 g iod (TT) trong 1 L methanol (TT). Vừa rót dung dịch thu được vào 400 ml dung dịch natri hydroxyd 1 M (TT), vừa khuấy. Thêm 150 ml nước và để yên 16 h. Lọc và đun sôi dưới sinh hàn đến khi hết iod. Chừng cất phân đoạn dung dịch thu được.
Aldehyd và keton: Không được quá 0,001 %.
2-Methoxyethanol
Ethylen glycol monomethyl ether
C3H8O2 = 76,10
Dùng loại tinh khiết dùng cho sắc ký.
Chất lỏng không màu.
Điểm sôi: Khoảng 125 °C.
Tỷ trọng ở 20 °C: Khoảng 0,93.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,406.
Methyl acetat
C3H6O2= 74,1
Dùng loại tinh khiết hóa học.
Tỷ trọng ở 20 °C: Khoảng 0,933.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,361.
Điểm sôi: 56 °C đến 58 °C.
Methylbenzothiazolon hydrazon hydroclorid
3- Methylbenzothiazol-2 (3H)-on hydrazon hydroclorid monohydrat C8H10ClN3S.H2O = 233,7
Bột kết tinh gần như trắng hoặc vàng.
Điểm chảy: Khoảng 270 °C.
Kiểm tra tính thích hợp để xác định aldehyd:
Dung dịch thử. Lấy 2 ml methanol không có aldehyd (TT), thêm 60 μl dung dịch propionaldehyd (TT) 0,1 % trong methanol không có aldehyd (TT) và 5 ml dung dịch methylbenzothiazolon hydrazon hydroclorid 0,4 %, trộn đều, để yên 30 min.
Dung dịch mẫu trắng. Chuẩn bị như dung dịch thử, không có dung dịch propionaldehyd.
Thêm 25,0 ml dung dịch sắt (III) clorid 0,2 % vào dung dịch thử và mẫu trắng và pha loãng thành 100,0 ml bằng aceton (TT) và trộn đều.
Độ hấp thụ (Phụ lục 4.1) của dung dịch thử ở bước sóng 660 nm không được nhỏ hơn 0,62.
(R)-(+)-α-Methylbenzyl isocyanat
(+)-(R)-α-methylbenzylisocyanat; (+)-[(1R)-1-lsocyanatoethyl]benzen;
(+)-(1R)-1 -phenylethyl isocyanat
C9H9NO = 147,2
Chất lỏng không màu, chứa ít nhất 99,0 % (R)-(+)-α-methylbenzyl isocyanat.
Tỷ trọng ở 20 °C: Khoảng 1,045.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,513.
Điểm sôi: Khoảng 55 °C đến 56 °C ở 2,5 mmHg.
Tinh khiết đồng phân đối quang: Không được nhỏ hơn 99,5 %.
Bảo quản ở nhiệt độ từ 2 °C đến 8 °C.
Methyl decanoat
Methyl caprat
C11H22O2 = 186,3
Dùng loại tinh khiết hóa học.
Chất lỏng không màu hay màu vàng.
Tỷ trọng ở 20 °C: 0,871 đến 0,876.
Chỉ số khúc xạ ở 20 °C: 1,425 đến 1,426.
Phải đáp ứng phép thử sau:
Tạp chất liên quan: Xác định bằng phương pháp sắc ký khí (Phụ lục 5.2).
Dùng các dung dịch sau:
Dung dịch (1): Dung dịch chế phẩm 0,002 % (kl/tt) trong carbon disulfid (TT).
Dung dịch (2): Dung dịch chế phẩm 0,2 % (kl/tt) trong carbon disulfid (TT).
Dung dịch (3): Carbon disulfid (TT).
Điều kiện sắc ký:
Cột thủy tinh (1,5 m ´ 4 mm) nhồi diatomit silan hóa (như Diatomit CQ là thích hợp) (100 đến 120 mesh) được bao bằng cao su (methyl) gum silicon 10 % (kl/kl) (như SE-30 là thích hợp). Duy trì nhiệt độ cột ở 150 °C và sử dụng tiền cột chứa bông thủy tinh silan hóa.
Tổng diện tích các pic phụ trong sắc ký đồ thu được của dung dịch (2) không được lớn hơn diện tích pic chính trong sắc ký đồ thu được của dung dịch (1).
4- Methylaminophenol sulphat
Metol
(C7H9NO)2.H2SO4= 344,4
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoáng 260 °C.
Methylen clorid
Dicloromethan
CH2Cl2 = 84,93
Dùng loại tinh khiết phân tích.
Chất lỏng dễ bay hơi, ngửi thấy ngọt.
Điểm sôi: Khoảng 40 °C.
Khối lượng riêng: Khoảng 1,32 g/ml.
Methylen clorid đã được acid hóa
Dichloromethan đã được acid hóa
Thêm 10 ml acid hydrochloric (TT) vào 100 ml methylen clorid (TT), lắc đều, để yên cho tách thành 2 lớp. Lấy lớp dưới để dùng.
4-Methylpentan-2-on
Methyl isobutyl keton
C6H12O = 100,2
Chất lỏng trong và không màu. Khó tan trong nước, có thể trộn với hầu hết các dung môi hữu cơ.
Tỷ trọng ở 20 °C: Khoảng 0,80.
Điểm sôi: Khoảng 115 °C.
Khoảng chưng cất: Chưng cất 100 ml chế phẩm, khoảng nhiệt độ để chưng cất được từ 1 ml đến 95 ml chế phẩm không được quá 4,0 °C.
Cắn sau khi bay hơi: Không được quá 0,01 %. Xác định bằng cách bốc hơi trong cách thủy và sấy khô ở nhiệt độ từ 100 °C đến 105 °C.
4-Methylpentan-2-on (TT1)
Methyl isobutyl keton TT1
Lắc 50 ml dung dịch mới cất 4-methylpentan-2-on (TT) với 0,5 ml dung dịch acid hydrocloric 25 % (TT) trong 1 min. Để yên cho tách lớp và dùng lớp trên. Chuẩn bị ngay trước khi dùng.
N-Methylpyrolidin
C5H11N = 85 2
Hàm lượng C5H11N: Không được ít hơn 97,0 %.
Điểm sôi: Khoảng 80 °C.
N-Methylpyrolidon
1 -Methylpyrolidin-2-on
C5H9NO = 99,1
Tỷ trọng ở 20 °C: Khoảng 1,028.
Điểm sôi: Khoảng 202 °C.
Điểm chảy: Khoảng -24 °C.
Muối natri của acid cromotropic
Dinatri 4,5-dihydroxynaphthalen-2,7-disulfonat dihydrat C10H6Na2O8S2.2H2O = 400,3
Dùng loại tinh khiết hóa học.
Bột màu trắng vàng. Tan trong nước, thực tế không tan trong ethanol.
Dung dịch acid cromotropic-acid sulfuric
Hòa tan 5 mg muối natri của acid cromotropic (TT) trong 10 ml hỗn hợp gồm 9 ml acid sulfuric (TT) và 4 ml nước.
Phải đáp ứng phép thử sau:
Độ nhạy với formaldehyd: Thêm 5 ml dung dịch formaldehyd (TT) có nồng độ 1 mg/ml vào 13 ml dung dịch, đun nóng trong cách thủy 30 min. Màu tím xuất hiện, so sánh với mẫu trắng được chuẩn bị tương tự, dùng nước thay cho dung dịch formaldehyd.
Naphtalen
C10H8 = 128,2
Dùng loại tinh khiết hóa học.
Tinh thể trắng.
Điểm chảy: Khoảng 81 °C.
1-Naphthylamin
Naphthylamin; 1-Aminonaphthalen
C10H9N = 143,2
Dùng loại tinh khiết hóa học.
Tinh thể gần như không màu hay bột kết tinh màu trắng.
Điểm chảy: Khoảng 51 °C.
Bảo quản tránh ánh sáng.
Dung dịch naphthylamin 0,001 %
Hòa tan 1 mg naphthylamin (TT) trong nước vừa đủ 100 ml.
N-(1-naphthyl)-ethylendiamin dihydroclorid
C12H14N2.2HCl = 259,2.
Dùng loại tinh khiết hóa học, có thể chứa methanol lúc kết tinh.
Bột trắng hay màu kem.
Điểm chảy: Không thấp hơn 188 °C.
Dung dịch N-(1-naphthyl)-ethylendiamin dihydroclorid 0,5 %
Hòa tan 0,5 g N-(1-naphthyl)-ethylendiamin dihydro-clorid (TT) trong nước vừa đủ 100 ml.
1- Naphtol
a-Naphtol
C10H8O = 144,17
Dùng loại tinh khiết phân tích.
Tinh thể không màu, có mùi phenol. Dễ tan trong ethanol, cloroform và ether, ít tan trong nước.
Điểm chảy: Khoảng 95 °C.
Dung dịch 1-naphtol
Hòa tan 0,1 g 1-naphtol (TT) trong 3 ml dung dịch natri hydroxyd 15 % (TT) và pha loãng thành 100 ml với nước.
Chỉ pha trước khi dùng.
2- Naphtol
β-Naphtol
C10H7OH = 144,17
Dùng loại tinh khiết phân tích.
Bột kết tinh trắng hay hồng nhạt. Khó tan trong nước, dễ tan trong ethanol, ether và dung dịch natri hydroxyd.
Điểm chảy: Khoảng 122 °C.
Dung dịch 2-naphtol
Hòa tan 5 g 2-naphtol (TT) mới kết tinh lại trong 40 ml dung dịch natri hydroxyd 2M và thêm nước vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch 2-naphtol trong kiềm
Hòa tan 0,20 g 2-naphtol (TT) trong 2 ml dung dịch natri hydroxyd 10 % (TT) và pha loãng thành 100 ml với nước.
Dung dịch chỉ pha khi dùng.
Natri
Na = 23,0
Dùng loại tinh khiết phân tích.
Kim loại mềm màu xám bạc. Phản ứng rất mạnh với nước.
Bảo quản trong ether dầu hỏa nhẹ hay parafin lỏng.
Natri acetat
CH3COONa.3H2O = 136,08
Dùng loại tinh khiết phân tích.
Tinh thể trong, không màu, chảy nước ở ngoài không khí. Rất dễ tan trong nước, tan trong ethanol.
Dung dịch natri acetat 40 %
Hòa tan 40 g natri acetat (TT) trong nước vừa đủ 100 ml.
Dung dịch không bảo quản được lâu.
Dung dịch natri acetat 20 %
Hòa tan 20 g natri acetat (TT) trong nước vừa đủ 100 ml.
Dung dịch không bảo quản được lâu.
Natri acetat khan
C2H3O2Na = 82,0
Tinh thể hoặc hạt không màu. Rất tan trong nước, hơi tan trong ethanol 96 %.
Mất khối lượng do làm khô: Không được quá 2,0 % (Phụ lục 9.6,105 °C).
Natri azid
NaN3= 65,01
Dùng loại tinh khiết hóa học, chứa không dưới 98,5 % NaN3.
Bột trắng.
Chú ý: Natri azid là một chất độc mạnh. Khi thực nghiệm với natri azid phải tiến hành trong tủ hút có độ thông hơi tốt, đeo găng tay bảo vệ.
Natri benzoat
C7H5NaO2 = 144,11
Dùng loại tinh khiết phân tích.
Natri bismuthat
NaBiO3= 280,0
Dùng loại tinh khiết phân tích, chứa không ít hơn 85 % NaBiO3.
Bột màu vàng tới nâu vàng, phân hủy chậm khi gặp ẩm hoặc nhiệt độ cao.
Natri butansulfonat
Natri 1-butansulfonat
C4H9NaO3S = 160,2
Dùng loại tinh khiết sắc ký.
Natri carbonat khan
Na2CO3 = 106,00
Dùng loại tinh khiết phân tích.
Bột màu trắng, dễ hút ẩm. Tan trong nước, không tan trong ethanol.
Khối lượng mất đi không được quá 1 % khi nung ở 300 °C.
Dung dịch natri carbonat bão hòa
Hòa tan natri carbonat khan (TT) trong nước đến bão hòa, lọc.
Dung dịch natri carbonat 30 %
Hòa tan 30 g natri carbonat khan (TT) trong nước vừa đủ 100 ml.
Dung dịch natri carbonat 20 %
Hòa tan 20 g natri carbonat khan (TT) trong nước vừa đủ 100 ml.
Dung dịch natri carbonat 10 %
Hòa tan 10 g natri carbonat khan (TT) trong nước vừa đủ 100 ml.
Dung dịch natri carbonat 5 %
Hòa tan 5 g natri carbonat khan (TT) trong nước vừa đủ 100 ml.
Natri citrat
C6H5Na3O7.2H2O = 294,10
Dùng loại tinh khiết phân tích.
Natri clorid
NaCl = 58,44
Dùng loại tinh khiết phân tích.
Dung dịch natri clorid bão hòa
Hòa tan 400 g natri clorid (TT) trong 1000 ml nước và để yên 24 h, thỉnh thoảng lắc. Lọc.
Dung dịch natri clorid 10%
Hòa tan 10 g natri clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch natri clorid 1 %
Hòa tan 1 g natri clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch natri clorid đẳng trương
Hòa tan 9,0 g natri clorid (TT) trong nước vừa đủ 1000 ml. Lọc và tiệt khuẩn.
Dung dịch natri clorid 2 M
Hòa tan 11,688 g natri clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch natri clorid - gelatin
Hòa tan 1 g gelatin (TT) va 10 g natri clorid (TT) trong 100 ml nước bằng cách đun nóng trên cách thủy ở nhiệt độ dưới 60 °C.
Dung dịch chỉ pha khi dùng.
Natri cobaltinitrit
Na3[Co(NO2)6] = 403,9
Dùng loại tinh khiết phân tích.
Bột màu vàng cam. Dễ tan trong nước, khó tan trong ethanol.
Dung dịch natri cobaltinitrit 10%
Hòa tan 10 g natri cobaltinitrit (TT) trong nước vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Natri dihydrophosphat
NaH2PO4.2H2O = 156,01
Dùng loại tinh khiết phân tích.
Tinh thể trắng hay không màu. Dễ tan trong nước, hầu như không tan trong ethanol.
Natri dihydrophosphat khan
Natri dihydro orthophosphat khan
NaH2PO4 = 120,0
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng. Tan trong nước, không tan trong ethanol.
Natri dithionit
Na2S2O4 = 174.
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng hay trắng xám.
Bảo quản trong đồ đựng kín.
Natri fluorid
NaF = 41,99
Dùng loại tinh khiết phân tích.
Tinh thể hay bột màu trắng. Tan trong nước, không tan trong ethanol.
Dung dịch natri fluorid 2,5%
Hòa tan 2,5 g natri fluorid (TT) trong nước vừa đủ 100 ml.
Natri heptansulfonat
C7H15NaO3S = 202,3
Dùng loại tinh khiết sắc ký, chứa không ít hơn 96,0 % C7H15NaO3S.
Khối tinh thể trắng hay gần như trắng. Dễ tan trong nước, tan trong methanol.
Natri hydrocarbonat
Natri bicarbonat
NaHCO3 = 84,01
Dùng loại tinh khiết phân tích.
Dung dịch natri bicarbonat 5 %
Hòa tan 5 g natri bicarbonat (TT) trong nước vừa đủ 100 ml.
Dung dịch natri hydrocarbonat 4,2%
Hòa tan 4,2 g natri hydrocarbonat (TT) trong nước vừa đủ 100 ml.
Dung dịch bão hòa natri hydrocarbonat
Hòa tan 15 g natri hydrocarbonat (TT) trong 100 ml nước, để yên 24 h, thỉnh thoảng lắc. Lọc, sử dụng dịch lọc.
Natri hydrosulfat
Natri bisulfat
NaHSO4 = 120,1
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 315 °C.
Natri hydroxyd
NaOH = 40,00
Dùng loại tinh khiết phân tích, chứa không ít hơn 97 % lượng kiềm toàn phần tính theo NaOH và không quá 2,0 % Na2CO3.
Phiến hay hạt màu trắng, dễ hút ẩm. Dễ tan trong nước và ethanol.
Dung dịch natri hydroxyd 40 % (Dung dịch natri hydroxyd 10 M)
Hòa tan 400 g natri hydroxyd (TT) trong nước, để nguội, pha loãng thành 1000 ml với nước. Để lắng và gạn lấy phần trong.
Dung dịch natri hydroxyd 30 %
Hòa tan 300 g natri hydroxyd (TT) trong nước, để nguội, pha loãng thành 1000 ml với nước. Để lắng và gạn lấy phần trong.
Dung dịch natri hydroxyd 20 % (Dung dịch natri hydroxyd 5 M)
Hòa tan 200 g natri hydroxyd (TT) trong nước, để nguội, pha loãng thành 1000 ml với nước. Để lắng và gạn lấy phần trong.
Dung dịch natri hydroxyd 15 %
Hòa tan 150 g natri hydroxyd (TT) trong nước, để nguội, pha loãng thành 1000 ml với nước. Để lắng và gạn lấy phần trong.
Dung dịch natri hydroxyd 10%
Hòa tan 100 g natri hydroxyd (TT) trong nước, để nguội, pha loãng thành 1000 ml với nước. Để lắng và gạn lấy phần trong.
Dung dịch natri hydroxyd 1 M
Hòa tan 40 g natri hydroxyd (TT) trong nước vừa đủ 1000 ml.
Dung dịch natri hydroxyd 2 M
Hòa tan 80 g natri hydroxyd (TT) trong nước vừa đủ 1000 ml.
Dung dịch natri hydroxyd loãng
Hòa tan 8,5 g natri hydroxyd (TT) trong nước vừa đủ 100 ml.
Dung dịch natri hydroxyd 5 %
Hòa tan 5 g natri hydroxyd (TT) trong nước vừa đủ 100 ml.
Dung dịch natri hydroxyd 2 %
Hòa tan 2 g natri hydroxyd (TT) trong nước vừa đủ 100 ml.
Dung dịch natri hydroxyd trong methanol (TT1)
Hòa tan 0,2 g natri hydroxyd (TT) trong 50 ml nước, để nguội và thêm 50 ml methanol (TT).
Dung dịch natri hydroxyd không có carbonat
Hòa tan natri hydroxyd (TT) vào nước không có carbon dioxyd (TT) để được dung dịch 50 %, để yên. Gạn lấy phần dịch trong ở trên, cẩn thận để tránh sự xâm nhập của carbon dioxyd.
Natri hypobromit (dung dịch)
Trộn 20 ml dung dịch natri hydroxyd 10 M (TT) với 500 ml nước, để lạnh trong nước đá. Thêm 5 ml dung dịch brom (TT), khuấy nhẹ đến khi thu được dung dịch trong.
Dung dịch chỉ pha khi dùng.
Natri hypoclorit (dung dịch)
Dùng loại tinh khiết hóa học, chứa từ 10 % đến 14 % Cl.
Dung dịch natri hypoclorit (3 % Cl) (Dung dịch natri hypoclorit mạnh)
Pha loãng dung dịch natri hypoclorit (TT) với nước để thu được dung dịch chứa từ 2,5 % đến 3 % Cl.
Định lượng: Cho lần lượt vào bình nón 50 ml nước, 1 g kali iodid (TT) và 12,5 ml dung dịch acid acetic 2 M (TT). Pha loãng 10,0 ml thuốc thử cần định lượng thành 100 ml với nước. Lấy 10,0 ml dung dịch thu được vào bình nón đã chuẩn bị ở trên và chuẩn độ bằng dung dịch natri thiosulfat 0,1 N (CĐ), dùng 1 ml dung dịch hồ tinh bột (TT) làm chỉ thị.
1 ml dung dịch natri thiosulfat 0,1 N (CĐ) tương đương với 3,546 mg Cl.
Bảo quản tránh ánh sáng.
Natri hypophosphit
Natri phosphinat monohydrat
NaH2PO2.H2O = 106,0
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng hay tinh thể không màu, dễ hút ẩm. Dễ tan trong nước, tan trong ethanol.
Dung dịch hypophosphit (Thuốc thử Tilé)
Hòa tan 20 g natri hypophosphit (TT) trong 40 ml nước. Đổ dung dịch thu được vào 180 ml acid hydrocloric đậm đặc (TT) và để yên 24 h. Khi các tinh thể natri clorid hình thành đã lắng cặn, gạn lấy phần nước. Dung dịch không được có màu.
Natri iodid
Nal = 149,9
Dùng loại tinh khiết phân tích.
Natri kali tartrat
Muối Seignette
C4H4O6NaK.4H2O = 282,23
Dùng loại tinh khiết phân tích.
Tinh thể hình lăng trụ không màu. Rất dễ tan trong nước.
Dung dịch natri kali tartrat 5 %
Hòa tan 5 g natri kali tartrat (TT) trong nước vừa đủ 100 ml.
Natri Iauryl sulfat
Natri dodecyl sulfat
C12H25NaO4S = 288,4
Dùng loại tinh khiết phân tích, chứa không ít hơn 99,0 % C12H25NaO4S.
Bột hay tinh thể màu trắng đến trắng ngà. Dễ tan trong nước, tan trong ethanol nóng.
Natri metabisulfit
Natri pyrosulfit
Na2S2O5 = 190,1
Dùng loại tinh khiết phân tích, chứa không ít hơn 95,0 % Na2S2O5.
Natri molybdat
Na2MoO4.2H2O = 242,0
Dùng loại tinh khiết hóa học.
Bột tinh thể màu trắng, mất nước kết tinh ở 100 °C. Tan trong nước.
Natri nitrit
NaNO2 = 69,00
Dùng loại tinh khiết phân tích, chứa không được ít hơn 97,0 % NaNO2.
Cốm hay tinh thể màu trắng ngà đến trắng. Tan trong nước, tan ít trong ethanol và ether.
Dung dịch natri nitrit 10%
Hòa tan 10 g natri nitrit (TT) trong nước vừa đủ 100 ml.
Pha ngay trước khi dùng.
Dung dịch natri nitrit 5 %
Hòa tan 5 g natri nitrit (TT) trong nước vừa đủ 100 ml.
Pha ngay trước khi dùng.
Dung dịch natri nitrit 1 %
Hòa tan 1 g natri nitrit (TT) trong nước vừa đủ 100 ml.
Pha ngay trước khi dùng.
Natri nitroprusiat
Natri nitroprussid
Na2[Fe(CN)5(NO)].2H2O = 297,95.
Dùng loại tinh khiết phân tích.
Bột hay tinh thể màu nâu đỏ. Dễ tan trong nước, khó tan trong ethanol.
Dung dịch natri nitroprusiat 1 %
Hòa tan 1 g natri nitroprusiat (TT) trong nước và thêm nước vừa đủ 100 ml.
Dung dịch natri nitroprusiat 0,5 %
Hòa tan 0,5 g natri nitroprusiat (TT) trong nước vừa đủ 100 ml.
Dung dịch natri nitroprusiat 2 %
Hòa tan 2 g natri nitroprusiat (TT) trong nước vừa đủ 100 ml.
Dung dịch natri nitroprusiat 5 %
Hòa tan 5 g natri nitroprusiat (TT) trong nước vừa đủ 100 ml.
Dung dịch natri nitroprusiat 10 %
Hòa tan 10 g natri nitropnisiat (TT) trong nước vừa đủ 100 ml.
Natri octansulfonat
Muối natri của acid octansulfonic
C8H17NaO3S = 216,3
Dùng loại tinh khiết sắc ký, chứa không ít hơn 98 % C8H17NaO3S.
Natri pentansulfonat
Natri 1-pentansulfonat
C5H11NaO3S = 174,2
Dùng loại tinh khiết sắc ký.
Tinh thể màu trắng. Tan trong nước.
Natri perclorat
NaClO4.H2O = 140,5
Dùng loại tinh khiết hóa học, chứa không ít hơn 99,0 % NaClO4.H2O.
Dễ chảy nước.
Natri periodat
Natri metaperiodat
NalO4 = 213,9
Dùng loại tinh khiết phân tích, chứa không ít hơn 99,0 % NalO4.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước, trong các acid vô cơ và acid acetic.
Dung dịch natri periodat 2,14 %
Hòa tan 2,14 g natri metaperiodat (TT) trong nước vừa đủ 100 ml.
Natri phosphat, tribasic
Trinatri orthophosphat; Trinatri phosphat dodeca-hydrat
Na3PO4.12H2O = 380,1
Dùng loại tinh khiết hóa học.
Natri pyrophosphat
Na4P2O7.10H2O = 446,1
Dùng loại tinh khiết phân tích.
Tinh thể không màu, hơi thăng hoa. Dễ tan trong nước.
Natri suIfat
Na2SO4.10H2O = 322,2
Dùng loại tinh khiết phân tích.
Dung dịch natri sulfat 10%
Hòa tan 10 g natri sulfat (TT) trong nước vừa đủ 100 ml.
Natri sulfat khan
Na2SO4 = 142,0
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng, dễ hút ẩm. Tan trong nước, không tan trong ethanol.
Mất khối lượng khi sấy khô ở 130 °C không được quá 0,5 %.
Natri sulfid
Na2S.9H2O = 240,19.
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng, dễ chảy nước. Rất dễ tan trong nước, tan ít trong ethanol, bị vàng trong quá trình bảo quản.
Dung dịch natri sulfid 2 %
Hòa tan 2 g natrisulfid (TT) trong nước, thêm 2-3 giọt glycerin (TT) và pha loãng thành 100 ml với nước.
Dung dịch natri sulfid
Hòa tan bằng cách làm nóng 12 g natri sulfid (TT) trong 45 ml hỗn hợp gồm 10 thể tích nước và 29 thể tích glycerin 85 % (TT), để nguội và pha loãng thành 100 ml với cùng hỗn hợp dung môi. Dung dịch phải không màu.
Dung dịch natri sulfid (TT1)
Hòa tan 5 g natri sulfid (TT) trong hỗn hợp 10 ml nước và 30 ml glycerin (TT). Hoặc hòa tan 5 g natri hydroxyd (TT) trong hỗn hợp 30 ml nước và 90 ml glycerin (TT), bão hòa 1/2 thể tích dung dịch thu được bằng hydrogen sulfid (TT), để nguội, rồi trộn với 1/2 thể tích còn lại.
Natri sulfit
Na2SO3.7H2O = 252,15.
Dùng loại tinh khiết hóa học, chứa không ít hơn 95,0 % Na2SO3.7H2O.
Tinh thể trắng trong. Tan trong nước, tan rất ít trong ethanol.
Dung dịch natri sulfit 40 %
Hòa tan 40 g natri sulfit (TT) trong nước vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch natri sulfit 20 %
Hòa tan 20 g natri sulfit (TT) trong nước vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Natri (+) -tartrat
C4H4O6Na2.2H2O = 230,1
Dùng loại tinh khiết phân tích.
Tinh thể hay cốm màu trắng. Rất dễ tan trong nước, thực tế không tan trong ethanol.
Natri tetraborat
Borax
Na2B4O7.10H2O = 381,4
Dùng loại tinh khiết phân tích.
Natri borohydrid
Natri tetrahydroborat
NaBH4 = 37,83
Tinh thể hút ẩm, dễ tan trong nước, tan trong ethanol.
Natri tetraphenylborat
(C6H5)4BNa = 342,2
Dùng loại tinh khiết hóa học.
Bột màu trắng hay hơi vàng. Dễ tan trong nước, methanol, ethanol khan và aceton.
Dung dịch natri tetraphenylborat 1 %
Hòa tan 1,0 g natri tetraphenylborat (TT) trong nước vừa đủ 100 ml.
Dùng trong vòng một tuần và lọc trước khi dùng nếu cần.
Natri thiosulfat
Na2S2O3.5H2O = 248,2
Dùng loại tinh khiết phân tích.
Cốm màu trắng hay tinh thể trong, không màu. Tan trong nước, tan ít trong ethanol.
Natri tungstat
Na2WO4.2H2O = 329,9
Dùng loại tinh khiết hóa học.
Bột tinh thể màu trắng. Tan trong nước, ít tan trong ethanol.
Nghệ
Thân rễ cây Nghệ (Curcuma longa L.), họ Gừng (Zingiberaceae).
Cồn nghệ
Lấy 20 g bột thân rễ đã sấy khô, ngâm 4 lần, mỗi lần với 100 ml nước. Mỗi lần đều gạn nước trong bỏ đi. Sấy khô cắn thu được ở 100°C đến 105 °C, rồi ngâm với 100 ml ethanol 90 % trong 3 ngày, lọc.
Nhôm clorid
Nhôm clorid hexahydrat
AlCI3.6H2O = 241,4
Dùng loại tinh khiết hóa học chứa không ít hơn 98,0 % AlCI3.6H2O.
Bảo quản trong đồ chứa kín.
Dung dịch nhôm clorid
Hòa tan 65 g nhôm clorid (TT) trong nước vừa đủ 100 ml, thêm 0,5 g than hoạt (TT), khuấy 10 min, lọc. Thêm dung dịch natri hydroxyd 1 % vào dịch lọc, khuấy liên tục, để điều chỉnh pH đến khoảng 1,5 (cần dùng khoảng 60 ml).
Dung dịch nhôm clorid 3 %
Hòa tan 3 g nhôm clorid (TT) trong vừa đủ 100 ml ethanol (TT).
Nhôm (Dây)
Al = 26,98
Dùng loại tinh khiết phân tích.
Nhôm nitrat
Al(NO3)3.9H2O = 375,1
Dùng loại tinh khiết phân tích.
Tinh thể dễ bị chảy nước.
Dung dịch nhôm nitrat 10%
Hòa tan 10 g nhôm nitrat (TT) trong nước vừa đủ 100 ml.
Nhôm - nickel hợp kim (Hợp kim Raney)
Chứa 48 % đến 52 % nhôm và 48 đến 52 % nickel.
Kim loại màu xám. Thực tế không tan trong nước, tan trong các acid vô cơ.
Trước khi dùng tán thành bột mịn.
Nhôm - nickel hợp kim không chứa halogen
Có chứa 48 % đến 52 % nhôm (Al; 26,98) và 48 % đến 52 % nickel (Ni; 58,71). Bột mịn màu xám. Thực tế không tan trong nước, tan trong các acid vô cơ và tạo thành muối của các acid đó.
Clorid: Không được quá 10 phần triệu.
Hòa tan 0,400 g chế phẩm trong 40 ml dung dịch hỗn hợp acid sulfuric - dung dịch acid nitric loãng (67: 33). Bay hơi dung dịch gần đến khô, hòa tan cắn thu được trong nước và pha loãng thành 20,0 ml với cùng dung môi.
Lấy 1/2 thể tích của dung dịch thu được, thêm 1,0 ml dung dịch bạc nitrat 0,1 M (TT). Sau 15 min, lọc và thêm 0,2 ml dung dịch natri clorid (TT) (có chứa 10 μg clorid/1 ml) vào dịch lọc. Sau 5 min, dung dịch thu được đục hơn hỗn hợp gồm 1/2 thể tích dung dịch còn lại và 1,0 ml dung dịch bạc nitrat 0,1 M (TT).
Nhựa trao đổi ion acid mạnh
Hạt nhựa acid mạnh, kích thước 0,3 mm đến 1,2 mm.
Hiệu suất: 4,5 - 5 mmol/g với hàm lượng nước 50 % đến 60 %.
Chuẩn bị cột:
Dùng ống thủy tinh (40 cm × 20 mm), phía dưới lót đĩa thủy tinh xốp và chiều cao nhựa nhồi khoảng 20 cm. Trộn nhựa với nước, đổ hỗn hợp vào cột sao cho không có bọt khí giữa các hạt nhựa. Khi sử dụng, chất lỏng phải luôn phủ trên bề mặt nhựa.
Nếu nhựa ở dạng quá acid, rửa với nước cho đến khi cần không quá 0,05 ml dung dịch natri hydroxyd 0,1 N (CĐ) để trung hòa 50 ml dịch rửa, dùng dung dịch da cam methyl (TT) làm chỉ thị. Nếu nhựa ở dạng natri hoặc cần phải hoàn nguyên, cho 100 ml hỗn hợp đồng thể tích dung dịch acid hydrocloric 7 M (TT) và nước chảy chậm qua cột, sau đó rửa cột như mô tả ở trên.
Nickel (II) sulfat
NiSO4.7H2O = 280,9
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu lục. Tan trong nước và ethanol.
Nicotinamid-adenin dinucleotid
NAD+; C21H27N7O14P2 = 663
Dùng loại tinh khiết hóa học.
Dung dịch nicotinamid-adenin dinucleotid
Hòa tan 40 mg nicotinamid-adenin dinucleotid (TT) trong nước vừa đủ 10 ml.
Pha ngay trước khi dùng.
Ninhydrin
C9H4O3.H2O = 178,15
Dùng loại tinh khiết phân tích.
Bột kết tinh trắng ngà. Tan trong nước và ethanol, ít tan trong cloroform và ether.
Điểm chảy: Khoảng 255 °C.
Dung dịch ninhydrin 2 %
Hòa tan 2 g ninhydrin (TT) trong nước vừa đủ 100 ml.
Dung dịch ninhydrin 0,25 %
Hòa tan 0,25 g ninhydrin (TT) trong nước vừa đủ 100 ml.
Dung dịch ninhydrin 0,1 %
Hòa tan 0,1 g ninhydrin (TT) trong nước vừa đủ 100 ml.
Dung dịch ninhydrin 0,2 %
Hòa tan 0,2 g ninhydrin (TT) trong 100 ml hỗn hợp gồm 95 thể tích n-butanol (TT) và 5 thể tích acid acetic 2 M (TT).
Dung dịch ninhydrin 0,3 % trong ethanol
Hòa tan 0,3 g ninhydrin (TT) trong ethanol (TT) vừa đủ 100 ml.
Dung dịch ninhydrin
Dung dịch ninhydrin (TT1)
Hòa tan 1,0 g ninhydrin (TT) trong 50 ml ethanol 96 % (TT), thêm 10 ml acid acetic băng (TT).
Dung dịch ninhydrin (TT2)
Hòa tan 3 g ninhydrin (TT) trong 100 ml dung dịch natri metabisulfit 4,55 %.
Thuốc thử ninhydrin - thiếc clorid
Hòa tan 0,2 g ninhydrin (TT) trong 4 ml nước nóng, thêm 5 ml dung dịch thiếc (II) clorid 0,16%, để yên 30 min, lọc và bảo quản ở nhiệt độ 2 °C đến 8 °C. Trước khi dùng, thêm vào 2,5 ml dung dịch thu được 5 ml nước và 45 ml isopropanol (TT), trộn đều.
p-Nitroanilin
C6H6N2O2 = 138,13
Dùng loại tinh khiết phân tích.
Bột kết tinh màu vàng đậm. Rất khó tan trong nước, dễ tan trong methanol, tan trong ethanol và ether.
Điểm chảy: Khoảng 148 °C.
Dung dịch p-nitroanilln diazo hóa
Hòa tan 0,4 g p-nitroanilin (TT) trong 60 ml dung dịch acid hydrocloric 1 M (TT) bằng cách đun nóng, làm lạnh đến 15 °C và nhỏ dung dịch natri nitrit 10 % (TT) đến khi 1 giọt hỗn hợp này làm xanh giấy hồ tinh bột có iodid (TT).
Chỉ pha trước khi dùng.
Thuốc thử diazo p-nitroanilin
Hòa tan 0,4 g p nitroanilin (TT) trong một hỗn hợp gồm 20 ml dung dịch acid hydrocloric loãng (TT) và 40 ml nước. Làm lạnh ở 15 °C và thêm dung dịch acid nitric 10 % (TT) cho đến khi một giọt dung dịch làm giấy hồ tinh bột có iodid chuyển thành màu xanh.
Nitrobenzaldehyd
2-Nitrobenzaldehyd
C7H5NO3 = 151,1
Dùng loại tinh khiết hóa học.
Tinh thể hình kim màu vàng, có mùi hạnh nhân.
Điểm chảy: Khoảng 42 °C.
Dung dịch nitrobenzaldehyd
Thêm 0,12 g bột mịn nitrobenzaldehyd (TT) vào 10 ml dung dịch natri hydroxyd 2 M (TT). Lắc đều. Để yên 10 min, thỉnh thoảng lắc, lọc.
Chỉ pha trước khi dùng.
Nitrobenzen
C6H5NO2 = 123,1
Dùng loại tinh khiết phân tích.
Chất lỏng màu vàng nhạt, có mùi hạnh nhân.
Điểm sôi: khoảng 211 °C.
Phải đạt thử nghiệm sau đây:
Dinitrobenzen: Thêm 5 ml aceton (TT), 5 ml nước và 5 ml dung dịch natri hydroxyd 10 M (TT) vào 0,1 ml nitrobenzen (TT), lắc và để yên. Lớp chất lỏng ở trên phải gần như không màu.
Nitrobenzoyl clorid
C7H4ClNO3= 185,6
Dùng loại tinh khiết phân tích.
Tinh thể màu vàng, mùi cay hắc.
Điểm chảy: Khoảng 73 °C.
Nitrogen
N2= 28,0
Dùng loại tinh khiết cho phòng thí nghiệm.
Nitrogen dùng cho sắc ký khí
Nitrogen có chứa không ít hơn 99,5 % (tt/tt) N2.
Nitrogen không có oxygen
Nitrogen được sục qua dung dịch kiềm pyrogalol (TT) để đuổi oxygen.
Nitromethan
CH3NO2 = 61,04
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Tỷ trọng ở 20 °C: Từ 1,132 đến 1,134.
Chỉ số khúc xạ ở 20 °C: Từ 1,381 đến 1,383.
Điểm sôi: Khoảng 102 °C.
Nước
Xem chuyên luận “Nước tinh khiết”.
Nước không có amoniac (Nước không có amoni)
Lấy 100 ml nước, thêm 0,1 ml acid sulfuric (TT) và cất (dùng dụng cụ cất như mô tả trong Phụ lục 6.8), bỏ 10 ml dịch cất đầu, lấy 50 ml dịch cất tiếp theo.
Nước không có carbon dioxyd
Đun sôi một lượng nước cần thiết trong 10 min, làm nguội đến 20 °C, tránh sự thâm nhập của carbon dioxyd từ không khí.
Phải đáp ứng phép thử sau: Thêm 1 giọt dung dịch phenolphtalein (TT) và 0,05 ml dung dịch kali hydroxyd 0,01 N (TT) vào 10 ml nước, hỗn hợp phải có màu hồng bền vững trong ít nhất 120 s.
Nước không có nitrat
Lấy 100 ml nước, thêm 5 mg kali permanganat (TT) và 5 mg bari hydroxyd (TT), cất (dùng dụng cụ cất như đã mô tả trong Phụ lục 6.8), bỏ 10 ml dịch cất đầu, lấy 50 ml dịch cất tiếp theo.
Nước không có các tiểu phân
Nước được lọc qua màng lọc có đường kính lỗ lọc 0,22 mm.
Nước dùng cho sắc ký
Nước khử ion, có điện trở suất không ít hơn 0,18 Mohm-m.
Nước trao đổi ion
Nước cất khử ion, có điện trở suất không ít hơn 18 Mohm-m.
Nước cất
Xem chuyên luận “Nước cất”.
Nước muối sinh lý
Là dung dịch natri clorid 0,9 %.
Oracet lam 2R
1 -Amino-4-(phenylamino)anthracen-9,10-dion
C20H14N2O2 = 314,3
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 174 °C.
Osmi tetroxyd
Acid osmic
OsO4 = 254,2
Khối kết tinh màu vàng hoặc tinh thể hình kim màu vàng nhạt, hút ẩm, nhạy cảm với ánh sáng. Tan trong nước và trong ethanol 96 %.
Bảo quản trong bao bì kín.
Dung dịch osmi tetroxyd
Hòa tan osmi tetroxyd (TT) trong dung dịch acid sulfuric 0,05 M (TT) để được dung dịch có nồng độ khoảng 0,25 %.
Paladi
Pd = 106,4
Dùng loại tinh khiết hóa học.
Pararosanilin hydroclorid
Basis red 9
C19H18ClN3 = 323,8
Dùng loại tinh khiết hóa học.
Bột kết tinh màu đỏ lam.
Điểm chảy: Khoảng 270 °C kèm phân hủy.
Dung dịch pararosanilin đã khử màu
Lấy 0,1 g pararosanilin hydroclorid (TT) vào bình nón nút mài, thêm 60 ml nước và 10 ml nước chứa 1,0 g natri sulfit khan (TT) hoặc 2,0 g natri sulfit (TT), hoặc 0,75 g natri metabisulfit (TT). Thêm từ từ 6 ml dung dịch acid hydrocloric 2 M (TT), khuấy liên tục. Đậy nắp bình, tiếp tục khuấy cho đến khi tan hoàn toàn, pha loãng thành 100 ml với nước. Để yên 12 h trước khi dùng.
Bảo quản tránh ánh sáng.
Penicilinase, dung dịch
Dung dịch penicilinase
Hòa tan 10 g casein thủy phân, 2,72 g kali dihydrophosphat (TT) và 5,88 g natri citrat (TT) trong 200 ml nước, chỉnh đến pH 7,2 bằng dung dịch natri hydroxyd 5 M (TT) và pha loãng thành 1000 ml bằng nước. Hòa tan 0,41 g magnesi sulfat (TT) trong 5 ml nước, thêm 1 ml dung dịch amoni sắt (II) sulfat (TT) 0,16 % và thêm nước vừa đủ 10 ml. Hấp tiệt trùng cả 2 dung dịch bằng nồi hấp autoclave. Để nguội, trộn đều 2 dung dịch và rót vào các bình nón thành lớp mỏng và cấy vào đó chủng vi khuẩn Bacillus cereus (NCTC 9946). Ủ các bình ở nhiệt độ từ 18 °C đến 37 °C đến khi quan sát thấy vi khuẩn phát triển thì ủ ở nhiệt độ 35 °C đến 37 °C trong 16 h, lắc liên tục để đảm bảo thông khí tối đa. Ly tâm và tiệt trùng lớp dịch ở trên bằng cách lọc qua màng lọc thích hợp.
1,0 ml dung dịch penicilinase phải thủy phân benzylpenicilin thành acid benzylpeniciloic với tốc độ thấp nhất là 500 mg/h ở 30 °C và pH 7,0, với điều kiện nồng độ benzylpenicilin không xuống thấp dưới mức cần để bão hòa enzym. Hằng số Michaelis đối với benzylpenicilin của penicilinase trong dung dịch penicinlinase xấp xỉ bằng 12 μg/ml.
Thử vô khuẩn: Phải đáp ứng yêu cầu Thử vô khuẩn (Phụ lục 13.7).
Bảo quản ở nhiệt độ từ 0 °C đến 2 °C và dùng trong vòng 2 đến 3 ngày. Nếu đông khô và đựng trong lọ kín có thể bảo quản trong vài tháng.
Pentan
n-Pentan
C5H12 = 72,15
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, dễ bay hơi.
Điểm sôi: Khoảng 36 °C.
Tỷ trọng ở 20 °C: Khoảng 0,63.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,359.
Pentan dùng cho phương pháp quang phổ
Phải đáp ứng phép thử sau:
Độ truyền quang: Không ít hơn 20 % ở 200 nm, 50 % ở 210 nm, 85 % ở 220 nm, 93 % ở 230 nm, 98 % ở 240 nm, dùng nước làm mẫu trắng.
Pentanol
Pentan-1-ol, n-Pentanol
C5H12O = 88,15
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Điểm sôi: Khoảng 137 °C.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,410.
Phèn chua
Nhôm kali sulfat dodecahydrat
AlK(SO4)2.12H2O = 474,4
Dùng loại tinh khiết phân tích.
Bột hay tinh thể màu trắng, trong. Dễ tan trong nước, không tan trong ethanol.
Dung dịch phèn chua 2 %
Hòa tan 2 g phèn chua (TT) trong nước vừa đủ 100 ml.
Phenazon
2,3-Dimethyl-1-phenyl-3-pyrazolin-5-on
C11H12N2O = 188,2
Dùng loại tinh khiết hóa học.
Bột kết tinh không màu.
Điểm chảy: Khoảng 112 °C.
Phenol
C6H6O = 94,11
Dùng loại tinh khiết phân tích.
Tinh thể dễ chảy nước, ăn da, có mùi đặc trưng.
Điểm đông đặc: Không thấp hơn 40 °C.
Điểm sôi: Khoảng 180 °C.
Dung dịch acid phenoldisulphonic (Xem acid phenoldisulphonic, dung dịch)
Phenoxyethanol
2-Phenoxyethanol
C8H10O2 = 138,2
Dùng loại tinh khiết hóa học.
Chất lỏng dạng dầu, không màu.
Tỷ trọng ở 20°C: Khoảng 1,11.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,537.
Điểm đông đặc: Không được nhỏ hơn 12 °C.
L-Phenylalanin
C9H11NO2 = 165,2
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng.
Điểm chảy: Khoảng 272 °C, kèm theo phân hủy.
Góc quay cực riêng ở 20 °C: Khoảng -34° (dùng dung dịch 2 % trong nước để đo).
Phải đạt phép thử sau:
Tính đồng nhất: Xác định bằng phương pháp sắc ký lớp mỏng (Phụ lục 5.4), tiến hành tránh ánh sáng. Bản mỏng silica gel G (TT), pha động: Nước - phenol (25: 75). Chấm lên bản mỏng 5 μl dung dịch chế phẩm 0,2 %, đặt bản mỏng trong bình bão hòa hơi pha động trước khi khai triển 12 h. Triển khai sắc ký đến khi dung môi đi được 12 cm từ vạch chấm sắc ký. Lấy bản mỏng ra, sấy khô và phun dung dịch ninhydrin 0,1 % trong butan-1-ol đã bão hòa nước (TT), sấy ở 100 °C đến 105 °C trong 10 min. Sắc ký đồ chỉ được có một vết chính.
p-Phenylendiamin dihydroclorid
1,4-Diaminobenzen dihydroclorid
C6H8N2.2HCl = 181,1
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng hoặc vàng nâu nhạt, chuyển màu hồng khi tiếp xúc với không khí. Dễ tan trong nước, ít tan trong ethanol và ether.
Phenylhydrazin hydroclorid
C6H5NH-NH2.HCl = 144,61
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng hay gần như trắng, chuyển sang nâu khi tiếp xúc với không khí. Dễ tan trong nước, tan trong ethanol.
Điểm chảy: Khoảng 245 °C kèm theo phân hủy.
Dung dịch phenylhydrazin - acid sulfuric
Hòa tan 65 mg phenylhydrazin hydroclorid (TT) đã được kết tinh lại trong ethanol 85 % trong hỗn hợp gồm 80 ml nước và 170 ml acid sulfuric (TT) vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch phenylhydrazin hydroclorid
Hòa tan 0,9 g phenylhydrazin hydroclorid (TT) trong 50 ml nước. Khử màu bằng than hoạt (TT) và lọc. Thêm vào dịch lọc 30 ml acid hydrocloric (TT) và pha loãng với nước thành 250 ml.
Phloroglucinol
Benzen-1,3,5-triol
C6H6O3.2H2O = 162,1
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng hay màu kem nhạt. Dễ tan trong nước, tan trong ethanol 96 %.
Điểm chảy: Khoảng 223 °C (Phụ lục 6.7, phương pháp 3).
Dung dịch phloroglucinol
Thêm 9 ml acid hydrocloric (TT) vào 1 ml dung dịch phloroglucinol 10% (kl/tt) trong ethanol 96 % (TT).
Phosphor pentoxyd
Diphosphor pentoxid
P2O5= 142,0
Sử dụng loại chuyên dùng cho bình hút ẩm.
Bột vô định hình màu trắng, chảy rữa khi hydrat hóa với nước và tỏa nhiệt.
Bảo quản trong đồ chứa kín.
Phthalaldehyd
C8H6O2= 134,1
Dùng loại tinh khiết hóa học.
Tinh thể màu vàng nhạt. Tan trong nước, ethanol và ether.
Điểm chảy: Khoảng 55 °C.
Thuốc thử phthalaldehyd
Hòa tan 2,47 g acid boric (TT) trong 75 ml nước, điều chỉnh pH đến 10,4 bằng dung dịch kali hydroxyd 45 % và thêm nước vừa đủ 100 ml (dung dịch A).
Hòa tan 1,0 g phthalaldehyd (TT) trong 5 ml methanol (TT), thêm 95 ml dung dịch A và 2 ml acid mer- captoacetic (TT), điều chỉnh pH đến 10,4 bằng dung dịch kali hydroxyd 45 %.
Bảo quản tránh ánh sáng và dùng trong vòng 3 ngày.
Piperidin
Hexahydropyridin
C5H11N = 85,2
Dùng loại tinh khiết hóa học.
Chất lỏng có tính kiềm, không màu đến hơi vàng. Trộn lẫn được với nước, ethanol 96 %, ether và dầu hỏa nhẹ.
Điểm sôi: Khoảng 106 °C.
Poly(dimethyl)siloxan
Silicone gum rubber (methyl)
Dùng loại tinh khiết hóa học dùng cho sắc ký khí.
Organosilicon polymer có bề ngoài là lớp gôm không màu, bán lỏng. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) không được có đỉnh hấp thụ tại 3053 cm-1 của nhóm vinyl, chuẩn bị mẫu đo bằng cách phân tán chất thử vào vài giọt carbon tetraclorid, tạo đĩa natri clorid để đo phổ.
Polyethylen glycol 300
Macrogol 300
Dùng loại tinh khiết hóa học.
Chất lỏng nhớt, khối lượng riêng khoảng 1,13 g/ml.
Chỉ số khúc xạ: Khoảng 1,465.
Độ nhớt ở 25 °C: Khoảng 80 mPa.s (Phụ lục 6.3, phương pháp 2).
Polyethylen glycol 1500
Macrogol 1500
Dùng loại tinh khiết hóa học.
Khối sáp màu trắng.
Điểm đông đặc: Khoảng 43 °C đến 48 °C.
Độ nhớt: Khoảng 70 mPas đối với dung dịch 50 % (kl/kl).
Polysorbat-80
Thuốc thử Tween-80
Dùng loại tinh khiết hóa học.
Chất lỏng sánh, màu vàng hoặc màu đỏ nhạt.
Khối lượng riêng: Khoảng 1,10 g/ml.
Propanol
Propan-1-ol, n-Propyl alcol
C3H8O = 60,1
Dùng loại tinh khiết phân tích.
Chất lỏng không màu.
Điểm sôi: Khoảng 97 °C.
Tỷ trọng ở 20 °C: 0,802 đến 0,806
Lượng cất được ở khoảng từ 96 °C đến 99 °C, không được ít hơn 95 %.
Propionaldehyd
Propanal
C3H6O = 58,1
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng -81 °C.
Điểm sôi: Khoảng 49 °C.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,365.
Tỷ trọng ở 20 °C: Khoảng 0,81.
Pyrazin-2-carbonitril
2-Cyanopyrazin
C5H3N3 = 105,1
Chất lỏng màu vàng nhạt, trong suốt.
Hàm lượng không được nhỏ hơn 99 %.
Điểm sôi: Khoảng 199 °C.
Pyridin
C5H5N = 79,10
Dùng loại tinh khiết phân tích.
Chất lỏng không màu, mùi đặc biệt khó chịu, rất hút ẩm. Hòa lẫn được với nước và ethanol.
Điểm sôi: Khoảng 115 °C.
Ghi chú: Nếu chỉ định dùng pyridin khan, dùng loại có hàm lượng nước không quá 0,01 % (kl/kl).
Pyridin-2-amin
2-Aminopyridin
C5H6N2 = 94,1
Tinh thể to, tan trong nước và trong ethanol 96 %.
Điểm sôi: Khoảng 210 °C.
Điểm chảy: Khoảng 58 °C.
Pyrocatechol
Catechol, Benzen-1,2-diol
C6H6O2= 110,1
Dùng loại tinh khiết hóa học.
Bột kết tinh trắng. Tan trong nước, aceton, ethanol và ether.
Điểm chảy: Khoảng 103 °C.
Pyrogalol
Benzen-1,2,3-triol
C6H6O3 = 126,1
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng, chuyển sang màu nâu khi tiếp xúc với không khí và ánh sáng. Tan trong nước, ethanol và ether, tan ít trong cloroform.
Điểm chảy: Khoảng 131 °C.
Dung dịch kiềm pyrogalol
Hòa tan 0,5 g pyrogalol (TT) trong 2 ml nước không có carbon dioxyd (TT). Hòa tan 12,0 g kali hydroxyd (TT) trong 8 ml nước không có carbon dioxyd (TT). Trộn hai dung dịch này ngay trước khi dùng.
Pyrrolidin
C4H9N = 71,1
Hàm lượng C4H9N: Không được dưới 99 %.
Điểm sôi: 87 °C đến 88 °C.
2-PyrroIidon
Pyrrolidin-2-on
C4H7NO = 85,1
Dùng loại tinh khiết hóa học.
Hàm lượng không được dưới 98,0 %.
Trên 25 °C ở trạng thái lỏng.
Tỷ trọng ở 25 °C: Khoảng 1,116.
Resorcin
Benzen-1,3-diol
C6H4(OH)2 = 110,1
Dùng loại tinh khiết phân tích.
Tinh thể hay bột kết tinh không màu. Tan trong nước, ethanol và ether.
Điểm chảy: Khoảng 111 °C.
Dung dịch resorcin
Hòa tan 50 g resorcin (TT) trong 50 ml nước, để yên 24 h, thình thoảng lắc, lọc.
Dung dịch resorcin 2 %
Hòa tan 2 g resorcin (TT) trong nước vừa đủ 100 ml.
Dung dịch resorcin 5 % trong ethanol
Hòa tan 5 g resorcin (TT) trong ethanol (TT) vừa đủ 100 ml.
Rhodamin B
Basic violet 10
C28H31CIN2O3 = 479,0
Dùng loại tinh khiết hóa học.
Tinh thể màu xanh lá hay bột màu tím đỏ. Dễ tan trong nước và ethanol.
Salicylaldehyd
2-Hydroxybenzaldehyd
C7H6O2= 122,1
Dùng loại tinh khiết hóa học.
Chất lỏng dầu, không màu với mùi đặc trưng.
Tỷ trọng ở 20 °C: Khoảng 1,167.
Chỉ sổ khúc xạ ở 20 °C: 1,574.
Điểm chảy: Khoảng -7 °C.
Điểm sôi: Khoảng 196 °C.
Salicylaldehyd azin
C14H12N2O2= 240,3
Hòa tan 0,30 g hydrazin sulfat (TT) trong 5 ml nước, thêm 1 ml acid acetic băng (TT) và 2 ml dung dịch salicylaldehyd 20 % trong propan-2-ol (TT) mới pha. Lắc đều, để yên cho đến khi tạo thành tủa màu vàng. Lắc với dicloromethan (TT) 2 lần, mỗi lần 15 ml, để yên cho tách lớp, làm khô các lớp dicloromethan bằng natri sulfat khan (TT), cho bay hơi đến khô. Kết tinh lại cắn thu được bằng hỗn hợp 60 thể tích toluen (TT) và 40 thể tích methanol (TT) bằng cách làm lạnh. Điểm chảy của tinh thể thu được, sau khi làm khô trên phosphor pentoxyd (TT) và ở áp suất 1,5 đến 2,5 kPa, khoảng 213 °C.
Phải đáp ứng phép thử sau:
Độ đồng nhất: Tiến hành như mô tả trong phép thử Hydrazin trong chuyên luận Povidon. Sắc ký đồ chỉ có một vết duy nhất.
Sắt (II) amoni sulfat
Amoni sắt (II) sulfat
(NH4)2Fe(SO4)2.6H2O = 392,1
Dùng loại tinh khiết phân tích.
Bảo quản tránh ánh sáng.
Sắt (III) amoni sulfat
Phèn sắt amoni; Amoni sắt (III) sulfat
FeNH4(SO4)2.12H2O = 482,2
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng tới tím nhạt. Dễ tan trong nước, thực tế không tan trong ethanol.
Dung dịch sắt (III) amoni sulfat 20 %
Hòa tan 20 g sắt (III) amoni sulfat (TT) trong 75 ml nước, thêm 10 ml dung dịch acid sulfuric 2,8 % và thêm nước vừa đủ 100 ml.
Dung dịch sắt (III) amoni sulfat
Hòa tan 30 g sắt (III) amoni sulfat (TT) trong 40 ml acid nitric (TT) và thêm nước vừa đủ 100 ml. Nếu dung dịch đục thì phải lọc hoặc ly tâm.
Dung dịch sắt (III) amoni sulfat 10%
Hòa tan 10 g sắt (III) amoni sulfat (TT) trong nước vừa đủ 100 ml.
Sắt (III) clorid
FeCI3.6H2O = 270,30
Dùng loại tinh khiết phân tích.
Khối kết tinh màu vàng nâu hay vàng cam, dễ chảy nước. Rất dễ tan trong nước, tan trong ethanol và ether.
Dung dịch sắt (III) clorid 20 %
Hòa tan 20 g sắt (III) clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch sắt (III) clorid 10,5 %
Hòa tan 10,5 g sắt (III) clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch sắt (III) clorid 5 %
Hòa tan 5 g sắt (III) clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch sắt (III) clorid 1,3 %
Hòa tan 1,3 g sắt (III) clorid (TT) trong nước vừa đủ 100 ml.
Dung dịch sắt (III) clorid trong ethanol
Hòa tan 1 g sắt (III) clorid (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Dung dịch sắt (III) clorid 5 % trong acid nitric 50 %
Hòa tan 5 g sắt (III) clorid (TT) trong acid nitric 50 % (TT) vừa đủ 100ml.
Dung dịch sắt (III) clorid - fericyanid - arsenit
Dung dịch A: Hòa tan 2,7 g sắt (III) clorid (TT) trong 100 ml dung dịch acid hydrocloric 2 M (TT).
Dung dịch B: Hòa tan 3,5 g kali fericyanid (TT) trong 100 ml nước.
Dung dịch C: Hòa tan 3,8 g arsen trioxyd (TT) trong 25 ml dung dịch natri hydroxyd 2 M (TT) nóng. Để nguội, thêm 50 ml dung dịch acid sulfuric 1 M (TT) và pha loãng 100 ml với nước.
Trước khi dùng, trộn đều 5 thể tích dung dịch A, 5 thể tích dung dịch B và 1 thể tích dung dịch C.
Sắt (III) sulfat pentahydrat
Sắt (III) sulphat pentahydrat; sắt sufat pentahydrat
Fe2(SO4)3.5H2O = 489,9
Bột màu trắng hoặc vàng.
Sắt (III) nitrat
Fe(NO3)3.9H2O = 404,0
Dùng loại tinh khiết phân tích.
Dung dịch sắt (III) nitrat 0,1 % trong acid nitric 0,1 %
Hòa tan 0,1 g sắt (III) nitrat (TT) trong dung dịch acid nitric 0,1 % vừa đủ 100 ml.
Sắt (II) sulfat
FeSO4.7H2O = 278,0
Dùng loại tinh khiết phân tích.
Dung dịch sắt (II) sulfat 8 %
Hòa tan 8,0 g sắt (II) sulfat (TT) trong nước mới đun sôi để nguội vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch sắt (II) sulfat 1,5 %
Hòa tan 1,5 g sắt (II) sulfat (TT) trong nước mới đun sôi để nguội vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch sắt (II) sulfat (TT1)
Hòa tan 3 g sắt (II) sulfat (TT) trong hỗn hợp gồm 3 ml dung dịch acid sulfuric 16 % (TT) và 3 ml nước không có carbon dioxyd (TT).
Dung dịch sắt (II) sulfat (TT2)
Hòa tan 0,45 g sắt (II) sulfat (TT) trong 50 ml dung dịch acid hydrocloric 0,1 M (TT) và pha loãng thành 100 ml bằng nước không có carbon dioxyd (TT).
Chỉ pha trước khi dùng.
Selen
Se = 78,96
Dùng loại tinh khiết phân tích.
Bột màu đỏ rất tối đến đen.
Điểm chảy: Khoảng 220 °C.
Silica gel F254
Bột trắng mịn, đồng nhất, kích thước hạt khoảng 15 mm, có chứa chất bám dính (gắn) thích hợp và khoảng 1,5 % chỉ thị huỳnh quang có cường độ cực đại ở 254 nm. Phải đáp ứng phép thử sau:
Huỳnh quang: Tiến hành bằng phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Dung môi khai triển: Hỗn hợp acid formic khan- propan-2-ol (1: 9).
Chấm riêng biệt lên bản mỏng 10 vết, từ 1 ml đến 10 ml dung dịch acid benzoic 0,1 % trong pha động. Triển khai sắc ký tới khi dung môi đi được 10 cm. Làm khô bản mỏng bằng luồng khí nóng và quan sát dưới ánh sáng từ ngoại ở bước sóng 254 nm. Các vết acid benzoic màu sẫm nằm ở 1/3 phía trên của sắc ký đồ và phát hiện được các vết từ 2 ml trở lên.
Silica gel G
Bột trắng mịn, đồng nhất, kích thước hạt trung bình 10-40 mm, có chứa khoảng 13 % CaSO4.1/2H2O. Hàm lượng CaSO4.1/2H2O:
Cân chính xác khoảng 0,25 g chế phẩm, thêm 3 ml dung dịch acid hydrocloric 2 M (TT) và 100 ml nước, lắc mạnh trong 30 min. Lọc, rửa cắn bằng nước. Gộp nước rửa vào dịch lọc và tiến hành định lượng calci bằng phương pháp chuẩn độ complexon (Phụ lục 10.5).
1 ml dung dịch trilon B 0,1 M (CĐ) tương đương với 14,51 mg CaSO4.1/2H2O.
Độ acid - kiềm:
Lắc 1 g chế phẩm với 10 ml nước không có carbon dioxyd (TT) trong 5 min, pH của hỗn dịch thu được vào khoảng 7.
Silica gel GF254
Bột trắng mịn, đồng nhất, kích thước hạt khoảng 15 mm, có chứa khoảng 13 % CaSO4.1/2 H2O và khoảng 1,5 % chỉ thị huỳnh quang có cường độ cực đại ở 254 nm. Phải đáp ứng yêu cầu về hàm lượng calci sulfat và độ acid - kiềm như mô tả trong chuyên luận “Silica gel G” và phép thử sau:
Huỳnh quang: Tiến hành bằng phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Dung môi khai triển: Hỗn hợp acid formic khan- propan-2-ol (1:9).
Chấm riêng biệt lên bản mỏng 10 vết, từ 1 ml đến 10 ml dung dịch acid benzoic 0,1 % trong pha động. Triển khai sắc ký tới khi dung môi đi được 10 cm. Làm khô bản mỏng bằng luồng khí nóng và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm. Các vết acid benzoic nằm ở 1/3 phía trên của sắc ký đồ và phát hiện được các vết từ 2 ml trở lên.
Silica gel H
Bột trắng mịn, đồng nhất, có kích thước hạt trung bình khoảng 15 mm.
Phải đáp ứng phép thử Độ acid - kiềm như mô tả trong chuyên luận "Silica gel G".
Silica gel HF254
Bột trắng mịn, đồng nhất, có kích thước hạt trung bình khoảng 15 mm, chứa 1,5 % chỉ thị huỳnh quang có cường độ cực đại ở 254 nm.
Phải đáp ứng phép thử Độ acid - kiềm như mô tả trong chuyên luận "Silica gel G" và phép thử Huỳnh quang như mô tả trong chuyên luận "Silica gel GF254".
Silica gel khan
Acid silicic vô định hình, polyme hóa, đã loại nước một phần, ở 20 °C hấp thụ lượng nước tương ứng với khoảng 30 % trọng lượng. Thực tế không tan trong nước, tan một phần trong các dung dịch natri hydroxyd. Chứa chỉ thị thích hợp để xác định trạng thái ẩm: màu thay đổi khi chế phẩm chuyển từ dạng khan sang dạng ngậm nước.
Silica Gel OC để tách các Chiral
Là silica gel dùng cho sắc ký được làm thành bột rất mịn (5 μm), có chứa các tiểu phân được bao bằng dẫn chất sau đây:
Silica Gel OD để tách các Chiral
Là silica gel dùng cho sắc ký được làm thành bột rất mịn (5 μm), có chứa các tiểu phân được bao bằng dẫn chất sau đây:
Silica Gel OJ để tách các Chiral
Là silica gel dùng cho sắc ký được làm thành bột rất mịn, có chứa các hạt hình cầu được bao bằng tris(4-methylbenzoat) celluose. Kích thước các tiểu phân được quy định trong chuyên luận riêng.
Squalan
2, 6, 10, 15, 19, 23-Hexamethyltetracosan
C30H62=422,8
Dùng loại tinh khiết sắc ký khí.
Chất lỏng sánh, không màu.
Tỷ trọng ở 20 °C: 0,811 đến 0,813.
Chỉ số khúc xạ ở 20 °C: 1,451 đến 1,453.
Sudan III
Sudan đỏ; 1-(4-phenylazophenyIazo)-2-naphthol
C22H16N4O = 352,4
Dùng loại tinh khiết hóa học.
Bột màu nâu đỏ.
Dung dịch sudan III
Dung dịch sudan III (TT) 0,5 % trong acid acetic khan (TT).
Sulfanilamid
4-Aminobenzenesulfonamid,
4-aminobenzenesulphonamid, sulphanilamid
C6H8N2O2S = 172,2
Bột trắng hoặc gần như trắng. Khó tan trong nước, dễ tan trong nước sôi, trong aceton và trong dung dịch acid loãng, dung dịch kiềm loãng; hơi tan trong ethanol 96 % và trong ether dầu hỏa có nhiệt độ sôi từ 50 °C đến 60 °C
Điểm chảy: Khoảng 165 °C.
Sulfur dioxyd
Sulphur dioxyd, sulfurous anhydrid
SO2 = 64,05
Khí không màu. Khi nén trở thành chất lỏng không màu.
Dung dịch Sulfur dioxyd
Dung dịch sulphur dioxyd, acid sulfurous, acid sulphurous
H2SO3 = 82,08
Dung dịch pha sẵn chứa 5 % SO2.1 ml dung dịch có khối lượng khoảng 1,03 g.
Tanin
Acid tanic
C76H52O46 = 1701
Dùng loại tinh khiết hóa học.
Bột vô định hình hay dạng vẩy lấp lánh, màu vàng tới nâu nhạt. Tan trong nước và ethanol.
Dung dịch tanin 5 %
Hòa tan 5 g tanin (TT) trong nước vừa đủ 100 ml.
Tetrabutylamoni bromid
(C4H9)4NBr = 322,4
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 102 °C đến 104 °C.
Tetrabutylamoni hydrosulfat
Tetrabutylamino hydrosulphat
C16H37NO4S = 339,5
Dùng loại tinh khiết phân tích.
Bột kết tinh mầu trắng.
Điểm chảy: 169 °C đến 173 °C.
Độ hấp thụ: Độ hấp thụ của dung dịch chế phẩm 5 % (kl/tt) trong khoảng 240 nm đến 300 nm (Phụ lục 4.1) không được lớn hơn 0,05.
pH của dung dịch chế phầm 1,7 % khoảng 1,5.
Tetrabutylamoni hydroxyd
C16H37NO.30H2O = 800
Dùng loại tinh khiết hóa học, chứa không ít hơn 98,0 % C16H37NO.30H2O.
Định lượng: Hòa tan 1 g trong 100 ml nước và chuẩn độ ngay bằng dung dịch acid hydrocloric 0,1 M (CĐ). Xác định điểm kết thúc bằng phương pháp chuẩn độ đo điện thế. 1 ml dung dịch acid hydrocloric 0,1 M (CĐ) tương đương với 80,0 mg C16H37NO.30H2O.
Tetrabutylamoni iodid
C16H36IN = 369,4
Dùng loại tinh khiết phân tích, chứa không ít hơn 98,0 % C16H36IN.
Bột kết tinh hay tinh thể màu trắng hay trắng ngà. Tan trong ethanol.
Phải đáp ứng các phép thử sau:
Tro sulfat: Không quá 0,02 % (Phụ lục 9.9).
Định lượng: Hòa tan 1,200 g chế phẩm trong 30 ml nước. Thêm 50,0 ml dung dịch bạc nitrat 0,1 N (CĐ) và 5 ml dung dịch acid nitric loãng (TT). Định lượng bạc nitrat thừa bằng dung dịch amoni thiocyanat 0,1 N (CĐ), dùng 2 ml dung dịch sắt (III) amoni sulfat (TT) làm chỉ thị.
1 ml dung dịch bạc nitrat 0,1 N (CĐ) tương ứng với 0,03694 g C16H36IN.
Tetrachloroethan
1,1,2,2-tetracloroethan
C2H2Cl4 = 167,9
Dùng loại tinh khiết hóa học
Tỷ trọng ở 20 °C: Khoảng 1,59.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,495.
Phần chưng cất được trong khoảng 145 °C đến 147 °C không được ít hơn 95 %.
n-Tetradecan
C14H30 = 198,4
Dùng loại tinh khiết hóa học, chứa không ít hơn 99,5 % C14H30.
Chất lỏng không màu.
Điểm sôi: Khoảng 253 °C.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,429.
Tỷ trọng ở 20 °C: Khoảng 0,76.
Điểm chảy: Khoảng - 5 °C.
Tetradecylamoni bromid
C40H84BrN = 659,0
Dùng loại tinh khiết sắc ký
Điểm chảy: Từ 88 °C đến 89 °C.
Tetraheptylamoni bromid
C28H60BrN = 490,7
Dùng loại tinh khiết sắc ký.
Điểm chảy: 89 °C đến 91 °C.
Tetrahydrofuran
Tetramethylen oxyd
C4H8O = 72,1.
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, dễ cháy.
Điểm sôi: Khoảng 66 °C.
Tỷ trọng ở 20 °C: Khoảng 0,89.
Chú ý: Chỉ cất nếu đạt phép thử sau:
Peroxyd: Cho 8 ml dung dịch hồ tinh bột có kali iodid (TT) vào ống nghiệm nút mài dung tích 12 ml, đường kính khoảng 1,5 cm, đổ đầy ống nghiệm với tetrahydrofuran cần thử. Đậy nút, lắc mạnh và để yên tránh ánh sáng trong 30 min. Không được có màu tạo thành.
Tetrahydrofuran dùng cho phương pháp quang phổ
Độ truyền quang: Không nhỏ hơn 20 % ở 255 nm, 80 % ở 270 nm và 98 % ở 310 nm, dùng nước làm mẫu trắng.
Tetramethylamoni hydrosulfat
C4H13NO4S = 171,2.
Dùng loại tinh khiết sắc ký.
Tetramethylamoni hydrosulfat dùng cho sắc ký lỏng phải thỏa mãn yêu cầu sau:
Độ truyền quang: Không được ít hơn 50 % ở 200 nm và 90 % ở 220 nm. Dùng dung dịch 0,005 M để xác định.
Tetramethylamoni hydroxyd pentahydrat
C4H13NO.5H2O = 181,2.
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 66 °C.
Dung dịch tetramethylamoni hydroxyd
Dùng loại tinh khiết hóa học, có chứa không ít hơn 10,0 % C4H13NO (kl/kl).
Chất lỏng trong, không màu hoặc có màu rất nhạt.
Định lượng: Lấy 1,0 g chế phẩm, thêm 50 ml nước và định lượng bằng dung dịch acid sulfuric 0,1 N (CĐ), dùng dung dịch đỏ methyl (TT) làm chỉ thị.
1 ml dung dịch acid sulfuric 0,1 N (CĐ) tương đương với 9,115 mg C4H13NO.
Dung dịch tetramethylamonl hydroxyd loãng
Pha loãng 10 ml dung dịch tetramethylamoni hydroxyd (TT) với ethanol không có aldehyd (TT) thành 100 ml. Dung dịch chứa khoảng 1 % C4H13NO (kl/tt).
Pha ngay trước khi dùng.
Tetramethylethylendiamin
Tetramethylethan-1,2-diamin;
N,N,N',N'-tetramethylethylendiamin
C6H16N2= 116,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Điểm sôi: Khoảng 121 °C.
Tỷ trọng ở 20 °C: Khoảng 0,78.
Tetrazoli lam
Muối tetrazoli lam; 3,3’-(3,3'- dimethoxy-4,4’-biphe-nylylen)bis(2,5-diphenyl-2H-tetrazoli clorid) C40H32Cl2N8O2 = 727,7.
Dùng loại tinh khiết hóa học.
Tinh thể màu vàng. Ít tan trong nước, dễ tan trong methanol, ethanol và cloroform.
Điểm chảy: Khoảng 245°C, kèm phân hủy.
Thạch trắng
Agar agar
Dùng loại tinh khiết vi sinh.
Phiến, sợi màu trắng hay trắng ngà, gần như trong suốt, hoặc bột không màu hay trắng ngà. Tan trong nước sôi, ít tan trong nước lạnh, nhưng trương nở thành khối sền sệt.
Theobromin
C7H8N4O2 = 180,2
Dùng loại tinh khiết phân tích.
Bột kết trắng, rất khó tan trong nước và ethanol, khó tan trong amoniac. Tan trong các dung dịch hydroxyd kiềm loãng và acid loãng.
Thiếc
Thiếc hạt
Sn = 118,7
Dùng loại hạt tinh khiết phân tích và đáp ứng phép thử sau:
Arsen: Không quá 10 phần triệu, Phụ lục 9.4.2, phương pháp A, dùng 1 g.
Thiếc (II) clorid
SnCl2.2H2O = 225,63
Dùng loại tinh khiết phân tích, chứa không ít hơn 97,0 % SnCl2.2H2O.
Tinh thể màu trắng. Tan trong nước, ethanol và dung dịch natri hydroxyd.
Dung dịch thiếc (II) clorid 4 %
Hòa tan 4 g thiếc (lI) clorid (TT) trong 4 ml acid hydro-cloric (TT), pha loãng thành 100 ml với nước.
Dung dịch chỉ pha khi dùng.
Dung dịch thiếc (II) clorid 40 % trong acid hydrocloric
Hòa tan 40 g thiếc (II) clorid (TT) trong acid hydrocloric (TT) vừa đủ 100 ml.
Dung dịch thiếc (II) clorid
Đun nóng 20 g thiếc với 85 ml acid hydrocloric (TT) đến khi không còn khí hydro bay ra.
Dung dịch thiếc (II) clorid AsT
Pha loãng gấp đôi dung dịch thiếc (II) clorid (TT) với acid hydrocloric (TT). Bốc hơi tới thể tích ban đầu và lọc qua giấy lọc mịn.
Phải đáp ứng phép thử sau:
Lấy 10 ml thuốc thử, thêm 6 ml nước và 10 ml acid hydrocloric (TT), lấy 16 ml, thêm 50 ml nước, 0,1 ml thuốc thử, 5 ml dung dịch kali iodid 0,1 M và 5 g kẽm không có arsen (TT). Tiến hành thử giới hạn arsen (Phụ lục 9.4.2). Vết thu được trên giấy thủy ngân (II) bromid (TT) không được đậm màu hơn vết khi lặp lại phép thử trên, nhưng thêm 1 ml dung dịch arsen mẫu 1 phần triệu As (TT).
Dung dịch thiếc (II) clorid (TT1)
Pha loãng 1 thể tích dung dịch thiếc (II) clorid (TT) bằng 10 thể tích acid hydrocloric 2 M (TT). Chỉ pha dung dịch trước khi dùng.
Dung dịch thiếc (II) clorid (TT2)
Lấy 8 g thiếc (II) clorid (TT), thêm 100 ml dung dịch acid hydrocloric 20 %. Lắc đến khi tan hoàn toàn, đun nóng trên cách thủy ở 50 °C nếu cần. Sục khí nitrogen (TT) vào dung dịch trong 15 min.
Chỉ pha dung dịch trước khi dùng.
Thioacetamid
CH3CSNH2 = 75,13
Dùng loại tinh khiết hóa học.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước và ethanol, tan ít trong ether.
Điểm chảy: Khoảng 113 °C.
Dung dịch thioacetamid 4 %
Hòa tan 4 g thioacetamid (TT) trong nước vừa đủ 100 ml.
Dung dịch thioacetamid
Thêm 1 ml hỗn hợp gồm 15 ml dung dịch natri hydroxyd 1 N (TT), 5 ml nước và 20 ml glycerin 85 % (TT) vào 0,2 ml dung dịch thioacetamid 4 % (TT), đun nóng trong cách thủy 20 s, làm lạnh và dùng ngay.
Thiodiglycol
Thiodiethylen glycol; 2,2’-thiodiethanol
C4H10O2S = 122,2
Dùng loại tinh khiết hóa học.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,522.
Thioure
NH2.CS.NH2 = 76,12
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể trắng. Tan trong nước và ethanol.
Điểm chảy: Khoảng 178 °C.
Dung dịch thioure 10%
Hòa tan 10 g thioure (TT) trong nước vừa đủ 100 ml.
L-Threonin
C4H9NO3= 119,1
Dùng loại tinh khiết dùng cho sắc ký.
Bột kết tinh màu trắng.
Góc quay cực riêng ở 20 °C: Khoảng -28° (dùng dung dịch 2 % trong nước để đo).
Phải đáp ứng phép thử sau:
Tính đồng nhất: Phương pháp sắc ký lớp mỏng (Phụ lục 5.4). Tiến hành tránh ánh sáng. Bản mỏng silica gel G (TT), pha động: Nước - phenol (25: 75). Chấm lên bản mỏng 5 μl dung dịch chế phẩm 0,2 %, đặt bản mỏng trong hơi pha động trước khi khai triển 12 h. Triển khai sắc ký đến khi dung môi đi được 12 cm từ đường chấm vết. Lấy bản mỏng ra, sấy khô và phun dung dịch ninhydrin 0,1 % trong butan-1-ol (TT) đã bão hòa nước và sấy ở 100 °C đến 105 °C trong 10 min. Sắc ký đồ chỉ được có một vết.
Thuốc thử Diazo
Dung dịch acid sulfanilic: Hòa tan 4,5 g acid sulfanilic (TT) và 45 ml acid hydrocloric (TT) trong nước vừa đủ 500 ml.
Cho 5 ml dung dịch acid sulfanilic vào bình định mức 100 ml được làm lạnh bên ngoài bằng nước đá, thêm 2,5 ml dung dịch natri nitrit 10 % (TT), để yên trong nước đá 5 min. Thêm 10 ml dung dịch natri nitrit 10% (TT) và nước vừa đủ đến vạch.
Bảo quản trong nước đá.
Thuốc thử Dragendorff
Dung dịch kali iodobismuthat
Dung dịch 1: Hòa tan 0,85 g bismuth nitrat base (TT) trong 40 ml nước và 10 ml acid acetic (TT).
Dung dịch 2: Hòa tan 8 g kali iodid (TT) trong 20 ml nước.
Trộn đồng thể tích dung dịch 1 và dung dịch 2. Thêm 100 ml nước và 20 ml acid acetic (TT) vào mỗi 10 ml hỗn hợp thu được.
Thuốc thử Erdmann
Trộn 20 ml acid sulfuric (TT) với 10 giọt hỗn hợp gồm 10 giọt acid nitric (TT) và 100 ml nước.
Thuốc thử Fehling
Dung dịch A: Hòa tan 34,66 g đồng sulfat (TT) trong nước đã acid hóa bằng 2 giọt đến 3 giọt acid sulfuric loãng (TT) vừa đủ 500 ml.
Dung dịch B: Hòa tan 173 g natri kali tartrat (TT) và 50 g natri hydroxyd (TT) trong 400 ml nước, làm nguội, thêm nước vừa đủ 500 ml.
Khi dùng, trộn đồng thể tích dung dịch A và dung dịch B.
Pha loãng 5 ml thuốc thử Fehling với 5 ml nước và đun sôi, dung dịch phải trong, không được xuất hiện vết tủa.
Thuốc thử iodoplatinat
Thêm 97 ml nước và 100 ml dung dịch kali iodid 6 % vào 3 ml dung dịch acid cloroplatinic 10%.
Thuốc thử kali iodobismuthat (TT1)
Hòa tan 100 g acid tartric (TT) trong 400 ml nước, thêm 8,5 g bismuth nitrat base (TT), lắc trong 1 h. Thêm 200 ml dung dịch kali iodid 40 %, lắc đều. Để yên 24 h và lọc.
Thuốc thử kali iodobismuthat loãng
Hòa tan 100 g acid tartric (TT) trong 500 ml nước, thêm 50 ml thuốc thử kali iodobismuthat (TT1), trộn đều.
Dung dịch kali iodobismuthat (TT2)
Phân tán 1,7 g bismuth oxynitrat (TT) và 20 g (+)-acid tartaric (TT) trong 40 ml nước. Thêm 40 ml dung dịch kali iodid 40 % (TT), khuấy trong 1 h và lọc. Dung dịch có thể giữ được trong vài ngày trong điều kiện tránh ánh sáng. Trộn đều 5 ml dung dịch trên với 15 ml nước ngay trước khi dùng.
Thuốc thử kẽm dithiol
Hòa tan 0,2 g toluen - 3,4 dithiol - kẽm phức hợp (TT) trong dung dịch natri hydroxyd 1 % (TT) chứa 0,25 ml ethanol 96 % (TT). Thêm 1 ml acid thioglycolic (TT) và dung dịch natri hydroxyd 1 % (TT) vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Thuốc thử Mayer
Dung dịch kali tetraiodomercurat
Hòa tan 1,358 g thủy ngân diclorid (TT) trong 60 ml nước, thêm 10 ml dung dịch kali iodid 50 % và thêm nước vừa đủ 100 ml.
Thuốc thử methylaminophenol - sulfit
Hòa tan 0,1 g 4-methylaminophenol sulfat (TT), 20 g natri metabisulfit (TT) và 0,5 g natri sulfit khan (TT) trong nước vừa đủ 100 ml.
Thuốc thử Molybdovanadic
Nghiền mịn 4 g amoni molybdat (TT) và 0,1 g amoni metavanadat (TT), hòa tan trong 70 ml nước, thêm 20 ml acid nitric (TT) và nước vừa đủ 100 ml.
Thuốc thử Nessler
Hòa tan 10 g kali iodid (TT) trong 10 ml nước, thêm từ từ dung dịch bão hòa thủy ngân diclorid (TT), khuấy liên tục, cho tới khi xuất hiện tủa đỏ bền vững. Thêm 30 g kali hydroxyd (TT), lắc cho tan, thêm 1 ml dung dịch bão hòa thủy ngân diclorid (TT). Pha loãng thành 200 ml với nước. Để yên và gạn lấy phần nước trong.
Cho 3 giọt thuốc thử vào 10 ml dung dịch amoni mẫu 2 phần triệu NH4 (TT), màu vàng cam phải xuất hiện ngay tức khắc.
Thuốc thử phosphomolybdotungstic
Thuốc thử Folin ciocalteau phenol
Hòa tan 100 g natri tungstat (TT) và 25 g natri molybdat (TT) trong 700 ml nước, thêm 100 ml acid hydrocloric (TT) và 50 ml acid phosphoric (TT), đun nóng hỗn hợp dưới ống sinh hàn ngược trong 10 h. Thêm 150 g lithi sulfat (TT), 50 ml nước và 0,2 ml brom (TT), đun sôi (khoảng 15 min) để đuổi brom thừa, để nguội, pha loãng thành 1000 ml với nước, lọc. Thuốc thử có thể có màu vàng. Nếu thuốc thử có màu xanh thì không được dùng. Xử lý bằng cách đun sôi với 0,2 ml brom (TT). Thận trọng khi đun sôi để đuổi brom thừa.
Bảo quản ở nhiệt độ từ 2 °C đến 8 °C.
Thuốc thử pyridin - anhydrld acetic
Hòa tan 25 g anhydric acetic (TT) trong pyridin khan (TT) vừa đủ 100 ml.
Thuốc thử Schweitzer
Hòa tan 10 g đồng sulfat (TT) trong 100 ml nước và thêm dung dịch natri hydroxyd vừa đủ để kết tủa đồng hydroxyd. Lọc lấy tủa, rửa với nước cho tới khi không cỏn phản ứng với sulfat. Hòa tan tủa còn ướt trong lượng tối thiểu amoniac.
Thuốc thử Smith
Dung dịch glycerin - acid acetic
Trộn đều 1 thể tích glycerin (TT), 1 thể tích dung dịch acid acetic 50 % (TT) và 1 thể tích nước.
Thuốc thử Stiasny
Dung dịch gồm formaldehyd - acid hydrocloric đậm đặc (2:1), thuốc thử pha khi dùng.
Thuốc thửTilé
Xem dung dịch hypophosphit.
Thủy ngân (II) acetat
Hg(CH3COO)2 = 318,68
Dùng loại thuốc thử tinh khiết.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước và ethanol.
Dung dịch thủy ngân (II) acetat 5 %
Hòa tan 5 g thủy ngân (II) acetat (TT) trong acid acetic khan (TT), để nguội, thêm acid acetic khan (TT) vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch thủy ngân (II) acetat
Hòa tan 3,19 g thủy ngân (II) acetat (TT) trong acid acetic khan (TT) vừa đủ 100 ml. Nếu cần, trung hòa với dung dịch acid percloric 0,1 M (CĐ), dùng dung dịch tím tinh thể (TT) làm chỉ thị.
Thủy ngân (II) bromid
HgBr2 = 360,4
Dùng loại tinh khiết phân tích.
Giấy tẩm thủy ngân (II) bromid
Nhúng giấy lọc màu trắng, loại 80g/m2 (Whatman No.1 là thích hợp) kích thước 20 cm × 15 mm đã được gập làm đôi vào một đĩa hình chữ nhật có chứa dung dịch thủy ngân (II) bromid (TT) 5 % trong ethanol (TT). Gạn bỏ dung dịch thừa và làm khô bằng cách treo giấy lên một sợi dây không phải bằng kim loại, ở nơi tránh ánh sáng. Cắt bỏ 1 cm ở chỗ nếp gấp và cạnh ngoài. Cắt thành những mẩu vuông có cạnh 15 cm hoặc thành những đĩa hình tròn có đường kính 15 mm. Bảo quản trong lọ thủy tinh nút kín có bọc giấy đen.
Thủy ngân diclorid
Thủy ngân (II) clorid
HgCl2 = 271,50
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước, ethanol, aceton và ether.
Dung dịch thủy ngân diclorid 5 %
Hòa tan 5 g thủy ngân diclorid (TT) trong nước vừa đủ 100 ml.
Thủy ngân (II) nitrat
Hg(NO3)2.H2O = 342,6
Dùng loại tinh khiết phân tích.
Bột kết tinh màu trắng hay trắng ngà. Dễ tan trong nước và acid loãng.
Dung dịch thủy ngân (II) nitrat
Hòa tan 40 g thủy ngân oxyd vàng (TT) trong hỗn hợp gồm 32 ml acid nitric (TT) và 15 ml nước.
Thủy ngân oxyd vàng
HgO = 216,6
Dùng loại tinh khiết hóa học.
Bột vô định hình màu vàng cam. Thực tế không tan trong nước và ethanol.
Thủy ngân (II) sulfat
HgSO4 = 296,7
Dùng loại tinh khiết hóa học.
Bột kết tinh hay cồm màu trắng, không mùi.
Dung dịch thủy ngân (II) sulfat
Trộn 5 g thủy ngân oxyd vàng (TT) với 40 ml nước, thêm 20 ml acid sulfuric (TT) và 40 ml nước, khuấy liên tục cho tới khi tan hoàn toàn.
Thủy ngân (II) thiocyanat
Hg(SCN)2 = 316,8
Dùng loại tinh khiết hóa học.
Bột kết tinh màu trắng. Rất khó tan trong nước, khó tan trong ethanol và ether, tan trong dung dịch natri clorid.
Dung dịch thủy ngân (II) thiocyanat
Hòa tan 0,3 g thủy ngân (II) thiocyanat (TT) trong ethanol (TT) vừa đủ 100 ml.
Dùng trong vòng một tuần.
Thymin
5-Methylpyrimidin-2,4(1H,3H)-dion
C5H6N2O2= 126,1
Dùng loại tinh khiết hóa học.
Thymidin
1-(2-Deoxy-β-D-erythro-pentofuranosyl)-5- methylpyrimidin-2,4(1H,3H)-dion
C10H14N2O5 = 242,2
Dùng loại tinh khiết hóa học.
Thymol
2- lsopropyl-5-methylphenol
C10H14O - 150,2
Dùng loại tinh khiết phân tích.
Tinh thể không màu.
Điểm chảy: Khoảng 50 °C.
Thymol dùng để pha dung dịch phun bản mỏng
Bột kết tinh hoặc tính thể màu trắng, có mùi thơm. Rất tan trong methanol và ethanol, thực tế không tan trong nước.
Định tính: Phương pháp quang phổ hấp thụ hồng ngoại, chuẩn bị mẫu đo dạng đĩa nén kali bromid. Phổ của chế phẩm có đỉnh hấp thụ ở 2960 cm-1, 1420 cm-1,1290 cm-1,1090 cm-1, và 810 cm-1.
Điểm chảy: Từ 49 °C đến 52 °C.
Các phenol khác: Lắc mạnh 1,0 g chế phẩm với 20 ml nước ấm trong 1 min và lọc. Thêm 1 giọt dung dịch sắt (III) clorid (TT) 27 % vào 5 ml dịch lọc. Dung dịch thu được cho màu xanh lá cây và màu xanh không được chuyển sang màu tím.
Thuốc thử thymol - acid sulfuric - methanol để phun bản mỏng
Hòa tan 1,5 g thymol dùng để pha dung dịch phun bản mỏng (TT) trong 100 ml methanol (TT), thêm 5,7 ml acid sulfuric (TT), trộn đều.
Titan
Ti = 47,9
Bột kim loại, sợi mảnh (đường kính nhỏ hơn 0,5 mm) hoặc hạt xốp chứa ít nhất 99 % Ti.
Điểm chảy: Khoảng 1668 °C.
Khối lượng riêng: 4,507 g/cm3.
Titan (III) clorid
Titan triclorid
TiCI3= 154,3
Dùng loại tinh khiết hóa học.
Tinh thể màu tím đỏ.
Điểm chảy: Khoảng 440 °C.
Bảo quản trong đồ chứa kín.
Dung dịch titan (III) clorid (Dung dịch titan triclorid)
Dùng loại chứa khoảng 15 % (kl/tt) TiCI3 trong dung dịch acid hydrocloric 10%.
Khối lượng riêng: Khoảng 1,2 g/ml.
Thuốc thử titan (III) clorid - acid sulfuric (Thuốc thử titan (III) triclorid - acid sulfuric)
Trộn cẩn thận 20 ml dung dịch titan (III) clorid (TT) với 13 ml acid sulfuric (TT). Thêm dung dịch hydrogen peroxyd 100 tt (TT) vừa đủ để dung dịch có màu vàng. Đun nóng cho đến khi khói trắng bay lên, để nguội. Pha loãng với nước. Lặp lại quá trình bốc hơi và thêm nước cho đến khi dung dịch không màu. Pha loãng với nước thành 100 ml.
Toluen
Methyl benzen
C6H5CH3 = 92,14
Dùng loại tinh khiết phân tích.
Chất lỏng trong, không màu, dễ cháy. Rất ít tan trong nước, hỗn hợp được với ethanol.
Điểm sôi: Khoảng 110 °C.
Tỷ trọng ở 20 °C: 0,865 đến 0,870.
Toluen-p-sulfonamid
p-Toluensulfonamid; Toluen sulfonamid;
4-Methylbenzensulfonamid
C7H9NO2S= 171,2
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 136 °C.
Phải đáp ứng các phép thử sau:
Độ đồng nhất: Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel G.
Dung môi khai triển: Cloroform - methanol - acid formic khan (90:8: 2).
Cách tiến hành: Chấm 5 ml dung dịch chế phẩm 0,015 % trong aceton (TT). Triển khai sắc ký đến khi dung môi đi được 15 cm. Lấy bản mỏng ra, làm khô bằng luồng không khí ấm và sấy 10 min ở 110 °C. Trên sắc ký đồ thu được phải có 4 vết, tách nhau rõ ràng. Đặt dưới đáy bình triển khai sắc ký khô một cốc chứa dung dịch kali permanganat 5 %, thêm đồng thể tích acid hydrocloric (TT). Đặt bản mỏng còn đang nóng vào bình, đậy nắp. Để bản mỏng tiếp xúc với hơi clor trong 2 min. Lấy bản mỏng ra, để dưới luồng khí lạnh cho đến khi hết clor dư và khu vực dưới vết chấm có màu lam rất nhạt khi nhỏ 1 giọt dung dịch hồ tinh bột có kali iodid (TT). Tránh để quá lâu dưới luồng khí lạnh. Phun dung dịch hồ tinh bột có kali iodid (TT) và để yên 5 min. Trên sắc ký đồ thu được chỉ có duy nhất một vết chính.
Toluensulfonyl ure
4-Methylbenzensulfonylure; 4-methylbenzensulphonylure;
p-toluensulfonylure; p-toluensulphonylure; (4- methylphenyl)sulfonylure; (4-methylphenyl)sulphonylure
C8H10N2O3S = 214,2
Bột kết tinh trắng hoặc gần như trắng.
Điểm chảy: Khoảng từ 196 °C đến 198 °C.
Tosylarginin methyl ester hydroclorid
Methyl N-tosyl-L-argininat hydrochlorid
C14H22N4O4S.HCl = 378,9
Dùng loại tinh khiết hóa học thích hợp cho định lượng trypsin.
Bột kết tinh màu trắng.
Điểm chảy: Khoảng 145 °C.
Góc quay cực riêng ở 20 °C: -12° đến -16°, dung dịch 4 % (kl/tt) trong nước.
Tributyl phosphat (tributyl orthophosphat)
C12H27O4P = 266,3
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Khối lượng riêng: Khoảng 0,98 g/ml.
Trước khi dùng, rửa 3 lần với dung dịch chứa 10 % natri clorid (TT) và 1 % dinatri hydrophosphat (TT), mỗi lần với lượng bằng 1/6 thể tích tributyl phosphat.
Triethanolamin
C6H15NO3 = 149,2
Dùng loại tinh khiết hóa học.
Chất lỏng nhớt, không màu, rất hút ẩm, hơi có mùi amoniac, chuyển sang màu nâu khi tiếp xúc với không khí và ánh sáng.
Tỷ trọng ở 20 °C: Khoảng 1,13.
Triethylamin
C6H15N = 101,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, có mùi amoniac.
Tỷ trọng ở 20 °C: Khoảng 0,727.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,401.
Điểm sôi: Khoảng 90 °C.
Triethylendiamin
1,4- Diazobicyclo [2,2,2] octan
C6H12N2= 112,17
Dùng loại tinh khiết hóa học.
Tinh thể dễ hút ẩm, thăng hoa ở nhiệt độ phòng. Dễ tan trong nước, aceton và ethanol.
Điểm chảy: Khoảng 158 °C.
Điểm sôi: Khoảng 174 °C.
Trilon B
Dinatri dihydro ethylendiamin tetraacetat; Natri edetat C10H14N2Na2O8.2H2O = 372,24
Dùng loại tinh khiết phân tích.
Bột kết tinh trắng. Tan trong nước, tan rất ít trong ethanol.
Trimethylclorosilan
Clorotrimethylsilan
C3H9CISi = 108,7
Dùng loại tinh khiết hóa học.
Chất lỏng không màu.
Tỷ trọng ở 20 °C: Khoảng 0,86.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,388.
Điểm sôi: Khoảng 57 °C.
Trimethylpentan
Iso-octan; 2,2,4-Trimethylpentan
C8H18 = 114,2
Dùng loại tinh khiết hóa học.
Chất lỏng không màu, dễ bắt lửa. Không tan trong nước, tan trong aceton, clorotorm và ether.
Tỷ trọng ở 20 °C: 0,691 đến 0,696.
Chỉ số khúc xạ 20 °C: 1,391 đến 1,393.
Phải đáp ứng các phép thử sau:
Khoảng chưng cất: Không ít hơn 95 % được chưng cất ở 98 °C đến 100 °C.
Trimethylpentan dùng cho phương pháp quang phổ
Độ truyền quang: Không nhỏ hơn 98 % trong khoảng 250 nm đến 420 nm, dùng nước làm mẫu trắng.
Triphenylmethanol
Triphenylcarbinol
C19H18O = 260,3
Dùng loại tinh khiết hóa học.
Triphenyl tetrazoli clorid
2,3,5- Triphenyl tetrazolium clorid; Muối tetrazolium
C19H15ClN4 = 334,8.
Dùng loại tinh khiết phân tích, chứa không ít hơn 98,0 % C19H15ClN4.
Bột màu kem hay vàng nhạt.
Điểm chảy: Khoảng 240 °C kèm phân hủy.
Định lượng: Hòa tan 1,0 g trong hỗn hợp gồm 5 ml acid nitric 2 M (TT) và 45 ml nước, thêm 50 ml dung dịch bạc nitrat 0,1 M (CĐ) và đun đến sôi. Để nguội, thêm 3 ml dibutyl phthalat (TT), lắc mạnh và định lượng bằng dung dịch amoni thiocyanat 0,1 M (CĐ), dùng 2 ml dung dịch amoni sắt (II) sulfat 10% (TT) làm chỉ thị.
1 ml dung dịch bạc nitrat 0,1 M (CĐ) tương đương với 33,48 mg C19H15ClN4.
Dung dịch triphenyl tetrazoli clorid
Hòa tan 0,5 g triphenyl tetrazoli clorid (TT) trong 100 ml ethanol không có aldehyd (TT).
Tris(hydroxymethyl)aminomethan
Tris(hydroxymethyl)methylamin; Trometamol; THAM
C4H11NO3=121,14.
Dùng loại tinh khiết hóa học.
Điểm chảy: Khoảng 170 °C.
Uracil
C4H4N2O2 = 112,1
Hàm lượng không được dưới 95,0 %.
Uranyl acetat
(CH3COO)2UO2.2H2O = 424,15
Dùng loại tinh khiết hóa học.
Bột kết tinh màu vàng.
Tan trong nước, tan ít trong ethanol.
Ure
CO(NH2)2 = 60,06
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể màu trắng. Tan trong nước, ethanol và benzen, thực tế không tan trong cloroform và ether.
Điểm chảy: Khoảng 133 °C.
Urotropin
Hexamin; Hexamethylentetramin
(CH2)6N4 = 140,2
Dùng loại tinh khiết phân tích.
Tinh thể màu trắng. Tan trong nước, ethanol và cloroform, tan ít trong ether.
Vàng clorid
Acid cloroauric; Hydrogen tetracloroaurat
HAuCI4 + nước
Dùng loại tinh khiết hóa học.
Khối màu nâu, dễ chảy nước.
Dung dịch vàng clorid (TT)
Hòa tan 1 g vàng clorid (TT) trong 35 ml nước.
Vanilin
4-Hydroxy-3-methoxy benzaldehyd
C8H8O3 =152,2
Dùng loại tinh khiết phân tích.
Bột kết tinh hay tinh thể hình kim trắng hoặc màu kem, có mùi thơm vani. Dễ tan trong ethanol, cloroform và ether, tan trong dầu và dung dịch natri hydroxyd.
Điểm chảy: Khoảng 81 °C, xác định bằng cách không sấy trước.
Dung dịch vanilin 1 % trong acid sulfuric
Hòa tan 1 g vanilin (TT) trong acid sulfuric đậm đặc (TT) vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Dung dịch vanilin 2 % trong acid sulfuric
Hòa tan 0,1 g vanilin (TT) trong 5 ml acid sulfuric (TT), pha dùng ngay.
Dung dịch vanilin 5 % trong acid sulfuric
Hòa tan 5 g vanilin (TT) trong acid sulfuric (TT) vừa đủ 100 ml.
Dung dịch vanilin trong acid hydrocloric
Hòa tan 0,2 g vanilin (TT) trong 10 ml acid hydrocloric đậm đặc (TT).
Dung dịch chỉ pha khi dùng.
Dung dịch vanilin trong ethanol 96 %
Thêm cẩn thận từng giọt 2 ml acid sulfuric (TT) vào 100 ml dung dịch vanilin 1 % trong ethanol 96 %.
Dung dịch vanilin trong acid sulfuric
Thuốc thử vanilin trong acid sulfuric
Hòa tan vanilin (TT) trong ethanol 96 % (TT) đến được dung dịch bão hòa. Lấy phần dung dịch bão hòa, thêm acid sulfuric (TT) đến khi có màu vàng.
Dung dịch vanilin-acid sulfuric
Thuốc thử vanilin-acid sulfuric
Trộn đồng lượng dung dịch vanilin (TT) 1 % trong ethanol 96 % (TT) và dung dịch acid sulfuric 5 % trong ethanol 96 %. Pha trước khi dùng.
Dung dịch vanilin trong acid phosphoric
Hòa tan 1 g vanilin (TT) trong 25 ml ethanol 96 % (TT), thêm 25 ml nước và 35 ml acid phosphoric (TT). Dùng trong 48 h.
Vinyl acetat
Ethenyl acetat
C4H6O2 = 86,1
Tỷ trọng ở 20 °C: Khoảng 0,930.
Điểm sôi: Khoảng 72 °C.
2- Vinylpyridin
C7H7N = 105,1
Dùng loại tinh khiết hóa học.
Chất lỏng màu vàng.
Tỷ trọng ở 20 °C: Khoảng 0,97.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,549.
1-Vinylpyrrolidin-2-on
1-Ethenylpyrrolidin-2-on
C6H9NO= 111,1
Hàm lượng không được dưới 99,0 %.
Chất lỏng trong, không màu.
Nước: Không được quá 0,1 % (Phụ lục 10.3). Dùng 2,5 g chế phẩm. Dùng dung môi là hỗn hợp 50 ml methanol khan (TT) và 10 ml butyrolacton (TT).
Định lượng: Phương pháp sắc ký khí (Phụ lục 5.2).
Cột silica nung chảy, dài 30 m, đường kính trong 0,5 mm, được phủ pha tĩnh macrogol 20 000.
Khí mang: Heli dùng cho sắc ký.
Nhiệt độ:
|
| Thời gian | Nhiệt độ |
| Cột | 0-5 | 35 |
| 5-15 | 35 → 85 | |
| Buồng tiêm |
| 250 |
| detector |
| 250 |
Detector: lon hỏa ngọn lửa.
Thể tích tiêm: 0,3 μl.
Điều chỉnh tốc độ dòng của khí mang để thời gian lưu của pic tương ứng với 1-vinylpyrrolidin-2-on khoảng 17 min.
Xanthydrol
9-Hydroxyxanthen; Xanthen-9-ol
C13H10O2= 198,2
Dùng loại tinh khiết hóa học chứa không ít hơn 90,0 % C13H10O2.
Bột màu trắng hay vàng nhạt.
Điểm chảy: Khoảng 123 °C.
Xanthydrol còn có dạng dung dịch trong methanol chứa 9,0 % đến 11,0 % C13H10O2.
Định lượng: Cân 0,3 g chế phẩm vào bình nón 250 ml, thêm 3ml methanol (TT) để hòa tan hay lấy 3 ml dung dịch. Thêm 50 ml acid acetic băng (TT) và từng giọt 25 ml dung dịch ure 2 %, lắc liên tục. Để yên 12 h, lọc lấy tủa qua phễu xốp thủy tinh (16 mm). Rửa tủa với 20 ml ethanol 96 % (TT), sấy ở 100 °C đến 105 °C và cân.
Mỗi g tủa tương ứng với 0,9429 g xanthydrol.
Bảo quản tránh ánh sáng. Nếu sử dụng dung dịch trong methanol, bảo quản trong các ống nhỏ, hàn kín và lọc nếu cần trước khi sử dụng.
Xanh methylen
C16H18N3CIS. 3H2O = 373,90
Bột kết tinh màu nâu sẫm hay lục sẫm, gần như không mùi. Dễ tan trong nước nóng.
Dung dịch xanh methylen 0,15 %
Hòa tan 0,15 g xanh methylen (TT) trong nước vừa đủ 100 ml.
Dung dịch xanh methylen 0,37 %
Hòa tan 0,37 g xanh methylen (TT) trong nước vừa đủ 100 ml.
Xanh tetrazolium
C40H32Cl2N8O2 = 727,7
Dùng loại tinh khiết hóa học.
Tinh thể màu vàng. Ít tan trong nước, dễ tan trong ethanol, methanol và cloroform.
Điểm chảy: Khoảng 245 °C kèm phân hủy.
Dung dịch xanh tetrazolium
Trộn 1 thể tích dung dịch xanh tetrazolium (TT) 0,2% trong methanol (TT) với 3 thể tích dung dịch natri hydroxyd (TT) 12% trong methanol (TT).
Dung dịch chỉ pha khi dùng.
Xylen
Dimethyl benzen
C6H4(CH3)2 = 106,17
Dùng loại tinh khiết phân tích.
Hỗn hợp của ortho, meta và para-xylen.
Chất lỏng trong, không màu, dễ bắt lửa.
Tỷ trọng ở 20 °C: Khoảng 0,867.
Chỉ số khúc xạ ở 20 °C: Khoảng 1,497.
Điểm sôi: Khoảng 138 °C.
Zirconyl nitrat
Công thức khoảng: ZrO(NO3)2 + nước.
Dùng loại tinh khiết hóa học, thường chứa khoảng 44,5 % ZrO2.
Dung dịch Zirconyl nitrat
Hòa tan 0,1 g zirconyl nitrat (TT) trong hỗn hợp gồm 60 ml acid hydrocloric (TT) và 40 ml nước.
2.1.2 CÁC CHẤT CHỈ THỊ
Calcon
Solochrom dark blue; Mordant black 17; Natri 2-Hydroxy-1-(2-hydroxy-1-naphthylazo)-naphthalen-4- sulfonat
C20H13N2NaO5S = 416,4
Dùng loại tinh khiết hóa học.
Bột màu đen nâu có ánh tím. Tan trong nước và ethanol.
Trong môi trường kiềm, tạo màu đỏ tía với ion calci. Khi không có mặt ion kim loại và dư thừa một lượng nhỏ trilon B, dung dịch có màu xanh lam.
Hỗn hợp calcon
Nghiền, trộn đều 0,1 g calcon (TT) với 9,9 g natri sulfat khan (TT).
Độ nhạy: Hòa tan 0,2 g hỗn hợp calcon (TT) trong 5 ml nước. Lấy 1 ml dung dịch thu được, thêm 50 ml nước, 10 ml dung dịch natri hydroxyd 1 N (CĐ) và 1 ml dung dịch magnesi sulfat 1 %, dung dịch có màu xanh lam. Thêm 0,1 ml dung dịch calci clorid 0,15 %, màu chuyển sang đỏ tía. Thêm 0,1 ml dung dịch trilon B 0,01 M (CĐ), màu lại chuyển sang màu xanh lam.
Da cam methyl
Helianthin; Natri 4'-dimethylaminoazobenzen-4-sutfonat C14H14N3NaO3S = 327,3
Dùng loại tinh khiết hóa học.
Bột hay tinh thể màu vàng cam. Dễ tan trong nước nóng, không tan trong ethanol.
Vùng chuyển màu: pH 3,0 (đỏ) đến pH 4,4 (vàng).
Dung dịch da cam methyl
Hòa tan 0,1 g da cam methyl (TT) trong 80 ml nước, thêm ethanol 96 % (TT) vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,1 ml dung dịch da cam methyl (TT), dung dịch có màu vàng. Khi thêm không quá 0,1 ml dung dịch acid hydrocloric 0,1 N (CĐ), màu phải chuyển sang đỏ.
Da cam xylenol
Tetranatri 3,3’- (3H - 2, 1 - benzoxathiol -3- yliden) bis [(6 - hydroxy -5- methyl -3, 1 - phenylen) methy-leneiminobisacetat] S, S- dioxid.
C31H28N2Na4O13S = 761,0
Dùng loại tinh khiết hóa học.
Bột kết tinh màu nâu đỏ. Dễ tan trong nước, không tan trong ethanol.
Trong các dung dịch kiềm, tạo màu tím với ion thủy ngân, chì, kẽm và một số ion kim loại khác. Khi không có mặt các ion kim loại và dư trilon B, dung dịch có màu vàng.
Hỗn hợp da cam xylenol
Nghiền, trộn đều 1 phần da cam xylenol (TT) với 99 phần kali nitrat (TT).
Thử độ nhạy: Thêm 50 mg hỗn hợp da cam xylenol (TT) vào hỗn hợp gồm 50 ml nước, 1 ml acid acetic 2 M (TT) và 0,05 ml dung dịch chì nitrat (TT). Thêm vừa đủ một lượng hexamin (TT) để làm chuyển màu từ vàng sang đỏ tím, thêm 0,1 ml dung dịch trilon B 0,1 M (CĐ), màu chuyển sang vàng.
Dung dịch da cam xylenol (TT)
Hòa tan 0,1 g da cam xylenol (TT) trong nước vừa đủ 100 ml và lọc nếu cần.
Diclorofluorescein
2,7-Diclorofluorescein; Acid 2-(2,7-Dicloro-6-hydroxy- 3-oxo-3H-xanthen-9-yl) benzoic
C20H10Cl2O5 = 401,2
Dùng loại tinh khiết hóa học.
Bột màu nâu vàng đến cam vàng. Ít tan trong nước, dễ tan trong ethanol và các dung dịch hydroxyd kiêm loãng, thực tế không tan trong ether.
Đen eriocrom T
Mordant black 11; Natri 2- hydroxy-1-[(1-hydroxy-naphth-2-yl)azo]-6-nitronaphthalen-4-sulfonat
C20H12N3NaO7S = 461,4
Dùng loại tinh khiết hóa học.
Bột màu đen nâu, có ánh kim loại. Tan trong nước và ethanol.
Trong môi trường kiềm, tạo màu đỏ với ion calci, magnesi, kẽm và một số kim loại khác. Khi không có mặt các ion kim loại và dư thừa một lượng nhỏ trilon B, dung dịch có màu xanh lam.
Dung dịch đen eriocrom T
Hòa tan 0,1 g đen eriocrom T (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Hỗn hợp đen eriocrom T
Nghiền, trộn đều 1 phần đen eriocrom T (TT) với 99 phần natri clorid (TT).
Độ nhạy: Hòa tan 0,05 g hỗn hợp đen eriocrom T (TT) trong 100 ml nước, dung dịch có màu tím nâu.
Thêm 0,3 ml dung dịch amoniac 10 % (TT), dung dịch chuyển thành màu xanh lam, thêm tiếp 0,1 ml dung dịch magnesi sulfat 1 %, màu phải chuyển sang tím.
Đỏ Congo
Dinatri (biphenyl-4,4’-diyl-bis-2,2’-azo)bis(1-aminonaphthalen-4-sulfonat)
C32H22N6Na2O6S2 = 697,0
Dùng loại tinh khiết hóa học.
Bột màu đỏ nâu. Tan trong nước.
Vùng chuyển màu: pH 3,0 (lam) đến pH 5,0 (hồng).
Dung dịch đỏ congo
Hòa tan 0,1 g đỏ congo (TT) trong 20 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,2 ml dung dịch đỏ congo (TT) và 0,3 ml dung dịch acid hydrocloric 0,1 N (CĐ), dung dịch có màu xanh lam. Khi thêm không quá 0,3 ml dung dịch natri hydroxyd 0,1 N (CĐ), màu phải chuyển sang hồng đỏ.
Đỏ cresol
o-Cresolsulfonphthalein; 4,4'-(3H-2,1-benzoxathiol-3-yliden) di-o-cresol S,S-dioxyd
C21H18O5S = 382,4
Dùng loại tinh khiết hóa học.
Bột kết tinh màu nâu đỏ. Ít tan trong nước, dễ tan trong ethanol và dung dịch natri hydroxyd.
Vùng chuyển màu: pH 7,0 (vàng) đến pH 8,8 (đỏ).
Dung dịch đỏ cresol
Hòa tan bằng cách đun nóng nhẹ 0,1 g đỏ cresol (TT) với hỗn hợp gồm 2,65 ml dung dịch natri hydroxyd 0,1 N (TT) và 20 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,1 ml dung dịch đỏ cresol (TT) và 0,15 ml dung dịch natri hydroxyd 0,02 N (CĐ), dung dịch có màu đỏ tía. Khi thêm không quá 0,15 ml dung dịch acid hydrocloric 0,02 N (CĐ), màu phải chuyển sang vàng.
Đỏ methyl
Acid 2-(4-dimethylaminophenylazo)benzoic
C15H15N3O2 = 269 3
Dùng loại tinh khiết hóa học.
Tinh thể màu tím hay bột màu đỏ sẫm. Thực tế không tan trong nước, tan trong ethanol và acid acetic. Vùng chuyển màu: pH 4,4 (đỏ) đến pH 6,0 (vàng).
Dung dịch đỏ methyl
Hòa tan 50 mg đỏ methyl (TT) trong hỗn hợp gồm 1,86 ml dung dịch natri hydroxyd 0,1 N (TT) và 50 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,05 ml dung dịch acid hydrocloric 0,02 N (CĐ) và 0,1 ml dung dịch đỏ methyl (TT), dung dịch có màu đỏ. Khi thêm không quá 0,1 ml dung dịch natri hydroxyd 0,02 N (CĐ), màu phải chuyển sang vàng.
Dung dịch hỗn hợp đỏ methyl
Hòa tan 0,1 g đỏ methyl (TT) và 0,05 g xanh methylen (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Đỏ phenol
Phenolsulfonphthalein; 4,4'-(3H-2,1-Benzoxathiol-3-yliden)diphenol S,S-dioxyd
C19H14O5S = 354,4
Dùng loại tinh khiết hóa học.
Bột màu đỏ. Rất khó tan trong nước, khó tan trong ethanol, dễ tan trong các dung dịch kiềm.
Vùng chuyển màu: pH 6,8 (vàng) đến pH 8,4 (đỏ).
Dung dịch đỏ phenol (TT1)
Hòa tan 0,1 g đỏ phenol (TT) trong hỗn hợp gồm 2,82 ml dung dịch natri hydroxyd 0,1 N (TT) và 20 ml ethanol
96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,1 ml dung dịch đỏ phenol (TT), dung dịch có màu vàng. Khi thêm không quá 0,1 ml dung dịch natri hydroxyd 0,02 N (CĐ), màu phải chuyển sang tím đỏ.
Dung dịch đỏ phenol (TT2)
Dung dịch 1: Hòa tan 33 mg đỏ phenol (TT) trong 1,5 ml dung dịch natrihydroxyd 2 M (TT), thêm nước vừa đủ 100 ml.
Dung dịch 2: Hòa tan 25 mg amoni sulfat (TT) trong 235 ml nước, thêm 105 ml dung dịch natri hydroxyd 2 M (TT) và 135 ml dung dịch acid acetic 2 M (TT).
Thêm 25 ml dung dịch 1 vào dung dịch 2. Nếu cần, điều chỉnh pH của hỗn hợp tới 4,7.
Đỏ tía bromocresol
3', 3-Dibromo-o-cresolsulfonphthalein; 4,4'-(3H-2,1 -Benzoxathiol-3-yliden)bis(6-bromo-o-cresol) S,S- dioxyd
C21H16Br2O6S = 540,2
Dùng loại tinh khiết hóa học.
Bột kết tinh màu hồng. Thực tế không tan trong nước, tan trong ethanol và các dung dịch kiềm loãng. Vùng chuyển màu: pH 5,2 (vàng) đến pH 6,8 (lam).
Dung dịch đỏ tía bromocresol
Hòa tan 0,05 g đỏ tía bromocresol (TT) trong hỗn hợp gồm 0,92 ml dung dịch natri hydroxyd 0,1 N (TT) và 20 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,2 ml dung dịch đỏ tía bromocresol (TT) và 0,05 ml dung dịch natri hydroxyd 0,02 N (CĐ), dung dịch có màu tím xanh. Khi thêm không quá 0,20 ml dung dịch acid hydrocloric 0,02 N (TT), màu phải chuyển sang vàng.
Đỏ tía cresol
m-Cresolsulfonphthalein
C21H18O5S = 382,4
Dùng loại tinh khiết hóa học.
Bột kết tinh màu lục nâu. Khó tan trong nước, tan trong methanol, ethanol và acid acetic băng.
Vùng chuyển màu: pH 1,2 (đỏ) đến pH 2,8 (vàng); pH 7,4 (vàng) đến pH 9,0 (đỏ tía).
Dung dịch đỏ tía cresol
Hòa tan 0,1 g đỏ tía cresol (TT) trong 13 ml dung dịch natri hydroxyd 0,01 N (TT), thêm nước vừa đủ 100 ml.
Đỏ tía phthalein
Metalphtalein;(1,3-Dihydro-3-oxo-isobenzofuran-1-yliden)bis[(6-hydroxy-5-methyl-3,1-phenylen) bis(methylen eimino)diacetic acid]
C32H32N2O12 + nước
Dùng loại tinh khiết hóa học.
Bột màu trắng kem đến nâu. Thực tế không tan trong nước, tan trong ethanol.
Độ nhạy: Hòa tan 10 mg đỏ tía phthalein (TT) trong 1 ml amoniac 13,5 M (TT), pha loãng thành 100 ml với nước. Lấy 5 ml dung dịch thu được, thêm 95 ml nước, 4 ml amoniac 13,5 M (TT), 50 ml ethanol 96 % (TT) và 0,1 ml dung dịch bari clorid 0,1 M (CĐ); dung dịch có màu tím lam. Thêm 0,15 ml dung dịch trilon B 0,1 M (CĐ), dung dịch mất màu.
Đỏ trung tính
Basic red 5; 2 - Methyl -3- amino -7- dimethy-lamino-phenazin hydroclorid
C15H17ClN4 = 288,8
Dùng loại tinh khiết hóa học.
Bột màu đỏ nhạt. Ít tan trong nước và ethanol.
Vùng chuyển màu: pH 6,8 (đỏ) đến pH 8,0 (da cam).
Dung dịch đỏ trung tính
Hòa tan 0,1 g đỏ trung tính (TT) trong ethanol 50 % vừa đủ 100 ml.
Eosin
Acid đỏ 87
C20H6Br4Na2O5 = 691,9
Dùng loại tinh khiết hóa học.
Bột màu đỏ. Dễ tan trong nước, ít tan trong ethanol.
Dung dịch eosin
Hòa tan 0,5 g eosin (TT) trong nước vừa đủ 100 ml.
Ethoxycrysoidin hydroclorid
4-[(4-Ethoxyphenyl]diazenyl]phenylen-1,3-diamin hydroclorid
C14H16N4O. HCl = 292,8
Dùng loại tinh khiết hóa học.
Bột màu đỏ. Tan trong nước và ethanol.
Dung dịch ethoxycrysoidin
Hòa tan 0,1 g ethoxycrysoidin hydroclorid (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Độ nhạy: Thêm 0,05 ml dung dịch brom 0,0167 M (TT) vào hỗn hợp gồm 0,05 ml dung dịch ethoxy- crysoidin hydroclorid (TT) và 5 ml dung dịch acid hydrocloric 2 M (TT). Màu của dung dịch chuyển từ đỏ sang vàng nhạt trong vòng 2 min.
Lam hydroxy naphtol
Muối dinatri Acid (1-(2-Naphtholazo-3,6-disulfonic acid)-2-naphthol-4-sulfonic.
C20H12N2O11S3Na2 = 598,50
Làm kết tinh tinh thể natri clorid trong dung dịch nồng độ 1 %.
Lục bromocresol
3’,3”,5’,5"-Tetrabromo-m-cresolsulfonphthalein; 4,4'- (3H-2,1-Benzoxathiol-3-yliden) bis (2,6-dibromo-m - cresol) S,S-dioxyd C21H14Br4O5S = 698,0
Dùng loại tinh khiết hóa học.
Bột màu trắng nâu hay vàng nhạt. Ít tan trong nước, tan trong ethanol và các dung dịch kiềm loãng.
Vùng chuyển màu: pH 3,6 (vàng) đến pH 5,2 (lam).
Dung dịch lục bromocresol (TT1)
Hòa tan 0,05 g lục bromocresol (TT) trong hỗn hợp gồm 0,72 ml dung dịch natri hydroxyd 0,1 N (TT) và 20 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,2 ml dung dịch lục bromocresol (TT), dung dịch có màu xanh lam. Khi thêm không quá 0,2 ml dung dịch acidhydrocloric 0,02 N (CĐ), màu phải chuyển sang vàng.
Dung dịch lục bromocresol (TT2)
Nghiền kỹ 0,2 g lục bromocresol (TT) với 2,8 ml dung dịch natri hydroxyd 0,1 N (TT), pha loãng thành 200 ml với nước và lọc nếu cần.
Dung dịch lục bromocresol - đỏ methyl (Dung dịch xanh bromocresol - đỏ methyl)
Hòa tan 0,15 g lục bromocresol (TT) và 0,1 g đỏ methyl (TT) trong 180 ml ethanol (TT), pha loãng với nước thành 200 ml.
Lục malachit
Victoria green; [4-[[4-Dimethylamino)phenyl]phenyl-methylen]cyclohexan-2,5-dien-1-yliden] dimethyl amoni clorid
C23H25ClN2 = 364,9
Dùng loại tinh khiết hóa học.
Tinh thể màu lục, có ánh kim loại. Rất dễ tan trong nước, tan trong ethanol và methanol.
Dung dịch 0,001 % (kl/tt) trong ethanol 96 % (TT) có hấp thụ cực đại ở 617 nm.
Dung dịch lục malachit 0,5%
Hòa tan 0,5 g lục malachit (TT) trong acid acetic khan (TT) vừa đủ 100 ml.
Magneson
Tím azo; 4-(4-nitrophenylazo)resorcinol
C12H9N3O4 = 259,2
Dùng loại tinh khiết chỉ thị hóa học. Khi dùng làm chỉ thị trong phép thử định lượng môi trường khan, phải chuyển từ màu cam (môi trường acid) sang màu hồng (môi trường trung tính) đến màu xanh lam (môi trường kiềm).
Thuốc thử magneson
Hòa tan magneson (TT) trong dung dịch natri hydroxyd 0,1 % (TT) để được dung dịch magneson 0,1 %.
Murexid
Muối monoamoni 5,5’-nitrilobis[pirimidin-2,4,6-(1H,3H,5H)trion] của acid purpuric.
C8H8N6O6. H2O = 302,20
Dùng loại tinh khiết hóa học.
Bột tinh thể màu đỏ nâu. Khó tan trong nước lạnh, tan trong nước nóng và ethanol. Tan trong các dung dịch kali hydroxyd và natri hydroxyd, cho màu xanh lam.
Dung dịch murexid
Hòa tan 0,25 g murexid (TT) trong nước vừa đủ 100 ml.
Dung dịch chỉ pha khi dùng.
Hỗn hợp murexid
Nghiền, trộn đều 0,25 g murexid (TT) với 25 g natri clorid (TT).
1 -Naphtholbenzein
a-Naphtholbenzein;Phenylbis(4-hydroxynaphthyl)-methanol
C27H20O3 = 392,5
Dùng loại tinh khiết hóa học.
Bột màu nâu đỏ. Thực tế không tan trong nước, tan trong ethanol và acid acetic băng.
Dung dịch 1-naphtholbenzein
Hòa tan 0,2 g 1-naphtholbenzein (TT) trong 100 ml acid acetic khan (TT).
Độ nhạy: Lấy 50 ml acid acetic khan (TT), thêm 0,25 ml dung dịch 1-naphtholbenzein (TT), dung dịch có màu vàng nâu. Khi thêm không quá 0,05 ml dung dịch acid percloric 0,1 N (CĐ), màu phải chuyển sang xanh lục.
O-Phenanthrolin
1,10- Phenanthrolin hydroclorid
C12H8N2. HCl. H2O = 234,7
Dùng loại tinh khiết hóa học.
Bột trắng hoặc gần như trắng. Dễ tan trong nước, tan trong ethanol.
Điểm chảy: Khoảng 215 °C kèm phân hủy.
Dung dịch feroin sulfat
Hòa tan 0,7 g sắt (II) sulfat (TT) và 1,76 g O- phenan- throlin (TT) trong 70 ml nước. Thêm nước vừa đủ 100 ml.
Phenolphtalein
3,3'-Bis(4-hydroxyphenyl)phthalid
C20H14O4 = 318,3
Dùng loại tinh khiết hóa học.
Bột màu trắng hay trắng ngà. Thực tế không tan trong nước, tan trong ethanol.
Vùng chuyển màu: pH 8,2 (không màu) đến pH 10,0 (đỏ).
Dung dịch phenolphtalein
Hòa tan 0,1 g phenolphtalein (TT) trong 80 ml ethanol 96 % (TT) và thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,1 ml dung dịch phenolphtalein (TT), dung dịch không màu. Khi thêm không quá 0,2 ml dung dịch natri hydroxyd 0,02 N (CĐ), phải xuất hiện màu hồng.
Dung dịch phenolphtalein (TT1)
Hòa tan 1 g phenolphtalein (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Quỳ
Là sắc tố màu chàm chiết từ các loại địa y như Rocella, Lecanora v.v....
Mảnh nhỏ màu lục thẫm. Tan trong nước và ethanol.
Vùng chuyển màu: đỏ (pH 5,0) đến xanh lam (pH 8,0).
Giấy quỳ xanh
Đun sôi 10 phần quỳ (TT) đã tán thành bột thô với 100 phần ethanol 96 % (TT) dưới sinh hàn trong 1 h. Gạn bỏ phần ethanol. Thêm hỗn hợp gồm 45 phần ethanol 96 % (TT) và 55 phần nước vào cắn. Để yên 2 ngày, gạn lấy phần dịch trong (dịch chiết A). Tẩm dịch chiết A vào các mảnh giấy lọc, để khô.
Độ nhạy: Nhúng mảnh giấy quỳ xanh, kích thước 10 mm x 60 mm, vào hỗn hợp gồm 100 ml dung dịch acid hydrocloric 0,002 N (TT) và 90 ml nước. Lắc nhẹ, giấy phải chuyển màu sáng đỏ trong vòng 45 s.
Giấy quỳ đỏ
Lấy dịch chiết A trong điều chế Giấy quỳ xanh, thêm từng giọt dung dịch acid hydrocloric 2 N (TT) cho đến khi màu xanh chuyển thành đỏ. Tẩm dung dịch thu được vào các mảnh giấy lọc, để khô.
Độ nhạy: Nhúng mảnh giấy quỳ đỏ, kích thước 10 mm x 60 mm, vào 100 ml dung dịch natri hydroxyd 0,002 N (TT). Lắc nhẹ, giấy phai chuyển màu sang xanh trong vòng 45 s.
Dung dịch quỳ
Đun sôi 25 g quỳ (TT) đã tán thành bột thô với 100 ml ethanol 90 % dưới sinh hàn trong 1 h. Gạn bỏ phần dịch trong, lập lại quá trình này 2 lần, mỗi lần với 75 ml ethanol 90 %. Hòa lượng quỳ đã được chiết vào 250 ml nước, lọc.
Thymolphtalein
3,3-Bis (4-hydroxy-5-isopropyl-2-methylphenyl) phthalid
C28H30O4 = 430,5
Dùng loại tinh khiết hóa học.
Bột màu trắng. Thực tế không tan trong nước, tan trong ethanol và các dung dịch kiềm loãng.
Vùng chuyển màu: Không màu (pH 9,3) đến xanh lam (pH 10,5).
Dung dịch thymolphtalein
Hòa tan 0,10 g thymolphtalein (TT) trong ethanol 96 % (TT) vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,2 ml dung dịch thymolphtalein (TT), dung dịch không màu. Khi thêm không quá 0,05 ml dung dịch natri hydroxyd 0,1 N (CĐ), phải xuất hiện màu xanh lam.
Tím pyrocatechin
Pyrocatechinsulfonphthalein; Tím catechol; 4,4'-(3H-2,1-Benzoxanthiol-3-yliden)dipyrocatechol S,S- dioxyd
C19H14O7S = 386,38
Dùng loại tinh khiết hóa học.
Bột kết tinh màu nâu đỏ với ánh kim loại. Dễ tan trong nước và ethanol.
Trong môi trường acid (pH 2-3) tạo màu xanh lam với ion bismuth và khi không có mặt ion này, dung dịch có màu vàng. Trong môi trường kiềm tạo màu lam lục với ion magnesi và ion kẽm, khi không có mặt các ion này, dung dịch có màu tím đỏ.
Dung dịch tím pyrocatechin
Hòa tan 0,1 g tím pyrocatechin (TT) trong nước vừa đủ 100 ml.
Tím tinh thể
Gentialviolet; Hexamethyl-p-rosanilin clorid
C25H30ClN3 = 408,0
Dùng loại tinh khiết hóa học.
Bột kết tinh hay tinh thể màu lục thẫm, có ánh kim loại. Tan trong nước, ethanol và acid acetic khan.
Khi dùng chuẩn độ trong môi trường khan, màu chuyển từ tím (kiềm) qua lục lam (trung tính) đến lục vàng (acid).
Dung dịch tím tinh thể
Hòa tan 0,50 g tím tinh thể (TT) trong acid acetic khan (TT) vừa đủ 100 ml.
Độ nhạy: Lấy 50 ml acid acetic khan (TT), thêm 0,1 ml dung dịch tím tinh thể (TT), dung dịch có màu tím.
Thêm 0,10 ml dung dịch acid peroloric 0,1 N (CĐ), màu của hỗn hợp phải chuyển sang lục lam.
Tinh bột
Bột rất mịn, không mùi, không vị.
Dùng loại tinh khiết.
Dung dịch hồ tinh bột
Nghiền 1 g tinh bột (TT) với 5 ml nước, rồi vừa đổ vừa khuấy vào 100 ml nước sôi. Đun sôi tiếp cho đến khi thu được chất lỏng chỉ hơi đục.
Pha trước khi dùng.
Độ nhạy: Lấy 5 ml dung dịch hồ tinh bột (TT), pha loãng thành 100 ml với nước, thêm 2 giọt dung dịch iod 0,1 N (CĐ), dung dịch phải có màu xanh lam.
Dung dịch hồ tinh bột có kali iodid
Hòa tan 0,5 g kali iodid (TT) vào 100 ml dung dịch hồ tinh bột (TT) mới pha.
Dung dịch này chỉ dùng trong 24 h.
Giấy hồ tinh bột có iodid
Tẩm ướt giấy lọc với dung dịch hồ tinh bột có kali iodid (TT), để khô ở chỗ tối không có hơi acid, cắt giấy thành những băng dài 50 mm, rộng 6 mm. Khi nhỏ 1 giọt dung dịch acid hydrocloric 0,1 N (CĐ) vào băng giấy chỉ thị, không được xuất hiện màu xanh lam ngay lập tức.
Bảo quản trong lọ thủy tinh màu nâu, nút mài.
Độ nhạy: Trộn 0,05 ml dung dịch natri nitrit 0,1 M (CĐ) và 4 ml acid hydrocloric (TT), thêm nước vừa đủ 100 ml. Nhỏ 0,05 ml dung dịch thu được lên băng giấy chỉ thị, phải xuất hiện màu xanh lam ngay tức khắc.
Vàng alizarin
4-Nitro-4'-oxyazobenzen-3'-natri carboxylat
C13H8N3NaO5 = 309,2
Dùng loại tinh khiết hóa học.
Bột kết tinh màu nâu đỏ hay nâu vàng. Ít tan trong nước và ethanol.
Vùng chuyển màu: pH 10,1 (vàng) đến pH 12,1 (tím hồng).
Dung dịch vàng alizarin
Đun cách thủy để hòa tan 0,1 g vàng alizarin (TT) đã tán nhỏ trong một ít nước, để nguội và thêm nước vừa đủ 100 ml.
Vàng metanil
Natri 3-[4-(phenylamino) phenylazo] benzensulphonat
C18H14N3NaO3S = 375,4
Dùng loại tinh khiết hóa học.
Bột màu vàng nâu. Tan trong nước và ethanol, rất ít tan trong ether.
Khi dùng để chuẩn độ trong môi trường khan, màu chuyển từ vàng (kiềm) sang đỏ tía (acid).
Dung dịch vàng metanil
Hòa tan 0,1 g vàng metanil (TT) trong methanol (TT) vừa đủ 100 ml.
Độ nhạy: Thêm 0,1 ml dung dịch vàng metanil (TT) vào 50 ml acid acetic khan (TT), dung dịch có màu đỏ hồng. Thêm 0,05 ml dung dịch acid percloric 0,1 M (CĐ), màu chuyển sang tím.
Vùng chuyển màu: pH 1,2 (đỏ) đến pH 2,3 (vàng cam).
Vàng titan
Thiazol yellow
Dinatri 2,2’-[(1-triazen-1,3-diyl)di-4,1-phenylen] bis-[6-methylbenzothiazol-7-sulfonat]
C28H19N5Na2O6S4 = 696,0
Dùng loại tinh khiết hóa học.
Bột màu nâu vàng. Dễ tan trong nước và ethanol.
Dung dịch vàng titan
Hòa tan 0,05 g vàng titan (TT) trong nước vừa đủ 100 ml.
Độ nhạy: Thêm 0,1 ml dung dịch vàng titan (TT) vào hỗn hợp gồm 10 ml nước, 0,2 ml dung dịch magnesi mẫu 10 phần triệu (TT) và 1,0 ml natri hydroxyd 1 M (CĐ). Dung dịch có màu hồng, thấy rõ khi so sánh với dung dịch đối chiếu được chuẩn bị tương tự, nhưng không có dung dịch magnesi mẫu 10 phần triệu (TT).
Giấy vàng titan
Tẩm giấy lọc bằng dung dịch vàng titan (TT), rồi để khô ở nhiệt độ phòng.
Xanh bromophenol
3,3'-5,5' Tetrabromophenolsulfonphthalein
4,4'-(3H-2,1-Benzoxathiol-3-yliden) bis (2,6-Dibromo-phenol) S,S-dioxyd
C19H10Br4O5S = 670,0
Dùng loại tinh khiết hóa học.
Bột màu vàng cam. Rất khó tan trong nước, khó tan trong ethanol, dễ tan các dung dịch kiềm hydroxyd loãng.
Vùng chuyển màu: pH 2,8 (vàng) đến pH 4,6 (tím lam).
Dung dịch xanh bromophenol
Hòa tan 0,1 g xanh bromophenol (TT) trong hỗn hợp gồm 1,5 ml dung dịch natri hydroxyd 0,1 N (TT) và 20 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 20 ml nước không có carbon dioxyd (TT), thêm 0,05 ml dung dịch xanh bromophenol (TT) và 0,05 ml dung dịch acid hydrocloric 0,1 N (CĐ), dung dịch có màu vàng. Khi thêm không quá 0,10 ml dung dịch natri hydroxyd 0,1 N (CĐ), màu phải chuyển sang tím lam.
Dung dịch xanh bromophenol trong ethanol
Hòa tan 0,1g xanh bromophenol (TT) trong ethanol 20 % vừa đủ 100 ml.
Dung dịch xanh bromophenol (TT1)
Hòa tan 50 mg xanh bromophenol (TT) bằng cách đun nóng nhẹ với 3,73 ml dung dịch natri hydroxyd 0,02 N (TT), pha loãng với nước vừa đủ 100 ml.
Xanh bromothymol
3,3' -Dibromothymolsulfonphthalein
4,4’-(3H-2,1-Benzoxathiol-3-yliden) bis (2-bromothymol) S,S-dioxyd
C27H28Br2O5S = 624
Dùng loại tinh khiết hóa học.
Bột kết tinh màu hồng đỏ hay nâu. Thực tế không tan trong nước, tan trong ethanol và các dung dịch kiềm loãng.
Vùng chuyển màu: pH 6,0 (vàng) đến pH 7,6 (xanh lam).
Dung dịch xanh bromothymol
Hòa tan 50 mg xanh bromothymol (TT) trong hỗn hợp gồm 4 ml dung dịch natri hydroxyd 0,02 N (TT) và 20 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,3 ml dung dịch xanh bromothymol (TT), dung dịch có màu vàng. Khi thêm không quá 0,10 ml dung dịch natri hydroxyd 0,02 N (CĐ), màu phải chuyển sang xanh lam.
Dung dịch xanh bromothymol (TT1)
Dung dịch chỉ thị BRP
Hòa tan 0,1 g xanh bromothymol (TT), 20 mg đỏ methyl (TT) và 0,2 g phenolphtalein (TT) trong ethanol 96 % (TT) vừa đủ 100 ml, lọc.
Dung dịch xanh bromothymol (TT2)
Dung dịch 1 % xanh bromothymol (TT) trong dimethylformamid (TT).
Xanh thymol
Thymolsulfonphthalein
4,4''-(3H-2,1-Benzoxathiol-3-yliden) dithymol S,S-dioxyd
C27H30O5S = 466,6
Dùng loại tinh khiết hóa học.
Bột kết tinh màu lam lục đến lục nâu. Khó tan trong nước, tan trong ethanol và các dung dịch kiềm loãng.
Vùng chuyển màu: pH 1,2 (đỏ) đến pH 2,8 (vàng); pH 8,0 (lục nâu) đến pH 9,6 (lam).
Dung dịch xanh thymol
Hòa tan 0,1 g xanh thymol (TT) trong hỗn hợp gồm 2,15 ml dung dịch natri hydroxyd 0,1 N (TT) và 20 ml ethanol 96 % (TT), thêm nước vừa đủ 100 ml.
Độ nhạy: Lấy 100 ml nước không có carbon dioxyd (TT), thêm 0,1 ml dung dịch xanh thymol (TT) và 0,2 ml dung dịch natri hydroxyd 0,02 N (CĐ), dung dịch có màu xanh lam. Khi thêm không quá 0,1 ml dung dịch acid hydrocloric 0,02 N (CĐ), phải chuyển sang vàng.
Dung dịch xanh thymol trong dimethylformamid
Hòa tan 1 g xanh thymol (TT) trong dimethylformamid (TT) vừa đủ 100 ml.
Dung dịch xanh thymol trong methanol
Hòa tan 0,3 g xanh thymol (TT) trong methanol (TT) vừa đủ 100 ml. Lọc, nếu cần.
Khoảng pH và màu chuyển của các chất chỉ thị
| Chỉ thị | Khoảng pH | Màu chuyển |
| Đỏ cresol | 0,2 đến 1,8 | Đỏ - Vàng |
|
| 7,0 đến 8,8 | Vàng - Đỏ |
| Xanh thymol | 1,2 đến 2,8 | Đỏ - Vàng |
| Tropeolin 00 | 1,3 đến 3,2 | Đỏ-Vàng |
| Da cam methyl | 3,0 đến 4,4 | Đỏ-Vàng |
| Xanh bromophenol | 2,8 đến 4,6 | Vàng - Tím lam |
| Đỏ congo | 3,0 đến 5,0 | Lam - Hồng |
| Lục bromocresol | 3,6 đến 5,2 | Vàng - Lam |
| Đỏ methyl | 4,4 đến 6,0 | Đỏ-Vàng |
| Đỏ tía bromocresol | 5,2 đến 6,8 | Vàng - Tím lam |
| Xanh bromothymol | 6,0 đến 7,6 | Vàng - Lam |
| Đỏ trung tính | 6,8 đến 8,0 | Đỏ - Da cam |
| Đỏ phenol | 6,8 đến 8,4 | Vàng - Tím đỏ |
| Xanh thymol | 8,0 đến 9,6 | Lục nâu - Lam |
| Phenolphtalein | 8,2 đến 10,0 | Không màu - Đỏ |
| Thymolphtalein | 9,3 đến 10,5 | Không màu - Lam |
| Vàng alizarin | 10,1 đến 12,1 | Vàng - Tím hồng |
2.2 CÁC DUNG DỊCH CHUẨN ĐỘ (CĐ)
Quy định chung
Dung dịch chuẩn độ là dung dịch có nồng độ chính xác, biết trước dùng trong phân tích định lượng thể tích.
Nồng độ của dung dịch chuẩn độ thường được biểu thị bằng:
Nồng độ đương lượng gam (N): Số đương lượng gam của chất tan trong 1000 ml dung dịch.
Nồng độ mol (M): số mol của chất tan trong 1000 ml dung dịch.
Tỷ số giữa nồng độ thực và nồng độ lý thuyết là hệ số hiệu chỉnh K, không được nằm ngoài giới hạn 1,00 ± 0,10. Nên dùng các dung dịch chuẩn độ với K trong khoảng từ 0,970 đến 1,030. Nồng độ dung dịch chuẩn độ được xác định với số lần chuẩn độ thích hợp và độ lệch chuẩn tương đối của các kết quả thu được không được quả 0,2 %.
Phương pháp chung để pha chế các dung dịch chuẩn độ
Đối với mỗi loại dung dịch chuẩn độ, phương pháp pha chế và chuẩn hóa những dung dịch ở các nồng độ hay được sử dụng nhất sẽ được mô tả dưới đây. Các dung dịch đậm đặc hơn được pha chế và chuẩn hóa bằng cách tăng lượng thuốc thử lên tương ứng. Các dung dịch nước có nồng độ loãng hơn được điều chế bằng cách pha loãng chính xác một dung dịch đậm đặc hơn với nước không có carbon dioxyd (TT). Hệ số hiệu chỉnh của những dung dịch này chính là hệ số hiệu chỉnh của dung dịch đã dùng để pha loãng. Các dung dịch nước có nồng độ mới nhỏ hơn 0,1 M phải được pha chế với nước không có carbon dioxyd (TT).
Khi pha chế các dung dịch kém bền vững như kali permanganat, natri thiosulfat, phải dùng nước mới đun sôi để nguội. Đối với dung dịch chuẩn độ được dùng trong định lượng mà điểm tương đương được xác định bằng phương pháp điện hóa thì kỹ thuật xác định điểm tương đương này cũng phải được dùng trong chuẩn hóa dung dịch đó.
Tất cả các dung dịch chuẩn độ phải được pha chế, chuẩn hóa và sử dụng ở nhiệt độ 25 °C. Nếu nhiệt độ khi định lượng khác với nhiệt độ lúc chuẩn hóa thì thể tích dung dịch chuẩn độ sẽ được hiệu chỉnh lại.
Dưới đây là phương pháp pha chế và chuẩn hóa các dung dịch chuẩn độ được dùng:
Pha chế từ chất chuẩn độ gốc
Cân chính xác một lượng chất chuẩn độ gốc tương ứng với lượng chất lý thuyết tính theo nồng độ và thể tích dung dịch chuẩn độ cần pha, hòa tan trong dung môi chỉ dẫn vừa đủ thể tích.
Pha gần đúng rồi chuẩn hóa bằng chất chuẩn độ gốc hoặc bằng dung dịch chuẩn độ có hệ số K đã biết
Chất chuẩn độ gốc: Các hóa chất loại tinh khiết phân tích dưới đây, sau khi làm khô trong những điều kiện chỉ dẫn, được dùng làm chất chuẩn độ gốc để xác định K của dung dịch chuẩn độ:
Acid benzoic (C7H6O2): Để 24 h trong bình hút ẩm chứa silicagel khan.
Acid sulfanilic (C6H7NO3S): Sấy ở 100 °C đến 105 °C đến khối lượng không đổi.
Arsen trioxyd (As2O3): Để 24 h trong bình hút ẩm chứa silicagel khan.
Kali bromat (KBrO3): Sấy ở 180 °C đến khối lượng không đổi.
Kali dicromat (K2Cr2O7): Sấy ở 150 °C đến khối lượng không đổi.
Kail hydrophtalat (C8H5O4K): Sấy ở 110 °C đến khối lượng không đổi.
Kali iodat (KIO3): Sấy ở 130 °C đến khối lượng không đổi.
Kẽm hạt (Zn): Sử dụng loại có hàm lượng Zn không ít hơn 99,9 %.
Natri carbonat khan (Na2CO3): Nung ở 270 °C đến 300 °C đến khối lượng không đổi.
Natri clorid (NaCI): Nung ở 300 °C đến khối lượng không đổi.
Cách xác định K:
Chuẩn hóa bằng chất chuẩn độ gốc: Cân chính xác một lượng chất chuẩn độ gốc, hòa tan trong dung môi chỉ dẫn, chuẩn độ bằng dung dịch chuẩn độ mới pha, tính K theo công thức:
![]() (1)
(1)
trong đó:
a là lượng chất chuẩn độ gốc đã cân (g);
T là độ chuẩn lý thuyết của chất chuẩn độ gốc (g/ml);
V là số ml dung dịch chuẩn độ đã dùng.
Chuẩn hóa bằng dung dịch chuẩn độ có hệ số K0 đã biết:
Trong trường hợp này, K được tính theo công thức:
![]() (2)
(2)
trong đó:
C0 là nồng độ lý thuyết của dung dịch chuẩn độ dùng để chuẩn hóa;
K0 là hệ số hiệu chỉnh của dung dịch chuẩn độ dùng để chuẩn hóa;
V0 là số ml dung dịch chuẩn độ dùng để chuẩn hóa đã dùng;
C là nồng độ lý thuyết của dung dịch chuẩn độ cần pha;
V là số ml dung dịch chuẩn độ cần xác định hệ số K đã dùng.
CHÚ THÍCH:
Nếu K nằm ngoài giới hạn quy định, cần pha loãng hay làm đậm đặc dung dịch. Khi cần pha loãng, tích (K - 1) nhân với 1000 là số ml dung môi cần thêm vào 1000 ml dung dịch. Trong trường hợp cần làm đậm đặc, tích (1 - K) nhân với số g hóa chất cần lấy để pha 1000 ml dung dịch là số g hóa chất phải thêm vào 1000 ml dung dịch. Sau khi thêm dung môi hay hóa chất, xác định lại K của dung dịch thu được.
Pha loãng những dung dịch chuẩn độ có nồng độ cao
Pha loãng môt cách chính xác dung dịch chuẩn độ có nồng độ cao thành dung dịch có nồng độ thấp hơn 2 lần, 5 lần, 10 lần v.v... bằng dung môi tương ứng. Hệ số hiệu chỉnh của dung dịch chuẩn độ pha được chính là hệ số hiệu chỉnh của dung dịch đã dùng để pha loãng. Những dung dịch chuẩn độ có nồng độ nhỏ hơn 0,1N chỉ được điều chế theo cách này ngay trước khi dùng.
Dùng ống chuẩn pha sẵn
Ống chuẩn pha sẵn (fixanal) chứa lượng hóa chất hay dung dịch hóa chất đủ để pha thành một thể tích dung dịch chuẩn độ quy định. Dùng dung môi pha chế theo chỉ dẫn ghi trên nhãn ống, thu được dung dịch chuẩn độ có K đã biết.
Cách pha chế một số dung dịch chuẩn độ
Dung dịch acid acetic 0,1 N
1 ml dung dịch chứa 0,00601 g acid acetic (C2H4O2).
Điều chế: Lấy 6,0 g acid acetic băng (TT) pha loãng với nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 25,0 ml dung dịch acid acetic đã điều chế, chuẩn độ bằng dung dịch natri hydroxyd 0,1N (CĐ), dùng 0,5 ml dung dịch phenolphtalein (TT) làm chỉ thị.
Hệ số hiệu chỉnh được tính theo công thức (2).
Dung dịch acid hydrocloric 1 N
1 ml dung dịch chứa 0,03646 g acid hydrocloric (HCl).
Điều chế: Lấy 85 ml acid hydrocloric đậm đặc (TT) pha loãng với nước vừa đủ 1000 ml.
Chuẩn độ: Cân chính xác khoảng 0,50 g chất chuẩn gốc natri carbonat, hòa tan trong 50 ml nước, thêm 2 giọt dung dịch da cam methyl (TT). Chuẩn độ bằng dung dịch acid hydrocloric đã điều chế cho đến khi chuyển sang màu hồng cam. Đun sôi 2 min, để nguội, chuẩn độ tiếp đến màu hồng cam.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,053 g/ml.
Dung dịch acid hydrocloric 0,5 N
1 ml dung dịch chứa 0,01823 g acid hydrocloric (HCl).
Điều chế: Lấy 42 ml acid hydrocloric đậm đặc (TT) pha loãng với nước vừa đủ 1000 ml.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch acid hydrocloric 1 N, dùng khoảng 0,25 g (cân chính xác) chất chuẩn gốc natri carbonat.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,0265 g/ml.
Dung dịch acid hydrocloric 0,1 N
1 ml dung dịch chứa 0,003646 g acid hydrocloric (HCl).
Điều chế: Lấy 8,5 ml acid hydrocloric đậm đặc (TT) pha loãng với nước vừa đủ 1000 ml.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch acid hydrocloric 1 N, dùng khoảng 0,10 g (cân chính xác) chất chuẩn gốc natri carbonat.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,0053 g/ml.
Dung dịch acid hydrocloric 0,1 N trong methanol
1 ml dung dịch chứa 0,003646 g acid hydrocloric (HCl).
Điều chế: Lấy 8,5 ml acid hydrocloric đậm đặc (TT) pha loãng với methanol (TT) vừa đủ 1000 ml.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch acid hydrocloric 0,1 N.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,0053 g/ml.
Dung dịch acid percloric 0,1 N
1 ml dung dịch chứa 0,01005 g acid percloric (HClO4).
Điều chế: Lấy 900 ml acid acetic băng (TT), thêm 8,5 ml acid percloric (TT), trộn đều và thêm 30 ml anhydrid acetic (TT). Thêm acid acetic băng (TT) vừa đủ 1000 ml. Lắc đều, để yên 24 h. Xác định hàm lượng nước theo Phụ lục 10.3 mà không thêm methanol, và nếu cần thì điều chỉnh hàm lượng nước trong khoảng từ 0,1 % đến 0,2 % bằng anhydrid acetic hoặc nước. Sau đó, để yên 24 h.
Chuẩn độ: Cân chính xác khoảng 0,35 g chất chuẩn gốc kali hydrophtalat, hòa tan trong 50 ml acid acetic khan (TT) trong một bình nón có nút mài, đun nóng nhẹ nếu cần. Thêm 0,05 ml dung dịch tím tinh thể (TT) và chuẩn độ bằng dung dịch acid percloric đã điều chế cho đến khi chuyển màu từ tím sang xanh lơ.
Song song tiến hành một mẫu trắng.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,02042 g/ml.
Ghi nhiệt độ khi tiến hành chuẩn hóa. Nếu nhiệt độ khi định lượng khác với nhiệt độ lúc chuẩn hóa dung dịch, thể tích dung dịch chuẩn độ acid percloric dùng trong định lượng được hiệu chỉnh theo công thức sau:
VC = V[1 + 0,0011(t1 - t2)]
trong đó:
t1 là nhiệt độ khi chuẩn hóa;
t2 là nhiệt độ khi định lượng:
V là thể tích đã dùng khi định lượng;
VC là thể tích hiệu chỉnh.
Các dung dịch acid percloric nồng độ loãng hơn được điều chế bằng cách pha loãng dung dịch acid percloric 0,1 N (CĐ) với acid acetic khan (TT).
Dung dịch acid sulfuric 1N
1 ml dung dịch chứa 0,04904 g acid sulfuric (H2SO4).
Điều chế: Rót cẩn thận 30 ml acid sulfuric (TT) vào 200 ml nước, lắc đều, làm nguội đến nhiệt độ phòng và pha loãng với nước vừa đủ 1000 ml, trộn đều.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch acid hydrocloric 1 N.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,053 g/ml.
Dung dịch acid sulfuric 0,5 N
1 ml dung dịch chứa 0,02452 g acid sulfuric (H2SO4).
Điều chế: Rót cẩn thận 15 ml acid sulfuric (TT) vào 200 ml nước, lắc đều, làm nguội đến nhiệt độ phòng và pha loãng với nước vừa đủ 1000 ml, trộn đều.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch acid hydrocloric 0,5 N.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,0265 g/ml.
Dung dịch acid sulfuric 0,1 N
1 ml dung dịch chứa 0,004904 g acid sulfuric (H2SO4).
Điều chế: Pha loãng 100,0 ml dung dịch acid sulfuric 1N (CĐ) với nước vừa đủ 1000 ml.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch acid hydrocloric 0,1N.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,0053 g/ml.
Dung dịch amoni ceri nitrat 0.1 M
1 ml dung dịch chứa 0,05482 g amoni ceri (IV) nitrat (NH4)2[Ce(NO3)6].
Lắc dung dịch chứa 56 ml acid sulfuric (TT) và 54,82 g amoni ceri (IV) nitrat (TT) trong 2 min và thêm nước 5 lần, mỗi lần 100 ml, lắc cẩn thận mỗi lần thêm. Sau đó thêm nước vừa đủ 1000 ml.
Sau 10 ngày, xác định nồng độ chính xác của dung dịch bằng cách như sau: Thêm 2 g kali iodid (TT) và 150 ml nước vào 25 ml dung dịch. Chuẩn độ ngay lập tức bằng dung dịch natri thiosulfat 0,1 M (CĐ), dùng 1 ml dung dịch hồ tinh bột làm chỉ thị.
1 ml dung dịch natri thiosulfat 0,1 M (TT) tương đương với 54,82 mg (NH4)2[Ce(NO3)6].
Bảo quản tránh ánh sáng.
Dung dịch amoni ceri nitrat 0.01 M
1 ml dung dịch chứa 0,005482 g amoni ceri (IV) nitrat (NH4)2[Ce(NO3)6].
Thêm 30 ml acid sulfuric (TT) vào 100 ml dung dịch amoni ceri (IV) nitrat 0,1 M (CĐ) trong khi đang làm lạnh và thêm nước vừa đủ 1000 ml.
Dung dịch amoni ceri sulfat 0,1 M (Dung dịch amoni và ceri sulfat 0,1 M)
1 ml dung dịch chứa 0,06326 g amoni ceri sulfat [(2(NH4)2SO4.Ce(SO4)2.2H2O)].
Điều chế: Hòa tan 65,0 g amoni ceri sulfat (TT) trong hỗn hợp 30 ml acid sulfuric (TT) và 500 ml nước, để nguội và thêm nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 25,0 ml dung dịch amoni ceri sulfat đã điều chế, thêm 2 g kali iodid (TT) và 150 ml nước. Chuẩn độ ngay lập tức bằng dung dịch natri thiosulfat 0,1 N (CĐ), dùng dung dịch hồ tinh bột (TT) làm chỉ thị.
Hệ số hiệu chỉnh được tính theo công thức (2).
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch amoni sulfocyanid 0,1 N (Dung dịch amoni thiocyanat 0,1 N)
1 ml dung dịch chứa 0,007612 g amoni sulfocyanid (NH4SCN).
Điều chế: Hòa tan 7,63 g amoni sulfocyanid (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 20,0 ml dung dịch bạc nitrat 0,1 N (CĐ), thêm 25 ml nước, 2 ml acid nitric 2 M (TT), 2 ml dung dịch sắt (III) amoni sulfat (TT) và chuẩn độ bằng dung dịch amoni sulfocyanid đã điều chế cho đến màu hồng cam.
Hệ số hiệu chỉnh được tính theo công thức (2).
Dung dịch bạc nitrat 0,1 N
1 ml dung dịch chứa 0,01699 g bạc nitrat (AgNO3).
Điều chế: Hòa tan 17,5 g bạc nitrat (TT) trong nước và pha loãng vừa đủ 1000 ml.
Chuẩn độ: Cân chính xác khoảng 0,12 g chất chuẩn gốc natri clorid, hòa tan trong 50 ml nước thêm vài giọt dung dịch kali cromat (TT). Chuẩn độ bằng dung dịch bạc nitrat đã điều chế đến khi xuất hiện tủa màu đỏ nhạt.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,005844 g/ml.
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch bari clorid 0,1 M
1 ml dung dịch chứa 0,02443 g bari clorid (BaCl2.2H2O)
Điều chế: Hòa tan 24,4 g bari clorid (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 10,0 ml dung dịch bari clorid đã điều chế, thêm 60 ml nước, 3 ml dung dịch amoniac 13,5 M (TT) và 0,5 mg đến 1,0 mg đỏ tía phtalein (TT). Chuẩn độ bằng dung dịch Trilon B 0,1 M (CĐ). Khi dung dịch bắt đầu nhạt màu, thêm 50 ml ethanol 96 % (TT) và chuẩn độ cho đến khi mất màu tím xanh.
Hệ số hiệu chỉnh được tính theo công thức (2).
Dung dịch bismuth nitrat 0,01 M
1 ml dung dịch chứa 0,004851 g bismuth nitrat pentahydrat [Bi(NO3)3.5H2O].
Hòa tan 4,86 g bismuth nitrat pentahydrat (TT) trong 60 ml dung dịch acid nitric loãng (TT) và pha loãng thành 1000.0 ml bằng nước.
Chuẩn độ: Lấy 25,0 ml dung dịch nitrat bismuth, thêm 50 ml nước và chuẩn độ bằng dung dịch natri edetat 0,01 M (CĐ) dùng 0,05 ml dung dịch xylenol da cam 0,1 % làm chỉ thị.
Dung dịch brom 0,1 N
1 ml dung dịch chứa 0,00799 g brom (Br2).
Điều chế: Hòa tan 2,7835 g chất chuẩn gốc kali bromat và 13 g kali bromid (TT) trong nước vừa đủ 1000ml.
Bảo quản dung dịch trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch ceri sulfat 0,1 M
1 ml dung dịch chứa 0,04043 g ceri sulfat [Ce(SO4)2. 4H2O].
Điều chế: Hòa tan 40,4 g ceri sulfat (TT) trong hỗn hợp gồm 50 ml acid sulfuric (TT) và 500 ml nước, để nguội và thêm nước đến vừa đủ 1000 ml.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch amoni ceri sulfat 0,1 M, dùng 25,0 ml dung dịch ceri sulfat đã điều chế.
Hệ số hiệu chỉnh được tính theo công thức (2).
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch chì nitrat 0,1 M
1 ml dung dịch chứa 0,03312 g chì nitrat [Pb(NO3)2].
Điều chế: Hòa tan 33,0 g chì nitrat (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 20,0 ml dung dịch chì nitrat đã điều chế, thêm 300 ml nước và chuẩn độ bằng phương pháp chuẩn độ complexon cho chì (Phụ lục 10.5).
Hệ số hiệu chỉnh được tính theo công thức (2).
Dung dịch chì nitrat 0,1 N (Dung dịch chì nitrat 0,05 M)
1 ml dung dịch chứa 0,01656 g chì nitrat [Pb(NO3)2].
Điều chế: Hòa tan 16,6 g chì nitrat (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 50,0 ml dung dịch chì nitrat đã điều chế, thêm 300 ml nước và chuẩn độ bằng phương pháp chuẩn độ complexon cho chì (Phụ lục 10.5).
Hệ số hiệu chỉnh được tính theo công thức (2).
Dung dịch iod 0,1 N
1 ml dung dịch chứa 0,01269 g iod (l2).
Điều chế: Hòa tan 20 g kali iodid (TT) trong 50 ml nước, thêm 12,7 g iod (TT), lắc cho tan và thêm nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 25,0 ml dung dịch iod đã điều chế, thêm 1 ml dung dịch acid acetic 2 M (TT) và 30 ml nước. Chuẩn độ ngay lập tức bằng dung dịch natri thiosulfat 0,1 N (CĐ), dùng dung dịch hồ tinh bột (TT) làm chỉ thị.
Hệ số hiệu chỉnh được tính theo công thức (2).
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch kali bromat 0,1 N (Dung dịch kali bromat 0,0167 M)
1 ml dung dịch chứa 0,002784 g kali bromat (KbrO3).
Điều chế: Hòa tan 2,7840 g chất chuẩn gốc kali bromat trong nước vừa đủ 1000 ml.
Dung dịch kali dicromat 0,1 N (Dung dịch kali dicromat 0,0167 M)
1 ml dung dịch chứa 0,004903 g kali dicromat (K2Cr2C7).
Điều chế: Hòa tan 4,9033 g chất chuẩn gốc kali dicromat trong nước vừa đủ 1000 ml.
Dung dịch kali hydrophtalat 0,1 M
1 ml dung dịch chứa 0,02042 g kali hydrophtalat (C8H5O4K).
Điều chế: Hòa tan 20,42 g chất chuẩn gốc kali hydro-phtalat trong khoảng 800 ml acid acetic khan (TT), đun trên cách thủy cho tới khi tan hoàn toàn, tránh ẩm. Làm lạnh tới 20 °C, thêm acid acetic khan (TT) vừa đủ 1000 ml.
Dung dịch kali hydroxyd 1 N
1 ml dung dịch chứa 0,05611 g kali hydroxyd (KOH).
Điều chế: Hòa tan 60 g kali hydroxyd (TT) trong nước không có carbon dioxyd (TT) vừa đủ 1000 ml.
Chuẩn độ: Lấy 25,0 ml dung dịch acid hydrocloric 1 N (CĐ) và chuẩn độ băng dung dịch kali hydroxyd đã điều chế, dùng 0,5 ml dung dịch phenolphtalein (TT) làm chỉ thị.
Hệ số hiệu chỉnh được tính theo công thức (2).
Dung dịch kali hydroxyd 0,1 N
1 ml dung dịch chứa 0,00561 g kali hydroxyd (KOH).
Điều chế: Hòa tan 6 g kali hydroxyd (TT) trong nước không có carbon dioxyd (TT) vừa đủ 1000 ml.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch kali hydroxyd 1 N, dùng 25,0 ml dung dịch acid hydro- done 0,1 N (CĐ).
Hệ số hiệu chỉnh được tính theo công thức (2).
Dung dịch kali hydroxyd 0,5 N trong ethanol
1 ml dung dịch chứa 0,02805 g kali hydroxyd (KOH).
Điều chế: Hòa tan 30 g kali hydroxyd (TT) trong 50 ml nước và thêm ethanol 96 % không có aldehyd (TT) vừa đủ 1000 ml. Để yên 24 h và gạn nhanh phần dung dịch trong sang lọ thủy tinh màu.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch kali hydroxyd 1 N, dùng 25,0 ml dung dịch acid hydro- done 0,5 N (CĐ).
Hệ số hiệu chỉnh được tính theo công thức (2).
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch kali hydroxyd 0,1 N trong ethanol
1 ml dung dịch chứa 0,00561 g kali hydroxyd (KOH).
Điều chế: Hòa tan 6 g kali hydroxyd (TT) trong 50 ml nước và thêm ethanol 96 % không có aldehyd (TT) vừa đủ 1000 ml.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch kali hydroxyd 0,1 N.
Hệ số hiệu chỉnh được tính theo công thức (2).
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch kali iodat 0,05 M
1 ml dung dịch chứa 0,0107 g kali iodat (KIO3).
Điều chế: Hòa tan 10,700 g chất chuẩn gốc kali iodat trong nước vừa đủ 1000 ml.
Dung dịch kali iodat 0,1 N (Dung dịch kali iodat 1/60 M)
1 ml dung dịch chứa 0,003567 g kali iodat (KIO3).
Điều chế: Hòa tan 3,5670 g chất chuẩn gốc kali iodat trong nước vừa đủ 1000 ml.
Dung dịch kali permanganat 0,1 N (Dung dịch kali permanganat 0,02 M)
1 ml dung dịch chứa 0,00316 g kali permanganat (KmnO4)
Điều chế: Hòa tan 3,20 g kali permanganat (TT) trong 1000 ml nước, đun nóng trên cách thủy 1 h, để nguội và lọc qua phễu thủy tinh xốp.
Chuẩn độ: Lấy 20,0 ml dung dịch kali permanganat đã điều chế, thêm 2 g kali iodid (TT) và 10 ml dung dịch acid sulfuric loãng (TT). Chuẩn độ bằng dung dịch natri thiosulfat 0,1 N (CĐ), dùng 1 ml dung dịch hồ tinh bột (TT) làm chỉ thị. Song song tiến hành một mẫu trắng.
Hệ số hiệu chỉnh tính theo công thức (2).
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch kẽm clorid 0,05 M
1 ml dung dịch chứa 0,006815 g kẽm clorid (ZnCl2).
Điều chế: Hòa tan 6,82 g kẽm clorid (TT) trong nước. Nếu cần, thêm từng giọt dung dịch acid hydrocloric 2 M (TT) cho đến khi hết đục và thêm nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 20,0 ml dung dịch kẽm clorid đã điều chế, thêm 5 ml dung dịch acid acetic 2 M (TT) và chuẩn độ bằng phương pháp chuẩn độ complexon cho kẽm (Phụ lục 10.5).
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch kẽm sulfat 0,1 M
1 ml dung dịch chứa 0,02875 g kẽm sulfat (ZnSO4.7H2O).
Điều chế: Hòa tan 29,0 g kẽm sulfat (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 20,0 ml dung dịch kẽm sulfat đã điều chế, thêm 5 ml dung dịch acid acetic 2 M (TT) và chuẩn độ bằng phương pháp chuẩn độ complexon cho kẽm (Phụ lục 10.5).
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch lithi methoxyd 0,1 M
1 ml dung dịch chứa 0,003797 g lithi methoxyd (CH3OLi).
Điều chế: Hòa tan từng lượng nhỏ 0,694 g lithi (TT) trong 150 ml methanol khan (TT), thêm toluen (TT) vừa đủ 1000 ml.
Xác định độ chuẩn ngay trước khi dùng theo cách sau: Lấy 10 ml dimethylformamid (TT), thêm 0,05 ml dung dịch xanh thymol 0,3% trong methanol (TT) và chuẩn độ bằng dung dịch lithi methoxyd đã điều chế tới màu xanh lam. Ngay lập tức thêm 0,2 g chất chuẩn gốc acid benzoic, lắc cho tan hoàn toàn và chuẩn độ bằng dung dịch lithi methoxyd đã điều chế cho tới khi màu xanh lam xuất hiện trở lại. Tránh carbon dioxyd từ môi trường xung quanh trong suốt quá trình chuẩn độ. Số ml dung dịch lithi methoxyd đã điều chế dùng cho lần chuẩn độ sau là thể tích dung dịch chuẩn độ đã dùng (V).
Hệ số hiệu chỉnh tính theo công thức (1), trong đó T = 0,01221 g/ml.
Nên bảo quản trong lọ có gắn buret tự động và thiết bị chống ẩm, carbon dioxyd.
Dung dịch magnesi clorid 0,1 M
1 ml dung dịch chứa 0,02033 g magnesl clorid (MgCl2.6H2O)
Điều chế: Hòa tan 20,33 g magnesi clorid (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Lấy chính xác 25,0 ml dung dịch magnesi clorid đã điều chế và chuẩn độ bằng phương pháp chuẩn độ complexon cho magnesi (Phụ lục 10.5).
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch magnesi sulfat 0,05 M
1 ml dung dịch chứa 0,01233 g magnesi sulfat (MgSO4.7H2O).
Điều chế: Hòa tan 12,5 g magnesi sulfat (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Lấy đúng 25,0 ml dung dịch magnesi sulfat đã điều chế và chuẩn độ bằng phương pháp complexon cho magnesi (Phụ lục 10.5).
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch natri arsenit 0,1 M
1 ml dung dịch chứa 0,01299 g natri arsenit (NaAsO2).
Điều chế: Hòa tan 4,946 g chất chuẩn gốc arsen trioxyd trong hỗn hợp gồm 20 ml dung dịch natri hydroxyd 10 M (TT) và 20 ml nước, pha loãng với nước thành 400 ml và thêm dung dịch acid hydrocloric 2 M (TT) cho đến khi trung tính với giấy quì. Hòa tan 2 g natri hydrocarbonat (TT) trong dung dịch thu được và thêm nước vừa đủ 500 ml.
Dung dịch natri hydroxyd 1N
1 ml dung dịch chứa 0,0400 g natri hydroxyd (NaOH).
Điều chế: Hòa tan 45,0 g natri hydroxyd (TT) trong 50 ml nước. Đậy kín bình bằng nút cao su, để yên qua đêm, gạn phần dung dịch trong ở trên và pha loãng với nước không có carbon dioxyd (TT) vừa đủ 1000 ml.
Chuẩn độ: Lấy 20,0 ml dung dịch natri hydroxyd đã điều chế, thêm loại chỉ thị sẽ dùng trong phép định lượng mà dung dịch natri hydroxyd 1 N được sử dụng làm dung dịch chuẩn độ và chuẩn độ bằng dung dịch acid hydrocloric 1 N (CĐ).
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch natri hydroxyd 0,5 N
1 ml dung dịch chứa 0,0200 g natri hydroxyd (NaOH).
Điều chế: Hòa tan 25 g natri hydroxyd (TT) trong 25 ml nước và tiến hành như mô tả trong Dung dịch natri hydroxyd 1 N.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch natri hydroxyd 1 N, chuẩn độ bằng dung dịch acid hydrocloric 0,5 N (CĐ).
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch natri hydroxyd 0,1 N
1 ml dung dịch chứa 0,0040 g natri hydroxyd (NaOH).
Điều chế: Hòa tan 4,50 g natri hydroxyd (TT) trong 5 ml nước và tiến hành như mô tả trong Dung dịch natri hydroxyd 1 N.
Chuẩn độ: Tiến hành như mô tả trong Dung dịch natri hydroxyd 1 N, chuẩn độ bằng dung dịch acid hydrocloric 0,1 N (CĐ).
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch natri hydroxyd 0,1 N trong ethanol
1 ml dung dịch chứa 0,0040 g natri hydroxyd (NaOH).
Điều chế: Thêm 3,3 g dung dịch natri hydroxyd 10 M (TT) vào 250 ml ethanol (TT).
Chuẩn độ: Hòa tan 0,2 g chất chuẩn gốc acid benzoic trong hỗn hợp gồm 10 ml ethanol 96 % (TT) và 2 ml nước. Chuẩn độ bằng dung dịch natri hydroxyd trong ethanol đã điều chế, dùng 0,2 ml dung dịch thymolphtalein (TT) làm chỉ thị.
Hệ số hiệu chỉnh được tỉnh theo công thức (1), trong đó T = 0,01221 g/ml.
Dung dịch natri methoxyd 0,1 M
1 ml dung dịch chứa 0,005402 g natri methoxyd (CH3ONa).
Điều chế: Lấy 175 ml methanol khan (TT), làm lạnh trong nước đá và thêm từng lượng nhỏ 2,5 g natri (TT) mới cắt. Khi natri tan hết, thêm toluen (TT) vừa đủ đến vạch, lắc đều.
Xác định độ chuẩn ngay trước khi dùng: Tiến hành như mô tả trong Dung dịch lithi methoxyd 0,1 N, chuẩn độ bằng dung dịch natri methoxyd đã điều chế.
Hệ số hiệu chỉnh tính theo công thức (1), trong đó T = 0,01221 g/ml.
Nên bảo quản trong lọ có gắn buret tự động và thiết bị chống ẩm, carbon dioxyd.
Dung dịch natri nitrit 0,1 M
1 ml dung dịch chứa 0,0069 g natri nitrit (NaNO2).
Điều chế: Hòa tan 7,30 g natri nitrit (TT) trong nước vừa đủ 1000 ml.
Xác định độ chuẩn ngay trước khi dùng theo cách sau: Hòa tan 0,3 g chất chuẩn gốc acid sulfanilic trong 50 ml dung dịch acid hydrocloric 2 M (TT), thêm 3 g kali bromid (TT), làm lạnh trong nước đá và chuẩn độ bằng dung dịch natri nitrit đã điều chế, xác định điểm tương đương bằng phương pháp chuẩn độ đo ampe (Phụ lục 10.1).
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,01732 g/ml.
Bảo quản trong lọ thủy tinh nâu, tránh ánh sáng.
Dung dịch natri periodat 0,1 M
1 ml dung dịch chứa 0,02139 g natri periodat (NalO4)
Điều chế: Hòa tan 21,4 g natri periodat (TT) trong khoảng 500 ml nước và thêm nước vừa đủ 1000 ml.
Chuẩn độ: Lấy 20,0 ml dung dịch natri periodat đã điều chế cho vào bình nón nút mài, thêm 5 ml acid percloric (TT), đậy nắp bình, lắc. Điều chỉnh tới pH 6,4 bằng dung dịch bão hòa natri hydrocarbonat (TT), thêm 10 ml dung dịch kali iodid (TT), đậy nắp bình, lắc và để yên 2 min. Chuẩn độ bằng dung dịch natri arsenit 0,1 M (CĐ) cho đến màu vàng nhạt, thêm 2 ml dung dịch hồ tinh bột (TT) và chuẩn độ chậm cho tới mất màu hoàn toàn.
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch natri thiosulfat 0,1 N
1 ml dung dịch chứa 0,02482 g natri thiosulfat (Na2S2O3.5H2O).
Điều chế: Hòa tan 25,0 g natri thiosuffat (TT) và 0,2 g natri carbonat (TT) trong nước không có carbon dioxyd (TT) vừa đủ 1000 ml.
Chuẩn độ: Lấy 20,0 ml dung dịch kali bromat 0,1 N (CĐ), thêm 40 ml nước, 10 ml dung dịch kali iodid (TT) và 5 ml dung dịch acid hydrocloric 7 M (TT). Chuẩn độ bằng dung dịch natri thiosulfat đã điều chế, dùng 1 ml dung dịch hồ tinh bột (TT) làm chỉ thị.
Hệ số hiệu chỉnh tính theo công thức (2).
Dung dịch tetrabutylamoni hydroxyd 0,1 M
1 ml dung dịch chứa 0,02595 g tetrabutylamoni hydroxyd (C16H37NO).
Điều chế: Hòa tan 40,0 g tetrabutylamoni iodid (TT) trong 90 ml methanol khan (TT), thêm 20 g bạc oxyd (TT) nghiền mịn và lắc mạnh trong 1 h. Ly tâm vài ml hỗn hợp và thử phản ứng iodid của dung dịch trong ở trên. Nếu phản ứng dương tính, thêm 2 g bạc oxyd (TT) và lắc 30 min. Lặp lại qui trình này cho đến khi hỗn hợp không còn iodid. Lọc hỗn hợp qua phễu lọc thủy tinh xốp, rửa bình và phễu lọc 3 lần, mỗi lần với 50 ml toluen (TT). Thêm dịch rửa vào dịch lọc và thêm toluen (TT) vừa đủ 1000 ml. Cho luồng khí nitrogen không có carbon dioxyd sục 5 min qua dung dịch.
Xác định độ chuẩn ngay trước khi dùng: Tiến hành như mô tả trong Dung dịch lithi methoxyd 0,1 N, chuẩn độ bằng dung dịch tetrabutylamoni hydroxyd đã điều chế.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,01221 g/ml.
Nên bảo quản trong lọ có gắn buret tự động và thiết bị chống ẩm, carbon dioxyd.
Dung dịch thủy ngân (II) nitrat 0,02 M
1 ml dung dịch chứa 0,006852 g thủy ngân (II) nitrat [Hg(NO3)2.H2O].
Điều chế: Hòa tan 6,85 g thủy ngân (II) nitrat (TT) trong 20 ml dung dịch acid nitric 1 M (TT) và thêm nước vừa đủ 1000 ml.
Chuẩn độ: Cân chính xác khoảng 0,15 g chất chuẩn gốc natri clorid cho vào bình định mức 100 ml, thêm 50 ml nước, lắc cho tan hoàn toàn và thêm nước vừa đủ 100 ml, lắc đều. Lấy 10,0 ml dung dịch thu được, thêm 40 ml nước. Chuẩn độ bằng dung dịch thủy ngân nitrat đã điều chế, xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2), dùng điện cực chỉ thị platin hoặc thủy ngân và điện cực so sánh thủy ngân - thủy ngân (I) sulfat.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,002338 g/ml.
Dung dịch Triton B 0,05 M (Dung dịch natri edetat 0,05 M)
1 ml dung dịch chứa 0,01861 g Trilon B (C10H14N2O8Na2.2H2O).
Điều chế: Hòa tan 18,8 g Trilon B (TT) trong nước vừa đủ 1000 ml.
Chuẩn độ: Cân chính xác khoảng 1,64 g chất chuẩn gốc kẽm hạt, hòa tan trong 20 ml dung dịch acid sulfuric loãng (TT), thêm nước vừa đủ 500 ml. Lấy 25,0 ml dung dịch thu được, thêm 5 ml dung dịch đệm amoniacpH 10,0 (TT), khoảng 0,1 g hỗn hợp chỉ thị đen eriocrom T (TT) và 70 ml nước. Chuẩn độ bằng dung dịch Trilon B đã điều chế cho đến khi chuyển màu sang xanh lam.
Hệ số hiệu chỉnh được tính theo công thức (1) trong đó T = 0,003269 g/ml.
Dung dịch Trilon B0,1 M (Dung dịch natri edetat 0,1 M)
1 ml dung dịch chứa 0,03722 g Trilon B (C10H14N2O8Na2.2H2O).
Điều chế: Hòa tan 37,5 g Trilon B (TT) trong nước vừa đủ 1000 ml. Chuẩn độ: Tiến hành như mô tả trong Dung dịch Trilon B 0,05 M.
Hệ số hiệu chỉnh được tính theo công thức (1), trong đó T = 0,00654 g/ml.
2.3 CÁC DUNG DỊCH ĐỆM
Các dung dịch đệm được pha trong nước không có carbon dioxyd (TT).
Dung dịch đệm acetat pH 4,5
Hòa tan 77,1 g amoni acetat (TT) trong nước, thêm 70 ml acid acetic băng (TT), thêm nước vừa đủ 1000ml.
Dung dịch đệm acetat pH 4,7
Hòa tan 136,1 g natri acetat (TT) trong 500 ml nước. Trộn 250 ml dung dịch thu được với 250 ml dung dịch acid acetic 2 M (TT). Lắc 2 lần với dung dịch dithizon 0,01 % trong cloroform (TT) mới pha và lọc. Lắc với carbon tetraclorid (TT) đến khi dịch chiết trở nên không màu, lọc lớp nước để loại bỏ hết carbon tetraclorid.
Dung dịch đệm acetat pH 5,0
Thêm 100 ml dung dịch kali hydroxyd 0,1 M (TT) và khoảng 250 ml nước vào 120 ml dung dịch acid acetic 0,6% (TT). Trộn đều, chỉnh pH tới 5,0 bằng dung dịch acid acetic 0,6% (TT) hoặc dung dịch kali hydroxyd 0,1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm acetat - edetat pH 5,5
Hòa tan 250 g amoni acetat (TT) và 15 g dinatri edetat (TT) trong 400 ml nước và thêm 125 ml acid acetic băng (TT).
Dung dịch đệm aceton
Hòa tan 8,15 g natri acetat (TT) và 42 g natri clorid (TT) trong nước, thêm 68 ml dung dịch acid hydrocloric 0,1 M (TT) và 150 ml aceton (TT), thêm nước vừa đủ 500 ml.
Dung dịch đệm amoni carbonat 0,1 M pH 10,3
Hòa tan 7,91 g amoni cacbonat (TT) trong 800 ml nước. Điều chỉnh pH bằng dung dịch natri hydroxyd loãng (TT), thêm nước vừa đủ 1000 ml.
Dung dịch đệm amoni clorid pH 9,5 (Đệm amoniac pH 9,5)
Hòa tan 33,5 g amoni clorid (TT) trong 150 ml nước, thêm 42 ml amoniac (TT), thêm nước vừa đủ 250 ml. Bảo quản trong đồ đựng bằng polyethylen.
Dung dịch đệm amoni clorid pH 10,4
Hòa tan 70 g amoni clorid (TT) trong 200 ml nước, thêm 330 ml amoniac (TT) và thêm nước vừa đủ 1000 ml. Điều chỉnh pH nếu cần tới 10,4 bằng amoniac (TT).
Dung dịch đệm borat kiềm
Dung dịch acid boric và kali clorid 0,2 N: Hòa tan 12,37 g acid boric (TT) và 14,91 g kali clorid (TT) trong nước vừa đủ 1000 ml.
Dung dịch natri hydroxyd 0,2 N: Pha như mô tả trong dung dịch chuẩn độ.
Cách pha: Lấy 50 ml dung dịch acid boric và kali clorid 0,2 N, thêm lượng dung dịch natri hydroxyd 0,2 N như ghi trong bảng và thêm nước vừa đủ 200,0 ml.
| pH | Số ml dung dịch natri hydroxyd 0,2 N |
| 8,0 | 3,9 |
| 8,2 | 6,0 |
| 8,4 | 8,6 |
| 8,6 | 11,8 |
| 8,8 | 15,8 |
| 9,0 | 20,8 |
| 9,2 | 26,4 |
| 9,4 | 32,1 |
| 9,6 | 36,9 |
| 9,8 | 40,6 |
| 10,0 | 43,7 |
Dung dịch đệm borat 0,0015 M pH 8,0
Hòa tan 0,572 g natri tetraborat (TT) và 2,94 g calci clorid (TT) trong 800 ml nước, điều chỉnh tới pH 8,0 bằng dung dịch acid hydrocloric 1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm borat pH 10,4
Hòa tan 24,64 g acid boric (TT) trong 900 ml nước, điều chỉnh tới pH 10,4 bằng dung dịch natri hydroxyd 40 % (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm citrat pH 5,0
Chuẩn bị dung dịch chưa acid citric 2,01 % và natri hydroxyd 0,80 %. Điều chỉnh pH bằng dung dịch acid hydrocloric loãng (TT).
Dung dịch đệm diethanolamin pH 10,0
Hòa tan 96,4 g diethanolamin (17) trong nước vừa đủ 400 ml. Thêm 0,5 ml dung dịch magnesi clorid 18,6 %, điều chỉnh pH tới 10,0 bằng dung dịch acid hydrocloric 1 M (TT), thêm nước vưa đủ 500 ml.
Dung dịch đệm đồng sulfat pH 4,0
Hòa tan 0,25 g đồng (II) sulfat (TT) và 4,5 g amoni acetat (TT) trong dung dịch acid acetic vừa đủ 100 ml.
Dung dịch đệm đồng sulfat pH 5,2
Hòa tan 15,22 g dinatri hydrophosphat khan (TT) trong nước vừa đủ 536 ml, thêm dung dịch acid citric 2,1 % đến khi pH trong khoảng từ 5,15 đến 5,25 (khoảng 464 ml). Trộn 985 ml dung dịch thu được với 15 ml dung dịch đồng (II) sulfat 0,393 % (TT).
Dung dịch đệm glycin
Hòa tan 42 g natri hydrocarbonat (TT) và 50 g kali hydrocarbonat (TT) trong 180 ml nước. Thêm vào dung dịch thu được một dung dịch chứa 37,5 g glycin (TT) và 15 ml amoniac trong 180 ml nước. Thêm nước vừa đủ 500 ml và khuấy đều cho tới khi thu được dung dịch đồng nhất.
Dung dịch đệm HEPES pH 7,5
Hòa tan 2,38 g acid 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethansulfonic trong khoảng 90 ml nước, điều chỉnh pH bằng dung dịch natri hydroxyd 20 % (TT) và thêm nước vừa đủ 100 ml.
Dung dịch đệm imidazol pH 6,5
Hòa tan 6,81 g imidazol (TT), 1,23 g magnesi sulfat (TT) và 0,73 g calci sulfat (TT) vào 752 ml dung dịch acid hydrocloric 0,1 M (CĐ). Điều chỉnh pH nếu cần và thêm nước vừa đủ 1000 ml.
Dung dịch đệm imidazol pH 7,3
Hòa tan 3,4 g imidazol (TT) và 5,8 g natri clorid (TT) trong nước, thêm 18,6 ml dung dịch acid hydrocloric 1 M (TT) và thêm nước vừa đủ 1000 ml. Điều chỉnh pH nếu cần.
Dung dịch đệm maleat pH 7,0
Hòa tan 10 g natri clorid (TT); 6,06 g tris(hydroxy-methyl) methylamin (TT) và 4,90 mg anhydrid maleic (TT) trong 900 ml nước. Điều chỉnh pH bằng dung dịch natri hydroxyd 17,0 % và thêm nước vừa đủ 1000 ml.
Bảo quản ở 2 °C đến 8 °C trong 3 ngày.
Dung dịch đệm natri acetat pH 4,5
Hòa tan 63 g natri acetat khan (TT) trong nước, thêm 90 ml dung dịch acid acetic 5 M (TT), điều chỉnh pH đến 4,5 và thêm nước vừa đủ 1000ml.
Dung dịch đệm natri acetat pH 6,0
Hòa tan 4,1 g natri acetat khan (TT) trong 1000 ml nước, điều chỉnh pH đến 6,0 bằng acid acetic băng (TT).
Dung dịch đệm natri citrat pH 7,8 (natri citrat 0,034 M, natri clorid 0,101 M)
Hòa tan 10 g natri citrat (TT) và 5,9 g natri clorid (TT) trong 900 ml, điều chỉnh pH bằng acid hydrocloric (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm pH 2,0
Hòa tan 6,57 g kali clorid (TT) trong nước, thêm 119 ml dung dịch acid hydrocloric 0,1 M (CĐ) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm pH 2,2
Trộn 6,7 ml acid phosphoric (TT) với 50 ml dung dịch 4 % (tt/tt) của dung dịch natri hydroxyd 2 M (TT), thêm nước vừa đủ 1000 ml.
Dung dịch đệm pH 2,5 (Đệm phosphat pH 2,5)
Hòa tan 100 g kali dihydrophosphat (TT) trong 800 ml nước, điều chỉnh tới pH 2,5 bằng acid hydrocloric (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm pH 2,5 số 1 (Đệm phosphat pH 2,5 số 1)
Thêm 5 ml dung dịch acid phosphoric 2 M (TT) vào 250 ml nước. Điều chỉnh tới pH 2,5 bằng dung dịch natri hydroxyd 2 M (TT) và pha loãng với nước vừa đủ 500 ml.
Dung dịch đệm pH 3,0
Hòa tan 21,0 g acid citric (TT) trong 200 ml dung dịch natri hydroxyd 1 M (TT), thêm nước vừa đủ 1000 ml. Pha loãng 40,3 ml dung dịch này thành 100 ml với dung dịch acid hydrocloric 0,1 M (TT).
Dung dịch đệm pH 3,7 (Đệm acetic - amoniac trong ethanol pH 3,7)
Thêm 60 ml ethanol 96 % (TT) và 20 ml nước vào 15 ml dung dịch acid acetic 5 M (TT). Điều chỉnh pH tới 3,7 bằng dung dịch amoniac 10 M (TT) và thêm nước vừa đủ 100 ml.
Dung dịch đệm pH 5,2
Hòa tan 1,02 g kali dihydrophthalat (TT) trong 30 ml dung dịch natri hydroxyd 0,1 M (CĐ) và thêm nước vừa đủ 100 ml.
Ở 20 °C dung dịch này có thể được sử dụng làm dung dịch pH chuẩn.
Dung dịch đệm pH 5,5
Hòa tan 54,4 g natri acetat (TT) trong 50 ml nước, làm nóng đến 35 °C nếu cần. Để nguội, thêm từ từ 10 ml acid acetic khan (TT), lắc đều và thêm nước vừa đủ 100 ml.
Dung dịch đệm pH 6,5
Hòa tan 60,5g dinatri hydrophosphat (TT) và 46 g kali dihydrophosphat (TT) trong nước, thêm 100 ml dung dịch dinatri edetat 0,02 M (TT) và 20 mg thủy ngân (II) clorid (TT) và thêm nước vừa đủ 100 ml.
Dung dịch đệm pH 7,0
Thêm vào 1000 ml dung dịch chứa dinatri hydro-phosphat 1,8 % và natri clorid 2,3 % một lượng vừa đủ (khoảng 280 ml) dung dịch chứa natri dihydro-phosphat 0,78 % và natri clorid 2,3 % để điều chỉnh pH. Hòa tan vào dung dịch thu được một lượng natri azid (TT) vừa đủ để được dung dịch có nồng độ 0,02 %.
Dung dịch đệm pH 7,2
Xem đệm phosphat pH 7,2.
Dung dịch đệm pH 7,4
Hòa tan 0,6 g kali dihydrophosphat (TT); 6,4 g dinatri hydrophosphat (TT) và 5,85 g natri clorid (TT) trong nước vừa đủ 1000 ml. Điều chỉnh pH nếu cần.
Dung dịch đệm pH 8,0
Xem đệm phosphat pH 8,0.
Dung dịch đệm pH 8,0 (TT1)
Hòa tan 20 g dikali hydrophosphat (TT) trong 900 ml nước, điều chỉnh pH bằng acid phosphoric (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 2,0
Hòa tan 8,95 g dinatri hydrophosphat khan (TT) và 3,40 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml. Điều chỉnh pH nếu cần bằng acid phosphoric (TT).
Dung dịch đệm phosphat pH 2,8
Hòa tan 7,8 natri dihydrophosphat khan (TT) trong 900 ml nước, điều chỉnh pH tới 2,8 bằng acid phosphoric (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 3,0
Trộn 0,7 ml acid phosphoric (TT) với 900 ml nước, điều chỉnh pH tới 3,0 bằng dung dịch natri hydroxyd 10 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat 0,1 M pH 3,0
Hòa tan 12 g natri dihydrophosphat khan (TT) trong nước, điều chỉnh pH bằng dung dịch acid phosphoric 2 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 3,0 (TT1)
Hòa tan 3,4 g kali dihydrophosphat (TT) trong 900 ml nước. Điều chỉnh pH đến 3,0 bằng acid phosphoric (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 3,2
Thêm 100 ml dung dịch acid phosphoric 0,25 % vào 900 ml dung dịch natri dihydrophosphat 0,4 %, điều chỉnh pH nếu cần.
Dung dịch đệm phosphat pH 3,2 (TT1)
Điều chỉnh pH của dung dịch dinatri hydrophosphat 3,58 % tới 3,2 bằng dung dịch acid phosphoric 2 M (TT). Pha loãng 100 ml dung dịch thu được thành 2000 ml với nước.
Dung dịch đệm phosphat pH 3,5
Hòa tan 68,0 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml, điều chỉnh pH bằng acid phosphoric (TT).
Dung dịch đệm phosphat 0,05 M pH 4,5
Hòa tan 6,8 g kali dihydrophosphat (TT) trong 1000 ml nước, pH của dung dịch bằng 4,5.
Dung dịch đệm phosphat pH 5,0
Hòa tan 2,72 g kali dihydrophospliat (TT) trong 800 ml nước. Điều chỉnh pH bằng dung dịch kali hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 5,5
Trộn 96,4 ml dung dịch kali dihydrophosphat 1,36 % (TT) với 3,6 ml dung dịch dinatri hydrophosphat 3,58 % (TT).
Dung dịch đệm phosphat pH 5,6
Trộn 94,4 ml dung dịch kali dihydrophosphat 0,908 % (TT) với 5,6 ml dung dịch dikali hydrophosphat 1,16 % (TT). Điều chỉnh pH nếu cần bằng một trong hai dung dịch thành phần.
Dung dịch đệm phosphat pH 5,8
Hòa tan 1,19 g dinatri hydrophosphat dihydrat (TT) và 8,25 g kali dihydro phosphat (TT) trong nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 6,0 (TT1)
Hòa tan 6,8 g natri dihydrophosphat (TT) trong nước vừa đủ 1000 ml và điều chỉnh pH bằng dung dịch natri hydroxyd 10 M (TT)
Dung dịch đệm phosphat pH 6,0 (TT2)
Thêm 28,5 ml dung dịch natri hydroxyd 0,2 M (TT) vào 250 ml dung dịch kali dihydrophosphat 0,2 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat 0,1 M pH 6,3
Hòa tan 15,6 g diatri dihydrophosphat (TT) trong 900 ml nước, điều chỉnh pH tới 6,3 bằng dung dịch natrri hydroxydo, 1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 6,4
Hòa tan 2,5 g dinatri hydrophosphat (TT); 2,5 g natri dihydrophosphat (TT) và 8,2g natri clorid (TT) trong 950 ml nước. Điều chỉnh pH nếu cần tới 6,4 bằng dung dịch natri hydroxyd 1 M (TT) hoặc dung dịch acid hydrocloric 1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat 0,1 M pH 6,5
Hòa tan 13,8 g natri dihydrophosphat monohydrat (TT) trong 900 ml nước. Điều chỉnh pH bằng dung dịch natri hydroxyd 40 % (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 6,8
Xem đệm citro-phosphat pH 6,8.
Dung dịch đệm phosphat pH 7,0
Xem đệm citro-phosphat pH 7,0.
Dung dịch đệm phosphat pH 7,0 (TT1)
Xem đệm phosphat pH 7,0 hỗn hợp.
Dung dịch đệm phosphat pH 7,0 (TT2)
Trộn 50 ml dung dịch kali dihydrophosphat 13,6 % (TT) với 29,5 ml dung dịch natri hydroxyd 1 M (TT), thêm nước đến vừa đủ 100 ml. Điều chỉnh pH từ 6,9 đến 7,1.
Dung dịch đệm phosphat pH 7,0 (TT3)
Hòa tan 5 g kali dihydrophosphat (TT) và 11 g dikali hydrophosphat (TT) trong 900 ml nước. Điều chỉnh pH tới 7,0 bằng dung dịch acid phosphoric 2 M (TT) hoặc dung dịch natri hydroxyd 2 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 7,0 (TT4)
Hòa tan 28,4 g dinatri hydrophosphat khan (TT) và 18,2 g kali dihydrophosphat (TT) trong nước vừa đủ 500 ml.
Dung dịch đệm phosphat pH 7,0 (TT5)
Hòa tan 28,4 g dinatri hydrophosphat khan (TT) trong 800 ml nước. Điều chỉnh pH bằng dung dịch acid phosphoric 30 % (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat 0,025 M pH 7,0
Trộn 1 thể tích dung dịch đệm phosphat 0,063 M pH 7,0 (TT) với 1,5 thể tích nước.
Dung dịch đệm phosphat 0,03 M pH 7,0
Hòa tan 5,2 g dikali hydrophosphat (TT) trong 900 ml nước, điều chỉnh pH tới 7,0 ± 0,1 bằng acid phosphoric (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat 0,05 M pH 7,0
Trộn 34 ml nước với 100 ml đệm phosphat hỗn hợp 0,067 M pH 7,0 (TT).
Dung dịch đệm phosphat 0,063 M pH 7,0
Hòa tan 5,18 g dinatri hydrophosphat khan (TT) và 3,65 g natri dihydrophosphat monohydrat (TT) trong 950 ml nước, chỉnh pH bằng acid phosphoric (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 7,2
Xem đệm citro-phosphat pH 7,2.
Dung dịch đệm phosphat pH 7,4
Thêm 250 ml dung dịch kali dihydrophosphat 0,2 M (TT) vào 393,4 ml dung dịch natri hydroxyd 0,1 M (TT).
Dung dịch đệm phosphat 0,33 M pH 7,5
Trộn 85 ml dung dịch dinatri hydrophosphat 11,93 % (TT) với 15 ml dung dịch kali dihydrophosphat 4,54 % (TT). Điều chỉnh pH nếu cần.
Dung dịch đệm phosphat 0,02 M pH 8,0
Trộn 50 ml dung dịch kali dihydrophosphat 0,2 M (TT) với 46,8 ml dung dịch natri hydroxyd 0,2 M (TT), thêm nước vừa đủ 500 ml.
Dung dịch đệm phosphat 0,1 M pH 8,0
Hòa tan 0,523 g kali dihydrophosphat (TT) và 16,73 g dikali hydrophosphat (TT) trong nước vừa đủ 1000 ml.
Dung dịch đệm phosphat 1 M pH 8,0
Hòa tan 136,1 g kali dihydrophosphat (TT) trong nước, điều chỉnh pH bằng dung dịch natri hydroxyd 1 M (TT), thêm nước vừa đủ 1000 ml.
Dung dịch đệm phosphat pH 9,0
Hòa tan 1,74 g kali dihydrophosphat (TT) trong 80 ml nước, điều chỉnh pH nếu cần bằng dung dịch kali hydroxyd 1 M (TT) và thêm nước vừa đủ 100 ml.
Dung dịch đệm phosphat-citrat pH 5,5
Trộn 56,85 ml dung dịch dinatri hydrophosphat khan 2,84 % (TT) và 43,15 ml dung dịch acid citric 2,1 % (TT).
Dung dịch đệm phosphat diethylamoni pH 6,0
Pha loãng 68 ml acid phosphoric (TT) thành 500 ml với nước. Thêm vào 25 ml dung dịch này 450 ml nước và 6 ml diethylamin (TT), điều chỉnh pH (nếu cần) trong khoảng 5,95 đến 6,05 bằng diethylamin (TT) hoặc acid phosphoric (TT) và thêm nước vừa đủ 500 ml.
Dung dịch đệm phosphat octylamin pH 3,0
Pha loãng 3,32 ml octylamin (TT) tới 1000 ml, điều chỉnh pH tới 3,0 bằng acid phosphoric (TT) và lọc qua màng lọc có kích thước không quá 0,5 μm.
Dung dịch đệm phtalat pH 4,4
Hòa tan 2,042 g kali hydrophtalat (TT) trong 50 ml nước, thêm 7,5 ml dung dịch natri hydroxyd 0,2 M (CĐ), thêm nước vừa đủ 200 ml. Ở 20 °C, dung dịch thu được có thể được dùng như dung dịch pH chuẩn.
Dung dịch đệm phtalat 0,5 M pH 6,4
Hòa tan 100 g kali hydrophthalat (TT) trong nước vừa đủ 1000 ml. Điều chỉnh pH nếu cần bằng dung dịch natri hydroxyd 10 M (TT).
Dung dịch đệm sucinat pH 4,6
Hòa tan 11,8 g acid sucinic (TT) trong hỗn hợp của 600 ml nước và 82 ml dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm sulfat pH 2,0
Dung dịch I: Hòa tan 132,1 g amoni sulfat (TT) trong nước vừa đủ 500 ml.
Dung dịch II: Thêm từ từ 14 ml acid sulfuric (TT) vào khoảng 400 ml nước, vừa thêm vừa khuấy và làm nguội. Để dung dịch nguội hoàn toàn, thêm nước vừa đủ 500 ml.
Trộn dung dịch I và II đồng thể tích, điều chỉnh pH nếu cần.
Dung dịch đệm tetrabutylamonl pH 7,0
Hòa tan 6,16 g amoni acetat (TT) trong hỗn hợp 15 ml dung dịch tetrabutylamoni hydroxyd 40 % và 185 ml nước. Điều chỉnh pH bằng acid nitric (TT).
Dung dịch đệm tris-acetat pH 8,5
Hòa tan 0,294 g calci clorid (TT) và 12,11 g tris(hydroxymethyl)methylamin (TT) trong nước. Điều chỉnh pH bằng dung dịch acid acetic 5 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm tris-EDTA BSA pH 8,4
Hòa tan 6,1 g tris(hydroxymethyl)methylamin (TT), 2,8 g dinatri edetat (TT), 10,2 g natri clorid (TT) và 10 g albumin bò trong nước. Điều chỉnh pH tới 8,4 bằng dung dịch acid hydrocloric 1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch đệm tris-glycin pH 8,3
Hòa tan 6g tris(hydroxymethyl)methylamin (TT) và 28,8 g glycin (TT) trong nước vừa đủ 1000 ml. Pha loãng 1 thể tích của dung dịch thu được thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch đệm tris-hydroclorid pH 8,3
Hòa tan 9,0 g tris(hydroxymethyl)aminomethan (TT) trong 2,9 lít nước. Điều chỉnh pH bằng dung dịch acid hydrocloric 1 M (TT), thêm nước vừa đủ 3 lít.
Dung dịch đệm tris-hydroclorid 1 M pH 6,8
Hòa tan 60,6 g tris(hydroxymethyl)methylamin (TT) trong 400 ml nước. Điều chỉnh pH bằng acid hydrocloric (TT), thêm nước vừa đủ 500 ml.
Dung dịch đệm tris-hydroclorid pH 8,0
Hòa tan 1,21 g tris(hydroxymethyl)aminomethan (TT) và 29,4 mg calci clorid (TT) trong nước. Điều chỉnh pH bằng dung dịch acid hydrocloric 1 M (TT), thêm nước vừa đủ 100 ml.
Dung dịch đệm tris-hydroclorid 1,5 M pH 8,8
Hòa tan 90,8 g tris(hydroxymethyl)methylamin (TT) trong 400 ml nước. Điều chỉnh pH bằng acid hydrocloric (TT), thêm nước vừa đủ 500 ml.
Dung dịch đệm tris(hydroxymethyl) aminomethan pH 7,4
Hòa tan 30,3 g tris(hydmxymethyl)aminomethan (TT) trong khoảng 200 ml nước. Thêm 183 ml dung dịch acid hydrocloric 1 M (TT), thêm nước vừa đủ 500 ml.
Chú ý: pH của dung dịch thu được là 7,7 - 7,8 ở nhiệt độ phòng và 7,4 ở 37 °C. Dung dịch bền trong vài tháng ở nhiệt độ 4 °C.
Dung dịch đệm tris(hydroxymethyl) aminomethan 1 M pH 8,5
Hòa tan 60,6 g tris(hyaroxymethyl)aminomethan (TT) trong 40 ml nước, điều chỉnh pH tới 8,5 bằng dung dịch acid hydrocloric 1 N (TT), thêm nước vừa đủ 500 ml.
Dung dịch đệm tris(hydroxymethyl) aminomethan- natri clorid pH 7,4
Hòa tan 0,1 g albumin bò trong hỗn hợp gồm 2 ml dung dịch đệm tris(hydroxymethyl) aminomethan pH 7,4 (TT) và 50 ml dung dịch natri clorid 5,84 mg/ml (TT), thêm nước vừa đủ 100 ml.
Dung dịch muối đệm pH 7,2
Hòa tan 8 g natri clorid (TT); 0,2g kali clorid (TT); 0,1g calci clorid khan (TT); 0,1 g magnesi clorid (TT); 3,18 g dinatri hydrophosphat (TT) và 0,2 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml.
Dung dịch salin đệm phosphat pH 6,4
Hòa tan 1,79 g dinatri hydrophosphat (TT), 1,36 g kali dihydrophosphat (TT) và 7,02 g natri clorid (TT) trong nước vừa đủ 1000 ml.
Dung dịch salin đệm phosphat pH 6,8
Hòa tan 1,0 g kali dihydrophosphat (TT), 2,0 g dikali hydrophosphat (TT) và 8,5 g natri clorid (TT) trong 900 ml nước, điều chỉnh pH nếu cần và thêm nước vừa đủ 1000 ml.
Dung dịch salin đệm phosphat - albumin pH 7,2
Hòa tan 10,75 g dinatri hydrophosphat (TT), 7,6 g natri clorid (TT) và 10 g albumin bò trong nước vừa đủ 1000 ml. Ngay trước khi sử dụng, điều chỉnh pH tới 7,2 bằng dung dịch natri hydroxyd 2 M (TT) hoặc dung dịch acid phosphoric 10 % (kl/kl) (TT).
Dung dịch salin đệm phosphat - albumin pH 7,2 (TT1)
Hòa tan 10,75 g dinatri hydrophosphat (TT), 7,6 g natri clorid (TT) và 1,0 g albumin bò trong nước vừa đủ 1000 ml. Ngay trước khi sử dụng, điều chỉnh pH tới 7,2 bằng dung dịch natri hydroxyd 2 M (TT) hoặc dung dịch acid phosphoric 10 % (kl/kl) (TT).
Dung dịch salin đệm phosphat pH 7,4
Hòa tan 2,38 g dinatri hydrophosphat (TT), 0,19 g kali dihydrophosphat (TT) và 8,0 g natri clorid (TT) trong nước vừa đủ 1000 ml, điều chỉnh pH nếu cần.
Đệm acid acetic - amoni acetat (Dung dịch đệm acid acetic - amoni acetat)
Hòa tan 77,1 g amoni acetat (TT) trong nước, thêm 57 ml acid acetic băng (TT) và thêm nước vừa đủ 1000 ml.
Đệm acetat pH 2,45
Trộn 200 ml dung dịch acid hydrocloric 1 M (TT) với 200 ml dung dịch natri acetat 1 M (TT) và thêm nước vừa đủ 1000 ml. Điều chỉnh tới pH 2,45 bằng dung dịch acid hydrocloric 1 M (TT) hoặc bằng dung dịch natri acetat 1M (TT) ngay trươc khi dùng.
Đệm acetat pH 2,8
Hòa tan 4 g natri acetat khan (TT) trong khoảng 840 ml nước, điều chỉnh tới pH 2,8 bằng acid acetic băng (TT) (dùng khoảng 155 ml), thêm nước vừa đủ 1000 ml.
Đệm acetat pH 3,4
Trộn 5 thể tích dung dịch natri acetat 0,1 M (TT) với 95 thể tích dung dịch acid acetic 0,1 M (TT).
Đệm acetat pH 3,5 (Dung dịch đệm acetat pH 3,5)
Hòa tan 25 g amoni acetat (TT) trong 25 ml nước, thêm 38 ml dung dịch acid hydroclorid 7 M (TT). Điều chỉnh tới pH 3,5 bằng dung dịch acid hydrocloric 2 M (TT) hoặc bằng dung dịch amoniac 6 M (TT) và thêm nước vừa đủ 100 ml.
Đệm acetat pH 3,7
Hòa tan 10 g natri acetat khan (TT) trong 300 ml nước, điều chỉnh tới pH 3,7 bằng acid acetic băng (TT) và thêm nước vừa đủ 1000 ml. Nếu cần thiết, điều chỉnh lại tới pH 3,7 bằng acid acetic băng (TT) hoặc bằng natri acetat khan (TT) trước khi dùng.
Đệm acetat pH 4,4 (Dung dịch đệm acetat pH 4,4)
Hòa tan 136 g natri acetat (TT) và 77 g amoni acetat (TT) trong nước, thêm nước vừa đủ 1000 ml. Thêm 250 ml acid acetic băng (TT), trộn đều.
Đệm acetat pH 4,6 (Dung dịch đệm acetat pH 4,6)
Hòa tan 5,4 g natri acetat (TT) trong 50 ml nước, thêm 2,4 g acid acetic băng (TT) và thêm nước vừa đủ 100 ml. Điều chỉnh pH nếu cần.
Đệm acetat pH 5,0
Hòa tan 13,6 g natri acetat (TT) và 6 ml acid acetic băng (TT) trong nước và thêm nước vừa đủ 1000 ml.
Đệm acetat pH 6,0 (Dung dịch đệm acetat pH 6,0)
Hòa tan 100 g amoni acetat (TT) trong 300 ml nước, thêm 4,1 ml acid acetic băng (TT). Nếu cần, điều chỉnh pH bằng dung dịch amoniac 10 M (TT) hay dung dịch acid acetic 5 M (TT), thêm nước vừa đủ 500 ml.
Đệm acid thiobarbituric-citrat
Hòa tan 5,0 g acid thiobarbituric (TT) trong 5 ml dung dịch natri hydroxyd 4 M (TT) và thêm nước vừa đủ 500 ml. Hòa tan 37 g natri citrat (TT) trong 32 ml acid hydrocloric (TT) và thêm nước vừa đủ 250 ml. Trộn hai dung dịch và điều chỉnh pH tới 2,0.
Đệm amoniac - amoni clorid
Hòa tan 67,5 g amoni clorid (TT) trong nước, thêm 570 ml amoniac (TT), thêm nước vừa đủ 1000 ml.
Đệm amoniac pH 10,0 (Dung dịch đệm amoni clorid pH 10,0)
Hòa tan 5,4 g amoni clorid (TT) trong 20 ml nước, thêm 35 ml dung dịch amoniac 10 M (TT) và thêm nước vừa đủ 100 ml.
Đệm amoniac pH 10,9 (Dung dịch đệm pH 10,9)
Hòa tan 67,5 g amoni clorid (TT) trong dung dịch amoniac 10 M (TT) vừa đủ 1000 ml.
Đệm amoniac pH 10,9 pha loãng
Pha loãng 2 ml đệm amoniac pH 10,9 (TT) thành 1000 ml với nước.
Đệm barbiton pH 7,4 (Dung dịch đệm barbital pH 7,4)
Trộn 50 ml dung dịch chứa natri acetat 1,944 % và natri barbiton 2,946 % với 50,5 ml dung dịch acid hydrocloric 0,1 M (TT), thêm 20 ml dung dịch natri clorid 8,5% (TT) và thêm nước vừa đủ 250 ml.
Đệm barbiton pH 8,4 (Dung dịch đệm barbital pH 8,4)
Hòa tan 8,25 g natri barbiton (TT) trong nước vừa đủ 1000 ml.
Đệm barbiton pH 8,6 (Dung dịch đệm barbital pH 8,6)
Hòa tan 1,38 g barbiton (TT), 8,76g natri barbiton (TT) và 0,38 g calci lactat (TT) trong nước vừa đủ 1000 ml.
Đệm borat pH 7,5 (Dung dịch đệm borat pH 7,5)
Hòa tan 2 5 g natri clorid (TT), 2,85 g natri tetraborat (TT) và 10,5 g acid boric (TT) trong nước vừa đủ 1000 ml. Điều chỉnh pH nếu cần. Bảo quản dung dịch từ 2 °C đến 8 °C.
Đệm borat pH 8,0
Thêrn 3,97 ml dung dịch natri hydroxyd 0,2 M (CĐ) vào 50 ml dung dịch chứa 0,6189 g acid boric (TT) và 0,7456 g kali clorid (TT), thêm nước vừa đủ 200 ml. Ở 20 °C dung dịch này có thể được sử dụng làm dung dịch pH chuẩn.
Đệm borat pH 9,0 (Dung dịch đệm pH 9,0)
Dung dịch A: Hòa tàn 6,18 g acid boric (TT) trong dung dịch kali clorid 0,1M vừa đủ 1000 ml.
Dung dịch B: Dung dịch natri hydroxyd 0,1 M (TT).
Trộn 1000 ml dung dịch A với 420 ml dung dịch B. Ở 20 °C dung dịch này có thể được sử dụng làm dung dịch pH chuẩn.
Đệm borat pH 9,6
Thêm 36,85 ml dung dịch natri hydroxyd 0,2 M (CĐ) vào 50 ml dung dịch chứa 0,6189 g acid boric (TT) và 0,7456 g kali clorid (TT), thêm nước vừa đủ 200 ml. Ở 20 °C dung dịch này có thể được sử dụng làm dung dịch pH chuẩn.
Đệm boric pH 9,0 (Dung dịch đệm pH 9,0)
Hòa tan 6,20 g acid boric (TT) trong 500 ml nước, điều chỉnh tới pH 9,0 bằng dung dịch natri hydroxyd 1 M (TT) (khoảng 41,5 ml) và thêm nước vừa đủ 1000 ml.
Đệm carbonat pH 9,7
Hòa tan 8,4 g natri hydrocarbonat (TT) và 10,6 g natri carbonat (TT) trong nước vừa đủ 500 ml.
Đệm citro - phosphat pH 4,5
Thêm vào 30 thể tích dung dịch dinatri hydrophosphat 0,2 M (TT) một lượng vừa đủ dung dịch acid citric 0,1 M (TT) để được pH 4,5 (khoảng 36 thể tích).
Đệm citro - phosphat pH 5,0
Trộn 48,5 ml dung dịch acid citric 0,1 M (TT) với lượng vừa đủ dung dịch dinatri hydrophosphat 0,2 M (TT) để được 100 ml.
Đệm citro - phosphat pH 6,0 (Dung dịch đệm phosphat pH 6,0)
Trộn 63,2 ml dung dịch dinatri hydrophosphat 7,15 % (TT) với 36,8 ml dung dịch acid citric 2,1 % (TT).
Đệm citro - phosphat pH 6,5
Trộn 29,0 ml dung dịch acid citric 0,1 M (TT) với lượng vừa đủ dung dịch dinatri hydrophosphat 0,2 M (TT) để được 100 ml.
Đệm citro - phosphat pH 6,8 (Dung dịch đệm phosphat pH 6,8)
Trộn 77,3 ml dung dịch dinatri hydrophosphat 7,15% (TT) với 22,7 ml dung dịch acid citric 2,1 % (TT).
Đệm citro - phosphat pH 7,0 (Dung dịch đệm phosphat pH 7,0)
Trộn 82,4 ml dung dịch dinatri hydrophosphat 7,15% (TT) với 17,6 ml dung dịch acid citric 2,1 % (TT).
Đệm citro - phosphat pH 7,2 (Dung dịch đệm phosphat pH 7,2)
Trộn 87 ml dung dịch dinatri hydrophosphat 7,15% (TT) với 13 ml dung dịch acid citric 2,1 % (TT).
Đệm citro - phosphat pH 7,6
Hòa tan 67,1 g dinatri hydrophosphat (TT) và 1,33 g acid citric (TT) trong nước vừa đủ 1000 ml.
Đệm clorid 0,1 M pH 2,0 (Dung dịch đệm pH 2,0)
Hòa tan 6,57 g kali clorid (TT) trong nước, thêm 119 ml dung dịch acid hydrocloric 0,1 M (CĐ) và thêm nước vừa đủ 1000 ml.
Đệm glycin pH 2,9
Hòa tan 6,0g glycin (TT) và 4,68 g natri clorid (TT) trong 10 lít nước. Điều chỉnh pH bằng dung dịch acid hydrocloric 1 M (TT) (khoảng 30 ml).
Đệm glycin pH 11,3
Trộn dung dịch chứa glycin 0,75 % và natri clorid 0,58 % với đồng thể tích dung dịch natri hydroxyd 0,1 M (TT), điều chỉnh pH nếu cần.
Đệm kali phtalat pH 5,6 (Dung dịch đệm kali biphtalat pH 5,6)
Hòa tan 4,085 g kali biphtalat (TT) trong nước vừa đủ 100 ml. Lấy 50 ml dung dịch thu được, thêm 38,8 ml dung dịch natri hydroxyd 0,2 N (CĐ) và thêm nước vừa đủ 200 ml.
Đệm hiệu chỉnh lực ion toàn phần
Hòa tan 58,5 g natri clorid (TT), 57 ml acid acetic băng (TT), 61,5 g natri acetat (TT) và 5,0 g acid (cyclohexylendinitril) tetraacetic (TT) trong nước vừa đủ 500 ml. Điều chỉnh tới pH 5,0 đến 5,5 bằng dung dịch natri hydroxyd 30 % (TT), thêm nước cất vừa đủ 1000 ml.
Đệm hiệu chỉnh lực ion toàn phần (TT1)
Dung dịch A: Hòa tan 210 g acid citric (TT) trong 400 ml nước, chỉnh pH tới 7,0 bằng amoniac đặc (TT), thêm nước vừa đủ 1000 ml.
Dung dịch B: Hòa tan 132 g amoni phosphat (TT) trong nước vừa đủ 1000 ml.
Dung dịch C: Thêm khoảng 200 ml amoniac (TT) vào hỗn dịch của 292 g acid (ethylendinitril) tetraacetic (TT) trong khoảng 500 ml nước để hòa tan. Điều chỉnh pH tới 6 - 7 bằng amoniac đậm đặc (TT) và thêm nước vừa đủ 1000 ml.
Trộn đồng thể tích ba dung dịch A, B và C rồi điều chỉnh pH tới 7,5 bằng amoniac đậm đặc (TT).
Đệm phosphat chuẩn 0,025 M
Hòa tan 3,40 g kali dihydrophosphat (TT) và 3,55 g dinatri hydrophosphat khan (TT), cả hai đã được sấy khô ở 110 °C đến 130 °C trong 2 h, trong nước vừa đủ 1000 ml.
Đệm phosphat hỗn hợp pH 4,0
Hòa tan 5,04 g dinatri hydrophosphat (TT) và 3,01 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml. Điều chỉnh pH bằng acid acetic băng (TT).
Đệm phosphat hỗn hợp pH 5,4
Hòa tan 1,76 g dinatri hydrpphosphat (TT) và 13,61 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml. Điều chỉnh pH nếu cần bằng dung dịch acid phosphoric 0,05 M (TT).
Đệm phosphat hỗn hợp pH 6,8
Hòa tan 28,80 g dinatri hydrophosphat (TT) và 11,45 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml.
Đệm phosphat hỗn hợp 0,2 M pH 6,8 (Dung dịch đệm phosphat pH 6,8 số 1)
Trọn 51 ml dung dịch kali dihydrophosphat 2,72 % (TT) với 49 ml dung dịch dinatri hydrophosphat 7,16% (TT) và điều chỉnh pH nếu cần. Bảo quản dung dịch ở nhiệt độ từ 2 °C đến 8 °C.
Đệm phosphat hỗn hợp pH 7,0
Hòa tan 0,50 g dinatri hydrophosphat khan (TT) và 0,301 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml.
Đệm phosphat hỗn hợp 0,067 M pH 7,0 (Dung dịch đệm phosphat 0,067 M pH 7,0)
Trộn 38,9 ml dung dịch kali dihydrophosphat 0,908 % (TT) với 61,1 ml dung dịch dinatri hydrophosphat 2,38 % (TT) và điều chỉnh pH nếu cần.
Đệm phosphat hỗn hợp 0,1 M pH 7,0 (Dung dịch đệm phosphat 0,1 M pH 7,0)
Hòa tan 1,361 g kali dihydrophosphat (TT) trong nước vừa đủ 100 ml. Chỉnh pH bằng dung dịch dinatri hydrophosphat 3,5 % (TT).
Đệm phosphat hỗn hợp pH 10
Thêm 6 ml dung dịch natri phosphat 0,25 M (TT) vào 100 ml dung dịch dinatri hydrophosphat 0,2 M (TT).
Đệm phosphat pH 3,0
Hòa tan 34 g kali dihydrophosphat (TT) trong 250 ml nước. Điều chỉnh pH đến 3,0 bằng acid phosphoric (TT).
Đệm phosphat pH 3,5 (Dung dịch đệm phosphat pH 3,5)
Hòa tan 68 g kali dihydrophosphat (TT) trong 1000 ml nước. Điều chỉnh pH đến 3,5 bằng acid phosphoric (TT).
Đệm phosphat pH 4,0
Hòa tan 6,8 g kali dihydrophosphat (TT) trong 700 ml nước. Điều chỉnh pH nếu cần bằng dung dịch acid phosphoric 10 % (TT), thêm nước vừa đủ 1000 ml.
Đệm phosphat pH 4,75
Pha loãng 100 ml dung dịch kali dihydrophosphat 0,5 M (TT) thành 800 ml với nước. Điều chỉnh pH tới 4,75 bằng dung dịch natri hydroxyd 0,1 M (TT) và thêm nước vừa đủ 1000 ml.
Đệm phosphat pH 4,9
Hòa tan 40 g natri dihydrophosphat (TT) và 1,2 g natri hydroxyd (TT) trong nước vừa đủ 100 ml. Điều chỉnh pH nếu cần bằng dung dịch acid sulfuric 1 M (TT) hoặc dung dịch natri hydroxyd 1 M (TT).
Đệm phosphat pH 6,0 (Dung dịch đệm phosphat hỗn hợp pH 6,0)
Hòa tan 2,0 g dikali hydrophosphat (TT) và 8,0 g kali dihydrophosphat (TT) trong nước vừa đủ 1000 ml. Điều chỉnh pH đến 6,0 bằng acid phosphoric (TT) hoặc bằng dung dịch kali hydroxyd 30 % (TT).
Đệm phosphat 0,2 M pH 7,5 (Dung dịch đệm phosphat 0,2 M pH 7,5)
Hòa tan 27,22 g kali dihydrophosphat (TT) trong 930 ml nước. Chỉnh pH đến 7,5 bằng dung dịch kali hydroxyd 30 % (TT) và thêm nước vừa đủ 1000 ml.
(Các) Đệm phosphat chuẩn
Các đệm phospnat chuẩn pH từ 5,8 đến 8,0 được pha bằng cách trộn 50 ml dung dịch kali dihydrophosphat 0,2 M (TT) với lượng dung dịch natri hydroxyd 0,2 N (CĐ) như chỉ dẫn trong Bảng 2.3 và thêm nước vừa đủ 200 ml.
Ở 20 °C các dung dịch này có thể được sử dụng làm các dung dịch pH chuẩn, nhưng không được dùng để chuẩn hóa máy đo pH.
Bảng - 2.3
| pH | Số ml dung dịch natri hydroxyd 0,2 N |
| 5,8 | 3,72 |
| 6,0 | 5,70 |
| 6,2 | 8,60 |
| 6,4 | 12,60 |
| 6,6 | 17,80 |
| 6,8 | 23,65 |
| 7,0 | 29,63 |
| 7,2 | 35,00 |
| 7,4 | 39,50 |
| 7,6 | 42,80 |
| 7,8 | 45,20 |
| 8,0 | 46,80 |
Đệm phtalat pH 3,6 (Dung dịch đệm pH 3,6)
Thêm 11,94 ml dung dịch acid hydrocloric 0,2 N (CĐ) vào 250 ml dung dịch kali hydrophtalat 0,2 M (TT), thêm nước vừa đủ 1000 ml. Ở 20 °C, dung dịch thu được có thể được dùng như dung dịch pH chuẩn.
Đệm tris-clorid pH 7,4 [Dung dịch đệm tris(hydroxy-methyl)aminomethan -natri clorid pH 7,4]
Hòa tan 8,77g natri clorid (TT) và 6,08,g tris(hydroxy-methyl)methylamin (TT) trong 500 ml nước. Thêm 10 g albumin bò. Điều chỉnh pH tới 7,4 bằng acid hydrocloric (TT) và thêm nước vừa đủ 1000 ml.
Đệm tris-clorid pH 7,5 [Dung dịch đệm tris(hydroxy-methyl)aminomethan pH 7,5]
Hòa tan 5,27g natri clorid (TT) và 7,27 g tris(hydroxy-methyl)methylamin (TT) trong nước. Điều chỉnh pH nếu cần và thêm nước vừa đủ 1000 ml.
Đệm tris-clorid pH 7,5 số 1 (Dung dịch đệm tris-hydroclorid 0,05 M pH 7,5)
Hòa tan 6,057 g tris(hydroxymethyl)methylamin (TT) trong nước. Điều chỉnh pH bằng acid hydrocloric (TT) nếu cần và thêm nước vừa đủ 1000 ml.
Đệm tris-clorid pH 8,1 [Dung dịch đệm tris(hydroxy-methyl)aminomethan pH 8,1]
Hòa tan 0,294 g calci clorid (TT) và 0,969 g tris(hydroxymethyl)methylamin (TT) trong nước. Điều chỉnh pH tới 8,1 bằng dung dịch acid hydrocloric 1 M (TT) và thêm nước vừa đủ 100 ml.
Đệm tris-clorid pH 8,6
Hòa tan 2,4 g natri clorid (TT) và 2,0 g tris(hydroxy-methyl)methylamin (TT) trong khoảng 100 ml nước. Điều chỉnh pH tới 8,6 bằng dung dịch natri hydroxyd 1 M (TT) hoặc dung dịch acid hydrocloric 1 M (TT) và thêm nước vừa đủ 200 ml.
Đệm tris-EDTA pH 8,4 [Dung dịch đệm tris(hydroxy-methyl)aminomethan EDTA pH 8,4]
Hòa tan 5,12 g natri clorid (TT), 3,03 g tris(hydroxy-methyl)methylamin (TT) và 1,4 g dinatri edetat (TT) trong 250 ml nước. Điều chỉnh pH tới 8,4 bằng acid hydrocloric (TT) và thêm nước vừa đủ 500 ml.
2.4 CÁC DUNG DỊCH MẪU
Các dung dịch sau đây được sử dụng làm mẫu so sánh trong các phép thử giới hạn tạp chất:
Dung dịch acetaldehyd mẫu 100 phần triệu C2H4O trong isopropanol
Hòa tan 1,0 g acetaldehyd (TT) trong isopropanol (TT) vừa đủ 100 ml. Pha loãng 5,0 ml dung dịch thu được thành 500,0 ml bằng isopropanol (TT) ngay trước khi sử dụng.
Dung dịch acetaldehyd mẫu 100 phần triệu C2H4O trong nước
Hòa tan 1,0 g acetaldehyd (TT) trong nước vừa đủ 100 ml. Pha loãng 5,0 ml dung dịch thu được thành 500,0 ml bằng nước ngay trước khi sử dụng.
Dung dịch amoni mẫu 100 phần triệu NH4
Pha loãng 10,0 ml dung dịch amoni clorid 0,0741 % (TT) thành 25,0 ml bằng nước.
Dung dịch amoni mẫu 2,5 phần triệu NH4
Pha loãng 1 thể tích dung dịch amoni clorid 0,0741 % (TT) thành 100 ml bằng nước.
Dung dịch amoni mẫu 1 phần triệu NH4
Pha loãng 1 thể tích dung dịch amoni mẫu 2,5 phần triệu NH4 (TT) thành 2,5 thể tích với nước ngay trước khi sử dụng.
Dung dịch antimony mẫu 100 phần triệu Sb
Hòa tan 0,274 g kali antimony tartrat (TT) trong 500 ml dung dịch acid hydrocloric 1 M (TT). Pha loãng dung dịch thu được thành 1000 ml với nước.
Dung dịch antimony mẫu 1 phần triệu Sb
Hòa tan 0,274 g kali antimony tartrat (TT) trong 20 ml dung dịch acid hydrocloric 7 M (TT). Pha loãng dung dịch thu được thành 100 ml với nước. Thêm vào 10 ml dung dịch này 200 ml dung dịch acid hydrocloric 7 M (TT) và pha loãng thành 1000 ml với nước. Thêm vào 100 ml dung dịch này 300 ml dung dịch acid hydrocloric 7 M (TT) và pha loãng thành 1000 ml với nước. Chuẩn bị các dung dịch pha loãng ngay trước khi sử dụng.
Dung dịch arsen mẫu 1000 phần triệu As
Hòa tan 0,330 g arsen trioxyd (TT) trong 5 ml dung dịch natri hydroxyd 2 M (TT) và thêm nước vừa đủ 250 ml.
Dung dịch arsen mẫu 10 phần triệu As
Pha loãng 1 thể tích dung dịch arsen mẫu 1000 phần triệu As (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch arsen mẫu 1 phần triệu As
Pha loãng 1 thể tích dung dịch arsen mẫu 10 phần triệu As (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch arsen mẫu 0,1 phần triệu As
Pha loãng 1 thể tích dung dịch arsen mẫu 1 phần triệu As (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch bari mẫu 1000 phần triệu Ba
Hòa tan 1,778 g bari clorid (TT) trong nước vừa đủ 1000 ml.
Dung dịch bari mẫu 50 phần triệu Ba
Pha loãng 1 thể tích dung dịch bari clorid 0,178 % (TT) thành 20 thể tích với nước ngay trước khi sử dụng.
Dung dịch bạc mẫu 5 phần triệu Ag
Pha loãng 1 thể tích dung dịch bạc nitrat 0,079 % (TT) thành 100 thể tích với nước.
Dung dịch bismuth mẫu 100 phần triệu Bi
Hòa tan 0,500 g bismuth (TT) tương ứng với 50 ml acid nitric (TT) và thêm nước vừa đủ 500 ml. Pha loãng 1 thể tích dung dịch thu được thành 10 thể tích với dung dịch acid nitric loãng (TT) ngay trước khi sử dụng.
Dung dịch cadmi mẫu 1000 phần triệu Cd
Hòa tan một lượng cadmi (TT) chứa 0,100 g Cd trong một lượng tối thiểu hỗn hợp đồng thể tích của acid hydrocloric (TT) với nước và thêm dung dịch acid hydrocloric 1 % (TT) vừa đủ 100 ml.
Dung dịch cadmi mẫu 10 phần triệu Cd
Pha loãng 1 thể tích dung dịch cadmi mẫu 1000 phần triệu Cd (TT) thành 100 thể tích với dung dịch acid hydrocloric 1 % (TT) ngay trước khi sử dụng.
Dung dịch calci mẫu 1000 phần triệu Ca
Hòa tan 0,624 g calci carbonat (TT) đã sấy khô ở 100 °C đến 105 °C đến khối lượng không đổi trong nước có chứa 3 ml dung dịch acid acetic 5 M (TT) và thêm nước vừa đủ 250 ml.
Dung dịch calci mẫu 400 phần triệu Ca
Hòa tan 1 g calci carbonat (TT) trong 23 ml dung dịch acid hydrocloric 1 M (TT) và thêm nước vừa đủ 100 ml. Pha loãng 1 thể tích dung dịch thu được thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch calci mẫu 100 phần triệu Ca trong nước
Pha loãng 1 thể tích dung dịch calci mẫu 1000 phần triệu Ca (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch calci mẫu 100 phần triệu Ca trong nước số 1
Pha loãng 1 thể tích dung dịch calci clorid khan chứa 0,2769 % CaCl2 trong dung dịch acid hydrocloric 2 M (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch calci mẫu 100 phần triệu Ca trong ethanol 96 %
Pha loãng 1 thể tích dung dịch calci mẫu 1000 phần triệu Ca (TT) thành 10 thể tích với ethanol 96 % (TT) ngay trước khi sử dụng.
Dung dịch calci mẫu 10 phần triệu Ca
Pha loãng 1 thể tích dung dịch calci mẫu 1000 phần triệu Ca (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch chì mẫu 1000 phần triệu Pb
Hòa tan 0,400 g chì (II) nitrat (TT) trong nước vừa đủ 250 ml.
Dung dịch chỉ mẫu 1000 phần triệu Pb trong acid nitric không có chì
Hòa tan 0,400 g chì (II) nitrat (TT) trong dung dịch acid nitric loãng không có chì (TT) vừa đủ 250 ml.
Dung dịch chì mẫu 100 phần triệu Pb
Pha loãng 1 thể tích dung dịch chì mẫu 1000 phần triệu Pb (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch chì mẫu 20 phần triệu Pb
Hòa tan 0,80 g chì (II) nitrat (TT) trong nước có chứa 2 ml acid nitric (TT) và thêm nước vừa đủ 250 ml. Pha loãng 1 thể tích dung dịch thu được thành 100 thể tích với nước ngay trước khi sử dụng
Dung dịch chì mẫu 10 phần triệu Pb
Pha loãng 1 thể tích dung dịch chì mẫu 100 phần triệu Pb (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch chì mẫu 10 phần triệu Pb trong acid nitric không có chì
Pha loãng 1 thể tích dung dịch chì mẫu 1000 phần triệu Pb trong acid nitric không có chì (TT) thành 100 thể tích với acid nitric không có chì (TT). Sử dụng trong vòng một tuần.
Dung dịch chì mẫu 2 phần triệu Pb
Pha loãng 1 thể tích dung dịch chì mẫu 100 phần triệu Pb (TT) thành 50 thể tích với nước ngay trước khi sử dụng.
Dung dịch chì mẫu 1 phần triệu Pb
Pha loãng 1 thể tích dung dịch chì mẫu 100 phần triệu Pb (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch chì mẫu 0,5 phần triệu Pb trong acid nitric không có chì
Pha loãng 1 thể tích dung dịch chì mẫu 10 phần triệu Pb trong acid nitric không có chì (TT) thành 20 thể tích với acid nitric không có chì (TT). Sử dụng trong vòng một ngày.
Dung dịch chì mẫu 0,1 phần triệu Pb
Pha loãng 1 thể tích dung dịch chì mẫu 1 phần triệu Pb (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch clorid mẫu 500 phần triệu Cl
Hòa tan 0,0824 g natri clorid (TT) đã sấy khô ở 100 °C đến 105 °C đến khối lượng không đổi trong nước vừa đủ 100 ml.
Dung dịch clorid mẫu 50 phần triệu Cl
Pha loãng 1 thể tích dung dịch clorid mẫu 500 phần triệu Cl (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch clorid mẫu 8 phần triệu Cl
Pha loãng 1 thể tích dung dịch natri clorid 0,132 % (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch clorid mẫu 5 phần triệu Cl
Pha loãng 1 thể tích dung dịch clorid mẫu 500 phần triệu Cl (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch cobalt mẫu 100 phần triệu Co
Hòa tan 0,494 g cobalt nitrat (TT) trong 500 ml dung dịch acid nitric 1 M (TT) và pha loãng dung dịch trong thu được thành 1000 ml với nước.
Dung dịch crom mẫu 1000 phần triệu Cr
Hòa tan 2,82 g kali cromat (TT) đã sấy khô đến khối lượng không đổi trong nước vừa đủ 1000 ml.
Dung dịch crom mẫu 100 phần triệu Cr
Hòa tan 0,283 g kali cromat (TT) đã sấy khô đến khối lượng không đổi trong nước vừa đủ 1000 ml.
Dung dịch crom mẫu 0,1 phần triệu Cr
Pha loãng 1 thể tích dung dịch crom mẫu 100 phần triệu Cr (TT) thành 1000 thể tích với nước ngay trước khi sử dụng.
Dung dịch digitoxin mẫu
Hòa tan 0,1250 g digitoxin (TT) trong acid acetic băng (TT) vừa đủ 100 ml. Pha loãng 4 ml này thành 100 ml với acid acetic băng (TT). Thêm 3 ml nước vào 25 ml dung dịch thu được, lắc đều.
Dung dịch đồng mẫu 1000 phần triệu Cu
Dung dịch đồng chuẩn (0,1 % Cu)
Hòa tan 0,393 g đồng sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch đồng mẫu 10 phần triệu Cu
Pha loãng 1 thể tích dung dịch đồng mẫu 1000 phần triệu Cu (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch đồng mẫu 0,1 phần triệu Cu
Pha loãng 1 thể tích dung dịch đồng mẫu 10 phần triệu Cu (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch fluorid mẫu 10 phần triệu F
Hòa tan 0,0442 g natri fluorid (TT) đã sấy 12 h ở 300 °C trong nước vừa đủ 100 ml. Pha loãng 1 thể tích dung dịch thu được thành 20 thể tích với nước ngay trước khi sử dụng. Bảo quản dung dịch đặc trong đồ đựng bằng polyethylen.
Dung dịch fluorid mẫu 1 phần triệu F
Pha loãng 1 thể tích dung dịch fluorid mẫu 10 phần triệu F (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch formaldehyd mẫu 5 phần triệu CH2O
Pha loãng 1 thể tích dung dịch chứa 3 g formaldehyd (TT) trong 1000 ml thành 200 thể tích với nước ngay trước khi sử dụng.
Dung dịch germani mẫu 100 phần triệu Ge
Hòa tan một lượng amoni hexafluorogermanat (TT) tương ứng với 0,307 g (NH4)2GeF6 trong dung dịch acid hydrofluoric 0,01 % (TT) và pha loãng dung dịch trong thu được thành 1000 ml với nước.
Dung dịch glucose mẫu
Hòa tan 0,1 g glucose (TT) trong dung dịch acid benzoic bão hòa trong nước (TT) vừa đủ 100 ml. Pha loãng 2 ml dung dịch thu được thành 100 ml với nước.
Dung dịch glucose mẫu chứa 20 mg glucose trong 1 ml.
Dung dịch glyoxal mẫu 20 phần triệu C2H2O2
Pha loãng một lượng dung dịch glyoxal (TT) chứa 0,200 g C2H2O2 thành 100 ml với ethanol tuyệt đối (TT). Pha loãng 1 thể tích dung dịch thu được thành 100 thể tích với ethanol tuyệt đối (TT) ngay trước khi sử dụng.
Dung dịch glyoxal mẫu 2 phần triệu C2H2O2
Pha loãng 1 thể tích dung dịch glyoxal mẫu 20 phần triệu C2H2O2 (TT) thành 10 thể tích với ethanol (TT).
Dung dịch hydrogen peroxyd mẫu 10 phần triệu H2O2
Pha loãng 10 ml dung dịch hydrogen peroxyd loãng (TT) thành 300 ml với nước. Pha loãng 10 ml dung dịch thu được thành 1000 ml với nước ngay trước khi sử dụng.
Dung dịch iod mẫu 20 phần triệu I
Hòa tan 0,026 g kali iodid (TT) trong nước vừa đủ 100 ml. Pha loãng 10 ml dung dịch thu được thành 100 ml với nước.
Dung dịch iod mẫu 10 phần triệu I
Hòa tan 0,131 g kali iodid (TT) trong nước vừa đủ 100 ml. Pha loãng 1 ml dung dịch thu được thành 100 ml với nước ngay trước khi sử dụng.
Dung dịch kali mẫu 2000 phần triệu K
Hòa tan 0,446 g kali sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch kali mẫu 600 phần triệu K
Hòa tan 1,144 g kali clorid (TT) đã sấy 3 h ở 100 °C đến 105 °C trong nước vừa đủ 1000 ml.
Dung dịch kali mẫu 100 phần triệu K
Pha loãng 1 thể tích dung dịch kali mẫu 2000 phần triệu K (TT) thành 20 thể tích với nước ngay trước khi sử dụng.
Dung dịch kali mẫu 20 phần triệu K
Pha loãng 1 thể tích dung dịch kali mẫu 100 phần triệu K (TT) thành 5 thể tích với nước ngay trước khi sử dụng.
Dung dịch kẽm mẫu 5 mg/ml Zn
Hòa tan 3,15 g kẽm oxyd (TT) trong 15 ml acid hydrocloric (TT) và thêm nước vừa đủ 500 ml.
Dung dịch kẽm mẫu 100 phần triệu Zn
Hòa tan 0,440 g kẽm sulfat (TT) trong nước có chứa 1 ml dung dịch acid acetic 5 M (TT) và thêm nước vừa đủ 100 ml. Pha loãng 1 thể tích dung dịch thu được thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch kẽm mẫu 25 phần triệu Zn
Pha loãng 25 ml dung dịch kẽm mẫu 100 phần triệu Zn (TT) thành 100 ml với nước ngay trước khi sử dụng.
Dung dịch kẽm mẫu 10 phần triệu Zn
Pha loãng 1 thể tích dung dịch kẽm mẫu 100 phần triệu Zn (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch kẽm mẫu 5 phần triệu Zn
Pha loãng 1 thể tích dung dịch kẽm mẫu 100 phần triệu Zn (TT) thành 20 thể tích với nước ngay trước khi sử dụng.
Dung dịch lithi mẫu 100 phần triệu Li
Hòa tan 0,6109 g lithi clorid (TT) trong nước vừa đủ 1000 ml.
Dung dịch magnesi mẫu 1000 phần triệu Mg
Hòa tan 1,010 g magnesi sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch magnesi mẫu 100 phần triệu Mg
Pha loãng 1 thể tích dung dịch magnesi mẫu 100 phần triệu Mg (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch magnesi mẫu 10 phần triệu Mg
Pha loãng 1 thể tích dung dịch magnesi mẫu 100 phần triệu Mg (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch mangan mẫu 100 phần triệu Mn
Hòa tan 0,308 g mangan sulfat (TT) trong 500 ml dung dịch acid nitric 1 M (TT) và pha loãng dung dịch trong thu được thành 1000 ml với nước.
Dung dịch natri mẫu 200 phần triệu Na
Pha loãng 1 thể tích dung dịch natri clorid 0,509 % (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch natri mẫu 50 phần triệu Na
Pha loãng 1 thể tích dung dịch natri mẫu 200 phần triệu Na (TT) thành 4 thể tích với nước ngay trước khi sử dụng.
Dung dịch nhôm mẫu 200 phần triệu Al
Hòa tan 0,352 g phèn chua (TT) trong dung dịch acid sulfuric 0,1 M (TT) vừa đủ 100 ml.
Dung dịch nhôm mẫu 100 phần triệu Al
Pha loãng 1 thể tích dung dịch nhôm clorid 0,8947 % (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch nhôm mẫu 10 phần triệu Al
Pha loãng 1 thể tích dung dịch nhôm nitrat 1,39 % (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch nhôm mẫu 2 phần triệu Al
Pha loãng 1 thể tích dung dịch nhôm mẫu 200 phần triệu Al (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch nickel mẫu 1000 phần triệu Ni
Hòa tan 0,478 g nickel (II) sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch nickel mẫu 10 phần triệu Ni
Pha loãng 1 thể tích dung dịch nickel mẫu 1000 phần triệu Ni (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch nickel mẫu 0,2 phần triệu Ni
Pha loãng 1 thể tích dung dịch nickel mẫu 10 phần triệu Ni (TT) thành 50 thể tích với nước ngay trước khi sử dụng.
Dung dịch nickel mẫu 0,1 phần triệu Ni
Pha loãng 1 thể tích dung dịch nickel mẫu 10 phần triệu Ni (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch nitrat mẫu 1000 phần triệu NO3
Hòa tan 0,163 g kali nitrat (TT) trong nước vừa đủ 100 ml.
Dung dịch nitrat mẫu 100 phần triệu NO3
Pha loãng 1 thể tích dung dịch nitrat mẫu 1000 phần triệu NO3 (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch nitrat mẫu 10 phần triệu NO3
Pha loãng 1 thể tích dung dịch nitrat mẫu 100 phần triệu NO3 (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch nitrat mẫu 2 phần triệu NO3
Pha loãng 1 thể tích dung dịch nitrat mẫu 10 phần triệu NO3 (TT) thành 5 thể tích với nước ngay trước khi sử dụng.
Dung dịch nitrit mẫu 20 phần triệu NO2
Hòa tan 0,6 g natri nitrit (TT) trong nước vừa đủ 100 ml và pha loãng 1 ml dung dịch thu được thành 200 ml với nước.
Dung dịch paladi mẫu 500 phần triệu Pd
Hòa tan 50 mg paladi (TT) trong 9 ml acid hydrocloric (TT) và thêm nước vừa đủ 100 ml.
Dung dịch paladi mẫu 20 phần triệu Pd
Hòa tan 0,333 g paladi clorid (TT) trong 2 ml acid hydrocloric (TT) đã được làm ấm và thêm hỗn hợp đồng thể tích của dung dịch acid hydrocloric 2 M (TT) và nước vừa đủ 1000 ml. Pha loãng 1 thể tích dung dịch thu được thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch paladi mẫu 0,5 phần triệu Pd
Pha loãng 1 thể tích dung dịch paladi mẫu 500 phần triệu Pd (TT) thành 1000 thể tích với hỗn hợp của 0,3 thể tích acid nitric (TT) và 99,7 thể tích nước.
Dung dịch platin mẫu 30 phần triệu Pt
Ngay trước khi dùng, pha loãng 10 lần dung dịch chứa 80 mg acid cloroplatinic (IV) (TT) trong 100 ml dung dịch acid hydrocloric 1 M (TT) bằng dung dịch acid hydrocloric 1 M (TT)
Dung dịch phosphat mẫu 500 phần triệu PO4
Hòa tan 0,0716 g kali dihydrophosphat (TT) trong nước vừa đủ thành 100 ml.
Dung dịch phosphat mẫu 200 phần triệu PO4
Pha loãng 10 thể tích dung dịch phosphat mẫu 500 phần triệu PO4 (TT) thành 25 thể tích với nước ngay trước khi sử dụng.
Dung dịch phosphat mẫu 100 phần triệu PO4
Pha loãng 1 thể tích dung dịch phosphat mẫu 500 phần triệu PO4 (TT) thành 5 thể tích với nước ngay trước khi sử dụng.
Dung dịch phosphat mẫu 5 phần triệu PO4
Pha loãng 1 thể tích dung dịch phosphat mẫu 500 phần triệu PO4 (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch sắt mẫu 1000 phần triệu Fe
Hòa tan 0,10 g sắt (TT) trong một lượng tối thiểu dung dịch đồng thể tích của acid hydrocloric (TT) với nước và thêm nước vừa đủ 100 ml.
Dung dịch sắt mẫu 250 phần triệu Fe
Pha loãng 1 thể tích dung dịch sắt (III) clorid hexahydrat 4,84 % (TT) trong dung dịch acid hydro-cloric 15 % (TT) thành 40 thể tích với nước ngay trước khi sử dụng.
Dung dịch sắt mẫu 200 phần triệu Fe
Hòa tan 0,863 g sắt (III) amoni sulfat (TT) trong nước có chứa 25 ml dung dịch acid sulfuric 1 M (TT) và thêm nước vừa đủ 500 ml.
Dung dịch sắt mẫu 20 phần triệu Fe
Pha loãng 1 thể tích dung dịch sắt mẫu 200 phần triệu Fe (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch sắt mẫu 10 phần triệu Fe
Hòa tan 7,022 g sắt (II) amoni sulfat (TT) trong nước có chứa 25 ml dung dịch acid sulfuric 1 M (TT) và thêm nước vừa đủ 1000 ml. Pha loãng 1 thể tích dung dịch thu được thành 100 thể tích với nước.
Dung dịch sắt mẫu 8 phần triệu Fe
Pha loãng 1 thể tích dung dịch chứa 80 mg sắt (TT) và 50 ml dung dịch acid hydrocloric 220 g/l (TT) trong 1000 ml thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch sắt mẫu 2 phần triệu Fe
Pha loãng 1 thể tích dung dịch sắt mẫu 20 phần triệu Fe (TT) thành 10 thể tích với nước ngay trước khi sử dụng.
Dung dịch sắt mẫu 1 phần triệu Fe
Pha loãng 1 thể tích dung dịch sắt mẫu 20 phần triệu Fe (TT) thành 20 thể tích với nước ngay trước khi sử dụng.
Dung dịch selen mẫu 100 phần triệu Se
Hòa tan 0,100 g selen (TT) trong 2 ml acid nitric (TT), bốc hơi đến khô. Hòa tan cắn trong 2 ml nước và bốc hơi đến khô, tiến hành tương tự 3 lần. Hòa tan cắn trong dung dịch acid hydrocloric 2 M (TT) vừa đủ 1000 ml.
Dung dịch selen mẫu 1 phần triệu Se
Pha loãng 25 ml dung dịch acid selenious 0,00654 % (kl/tt) thành 1000 ml với nước ngay trước khi sử dụng.
Dung dịch stronti mẫu 1 phần trăm Sr
Thấm ướt 1,6849 g stronti carbonat (TT) với nước, thêm cẩn thận acid hydrocloric (TT) cho tới khi chất rắn tan hoàn toàn và không có bọt khí, thêm nước tới vừa đủ 100 ml.
Dung dịch sulfat mẫu 1000 phần triệu SO4
Hòa tan 0,181 g kali sulfat (TT) trong nước vừa đủ 100 ml.
Dung dịch sulfat mẫu 10 phần triệu SO4
Pha loãng 1 thể tích dung dịch sulfat mẫu 1000 phần triệu SO4 (TT) thành 100 thể tích với nước ngay trước khi sử dụng.
Dung dịch sulfat mẫu 1000 phần triệu SO4 trong ethanol
Hòa tan 0,181 g kali sulfat (TT) trong ethanol 30 % (TT) vừa đủ 100 ml.
Dung dịch sulfat mẫu 10 phần triệu SO4 trong ethanol
Pha loãng 1 thể tích dung dịch sulfat mẫu 1000 phần triệu SO4 trong ethanol (TT) thành 100 thể tích với ethanol 30 % (TT) ngay trước khi sử dụng.
Dung dịch sulfit mẫu 80 phần triệu SO2
Hòa tan 3,150 g natri sulfit khan (TT) trong nước mới cất vừa đủ 100 ml. Pha loãng 0,5 ml thành 100 ml với nước mới cất.
Dung dịch sulfit mẫu 1,5 phần triệu SO2
Pha loãng 5 ml dung dịch natri metabisulfit 0,152% (TT) thành 100 ml với nước. Thêm 4 ml dung dịch natri hydroxyd 0,1 M (CĐ) vào 3 ml dung dịch thu được và thêm nước vừa đủ 100 ml.
Dung dịch thali mẫu 10 phần triệu TI
Pha loãng 10 ml dung dịch natri thali (I) sulfat 0,01235 % (TT) trong dung dịch natri clorid 0,9% (TT) thành 100 ml với cùng dung môi.
Dung dịch thiếc mẫu 5 phần triệu Sn
Hòa tan 0,5 g thiếc (TT) trong hỗn hợp của 5 ml nước với 25 ml acid hydrocloric (TT) và thêm nước vừa đủ 1000 ml. Pha loãng 1 thể tích dung dịch thu được thành 100 thể tích với dung dịch acid hydrocloric 2,5% (TT) ngay trước khi sử dụng.
Dung dịch thiếc mẫu 0,1 phần triệu Sn
Pha loãng 1 thể tích dung dịch thiếc mẫu 5 phần triệu Sn (TT) thành 50 thể tích với nước ngay trước khi sử dụng.
Dung dịch titan mẫu 100 phần triệu Ti
Hòa tan 0,1 g titan (TT) trong 100 ml acid hydrocloric (TT), thêm nước vừa đủ 150 ml, đun nóng nếu cần. Làm nguội rồi thêm nước vừa đủ 1000 ml.
Dung dịch vanadi mẫu 1 g/l V
Hòa tan 0,230 g amoni vanadat (TT) trong nước vừa đủ 100 ml.
Dung dịch zirconi 1 g/l Zr
Hòa tan 0,293 g zirconyl nitrat (TT) trong trong hỗn hợp của 8 thể tích nước với 2 thể tích acid hydrocloric (TT) vừa đủ 100 ml.
2.5 CÁC CHẤT ĐỐI CHIẾU (ĐC)
Chất đối chiếu là chất đồng nhất đã được xác định là đúng để dùng trong các phép thử đã được quy định về hóa học, vật lý và sinh học. Trong các phép thử đó các tính chất của chất đối chiếu được so sánh với các tính chất của chất cần thử. Chất đối chiếu phải có độ tinh khiết phù hợp với mục đích sử dụng.
Chất đối chiếu được dùng trong các phép thử sau:
Định tính bằng phương pháp quang phổ hấp thụ hồng ngoại.
Định lượng bằng phương pháp quang phổ hấp thụ tử ngoại và khả kiến, quang phổ huỳnh quang.
Các phép thử định tính tạp chất và định lượng bằng phương pháp sắc ký.
Định lượng bằng phương pháp vi sinh vật.
Các phép chuẩn độ đo thể tích, phân tích khối lượng.
Các phép thử sinh học.
Một số phép thử khác có hướng dẫn trong các chuyên luận riêng.
Cách sử dụng chất đối chiếu
Để đáp ứng mục đích sử dụng, chất đối chiếu phải được bảo quản, theo dõi và sử dụng đúng. Theo quy định thông thường, chất đối chiếu phải được đựng trong bao bì gốc, kín, có nhãn rõ ràng, được thường xuyên bảo quản ở nhiệt độ thấp, tránh ánh sáng và ẩm; nếu cần có điều kiện bảo quản đặc biệt khác thì có hướng dẫn trên nhãn.
Trước khi mở bao gói để dùng, chất đối chiếu cần được để một thời gian để đạt tới nhiệt độ phòng thí nghiệm.
Các phép thử được tiến hành đồng thời trên mẫu thử và mẫu đối chiếu đã được chuẩn bị trong cùng điều kiện ghi trong chuyên luận.
Nếu trên nhãn của chất đối chiếu không có chỉ dẫn phải làm khô và nếu trong phép thử riêng của chuyên luận không chỉ định phải làm khô thì có thể sử dụng ngay chất đối chiếu mà không phải làm khô. Trong trường hợp này cần hiệu chỉnh lại lượng cân của chất đối chiếu do khối lượng bị giảm khi làm khô hoặc do hàm lượng nước. Mất khối lượng do làm khô hoặc hàm lượng nước được xác định theo hướng dẫn ở chuyên luận của chất thuốc tương ứng. Khi chất đối chiếu có nước kết tinh, có thể có hướng dẫn đặc biệt trong một phép thử riêng.
Nếu không thể thực hiện được việc xác định hàm lượng nước của chất đối chiếu bằng phương pháp chuẩn độ (phương pháp Karl Fischer) và nếu chuyên luận thuốc tương ứng không có phép thử mất khối lượng do làm khô, thì nên làm khô chất đối chiếu trên một chất hút ẩm thích hợp để chuyển lượng cân của chất đối chiếu thành chất khan, trừ khi có hướng dẫn khác.
Khi đọc thấy chữ "chuẩn” xuất hiện sau tên chất được ghi trong các phép thử hay phép định lượng, có nghĩa là phải sử dụng chất đối chiếu.
Bộ tiêu chuẩn quốc gia về thuốc này cho phép sử dụng các chất chuẩn Dược điển Việt Nam, các dược liệu chuẩn được thiết lập, bảo quản và phân phối tại Viện Kiểm nghiệm thuốc Trung ương (48 Hai Bà Trưng, Quận Hoàn Kiếm, TP. Hà Nội) hoặc Viện Kiểm nghiệm thuốc TP; Hồ Chí Minh (200 Cô Bắc, Quận 1, TP. Hồ Chí Minh) theo sự phân công của Bộ Y tế. Những chất đối chiếu quốc tế, khu vực hay quốc gia khác được sử dụng theo quy định hiện hành.
PHỤ LỤC 3
(Quy định)
3.1 CÂN VÀ XÁC ĐỊNH KHỐI LƯỢNG
Xác định khối lượng là bước thường xuyên trong quy trình phân tích. Cân là thiết bị phòng thí nghiệm cần thiết trong hầu hết các phép phân tích. Tuy nhiên, phép cân lại là nguồn phổ biến gây sai số khó phát hiện.
Quy trình mô tả dưới đây áp dụng trực tiếp trên các cân điện tử.
Quy trình xác định khối lượng có thể chia thành ba bước cơ bản: Chuẩn bị cân, kiểm tra cân và cân xác định khối lượng.
3.1.1. Chuẩn bị cân
Bước ban đầu là chọn, trang bị các dụng cụ thí nghiệm thích hợp, như các đồ đựng vật cân (giấy cân, phễu cân...), bình chứa, kẹp gắp, pipet, thìa có kích cỡ phù hợp...
Sử dụng đồ đựng có kích cỡ không vượt quá tải trọng của cân và phải đảm bảo sạch, khô.
Chuẩn bị các mẫu cân và các hóa chất cần thiết, nếu có yêu cầu, pha các dung dịch, thuốc thử.
Mẫu cân có thể yêu cầu nghiền mịn hoặc sấy khô. Một vài mẫu cân phải được làm nóng hoặc bảo quản trong thiết bị làm lạnh. Trước khi cân, mẫu cân phải đảm bảo được cân bằng nhiệt với nhiệt độ của cân. Để tránh sự ngưng tụ hơi ẩm, các mẫu cân làm lạnh phải được để tới nhiệt độ phòng trước khi mở nắp bình đựng.
3.1.2. Kiểm tra cân
Cân phải được kiểm tra trước khi tiến hành cân để loại các sai số có thể xảy ra. Người sử dụng cân cần kiểm tra môi trường cân, hiệu chuẩn cân và kiểm tra độ không đảm bảo đo của cân.
Môi trường cân
Cân phải được đặt ở vị trí thích hợp có mức rung động đủ thấp, không có luồng khí và có nguồn điện ổn định.
Cân và khu vực làm việc chung quanh phải được giữ gọn gàng và sạch sẽ. Nên dùng bàn chải lông mịn làm sạch đĩa cân trước khi cân.
Khi cân được di chuyển đến nơi khác, cần phải để cân ổn định tới nhiệt độ của môi trường mới và phải hiệu chuẩn lại cân.
Hiệu chuẩn cân
Mở nguồn và để cân ổn định trong ít nhất 1 h trước khi tiến hành hiệu chuẩn (các cân vi phân tích cần để ổn định 24 h để đạt sự cân bằng).
Các cân phân tích điện tử có hệ thống nội chuẩn dựa trên tải trọng xác định.
Sự hiệu chuẩn được tiến hành trong điều kiện nhiệt độ phòng.
Độ không đảm bảo đo của cân
Cách giảm độ trôi
Độ trôi là một trong những sai số thường gặp và cũng là yếu tố có thể làm giảm hoặc loại bỏ dễ dàng nhất.
Kiểm tra mẫu thử, cân phân tích và môi trường phòng thí nghiệm để loại bỏ các nguyên nhân sau đây có thể gây sai số:
Cửa buồng cân mở.
Nhiệt độ của cân và mẫu cân không đồng nhất.
Mẫu cân tăng hoặc giảm khối lượng.
Cân mới được di chuyển nhưng chưa được để cân bằng với môi trường hoặc chưa được hiệu chuẩn lại.
Có các luồng gió trong phòng thí nghiệm.
Nhiệt độ trong phòng thí nghiệm thay đổi.
Cân không đặt thật thăng bằng.
Các hoạt động trong phòng thí nghiệm gây rung động.
Ảnh hưởng của các bộ phận cơ học xảy ra trong suốt quá trình cân.
Hiện tượng trễ cơ học
Hiện tượng trễ của cân là do sự căng quá mức các lò xo, chủ yếu gây bởi việc cân quá tải hoặc bởi đánh rơi vật nào đó lên đĩa cân.
Quy trình đảm bảo chất lượng cho sự đo độ trôi của cân
Sau một khoảng thời gian vận hành cân, độ trôi của cân được kiểm tra bằng việc cân khối lượng quả cân chuẩn đã được nối chuẩn. Việc kiểm tra này nên thực hiện sau khi cân đã được hiệu chuẩn ở nhiệt độ phòng thí nghiệm, trước khi cân mẫu đầu tiên trong ngày hoặc sau một sự cố có thể làm sai lệch cân (mất nguồn điện, di chuyển cân đến vị trí khác...).
Quả cân chuẩn phải có khối lượng hằng định và không vượt quá mức trọng tải của cân. Mỗi cân nên được cung cấp một quả cân chuẩn, đựng trong hộp bảo vệ và đặt cạnh cân.
Cần thực hiện những quy trình sau đây để làm giảm các sai số cân và khả năng sai số do độ trôi:
Đảm bảo nối nguồn điện cho cân, kiểm tra bọt nước thăng bằng của cân.
Hiệu chuẩn cân phân tích hoặc cân vi phân tích.
Hàng ngày, người đầu tiên sử dụng cân phải kiểm tra cân bằng cách cân khối lượng quả cân chuẩn, ghi chép khối lượng này vào sổ theo dõi để so sánh với các lần cân trước. Nếu sự chênh lệch lớn hơn giới hạn cho phép của cân phân tích và cân vi phân tích, phải báo cáo để sửa chữa.
(Quả cân chuẩn phải được bảo quản, tránh sự tiếp xúc với những chất gây ô nhiễm trong không khí, có thể làm sạch bằng một miếng vải gạc được làm ẩm bằng một lượng nhỏ dung môi thích hợp như ether ethylic).
Cân phân tích
Chọn một quả cân chuẩn có khối lượng phù hợp để kiểm tra cân phân tích. Nếu có thể, cài đặt cân để đọc tới số thập phân thứ 5. Vận hành cân theo hướng dẫn của nhà sản xuất.
Dùng kẹp gắp quả cân chuẩn đặt cẩn thận lên đĩa cân, tránh để rơi xuống đĩa cân. Đặt quả cân vào chính giữa đĩa cân để loại trừ độ lệch góc. Nếu không có sự trôi, khối lượng của quả cân chuẩn sẽ hằng định.
Định kỳ sử dụng quả cân chuẩn cố định sẽ phát hiện được độ trôi của cân (nếu có). Việc kiểm tra độ trôi được thực hiện bằng cách cân ở những vị trí nhạy nhất. Mức dao động khối lượng thu được không được vượt quá ± 0,2 mg. Ví dụ: Với quả cân 20 g, nếu giá trị trung bình đọc được là 19,9984 g, khối lượng dao động từ 19,9982 đến 19,9986 g là chấp nhận được. Vì vậy phải đọc kết quả nhiều lần trước khi thiết lập dung sai.
Cân vi phân tích
Tiến trình như hướng dẫn cho cân phân tích nhưng dùng một quả cân chuẩn thích hợp cho loại cân này.
VÍ DỤ: Có thể dùng quả cân chuẩn 100 mg cho cân có mức trọng tải là 150 mg hoặc có thể dùng quả cân chuẩn 10 mg cho một cân siêu vi phân tích có mức trọng tải là 15 mg.
(Người sử dụng cân phải biết trọng tải tối đa của cân để chọn quả cân chuẩn thích hợp).
Cân biểu thị khối lượng bằng miligam. Ghi chép khối lượng ngay sau khi kết quả đọc ổn định một vài giây. Sự biến thiên khối lượng phải trong giới hạn phù hợp với tiêu chuẩn kỹ thuật của nhà sản xuất nhưng không được lớn hơn 0,1 % lượng mẫu cân.
VÍ DỤ: Nếu cân các mẫu 10 mg, sự biến thiên khối lượng của quả cân chuẩn phải không vượt quá 0,01 mg.
3.1.3. Cân xác định khối lượng mẫu
Trong bước cuối này, chọn phần thập phân của kết quả cân theo yêu cầu của quy trình phân tích. Phần lớn các phép phân tích trong dược phẩm sử dụng một lượng nhỏ mẫu thử, nên yêu cầu giá trị đọc trên cân cài đặt tới số thập phân thứ năm để đạt độ đúng cần thiết.
Số thập phân thứ tư thích hợp hơn với các khối lượng mẫu cân gần bằng gam.
Không cân mẫu quá lâu để tránh những thay đổi gây ra bởi sự tương tác với nước trong không khí và khí carbonic.
Giới hạn trọng tải
Chọn cân phù hợp với khối lượng phải cân và độ đúng cần thiết. Mỗi cân có một giới hạn trọng tải, không nên vượt quá. Mỗi nhà sản xuất cân đã xác định trọng tải tối đa của cân và giới hạn này thay đổi với từng loại cân. Người sử dụng nên biết giới hạn trọng tải để tránh làm hỏng cân.
Dụng cụ chứa mẫu
Những dụng cụ chứa mẫu cân thông thường là lọ cân, thuyền cân (phễu cân), giấy cân, bình nón... Dụng cụ chứa mẫu thích hợp phụ thuộc vào khối lượng và trạng thái mẫu cân (lỏng, rắn, bột...). Phải chọn dụng cụ chứa mẫu thích hợp cho từng mẫu cân. Khối lượng dụng cụ chứa cộng khối lượng mẫu cần cân phải không vượt quá trọng tải tối đa của cân. Kích cỡ, hình dáng của dụng cụ chứa phải phù hợp với đĩa cân và không gian buồng cân để không ảnh hưởng đến các thao tác cân. Quan trọng là dụng cụ chứa mẫu phải sạch và khô.
Cần sử dụng găng tay, kẹp gắp và các dụng cụ để lấy các dụng cụ chứa vì mồ hôi từ tay có thể ảnh hưởng tới khối lượng cân.
Thuyền cân thường được sử dụng nhiều nhất vì nó vừa là dụng cụ chứa vừa là phễu giúp chuyển dễ dàng mẫu cân vào bình. Thuyền cân có nhiều cỡ khác nhau, nên chọn cỡ phù hợp cho từng trường hợp.
Giấy cân thường sử dụng để cân chất rắn. Giấy cân được cầm bằng tay, phải cẩn thận để tránh làm rơi, đổ mẫu cân.
Các phương pháp cân
Những phương pháp sau đây thường được sử dụng và cho kết quả phân tích tốt.
Phương pháp 1
Cân trừ bì: Đặt dụng cụ chứa rỗng lên chính giữa đĩa cân và nhấn phím cân trừ bì để khối lượng của dụng cụ chứa không hiển thị nữa (giá trị 0). Cho mẫu cân vào dụng cụ chứa và ghi nhận khối lượng mẫu cân. Chuyển lượng mẫu cân vào bình định mức rồi cân lại dụng cụ chứa ban đầu. (Chú ý không thay đổi sự cài đặt cân trừ bì của cân giữa 2 lần cân này). Khối lượng đọc được ở lần cân thứ 2 chính là lượng mẫu thử không chuyển hết vào bình định mức (còn dính lại trên dụng cụ chứa mẫu cân) và được trừ vào khối lượng mẫu cân đọc được ở lần cân thứ nhất để xác định khối lượng mẫu thử đã được chuyển vào bình.
Phương pháp 2
Cân không trừ bì: Cho mẫu thử vào dụng cụ chứa và đặt lên chính giữa đĩa cân. Ghi nhận khối lượng cân và chuyển mẫu thử vào bình định mức, cân lại dụng cụ chứa ban đầu. Khối lượng đọc được ở lần cân thứ 2 là tổng khối lượng của dụng cụ chứa mẫu và lượng mẫu cân không được chuyển hết vào bình (còn dính lại trên dụng cụ chứa mẫu cân). Hiệu của khối lượng cân ban đầu và khối lượng cân lần 2 là khối lượng mẫu thử đã chuyển vào bình định mức.
Phương pháp 3
Phương pháp này được xem như là phương pháp chuyển toàn lượng. Mẫu cân được cho vào dụng cụ chứa đã cân bì, xác định khối lượng mẫu cân bởi sự chênh lệch giữa khối lượng cân đọc được và khối lượng cân bì. Toàn bộ lượng mẫu thử được chuyển hết vào bình bằng cách dùng một dung môi để hòa tan mẫu và tráng dụng cụ chứa mẫu.
Quy trình xử lý mẫu cân an toàn
Các thao tác an toàn khi cân: Người tiến hành cân cần đọc kỹ các thông báo về mẫu thử, các tài liệu an toàn về hóa chất, nguyên vật liệu... trước khi cân.
Những nguyên liệu, hóa chất nguy hiểm (chất độc, chất ăn mòn, chất dễ cháy, chất nổ, chất gây dị ứng, chất phóng xạ...) phải đặt ở nơi riêng biệt, kín, có hệ thống lọc khí thích hợp.
Sử dụng khẩu trang che mũi và miệng để ngăn ngừa việc hít các bụi hóa chất, cần dùng găng tay để tránh sự tiếp xúc với da và cũng để ngăn ngừa hơi ẩm, mồ hôi bám trên dụng cụ chứa khi cân mẫu. Trong suốt quá trình cân, người tiến hành cân phải tiếp xúc với các chất liệu nguyên chất ở nồng độ cao, do đó phải luôn luôn quan tâm đến biện pháp bảo đảm an toàn.
Quy trình cân được thực hiện với nhiều loại mẫu thử khác nhau như chất rắn, bột mịn, chất lỏng (nhớt hoặc không nhớt, bay hơi hoặc không bay hơi)..., nên mỗi loại mẫu thử yêu cầu những thao tác riêng biệt.
Cân chất rắn
Chất rắn có 2 dạng: Dạng khối lượng lớn có bột hay không có bột trên bề mặt và dạng bột mịn hay những tinh thể nhỏ.
Khi cân chất rắn có khối lượng lớn và có bột trên bề mặt, dùng giấy cân để bảo vệ cân, còn không có bột trên bề mặt, trơ, có thể đặt trực tiếp lên đĩa cân (ví dụ: viên bao).
Mẫu chất rắn phải lấy bằng kẹp gắp, không lấy bằng tay.
Hiện tượng tích điện:
Dạng bột mịn có xu hướng tích điện, làm các tiểu phân bay xung quanh. Hiện tượng này phải được loại bỏ trước khi cân. Có thể dùng thiết bị chống tích điện để làm giảm đến mức tối thiểu vấn đề này. Hiện tượng tích điện phụ thuộc vào độ ẩm tương đối của phòng thí nghiệm. Trong điều kiện nào đó, có thể sự tích điện được tạo ra bởi loại quần áo mà người thao tác cân đang mặc. Điện tích này khi phóng điện gây những sai số lớn về khối lượng.
Quy trình cân:
Đặt dụng cụ chứa mẫu lên đĩa cân, đóng cửa buồng cân và cân theo chỉ dẫn “Các phương pháp cân”. Cẩn thận thêm mẫu cân dạng bột bằng một thìa xúc vào dụng cụ chứa cho tới khi đạt lượng mong muốn. Thao tác cẩn thận để tránh rơi vãi ra ngoài. Đóng cửa buồng cân và ghi khối lượng mẫu cân ngay khi cân ổn định.
Sự rơi vãi:
Nếu chất rắn bị rơi vãi ra ngoài, lấy dụng cụ chứa ra, loại bỏ hoàn toàn các chất liệu rơi đổ trên cân, làm sạch bên trong, bên ngoài buồng cân, bàn cân, tránh để người sau tiếp xúc với các chất liệu rơi vãi.
(Chú ý: Không cho trở lại lọ chứa mẫu các mẫu cân dư).
Cân chất lỏng
Chất lỏng có thể bay hơi hoặc không bay hơi và nhớt hoặc không nhớt. Mỗi loại có những chú ý riêng biệt.
Cân theo hướng dẫn “Các phương pháp cân”, chú ý thêm các phần sau:
Chất lỏng luôn luôn được cân vào đồ đựng (chứa) có nắp để tránh sự bay hơi. Tốt nhất chất lỏng nên được cho vào bình cân ở bên ngoài cân để tránh đổ tràn trên cân.
(Chú ý: chất lỏng đổ tràn trong buồng cân có thể gây những hư hỏng nghiêm trọng cho cân và khó lau chùi sạch).
Chất lỏng không nhớt có thể được lấy bằng một pipet Pasteur có gắn một quả bóp cao su nhỏ như ống nhỏ giọt. Chất lỏng được rót vào bình, đậy nắp và đặt lên cân.
Một lượng nhỏ chất lỏng nhớt có thể được lấy bằng cách chạm một đũa thủy tinh tới bề mặt chất lỏng rồi cẩn thận chạm que này tới thành bình cân mẫu để một ít chất lỏng được chuyển qua.
Cân các chất liệu ăn mòn
Nhiều hóa chất có tính ăn mòn, như các muối. Các chất liệu có tính ăn mòn không nên để rơi đổ trên đĩa cân hoặc bên trong buồng cân. Cẩn thận khi cân các chất liệu này.
3.1.4. Kết luận
Thực hiện chặt chẽ theo các quy trình chỉ dẫn trên, nhân viên phòng thí nghiệm có thể loại bỏ nhiều sai số trong quá trình tiến hành cân. Tuy nhiên, phải có cán bộ kỹ thuật có chuyên môn bảo trì và hiệu chuẩn cân định kỳ. Phải dùng các quả cân chuẩn đã được liên kết với chuẩn quốc gia. Điều này là rất quan trọng.
3.2 NHIỆT KẾ
Các thiết bị đo nhiệt độ thích hợp cho các thử nghiệm trong Bộ tiêu chuẩn quốc gia về thuốc phải đạt các yêu cầu kỹ thuật trong hệ thống nhiệt kế chuẩn quốc gia.
Các thiết bị đo nhiệt độ có thể là nhiệt kế thủy tinh - chất lỏng, nhiệt kế hiện số hoặc nhiệt kế đồng hồ. Nhiệt kế hiện số hoặc nhiệt kế đồng hồ gồm một đầu dò nhiệt độ. Đầu dò được gắn với một đồng hồ đo có khả năng chuyển tín hiệu ôm hoặc milivôn sang đại lượng nhiệt độ. Phần đầu dò nhiệt độ được nhúng ngập trong môi trường cần đo được làm bằng chất liệu trơ.
Việc chuẩn hóa nhiệt kế được thực hiện định kỳ theo tần suất xác định nối với thang nhiệt độ chuẩn quốc gia. Cần chú ý điều kiện, thiết bị cụ thể được sử dụng để chọn thiết bị đo nhiệt độ phù hợp.
Các nhiệt kế thủy tinh - chất lỏng được chuẩn hóa theo 3 cách: Nhúng sâu toàn phần, nhúng từng phần hoặc nhúng sâu hoàn toàn. Vì vậy khi sử dụng các nhiệt kế này cần tuân thủ theo điều kiện đã được chuẩn hóa. Việc chuẩn hóa các nhiệt kế được thực hiện định kỳ theo tần suất xác định nối với thang nhiệt độ chuẩn quốc gia.
Việc chuẩn hóa nhiệt kế thủy tinh - chất lỏng loại nhúng sâu toàn phần được thực hiện bằng cách nhúng sâu nhiệt kế vào môi trường đo tới phần trên của cột chất lỏng, phần còn lại của thân nhiệt kế ở nhiệt độ phòng.
Việc chuẩn hóa nhiệt kế thủy tinh - chất lỏng loại nhúng từng phần được thực hiện bằng cách nhúng nhiệt kế ở một độ sâu cố định, phần thân nhiệt kế còn lại ở nhiệt độ phòng. Loại nhiệt kế này được khắc vạch quy định độ sâu được nhúng trong môi trường đo nhiệt độ.
Việc chuẩn hóa nhiệt kế thủy tinh - chất lỏng loại nhúng sâu hoàn toàn được thực hiện bằng cách cho toàn bộ nhiệt kế vào môi trường cần đo nhiệt độ.
Nếu cần sử dụng trong những điều kiện khác với điều kiện chuẩn hóa thì cần phải hiệu chỉnh để có giá trị đo đúng.
3.3 DỤNG CỤ ĐO THỂ TÍCH
Hầu hết các dụng cụ đo thể tích dùng trong phân tích được chuẩn hóa dung tích ở 20 °C, mặc dù nhiệt độ phòng thí nghiệm và nhiệt độ quy định cho các phép thử và định lượng thường là 25 °C. Sự khác nhau này không quan trọng, miễn là nhiệt độ phòng thí nghiệm tương đối hằng định. Khi cần tiến hành thử nghiệm đúng ở 20 °C thì yêu cầu này được ghi trong chuyên luận riêng.
Để giảm thiểu sai số phép đo thể tích, dụng cụ đo thể tích, nguyên vật liệu và dung môi dùng pha chế dung dịch, khu vực pha chế và việc điều chỉnh thể tích cuối cùng phải được duy trì ở cùng một nhiệt độ.
Để đạt được độ chính xác yêu cầu trong nhiều phương pháp định lượng có các phép đo thể tích chính xác, cần lựa chọn và dùng các dụng cụ đo thể tích thích hợp. Chọn kích cỡ của buret phù hợp sao cho thể tích dung dịch chuẩn độ không ít hơn 30 % thể tích danh định của buret. Muốn đo thể tích một dung dịch chuẩn độ dưới 10 ml thì phải dùng buret 10 ml hoặc micro buret.
Dung sai thể tích đối với các loại bình định mức, các pipet chính xác có bầu, pipet chia độ và các buret là dung sai đã được Tổ chức Tiêu chuẩn hóa quốc tế (ISO) thừa nhận và được ghi trong các bảng kèm theo dưới đây. Sử dụng dụng cụ đo thể tích loại A trừ khi có chỉ dẫn trong chuyên luận riêng.
Khi sử dụng các pipet chính xác có bầu và pipet chia độ được chuẩn hóa theo kiểu đổ ra “TD” (to deliver) phải giữ pipet thẳng đứng và đầu pipet phải chạm vào thành bình hứng rồi cho chất lỏng chảy ra.
Các số đọc thể tích trên buret phải được lượng giá tới 0,01 ml đối với buret 25 ml và 50 ml, và tới 0,005 ml đối với buret 5 ml và 10 ml.
Trong các trường hợp đặc biệt như đo thể tích chất lỏng nhớt như sirô, dùng pipet được chuẩn hóa theo kiểu đổ vào “TC” (to contain) hoặc bình định mức. Sau khi đã đổ chất lỏng ra, phải rửa sạch thành trong của dụng cụ và gộp nước rửa vào phần chất lỏng đã lấy.
Bình định mức
| Dung tích (ml) | Sai số cho phép (ml) |
| 5 | ± 0,025 |
| 10 | ± 0,025 |
| 25 | ± 0,04 |
| 50 | ± 0,06 |
| 100 | ± 0,10 |
| 200 | ± 0,15 |
| 250 | ± 0,15 |
| 500 | ± 0,25 |
| 1000 | ± 0,40 |
| 2000 | ± 0,60 |
Pipet chính xác có bầu (kiểu đổ ra)
| Dung tích (ml) | Sai số cho phép (ml) |
| 0,5 | ± 0,005 |
| 1 | ± 0,007 |
| 2 | ± 0,01 |
| 5 | ± 0,015 |
| 10 | ± 0,02 |
| 20 | ± 0,03 |
| 25 | ± 0,03 |
| 50 | ± 0,05 |
| 100 | ± 0,08 |
| 200 | ± 0,10 |
Pipet chia độ
| Dung tích (ml) | Sai số cho phép (ml) |
| 1 | ± 0,006 |
| 2 | ± 0,01 |
| 5 | ± 0,03 |
| 10 | ± 0,05 |
| 25 | ± 0,1 |
Buret
| Thể tích xác định (ml) | Phân độ (ml) | Sai số cho phép (ml) |
| 10 (micro buret) | 0,02 | ± 0,02 |
| 25 | 0,10 | ± 0,03 |
| 50 | 0,10 | ± 0,05 |
3.4 PHỄU LỌC THỦY TINH XỐP
Bảng 3.4.1 - Đường kính lỗ xốp của phễu lọc thủy tinh xốp
| Đường kính lỗ xốp (µm) | Cách dùng chính |
| Nhỏ hơn 2,5 | Lọc vi khuẩn |
| 4 đến 10 | Lọc cực mịn, phân tách các vi sinh vật có đường kính lớn. |
| 10 đến 40 | Lọc dùng trong phân tích, lọc thủy ngân rất mịn, phân tán rất nhỏ các khí. |
| 40 đến 100 | Lọc mịn, lọc thủy ngân, phân tán nhỏ các khí. |
| 100 đến 160 | Lọc các chất thô, phân tán và rửa khí, làm giá lọc cho các vật liệu lọc khác. |
| 160 đến 500 | Lọc các chất rất thô, phân tán và rửa khí. |
Bảng 3.4.2 - Bảng so sánh độ xốp của các phễu lọc xốp
| Số độ xốp | Đường kính tối đa của các lỗ xốp (µm) | Đức | Pháp | Anh |
| 1,6 | dưới 1,6 | 5f | - | - |
| - | 1 đến 2,5 | 5 | - | 5 |
| 4 | 1,6 đến 4 | - | - | - |
| - | 4 đến 6 | - | 5 | - |
| 10 | 4 đến 10 | 4f | - | 4 |
| 16 | 10 đến 16 | 4 | 4 | - |
| 40 | 16 đến 40 | 3 | 3 | 3 |
| - | 40 đến 50 | - | - | 2 |
| 100 | 40 đến 100 | 2 | 2 | - |
| - | 100 đến 120 | - | - | 1 |
| 160 | 100 đến 160 | 1 | 1 | - |
| - | 150 đến 200 | 0 | 0 | - |
| 250 | 160 đến 250 | - | - | - |
| - | 200 đến 500 | - | 00 | - |
(Những giới hạn chỉ dẫn là gần đúng)
3.5 CỠ BỘT VÀ RÂY
Bột
Các cỡ bột được quy định dựa vào các số của rây. Trừ khi có chỉ dẫn khác, khi quy định dùng một rây để xác định cỡ bột thì không được có dưới 97 % khối lượng thuốc bột qua được cỡ rây đó. Khi quy định dùng hai rây để xác định cỡ bột thì để một rây lên trên rây kia và tiến hành rây; không được có dưới 95 % khối lượng thuốc bột qua rây có số rây cao hơn và không được quá 40% khối lượng thuốc bột qua rây có số rây thấp hơn.
Người ta dùng những ký hiệu sau đây để quy định các cỡ bột:
Bột thô (1400/355) là bột mà không ít hơn 95 % phần tử qua được rây số 1400 và không quá 40 % qua được rây số 355.
Bột nửa thô (710/250) là bột mà không ít hơn 95 % phần tử qua được rây số 710 và không quá 40 % qua được rây số 250.
Bột nửa mịn (355/180) là bột mà không ít hơn 95 % phần tử qua được rây số 355 và không quá 40 % qua được rây số 180.
Bột mịn (180/125) là bột mà không ít hơn 95 % phần tử qua được rây số 180 và không quá 40 % qua được rây số 125.
Bột rất mịn (125/ 90) là bột mà không ít hơn 95 % phần tử qua được rây số 125 và không quá 40 % qua được rây số 90.
Rây
Lưới rây có thể dệt bằng sợi kim loại hoặc sợi các vật liệu khác thích hợp và dệt thành những mắt vuông. Lưới của rây dùng để rây bột thuốc được phân loại bằng những con số, chúng biểu thị kích thước lỗ rây quy định tính bằng µm. Vật liệu để làm lưới rây không được tạo ra một phản ứng nào với những bột đem rây. Khi rây, tránh kéo dài thời gian vì sẽ làm tăng độ mịn của bột. Khi không dùng vào mục đích phân tích, có thể dùng rây có mắt tròn, có đường kính trong bằng 1,25 lần chiều rộng mắt vuông của rây có cỡ tương ứng.
Đối với bột thô hoặc nửa thô thì lấy 25 g tới 100 g bột để thử. Cho vào rây thích hợp, lắc rây theo chiều ngang quay tròn ít nhất 20 min và rây tới khi xong. Cân đúng số lượng còn lại ở trên rây và số thu được trong hộp hứng.
Đối với bột nửa mịn, mịn hay rất mịn thì tiến hành như bột thô, nhưng mẫu bột lấy để thử không quá 25 g và lắc rây ít nhất 30 min rồi rây tới khi xong.
Trường hợp phải rây những chất có dầu hay những bột khác có xu hướng bít mắt rây thì trong quá trình rây thỉnh thoảng chải cẩn thận mắt rây, tách rời những đống tụ lại khi rây.
Bảng 3.5 - Cỡ rây (kim loại)
| Số rây* | Cỡ mắt rây (mm) | Đường kính sợi dây (mm) |
| 2000 | 2,000 | 0,900 |
| 1400 | 1,400 | 0,710 |
| 710 | 0,710 | 0,450 |
| 500 | 0,500 | 0,315 |
| 355 | 0,355 | 0,224 |
| 250 | 0,250 | 0,160 |
| 180 | 0,180 | 0,125 |
| 150 | 0,150 | 0,100 |
| 125 | 0,125 | 0,090 |
| 90 | 0,090 | 0,063 |
| 75 | 0,075 | 0,050 |
| 45 | 0,045 | 0,032 |
* Số rây biểu thị kích thước đo bằng µm của mắt rây.
Những quy định mắt rây bằng sợi kim loại chủ yếu được lựa chọn trong tiêu chuẩn ISO 565 -1975.
GHI CHÚ: Đối với dược liệu có khi cần dùng các cỡ rây to hơn so với bảng cỡ rây trên, trong trường hợp này chỉ cần nêu rõ kích thước của cỡ mắt rây.
3.6 RỬA DỤNG CỤ THỦY TINH
Độ sạch của dụng cụ thủy tinh đem dùng có ảnh hưởng rất lớn đến kết quả của một phép thử hoặc một phép định lượng. Các dụng cụ thủy tinh như cốc vại có mỏ, buret, pipet, bình nón, bình cầu v.v... đều phải thật sạch, đặc biệt là khi được dùng để định lượng bằng phương pháp vi sinh vật, thử chất gây sốt, hoặc khi dùng để lấy một thể tích nhỏ chất lỏng hay dung dịch.
Một trong những chất làm sạch dụng cụ thủy tinh tốt nhất là acid nitric đun nóng. Hỗn hợp acid cromic cũng là tác nhân làm sạch rất tốt dùng để loại sạch chất hữu cơ khỏi bề mặt thủy tinh mà không phải đun nóng. Hỗn hợp này được pha chế bằng cách hòa tan 200 g natri dicromat (TT) hoặc kali dicromat (TT) vào khoảng 100 ml nước, làm lạnh trong nước đá rồi thêm từ từ 1500 ml acid sulfuric (TT), vừa thêm vừa khuấy. Việc pha chế phải được thực hiện trong cốc vại bằng thủy tinh boro-silicat và cần đeo kính bảo hộ khi thêm acid. Hỗn hợp acid cromic là chất rất ăn mòn và hút nước, vì thế, phải được bảo quản trong những bình thủy tinh có nút mài và để ở nơi an toàn. Khi để yên, tinh thể acid cromic có thể được tạo thành, tách ra khỏi hỗn hợp, khi đó cần gạn để loại đi. Nếu hỗn hợp acid cromic có màu xanh thì không dùng nữa.
Dụng cụ thủy tinh được xử lý bằng hỗn hợp acid cromic cần phải rửa bằng nước rất nhiều lần để tránh acid cromic bị hút bám lên bề mặt. Không dùng hỗn hợp này để làm sạch những bình đựng dùng cho các phép đo quang học.
Mặc dù hỗn hợp acid cromic là chất làm sạch rất tốt, nhưng nó cũng được khuyến cáo không sử dụng ở nhiều phòng thí nghiệm vì acid cromic có trong danh sách chất thải độc hại nguy hiểm của cơ quan bảo vệ môi trường Mỹ (EPA).
Để rửa sạch dụng cụ thủy tinh, người ta còn có thể dùng những dung dịch tẩy rửa tổng hợp hoặc những hóa chất tẩy có tính kiềm như trinatri phosphat; dùng những chất này cũng có yêu cầu phải rửa nhiều lần bằng nước.
Tất cả dụng cụ thủy tinh cuối cùng đều được tráng bằng nước cất và làm khô trước khi dùng.
PHỤ LỤC 4
(Quy định)
4.1 PHƯƠNG PHÁP QUANG PHỔ HẤP THỤ TỬ NGOẠI VÀ KHẢ KIẾN
Phương pháp quang phổ hấp thụ tử ngoại và khả kiến còn được gọi là phương pháp quang phổ hấp thụ điện tử, là một trong các phương pháp phân tích dựa trên sự hấp thụ bức xạ điện từ.
Xác định độ hấp thụ
Độ hấp thụ A của một dung dịch là logarit thập phân của nghịch đảo độ truyền quang T khi cho ánh sáng đơn sắc đi qua. Nó được biểu thị bằng phương trình:
![]()
trong đó:
I: cường độ ánh sáng đơn sắc sau khi đã truyền qua dung dịch;
l0: cường độ ánh sáng đơn sắc tới;
T: độ truyền quang.
Ngoại trừ sự có mặt của các yếu tố lý - hóa học khác, độ hấp thụ A tỷ lệ với độ dài quang trình d của ánh sáng truyền qua dung dịch (bề dày lớp dung dịch) và nồng độ c của dung dịch chất khảo sát. Sự phụ thuộc này được biểu thị bằng phương trình:
A = ɛ × c × d
trong đó:
ɛ: độ hấp thụ mol;
d: được biểu thị bằng cm;
c: biểu thị bằng mol/lít.
Độ hấp thụ của dung dịch chất tan ở nồng độ 1% (kl/tt) hay 10 g/l trong một cốc đo có chiều dày 1 cm và đo ở một bước sóng xác định là độ hấp thụ riêng của chất tan và được ký hiệu là A (1 %, 1 cm). Độ hấp thụ riêng của chất tan được tính bằng công thức:
![]()
trong đó:
M: khối lượng phân tử của chất thử.
Độ hấp thụ riêng của một chất trong một dung môi xác định và đo ở một bước sóng xác định là một đặc tính của chất đó. Trừ những chỉ dẫn khác trong chuyên luận riêng, người ta đo độ hấp thụ ở độ dài sóng quy định và sử dụng quang trình dài 1 cm ở 19 °C đến 21 °C.
Ngoại trừ các chỉ dẫn trong chuyên luận riêng, các phép đo được tiến hành so sánh với dung môi hoặc với hỗn hợp dung môi đã dùng để chuẩn bị mẫu thử. Độ hấp thụ của dung môi hoặc hỗn hợp dung môi đo đối chiếu với không khí không được vượt quá 0,4 và tốt nhất là dưới 0,2. Khi vẽ phổ hấp thụ, đặt độ hấp thụ hoặc hàm số của độ hấp thụ trên trục tung và đặt độ dài sóng hoặc hàm số của độ dài sóng trên trục hoành.
Khi một chuyên luận riêng đưa ra một trị số riêng lẻ cho vị trí của một cực đại, điều này được hiểu là trị số khảo sát được có thể lệch không quá ± 2 nm.
Thiết bị
Máy quang phổ thích hợp dùng cho việc đo phổ vùng tử ngoại và khả kiến bao gồm một hệ thống quang học có khả năng cung cấp ánh sáng đơn sắc trong dải từ 200 nm đến 800 nm và một thiết bị phù hợp để đo độ hấp thụ. Hai cóng đo dùng cho dung dịch thử và dung dịch đối chiếu cần phải có đặc tính quang học như nhau. Khi đo trên máy tự ghi hai chùm tia, cốc đựng dung dịch đối chiếu được đặt ở bên có chùm tia đối chiếu đi qua.
Kiểm tra độ dài sóng
Kiểm tra thang độ dài sóng bằng cách sử dụng cực đại hấp thụ của dung dịch holmi perclorat. Vạch phổ của đèn nguồn hydro hoặc đèn deuterium và các vạch phổ của đèn hơi thủy ngân được chỉ ra ở dưới đây:
Giới hạn sai lệch cho phép là ± 1 nm ở vùng tử ngoại và ± 3 nm ở vùng khả kiến.
| 241,15 nm (Ho) | 404,66 nm (Hg) |
| 253,70 nm (Hg) | 435,83 nm (Hg) |
| 287,15 nm (Ho) | 486,00 nm (Dβ) |
| 302,25 nm (Hg) | 486,10 nm (Hβ) |
| 313,16 nm (Hg) | 536,30 nm (Ho) |
| 334,15 nm (Hg) | 546,07 nm (Hg) |
| 361,50 nm (Ho) | 576,96 nm (Hg) |
| 365,48 nm (Hg) | 579,07 nm (Hg) |
Kiểm tra độ hấp thụ
Kiểm tra độ hấp thụ của dung dịch kali dicromat ở các độ dài sóng chỉ ra ở Bảng 4.1.1. Trong bảng mỗi độ dài sóng có một giá trị chính xác của độ hấp thụ riêng A (1%, 1 cm) và các giới hạn cho phép. Dung sai độ hấp thụ là 0,01.
Để kiểm tra độ hấp thụ ở bước sóng 235 nm, 257 nm, 313 nm và 350 nm, dùng một dung dịch kali dicromat được chuẩn bị theo cách sau: Hòa tan 57,0 mg đến 63,0 mg kali dicromat đã được sấy ở 130 °C đến khối lượng không đổi trong một lượng dung dịch acid sulfuric 0,005 M để tạo thành vừa đủ 1000 ml dung dịch. Để kiểm tra độ hấp thụ ở bước sóng 430 nm, hòa tan 57,0 mg đến 63,0 mg kali dicromat đã được sấy ở 130 °C đến khối lượng không đổi trong một lượng dung dịch acid sulfuric 0,005 M để tạo thành vừa đủ 100 ml dung dịch.
Bảng 4.1.1
| Độ dài sóng (nm) | A (1%, 1 cm) | Giới hạn cho phép |
| 235 | 124,5 | 122,9 - 126,2 |
| 257 | 144,5 | 142,8 - 146,2 |
| 313 | 48,6 | 47,0 - 50,3 |
| 350 | 107,3 | 105,6 - 109,0 |
| 430 | 15,9 | 15,7 - 16,1 |
Giới hạn ánh sáng lạc
Ánh sáng lạc có thể được phát hiện ở độ dài sóng đã cho bằng kính lọc thích hợp, hoặc dung dịch hóa chất thích hợp, ví dụ độ hấp thụ của dung dịch kali clorid 1,2 % (TT) trong một cốc đo có quang trình dài 1 cm phải tăng rõ rệt giữa bước sóng 220 nm và bước sóng 200 nm và phải lớn hơn 2,0 ở độ dài sóng 198 nm khi so sánh với nước cất.
Độ phân giải (cho phân tích định tính)
Khi có quy định trong chuyên luận, tiến hành kiểm tra độ phân giải của máy như sau: Ghi phổ của dung dịch toluen 0,02 % (tt/tt) trong hexan (TT). Giá trị tối thiểu của tỷ số giữa độ hấp thụ ở cực đại 269 nm và độ hấp thụ ở cực tiểu 266 nm được quy định trong chuyên luận riêng.
Độ rộng khe phổ (cho phân tích định lượng)
Để tránh sai số gây ra bởi độ rộng khe phổ, khi sử dụng một máy có độ rộng khe phổ thay đổi ở độ dài sóng đã chọn thì độ rộng khe phổ phải nhỏ so với nửa độ rộng của băng hấp thụ. Nhưng độ rộng này phải đủ lớn để thu được giá trị cao của cường độ ánh sáng tới l0 và việc thu hẹp độ rộng khe phổ sau đó phải không làm cho độ hấp thụ đọc được tăng lên.
Cốc đo (cóng đo)
Dung sai về độ dài quang trình của cốc đo là ± 0,005 cm. Khi đã được nạp đầy cùng một dung môi, các cốc đo có ý định sử dụng để chứa dung dịch mẫu thử và để chứa dung dịch mẫu trắng phải có độ truyền quang như nhau. Nếu tiêu chuẩn này không được đáp ứng thì cần có sự hiệu chỉnh thích hợp. Các cốc đo cần được làm sạch và thao tác thận trọng.
Phương pháp phổ đạo hàm
Phương pháp phổ đạo hàm là sự chuyển dạng của phổ hấp thụ sang phổ đạo hàm bậc một, bậc hai hoặc bậc cao hơn. Phổ đạo hàm bậc một là đồ thị của gradient đường cong hấp thụ (tốc độ của sự thay đổi độ hấp thụ theo độ dài sóng, dA/dλ) theo độ dài sóng. Phổ đạo hàm bậc hai là đồ thị của độ cong của phổ hấp thụ (d2A/ dλ2) theo độ dài sóng. Nếu độ hấp thụ tuân theo định luật Lambert Beer, đạo hàm bậc hai tại bất kỳ độ dài sóng λ nào cũng liên quan tới nồng độ bởi phương trình sau:
![]()
trong đó:
A là độ hấp thụ ở bước sóng λ;
A (1 %, 1cm) là độ hấp thụ riêng ở bước sóng λ;
c là nồng độ của chất hấp thụ biểu thị dưới dạng % (kl/tt);
d là bề dày của lớp chất hấp thụ tính bằng cm.
Thiết bị
Là một máy quang phổ đáp ứng các yêu cầu về kiểm tra độ dài sóng, kiểm tra độ hấp thụ đã nói ở trên và được ghép nối thêm một modul vi phân trở - tụ cho tín hiệu tương tự, hoặc một vi phân kế kỹ thuật số, hoặc một thiết bị khác thích hợp, tạo ra được phổ đạo hàm bậc hai. Máy được sử dụng theo sự hướng dẫn của hãng sản xuất.
Một vài phương pháp tạo phổ đạo hàm bậc hai đưa đến sự trôi sóng so với vị trí của độ dài sóng ở phổ bậc không, điều này cần được tính đến khi cần thiết. Trừ khi có những chỉ dẫn khác trong chuyên luận riêng, độ rộng khe phổ của máy phải được điều chỉnh như chỉ dẫn trong mục độ rộng khe phổ ở trên. Các cóng đo phải đáp ứng các yêu cầu quy định trong mục Cóng đo.
Nhiệt độ của tất cả các dung dịch dùng trong phép thử không được khác nhau quá 0,5 °C.
Độ phân giải
Khi được quy định trong chuyên luận riêng, ta ghi phổ đạo hàm bậc hai của dung dịch toluen 0,020 % (tt/tt) trong methanol (TT) trong dải độ dài sóng từ 255 nm đến 275 nm, dùng methanol (TT) trong cóng đối chiếu. Phổ phải thấy rõ một cực trị âm nhỏ ở giữa hai cực trị âm lớn trong khoảng 261 nm và 268 nm như Hình 4.1.1.
Trừ khi có chỉ dẫn khác trong chuyên luận riêng, tỷ lệ A/B (xem Hình 4.1.1) không được nhỏ hơn 0,2.
Hình 4.1.1
4.2 PHƯƠNG PHÁP QUANG PHỔ HỒNG NGOẠI
Máy quang phổ
Máy quang phổ hồng ngoại dùng để ghi phổ trong vùng từ 4000 cm-1 đến 650 cm-1 (từ 2,5 µm đến 15,4 µm) hoặc trong một vài trường hợp tới 200 cm-1 (50 µm). Máy quang phổ chuyển đổi Fourier sử dụng bức xạ đa sắc và tính toán phổ theo dải tần số từ các dữ liệu gốc bằng chuyển đổi Fourier. Các máy quang phổ với hệ thống quang học tạo bức xạ đơn sắc trong vùng đo cũng có thể được sử dụng. Thông thường phổ được xem là hàm số của độ truyền quang T; T là tỷ số của cường độ bức xạ truyền qua và cường độ bức xạ tới.
Độ hấp thụ ánh sáng (A) là logarit thập phân của nghịch đảo độ truyền quang.
![]()
T là l/lo;
lo là cường độ ánh sáng tới;
I là cường độ ánh sáng truyền qua.
Chuẩn bị mẫu
Để ghi phổ truyền quang hay phổ hấp thụ, chất thử được chuẩn bị theo một trong các phương pháp sau:
Chất lỏng
Đo phổ của một chất lỏng dưới dạng phim giữa 2 tấm phẳng trong suốt đối với bức xạ hồng ngoại hay trong cốc đo có bề dầy thích hợp và cũng trong suốt đối với bức xạ hồng ngoại.
Các chất lỏng hay chất rắn chuẩn bị dưới dạng dung dịch
Chuẩn bị dung dịch thử với dung môi thích hợp. Chọn nồng độ và bề dày của cốc đo thích hợp để có thể ghi được một phổ tốt. Nói chung, có thể thu được kết quả tốt với nồng độ từ 1,0 % đến 10 % và bề dầy của cốc đo từ 0,5 mm đến 0,1 mm.
Các chất rắn
Đo phổ của chất rắn được phân tán trong một chất lỏng (bột nhão) hay trong một chất rắn thích hợp (đĩa halid). Nếu có quy định trong chuyên luận riêng, làm một phim mỏng từ khối chất rắn nóng chảy giữa hai tấm phẳng trong suốt với bức xạ hồng ngoại.
Bột nhão
Nghiền một lượng nhỏ chất thử với một lượng tối thiểu parafin lỏng hoặc chất lỏng khác phù hợp. Dùng từ 5 mg đến 10 mg chất thử là vừa đủ để tạo một bột nhão phù hợp. Ép bột nhão giữa hai tấm phẳng trong suốt với bức xạ hồng ngoại.
Viên nén (đĩa halid)
Trừ khi có chỉ dẫn khác, nghiền 1 mg đến 2 mg chất thử với 300 mg đến 400 mg bột mịn kali bromid (IR) hoặc kali clorid (IR) đã sấy khô. Lượng này thường đủ để tạo một viên nén có đường kính 10 mm đến 15 mm và cho phổ có cường độ phù hợp. Nghiền hỗn hợp cẩn thận và rải đều nó trong một khuôn thích hợp. Nén khuôn có hỗn hợp chất thử tới áp suất khoảng 800 MPa trong điều kiện chân không. Một vài yếu tố có thể tạo nên viên nén không tốt như: Nghiền không kỹ hay nghiền quá kỹ, độ ẩm, các tạp chất trong môi trường phân tán. Viên nén không đạt yêu cầu nếu kiểm tra bằng mắt thấy viên nén không đồng nhất và không trong suốt hay độ truyền quang ở khoảng 2000 cm-1 (5 µm) nhỏ hơn 60 % khi không có băng hấp thụ đặc hiệu ở vùng này và không có bù trừ bên tia đối chiếu, trừ khi có chỉ dẫn khác.
Các chất khí
Ghi phổ các khí trong cốc đo trong suốt với bức xạ hồng ngoại và có quang trình 50 mm đến 100 mm. Đối với các chất khí có hệ số hấp thụ hồng ngoại nhỏ hoặc có ở dạng vết, ta dùng các cốc đo có hệ thống gương, tạo nên sự phản xạ bên trong cốc nhiều lần và do đó có thể đạt quang trình 1 mét hoặc thậm chí đến 10 mét hoặc hơn nữa.
Tạo chân không trong cốc đo và nạp đầy khí đến áp suất yêu cầu nhờ một nút xoay hoặc một van kim với đường chuyển khí thích hợp nối cốc đo với bình chứa khí cần khảo sát. Nếu cần điều chỉnh áp lực trong cốc đo đến áp suất khí quyển, dùng một khí trong suốt với bức xạ hồng ngoại, ví dụ khí nitrogen hay argon. Để tránh ảnh hưởng sự hấp thụ của hơi nước, carbon dioxyd hoặc những khí khác có mặt trong bầu khí quyển, đặt một cốc đo tương tự như cốc đo đựng khí cần khảo sát bên tia đối chiếu. Cốc đo này đã được hút chân không hoặc được nạp khí trong suốt với bức xạ hồng ngoại.
Ghi phổ bằng phản xạ toàn phần suy giảm
Đặt mẫu thử tiếp xúc trực tiếp với lăng kính phản xạ toàn phần suy giảm.
Ghi phổ bằng phản xạ nhiều lần
Khi có chỉ định trong chuyên luận riêng, chuẩn bị chất thử theo các phương pháp sau:
Dung dịch
Hòa tan chất thử trong dung môi thích hợp theo những điều kiện đã được quy định trong chuyên luận riêng. Làm bay hơi dung dịch trên tấm thali bromo-iodid hoặc tấm phẳng khác phù hợp.
Chất rắn
Đặt chất thử trên tấm thali bromo-iodid hoặc trên tấm phẳng khác phù hợp sao cho có sự tiếp xúc đồng đều.
Định tính
Định tính bằng các chất đối chiếu hóa học
Chuẩn bị chất thử và chất đối chiếu theo cùng một quy trình và ghi phổ từ 4000 cm-1 đến 650 cm-1 (2,5 µm đến 15,4 µm) trong những điều kiện như nhau. Cực tiểu độ truyền qua (cực đại hấp thụ) trong phổ của chất thử và chất đối chiếu phải tương ứng về vị trí và cường độ.
Khi phổ của chất thử và của chất đối chiếu ở dạng rắn có sự khác nhau về vị trí của cực tiểu độ truyền qua (cực đại hấp thụ), phải xử lý chất thử và chất đối chiếu theo cùng một cách sao cho chúng kết tinh hay tạo thành cùng dạng, hay tiến hành như mô tả trong chuyên luận riêng rồi mới ghi phổ.
Định tính bằng phổ đối chiếu
Kiểm tra độ phân giải của máy
Ghi phổ của phim polystyren có bề dày 35 µm. Hiệu số x (Hình 4.2) giữa phần trăm truyền quang tại cực đại truyền qua A ở 2870 cm-1 (3,48 µm) và tại cực tiểu truyền qua B ở 2851 cm-1 (3,51 µm) phải lớn hơn 18. Hiệu số y giữa phần trăm truyền quang ở cực đại truyền qua c tại 1589 cm-1 (6,29 µm) và ở cực tiểu độ truyền qua D tại 1583 cm-1 (6,32 µm) phải lớn hơn 10.
Hình 4.2 - Phổ của polystyren dùng để kiểm tra độ phân giải
Kiểm tra thang số sóng
Có thể kiểm tra thang số sóng bằng cách dùng phim polystyren. Phim này có các cực tiểu truyền qua (cực đại hấp thụ) ở các số sóng (tính bằng cm-1) như Bảng 4.2.
Bảng 4.2 - Cực tiểu độ truyền qua của phim polystyren và các giới hạn cho phép
| 3060,0 (± 1,5) cm-1 2849,5 (± 2,0) cm-1 1942,9 (± 1,5) cm-1 1601,2 (± 1,0) cm-1 1583,0 (± 1,0) cm-1 1154,5 (± 1,0) cm-1 1028,3 (± 1,0) cm-1 |
Phương pháp:
Chuẩn bị chất thử theo chỉ dẫn đi kèm với phổ đối chiếu. Tiến hành trong điều kiện giống như khi kiểm tra độ phân giải, ghi phổ của chất thử và chồng phổ lên các băng của polystyren ở 2849,5 cm-1 (3,51 µm), 1601,2 cm-1 (6,25 µm) và 1028,3 cm-1 (9,72 µm). So sánh phổ chất thử với phổ đối chiếu và với các băng của polystyren như chỉ dẫn trên. Dùng các vị trí của băng polystyren để làm chuẩn, vị trí của các băng có giá trị trong phổ của chất thử và phổ đối chiếu phải tương ứng trong khoảng 0,5 % thang số sóng. Cường độ tương đối của các băng phải phù hợp giữa 2 phổ.
Phương pháp quang phổ cận hồng ngoại
Phương pháp quang phổ cận hồng ngoại là kỹ thuật đặc biệt hữu ích để định tính các chất hữu cơ. Mặc dù các phổ chỉ giới hạn trong các cộng hưởng của C - H, N - H, O - H và S - H, thường chúng cho các thông tin có giá trị. Tuy vậy, các phổ phụ thuộc vào một số thông số như kích thước hạt, tính đa hình, vết dung môi, độ ẩm... mà những thông số này không thể luôn được kiểm soát. Vì lý do này, không thể so sánh trực tiếp phổ của chất thử với phổ đối chiếu mà cần xử lý dữ liệu bằng phương pháp toán học đã được thẩm định.
4.3 PHƯƠNG PHÁP QUANG PHỔ HUỲNH QUANG
Phương pháp quang phổ huỳnh quang là phương pháp phân tích dựa trên phép đo cường độ huỳnh quang phát ra từ một chất hóa học khi nó được kích thích do hấp thụ bức xạ tử ngoại, khả kiến hoặc các bức xạ điện từ khác. Trong phương pháp này, cường độ huỳnh quang của chất thử được so sánh với cường độ huỳnh quang của chất chuẩn đo trong cùng điều kiện.
Máy
Vận hành máy quang phổ huỳnh quang theo chỉ dẫn của hãng sản xuất máy.
Phương pháp tiến hành
Chuẩn bị dung dịch như mô tả trong chuyên luận, chuyển dung dịch vào cốc đo của máy quang phổ huỳnh quang và chiếu vào nó bức xạ kích thích với bước sóng đơn sắc đã được ghi trong chuyên luận. Đo cường độ huỳnh quang của dung dịch ở bước sóng phát huỳnh quang được ghi trong chuyên luận.
Để định lượng, đưa vào máy cốc đựng dung môi hoặc hỗn hợp dung môi dùng để hòa tan mẫu thử và điều chỉnh máy về điểm "0". Đưa dung dịch chuẩn đã được chuẩn bị như mô tả trong chuyên luận vào máy và điều chỉnh độ nhạy của máy để kết quả đọc trên thang đo lớn hơn 50. Nếu thay đổi khe sáng khi điều chỉnh lần thứ hai thì phải đặt lại điểm 0 và cường độ huỳnh quang của chuẩn cũng phải được đo lại. Cuối cùng, đưa mẫu thử vào máy, đo cường độ huỳnh quang và tính nồng độ của dung dịch thử theo công thức sau:
Cx = (lx × Cs) / ls
trong đó:
Cs và Cx lần lượt là nồng độ của dung dịch chuẩn và dung dịch thử;
ls và lx lần lượt là cường độ huỳnh quang của dung dịch chuẩn và dung dịch thử.
Khi cường độ huỳnh quang không thật sự tỷ lệ thuận với nồng độ, việc định lượng có thể được thực hiện dựa trên đồ thị chuẩn.
Trong một số trường hợp, phép định lượng có thể được so sánh với một mẫu chuẩn đối chiếu có bản chất hóa học khác mẫu thử (Ví dụ: thủy tinh phát huỳnh quang hoặc dung dịch một chất phát huỳnh quang khác với mẫu thử). Trong trường hợp này, nồng độ của chất thử phải được xác định bằng đồ thị chuẩn đo trong cùng điều kiện với mẫu thử.
4.4 PHƯƠNG PHÁP QUANG PHỔ NGUYÊN TỬ PHÁT XẠ VÀ HẤP THỤ
Các phương pháp quang phổ nguyên tử phát xạ và hấp thụ được sử dụng để xác định nồng độ của các nguyên tố bằng phép đo cường độ phát xạ hoặc hấp thụ ánh sáng ở bước sóng đặc trưng bởi hơi nguyên tử của nguyên tố được hóa hơi từ chất cần phân tích, ví dụ, bằng cách đưa một dung dịch của chất phân tích vào ngọn lửa.
Phương pháp quang phổ phát xạ nguyên tử
Phương pháp quang phổ phát xạ nguyên tử là phương pháp xác định nồng độ của một nguyên tố trong một chất bằng cách đo cường độ của một trong các vạch phát xạ của hơi nguyên tử của nguyên tố được hóa hơi từ chất đó. Việc xác định nồng độ nguyên tố được tiến hành ở bước sóng tương ứng với các vạch phát xạ này.
Thiết bị
Máy quang phổ phát xạ nguyên tử bao gồm các bộ phận chủ yếu là bộ phận hóa hơi nguyên tử của các nguyên tố cần xác định (ngọn lửa, plasma, hồ quang điện v.v...), bộ đơn sắc hóa và bộ phận phát hiện. Nếu bộ phận hóa hơi là ngọn lửa, nước là dung môi được lựa chọn để pha dung dịch thử và dung dịch chuẩn. Tuy nhiên, dung môi hữu cơ cũng được sử dụng nếu đảm bảo chắc chắn rằng nó không ảnh hưởng đến độ ổn định của ngọn lửa.
Phương pháp tiến hành
Vận hành máy quang phổ phát xạ nguyên tử theo như chỉ dẫn của hãng sản xuất về cách đặt bước sóng quy định. Đưa dung dịch mẫu trắng vào bộ phận hóa hơi nguyên tử và hiệu chỉnh tín hiệu đọc được về "0". Đưa dung dịch chuẩn đặc nhất vào buồng hóa hơi nguyên tử và hiệu chỉnh độ nhạy của máy để có trị giá đọc thích hợp.
Việc định lượng được thực hiện bằng cách so sánh các dung dịch thử chưa biết nồng độ với các dung dịch chuẩn của nguyên tố cần xác định bằng phương pháp xác định trực tiếp (phương pháp 1) hoặc bằng phương pháp thêm chuẩn (phương pháp 2).
Sử dụng phương pháp 1, trừ khi có chỉ dẫn khác.
Phương pháp 1: Phương pháp xác định trực tiếp
Chuẩn bị dung dịch chất để thử (dung dịch thử) như được mô tả trong chuyên luận riêng sao cho nồng độ của nguyên tố cần xác định nằm trong khoảng nồng độ của các dung dịch chuẩn. Chuẩn bị không ít hơn ba dung dịch chuẩn của nguyên tố cần xác định có nồng độ nằm trong khoảng phụ thuộc tuyến tính giữa nồng độ và cường độ vạch phát xạ của nguyên tố cần phân tích. Bất kỳ thuốc thử nào được dùng trong việc chuẩn bị các dung dịch thử đều phải được thêm vào các dung dịch chuẩn và dung dịch mẫu trắng ở cùng nồng độ. Đưa dung dịch mẫu trắng vào máy, điều chỉnh tín hiệu đọc được về "0". Đưa lần lượt dung dịch thử và từng dung dịch chuẩn vào máy, ít nhất mỗi dung dịch làm ba lần, ghi lại kết quả đọc ổn định. Rửa máy bằng dung dịch mẫu trắng sau mỗi lần đo cho đến khi tín hiệu trở về giá trị đọc ban đầu của mẫu trắng. Từ cường độ vạch phát xạ đọc được của các dung dịch chuẩn, thiết lập đường chuẩn biểu thị sự thay đổi cường độ vạch phát xạ theo nồng độ nguyên tố cần xác định và từ đường chuẩn này xác định nồng độ nguyên tố trong dung dịch thử.
Phương pháp 2: Phương pháp thêm chuẩn
Cho vào ít nhất ba bình định mức có dung tích như nhau, các thể tích bằng nhau của dung dịch chất đem thử (dung dịch thử) được chuẩn bị như chỉ dẫn trong chuyên luận. Thêm vào tất cả các bình, trừ một bình, các thể tích lớn dần của dung dịch chuẩn có nồng độ đã biết của nguyên tố cần xác định sao cho nồng độ của nguyên tố này trong các bình đều nằm trong khoảng tuyến tính giữa nồng độ và cường độ phát xạ. Pha loãng các dung dịch có chứa trong bình đến vừa đủ thể tích bằng dung môi. Đưa dung dịch mẫu trắng (dung môi) vào máy, điều chỉnh tín hiệu đọc được về "0". Đưa lần lượt từng dung dịch thử và từng dung dịch chuẩn vào máy, ít nhất mỗi dung dịch làm ba lần, ghi lại kết quả đọc ổn định. Rửa máy bằng dung dịch mẫu trắng sau mỗi lần đo cho đến khi tín hiệu trở về giá trị đọc ban đầu của mẫu trắng. Tính phương trình hồi quy của đường chuẩn bằng cách dùng phương pháp bình phương tối thiểu và từ đường thẳng này, xác định nồng độ của nguyên tố cần định lượng trong dung dịch thử. Một cách khác, vẽ đồ thị biểu diễn sự liên quan giữa giá trị đọc được và lượng thêm vào của nguyên tố cần xác định. Nối các điểm trên đồ thị và kéo dài về phía trái cho đến khi nó gặp trục nồng độ. Khoảng cách giữa điểm này và điểm giao nhau của trục tọa độ là nồng độ của nguyên tố cần xác định trong dung dịch thử.
Phương pháp quang phổ hấp thụ nguyên tử
Phương pháp quang phổ hấp thụ nguyên tử là phương pháp xác định nồng độ của nguyên tố trong một chất bằng cách đo độ hấp thụ bức xạ bởi hơi nguyên tử tự do của nguyên tố đó được hóa hơi từ chất thử. Việc định lượng được tiến hành ở bước sóng của một trong những vạch hấp thụ của nguyên tố cần xác định.
Thiết bị
Máy quang phổ hấp thụ nguyên tử chủ yếu bao gồm nguồn bức xạ, bộ phận hóa hơi nguyên tử của nguyên tố cần xác định (ngọn lửa, lò graphit v.v...), bộ đơn sắc hóa và bộ phận phát hiện.
Phương pháp đưa các chất vào để phân tích phụ thuộc vào kỹ thuật nguyên tử hóa mẫu. Nếu nguyên tử hóa bằng ngọn lửa, các chất sẽ được hóa hơi và nước là dung môi được chọn để chuẩn bị các dung dịch chuẩn và thử, tuy nhiên các dung môi hữu cơ cũng có thể được sử dụng nếu đảm bảo chắc chắn là dung môi không ảnh hưởng đến độ ổn định của ngọn lửa. Nếu nguyên tử hóa không ngọn lửa (sử dụng lò graphit), các chất đưa vào có thể được hòa tan trong nước hoặc trong dung môi hữu cơ. Quá trình hóa hơi nguyên tử cũng có thể được tạo ra bên ngoài máy quang phổ, ví dụ, phương pháp hóa hơi lạnh cho thủy ngân hoặc một số hydrid. Đối với thủy ngân, nguyên tử được hóa hơi bằng sự khử hóa học và hơi nguyên tử được dẫn bằng một dòng khí trơ đến một cốc đo đặt trên trục quang của máy. Các hydrid được dẫn bằng hỗn hợp khí đốt hoặc bằng khí trơ đến một cốc đo đã được đốt nóng, tại đây chúng được nguyên tử hóa.
Phương pháp tiến hành
Vận hành máy quang phổ hấp thụ nguyên tử theo các chỉ dẫn của hãng sản xuất máy về cách đặt bước sóng quy định. Đưa dung dịch mẫu trắng vào buồng hóa hơi nguyên tử và hiệu chỉnh kết quả sao cho độ hấp thụ đọc được trên máy bằng "0". Đưa dung dịch chuẩn có nồng độ lớn nhất vào máy và hiệu chỉnh độ nhạy để đạt được một phép đọc độ hấp thụ thích hợp.
Việc định lượng được thực hiện bằng cách so sánh với các dung dịch chuẩn đã biết nồng độ của nguyên tố cần xác định bằng phương pháp xác định trực tiếp (phương pháp 1) hoặc bằng phương pháp thêm chuẩn (phương pháp 2).
Sử dụng phương pháp 1 trừ khi có chỉ dẫn khác.
Phương pháp 1: Phương pháp xác định trực tiếp
Chuẩn bị dung dịch chất để thử (dung dịch thử) như được mô tả trong chuyên luận riêng sao cho nồng độ của nguyên tố cần xác định nằm trong khoảng nồng độ của các dung dịch chuẩn. Chuẩn bị không ít hơn ba dung dịch chuẩn của nguyên tố cần xác định có nồng độ nằm trong khoảng tuyến tính giữa nồng độ và độ hấp thụ của nguyên tố cần phân tích. Bất kỳ thuốc thử nào được dùng trong việc chuẩn bị các dung dịch thử đều phải được thêm vào các dung dịch chuẩn và dung dịch mẫu trắng ở cùng nồng độ. Đưa dung dịch mẫu trắng vào máy, điều chỉnh tín hiệu đọc được về "0". Đưa lần lượt dung dịch thử và từng dung dịch chuẩn vào máy, ít nhất mỗi dung dịch làm ba lần, ghi lại kết quả đọc ổn định. Rửa máy bằng dung dịch mẫu trắng sau mỗi lần đo cho đến khi tín hiệu trở về giá trị đọc ban đầu của mẫu trắng. Nếu sử dụng kỹ thuật nguyên tử hóa không ngọn lửa, lò graphit phải được đốt lại giữa các lần phân tích. Từ cường độ vạch hấp thụ đọc được của các dung dịch chuẩn, thiết lập đường chuẩn biểu thị sự thay đổi cường độ vạch hấp thụ theo nồng độ nguyên tố cần xác định và từ đường chuẩn này xác định nồng độ nguyên tố trong dung dịch thử.
Phương pháp 2: Phương pháp thêm chuẩn
Cho vào ít nhất ba bình định mức có dung tích như nhau, các thể tích bằng nhau của dung dịch chất đem thử (dung dịch thử) được chuẩn bị như chỉ dẫn trong chuyên luận. Thêm vào tất cả các bình, trừ một bình, các thể tích lớn dần của dung dịch chuẩn có nồng độ đã biết của nguyên tố cần xác định sao cho nồng độ của nguyên tố cần xác định trong các bình đều nằm trong khoảng tuyến tính giữa nồng độ và cường độ hấp thụ. Pha loãng các dung dịch có chứa trong bình đến vừa đủ thể tích bằng dung môi.
Đưa dung dịch mẫu trắng vào máy, điều chỉnh tín hiệu đọc được về "0". Đưa lần lượt dung dịch thử và từng dung dịch chuẩn vào máy, ít nhất mỗi dung dịch ba lần, ghi lại kết quả đọc ổn định. Rửa máy bằng dung dịch mẫu trắng sau mỗi lần đo cho đến khi tín hiệu trở về giá trị đọc ban đầu của mẫu trắng. Nếu sử dụng kỹ thuật nguyên tử hóa không ngọn lửa, lò graphit phải được đốt lại giữa các lần phân tích.
Tính phương trình hồi quy của đường chuẩn bằng cách dùng phương pháp bình phương tối thiểu và từ đường thẳng này, xác định nồng độ của nguyên tố cần định lượng trong dung dịch thử. Một cách khác, vẽ đồ thị biểu diễn sự liên quan giữa giá trị đọc được và lượng thêm vào của nguyên tố cần xác định. Nối các điểm trên đồ thị và kéo dài về phía trái cho đến khi nó gặp trục nồng độ. Khoảng cách giữa điểm này và điểm giao nhau của trục tọa độ là nồng độ của nguyên tố cần xác định trong dung dịch thử.
PHỤ LỤC 5 CÁC KỸ THUẬT TÁCH SẮC KÝ
Các kỹ thuật tách sắc ký là những phương pháp tách trong đó các thành phần của mẫu được phân bố vào hai pha: một pha tĩnh và một pha động. Pha tĩnh có thể là một chất rắn, cũng có thể là một chất lỏng được giữ trên một chất rắn hay một gel. Pha tĩnh có thể được nhồi vào cột, hoặc trải thành một lớp, hay phân tán thành một lớp phim v.v... Pha động có thể là chất khí, chất lỏng hay chất lưu siêu tới hạn (còn được gọi là chất lỏng siêu tới hạn). Sự tách sắc ký có thể dựa trên các cơ chế khác nhau như hấp phụ, phân bố khối lượng (hay phân chia), trao đổi ion, hoặc dựa trên sự khác nhau về tính chất lý hóa của các phân tử như kích thước, khối lượng, thể tích v.v...
Phụ lục này chỉ đề cập đến các định nghĩa và phép tính các thông số thông thường trong kỹ thuật sắc ký, các yêu cầu thường được áp dụng cho sự phù hợp của hệ thống sắc ký. Nguyên lý, thiết bị và phương pháp tách được trình bày trong các phép thử chung sau đây:
• Phương pháp sắc ký giấy (Phụ lục 5.1)
• Phương pháp sắc ký khí (Phụ lục 5.2)
• Phương pháp sắc ký lỏng (Phụ lục 5.3)
• Phương pháp sắc ký lớp mỏng (Phụ lục 5.4)
• Phương pháp sắc ký rây phân tử (Phụ lục 5.5)
Định nghĩa
Các định nghĩa sau đây được dùng trong các chuyên luận để tính toán các thông số về sự phù hợp của hệ thống và các tiêu chuẩn chấp nhận. Một số thiết bị có phần mềm của nhà sản xuất dùng để tính toán một vài thông số như tỷ số tín hiệu trên nhiễu và độ phân giải có thể được sử dụng. Trong trường hợp này, người sử dụng có trách nhiệm đảm bảo rằng các phương pháp dùng trong phần mềm đó phù hợp với yêu cầu của bộ tiêu chuẩn này; nếu không, phải điều chỉnh cho phù hợp.
Sắc đồ
Sắc đồ là một đồ thị hay một cách trình bày khác mô tả sự thay đổi đáp ứng của detector (hay nồng độ của chất hay một đại lượng khác làm thước đo nồng độ của chất) theo thời gian, thể tích hay khoảng cách. Một sắc đồ lý tưởng có dạng một chuỗi các pic kiểu Gauss trên một đường nền (Hình 5.1).
Pic
Pic, hay còn gọi là đỉnh, là phần sắc đồ ghi lại đáp ứng của detector khi một chất (hoặc 2 hay nhiều chất không tách ra khỏi nhau) được rửa giải khỏi cột.
Pic có thể được xác định bằng diện tích pic, hoặc chiều cao pic (h) và chiều rộng pic ở nửa chiều cao (wh), hoặc chiều cao pic (h) và chiều rộng pic giữa các điểm uốn (wi). Ở các pic có dạng hình Gauss (Hình 5.1), có mối quan hệ biểu thị bằng phương trình sau:
wh = 1,18 × wi
Thời gian lưu (tR)
Là thời gian cần thiết để rửa giải một chất (Hình 5.1, đường nền biểu thị bằng phút).
Thể tích lưu (VR)
Là thể tích pha động cần thiết để rửa giải một chất. Thể tích lưu có thể tính được từ thời gian lưu và tốc độ dòng (ml/min) theo công thức:
VR = tR x F
trong đó:
F là lưu lượng dòng (hay tốc độ dòng) của pha động (ml/min).
Hình 5.1
Thời gian chết (tM) (hold-up time)
Là thời gian cần thiết để rửa giải một chất không bị lưu giữ (Hình 5.1, đường nền biểu thị bằng phút). Trong phương pháp sắc ký rây phân tử (xem Hình 5.2), thời gian chết được ký hiệu là t0.
Thể tích chết (VM) (hold-up volume)
Là thể tích pha động cần thiết để rửa giải một chất không bị lưu giữ. Nó có thể được tính từ thời gian chết (tw) và tốc độ dòng F (số ml pha động trong 1 min) theo công thức sau:
VM = tM x F
Trong phương pháp Sắc ký rây phân tử (xem Hình 5.2), thể tích chết được ký hiệu là V0.
Hình 5.2
Hệ số phân bố khối lượng
Hệ số phân bố khối lượng (Dm) hay còn gọi là thừa số dung lượng (k'), hoặc thừa số lưu giữ (k), được định nghĩa và tính theo công thức sau:
![]()
trong đó:
Kc là hằng số phân bố hay còn gọi là hệ số phân bố cân bằng;
VS là thể tích pha tĩnh;
VM là thể tích pha động;
ms là lượng chất trong pha tĩnh;
mM là lượng chất trong pha động.
Thừa số lưu giữ k của một chất có thể được xác định trên sắc đồ theo công thức:
![]()
Thời gian toàn bộ của pha động (t1)
Trong phương pháp sắc ký rây phân tử, thời gian toàn bộ của pha động là thời gian lưu của một chất mà kích thước phân tử nhỏ hơn kích thước các lỗ nhỏ nhất của gel (Hình 5.2).
Thể tích toàn bộ của pha động (Vt)
Trong phương pháp sắc ký rây phân tử, thể tích toàn bộ của pha động là thể tích lưu của một chất mà kích thước phân tử nhỏ hơn kích thước các lỗ nhỏ nhất của gel. Nó có thể được tính từ thời gian toàn bộ của pha động (t1) và tốc độ dòng F (ml/min) theo công thức sau:
Vt = t1 × F
Thời gian lưu của chất không bị lưu giữ (t0)
Trong phương pháp sắc ký rây phân tử, thời gian lưu của chất không bị lưu giữ là thời gian lưu của một chất mà kích thước phân tử lớn hơn kích thước các lỗ lớn nhất của gel (Hình 5.2).
Thể tích lưu của chất không bị lưu giữ (V0)
Trong phương pháp sắc ký rây phân tử, thể tích lưu của chất không bị lưu giữ là thể tích lưu của một chất mà kích thước phân tử lớn hơn kích thước các lỗ lớn nhất của gel. Có thể tính thể tích lưu của chất không bị lưu giữ từ thời gian lưu của chất không bị lưu giữ (t0) và tốc độ dòng F (ml/min) theo công thức sau:
Vo = to × F
Hằng số phân bố (K0)
Trong phương pháp sắc ký rây phân tử, đặc tính rửa giải của một chất có thể được biểu thị dưới dạng hằng số phân bố (còn gọi là hệ số phân bố) K0 và được tính theo công thức:
![]()
Thừa số chậm (RF)
Thừa số chậm RF, hay còn gọi là hệ số di chuyển Rf, được dùng trong sắc ký trên mặt phẳng (sắc ký lớp mỏng, sắc ký giấy) là tỷ số giữa khoảng cách từ điểm chấm mẫu đến tâm của vết sắc ký và khoảng cách di chuyển của dung môi tính từ điểm chấm mẫu (Hình 5.3).
![]()
trong đó:
b là quãng đường di chuyển của chất phân tích;
a là quãng đường di chuyển của tuyến dung môi.
Hình 5.3
Số đĩa lý thuyết (N)
Tùy thuộc vào kỹ thuật sử dụng, hiệu năng của cột (hay hiệu lực biểu kiến của cột) biểu thị dưới dạng số đĩa lý thuyết, có thể tính được từ những dữ liệu thu được dưới điều kiện đẳng nhiệt, đẳng dòng hoặc đẳng mật độ (isodense), theo công thức sau, trong đó các giá trị tR và wh có cùng đơn vị đo:
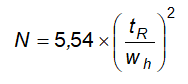
trong đó:
tR là thời gian lưu của pic tương ứng với chất phân tích;
wh là chiều rộng pic ở nửa chiều cao pic.
Số đĩa lý thuyết thay đổi theo chất, theo cột, nhiệt độ cột, pha động và thời gian lưu.
Thể tích lưu trú (D) (dwell volume)
Thể tích lưu trú (còn gọi là thể tích trễ gradient) là thể tích giữa điểm mà các chất rửa giải của pha động gặp nhau và đỉnh của cột. Nó có thể được xác định bằng cách sau:
Cột: Thay cột sắc ký bằng một ống mao quản thích hợp (ví dụ ống 1 m × 0,12 mm).
Pha động: Pha động A: Nước cất;
Pha động B: Dung dịch aceton 0,1 % (tt/tt);
| Thời gian (min) | Pha động A (% tt/tt) | Pha động B (% tt/tt) |
| 0 - 20 | 100 → 0 | 0 → 100 |
| 20 - 30 | 0 | 100 |
Tốc độ dòng: Đặt tốc độ dòng sao cho có áp suất ngược thích hợp (back-pressure) (ví dụ 2 ml/min).
Phát hiện: Quang phổ tử ngoại ở 265 nm.
Xác định thời điểm (t0,5) tính bằng phút khi độ hấp thụ tăng lên đạt 50 % (Hình 5.4).
D = tD × F
Trong đó:
tD = t0,5 - 0,5tG (tính bằng min);
tG là thời gian gradient đã xác định trước (= 20 min);
F là tốc độ dòng (tính bằng ml/min).
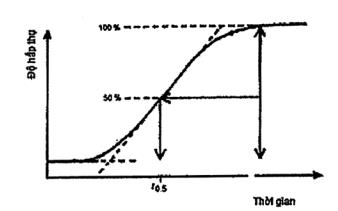
Hình 5.4
Hệ số đối xứng (As)
Hệ số đối xứng của một pic (Hình 5.5) được tính theo công thức sau:
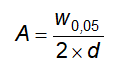
trong đó:
w0,05 là chiều rộng của pic ở 1/20 chiều cao của pic;
d là khoảng cách tính từ đường thẳng đứng đi qua đỉnh pic đến cạnh phía trước của pic ở 1/20 chiều cao của pic.
Khi As = 1,0 thì pic hoàn toàn đối xứng. Khi As > 1,0 thì pic bị kéo đuôi. Khi As < 1,0 thì pic bị doãng về phía trước.
Độ phân giải (Rs)
Độ phân giải (Rs) giữa hai pic của hai chất (Hình 5.5) được tính theo công thức sau:
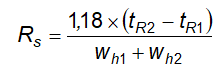
trong đó:
tR2 > tR1
tR1, tR2 là thời gian lưu của các pic tương ứng;
wh1, wh2 là chiều rộng pic ở nửa chiều cao pic.
Hình 5.5
Trong phương pháp định lượng bằng sắc ký trên mặt phẳng dùng phép đo mật độ, khoảng cách di chuyển được dùng thay cho thời gian lưu và độ phân giải giữa các pic của 2 chất có thể được tính theo công thức sau:
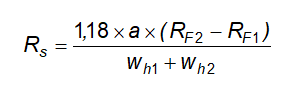
trong đó:
RF1, RF2 là thừa số chậm của các pic tương ứng;
wh1, wh2 là chiều rộng pic ở nửa chiều cao pic;
a là khoảng cách di chuyển của tuyến dung môi.
Tỷ số đỉnh - hõm (p/v)
Tỷ số đỉnh - hõm (p/v) có thể được dùng như một thông số về tính phù hợp của hệ thống trong phép thử các tạp chất liên quan khi hai pic không được tách đến đường nền (Hình 5.6)
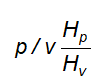
trong đó:
Hp là chiều cao của đỉnh pic nhỏ so với đường nền ngoại suy;
Hv là chiều cao của đáy hõm tách hai pic lớn và nhỏ so với đường nền ngoại suy.
Hình 5.6
Độ lưu giữ tỷ đối (r)
Độ lưu giữ tỷ đối (r) có thể ước lượng theo công thức sau:
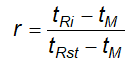
trong đó:
tRi là thời gian lưu của pic chất được quan tâm;
tRst là thời gian lưu của pic chất đối chiếu (thường là pic tương ứng với chất cần thử); tM là thời gian chết.
Độ lưu giữ tỷ đối chưa hiệu chỉnh (rG) được tính theo công thức:
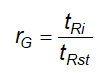
Trừ khi có hướng dẫn khác, các giá trị độ lưu giữ tỷ đối cho trong các chuyên luận là độ lưu giữ tỷ đối chưa hiệu chỉnh.
Trong sắc ký trên mặt phẳng, các thừa số chậm RFst và RFi được dùng thay cho các thời gian lưu tRst và tRi
Tỷ số tín hiệu trên nhiễu (S/N)
Nhiễu có ảnh hưởng đến độ chụm của phép định lượng. Tỷ số tín hiệu trên nhiễu được tính theo công thức:
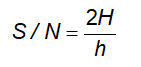
trong đó:
H là chiều cao pic (Hình 5.7) tương ứng với chất được khảo sát trên sắc đồ thu được với dung dịch chất đối chiếu đã định; chiều cao này đo từ đỉnh pic đến đường nền ngoại suy được xác định trên một khoảng bằng ít nhất 5 lần chiều rộng của pic ở nửa chiều cao pic;
h là khoảng dao động của nhiễu đường nền trên sắc đồ thu được sau khi tiêm một mẫu trắng; khoảng dao động này được xác định trên một quãng đường bằng ít nhất 5 lần chiều rộng của pic ở nửa chiều cao pic trên sắc đồ thu được khi tiêm dung dịch chất đối chiếu đã định và nếu có thể thì quãng đường này phân bổ đều hai bên vị trí của pic của chất khảo sát.
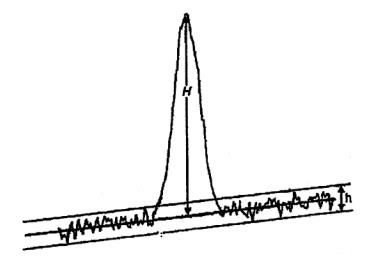
Hình 5.7
Độ lặp lại
Độ lặp lại của đáp ứng được biểu thị dưới dạng phần trăm độ lệch chuẩn tương đối (RSD %) của một dãy kết quả của ít nhất 3 lần tiêm hoặc nạp liên tiếp dung dịch chất đối chiếu, và được tính theo công thức:
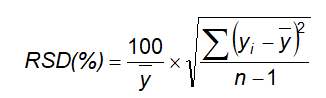
trong đó:
yi là các giá trị cá thể dưới dạng diện tích pic, hay chiều cao pic, hoặc tỷ số các diện tích trong phương pháp nội chuẩn;
![]() là trung bình của các giá trị cá thể;
là trung bình của các giá trị cá thể;
n là số các giá trị cá thể.
Tính phù hợp của hệ thống
Các bộ phận khác nhau của thiết bị sắc ký cần phải được kiểm tra hiệu chỉnh để có thể đạt được hiệu năng theo yêu cầu khi tiến hành phép thử hay định lượng.
Các phép thử tính phù hợp của hệ thống là phần không thể thiếu của một phương pháp và được dùng để đảm bảo hệ thống sắc ký có hiệu năng phù hợp. Hiệu lực biểu kiến, thừa số lưu giữ (hệ số phân bố khối lượng), độ phân giải, độ lưu giữ tỷ đối và hệ số đối xứng là những thông số thường được dùng để đánh giá hiệu năng của cột. Các yếu tố có thể ảnh hưởng đến đặc tính sắc ký bao gồm:
Thành phần, nồng độ ion, nhiệt độ và pH biểu kiến của pha động;
Tốc độ dòng, kích thước cột, nhiệt độ cột và áp suất;
Các đặc tính của pha tĩnh bao gồm loại chất mang, độ xốp (cỡ lỗ), cỡ hạt, kiểu các hạt, diện tích bề mặt riêng;
Pha đảo và các pha tĩnh có biến đổi bề mặt khác như biến đổi hóa học (được biểu thị bởi end- capping, carbon loading v.v...].
Các yêu cầu sau đây và các yêu cầu bổ sung trong chuyên luận cụ thể phải được đáp ứng, trừ khi có chỉ dẫn khác:
• Trong phép thử các tạp chất liên quan hay đinh lượng, hệ số đối xứng của pic chính trên sắc ký đồ thu được từ dung dịch đối chiếu phải trong khoảng từ 0,8 đến 1,5, trừ khi có chỉ dẫn khác;
• Trong phép định lượng hoạt chất mà giá trị xác định là 100 % chất tinh khiết, độ lệch chuẩn tối đa được phép (RSD(%)max) cho các lần tiêm lặp lại của dung dịch đối chiếu được tính theo công thức sau:
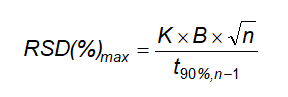
trong đó:
K là một hằng số (= 0,349) thu được từ biểu thức
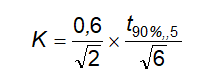
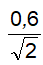 mà biểu thị độ lệch chuẩn tương đối yêu cầu cho 6 lần tiêm với B = 1,0;
mà biểu thị độ lệch chuẩn tương đối yêu cầu cho 6 lần tiêm với B = 1,0;
B là giới hạn trên được cho trong chuyên luận riêng trừ đi 100 %;
n là số lần tiêm lặp lại dung dịch đối chiếu (3 ≤ n ≤ 6);
t90 %,n-1 là giá trị t-Student ở mức xác xuất 90 % (hai phía) với bậc tự do n -1.
Nếu không có chỉ dẫn gì khác, độ lệch chuẩn tương đối được phép tối đa không được vượt quá giá trị tương ứng ghi trong Bảng 5.1. Yêu cầu này không áp dụng cho phép thử các tạp chất liên quan.
Bảng 5.1 - Yêu cầu về độ lặp lại
|
| Số lần tiêm lặp lại | |||
|
| 3 | 4 | 5 | 6 |
| B (%) | Độ lệch chuẩn tương đối được phép tối đa | |||
| 2,0 | 0,41 | 0,59 | 0,73 | 0,85 |
| 2,5 | 0,52 | 0,74 | 0,92 | 1,06 |
| 3,0 | 0,62 | 0,89 | 1,10 | 1,27 |
• Trong phép thử các tạp chất liên quan, giới hạn định lượng (tương ứng với tỷ số tín hiệu trên nhiễu bằng 10) phải bằng hoặc nhỏ hơn giới hạn được bỏ qua.
Yêu cầu về tính phù hợp của hệ thống là đòi hỏi cho quá trình tiến hành sắc ký. Tùy thuộc vào các yếu tố khác nhau như tần suất sử dụng và kinh nghiệm với hệ thống sắc ký, người phân tích sẽ lựa chọn một quy trình kiểm chứng thích hợp về tính phù hợp này.
Điều chỉnh các điều kiện sắc ký
Mức độ điều chỉnh các thông số khác nhau để thỏa mãn tính phù hợp của hệ thống sắc ký mà không làm thay đổi căn bản phương pháp được liệt kê dưới đây. Việc điều chỉnh các điều kiện rửa giải gradient thì khắt khe hơn so với rửa giải đẳng dòng vì nó có thể dẫn tới sự chuyển dịch các pic đến một bước khác của quá trình gradient. Điều này dẫn đến việc xác định không đúng vị trí các pic, làm che lấp pic hoặc làm chuyển dịch pic tới mức chất phân tích được rửa giải ra sau thời gian đã định. Những thay đổi khác với những điểm đã được quy định đòi hỏi phải thẩm định lại phương pháp. Những điều kiện sắc ký được mô tả đều đã được thẩm định khi biên soạn xây dựng chuyên luận.
Các phép thử tính thích hợp của hệ thống được đưa vào để đảm bảo yêu cầu chia tách cho phép thử hay phép định lượng đạt chất lượng mong muốn. Tuy nhiên, vì pha tĩnh chỉ được mô tả một cách chung chung, và trên thị trường lại có nhiều loại pha tĩnh có những đặc tính sắc ký khác nhau, nên có thể cần điều chỉnh một số điều kiện sắc ký để đạt yêu cầu của tính phù hợp hệ thống. Đặc biệt trong phương pháp sắc ký pha đảo, việc thay đổi các thông số sắc ký không phải lúc nào cũng cho kết quả thỏa đáng. Trong trường hợp này, có thể phải thay cột khác cùng loại, ví dụ cột silica gel gắn C18 khác mà có thể cho kết quả mong muốn. Với các thông số quan trọng, việc điều chỉnh được xác định rõ ràng trong chuyên luận để đảm bảo tính phù hợp của hệ thống.
Sắc ký lớp mỏng và sắc ký giấy
Thành phần của pha động: Lượng thành phần dung môi nhỏ có thể điều chỉnh ± 30 % nồng độ tương đối hay ± 2 % nồng độ tuyệt đối tùy theo khoảng giá trị nào lớn hơn. Ví dụ với dung môi thành phần nhỏ là 10 % của pha động thì thay đổi 30 % tương đối nghĩa là nồng độ trong khoảng 7 % đến 13 % trong khi thay đổi 2 % nồng độ tuyệt đối nghĩa là nồng độ trong khoảng 8 % đến 12 %: như vậy, khoảng theo nồng độ tương đối lớn hơn; nếu thành phần dung môi nhỏ là 5 % thì thay đổi 30 % tương đối nghĩa là trong khoảng 3,5 % đến 6,5 % và thay đổi 2 % nồng độ tuyệt đối nghĩa là trong khoảng 3 % đến 7 %, trong trường hợp này khoảng theo nồng độ tuyệt đối lớn hơn. Không thành phần dung môi nào được thay đổi lớn hơn 10 % nồng độ tuyệt đối.
pH của thành phần nước trong pha động: Chỉ điều chỉnh thay đổi ± 0,2 đơn vị pH trừ khi có chỉ dẫn khác trong chuyên luận, hoặc thay đổi ± 1,0 đơn vị pH khi nghiên cứu các chất không ion hóa.
Nồng độ muối trong thành phần đệm của pha động được thay đổi ± 10 %.
Thể tích mẫu chấm sắc ký điều chỉnh trong khoảng 10 % đến 20 % của thể tích quy định nếu sử dụng bản mỏng cỡ hạt nhỏ (2 µm đến 10 µm).
Khoảng dịch chuyển của tuyến dung môi không được dưới 50 mm và với bản mỏng hiệu năng cao thì không được dưới 30 mm.
Sắc ký lỏng rửa giải đẳng dòng
Thành phần pha động: Lượng thành phần dung môi nhỏ có thể điều chỉnh ± 30 % nồng độ tương đối hay ± 2 % nồng độ tuyệt đối tùy theo khoảng giá trị nào lớn hơn (xem ví dụ ở trên); Không thành phần dung môi nào được thay đổi lớn hơn 10 % nồng độ tuyệt đối.
pH của thành phần nước trong pha động: Chỉ điều chỉnh thay đổi ± 0,2 đơn vị pH trừ khi có chỉ dẫn khác trong chuyên luận, hoặc thay đổi ± 1,0 đơn vị pH khi nghiên cứu các chất không ion hóa.
Nồng độ muối trong thành phần đệm của pha động được thay đổi ± 10 %.
Tốc độ dòng: ± 50 %; có thể chấp nhận một sự điều chỉnh lớn hơn khi thay đổi kích thước cột (xem công thức ở dưới).
Các thông số cột:
Pha tĩnh:
Không thay đổi bản chất của nhóm thế trong pha tĩnh (ví dụ không thay C18 bằng C8);
Kích thước hạt: cho phép giảm tối đa 50 %; không được phép tăng.
Kích thước cột:
Chiều dài: ± 70 %;![]()
Đường kính trong: ± 25 %.
Khi kích thước cột thay đổi, tốc độ dòng có thể phải điều chỉnh theo công thức sau:
![]()
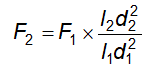
trong đó:
F1 là tốc độ dòng được chỉ ra trong chuyên luận, tính theo ml/min;
F2 là tốc độ dòng hiệu chỉnh, tính theo ml/min;
l1 là chiều dài cột ghi trong chuyên luận, tính bằng mm;
l2 là chiều dài cột được sử dụng, tính bằng mm;
d1 là đường kính trong của cột ghi trong chuyên luận, tính bằng mm;
d2 là đường kính trong của cột được sử dụng, tính bằng mm.
Nhiệt độ: Được điều chỉnh ± 10 °C khi có quy định nhiệt độ tiến hành sắc ký, trừ khi có chỉ dẫn khác.
Bước sóng của detector phát hiện: Không được điều chỉnh.
Thể tích tiêm: Có thể giảm đi miễn là khả năng phát hiện và độ lặp lại của pic cần xác định thỏa mãn yêu cầu; không cho phép tăng thể tích tiêm.
Sắc ký lỏng rửa giải gradient
Việc điều chỉnh các điều kiện cho hệ thống sắc ký rửa giải gradient đòi hỏi cần cẩn thận hơn so với hệ thống sắc ký đẳng dòng.
Thành phần của pha động dùng rửa giải gradient: Những điều chỉnh nhỏ về thành phần của pha động và chương trình dung môi có thể được chấp nhận, miễn là:
Các yêu cầu về tính phù hợp của hệ thống được thỏa mãn;
Các pic chính được rửa giải trong vòng ± 15 % của thời gian lưu quy định;
Thành phần cuối cùng của pha động không yếu hơn về lực rửa giải so với thành phần quy định.
Khi không thể đạt được thỏa mãn các yêu cầu về tính phù hợp của hệ thống thì nên xem xét về thể tích lưu trú (dwell volume) hoặc thay đổi cột tách.
Thể tích lưu trú (Dwell volume): cấu hình của thiết bị dùng có thể làm thay đổi đáng kể độ phân giải, thời gian lưu và thời gian lưu tỷ đối được quy định; đó là do thể tích lưu trú quá lớn. Các chuyên luận thường đưa vào một giai đoạn đẳng dòng trước khi bắt đầu chương trình gradient để làm thích ứng với các thời điểm gradient; điều này giúp xem xét sự khác nhau giữa thể tích lưu trú của hệ thống thiết bị được sử dụng để phát triển phương pháp và hệ thống đang được sử dụng. Người sử dụng có trách nhiệm điều chỉnh khoảng thời gian của giai đoạn đẳng dòng cho phù hợp với thiết bị được sử dụng. Nếu thể tích lưu trú được dùng khi xây dựng chuyên luận được chỉ ra trong chuyên luận, các thời điểm (t, min) ghi trong bảng chương trình dung môi có thể được thay thế bằng các thời điểm điều chỉnh (tc min) và được tính bằng công thức sau:
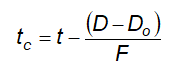
trong đó:
D là thể tích lưu trú, tính bằng ml;
D0 là thể tích lưu trú khi phát triển phương pháp, tính bằng ml;
F là tốc độ dòng tính bằng ml/min.
Giai đoạn đẳng dòng bổ sung cho mục đích trên đây có thể bỏ qua nếu các số liệu thẩm định để áp dụng phương pháp không có giai đoạn này.
pH của thành phần nước trong pha động không được phép điều chỉnh.
Nồng độ muối trong thành phần đệm của pha động cũng không được phép điều chỉnh.
Tốc độ dòng: Có thể điều chỉnh tốc độ dòng khi kích thước cột tách thay đổi (xem công thức ở dưới).
Các thông số về cột:
Pha tĩnh:
Không thay đổi bản chất của nhóm thế trong pha tĩnh (ví dụ không thay C18 bằng C8);
Kích thước tiểu phân không được thay đổi.
Kích thước cột:
Chiều dài: cho phép ± 70 %;
Đường kính trong: cho phép ± 25 %.
Khi kích thước cột tách thay đổi, tốc độ dòng có thể phải điều chỉnh dựa theo công thức sau:
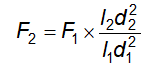
trong đó:
F1 là tốc độ dòng ghi trong chuyên luận, tính bằng ml/min;
F2 là tốc độ dòng được điều chỉnh, tính bằng ml/min;
l1 là chiều dài cột ghi trong chuyên luận, tính bằng mm;
l2 là chiều dài cột được sử dụng, tính bằng mm;
d1 là đường kính trong của cột ghi trong chuyên luận, tính bằng mm;
d2 là đường kính trong của cột được sử dụng, tính bằng mm.
Nhiệt độ: Được điều chỉnh ± 5 °C, khi nhiệt độ vận hành sắc ký được quy định trong chuyên luận, trừ khi có chỉ dẫn khác.
Bước sóng phát hiện của detector không được điều chỉnh.
Thể tích tiêm có thể giảm đi miễn là khả năng phát hiện và độ lặp lại của pic cần xác định thỏa mãn yêu cầu; không cho phép tăng thể tích tiêm.
Sắc ký khí
Các thông số về cột:
Pha tĩnh:
Kích thước tiểu phân: có thể giảm tối đa 50 %; không được phép tăng lên (cột nhồi);
Bề dầy lớp phim: cho phép thay đổi từ -50 % đến +100 % (cột mao quản).
Kích thước cột:
Chiều dài: ± 70 %;
Đường kính trong: ± 50 %.
Tốc độ dòng: ± 50 %.
Nhiệt độ: ± 10 %.
Thể tích tiêm và thể tích chia dòng: có thể được điều chỉnh, miễn là khả năng phát hiện và độ lặp lại là thỏa mãn yêu cầu.
Sắc ký lỏng siêu tới hạn
Thành phần pha động: Đối với cột nhồi, lượng hợp phần dung môi nhỏ có thể được điều chỉnh ± 30 % tương đối hay ± 2 % tuyệt đối, tùy theo khoảng giá trị nào lớn hơn; không cho phép thay đổi đối với hệ thống cột mao quản.
Bước sóng phát hiện của detector không được thay đổi.
Các thông số về cột:
Pha tĩnh:
Kích thước tiểu phân: có thể giảm tối đa 50 %; không được phép tăng lên (cột nhồi);
Kích thước cột:
Chiều dài: ± 70 %;
Đường kính trong: ± 25 % cho cột nhồi và ± 50 % cho cột mao quản.
Tốc độ dòng: ± 50 %.
Nhiệt độ cho phép ± 5 °C, khi nhiệt độ vận hành sắc ký được quy định trong chuyên luận.
Thể tích tiêm có thể được điều chỉnh, miễn là khả năng phát hiện và độ lặp lại là thỏa mãn yêu cầu; không được phép tăng lên.
Định lượng
Các pic của dung môi và thuốc thử hoặc xuất hiện từ pha động hay nền mẫu thì bỏ qua trong quá trình định lượng.
Độ nhạy của detector: Độ nhạy của detector là tín hiệu đầu ra tính trên một đơn vị nồng độ hay một đơn vị khối lượng chất có trong pha động đi vào detector. Hệ số đáp ứng tương đối của detector, thường được gọi là hệ số đáp ứng, biểu thị độ nhạy của một detector đối với một chất chuẩn. Hệ số hiệu chỉnh là nghịch đảo của hệ số đáp ứng.
Phương pháp chuẩn ngoại: Nồng độ các thành phần cần phân tích được xác định bằng cách so sánh (các) đáp ứng pic của dung dịch thử đối với đáp ứng pic của dung dịch chất đối chiếu.
Phương pháp chuẩn nội: Chất chuẩn nội phải là một chất có thể tách khỏi chất khảo sát. Thêm cùng lượng chuẩn nội vào dung dịch thử và dung dịch chất đối chiếu. Chất chuẩn nội không được có phản ứng với chất khảo sát, phải bền và không chứa tạp có thời gian lưu tương tự thời gian lưu chất khảo sát. Nồng độ chất khảo sát được xác định dựa vào việc so sánh tỷ số diện tích pic (hay chiều cao pic) giữa chất khảo sát và chuẩn nội trong dung dịch thử với tỷ số diện tích pic (hay chiều cao pic) giữa chất khảo sát và chuẩn nội trong dung dịch chất đối chiếu.
Phương pháp chuẩn hóa: Còn gọi là quy trình chuẩn hóa. Hàm lượng phần trăm của một hay nhiều thành phần trong mẫu thử được tính bằng cách xác định diện tích một pic hay nhiều pic dưới dạng % của tổng diện tích tất cả các pic trừ các pic của dung môi hay thuốc thử và các pic ở mức hay dưới mức có thể bỏ qua.
Phương pháp đường chuẩn: Còn gọi là quy trình hiệu chuẩn. Xác định mối quan hệ giữa tín hiệu đo được hay đánh giá được (y) và lượng (nồng độ, khối lượng, v.v...) của chất (x) rồi tính hàm hiệu chuẩn (phương trình đường chuẩn). Tính kết quả phân tích từ tín hiệu của chất phân tích (được đo hay được đánh giá) theo phương pháp hàm số ngược.
Trong các phép thử tạp chất liên quan, người ta dùng hoặc phương pháp chuẩn ngoại với một dung dịch thử được pha loãng để so sánh, hoặc dùng phương pháp chuẩn hóa, khi hệ số đáp ứng nằm ngoài khoáng 0,8 đến 1,2 thì phải áp dụng những hệ số hiệu chỉnh được chỉ ra trong chuyên luận.
Khi trong chuyên luận có quy định phép thử các tạp chất liên quan là tổng các tạp chất hay khi phải định lượng một tạp chất thì điều quan trọng là phải chọn một ngưỡng thích hợp và các điều kiện thích hợp cho việc lấy tích phân các diện tích pic. Trong các phép thử như vậy thì ngưỡng bỏ qua (nghĩa là các diện tích pic bằng hoặc dưới ngưỡng này không phải tính đến) thường là 0,05 %. Như vậy, ngưỡng đặt cho hệ thống thu thập dữ liệu ít nhất phải là nửa ngưỡng bỏ qua. Việc lấy tích phân các diện tích pic của các tạp không được tách hoàn toàn khỏi pic chính nên được thực hiện theo phương pháp ngoại suy từ hõm tới hõm (vạch tiếp tuyến).
5.1 PHƯƠNG PHÁP SẮC KÝ GIẤY
Phương pháp sắc ký giấy, cũng như phương pháp sắc ký lớp mỏng, được áp dụng trong kiểm nghiệm chất lượng thuốc để định tính, thử tinh khiết và đôi khi để bán định lượng và định lượng.
Sự tách các chất bằng phương pháp sắc ký giấy dựa chủ yếu trên sự khác nhau về hệ số phân bố của chúng giữa hai pha lỏng: Một pha tĩnh và một pha động. Pha tĩnh ở đây là nước có sẵn trong sợi celulose của giấy, hoặc thành phần thân nước từ hỗn hợp dung môi của pha động được hút chọn lọc vào giấy. Pha động là một hệ dung môi thích hợp cho sự tách đã được quy định trong các chuyên luận.
Mức độ di chuyển của một chất được đặc trưng bởi hệ số di chuyển (Rf) và tính bằng tỷ lệ giữa khoảng cách di chuyển của chất đó và khoảng cách di chuyển của dung môi:
Rf = a/b.
trong đó:
a là khoảng cách di chuyển của chất phân tích;
b là khoảng cách di chuyển của dung môi tính từ điểm chấm mẫu.
Giá trị Rf bao giờ cũng nhỏ hơn 1.
Trong trường hợp sắc ký liên tục không còn xác định được giới tuyến của dung môi, người ta dùng hệ số di chuyển Rr. Rr là tỷ số giữa khoảng cách di chuyển của chất phân tích và khoảng cách di chuyển của chất dùng làm chuẩn so sánh. Giá trị Rr có thể nhỏ hơn hay lớn hơn 1.
Cách tiến hành
Chuẩn bị bình sắc ký: Bình sắc ký là những bình thủy tinh hình trụ hoặc hình hộp dẹt, có kích thước thích hợp, có nắp đậy kín. Nắp có lỗ ở giữa để lắp bình gạn có khóa đựng dung môi. Trong bình có các giá để máng đựng dung môi và treo giấy sắc ký được thiết kế thích hợp cho kiểu sắc ký đi lên hay sắc ký đi xuống.
Chuẩn bị dung môi: Thành phần và tỷ lệ các dung môi được quy định trong các chuyên luận. Việc trộn các thành phần được tiến hành trong bình gạn, sau khi lắc đều, để yên. Nếu có tách lớp thì gạn lấy lớp pha thân nước, thường là lớp dưới, làm dung môi pha tĩnh để bão hòa giấy sắc ký sau khi đã chấm các chất phân tích, còn lớp trên dùng làm dung môi pha động. Nếu dung môi không tách lớp thì dùng chính dung môi đó để bão hòa giấy sắc ký.
Chuẩn bị giấy: Giấy sắc ký là loại giấy đặc biệt dành riêng cho sắc ký, có bề dày thích hợp. Tùy theo kích thước của bình và số lượng các vết cần chấm mà cắt những khổ giấy hình chữ nhật thích hợp, chiều dài dọc theo thớ giấy. Chiều rộng phải nhỏ hơn chiều dài của máng dung môi nhưng không được nhỏ hơn 2,5 cm. Bề dài phải tính sao cho khi treo giấy vào bình không được để đầu dưới chạm vào máng dung môi trong khi bão hòa dung môi. Trường hợp bình sắc ký có hình chuông úp thì giấy phải cuộn tròn và cố định bằng móc thủy tinh hoặc khâu bằng chỉ. Đường kính của cuộn giấy phải nhỏ hơn đường kính của đĩa để giấy nhằm tránh giấy chạm vào thành dĩa.
Chấm sắc ký: Kẻ một đường chì mảnh song song với mép giấy (đường vạch để chấm sắc ký) theo chiều rộng và cách mép giấy 3 cm đối với sắc ký đi lên để sao cho các vết chấm không bị ngập vào dung môi, 6 cm đối với sắc ký đi xuống để vạch chấm không trùng với đũa thủy tinh đỡ giấy mà thấp hơn 2 cm khi đặt giấy treo vào máng ở phần trên của bình.
Dùng micropipet chia độ tới 0,001 ml đến 0,002 ml hoặc mao quản có dung tích xác định (2 µl; 5 µl; 10 µl) để chấm các dung dịch chất cần sắc ký thành vết có đường kính không quá 8 mm. Muốn giữ đường kính của vết chấm nhỏ, phải chấm nhanh nhiều lần, đợi giọt trước khô mới chấm tiếp.
Lượng chấm và nồng độ dung dịch chất thử được quy định trong chuyên luận.
Khi chấm nhiều vết trên một dải giấy thì vết chấm nọ phải cách vết chấm kia ít nhất là 30 mm.
Triển khai sắc ký
Phương pháp sắc ký đi lên: Rót dung môi làm pha động vào máng dung môi để được một lớp dung môi cao 2,5 cm, và trong trường hợp pha tĩnh được chỉ định (là phần tách lớp phía dưới khi trộn hỗn hợp dung môi) thì rót pha tĩnh vào khe giữa máng dung môi và thành bình sắc ký. Đậy kín nắp bình, để yên trong 24 h ở nhiệt độ 20 °C đến 25 °C và duy trì ở nhiệt độ này trong quá trình tiếp theo. Sau đó treo giấy đã chuẩn bị vào bình, đậy nắp, để yên tiếp 1 h 30 min. Tiếp theo dùng tay vặn ở ngoài bình để hạ tờ giấy sắc ký vào máng dung môi sao cho vạch chấm không ngập vào dung môi. Triển khai sắc ký đến một thời gian hoặc một khoảng cách quy định trong chuyên luận. Lấy giấy ra, đánh dấu ngay giới tuyến dung môi và để khô ngoài không khí hoặc làm khô bằng không khí nóng của máy quạt sấy. Phải chú ý tránh ánh sáng trong suốt quá trình triển khai. Làm hiện vết bằng cách phun thuốc thử màu thích hợp, hoặc soi dưới đèn tử ngoại theo quy định của chuyên luận. Tính giá trị Rf và đánh giá kết quả.
Phương pháp sắc ký đi xuống: Cũng tiến hành tương tự như sắc ký đi lên, chỉ khác các điểm sau đây: Rót dung môi làm pha tĩnh vào đáy bình, tạo một lớp cao khoảng 2 cm; đặt giấy vào máng dung môi ở phần trên bình, dùng một đũa thủy tinh để chèn giấy, gác đầu giấy treo qua đũa thủy tinh để không cho giấy chạm vào mép máng và chạm vào thành bình. Sau thời gian để bão hòa bình và giấy theo quy định như trên, rót pha động vào máng đựng dung môi ở phía trên. Thường sắc ký đi xuống được áp dụng cho các hỗn hợp khó tách và phải chạy sắc ký liên tục nên phải tính kết quả theo Rr.
5.2 PHƯƠNG PHÁP SẮC KÝ KHÍ
Sắc ký khí là một phương pháp tách dựa trên sự phân bố khác nhau của các chất giữa hai pha không trộn lẫn vào nhau, trong đó pha động là chất khí (khí mang) đi qua pha tĩnh chứa trong cột. Sắc ký khí được áp dụng để tách những chất hoặc dẫn xuất của chúng mà có thể hóa hơi ở nhiệt độ phân tích. Phương pháp sắc ký khí dựa trên cơ chế hấp phụ, phân bố hoặc loại theo kích thước (dùng rây phân tử).
Thiết bị
Thiết bị gồm nguồn cung cấp khí, bộ phận tiêm mẫu, cột sắc ký chứa trong buồng ổn nhiệt, hệ thống phát hiện và ghi sắc đồ. Khí mang đi qua cột với tốc độ và áp suất có kiểm soát và đi qua detector. Nhiệt độ của cột hoặc được giữ ở một giá trị không đổi hoặc thay đổi theo một chương trình quy định trong chuyên luận riêng.
Sự đặc thù về thiết kế của máy sắc ký khí có thể yêu cầu phải thay đổi điều kiện sắc ký đã ghi trong chuyên luận riêng. Trong những trường hợp có thay đổi như thế, phải cho những kết quả tương tự. Nếu cần, điều chỉnh tốc độ dòng khí mang để cải thiện chất lượng sắc ký đồ hoặc để thay đổi thời gian lưu của các pic quan tâm.
Bộ phận tiêm mẫu
Tiêm mẫu trực tiếp: Là kiểu tiêm mẫu thường dùng trừ khi có chỉ dẫn khác, có thể tiêm mẫu trực tiếp vào đầu cột bằng bơm tiêm (syringe) hoặc dùng van tiêm mẫu hoặc đưa vào buồng hóa hơi có thể có gắn thêm bộ chia dòng.
Tiêm pha hơi: Có thể thực hiện bằng hệ thống tiêm mẫu head-space tĩnh hay động.
Hệ thống tiêm mẫu headspace động (kỹ thuật bẫy và làm sạch): Bao gồm thiết bị nhúng chìm vào dung dịch cho các chất bay hơi bám vào cột hấp phụ ở nhiệt độ thấp. Các chất lưu giữ được giải hấp phụ vào pha động bằng cách làm nóng nhanh cột hấp phụ.
Hệ thống tiêm mẫu headspace tĩnh: Bao gồm buồng ổn nhiệt mẫu có kiểm soát nhiệt độ trong đó đặt lọ đựng mẫu chứa mẫu rắn hoặc lỏng trong thời gian cố định để cho các thành phần bay hơi đạt được trạng thái cân bằng giữa pha hơi và pha lỏng hoặc rắn. Sau khi đạt được trạng thái cân bằng, một lượng mẫu khí xác định trước được đưa vào máy sắc ký khí.
Pha tĩnh
Pha tĩnh chứa trong cột có thể là:
• Cột mao quản làm bằng silica nung chảy, pha tĩnh phủ trên thành cột.
• Cột nhồi chứa hạt chất mang, trơ tẩm pha tĩnh lỏng.
• Cột nhồi chứa pha tĩnh rắn.
Cột mao quản có đường kính trong từ 0,1 mm đến 0,53 mm và dài từ 5 m đến 60 m. Pha tĩnh là một lớp phim bề dày từ 0,1 µm đến 5,0 µm có thể gắn kết về hóa học vào bề mặt bên trong.
Cột nhồi làm bằng thủy tinh hay kim loại, thông thường dài từ 1 m đến 3 m, đường kính trong từ 2 mm đến 4 mm. Pha tĩnh thường là chất polymer xốp hoặc chất mang rắn phủ pha tĩnh lỏng.
Trong phân tích các hợp chất phân cực ở cột nhồi với pha tĩnh có dung lượng thấp, tính phân cực thấp, chất mang phải trơ để tránh làm cho pic không đối xứng. Hoạt tính chất mang có thể được giảm đi bằng cách silan hóa trước khi phủ pha tĩnh lên. Thường dùng diatomit nung, rửa với acid. Các chất mang thường có các kích thước hạt khác nhau, thường sử dụng nhất các hạt từ 150 µm đến 180 µm và 125 µm đến 150 µm.
Pha động
Thời gian lưu và hiệu lực cột phụ thuộc vào tốc độ dòng khí mang: Thời gian lưu tỷ lệ thuận với chiều dài cột và độ phân giải tỷ lệ thuận với căn bậc hai chiều dài cột. Đối với cột nhồi, lưu lượng khí mang tĩnh bằng ml/min ở áp suất khí quyển và nhiệt độ phòng. Lưu lượng đo ở đầu ra của detector bằng dụng cụ đo cơ học có chia độ hoặc ống đo tốc độ dòng dùng bọt xà phòng ở nhiệt độ cột đang phân tích. Tốc độ dài của dòng khí mang đi qua cột nhồi tỷ lệ nghịch với căn bậc hai đường kính trong của cột ở một thể tích dòng xác định. Lưu lượng 60 ml/min trong cột đường kính trong 4 mm và lưu lượng 15 ml/min trong cột đường kính trong 2 mm cùng cho tốc độ dòng như nhau, do đó thời gian lưu tương đương nhau.
Thường dùng heli và nitơ làm khí mang cho cột nhồi và nitơ, heli và hydro cho cột mao quản.
Detector
Thường dùng detector ion hóa ngọn lửa, ngoài ra còn dùng detector dẫn nhiệt, nitrogen-phosphor (electron nhiệt hay nhiệt điện tử), cộng kết điện tử, phổ khối, phổ hồng ngoại FTIR và các loại detector khác tùy theo mục đích phân tích.
Các đại lượng đặc trưng cho quá trình sắc ký: Xem Phụ lục 5. Các kỹ thuật tách sắc ký.
Phương pháp tiến hành
Ổn định cột, buồng tiêm và detector ở nhiệt độ và tốc độ dòng khí mang quy định trong chuyên luận cho đến khi đường nền ổn định.
Chuẩn bị các dung dịch thử và đối chiếu theo quy định của chuyên luận. Các dung dịch phải không được có các tiểu phân rắn. Dùng dung dịch chuẩn để xác định các điều kiện đo phù hợp đặt trên thiết bị và thể tích dung dịch mẫu tiêm cần dùng để tạo ra một đáp ứng phù hợp. Tiến hành các lần tiêm lặp lại để kiểm tra độ lặp lại của đáp ứng và kiểm tra số đĩa lý thuyết, nếu có yêu cầu.
Tiêm các dung dịch thử và ghi lại sắc đồ thu được. Tiến hành tiêm lại nhiều lần để kiểm tra độ lặp lại của đáp ứng. Xác định diện tích pic của các thành phần cần quan tâm khi không có chỉ dẫn khác. Cũng có thể xác định chiều cao của pic tương ứng với các thành phần chất cần quan tâm khi hệ số đối xứng từ 0,80 đến 1,20. Từ những giá trị thu được tính ra hàm lượng của một thành phần hay nhiều thành phần có trong mẫu đem thử.
Khi dùng một chất làm chuẩn nội, pic của chất này không được che phủ pic của mẫu thử. Nếu có pic bị ảnh hưởng cần phải có sự bổ chính thích hợp. Trong những áp dụng có yêu cầu chương trình nhiệt độ, phải tính kết quả theo diện tích pic.
Trừ những chỉ dẫn khác quy định trong chuyên luận riêng, dùng nitrogen làm khí mang và detector ion hóa ngọn lửa. Thỉnh thoảng cần tiến hành xác định bằng kỹ thuật tiêm on-column, trong đó mẫu được tiêm trực tiếp vào chất nhồi cột không sử dụng bộ phận nạp mẫu gia nhiệt. Khi mẫu thử là chất không bay hơi được tiêm vào cột, nên dùng tiền cột phù hợp, loại có thể thay đổi được.
Thuốc thử
Các thuốc thử và các dung môi sử dụng trong việc chuẩn bị các dung dịch khảo sát cần có chất lượng phù hợp cho phân tích trên sắc ký khí. Có rất nhiều chất hóa học được dùng làm pha tĩnh như các polyethylen glycol, các ester và các amid có phân tử lượng cao, các hydrocarbon, các silicon dạng gôm hay dạng lỏng (polysiloxan đã được thế bằng nhóm methyl, phenyl, nitril, vinyl hoặc fluoroalkyl, hoặc hỗn hợp của chúng) và các vi hạt rỗng cấu tạo đa nhân thơm liên kết chéo. Pha tĩnh phù hợp khi nồng độ của nó, bản chất và bậc của chất mang rắn theo đúng quy định của chuyên luận riêng. Cột phải luyện theo chỉ dẫn của nhà sản xuất. Trong đa số các trường hợp, tên thương mại của cột ghi trong chuyên luận là phù hợp cho mục đích phân tích nhưng có thể dùng cột có tên thương mại khác tương đương.
Chuẩn nội
Các thuốc thử được dùng làm chuẩn nội không được chứa bất kỳ tạp chất nào gây ra các pic có ảnh hưởng tới việc xác định các thành phần của mẫu thử đã mô tả trong chuyên luận riêng.
Chuẩn hóa
Trong một số trường hợp, cần tiến hành xác định bằng phương pháp quy về 100 % để đánh giá một hoặc nhiều thành phần hoặc các tạp chất liên quan. Trong những trường hợp này, tổng diện tích của một pic hay các pic của các thành phần hoặc các tạp chất liên quan được biểu thị dưới dạng tỷ lệ phần trăm so với tổng diện tích của tất cả các pic thu được từ mẫu đem thử. Trong quá trình xác định, cần sử dụng bộ khuếch đại để mở rộng thang đo và bộ tính tích phân tự động.
Thể tích tiêm
Khi thể tích tiêm không quy định trong chuyên luận riêng, nên chọn một thể tích phù hợp để áp dụng. Chọn thể tích tiêm phụ thuộc vào đáp ứng của phép phân tích, detector sử dụng, hiệu lực cột và toàn bộ hiệu năng của hệ thống sắc ký. Khi không có chỉ dẫn, thường dùng thể tích tiêm là 1 µl, tuy nhiên cần kiểm tra sự thích hợp trong điều kiện cụ thể.
Pic phụ (pic thứ cấp)
Có thể cần chất đối chiếu để xác định pic phụ. Pic phụ là một pic có trên sắc đồ nhưng không phải là pic chính hay pic chuẩn nội hoặc pic của dung môi và các thuốc thử tạo dẫn xuất.
Sắc ký khí head-space
Sắc ký khí head-space là phương pháp đặc biệt thích hợp để tách và xác định các chất bay hơi hiện diện trong mẫu rắn hay lỏng. Phương pháp dựa trên sự phân tích pha hơi sau khi đạt được trạng thái cân bằng với pha rắn hoặc pha lỏng.
Thiết bị
Máy sắc ký khí kết nối với bộ phận nạp mẫu tự động có thiết bị kiểm soát nhiệt độ và áp suất. Trong trường hợp cần thiết, có thể gắn thêm bộ phận đuổi dung môi.
Đưa mẫu vào lọ đựng mẫu có nắp đậy gắn với hệ thống van có thể cho phép khí mang đi qua. Đặt lọ đựng mẫu trong buồng ổn nhiệt ở nhiệt độ tùy theo bản chất của mẫu và duy trì nhiệt độ này trong thời gian sao cho đạt được trạng thái cân bằng giữa pha rắn hoặc lỏng và pha hơi.
Đưa khí mang vào lọ đựng mẫu và sau thời gian quy định, mở van tương ứng để khí khuếch tán vào cột sắc ký mang theo các chất bay hơi cần phân tích.
Nếu không có bộ phận nạp mẫu tự động, có thể dùng bơm tiêm (syringe) mẫu khí đưa mẫu trực tiếp vào cột sau khi đạt được trạng thái cân bằng trong buồng ổn nhiệt đặt riêng, cần lưu ý tránh những yếu tố làm thay đổi trạng thái cân bằng.
Phương pháp tiến hành
Dùng các dung dịch đối chiếu để xác định tính thích hợp của cài đặt thiết bị để cho đáp ứng đạt yêu cầu.
Định lượng trực tiếp
Lần lượt cho mẫu thử và mẫu đối chiếu vào các lọ đựng mẫu đồng nhất như quy định trong chuyên luận riêng, tránh tiếp xúc giữa mẫu thử và thiết bị lấy mẫu. Đậy kín lọ và đặt trong buồng ổn nhiệt đã cài nhiệt độ và áp suất như trong chuyên luận riêng. Sau khi đạt được trạng thái cân bằng, tiến hành sắc ký theo điều kiện quy định trong chuyên luận riêng.
Phương pháp thêm chuẩn
Lần lượt cho cùng một thể tích mẫu thử vào các lọ đựng mẫu đồng nhất. Thêm vào mỗi lọ một lượng thích hợp dung dịch đối chiếu của chất cần xác định có hàm lượng đã biết trước để có một dãy dung dịch có nồng độ tăng dần một cách đều đặn.
Đậy kín lọ và đặt trong buồng ổn nhiệt đã đặt nhiệt độ và áp suất như trong chuyên luận riêng. Sau khi đạt được trạng thái cân bằng, tiến hành sắc ký theo điều kiện quy định trong chuyên luận riêng.
Lập phương trình tuyến tính của đồ thị bằng phương pháp bình phương tối thiểu và tìm được từ đó nồng độ của chất cần xác định trong dung dịch. Theo một cách khác, vẽ đồ thị trung bình các nồng độ xác định được theo lượng dung dịch đối chiếu thêm vào. Kéo dài đường nối các điểm trên đồ thị cho đến khi gặp trục nồng độ. Khoảng cách giữa điểm này và giao điểm của các trục thể hiện nồng độ chất cần xác định trong dung dịch.
Phương pháp rút ra liên tục
Nếu được quy định, phương pháp rút ra liên tục được mô tả đầy đủ trong chuyên luận riêng.
5.3 PHƯƠNG PHÁP SẮC KÝ LỎNG
Sắc ký lỏng là phương pháp tách sắc ký các chất dựa trên sự phân bố khác nhau của chúng giữa hai pha không trộn lẫn, trong đó pha động là một chất lỏng chảy qua pha tĩnh chứa trong cột.
Sắc ký lỏng được tiến hành chủ yếu dựa trên cơ chế hấp phụ, phân bố khối lượng, trao đổi ion, loại trừ theo kích thước hoặc tương tác hóa học tập thể.
Thiết bị
Thiết bị bao gồm một hệ thống bơm, bộ phận tiêm mẫu, cột sắc ký (bộ phận điều khiển nhiệt độ có thể được sử dụng nếu cần thiết), detector và một hệ thống thu dữ liệu (hay một máy tích phân hoặc một máy ghi đồ thị). Pha động được cung cấp từ một hoặc vài bình chứa và chảy qua cột, thông thường với tốc độ không đổi và sau đó chạy qua detector.
Hệ thống bơm
Hệ thống bơm trong sắc ký lỏng phải giữ cho pha động luôn chảy với một lưu lượng không đổi. Những biến đổi áp suất sẽ được giảm thiểu, ví dụ cho dung môi chạy qua một thiết bị giảm xung, ống dẫn và hệ thống nối phải là loại chịu được áp suất sinh ra do hệ thống bơm. Các bơm có thể được lắp với thiết bị loại bỏ bọt khí.
Hệ thống điều khiển bằng bộ vi xử lý có khả năng cung cấp pha động hoặc hằng định (rửa giải đẳng dòng) hoặc thay đổi tỷ lệ thành phần (rửa giải gradient) theo một chương trình xác định. Trong trường hợp rửa giải gradient, hệ thống bơm lấy các dung môi từ một vài bình chứa và các dung môi có thể được trộn lẫn ở áp suất thấp hoặc áp suất cao.
Bộ phận tiêm mẫu
Dung dịch mẫu thử được đưa vào dòng pha động hoặc vào vị trí gần đầu hoặc đầu cột nhờ một bộ phận tiêm mẫu có khả năng hoạt động ở áp suất cao. Có thể dùng vòng chứa mẫu thử, có thể tích cố định hoặc thiết bị có thể tích thay đổi, có thể vận hành bằng tay hoặc tự động. Khi tiêm mẫu bằng tay có thể gây ra sai số do thể tích tiêm vào vòng chứa mẫu không đủ.
Pha tĩnh
Có nhiều loại pha tĩnh có thể được sử dụng trong sắc ký lỏng, bao gồm:
• Silica (silic dioxyd), nhôm oxyd hoặc than graphit xốp thường được dùng trong sắc ký pha thuận mà quá trình phân tách dựa trên sự khác nhau về khả năng hấp phụ hoặc (và) phân bố khối lượng;
• Nhựa hoặc polymer có chứa các nhóm chức acid hoặc base, sử dụng trong sắc ký trao đổi ion mà trong đó sự chia tách được thực hiện dựa trên sự cạnh tranh giữa các ion cần tách và các ion trong pha động;
• Silica xốp hoặc polymer, sử dụng trong sắc ký rây phân tử, ở đó sự chia tách dựa trên sự khác nhau về kích thước phân tử, tương ứng với sự loại trừ không gian;
• Rất nhiều chất mang biến đổi hóa học được chế tạo từ polymer, silica gel hoặc than graphit xốp được dùng trong sắc ký lỏng pha đảo mà ở đó sự chia tách về nguyên tắc cơ bản dựa trên sự phân bố phân tử các chất giữa pha động và pha tĩnh;
• Pha tĩnh loại biến đổi hóa học đặc biệt, ví dụ dẫn xuất của celulose hoặc amylose, protein hoặc peptid, cyclodextrin v.v... dùng để tách các đồng phân đổi quang (sắc ký đổi quang).
Phần lớn sự chia tách dựa trên cơ chế phân bố, sử dụng silica biến đổi hóa học làm pha tĩnh và các dung môi phân cực làm pha động. Bề mặt của chất mang, ví dụ như các nhóm silanol của silica được phản ứng với các thuốc thử silan khác nhau tạo thành các dẫn xuất silyl có liên kết cộng hóa trị, che phủ một số lượng khác nhau các vị trí hoạt động trên bề mặt chất mang. Bản chất của các pha liên kết là tham số quan trọng để xác định các tính chất tách của hệ sắc ký.
Các pha liên kết dùng phổ biến là:
Octyl = Si-(CH2)7-CH3 C8
Octadecyl = Si-(CH2)17-CH3 C18
Phenyl = Si-(CH2)n-(C6H5) C6H5
Cyanopropyl = Si-(CH2)3-CN CN
Aminopropyl = Si-(CH2)3-NH2 NH2
Diol = Si-(CH2)3-OCH(OH)-CH2-OH
Trừ khi có tiêu chuẩn riêng của nhà sản xuất, thông thường các cột sắc ký pha đảo dựa trên silica được coi là ổn định đối với pha động có pH từ 2,0 tới 8,0. Cột chứa than graphit xốp hoặc các hạt vật liệu polymer như styren - divinylbenzen copolymer ổn định ở một khoảng pH rộng hơn.
Phân tích sử dụng sắc ký pha thuận với pha tĩnh là silica không bị biến đổi, than graphit xốp hoặc silica biến đổi hóa học làm cho phân cực (ví dụ cyanopropyl hoặc diol) và pha động không phân cực được sử dụng trong một số trường hợp.
Đối với sự tách nhằm mục đích phân tích, kích thước hạt của pha tĩnh phổ biến nhất từ 3 µm đến 10 µm. Các hạt có thể hình cầu hoặc không có hình dạng nhất định, có độ xốp khác nhau và diện tích bề mặt đặc hiệu. Những tham số này cấu thành biểu hiện sắc ký của từng pha tĩnh cụ thể. Trong trường hợp pha đảo, các yếu tố bổ sung như bản chất của pha tĩnh, mức độ liên kết, ví dụ như độ dài mạch carbon liên kết, hoặc các nhóm hoạt động bề mặt của pha tĩnh có được che phủ hết hay không. Sự kéo đuôi pic, đặc biệt của các chất base, có thể xảy ra khi có mặt các nhóm silanol bề mặt của silica.
Cột được làm bằng thép không gỉ trừ khi có chỉ dẫn khác trong chuyên luận riêng, có chiều dài và đường kính trong (ϕ) khác nhau được sử dụng cho phân tích sắc ký. Cột với đường kính trong nhỏ hơn 2 mm thường được coi là vi cột. Nhiệt độ của pha động và cột phải được giữ ổn định trong suốt thời gian phân tích. Phần lớn quá trình tách được thực hiện ở nhiệt độ phòng, nhưng cột có thể được làm nóng nhằm thu được hiệu quả cao hơn. Tuy nhiên, nhiệt độ cột cũng không được phép vượt quá 60 °C vì khả năng phân hủy của pha tĩnh hoặc sự thay đổi thành phần của pha động có thể xảy ra.
Pha động
Đối với sắc ký pha thuận, thường sử dụng dung môi ít phân cực. Sự có mặt của nước trong pha động phải được hạn chế và kiểm tra chặt chẽ nhằm thu được kết quả tái lặp lại. Đối với sắc ký lỏng pha đảo, sử dụng pha động chứa nước, có hoặc không có dung môi hữu cơ.
Các thành phần của pha động thường được lọc nhằm loại bỏ các tiểu phân lớn hơn 0,45 µm. Pha động chứa nhiều thành phần được chuẩn bị bằng cách đong các thể tích quy định (trừ khi có chỉ định về khối lượng) của các thành phần riêng lẻ rồi sau đó trộn lẫn với nhau. Ngoài ra, dung môi cũng có thể được cấp qua các bơm riêng lẻ, điều khiển bằng các van chia tỷ lệ, để có thể trộn lẫn theo các tỷ lệ mong muốn. Dung môi thường được loại khí trước khi bơm bằng cách sục khí heli, lắc siêu âm hoặc sử dụng hệ thống lọc màng lọc/chân không trực tuyến nhằm tránh sự tạo bọt khí trong cốc đo của detector.
Dung môi dùng để chuẩn bị pha động thường không được chứa các chất làm ổn định và phải trong suốt (không hấp thụ quang) ở vùng bước sóng phát hiện, nếu như sử dụng detector tử ngoại. Dung môi và những thành phần khác được dùng phải có chất lượng phù hợp. Khi cần điều chỉnh pH chỉ thực hiện với thành phần nước của pha động mà không điều chỉnh với hỗn hợp. Nếu sử dụng dung dịch đệm, cần phải rửa hệ thống bằng hỗn hợp nước và dung môi hữu cơ (5 % tt/tt) nhằm ngăn chặn sự kết tinh muối sau khi kết thúc quá trình sắc ký.
Pha động có thể chứa những thành phần khác, ví dụ một ion trái dấu trong sắc ký tạo cặp ion hoặc một chất chọn lọc đối quang trong trường hợp sắc ký sử dụng pha tĩnh không chọn lọc đối quang.
Detector
Detector hấp thụ tử ngoại/khả kiến gồm cả detector chuỗi diod là được sử dụng phổ biến nhất. Detector huỳnh quang, detector khúc xạ vi sai, detector điện hóa, detector khối phổ, detector tán xạ ánh sáng bay hơi, detector phóng xạ hoặc các loại detector đặc biệt khác cũng có thể được sử dụng.
Các đại lượng đặc trưng cho quá trình sắc ký
Xem phần chung Phụ lục 5. Các kỹ thuật tách sắc ký.
Phương pháp tiến hành
Làm cân bằng cột với pha động và tốc độ dòng theo quy định, ở nhiệt độ phòng hoặc nhiệt độ quy định trong chuyên luận riêng, cho đến khi đường nền ổn định. Chuẩn bị các dung dịch chuẩn và dung dịch thử theo yêu cầu. Các dung dịch phải không được có các tiểu phân rắn.
Hiệu năng
Quy định về sự phù hợp của hệ thống được mô tả trong Phụ lục 5. Các kỹ thuật tách sắc ký. Xác định và điều chỉnh các thông số của hệ thống sắc ký có thể được thực hiện nhằm đáp ứng các yêu cầu về sự phù hợp của hệ thống cũng được trình bày trong Phụ lục này.
Thành phần và tốc độ dòng của pha động được quy định trong chuyên luận riêng. Pha động là hỗn hợp dung môi được đuổi khí bằng bơm chân không hoặc bằng một thiết bị đuổi khí khác phù hợp nhưng không ảnh hưởng đến thành phần của hỗn hợp.
Trong quá trình định lượng, khi trong chuyên luận riêng không quy định dùng chuẩn nội, nên sử dụng bộ phận tiêm mẫu có thể tích cố định. Trong một số trường hợp ngoại lệ, khi trong chuyên luận chỉ dẫn tính theo chiều cao pic thì không cần quan tâm đến hệ số đối xứng.
Cột sắc ký thường được làm bằng thép không gỉ có kích thước (chiều dài × đường kính trong) được quy định trong chuyên luận riêng. Trong chuyên luận riêng, khi pha tĩnh được ký hiệu bằng một chữ cái thì tra cứu ở phần Nguyên vật liệu nêu ở dưới đây. Đường kính danh nghĩa của hạt pha tĩnh được để trong ngoặc đơn ngay sau ký hiệu chữ cái cụ thể. Nếu không có chỉ dẫn khác trong chuyên luận riêng, quá trình sắc ký được tiến hành ở điều kiện nhiệt độ không đổi trong môi trường phòng thí nghiệm. Khi sử dụng các pha động có pH cao với cột có bản chất là silica nên sử dụng một tiền cột ở trước cột phân tích.
Trừ khi có các chỉ dẫn khác trong chuyên luận riêng, hệ thống detector gồm một detector đo quang gắn với một cốc đo có thể tích nhỏ (khoảng 10 µl là phù hợp). Phải đặt bước sóng theo chỉ dẫn trong chuyên luận riêng.
Khi dùng một máy sắc ký có thiết kế riêng biệt có thể phải thay đổi các điều kiện sắc ký đã ghi trong chuyên luận riêng. Trong trường hợp này, người phân tích cần đảm bảo rằng những thay đổi đó cho kết quả tương đương.
Thể tích tiêm
Khi thể tích tiêm không quy định trong chuyên luận riêng, nên chọn một thể tích tiêm phù hợp để áp dụng. Chọn thể tích tiêm phụ thuộc vào đáp ứng của phép phân tích, detector sử dụng, hiệu lực cột và toàn bộ hiệu năng của hệ thống sắc ký. Khi không có chỉ dẫn, thường dùng thể tích tiêm là 20 µl, tuy nhiên cần kiểm tra sự thích hợp trong điều kiện cụ thể.
Pic phụ (Pic thứ cấp)
Có thể cần chất đối chiếu để xác định pic phụ. Pic phụ là một pic có trên sắc ký đồ nhưng không phải là pic chính hay pic của chuẩn nội hoặc pic của dung môi hay của các thuốc thử tạo dẫn xuất.
Nguyên vật liệu
Các dung môi và thuốc thử dùng để pha các dung dịch sử dụng trong phân tích phải có chất lượng thích hợp cho sắc ký lỏng.
Khi chuyên luận riêng quy định pha tĩnh được gán với một chữ cái (A hoặc B hoặc C) là muốn nói tới các pha tĩnh như mô tả dưới đây:
Pha tĩnh A, hạt silica;
Pha tĩnh B, hạt silica được biến đổi hóa học, gắn với nhóm octylsilyl (C8);
Pha tĩnh C, hạt silica được biến đổi hóa học, gắn với nhóm octadecylsilyl (C18).
5.4 PHƯƠNG PHÁP SẮC KÝ LỚP MỎNG
Phương pháp sắc ký lớp mỏng được dùng để định tính, thử tinh khiết và đôi khi để bán định lượng hoặc định lượng hoạt chất thuốc.
Sắc ký lớp mỏng là một kỹ thuật tách các chất được tiến hành khi cho pha động di chuyển qua pha tĩnh trên đó đã chấm hỗn hợp các chất cần tách. Pha tĩnh là chất hấp phụ được chọn phù hợp theo từng yêu cầu phân tích, được trải thành lớp mỏng đồng nhất và được cố định trên các phiến kính hoặc phiến kim loại. Pha động là một hệ dung môi đơn hoặc đa thành phần được trộn với nhau theo tỷ lệ quy định trong từng chuyên luận. Trong quá trình di chuyển qua lớp hấp phụ, các cấu tử trong hỗn hợp mẫu thử được di chuyển trên lớp mỏng, theo hướng pha động, với những tốc độ khác nhau. Kết quả thu được là một sắc ký đồ trên lớp mỏng. Cơ chế của sự tách có thể là cơ chế hấp phụ, phân bố, trao đổi ion, sàng lọc phân tử hay sự phối hợp đồng thời của nhiều cơ chế tùy thuộc vào tính chất của chất làm pha tĩnh và dung môi làm pha động.
Đại lượng đặc trưng cho mức độ di chuyển của chất phân tích là hệ số di chuyển Rf được tính bằng tỷ lệ giữa khoảng dịch chuyển của chất thử và khoảng dịch chuyển của dung môi:
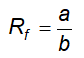
trong đó:
a là khoảng cách di chuyển của chất phân tích;
b là khoảng cách di chuyển của dung môi tính từ điểm chấm mẫu.
Rf: Chỉ có giá trị từ 0 đến 1.
Ngoài ra, khi sắc ký liên tục không xác định được tuyến dung môi, vị trí vết chất thử trên sắc đồ có thể xác định bằng hệ số dịch chuyển tương đối Rr. Hệ số dịch chuyển tương đối Rr được xác định bằng tỷ số giữa khoảng cách dịch chuyển của vết chất thử và khoảng cách dịch chuyển của vết chất chuẩn đối chiếu được sắc ký trong cùng điều kiện và trên cùng bản mỏng với mẫu thử:
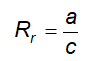
trong đó:
a là khoảng cách từ điểm xuất phát đến tâm của vết mẫu thử;
c là khoảng cách từ điểm xuất phát đến tâm của vết chất chuẩn.
Giá trị Rr có thể lớn hay nhỏ hơn 1.
Cách tiến hành
Dụng cụ
Bình triển khai, thường bằng thủy tinh trong suốt có kích thước phù hợp với các phiến kính cần dùng và có nắp đậy kín.
Đèn tử ngoại, phát các bức xạ có bước sóng ngắn 254 nm và bước sóng dài 365 nm.
Dụng cụ để phun thuốc thử.
Tủ sấy điều nhiệt để hoạt hóa và sấy bản mỏng và sắc ký đồ, hoặc để sấy nóng đối với một số phản ứng phát hiện.
Tủ hút hơi độc.
Máy sấy dùng để sấy khô sắc ký đồ và cho phép chấm nhanh nhiều lần những dung dịch pha loãng chất cần phân tích.
Một máy ảnh tốt có thể chụp lưu giữ sắc ký đồ ở ánh sáng ban ngày với khoảng cách 30 cm đến 50 cm. Tủ lạnh để bảo quản những thuốc thử dễ hỏng.
Micropipet nhiều cỡ từ 1 µl, 2 µl, 5 µl, 10 µl đến 20 µl, các ống mao quản hoặc dụng cụ thích hợp.
Bản mỏng tráng sẵn chất hấp phụ có chất phát quang thích hợp.
Trường hợp phòng thí nghiệm không có điều kiện trang bị các loại bản mỏng tráng sẵn thì tự chuẩn bị lấy bản mỏng với các dụng cụ sau đây:
Các tấm kính phẳng có kích thước phù hợp đã được xử lý trước bằng hóa chất rồi rửa sạch bằng nước và sấy khô.
Thiết bị trải chất hấp phụ lên tấm kính thành một lớp mỏng đồng đều, có chiều dày thích hợp.
Giá để xếp các tấm kính đã trải.
Chuẩn bị bản mỏng
Sắp xếp các bản mỏng và chuẩn bị thiết bị: Các phiến kính phải được lau chùi cẩn thận và tẩy sạch hoàn toàn các chất béo bằng cách ngâm trong dung dịch sulfocromic. Sau đó, rửa dưới tia nước máy rồi tráng nước cất và làm khô trên giá ở nhiệt độ thường hay trong tủ sấy. Đặt lên khung trải 5 phiến kính 20 cm × 20 cm (hay phiến kính 10 cm × 20 cm) dày bằng nhau. Điều chỉnh độ dày của lớp hấp phụ bằng các thông số trên thiết bị trải.
Điều chế vữa của chất hấp phụ: Chất hấp phụ được chọn lọc sao cho phù hợp với yêu cầu phân tích như: Silica gel G, kieselguhr, celulose, nhôm oxyd, trong số đó silica gel G được dùng thông dụng nhất. Trộn 25 g silica gel G với 50 ml nước cất và nhào trong cối hoặc lắc mạnh trong bình nón có dung tích 200 ml đến 250 ml, nút kín, trong 30 s đến 45 s. Dịch treo tạo được ở dạng lỏng và đồng nhất. Rót ngay vào thiết bị trải đã điều chỉnh độ dày cho bản mỏng khoảng 0,25 mm (nếu không có chỉ dẫn trong chuyên luận riêng). Sau khi rót dịch treo vào thiết bị trải, lật tay đòn 180° về phía trái và chờ cho đến khi dịch treo chảy ra thi bắt đầu trải. Khi đã trải đến phiến cuối cùng lật tay đòn về phía phải để giữ cho dịch treo không chảy xuống.
Để nguyên các phiến kính tại chỗ khoảng 10 min tới khi mặt trên hết bóng, hoặc để khô tự nhiên tại nhiệt độ phòng.
Hoạt hóa: Cho các bản mỏng đã khô mặt vào tủ sấy và sấy ở 105 °C đến 110 °C trong 30 min (nếu không có chỉ dẫn ở chuyên luận riêng). Để nguội rồi bảo quản trong bình hút ẩm. Khi dùng, nếu cần thì hoạt hóa lại bằng cách sấy ở 105 °C đến 110 °C trong 1 h rồi cạo bỏ một dải chất hấp phụ dọc hai bên cạnh của tấm kính.
Chuẩn bị bình khai triển: Các bình khai triển thường là bình thủy tinh, hình hộp hay hình trụ, có nắp đậy kín, kích thước thay đổi tùy theo yêu cầu của các bản mỏng sử dụng. Bão hòa hơi dung môi vào khí quyển trong bình bằng cách lót giấy lọc xung quanh thành trong của bình, rồi rót một lượng vừa đủ dung môi vào bình, lắc rồi để giấy lọc thấm đều dung môi. Lượng dung môi sử dụng sao cho sau khi thấm đều giấy lọc còn lại một lớp dày khoảng 5 mm đến 10 mm ở đáy bình. Đậy kín nắp bình và để yên 1 h ở nhiệt độ 20 °C đến 25 °C. Muốn thu được những kết quả lặp lại, ta chỉ nên dùng những dung môi thật tinh khiết, loại dùng cho sắc ký. Những dung môi dễ bị biến đổi về hóa học chỉ nên pha trước khi dùng. Nếu sử dụng những hệ pha động phức tạp phải chú ý đến những thành phần dễ bay hơi làm thay đổi thành phần của hệ pha động dẫn đến hiện tượng không lặp lại của trị số Rf.
Chấm chất phân tích lên bản mỏng: Lượng chất hoặc hỗn hợp chất đưa lên bản mỏng có ý nghĩa quan trọng đối với hiệu quả tách sắc ký, đặc biệt ảnh hưởng rất lớn đến trị số Rf. Lượng chất quá lớn làm cho vết sắc ký lớn và kéo dài, khi đó, vết của các chất có trị số Rf gần nhau sẽ bị chồng lấp. Lượng chất nhỏ quá có thể không phát hiện được do độ nhạy của thuốc thử không đủ (thông thường độ nhạy của các thuốc thử trên 0,005 µg). Lượng mẫu thông thường cần đưa lên bản mỏng là 0,1 µg đến 50 µg ở dạng dung dịch trong dung môi thích hợp. Thể tích dung dịch từ 0,001 ml đến 0,005 ml đối với trường hợp đưa mẫu lên bản mỏng dưới dạng điểm và từ 0,1 ml đến 0,2 ml khi đưa mẫu lên bản mỏng dưới dạng vạch như trong trường hợp sắc ký điều chế. Đối với các dung dịch có nồng độ rất loãng thì có thể làm giàu trực tiếp trên bản mỏng bằng cách chấm nhiều lần ở cùng một vị trí và sấy khô sau mỗi lần chấm.
Đường xuất phát phải cách mép dưới của bản mỏng 1,5 cm đến 2 cm và cách bề mặt dung môi từ 0,8 cm đến 1 cm. Các vết chấm phải nhỏ, có đường kính 2 mm đến 6 mm và cách nhau 15 mm. Các vết ở bìa phải cách bờ bên của bản mỏng ít nhất 1 cm để tránh hiệu ứng bờ. Khi làm sắc ký lớp mỏng bán định lượng, độ chính xác của kết quả phân tích phụ thuộc rất nhiều vào độ chính xác của lượng chất thử đưa lên bản mỏng, tức là thể tích dung dịch chấm lên bản mỏng. Do đó, với những trường hợp phân tích bản định lượng phải dùng các mao quản định mức chính xác. Khi không cần định lượng dùng micropipet hoặc ống mao quản thường.
Triển khai sắc ký: Đặt bản mỏng vào bình triển khai. Chú ý lượng dung môi dùng sao cho để các vết chấm phải ở trên bề mặt của lớp dung môi. Đậy kín bình và để triển khai ở nhiệt độ từ 20 °C đến 25 °C và tránh ánh mặt trời. Nếu không có quy định trong chuyên luận riêng, triển khai sắc ký đến khi dung môi đi được khoảng 3/4 bản mỏng, lấy bản mỏng ra khỏi bình, đánh dấu mức dung môi, làm bay hơi dung môi còn đọng lại trên bản mỏng rồi hiện vết theo chỉ dẫn trong chuyên luận riêng. Quan sát các vết xuất hiện, tính giá trị Rf hoặc Rr và tiến hành định tính, phát hiện tạp chất hoặc định lượng như quy định trong chuyên luận riêng.
CHÚ Ý: Khi phân tích bằng sắc ký lớp mỏng luôn phải chấm chất đối chiếu bên cạnh vết chất thử với lượng tương tự như dự đoán có trong vết chất thử để so sánh kết quả về màu sắc, kích thước và giá trị Rf hoặc Rr trong cùng điều kiện.
Hiệu năng của bản mỏng được kiểm tra như sau: Chấm một thể tích thích hợp (10 µl) cho lớp mỏng thông thường và 1 µl đến 2 µl cho loại bản mỏng có kích thước hạt mịn dung dịch thử hiệu năng lớp mỏng gồm lục bromocresol, da cam methyl, đỏ methyl và sudan G đỏ (thuốc thử). Tiến hành sắc ký với hệ dung môi methanol - toluen (20 : 80). Kết quả trên sắc ký đồ phải có 04 vết tách rõ ràng có Rf như sau: Lục bromocresol: < 0,15; da cam methyl: 0,1 đến 0,25, đỏ methyl: 0,35 đến 0,55 và sudan G đỏ: 0,75 đến 0,98.
5.5 PHƯƠNG PHÁP SẮC KÝ RÂY PHÂN TỬ
Sắc ký rây phân tử là phương pháp chia tách các phân tử trong dung dịch dựa trên kích thước của chúng. Trong trường hợp pha động là một dung môi hữu cơ, kỹ thuật được gọi là sắc ký thấm qua gel, còn trường hợp pha động là nước thì kỹ thuật được gọi là sắc ký lọc trên gel. Mẫu được đưa vào cột chứa đầy gel hoặc một loại vật liệu xốp, và được pha động dẫn chạy qua cột. Sự chia tách theo kích thước được thực hiện nhờ sự trao đổi lặp đi lặp lại các phân tử chất tan giữa dung môi pha động và cùng dung môi đó được pha tĩnh giữ ở trong các lỗ xốp của gel. Khoảng kích thước lỗ của vật liệu nhồi trong cột sẽ xác định khoảng kích thước phân tử được chia tách qua quá trình sắc ký.
Những phân tử có kích thước đủ nhỏ để có thể đi vào trong tất cả khoảng không gian của lỗ xốp được rửa giải trong thể tích thấm tổng Vt. Các phân tử có kích thước lớn hơn kích thước lỗ xốp lớn nhất chỉ di chuyển được dọc theo cột qua các khoảng trống giữa các hạt vật liệu nhồi mà không bị giữ lại, được rửa giải trong thể tích loại trừ Vo (thể tích trống). Sự chia tách theo kích thước phân tử xảy ra giữa thể tích trống và tổng thể tích thấm, chia tách hiệu quả thường được thực hiện ở hai phần ba đầu tiên của khoảng này.
Thiết bị
Thiết bị bao gồm một cột sắc ký có kích thước phù hợp, nếu cần thiết phải khống chế nhiệt độ, được nhồi đầy vật liệu chia tách có thể phân loại theo các khoảng kích thước phân tử phù hợp. Kích thước của cột (chiều cao × đường kính trong) được quy định ở chuyên luận riêng. Pha động chạy qua cột ở tốc độ không đổi dưới tác dụng của trọng lực hoặc nhờ một bơm. Một đầu của cột được nối với một thiết bị phù hợp để cấp mẫu theo kiểu bộ phận điều chỉnh lưu lượng dòng, một bơm tiêm đi qua vách hoặc một van tiêm và có thể được nối với một bơm điều khiển lưu lượng dòng dung dịch rửa giải. Một cách khác là mẫu có thể được tiêm vào trực tiếp trên bề mặt lớp gel hoặc dưới dung dịch rửa giải nếu như mẫu có tỷ trọng lớn hơn dung dịch rửa giải.
Đường ra của cột được nối với một detector có gắn với một máy ghi tự động để theo dõi nồng độ tương đối của các thành phần trong mẫu. Detector thường được thiết kế dựa trên các tính chất hấp thụ quang, khúc xạ ánh sáng hay phát quang. Bộ phận thu hứng phân đoạn tự động có thể được lắp trong trường hợp cần thiết. Vật liệu trong cột có thể là một chất mang mềm như gel nở hay một chất mang cứng cấu tạo từ thủy tinh, silic hoặc một hợp chất cao phân tử liên kết chéo tương thích dung môi. Chất mang cứng thường đòi hỏi hệ thống điều áp để tăng tốc độ phân tách. Pha động thường được chọn theo loại mẫu, môi trường phân tách và phương pháp phát hiện.
Vật liệu nhồi cần được xử lý và được nạp vào cột như mô tả trong chuyên luận hoặc theo hướng dẫn của nhà sản xuất.
Hiệu năng
Hệ thống sắc ký phải thỏa mãn quy định về sự phù hợp được mô tả trong phần Các kỹ thuật tách sắc ký (Phụ lục 5). Việc điều chỉnh các thông số của hệ sắc ký có thể được thực hiện nhằm đáp ứng các yêu cầu về sự phù hợp của hệ thống cũng được trình bày trong phụ lục này.
Xác định tỷ lệ thành phần tương đối
Tiến hành phương pháp theo các điều kiện ghi trong chuyên luận riêng. Nếu có thể, theo dõi quá trình rửa giải một cách liên tục và ghi lại diện tích của các pic tương ứng. Nếu mẫu được xác định dựa vào tính chất hóa lý mà tất cả các thành phần của mẫu đều cho một đáp ứng tương đương (ví dụ cùng có một hệ số hấp thụ riêng), thì lượng tương đối của mỗi thành phần có thể được xác định bằng cách tính tỷ lệ diện tích mỗi pic thu được trên tổng diện tích pic của những thành phần quan tâm. Nếu sự đáp ứng khác nhau, tính tỷ lệ thành phần tương đối bằng cách dựa vào đường chuẩn của các dung dịch chuẩn được ghi trong chuyên luận riêng hoặc bằng phương pháp khác được ghi trong chuyên luận riêng.
Xác định khối lượng phân tử
Thực hiện phương pháp với dung dịch thử và các dung dịch chuẩn theo quy trình ghi trong chuyên luận riêng. Xây dựng một đồ thị biểu thị sự phụ thuộc giữa thể tích lưu của các chất chuẩn dùng cho hiệu chuẩn và hàm logarit của khối lượng phân tử của chúng. Đường chuẩn thường xấp xỉ với một đường thẳng nằm giữa giới hạn loại trừ và giới hạn thấm tổng thể. Khối lượng phân tử của thành phần quan tâm có thể được ước lượng từ đường chuẩn. Việc xây dựng đường chuẩn chỉ có giá trị đối với một hệ cụ thể trong những điều kiện thí nghiệm xác định.
Sự phân bố polymer theo kích thước phân tử
Sắc ký rây phân tử có thể được sử dụng để xác định sự phân bố các polymer theo kích thước phân tử. Tuy nhiên, việc so sánh mẫu sẽ chỉ có ý nghĩa nếu kết quả thu được là cùng một điều kiện thí nghiệm.
Nguyên liệu sử dụng để xây dựng đường chuẩn và phương pháp xác định sự phân bố dựa theo kích thước phân tử của các polymer được ghi trong chuyên luận riêng.
Phân bố khối lượng phân tử trong dextran
Kiểm tra bằng phương pháp sắc ký rây phân tử.
Dung dịch thử: Hòa tan 0,200 g mẫu thử vào pha động và pha loãng tới 10 ml bằng pha động.
Dung dịch đánh dấu: Hòa tan 5 mg dextrose chuẩn và 2 mg chất đối chiếu hóa học dextran Vo trong 1 ml pha động.
Dung dịch hiệu chuẩn: Hòa tan riêng rẽ 15 mg các chất đối chiếu hóa học dextran 4, dextran 10 dùng cho hiệu chuẩn và 20 mg các chất đối chiếu hóa học dextran 40, dextran 70, dextran 250 dùng cho hiệu chuẩn trong 1 ml pha động.
Dung dịch kiểm tra tính phù hợp của hệ thống:
Hòa tan 20 mg chất đối chiếu dextran 40 dùng cho chuẩn hiệu năng (đối với dextran 40) hoặc 20 mg dextran 60/70 dùng cho chuẩn hiệu năng (đối với dextran 60 và dextran 70) vào 1 ml pha động.
Điều kiện sắc ký:
Cột (0,3 m × 10 mm) nhồi agarose loại liên kết chéo dùng trong sắc ký hoặc một dãy các cột (0,3 m × 10 mm) nhồi gel polyether hydroxyl hóa dùng trong sắc ký.
Pha động: Dung dịch chứa 7 g natri sulfat khan và 1 g clorobutanol trong 1 lít nước.
Tốc độ dòng: 0,5 ml/min đến 1 ml/min, giữ ổn định với sai số ± 1 %/h.
Detector khúc xạ ánh sáng vi sai.
Buồng tiêm mẫu dung tích 100 µl đến 200 µl.
Duy trì hệ thống hoạt động ở nhiệt độ ổn định (± 0,1 °C).
Chuẩn hóa hệ thống sắc ký
Tiến hành tiêm lặp lại vài lần một thể tích nhất định dung dịch đánh dấu. Trên sắc ký đồ sẽ xuất hiện 2 pic, pic đầu tương ứng chất đối chiếu dextran Vo và pic thứ 2 là của dextrose. Từ thể tích rửa giải của pic tương ứng với dextran Vo, tính thể tích trống Vo và từ pic tương ứng với dextrose, tính được thể tích thấm tổng Vt.
Tiêm một thể tích nhất định các dung dịch chuẩn. Vẽ cẩn thận đường nền của mỗi sắc ký đồ. Phân chia mỗi sắc ký đồ thành p phần (ít nhất là 60) bằng những đường gạch thẳng dọc bằng nhau (tương ứng với những thể tích rửa giải).
Trong mỗi phần, i tương ứng với một thể tích rửa giải Vi, đo chiều cao (yi) của đường sắc ký đồ phía trên đường nền và tính hệ số phân bố Ki theo công thức:
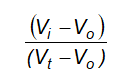 (1)
(1)
trong đó:
Vo là thể tích trống của cột, được xác định bởi pic tương ứng với chất đối chiếu hóa học dextranVo trong sắc ký đồ thu được từ dung dịch đánh dấu;
Vt là thể tích thấm tổng của cột, được xác định bởi pic tương ứng với dextrose trong sắc ký đồ thu được từ dung dịch đánh dấu;
Vi là thể tích rửa giải của phần i trong sắc ký đồ thu được với mỗi dung dịch chuẩn.
Tiến hành hiệu chuẩn sử dụng một trong những phương pháp dưới đây:
Vẽ đường cong chuẩn
Với mỗi chất đối chiếu hóa học dextran dùng cho hiệu chuẩn, tính hệ số phân bố Kmax tương ứng với chiều cao cực đại của đường sắc ký, sử dụng công thức (1). Trên giấy kẻ ô bán logarit đặt các giá trị của Kmax (trục x) tương ứng với khối lượng phân tử ghi trên nhãn của mỗi chất dextran hiệu chuẩn và của dextrose; những giá trị này được lấy tại điểm cao nhất của đường sắc ký (Mmax). Vẽ đường cong chuẩn đầu tiên qua những điểm thu được, ngoại suy từ điểm Kmax thu được với chất đối chiếu hóa học dextran 250 dùng cho hiệu chuẩn tới giá trị K thấp nhất thu được từ chất đối chiếu hóa học này (xem hình 5.5.1). Sử dụng đường cong chuẩn đầu tiên này, chuyển tất cả các giá trị Ki ở mỗi sắc ký đồ sang khối lượng phân tử tương ứng Mi để thu được sự phân bố khối lượng phân tử. Tính khối lượng phân tử trung bình Mw cho mỗi loại dextran đối chiếu hóa học sử dụng phương trình (3) dưới đây. Nếu giá trị thu được đối với Mw không khác hơn 5 % so với giá trị công bố của mỗi dextran và mức khác biệt trung bình là ± 3 %, đường cong chuẩn được chấp nhận. Nếu không đạt yêu cầu đó, chuyển dịch đường cong chuẩn dọc theo trục y và lặp lại tiến trình này cho tới khi các giá trị Mw thu được và giá trị công bố không khác nhau lớn hơn 5 %.
Hình 5.5.1-Ví dụ về đường cong chuẩn. Đường chấm chấm tương ứng với phần đường cong ngoại suy. Đường nằm ngang ở dưới cùng của đồ thị thể hiện độ rộng và vị trí của đường sắc ký thu được tương ứng với mỗi dextran chuẩn.
Tính qua đường cong chuẩn
Tính từ phương trình (2) và (3) dưới đây, sử dụng phương pháp thích hợp1, các giá trị b1, b2, b3, b4 và b5 cho phép thu được giá trị Mw nằm trong khoảng 5 % của tất cả những giá trị công bố từ mỗi dextran chuẩn và khối lượng phân tử của dextrose nằm trong khoảng 180 ± 2.
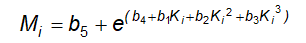 (2)
(2)
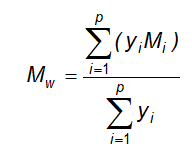 (3)
(3)
trong đó:
p là số phân đoạn chia trong sắc ký đồ;
yi là chiều cao của đường sắc ký phía trên đường nền trong phân đoạn i;
Mi là khối lượng phân tử trong phân đoạn i.
Tính phù hợp của hệ thống
Tiêm một thể tích nhất định dung dịch thử tính phù hợp hệ thống thích hợp.
Khối lượng phân tử trung bình của chất đối chiếu hóa học dextran dùng cho thử hiệu năng
Tính khối lượng phân tử trung bình Mw như đã trình bày trong phần hiệu chuẩn hệ sắc ký, sử dụng đường cong chuẩn hoặc các giá trị thu được ở trên đối với b1, b2, b3, b4, b5. Phép thử chỉ có giá trị khi Mw nằm trong khoảng 41 000 đến 47 000 (chất đối chiếu hóa học dextran 40 dùng cho chuẩn hiệu năng) và 67 000 đến 75 000 (chất đối chiếu hóa học dextran 60/70 dùng cho chuẩn hiệu năng).
Khối lượng phân tử trung bình của dextran phân đoạn cao 10%
Tính Mw đối với dextran phân đoạn cao 10 % được rửa giải qua phần thứ n sử dụng công thức:
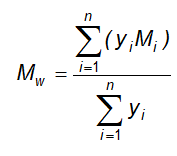 (4)
(4)
Với n được xác định như sau:
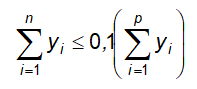 (5)
(5)
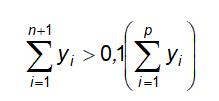 (6)
(6)
trong đó:
p là số phần chia trong sắc ký đồ;
yi là chiều cao đường sắc ký phía trên đường nền trong phần i;
Mi là khối lượng phân tử của phần i.
Phép thử chỉ có giá trị khi Mw của dextran phân đoạn cao nằm trong khoảng: 110 000 đến 130 000 (chất đối chiếu hóa học dextran 40 dùng cho hiệu chuẩn) và 190 000 đến 230 000 (chất đối chiếu hóa học dextran 60/70 dùng cho hiệu chuẩn).
Khối lượng phân tử trung bình của dextran phân đoạn thấp 10%
Tính Mw đối với dextran phân đoạn thấp 10 % rửa giải trong và sau phần thứ m theo công thức sau:
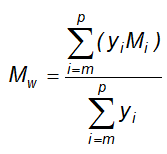 (7)
(7)
trong đó:
m được xác định như sau:
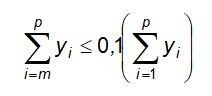 (8)
(8)
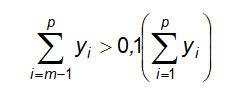 (9)
(9)
p là số phần chia trong sắc ký đồ;
yi là chiều cao đường sắc ký phía trên đường nền trong phần i;
Mi là khối lượng phân tử của phần i.
Phép thử chỉ có giá trị khi Mw của dextran phân đoạn thấp 10 % là: 6 000 đến 8 500 (chất đối chiếu hóa học dextran 40 dùng cho chuẩn hiệu năng) và 7 000 đến 11 000 (chất đối chiếu hóa học dextran 60/70 dùng cho chuẩn hiệu năng).
Sự phân bố khối lượng phân tử của dextran được phân tích
Tiêm một thể tích nhất định dung dịch (1) và tính của phân bố tổng khối lượng phân tử, Mw của dextran phân đoạn cao 10 % và Mw của dextran phân đoạn thấp 10 % như chỉ dẫn trong phần thử tính phù hợp của hệ thống.
1Phương pháp lặp như phương pháp Gauss - Newton cải tiến bởi Hartley là phù hợp [Xem O Hartley, Tecnometrics, 3 (1961) and G Nilsson and K Nilsson, J.Chromat. 101, 137 (1974)]. Một chương trình đường cong thích hợp xử lý tương quan hồi quy không tuyến tính chạy trên máy tính điện tử có thể được sử dụng.
5.6 PHƯƠNG PHÁP ĐIỆN DI
Điện di là hiện tượng liên quan đến sự di chuyển của các protein, các colloid, các phân tử, hay các tiểu phân tích điện hòa tan hoặc phân tán trong dung dịch điện giải khi có dòng điện đi qua.
Điện di được chia thành hai loại tùy theo thiết bị sử dụng: Điện di dung dịch tự do hay điện di với sự chuyển dịch của các lớp biên và điện di vùng.
Trong phương pháp điện di dung dịch tự do, dung dịch đệm của các protein trong một ống hình chữ U dưới tác dụng của dòng điện sẽ hình thành nên các lớp protein có linh độ giảm dần. Chỉ phần di chuyển nhanh nhất của protein sẽ tách rõ rệt khỏi các protein khác, kiểm tra sự dịch chuyển của các lớp biên bằng hệ thống quang học sẽ cung cấp các dữ liệu để tính toán linh độ điện di và các thông tin về mặt định tính và định lượng của thành phần hỗn hợp protein.
Trong điện di vùng, mẫu được đưa vào dưới dạng một dải hẹp hay điểm trong cột, trên tấm phẳng, hay trên một lớp phim chứa đệm. Sự di chuyển của các thành phần trong các dải hẹp cho phép tách hoàn toàn chúng. Có thể tránh sự chồng lấn giữa các dải hẹp do sự đối lưu nhiệt bằng cách đưa chất điện giải vào các nền (matrix) xốp như chất rắn dạng bột, hay nguyên liệu dạng sợi như giấy, hay gel như tinh bột, agar hoặc polyacrylamid.
Các phương pháp điện di vùng có ứng dụng rộng rãi. Điện di gel, đặc biệt là điện di đĩa được sử dụng để tách protein do khả năng cho độ phân giải cao.
Sự di chuyển điện di của các tiểu phân của một chất nhất định phụ thuộc vào tính chất của tiểu phân mà chủ yếu là điện tích, kích thước hay khối lượng phân tử và hình dạng của các tiểu phân cũng như các tính chất và các thông số thực nghiệm của hệ thống. Những thông số này bao gồm: pH, nồng độ ion, độ nhớt và nhiệt độ của chất điện giải, mật độ hoặc độ liên kết chéo của các matrix nền như gel và điện thế áp đặt.
Ảnh hưởng của điện tích, kích thước tiểu phân, độ nhớt của chất điện giải và biến thiên điện thế
Các tiểu phân tích điện sẽ di chuyển về phía điện cực ngược dấu với chúng, sự di chuyển của các phân tử tích cả điện dương và điện âm phụ thuộc vào điện tích mạng. Tốc độ di chuyển tỷ lệ thuận với độ lớn của điện tích mạng trên tiểu phân và tỷ lệ nghịch với kích thước tiểu phân, mà kích thước tiểu phân lại có liên quan trực tiếp với phân tử lượng.
Những tiểu phân hình cầu lớn, khi định luật Stoke có giá trị, linh độ điện di µo sẽ tỷ lệ nghịch với bán kính của tiểu phân theo phương trình:
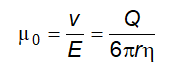
Trong đó v là vận tốc của tiểu phân, E là thế đặt lên chất điện giải, Q là điện tích của tiểu phân, r là bán kính tiểu phân và ƞ là độ nhớt của dung dịch chất điện giải.
Phương trình này chỉ có giá trị giới hạn ở độ pha loãng nhất định và không có sự hiện diện của các nền như giấy hoặc gel.
Các ion và peptid có trọng lượng phân tử lớn hơn 5000, đặc biệt khi có mặt môi trường nền, không tuân theo định luật Stoke và linh độ điện di của chúng được biểu thị bằng phương trình:
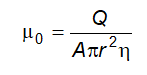
Trong đó A là thừa số hình dạng, thay đổi trong khoảng từ 4 - 6, biểu thị sự phụ thuộc tỷ lệ nghịch của linh độ điện di đối với bình phương bán kính tiểu phân, về mặt khối lượng phân tử, linh độ điện di sẽ tỷ lệ nghịch với căn bậc ba của bình phương khối lượng phân tử.
Ảnh hưởng của pH
Hướng và tốc độ di chuyển của phân tử có nhiều nhóm chức có thể bị ion hóa như acid amin và protein sẽ phụ thuộc vào pH của dung dịch chất điện giải. Ví dụ: Linh độ điện di của acid amin đơn giản như glycin sẽ thay đổi theo giá trị pH gần đúng như trong Hình 5.6.1.
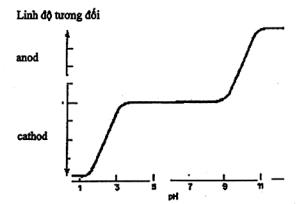
Hình 5.6.1
Giá trị pKa bằng 2,2 và 9,9 trùng với điểm uốn trên biểu đồ. Do nhóm chức bị ion hóa 50 % ở giá trị pH = pKa, nên linh độ điện di ở những điểm này sẽ bằng 1/2 giá trị quan sát được khi cation và anion bị ion hóa hoàn toàn lần lượt ở pH rất thấp và pH rất cao. lon lưỡng cực tồn tại ở khoảng pH trung gian là những chất trung hòa về điện và có linh độ điện di bằng 0.
Ảnh hưởng của lực ion và nhiệt độ
Linh độ điện di sẽ giảm khi tăng lực ion của chất điện giải nền. Hoạt độ ion µ được xác định theo phương trình:
Trong đó Ci là nồng độ ion (mol/l), Zi là hóa trị và tổng được tính với tất cả các ion trong dung dịch. Đối với dung dịch đệm có cả anion và cation hóa trị 1 thì lực ion được xác định bằng nồng độ mol.
Lực ion của chất điện giải dùng trong điện di nói chung nằm trong dãy từ khoảng 0,01 đến 0,10. Hoạt độ ion thích hợp phụ thuộc một phần vào thành phần của mẫu thử, do đó dung lượng đệm phải đủ lớn để duy trì pH hằng định trên diện tích của các dải hợp phần. Các dải hợp phần trở nên sắc nét hơn và hẹp hơn khi lực ion tăng.
Nhiệt độ ảnh hưởng gián tiếp lên linh độ, vì độ nhớt ƞ của chất điện giải nền phụ thuộc vào nhiệt độ. Độ nhớt của nước sẽ giảm theo tỷ lệ khoảng 3 %/ °C trong vùng từ 0 °C đến 5 °C và tỷ lệ giảm này sẽ thấp hơn khi ở nhiệt độ phòng. Bởi vậy, linh độ điện di sẽ tăng cùng với sự tăng nhiệt độ của chất điện giải.
Nhiệt giải phóng là đáng kể khi dòng điện chạy qua chất điện giải. Nhiệt này tăng cùng với sự tăng thế đặt và lực ion tăng, ở các thiết bị lớn, cho dù có bộ phận làm lạnh, nhiệt giải phóng vẫn tạo nên sự biến thiên về nhiệt độ ở bể điện di, dẫn đến làm biến dạng các dải phân cách. Bởi vậy, cần quan tâm thiết kế các dụng cụ đặc biệt tính đến lực ion và thế đặt.
Ảnh hưởng của môi trường nền, dòng điện thẩm
Khi dòng điện chạy qua chất điện giải chứa trong ống thủy tinh hoặc chứa giữa các bản thủy tinh hoặc bản plastic thì xuất hiện dòng chất điện giải hướng về phía đầu một điện cực, gọi là dòng điện thẩm. Dòng điện thẩm được hình thành do điện tích trên bề mặt của thành dụng cụ phát sinh do quá trình ion hóa các nhóm chức trong vật liệu hay do các ion hấp phụ trên thành buồng điện di chứa các chất điện giải. Hiệu ứng này thường tăng khi buồng điện di chứa các chất xốp như gel, được sử dụng để làm ổn định cho chất điện giải nền và đề phòng hiện tượng xáo trộn các dải phân tách do sự đối lưu nhiệt và hiện tượng khuếch tán. Dung dịch ngay sát trên bề mặt sẽ hình thành điện tích bằng nhưng ngược dấu với điện tích trên bề mặt và điện trường đi qua buồng điện di sẽ tạo nên sự chuyển động của dung dịch về phía điện cực ngược dấu.
Các chất thường được sử dụng làm môi trường nền trong điện di vùng sẽ tạo điện tích âm trên bề mặt, dòng điện thẩm của chất điện giải sẽ di chuyển về phía cathod. Vì vậy tất cả các vùng, bao gồm cả các chất trung tính sẽ di chuyển về phía cathod trong quá trình điện di.
Mức độ thẩm thấu sẽ thay đổi khi các chất nền thay đổi. Độ thẩm thấu thay đổi đáng kể đối với gel agar và thay đổi không đáng kể đối với gel polyacrylamid.
Sự rây phân tử
Khi không có sự hiện diện của môi trường nền hay trong trường hợp môi trường rất xốp thì sự tách điện di của các phân tử là do sự khác nhau về tỷ lệ của điện tích với kích thước phân tử. Nếu có sự hiện diện của chất nền, sự khác nhau về khả năng hấp phụ hay ái lực của phân tử đối với môi trường tạo nên hiệu ứng sắc kí có thể làm tăng hiệu quả tách.
Hiệu ứng rây phân tử sẽ xuất hiện nếu môi trường nền là các loại gel có sự phân nhánh nhiều đến mức tạo thành các lỗ xốp để tách các phân tử theo kích thước. Hiệu ứng này tương đương hiệu ứng thu được trong quá trình tách thẩm thấu gel hay sắc ký rây phân tử, nhưng trong điện di gel hiệu ứng này chồng lên quá trình tách điện di. Có thể nhận thấy sự rây phân tử do cản trở không gian đối với sự di chuyển của các phân tử lớn. Các phân tử nhỏ di chuyển qua các lỗ xốp có kích thước lớn, nên sự điện di qua gel không bị cản trở. Khi kích thước phân tử tăng, một số ít hơn các lỗ xốp cho phép các phân tử đi qua, làm chậm sự di chuyển của các phân tử có trọng lượng phân tử lớn.
Điện di gel
Quá trình sử dụng một gel như agar (thạch), tinh bột hay polyacrylamid làm môi trường nền được hiểu theo nghĩa rộng là điện di gel. Phương pháp này đặc biệt thuận lợi cho sự tách các protein. Sự tách này đạt được tùy thuộc vào tỷ lệ điện tích đối với kích thước phân tử đi đôi với hiệu ứng rây phân tử phụ thuộc chủ yếu vào khối lượng phân tử.
Gel polyacrylamid được sử dụng nhiều do có một số thuận lợi. Nó có đặc tính ít hấp phụ và tạo nên hiệu ứng dòng điện thẩm không đáng kể. Các gel với dải rộng các lỗ xốp có kích thước khác nhau có thể được điều chế bằng cách thay đổi nồng độ gel tổng cộng (dựa trên monomer kết hợp với các yếu tố tạo mạch nhánh) và phần trăm của chất tạo mạch nhánh dùng để tạo gel. Lượng các chất được biểu thị bằng phương trình sau:
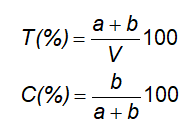
Trong đó T là nồng độ gel tổng cộng (%), C là phần trăm tác nhân tạo mạch nhánh dùng để tạo gel, V là thể tích (mL) của đệm dùng điều chế gel, a và b là khối lượng (g) của monomer (acrylamid) và tác nhân tạo mạch nhánh (thường dùng N,N'-methylenbisacrylamid) dùng điều chế gel. Các gel thích hợp có nồng độ trong khoảng 3 % - 30 %. Tổng tác nhân tạo mạch nhánh thường chiếm khoảng 1/10 đến 1/20 lượng monomer (C = từ 10 % đến 5 %), tỷ lệ phần trăm nhỏ hơn được dùng cho giá trị cao hơn của T.
Trong quá trình điều chế gel, bể của dụng cụ điện di chứa dung dịch nước của monomer và tác nhân tạo mạch nhánh, thường được cân bằng bằng dung dịch đệm đến pH mong muốn trong những lần chạy cuối và được polymer hóa tại chỗ bằng quá trình gốc tự do. Quá trình polymer hóa được kích hoạt bởi quá trình hóa học, thông thường sử dụng amoni persulfat kết hợp với N,N,N',N'-tetramethylendiamin hay quang hóa hỗn hợp riboflavin và N,N,N',N'-tetramethylendiamin. Quá trình polymer hóa bị cản trở bởi các phân tử oxy và môi trường acid. Lựa chọn thành phần của gel và điều kiện polymer hóa phải bảo đảm chất lượng của gel.
Thiết bị cho điện di gel
Nói chung bể hay môi trường để thực hiện quá trình điện di có thể được đặt theo phương thẳng đứng hay nằm ngang tùy theo thiết kế của thiết bị. Để so sánh sự tách có thể thực hiện trong một số ống riêng biệt hay đưa các mẫu khác nhau vào các hố kề bên nhau vào khuôn hay cắt vào phiến của gel. Phiến gel thẳng đứng như mô tả trong Hình 5.6.2 thuận lợi cho sự so sánh trực tiếp một số mẫu.
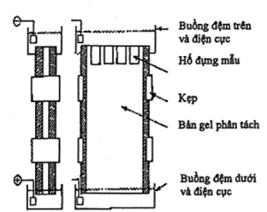
Hình 5.6.2 - Thiết bị điện di gel phiến thẳng đứng
Tốt nhất là sự so sánh các mẫu trong cùng một bản gel, như vậy sẽ đồng nhất hơn về mặt thành phần so với các khuôn gel trong nhiều bể điện di.
Đặc tính của nhiều loại thiết bị là phải hàn kín buồng đệm phía dưới với chân đế của bể điện di, cho phép mức đệm trong buồng đệm phía dưới bằng mức đệm trong buồng đệm phía trên để loại trừ áp suất thủy tĩnh trên gel. Ngoài ra có một vài bộ phận tuần hoàn làm lạnh cả hai mặt bể điện di gel.
Để điều chế gel, buồng gel được đậy bằng nắp thích hợp có chứa dung dịch monomer, tác nhân tạo mạch nhánh và chất xúc tác. Một cái lược có răng với kích thước thích hợp được đặt lên phía trên và cho phép quá trình polymer hóa xảy ra hoàn toàn. Sau khi lấy lược ra sẽ tạo thành các hố trên bản gel được polymer hóa.
Trong điện di gel đơn giản, chỉ sử dụng một dung dịch đệm cho vào buồng đệm phía trên và phía dưới giống nhau. Sau khi cho dung dịch đệm vào buồng đệm, mẫu được hòa tan trong nước đường hay dung dịch đặc và hơi nhớt khác để đề phòng sự khuếch tán. Sau đó mẫu được đưa vào đáy các hố đựng mẫu bằng bơm tiêm hay micropipet và bắt đầu thực hiện quá trình điện di.
Điện di đĩa
Điện di gel polyacrylamid sử dụng một dãy không liên tục các đệm và cũng thường sử dụng nhiều lớp gel không liên tục, được gọi là điện di đĩa. Tên của phương pháp xuất phát từ dạng hình dạng đĩa của các dải hẹp được tạo thành. Do tạo thành các dải hẹp, nên kỹ thuật này có độ phân giải đặc biệt cao và thường được sử dụng trong phân tích đặc tính của hỗn hợp protein và phát hiện tạp chất mà có thể có linh độ gần thành phần chính.
Cơ sở của điện di đĩa được trình bày trong phần sau có liên quan đến các hệ anion thích hợp cho việc phân tách protein mang điện tích âm. Để hiểu được điện di đĩa cần phải có kiến thức chung về điện di và dụng cụ đã được mô tả.
Cơ sở của điện di đĩa
Độ phân giải cao thu được trong điện di đĩa phụ thuộc vào việc sử dụng hệ đệm không liên tục về pH và thành phần. Thường phối hợp hai hoặc ba gel khác nhau về tỷ trọng.
Hệ thống điển hình được mô tả trong Hình 5.6.3.
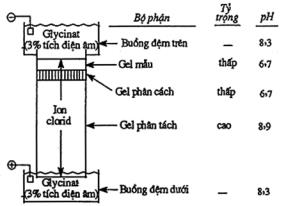
Hình 5.6.3 - Thuật ngữ, pH đệm và thành phần đệm cho điện di dĩa acrylamid
Gel phân tách có chiều cao vài cm với tỷ trọng cao (T = 10 % - 30 %) được polymer hóa trong đệm tris-clorid trong bể của thiết bị. Trong quá trình polymer hóa đệm được phủ lên một lớp nước mỏng để đề phòng hiện tượng tạo thành mặt khum trên bề mặt của gel. Lớp nước phủ lên sau đó được loại đi, lớp gel mỏng tỷ trọng thấp (T = 3 %), có bề dày 3 mm đến 10 mm được gọi là khoảng trống hay gel phân cách, được polymer hóa trong đệm tris-clorid trên mặt của gel phân tách. Lớp nước phủ phía trên lại tiếp tục được sử dụng để tạo mặt phẳng. Mẫu được trộn với một lượng nhỏ dung dịch monomer dùng để tạo gel phân cách, đổ lên mặt của gel phân cách và được polymer hóa. pH của gel phân tách thường là 8,9, trong khi đó pH của gel phân cách và gel chứa mẫu là 6,7. Tất cả ba loại gel này được điều chế dùng clorid làm anion.
Dung dịch đệm chứa trong buồng chứa đệm phía trên và phía dưới có pH là 8,3 và được điều chế từ tris và glycin. Ở pH này khoảng 3 % phân tử glycin mang điện tích âm.
Khi đặt điện thế lên hệ thống, lớp tiếp giáp glycinat-clorid sẽ di chuyển theo hướng đi xuống và về phía anod. Lớp tiếp giáp bắt đầu ở vị trí lớp tiếp xúc của buồng chứa đệm phía trên và đỉnh của lớp gel chứa mẫu. Tại đây ion clorid do có kích thước nhỏ nên sẽ di chuyển nhanh hơn bất kỳ protein nào hiện diện trong mẫu. pH của lớp gel mẫu và lớp gel phân cách được chọn phải thấp hơn giá trị pKa cao của glycin khoảng 3 đơn vị. Do vậy, khi băng qua các lớp gel này, chỉ có khoảng 0,1 % phân tử glycin tích điện âm. Cho nên, glycin sẽ di chuyển chậm hơn clorid. Khuynh hướng dịch chuyển nhanh hơn của clorid khỏi glycinat làm giảm nồng độ nơi tiếp giáp, tạo nên sự giảm thế nhiều hơn ở lớp tiếp giáp, điều này làm cho glycinat bắt kịp ion clorid. Trong điều kiện này, lớp tiếp giáp rất hẹp được duy trì, và khi đó nó di chuyển qua lớp gel mẫu thử và lớp gel phân cách, các protein trong mẫu thử ở lớp tiếp giáp sắp xếp thành lớp rất mỏng theo thứ tự về linh độ. Quá trình này là nguồn gốc của việc các đĩa được phân tách.
Các protein được sắp xếp theo trình tự khi tiếp xúc với gel phân tách tỷ trọng cao, chúng sẽ di chuyển chậm lại do quá trình rây phân tử. pH cao hơn trong gel phân tách làm cho glycinat di chuyển nhanh hơn, đến mức độ làm cho lớp tiếp xúc của các đệm không liên tục sẽ vượt qua các protein và cuối cùng sẽ tiếp xúc với đáy của gel phân tách. Trong chu trình này, các đĩa protein tiếp tục phân tách bằng quá trình điện di và quá trình rây phân tử trong gel phân tách. Kết thúc quá trình tách, pH của gel phân tách sẽ tăng từ giá trị ban đầu là 8,9 lên khoảng pH 9,5.
Linh độ tương đối
Bromophenol xanh thường được sử dụng như là một chuẩn để tính toán linh độ tương đối của các dải phân tách và để quan sát các quá trình tách. Nó có thể được thêm vào trong hố đựng mẫu hoặc trộn lẫn với mẫu, hoặc đơn giản được thêm vào dung dịch đệm ở buồng chứa đệm phía trên.
Linh độ tương đối MB được tính toán như sau:
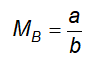
a là khoảng cách từ mốc xuất phát đến vùng mẫu và b là khoảng cách từ mốc xuất phát đến vùng bromo-phenol xanh.
Quan sát các vùng phân tách
Do polyacrylamid trong suốt, nên các dải protein có thể được quan sát bằng đo mật độ quang với ánh sáng UV. Các vùng có thể cố định bằng cách nhúng vào các dung dịch làm tủa protein như acid phospho-tungstic hoặc acid tricloracetic 10 %. Các thuốc thử hiện màu được sử dụng bao gồm naphthalen đen (amido đen) và Coomassie lam sáng R250. Những vùng được cố định hay có mầu có thể được quan sát một cách thuận lợi và được chụp ảnh với ánh sáng truyền qua từ nguồn phát tia X.
Những điều an toàn cần chú ý
Thế sử dụng trong điện di có thể gây sốc chết người. Nguy cơ này sẽ tăng do sử dụng dung dịch đệm nước và điều kiện môi trường làm việc ẩm ướt.
Bộ phận cung cấp nguồn của thiết bị cần được bao lại trong các lồng kim loại có dây nối đất hoặc cần được chế tạo bằng các vật liệu cách điện, các lồng kim loại này cần có chốt tự cắt nguồn khi mở nắp thiết bị.
Vì sự nguy hiểm của thế cao áp, nhân viên phòng thí nghiệm cần làm quen với thiết bị trước khi sử dụng chúng.
5.7 PHƯƠNG PHÁP ĐIỆN DI MAO QUẢN
Điện di là hiện tượng di chuyển của tiểu phân tích điện hòa tan hay phân tán trong chất điện giải khi có dòng điện đi qua. Cation di chuyển về phía cực âm (cathod), anion di chuyển về phía cực dương (anod). Các phần tử không tích điện không bị hút về phía hai điện cực.
Trong phương pháp điện di mao quản (CE) việc sử dụng mao quản để làm kênh di chuyển cho phép thực hiện quá trình tách điện di trên một thiết bị có thể so sánh với thiết bị của sắc ký lỏng hiệu năng cao. Tuy nhiên, vẫn còn những khác biệt rõ rệt về cách vận hành, những điểm thuận lợi và không thuận lợi so với sắc ký lỏng hiệu năng cao. Quá trình vận hành của CE điển hình với mao quản mà mặt trong thành mao quản không có lớp bao và mao quản chứa dung dịch “đệm làm việc”, thì nhóm silanol hiện diện trên thành phía trong của mao quản thủy tinh sẽ giải phóng ion hydrogen vào dung dịch đệm và bể mặt thành mao quản sẽ tích điện âm ngay cả ở pH rất thấp. Cation hay các chất hòa tan tích điện dương một phần trong môi trường bị hút tĩnh điện vào thành mao quản tích điện âm tạo nên một lớp điện tích kép. Quá trình điện di bắt đầu bằng cách đặt thế trên chiều dài cột mao quản làm cho phần dung dịch của lớp điện tích kép di chuyển về phía đầu cathod của mao quản kéo theo khối dung dịch. Sự chuyển động của khối dung dịch dưới tác dụng của lực điện trường được gọi là dòng điện thẩm (electroosmotic flow - EOF). Mức độ ion hóa của nhóm silanol trên thành mao quản phụ thuộc chủ yếu vào pH của dung dịch đệm làm việc và các chất điều chỉnh được thêm vào chất điện giải. Ở pH thấp, nhóm silanol nói chung có độ ion hóa thấp và dòng EOF nhỏ. Ở pH cao hơn, nhóm silanol bị ion hóa nhiều hơn và dòng EOF tăng. Trong một số trường hợp, các dung môi hữu cơ như methanol hay acetonitril được cho thêm vào dung dịch đệm để làm tăng độ tan của các chất hòa tan và các chất phụ gia hay để ảnh hưởng đến mức độ ion hóa của mẫu. Nói chung, việc thêm các chất điều chỉnh hữu cơ sẽ làm giảm dòng EOF. Detector được đặt về phía đầu cathod của mao quản. Dòng EOF thường lớn hơn linh độ điện di. Do vậy, ngay cả anion cũng bị đẩy về phía cathod và detector. Khi sử dụng đệm phosphat pH 7,0 ở mao quản không có lớp bao, thông thường trình tự xuất hiện các chất tan trong điện di đồ là các cation, các chất trung tính và các anion.
Hiện nay, có 5 loại chủ yếu của điện di mao quản:
Điện di mao quản vùng (capillary zone electro-phoresis - CZE) còn gọi là điện di dung dịch tự do hay điện di mao quản dòng tự do.
Sắc ký mixen điện động, còn gọi là sắc ký điện động mixen (micellar electrokinetic chromatography - MEKC).
Điện di mao quản gel (capillary gel electrophoresis - CGE).
Điện di mao quản hội tụ đẳng điện (capillary iso-electric focusing - CIEF)
Điện di mao quản đẳng tốc (capillary isotachophoresis - CITP).
Trong điện di mao quản vùng, quá trình tách được kiểm soát bằng sự khác nhau về linh độ tương đối của từng thành phần trong mẫu thử hoặc dung dịch thử. Linh độ là hàm số của điện tích chất phân tích và kích thước trong điều kiện nhất định của phương pháp. Chúng được tối ưu hóa bằng cách kiểm soát các thành phần của đệm, pH và lực ion.
Trong sắc ký mixen điện động, các chất hoạt động bề mặt được thêm vào dung dịch đệm làm việc ở nồng độ lớn hơn nồng độ mixen tới hạn. Các chất phân tích có thể phân bố trong pha tĩnh giả do mixen tạo thành. Kỹ thuật này thường được ứng dụng để tách các chất trung tính và các ion.
Trong điện di mao quản gel, tương tự như lọc gel, sử dụng các mao quản chứa gel để tách các phân tử, dựa trên cơ sở sự khác nhau tương đối về khối lượng phân tử hay kích thước phân tử. Nó được dùng chủ yếu để tách các protein, peptid và các oligomer. Các gel có ưu điểm là làm giảm dòng EOF và đồng thời làm giảm một cách đáng kể sự hấp phụ protein trên thành phía trong của mao quản, do đó làm giảm đáng kể hiệu ứng bất đối của pic.
Trong điện di mao quản hội tụ đẳng điện, các chất được tách trên cơ sở sự khác nhau tương đối về điểm đẳng điện. Nó được thực hiện bằng cách biến đổi pH đệm để thu được các vùng mẫu thử ở trạng thái bền (pH thấp ở anod và pH cao ở cathod). Quá trình biến đổi được xác lập bằng cách đặt thế vào mao quản chứa hỗn hợp thành phần của các chất mang bao gồm các chất lưỡng cực có giá trị pl khác nhau.
Trong điện di mao quản đẳng tốc sử dụng hai đệm bao quanh các vùng chất phân tích. Cả anion và cation có thể tách thành những vùng rõ rệt. Ngoài ra, nồng độ chất phân tích như nhau trong mỗi vùng. Như vậy, chiều dài của mỗi vùng sẽ tỷ lệ thuận với lượng chất phân tích.
Hai kỹ thuật điện di mao quản thường được sử dụng nhiều nhất là điện di mao quản vùng và sắc ký mixen điện động. Chúng được đề cập một cách tóm tắt trong các phần sau.
Nguyên lý của điện di mao quản vùng
Điện di mao quản vùng sử dụng nguyên lý của điện di và điện thẩm để tách các tiểu phân tích điện.
Linh độ điện di của ion (µEP), được biểu diễn bằng phương trình:
µEP = q/(6πƞr)
Trong đó q là điện tích của ion, ƞ là độ nhớt dung dịch và r là bán kính của ion hydrat hóa. Từ mối liên hệ này suy ra, chất phân tích kích thước nhỏ, điện tích lớn có linh độ lớn. Chất phân tích kích thước lớn, điện tích nhỏ có linh độ thấp.
Tốc độ di chuyển (vEP), tính bằng cm/s, được biểu thị bằng phương trình:
vEP = µEP (V / L)
Trong đó µEP là linh độ điện di, V là điện thế đặt và L là chiều dài (cm) tổng cộng của mao quản.
Tốc độ của EOF (vEO), tính bằng cm/s, được biểu diễn bằng phương trình:
vEO = µEO (V / L)
Trong đó µEO là linh độ của EOF (hệ số của dòng EOF) và các ký hiệu khác được định nghĩa như trên. Thời gian (t), tính bằng giây, cần thiết cho chất tan di chuyển trên toàn bộ chiều dài hiệu dụng (l) của mao quản (từ đầu vào đến detector), t được biểu thị bằng hệ thức sau:
t=l/[E(µEP + µEO)]=lL/[V(µEP + µEO)]
Trong đó E là cường độ điện trường và các ký hiệu khác được định nghĩa như ở trên.
Hiệu lực của hệ thống điện di có thể liên quan đến linh độ, dòng EOF và được biểu thị bằng số đĩa lý thuyết (N), được tính bằng phương trình:
N = (µEP + µEO)V/2D
Trong đó D là hệ số khuếch tán của chất hòa tan và các ký hiệu khác được định nghĩa như trên.
Độ phân giải (R) của hai chất hòa tan được rửa giải kế tiếp nhau, có thể xác định bằng phương trình:
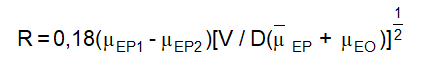
Trong đó: µEP1 và µEP2 là linh độ của hai chất hòa tan;
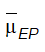 là linh độ trung bình của hai chất tan và các ký hiệu khác được định nghĩa như trên.
là linh độ trung bình của hai chất tan và các ký hiệu khác được định nghĩa như trên.
Nguyên lý của sắc ký mixen điện động
Trong MEKC, môi trường chất điện giải nền có chứa chất hoạt động bề mặt ở nồng độ cao hơn nồng độ mixen tới hạn (CMC). Trong môi trường nước, các chất hoạt động bề mặt tự ngưng kết và hình thành mixen gồm nhóm có đầu thân nước tạo nên một lớp áo bên ngoài và nhóm có đuôi không thân nước tạo nhân không phân cực ở bên trong mixen để cho các chất tan có thể phân bố. Nói chung, các mixen có những anion trên bề mặt ngoài, dưới tác dụng của điện thế áp đặt chúng di chuyển theo hướng ngược lại so với dòng EOF. Kiểu phân bố này tương tự với phân bố trong chiết xuất với dung môi hay trong sắc ký lỏng hiệu năng cao pha đảo. Sự phân bố khác nhau của các phân tử trung tính vào pha động nước và pha “tĩnh giả” mixen là cơ sở duy nhất cho quá trình tách. Dung dịch đệm và các mixen tạo thành hệ thống hai pha, chất tan có thể phân bố giữa hai pha này.
Hệ thống mixen thích hợp cho MEKC phải đáp ứng các tiêu chuẩn sau: Chất hoạt động bề mặt tan tốt trong đệm và dung dịch mixen đồng nhất và trong suốt đối với detector UV. Chất hoạt động bề mặt phổ biến nhất cho MEKC là natri dodecyl sulfat (chất hoạt động bề mặt anion). Những chất hoạt động khác bao gồm cetyltrimethylamoni bromid (chất hoạt động bề mặt cation) và các muối mật (chất hoạt động bề mặt chiral). Độ chọn lọc của hệ thống MEKC chủ yếu tùy thuộc vào bản chất của chất hoạt động bề mặt. Dung môi hữu cơ thường thêm vào trong đệm MEKC để điều chỉnh hệ số dung lượng, tương tự như trong tách bằng sắc ký lỏng pha đảo. MEKC có thể sử dụng để tách các đồng phân đối quang. Để tách các đồng phân, trong dung dịch đệm được thêm vào các chất phụ gia chiral hoặc chất hoạt động bề mặt chiral như muối mật.
Kiến thức chung về sắc ký cột cổ điển sẽ hỗ trợ cho sự hiểu biết nguyên lý MEKC. Tuy vậy, trong MEKC, các mixen không thuần túy là pha tĩnh. Bởi vậy, lý thuyết sắc ký cột cần thiết phải được điều chỉnh. Sự điều chỉnh cơ bản đối với nguyên lí của MEKC là bản chất xác định của cửa sổ tách đối với các phân tử trung tính.
Thời gian lưu (tR) đối với các chất trung tính được biểu diễn bằng phương trình sau:
tR = (1 + k’)t0 /[1 + (t0 /tMC)]
Trong đó, t0 là thời gian yêu cầu đối với chất không lưu giữ đi qua chiều dài hiệu dụng của cột mao quản, tMC là thời gian cần để mixen đi qua mao quản, k' là hệ số dung lượng và tR luôn luôn ở giữa giá trị t0 và tMC.
Hệ số dung lượng (k') đối với các chất trung tính được tính theo phương trình:
K’ = (tR / t0 -1)/(1-tR/tMC)
Trong đó các ký hiệu được định nghĩa như trên.
Để áp dụng cho mục đích thực tiễn, k' được tính bằng phương trình:
k' = tR / t0 -1
Trong đó tR là thời gian được đo từ thời điểm đặt thế (hoặc điểm bắt đầu tiêm mẫu) đến thời điểm ứng với đỉnh pic tối đa, t0 được đo từ điểm đặt thế (hoặc điểm bắt đầu tiêm mẫu) đến điểm biên phía trước của pic dung môi hay đến tín hiệu của chất không lưu giữ. k' trong MEKC có ý nghĩa nhất định và là đặc trưng của một chất tan nhất định trong hệ MEKC. Những bàn luận khác của k' được trình bày trong phần tính phù hợp hệ thống trong mục các thông số làm việc.
Độ phân giải (Rs) đối với các chất trung tính được tính bằng phương trình:
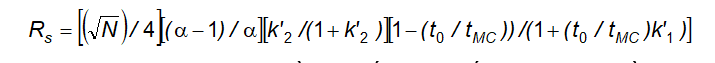
Trong đó α là độ chọn lọc, bằng tỷ số của k'2 đối với k'1 trong điều kiện cụ thể cho việc tách hai chất tan. Nếu hai chất tan rửa giải gần nhau (α≤ 1,1), có thể dùng một trong hai trị giá k’. Phương trình chứng tỏ rằng, cũng như với sắc ký kinh điển, độ phân giải trong MEKC có thể được cải thiện qua việc kiểm soát hiệu lực của hệ thống, độ chọn lọc, sự lưu giữ và bản chất hóa học của các chất hoạt động bề mặt. Hạng từ cuối cùng của phương trình chính là khoảng rửa giải giới hạn. Mặc dầu MEKC đặc biệt hữu hiệu trong tách các chất trung tính, kỹ thuật này cũng có thể được áp dụng cho việc tách các chất tan tích điện. Qui trình sau đây liên quan đến sự phối hợp cơ chế tách giữa sắc ký và điện di. Tương tác giữa chất tan tích điện và mixen có thể được sử dụng để tối ưu hóa sự tách. Cặp ion có thể hình thành nếu như điện tích trên chất hoạt động bề mặt và chất tan là trái dấu nhau. Trong trường hợp ngược lại, chất hoạt động bề mặt và chất tan đẩy nhau. Sự khác biệt này có thể ảnh hưởng có ý nghĩa đến sự tách của các phân tử tích điện.
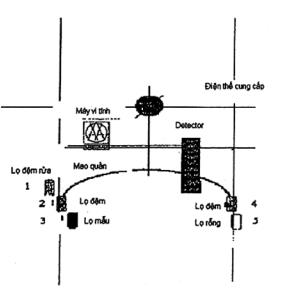
Hình 5.7 - Cấu hình thiết bị CE điển hình
Các điều chú ý đối với thiết bị
Hệ thống CE điển hình (Hình 5.7) bao gồm một mao quản bằng silica nung chảy có đường kính trong từ 50 µm đến 100 µm và chiều dài từ 20 cm đến 100 cm. Các đầu mao quản được nhúng vào các bình điện giải riêng biệt. Nguồn cung cấp điện một chiều có khả năng tạo điện áp cao từ 0 kV đến 30 kV. Thêm vào đó một detector, bộ phận tiêm mẫu tự động và thiết bị ghi dữ liệu là đủ một hệ thiết bị CE. Hệ thống cung cấp dung dịch đệm tự động và hệ thống kiểm soát bằng máy vi tính, ghi nhận dữ liệu hay kiểm soát nhiệt độ cho cả mao quản và bộ phận tiêm mẫu tự động có thể thấy ở các thiết bị có mặt trên thị trường.
Những điều cần quan tâm đầu tiên đối với thiết bị là kiểu mao quản, cấu hình, cách tiêm mẫu, nguồn cung cấp điện và kiểu detector.
Các loại mao quản và cấu hình thiết bị
Mao quản dùng trong CZE thường được làm bằng silica nung chảy và không có lớp bao mặt trong mao quản. Một vài thiết bị có cấu hình với cột mao quản để tự do, nghĩa là mao quản không đưa vào bên trong hộp chứa. Trong hầu hết các thiết bị trên thị trường, mao quản được đặt vào trong một hộp chứa. Cả hai cấu hình đều có những ưu và nhược điểm riêng. Điều quan trọng là khả năng của thiết bị có thể tương thích được các loại mao quản khác nhau với chiều dài và đường kính mao quản khác nhau. Mao quản với chất bao mặt trong khác nhau hiện nay cũng có mặt trên thị trường. Bởi vậy, khả năng tương thích với các loại cột khác nhau rất quan trọng. Các chất bao mặt trong mao quản có thể được sử dụng để thay đổi cường độ hay hướng của dòng EOF hay làm giảm sự hấp phụ của mẫu. Nếu sử dụng mao quản có chất bao mặt trong, phải có ghi các chi tiết về chất bao và nhà cung cấp trong phương pháp. Mao quản từ nhà cung cấp khác cũng có thể được sử dụng nếu chúng thích hợp.
Kỹ thuật đưa mẫu và tiêm mẫu
Cách đưa mẫu vào trong mao quản bao gồm tiêm kiểu di chuyển điện (kiểu điện động), và tiêm áp suất âm và tiêm áp suất dương (kiểu thủy tĩnh).
Để tiêm kiểu di chuyển điện, dung dịch mẫu được đưa vào mao quản bằng cách nhúng mao quản và điện cực vào trong lọ mẫu và đặt lên đó một điện thế cao trong thời gian ngắn. Mẫu đi vào mao quản bằng sự phối hợp của điện di và dòng EOF. Bởi vậy, các chất phân tích có linh độ khác nhau đi vào trong mao quản ở mức độ khác nhau. Độ dẫn của mẫu thử và chất chuẩn cũng ảnh hưởng đến dòng EOF và thể tích tiêm.
Tiêm áp suất âm là đặt áp suất âm ở đầu mao quản có detector và hút dung dịch mẫu vào đầu tiêm của mao quản. Tiêm áp suất dương là tạo áp suất lên lọ chứa mẫu, đẩy mẫu vào trong mao quản. Tiêm áp suất có thể đưa tất cả các thành phần của mẫu vào trong mao quản ở cùng một mức độ, nói chung là cho độ tái lặp tốt nhất và là cách tiêm thường được áp dụng nhất. Thể tích mẫu phụ thuộc vào chiều dài và đường kính trong của mao quản và điện thế hay áp suất áp đặt. Thể tích mẫu đặc trưng được tiêm vào trong mao quản khoảng từ 1 nl đến 20 nl.
Mỗi một phương pháp tiêm mẫu có những ưu và nhược điểm riêng, tùy thuộc vào thành phần của mẫu, kiểu tách và áp dụng phương pháp. Không có kiểu tiêm mẫu nào nói trên có độ tái lặp như bộ tiêm mẫu của máy sắc ký lỏng hiệu năng cao hiện đang có mặt trên thị trường. Tùy theo hoàn cảnh cụ thể, cần thiết phải sử dụng chất chuẩn nội cho các phương pháp cụ thể khi cần sự tiêm mẫu chính xác.
Cung cấp nguồn điện
Hầu hết các thiết bị CE có mặt trên thị trường có nguồn điện một chiều, nó có khả năng tạo nguồn điện thay đổi từ từ hay từng nấc đảm bảo duy trì được điện thế mong muốn một cách ổn định. Điều này sẽ đảm bảo có được đường nền tương đối phẳng.
Một đặc điểm thiết yếu khác của nguồn điện là khả năng sử dụng nguồn này để đưa mẫu vào đầu cathod hay anod của mao quản. Bởi vì trên thực tế không thể thay đổi vị trí detector tử đầu này đến đầu kia của thiết bị, mà chỉ có thể ấn định đầu tiêm mẫu ở phía cathod hay anod.
Các kiểu detector
Nói chung hệ thống CE thường ghép với các detector UV-VIS và detector huỳnh quang cảm ứng laser. Detector quét phổ UV hay detector dãy diod có thể thấy ở nhiều thiết bị CE hiện có mặt trên thị trường. Sự ghép nối CE với phổ khối cho phép thu được thông tin về cấu trúc kết hợp các dữ liệu điện di.
Phát hiện bằng detector huỳnh quang sẽ làm tăng độ nhạy phát hiện đối với mẫu chứa một lượng rất nhỏ chất cần phân tích có khả năng hấp thụ UV. Việc ghép một nhóm huỳnh quang với những chất không hấp thụ UV có thể là có ích. Các chất không hấp thụ UV hoặc không phát quang có thể được phát hiện bằng cách thêm trực tiếp vào dung dịch đệm các chất mang màu hay chất có huỳnh quang: Các chất không hấp thụ được phát hiện nhờ sự mất tín hiệu so với chất nền hấp thụ hay huỳnh quang. Detector đo độ dẫn và đo amper có thể được sử dụng, nhưng nói chung chúng không thông dụng ở các thiết bị có mặt trên thị trường.
Những điểm cần chú ý khi phân tích mẫu
Một vài thông số như kích thước mao quản, điện thế, lực ion và pH được tối ưu hóa để đạt độ phân giải và sự tách phù hợp. Cần tránh sự thay đổi nhiệt độ vì sẽ ảnh hưởng đến độ nhớt của dung dịch đệm và ảnh hưởng cả đến dòng EOF và linh độ của chất tan.
Kích thước mao quản
Những thay đổi về đường kính và chiều dài mao quản có thể ảnh hưởng đến độ phân giải điện di. Chiều dài mao quản tăng sẽ kéo dài thời gian di chuyển, thông thường sẽ tăng độ phân giải và làm giảm dòng điện. Đường kính mao quản tăng thường làm tăng dòng điện và tạo ra gradient nhiệt độ bên trong cột, dẫn đến làm giảm độ phân giải. Ngược lại, giảm đường kính trong của mao quản sẽ làm giảm sinh nhiệt và cho độ phân giải tốt hơn. Tuy nhiên, ở mao quản có đường kính lớn hơn sẽ có thuận lợi là tiêm được lượng mẫu nhiều hơn và cải thiện được tỷ lệ tín hiệu trên nhiễu.
Ảnh hưởng điện thế
Khi đặt một điện thế cao hơn, xảy ra sự tăng sinh nhiệt của dung dịch đệm làm việc bên trong mao quản vì có dòng điện đi qua dung dịch đệm. Hiệu ứng nhiệt này còn gọi là nhiệt Jun cần phải được kiểm soát vì trở kháng, hằng số điện môi và độ nhớt phụ thuộc vào nhiệt độ và làm thay đổi tốc độ dòng EOF và linh độ của chất tan.
Nói chung, sự tăng điện thế sẽ làm tăng hiệu lực tách và độ phân giải đến điểm mà nhiệt Jun không phát tán được. Độ phân giải tối đa có thể đạt được bằng cách duy trì điện thế ở dưới mức xảy ra sự phát nhiệt Jun và sự khuếch tán trở thành yếu tố giới hạn.
Ảnh hưởng của lực ion
Kiểm soát lực ion cho phép điều chỉnh độ phân giải, hiệu lực tách và độ nhạy. Tăng lực ion nói chung sẽ cải thiện độ phân giải, hiệu lực tách, và hình dạng pic. Độ nhạy có thể được cải thiện do có sự tụ hợp tốt hơn. Tuy nhiên, bởi vì dòng điện sinh ra tỷ lệ thuận với nồng độ đệm, nhiệt sinh ra nhiều hơn khi lực ion của đệm tăng, do đó cần hạn chế tăng lực ion.
Ảnh hưởng của pH
Độ phân giải, độ nhạy và hình dạng pic có thể thay đổi một cách rõ rệt khi có sự thay đổi pH vì thông số này ảnh hưởng đến mức ion hóa của chất tan và dòng EOF. Dòng EOF cao ở pH cao và thấp ở pH thấp đối với cột mao quản bằng silica nung chảy không có lớp bao mặt trong.
Các thông số vận hành
Các bước cơ bản trong quá trình vận hành hệ thống CE là thiết lập hệ thống, quy trình rửa cột mao quản, chuẩn bị mẫu, thử tính phù hợp hệ thống, phân tích mẫu, xử lý dữ liệu và tắt hệ thống.
Thiết lập hệ thống
Cần quan tâm đến khả năng tách, độ phân giải, lực ion của dung dịch đệm và ảnh hưởng pH để chọn cột có kích thước và lớp bao bề mặt cho phù hợp. Điều chế dung dịch đệm có thành phần, lực ion và pH thích hợp, loại khí nếu cần thiết và lọc qua lọc thích hợp.
Tất cả dung môi, bao gồm cả nước cần dùng loại tinh khiết dùng cho sắc ký lỏng hay CE.
Quy trình rửa mao quản
Sự ổn định về thời gian di chuyển và độ phân giải có thể thu được nếu tuân theo quy trình rửa cột mao quản nhất định. Quy trình rửa và cân bằng mao quản phải phù hợp với các chất phân tích, các nền mẫu (matrix) và phương pháp phân tích. Bởi vậy, những qui trình này được xây dựng như là một phần của phương pháp và được quy định trong từng chuyên luận riêng. Rửa cột có thể dùng dung dịch acid phosphoric 0,1 M, nước và dung dịch natri hydroxyd 0,1 M. Trước khi bắt đầu quá trình phân tích mẫu, cột mao quản có thể được rửa với năm lần thể tích cột bằng dung dịch đệm làm việc dùng cho quá trình phân tích. Khi thay đổi thành phần đệm, nên rửa cột mao quản với năm lần thể tích cột bằng dung dịch đệm làm việc mới để làm sạch đệm của lần phân tích trước. Khi sử dụng cột mao quản mới bằng silica nung chảy không có lớp bao, thông thường yêu cầu phải hoạt hóa các nhóm silanol trên bề mặt mao quản. Quá trình này bao gồm việc tráng rửa kỹ với dung dịch natri hydroxyd. Đối với các loại mao quản có lớp bao bên trong, quy trình rửa cột cần theo sự hướng dẫn của nhà sản xuất vì rửa cột không đúng cách có thể làm phá hủy lớp bao bên trong cột. Cột có thể được chỉ định chuyên dùng cho những loại đệm hay cho những phương pháp phân tích cụ thể để đề phòng nhiễm chéo.
Tiến hành phân tích mẫu
Để có được độ phân giải, độ nhạy và sự tách tốt với pic có hình dạng đẹp cần phải lựa chọn cột mao quản, chất điện giải và qui trình tiêm mẫu phù hợp. Khi cần thì sử dụng nội chuẩn để có được độ chính xác khi tiêm mẫu. Khi lựa chọn nội chuẩn cần quan tâm đến khả năng tách khỏi chất cần phân tích. Sự hoàn hảo của hệ thống có thể được cải thiện bằng cách rửa mao quản giữa các lần tiêm và cung cấp đệm mới cho hai lọ đệm dùng áp đặt thế trong quá trình tách (lọ 2 và 4 trong Hình 5.7). Có thể tiêm lặp lại từ cùng lọ mẫu thử miễn là tránh được sự nhiễm chéo. Nếu có sự nhiễm chéo xảy ra, nên rửa đầu mao quản nơi tiêm mẫu bằng cách nhúng nhanh nó vào trong một lọ có chứa đệm trước khi nhúng vào trong lọ chứa mẫu hay lọ chứa chất điện giải.
Các thông số vận hành được quy định cụ thể trong từng chuyên luận nhằm làm giảm ảnh hưởng của điện thế, lực ion và pH. Thiết bị được lắp đặt để phân tích mẫu với cột mao quản và điều kiện tiêm mẫu phù hợp nằm trong khoảng tuyến tính động học của detector. Sự chính xác về độ di chuyển có thể đạt được bằng cách chọn chất pha loãng mẫu, chất điện giải và điều kiện xử lý cột mao quản phù hợp. Khi tiến hành phân tích chú ý tránh hiện tượng đưa lượng mẫu quá tải vào cột vì điều này sẽ làm giảm hiệu lực và độ tái lặp.
Tính phù hợp của hệ thống
Các thông số được đo đạc có thể bao gồm độ lặp của bộ phận tiêm mẫu, độ chọn lọc và hiệu lực của hệ thống, và sự đối xứng của pic. Độ phân giải giữa các chất phân tích và các chất khác có thể được xác định bằng cách sử dụng các chất chuẩn hỗn hợp.
Các thông số tiêu biểu dùng để xác định tính phù hợp hệ thống bao gồm độ lệch chuẩn tương đối (RSD), hệ số dung lượng (k'), số đĩa lý thuyết (N), độ nhạy (giới hạn phát hiện và giới hạn định lượng), số đĩa lý thuyết tính trên 1 mét (n), hệ số đối xứng (T) và độ phân giải (R).
Hình dạng pic được kiểm tra chặt chẽ, pic lý tưởng phải đối xứng, không có vai và không kéo đuôi. Nếu những điều kiện này không đạt, cần phải tiến hành điều chỉnh trước khi tiến hành phân tích. Việc tích phân pic cũng phải được kiểm tra chặt chẽ để đảm bảo tín hiệu đáp ứng của pic được định lượng chính xác.
Tiêm lặp lại dung dịch chuẩn với nồng độ biết trước để xác định độ tái lặp của hệ thống CE. Dữ liệu tiêm lặp lại từ 5 lần hoặc nhiều hơn được sử dụng để tính toán RSD. Trừ khi có quy định trong chuyên luận riêng, độ lệch chuẩn tương đối cho sự tiêm lặp lại không được quá 3,0 %. Giá trị của độ chính xác nhỏ nhất có thể được quy định riêng trong phương pháp CE, đặc biệt khi xác định các thành phần dạng vết. Việc tính toán các thông số điện di trong MEKC, cũng như các phương pháp khác của CE có thể cần đến sự kết hợp các mối quan hệ giữa sắc ký và điện di. Vì vậy, hệ số dung lượng (k') cho sự di chuyển của chất trung tính trong MEKC có thể tính bằng phương trình:
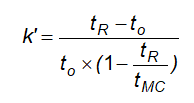
Trong đó tR, t0, và tMC lần lượt là thời gian di chuyển của chất cần phân tích, của dung dịch (EOF) và của mixen.
Số đĩa lý thuyết (N), là thước đo hiệu lực của hệ thống và được tính bằng phương trình:
N = 16(tR/W)2 hay N = 5,54(tR/W1/2)2
Trong đó W là độ rộng pic của chất cần phân tích đo được ở đường nền, W1/2 là độ rộng pic của chất cần phân tích đo được ở phân nửa chiều cao của pic và tR là thời gian di chuyển của chất phân tích.
Số đĩa lý thuyết tính trên mét (n) là thước đo hiệu lực của cột mao quản được xem như là hàm số về độ rộng pic ở đường nền và có thể tính toán bằng công thức:
n = 1600(tR/W)2/L
Trong đó L tính bằng cm là chiều dài tổng cộng của mao quản, các ký kiệu khác được định nghĩa như trên. Hệ số đối xứng pic (T) của chất cần phân tích là thước đo sự đối xứng của pic, nó thể hiện độ lệch của pic so với pic đối xứng lý tưởng. Hệ số này được tính bằng phương trình:
T = W0,05/2f
Trong đó W0,05 là chiều rộng pic đo ở 5 % chiều cao của pic tính từ đường nền, f là chiều rộng nửa bên trái của pic đo ở 5 % chiều cao pic tính từ đường nền. Hệ số 1,0 chỉ rằng pic là đối xứng. Nếu các hiệu ứng phân tán điện di xảy ra, thì chúng có thể tạo ra pic không đối xứng. Điều này có thể xảy ra khi dùng nồng độ mẫu thử cao như trong phân tích tạp chất. Trường hợp pic bất đối xứng cũng có thể chấp nhận được miễn là đạt được độ lặp và chúng không ảnh hưởng đến độ chọn lọc về tách.
Hệ số phân giải (R) đo khả năng của hệ thống CE tách các chất cần phân tích di chuyển kế tiếp nhau. Độ phân giải được xác định cho tất cả các chất cần phân tích, với pH của đệm được điều chỉnh nếu cần thiết để đáp ứng các yêu cầu về tính phù hợp hệ thống. Nó có thể được tính bằng phương trình:
R = 2(t2 - t1)/(W1+W2)
Trong đó t2 và t1 lần lượt là thời gian di chuyển (đo được ở cực đại của pic) của pic đi sau và pic trước.
W2 và W1 lần lượt là chiều rộng của hai pic đo được ở đường nền.
Phân tích mẫu
Sau khi xác định tính phù hợp hệ thống, có thể tiêm dung dịch mẫu thử và mẫu chuẩn. Mẫu chuẩn có thể tiêm trước hay sau mẫu thử và tiêm xen kẽ trong suốt quá trình phân tích.
Xử lý dữ liệu
Diện tích pic được chuẩn hóa theo thời gian thường được sử dụng trong định lượng. Các dữ liệu được xác định bằng cách chia diện tích pic đã tích phân được cho thời gian di chuyển của chất phân tích. Điều này làm giảm sai số do trong CE, không giống như trong sắc ký lỏng, mỗi chất cần phân tích di chuyển qua detector với một tốc độ khác nhau. Trừ khi sự chuẩn hóa này được thực hiện, những chất di chuyển chậm hơn sẽ có diện tích lớn bất thường so với diện tích của những thành phần di chuyển nhanh.
Tắt hệ thống
Sau khi phân tích, cột mao quản được rửa theo sự hướng dẫn được quy định riêng trong từng chuyên luận hay theo khuyến cáo của nhà sản xuất. Ví dụ: Cột mao quản có thể được rửa bằng nước cất để loại các thành phần đệm và sau đó được nạp không khí hoặc khí nitrogen lấy từ lọ rỗng. Thông thường, lọ đích và lọ nguồn (lọ 4 và lọ 2 trong hình 5.7) là các lọ được bỏ dung dịch đệm đi và được tráng kỹ bằng nước khử khoáng.
PHỤ LỤC 6
(Quy định)
6.1 XÁC ĐỊNH CHỈ SỐ KHÚC XẠ
Chỉ số khúc xạ (n) của một chất so với không khí là tỷ lệ giữa sin của góc tới và sin của góc khúc xạ của chùm tia sáng truyền từ không khí vào chất đó.
Chỉ số khúc xạ thay đổi theo bước sóng ánh sáng được dùng để đo và nhiệt độ. Chỉ số khúc xạ có giá trị để định tính và đánh giá sơ bộ mức độ tinh khiết của mẫu đo.
Nếu không có chỉ dẫn gì khác, chỉ số khúc xạ được đo ở 20 °C ± 0,5 °C với tia sáng có bước sóng tương ứng với vạch D của natri (589,3 nm), ký hiệu ![]() .
.
Máy
Khúc xạ kế dùng để xác định góc tới hạn của môi trường. Phần chủ yếu của khúc xạ kế là một lăng kính có chỉ số khúc xạ biết trước đặt tiếp xúc với môi trường được khảo sát.
Hầu hết khúc xạ kế được thiết kế để sử dụng nguồn sáng trắng, khi sử dụng nguồn sáng trắng, khúc xạ kế được trang bị hệ thống bổ chính và được hiệu chuẩn lại để cho kết quả đọc tương ứng với vạch D của đèn natri.
Thang đo chỉ số khúc xạ phải đọc được các giá trị với ít nhất 3 số lẻ thập phân.
Nhiệt kế chia độ tới 0,5 °C hoặc nhỏ hơn.
Để đạt được độ chính xác, cần thiết phải hiệu chuẩn lại máy với các chất chuẩn do nhà sản xuất cung cấp hay bằng cách xác định chỉ số khúc xạ của nước cất tại 25 °C là 1,3325 và tại 20 °C là 1,3330.
6.2 XÁC ĐỊNH CHỈ SỐ pH
pH là một số biểu thị quy ước nồng độ ion hydrogen của dung dịch nước. Trong thực hành, định nghĩa trên là một định nghĩa thực nghiệm. pH của một dung dịch liên quan với pH của một dung dịch đối chiếu theo biểu thức sau:
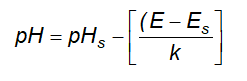
trong đó:
E là điện thế, tính bằng von, của pin chứa dung dịch được khảo sát;
Es là điện thế, tính bằng von, của pin chứa dung dịch đã biết pH (dung dịch đối chiếu);
pHs là pH của dung dịch đối chiếu;
k là hệ số có giá trị thay đổi theo nhiệt độ ghi ở Bảng 6.2.1.
Bảng 6.2.1 - Giá trị của k ở các nhiệt độ khác nhau
| Nhiệt độ (°C) | k(V) |
| 15 | 0,0572 |
| 20 | 0,0582 |
| 25 | 0,0592 |
| 30 | 0,0601 |
| 35 | 0,0611 |
Máy
Trị số pH của một dung dịch được xác định bằng cách đo thế hiệu giữa điện cực chỉ thị nhạy cảm với ion hydrogen (thường là điện cực thủy tinh) và một điện cực so sánh (ví dụ điện cực calomel bão hòa). Máy đo là một điện thế kế có trở kháng đầu vào gấp ít nhất 100 lần trở kháng của các điện cực sử dụng. Nó thường được phân độ theo đơn vị pH và có độ nhạy đủ để phát hiện được những thay đổi cỡ 0,05 đơn vị pH hoặc ít nhất 0,003 V. Các điện cực thủy tinh phù hợp và các kiểu máy đo pH kể cả máy đo pH hiện số đều phải đáp ứng yêu cầu trên.
Vận hành máy đo pH và hệ thống điện cực theo sự chỉ dẫn của hãng sản xuất.
Tất cả các phép đo đều cần phải tiến hành trong cùng một điều kiện nhiệt độ khoảng từ 20 °C đến 25 °C, trừ những trường hợp có quy định khác trong chuyên luận riêng.
Hiệu chuẩn máy: Dùng dung dịch đệm chuẩn D ghi trong Bảng 6.2.2 là chuẩn thứ nhất, đo và chỉnh máy để đọc được trị số pH của chuẩn ghi ở bảng tương ứng với nhiệt độ của dung dịch.
Dùng một dung dịch đệm chuẩn thứ hai (chọn một trong các dung dịch quy định ghi ở Bảng 6.2.2) để chỉnh thang đo.
Trị số pH đo được của dung dịch đệm chuẩn thứ ba, dung dịch có trị số pH nằm giữa trị số pH của đệm chuẩn thứ nhất và thứ hai, phải không được sai khác nhiều hơn 0,05 đơn vị pH so với trị số pH tương ứng ghi trong Bảng 6.2.2.
Phương pháp đo
Nhúng các điện cực vào trong dung dịch cần khảo sát và đo trị số pH ở cùng nhiệt độ đo của các dung dịch đệm chuẩn khi hiệu chuẩn máy.
Khi máy được dùng thường xuyên, việc kiểm tra thang đo pH phải được thực hiện định kỳ. Nếu máy không thường xuyên dùng, việc kiểm tra cần thực hiện trước mỗi phép đo.
Tất cả các dung dịch và dịch treo của chế phẩm khảo sát và các dung dịch đệm chuẩn phải được pha chế với nước không có carbon dioxyd (TT).
Khi đo các dung dịch có pH trên 10,0 phải đảm bảo rằng điện cực thủy tinh đang dùng là phù hợp, chịu được các điều kiện kiềm và cần áp dụng hệ số điều chỉnh trong phép đo.
Sau cùng đo lại trị số pH của dung dịch đệm chuẩn dùng để hiệu chuẩn máy và điện cực. Nếu sự khác nhau giữa lần đọc này và trị số gốc của dung dịch đệm chuẩn ấy lớn hơn 0,05 thì các phép đo phải làm lại.
Các dung dịch đệm chuẩn
Dung dịch đệm A: Hòa tan 12,61 g kali tetraoxalat (TT) trong nước không có carbon dioxyd (TT) vừa đủ để có 1000 ml dung dịch (0,05 M).
Dung dịch đệm B: Lắc kỹ một lượng thừa kali hydro (+)-tartrat (TT) với nước không có carbon dioxyd (TT) ở 25 °C. Lọc hoặc để lắng gạn. Pha ngay trước khi dùng.
Dung dịch đệm C: Hòa tan 11,41 g kali dihydrocitrat (TT) trong nước không có carbon dioxyd (TT) vừa đủ để có 1000 ml dung dịch (0,05 M). Pha ngay trước khi dùng.
Dung dịch đệm D: Hòa tan 10,13 g kali hydrophtalat (TT) (đã sấy khô trước ở 110 °C ± 2 °C trong 1 h) trong nước không có carbon dioxyd (TT) vừa đủ để có 1000 ml dung dịch (0,05 M).
Dung dịch đệm E: Hòa tan 3,39 g kali dihydrophosphat (TT) và 3,53 g dinatri hydrophosphat khan (TT) (cả hai đã được sấy khô trước ở 120 °C ± 2 °C trong 2 h) trong nước không có carbon dioxyd (TT) vừa đủ để có 1000 ml dung dịch (0,025 M cho mỗi muối).
Dung dịch đệm F: Hòa tan 1,18 g kali dihydrophosphat (TT) và 4,30 g dinatri hydrophosphat khan (TT) (cả hai đã được sấy khô trước ở 120 °C ± 2 °C trong 2 h) trong nước không có carbon dioxyd (TT) vừa đủ để có 1000 ml dung dịch (0,0087 M và 0,0303 M cho mỗi muối theo thứ tự kể trên).
Dung dịch đệm G: Hòa tan 3,80 g natri tetraborat (TT) trong nước không có carbon dioxyd (TT) vừa đủ để có 1000 ml dung dịch (0,01 M). Bảo quản tránh carbon dioxyd của không khí.
Dung dịch đệm H: Hòa tan 2,64 g natri carbonat khan (TT) và 2,09 g natri hydrocarbonat (TT) trong nước không có carbon dioxyd (TT) vừa đủ để có 1000 ml dung dịch (0,025 M cho mỗi muối).
Dung dịch I: Lắc kỹ một lượng dư calci hydroxyd (TT) trong nước không có carbon dioxyd (TT) và để yên ở 25 °C, gạn lấy phần dịch trong. Bảo quản tránh carbon dioxyd.
Bảng 6.2.2- pH của dung dịch đệm chuẩn ở nhiệt độ khác nhau
| Nhiệt độ | Dung dịch đệm | ||||||||
| t° | A | B | C | D | E | F | G | H | I |
| 15° | 1,67 | - | 3,80 | 4,00 | 6,90 | 7,45 | 9,28 | 10,12 | 12,81 |
| 20° | 1,68 | - | 3,79 | 4,00 | 6,88 | 7,43 | 9,23 | 10,06 | 12,63 |
| 25° | 1,68 | 3,56 | 3,78 | 4,01 | 6,87 | 7,41 | 9,18 | 10,01 | 12,45 |
| 30° | 1,68 | 3,55 | 3,77 | 4,02 | 6,85 | 7,40 | 9,14 | 9,97 | 12,29 |
| 35° | 1,69 | 3,55 | 3,76 | 4,02 | 6,84 | 7,39 | 9,10 | 9,93 | 12,13 |
| ΔpH/Δt | + 0,001 | -0,0014 | - 0,0022 | + 0,0012 | - 0,0028 | - 0,0028 | - 0,0082 | - 0,0096 | 0,034 |
ΔpH/Δt là độ lệch pH trên 1 °C.
6.3 XÁC ĐỊNH ĐỘ NHỚT CỦA CHẤT LỎNG
Độ nhớt của chất lỏng là một đặc tính của chất lỏng liên quan chặt chẽ đến lực ma sát nội tại cản lại sự di động tương đối của các lớp phân tử trong lòng chất lỏng đó.
Độ nhớt động lực hay độ nhớt tuyệt đối, ký hiệu là ƞ, là lực tiếp tuyến trên một đơn vị diện tích bề mặt, được biết như một ứng suất trượt t (biểu thị bằng pascal), cần thiết để di chuyển một lớp chất lỏng 1 m2 song song với mặt phẳng trượt ở tốc độ (v) là 1 m/s so với lớp chất lỏng song song ở một khoảng cách (X) là 1 m.
Tỷ lệ dv/dx là gradient vận tốc cho tốc độ trượt D, biểu thị là nghịch đảo của giây (s-1) và ƞ = t/D.
Đơn vị của độ nhớt động lực là pascal giây (Pa∙s) hoặc newton giây trên mét vuông (N.s/ m2) và ước số hay dùng là milipascal giây (mPa∙s). Ngoài ra người ta còn dùng đơn vị độ nhớt cơ bản là poise (P) và ước số hay dùng là centipoise (cP).
1 Pa∙s = 1000 mPa∙s = 1 N s/m2
1 P = 0,1 Pa∙s = 100 cP = 100 mPa∙s
Độ nhớt động lực của nước cất ở 20 °C xấp xỉ bằng 1 centipoise.
Độ nhớt động học (v) là tỷ số giữa độ nhớt động lực và khối lượng riêng (ρ) của chất lỏng (biểu thị bằng kg/m3), cả hai đều được xác định ở cùng nhiệt độ t.
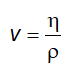
Đơn vị độ nhớt động học là m2/s, ước số là mm2/s. Ngoài ra người ta còn dùng đơn vị độ nhớt động học là Stocker (St) và ước số hay dùng là centistocker (cSt).
1 St = 10-4 m2/s
1 cSt = 10-6m2/s = 1 mm2/s
Độ nhớt động học của nước cất ở 20 °C xấp xỉ bằng 1 cSt.
Khi tính độ nhớt động học của một chất lỏng theo Stocker hay centistocker, đi từ độ nhớt động lực tính theo poise hay centipoise thì khối lượng riêng của chất lỏng đó phải tính theo g/cm3.
Độ nhớt thay đổi rõ rệt khi nhiệt độ thay đổi. Nhiệt độ tăng thì độ nhớt giảm và ngược lại. Vì vậy, phải xác định độ nhớt của chất lòng ở nhiệt độ ổn định, dao động không quá ± 0,1 °C.
Phương pháp xác định độ nhớt của chất lỏng
Phương pháp I: Phương pháp đo thời gian chảy của chất lỏng qua ống mao quản
Nhiều nhớt kế mao quản với những kích thước khác nhau, thích hợp cho việc xác định độ nhớt của các chất lỏng khác nhau. Mỗi loại có một hằng số dụng cụ (k) riêng. Trong số những nhớt kế mao quản, nhớt kế Ostwald thường hay được sử dụng nhất (Hình 6.3.1).
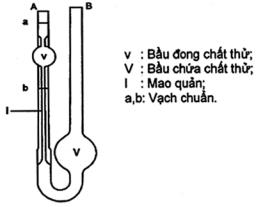
Hình 6.3.1 - Nhớt kế Ostwald.
Cách xác định độ nhớt bằng nhớt kế Ostwald
Dùng pipet dài để chuyển qua miệng ống B chất lỏng cần xác định độ nhớt đã được ổn định nhiệt độ ở 20 °C ± 0,1 °C (trừ khi có chỉ dẫn khác) vào bầu chứa V, sao cho không dính hoặc chỉ dính rất ít chất lỏng đem thử vào thành ống B ở phía trên bầu V. Đặt nhớt kế thẳng đứng và chìm hết bầu V trong môi trường điều nhiệt ở nhiệt độ 20 °C ± 0,1 °C (trừ khi có chỉ dẫn khác) trong 30 min. Sau đó dùng quả bóp cao su (phụ kiện của dụng cụ đo độ nhớt) thổi từ miệng ống B để chất lỏng dâng lên quá ngấn chuẩn a thì ngừng bơm, bỏ quả bóp cao su khỏi miệng ống B để chất lỏng đem thử chảy tự do về bầu V. Ghi thời gian cần thiết để vòng khúm dưới của chất lỏng đem thử chuyển dịch từ ngấn a đến ngấn b. Làm như vậy 5 lần, lấy trung bình cộng của các kết quả đo được làm thời gian t cần xác định. Sai số các kết quả đo không vượt quá 0,5 %. Để đỡ mắc sai số lớn, cần chọn nhớt kế thích hợp sao cho thời gian t không được dưới 200 giây.
Tính độ nhớt lực học ƞ hoặc độ nhớt động học v lần lượt theo công thức sau:
ƞ = k × ρ × t (1)
v = k × t (2)
trong đó:
ƞ: độ nhớt động lực (mPa∙s hoặc cP);
v: độ nhớt động học (mm2/s hoặc cSt);
k: hằng số dụng cụ đo;
ρ: khối lượng riêng của chất lỏng đem thử (g/cm3);
t: thời gian chảy (s).
Khi không có giá trị k, có thể tự xác định hằng số k bằng cách dùng một chất lỏng đã biết trước độ nhớt ƞ hoặc v và tính k theo công thức (1) hoặc (2). Nhớt kế đã bị sửa chữa thì khi sử dụng lại, phải được chuẩn lại. Nếu dùng nhớt kế mao quản với máy đo tự động thì vận hành máy theo quy trình hướng dẫn của hãng sản xuất máy. Thời gian cần thiết để chất lỏng đem thử chuyển dịch từ ngấn a đến ngấn b sẽ được tự động ghi lại.
Phương pháp II: Phương pháp đo thời gian rơi của trái cầu
Phương pháp này thích hợp cho các chất lỏng trong suốt và có độ nhớt cao (từ 8 poise đến 1000 poise).
Dụng cụ: (Hình 6.3.2) Gồm 1 ống thử a, dài 30 cm, đường kính trong là 2 cm ± 0,05 cm. Trên thành ống có khắc 5 ngấn vòng quanh, mỗi ngấn cách nhau 5 cm. Ống thử a được đặt trong bình điều nhiệt b có chứa nước, phía trên có nắp đậy. Trên nắp có các lỗ để đặt nhiệt kế chia độ đến 0,1 °C, que khuấy c và phễu g. Trái cầu là những viên bi bằng thép có đường kính 0,15 cm. Viên bi sẽ được thả vào ống thử a qua một ống nhỏ d có đường kính trong là 0,3 cm. Ống d có một lỗ ngang cao hơn mực chất thử trong ống a, đầu dưới của ống d ở ngang ngấn trên cùng của ống a và thấp hơn mặt chất thử 3 cm.
Khi không có dụng cụ như trên có thể dùng các nhớt kế khác có tính năng tương tự, đảm bảo cung cấp giá trị độ nhớt với độ chính xác và độ đúng như những nhớt kế đã mô tả ở trên (ví dụ: Nhớt kế Hoppler).
Phương pháp đo: Đổ chất lỏng cần xác định độ nhớt vào ống thử a sao cho mực chất lỏng cao hơn đầu dưới của ống d 3 cm. Đặt các viên bi vào một ống thử nghiệm nhỏ lồng qua lỗ đặt phễu g. Giữ chất lỏng cần thử và các viên bi trong môi trường điều nhiệt có nhiệt độ ở 20 °C ± 0,1 °C trong 30 min (trừ khi có chỉ dẫn khác). Sau đó thả từng viên bi vào trong chất lỏng đem thử qua ống d. Ghi thời gian rơi của 5 viên bi từ ngấn thứ hai đến ngấn thứ năm (15 cm). Lấy trung bình cộng của 5 lần đo này làm thời gian t cần xác định.
Tính độ nhớt động lực ƞ của chất lỏng đem thử theo công thức:
ƞ= k × t (ρK - ρ)
trong đó:
ƞ: độ nhớt động lực (mPa∙s hoặc cP);
k: hằng số của viên bi;
ρK: khối lượng riêng của viên bi (g/cm3);
ρ: khối lượng riêng của chất lỏng đem thử (g/cm3);
T: thời gian rơi của viên bi (s).
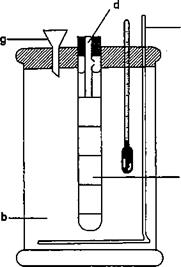
Hình 6.3.2 - Dụng cụ đo độ nhớt bằng cách đo thời gian rơi của trái cầu
Phương pháp III
Thiết bị thường dùng là loại nhớt kế quay, dựa trên việc đo lực trượt trong môi trường lỏng được đặt giữa hai ống hình trụ đồng trục, một ống được quay nhờ môtơ, còn ống kia quay được do sự quay của ống thứ nhất tác động vào. Dưới những điều kiện như vậy, độ nhớt (hay độ nhớt biểu kiến) trở thành phép đo (M) độ lệch của góc đối với ống hình trụ thứ 2, tương ứng với momen lực, biểu thị bằng N.m. Đối với lớp chất lỏng rất mỏng, độ nhớt động lực ƞ, biểu thị bằng Pa∙S được tính theo công thức:
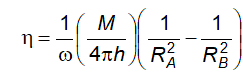
trong đó:
h: độ cao tính bằng m của ống hình trụ thứ 2 được nhúng trong chất lỏng;
RA và RB: đường kính tính bằng m của hai ống hình trụ (RA nhỏ hơn RB);
ω: vận tốc góc tính bằng rad/s.
Hằng số k của máy được tính ở các tốc độ quay khác nhau bằng cách sử dụng các chất lỏng có độ nhớt chuẩn.
Các máy luôn được cung cấp một bảng có các hằng số liên quan đến diện tích bề mặt của các ống trụ được dùng và tốc độ quay của chúng. Độ nhớt được tính theo công thức:
ƞ = k(M/ω)
Cách đo: Đo độ nhớt theo sự chỉ dẫn cách vận hành nhớt kế quay. Nhiệt độ đo được chỉ dẫn trong chuyên luận. Nếu không thể đặt được tốc độ trượt chính xác như chỉ dẫn thì đặt tốc độ trượt cao hơn một chút và thấp hơn một chút sau đó dùng phương pháp nội suy để tính độ nhớt.
6.4 XÁC ĐỊNH GÓC QUAY CỰC VÀ GÓC QUAY CỰC RIÊNG
Góc quay cực của một chất là góc của mặt phẳng phân cực bị quay đi khi ánh sáng phân cực đi qua chất đó nếu là chất lỏng, hoặc qua dung dịch chất đó nếu là chất rắn.
Chất làm quay mặt phẳng phân cực theo cùng chiều kim đồng hồ được gọi là chất hữu tuyền, ký hiệu là (+). Chất làm quay mặt phẳng phân cực ngược chiều kim đồng hồ được gọi là chất tả tuyền, ký hiệu là (-).
Nếu không có hướng dẫn riêng, góc quay cực α được xác định ở nhiệt độ 20 °C và với chùm tia đơn sắc có bước sóng ứng với vạch D (589,3 nm) của đèn natri qua lớp chất lỏng hay dung dịch có bề dày 1 dm.
Góc quay cực riêng  của một chất lỏng là góc quay cực đo được khi chùm ánh sáng D truyền qua lớp chất lỏng đó có bề dày là 1 dm ở 20 °C chia cho tỷ trọng tương đối của chất ở cùng nhiệt độ.
của một chất lỏng là góc quay cực đo được khi chùm ánh sáng D truyền qua lớp chất lỏng đó có bề dày là 1 dm ở 20 °C chia cho tỷ trọng tương đối của chất ở cùng nhiệt độ.
Góc quay cực riêng  , của một chất rắn là góc quay cực đo được khi chùm ánh sáng D truyền qua lớp dung dịch có bề dày là 1 dm và có nồng độ là 1 g/ml, ở 20 °C. Góc quay cực riêng của chất rắn luôn được biểu thị cùng với dung môi và nồng độ dung dịch đo.
, của một chất rắn là góc quay cực đo được khi chùm ánh sáng D truyền qua lớp dung dịch có bề dày là 1 dm và có nồng độ là 1 g/ml, ở 20 °C. Góc quay cực riêng của chất rắn luôn được biểu thị cùng với dung môi và nồng độ dung dịch đo.
Phân cực kế
Các phân cực kế thường dùng nguồn sáng là đèn hơi natri hay hơi thủy ngân. Trong một số trường hợp cần thiết phải sử dụng phân cực kế quang điện cho phép đo ở các bước sóng riêng biệt. Phân cực kế phải cho phép đọc chính xác tới gần 0,01°. Thang đo phải thường xuyên được kiểm định bằng các bản thạch anh chuẩn. Độ tuyến tính của thang đo phải được kiểm tra bằng các dung dịch sucrose.
Cách tiến hành
Xác định góc quay cực của chất thử ở nhiệt độ 19,5 °C đến 20,5 °C, nếu không có chỉ dẫn gì khác trong chuyên luận riêng dùng tia D của ánh sáng đèn natri phân cực. Có thể đo ở nhiệt độ khác nếu chuyên luận riêng chỉ ra cách hiệu chỉnh nhiệt độ cho góc quay cực đo được. Tiến hành đo ít nhất 5 lần và lấy giá trị trung bình. Xác định điểm "0" của phân cực kế với ống đo rỗng khi đo chất lỏng và với ống đo chứa đầy dung môi khi đo dung dịch chất rắn.
Tính góc quay cực riêng theo các biểu thức sau:
Cho chất lỏng:
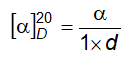
Cho chất rắn:
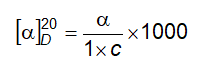
trong đó:
α là góc quay cực đo được;
I là chiều dài ống đo của phân cực kế, tính bằng dm;
d là tỷ trọng tương đối của chất lỏng;
c là nồng độ của chất thử (rắn) trong dung dịch (g/l);
Căn cứ vào góc quay cực đo được, có thể tính nồng độ g/l của chất thử trong dung dịch theo biểu thức:
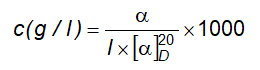
hoặc nồng độ % (kl/kl) của chất thử trong dung dịch theo biểu thức:
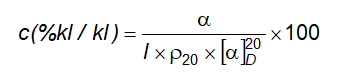
Trong đó:
ρ20 là khối lượng riêng (g/cm3) của dung dịch ở 20 °C.
6.5 XÁC ĐỊNH KHỐI LƯỢNG RIÊNG VÀ TỶ TRỌNG
Khối lượng riêng
Khối lượng riêng của một chất ở nhiệt độ t (ρt) là khối lượng một đơn vị thể tích của chất đó, xác định ở nhiệt độ t:
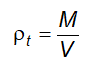
trong đó:
M là khối lượng của chất, xác định ở nhiệt độ t;
V là thể tích chết, xác định ở nhiệt độ t, được tính từ khối lượng nước cân trong không khí ở nhiệt độ t.
Khối lượng của 1 lít nước, có tính đến ảnh hưởng của sức đẩy của không khí, ở nhiệt độ xác định được đưa ra trong bảng sau.
| Nhiệt độ (°C) | Khối lượng của một lít nước (g) |
| 20 | 997,18 |
| 25 | 996,02 |
| 30 | 994,62 |
Trong hệ đơn vị quốc tế SI, đơn vị của khối lượng riêng là kg/m3. Trong ngành dược thường xác định khối lượng riêng ở nhiệt độ 20 °C (ρ20) có tính đến ảnh hưởng của sức đẩy của không khí (tức là quy về giá trị xác định trong chân không) và dùng đơn vị kg/l hoặc g/ml.
Tỷ trọng tương đối
Tỷ trọng tương đối ![]() của một chất là tỷ số giữa khối lượng của một thể tích cho trước của chất đó, và khối lượng của cùng thể tích nước cất, tất cả đều cân ở 20 °C.
của một chất là tỷ số giữa khối lượng của một thể tích cho trước của chất đó, và khối lượng của cùng thể tích nước cất, tất cả đều cân ở 20 °C.
Tỷ trọng biểu kiến
Đại lượng “tỷ trọng biểu kiến” được dùng trong các chuyên luận ethanol, ethanol 96 % và loãng hơn..., là khối lượng cân trong không khí của một đơn vị thể tích chất lỏng. Tỷ trọng biểu kiến được biểu thị bằng đơn vị kg/m3 và được tính toán theo biểu thức sau đây:
Tỷ trọng biểu kiến = 997,2 × 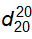
Trong đó:
![]() là tỷ trọng tương đối của chất thử.
là tỷ trọng tương đối của chất thử.
997,2 là khối lượng cân trong không khí của 1 m3 nước, tính bằng kg.
Xác định tỷ trọng tương đối 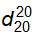 của một chất lỏng
của một chất lỏng
Xác định tỷ trọng của một chất lỏng theo phương pháp được chỉ rõ trong chuyên luận. Nếu chuyên luận không chỉ rõ dùng phương pháp nào để xác định tỷ trọng một chất lỏng, có thể dùng một trong các phương pháp sau đây:
Phương pháp dùng picnomet: Cân chính xác picnomet rỗng, khô và sạch. Đổ vào picnomet mẫu thử đã điều chỉnh nhiệt độ thấp hơn 20 °C, chú ý không để có bọt khí. Giữ picnomet ở nhiệt độ 20 °C trong khoảng 30 min. Dùng một băng giấy lọc để thấm hết chất lỏng thừa trên vạch mức, làm khô mặt ngoài của picnomet, cân rồi tính khối lượng chất lỏng chứa trong picnomet. Tiếp đó đổ mẫu thử đi, rửa sạch picnomet, làm khô bằng cách tráng ethanol rồi tráng aceton, thổi không khí nén hoặc không khí nóng đuổi hết hơi aceton, sau đó xác định khối lượng nước cất chứa trong picnomet ở nhiệt độ 20 °C như làm với mẫu thử. Tỷ số giữa khối lượng mẫu thử và khối lượng nước cất thu được là tỷ trọng 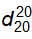 cần xác định.
cần xác định.
Phương pháp này cho kết quả với 4 chữ số lẻ thập phân.
Phương pháp dùng cân thủy tĩnh Mohr-Westphal: Đặt cân trên mặt phẳng nằm ngang. Mắc phao vào đòn cân, đặt phao chìm trong nước cất ở nhiệt độ 20 °C và chỉnh thăng bằng bằng các con mã đặt ở các vị trí thích hợp, thu được giá trị M. Lấy phao ra, thấm khô rồi đặt lại phao chìm trong chất lỏng cần xác định tỷ trọng, ở cùng nhiệt độ 20 °C, chú ý sao cho phần dây treo chìm trong chất lỏng một đoạn bằng đoạn đã chìm trong nước cất. Chỉnh lại thăng bằng bằng các con mã đặt ở vị trí thích hợp, thu được giá trị M1. Tỷ số M1/M là tỷ trọng 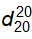 cần xác định.
cần xác định.
Phương pháp này cho kết quả với 3 chữ số lẻ thập phân.
Phương pháp dùng tỷ trọng kế: Lau sạch tỷ trọng kế bằng ethanol hoặc ether. Dùng đũa thủy tinh trộn đều chất lỏng cần xác định tỷ trọng. Đặt nhẹ nhàng tỷ trọng kế vào chất lỏng đó sao cho tỷ trọng kế không chạm vào thành và đáy của dụng cụ đựng chất thử. Chỉnh nhiệt độ tới 20 °C và khi tỷ trọng kế ổn định, đọc kết quả theo vòng khum dưới của mức chất lỏng. Đối với chất lỏng không trong suốt, đọc theo vòng khum trên.
Phương pháp này cho kết quả với 2 hoặc 3 chữ số lẻ thập phân.
Xác định tỷ trọng 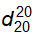 của mỡ, sáp, nhựa, nhựa thơm
của mỡ, sáp, nhựa, nhựa thơm
Dùng picnomet: Đầu tiên cân chính xác picnomet rỗng (M1) rồi cân picnomet đổ đầy nước cất ở 20 °C (M4). Sau đó đổ nước đi, làm khô picnomet, dùng ống hút hoặc phễu cuống nhỏ, cho vào trong picnomet mẫu thử đã được đun chảy khoảng 1/3 đến 1/2 thể tích của picnomet. Để 1 h trong nước nóng, không đậy nút. Làm nguội đến 20 °C, đậy nút. Lau khô mặt ngoài picnomet rồi lại cân (M2). Cuối cùng thêm nước cất đến vạch, lau khô mặt ngoài picnomet rồi lại cân (M3). Chú ý không được để bọt khí còn lại giữa lớp nước và mẫu thử.
Tính kết quả theo công thức:
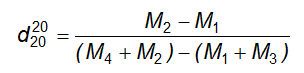
6.6 XÁC ĐỊNH NHIỆT ĐỘ ĐÔNG ĐẶC
Nhiệt độ đông đặc là nhiệt độ cao nhất giữ nguyên không đổi trong quá trình chuyển từ trạng thái lỏng sang trạng thái rắn.
Dụng cụ
Một ống nghiệm (a) 150 mm × 25 mm đặt vào bên trong một ống nghiệm lớn (b) khoảng 160 mm × 40 mm. Hai ống nghiệm không chạm vào nhau và cách nhau một lớp không khí. Ống nghiệm bên trong được đậy bằng một nút có mang một que khuấy và một nhiệt kế (dài khoảng 175 mm, chia độ tới 0,2 °C), được cố định sao cho bầu nhiệt kế cách đáy ống khoảng 15 mm. Que khuấy được làm bằng một đũa thủy tinh hoặc bằng thép, đầu dưới uốn thành một vòng tròn có đường kính khoảng 18 mm, vuông góc với que khuấy.
Ống nghiệm (a) và ống bọc nó (b) được giữ ở giữa một cốc hình vại (c) có dung tích 1 lít, chứa một chất lỏng làm lạnh thích hợp. Mức chất lỏng làm lạnh cách miệng ống khoảng 20 mm. Một nhiệt kế bổ trợ được giữ trong chất lỏng làm lạnh.
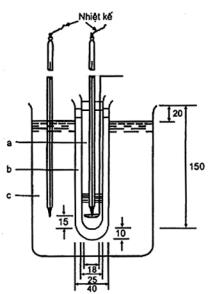
Hình 6.6 - Dụng cụ xác định nhiệt độ đông đặc
(kích thước tính bằng mm)
Cách tiến hành
Lấy một lượng chế phẩm (đã được làm nóng chảy nếu cần thiết), cho vào ống nghiệm (a) của dụng cụ sao cho bầu thủy ngân của nhiệt kế ngập trong lớp chế phẩm và xác định sơ bộ khoảng nhiệt độ đông đặc bằng cách làm lạnh nhanh. Đặt ống nghiệm (a) đã chứa chế phẩm vào trong một bể cách thủy có nhiệt độ trên nhiệt độ động đặc dự kiến khoảng 5 °C cho đến khi các tinh thể chế phẩm chảy hoàn toàn. Làm đầy cốc (c) bằng nước hoặc dung dịch bão hòa natri clorid ở nhiệt độ thấp hơn nhiệt độ đông đặc dự kiến khoảng 5 °C. Lồng ống nghiệm (a) vào ống nghiệm (b) và đặt vào cốc (c). Khuấy liên tục, nhẹ nhàng và cứ 30 s lại đọc nhiệt độ trên nhiệt kế một lần. Lúc đầu nhiệt độ hạ thấp dần, rồi giữ nguyên một thời gian hoặc tăng một chút, rồi giữ nguyên không đổi trong suốt thời gian đông.
Để tránh hiện tượng chậm đông, khi gần đến điểm đông, có thể cho vào ống nghiệm một tinh thể nhỏ chế phẩm đã có sẵn, hoặc cọ nhẹ thành trong của ống nghiệm.
6.7 XÁC ĐỊNH NHIỆT ĐỘ NÓNG CHẢY, KHOẢNG NÓNG CHẢY VÀ ĐIỂM NHỎ GIỌT
Khoảng nóng chảy (gọi tắt là khoảng chảy) của một chất là khoảng nhiệt độ đã hiệu chỉnh, kể từ khi chất rắn bắt đầu nóng chảy và xuất hiện những giọt chất lỏng đầu tiên, đến khi chất rắn chuyển hoàn toàn sang trạng thái lỏng.
Nhiệt độ nóng chảy (gọi tắt là điểm chảy) của một chất là nhiệt độ đã hiệu chỉnh, tại đó hạt chất rắn cuối cùng của chất thử nghiệm chuyển thành trạng thái lỏng, bắt đầu biến màu, hóa than hoặc sủi bọt. Khi phải xác định khoảng chảy, nếu nhiệt độ bắt đầu hoặc nhiệt độ kết thúc nóng chảy không xác định rõ ràng, ta có thể chỉ xác định nhiệt độ kết thúc, hoặc nhiệt độ bắt đầu nóng chảy. Nhiệt độ này phải nằm trong giới hạn quy định trong chuyên luận riêng của chế phẩm.
Để xác định khoảng chảy và điểm chảy, tùy theo tính chất lý học của từng chất, áp dụng một trong các phương pháp 1, 2 hay 3, còn để xác định điểm nhỏ giọt thì sử dụng phương pháp 4.
Phương pháp 1
(Áp dụng cho các chất rắn dễ nghiền nhỏ).
Dụng cụ
Một bình thủy tinh chịu nhiệt, trong bình chứa một chất lỏng thích hợp, thường dùng là dầu parafin, hoặc để xác định ở nhiệt độ cao dùng dầu Silicon, lượng chất lỏng đủ để nhúng chìm được nhiệt kế và mẫu thử sao cho bầu thủy ngân cách đáy bình 2 cm.
Một dụng cụ khuấy có khả năng duy trì sự đồng nhất về nhiệt độ trong chất lỏng.
Một nhiệt kế đã được hiệu chuẩn và chia độ đến 0,5 °C, có khoảng nhiệt độ đo từ thấp nhất đến cao nhất không được quá 100 °C.
Ống mao quản, hàn kín một đầu, dài 6 cm đến 8 cm, đường kính trong 1,0 mm ± 0,1 mm, thành ống dày khoảng 0,10 mm đến 0,15 mm.
Nguồn nhiệt, có thể điều chỉnh được.
Dụng cụ có thể được hiệu chuẩn với các chất chuẩn có điểm chảy được chứng nhận của Tổ chức Y tế Thế giới hay những chất thích hợp khác.
Cách xác định
Nghiền thành bột mịn chất thử đã làm khô 24 h ở áp suất 1,5 kPa đến 2,5 kPa trong bình với chất hút ẩm thích hợp, hoặc sấy khô 2 h ở 100 °C đến 105 °C, trừ trường hợp có chỉ dẫn trong chuyên luận riêng.
Cho bột vào ống mao quản, lèn bột bằng cách gõ nhẹ ống mao quản xuống mặt phẳng cứng để có một lớp chế phẩm cao 4 mm đến 6 mm.
Đun nóng bình đựng chất lỏng đến khi nhiệt độ thấp hơn điểm chảy dự kiến của chất thử khoảng 10 °C, điều chỉnh nhiệt độ sao cho nhiệt độ tăng 1 °C trong 1 min (trừ trường hợp có chỉ dẫn trong chuyên luận riêng), hoặc cho nhiệt độ tăng 3 °C trong 1 min khi thử các chất không bền vì nhiệt.
Khi nhiệt độ đạt thấp hơn điểm chảy dự kiến khoảng 5 °C, lấy nhiệt kế ra, nhanh chóng buộc ống mao quản có chế phẩm vào nhiệt kế, sao cho lớp chế phẩm ngang với phần giữa bầu thủy ngân của nhiệt kế. Đặt lại nhiệt kế vào bình.
Nhiệt độ mà tại đó nhìn thấy cột chất thử xẹp xuống, so sánh với một điểm nào đó trên thành ống, được xác định là điểm bắt đầu nóng chảy và nhiệt độ mà tại đó chất thử trở thành chất lỏng hoàn toàn, được xác định là điểm cuối của sự chảy hay điểm chảy.
Hiệu chỉnh nhiệt độ quan sát được nếu nhiệt kế đo có sai số với nhiệt kế chuẩn và sự khác biệt nếu có giữa nhiệt độ của đoạn cột thủy ngân ở ngoài chất lỏng trong điều kiện thí nghiệm và của đoạn cột thủy ngân ngoài chất lỏng trong điều kiện hiệu chuẩn. Nhiệt độ của đoạn cột thủy ngân ở ngoài chất lỏng được xác định bằng cách đặt bầu thủy ngân của nhiệt kế phụ ở điểm giữa của phần cột thủy ngân lộ ra ngoài chất lỏng của nhiệt kế chính.
Tính nhiệt độ đã hiệu chỉnh theo công thức sau:
T hiệu chỉnh = T + 0.00016N (Ts - t)
trong đó:
T là nhiệt độ đọc trên nhiệt kế chính;
Ts là nhiệt độ trung bình của đoạn cột thủy ngân ở ngoài chất lỏng của nhiệt kế chính trong điều kiện chuẩn hoá;
t là nhiệt độ của đoạn cột thủy ngân ở ngoài chất lỏng đọc trên nhiệt kế phụ tại điểm chảy;
N là số khoảng chia độ (°C) của đoạn cột thủy ngân của nhiệt kế chính ở ngoài chất lỏng.
Phương pháp 2
(Áp dụng cho các chất không nghiền thành bột được như mỡ, sáp, acid béo, parafin rắn, lanolin và chất tương tự, không tan trong nước):
Dụng cụ
Dùng dụng cụ như phương pháp 1, riêng ống mao quản hở hai đầu, dài 80 mm đến 100 mm, đường kính trong 0,8 mm đến 1,2 mm, thành dày 0,1 mm đến 0,3 mm.
Cách xác định
Cẩn thận làm chảy chất thử ở nhiệt độ thấp tối thiểu, cắm ống mao quản vào chất thử sao cho lấy được một cột chất thử cao 10 mm không có bọt. Dùng khăn sạch lau sạch phía ngoài mao quản, để nguội ở 10 °C hay thấp hơn trong 24 h, hoặc để tiếp xúc với nước đá ít nhất 2 h.
Đổ nước cất vào cốc thủy tinh đến chiều cao 6 cm. Treo nhiệt kế vào giá và nhúng vào chính giữa cốc nước. Đun nóng cốc nước đến nhiệt độ thấp hơn điểm chảy dự kiến 10 °C, lấy nhiệt kế ra, nhanh chóng buộc mao quản vào nhiệt kế bằng phương tiện thích hợp sao cho cột chất thử ngang với phần giữa bầu thủy ngân của nhiệt kế, điều chỉnh nhiệt kế để đầu dưới của ống mao quản cách đáy cốc 1 cm. Điều chỉnh tốc độ gia nhiệt không quá 1 °C trong một phút. Trong lúc đó luôn khuấy nước nhẹ nhàng và quan sát cột chất thử, khi cột chất thử bắt đầu dâng lên trong ống mao quản thì đọc nhiệt độ. Kết quả đọc giữa 2 lần đo liên tiếp không được chênh lệch nhau quá 0,5 °C. Lấy kết quả trung bình. Nhiệt độ này được coi là điểm chảy và phải nằm trong khoảng nóng chảy quy định của chuyên luận.
Phương pháp 3
(Áp dụng cho chất rắn dễ nghiền nhỏ, giống như phương pháp 1).
Dụng cụ
Một khối kim loại (như đồng) không bị ăn mòn bởi chất thử nghiệm, khả năng truyền nhiệt tốt, bề mặt trên được đánh bóng cẩn thận. Khối kim loại này được đun nóng toàn khối và đồng đều bằng một dụng cụ như đèn ga nhỏ điều chỉnh được hay dụng cụ đun nóng bằng điện có thể điều chỉnh chính xác. Khối kim loại có một khoang hình ống nằm song song, bên dưới và cách bề mặt đánh bóng khoảng 3 mm, có kích thước thích hợp để chứa được một nhiệt kế thủy ngân, đặt ở vị trí giống vị trí khi hiệu chuẩn. Dụng cụ được hiệu chuẩn bằng một chất chuẩn thích hợp đã ghi trong phương pháp 1.
Cách xác định
Đun nóng khối kim loại với một tốc độ thích hợp tới nhiệt độ dưới điểm nóng chảy dự kiến khoảng 10 °C thì điều chỉnh nhiệt độ tăng 1 °C/min, tại những khoảng thời gian đều nhau, thả một vài hạt chất thử đã được làm khô bằng phương pháp thích hợp hay theo sự chỉ dẫn trong chuyên luận và nghiền thành bột mịn lên bề mặt khối kim loại gần vị trí của bầu thủy ngân của nhiệt kế, lau sạch bề mặt sau mỗi lần thử nghiệm. Ghi lại nhiệt độ mà tại đó chất thử tan chảy lần đầu tiên ngay khi nó chạm tới bề mặt kim loại (t1) và ngừng đun ngay. Trong khi để nguội dần, lại thả một vài hạt chất thử trong khoảng thời gian đều đặn, lau sạch bề mặt sau mỗi lần thử. Ghi lại nhiệt độ mà tại đó chất thử ngừng tan chảy ngay khi tiếp xúc với bề mặt tấm kim loại (t2). Điểm chảy tức thời được tính theo công thức:
(t1 + t2)/2
Phương pháp 4
Xác định điểm nhỏ giọt (áp dụng cho vaselin và những chất tương tự).
Điểm nhỏ giọt là nhiệt độ mà tại đó giọt đầu tiên của chất thử nóng chảy rơi xuống từ một cái chén nhỏ trong điều kiện xác định dưới đây:
Dụng cụ (Hình 6.7)
Một ống kim loại (A) bao bọc nhiệt kế thủy ngân và được vặn vào một ống kim loại thứ hai (B), một chén nhỏ bằng kim loại (F) được gắn cố định vào phần dưới của ống (B) bằng hai cái đai xiết chặt (E), vị trí chính xác của chén được xác định bằng hai giá đỡ (D) dài 2 mm và cũng dùng để giữ cho nhiệt kế ở chính giữa. Có một lỗ (C) xuyên qua thành của ống kim loại thứ 2 (B), được dùng để cân bằng áp suất.
Bề mặt lỗ thoát ở đáy chén phải phẳng và vuông góc với thành trong của nó. Nhiệt kế có đường kính trong của bầu thủy ngân từ 3,3 mm đến 3,7 mm, dài 5,7 mm đến 6,3 mm, được chuẩn hóa từ 0 °C đến 110 °C chia vạch 1 mm tương ứng 1 °C. Dụng cụ được đặt theo chiều dọc của một ống có kích thước khoảng 20 cm × 4 cm, được cố định bằng một cái nút và nhiệt kế xuyên qua nút theo một đường rãnh. Miệng lỗ thoát của chén cách đáy của ống khoảng 15 mm. Ống được nhúng vào một cái cốc vại có dung tích 1 lít, chứa nước sao cho đáy của ống cách đáy của cốc khoảng 25 mm, mặt thoáng của nước bằng với đầu trên của ống kim loại thứ nhất (A). Một dụng cụ khuấy được dùng để giữ cho nhiệt độ của nước đồng nhất.
Cách xác định
Nếu không có chỉ dẫn gì khác trong chuyên luận, đổ chất thử tới miệng chén mà không làm nóng chảy nó, dùng thìa xúc hóa chất để loại bỏ những phần dư ở miệng và đáy chén. Ấn chén vào vị trí của nó trong lớp vỏ kim loại (B) cho tới khi chạm tới giá đỡ, dùng thìa xúc bớt chất thử do nhiệt kế đẩy ra. Đun nóng cốc nước, khi nhiệt độ đạt tới thấp hơn nhiệt độ nóng chảy hay nhỏ giọt khoảng 10 °C thì điều chỉnh tốc độ tăng nhiệt độ khoảng 1 °C/min và ghi nhiệt độ giọt chất lỏng đầu tiên rơi xuống. Tiến hành ít nhất 3 lần, mỗi lần thử với mẫu thử mới. Sự khác biệt giữa các lần đọc không quá 3 °C. Nhiệt độ trung bình của 3 lần xác định là điểm chảy hay điểm nhỏ giọt của mẫu thử.
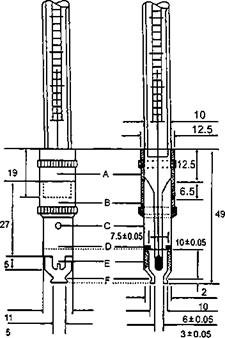
Hình 6.7- Dụng cụ xác định điểm nhỏ giọt
(kích thước tính bằng mm)
6.8 XÁC ĐỊNH NHIỆT ĐỘ SÔI VÀ KHOẢNG CHƯNG CẤT
Nhiệt độ sôi của một chất lỏng là nhiệt độ đã hiệu chỉnh, tại đó áp suất hơi của chất lỏng đạt tới 101,3 kPa.
Xác định nhiệt độ sôi
Dụng cụ (Hình 6.8)
Bình chưng cất: Bình chưng cất đáy tròn, bằng thủy tinh chịu nhiệt có dung tích 50 ml đến 60 ml, cổ bình cao 10 cm đến 12 cm, đường kính trong của cổ bình từ 14 mm đến 16 mm, ở khoảng giữa chiều cao của cổ bình có một ống nhánh dài 10 cm đến 12 cm, đường kính trong 5 mm và tạo với phần dưới của cổ bình một góc 70° đến 75°.
Ống ngưng ruột thẳng: Một ống thủy tinh thẳng dài 55 cm đến 60 cm, phần được cung cấp nước lạnh dài khoảng 40 cm. Ở cuối ống ngưng lắp một ống nối cong để dẫn chất lỏng cất được vào bình hứng.
Bình hứng: Là một ống đong thích hợp có dung tích 25 ml đến 50 ml, chia độ đến 0,5 ml.
Nguồn nhiệt có thể điều chỉnh được cường độ đốt nóng, có thể dùng bếp khí đốt, đèn cồn. Khi dùng bếp khí đốt hay đèn cồn phải dùng một lưới thép có phủ chất chịu nhiệt hình vuông, mỗi chiều dài 14 cm đến 16 cm, ở giữa có một lỗ tròn sao cho khi đặt bình cất vào, phần lọt xuống dưới lưới thép có phủ chất chịu nhiệt có dung tích 3 ml đến 4 ml.
Nhiệt kế đã hiệu chuẩn chia độ đến 0,5 °C hoặc tới 0,2 °C. Lắp nhiệt kế ở đúng giữa cổ bình và để đáy của bầu thủy ngân ngang với mức thấp nhất của cổ bình.
Cách xác định
Cho vào bình cất 20 ml chất thử, thêm vào bình cất vài viên bi thủy tinh hay đá bọt. Đun nóng bình nhanh cho đến sôi. Đọc nhiệt độ khi giọt chất lỏng đầu tiên hứng được.
Hiệu chỉnh nhiệt độ đọc được với áp suất khí quyển bằng công thức sau:
t1 = t2+k (101,3-b)
trong đó:
t1 là nhiệt độ đã được hiệu chỉnh;
t2 là nhiệt độ đọc được ở áp suất b;
b là áp suất không khí tại thời điểm thử nghiệm (tính theo kPa);
k là hệ số hiệu chỉnh có giá trị như ở bảng dưới đây:
| Nhiệt độ sôi (°C) | Hệ số hiệu chỉnh |
| Dưới 100 °C | 0,30 |
| Trên 100 °C tới 140 °C | 0,34 |
| Trên 140 °C tới 190 °C | 0,38 |
| Trên 190 °C tới 240 °C | 0,41 |
| Trên 240 °C | 0,45 |
Xác định khoảng chưng cất
Khoảng chưng cất của một chất lỏng là khoảng nhiệt độ được hiệu chỉnh ở áp suất 101,3 kPa, trong khoảng đó một chất lỏng hay một phần xác định chất lỏng chưng cất được trong điều kiện dưới đây: Dụng cụ
Dụng cụ giống như dụng cụ xác định điểm sôi, chỉ khác là bình cất có dung tích 200 ml và nhiệt kế lắp sao cho đầu trên của bầu thủy ngân thấp hơn thành dưới của ống nhánh 5 mm. Nhiệt kế được chia vạch tới 0,2 °C và có khoảng thang đo được 50 °C. Trong khi chưng cất bình và cổ bình được bảo vệ để tránh gió lùa bằng một tấm màn chắn thích hợp. Hứng dịch cất vào một ống đong 50 ml chia độ tới 1 ml. Làm lạnh ống ngưng bằng nước tuần hoàn đối với các chất chưng cất dưới 150 °C.
Cách xác định
Cho vào bình cất 50 ml chất lỏng thử nghiệm, thêm vài viên đá bọt. Đun bình sao cho chất lỏng sôi nhanh và ghi nhiệt độ tại đó giọt chất lỏng đầu tiên cất được nhỏ vào ống đong. Điều chỉnh nguồn nhiệt để có tốc độ cất 2 ml/min đến 3 ml/min và ghi lại nhiệt độ mà tất cả chất thử hay một phần quy định chất thử ở 20 °C được chưng cất hết.
Hiệu chỉnh nhiệt độ đọc được với áp suất khí quyển bằng công thức sau: t1 = t2 + k (101,3-b)
trong đó:
t1 là nhiệt độ đã được hiệu chỉnh;
t2 là nhiệt độ đọc được ở áp suất b;
b là áp suất không khí tại thời điểm thử nghiệm (tính theo kPa);
k là hệ số hiệu chỉnh ở bảng trong mục xác định điểm sôi, nếu không có hệ số nào khác được quy định trong chuyên luận riêng.
Hình 6.8 - Dụng cụ xác định điểm sôi, khoảng chưng cất
(kích thước tính bằng mm).
6.9 XÁC ĐỊNH ĐỘ THẨM THẤU
Xác định độ thẩm thấu là một phương pháp đo nồng độ thẩm thấu của dung dịch mẫu thử từ phép đo của độ hạ băng điểm.
Khi một dung dịch và một dung môi tinh khiết được ngăn cách nhau bởi một màng bán thấm, thì dung môi có thể đi qua tự do nhưng chất tan thì không thể, một phần của dung môi tinh khiết sẽ đi qua màng sang bên ngăn của dung dịch. Sự chênh lệch áp suất giữa hai ngăn đồng thời với việc di chuyển của dung môi qua màng được định nghĩa là áp suất thẩm thấu. Áp suất thẩm thấu là một đặc tính vật lý phụ thuộc vào tổng các dạng tiểu phân có mặt, bao gồm cả các phân tử trung hòa và các ion, và không phụ thuộc vào bản chất chất tan. Các đặc tính của dung dịch như áp suất thẩm thấu, độ hạ băng điểm, độ tăng điểm sôi... không phụ thuộc vào bản chất của chất tan mà phụ thuộc vào tổng số của tất cả các dạng tiểu phân được gọi là các đặc tính phụ thuộc số lượng của dung dịch.
Áp suất thẩm thấu của một dung dịch polymer có thể đo trực tiếp như là sự chênh lệch áp suất thủy tĩnh giữa hai ngăn được cách nhau bởi một màng bán thấm, ví dụ màng celulose. Tuy nhiên, nó không áp dụng đối với dung dịch có chứa các phần tử nhỏ có thể đi qua màng bán thấm. Mặc dù áp suất thẩm thấu của một dung dịch như vậy không thể đo được trực tiếp, hướng và mức độ của việc di chuyển dung môi qua màng sinh học có thể được dự đoán trước từ tổng số tất cả các tiểu phân có mặt khi dung dịch được đặt dưới các điều kiện sinh lý. Các đặc tính phụ thuộc số lượng khác của dung dịch như độ hạ băng điểm, độ tăng điểm sôi, độ hạ áp suất hơi... có thể thu được trực tiếp bằng cách theo dõi sự thay đổi của nhiệt độ và áp suất.... Những đặc tính này phụ thuộc vào tổng các dạng ion và phân tử trung hòa trong dung dịch cũng như áp suất thẩm thấu và nồng độ các hạt phân tử được định nghĩa như là nồng độ thẩm thấu. Nồng độ thẩm thấu có thể được định nghĩa bằng hai cách, một là nồng độ dựa trên khối lượng (độ thẩm thấu ξm, mol/kg) và cách khác là nồng độ dựa trên thể tích (độ thẩm thấu ξc, mol/l). Trong thực tế, cách thứ hai thuận tiện hơn.
Phương pháp đo độ hạ băng điểm thường được dùng để xác định nồng độ thẩm thấu, trừ khi có chỉ dẫn khác. Phương pháp dựa trên sự phụ thuộc tuyến tính của độ hạ băng điểm ΔT (°C) với độ thẩm thấu (mol/kg) được biểu thị theo phương trình sau:
ΔT = K .ξm
Trong đó: K là hằng số của độ hạ băng điểm mol và bằng 1,86 °C kg/mol, khi dung môi là nước. Vì hằng số K được định nghĩa dựa trên nồng độ phân tử gam, nồng độ thẩm thấu mol có thể thu được từ phương trình trên. Trong khoảng nồng độ thẩm thấu loãng, độ thẩm thấu ξm (mol/kg) có thể coi bằng độ thẩm thấu ξc (mol/l). Bởi vậy, độ thẩm thấu thông thường (mol/l) và đơn vị osmol (Osm) được chấp nhận trong phương pháp thử này.
Một Osm có nghĩa là có một số lượng tiểu phân bằng với số Avogadro (6,022 × 1023/mol) chứa trong 1 lít dung dịch. Nồng độ thẩm thấu thường được biểu thị bằng đơn vị miliosmol (mOsm, mosmol/l).
Thiết bị
Thẩm thấu kế gồm một cốc đo chứa một thể tích xác định của dung dịch mẫu thử, một bộ phận giữ cốc đo, một bể làm lạnh có bộ phận điều chỉnh nhiệt độ và một nhiệt điện trở để phát hiện nhiệt độ.
Cách tiến hành
Một thể tích xác định của dung dịch thử được đưa vào cốc đo theo quy định đối với từng loại thiết bị. Thiết bị trước tiên phải được hiệu chuẩn bằng phương pháp hiệu chuẩn hai điểm dùng các dung dịch chuẩn.
Để hiệu chuẩn, chọn hai dung dịch chuẩn khác nhau để nồng độ thẩm thấu của dung dịch thử nằm trong khoảng đó. Ngoài các dung dịch chuẩn được cho trong bảng dưới đây, nước có thể được dùng như là một dung dịch chuẩn (0 mOsm) để đo các dung dịch có độ thẩm thấu thấp (0 mOsm đến 100 mOsm). Sau khi rửa cốc đựng mẫu và nhiệt điện trở như chỉ dẫn trong từng thiết bị riêng, đo độ hạ băng điểm của dung dịch thử. Dùng phương trình đã nêu trên để tính độ thẩm thấu của dung dịch.
Trong trường hợp dung dịch có độ thẩm thấu lớn hơn 1000 mOsm, cần hòa loãng mẫu bằng cách thêm nước cất. Đo độ thẩm thấu của dung dịch pha loãng như chỉ dẫn ở trên. Trong trường hợp này, cần chỉ rõ độ thẩm thấu đã được tính đối với mẫu thử là độ thẩm thấu biểu kiến thu được bằng phương pháp pha loãng. Khi áp dụng phương pháp pha loãng, độ pha loãng nên được chọn để độ thẩm thấu đo được gần với độ thẩm thấu của dung dịch nước muối sinh lý.
Trong trường hợp mẫu thử ở trạng thái rắn như là thuốc đông khô, chuẩn bị dung dịch thử bằng cách hòa tan chất rắn trong dung môi thích hợp như chỉ dẫn.
Sự thích hợp của thiết bị
Sau khi hiệu chuẩn thiết bị, phép thử độ thích hợp phải được làm bằng cách lặp lại phép đo độ thẩm thấu đối với một dung dịch chuẩn không ít hơn 6 lần. Trong khi làm phép thử này nên chọn độ thẩm thấu của dung dịch thử và dung dịch chuẩn gần giống nhau.
Trong phép thử này, độ lặp lại của giá trị đo và độ lệch trung bình nên nhỏ hơn 2,0 % và 3,0 % tương ứng.
Chuẩn bị dung dịch chuẩn để hiệu chuẩn thẩm thấu kế
Cân chính xác một lượng natri clorid chuẩn (đã được nung trước ở 500 °C đến 650 °C trong 40 min đến 50 min và để nguội trong bình hút ẩm) theo chỉ dẫn trong Bảng 6.9 và hòa tan natri clorid trong chính xác 100 g nước để làm dung dịch chuẩn
Bảng 6.9 - Lượng natri clorid để pha dung dịch chuẩn
| Dung dịch chuẩn để hiệu chuẩn thẩm thấu kế (mosmol/kg) | Lượng natri clorid (g) |
| 100 | 0,309 |
| 200 | 0,626 |
| 300 | 0,946 |
| 400 | 1,270 |
| 500 | 1,593 |
| 700 | 2,238 |
| 1000 | 3,223 |
Tỷ số thẩm thấu
Trong phương pháp thử này tỷ số thẩm thấu được định nghĩa là tỷ số độ thẩm thấu của dung dịch thử đối với dung dịch natri clorid đẳng trương. Tỷ số có thể được dùng để đo độ đẳng trương của dung dịch thử. Vì độ thẩm thấu của dung dịch natri clorid đẳng trương (NaCI 0,900 g/100 ml) Cs(mOsm) được coi như là hằng số (286 mOsm), độ thẩm thấu của dung dịch thử là CT (mOsm), tỷ số thẩm thấu được tính theo phương trình:
Tỷ số thẩm thấu = 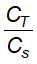
Cs: 286 mOsm
Khi phép đo được tiến hành bằng phương pháp pha loãng, vì mẫu thử có độ thẩm thấu lớn hơn 1000 mOsm, độ thẩm thấu biểu kiến của dung dịch mẫu CT có thể được tính theo phương trình: n x C'T = CT
trong đó:
n: độ pha loãng;
C'T: độ thẩm thấu đo được của dung dịch loãng.
Trong phép tính này, đã chấp nhận giả thiết có sự tương quan tuyến tính giữa độ thẩm thấu và nồng độ dung dịch. Bởi vậy, khi có bước pha loãng, cần phải chỉ rõ độ pha loãng là n lần.
6.10 XÁC ĐỊNH ĐIỆN DẪN SUẤT
Điện dẫn suất (còn gọi là Độ dẫn điện riêng) là giá trị nghịch đảo của điện trở suất.
Một dây dẫn có độ dài L (cm) và thiết diện S (cm2) thì điện trở của nó được tính theo công thức:
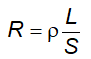
trong đó:
ρ: điện trở suất.
Đơn vị của điện trở R là ôm (Ω), của điện trở suất ρ là ôm.cm (Ω.cm).
Đơn vị của độ dẫn điện là simen (S). Simen là nghịch đảo của ôm: 1 S = 1/Ω.
Điện dẫn suất là nghịch đảo của ρ, có thứ nguyên là Ω-1. cm-1 hoặc s/cm (S.cm-1).
Điện dẫn suất K (kappa) của một dung dịch được biểu thị bằng độ dẫn điện của một lớp dung dịch giữa 2 mặt đối nhau của một khối lập phương có cạnh 1 cm.
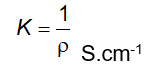
Ngoài đơn vị simen trên centimét, S.cm-1, điện dẫn suất K còn được biểu thị bằng microsimen trên centimét, µS.cm-1.
Nếu không có chỉ dẫn riêng, nhiệt độ để xác định K là 25 °C.
Thiết bị và cách tiến hành mô tả dưới đây được áp dụng để đo các mẫu có điện dẫn suất lớn hơn 10 µS.cm-1. Việc đo điện dẫn suất của nước được đề cập trong các chuyên luận tương ứng.
Thiết bị
Thiết bị đo (là độ dẫn điện kế hay điện trở kế) được dùng để đo điện trở của cột chất lỏng giữa các điện cực của tế bào đo được nhúng trong chất lỏng cần đo. Thiết bị này dùng dòng điện xoay chiều để tránh ảnh hưởng của sự phân cực tại điện cực và được trang bị một đầu dò nhiệt độ và bộ phận bổ chính nhiệt độ. Tế bào đo gồm hai điện cực platin song song được phủ lớp bột đen platin, cách nhau một khoảng cách L; mỗi điện cực có diện tích bề mặt S. Cả hai điện cực được bảo vệ bằng một ống thủy tinh.
Một số loại tế bào đo khác với mô tả trên cũng có thể được sử dụng.
Hằng số C của tế bào đo được biểu thị bằng cm-1 theo phương trình:
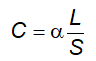
trong đó:
α: hệ số không có thứ nguyên, đặc trưng cho thiết kế của tế bào.
Thuốc thử dùng để xác định hằng số tế bào
Chuẩn bị 3 dung dịch chuẩn kali clorid có chứa lần lượt 0,7455 g, 0,0746 g và 0,0149 g kali clorid (TT) trong 1000 g dung dịch, dùng nước không có carbon dioxyd (TT) được điều chế từ nước cất có điện dẫn suất không quá 2 µS.cm-1.
Điện dẫn suất và điện trở suất của 3 dung dịch này ở 20 °C cho trong Bảng 6.10.
Bảng 6.10 - Điện dẫn suất và điện trở suất của các dung dịch kali clorid
| Nồng độ | Điện dẫn suất | Điện trở suất |
| (g/1000 g) | (µS.cm-1) | (Ω.cm) |
| 0,7455 | 1330 | 752 |
| 0,0746 | 133,0 | 7519 |
| 0,0149 | 26,6 | 37594 |
Nếu điện dẫn suất được xác định ở nhiệt độ khác 20 °C, dùng phương trình sau để hiệu chỉnh điện dẫn suất của các dung dịch kali clorid cho trong bảng 6.10. Phương trình này chỉ có giá trị khi nhiệt độ trong khoảng 15 °C đến 25 °C
KT = K20 [1 + 0,021 (T-20)]
trong đó:
T: nhiệt độ quy định trong chuyên luận;
KT: điện dẫn suất của kali clorid ở T °C;
K20: điện dẫn suất của kali clorid ở 20 °C.
Cũng có thể sử dụng những dung dịch chuẩn khác được xác nhận cung cấp trên thị trường, nhất là đối với các tế bào đo có hằng số 0,1 cm-1.
Cách tiến hành
Xác định hằng số tế bào
Chọn một tế bào đo thích hợp với tính chất và độ dẫn điện của dung dịch thử.
Điện dẫn suất của dung dịch cần đo càng cao thì cần chọn tế bào đo có hằng số c càng cao (có ρ thấp) để giá trị R đo được càng lớn càng tốt đối với máy đo. Các tế bào đo thường dùng có hằng số theo thứ tự là 0,1 cm-1; 1 cm-1 và 10 cm-1.
Dùng một dung dịch chuẩn, ví dụ, dung dịch kali clorid nồng độ thích hợp, để xác định hằng số tế bào. Nên chọn dung dịch chuẩn có điện dẫn suất gần với giá trị điện dẫn suất dự kiến của dung dịch cần đo. Rửa tế bào vài lần với nước cất và ít nhất hai lần với dung dịch chuẩn kali clorid trên. Đo điện trở của tế bào với dung dịch kali clorid ở nhiệt độ 25 °C ± 1 °C, hoặc ở nhiệt độ quy định trong chuyên luận. Hằng số c (cm-1) của tế bào đo được tính bởi công thức sau:
c = RKCl x KKCl
trong đó:
RKCl: điện trở đo được của dung dịch chuẩn kali clorid tính bằng mega ôm (MΩ);
KKCl: điện dẫn suất của dung dịch chuẩn kali clorid tính bằng µS.cm-1;
Hằng số C đo được của tế bào phải nằm trong khoảng ± 5 % của giá trị đã cho.
Xác định điện dẫn suất của dung dịch thử
Sau khi hiệu chuẩn máy với một trong số các dung dịch chuẩn, rửa tế bào đo vài lần với nước cất và ít nhất hai lần với dung dịch mẫu thử ở 25 °C ± 1 °C, hoặc ở nhiệt độ quy định trong chuyên luận. Tiến hành tiếp theo các phép đo điện trở như quy định trong chuyên luận; từ điện trở đo được và hằng số tế bào xác định được ở trên, tính ra điện dẫn suất của dung dịch mẫu thử.
6.11 XÁC ĐỊNH KHỐI LƯỢNG RIÊNG CỦA CHẤT RẮN
Khối lượng riêng có liên quan đến sự phân bố không gian của chất trong một nguyên liệu.
Khối lượng riêng của chất rắn thường được biểu thị bằng số g/cm3, khác với chất lỏng, khối lượng riêng thường được biểu thị bằng g/ml ở một nhiệt độ quy định.
Khối lượng riêng của một tiểu phân chất rắn có thể có các giá trị khác nhau phụ thuộc vào phương pháp đo thể tích tiểu phân đó. Cần phân biệt ba loại khối lượng riêng khác nhau:
Khối lượng riêng thực của một chất là khối lượng trung bình tính trên một đơn vị thể tích, không tính đến tất cả các khoảng trống không có vai trò cơ bản trong việc sắp xếp các phân tử. Đó là một đặc tính bên trong của chất, bởi vậy nó không phụ thuộc vào phương pháp xác định. Khối lượng riêng thực của một tinh thể hoàn chỉnh có thể được xác định từ kích thước và cấu trúc của đơn vị cấu tạo.
Khối lượng riêng đo bằng picnomet được đo bằng picnomet sử dụng khí. Đó là một phép đo khối lượng riêng thuận tiện đối với các bột dược phẩm. Trong một picnomet khí, thể tích bị chiếm chỗ bởi một khối lượng bột đã biết được xác định bằng cách đo thể tích của khí được thay thế vào chỗ của bột. Tỷ số giữa khối lượng và thể tích là khối lượng riêng đo bằng picnomet.
Khối lượng riêng đo bằng picnomet bằng khối lượng riêng thực trừ khi nguyên liệu có chứa những khoảng trống mà chất khí không thể thâm nhập vào được hoặc các lỗ hổng bị bịt kín mà khí dùng trong phương pháp này không thể lọt vào được.
Khối lượng riêng hạt tính cả những lỗ hổng hở nhỏ hơn một kích cỡ giới hạn vào thể tích của tiểu phân. Giới hạn kích cỡ phụ thuộc vào phương pháp đo. Một kỹ thuật đo chung là đo độ xốp bằng thủy ngân, tại đó kích cỡ lỗ giới hạn phụ thuộc vào áp suất khuếch tán tối đa. Vì thể tích lỗ hổng được tính thêm vào, nên khối lượng riêng hạt không bao giờ lớn hơn khối lượng riêng thực.
Một khái niệm có liên quan là khối lượng riêng khí động lực, đó là khối lượng riêng của tiểu phân với một thể tích được xác định bằng sự bao bọc tiểu phân bằng khí động lực trong một dòng chảy. Cả hai loại lỗ hổng kín và hở đều được tính đến nhưng các lỗ hở được làm đầy bằng chất lỏng tràn vào. Bởi vậy, khối lượng riêng khí động lực phụ thuộc vào khối lượng riêng của chất lỏng dùng trong phép thử nếu tiểu phân là xốp.
Tóm lại, khối lượng riêng đo bằng picnomet khí và khối lượng riêng thực đều chỉ khối lượng riêng, nếu cần có thể phân biệt dựa vào phương pháp đo.
Khối lượng riêng của một nguyên liệu phụ thuộc vào trạng thái tập hợp phân tử. Đối với chất khí và chất lỏng, khối lượng riêng chỉ phụ thuộc vào nhiệt độ và áp suất. Đối với chất rắn, khối lượng riêng sẽ rất khác nhau tùy thuộc vào cấu trúc kết tinh và độ kết tinh.
Nếu chất rắn ở thể vô định hình, khối lượng riêng của nó có thể phụ thuộc vào quá trình tinh chế và xử lý. Bởi vậy, không giống như chất lỏng, khối lượng riêng của hai chất rắn tương đương về mặt hóa học có thể khác nhau và sự khác nhau này phản ánh sự không giống nhau về cấu trúc trạng thái rắn. Khối lượng riêng của các hạt thành phần là một đặc tính lý học quan trọng của bột dược phẩm.
Ngoài các định nghĩa trên về khối lượng riêng của tiểu phân, còn có khối lượng riêng toàn khối bao gồm cả thể tích trống giữa các hạt được tạo thành trong khối bột. Do đó, khối lượng riêng toàn khối phụ thuộc vào cả hai: Khối lượng riêng của các tiểu phân bột và sự sắp xếp của các tiểu phân bột.
Đo khối lượng riêng bằng picnomet khí
Đo bằng picnomet khí là một phương pháp thuận tiện và thích hợp để xác định khối lượng riêng của các tiểu phân bột.
Một loại picnomet khí đơn giản được mô tả như trong Hình 6.11.
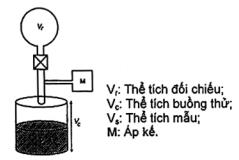
Hình 6.11 - Sơ đồ picnomet khí
Mẫu thử có khối lượng w và thể tích Vs được đặt trong một buồng thử kín có thể tích của buồng rỗng là Vc. Áp suất đối chiếu của hệ thống là Pr được xác định bằng áp kế khi mở van nối thể tích đối chiếu với buồng thử. Sau đó van được đóng để tách riêng thể tích đối chiếu Vr khỏi buồng thử. Buồng thử được nén với một áp suất ban đầu Pi. Sau đó van lại được mở để nối thể tích đối chiếu Vr với buồng thử và áp suất cuối cùng sẽ giảm xuống còn Pf. Thể tích của mẫu thử Vs được tính theo công thức:
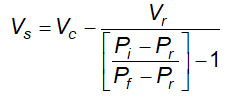 (1)
(1)
Khối lượng riêng ρ được tính theo phương trình:
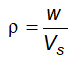 (2)
(2)
Các chi tiết thiết kế thiết bị có thể khác nhau, nhưng tất cả các picnomet khí đều dựa trên việc đo sự thay đổi áp suất khi một thể tích đối chiếu được thêm vào hoặc giảm đi ở buồng thử.
Khối lượng riêng đo được là khối lượng riêng trung bình của các tiểu phân bột. Kết quả đo khối lượng riêng này sẽ mắc sai số nếu chất khí trong phép thử bị hấp phụ vào bột hoặc có các tạp chất bay hơi thoát ra ngoài trong quá trình đo. Sự hấp phụ chất khí có thể loại bỏ được bằng cách chọn loại khí thử thích hợp. Khí heli nói chung hay được chọn cho phép đo này. Các tạp chất bay hơi có trong bột có thể được loại ra khỏi bột bằng cách dùng khí heli áp suất không đổi trước khi đo. Đôi khi bột có thể được khử khí dưới chân không.
Hai lần đọc kết quả liên tiếp của thể tích mẫu thử phải nằm trong khoảng 0,2 % nếu các chất lạ bay hơi không có ảnh hưởng đến phép đo. Vì các chất bay hơi có thể thoát ra trong lúc đo, nên khối lượng của mẫu thường cân sau khi đo thể tích bằng picnomet.
Phải đảm bảo rằng thể tích đối chiếu và thể tích hiệu chuẩn dùng cho picnomet khí đã được xác định bằng một quy trình thích hợp. Khí dùng trong phép thử là heli trừ khi một loại khí khác được quy định trong chuyên luận riêng. Nhiệt độ picnomet khí nên duy trì trong khoảng 15 °C và 30 °C và không nên thay đổi quá 2 °C trong cả quá trình đo.
Cho mẫu thử đã được chuẩn bị như chỉ dẫn trong chuyên luận riêng vào buồng đo. Dùng một lượng bột như hướng dẫn đối với picnomet. Đậy kín buồng thử, đẩy qua hệ thống picnomet dòng khí thử như trong hướng dẫn của nhà sản xuất. Nếu mẫu phải khử khí dưới chân không, làm như quy định trong chuyên luận riêng và hướng dẫn sử dụng của picnomet.
Lặp lại phép đo trên cùng mẫu bột cho đến khi các kết quả đo liên tiếp của thể tích mẫu Vs khớp trong khoảng 0,2 %. Cân lượng bột có trong cốc. Tính khối lượng riêng ρ của mẫu theo phương trình (2) ở trên.
PHỤ LỤC 7
(Quy định)
7.1 XÁC ĐỊNH CHỈ SỐ ACETYL
Chỉ số acetyl là số miligam kali hydroxyd cần thiết để trung hòa acid acetic được giải phóng sau khi thủy phân 1 gam chế phẩm đã acetyl hóa.
Cách xác định
a) Xác định chỉ số xà phòng hóa của chế phẩm theo Phụ lục 7.7.
b) Acetyl hóa chế phẩm theo phương pháp sau: Lấy 10 g chế phẩm cho vào bình Kjeldahl dung tích 200 ml, thêm 20 ml anhydrid acetic (TT). Lắp ống sinh hàn ngược dùng nguồn làm lạnh là không khí. Đặt bình trên tấm lưới phủ chất chịu nhiệt có khoét một lỗ tròn đường kính 4 cm. Đun sôi nhẹ trong 2 h bằng ngọn lửa nhỏ có chiều cao không quá 25 mm và được điều chỉnh để lửa không chạm vào đáy bình. Để nguội, rót vào một cốc to chứa sẵn 600 ml nước, thêm 0,2 g đá bọt (TT) và đun sôi 30 min. Làm nguội, chuyển sang bình gạn và gạn bỏ lớp dưới. Rửa chế phẩm đã acetyl hóa ít nhất 3 lần, mỗi lần với 50 ml dung dịch natri clorid bão hòa (TT) ấm, cho đến khi nước rửa không còn phản ứng acid với giấy quỳ. Cuối cùng, lắc với 20 ml nước ấm và loại thật kiệt nước. Rót chế phẩm đã acetyl hóa vào một bát sứ nhỏ, thêm 1 g natri sulfat khan (TT), khuấy kỹ rồi lọc qua giấy lọc khô gấp nếp.
c) Xác định chỉ số xà phòng hóa của chế phẩm đã acetyl hóa theo Phụ lục 7.7.
d) Tính chỉ số acetyl của chế phẩm theo công thức sau:
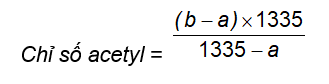
trong đó:
a: chỉ số xà phòng hóa của chế phẩm chưa acetyl hóa;
b: chỉ số xà phòng hóa của chế phẩm đã acetyl hóa.
7.2 XÁC ĐỊNH CHỈ SỐ ACID
Chỉ số acid là số miligam kali hydroxyd cần thiết để trung hòa các acid tự do chứa trong 1 gam chế phẩm.
Cách xác định
Nếu không có chỉ dẫn khác trong chuyên luận riêng, cân chính xác khoảng 10 g chế phẩm, thêm 50 ml hỗn hợp đồng thể tích ethanol 96 % (TT) và ether (TT) đã được trung hòa trước với dung dịch kali hydroxyd 0,1 N (CĐ) hoặc natri hydroxyd 0,1 N, dùng 0,5 ml dung dịch phenolphtalein (TT1) làm chỉ thị. Lắc để chế phẩm tan hoàn toàn. Nếu chế phẩm khó tan, có thể đun hồi lưu trên cách thủy. Chuẩn độ bằng dung dịch kali hydroxyd 0,1 N (CĐ), lắc liên tục cho đến khi xuất hiện màu hồng bền vững trong 15 s.
Tính chỉ số acid của chế phẩm theo công thức sau:
Chỉ số acid = 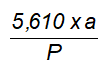
trong đó:
a: số ml dung dịch kali hydroxyd 0,1 N (CĐ) đã dùng;
P: lượng chế phẩm đem thử (g).
7.3 XÁC ĐỊNH CHỈ SỐ ESTER
Chỉ số ester là số miligam kali hydroxyd cần thiết để xà phòng hóa các ester có trong 1 gam chế phẩm.
Cách xác định
a) Xác định chỉ số xà phòng hóa của chế phẩm theo Phụ lục 7.7.
b) Xác định chỉ số acid của chế phẩm theo Phụ lục 7.2.
Tính chỉ số ester bằng cách lấy chỉ số xà phòng hóa trừ đi chỉ số acid của chế phẩm.
7.4 XÁC ĐỊNH CHỈ SỐ HYDROXYL
Chỉ số hydroxyl là số miligam kali hydroxyd cần thiết để trung hòa acid tổ hợp do acyl hóa 1 gam chế phẩm.
Cách xác định
Sử dụng phương pháp A, trừ khi có chỉ dẫn khác trong chuyên luận riêng.
Phương pháp A
Nếu không có chỉ dẫn khác trong chuyên luận riêng, cân chính xác một lượng chế phẩm (theo chỉ dẫn trong Bảng 7.4) cho vào bình acetyl hóa dung tích 150 ml có lắp ống sinh hàn ngược dùng nguồn làm lạnh là không khí, thêm lượng thuốc thử pyridin - anhydrid acetic (TT) tương ứng. Đun sôi 1 h trong cách thủy, điều chỉnh để mực nước cao hơn mực chất lỏng trong bình khoảng 2,5 cm. Lấy bình cùng ống sinh hàn ra khỏi nồi cách thủy, để nguội, dùng 5 ml nước rửa ống sinh hàn. Nếu hỗn hợp bị đục, thêm pyridin (TT) vừa đủ để làm trong. Ghi lại số ml pyridin (TT) đã thêm. Lắc và đun lại trong cách thủy 10 min nữa, rồi lấy ra, để nguội. Rửa ống sinh hàn và thành bình với 5 ml ethanol 96 % (TT) đã trung tính trước với dung dịch phenolphtalein (TT1). Chuẩn độ bằng dung dịch kali hydroxyd 0,5 N trong ethanol (CĐ), dùng 0,2 ml dung dịch phenolphtalein (TT1). Song song tiến hành một mẫu trắng.
Tính chỉ số hydroxyl của chế phẩm theo công thức sau:
Chỉ số hydroxyl =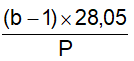 + chỉ số acid
+ chỉ số acid
trong đó:
a: số ml dung dịch kali hydroxyd 0,5 N trong ethanol (CĐ) đã dùng trong mẫu thử;
b: số ml dung dịch kali hydroxyd 0,5 N trong ethanol (CĐ) đã dùng trong mẫu trắng;
P: lượng chế phẩm đem thử (g).
Bảng 7.4 - Lượng chế phẩm và lượng thuốc thử tương ứng
| Chỉ số hydroxyl | Lượng chế phẩm cần lấy (g) | Lượng thuốc thử Pyridin - anhydrid acetic (ml) |
| 10 - 100 | 2,0 | 5,0 |
| 100 - 150 | 1,5 | 5,0 |
| 150 - 200 | 1,0 | 5,0 |
| 200 - 250 | 0,75 | 5,0 |
| 250 - 300 | 0,60 hay 1,20 | 5,0 hay 10,0 |
| 300 - 350 | 1,0 | 10,0 |
| 350 - 700 | 0,75 | 15,0 |
| 700 - 950 | 0,5 | 15,0 |
Phương pháp B
Cho lượng chế phẩm đã chỉ dẫn vào bình nón nút mài hoàn toàn khô dung tích 5 ml, thêm 2 ml thuốc thử anhydrid propionic (TT). Đậy nút và lắc nhẹ nhàng cho đến khi chế phẩm tan hoàn toàn. Để yên trong 2 h. Nếu không có chỉ dẫn khác, mở nắp, cho cả bình vào một bình nón rộng miệng dung tích 500 ml, có chứa sẵn 25,0 ml dung dịch anilin trong cyclohexan (TT) và 30 ml acid acetic băng (TT). Lắc tròn, để yên 5 min và chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ) cho đến màu xanh ngọc, dùng 0,05 ml dung dịch tím tinh thể (TT) làm chỉ thị. Song song tiến hành một mẫu trắng.
Tính chỉ số hydroxyl của chế phẩm theo công thức sau:
Chỉ số hydroxyl = 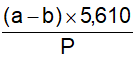
trong đó:
a là số ml dung dịch acid percloric 0,1 N (CĐ) đã dùng trong mẫu thử;
b là số ml dung dịch acid percloric 0,1 N (CĐ) đã dùng trong mẫu trắng;
P là lượng chế phẩm đem thử (g).
Xác định hàm lượng (%) nước trong chế phẩm (A) theo Phụ lục 10.3. Tính chỉ số hydroxyl hiệu chỉnh theo công thức sau:
Chỉ số hydroxyl hiệu chỉnh = Chỉ số hydroxyl - 31,1 x A
7.5 XÁC ĐỊNH CHỈ SỐ IOD
Chỉ số iod là số gam halogen, tính theo iod, kết hợp với 100 g chế phẩm trong những điều kiện quy định.
Cách xác định
Sử dụng phương pháp iod bromid, trừ khi có chỉ dẫn khác trong chuyên luận riêng.
Phương pháp iod bromid
Nếu không có chỉ dẫn khác trong chuyên luận riêng, lấy lượng chế phẩm theo chỉ dẫn trong Bảng 7.5 để thử.
Bảng 7.5 - Lượng chế phẩm cần lấy
| Chỉ số iod | Lượng chế phẩm cần lấy (g) |
| Dưới 20 | 1,0 |
| 20 - 60 | 0,5 đến 0,25 |
| 60 - 100 | 0,25-0,15 |
| Trên 100 | 0,15-0,10 |
Cân lượng chế phẩm đã chỉ dẫn, cho vào bình nón nút mài đã được làm khô hoặc được tráng bằng acid acetic băng (TT), nếu không chỉ dẫn khác, hòa tan trong 15 ml cloroform (TT). Thêm từ từ 25,0 ml dung dịch iod bromid (TT), đậy nút, nếu không có chỉ dẫn khác, để yên trong chỗ tối 30 min, thỉnh thoảng lắc. Thêm 10 ml dung dịch kali iodid 10% (TT) và 100 ml nước cất. Chuẩn độ bằng dung dịch natri thiosulfat 0,1 N (CĐ), lắc mạnh cho đến khi dung dịch có màu vàng nhạt, thêm 5 ml dung dịch hồ tinh bột (TT) và tiếp tục chuẩn độ chậm cho đến khi dung dịch mất mầu. Song song tiến hành một mẫu trắng.
Tính chỉ số iod của chế phẩm theo công thức sau:
Chỉ số iod = ![]()
trong đó:
a: số ml dung dịch natri thiosulfat 0,1 N (CĐ) đã dùng trong mẫu thử;
b: số ml dung dịch natri thiosulfat 0,1 N (CĐ) dùng trong mẫu trắng;
P: lượng chế phẩm đem thử (g).
Phương pháp iod monoclorid
Nếu chuyên luận quy định dùng bình định lượng iod, trừ khi có chỉ dẫn khác, dùng loại bình dung tích 250 ml và đáp ứng các tiêu chuẩn quốc gia.
Hòa tan lượng chế phẩm đã chỉ dẫn với 10 ml dicloromethan (TT) trong một bình định lượng iod khô. Thêm 20 ml dung dịch iod monoclorid (TT). Đậy nút đã được làm ướt với dung dịch kali iodid 10 % (TT), để yên 30 min trong chỗ tối ở nhiệt độ 15 °C đến 25 °C. Nhỏ 15 ml dung dịch kalilodid 10% (TT) lên miệng bình và cẩn thận mở nút. Rửa nút, miệng và thành bình với 100 ml nước, lắc đều. Chuẩn độ bằng dung dịch natri thiosulfat 0,1 N (CĐ), lắc mạnh cho đến khi dung dịch có màu vàng nhạt, thêm 0,5 ml dung dịch hồ tinh bột (TT) và tiếp tục chuẩn độ chậm cho đến khi dung dịch mất mầu. Song song tiến hành một mẫu trắng.
Tính chỉ số iod của chế phẩm theo công thức sau:
Chỉ số iod = ![]()
trong đó:
a: số ml dung dịch natri thiosulfat 0,1 N (CĐ) đã dùng trong mẫu thử;
b: số ml dung dịch natri thiosulfat 0,1 N (CĐ) dùng trong mẫu trắng;
P: lượng chế phẩm đem thử (g).
Có thể tính lượng chế phẩm đem thử (g) bằng cách lấy 20 chia cho chỉ số iod lý thuyết cao nhất của chế phẩm. Nếu mẫu thử tiêu thụ quá nửa lượng thuốc thử iod monoclorid cho vào, phải làm lại thí nghiệm với lượng chế phẩm nhỏ hơn.
7.6 XÁC ĐỊNH CHỈ SỐ PEROXYD
Chỉ số peroxyd là số mill đương lượng gam oxygen hoạt tính biểu thị lượng peroxyd chứa trong 1000 g chế phẩm khi được xác định theo các phương pháp dưới đây.
Cách xác định
Sử dụng phương pháp A, khi chuyên luận riêng không quy định phương pháp thử.
Phương pháp A
Cân 5,00 g chế phẩm cho vào bình nón nút mài dung tích 250 ml, thêm 30 ml hỗn hợp gồm 3 thể tích acid acetic băng (TT) và 2 thể tích cloroform (TT), lắc cho tan và thêm 0,5 ml dung dịch kali iodid bão hòa (TT). Lắc đúng 1 min, thêm 30 ml nước. Chuẩn độ chậm bằng dung dịch natri thiosulfat 0,01 N (CĐ), liên tục lắc mạnh, cho đến khi màu vàng gần như biến mất. Thêm 0,5 ml dung dịch hồ tinh bột (TT) và tiếp tục chuẩn độ, lắc mạnh, đến khi dung dịch mất màu.
Song song tiến hành một mẫu trắng. Lượng dung dịch natri thiosulfat 0,01 N (CĐ) đã dùng trong mẫu trắng không được vượt quá 0,1 ml.
Chỉ số peroxyd của chế phẩm được tỉnh theo công thức sau:
Chỉ số peroxyd = ![]()
trong đó:
a: số ml dung dịch natri thiosulfat 0,01 N (CĐ) đã dùng trong mẫu thử;
b: số ml dung dịch natri thiosulfat 0,01 N (CĐ) đã dùng trong mẫu trắng;
P: lượng chế phẩm đem thử (g).
Phương pháp B
Tiến hành thử trong điều kiện tránh ánh sáng.
Cân chính xác một lượng chế phẩm (theo chỉ dẫn trong Bảng 7.6.) cho vào bình nón nút mài dung tích 250 ml, thêm 50 ml hỗn hợp gồm 3 thể tích acid acetic băng (TT) và 2 thể tích trimethylpentan (TT). Đậy nút, lắc cho tan và thêm 0,5 ml dung dịch kali iodid bão hòa (TT). Đậy nút, để yên dung dịch trong (60 ± 1) s. Tiếp tục lắc mạnh, rồi thêm 30 ml nước. Chuẩn độ chậm bằng dung dịch natri thiosulfat 0,01 N (CĐ), liên tục lắc mạnh cho đến khi màu vàng gần như biến mất. Thêm 0,5 ml dung dịch hồ tinh bột (TT) và tiếp tục chuẩn độ, liên tục lắc mạnh, nhất là gần điểm kết thúc để giải phóng hết iod ra khỏi lớp dung môi. Thêm từng giọt dung dịch natri thiosulfat 0,01 N (CĐ) cho đến khi mất màu lam.
Bảng 7.6 - Lượng chế phẩm cần lấy
| Chỉ số peroxyd | Lượng chế phẩm cần lấy (g) |
| 0 - 12 | 2,00 - 5,00 |
| 12 - 20 | 1,20 - 2,00 |
| 20 - 30 | 0,80 - 1,20 |
| 30 - 50 | 0,50 - 0,80 |
| 50 - 90 | 0,30 - 0,50 |
Tùy thuộc vào lượng dung dịch natri thiosulfat 0,01 N (CĐ) đã sử dụng, nếu cần có thể thay thế bằng dung dịch natri thiosulfat 0,1 N (CĐ).
Ghi chú: Đối với chỉ số peroxyd ≥ 70, khi chuẩn độ đến dung dịch có màu vàng nhạt, chờ 15 s đến 30 s mới thêm dung dịch hồ tinh bột (TT), do trimethylpentan có xu hướng nổi lên bề mặt lớp nước và đây là thời gian cần thiết để trộn dung môi với dung dịch định lượng, đồng thời giải phóng hết iod. Nên dùng dung dịch natri thiosulfat 0,1 N (CĐ) khi chỉ số peroxyd lớn hơn 150. Có thể cho lượng nhỏ dung dịch chất nhũ hóa cân bằng ưa nước/ưa dầu cao (như polysorbat 60) 0,5 % đến 1,0 % (kl/kl) vào hỗn hợp phản ứng để làm chậm sự tách pha và đẩy nhanh thời gian giải phóng iod.
Song song tiến hành một mẫu trắng. Nếu lượng dung dịch natri thiosulfat dùng trong mẫu trắng vượt quá 0,1 ml, thay các thuốc thử tinh khiết và tiến hành thử nghiệm lại.
Chỉ số peroxyd của chế phẩm được tính theo công thức sau:
Chỉ số peroxyd = ![]()
trong đó:
a: số ml dung dịch natri thiosulfat đã dùng trong mẫu thử;
b: số ml dung dịch natri thiosulfat đã dùng trong mẫu trắng;
c: nồng độ đương lượng của dung dịch natri thiosulfat;
P: lượng chế phẩm đem thử (g).
7.7 XÁC ĐỊNH CHỈ SỐ XÀ PHÒNG HÓA
Chỉ số xà phòng hóa là số mill gam kali hydroxyd cần thiết để trung hòa các acid tự do và để xà phòng hóa các este chứa trong 1 g chất thử.
Cách xác định
Nếu không có chỉ dẫn khác trong chuyên luận riêng, lấy lượng chế phẩm theo chỉ dẫn trong bảng 7.7 để thử.
Bảng 7.7 - Lượng chế phẩm cần lấy
| Chỉ số xà phòng hóa | Lượng chế phẩm cần lấy (g) |
| 3 - 10 | 12,0 - 15,0 |
| 10 - 40 | 8,0 - 12,0 |
| 40 - 60 | 5,0 - 8,0 |
| 60 - 100 | 3,0 - 5,0 |
| 100 - 200 | 2,5 - 3,0 |
| 200 - 300 | 1,0 - 2,0 |
| 300 - 400 | 0,5 - 1,0 |
Cân chính xác lượng chế phẩm đã chỉ dẫn cho vào bình nón nút mài dung tích 250 ml. Thêm 25,0 ml dung dịch kali hydroxyd 0,5 N trong ethanol (CĐ) và vài viên bi thủy tinh. Lắp ống sinh hàn ngược và, trừ khi có chỉ dẫn khác, đun sôi 30 min trên cách thủy, thỉnh thoảng lắc. Thêm 1 ml dung dịch phenolphtalein (TT1) và chuẩn độ ngay (khi dung dịch còn đang nóng) bằng dung dịch acid hydrocloric 0,5 N (CĐ). Song song tiến hành một mẫu trắng.
Chỉ số xà phòng hóa của chế phẩm được tính theo công thức sau:
Chỉ số xà phòng hóa = ![]()
trong đó:
a: số ml dung dịch kali hydroxyd 0,5 N trong ethanol (CĐ) đã dùng trong mẫu trắng;
b: số ml dung dịch kali hydroxyd 0,5 N trong ethanol (CĐ) đã dùng trong mẫu thử;
P: lượng chế phẩm đem thử (g).
7.8 XÁC ĐỊNH CHẤT KHÔNG BỊ XÀ PHÒNG HÓA
Chất không bị xà phòng hóa, được tính bằng tỷ lệ phần trăm (kl/kl), là phần không bay hơi ở 100 °C đến 105 °C thu được khi chiết chế phẩm đã được xà phòng hóa với một dung môi hữu cơ.
Cách xác định
Dùng phương pháp 1, trừ khi có chỉ dẫn khác trong chuyên luận riêng.
Phương pháp 1
Cân chính xác khoảng 2,0 g đến 2,5 g chế phẩm cho vào bình dung tích 250 ml, thêm 25 ml dung dịch kali hydroxyd 0,5 N trong ethanol và đun sôi 1 h dưới ống sinh hàn ngược trên cách thủy, thỉnh thoảng lắc. Chuyển dung dịch sang bình gạn, tráng rửa bình với 50 ml nước và khi dung dịch còn đang ấm, chiết bằng cách lắc mạnh 3 lần, mỗi lần với 50 ml ether không có peroxyd (TT), dùng 50 ml ether đầu để tráng rửa bình. Tập trung dịch chiết ether vào một bình gạn khác chứa sẵn 20 ml nước. Nếu dịch chiết ether có chứa các tiểu phân rắn lơ lửng, lọc qua giấy lọc loại không có chất béo, rồi rửa giấy lọc bằng ether không có peroxyd (TT). Lắc nhẹ bình gạn trong vài phút, để yên cho tách lớp và loại bỏ lớp nước. Rửa dịch chiết ether bằng cách lắc mạnh 2 lần, mỗi lần với 20 ml nước và sau đó 3 lần, mỗi lần với 20 ml dung dịch kali hydroxyd 0,5 N (TT), lắc mạnh và rửa với 20 ml nước sau mỗi lần xử lý. Cuối cùng rửa nhiều lần, mỗi lần với 20 ml nước, cho đến khi nước rửa không còn phản ứng kiềm với dung dịch phenolphtalein (TT1). Chuyển dịch chiết ether vào một bình đã cân bì và tráng bình gạn bằng ether không có peroxyd (TT). Cất bay hơi ether, rồi thêm 3 ml aceton (TT). Dùng luồng không khí nhẹ nhàng đuổi hết dung môi ra khỏi bình được ngâm ngập trong nước đang sôi, để nghiêng và quay tròn. Sấy bình đến khối lượng không đổi ở nhiệt độ không quá 80 °C.
Hòa tan cắn trong bình vơi 10 ml ethanol 96 % (TT) mới đun sôi và đã được trung tính với dung dịch phenolphtalein (TT1). Chuẩn độ bằng dung dịch natri hydroxyd 0,1 N trong ethanol (CĐ) với chỉ thị là dung dịch phenolphtalein (TT1).
Nếu lượng dung dịch natri hydroxyd 0,1 N trong ethanol (CĐ) tiêu thụ không quá 0,1 ml, khối lượng cắn cân được là lượng chất không bị xà phòng hóa. Tỉnh tỷ lệ phần trăm chất không bị xà phòng hóa có trong chế phẩm.
Nếu lượng dung dịch natri hydroxyd 0,1 N trong ethanol (CĐ) tiêu thụ quá 0,1 ml, phải làm lại thí nghiệm.
Phương pháp 2
Cân chính xác lượng chế phẩm theo chỉ dẫn (P g) cho vào một bình dung tích 250 ml, thêm 50 ml dung dịch kali hydroxyd 2 M trong ethanol (TT) và đun 1 h dưới ống sinh hàn ngược trên cách thủy, thỉnh thoảng lắc. Làm nguội tới nhiệt độ dưới 25 °C và chuyển dung dịch sang một bình gạn, tráng rửa với 100 ml nước, rồi chiết cẩn thận 3 lần, mỗi lần với 100 ml ether không có peroxyd (TT). Tập trung dịch chiết ether vào một bình gạn khác chứa sẵn 40 ml nước, lắc nhẹ vài phút, để yên cho tách lớp và loại bỏ lớp nước. Rửa dịch chiết ether 2 lần, mỗi lần với 40 ml nước và sau đó 3 lần, mỗi lần với 40 ml dung dịch kali hydroxyd 3 % (TT), rửa với 40 ml nước sau mỗi lần xử lý. Cuối cùng rửa nhiều lần, mỗi lần với 40 ml nước, cho đến khi nước rửa không còn phản ứng kiềm với dung dịch phenolphtalein (TT1). Chuyển dung dịch ether vào bình đã cân bì, rửa bình gạn với ether không có peroxyd (TT). Cất cẩn thận cho bay hơi hết ether, thêm 6 ml aceton (TT) vào cắn. Dùng luồng khí nhẹ nhàng đuổi hết dung môi ra khỏi bình và sấy khô cắn đến khối lượng không đổi ở nhiệt độ 100 °C đến 105 °C. Để nguội bình trong bình hút ẩm và cân xác định khối lượng cắn (A g).
Tỷ lệ phần trăm chất không bị xà phòng hóa trong chế phẩm = 100 A/P
Hòa tan cắn trong 20 ml ethanol 96 % (TT) đã được trung hòa trước với dung dịch phenolphtalein (TT) và chuẩn độ bằng dung dịch natri hydroxyd 0,1 N trong ethanol (CĐ). Nếu lượng dung dịch natri hydroxyd 0,1 N trong ethanol (CĐ) tiêu thụ quá 0,2 ml, cắn đã cân không được coi là chất không bị xà phòng hóa và phải làm lại thí nghiệm.
7.9 XÁC ĐỊNH LƯU HUỲNH DIOXYD
Áp dụng phương pháp 1, trừ khi có chỉ dẫn khác.
Phương pháp 1
Dụng cụ: Bình cầu dung tích 1000 ml đến 1500 ml được lắp với sinh hàn ngược. Đầu trên của sinh hàn nối liên tiếp với hai ống hấp thụ, mỗi ống chứa 10 ml dung dịch hydrogen peroxyd 20 thể tích (TT) mới được trung hòa với dung dịch natri hydroxyd 0,1 N (CĐ), dùng dung dịch xanh bromophenol (TT) làm chỉ thị. Bình được lắp một vòi sục khí với đầu gần chạm đáy bình.
Cách tiến hành:
Cho 500 ml nước và 20 ml acid hydrocloric (TT) vào bình. Sục luồng khí nitơ hay carbon dioxyd đã đi qua dung dịch natri carbonat 10% (TT), đun từ từ dung dịch trong bình cho đến sôi. Duy trì luồng khí sục, để dung dịch sôi trong khoảng 10 min, làm nguội bằng cách nhúng bình từ từ vào nước. Mở nắp, cho nhanh lượng chế phẩm đã cân (50 g đến 100 g) vào bình, đun nhẹ và để sôi trong 45 min. Tháo hai ống hấp thụ trước khi ngừng sục khí. Gộp dung dịch trong hai ống hấp thụ và chuẩn độ bằng dung dịch natri hydroxyd 0,1 N (CĐ).
1 ml dung dịch natri hydroxyd 0,1 N (CĐ) tương đương với 3,203 mg SO2.
Lặp lại phép thử trên, nhưng không có chế phẩm. Dung dịch trong ống hấp thụ vẫn phải trung tính.
Phương pháp 2
Cho 150 ml nước vào bình A (Hình 7.9.) và sục khí carbon dioxyd 15 min qua toàn bộ hệ thống với tốc độ 100 ml/min. Lấy 10 ml dung dịch hydrogen peroxyd loãng (TT), thêm 0,15 ml dung dịch xanh bromophenol trong ethanol (TT). Thêm dung dịch natri hydroxyd 0,1 N (CĐ) cho đến khi xuất hiện màu tím xanh (không vượt quá điểm tương đương) và chuyển dung dịch thu được vào ống nghiệm D.
Duy trì luồng khí sục carbon dioxyd, tháo bình gạn B ra và dùng 100 ml nước chuyển khoảng 25,0 g chế phẩm vào bình A. Qua bình gạn, cho 80 ml dung dịch acid hydrocloric 10 % (TT) vào bình A và đun sôi 1 h. Mở khóa bình gạn, ngừng sục khí, dừng đun nóng và nước sinh hàn. Dùng một ít nước chuyển dung dịch trong ống nghiệm D sang bình nón miệng rộng dung tích 200 ml. Đun 15 min trên cách thủy và để nguội. Thêm 0,1 ml dung dịch xanh bromophenol trong ethanol (TT) và chuẩn độ bằng dung dịch natri hydroxyd 0,1 N (CĐ) cho đến khi chuyển màu từ vàng sang xanh tím.
Song song tiến hành một mẫu trắng.
Hàm lượng lưu huỳnh dioxyd (phần triệu) được tính theo công thức sau:
32030 = 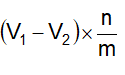
trong đó:
V1: số ml dung dịch natri hydroxyd 0,1 N (CĐ) đã dùng trong mẫu thử;
V2: số ml dung dịch natri hydroxyd 0,1 N (CĐ) đã dùng trong mẫu trắng;
n: nồng độ đương lượng thực của dung dịch natri hydroxyd 0,1 N (CĐ);
m: lượng chế phẩm đem thử (g).
Hình 7.9 - Dụng cụ xác định lưu huỳnh dioxyd
7.10 XÁC ĐỊNH CÁC CHẤT OXY HÓA
Cách xác định
Nếu không có chỉ dẫn khác trong chuyên luận riêng, cân chính xác khoảng 4,0 g chế phẩm cho vào bình nón nút mài dung tích 125 ml, thêm 50,0 ml nước. Đậy nút và lắc 5 min. Chuyển sang ống nút mài dung tích 50 ml và ly tâm. Lấy 25,0 ml dung dịch trong ở trên cho vào bình nón nút mài dung tích 125 ml, thêm 1 ml acid acetic băng (TT) và 0,5 g đến 1,0 g kali iodid (TT). Đậy nút, lắc nhẹ và để yên 25 min đến 30 min trong chỗ tối. Thêm 1 ml dung dịch hồ tinh bột (TT) và định lượng bằng dung dịch natri thiosulfat 0,002 N (CĐ) cho đến khi dung dịch mất màu. Song song tiến hành một mẫu trắng.
1 ml dung dịch natri thiosulfat 0,002 N (CĐ) tương ứng với 34 μg chất oxy hóa, tính theo H2O2.
Hàm lượng chất oxy hóa, tính theo H2O2, không được quá 0,002 % (kl/kl), trừ khi có chỉ dẫn khác trong chuyên luận riêng.
7.11 XÁC ĐỊNH CARBON HỮU CƠ TOÀN PHẦN TRONG NƯỚC DÙNG CHO NGÀNH DƯỢC
Lượng carbon hữu cơ toàn phần (TOC) có trong nước dùng cho ngành dược được xác định bằng một phép định lượng gián tiếp các chất hữu cơ. Việc định lượng TOC cũng có thể được dùng để kiểm soát quá trình thực hiện các công đoạn khác nhau trong sản xuất thuốc.
Có nhiều phương pháp xác định TOC. Bởi vậy, thay vì đưa ra một kỹ thuật cụ thể, chuyên luận chung này chỉ mô tả các quy trình được sử dụng để đánh giá phương pháp đã chọn và xử lý kết quả trong các phép thử giới hạn. Một dung dịch chuẩn được phân tích sau những khoảng thời gian thích hợp, tùy thuộc vào tần suất đo. Dung dịch chuẩn này được pha từ một chất dễ bị oxy hóa (như đường trắng) có nồng độ được điều chỉnh đủ để thiết bị phát hiện được và tương ứng với giới hạn TOC cần đo. Độ thích hợp của hệ thống được xác định bằng việc phân tích dung dịch một chất khó bị oxy hóa hơn (như 1,4 - benzoquinon).
Nói chung, các kiểu thiết bị dùng để xác định TOC trong nước dùng cho ngành dược đều hoạt động theo nguyên lý oxy hóa hoàn toàn các phân tử chất hữu cơ trong mẫu nước thử thành carbon dioxyd, sau đó định lượng carbon dioxyd tạo thành và dùng kết quả thu được để tính hàm lượng carbon trong nước.
Thiết bị sử dụng phải phân biệt được carbon hữu cơ và carbon vô cơ (thường tồn tại ở dạng carbonat). Khả năng phân tách phải hiệu quả bằng cách xác định lượng carbon vô cơ rồi tính lượng carbon hữu cơ bằng cách trừ nó khỏi hàm lượng carbon toàn phần. Cũng có thể loại sạch carbon vô cơ khỏi mẫu thử trước khi oxy hóa. Trong trường hợp này có thể có những phân tử carbon hữu cơ cũng bị loại theo, tuy nhiên, những chất hữu cơ đó tồn tại rất ít trong nước dùng cho ngành dược.
Thiết bị
Sử dụng loại thiết bị đã được hiệu chỉnh gắn trên dây chuyền hoặc đặt riêng rẽ. Kiểm tra độ thích hợp của hệ thống sau những khoảng thời gian thích hợp như mô tả ở dưới đây. Thiết bị phải có giới hạn phát hiện ≤ 0,05 mg carbon/l.
Nước TOC
Sử dụng nước tinh khiết cao đạt các tiêu chuẩn sau:
Điện dẫn suất: Không quá 1,0 μS.cm-1 ở 25 °C.
TOC: Không quá 0,1 mg/l.
Tùy thuộc vào loại thiết bị sử dụng, hàm lượng kim loại nặng và đồng có thể phải được khống chế. Cần tuân theo các hướng dẫn của nhà sản xuất thiết bị.
Chuẩn bị đồ thủy tinh
Sử dụng đồ thủy tinh được rửa sạch cẩn thận bằng phương pháp thích hợp để loại bỏ các chất hữu cơ. Cuối cùng, dùng nước TOC để tráng lại.
Dung dịch chuẩn
Hòa tan đường trạng, đã được sấy khô 3 h ở 105 °C, trong nước TOC để thu được dung dịch chứa 1,19 mg đường trắng/l (0,50 mg carbon/l).
Dung dịch thử
Thực hiện tất cả các biện pháp cần thiết để tránh ô nhiễm. Lấy đầy nước cần thử vào một bình kín. Kiểm tra nước càng nhanh càng tốt để giảm thiểu ô nhiễm do đồ chứa và nút đậy của nó.
Dung dịch kiểm tra độ thích hợp của hệ thống
Hòa tan 1,4-benzoquinon trong nước TOC để thu được dung dịch có nồng độ 0,75 mg 1,4-benzoquinon/l (0,50 mg carbon/l).
Kiểm soát nước TOC
Sử dụng nước TOC thu được vào cùng thời điểm lấy để pha chế dung dịch chuẩn và dung dịch kiểm tra độ thích hợp của hệ thống.
Các dung dịch kiểm tra
Ngoài việc kiểm soát nước TOC, chuẩn bị các dung dịch trắng thích hợp hay các dung dịch cần thiết khác để hiệu chỉnh đường nền theo hướng dẫn của nhà sản xuất thiết bị. Tiến hành thử các mẫu trắng thích hợp để chỉnh thiết bị về không.
Độ thích hợp của hệ thống
Tiến hành thử và ghi lại đáp ứng của các dung dịch sau: Nước TOC (RN), dung dịch chuẩn (RC) và dung dịch kiểm tra độ thích hợp của hệ thống (RTH). Tính tỷ lệ phần trăm hiệu quả đáp ứng theo công thức sau:
![]()
Hệ thống được coi là thích hợp nếu RE nằm trong khoảng 85,0 % đến 115,0 %.
Cách tiến hành
Tiến hành kiểm tra mẫu nước thử và ghi lại đáp ứng (RT). Mẫu nước thử được coi là đạt yêu cầu khi RT ≤ RC - RN.
Có thể áp dụng phương pháp này khi sử dụng thiết bị đặt trong dây chuyền đã được hiệu chỉnh đúng mức và có độ thích hợp hệ thống chấp nhận được. Vị trí đặt thiết bị phải được lựa chọn để đảm bảo các đáp ứng là đại diện cho nước được sử dụng.
PHỤ LỤC 8
(Quy định)
8.1 CÁC PHẢN ỨNG ĐỊNH TÍNH
Acetat
A. Đun nóng chế phẩm với cùng một lượng acid oxalic (TT). Hơi xông ra có mùi acid acetic.
B. Hòa tan khoảng 30 mg chế phẩm trong 3 ml nước hoặc lấy 3 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm lần lượt 0,25 ml dung dịch lanthan nitrat 5 % (TT), 0,1 ml dung dịch iod 0,1 N và 0,05 ml dung dịch amoniac 2 M (TT) và đun nóng hỗn hợp cẩn thận đến sôi, sau vài phút xuất hiện tủa màu xanh lam hay màu xanh lam thẫm.
Nhóm acetyl
Lấy một ống nghiệm (kích thước 18 mm x 180 mm), cho vào khoảng 15 mg chế phẩm hay một lượng theo chỉ dẫn trong chuyên luận và 0,15 ml acid phosphoric (TT). Đậy ống nghiệm bằng một nút có mang bên trong một ống nghiệm nhỏ hơn (kích thước 10 mm x 100 mm), ống nghiệm này chứa nước để làm sinh hàn. Cho một giọt dung dịch lanthan nitrat 5 % (TT) bám vào thành đáy ngoài của ống nghiệm nhỏ (sao cho giọt thuốc thử này không bị rơi xuống ống nghiệm chứa chế phẩm trong suốt quá trình thí nghiệm, nếu rơi phải làm lại thí nghiệm từ đầu). Đặt thiết bị trong cách thủy sôi 5 min (trừ trường hợp chế phẩm khó thủy phân), sau đó lấy ống nghiệm nhỏ ra, gạt chất lỏng thu được ở đáy ngoài ống nghiệm vào một tấm sứ trắng đã chứa sẵn 0,05 ml dung dịch iod 0,02 N (TT), thêm vào cạnh đó 0,05 ml dung dịch amoniac 2 M (TT). Sau 1 min đến 2 min, ở vòng tiếp xúc giữa hai dung dịch xuất hiện màu xanh lam dần dần thẫm lên và bền trong một thời gian ngắn.
Đối với các chế phẩm khó thủy phân, tiến hành theo chỉ dẫn ở trên, nhưng đun hỗn hợp từ từ tới sôi trên ngọn lửa, thay cho việc đun trong cách thủy.
Alcaloid
Hòa tan vài mg chế phẩm hoặc một lượng chế phẩm theo chỉ dẫn trong chuyên luận trong 5 ml nước, thêm dung dịch acid hydrocloric 2 M (TT) đến khi có phản ứng acid, thêm 1 ml thuốc thử Dragendorff (TT), xuất hiện tủa màu cam hay đỏ cam.
Amin thơm bậc nhất
Acid hóa một lượng dung dịch chế phẩm theo chỉ dẫn trong chuyên luận bằng dung dịch acid hydrocloric 2 M (TT) hoặc hòa tan 0,1 g chế phẩm trong 2 ml dung dịch acid hydrocloric 2 M (TT). Thêm 0,2 ml dung dịch natri nitrit 10 % (TT), sau 1 đến 2 min thêm 1 ml dung dịch 2-naphthol trong kiềm (TT). Hỗn hợp có màu cam thẫm hay màu đỏ và thường có tủa cùng màu.
Amoni (muối)
A. Hòa tan khoảng 0,02 g chế phẩm trong 2 ml nước hoặc lấy một lượng dung dịch theo chỉ dẫn trong chuyên luận, thêm 2 ml dung dịch natri hydroxyd 2 M (TT), đun nóng, sẽ xông ra khí có mùi đặc biệt và làm xanh giấy quỳ đỏ đã thấm nước.
B. Hòa tan khoảng 10 mg chế phẩm trong 5 ml nước hay một lượng dung dịch theo chỉ dẫn trong chuyên luận, thêm 0,2 ml thuốc thử Nessler (TT), hỗn hợp có màu vàng hay tủa vàng nâu.
Arseniat
A. Đun cách thủy 5 ml dung dịch chế phẩm 5 % hoặc 5 ml dung dịch chế phẩm theo chỉ dẫn trong chuyên luận với cùng thể tích dung dịch hypophosphit (TT) (thuốc thử Tilé), sẽ cho tủa màu nâu.
B. Hòa tan khoảng 0,10 g chế phẩm trong 2 ml nước hoặc lấy 2 ml dung dịch theo chỉ dẫn trong chuyên luận, thêm 1 ml dung dịch bạc nitrat 2 % (TT) sẽ tạo thành tủa màu nâu đỏ, tủa này tan khi thêm một lượng acid nitric loãng (TT) hoặc một lượng dung dịch amoniac (TT).
C. Hòa tan khoảng 0,20 g chế phẩm trong 5 ml nước hoặc lấy 5 ml dung dịch theo chỉ dẫn trong chuyên luận, thêm 5 ml dung dịch amoni clorid (TT), 1 ml dung dịch amoniac (TT) và vài giọt dung dịch magnesi sulfat 5 % (TT) sẽ cho tủa kết tinh trắng.
Arsenit
A. Phản ứng A trong mục arseniat.
B. Hòa tan khoảng 0,10 g chế phẩm trong 2 ml nước hoặc lấy 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch bạc nitrat 2 % (TT) sẽ tạo thành tủa màu trắng hơi vàng, tủa này tan khi thêm một lượng dung dịch amoniac (TT) hoặc một lượng acid nitric loãng (TT).
c. Hòa tan 0,1 g chế phẩm trong 2 ml nước, hoặc lấy 2 ml dung dịch chế phẩm theo chỉ dẫn trong chuyên luận, thêm 1 ml dung dịch đồng sulfat (TT), tủa màu xanh lục sẽ được tạo thành, tách tủa và đun với dung dịch natri hydroxyd (TT), tủa sẽ chuyển thành màu nâu đỏ.
Bạc (muối)
A. Hòa tan khoảng 10 mg chế phẩm trong 5 ml nước hoặc lấy một lượng dung dịch theo chỉ dẫn trong chuyên luận, thêm 3 giọt dung dịch acid hydrocloric 10% (TT) sẽ xuất hiện tủa trắng lổn nhổn, tủa này không tan trong dung dịch acid nitric loãng (TT) nhưng tan trong dung dịch amoniac 6 M (TT).
B. Hòa tan 20 mg chế phẩm trong 5 ml nước hoặc lấy một lượng dung dịch theo chỉ dẫn trong chuyên luận, thêm 2 ml dung dịch amoniac 6 M (TT) và vài giọt formaldehyd (TT), đun nóng dung dịch sẽ có tủa bạc kim loại bám vào thành ống nghiệm (phản ứng tráng gương).
Barbiturat
A. Lấy khoảng 5 mg chế phẩm, hòa tan trong 3 ml dung dịch cobalt (II) acetat 0,2 % trong methanol (TT), thêm 20 mg đến 30 mg natri tetraborat (TT) đã được tán mịn, đun sôi sẽ xuất hiện màu xanh tím.
B. Barbiturat có hydro ở nhóm NH không bị thay thế: Hòa tan khoảng 5 mg chế phẩm trong 3 ml methanol (TT), thêm 0,1 ml dung dịch có chứa 10 % cobalt (II) nitrat (TT) và 10 % calci clorid (TT). Trộn đều, vừa lắc vừa thêm 0,1 ml dung dịch natri hydroxyd 2 M (TT) sẽ xuất hiện màu và tủa xanh tím.
Bari (muối)
A. Lấy một lượng dung dịch muối bari như chỉ dẫn trong chuyên luận, thêm vài giọt acid sulfuric loãng (TT) sẽ xuất hiện tủa trắng, tủa này không tan trong dung dịch acid hydrocloric 10% (TT) và acid nitric loãng (TT).
B. Dùng một dây bạch kim hoặc đũa thủy tinh lấy một lượng chất thử đốt trên ngọn lửa không màu, ngọn lửa sẽ nhuộm thành màu xanh lục hơi vàng, khi nhìn qua kính thủy tinh màu lục ngọn lửa sẽ có màu xanh lam.
Benzoat
A. Lấy 1 ml dung dịch trung tính 10 % chế phẩm trong nước hoặc một lượng chế phẩm theo chỉ dẫn trong chuyên luận, thêm 0,5 ml dung dịch sắt (III) clorid 10,5 % (TT) sẽ xuất hiện tủa màu vàng thẫm, tủa này tan trong ether (TT).
B. Lấy khoảng 0,2 g chế phẩm hoặc một lượng chế phẩm theo chỉ dẫn trong chuyên luận vào một ống nghiệm, làm ẩm bằng 0,2 ml đến 0,3 ml acid sulfuric (TT) và đun nóng nhẹ ở đáy ống nghiệm, sẽ có tinh thể màu trắng thăng hoa bám ở thành trong của ống.
C. Hòa tan 0,5 g chế phẩm trong 10 ml nước hoặc dùng 10 ml dung dịch theo chỉ dẫn trong chuyên luận, thêm 0,5 ml acid hydrocloric (TT), sẽ cho tủa. Kết tinh lại tủa trong nước ấm rồi làm khô dưới áp suất giảm, tủa thu được có nhiệt độ nóng chảy từ 120 °C đến 124 °C (Phụ lục 6.7).
Bismuth (muối)
A. Thêm 10 ml dung dịch acid hydrocloric 10% (TT) vào 0,5 g chế phẩm hoặc dùng 10 ml dung dịch theo chỉ dẫn trong chuyên luận. Đun sôi trong 1 min, để nguội rồi lọc nếu cần. Thêm 20 ml nước vào 1 ml dung dịch thu được ở trên, xuất hiện tủa trắng hay hơi vàng, tủa này sẽ chuyển thành nâu khi thêm 0,05 ml đến 0,1 ml dung dịch natri sulfid (TT).
B. Thêm 10 ml dung dịch acid nitric 2 M (TT) vào 40 mg đến 50 mg chế phẩm hoặc dùng 10 ml chế phẩm đã được xử lý theo chỉ dẫn trong chuyên luận. Đun sôi trong 1 min, để nguội rồi lọc nếu cần. Thêm 2 ml dung dịch thioure 10 % (TT) vào 5 ml dịch lọc thu được ở trên, xuất hiện màu vàng da cam hay tủa da cam. Thêm 4 ml dung dịch natri fluorid 2,5 % (TT), dung dịch không được mất màu trong vòng 30 min.
Borat
A. Hòa tan khoảng 0,1 g chế phẩm vào 0,1 ml acid sulfuric (TT) trong một chén sứ, thêm 3 ml methanol (TT), trộn đều rồi châm lửa vào hỗn hợp, hỗn hợp cháy với ngọn lửa màu lục.
B. Lấy 5 ml dung dịch chế phẩm 10 %, thêm 0,5 ml dung dịch acid hydrocloric loãng (TT) và 0,5 ml cồn nghệ (TT), sẽ cho màu nâu; thêm 1 ml dung dịch natri hydroxyd 10 % (TT), màu nâu chuyển thành lam hay lục.
Bromid
A. Hòa tan một lượng chế phẩm có chứa khoảng 3 mg ion bromid trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Acid hóa bằng acid nitric loãng (TT) và thêm 0,4 ml dung dịch bạc nitrat 4 % (TT). Lắc và để yên, có tủa màu vàng nhạt lổn nhổn tạo thành. Lọc lấy tủa, rửa tủa ba lần, mỗi lần với 1 ml nước. Phân tán tủa trong 2 ml nước, thêm 1,5 ml dung dịch amoniac 10 M (TT), tủa khó tan.
B. Hòa tan một lượng chế phẩm có chứa khoảng 5 mg ion bromid trong 2 ml nước hoặc lấy một lượng dung dịch theo chỉ dẫn trong chuyên luận, acid hóa dung dịch bằng acid sulfuric loãng (TT), thêm 1 ml nước clor (TT) và 2 ml cloroform (TT), lắc. Lớp cloroform sẽ có màu đỏ đến màu nâu đỏ.
Carbonat và hydrocarbonat
A. Cho vào ống nghiệm khoảng 0,1 g chế phẩm, thêm 2 ml nước hoặc lấy 2 ml dung dịch chế phẩm đã được chỉ dẫn trong chuyên luận. Thêm 2 ml dung dịch acid acetic 2 M (TT). Đậy ngay nút đã lắp sẵn một ống thủy tinh nhỏ uốn góc. Đun nhẹ ống nghiệm, khí thoát ra được dẫn vào một ống nghiệm khác có chứa sẵn 5 ml dung dịch bão hòa calci hydroxyd (TT), sao cho đầu ống thủy tinh nhỏ phải ngập trong dung dịch này, tủa trắng tạo thành, tủa này tan trong acid hydrocloric (TT) quá thừa.
B. Cho vào ống nghiệm khoảng 0,1 g chế phẩm, thêm 2 ml nước hoặc lấy 2 ml dung dịch chế phẩm đã được chỉ dẫn trong chuyên luận. Thêm 2 ml dung dịch magnesi sulfat (TT), nếu là carbonat sẽ cho tủa trắng, nếu là hydrocarbonat sẽ không tạo tủa nhưng khi đun sôi cũng cho tủa trắng.
Calci (muối)
A. Lấy khoảng 20 mg chế phẩm hoặc một lượng chế phẩm theo chỉ dẫn trong chuyên luận, hòa tan trong 5 ml dung dịch acid acetic 5 M (TT), thêm 0,5 ml dung dịch kali ferocyanid (TT), dung dịch vẫn trong, thêm khoảng 50 mg amoni clorid (TT), tạo thành tủa kết tinh trắng.
B. Thêm vào dung dịch chế phẩm theo chỉ dẫn trong chuyên luận vài giọt dung dịch amoni oxalat 4 % (TT), tạo thành tủa trắng, tủa này ít tan trong dung dịch acid acetic 6 M (TT), nhưng tan trong acid hydrocloric (TT).
Chì (muối)
A. Hòa tan 0,1 g chế phẩm trong 1 ml dung dịch acid acetic 5 M (TT) hoặc lấy 1 ml dung dịch theo chỉ dẫn trong chuyên luận, thêm 2 ml dung dịch kali cromat 5 % (TT), tủa vàng sẽ tạo thành, tủa này tan trong dung dịch natri hydroxyd 10 M (TT).
B. Hòa tan 50 mg chế phẩm trong 1 ml dung dịch acid acetic 5 M (TT) hoặc 1 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 10 ml nước và 0,2 ml dung dịch kali iodid 10 % (TT), tủa màu vàng sẽ tạo thành; đun sôi 1 đến 2 min cho tủa tan ra, để nguội, tủa lại xuất hiện có dạng những mảnh vàng lấp lánh.
Citrat
A. Hòa tan khoảng 0,1 g chế phẩm trong 2 ml nước, nếu cần có thể trung tính hóa dung dịch bằng amoniac (TT), hoặc dùng một lượng dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch calci clorid 10 % (TT), dung dịch vẫn trong. Đun sôi dung dịch sẽ xuất hiện tủa trắng, tủa này tan trong dung dịch acid acetic 6 M (TT).
B. Hòa tan một lượng chế phẩm tương đương với khoảng 50 mg acid citric trong 5 ml nước hoặc lấy 5 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 0,5 ml acid sulfuric (TT), 1 ml dung dịch kali permanganat (TT) và đun nóng đến khi mất màu. Thêm 0,5 ml dung dịch natri nitroprusiat 10 % trong dung dịch acid sulfuric 1 M, 4 g acid sulfamic (TT) và từng giọt amoniac (TT) đến khi acid sulfamic tan hết, khi cho thừa amoniac, xuất hiện màu tím chuyển dần thành xanh tím.
Clorat
A. Thêm vài giọt acid sulfuric (TT) vào 0,1 g chế phẩm, có tiếng nổ lép bép và khí màu vàng lục bay ra.
B. Hòa tan khoảng 0,05 g chế phẩm trong 5 ml nước, thêm 0,5 ml dung dịch bạc nitrat 2 % (TT), không xuất hiện tủa. Khi thêm 2 giọt đến 3 giọt dung dịch natri nitrit (TT) và 2 giọt đến 3 giọt acid nitric loãng (TT), sẽ có tủa trắng tạo thành, tủa này tan trong dung dịch amoniac 6 M (TT).
Clorid
A. Hòa tan một lượng chế phẩm tương ứng với 2 mg ion clorid trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Acid hóa bằng dung dịch acid nitric 2 M (TT), thêm 0,4 ml dung dịch bạc nitrat 2 % (TT). Lắc và để yên, có tủa màu trắng, lổn nhổn tạo thành. Lọc lấy tủa, rửa tủa 3 lần, mỗi lần với 1 ml nước, phân tán tủa trong 2 ml nước và thêm 1,5 ml dung dịch amoniac 10 M, tủa tan dễ dàng.
B. Cho vào ống nghiệm một lượng chế phẩm tương ứng khoảng từ 10 đến 20 mg ion clorid hay một lượng theo chỉ dẫn trong chuyên luận. Thêm 1 ml kali permanganat 5 % (TT) và 1 ml acid sulfuric (TT), đun nóng, sẽ giải phóng khí clor có mùi đặc biệt, khí này làm xanh giấy tẩm hồ tinh bột có kali iodid (TT) đã thấm nước.
Đồng (muối)
A. Hòa tan khoảng 10 mg chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận, thêm vài giọt dung dịch kali ferocyanid (TT), sẽ có tủa màu nâu đỏ không tan trong acid acetic (TT).
B. Hòa tan khoảng 10 mg chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm vài giọt dung dịch amoniac (TT), sẽ có tủa màu xanh, tủa này tan trong thuốc thử quá thừa tạo thành dung dịch màu xanh lam thẫm.
Ester
Lấy khoảng 30 mg chế phẩm, hoặc một lượng chế phẩm theo chỉ dẫn trong chuyên luận, thêm 0,5 ml dung dịch chứa 7 % hydroxylamin hydroclorid (TT) trong methanol (TT) và 0,5 ml dung dịch kali hydroxyd 10 % trong ethanol (TT). Đun sôi, để nguội, acid hóa dung dịch bằng dung dịch acid hydrocloric 2 M (TT), rồi thêm vài giọt dung dịch sắt (III) clorid 1 %, sẽ xuất hiện màu đỏ hay màu đỏ có ánh lam.
lodid
A. Hòa tan một lượng chế phẩm tương ứng khoảng 4 mg ion iodid trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Add hóa bằng dung dịch acid nitric 2 M (TT), thêm 0,4 ml dung dịch bạc nitrat 4 % (TT). Lắc và để yên, có tủa màu vàng nhạt, lổn nhổn tạo thành. Lọc lấy tủa, rửa tủa 3 lần, mỗi lần với 1 ml nước. Phân tán tủa trong 2 ml nước, thêm 1,5 ml dung dịch amoniac 10 M (TT), tủa không tan.
B. Hòa tan một lượng chế phẩm tương ứng với 4 mg ion iodid trong 2 ml nước, hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm vài giọt dung dịch sắt (III) clorid 3 % (TT), 2 giọt acid hydrocloric (TT), 1 ml cloroform (TT) và lắc, để yên. Lớp cloroform có màu tím.
Kali (muối)
A. Hòa tan 0,1 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch natri carbonat 10 % (TT) rồi đun nóng, không tạo thành tủa. Thêm vào trong lúc nóng 0,05 ml dung dịch natri sulfid (TT), không tạo thành tủa. Làm nguội trong nước đá và thêm 1,5 ml dung dịch acid tartric 20 % (TT) và để yên, tạo thành tủa kết tinh màu trắng.
B. Hòa tan khoảng 40 mg chế phẩm trong 1 ml nước hoặc dùng 1 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch acid acetic 2 M (TT) và 1 ml dung dịch natri cobaltinitrit 10 % (TT) mới pha, tạo thành tủa màu vàng hay da cam.
Kẽm (muối)
Hòa tan 0,1 g chế phẩm trong 5 ml nước hoặc dùng 5 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 0,2 ml dung dịch natri hydroxyd 10 M (TT), tạo thành tủa trắng, tủa này tan khi tiếp tục thêm 2 ml natri hydroxyd 10 M (TT). Thêm 10 ml dung dịch amoni clorid 10 % (TT), dung dịch vẫn trong, thêm 0,1 ml dung dịch natri sulfid (TT), tủa bông trắng được tạo thành.
Lactat
Hòa tan một lượng chế phẩm tương ứng khoảng 5 mg acid lactic trong 5 ml nước, hoặc lấy 5 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml nước brom (TT) và 0,5 ml dung dịch acid sulfuric 1 M (TT). Đun trong cách thủy đến khi mất màu, thỉnh thoảng khuấy bằng đũa thủy tinh. Thêm 4 g amoni sulfat (TT) và trộn đều. Thêm từng giọt theo thành ống 0,2 ml dung dịch có chứa 10 % natri nitroprusiat (TT) trong acid sulfuric 1 M (chú ý không trộn) và 1 ml amoniac đậm đặc (TT). Để yên 30 min, sẽ xuất hiện một vòng màu lục giữa bề mặt của hai chất lỏng.
Magnesi (muối)
Hòa tan khoảng 15 mg chế phẩm trong 2 ml nước hoặc lấy 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch amoniac 6 M (TT), tạo thành tủa trắng, tủa này tan khi thêm 1 ml dung dịch amoni clorid (TT). Thêm 1 ml dung dịch dinatri hydrophosphat 9 %, sẽ có tủa kết tinh màu trắng.
Natri (muối)
A. Hòa tan 0,1 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch đã được chỉ dẫn trong chuyên luận. Thêm 2 ml dung dịch kali carbonat 15% (TT) và đun đến sôi. Không được có tủa tạo thành. Thêm 4 ml dung dịch kali pyroantimonat (TT) và đun đến sôi. Để nguội trong nước đá, nếu cần cọ thành cốc bằng đũa thủy tinh. Tủa trắng được tạo thành.
B. Dùng một dây bạch kim hay đũa thủy tinh, lấy một hạt chất thử hay một giọt dung dịch chế phẩm, đưa vào ngọn lửa không màu, ngọn lửa sẽ nhuộm thành màu vàng.
Nhôm (muối)
Hòa tan khoảng 0,1 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm từng giọt dung dịch amoniac (TT) cho tới khi tạo tủa keo trắng, tủa này chuyển thành đỏ khi thêm vài giọt dung dịch alizarin S (TT).
Nitrat
A. Hòa tan 0,1 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm từ từ 2 ml acid sulfuric (TT), trộn và để nguội. Cho cẩn thận (không trộn) dọc theo thành ống nghiệm 1 ml dung dịch sắt (II) sulfat 1,5 % (TT). Ở miền tiếp giáp giữa hai chất lỏng có một vòng màu nâu.
B. Trộn 0,1 ml nitrobenzen (TT) với 0,2 ml acid sulfuric (TT), thêm một lượng chế phẩm đã tán nhỏ tương ứng với khoảng 1 mg ion nitrat hoặc một lượng dung dịch theo chỉ dẫn trong chuyên luận, để yên 5 min, làm lạnh trong nước đá vài min, vừa lắc vừa thêm từ từ 5 ml nước, sau đó 5 ml dung dịch natri hydroxyd 10 M (TT) và 5 ml aceton (TT), lắc rồi để yên. Lớp chất lỏng ở trên có màu tím thẫm.
Oxalat
A. Hòa tan khoảng 0,1 g chế phẩm trong 5 ml nước hoặc dùng 5 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 0,5 ml dung dịch calci clorid 10 % (TT), tủa màu trắng tạo thành, tủa tan trong acid vô cơ, không tan trong dung dịch acid acetic 6 M (TT).
B. Hòa tan khoảng 0,1 g chế phẩm trong 5 ml nước hoặc dùng 5 ml dung dịch theo chỉ dẫn trong chuyên luận. Acid hóa dung dịch bằng dung dịch acid sulfuric 10 % (TT), dung dịch này sẽ làm mất màu dung dịch kali permanganat 5 % (TT) khi đun nóng.
Peroxyd
A. Nhỏ 1 giọt dung dịch chế phẩm vào 10 ml dung dịch acid sulfuric 2 % (TT), thêm 2 ml ether (TT) và 1 giọt dung dịch kali dicromat 5 % (TT), lắc, lớp ether có màu xanh.
B. Lấy 1 ml dung dịch chế phẩm, acid hóa nhẹ bằng dung dịch acid sulfuric 10 % (TT), thêm từng giọt dung dịch kali iodid (TT), sẽ giải phóng iod, làm xanh hồ tinh bột (TT).
Phosphat
A. Hòa tan khoảng 50 mg chế phẩm trong 3 ml nước hoặc dùng 3 ml dung dịch theo chỉ dẫn trong chuyên luận. Acid hóa dung dịch bằng acid nitric loãng (TT), thêm 2 ml dung dịch amoni molybdat (TT) sẽ hiện tủa màu vàng.
B. Hòa tan khoảng 0,1 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Trung tính hóa dung dịch bằng dung dịch acid nitric loãng (TT) hoặc dung dịch natri hydroxyd (TT). Thêm 1 ml dung dịch bạc nitrat 4 % (TT) sẽ tạo tủa màu vàng, màu của tủa không thay đổi khi đun sôi, tủa tan khi thêm dung dịch amoniac 10 M (TT).
Salicylat
A. Lấy 1 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 0,5 ml dung dịch sắt (III) clorid 10,5 % (TT), xuất hiện màu tím, màu không bị mất khi thêm 0,1 ml dung dịch acid acetic 5 M (TT).
B. Hòa tan khoảng 0,5 g chế phẩm trong 10 ml nước hoặc dùng 10 ml dung dịch theo chỉ dẫn của chuyên luận. Thêm 0,5 ml acid hydrocloric (TT), sẽ xuất hiện tủa kết tinh màu trắng. Lọc lấy tủa, rửa tủa bằng nước đến phản ứng trung tính với giấy quỳ và làm khô trong bình hút ẩm có chứa acid sulfuric (TT). Nhiệt độ nóng chảy của tủa từ 156 °C đến 161 °C (Phụ lục 6.7).
Sắt (II) (muối)
Hòa tan một lượng chế phẩm tương ứng với 10 mg sắt (II) trong 1 ml nước, hoặc dùng 1 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch kali fericyanid 5 % (TT), tủa màu xanh lam tạo thành, tủa này không tan trong dung dịch acid hydrocloric 2 M (TT).
Sắt (III) (muối)
A. Hòa tan một lượng chế phẩm có chứa khoảng 0,1 mg sắt (III) trong 3 ml nước hoặc dùng 3 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch acid hydrocloric 2 M (TT) và 1 ml dung dịch kali thiocyanat (TT), dung dịch có màu đỏ. Dùng 2 ống nghiệm, cho vào mỗi ống 1 ml dung dịch thu được ở trên. Một ống cho thêm 5 ml alcol amylic (TT) hoặc ether (TT), lắc và để yên, lớp dung môi có màu hồng. Cho vào ống nghiệm kia 3 ml dung dịch thủy ngân (II) clorid 5 % (TT), màu đỏ sẽ biến mất.
B. Hòa tan một lượng chế phẩm tương ứng với 1 mg sắt (III) trong 1 ml nước hoặc dùng 1 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch kali ferocyanid (TT) tủa màu xanh lam tạo thành, tủa này không tan trong dung dịch acid hydrocloric 2 M (TT).
Silicat
Trong một chén nung bằng chì hay bằng bạch kim, dùng một sợi dây đồng trộn đều một lượng chất để thử với 10 mg natri fluorid (TT) và 0,2 ml acid sulfuric đậm đặc (TT). Đậy chén bằng một nắp nhựa dẻo trong, mỏng; dưới nắp nhựa có một giọt nước. Đun nóng nhẹ, sẽ thấy xuất hiện một vòng màu trắng xung quanh giọt nước.
Stibi (antimony, hợp chất)
Hòa tan bằng cách đun nóng nhẹ khoảng 10 mg chế phẩm trong 10 ml dung dịch natri kali tartrat 5 % và để nguội. Lấy 2 ml dung dịch này hoặc 2 ml dung dịch chế phẩm theo chỉ dẫn trong chuyên luận, nhỏ vào đó từng giọt dung dịch natri sulfid (TT), tủa màu đỏ cam tạo thành, tủa này sẽ tan khi thêm dung dịch natri hydroxyd 2 M (TT).
Sulfat
A. Hòa tan 45 mg chế phẩm trong 5 ml nước hoặc dùng 5 ml dung dịch theo chỉ dẫn trong chuyên luận, thêm 1 ml dung dịch acid hydrocloric 2 M (TT) và 1 ml dung dịch bari clorid 5 % (TT), sẽ có tủa màu trắng tạo thành.
B. Thêm 0,1 ml dung dịch iod 0,1 N vào hỗn dịch thu được ở phản ứng trên, hỗn dịch có màu vàng (phân biệt với sulfit và dithionit) nhưng mất màu khi thêm từng giọt dung dịch thiếc (II) clorid (TT) (phân biệt với iodat). Đun sôi hỗn hợp, không được tạo thành tủa có màu (phân biệt với selenat và tungstat).
Sulfid
Hòa tan 0,1 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch acid hydrocloric 10 % (TT), khí bay ra có mùi của hydrosulfid, khí này sẽ làm nâu hoặc đen giấy tẩm dung dịch chì acetat (TT) đã thấm nước.
Bisulfit và sulfit
A. Hòa tan khoảng 0,1 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch acid sulfuric 10% (TT). Đun nóng nhẹ, sẽ có khí lưu huỳnh dioxyd bay ra và làm đen giấy tẩm dung dịch thủy ngân (II) nitrat (TT).
B. Nhỏ từng giọt dung dịch iod 0,1 N (TT) vào dung dịch chế phẩm, màu của dung dịch iod sẽ mất.
Tartrat
A. Hòa tan khoảng 15 mg chế phẩm trong 5 ml nước hoặc dùng 5 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 0,05 ml dung dịch sắt (II) sulfat 1 % và 0,05 ml dung dịch hydrogen peroxyd 10 tt (TT); màu vàng không bền vững được tạo thành, sau khi màu vàng biến mất, thêm từng giọt dung dịch natri hydroxyd 2 M (TT), xuất hiện màu tím hay đỏ tía.
B. Lấy 0,1 ml dung dịch chế phẩm chứa khoảng 15 mg acid tartric trong 1 ml hoặc dùng 0,1 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 0,1 ml dung dịch kali bromid 10 % (TT), 0,1 ml dung dịch resorcin 2 % (TT) và 3 ml acid sulfuric (TT). Đun trên cách thủy 5 đến 10 min sẽ xuất hiện màu xanh lam thẫm. Để nguội và rót dung dịch vào nước, màu chuyển thành đỏ.
Thiosulfat
A. Hòa tan 0,10 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm vài giọt dung dịch iod 0,1 N (TT), màu của iod biến mất.
B. Hòa tan 0,10 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm vài giọt dung dịch acid hydrocloric 10 % (TT), có tủa màu trắng dần chuyển thành màu vàng và có khí lưu huỳnh dioxyd bay ra làm đen giấy tẩm dung dịch thủy ngân (II) nitrat (TT).
c. Hòa tan 0,10 g chế phẩm trong 2 ml nước hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm 1 ml dung dịch bạc nitrat 4 % (TT), tủa màu trắng xuất hiện, chuyển nhanh sang màu vàng rồi dần dần sang màu đen.
Thủy ngân (muối)
A. Nhỏ 2 giọt đến 3 giọt dung dịch thử lên một miếng phôi đồng (TT) đã được cọ sạch, sẽ xuất hiện vết hoen màu xám tối; vết này sáng ra khi được lau nhẹ. Đốt nóng phôi đồng đó trong ống nghiệm thì vết hoen sẽ mất.
B. Hòa tan khoảng 0,10 g chế phẩm trong 2 ml nước, nếu là thủy ngân oxyd thì hòa tan trong 2 ml dung dịch acid hydrocloric 10 % (TT), hoặc dùng 2 ml dung dịch theo chỉ dẫn trong chuyên luận. Thêm từng giọt dung dịch kali iodid (TT) sẽ cho tủa màu đỏ, tủa tan trong thuốc thử quá thừa [đối với muối thủy ngân (II)].
Nếu là muối thủy ngân (I), khi thêm từng giọt dung dịch kali iodid (TT) sẽ cho tủa màu vàng, sau đó chuyển sang màu lục.
Nhóm xanthin
Trộn vài miligam chế phẩm hoặc lấy một lượng chế phẩm theo chỉ dẫn trong chuyên luận với 0,1 ml dung dịch hydrogen peroxyd 100 tt (TT) và 0,3 ml dung dịch acid hydrocloric 2 M (TT), đun trên cách thủy tới khô, sẽ có cắn màu đỏ hơi vàng, thêm 0,1 ml dung dịch amoniac 2 M (TT), màu của cắn sẽ chuyển thành tím đỏ.
8.2 ĐỊNH TÍNH CÁC PENICILIN
Phương pháp sắc ký lớp mỏng
Tiến hành phương pháp sắc ký lớp mỏng, bản mỏng (pha tĩnh) là silicagel H đã được silan hoá.
Chấm riêng biệt lên bản mỏng 2 μl mỗi dung dịch sau:
Dung dịch 1: Dung dịch thử.
Dung dịch 2 và 3: Các dung dịch có chứa các chất đối chiếu tương ứng trong Bảng 8.2.
Bảng 8.2 - Định tính các penicilin bằng phương pháp sắc ký lớp mỏng
| Tên mẫu | Dung môi để hòa tan | Nồng độ% | Chất đối chiếu trong dung dịch | Chất đối chiếu trong dung dịch 3 | Pha động | Yêu cầu đối với phép thử |
| Amoxycilin natri | A | 0,25 | Amoxycilin trihydrat | Amoxycilin trihydrat + Ampicilin trihydrat | B | A |
| Amoxycilin trihydrat | A | 0,25 | Amoxycilin trihydrat | Amoxycilin trihydrat + Ampicilin trihydrat | B | A |
| Ampicilin | A | 0,25 | Ampicilin khan | Ampicilin khan + Amoxycilin trihydrat | B | A |
| Ampicilin natri | A | 0,25 | Ampicilin trihydrat | Ampicilin trihydrat + Amoxycilin trihydrat | B | A |
| Ampicilin trihydrat | A | 0,25 | Ampicilin trihydrat | Ampicilin trihydrat + Amoxycilin trihydrat | B | A |
| Benzathin penicillin | c | 0,5 | Benzathin benzyl- penicillin |
| A* | B |
| Benzylpenicilin kali | D | 0,5 | Benzylpenicilin kali | Benzylpenicilin kali +Phenoxymethylpeni- cilin kali | A | A |
| Benzylpenicilin natri | D | 0,5 | Benzylpenicilin natri | Benzylpenicilin natri +Phenoxymethylpeni- cilin kali | A | A |
| Cloxacilin natri | D | 0,5 | Cloxacilin natri | Cloxacilin natri + Dicloxacilin natri + Flucloxacilin natri | A | C |
| Dicloxacilin natri | D | 0,5 | Dicloxacilin natri | Cloxacilin natri + Dicloxacilin natri + Flucloxacilin natri | A | C |
| Flucloxacilin natri | D | 0,5 | Flucloxacilin natri | Cloxacilin natri + Dicloxacilin natri + Flucloxacilin natri | A | C |
| Phenoxymethyl -penicilin | E* | 0,5 | Phenoxymethylpenicilin | Phenoxymethylpenicilin kali + Benzylpenicilin kali | A | A |
| Phenoxymethyl -penicilin kali | D | 0,5 | Phenoxymethylpenicilin kali | Phenoxymethylpenicilin kali + Benzylpenicilin kali | A | A |
| Procain penlcilin | E | 0,5 | Procain benzylpenicilin |
| A* | B |
Sau khi triển khai sắc ký, để khô bản mỏng ngoài không khí, đặt bản mỏng vào bình có hơi iod cho đến khi xuất hiện các vết. Quan sát các vết dưới ánh sáng ban ngày. Trên sắc ký đồ, vết chính thu được của dung dịch (1) phải tương ứng về vị trí, màu sắc và kích thước với vết thu được của dung dịch (2). Đối với benzathin penicilin và procain penicilin, dung dịch 1 phải có 2 vết chính tương ứng về vị trí, màu sắc và kích thước với 2 vết chính trên sắc ký đồ của dung dịch 2. Phép thử chỉ có giá trị khi đạt các yêu cầu được chỉ ra trong Bảng 8.2.
Các hệ dung môi khai triển (Pha động)
A: Hỗn hợp của dung dịch amoni acetat 15,4 % (TT) và aceton (TT) (70: 30), được điều chỉnh tới pH 5,0 bằng acid acetic khan (TT) nếu như không có các chỉ dẫn khác trong bảng [A*: Điều chỉnh pH của pha động tới 7,0 bằng amoniac (TT)].
B: Hỗn hợp của dung dịch amoni acetat 15,4 % (TT) và aceton (TT) (90: 10) được điều chỉnh tới pH 5,0 bằng acid acetic khan (TT).
Dung môi
A: Dung dịch natri hydrocarbonat 4,2 %.
B: Hỗn hợp aceton - methanol (1:1).
C: Methanol.
D: Nước.
E: Aceton (E*: Dùng nước là dung môi cho dung dịch 3)
Yêu cầu đối với phép thử
A: Phép thử chỉ có giá trị khi trên sắc ký đồ dung dịch (3) tách ra 2 vết rõ ràng, riêng biệt.
B: Phép thử chỉ có giá trị khi trên sắc ký đồ dung dịch (2) tách ra 2 vết rõ ràng, riêng biệt.
C: Phép thử chỉ có giá trị khi trên sắc ký đồ dung dịch (3) tách ra 3 vết rõ ràng, riêng biệt.
8.3 PHẢN ỨNG MÀU CỦA CÁC PENICILIN VÀ CEPHALOSPORIN
A. Cho 2 mg chế phẩm vào một ống nghiệm (kích thước 15 cm x 1,5 cm), làm ẩm với 0,05 ml nước, thêm 2 ml acid sulfuric (TT). Lắc đều hỗn hợp và quan sát màu của dung dịch. Đặt ống nghiệm trong cách thủy sôi trong một phút và lại quan sát màu. Dung dịch có màu tương ứng trong Bảng 8.3.
B. Lặp lại cách thử như phản ứng A, nhưng thay 2 ml acid sulfuric (TT) bằng 2 ml dung dịch formaldehyd trong acid sulfuric (TT). Dung dịch có màu tương ứng trong Bảng 8.3.
Bảng 8.3 - Phản ứng màu của các penicilin và cephalosporin
| Tên mẫu | Acid sulfuric (TT) | Acid sulfuric (TT) sau 1 min ở 100°C | Dung dịch formaldehyd trong acid sulfuric (TT) | Dung dịch formaldehyd trong acid sulfuric (TT) sau 1 min ở 100 °C |
| Amoxicilin natri |
|
| Hầu như không màu | Vàng sẫm |
| Amoxicilin trihydrat |
|
| Hầu như không màu | Vàng sẫm |
| Ampicilin |
|
| Hầu như không màu | Vàng sẫm |
| Ampicilin natri |
|
| Hầu như không màu | Vàng sẫm |
| Ampicilin trihydrat |
|
| Hầu như không màu | Vàng sẫm |
| Benzathin penicilin |
|
| Hầu như không màu | Nâu đỏ |
| Benzylpenicilin kali |
|
| Hầu như không màu | Nâu đỏ |
| Benzylpenicilin natri |
|
| Hầu như không màu | Nâu đỏ |
| Cefaclor |
|
| Hầu như không màu | Vàng nâu |
| Cefadroxil |
|
| Vàng | Da cam |
| Cefixim |
|
| Vàng | Da cam |
| Cefotaxim natri |
|
| Vàng sáng | Nâu |
| Cefradin |
|
| Vàng nhạt | Vàng sẫm |
| Cephalexin |
|
| Vàng nhạt | Vàng sẫm |
| Cephaloridin | Vàng nhạt | Hầu như không màu | Đỏ | Đỏ nâu |
| Cephalothin natri | Vàng (màu chuyển nhanh) | Hồng nâu | Đỏ | Đỏ nâu |
| Cloxacilin natri |
|
| Vàng xanh lá nhạt | Vàng |
| Dicloxacilin natri |
|
| Vàng xanh lá nhạt | Vàng |
| Flucloxacilin natri |
|
| Vàng xanh lá nhạt | Vàng |
| Phenoxymethyl penicilin |
|
| Nâu đỏ | Nâu đỏ sẫm |
| Phenoxymethyl penicilin kali |
|
| Nâu đỏ | Nâu đỏ sẫm |
| Procain penicilin | Hầu như không màu | Hầu như không màu | Hầu như không màu | Nâu đỏ |
PHỤ LỤC 9
(Quy định)
9.1 ỐNG NGHIỆM DÙNG TRONG CÁC PHÉP THỬ SO SÁNH
Ống Nessler
Khi các phép thử yêu cầu sử dụng ống Nessler thì các ống Nessler được sử dụng phải đạt các yêu cầu sau:
Ống Nessler phải đạt các yêu cầu về tiêu chuẩn của ống Nessler do nhà sản xuất đưa ra. Ống được làm từ thủy tinh trung tính trong suốt và có thể tích danh định 50 ml. Chiều cao của ống khoảng 15 cm, chiều cao đo bên ngoài ống từ đáy đến vạch 50 ml là 11,0 cm đến 12,4 cm, độ dày của thành ống từ 1,0 mm đến 1,5 mm, độ dày của đáy ống từ 1,0 mm đến 3,0 mm. Chiều cao đo bên ngoài ống từ đáy đến vạch 50 ml đối với các ống dùng trong một phép thử không được sai khác quá 1 mm.
Ống nghiệm dùng cho các phép thử so sánh
Nếu không có chỉ dẫn gì khác, các ống nghiệm dùng trong các phép thử so sánh là những ống thích hợp được làm bằng thủy tinh không màu, có đường kính trong đồng nhất. Đáy trong suốt và phẳng. Khi quan sát, nhìn cột chất lỏng theo trục thẳng đứng từ trên xuống trên nền trắng hoặc nền đen (nếu cần thiết) dưới ánh sáng khuyếch tán ban ngày.
Thường dùng những ống có đường kính trong 16 mm. Có thể dùng những ống đường kính lớn hơn nhưng thể tích chất lỏng đem thử phải tăng lên sao cho chiều cao của lớp chất lỏng không thấp hơn chiều cao của lớp chất lỏng khi thử bằng nhưng ống có đường kính trong bằng 16 mm.
9.2 XÁC ĐỊNH ĐỘ TRONG CỦA DUNG DỊCH
Độ trong của các dung dịch được xác định bằng cách so sánh các dung dịch đó với các hỗn dịch đối chiếu.
Dung dịch hydrazin sulfat: Hòa tan 1,0 g hydrazin sulfat (TT) trong nước, pha loãng với nước thành 100,0 ml và để yên trong thời gian 4 h đến 6 h.
Dung dịch hexamethylentetramin: Trong một bình nón thủy tinh nút mài dung tích 100 ml, hòa tan 2,5 g hexamethylentetramin trong 25,0 ml nước.
Hỗn dịch đục gốc
Thêm 25,0 ml dung dịch hydrazin sulfat vào 25,0 ml dung dịch hexamethylentetramin, lắc kỹ và để yên trong 24 h.
Nếu được bảo quản trong lọ thủy tinh tốt (không có khuyết tật ở bề mặt) thì hỗn dịch đục gốc bền vững trong vòng 2 tháng. Hỗn dịch này phải không được bám dính vào thủy tinh và phải được lắc kỹ trước khi dùng.
Chuẩn đục: Pha loãng 15,0 ml hỗn dịch đục gốc thành 1000,0 ml với nước.
Chuẩn đục được chuẩn bị ngay trước khi dùng và có thể bảo quản tối đa trong vòng 24 h.
Hỗn dịch đối chiếu
Các hỗn dịch đối chiếu từ I tới IV được chuẩn bị như chỉ dẫn trong Bảng 9.2.
Mỗi hỗn dịch phải được trộn kỹ và lắc trước khi sử dụng.
Bảng 9.2 - Các hỗn dịch đối chiếu
|
| I | II | III | IV |
| Chuẩn đục (ml) | 5,0 | 10,0 | 30,0 | 50,0 |
| Nước | 95,0 | 90,0 | 70,0 | 50,0 |
Cách thử
Việc so sánh được tiến hành trong các ống nghiệm giống nhau, bằng thủy tinh trung tính, trong, không màu, đáy bằng, có đường kính trong khoảng từ 15 mm đến 25 mm. Chiều dày của lớp dung dịch thử và của hỗn dịch đối chiếu là 40 mm. Hỗn dịch đối chiếu sau khi pha 5 min phải được so sánh ngay với dung dịch cần thử bằng cách quan sát chất lỏng từ trên xuống trong các ống nghiệm trên nền đen dưới ánh sáng khuếch tán ban ngày. Ánh sáng khuếch tán phải phù hợp để có thể phân biệt được hỗn dịch đối chiếu I với nước cất và với hỗn dịch đối chiếu II.
Cách đánh giá kết quả
Một chất lỏng được coi như trong nếu nó tương đương với độ trong của nước cất hay của dung môi đã dùng khi thử nghiệm trong những điều kiện như đã mô tả, hoặc nếu chất lỏng đó hơi đục nhẹ thì cũng không được đục quá hỗn dịch đối chiếu I.
Các yêu cầu khác nhau về độ đục được biểu thị theo hỗn dịch đối chiếu I, II, III, và IV.
9.3 XÁC ĐỊNH MÀU SẮC CỦA DUNG DỊCH
Việc xác định màu sắc của dung dịch trong phạm vi nâu - vàng - đỏ được tiến hành theo một trong hai phương pháp dưới đây, tùy theo chỉ dẫn trong chuyên luận. Một dung dịch được coi là không màu nếu nó giống như nước cất hay dung môi dùng để pha dung dịch đó, hoặc có màu không thẫm hơn dung dịch màu đối chiếu Ng.
Phương pháp 1
Dùng những ống thủy tinh trung tính, không màu, trong suốt và giống hệt nhau, có đường kính ngoài 12 mm để so sánh 2,0 ml dung dịch thử với 2,0 ml nước cất, hoặc dung môi, hoặc dung dịch màu đối chiếu (Bảng 9.3.2a tới 9.3.2e) theo chỉ dẫn trong chuyên luận. Quan sát màu của dung dịch theo chiều ngang ống nghiệm, dưới ánh sáng khuếch tán, trên nền trắng.
Phương pháp 2
Dùng những ống thủy tinh trung tính, đáy bằng, không màu, trong suốt, giống hệt nhau và có đường kính trong từ 15 mm đến 25 mm để so sánh lớp dung dịch thử với nước cất, hoặc dung môi, hoặc dung dịch màu đối chiếu (Bảng 9.3.2 a tới Bảng 9.3.2 e) theo chỉ dẫn trong chuyên luận, bề dày của lớp chất lỏng là 40 mm. Quan sát màu của dung dịch dọc theo trục ống, dưới ánh sáng khuếch tán trên nền trắng.
Pha chế các dung dịch gốc
Dung môi A: 25 ml acid hydrocloric (TT) hòa tan trong 975 ml nước cất.
Dung dịch gốc màu vàng: Hòa tan 46 g sắt (III) clorid (TT) trong 900 ml dung môi A, cho vừa đủ dung môi A thành 1000 ml. Chuẩn độ rồi điều chỉnh bằng dung môi A để có dung dịch chứa 45 mg FeCI3.6H2O trong 1 ml. Bảo quản tránh ánh sáng.
Chuẩn độ: Cho vào một bình nón dung tích 250 ml có nút mài 10 ml dung dịch gốc màu vàng, 15 ml nước, 5 ml acid hydrocloric (TT) và 4 g kali iodid (TT). Đậy nút, lắc đều rồi để yên 15 min ở chỗ tối. Thêm vào bình 100 ml nước và chuẩn độ iod giải phóng bằng dung dịch natri thiosulfat 0,1 N (CĐ), chỉ thị là 0,5 ml dung dịch hồ tinh bột (TT) cho vào lúc gần cuối chuẩn độ.
1 ml dung dịch natri thiosulfat 0,1 N (CĐ) tương đương với 27,03 mg FeCI3.6H2O.
Dung dịch gốc màu đỏ: Hòa tan 60 g cobalt (II) clorid (TT) trong 900 ml dung môi A, cho vừa đủ dung môi A thành 1000 ml. Chuẩn độ, rồi điều chỉnh bằng dung môi A để có dung dịch chứa 59,5 mg CoCI2.6H2O trong 1 ml.
Chuẩn độ: Cho vào một bình nón dung tích 250 ml có nút mài 5 ml dung dịch gốc màu đỏ, 5 ml nước oxy già 10 thể tích và 10 ml dung dịch natri hydroxyd 30 % (TT). Đun sôi nhẹ trong 10 min, để nguội rồi thêm 60 ml dung dịch acid sulfuric 1 M (TT) và 2 g kali iodid (TT). Đậy bình và lắc nhẹ cho tan tủa, chuẩn độ iod giải phóng bằng dung dịch natri thiosulfat 0,1 N (CĐ) với chỉ thị là 0,5 ml dung dịch hồ tinh bột (TT) cho vào lúc gần cuối chuẩn độ. Dung dịch chuyển thành màu hồng khi chuẩn độ đến điểm tương đương.
1 ml dung dịch natri thiosulfat 0,1 N (CĐ) tương đương với 23,79 mg CoCl2.6H2O.
Dung dịch gốc màu xanh: Hòa tan 63 g đồng (II) sulfat (TT) trong 900 ml dung môi A, cho vừa đủ dung môi A thành 1000 ml. Chuẩn độ, rồi điều chỉnh bằng dung môi A để có dung dịch chứa 62,4 mg CuSO4.5H2O trong 1 ml.
Chuẩn độ: Cho vào một bình nón dung tích 250 ml có nút mài 10 ml dung dịch gốc màu xanh, 50 ml nước, 12 ml dung dịch acid acetic 2 M (TT) và 3 g kali iodid (TT). Chuẩn độ iod giải phóng bằng dung dịch natri thiosulfat 0,1 N (CĐ) đến khi có màu nâu nhạt, chỉ thị là 0,5 ml dung dịch hồ tinh bột (TT) cho vào lúc gần cuối chuẩn độ.
1 ml dung dịch natri thiosulfat 0,1 N (CĐ) tương tương với 24,97 mg CuSO4.5H2O.
Pha chế các dung dịch màu chuẩn
Dùng 3 dung dịch gốc để pha 5 dung dịch màu chuẩn theo Bảng 9.3.1
Bảng 9.3.2 d - Dung dịch màu đối chiếu VL
| Dung dịch màu đối chiếu | Dung dịch màu chuẩn VL (ml) | Dung dịch acid hydrocloric 1 % (ml) |
| VL1 | 25,0 | 75,0 |
| VL2 | 15,0 | 85,0 |
| VL3 | 8,5 | 91,5 |
| VL4 | 5,0 | 95,0 |
| VL5 | 3,0 | 97,0 |
| VL6 | 1,5 | 98,5 |
| VL7 | 0,75 | 99,25 |
Bảng 9.3.2 e - Dung dịch màu đối chiếu Đ
| Dung dịch màu đối chiếu | Dung dịch màu chuẩn Đ (ml) | Dung dịch acid hydrocloric 1 % (ml) |
| Đ1 | 100,0 | 0,0 |
| Đ2 | 75,0 | 25,0 |
| Đ3 | 50,0 | 50,0 |
| Đ4 | 37,5 | 62,5 |
| Đ5 | 25,0 | 75,0 |
| Đ6 | 12,5 | 87,5 |
| Đ7 | 5,0 | 95,0 |
Bảo quản
Với phương pháp 1, các dung dịch màu đối chiếu cần được bảo quản trong những ống thủy tinh trung tính, không màu, trong suốt có đường kính ngoài 12 mm, được hàn kín và tránh ánh sáng.
Với phương pháp 2, các dung dịch màu đối chiếu phải được chuẩn bị ngay trước khi dùng từ dung dịch màu chuẩn.
9.4 XÁC ĐỊNH GIỚI HẠN CÁC TẠP CHẤT
9.4.1 Amoni
Dùng phương pháp A, trừ khi có chỉ dẫn khác trong chuyên luận.
Phương pháp A
Hòa tan một lượng chế phẩm thử như chỉ dẫn trong chuyên luận với 14 ml nước trong một ống nghiệm có nút mài, kiềm hóa nếu cần bằng dung dịch natri hydroxyd 2 M (TT), thêm nước vừa đủ 15 ml. Thêm 0,3 ml dung dịch kali tetraiodomercurat kiềm (TT), lắc đều rồi để yên 5 min. So sánh màu tạo thành trong ống thử với màu mẫu được chuẩn bị đồng thời trong cùng điều kiện như dung dịch thử.
Màu mẫu: Lấy chính xác 10 ml dung dịch amoni mẫu 1 phần triệu NH4 (TT) cho vào một ống nghiệm, pha loãng với nước thành 15 ml. Thêm 0,3 ml dung dịch kali tetraiodomercurat kiềm (TT), lắc đều rồi để yên 5 min.
Khi so màu, quan sát dọc theo trục ống nghiệm, dưới ánh sáng khuếch tán trên nền trắng. Màu vàng xuất hiện trong ống thử không được đậm hơn màu trong ống mẫu.
Phương pháp B
Cho một lượng chế phẩm (theo chỉ dẫn trong chuyên luận riêng) đã nghiền mịn vào một bình 25 ml có nút đậy bằng polyethylen và hòa tan hoặc phân tán trong 1 ml nước. Thêm 0,3 g magnesi oxyd nặng (TT). Đậy bình ngay sau khi đã đặt xuống dưới nắp polyethylen một mẩu giấy tẩm mangan bạc (TT) hình vuông có cạnh 5 mm đã được làm ẩm bằng vài giọt nước. Lắc xoay tròn bình, tránh chất lỏng bắn lên và để yên ở 40 °C trong 30 min.
Màu xám nếu xuất hiện trên mẩu giấy tẩm mangan bạc ở bình thử không được đậm hơn ở bình mẫu được chuẩn bị đồng thời trong cùng điều kiện với lượng dung dịch amoni mẫu 1 phần triệu NH4 (TT) đã được chỉ dẫn trong chuyên luận, 1 ml nước và 0,3 g magnesi oxyd nặng (TT).
9.4.2 Arsen
Dùng phương pháp A, trừ khi có chỉ dẫn khác trong chuyên luận.
Phương pháp A
Dụng cụ: Bộ dụng cụ thử arsen (Hình 9.4.2) gồm một bình nón nút mài cỡ 100 ml được đậy bằng nút thủy tinh mài, xuyên qua nút có một ống thủy tinh dài khoảng 200 mm, đường kính trong là 5 mm. Phần dưới của ống thủy tinh được kéo nhỏ lại để có đường kính trong là 1,0 mm và cách đầu ống 15 mm có một lỗ trên thành ống với đường kính 2 mm đến 3 mm. Khi gắn ống thủy tinh vào nút thì lỗ này phải ở cách mặt dưới của nút ít nhất là 3 mm. Đầu trên của ống thủy tinh có một dĩa tròn phẳng, mặt phẳng của đĩa vuông góc với trục ống.
Một ống thủy tinh thứ hai dài 30 mm, có cùng đường kính và cũng có đĩa tròn phẳng tương tự như ống thứ nhất, đặt tiếp xúc mặt đĩa tròn với ống thứ nhất và được giữ chặt với ống thứ nhất bằng 2 dây lò xo.
Tiến hành: Cho xuống đầu thấp của ống thủy tinh dài khoảng 50 mg đến 60 mg bông tẩm chì acetat (TT) hoặc một miếng gạc cotton bọc một mẩu giấy tẩm chì acetat (TT) có khối lượng 50 mg đến 60 mg. Đặt một miếng giấy tẩm thủy ngân (II) bromid (TT), hình tròn hay hình vuông, có kích thước đủ để phủ kín lỗ tròn giữa 2 ống thủy tinh (15 mm x 15 mm), giữ chặt 2 ống thủy tinh bằng 2 dây lò xo.
Cho vào bình nón một lượng chế phẩm thử theo chỉ dẫn trong chuyên luận. Hòa tan hoặc pha loãng (nếu chế phẩm thử là dung dịch) với nước thành 25 ml. Thêm 15 ml acid hydrocloric (TT), 0,1 ml dung dịch thiếc (II) clorid AsT (TT) và 5 ml dung dịch kali iodid 16,6 % (TT). Để yên 15 min rồi thêm 5 g kẽm không có arsen (TT). Đậy ngay bình nón bằng nút đã lắp sẵn giấy thử ở trên và ngâm bình trong nước ở nhiệt độ sao cho khí được giải phóng đều đặn.
Song song tiến hành một mẫu so sánh trong cùng điều kiện, dùng 1 ml dung dịch arsen mẫu 1 phần triệu As (TT) hòa loãng với nước thành 25 ml thay cho chế phẩm thử.
Sau ít nhất 2 h lấy các miếng giấy tẩm thủy ngân (II) bromid (TT) ra so sánh các vết màu. Vết màu nếu có trên miếng giấy của bình thử phải không được đậm hơn vết màu trên miếng giấy của bình mẫu.
Phương pháp B
Lấy một lượng chế phẩm thử theo chỉ dẫn trong chuyên luận cho vào một ống nghiệm chứa 4 ml acid hydrocloric (TT) và khoảng 5 mg kali iodid (TT), thêm 3 ml dung dịch hypophosphit (TT). Đun cách thủy hỗn hợp trong 15 min, thỉnh thoảng lắc.
Tiến hành song song một mẫu so sánh trong cùng điều kiện, thay chế phẩm thử bằng 0,5 ml dung dịch arsen mẫu 10 phần triệu As (TT).
So sánh màu trong hai ống. Màu trong ống thử không được đậm hơn màu trong ống so sánh.
Hình 9.4.2 - Dụng cụ thử arsen
(Kích thước tính bằng mm)
9.4.3 Calci
Các dung dịch dùng trong phép thử này phải được chuẩn bị với nước cất.
Thêm 1 ml dung dịch amoni oxalat 4 % (TT) vào 0,2 ml dung dịch calci mẫu 100 phần triệu Ca trong ethanol 96 % (TT). Sau 1 min, thêm hỗn hợp gồm 1 ml dung dịch acid acetic 2 M (TT) và 15 ml dung dịch chế phẩm thử như chỉ dẫn trong chuyên luận và lắc.
Sau 15 min, so sánh độ đục tạo thành trong ống thử với độ đục mẫu được chuẩn bị đồng thời trong cùng điều kiện nhưng thay dung dịch chế phẩm thử bằng hỗn hợp gồm 10 ml dung dịch calci mẫu 10 phần triệu Ca (TT) và 5 ml nước.
Ống thử phải không được đục hơn ống mẫu.
9.4.4 Chì trong đường
Tiến hành theo phương pháp quang phổ hấp thụ nguyên tử (Phụ lục 4.4, phương pháp 2), dùng ngọn lửa acetylen - không khí, đèn cathod rỗng chì và các dung dịch sau:
Dung dịch thử: Hòa tan 20,0 g chế phẩm thử trong dung dịch acid acetic 1 M (TT) để được 100,0 ml. Thêm 2,0 ml dung dịch bão hòa amoni pyrolidin dithiocarbamat (TT) và 10,0 ml 4-methylpentan-2-on (TT), lắc trong 30 s, tránh ánh sáng. Để yên cho tách lớp và lấy lớp 4-methylpentan-2-on.
Các dung dịch đối chiếu: Chuẩn bị 3 dung dịch đối chiếu trong cùng điều kiện như dung dịch thử, bằng cách thêm riêng biệt 0,5 ml, 1,0 ml, 1,5 ml dung dịch chì mẫu 10 phần triệu Pb (TT) vào mỗi bình đã có 20,0 g chế phẩm thử.
Chuẩn bị một mẫu trắng trong cùng điều kiện giống như dung dịch thử nhưng không có chế phẩm thử.
Đo độ hấp thụ của dung dịch thử, 3 dung dịch đối chiếu và mẫu trắng ở cực đại 283,3 nm, dùng dung dịch trắng để hiệu chỉnh điểm 0 của máy. Vẽ đường biểu diễn sự phụ thuộc giữa độ hấp thụ với nồng độ chì trong các dung dịch và xác định hàm lượng của chì trong chế phẩm thử.
Hàm lượng chì không được lớn hơn 0,5 phần triệu trừ khi có chỉ dẫn khác.
9.4.5 Clorid
Chuẩn bị dung dịch thử như chỉ dẫn trong chuyên luận, cho vào một ống nghiệm, pha loãng với nước đến 15 ml. Thêm 1 ml dung dịch acid nitric 2 M (TT) và 1 ml dung dịch bạc nitrat 2 % (TT). Để yên 5 min, tránh ánh sáng. So sánh độ đục tạo thành trong ống thử với độ đục trong ống mẫu được chuẩn bị đồng thời trong cùng điều kiện nhưng thay dung dịch thử bằng hỗn hợp gồm 10 ml dung dịch clorid mẫu 5 phần triệu Cl (TT) và 5 ml nước. Quan sát dọc theo trục ống nghiệm, dưới ánh sáng khuếch tán trên nền đen. Ống thử không được đục hơn ống mẫu.
9.4.6 Fluorid
Dùng thiết bị cất như mô tả trong Hình 9.4.6 bao gồm một ống nghiệm nút mài nối với một ống ngưng ruột thẳng. Ống nghiệm được đặt trong một bộ phận đun bằng thủy tinh chịu nhiệt kín có nối với một ống sinh hàn và nhiệt kế.
Hình 9.4.6 - Dụng cụ thử giới hạn fluorid
(Kích thước tính bằng mm)
Cân và chuyển một lượng chế phẩm như chỉ dẫn trong chuyên luận vào ống nghiệm, thêm 0,1 g cát đã được rửa bằng acid và 20 ml acid sulfuric 50 % (tt/tt). Rót tetracloroethan (TT) vào bình đun, đun nóng và giữ ở nhiệt độ sôi của tetracloroethan (146 °C). Chưng cất và hứng dịch cất vào bình định mức 100 ml có chứa sẵn 0,3 ml dung dịch natri hydroxyd 0,1 N (TT) và 0,1 ml dung dịch phenolphtalein (TT). Duy trì thể tích ở trong ống nghiệm là 20 ml trong suốt quá trình cất, và giữ cho dịch trong bình cất luôn luôn kiềm bằng cách thêm dung dịch natri hydroxyd 0,1 N (TT) nếu cần. Thêm nước vào bình hứng dịch cất vừa đủ 100 ml, được dung dịch thử. Chuẩn bị mẫu chuẩn với cùng điều kiện như mẫu thử nhưng thay chế phẩm bằng 5 ml dung dịch fluorid chuẩn 10 phần triệu F (TT). Lấy 2 ống nghiệm nút mài, thêm riêng rẽ 20 ml dung dịch thử và dung dịch chuẩn và thêm 5 ml thuốc thử acid aminomethylalizarindiacetic (TT) vào mỗi dung dịch, sau 20 min nếu dung dịch thử có màu xanh (ban đầu là màu đỏ) thì không được đậm hơn màu của dung dịch đối chiếu.
9.4.7 Kali
Thêm 2 ml dung dịch natri tetraphenylborat 1 % (TT) mới pha vào 10 ml dung dịch chế phẩm thử đã chỉ dẫn trong chuyên luận, để yên 5 min. So sánh độ đục tạo thành trong ống thử với độ đục trong ống mẫu được chuẩn bị đồng thời trong cùng điều kiện một hỗn hợp của 5 ml dung dịch kali mẫu 20 phần triệu K (TT) và 5 ml nước.
Độ đục trong ống thử không được đậm hơn độ đục trong ống mẫu.
9.4.8 KIM LOẠI NẶNG
Các phương pháp sau đây yêu cầu dùng dung dịch thioacetamid (TT). Có thể dùng dung dịch natri sulfid (TT) (0,1 ml) để thay thế. Vì các phương pháp dưới đây được xây dựng dựa trên sử dụng dung dịch thioacetamid (TT) nên nếu sử dụng dung dịch natrisulfid (TT) thay thế thì phải chuẩn bị thêm dung dịch kiểm tra khi thử theo phương pháp 1, 2 và 8. Dung dịch kiểm tra được chuẩn bị từ lượng chế phẩm giống như lượng chế phẩm dùng để chuẩn bị dung dịch thử và thêm thể tích dung dịch chì mẫu giống như thể tích dung dịch chì mẫu dùng để chuẩn bị dung dịch đối chiếu. Phép thử chỉ có giá trị khi dung dịch kiểm tra có màu đậm ít nhất là bằng màu của dung dịch đối chiếu.
Phươmg pháp 1
Dung dịch thử: 12 ml dung dịch chế phẩm được chuẩn bị như chỉ dẫn trong chuyên luận.
Dung dịch đối chiếu: Hỗn hợp gồm 10 ml dung dịch chì mẫu 1 phần triệu Pb (TT) hoặc dung dịch chì mẫu 2 phần triệu Pb (TT) tùy theo chỉ dẫn trong chuyên luận và 2 ml dung dịch thử.
Dung dịch mẫu trắng: Hỗn hợp gồm 10 ml nước và 2 ml dung dịch thử.
Thêm 2 ml dung dịch đệm acetat pH 3,5 (TT) vào mỗi dung dịch trên. Lắc đều và thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT), lắc ngay. Quan sát các dung dịch sau 2 min.
Tính thích hợp của phép thử: Dung dịch đối chiếu phải có nàu nâu nhạt khi so sánh với dung dịch mẫu trắng.
Đánh giá kết quả: Màu nâu của dung dịch thử, nếu có, không được đậm hơn màu của dung dịch đối chiếu.
Nếu như khó đánh giá kết quả, lọc các dung dịch qua màng lọc thích hợp (Kích thước lỗ 0,45 µm). Tiến hành lọc chậm và đều với áp lực lên piston nhẹ nhàng và liên tục. So sánh màu sắc của các vết trên màng lọc thu được từ các dung dịch.
Phương pháp 2
Dung dịch thử: 12 ml dung dịch chế phẩm được chuẩn bị như chỉ dẫn trong chuyên luận, dùng dung môi hữu cơ có chứa một tỷ lệ nước tối thiểu [Ví dụ như 1,4-dioxan (TT) hoặc aceton (TT) có chứa 15 % nước (tt/tt)].
Dung dịch đối chiếu: Hỗn hợp gồm 10 ml dung dịch chì mẫu (1 hay 2 phần triệu Pb tùy theo chỉ dẫn trong chuyên luận) và 2 ml dung dịch thử.
Dung dịch ion chì mẫu 1 hoặc 2 phần triệu Pb được pha chế bằng cách pha loãng dung dịch chì mẫu 100 phần triệu Pb (TT) với dung môi được dùng để pha dung dịch thử.
Dung dịch mẫu trắng: Hỗn hợp gồm 10 ml dung môi được dùng để pha dung dịch thử và 2 ml dung dịch thử.
Thêm 2 ml dung dịch đệm acetat pH 3,5 (TT) vào mỗi dung dịch trên. Lắc đều và thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT), lắc đều ngay. Quan sát các dung dịch sau 2 min.
Tính thích hợp của phép thử: Dung dịch đối chiếu phải có nàu nâu nhạt khi so sánh với dung dịch mẫu trắng.
Đánh giá kết quả: Màu nâu của dung dịch thử, nếu có, không được đậm hơn màu của dung dịch đối chiếu.
Nếu như khó đánh giá kết quả, lọc các dung dịch qua màng lọc thích hợp (Kích thước lỗ 0,45 μm). Tiến hành lọc chậm và đều với áp lực lên piston nhẹ nhàng và liên tục. So sánh màu sắc của các vết trên màng lọc thu được từ các dung dịch.
Phương pháp 3
Dung dịch thử: Lấy một lượng chế phẩm như chỉ dẫn trong chuyên luận (không quá 2 g) cho vào một chén nung sứ. Thêm 4 ml dung dịch magnesi sulfat 25 % trong acid sulfuric 1 M (TT). Trộn đều bằng một đũa thủy tinh nhỏ rồi đun nóng cẩn thận. Nếu hỗn hợp là một chất lỏng thì làm bay hơi từ từ trên cách thủy đến khô. Đốt dần dần để chế phẩm cháy hết và tiếp tục đốt cho đến khi thu được cắn có màu gần trắng hay ít nhất là xám nhạt. Tiến hành nung ở nhiệt độ không quá 800 °C. Để nguội. Làm ẩm cắn bằng vài giọt dung dịch acid sulfuric 1 M (TT). Bốc hơi, nung lại và để nguội. Toàn bộ thời gian nung không được quá 2 h. Hòa tan cắn trong dung dịch acid hydrocloric 2 M (TT) 2 lần, mỗi lần dùng 5 ml. Thêm 0,1 ml dung dịch phenolphtalein (TT), cho từng giọt dung dịch amoniac đậm đặc (TT) đến khi có màu hồng. Để nguội, thêm acid acetic băng (TT) đến khi dung dịch mất màu và thêm thừa 0,5 ml nữa. Lọc nếu cần và pha loãng dung dịch với nước thành 20 ml.
Dung dịch đối chiếu: Tiến hành theo chỉ dẫn ở phần dung dịch thử, dùng một thể tích dung dịch chì mẫu 10 phần triệu Pb (TT) như đã ghi trong chuyên luận thay cho chế phẩm. Thêm 2 ml dung dịch thử vào 10 ml dung dịch thu được.
Dung dịch kiểm tra: Tiến hành theo chỉ dẫn ở phần dung dịch thử, dùng lượng chế phẩm như lượng dùng để chuẩn bị dung dịch thử và thể tích dung dịch chì mẫu 10 phần triệu Pb (TT) như thể tích dùng để chuẩn bị dung dịch đối chiếu. Thêm 2 ml dung dịch thử vào 10 ml dung dịch thu được.
Dung dịch mẫu trắng: Hỗn hợp gồm 10 ml nước và 2 ml dung dịch thử.
Lấy 12 ml của mỗi dung dịch trên, thêm 2 ml dung dịch đệm acetat pH 3,5 (TT). Lắc đều và thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT), lắc đều ngay. Quan sát các dung dịch sau 2 min.
Tính thích hợp của phép thử: Dung dịch đối chiếu phải có nàu nâu nhạt khi so sánh với dung dịch mẫu trắng và dung dịch kiểm tra phải có màu ít nhất là đậm bằng màu của dung dịch đối chiếu.
Đánh giá kết quả: Màu nâu của dung dịch thử, nếu có, không được đậm hơn màu của dung dịch đối chiếu.
Nếu như khó đánh giá kết quả, lọc các dung dịch qua màng lọc thích hợp (Kích thước lỗ 0,45 μm). Tiến hành lọc chậm và đều với áp lực lên piston nhẹ nhàng và liên tục. So sánh màu sắc của các vết trên màng lọc thu được từ các dung dịch.
Phương pháp 4
Dung dịch thử: Trộn đều một lượng chế phẩm như chỉ dẫn trong chuyên luận với 0,5 g magnesi oxyd (TT) trong một chén sứ. Nung đỏ hỗn hợp cho đến khi thu được một khối đồng nhất màu trắng hay trắng xám. Nếu sau khi nung 30 min mà hỗn hợp vẫn có màu thì để nguội, dùng đũa thủy tinh trộn đều rồi nung lại. Nếu cần, lặp lại thao tác đó. Cuối cùng nung ở 800 °C trong 1 h. Hòa tan cắn trong hỗn hợp đồng thể tích của dung dịch acid hydrocloric 2 M (TT) và nước, 2 lần, mỗi lần dùng 5 ml. Thêm 0,1 ml dung dịch phenolphtalein (TT), cho từng giọt dung dịch amoniac đậm đặc (TT) đến khi có màu hồng. Để nguội, thêm acid acetic băng (TT) đến khi dung dịch mất màu và thêm thừa 0,5 ml nữa. Lọc nếu cần và pha loãng dung dịch với nước thành 20 ml.
Dung dịch đối chiếu: Lấy thể tích dung dịch chì mẫu 10 phần triệu Pb (TT) như đã ghi trong chuyên luận, làm khô ở 100 °C đến 105 °C, thay cho chế phẩm và tiến hành theo chỉ dẫn ở phần dung dịch thử. Thêm 2 ml dung dịch thử vào 10 ml dung dịch thu được.
Dung dịch kiểm tra: Lấy lượng chế phẩm như lượng dùng để chuẩn bị dung dịch thử và thể tích dung dịch chì mẫu 10 phần triệu Pb (TT) như thể tích dùng để chuẩn bị dung dịch đối chiếu, làm khô ở 100 °C đến 105 °C và tiến hành theo chỉ dẫn ở phần dung dịch thử. Thêm 2 ml dung dịch thử vào 10 ml dung dịch thu được.
Dung dịch mẫu trắng: Hỗn hợp gồm 10 ml nước và 2 ml dung dịch thử.
Lấy 12 ml của mỗi dung dịch trên, thêm 2 ml dung dịch đệm acetat pH 3,5 (TT). Lắc đều và thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT), lắc đều ngay. Quan sát các dung dịch sau 2 min.
Tính thích hợp của phép thử: Dung dịch đối chiếu phải có nàu nâu nhạt khi so sánh với dung dịch mẫu trắng và dung dịch kiểm tra phải có màu ít nhất là đậm bằng màu của dung dịch đối chiếu.
Đánh giá kết quả: Màu nâu của dung dịch thử, nếu có, không được đậm hơn màu của dung dịch đối chiếu.
Nếu như khó đánh giá kết quả, lọc các dung dịch qua màng lọc thích hợp (kích thước lỗ 0,45 μm). Tiến hành lọc chậm và đều với áp lực lên piston nhẹ nhàng và liên tục. So sánh màu sắc của các vết trên màng lọc thu được từ các dung dịch.
Phương pháp 5
Dung dịch thử: Hòa tan một lượng chế phẩm như chỉ dẫn trong chuyên luận trong 30 ml nước (TT) hay trong thể tích như chỉ dẫn.
Dung dịch đối chiếu: Nếu không có chỉ dẫn khác, pha loãng thể tích dung dịch chì mẫu 1 phần triệu Pb (TT) như chỉ dẫn trong chuyên luận thành thể tích bằng thể tích của dung dịch thử.
Chuẩn bị một thiết bị lọc bằng cách lắp thân một bơm tiêm, không có piston, dung tích 50 ml với một giá đỡ có chứa, trên mặt đĩa, một màng lọc (Kích thước lỗ 3 μm) và phía trên có màng lọc phụ (Hình 9.4.8).
Chuyển dung dịch thử vào bơm tiêm, lắp piston, ấn nhẹ và đều đến khi dung dịch được lọc hết. Tháo giá đỡ và lấy màng lọc phụ ra, kiểm tra xem màng lọc có bị nhiễm bẩn không, nếu cần thì thay màng lọc và lọc lại trong cùng điều kiện.
Thêm 2 ml dung dịch đệm acetat pH 3,5 (TT) vào toàn bộ hay một phần dịch lọc như chỉ dẫn ở chuyên luận, lắc đều và thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT), lắc đều ngay và để yên 10 min. Lọc lại như chỉ dẫn ở trên nhưng đảo trật tự màng lọc, dung dịch qua màng lọc rồi mới qua màng lọc phụ. Quá trình lọc phải được thực hiện từ từ và đều bằng cách ấn piston nhẹ nhàng và liên tục. Sau khi lọc xong lấy màng lọc ra, làm khô bằng cách ép trên giấy lọc.
Song song tiến hành như chỉ dẫn ở trên với dung dịch đối chiếu.
Hình 9.4.8 - Dụng cụ thử giới hạn kim loại nặng
(Kích thước tính bằng mm)
Đánh giá kết quả: Màu của vết thu được từ dung dịch thử không được đậm màu hơn màu của vết thu được từ dung dịch đối chiếu.
Phương pháp 6
Dung dịch thử: Lấy một lượng chế phẩm như chỉ dẫn trong chuyên luận vào một bình Kjeldahl khô và sạch dung tích 100 ml (có thể dùng bình dung tích 300 ml nếu phản ứng tạo bọt nhiều). Giữ bình nghiêng một góc 45°. Nếu chế phẩm là chất rắn, thêm vừa đủ một thể tích hỗn hợp acid gồm 8 ml acid sulfuric (TT) và 10 ml acid nitric (TT) để làm ẩm toàn bộ mẫu. Nếu chế phẩm là chất lỏng, thêm vài ml hỗn hợp acid. Đun nóng nhẹ đến khi phản ứng bắt đầu, đợi phản ứng giảm bớt và tiếp tục thêm từng phần hỗn hợp acid, đun nóng sau mỗi lần thêm, đến khi hết 18 ml hỗn hợp acid. Tăng nhiệt độ và đun sôi nhẹ đến khi dung dịch thẫm màu. Để nguội, thêm 2 ml acid nitric (TT) và đun nóng đến khi dung dịch thẫm màu. Tiếp tục đun và thêm acid nitric (TT) đến khi dung dịch không còn thẫm màu thêm nữa. Đun nóng mạnh đến khi có khói trắng, dày đặc tạo thành. Để nguội, thêm cẩn thận 5 ml nước, đun sôi nhẹ đến khi có khói trắng, dày đặc tạo thành và tiếp tục đun đến khi còn 2 đến 3 ml. Để nguội, thêm cẩn thận 5 ml nước và quan sát màu của dung dịch. Nếu dung dịch có màu vàng, thêm cẩn thận 1 ml dung dịch hydrogen peroxyd đậm đặc (TT) và tiếp tục đun đến khi có khói trắng, dày đặc tạo thành và dung dịch còn khoảng 2 đến 3 ml. Nếu dung dịch vẫn có màu vàng, lặp lại bước thêm 5 ml nước và 1 ml dung dịch hydrogen peroxyd đậm đặc (TT) đến khi dung dịch không màu. Để nguội, pha loãng cẩn thận với nước và tráng vào ống Nessler dung tích 50 ml sao cho thể tích dung dịch thu được không vượt quá 25 ml. Điều chỉnh pH dung dịch đến 3,0 - 4,0 bằng amoniac (TT) (có thể dùng dung dịch amoniac 6 M (TT) để điều chỉnh khi đến gần khoảng pH yêu cầu), dùng chỉ thị ngoại là giấy chỉ thị có khoảng chỉ thị pH hẹp. Thêm nước để được 40 ml và trộn đều. Thêm 2 ml dung dịch đệm acetat pH 3,5 (TT). Lắc đều và thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT). Lắc đều ngay. Pha loãng với nước thành 50 ml, lắc đều.
Dung dịch mẫu (đối chiếu): Tiến hành đồng thời như dung dịch thử, thay chế phẩm bằng thể tích dung dịch ion chì mẫu 10 phần triệu Pb (TT) như chỉ dẫn trong chuyên luận.
Dung dịch kiểm tra: Tiến hành như dung dịch thử, dùng lượng chế phẩm như lượng dùng để chuẩn bị dung dịch thử và thể tích dung dịch chì mẫu 10 phần triệu Pb (TT) như thể tích dùng để chuẩn bị dung dịch đối chiếu.
Dung dịch mẫu trắng: Tiến hành như dung dịch thử nhưng không có chế phẩm.
Sau 2 min quan sát các dung dịch dọc theo chiều dải ống nghiệm trên nền trắng.
Tính thích hợp của phép thử: Dung dịch đối chiếu phải có nàu nâu nhạt khi so sánh với dung dịch mẫu trắng và dung dịch kiểm tra phải có màu ít nhất là đậm bằng màu của dung dịch đối chiếu.
Đánh giá kết quả: Màu nâu của dung dịch thử, nếu có, không được đậm hơn màu của dung dịch đối chiếu.
Nếu như khó đánh giá kết quả, lọc các dung dịch qua màng lọc thích hợp (Kích thước lỗ 0,45 μm). Tiến hành lọc chậm và đều với áp lực lên piston nhẹ nhàng và liên tục. So sánh màu sắc của các vết trên màng lọc thu được từ các dung dịch.
Phương pháp 7
Chú ý: Khi dùng thiết bị phá mẫu áp suất cao phải theo đúng hướng dẫn vận hành và chú ý về an toàn do nhà sản xuất đưa ra. Thiết lập chu kỳ phá mẫu tùy thuộc kiểu lò vi sóng (Ví dụ kiểu lò kiểm soát năng lượng, kiểu lò kiểm soát nhiệt độ hoặc lò áp suất cao). Chu kỳ này phải theo hướng dẫn của nhà sản xuất. Chu kỳ phá mẫu là phù hợp nếu thu được dung dịch trong.
Dung dịch thử: Lấy một lượng chế phẩm như chỉ dẫn trong chuyên luận (không quá 0,5 g) vào một cốc phù hợp, sạch. Thêm lần lượt 2,7 ml acid sulfuric (TT), 3,3 ml acid nitric (TT) và 2,0 ml dung dịch hydrogen peroxyd đậm đặc (TT) vừa thêm vừa khuấy đều bằng máy khuấy từ, để từng thuốc thử phản ứng với chế phẩm trước khi thêm thuốc thử tiếp theo. Chuyển hỗn hợp vào bình phá mẫu chịu áp suất cao (bằng fluoropolyme hoặc thạch anh), bình phải được làm khô trước khi cho mẫu vào.
Dung dịch đối chiếu: Tiến hành như dung dịch thử, thay chế phẩm bằng thể tích dung dịch ion chì mẫu 10 phần triệu Pb (TT) như chỉ dẫn trong chuyên luận.
Dung dịch kiểm tra: Tiến hành như dung dịch thử, dùng lượng chế phẩm như lượng dùng để chuẩn bị dung dịch thử và thể tích dung dịch chì mẫu 10 phần triệu Pb (TT) như thể tích dùng để chuẩn bị dung dịch đối chiếu
Dung dịch mẫu trắng: Tiến hành như dung dịch thử nhưng không có chế phẩm.
Đậy bình phá mẫu và đặt vào trong lò vi sóng. Tiến hành phá mẫu bằng cách sử dụng nối tiếp 2 chương trình tách biệt thích hợp. Thiết lập các chương trình theo một số bước để có thể kiểm soát phản ứng, theo dõi áp suất, nhiệt độ hoặc năng lượng tùy theo kiểu lò vi sóng sử dụng. Sau khi áp dụng chương trình đầu tiên, để nguội các bình phá mẫu trước khi mở. Thêm vào mỗi bình 2 ml dung dịch hydrogen peroxyd đậm đặc (TT) và áp dụng chương trình thứ hai. Sau khi áp dụng chương trình thứ hai, để nguội các bình phá mẫu trước khi mở. Nếu cần thiết để có được dung dịch trong, lặp lại bước thêm dung dịch hydrogen peroxyd đậm đặc (TT) và chương trình phá mẫu thứ hai.
Để nguội, pha loãng cẩn thận với nước và tráng vào một bình nón sao cho thể tích dung dịch thu được không vượt quá 25 ml. Điều chỉnh pH dung dịch đến 3,0 - 4,0 bằng amoniac (TT) (có thể dùng dung dịch amoniac 6 M (TT) để điều chỉnh khi đến gần khoảng pH yêu cầu), dùng chỉ thị ngoại là giấy chỉ thị có khoảng chỉ thị pH hẹp. Để tránh làm nóng dung dịch, ngâm dung dịch trong nước đá và dùng máy khuấy từ. Pha loãng thành 40 ml với nước và trộn đều. Thêm 2 ml dung dịch đệm acetat pH 3,5 (TT). Lắc đều và thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT). Lắc đều ngay. Pha loãng với nước thành 50 ml, lắc đều và để yên 2 min.
Lọc các dung dịch qua màng lọc thích hợp (Kích thước lỗ 0,45 µm). Tiến hành lọc chậm và đều với áp lực lên piston nhẹ nhàng và liên tục. So sánh màu sắc của các vết trên màng lọc thu được từ các dung dịch trên.
Tính thích hợp của phép thử: Vết thu được từ dung dịch đối chiếu phải có màu nâu khi so sánh với vết thu được từ dung dịch mẫu trắng và vết thu được từ dung dịch kiểm tra phải có màu ít nhất là đậm bằng màu của vết thu được tử dung dịch đối chiếu.
Đánh giá kết quả: Màu nâu của vết thu được từ dung dịch thử không được đậm hơn màu của vết thu được từ dung dịch đối chiếu.
Phương pháp 8
Dung dịch thử: Hòa tan một lượng chế phẩm theo chỉ dẫn trong chuyên luận trong 20 ml dung môi hay hỗn hợp dung môi được chỉ dẫn trong chuyên luận.
Dung dịch đối chiếu: Pha loãng thể tích dung dịch chì mẫu 10 phần triệu Pb (TT) theo chỉ dẫn trong chuyên luận thành 20 ml với dung môi để pha dung dịch thử.
Dung dịch mẫu trắng: 20 ml dung môi để pha dung dịch thử.
Thêm 2 ml dung dịch đệm acetat pH 3,5 (TT) vào mỗi dung dịch trên. Lắc đều (Trong một vài trường hợp có tủa tạo thành, khi đó chuyên luận riêng có hướng dẫn hòa tan lại trong thể tích xác định của dung môi cho sẵn). Thêm dung dịch thu được vào 1,2 ml dung dịch thioacetamid (TT). Lắc ngay và để yên 2 min. Lọc các dung dịch qua màng lọc thích hợp (Kích thước lỗ 0,45 μm). So sánh các vết trên màng lọc thu được từ các dung dịch trên.
Tính thích hợp của phép thử: Vết thu được từ dung dịch đối chiếu phải có màu đen nâu so với vết thu được từ dung dịch mẫu trắng.
Đánh giá kết quả: Màu đen nâu của vết thu được từ dung dịch thử không được đậm hơn màu của vết thu được từ dung dịch đối chiếu.
9.4.9 Nhôm
Chuyển dung dịch chế phẩm thử đã chỉ dẫn trong chuyên luận vào một bình gạn và chiết lần lượt với 20 ml, 20 ml và 10 ml dung dịch 8-hydroxyquinolin 0,5 % trong cloroform (TT). Gộp các dịch chiết cloro- form và pha loãng tới 50,0 ml bằng cloroform (TT) (dung dịch thử).
Trừ khi có chỉ dẫn khác, chuẩn bị dung dịch trắng và dung dịch chuẩn trong cùng điều kiện như dung dịch thử.
Dung dịch trắng là hỗn hợp 10 ml dung dịch đệm acetat pH 6,0 và 100 ml nước.
Dung dịch chuẩn là hỗn hợp 2 ml dung dịch nhôm mẫu 2 phần triệu AI (TT), 10 ml dung dịch đệm acetat pH 6,0 và 98 ml nước.
Đo huỳnh quang của dung dịch thử (l1), dung dịch chuẩn (l2) và dung dịch trắng (l3) (Phụ lục 4.3) với bước sóng kích thích là 392 nm và một kính lọc phụ có dài truyền quang tập trung ở 518 nm, hoặc đặt một thiết bị làm đơn sắc ánh sáng để truyền quang ở chính bước sóng đó.
Cường độ huỳnh quang của dung dịch thử (I1 -I3) không được lớn hơn của dung dịch chuẩn (l2-I3).
9.4.10 Nickel trong polyol
Tiến hành theo phương pháp II của quang phổ hấp thụ nguyên tử (Phụ lục 4.4), dùng ngọn lửa acetylen - không khí, đèn cathod rỗng nickel và các dung dịch sau:
Dung dịch thử: Hòa tan 20,0 g chế phẩm thử trong dung dịch acid acetic 1 M (TT) và pha loãng thành 100,0 ml với cùng dung môi. Thêm 2,0 ml dung dịch bão hòa amoni pyrolidin dithiocarbamat (TT) (khoảng 1 %) và 10,0 ml 4-methylpentan-2-on (TT) lắc trong 30 s, tránh ánh sáng. Để yên cho tách lớp và lấy lớp methylpentanon.
Dung dịch đối chiếu: Chuẩn bị 3 dung dịch đối chiếu trong cùng điều kiện như dung dịch thử bằng cách thêm riêng biệt 0,5 ml; 1,0 ml; 1,5 ml dung dịch nickel mẫu 10 phần triệu Ni (TT) vào mỗi bình đã có 20,0 g chế phẩm thử.
Chuẩn bị một mẫu trắng trong cùng điều kiện giống như dung dịch thử, nhưng không có chế phẩm thử và dùng dung dịch này để hiệu chỉnh điểm “0” của máy.
Đo độ hấp thụ của dung dịch thử, 3 dung dịch đối chiếu và mẫu trắng ở cực đại 232,0 nm. Vẽ đường cong biểu diễn sự phụ thuộc giữa độ hấp thụ với nồng độ nickel trong các dung dịch và xác định hàm lượng của nickel trong chế phẩm thử.
Hàm lượng nickel không lớn hơn 1 phần triệu, trừ khi có chỉ dẫn khác.
9.4.11 Kim loại nặng trong dược liệu và trong dầu béo
Tiến hành bằng phương pháp quang phổ hấp thụ nguyên tử (Phụ lục 4.4).
Chú ý: Khi dùng thiết bị phá mẫu áp suất cao và lò vi sóng dùng trong phòng thí nghiệm, phải thành thạo các thao tác an toàn và vận hành thiết bị mà nhà sản xuất đưa ra.
Thiết bị
Bao gồm các bộ phận sau:
Bình phá mẫu bằng polytetrafluoroethylen thể tích 120 ml có nắp đậy kín, có van điều chỉnh áp suất bên trong và một ống bằng polytetrafluoroethylen để xả khí.
Một hệ thống giữ các bình phá mẫu kín với cùng một lực xoắn.
Lò vi sóng với tần số 2450 MHz có công suất điều chỉnh được từ 0 đến (630 ± 70) W, nối với một máy tính có phần mềm điều khiển chương trình. Vách lò phủ một lớp polytetrafluoroethylen. Lò có quạt hút thay đổi được tốc độ, có hệ thống đĩa quay và ống hút để xả khói.
Máy quang phổ hấp thụ nguyên tử dùng đèn cathod rỗng là nguồn phát xạ và đèn deuterium hiệu chỉnh đường nền. Máy được nối với:
a) Bộ hóa hơi nguyên tử không ngọn lửa lò graphit đối với cadimi, đồng, chì, sắt, nickel và kẽm.
b) Bộ hóa hơi hydrid đối với arsen và bộ phận hóa hơi lạnh cho thủy ngân, hoặc một bộ hóa hơi khác có tính năng phù hợp cho 2 nguyên tố này.
Tiến hành
Nếu dùng trang thiết bị khác với mô tả ở trên thì cần điều chỉnh các thông số thiết bị cho phù hợp.
Rửa sạch các đồ đựng bằng thủy tinh và dụng cụ thí nghiệm bằng dung dịch acid nitric (TT) 10 g/l trước khi dùng.
Dung dịch thử: Cân và chuyển vào bình phá mẫu một lượng chế phẩm theo chỉ dẫn trong chuyên luận (khoảng 0,5 g bột dược liệu hoặc dầu béo). Thêm 6 ml acid nitric không có kim loại nặng (TT) và 4 ml acid hydrocloric không có kim loại nặng (TT). Đậy kín bình.
Đặt bình phá mẫu vào lò vi sóng. Tiến hành phá mẫu theo 3 bước như sau (dùng cho 7 bình chứa mẫu thử): 80 % công suất ở 15 min đầu, 100 % công suất ở 5 min tiếp theo và 80 % ở 20 min cuối.
Để nguội các bình ngoài không khí, thêm vào mỗi bình 4 ml acid sulfuric không có kim loại nặng (TT). Lặp lại các bước phá mẫu như trên một lần nữa. Sau khi để nguội, mở bình phá mẫu ra, thu được dung dịch trong và không màu. Chuyển toàn bộ dung dịch vào bình định mức 50 ml, tráng bình phá mẫu 2 lần, mỗi lần với 15 ml nước và tập trung dịch tráng vào bình định mức trên. Thêm 1,0 ml dung dịch magnesi nitrat (TT) 1 % và 1,0 ml dung dịch amoni dihydrophosphat (TT) 10 %, thêm nước vừa đủ 50 ml.
Dung dịch mẫu trắng: Trộn 6 ml acid nitric không có kim loại nặng (TT) và 4 ml acid hydrocloric không có kim loại nặng (TT) vào bình phá mẫu và tiến hành phá mẫu như đối với dung dịch thử.
Arsen
Dung dịch mẫu: Lấy 19,0 ml dung dịch thử hoặc trắng chuẩn bị như trên, thêm 1 ml dung dịch kali iodid (TT) 20 %. Để yên dung dịch ở nhiệt độ phòng trong 50 min hoặc ở 70 °C trong 4 min.
Acid: Acid hydrocloric không có kim loại nặng (TT).
Dung dịch khử: Dung dịch natri borohydrid (TT) 0,6 % trong dung dịch natri hydroxyd (TT) 0,5 %.
Áp dụng các thông số kỹ thuật trong Bảng 9.4.11.2.
Bảng 9.4.11.1 - Các thông số kỹ thuật để tiến hành phép thử
|
|
| Cd | Cu | Fe | Ni | Pb | Zn |
| Bước sóng | nm | 228,8 | 324,8 | 248,3 | 232 | 283,5 | 213,9 |
| Độ rộng khe | nm | 0,5 | 0,5 | 0,2 | 0,2 | 0,5 | 0,5 |
| Cường độ đèn | mA | 6 | 7 | 5 | 10 | 5 | 7 |
| Nhiệt độ tro hóa | °C | 800 | 800 | 800 | 800 | 800 | 800 |
| Nhiệt độ nguyên tử hóa | °C | 1800 | 2300 | 2300 | 2500 | 2200 | 2000 |
| Hiệu chỉnh đường nền |
| có | không | không | không | không | không |
| Tốc độ khí nitrogen | l/min | 3 | 3 | 3 | 3 | 3 | 3 |
Bảng 9.4.11.2 - Các thông số kỹ thuật để tiến hành phép thử
|
|
| As | Hg |
| Bước sóng | nm | 193,7 | 253,7 |
| Độ rộng khe | nm | 0,2 | 0,5 |
| Cường độ đèn | mA | 10 | 4 |
| Tốc độ acid | ml/min | 1,0 | 1,0 |
| Tốc độ dung dịch khử | ml/min | 1,0 | 1,0 |
| Tốc độ dung dịch mẫu | ml/min | 7,0 | 7,0 |
| Buồng đo |
| Thạch anh (làm nóng) | Thạch anh (không làm nóng) |
| Hiệu chỉnh đường nền |
| Không | Không |
| Tốc độ khí nitrogen | l/min | 0,1 | 0,1 |
Định lượng cadimi, đồng, sắt, chì, nickel và kẽm
Xác định hàm lượng cadimi, đồng, sắt, chì, nickel, kẽm bằng phương pháp thêm chuẩn (Phụ lục 4.4), dùng các dung dịch đối chiếu của mỗi kim loại nặng và áp dụng các thông số kỹ thuật như Bảng 9.4.11.1. Độ hấp thụ của dung dịch mẫu trắng được tự động trừ vào độ hấp thụ của dung dịch thử.
Định lượng arsen và thủy ngân
Xác định hàm lượng arsen và thủy ngân trong mẫu dựa vào các dung dịch chuẩn arsen hoặc thủy ngân đã biết nồng độ bằng phương pháp xác định trực tiếp (Phụ lục 4.4) với hệ thống hóa hơi hydrid đối với arsen và hóa hơi lạnh đối với thủy ngân, hoặc với một thiết bị hóa hơi khác phù hợp.
Thủy ngân
Dung dịch mẫu: Dung dịch thử hoặc dung dịch trắng chuẩn bị như trên.
Acid: Dung dịch chứa 515 g/l acid hydrocloric không có kim loại nặng (TT).
Dung dịch khử: Dung dịch thiếc (II) clorid 1 % trong dung dịch acid hydrocloric loãng không có kim loại nặng (TT).
Áp dụng các thông số kỹ thuật trong Bảng 9.4.11.2.
9.4.12. Phosphat
Thêm 4 ml dung dịch sufomolybdic (TT) vào 100 ml dung dịch đã được chuẩn bị, nếu cần thì trung hòa như chỉ dẫn. Lắc và thêm 0,1 ml dung dịch thiếc (II) clorid (TT1). Chuẩn bị dung dịch chuẩn trong cùng điều kiện, dùng 2 ml dung dịch phosphat mẫu 5 phần triệu PO4 (TT) và 98 ml nước. Sau 10 min, lấy mỗi dung dịch 20 ml và so sánh màu. Màu trong ống thử phải không được đậm hơn màu trong ống chuẩn.
9.4.13 Sắt
Hòa tan một lượng chế phẩm thử quy định trong nước rồi pha loãng với nước thành 10 ml, hoặc lấy 10 ml dung dịch chế phẩm thử như chỉ dẫn trong chuyên luận cho vào một ống Nessler. Thêm 2 ml dung dịch acid citric 20 % và 0,1 ml acid mercaptoacetic (TT). Lắc đều, kiềm hóa bằng dung dịch amoniac 10 M (TT) và pha với nước thành 20 ml. Để yên 5 min.
Chuẩn bị một dung dịch chuẩn trong cùng điều kiện, dùng 10 ml dung dịch sắt mẫu 1 phần triệu Fe (TT) thay cho dung dịch chế phẩm thử.
Màu hồng tạo thành trong dung dịch thử không được đậm hơn màu trong dung dịch chuẩn.
9.4.14 Sulfat
Các dung dịch dùng trong phép thử này phải được chuẩn bị trong nước cất.
Thêm 1 ml dung dịch bari clorid 25 % vào 1,5 ml dung dịch sulfat mẫu 10 phần triệu SO4, lắc và để yên 1 min. Thêm 15 ml dung dịch chế phẩm thử đã được chỉ dẫn trong chuyên luận hoặc thêm một lượng chế phẩm thử quy định đã hòa tan trong 15 ml nước, và 0,5 ml dung dịch acid acetic 5 M (TT). Để yên 5 min.
Độ đục tạo thành trong ống thử không được đậm hơn trong ống chuẩn được chuẩn bị đồng thời trong cùng điều kiện, nhưng dùng 15 ml dung dịch sulfat mẫu 10 phần triệu SO4 (TT) thay cho dung dịch chế phẩm thử.
9.4.15 Magnesi
Lấy 10 ml dung dịch thử được pha như chỉ dẫn của chuyên luận, thêm 0,1 g natri tetraborat (TT). Nếu cần thì điều chỉnh pH của dung dịch tới khoảng từ 8,8 đến 9,2 bằng dung dịch acid hydrocloric 2 M hoặc dung dịch natri hydroxyd 2 M. Lắc 2 lần, mỗi lần với 5 ml dung dịch 8-hydroxyquinolin 0,1 % trong cloroform (TT), mỗi lần lắc 1 min, để yên cho tách lớp rồi gạn bỏ lớp cloroform phía dưới. Thêm vào lớp nước 0,4 ml n-butylamin (TT) và 0,1 ml triethanolamin (TT). Nếu cần thì điều chỉnh pH tới khoảng từ 10,5 đến 11,5. Thêm 4 ml dung dịch 8-hydroxyquinolin 0,1 % trong cloroform (TT), lắc 1 min rồi để yên cho tách lớp. Nếu lớp cloroform có màu thì không được đậm hơn màu mẫu thu được khi tiến hành như trên với 1 ml dung dịch magnesi mẫu 10 phần triệu Mg (TT) và 9 ml nước.
9.4.16 Magnesi và kim loại kiềm thổ
Lấy 200 ml nước, thêm 0,1 g hydroxylamin hydroclorid (TT); 10 ml dung dịch đệm amoniac pH 10,0 (TT); 1 ml dung dịch kẽm sulfat 0,1 M (CĐ) và khoảng 15 mg hỗn hợp đen eriocrom T (TT). Đun nóng tới 40 °C và chuẩn độ với dung dịch trilon B 0,01 M (CĐ) đến khi màu tím chuyển hẳn sang xanh. Thêm vào dung dịch một lượng chế phẩm đã được hòa tan trong 100 ml nước, hoặc một thể tích dung dịch chế phẩm, như chỉ dẫn trong chuyên luận. Nếu màu dung dịch chuyển sang tím, thì chuẩn độ tiếp với dung dịch trilon B 0,01 M (CĐ) đến khi màu hoàn toàn trở lại xanh. Thể tích dung dịch trilon B 0,01 M (CĐ) dùng trong lần chuẩn độ thứ hai không được quá lượng quy định.
9.5 XÁC ĐỊNH GIỚI HẠN CARBON MONOXYD TRONG KHÍ Y TẾ
Thiết bị
Thiết bị (Hình 9.5) gồm các phần sau được mắc nối tiếp với nhau:
U1: Ống hình chữ U chứa silica gel khan được tẩm crom (VI) oxyd (TT).
F1: Bình rửa chứa 100 ml dung dịch kali hydroxyd 40 % (TT).
U2: Ống hình chữ U chứa các hạt kali hydroxyd (TT) (ống này không cần khi thử carbon dioxyd).
U3: Ống hình chữ U chứa phosphor pentoxyd được phân tán trên đá bọt.
U4: Ống hình chữ U chứa 30 g các hạt iod pentoxyd kết tinh lại đã được sấy trước ở 200 °C và duy trì ở 120 °C trọng suốt quá trình thử. lod pentoxyd được nhồi trong một ống thành các cột 1 cm được phân cách bằng các cột bông thủy tinh 1 cm để được một đoạn hữu hiệu dài 5 cm.
F2: Ống phản ứng chứa 2,0 ml dung dịch kali iodid 16,6% (TT) và 0,15 ml dung dịch hồ tinh bột (TT).
Tiến hành
Rửa thiết bị bằng 5,0 L khí argon và nếu cần, làm mất màu của dung dịch iodid bằng cách thêm lượng ít nhất dung dịch natri thiosulfat 0,002 N (CĐ) mới pha. Tiếp tục rửa đến khi lượng dung dịch natri thiosulfat 0,002 N (CĐ) cần dùng không quá 0,045 ml sau khi cho 5,0 L khí argon chạy qua.
Cho khí cần kiểm tra chạy qua thiết bị, dùng thể tích và tốc độ dòng theo quy định trong chuyên luận riêng. Rửa vết iod giải phóng vào bình phản ứng bằng cách cho 1,0 L khí argon chạy qua. Chuẩn độ iod giải phóng bằng dung dịch natri thiosulfat 0,002 N (CĐ).
Tiến hành mẫu trắng trong cùng điều kiện, dùng thể tích argon theo quy định trong, chuyên luận riêng. Thể tích dung dịch natri thiosulfat 0,002 N (CĐ) chênh lệch giữa hai lần chuẩn độ không được quá giới hạn quy định trong chuyên luận riêng.
Hình 9.5 - Thiết bị thử giới hạn carbon monoxyd
(Kích thước tính bằng mm)
9.6 XÁC ĐỊNH MẤT KHỐI LƯỢNG DO LÀM KHÔ
Mất khối lượng do làm khô là sự giảm khối lượng của mẫu thử biểu thị bằng phần trăm (khối lượng/khối lượng) khi được làm khô trong điều kiện xác định ở mỗi chuyên luận. Phương pháp này dùng để xác định hàm lượng nước, một phần hoặc toàn bộ lượng nước kết tinh và lượng chất dễ bay hơi khác trong mẫu thử.
Việc xác định mất khối lượng do làm khô không được làm thay đổi tính chất lý hóa cơ bản của mẫu thử, vì vậy mỗi chuyên luận riêng sẽ có quy định cách làm khô theo một trong các phương pháp sau đây:
a) Trong bình hút ẩm. Tiến hành làm khô trong bình hút ẩm với những chất hút nước như phosphor pentoxyd, silica gel v.v...
b) Trong chân không. Tiến hành làm khô ở điều kiện áp suất từ 1,5 kPa đến 2,5 kPa có mặt chất hút ẩm phosphor pentoxyd và ở nhiệt độ phòng.
c) Trong chân không ở điều kiện nhiệt độ xác định. Tiến hành làm khô ở điều kiện áp suất từ 1,5 kPa đến 2,5 kPa có mặt chất hút ẩm phosphor pentoxyd và trong điều kiện nhiệt độ quy định trong chuyên luận riêng.
d) Trong tủ sấy ở điều kiện nhiệt độ xác định. Tiến hành làm khô trong tủ sấy ở điều kiện nhiệt độ quy định trong chuyên luận riêng.
e) Trong chân không hoàn toàn. Tiến hành làm khô trong điều kiện áp suất không quá 0,1 kPa có mặt chất hút ẩm phosphor pentoxyd và ở điều kiện nhiệt độ quy định trong chuyên luận riêng.
Cách tiến hành
Dùng dụng cụ dùng để sấy bằng thủy tinh rộng miệng đáy bằng có nắp mài làm bì đựng mẫu thử; làm khô bì trong thời gian 30 min theo phương pháp và điều kiện quy định trong chuyên luận rồi cân để xác định khối lượng bì. Cân ngay vào bì này một lượng chính xác mẫu thử bằng khối lượng quy định trong chuyên luận với sai số ± 10 %. Nếu không có chỉ dẫn gì đặc biệt thì lượng mẫu thử được dàn mỏng thành lớp có độ dày không quá 5 mm. Nếu mẫu thử có kích thước lớn thì phải nghiền nhanh tới kích thước dưới 2 mm trước khi cân. Tiến hành làm khô trong điều kiện quy định của chuyên luận. Nếu dùng phương pháp sấy thì nhiệt độ thực cho phép chênh lệch ± 2 °C so với nhiệt độ quy định. Sau khi sấy phải làm nguội tới nhiệt độ phòng cân trong bình hút ẩm có silica gel rồi cân ngay. Nếu chuyên luận không quy định thời gian làm khô có nghĩa là phải làm khô đến khối lượng không đổi, tức là sự chênh lệch khối lượng sau khi sấy thêm 1 h trong tù sấy hoặc 6 h trong bình hút ẩm so với lần sấy trước đó không quá 0,5 mg.
Nếu mẫu thử bị chảy ở nhiệt độ thấp hơn nhiệt độ sấy quy định thì trước khi đưa lên nhiệt độ đó, cần duy trì từ 1 đến 2 h ở nhiệt độ thấp hơn nhiệt độ nóng chảy của mẫu thử từ 5 °C đến 10 °C.
Nếu mẫu thử ở dạng nang hoặc viên bao thì phải bỏ vỏ (lấy không ít hơn 4 viên) và nghiền nhanh tới kích thước dưới 2 mm rồi lấy lượng bột viên như chỉ dẫn trong chuyên luận riêng.
Nếu mẫu thử là dược liệu, khi chuyên luận riêng không có chỉ dẫn gì đặc biệt thi tiến hành sấy trong tủ sấy ở áp suất thường. Dược liệu phải được làm thành mảnh nhỏ đường kính không quá 3 mm; lượng đem thử từ 2 g đến 5 g; chiều dày lớp mẫu thử đem sấy là 5 mm và không quá 10 mm đối với dược liệu có cấu tạo xốp. Nhiệt độ và thời gian sấy theo yêu cầu của chuyên luận riêng.
9.7 XÁC ĐỊNH TRO KHÔNG TAN TRONG ACID
Nếu không có hướng dẫn khác trong chuyên luận thì dùng phương pháp 1.
Phương pháp 1
Cho 25 ml dung dịch acid hydrocloric 2 M (TT) vào tro toàn phần, đun sôi 5 min, lọc để tập trung những chất không tan vào một phễu thủy tinh xốp đã cân bì, hoặc vào một giấy lọc không tro, rửa bằng nước nóng rồi đem nung ở 500 °C đến khối lượng không đổi. Tính tỷ lệ phần trăm của tro không tan trong acid so với dược liệu đã làm khô trong không khí.
Phương pháp 2
Cho vào chén nung chứa tro toàn phần hay tro sulfat (nếu trong chuyên luận riêng không có chỉ dẫn khác) 15 ml nước và 10 ml acid hydrocloric (TT). Đậy chén bằng một mặt kính đồng hồ, đun sôi cẩn thận 10 min rồi để nguội. Rửa mặt kính đồng hồ với 5 ml nước nóng rồi cho vào chén nung. Tập trung chất không tan vào một phễu lọc thủy tinh xốp đã cân bì hoặc vào một giấy lọc không tro, rửa bằng nước nóng tới khi dịch lọc cho phản ứng trung tính. Làm khô rồi nung tới đỏ tối, để nguội trong bình hút ẩm rồi cân. Nung tiếp tới khi giữa 2 lần cân khối lượng chênh lệch nhau không vượt quá 1 mg. Tính tỷ lệ phần trăm của tro không tan trong acid so với dược liệu đã được làm khô trong không khí.
9.8 XÁC ĐỊNH TRO TOÀN PHẦN
Nếu không có hướng dẫn khác trong chuyên luận thì áp dụng phương pháp 1.
Phương pháp 1
Với mẫu thử là dược liệu, thuốc từ dược liệu: Cho 2 g đến 3 g bột mẫu thử vào một chén sứ hoặc chén platin đã nung và cân bì. Nung ở nhiệt độ không quá 450 °C tới khi không còn carbon, làm nguội rồi cân. Bằng cách này mà tro chưa loại được hết carbon thì dùng một ít nước nóng cho vào khối chất đã than hóa, dùng đũa thủy tinh khuấy đều, lọc qua giấy lọc không tro. Rửa đũa thủy tinh và giấy lọc, tập trung nước rửa vào dịch lọc. Cho giấy lọc và cắn vào chén nung rồi nung đến khi thu được tro màu trắng hoặc gần như trắng. Tập trung dịch lọc vào cắn trong chén nung, đem bốc hơi đến khô rồi nung ở nhiệt độ không quá 450 °C đến khi khối lượng không đổi. Tính tỷ lệ phần trăm của tro toàn phần theo dược liệu đã làm khô trong không khí.
Với các mẫu thử khác: Cũng thực hiện như trên nhưng chỉ dùng 1 g mẫu thử nếu không có chỉ dẫn gì khác trong chuyên luận.
Phương pháp 2
Lấy một chén sứ hoặc chén platin nung tới đỏ trong 30 min. Để nguội trong bình hút ẩm rồi cân. Nếu trong chuyên luận riêng không có hướng dẫn gì khác thì lấy 1 g mẫu thử rải đều vào chén nung, sấy 1 h ở 100 °C đến 105 °C rồi đem nung trong lò nung ở 600 °C ± 25 °C. Sau mỗi lần nung, lấy chén nung cùng cắn tro đem làm nguội trong bình hút ẩm rồi cân. Trong quá trình thao tác không được để tạo thành ngọn lửa. Nếu sau khi đã nung lâu mà vẫn chưa loại hết carbon của tro thì dùng nước nóng để lấy cắn rã, lọc qua giấy lọc không tro rồi lại nung cắn và giấy lọc trong chén nung. Tập trung dịch lọc vào tro ở trong chén, làm bốc hơi cẩn thận tới khô rồi nung đến khối lượng không đổi.
9.9 XÁC ĐỊNH TRO SULFAT
Áp dụng một trong các phương pháp sau đây nếu trong chuyên luận riêng không có hướng dẫn khác.
Phương pháp 1
Nung một chén sứ hoặc chén platin tới đỏ trong 10 min, để nguội trong bình hút ẩm rồi cân. Nếu không có chỉ dẫn gì đặc biệt trong chuyên luận riêng thì cho 1 g mẫu thử vào chén nung, làm ẩm với acid sulfuric (TT), đốt cẩn thận rồi lại làm ẩm với acid sulfuric (TT) và nung ở khoảng 800 °C. Làm nguội rồi cân. Nung lại 15 min, làm nguội rồi cân nhắc lại. Lặp lại quá trình này cho đến khi hai lần cân liên tiếp, khối lượng không chênh lệch nhau quá 0,5 mg.
Phương pháp 2
Nung một chén nung sứ hoặc platin ở 600 °C ± 50 °C trong 30 min, để nguội trong bình hút ẩm rồi cân. Cho vào chén nung một lượng mẫu thử như chỉ dẫn trong chuyên luận và cân. Làm ẩm mẫu bằng một lượng nhỏ acid sufuric (TT) (khoảng 1 ml), đốt nóng ở mức độ nhẹ nhất có thể đến khi mẫu hóa tro hoàn toàn. Để nguội, làm ẩm cắn bằng một lượng nhỏ acid sufuric (TT), đốt nóng nhẹ đến khi bay hết khói trắng và nung ở 600 °C ± 50 °C đến khi cắn thành tro hoàn toàn. Trong khi đốt và nung không được để tạo thành ngọn lửa. Để nguội trong bình hút ẩm, cân và tính khối lượng của cắn. Nếu khối lượng cắn vượt ngoài giới hạn cho phép thì lại làm ẩm cắn bằng acid sufuric (TT) và nung như trên đến khối lượng không đổi nếu không có chỉ dẫn gì khác.
Lượng mẫu thử thường dùng (từ 1 g đến 2 g) được tính từ giới hạn tro sulfat đã qui định sao cho khối lượng tro sulfat (khoảng 1 mg) có thể cân được để đảm bảo độ chính xác.
9.10 XÁC ĐỊNH TRO TAN TRONG NƯỚC
Đun sôi tro toàn phần (Phụ lục 9.8) với 25 ml nước trong 5 min. Tập trung những chất không tan vào một phễu thủy tinh xốp, hoặc vào trong một chén lọc thủy tinh xốp, hoặc trên một giấy lọc không tro đã cân bì trước, rửa bằng nước nóng rồi nung 15 min ở nhiệt độ không quá 450 °C. Để nguội rồi cân để xác định khối lượng cần không tan trong nước. Lấy khối lượng tro toàn phần trừ đi khối lượng cắn không tan trong nước, được khối lượng tro tan trong nước.
Tính tỷ lệ phần trăm tro tan trong nước so với dược liệu đã làm khô trong không khí.
PHỤ LỤC 10
(Quy định)
10.1 PHƯƠNG PHÁP CHUẨN ĐỘ ĐO AMPE
Trong chuẩn độ đo ampe, điểm kết thúc được xác định bằng cách quan sát sự biến đổi của cường độ dòng điện đo giữa hai điện cực (một điện cực chỉ thị và một điện cực so sánh hoặc hai điện cực chỉ thị) nhúng trong dung dịch khảo sát và duy trì một hiệu điện thế không đổi. Cường độ dòng điện này là một hàm số mà biến số là lượng dung dịch chuẩn độ thêm vào.
Thế của điện cực đo cần phải đủ để đảm bảo dòng khuếch tán đối với chất điện hoạt.
Máy
Máy gồm có một nguồn cung cấp điện thế điều chỉnh được và một đồng hồ đo microampe nhạy. Hệ thống để phát hiện nói chung gồm một điện cực chỉ thị (ví dụ: điện cực platin, điện cực giọt thủy ngân, điện cực có đĩa quay hoặc điện cực than) ghép đôi với một điện cực so sánh như điện cực calomel hoặc điện cực bạc - bạc clorid.
Có thể dùng máy có 3 điện cực, máy này ngoài điện cực chỉ thị và điện cực so sánh còn có điện cực bổ trợ phân cực.
Cách tiến hành
Chỉnh thế của điện cực chỉ thị theo các chỉ dẫn trong chuyên luận riêng. Lập đồ thị biểu diễn cường độ dòng ban đầu và các giá trị cường độ dòng khác thu được trong khi chuẩn độ, dưới dạng một hàm số với biến số là lượng dung dịch chuẩn độ thêm vào. Khi tổng lượng dung dịch chuẩn độ thêm vào đạt xấp xỉ 80 % thể tích lý thuyết dung dịch chuẩn độ cần dùng để đạt điểm tương đương, thêm dung dịch chuẩn độ không ít hơn 3 lần liên tiếp để tiến dần tới điểm tương đương. Các giá trị thu được này cần phải ở trên một đường thẳng. Tiếp tục thêm dung dịch chuẩn độ đến quá điểm tương đương và lại thêm không ít hơn ba lần liên tiếp dung dịch chuẩn độ nữa, các giá trị thu được lần này phải ở trên một đường thẳng khác. Điểm kết thúc của phép chuẩn độ là giao điểm của hai đường thẳng nói trên.
Trong trường hợp chuẩn độ đo ampe bằng hai điện cực chỉ thị nên ghi lại toàn bộ đường cong chuẩn độ và dùng đồ thị này để xác định điểm tương đương.
|
|
|
| A: Nguồn cung cấp dòng có thể không đổi B: Micro -ampe kế C: Điện thế kế D: Dung dịch chuẩn độ E: Điện cực bổ trợ F: Điện cực chỉ thị G: Điện cực so sánh H: Khuấy từ |
Hình 10.1 - Sơ đồ chuẩn độ đo Ampe
10.2 PHƯƠNG PHÁP CHUẨN ĐỘ ĐO ĐIỆN THẾ
Trong chuẩn độ đo điện thế, điểm kết thúc của chuẩn độ được xác định bằng cách quan sát sự biến đổi hiệu số điện thế đo được giữa hai điện cực (một điện cực chỉ thị và một điện cực so sánh hoặc hai điện cực chỉ thị) nhúng trong dung dịch khảo sát. Sự biến đổi này như là một hàm số mà biến số là lượng dung dịch chuẩn độ thêm vào. Điện thế thường được đo ở cường độ dòng điện bằng không hoặc thực tế bằng không.
Máy
Máy được sử dụng (máy đo thế đơn hoặc máy có tổ hợp mạch điện tử) gồm có một von kế cho phép đọc được tới 1 mV.
Tùy theo bản chất của hợp chất cần định lượng mà chọn điện cực chỉ thị, có thể là điện cực thủy tinh hoặc điện cực kim loại (như platin, vàng, bạc, thủy ngân....). Điện cực so sánh thường là điện cực calomel hoặc điện cực bạc - bạc clorid.
Trong chuẩn độ acid kiềm, trừ trường hợp có những chỉ dẫn khác của chuyên luận riêng, người ta thường dùng điện cực kép thủy tinh - calomel hoặc thủy tinh - bạc - bạc clorid.
Cách tiến hành
Lập đồ thị biểu diễn những biến đổi về điện thế tương ứng với lượng dung dịch chuẩn độ thêm vào cho đến và vượt điểm tương đương giả định. Điểm kết thúc của chuẩn độ tương ứng với điểm mà ở đó có sự thay đổi đột biến về điện thế.
10.3 ĐỊNH LƯỢNG NƯỚC
Nếu không có chỉ dẫn khác trong chuyên luận riêng, áp dụng Phương pháp 1 và định lượng trực tiếp.
Phương pháp 1
Nguyên tắc
Phương pháp định lượng nước này dựa trên phản ứng toàn lượng của nước với lưu huỳnh dioxyd và iod trong dung môi khan chứa một chất base hữu cơ thích hợp.
Dung môi hữu cơ thông dụng là methanol khan nước, cũng có khi được thay bằng dung môi hữu cơ khác thích hợp để hòa tan chế phẩm. Chất base hữu cơ là pyridin, nhưng hiện nay đã dùng những chất base hữu cơ khác để thay thế như imidazol, 2-methylaminopyridin.
Thiết bị
Hiện nay có nhiều loại dụng cụ, nhưng nguyên tắc đều phải cấu tạo sao cho thao tác thuận tiện và tránh ẩm. Dụng cụ gồm có một cốc chuẩn độ dung tích khoảng 60 ml, có nắp gắn điện cực kép platin, một ống dẫn khí nitrogen, có lỗ cắm với buret và lỗ cắm ống thông hơi chứa chất hút ẩm. Chế phẩm được đưa vào bình chuẩn độ qua lỗ trên nắp hoặc miệng bên cạnh có nút mài. Trong quá trình chuẩn độ, khuấy bằng máy khuấy từ hoặc bằng luồng khí nitrogen khô đi qua dung dịch. Điểm kết thúc phản ứng được xác định bằng điện kế gắn trong mạch có biến trở 2000 Ω, nối với một nguồn pin 1,5 V. Lúc bắt đầu kim điện kế chỉ điểm không, vì dòng điện chạy qua 2 điện cực platin không đáng kể. Khi nhỏ thuốc thử Karl Fischer vào dung dịch, do hiện tượng khử cực nên kim điện kế lệch đi nhưng lập tức trở về vị trí ban đầu, chỉ khi đến điểm kết thúc thì một giọt thuốc thử thừa sẽ làm kim lệch đi và duy trì ít nhất 30 s.
Thuốc thử
Thuốc thử Karl Fischer gốc gồm 4 thành phần chính là lưu huỳnh dioxyd, iod, pyridin hoặc một base hữu cơ khác và methanol pha thành một dung dịch, hoặc hai dung dịch. Trường hợp pha thành hai dung dịch thì dung dịch A chứa lưu huỳnh dioxyd và pyridin pha trong methanol khan, dung dịch B chứa iod pha trong methanol khan. Trước khi dùng 1 h, trộn đều 1 thể tích dung dịch A với 1 thể tích dung dịch B, sau đó xác định đương lượng nước của thuốc thử. Lượng thuốc thử thu được chỉ dùng trong ngày. Hiện nay trên thị trường vừa có các loại thuốc thử như trên, vừa có loại đã thay pyridin và methanol bằng chất kiềm hữu cơ khác và dung môi hữu cơ khác. Do vậy, cần kiểm tra kỹ thành phần thuốc thử và cách sử dụng cho đúng mục đích. Các thuốc thử Karl Fischer và dung môi dùng trong phương pháp đều phải khan nước, bao quản trong lọ màu, tránh ánh sáng, chống ẩm và phải xác định lại đương lượng nước trước khi dùng.
Xác định đương lượng nước của thuốc thử
Đương lượng nước của thuốc thử Karl Fischer dễ bị thay đổi theo thời gian nên trước khi dùng phải xác định lại và phải đạt tối thiểu 3,5 mg nước cho 1 ml thuốc thử. Có thể xác định theo 2 cách:
a. Áp dụng xác định hàm lượng nước nhỏ hơn 1 %
Dùng một hóa chất có hàm lượng nước kết tinh xác định, sau khi đã sấy ở nhiệt độ quy định đến khối lượng không đổi để loại hết ẩm, cho tác dụng với thuốc thử rồi tính ra đương lượng. Thường dùng natri tartrat dihydrat (C4H4Na2O6.2H2O).
Cách tiến hành:
Cho một lượng methanol khan (TT) hoặc dung môi thích hợp dùng cho thuốc thử Karl Fischer vào cốc chuẩn độ đủ ngập điện cực platin rồi chuẩn độ bằng thuốc thử Karl Fischer đến điểm dừng. Cho nhanh khoảng từ 250 mg đến 350 mg natri tartrat dihydrat đã cân chính xác vào cốc và chuẩn độ bằng thuốc thử Karl Fischer đến điểm kết thúc, tính hệ số đương lượng nước F (tính bằng mg nước/ml thuốc thử) của thuốc thử theo công thức:
F = 2 x (18,02/230,08) X (W/V)
trong đó:
| 18,02 và 230,08: | khối lượng phân tử của nước và của natri tartrat dihydrat; |
| W: | khối lượng natri tartrat dihydrat (tính bằng mg); |
| V: | thể tích của thuốc thử Karl Fischer đã dùng (tính bằng ml). |
b. Áp dụng xác định hàm lượng nước lớn hơn hoặc bằng 1 %.
Dùng nước tinh khiết đã chưng cất đạt tiêu chuẩn làm chất chuẩn hòa vào methanol khan (TT) hoặc dung môi thích hợp loại dùng cho thuốc thử Karl Fischer rồi dùng thuốc thử Karl Fischer để chuẩn độ.
Cách tiến hành:
Cho 25 ml methanol khan (TT) vào cốc chuẩn độ, chuẩn độ bằng thuốc thử Karl Fischer đến điểm kết thúc. Thêm nhanh khoảng 50 mg nước tinh khiết đã cân chính xác vào cốc chuẩn độ trên và chuẩn độ bằng thuốc thử Karl Fischer đến điểm kết thúc. Tính hệ số đương lượng nước F (tính bằng mg nước/ml thuốc thử) của thuốc thử theo công thức:
F = W/V
trong đó:
W: khối lượng nước (tính bằng mg);
V: thể tích thuốc thử Karl Fischer đã dùng (tính bằng ml).
Định lượng
Chuẩn bị mẫu thử: Nếu không có chỉ dẫn gì khác trong chuyên luận riêng, cân hoặc lấy chính xác một lượng chế phẩm ước lượng chứa khoảng 10 mg đến 50 mg nước đem định lượng. Thao tác phải nhanh và thực hiện trong phòng có độ ẩm thấp để tránh ẩm ở ngoài ảnh hưởng đến chất phân tích.
Phương pháp định lượng trực tiếp
Nếu không có chỉ dẫn gì khác trong chuyên luận riêng, cho khoảng 20 ml methanol khan (TT) hoặc dung môi thích hợp dùng cho thuốc thử Karl Fischer vào cốc chuẩn độ, chuẩn độ bằng thuốc thử Karl Fischer đến điểm dừng. Cho nhanh một lượng chế phẩm đã chỉ dẫn trong chuyên luận riêng vào cốc chuẩn độ, đóng nút ngay, khuấy đều để phản ứng tác dụng trong khoảng 1 min rồi tiếp tục chuẩn độ bằng thuốc thử Karl Fischer đến điểm dừng. Tính hàm lượng nước X (tính bằng mg) của chế phẩm theo công thức:
X = N x F
trong đó:
N: thể tích thuốc thử Karl Fischer đã dùng cho lần chuẩn độ sau khi cho chế phẩm (tính bằng ml);
F: hệ số đương lượng nước của thuốc thử Karl Fischer (tính bằng mg/ml).
Phương pháp định lượng gián tiếp
Dung dịch nước chuẩn: Pha loãng 2 ml nước tinh khiết với methanol khan (TT) hoặc dung môi thích hợp thành 1000 ml. Lấy chính xác 25,0 ml dung dịch này cho vào cốc định lượng và chuẩn độ bằng thuốc thử Karl Fischer vừa mới xác định độ chuẩn. Tính hàm lượng nước W (tính bằng mg/ml) của dung dịch nước chuẩn theo công thức:
W = V x F/25
trong đó:
V: thể tích thuốc thử Karl Fischer đã dùng (ml);
F: hệ số đương lượng nước của thuốc thử Karl Fischer (tính bằng mg/ml).
Cách tiến hành: Nếu không có chỉ dẫn gì khác trong chuyên luận riêng, cho một lượng methanol khan (TT), hoặc dung môi được chỉ dẫn trong chuyên luận riêng vào cốc định lượng vừa đủ ngập điện cực, chuẩn độ bằng thuốc thử Kart Fischer đến điểm kết thúc. Cho nhanh một lượng chế phẩm đã chỉ dẫn trong chuyên luận riêng vào cốc, đóng nút ngay, thêm tiếp một lượng chính xác thuốc thử Karl Fischer vào cốc sao cho thừa khoảng 1 ml, hoặc theo một thể tích đã chỉ dẫn trong chuyên luận riêng. Đóng nút để yên 1 min, tránh ánh sáng, thỉnh thoảng khuấy. Chuẩn độ phần thuốc thử Kail Fischer thừa bằng dung dịch nước chuẩn vừa mới pha ở trên. Tính hàm lượng nước A (tỉnh bằng mg) có trong chế phẩm theo công thức:
A = F x V1 - W x V2
trong đó:
F: đương lượng nước của thuốc thử Karl Fischer (tính bằng mg/ml);
V1: thể tích thuốc thử Kart Fischer đã thêm vào (ml);
V2: thể tích dung dịch nước chuẩn đã dùng (ml);
W: hàm lượng nước của dung dịch nước chuẩn ở trên (tính bằng mg/ml).
Chú ý:
Cần phải kiểm tra xem chất thử có tương kỵ với thuốc thử Karl Fischer không trước khi áp dụng phương pháp này. Những chất có khả năng phản ứng với một hay nhiều thành phần của thuốc thử như acid ascorbic, các mercaptan, các sulfid, các muối hydrocarbonat và carbonat kiềm, các oxyd và hydrat của oxyd kim loại... không áp dụng được phương pháp này. Đối với các aldehyd và ceton, hiện nay đã có loại thuốc thử dành riêng để định lượng nước trong các chất này.
Những dung môi hữu cơ sau đây có thể dùng thay thế methanol trong thuốc thử Karl Fischer khi chất thử không tan trong methanol: Cloroform, methyl celosolve, diethylen glycol monoethyl ether. Trước khi sử dụng phải làm khan bằng zeolit đạt tiêu chuẩn cho định lượng nước.
Những chất base hữu cơ sau đây có thể thay pyridin trong thuốc thử Karl Fischer: Imidazol, 2-methyl- aminopyridin. Phải kiểm tra hàm lượng nước trước khi dùng.
Phương pháp 2 (Phương pháp chuẩn độ đo điện tích)
Nguyên tắc
Phương pháp chuẩn độ đo điện tích có nguyên tắc tương tự phương pháp 1 tức là dựa trên phản ứng toàn lượng của nước với lưu huỳnh dioxyd và iod trong dung môi khan chứa một chất base hữu cơ thích hợp. Tuy nhiên, khác với phương pháp 1, iod được tạo ra bằng cách oxy hóa ion iodid tại buồng phản ứng điện hóa. lod tạo thành ở anod phản ứng ngay với nước và lưu huỳnh dioxyd có trong buồng phản ứng. Hàm lượng nước trong chế phẩm tỷ lệ thuận với lượng điện tích tăng thêm cho đến khi kết thúc chuẩn độ. Điểm kết thúc đạt được khi toàn bộ nước phản ứng hết và iod dư xuất hiện. 1 mol iod tương ứng với 1 mol nước, 10,71 C điện tích tương ứng với 1 mg nước.
Độ ẩm bị loại khỏi hệ thống bằng quy trình trước điện phân. Các lần định lượng có thể thực hiện nối tiếp nhau trong cùng một dung dịch thuốc thử, trong các điều kiện sau:
• Mỗi thành phần của hỗn hợp thử không tương kỵ lẫn nhau,
• Không có phản ứng nào khác xảy ra,
• Thuốc thử điện phân phải đủ về thể tích và có đủ khả năng trung hòa nước.
Phương pháp chuẩn độ đo điện tích chỉ áp dụng được với những mẫu có hàm lượng nước nhỏ, khoảng từ 10 μg đến 10 mg nước là phù hợp.
Độ đúng và độ chính xác của phương pháp chủ yếu phụ thuộc vào mức độ loại bỏ được độ ẩm của môi trường ra khỏi hệ thống. Việc kiểm soát hệ thống phải được theo dõi bằng cách đo độ trôi của đường nền.
Thiết bị
Thiết bị bao gồm một buồng phản ứng, điện cực, và khuấy từ. Buồng phản ứng có một ngăn anod rộng và một ngăn cathod nhỏ hơn. Có thể có vách ngăn phân cách hai ngăn điện cực tùy theo thiết kế. Mỗi ngăn có một điện cực platin. Chất lỏng hoặc mẫu thử đã hòa tan được đưa vào buồng phản ứng bằng một xi lanh qua nắp septum. Có thể áp dụng kỹ thuật bay hơi trong đó mẫu thử được làm nóng lên trong một ống đựng mẫu (buồng sấy) và hơi nước từ mẫu được đưa đến buồng phản ứng nhờ một dòng khí trơ khô. Phải tránh việc đưa mẫu rắn vào buồng phản ứng. Tuy nhiên, nếu không có cách nào khác thì việc đưa mẫu rắn vào phải qua một cổng nạp mẫu kín, phải áp dụng các biện pháp thích hợp để tránh đưa thêm độ ẩm của môi trường vào, ví dụ phải làm việc trong hộp có lồng găng tay và trong môi tường khí trơ khô. Quy trình thử nghiệm được theo dõi bằng một thiết bị điện tử phù hợp, có hiển thị kết quả.
Cách tiến hành
Đổ đầy các ngăn của buồng phản ứng bằng thuốc thử điện phân dùng trong vi định lượng nước theo hướng dẫn của nhà sản xuất và tiến hành chuẩn độ đến điểm kết thúc ổn định. Đưa lượng mẫu theo chỉ dẫn vào buồng phản ứng và khuấy 30 s, nếu không có chỉ dẫn khác trong chuyên luận riêng, chuẩn độ đến khi đạt được điểm kết thúc ổn định. Trường hợp phải dùng kỹ thuật bay hơi, đưa lượng mẫu như chỉ dẫn vào buồng sấy và làm nóng. Sau khi hơi nước bay hết sang buồng phản ứng, quá trình chuẩn độ bắt đầu. Đọc kết quả trên máy và tính phần trăm nước có trong chế phẩm, nếu cần. Thực hiện mẫu trắng tùy theo loại mẫu hoặc cách chuẩn bị mẫu.
Kiểm tra độ đúng
Giữa hai lần chuẩn độ, đưa một lượng nước được cân chính xác tương đương với lượng nước trong một lần chuẩn độ mẫu thử, có thể dùng nước hoặc dung dịch chuẩn dùng cho vi định lượng nước, tiến hành chuẩn độ. Tỷ lệ tìm thấy phải từ 97,5 % đến 102,5 % trong trường hợp lượng nước thêm vào khoảng 1000 μg; từ 90,0 % đến 110,0 % trong trường hợp lượng nước thêm vào khoảng 100 μg.
10.4 PHƯƠNG PHÁP CHUẨN ĐỘ BẰNG NITRIT
Chuẩn độ bằng nitrit là phương pháp định lượng thể tích, trong đó dung dịch chuẩn độ là dung dịch natri nitrit.
Phương pháp này được dùng chủ yếu để định lượng các chế phẩm có chứa nhóm amin thơm bậc nhất hoặc những chế phẩm khác mà qua biến đổi hóa học chuyển được thành hợp chất có nhóm amin thơm bậc nhất.
Cách tiến hành
Cân chính xác một lượng chế phẩm tương ứng với khoảng 0,002 phân tử gam hoạt chất (hoặc theo chỉ dẫn trong chuyên luận riêng) hòa tan trong 20 ml acid hydrocloric (TT), và 50 ml nước. Cho thêm 2 g kali bromid (TT), làm lạnh trong nước đá rồi chuẩn độ bằng dung dịch natri nitrit 0,1 M (CĐ) (phải để đầu nhỏ dung dịch chuẩn độ của buret ngập dưới mặt dung dịch cần chuẩn độ). Trong quá trình chuẩn độ phải lắc bình liên tục, nhẹ nhàng sao cho không tạo ra dòng xoáy không khí trong dung dịch (tốt nhất là dùng máy khuấy từ). Nhỏ dung dịch chuẩn độ với tốc độ lúc đầu chừng 2 ml trong một phút, đến trước điểm tương đương khoảng 1 ml thì nhỏ từng 0,1 ml một và để yên ít nhất một phút sau mỗi lần thêm dung dịch.
Điểm kết thúc được xác định bằng phương pháp đo điện (theo hướng dẫn trong các Phụ lục 10.1, Phương pháp chuẩn độ đo ampe hoặc Phụ lục 10.2, Phương pháp chuẩn độ đo điện thế) hoặc bằng chỉ thị màu thích hợp.
10.5 PHƯƠNG PHÁP CHUẨN ĐỘ COMPLEXON
Nhôm (AI)
Lấy một lượng dung dịch như chỉ dẫn trong chuyên luận riêng cho vào một bình nón 500 ml. Thêm 25 ml dung dịch Trilon B 0,1 M (CĐ) và 10 ml hỗn hợp đồng thể tích của dung dịch amoni acetat 2 M và acid acetic 2 M (TT). Đun sôi dung dịch 2 min, để nguội, thêm 50 ml ethanol (TT) và 3 ml dung dịch dithizon 0,025 % (kl/tt) mới pha trong ethanol (TT) rồi chuẩn độ dung dịch Trilon B 0,1 M thừa bằng dung dịch kẽm sulfat 0,1 M (CĐ) đến khi màu của dung dịch chuyển từ xanh sang tím đỏ.
1 ml dung dịch Trilon B 0,1 M (CĐ) tương đương với 2,698 mg AI.
Bismuth (Bi)
Nếu không có chỉ dẫn gì khác, pha loãng chế phẩm với nước để thành 250 ml. Vừa lắc vừa thêm từng giọt amoniac 13,5 M (TT) cho đến khi tủa bắt đầu xuất hiện. Thêm 0,5 ml acid nitric (TT) và đun nóng đến 70 °C, duy trì nhiệt độ này cho đến khi dung dịch trong suốt. Thêm khoảng 50 mg hỗn hợp da cam xylenol (TT) rồi chuẩn độ bằng dung dịch Trilon B 0,1 M (CĐ) đến khi màu của dung dịch chuyển từ tím hồng sang vàng.
1 ml dung dịch Trilon B 0,1 M (CĐ) tương đương với 20,90 mg Bi.
Calci (Ca)
Lấy một lượng dung dịch chế phẩm như chỉ dẫn trong chuyên luận cho vào một bình nón 500 ml rồi pha loãng với nước thành 300 ml. Thêm 6 ml dung dịch natri hydroxyd 10 M (TT) và khoảng 200 mg hỗn hợp calcon (TT) rồi chuẩn độ bằng dung dịch Trilon B 0,1 M (CĐ) đến khi màu của dung dịch chuyển từ tím sang xanh hoàn toàn.
1 ml dung dịch Trilon B 0,1 M (CĐ) tương đương với 4,008 mg Ca.
Chì (Pb)
Lấy một lượng dung dịch chế phẩm như chỉ dẫn trong chuyên luận cho vào một bình 500 ml rồi pha loãng với nước thành 200 ml. Thêm khoảng 50 mg hỗn hợp da cam xylenol (TT) và một lượng hexamin (TT) vừa đủ để thu được dung dịch có màu hồng tím rồi chuẩn độ bằng dung dịch Trilon B 0,1 M (CĐ) đến khi dung dịch chuyển sang màu vàng.
1 ml dung dịch Trilon B0,1 M (CĐ) tương đương với 20,72 mg Pb.
Magnesi (Mg)
Cho vào một bình nón 500 ml một lượng dung dịch chế phẩm như chỉ dẫn trong chuyên luận và pha loãng với nước thành 300 ml hoặc hòa tan một lượng chế phẩm như chỉ dẫn trong chuyên luận trong 5 ml đến 10 ml nước hoặc trong một lượng nhỏ dung dịch acid hydrocloric 2 M (TT) rồi pha loãng với nước thành 50 ml. Thêm 10 ml dung dịch đệm amoniac pH 10,0 (TT) và khoảng 50 mg hỗn hợp đen eriocrom T (TT) vào dung dịch cuối cùng. Đun nóng dung dịch đến 40 °C và chuẩn độ ở nhiệt độ đó bằng dung dịch Trilon B 0,1 M (CĐ) đến khi màu của dung dịch chuyển từ tím sang xanh lam hoàn toàn.
1 ml dung dịch Trilon B 0,1 M (CĐ) tương đương với 2,431 mg Mg.
Kẽm (Zn)
Cho vào một bình nón 500 ml một lượng dung dịch như đã chỉ dẫn trong chuyên luận rồi pha loãng với nước thành 200 ml. Thêm khoảng 50 mg hỗn hợp da cam xylenol (TT) và một lượng hexamin (TT) vừa đủ để làm dung dịch chuyển sang màu hồng tím. Thêm 2 g hexamin (TT) nữa rồi chuẩn độ bằng dung dịch Trilon B 0,1 M (CĐ) đến khi màu của dung dịch chuyển sang vàng.
1 ml dung dịch Trilon B 0,1 M (CĐ) tương đương với 6,54 mg Zn.
10.6 PHƯƠNG PHÁP CHUẨN ĐỘ TRONG MÔI TRƯỜNG KHAN
Chuẩn độ trong môi trường khan là phương pháp chuẩn độ acid và base yếu hoặc những muối của chúng trong môi trường không phải là nước.
Phương pháp 1
(Áp dụng cho base và muối của chúng)
Hòa tan một lượng chế phẩm như chỉ dẫn trong chuyên luận riêng trong một thể tích thích hợp acid acetic khan (TT) đã được trung tính hóa trước với chỉ thị quy định trong chuyên luận riêng, nếu cần thiết có thể làm ấm hay làm lạnh, hoặc chuẩn bị một dung dịch như chỉ dẫn trong chuyên luận riêng.
Khi chế phẩm là muối của acid hydrocloric hoặc acid hydrobromic thì thêm 15 ml dung dịch thủy ngân (II) acetat 5 % (TT) trước khi trung tính dung môi, trừ khi có những chỉ dẫn khác trong chuyên luận riêng.
Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ) đến khi có sự chuyển màu của chỉ thị, điều này tương ứng với giá trị tuyệt đối cao nhất của dE/dV trong chuẩn độ đo thế của chế phẩm thử, ở đây E là thế điện động và V là thể tích dung dịch chuẩn độ (Phụ lục 10.2).
Việc trung tính hóa dung dịch thủy ngân (II) acetat và chuẩn hóa dung dịch chuẩn độ cũng phải dùng cùng một chỉ thị được quy định trong chuyên luận riêng cho chuẩn độ chế phẩm.
Khi nhiệt độ t2 của dung dịch chuẩn độ ở thời điểm định lượng khác với nhiệt độ t1 của dung dịch chuẩn độ lúc được chuẩn hóa thì tính kết quả định lượng căn cứ vào thể tích dung dịch chuẩn độ hiệu chỉnh.
V(h) = V(c) x [1 + 0,0011 (t1 -t2)]
trong đó:
V(h): thể tích dung dịch chuẩn độ hiệu chỉnh;
V(c): thể tích dung dịch chuẩn độ đã dùng.
Tiến hành chuẩn độ mẫu trắng khi cần thiết.
Phương pháp 2
(Áp dụng cho acid yếu)
Dung dịch chuẩn độ, dung môi và chỉ thị được chỉ dẫn trong chuyên luận riêng.
Bảo vệ dung dịch thử và dung dịch chuẩn độ khỏi sự thâm nhập của carbon dioxyd và độ ẩm của không khí trong suốt quá trình chuẩn độ.
Hòa tan chế phẩm trong một thể tích thích hợp dung môi đã trung tính hóa trước với chỉ thị quy định, nếu cần có thể làm ấm hay lạnh, hoặc chuẩn bị một dung dịch chế phẩm như đã chỉ dẫn trong chuyên luận riêng.
Chuẩn độ cho đến khi có sự chuyển màu của chỉ thị, điều này tương ứng với giá trị tuyệt đối cao nhất của dE/dV trong chuẩn độ đo thế của chế phẩm, ở đây E là thế điện động và V là thể tích dung dịch chuẩn độ (Phụ lục 10.2).
Dung dịch chuẩn độ được chuẩn hóa bằng cách sử dụng dung môi và chỉ thị giống như đã sử dụng cho chuẩn độ chế phẩm.
Tiến hành chuẩn độ mẫu trắng khi cần thiết.
10.7 ĐỊNH LƯỢNG CÁC KHÁNG SINH HỌ PENICILIN BẰNG PHƯƠNG PHÁP ĐO IOD
Phương pháp sau đây được áp dụng để định lượng phần lớn thuốc kháng sinh họ penicillin trong Dược điển và những dạng bào chế của chúng mà phép chuẩn độ đo iod là đặc biệt thích hợp. Tiến hành thí nghiệm ở nhiệt độ 25 °C ± 2 °C.
Dung dịch chuẩn
Cân chính xác một lượng thích hợp chất chuẩn quy định trong từng chuyên luận, hòa tan vào dung môi đã được ghi trong Bảng 10.7 và pha loãng với cùng dung môi đó để được một dung dịch có nồng độ ở khoảng nồng độ quy định trong Bảng 10.7.
Dung dịch thử
Nếu không có chỉ định gì khác trong chuyên luận riêng, cân chính xác một lượng mẫu thử thích hợp hòa tan trong dung môi ghi trong Bảng 10.7 và pha loãng với dung môi đó để được một dung dịch có nồng độ ở vào khoảng nồng độ ghi trong Bảng 10.7.
Phương pháp tiến hành
Làm mất hoạt tính và chuẩn độ: Hút riêng rẽ 2,0 ml dung dịch chuẩn và 2,0 ml dung dịch thử vào 2 bình nón nút mài dung tích 125 ml tương ứng. Thêm vào mỗi bình 2,0 ml dung dịch natri hydroxyd 1,0 N (TT), lắc đều và để yên 15 min. Tiếp tục cho vào mỗi bình 2,4 ml dung dịch acid hydrocloric 1,0 N (TT), thêm 10,0 ml dung dịch iod 0,01 N (CĐ), đậy ngay nút bình và để yên 15 min. Chuẩn độ bằng dung dịch natri thiosulfat 0,01 N (CĐ) đến gần điểm kết thúc, thêm 1 giọt dung dịch hồ tinh bột (TT) và tiếp tục chuẩn độ đến khi mất màu xanh.
Mẫu trắng: Hút 2,0 ml dung dịch chuẩn cho vào một bình nón nút mài, thêm 10,0 ml dung dịch iod 0,01 N (CĐ). Nếu dung dịch chuẩn là amoxicilin hay ampicilin thì thêm ngay lập tức 0,12 ml dung dịch acid hydrocloric 1,0 N (TT). Chuẩn độ ngay bằng dung dịch natri thiosulfat 0,01 N (CĐ) đến gần điểm kết thúc, thêm 1 giọt dung dịch hồ tinh bột (TT) và tiếp tục chuẩn độ đến khi mất màu xanh. Cũng làm như vậy đối với bình có chứa 2,0 ml dung dịch thử.
Tính toán: Tính đương lượng F [số microgam (hay đơn vị) kháng sinh chuẩn tương ứng với 1 ml dung dịch natri thiosulfat 0,01 N (CĐ)] bằng công thức:
![]()
trong đó:
C: nồng độ chất chuẩn tính bằng mg trong 1 ml của dung dịch chuẩn;
P: hoạt lực tính bằng microgam (hay đơn vị) trong 1 mg của chất chuẩn;
B: thể tích tính bằng ml của dung dịch natri thiosulfat 0,01 N (CĐ) tiêu thụ trong mẫu trắng của dung dịch chuẩn;
I: thể tích tính bằng ml của dung dịch natri thiosulfat 0,01 N (CĐ) tiêu thụ trong phép thử làm mất hoạt tính và chuẩn độ của dung dịch chuẩn;
Bảng 10.7 - Những dung môi và nồng độ cuối cùng của dung dịch kháng sinh
| Chất kháng sinh | Dung môi | Nồng độ cuối cùng |
| Amoxicilin | Nước | 1,0 mg/ml |
| Ampicilin | Nước | 1,25mg/ml |
| Ampicilin natri | Đệm phosphat pH 6,0 | 1,25 mg/ml |
| Cloxacilin natri | Nước | 1,25 mg/ml |
| Dicloxacilin natri | Đệm phosphat pH 6,0 | 1,25 mg/ml |
| Methicilin natri | Đệm phosphat pH 6,0 | 1,25 mg/ml |
| Oxacilin natri | Đệm phosphat pH 6,0 | 1,25 mg/ml |
| Penicilin G kali | Đệm phosphat pH 6,0 | 2000 đơn vị/ml |
| Penicilin G natri | Đệm phosphat pH 6,0 | 2000 đơn vị/ml |
| Penicilin V kali | Đệm phosphat pH 6,0 | 2000 đơn vị/ml |
10.8 ĐỊNH LƯỢNG CÁC STEROID BẰNG TETRAZOLIUM
Phương pháp này được áp dụng để định lượng các steroid chứa các nhóm chức có tính khử.
Các sản phẩm của phản ứng màu này có khuynh hướng hấp phụ lên bề mặt của thủy tinh. Để tránh sự sai lệch kết quả, nên xử lý các bình phản ứng thủy tinh bằng cách để chúng chứa chính các sản phẩm của phản ứng màu này trước khi dùng. Nên giữ các bình thủy tinh đã được xử lý để dùng riêng cho phép định lượng này và chỉ rửa bình bằng nước giữa các lần định lượng. Phương pháp này được tiến hành trong điều kiện tránh ánh sáng.
Dung dịch thử
Trừ khi có chỉ dẫn cụ thể trong chuyên luận riêng, hòa tan một lượng chế phẩm trong ethanol không có aldehyd (TT) để thu được một dung dịch thử có nồng độ từ 30 đến 35 μg/ml.
Dung dịch chuẩn
Chuẩn bị một dung dịch chất chuẩn tương ứng trong ethanol không có aldehyd (TT) có nồng độ tương đương với nồng độ của dung dịch thử.
Tiến hành
Lấy chính xác 10 ml mỗi dung dịch thử và dung dịch chuẩn cho vào hai bình định mức 25 ml riêng biệt và cho 10 ml ethanol không có aldehyd (TT) vào một bình định mức 25 ml thứ ba. Lần lượt thêm vào các bình 2 ml dung dịch triphenyltetrazolium clorid (TT), 2 ml dung dịch tetramethylamoni hydroxyd loãng (TT). Đậy bình, trộn đều bằng cách lắc xoay tròn nhẹ nhàng và ngâm các bình phản ứng này trong cách thủy ở 30°C trong 1 h, trừ khi có quy định cụ thể trong chuyên luận riêng. Làm lạnh nhanh, thêm ethanol không có aldehyd (TT) đến định mức 25 ml. Lắc đều và đo ngay độ hấp thụ ánh sáng của các dung dịch thu được trong hai bình đầu (theo thứ tự khi cho thuốc thử) ở cực đại 485 nm (Phụ lục 4.1), trong cốc đo có nắp đậy, lấy dung dịch thu được trong bình thứ ba làm mẫu trắng.
10.9 ĐỊNH LƯỢNG NITROGEN TRONG HỢP CHẤT HỮU CƠ
Nitrogen trong hợp chất hữu cơ được định lượng dưới dạng amoniac trong amoni sulfat thu được khi vô cơ hóa các hợp chất hữu cơ có chứa nitrogen với acid sulfuric.
Áp dụng phương pháp I nếu không có chỉ dẫn khác.
Phương pháp I
Dụng cụ
Bộ dụng cụ định lượng nitrogen có thể được chế tạo nguyên bộ chuyên dùng cất amoniac hoặc được lắp ghép từ các dụng cụ thủy tinh cần thiết với nhau sao cho đảm bảo đủ các bộ phận và yêu cầu như Hình 10.9. Các phần bằng cao su của thiết bị nên được xử lý bằng cách đun sôi 10 min đến 30 min trong dung dịch natri hydroxyd 1 M (TT), tiếp theo 30 min đến 60 min trong nước, cuối cùng rửa lại bằng nước trước khi dùng.
A. Bình Kjeldahl để vô cơ hóa mẫu thử và thực hiện phản ứng.
B. Bình cầu để cung cấp hơi nước,
C. Khóa an toàn.
D. Phễu cấp nước vào bình B.
E. Ống dẫn hơi nước từ bình B sang bình phản ứng A.
F. Phễu cấp dung dịch kiềm vào bình phản ứng A.
G. Ống nối bằng cao su có kẹp khóa.
H. Lỗ nhỏ có đường kính bằng đường kính ống cấp hơi.
J. Ống sinh hàn.
K. Bình hứng dịch cất được.
Hình 10.9 - Dụng cụ định lượng nitrogen
(Đơn vị đo tính bằng milimét)
Cách tiến hành
Nếu chuyên luận riêng không có chỉ dẫn gì đặc biệt thì tiến hành như sau:
a. Vô cơ hóa
Lấy chính xác một lượng mẫu thử có chứa khoảng 5 mg nitrogen cho vào bình Kjeldahl A, thêm 1 g hỗn hợp kali sulfat (TT) hoặc natri sulfat khan (TT) và đồng sulfat (TT) (tỷ lệ 10: 1) đã được tán nhỏ, 7 ml acid sulfuric đậm đặc (TT) và vài viên bi thủy tinh. Đậy bình bằng một phễu có cuống dài. Đặt bình nghiêng 45° trên ngọn lửa nhỏ để hỗn hợp nóng lên từ từ rồi tăng dần nhiệt độ tới khi sôi và tiếp tục đun tới khi chất lỏng trong bình có màu lục tươi và không còn những đốm đen của chất hóa than trên thành bình. Nếu chất lỏng trong bình chưa chuyển sang màu lục tươi, có thể thêm 1 ml đến 2 ml dung dịch hydrogen peroxyd 100 tt (TT) khi đã để nguội và đun tiếp đến khi thu được màu này. Đun thêm 30 min nữa, để nguội.
b. Chuẩn bị cất
Thêm 20 ml nước cất vào bình Kjeldahl đã vô cơ hóa, để nguội trở lại rồi lắp bình này vào bộ dụng cụ cắt đã được làm sạch trước bằng cách cho hơi nước chạy qua. Nếu bộ dụng cụ cất amoniac có bình phản ứng A gắn liền thì dùng 20 ml nước cất để chuyển hỗn hợp từ bình Kjeldahl vào bình phản ứng. Cho nước cất vào khoảng 2/3 bình cấp hơi nước B, thêm vài giọt acid sulfuric (TT) để acid hóa chống sự thâm nhập của amoniac từ không khí.
Lấy chính xác 30,0 ml dung dịch acid sulfuric 0,02 N (CĐ) và 2 giọt dung dịch hỗn hợp đỏ methyl (TT) cho vào bình hứng K.
Lắp nối các bộ phận với nhau như hình vẽ sao cho tạo thành một hệ thống kín, đầu cuối của ống sinh hàn thu dịch cất được phải ngập sâu trong dung dịch của bình hứng K.
c. Tiến hành cất
Khi việc chuẩn bị cất đã hoàn tất, cho nước lạnh chảy qua ống sinh hàn và bắt đầu đun nước trong bình B. Khi nước sôi thì cho vào bình phản ứng A qua phễu F từng ít một 30 ml dung dịch natri hydroxyd 40 % (TT) bằng cách sau: đổ hết lượng kiềm này vào phễu F và dùng kẹp G điều chỉnh cho dung dịch kiềm chảy xuống từ từ, khi xong khóa chặt kẹp lại. Tiếp tục cất đến khi hứng được khoảng 100 ml dịch cất. Hạ thấp bình hứng và rửa đầu sinh hàn với một ít nước cất.
d. Chuẩn độ
Chuẩn độ acid sulfuric thừa trong bình hứng dịch cất bằng dung dịch natri hydroxyd 0,02 N (CĐ) tới khi chuyển sang màu vàng, ghi số ml dung dịch natri hydroxyd 0,02 N (CĐ) đã dùng (a ml).
Tiến hành song song một mẫu trắng theo trình tự trên. Ghi số ml dung dịch natri hydroxyd 0,02 N (CĐ) đã dùng (b ml).
Tính kết quả: Lượng nitrogen trong mẫu thử (X) tính bằng gam theo công thức:
X = (b - a) × 0,00028
Trường hợp mẫu thử có lẫn nitrat và nitrit:
Tiến hành tương tự như trên nhưng giai đoạn vô cơ hóa cần tiến hành loại trừ ảnh hưởng của nitrat và nitrit như sau:
Sau khi cho mẫu thử vào bình Kjeldahl A, thêm 10 ml acid sulfuric (TT) đã hòa tan 0,4 g acid salicylic (TT), lắc đều và để yên 30 min (thỉnh thoảng lắc đều). Thêm 2 g natri thiosulfat (TT), lắc đều, thêm 200 mg bột đồng sulfat khan (TT) rồi tiến hành vô cơ hóa như trên bắt đầu từ "Đặt bình nghiêng 45°..."
Phương pháp II: Định lượng protein trong các chế phẩm máu
Đối với các chế phẩm máu khô, chuẩn bị dung dịch chế phẩm như chỉ dẫn trong chuyên luận riêng.
Lấy một thể tích chế phẩm tương ứng với 0,1 g protein, pha loãng thành 20 ml bằng dung dịch natri clorid 0,9 % (TT). Lấy 2 ml dung dịch thu được chuyển vào ống nghiệm dung tích 75 ml, thêm 2 ml dung dịch chứa 75 % acid sulfiric không có nitrogen (TT), 4,5 % kali sulfat (TT) và 0,5 % đồng sulfat (TT), trộn đều và đậy ống nghiệm một cách lỏng lẻo. Đun nóng từ từ đến sôi và tiếp tục đun sôi mạnh trong khoảng 1,5 h và để nguội. Nếu dung dịch thu được không trong thì thêm 0,25 ml dung dịch dung dịch hydrogen peroxyd 20 tt và đun tiếp đến khi thu được dung dịch trong và để nguội. Trong quá trình đun, chú ý không để phần trên của ống nghiệm bị nóng quá.
Chuyển dung dịch thu được vào bộ dụng cụ cất, dùng nước tráng rửa 3 lần, mỗi lần 3 ml. Thêm vào bình cất 10 ml dung dịch natri hydroxyd 10 M (TT) và cất nhanh trong vòng 4 min. Hứng dịch cất vào trong hỗn hợp 5 ml dung dịch acid boric bão hòa (TT) và 5 ml nước, giữ đầu cuối của ống sinh hàn ngập trong dịch hứng. Hạ thấp bình hứng để đầu ống sinh hàn không còn ngập trong dịch hứng và tiếp tục cất thêm 1 min nữa. Chuẩn độ bằng dung dịch acid hydrocloric 0,02 N (CĐ) dùng dung dịch hỗn hợp đỏ methyl (TT) làm chỉ thị (V1).
Lấy một thể tích chế phẩm tương ứng với 0,1 g protein, thêm 12 ml dung dịch natri clorid 0,9 % (TT), thêm 2 ml dung dịch natri molybdat 7,5 % và 2 ml hỗn hợp acid sulfiric không có nitrogen - nước (1: 30). Lắc đều, để yên 15 min và thêm nước vừa đủ 20 ml. Lắc và ly tâm. Lấy 2 ml dịch ly tâm ở trên chuyển vào ống nghiệm dung tích 75 ml và tiếp tục tiến hành như trên, bắt đầu từ: “thêm 2 ml dung dịch chứa 75 % acid sulfide không có nitrogen...” (V2). Tính hàm lượng protein trong chế phẩm (X mg/ml) áp dụng công thức sau (cần tính đến hệ số pha loãng ban đầu):
X = 6,25 × 0,280 (V1 - V2)
10.10 ĐỊNH LƯỢNG VITAMIN A
Hoạt lực của vitamin A được tính theo đơn vị quốc tế (ký hiệu IU). 1 IU vitamin A tương đương với 0,300 μg retinol; 0,344 μg retinyl acetat; 0,359 μg retinyl propionat hoặc 0,550 μg retinyl palmitat.
Tiến hành định lượng nhanh nhất có thể trong điều kiện tránh ánh sáng, không khí và các chất oxy hóa, các chất xúc tác sự oxy hóa (như đồng, sắt), acid và tránh đun nóng (với vitamin A có nguồn gốc tự nhiên), tránh đun nóng kéo dài với retinol tổng hợp đậm đặc dạng bột và dạng dầu. Sử dụng các dung dịch mới pha.
Tùy theo đặc điểm của vitamin A trong các thuốc và nguyên liệu làm thuốc mà áp dụng 1 trong 5 phương pháp dưới đây để định lượng.
Phương pháp 1 (Phương pháp đo quang trực tiếp)
Có thể áp dụng đối với nguyên liệu là các ester của retinol đậm đặc dạng dầu với hàm lượng lớn (≥ 5000 IU/g).
Cân chính xác đến 0,1 % (so với lượng cân) khoảng từ 25 mg đến 100 mg chế phẩm và đem hòa tan trong 5 ml pentan (TT), pha loãng trong 2-propanol (TT1) để được dung dịch chứa chính xác khoảng 10 IU đến 15 IU vitamin A trong 1 ml.
Xác định độ hấp thụ cực đại của dung dịch đo, nếu cực đại hấp thụ nằm trong khoảng bước sóng từ 325 nm đến 327 nm thì đo độ hấp thụ của dung dịch tại các bước sóng 300 nm, 326 nm, 350 nm và 370 nm trong cốc đo dày 1 cm, dùng 2-propanol (TT1) làm mẫu trắng, đo 2 lần lấy giá trị trung bình làm kết quả đo. Tính tỷ lệ độ hấp thụ tại mỗi bước sóng so với độ hấp thụ tại bước sóng 326 nm (Aλ/A326). Nếu tỷ lệ Aλ/A326 không lớn hơn các giá trị ghi dưới đây:
0,60 ở λ = 300 nm
0,54 ở λ = 350 nm
0,14 ở λ = 370 nm
thì tính kết quả hàm lượng vitamin A trong mẫu thử (IU/g) theo công thức:
![]()
trong đó:
A326: độ hấp thụ tại bước sóng 326 nm.
m: lượng chế phẩm đem thử (g).
V: lượng thể tích dung dịch thu được sau khi pha loãng đến nồng độ 10 IU/ml đến 15 IU/ml để đem đo (ml).
1900: hệ số chuyển đổi độ hấp thụ riêng của ester retinol thành IU/g.
Nếu cực đại hấp thụ nằm ngoài dải sóng từ 325 nm đến 327 nm hoặc có một giá trị Aλ/A326 lớn hơn giá trị qui định thì kết quả không có giá trị, tiến hành định lượng theo phương pháp 4.
Phương pháp 2 (Phương pháp đo quang sau khi chiết tách vitamin A)
Có thể áp dụng cho các nguyên liệu vitamin A có nguồn gốc tự nhiên và đa số các dạng thuốc chứa vitamin A: Thuốc nang, viên nén, thuốc bột, thuốc mỡ, dầu gan cá...
Lấy một lượng chế phẩm chứa khoảng 50 000 IU vitamin A vào bình nút mài, thêm 3 ml dung dịch kali hydroxyd (TT) 50 % mới pha, 30 ml ethanol (TT), đun sôi 30 min trên cách thủy có lắp ống sinh hàn hồi lưu, dưới dòng khí nitrogen không có oxygen. Làm nguội nhanh rồi dùng 30 ml nước cất chuyển hết hỗn hợp sang bình gạn. Chiết 4 lần mỗi lần với 50 ml ether (TT) và bỏ lớp dưới khi tách lớp hoàn toàn. Tập trung dịch chiết lại rồi rửa dịch chiết 4 lần, mỗi lần 50 ml nước cất, (chú ý lắc rất nhẹ nhàng ở 2 lần đầu để tránh tạo thành nhũ tương). Làm bay hơi dịch chiết ether trên cách thủy dưới dòng khí nitrogen không có oxygen hoặc cất quay chân không ở nhiệt độ không quá 30 °C đến hết dung môi. Hòa tan cắn trong một lượng 2-propanol (TT) vừa đủ để thu được dung dịch chứa 10 IU đến 15 IU vitamin A trong 1 ml. Đo độ hấp thụ của dung dịch thu được ở các bước sóng 300 nm, 310 nm, 325 nm, và 334 nm trong cốc dày 1 cm với mẫu trắng là 2-propanol (TT), sau đó xác định bước sóng có hấp thụ cực đại. Nếu cực đại hấp thụ nằm trong dải sóng từ 323 nm đến 327 nm và tỷ lệ A300/A325 ≤ 0,73 thì kết quả được tính như sau:
• Tính độ hấp thụ hiệu chỉnh A325(K): A325(K) = 6,815A325 - 2,555A310 - 4,260A334
• Nếu A325(K)/A325 ≥ 0,970 thì tính kết quả hàm lượng vitamin A trong mẫu thử (IU/g) theo công thức sau:
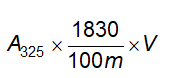
• Nếu A325(K)/A325 < 0,970 thì tính kết quả hàm lượng vitamin A trong mẫu thử (IU/g) theo công thức sau:
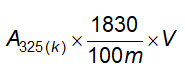
trong đó:
A325: độ hấp thụ quang tại bước sóng 325nm;
m: lượng chế phẩm đem thử (g);
V: lượng thể tích dung dịch thu được sau khi hòa tan cắn đến nồng độ 10 IU đến 15 IU vitamin A để đem đo (ml);
1830: hệ số chuyển đổi độ hấp thụ riêng của ester retinol thành IU.
Nếu cực đại hấp thụ nằm ngoài khoảng 323 nm đến 327 nm hoặc tỷ lệ A300/A325 > 0,73 thì phải tiến hành theo phương pháp 5.
Chú ý:
Trong cách tiến hành của phương pháp 2 nếu chế phẩm ở dạng thuốc nang, viên nén hoặc dạng bào chế thể rắn khác, sau khi đã bỏ vỏ, nghiền thành bột mà vẫn không xà phòng hóa được (do ở dạng vi nang) thì phải xử lý trước bằng cách đun nóng với 10 ml nước cất trên cách thủy 5 min, dùng đũa thủy tinh nghiền nhỏ các chất rắn còn lại và giữ nóng 5 min nữa.
Khi tiến hành xà phòng hóa, nếu không có khí nitrogen có thể thay thế bằng cách cho chất chống oxy hóa vào bình phản ứng như 3 ml dung dịch hydroquinon 1 % trong ethanol, hoặc 30 mg BHT [butyl hydroxytoluen (TT)].
Để chiết vitamin A có thể thay ether (TT) bằng ether dầu hỏa (TT) hoặc n-hexan (TT).
Phương pháp 3 (Phương pháp sắc ký lỏng trực tiếp)
Có thể áp dụng đối với các nguyên liệu vitamin A tổng hợp, các thành phẩm chứa vitamin A tổng hợp mà trong thành phần chỉ chứa vitamin A ở một dạng ester đồng nhất hoặc dạng retinol.
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Methanol - ethyl acetat - nước (90 : 7 : 3).
Dung dịch thử: Lấy chính xác một lượng chế phẩm có chứa khoảng 4000 IU vitamin A cho vào bình định mức dung tích 100 ml, thêm ethanol (TT), lắc kỹ rồi thêm ethanol (TT) đến định mức, lắc đều, lọc.
Dung dịch chuẩn: Pha chất chuẩn vitamin A (dạng giống với dung dịch thử: retinol, retinyl acetat, retinyl palmitat...) trong ethanol (TT) để thu được dung dịch chuẩn có nồng độ vitamin A chính xác khoảng 40 IU/ml.
Điều kiện sắc ký:
Cột kích thước 25 cm × 4 mm được nhồi pha tĩnh C (5 - 10 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 325 nm.
Tốc độ dòng: 1,5 ml/min đến 2,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống sắc ký: Tiến hành sắc ký với dung dịch chuẩn, độ lệch chuẩn tương đối của diện tích pic thu được trên sắc ký đồ của 6 lần tiêm lặp lại mẫu chuẩn (RSD) không được lớn hơn 2,0 %.
Tiến hành sắc ký lần lượt với dung dịch chuẩn và dung dịch thử.
Cách tính kết quả:
Hàm lượng vitamin A trong mẫu thử (IU/g) được tính theo công thức:
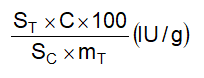
trong đó:
ST và SC: diện tích của pic vitamin A trên sắc đồ của thử và chuẩn;
mT: lượng cân mẫu thử (g);
C: nồng độ dung dịch chuẩn đem tiêm sắc ký (IU/ml).
Chú ý
Trong việc chuẩn bị dung dịch thử, nếu chế phẩm ở dạng thuốc nang, viên nén hoặc dạng bào chế thể rắn khác, sau khi bỏ vỏ, nghiền thành bột, mà vitamin A vẫn không hòa tan được trong ethanol (do ở dạng vi nang) thì phải xử lý trước bằng cách đun nóng với 10 ml nước cất trên cách thủy 5 min, dùng đũa thủy tinh nghiền nhỏ các khối rắn còn lại và giữ nóng thêm 5 min nữa rồi mới cho ethanol vào để hòa tan.
Nếu mẫu thử có nhiều tá dược dạng dầu thì nên thêm 2 ml ethyl acetat (TT) hoặc 2 ml n-hexan (TT) và lắc kỹ trước khi cho ethanol vào để hòa tan.
Nếu trên sắc đồ thu được ngoài pic của dạng vitamin A cần đo còn có pic của dạng vitamin A khác thì phải tiến hành theo phương pháp 2 hoặc 5.
Phương pháp 4 (Phương pháp sắc ký lỏng sau khi thủy phân)
Có thể áp dụng đối với nguyên liệu vitamin A tổng hợp dạng bột, dạng dầu và dạng nhũ tương.
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Methanol - nước (95: 5).
Dung dịch thử (1):
Đối với nguyên liệu dạng bột: Cân 0,200 g chế phẩm vào bình định mức 100 ml. Thêm khoảng 20 mg đến 30 mg bromelain (TT), 5,0 ml nước và 0,15 ml 2-propanol (TT). Đun trong cách thủy ở 60 °C đến 65 °C trong 5 min và thỉnh thoảng khuấy. Thêm 40 ml dung dịch tetrabutylamoni hydroxyd 0,1 M trong 2-propanol (TT). Khuấy nhẹ và để thủy phân trong cách thủy ở 60 °C đến 65 °C trong 10 min, thỉnh thoảng khuấy. Đảm bảo chế phẩm được làm ướt hoàn toàn. Để nguội đến nhiệt độ phòng rồi pha loãng thành 100,0 ml bằng 2-propanol (TT) có chứa 0,1 % butyl hydroxytoluen (TT). Lắc đều cẩn thận tránh tạo bọt khí. Dung dịch có thể vẩn đục.
Đối với nguyên liệu dạng dầu hoặc nhũ tương: Cân 0,100 g chế phẩm vào bình định mức 100 ml. Hòa tan ngay trong 5 ml pentan (TT). Thêm 40 ml dung dịch tetrabutylamoni hydroxyd 0,1 M trong 2-propanol. Khuấy nhẹ và để thủy phân trong cách thủy ở 60 °C đến 65 °C trong 10 min, thỉnh thoảng khuấy nhẹ. Để nguội đến nhiệt độ phòng, rồi pha loãng thành 100,0 ml bằng 2-propanol (TT) có chứa 0,1 % butyl hydroxytoluen (TT). Lắc cẩn thận tránh tạo bọt khí.
Dung dịch thử (2): Pha loãng dung dịch thử (1) bằng 2-propanol (TT) để thu được dung dịch có nồng độ 100 IU/ml.
Lọc trước khi tiêm đối với dung dịch thử được chuẩn bị từ nguyên liệu dạng bột.
Dung dịch chuẩn (1): Cân chính xác 0,100 g retinyl acetat chuẩn (hàm lượng khoảng 1 000 000 IU/g) vào bình định mức 100 ml và tiến hành như dung dịch thử (1) đối với nguyên liệu dạng dầu hoặc nhũ tương.
Dung dịch chuẩn (2): Pha loãng 5,0 ml dung dịch chuẩn (1) thành 50,0 ml bằng 2-propanol (TT). Lắc cẩn thận tránh tạo bọt khí.
Điều kiện sắc ký:
Cột kích thước 12,5 cm × 4 mm được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 325 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch thử (2) và dung dịch chuẩn (2) với thời gian gấp 1,5 lần thời gian lưu của retinol.
Thời gian lưu của retinol khoảng 3 min.
Cách tính kết quả:
Hàm lượng vitamin A trong mẫu thử (IU/g) được tính theo công thức:
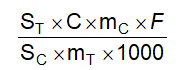
trong đó:
ST, SC: diện tích pic của retinol trên sắc đồ của dung dịch thử và dung dịch chuẩn.
mC, mT: lượng cân mẫu chuẩn, thử (mg).
C: hàm lượng retinyl acetat chuẩn được xác định theo phương pháp 1 (IU/g).
F: độ pha loãng của dung dịch thử.
Phương pháp 5 (Phương pháp sắc ký lỏng sau khi chiết tách vitamin A)
Có thể áp dụng đối với các chế phẩm phức tạp và có chứa vitamin A với hàm lượng thấp không thực hiện được 1 trong 4 phương pháp trên.
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Methanol - nước (97: 3).
Dung dịch thử: Lấy một lượng chế phẩm chứa khoảng 2000 IU vitamin A vào bình cầu nút mài, thêm 5 ml dung dịch acid ascorbic (TT) 10 % mới pha và 10 ml dung dịch kali hydroxyd (TT) 80 % mới pha, 100 ml ethanol 96 % (TT), đun sôi 15 min trên cách thủy có lắp ống sinh hàn hồi lưu, thêm 100 ml dung dịch natri clorid (TT) 1 % và để nguội. Chuyển dung dịch vào bình chiết dung tích 500 ml, tráng rửa bình nút mài bằng 75 ml dung dịch natri clorid (TT) 1 % và rửa tiếp với 150 ml dung dịch đồng thể tích ether dầu hỏa (40 °C đến 60 °C) và ether (TT), chuyển hết dịch tráng rửa vào bình chiết ở trên. Lắc 1 min, khi tách lớp hoàn toàn, bỏ lớp dưới, rửa lớp trên lần đầu với 50 ml dung dịch kali hydroxyd (TT) 3 % trong dung dịch 10 % (tt/tt) ethanol 96 % (TT), sau đó rửa 3 lần, mỗi lần với 50 ml dung dịch natri clorid (TT) 1 %. Lọc nhanh lớp trên qua giấy lọc có chứa 5 g natri sulfat khan (TT) vào bình cầu 250 ml thích hợp để lắp vào dụng cụ cất quay. Rửa bình chiết bằng 10 ml hỗn hợp dung môi dùng để chiết mới pha [dung dịch đồng thể tích ether dầu hỏa (nhiệt độ sôi 40 °C- 60 °C) và ether (TT)], lọc và gộp các dịch lọc lại. Cất quay ở nhiệt độ không quá 30 °C dưới áp suất giảm (đuổi nước) và đóng đầy với khí nitrogen khi bay hơi xong. Hoặc có thể bay hơi dung môi dưới luồng khí nitrogen ở nhiệt độ không quá 30 °C. Hòa tan cắn trong 2-propanol (TT) và chuyển vào bình định mức 25 ml, pha loãng thành 25 ml với cùng dung môi, đun nóng nhẹ hay siêu âm nếu cần.
Dung dịch chuẩn gốc: Hòa tan retinyl acetat chuẩn trong 2-propanol (TT1) để thu được dung dịch có nồng độ khoảng 1000 IU vitamin A trong 1 ml. Nồng độ chính xác của dung dịch chuẩn gốc được xác định bằng phương pháp quang phổ hấp thụ tử ngoại và khả kiến (Phụ lục 4.1). Pha loãng dung dịch chuẩn gốc bằng 2-propanol (TT1) tới nồng độ 10 đến 15 IU trong 1 ml rồi đo độ hấp thụ ở bước sóng 326 nm trong cốc đo dày 1 cm, dùng 2-propanol (TT1) làm mẫu trắng.
Tính nồng độ vitamin A trong dung dịch chuẩn gốc (IU/ml) tính theo retinyl acetat theo công thức:
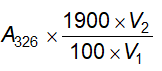
trong đó:
A326: độ hấp thụ quang của dung dịch ở 326 nm;
V1: thể tích dung dịch chuẩn gốc đem pha loãng (ml);
V2: thể tích dung dịch loãng đã pha (ml).
1900: hệ số chuyển đổi độ hấp thụ riêng của ester retinol thành IU.
Dung dịch chuẩn: Lấy chính xác 2 ml dung dịch chuẩn gốc đem xử lý hoàn toàn như mẫu thử. Xác định nồng độ chính xác của dung dịch chuẩn bằng phương pháp quang phổ hấp thụ tử ngoại và khả kiến (Phụ lục 4.1). Pha loãng dung dịch chuẩn bằng 2-propanol (TT1) tới nồng độ 10 IU đến 15 IU trong 1 ml rồi đo độ hấp thụ ở bước sóng 325 nm trong cốc đo dày 1 cm, dùng 2-propanol (TT1) làm mẫu trắng. Tính nồng độ all-trans-retinol trong dung dịch chuẩn (IU/ml) theo công thức:
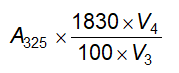
trong đó:
A325: độ hấp thụ của dung dịch ở 325 nm;
V3: thể tích dung dịch chuẩn đem pha loãng (ml);
V4: thể tích dung dịch đã pha loãng (ml).
1830: hệ số chuyển đổi độ hấp thụ riêng của all-trans-retinol thành IU.
Điều kiện sắc ký:
Cột kích thước 25 cm × 4 mm được nhồi pha tĩnh C (5 - 10 μm).
Tốc độ dòng: 1 ml/min.
Detector quang phổ tử ngoại đặt ở bước sóng 325 nm.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký 3 lần với mỗi dung dịch thử và dung dịch chuẩn.
Thời gian lưu của all-trans-retinol khoảng (5 ± 1) min.
Kết quả được chấp nhận nếu đáp ứng các yêu cầu sau:
Trên sắc ký đồ của dung dịch thử có pic tương ứng với pic của all-trans-retinol trên sắc ký đồ của dung dịch chuẩn.
Khi dùng phương pháp thêm chuẩn vào dung dịch thử thì phần trăm thu hồi retinol acetat chuẩn phải lớn hơn 95 % và phần trăm thu hồi all-trans-retinol trong dung dịch chuẩn được xác định bằng phương pháp đo quang phải lớn hơn 95 % so với nồng độ tính từ dung dịch chuẩn gốc.
Tính kết quả:
Hàm lượng vitamin A trong mẫu thử (IU/g) được tính theo công thức:
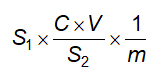
trong đó:
S1: diện tích pic của all-trans-retinol trên sắc đồ của dung dịch thử.
S2: diện tích pic của all-trans-retinol trên sắc đồ của dung dịch chuẩn.
C: nồng độ retinyl acetat trong dung dịch chuẩn gốc (IU/ml).
m: lượng chế phẩm đem xử lý thành dung dịch thử (g).
V: thể tích dung dịch chuẩn gốc đã đem xử lý (ml).
Kết quả được chấp nhận nếu đáp ứng các yêu cầu sau:
Trên sắc ký đồ của dung dịch thử có pic tương ứng với pic của all-trans-retinol trên sắc ký đồ của dung dịch chuẩn.
Khi dùng phương pháp thêm chuẩn vào dung dịch thử thì phần trăm thu hồi retinol acetat chuẩn phải lớn hơn 95 % và phần trăm thu hồi all-trans-retinol trong dung dịch chuẩn được xác định bằng phương pháp đo quang phải lớn hơn 95 % so với nồng độ tính từ dung dịch chuẩn gốc.
Tính kết quả:
Hàm lượng vitamin A trong mẫu thử (IU/g) được tính theo công thức:
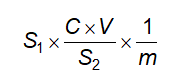
trong đó:
S1: diện tích pic của all-trans-retinol trên sắc đồ của dung dịch thử.
S2: diện tích pic của all-trans-retinol trên sắc đồ của dung dịch chuẩn.
C: nồng độ retinyl acetat trong dung dịch chuẩn gốc (IU/ml).
m: lượng chế phẩm đem xử lý thành dung dịch thử (g).
V: thể tích dung dịch chuẩn gốc đã đem xử lý (ml).
10.11 PHƯƠNG PHÁP PHÂN TÍCH ACID AMIN
Phương pháp phân tích acid amin là phương pháp xác định thành phần và hàm lượng các acid amin trong các protein và peptid, cũng như trong các chế phẩm thuốc. Phương pháp phân tích acid amin được áp dụng để định tính và định lượng các protein và peptid dựa trên các cấu tử acid amin tạo ra chúng, để giúp việc xác định cấu trúc của các protein và peptid cũng như việc xác định cách phân đoạn của chúng nhằm thiết lập giản đồ peptid và để phát hiện các acid amin không điển hình có thể có mặt trong một protein hoặc peptid.
Trước khi phân tích acid amin trong protein hoặc peptid, cần thiết phải thủy phân protein hoặc peptid thành các acid amin. Sau đó tiến hành phân tách các acid amin thu được giống như khi ta phân tách các acid amin tự do có trong các chế phẩm thuốc. Điều quan trọng là chúng phải được biến đổi thành các dẫn chất thích hợp cho việc phân tách và phát hiện.
Thiết bị
Các phương pháp để phân tích acid amin thường dựa trên việc tách các acid amin có trong mẫu thử bằng phương pháp sắc ký. Các thiết bị sắc ký tự động thường chiếm lợi thế. Thiết bị phân tích acid amin điển hình là một máy sắc ký lỏng áp suất thấp hoặc áp suất cao, có khả năng thực hiện chương trình dung môi để tách các acid amin khi qua cột sắc ký. Máy cần có thêm thiết bị tạo dẫn chất acid amin sau cột, trừ khi mẫu thử được tạo thành dẫn chất trước cột. Để phát hiện kết quả, thường dùng detector hấp thụ tử ngoại - khả kiến hoặc detector huỳnh quang, tùy thuộc vào cách tạo dẫn chất đã áp dụng. Một thiết bị tích phân cho phép chuyển đổi tín hiệu tương tự đi từ detector ra và cho phép định lượng.
Thường sử dụng các máy chuyên dụng để phân tích acid amin.
Chú ý
Nhiễu đường nền luôn là mối quan tâm của người tiến hành phân tích acid amin. Để khắc phục, phải dùng các thuốc thử có độ tinh khiết cao (ví dụ acid hydrocloric có độ tinh khiết thấp có thể gây nhiễm glycin). Thông thường cách một vài tuần lại phải thay mới các thuốc thử. Chỉ dùng các dung môi dùng cho sắc ký lỏng. Lọc lại các dung môi trước khi dùng để giảm thiểu sự nhiễm khuẩn và các tạp chất. Đậy kín các bình đựng dung môi. Không để các thiết bị phân tích acid amin tiếp xúc trực tiếp với tia sáng mặt trời.
Chất lượng thực hành phòng thí nghiệm có thể quyết định chất lượng phân tích acid amin. Giữ phòng thí nghiệm sạch sẽ. Đặt thiết bị phân tích trong phòng thí nghiệm tại chỗ riêng biệt, ít bị ảnh hưởng của các hoạt động khác. Định kỳ rửa sạch và chuẩn hóa lại các pipet. Bảo quản đầu pipet trong hộp đậy kín. Không được cầm đầu piped bằng tay trần, phải mang găng tay bằng cao su không xoa bột talc hoặc loại khác có chất lượng tương đương. Hạn chế số lần mở và đóng bình đựng mẫu thử vì bụi có thể làm gia tăng kết quả về hàm lượng các chất glycin, serin và alanin.
Cần bảo dưỡng tốt thiết bị phân tích acid amin để có kết quả chấp nhận được. Nếu thiết bị được dùng thường ngày thì hàng ngày phải kiểm tra độ rò rỉ dung môi của thiết bị, độ ổn định của đèn và detector, độ phân giải của cột.
Định kỳ làm sạch hoặc thay thế các kính lọc của thiết bị và các linh phụ kiện cần bảo dưỡng khác.
Các chất đối chiếu
Trên thị trường có sẵn các acid amin chuẩn để dùng trong phân tích acid amim; chúng thường là dung dịch hỗn hợp các acid amin chuẩn trong nước. Khi cần xác định thành phần acid amin trong một mẫu thử, các protein hoặc peptid chuẩn phải được phân tích song song với mẫu thử để kiểm tra sự toàn vẹn của thử nghiệm. Trong trường hợp này, chuẩn protein được sử dụng là albumin huyết thanh bò tinh khiết cao.
Chuẩn hóa thiết bị
Việc chuẩn hóa thiết bị phân tích acid amin được thực hiện bằng cách phân tích một chuẩn acid amin, gồm hỗn hợp các acid amin chuẩn đã biết trước hàm lượng của từng chất, để xác định hệ số đáp ứng và khoảng tuyến tính ứng với mỗi một acid amin chuẩn đã thử.
Pha loãng mẫu chuẩn acid amin thành nhiều dung dịch có nồng độ khác nhau, nằm trong khoảng tuyến tính dự đoán trước của các acid amin có trong mẫu chuẩn. Tiến hành phân tích nhiều lần với mỗi nồng độ. Biểu thị kết quả thu được trên biểu đồ thể hiện tương quan giữa diện tích pic thu được ứng với mỗi nồng độ của acid amin đã thử. Nhờ biểu đồ này, có thể xác định được khoảng tuyến tính của mỗi acid amin, tại đó, các diện tích pic thu được có tương quan xấp xỉ tuyến tính với các nồng độ của các acid amin đã thử. Khi phân tích acid amin, để có kết quả đúng và lặp lại, điều quan trọng là phải pha và thử nghiệm trên các mẫu thử có nồng độ nằm trong khoảng tuyến tính tương ứng với kỹ thuật phân tích đang áp dụng.
Để xác định hệ số đáp ứng cho mỗi acid amin, ta phân tích từ 4 nồng độ đến 6 nồng độ của acid amin chuẩn tương ứng. Hệ số đáp ứng tính được là giá trị trung bình của diện tích pic (hoặc của chiều cao pic) ứng với nồng độ 1 nanomol của dung dịch acid amin chuẩn. Thiết lập một dãy chuẩn hóa bao gồm hệ số đáp ứng tương ứng của các acid amin và sử dụng dãy này để tính nồng độ (nanomol) của mỗi acid amin có trong mẫu thử bằng cách chia diện tích pic (hoặc chiều cao pic) thu được của acid amin đó cho hệ số đáp ứng tương ứng có trong dãy chuẩn hóa.
Trong việc phân tích thường ngày, khi dùng dãy chuẩn hóa, ta chỉ cần xác định một diện tích pic là đủ. Tuy nhiên, dãy chuẩn hóa này phải thường xuyên được thử lại bằng các phân tích kiểm tra và được cập nhật để bảo đảm tính toàn vẹn của nó.
Độ lặp lại
Muốn có các kết quả phân tích acid amin có chất lượng ổn định tại một phòng thí nghiệm phân tích, cần quan tâm đến độ lặp lại của phép định lượng. Cần có một hệ thống thiết bị phân tích các acid amin có khả năng cung cấp các giá trị lặp lại của thời gian lưu của pic (để định tính) và các giá trị lặp lại của diện tích pic (để định lượng). Cách xác định tiêu biểu độ lặp lại bao gồm việc pha chế một dung dịch chuẩn các acid amin, rồi tiến hành đo mẫu đó nhiều lần (6 lần hoặc nhiều hơn). Sau đó, tính độ lệch chuẩn của các giá trị thời gian lưu và độ lệch chuẩn các giá trị diện tích pic đã được tích phân, ứng với mỗi acid amin đã được phân tích. Việc xác định độ lặp lại còn được mở rộng bằng cách nhiều người phân tích khác nhau cùng tiến hành xác định độ lặp lại đó trong nhiều ngày. Người ta còn thực hiện việc định lượng nhiều dung dịch có nồng độ khác nhau của chuẩn gốc để xác định sự biến thiên do việc pha chế mẫu thử. Thường người ta phân tích thành phần acid amin của một protein chuẩn (ví dụ albumin huyết thanh bò) để đánh giá độ lặp lại. Nhờ việc xác định độ lệch chuẩn, ta có thể thiết lập các giới hạn phân tích để đạt kết quả tốt, với độ lệch chuẩn thấp nhất. Để giảm bớt sai số trong phân tích, nhiều yếu tố cần được quan tâm và xem xét đầy đủ như: cách chuẩn bị mẫu thử, nhiễu đường nền do chất lượng của các thuốc thử, việc thực hành thí nghiệm, tính năng và việc bảo dưỡng máy móc thiết bị, các dữ liệu phân tích và cách biện giải và cuối cùng là việc thực thi thành thạo của người làm phân tích.
Chuẩn bị mẫu thử
Muốn có kết quả phân tích acid amin đúng, phải dùng các mẫu thử protein và peptid đã tinh chế. Các tạp chất như các muối, ure, chất tẩy rửa… có thể gây nhiễu, nên cần phải loại khỏi mẫu thử trước khi tiến hành phân tích. Phương pháp điều chế dẫn chất sau cột sắc ký không bị nhiễm bởi các tạp chất ở mức độ lớn như khi điều chế dẫn chất trước cột sắc ký. Nên giảm số thao tác trên mẫu thử để giảm nhiễu đường nền, tăng kết quả tìm thấy và giảm công lao động. Các cách thông thường để loại tạp chất trong mẫu thử protein bao gồm:
Tách bằng sắc ký lỏng cao áp pha đảo, thu protein bằng một dung môi bay hơi có chứa một lượng thích hợp thành phần hữu cơ rồi làm khô bằng ly tâm chân không;
Thẩm tách loại bỏ tạp chất;
Ly tâm siêu lọc;
Kết tủa protein bằng một dung môi hữu cơ (ví dụ aceton);
Lọc qua gel.
Chất chuẩn nội
Cần dùng một chất chuẩn nội để kiểm soát những mất mát và biến đổi lý hóa học xảy ra trong quá trình phân tích acid amin. Do đó, trước khi tiến hành thủy phân phải thêm một lượng chính xác chất chuẩn nội vào dung dịch protein cần phân tích. Lượng chuẩn nội tìm thấy được sẽ là một thông số chung cho lượng tìm thấy được của các acid amin có trong protein. Tuy nhiên các acid amin tự do và các acid amin liên kết trong cấu trúc protein không giống nhau về tốc độ thủy phân hoặc phân hủy. Vì vậy việc dùng chất chuẩn nội để hiệu chỉnh sự mất mát trong quá trình thủy phân có thể cho kết quả không đáng tin cậy. Cần chú ý điều này khi biện giải kết quả phân tích. Ta còn có thể thêm chất chuẩn nội vào hỗn hợp các acid amin sau khi đã được thủy phân để hiệu chỉnh những sai lệch về kết quả phân tích gây ra do các sai lệch khi tiêm mẫu, do thay đổi độ ổn định của thuốc thử cũng như tốc độ dòng của dung môi. Chất chuẩn nội lý tưởng là một acid amin bậc nhất nhân tạo, có sẵn trên thị trường với giá rẻ. Chất này phải bền vững trong quá trình thủy phân, có hệ số đáp ứng tuyến tính với nồng độ và phải được rửa giải cho một pic có thời gian lưu duy nhất và được phân giải tốt với các pic tương ứng với các acid amin khác. Các chất chuẩn nội thường được dùng bao gồm norleucin, nitrotyrosin và acid α-aminobutyric.
Thủy phân protein
Cần thiết phải tiến hành thủy phân các mẫu thử protein và peptid trước khi phân tích các acid amin tạo thành. Để tránh sai kết quả, các đồ thủy tinh dùng trong thủy phân phải hết sức sạch. Dấu vân tay trên ống thủy phân hoặc bột tách ra từ bao tay cũng có thể làm nhiễm bẩn mẫu thử. Để rửa sạch các ống thủy tinh dùng trong thủy phân (ống thủy phân), ta đun sôi chúng 1 h trong dung dịch acid hydrocloric 1 M hoặc ngâm chúng trong acid nitric đậm đặc hoặc trong hỗn hợp đồng thể tích acid hydrocloric và dung dịch nitric đậm đặc. Sau đó tráng sạch bằng nước cất tinh khiết cao, tiếp đến bằng methanol dùng trong sắc ký lỏng cao áp. Cuối cùng sấy qua đêm trong tủ sấy rồi bảo quản kín cho đến khi dùng. Cũng có thể nung đồ thủy tinh dùng để thủy phân ở nhiệt độ 500 °C trong 4 h để tránh nhiễm bẩn. Cũng có thể sử dụng loại dụng cụ thích hợp, chỉ dùng một lần rồi bỏ.
Thủy phân bằng acid là phương pháp thông dụng nhất để thủy phân một mẫu thử protein trước khi tiến hành phân tách các acid amin tạo ra mẫu thử đó. Thủy phân bằng acid có thể cho kết quả phân tích thay đổi vì một số acid amin bị phân hủy một phần hay hoàn toàn. Cụ thể là: Tryptophan bị phân hủy, serin và threonin bị phân hủy một phần, methionin có thể bị oxy hóa còn cystein thì chuyển đổi thành cystin (tuy nhiên lượng cystin tìm thấy được thường thấp hơn thực tế vì một phần bị phân hủy hoặc bị khử trở lại thành cystein). Có thể giảm thiểu sự phân hủy do bị oxy hóa bằng cách tiến hành thủy phân trong môi trường chân không thích hợp (áp suất thấp hơn 200 μm thủy ngân hoặc 26,7 Pa) hoặc bằng cách nạp một khí trơ (argon) vào khoảng trống phía trên của bình phản ứng. Các đường nối peptid giữa isoleucin - isoleucin, valin - valin, isoleucin - valin và valin - isoleucin chỉ có một phần được cắt đứt để giải phóng acid amin thôi. Asparagin và glutamin đều bị khử amid để thành acid aspartic và acid glutamic tương ứng. Vì tryptophan, asparagin và glutamin bị mất mát trong khi thủy phân bằng acid nên ta chỉ còn định lượng được 17 acid amin. Một số kỹ thuật thủy phân mô tả sau đây sẽ đề cập cách khắc phục các trở ngại nêu trên. Tuy nhiên một số kỹ thuật (từ số 4 đến số 11) lại có thể biến đổi một số acid amin khác. Vì vậy cần cân nhắc lợi hại khi chọn kỹ thuật thủy phân protein/peptid và phải thực nghiệm thích hợp trước khi chọn một kỹ thuật thủy phân khác kỹ thuật thủy phân bằng acid.
Thường hay dùng phương pháp phân tích acid amin theo chương trình thời gian thủy phân (tức là phân tích acid amin ở các thời điểm thủy phân là 24 h, 48 h và 72 h) để xác định nồng độ ban đầu của các acid amin dễ bị phân hủy hoặc chậm bị phân tách khi thủy phân: Vẽ đồ thị biểu thị sự liên quan giữa nồng độ thu được của acid amin dễ bị phân hủy (ví dụ serin, threonin) với thời gian thủy phân rồi ngoại suy tới điểm gốc thời gian, ta sẽ được nồng độ ban đầu (nồng độ ở gốc thời gian) của acid amin dễ bị phân hủy đó. Áp dụng phương pháp chương trình thời gian thủy phân này cho các acid amin chậm được phân tách (ví dụ isoleucin và valin), trên đồ thị sẽ có một đoạn bằng (đoạn thẳng nằm ngang) ứng với nồng độ của acid amin cần phân tích đó. Nếu thời gian thủy phân quá dài, nồng độ của các acid amin cần phân tích sẽ bắt đầu giảm, chứng tỏ acid amin đó đã bị phân hủy do điều kiện thủy phân.
Một cách khác của phân tích acid amin theo chương trình thời gian thủy phân được chấp thuận là cho một chuẩn acid amin trải qua cùng các điều kiện như khi thủy phân mẫu thử protein/peptid. Acid amin chuẩn, ở thể tự do, có thể sẽ không có kết quả hoàn toàn tiêu biểu cho tốc độ phân hủy của các acid amin dễ bị phân hủy có trong mẫu thử protein/peptid. Điều này đặc biệt đúng đối với các liên kết peptid chậm bị phân tách (như liên kết isoleucin - valin). Tuy nhiên cách phân tích này sẽ cho phép giải thích một vài trường hợp phân hủy acid amin trong mẫu thử.
Cách thủy phân bằng acid trong lò vi sóng cũng đã được sử dụng, cho kết quả nhanh chóng nhưng đòi hỏi thiết bị đặc biệt và cần thận trọng đặc biệt. Phải xác định được các điều kiện thủy phân trong lò vi sóng tối ưu cho mỗi mẫu thử protein/peptid riêng biệt. Thủy phân trong lò vi sóng chỉ cần thời gian ít phút, nhưng nếu chỉ thêm hoặc bớt một phút so với yêu cầu thôi cũng sẽ có thể có kết quả sai lệch (hoặc do các acid amin dễ bị phân hủy sẽ bị phân hủy hoặc do mẫu thử chưa được thủy phân hoàn toàn).
Cách thủy phân hoàn toàn bằng một hỗn hợp các men protease cũng được áp dụng nhưng phức tạp, đòi hỏi phải được kiểm soát chặt chẽ và thường được áp dụng nhiều để phân tích các peptid hơn là các protein.
Khi tiến hành phân tích lần đầu một mẫu thử protein mới, cần phải làm thực nghiệm để xác định các điều kiện tối ưu về thời gian và nhiệt độ thủy phân.
Kỹ thuật thủy phân 1
Thủy phân bằng acid hydrocloric có chứa một lượng phenol là kỹ thuật thông dụng nhất để thủy phân mẫu thử protein/peptid trước khi tiến hành phân tích acid amin. Thêm phenol vào môi trường thủy phân cốt để ngăn ngừa hiện tượng halogen hóa tyrosin.
Dung dịch thủy phân: Dung dịch acid hydrocloric 6 M chứa từ 0,1 % đến 1 % phenol.
Thủy phân pha lỏng: Cho vào ống thủy phân mẫu thử protein/peptid rồi làm khô (để loại nước, tránh pha loãng dung dịch thủy phân). Cứ 500 μg mẫu thử đông khô, ta thêm 200 μl dung dịch thủy phân. Làm lạnh ống thử trong aceton đông băng khan. Hàn ống thủy phân ở chân không. Thủy phân mẫu thử trong 24 h ở 110 °C và trong chân không hoặc khí trơ để tránh oxy hóa. Nếu lo ngại mẫu thử chưa được thủy phân hoàn toàn, cần nghiên cứu kéo dài thêm thời gian thủy phân (ví dụ trong 48 h và 72 h).
Thủy phân pha hơi: Đây là một trong các kỹ thuật thủy phân thông dụng nhất được ưa dùng để làm vi phân tích, khi chỉ có một lượng nhỏ mẫu thử. Kỹ thuật này cũng cho phép giảm thiểu việc mẫu thử bị nhiễm bẩn bởi dung dịch thủy phân. Đặt ống thủy phân chứa mẫu thử đã làm khô trong một ống thử to hơn, ống này chứa một lượng thích hợp dung dịch thủy phân và như vậy ngăn không cho mẫu thử tiếp xúc trực tiếp với dung dịch thủy phân. Hút chân không (áp suất thấp hơn 200 μm thủy ngân hoặc 26,7 Pa), hoặc bơm một khí trơ vào phần trên của ống thử. Hàn ống lớn và thủy phân ở 110 °C trong 24 h. Hơi acid sẽ thủy phân mẫu thử và lượng acid ngưng tụ trong ống thủy phân chứa mẫu thử là tối thiểu. Sau khi thủy phân xong, sấy khô mẫu thử trong chân không để loại bỏ acid dư thừa.
Kỹ thuật thủy phân 2
Giảm hiện tượng oxy hóa tryptophan trong khi thủy phân bằng cách dùng acid mercaptoethansulfonic (MESA) làm acid khử.
Dung dịch thủy phân: Dung dịch MESA 2,5 M.
Thủy phân pha hơi: Làm khô 1 μg đến 100 μg mẫu thử protein/peptid trong ống thủy phân. Đặt ống thủy phân trong một ống lớn hơn, chứa khoảng 200 μl dung dịch thủy phân. Hàn ống lớn trong chân không (áp suất khoảng 50 μm thủy ngân hoặc 6,7 Pa). Thủy phân ở 170 °C đến 185 °C trong khoảng 12,5 min. Sau khi thủy phân xong làm khô ống thủy phân ở chân không trong 15 min để loại acid dư thừa.
Kỹ thuật thủy phân 3
Ngăn ngừa hiện tượng oxy hóa tryptophan bằng cách dùng acid thioglycolic (TGA) làm acid khử.
Dung dịch thủy phân: Dung dịch acid hydrocloric 7 M chứa 1 % phenol, 10 % acid trifluoroacetic và 20 % acid thioglycolic.
Thủy phân pha hơi: Làm khô từ 10 đến 50 μg mẫu thử protein/peptid trong một ống thủy phân. Đặt ống thủy phân trong một ống lớn hơn, chứa khoảng 200 μl dung dịch thủy phân. Hàn ống lớn trong chân không (áp suất khoảng 50 μm thủy ngân hoặc 6,7 Pa). Thủy phân mẫu thử ở 166 °C trong khoảng 15 min đến 30 min. Sau khi thủy phân xong, làm khô ống thủy phân trong chân không trong 5 min để loại acid dư thừa.
Lượng tryptophan tìm thấy có thể phụ thuộc vào lượng mẫu lấy thử.
Kỹ thuật thủy phân 4
Oxy hóa cystein/cystin và methionin bằng acid performic trước khi thủy phân mẫu thử.
Dung dịch oxy hóa: Dùng acid performic mới pha bằng cách trộn đều 1 thể tích hydrogen peroxyd 30 % (TT) với 9 thể tích acid formic khan (TT), rồi để ở nhiệt độ phòng trong 1 h.
Tiến hành: Hòa tan mẫu thử protein/peptid trong 20 μl acid formic khan (TT) và để ở 50 °C trong 5 min. Sau đó thêm 100 μl dung dịch oxy hóa. Để phản ứng xảy ra trong 10 min đến 30 min. Cystein sẽ chuyển thành acid cysteic, còn methionin thành methionin sulfon. Ly tâm chân không để loại thuốc thử thừa, rồi thủy phân mẫu thử đã được oxy hóa theo kỹ thuật 1 hoặc 2 nói trên.
Kỹ thuật 4 này có thể làm biến đổi tyrosin khi môi trường có muối halid.
Kỹ thuật thủy phân 5
Oxy hóa cystein/cystin trong quá trình thủy phân pha lỏng bằng natri azid.
Dung dịch thủy phân: Thêm vào dung dịch acid hydrocloric 6 M (TT) có chứa 0,2 % phenol một lượng natri azid (TT) để có nồng độ 0,2 %. Phenol có trong dung dịch thủy phân ngăn ngừa sự halogen hóa của tyrosin.
Thủy phân pha lỏng: Thủy phân mẫu thử protein/ peptid ở 110 °C trong 24 h. Trong quá trình thủy phân, cystein/cystin trong mẫu thử chuyển thành acid cysteic bởi tác dụng của natri azid có trong dung dịch thủy phân. Kỹ thuật 5 này cho kết quả tìm thấy của tyrosin tốt hơn kỹ thuật 4 nhưng lại không định lượng được methionin. Methionin chuyển thành hỗn hợp của methionin với 2 dẫn chất oxy hóa là methionin sulfoxid và methionin sulton.
Kỹ thuật thủy phân 6
Oxy hóa cystein/cystin bằng dimethyl sulfoxid (DMSO) (TT).
Dung dịch thủy phân: Thêm vào dung dịch acid hydrocloric 6 M, chứa 0,1 % đến 1 % phenol, một lượng DMSO để được dung dịch nồng độ DMSO 2 % (tt/tt).
Thủy phân pha hơi: Tiến hành thủy phân mẫu thử protein/peptid ở khoảng 110 °C trong 24 h. Trong quá trình thủy phân, cystein/cystin có trong mẫu thử sẽ bị DMSO có trong dung dịch thủy phân chuyển thành acid cysteic. Để hạn chế sự bất ổn định của kết quả thu được và để chỉnh lý những sai lệch do sự phân hủy từng phần, người ta khuyên nên xác định kết quả tìm thấy acid cysteic sau khi đã thủy phân oxy hóa các mẫu protein chuẩn chứa từ 1 mol đến 8 mol cystein. Các hệ số đáp ứng thu được từ các dung dịch thủy phân protein/peptid thường thấp hơn khoảng 30 % so với hệ số đáp ứng thu được từ các chuẩn acid cysteic không qua thủy phân.
Vì histidin, methionin, tyrosin và tryptophan đều bị biến đổi nên kỹ thuật 6 này không cho kết quả phân tích đầy đủ thành phần cấu tạo của protein/peptid.
Kỹ thuật thủy phân 7
Khử oxy và alkyl hóa cystein/cystin bằng phản ứng pyridylethyl hóa ở pha hơi.
Dung dịch khử: Cho vào một bình thích hợp: 83,3 μl pyridin (TT), 16,7 μl 4-vinylpyridin, 16,7 μl tributyl phosphin và 83,3 μl nước cất rồi trộn đều.
Tiến hành: Cho vào ống thủy phân một lượng mẫu thử protein/peptid (trong khoảng từ 1 μg đến 100 μg). Đặt ống thủy phân vào một ống lớn hơn đã có sẵn dung dịch khử. Hàn kín ống lớn ở chân không (áp suất khoảng 50 μm thủy ngân hoặc 6,7 Pa) rồi làm nóng ở 100 °C trong 5 min. Sau đó lấy ống thủy phân ra, làm khô trong bình hút ẩm chân không trong 15 min để loại bỏ thuốc thử dư thừa. Cuối cùng thủy phân mẫu thử đã được pyridylethyl hóa, theo một trong các kỹ thuật thủy phân mô tả ở trên. Song song, tiến hành pyridylethyl hóa trong cùng điều kiện một mẫu chuẩn protein chứa 1 mol đến 8 mol cystein để xác định giá trị tìm thấy của pyridylethyl cystein.
Chú ý: Việc kéo dài thời gian phản ứng pyridylethyl hóa sẽ gây biến đổi các nhóm α-amino và ɛ- amino của lysin có trong mẫu thử protein/peptid.
Kỹ thuật thủy phân 8
Khử oxy và alkyl hóa cystein/cystin bằng phản ứng pyridylethyl hóa ở pha lỏng.
Các dung dịch gốc: Pha chế và lọc 3 dung dịch trong nước sau đây:
Dung dịch gốc A: Dung dịch Tris-hydroclorid 1 M (pH 8,5) chứa 4 mM dinatri edetat.
Dung dịch gốc B: Dung dịch guanidin hydroclorid 8 M.
Dung dịch gốc C: Dung dịch 2-mercaptoethanol 10 %.
Dung dịch khử: Pha hỗn hợp gồm 1 thể tích dung dịch gốc A và 3 thể tích dung dịch gốc B để có dung dịch đệm guanidin hydroclorid 6 M trong Tris-hydroclorid 0,25 M.
Tiến hành: Hòa tan khoảng 10 μg mẫu thử protein/peptid trong 50 μl dung dịch khử, rồi thêm 2,5 μl dung dịch gốc C. Để 2 h ở nhiệt độ phòng, trong khí nitrogen hoặc argon và ở chỗ tối. Để thực hiện phản ứng pyridylethyl hóa, thêm vào khoảng 2 μl 4-vinylpyridin và để yên 2 h trong tối, ở nhiệt độ phòng. Sau đó, loại tạp bằng cách tách bằng sắc ký lỏng cao áp pha đảo. Làm khô mẫu thử protein/peptid sau khi qua sắc ký bằng cách ly tâm chân không rồi tiến hành thủy phân bằng acid.
Kỹ thuật thủy phân 9
Khử oxy và alkyl hóa cystein/cystin bằng phản ứng carboxymethyl hóa ở pha lỏng.
Các dung dịch gốc: Pha như ở kỹ thuật thủy phân 8.
Dung dịch carboxymethyl hóa: Dung dịch iodoacetamid 10% trong ethanol 96 %.
Dung dịch đệm: Dùng dung dịch khử của kỹ thuật thủy phân 8.
Tiến hành: Hòa tan mẫu thử protein/peptid trong 50 μl dung dịch đệm, thêm khoảng 2,5 μl dung dịch gốc C. Bảo quản 2 h trong khí nitrogen hoặc argon ở nhiệt độ phòng và trong tối. Thêm một lượng dung dịch carboxymethyl hóa gấp 1,5 lần tổng lượng các thiol có trong mẫu thử theo lý thuyết. Nếu không biết hàm lượng các thiol trong mẫu thử thì cứ 20 nanomol protein dùng 5 μl dung dịch iodoacetamid 100 mM. Để tiếp 30 min trong tối ở nhiệt độ phòng. Sau đó thêm lượng dư 2- mercaptoethanol để dừng phản ứng. Loại tạp và thu phần protein/peptid bằng cách phân tách trên sắc ký lỏng pha đảo. Làm khô phần protein/peptid thu được bằng ly tâm chân không trước khi thủy phân bằng acid.
Chất S-carboxyamidomethylcystein mới tạo thành được chuyển đổi thành S-carboxymethylcystein trong quá trình thủy phân.
Kỹ thuật thủy phân 10
Cystein/cystin tác dụng với acid dithiodiglycolic hoặc acid dithiodipropionic để cho một disulfid hỗn tạp. Tùy theo yêu cầu về độ phân giải của kỹ thuật phân tích acid amin được áp dụng mà chọn dùng acid dithiodiglycolic hoặc acid dithiodipropionic.
Dung dịch khử: Dung dịch acid dithiodiglycolic (hoặc acid dithiodipropionic) 1 % trong dung dịch NaOH 0,2 M.
Tiến hành: Cho vào ống thủy phân khoảng 20 μg mẫu thử protein/peptid. Thêm 5 μl dung dịch khử và 10 μl isopropanol. Ly tâm chân không để loại pha lỏng rồi thủy phân mẫu thử theo kỹ thuật thủy phân 1. Kỹ thuật 10 này có lợi là các thành phần acid amin khác trong mẫu thử không bị ảnh hưởng của phản ứng và không cần loại tạp trước khi thủy phân.
Kỹ thuật thủy phân 11
Trong quá trình thủy phân bằng acid, asparagin và glutamin được chuyển đổi thành acid aspartic và acid glutamic tương ứng. Asparagin và acid aspartic được biểu thị bằng một đại lượng chung Asx, còn glutamin và acid glutamic bằng Glx. Trái lại, khi thủy phân bằng acid với sự có mặt của thuốc thử bis (1,1-trifluoroacetoxy)iodobenzen (BTI), asparagin và glu-tamin bị tác dụng và chuyển tương ứng thành acid diaminopropionic và acid diaminobutyric. Nhờ đó, ta xác định được asparagin và glutamin trong protein/peptid ngay khi có mặt của acid aspartic và acid glutamic.
Các dung dịch khử: Pha chế và lọc 3 dung dịch sau:
Dung dịch A: Dung dịch acid trifluoroacetic 10 mM.
Dung dịch B: Dung dịch chứa guanidin hydroclorid 5 M và acid trifluoroacetic 10 mM.
Dung dịch C: Dung dịch mới pha BTI 3,6% trong dimethylformamid.
Tiến hành: Cho vào một ống thủy phân sạch khoảng 200 μg mẫu thử protein/peptid, 2 ml dung dịch A hoặc dung dịch B và 2 ml dung dịch C. Hàn ống thủy phân trong chân không. Để 4 h ở nhiệt độ 60 °C, trong tối. Sau đó thẩm tách mẫu thử bằng nước cất để loại bỏ thuốc thử dư thừa. Chiết mẫu thử đã thẩm tách 3 lần bằng 3 thể tích tương đương butyl acetat, rồi làm đông khô.
Cuối cùng thủy phân mẫu thử đông khô theo các kỹ thuật thủy phân đã nói ở trên.
Các acid α,β-diaminopropionic và α,γ- diaminobutyric đều không được phân giải rõ ràng với lysin có trong mẫu thử, khi phân tách bằng sắc ký trao đổi ion. Vì vậy, khi dùng sắc ký trao đổi ion để tách các acid amin, hàm lượng của asparagin và của glutamin có mặt trong mẫu thử được xác định bằng hiệu số giữa hàm lượng của acid aspactic và của acid glutamic tương ứng thu được khi thủy phân acid không có BTI và hàm lượng của acid aspartic và của acid glutamic tương ứng thu được khi thủy phân có BTI.
Hàm lượng của threonin, methionin, cystein, tyrosin và histidin có thể bị sai lệch khi thủy phân bằng acid có BTI. Vì vậy, muốn có giá trị đúng của các hàm lượng này, mẫu thử phải được thủy phân bằng acid, không có thuốc thử BTI.
Tách và phát hiện các acid amin
Có nhiều kỹ thuật để tách và phát hiện các acid amin. Lựa chọn kỹ thuật nào phụ thuộc vào yêu cầu về độ nhạy của phép định lượng. Nhìn chung, khoảng một nửa các phép phân tích acid amin dựa trên việc tách thành acid amin tự do bằng sắc ký lỏng trao đổi ion rồi tạo dẫn chất sau cột phân tách. Kỹ thuật tạo dẫn chất sau cột có thể áp dụng cho các mẫu thử có một lượng nhỏ chất đệm (như các muối và urê) và thường cần từ 5 μg đến 10 μg mẫu thử protein cho một lần phân tích.
Các kỹ thuật còn lại, bao gồm việc tạo các dẫn chất trước cột rồi tách chúng bằng sắc ký lỏng cao áp pha đảo. Các kỹ thuật tạo dẫn chất trước cột rất nhạy và thường chỉ cần từ 0,5 μg đến 1,0 μg mẫu thử protein/peptid cho mỗi lần phân tích, nhưng lại có thể bị ảnh hưởng bởi các muối đệm có trong mẫu thử. Mặt khác, kỹ thuật tạo dẫn chất trước cột còn có thể cho nhiều dẫn chất khác nhau của cùng một acid amin, do đó gây trở ngại cho việc biện giải kết quả phân tích. So với kỹ thuật tạo dẫn chất trước cột, thường kỹ thuật tạo dẫn chất sau cột ít bị ảnh hưởng bởi các biến thiên trong việc thực hiện phép định lượng hơn.
Có thể dùng các kỹ thuật sau đây để phân tích định lượng các acid amin.
Thiết bị và thuốc thử đều có sẵn trên thị trường. Hơn nữa, hiện có nhiều cải tiến về các mặt như pha chế thuốc thử, cách tiến hành phản ứng, thiết bị sắc ký dùng trong các kỹ thuật này, v.v.... Các thông số đặc trưng có thể thay đổi tùy thuộc vào thiết bị đã dùng và cách tiến hành phân tích. Nhiều phòng thí nghiệm còn áp dụng đồng thời nhiều kỹ thuật chứ không chỉ một, để tận dụng lợi ích của mỗi kỹ thuật đã áp dụng. Trong các kỹ thuật sau đây, tín hiệu tương tự được hiển thị trên máy ghi và diện tích các pic được tích phân để định lượng.
Kỹ thuật 1: Tạo dẫn chất sau cột với thuốc thử ninhydrin
Sắc ký trao đổi ion tạo dẫn chất sau cột với thuốc thử ninhydrin là một trong các kỹ thuật thông dụng nhất để phân tích định lượng các acid amin. Theo nguyên tắc, sắc ký trao đổi cation, dùng lithi làm đối ion trong pha động được áp dụng để phân tích các mẫu thử sinh học phức tạp; trái lại, sắc ký trao đổi cation, dùng natri làm đối ion trong pha động, nhanh hơn, được dùng với các mẫu thử đơn giản hơn, gồm hỗn hợp các acid amin đã được thủy phân từ protein/peptid ra (tiêu biểu gồm 17 acid amin khác nhau). Việc phân tích acid amin trên cột trao đổi ion được thực hiện thông qua sự phối hợp giữa thay đổi pH và thay đổi lực ion của pha động (chương trình dung môi). Chương trình nhiệt độ cũng thường được áp dụng để tăng nhanh sự phân tách.
Các acid amin bậc nhất tác dụng với ninhydrin cho hợp chất màu tím, hấp thụ cực đại ở bước sóng 570 nm. Trái lại, các acid amin bậc 2 (các imino acid) như prolin, tác dụng với ninhydrin cho hợp chất màu vàng, hấp thụ cực đại ở 440 nm. Vì vậy ta phát hiện các dẫn chất của acid amin thu được sau cột ở các bước sóng 440 nm và 570 nm.
Đối với đa số các dẫn chất acid amin, giới hạn phát hiện được xác định là 10 picomol; đối với dẫn chất của prolin, giới hạn đó là 50 picomol. Khoảng tuyến tính là 20 picomol đến 500 picomol, với hệ số tương quan r > 0,999. Để có kết quả tốt, dùng một lượng mẫu thử lớn hơn 1 μg để thủy phân trước khi phân tích các acid amin trong protein/peptid là thích hợp nhất.
Kỹ thuật 2: Tạo dẫn chất sau cột với thuốc thử OPA
Thuốc thử ortho-phthalaldehyd (OPA) tác dụng với các amin bậc nhất, khi có mặt một hợp chất thiol, cho các hợp chất isoindol phát huỳnh quang. OPA không tác dụng với các amin bậc 2 (các imino acid như prolin) để có hợp chất phát huỳnh quang; tuy nhiên, nếu oxy hóa chúng bằng natri hypoclorid hoặc cloramin T thì phản ứng với OPA sẽ xảy ra. Do đó, để phân tích các acid amin trong mẫu thử protein/peptid bằng kỹ thuật này, ta phải tách các acid amin tự do bằng sắc ký trao đổi cation mạnh rồi oxy hóa sau cột bằng natri hypoclorid hoặc cloramin T và tiếp đó tạo dẫn chất phát huỳnh quang bằng thuốc thử OPA với sự có mặt của một hợp chất thiol như N-acetyl-L-cystein hoặc 2-mercaptoethanol. Các acid amin bậc nhất không bị ảnh hưởng bởi sự oxy hóa nói trên.
Việc tách các acid amin bằng sắc ký lỏng trao đổi ion được thực hiện nhờ sự phối hợp giữa việc thay đổi pH và thay đổi lực ion của pha động (chương trình dung môi). Sau khi được phân tách, các dẫn chất phát huỳnh quang sẽ được phát hiện với bước sóng kích thích ở 348 nm và bước sóng phát quang ở 450 nm.
Giới hạn phát hiện được xác định ở mức một vài chục picomol, đối với đa số các dẫn chất OPA của acid amin. Khoảng tuyến tính là từ vài picomol đến vài chục nanomol. Để có kết quả phân tích tốt, lấy mẫu thử protein/peptid lớn hơn 500 ng để thủy phân.
Kỹ thuật 3: Tạo dẫn chất trước cột với thuốc thử PITC
Thuốc thử phenylisothiocyanat (PITC) tác dụng với các acid amin để thành dẫn chất phenylthiocarbamyl (PTC) hấp thụ mạnh bức xạ ở bước sóng 254 nm. Tách các dẫn chất PTC của acid amin, đã được tạo trước cột, bằng sắc ký lỏng cao áp pha đảo, dùng cột octadecylsilyl (ODS) và phát hiện ở bước sóng 254 nm.
Việc phân tách các dẫn chất acid amin bằng sắc ký lỏng cao áp pha đảo được thực hiện nhờ việc phối hợp giữa các thay đổi về nồng độ acetonitril và về lực ion trong pha động.
Đối với đa số các dẫn chất PTC-acid amin, giới hạn phát hiện được xác định là 1 picomol. Khoảng tuyến tính là 20 picomol đến 500 picomol với hệ số tương quan r > 0,999. Để có kết quả phân tích tốt, lấy lượng mẫu thử lớn hơn 500 ng để thủy phân.
Chú ý: Sau khi đã loại bỏ trong chân không thuốc thử dư, các dẫn chất PTC-acid amin có thể bảo quản khô và đông băng trong vài tuần lễ mà không có sự phân hủy nào đáng kể. Nếu dung dịch để tiêm vào máy sắc ký được bảo quản lạnh, sau 3 ngày cũng chưa thấy suy giảm đáp ứng sắc ký nào đáng kể xảy ra.
Kỹ thuật 4: Tạo dẫn chất trước cột với thuốc thử AQC
Thuốc thử 6-aminoquinolyl-N-hydroxysuccinimidyl car-bamat (AQC) tác dụng với các acid amin tạo thành các dẫn chất urê bất đối xứng, ổn định, phát huỳnh quang (các dẫn chất AQC-acid amin) và được phân tách bằng sắc ký lỏng cao áp pha đảo. Do đó, ta tạo dẫn chất AQC trước cột rồi tách chúng bằng sắc ký lỏng cao áp pha đảo, dùng cột ODS và phát hiện bằng detector huỳnh quang, ở bước sóng kích thích 250 nm và bước sóng phát quang ở 395 nm. Việc phân tách được thực hiện nhờ việc phối hợp sự thay đổi nồng độ acetonitril và thay đổi lực ion của pha động (chương trình dung môi). Tiêm trực tiếp hỗn hợp mẫu thử và thuốc thử vào máy sắc ký. Không có nhiễu đáng kể gây ra bởi 6-aminoquinolin, sản phẩm thứ cấp chủ yếu phát huỳnh quang của thuốc thử. Thuốc thử dư được thủy phân nhanh chóng (nửa đời < 15 s) thành các chất 6-aminoquinolin, N-hydroxysuccinimid và carbon dioxyd và sau 1 min, không còn phản ứng tạo dẫn chất xảy ra.
Diện tích pic của các dẫn chất AQC-acid amin không thay đổi chủ yếu trong thời gian 1 tuần, ở nhiệt độ phòng. Nhờ tính ổn định này, có thể tiến hành phân tích qua đêm trên các máy tự động được.
Giới hạn phát hiện được xác định ở vào khoảng từ 40 femtomol đến 320 femtomol, tính cho mỗi acid amin, trừ cystein. Giới hạn phát hiện của cystein xấp xỉ bằng 800 femtomol. Khoảng tuyến tính là từ 2,5 icromol đến 200 micromol, với hệ số tương quan r > 0,999. Có thể thu được kết quả phân tích tốt với lượng mẫu protein/peptid lấy thử thấp tới 30 ng.
Kỹ thuật 5: Tạo dẫn chất trước cột với thuốc thử OPA
Thuốc thử orthophthalaldehyd (OPA), khi có mặt một hợp chất thiol (có thể dùng 2-mercaptoethanol hoặc acid 3-mercaptopropionic), sẽ tác dụng với các amin bậc nhất để cho một hợp chất isoindol phát huỳnh quang mạnh. Bản thân thuốc thử OPA không phát huỳnh quang, nên không gây trở ngại. Mặt khác, vì OPA dễ tan và ổn định trong nước đồng thời cho phản ứng nhanh nên có thể tạo dẫn chất và phân tích mẫu thử một cách tự động, dùng thiết bị tự nạp mẫu thử để trộn lẫn mẫu thử với thuốc thử. Tuy nhiên, OPA không cho phản ứng với các amin bậc 2, nên kỹ thuật 5 này không áp dụng được để phân tích các acid amin có hóa chức amin bậc 2 như prolin. Để khắc phục, phải phối hợp kỹ thuật 5 này với kỹ thuật 7 hoặc 8 mô tả sau đây.
Vì dẫn chất OPA-acid amin không bền vững nên sau khi tạo dẫn chất trước cột, phải tiến hành phân tích ngay trên sắc ký lỏng cao áp pha đảo và phát hiện kết quả bằng detector huỳnh quang ở bước sóng kích thích 348 nm và bước sóng phát quang 450 nm.
Giới hạn phát hiện bằng huỳnh quang thấp tới 50 femtomol đã được báo cáo. Tuy nhiên, giới hạn phát hiện thực tế của phép phân tích là 1 picomol.
Kỹ thuật 6: Tạo dẫn chất trước cột bằng thuốc thử DABS-CI
Thuốc thử dimethylamino-azobenzensulfonyl clorid (DABS-CI) là một thuốc thử cho màu để phát hiện acid amin. Các dẫn chất tạo thành (DABS-acid amin) rất bền vững và hấp thụ cực đại ở bước sóng 436 nm. Tạo các dẫn chất này trước cột rồi tách chung bằng sắc ký lỏng cao áp pha đảo, dùng cột ODS và phát hiện kết quả ở bước sóng 436 nm.
Kỹ thuật này có thể phân tích cả các acid amin bậc nhất và acid amin bậc hai (như prolin) với cùng độ nhạy như nhau. Còn có thể định lượng đồng thời thành phần tryptophan có trong mẫu thử protein/peptid sau khi đã thủy phân mẫu thử bằng các acid sulfonic như: acid mercaptoethansulfonic, acid p-toluensulfonic hoặc acid methansulfonic như đã mô tả ở kỹ thuật thủy phân 2 ở trên. Các thành phần khác dễ hỏng vì acid như asparagin và glutamin cũng được phân tích sau khi đã làm phản ứng chuyển đổi thành acid diaminopropionic và acid diaminobutyric tương ứng bằng phản ứng với thuốc thử BTI đã mô tả ở kỹ thuật thủy phân 11 ở trên.
Trong kỹ thuật này, không dùng norleucin nhân tạo để làm chuẩn nội, vì nó được rửa giải ra tại vùng dày đặc các pic của các acid amin bậc nhất trên sắc đồ. Có thể dùng nitrotyrosin làm chuẩn nội vì nó được rửa giải ra tại vùng có ít pic trên sắc đồ.
Giới hạn phát hiện các DABS-acid amin ở khoảng 1 picomol. Lượng nhỏ từ 2 đến 5 picomol của một DABS-acid amin nào đó cũng có thể cho kết quả định lượng đáng tin cậy, và chỉ cần dùng 10 ng đến 30 ng protein thủy phân đã được dẫn chất hóa cho mỗi lần phân tích.
Kỹ thuật 7: Tạo dẫn chất trước cột với thuốc thử FMOC-CI
Thuốc thử 9-fluorenylmethyl cloroformat (FMOC-CI) tác dụng với acid amin bậc nhất và acid amin bậc hai để tạo thành các dẫn chất FMOC-acid amin phát huỳnh quang mạnh.
Phản ứng xảy ra nhẹ nhàng trong môi trường nước, trong 30 s. Các dẫn chất được tạo thành đều bền vững, chỉ riêng dẫn chất của histidin là có biểu hiện phân hủy nào đó. Mặc dù bản thân thuốc thử và các sản phẩm phụ của phản ứng đều phát huỳnh quang nhưng ta có thể loại chúng mà không làm mất mát các dẫn chất FMOC-acid amin.
Sau khi tạo dẫn chất trước cột, tiến hành tách các FMOC-acid amin bằng sắc ký lỏng cao áp pha đảo, dùng cột ODS, với chương trình gradient dung môi, biến thiên tuyến tính từ hỗn hợp gồm 10 thể tích acetonitril, 40 thể tích methanol và 50 thể tích đệm acid acetic đến hỗn hợp gồm 50 thể tích acetonitril, 50 thể tích đệm acid acetic. Phát hiện kết quả bằng detector huỳnh quang ở bước sóng kích thích 260 nm và bước sóng phát quang 313 nm. Có thể phân tích được 20 dẫn chất acid amin trong vòng 20 min.
Giới hạn phát hiện vào cỡ femtomol. Đa số các acid amin có khoảng tuyến tính từ 0,1 micromol đến 50 micromol.
Kỹ thuật 8: Tạo dẫn chất trước cột bằng thuốc thử NBD-F
Thuốc thử 7-fluoro-4-nitrobenzo-2-oxa-1,3-diazol (NBD-F) tác dụng với cả acid amin bậc nhất và acid amin bậc 2, ở nhiệt độ 60 °C, trong 5 min, để cho các dẫn chất NBD-acid amin phát huỳnh quang mạnh. Sau khi tạo dẫn chất trước cột, tách chúng bằng sắc ký lỏng cao áp pha đảo, dùng cột ODS và chương trình gradient dung môi gồm acetonitril và hỗn hợp đệm trong nước. 17 dẫn chất acid amin được tách ra trong vòng 35 min. Có thể dùng acid ɛ-aminocaproic làm chất chuẩn nội, vì được rửa giải ở vùng ít pic trên sắc đồ. Phát hiện kết quả bằng detector huỳnh quang, ở bước sóng kích thích 480 nm và bước sóng phát quang ở 530 nm.
Kỹ thuật này có độ nhạy gần tương tự như độ nhạy của kỹ thuật tạo dẫn chất trước cột với thuốc thử OPA (kỹ thuật 5) đối với các acid amin, trừ prolin không phân tích được bằng kỹ thuật 5 vì không phản ứng với thuốc thử OPA. Như vậy, kỹ thuật 8 này ích lợi hơn kỹ thuật 5.
Giới hạn phát hiện mỗi acid amin ở khoảng 10 femtomol và lượng mẫu thử protein/peptid thủy phân cần lấy để tạo dẫn chất trước cột ở khoảng 1,5 mg.
Tính kết quả
Khi xác định hàm lượng các acid amin trong một mẫu thử protein/peptid, cần nhớ rằng, trong quá trình thủy phân bằng acid, tryptophan và cystein bị phá hủy, serin và threonin bị phá hủy một phần, valin và isoleucin không được tách hoàn toàn, methionin có thể bị oxy hóa và một vài acid amin như glycin và serin thường bị nhiễu. Tiến hành phân tích ở môi trường chân không thích hợp (áp suất thấp hơn 200 μm thủy ngân hoặc 26,7 Pa) hoặc ở môi trường có khí trơ (như argon) khi thủy phân pha hơi, có thể giảm mức phân hủy do bị oxy hóa. Các kết quả định lượng cystein, tryptophan, threonin, isoleucin, valin, methionin, glycin và serin trong một mẫu thử protein/peptid đã thủy phân có thể thay đổi và do đó thường cần tiến hành xem xét đánh giá bổ sung.
Tính tỷ lệ phần trăm hàm lượng của một loại acid amin có trong mẫu thử protein/peptid
Đó là số lượng nanomol của một loại acid amin có trong 100 nanomol của toàn thể các acid amin có trong mẫu thử protein/peptid. Tỷ lệ này có ích trong việc đánh giá các dữ liệu thu được khi phân tích các acid amin trong một protein mà ta chưa biết khối lượng phân tử. Nó giúp củng cố kết quả định tính một protein/peptid chưa biết bằng cách so sánh tỷ lệ phần trăm hàm lượng mỗi loại acid amin trong mẫu thử chưa biết với tỷ lệ phần trăm hàm lượng của mỗi loại acid amin tương ứng có trong các protein/peptid đã biết.
Tính tỷ lệ phần trăm hàm lượng của một acid amin trong protein/peptid theo công thức sau:
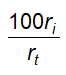
trong đó:
ri: đáp ứng (hàm lượng) tính theo nanomol của acid amin i;
rt: đáp ứng (hàm lượng) tính theo nanomol của tất cả các acid amin thu được trên sắc đồ.
Mẫu thử protein/peptid chưa biết
Xác định khối lượng Qi (tính theo microgam) của mỗi loại acid amin có mặt trong mẫu protein/peptid chưa biết
Tính bằng công thức sau:
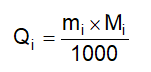
trong đó:
Qi: khối lượng (tính theo microgam) của acid amin i có trong mẫu thử;
mi: hàm lượng (tính theo nanomol) của tất cả các acid amin i tìm thấy trên sắc đồ;
Mi: phân tử lượng trung bình (tính theo gam) của acid amin i, đã được hiệu chỉnh về khối lượng H2O bị loại khi tạo thành liên kết peptid.
Ước lượng tổng khối lượng của mẫu thử protein/peptid:
Tổng khối lượng ∑Qi của các loại acid amin tìm thấy cho phép ta ước lượng khối lượng của protein/peptid đem thử sau khi đã hiệu chỉnh về khối lượng mất mát do có sự phân hủy từng phần hoặc toàn phần của một số acid amin dễ bị phân hủy trong quá trình thủy phân.
Xác định số lượng của mỗi loại acid amin tham gia cấu tạo mẫu thử protein/peptid chưa biết. Nếu xác định được phân tử lượng Mp của protein/peptid đem thử (ví dụ bằng khối phổ), ta tính số lượng của mỗi loại acid amin i theo công thức sau:
trong đó:
mi: hàm lượng tính theo nanomol của acid amin i tìm thấy trong mẫu thử;
Qp: khối lượng, tính theo microgam, của protein đem thử;
Mp: phân tử lượng của protein đem thử (tính theo gam).
Mẫu thử protein/peptid đã biết phân tử lượng và thành phần cấu tạo
Khi phân tích acid amin, một số acid amin cho kết quả tìm thấy tốt, trái lại, một số cho kết quả tìm thấy không sử dụng được vì hoặc bị phân hủy một phần hay toàn phần (ví dụ: tryptophan, cystein, threonin, serin, methionin), hoặc liên kết peptid không được tách hoàn toàn (như liên kết của isoleucin, của valin), hoặc bị nhiễu bởi một số acid amin tự do (như bởi glycin và serin). Các acid amin cho kết quả tìm thấy tốt điển hình là aspartat-asparagin, glutamat-glutamin, alanin, leucin, phenylalanin, lysin và arginin. Danh sách này có thể thay đổi tùy thuộc vào hệ thống phân tích đã dùng. Các acid amin cho kết quả tìm thấy tốt đại diện cho protein, do đó ta lợi dụng chúng để xác định hàm lượng protein và số lượng của mỗi loại acid amin (còn gọi là thành phần của acid amin) có trong mẫu thử.
Xác định hàm lượng protein trong mẫu thử
Chia hàm lượng (tính theo nanomol) của mỗi loại acid amin có giá trị tìm thấy tốt cho số lượng dự đoán của acid amin đó trong protein để có hàm lượng protein tính theo loại acid amin đó. Tính giá trị trung bình của tất cả các hàm lượng protein tính được theo từng loại acid amin có giá trị tìm thấy tốt của mẫu thử. Các hàm lượng protein tính theo từng loại acid amin riêng rẽ phải được phân bố đồng đều xung quanh giá trị trung bình của hàm lượng protein mới tính được ở trên. Phải loại bỏ các giá trị hàm lượng protein riêng rẽ của từng acid amin quá sai lệch với giá trị trung bình đã tính được. Thường các giá trị sai lệch quá 5 % phải loại bỏ. Khi đó, phải tính lại giá trị trung bình của hàm lượng protein mới, dựa trên các giá trị còn lại (không bị loại bỏ) của hàm lượng protein riêng rẽ tính theo từng loại acid amin còn lại.
Xác định số lượng của từng loại acid amin trong mẫu thử
Chia hàm lượng của mỗi loại acid amin cho giá trị trung bình của hàm lượng protein đã tính ở trên, ta được số lượng của loại acid amin đó (tức thành phần acid amin) trong mẫu thử.
Tính sai số tương đối (theo tỷ lệ phần trăm) về thành phần trong mẫu thử
Áp dụng công thức sau để tính sai số tương đối về thành phần đối với một loại acid amin i:
![]()
trong đó:
mi: hàm lượng, tính theo nanomol, xác định bằng thực nghiệm, của loại acid amin i có trong mẫu thử;
mis: hàm lượng (tính theo nanomol) đã biết của loại acid amin i có trong mẫu thử.
Giá trị sai số tương đối trung bình về thành phần của mẫu thử là giá trị trung bình của tất cả các sai số tương đối về thành phần tính theo từng loại acid amin riêng rẽ, trừ tryptophan và cystein. Giá trị sai số tương đối trung bình về thành phần của một mẫu thử cung cấp thông tin quan trọng về độ ổn định của phép phân tích theo thời gian. Sự phù hợp giữa giá trị thành phần acid amin trong mẫu thử, tìm thấy bằng thực nghiệm với giá trị thành phần acid amin đã biết trước của protein đem thử có thể giúp cho việc củng cố kết quả định tính và xác định độ tinh khiết của protein trong mẫu thử.
10.12 XÁC ĐỊNH HÀM LƯỢNG ETHANOL
Sử dụng phương pháp 1 hoặc phương pháp 2 trừ khi có những chỉ dẫn khác trong chuyên luận riêng.
Phương pháp 1
Tiến hành phương pháp sắc ký khí (Phụ lục 5.2) sử dụng các dung dịch sau:
Dung dịch 1: Chứa 5 % (tt/tt) ethanol (TT) và 5 % (tt/tt) propan 1-ol (TT) (chuẩn nội).
Dung dịch 2: Hòa loãng một thể tích chế phẩm thử bằng nước để được dung dịch có nồng độ ethanol từ 4,0 % đến 6,0 % (tt/tt).
Dung dịch 3: Chuẩn bị như dung dịch 2 nhưng thêm vừa đủ chất chuẩn nội để tạo ra nồng độ cuối cùng của chuẩn nội trong dung dịch này là 5,0 % (tt/tt).
Quá trình sắc ký có thể thực hiện bằng cách dùng cột (1,5 m × 4 mm) đã nhồi hạt xốp polymer (100 mesh đến 120 mesh) (Porapak Q hoặc Chromosorb 101 đều thích hợp) với nhiệt độ lò cột đặt ở 150 °C, nhiệt độ buồng tiêm và detector ở 170 °C.
Tính hàm lượng phần trăm ethanol căn cứ vào các diện tích pic ethanol trong sắc ký đồ thu được từ dung dịch 1 và dung dịch 3.
Phương pháp 2
Với những chế phẩm thử mà trong đó theo quy định của chuyên luận riêng đã sử dụng cồn công nghiệp, xác định hàm lượng ethanol giống như phương pháp 1 nhưng chuẩn bị dung dịch 2 bằng cách hòa loãng một thể tích chế phẩm thử với nước để tạo một dung dịch có nồng độ tổng cộng của methanol và ethanol từ 4,0 % đến 6,0 % (tt/tt).
Xác định hàm lượng methanol bằng cách thực hiện quá trình sắc ký như đã mô tả ở phương pháp 1 nhưng chuẩn bị các dung dịch như sau:
Dung dịch 1: Chứa 0,25 % (tt/tt) methanol (T) và 0,25 % (tt/tt) propan 1-ol (TT).
Dung dịch 2: Hòa loãng một thể tích chế phẩm thử bằng nước để được dung dịch có nồng độ methanol từ 0,2 % đến 0,3 % (tt/tt).
Dung dịch 3: Chuẩn bị như dung dịch 2 nhưng thêm vừa đủ chất chuẩn nội để có nồng độ cuối cùng của chuẩn nội trong dung dịch này là 0,25 %.
Tổng lượng ethanol và methanol phải ở trong giới hạn quy định của chuyên luận riêng và tỷ số giữa lượng methanol và lượng ethanol tìm được phải tương ứng với tỷ lệ này của cồn công nghiệp đã sử dụng.
Phương pháp 3
Phương pháp này chỉ dùng để khảo sát những chế phẩm thuốc dạng lỏng, chắc chắn chứa ethanol đồng thời với các chất tan khác, nhưng các chất này phải được tách khỏi ethanol khi cất.
Khi cất nếu trong mẫu thử, ngoài ethanol và nước, còn có lẫn các chất bay hơi khác thì các hướng dẫn thích hợp sẽ được quy định trong chuyên luận riêng.
Hàm lượng ethanol chứa trong một chất lỏng được biểu thị bằng số thể tích ethanol trong 100 thể tích chất lỏng ấy ở 20 °C ± 0,1 °C và được hiểu là phần trăm ethanol tính theo thể tích/thể tích.
Hàm lượng cũng có thể được biểu thị bằng gam ethanol cho 100 g chất lỏng và được hiểu là phần trăm ethanol tính theo khối lượng/khối lượng.
Liên quan giữa tỷ trọng ở 20 °C ± 0,1 °C, tỷ trọng tương đối (đã hiệu chỉnh ở chân không) và hàm lượng ethanol của hỗn hợp nước và ethanol được ghi trong Bảng của Tổ chức Quốc tế về Đo lường hợp pháp (1972) *Khuyến cáo Quốc tế số 22**.
[*International Organisation for Legal Metrology (1972).
** International Recommendation (No 22)].
Thiết bị
Thiết bị (Hình 10.12) bao gồm một bình cầu thủy tinh đáy tròn (A) gắn với một đầu cất có bộ phận bẫy hơi (B), bộ phận này được nối với một ống sinh hàn (C) đặt thẳng đứng. Tiếp theo là một ống dẫn gắn vào phần thấp của ống sinh hàn để dẫn dịch cất vào bình hứng (D). Bình hứng có vạch định mức dung tích 100 ml hoặc 250 ml. Trong suốt quá trình cất, bình hứng này được nhúng trong cốc (E) chứa hỗn hợp đá và nước đá. Ngoài ra còn có thêm một đĩa có khoét thủng một lỗ tròn đường kính khoảng 6 cm đặt dưới bình cất để bảo hiểm.
Hình 10.12 - Dụng cụ để xác định hàm lượng ethanol
(Kích thước tính bằng mm)
Xác định bằng lọ đo tỷ trọng (Pycnomet)
Đong chính xác 25 ml chế phẩm ở 20 °C ± 0,1 °C chuyển vào bình cất. Pha loãng với 100 ml đến 150 ml nước cất và thêm vài hạt đá bọt. Lắp hệ thống chưng cất. Chưng cất và hứng không dưới 90 ml dịch cất vào bình định mức 100 ml. Điều chỉnh nhiệt độ dịch cất tới 20 °C ± 0,1 °C, pha loãng đến 100 ml bằng nước cất ở 20 °C ± 0,1 °C. Xác định tỷ trọng tương đối ở 20 °C ± 0,1 °C bằng lọ đo tỷ trọng (Phụ lục 6.5).
Hàm lượng ethanol tính bằng phần trăm theo thể tích bằng 4 lần trị số chỉ ra trong cột 3 ở Bảng 10.12. (Liên quan giữa tỷ trọng, tỷ trọng tương đối và hàm lượng ethanol trong dung dịch). Biểu thị kết quả với một chữ số ở phần thập phân.
Xác định bằng tỷ trọng kế (Phụ lục 6.5)
Đong chính xác 50 ml chế phẩm ở 20 °C ± 0,1 °C chuyển vào bình cất. Pha loãng với 200 ml đến 250 ml nước cất và thêm vài hạt đá bọt. Lắp hệ thống chưng cất. Chưng cất và hứng không dưới 180 ml dịch cất vào bình định mức 250 ml. Điều chỉnh nhiệt độ dịch cất tới 20 °C ± 0,1 °C; pha loãng đến 250 ml bằng nước cất ở 20 °C ± 0,1 °C.
Chuyển dịch cất vào một ống đong hình trụ có đường kính lớn hơn đường kính bầu của tỷ trọng kế ít nhất 6 mm. Nếu thể tích dịch cất không đủ, tăng lượng mẫu gấp đôi và pha loãng dịch cất đến 500 ml bằng nước cất ở 20 °C ± 0,1 °C.
Hàm lượng ethanol tính bằng phần trăm theo thể tích bằng 5 lần trị số chỉ ra trong cột 3 ở Bảng 10.12 (Liên quan giữa tỷ trọng, tỷ trọng tương đối và hàm lượng ethanol trong dung dịch). Biểu thị kết quả ở dạng có một chữ số ở phần thập phân.
Bảng 10.12 - Liên quan giữa tỷ trọng, tỷ trọng tương đối và hàm lượng ethanol
| ρ 20 (kg.m-3) | Tỷ trọng tương đối của dịch cất đo trong không khí ở 20 °C ( | Hàm lượng ethanol tính theo % (tt/tt) ở 20 °C | ρ 20 (kg.m-3) | Tỷ trọng tương đối của dịch cất đo trong không khí ở 20 °C ( | Hàm lượng ethanol tính theo % (tt/tt) ở 20 °C |
| 1 | 2 | 3 | 1 | 2 | 3 |
| 968,0 | 0,9697 | 25,09 | 983,5 | 0,9853 | 11,02 |
| 968,5 | 0,9702 | 24,64 | 984,0 | 0,9858 | 10,60 |
| 969,0 | 0,9707 | 24,19 | 984,5 | 0,9863 | 10,18 |
| 969,5 | 0,9712 | 23,74 | 985,0 | 0,9868 | 9,76 |
| 970,0 | 0,9717 | 23,29 | 985,5 | 0,9873 | 9,35 |
| 970,5 | 0,9722 | 22,83 | 986,0 | 0,9878 | 8,94 |
| 971,0 | 0,9727 | 22,37 | 986,5 | 0,9883 | 8,53 |
| 971,5 | 0,9733 | 21,91 | 987,0 | 0,9888 | 8,13 |
| 972,0 | 0,9738 | 21,45 | 987,5 | 0,9893 | 7,73 |
| 972,5 | 0,9743 | 20,98 | 988,0 | 0,9898 | 7,34 |
| 973,0 | 0,9748 | 20,52 | 988,5 | 0,9903 | 6,95 |
| 973,5 | 0,9753 | 20,05 | 989,0 | 0,9908 | 6,56 |
| 974,0 | 0,9758 | 19,59 | 989,5 | 0,9913 | 6,17 |
| 974,5 | 0,9763 | 19,12 | 990,0 | 0,9918 | 5,79 |
| 975,0 | 0,9768 | 18,66 | 990,5 | 0,9923 | 5,42 |
| 975,5 | 0,9773 | 18,19 | 991,0 | 0,9928 | 5,04 |
| 976,0 | 0,9778 | 17,73 | 991,5 | 0,9933 | 4,67 |
| 976,5 | 0,9783 | 17,25 | 992,0 | 0,9938 | 4,30 |
| 977,0 | 0,9788 | 16,80 | 992,5 | 0,9943 | 3,94 |
| 977,5 | 0,9793 | 16,34 | 993,0 | 0,9948 | 3,58 |
| 978,0 | 0,9798 | 15,88 | 993,5 | 0,9953 | 3,22 |
| 978,5 | 0,9803 | 15,43 | 994,0 | 0,9958 | 2,86 |
| 979,0 | 0,9808 | 14,97 | 994,5 | 0,9963 | 2,51 |
| 979,5 | 0,9813 | 14,52 | 995,0 | 0,9968 | 2,16 |
| 980,0 | 0,9818 | 14,07 | 995,5 | 0,9973 | 1,82 |
| 980,5 | 0,9823 | 13,63 | 996,0 | 0,9978 | 1.47 |
| 981,0 | 0,9828 | 13,18 | 996,5 | 0,9983 | 1,13 |
| 981,5 | 0,9833 | 12,74 | 997,0 | 0,9988 | 0,80 |
| 982,0 | 0,9838 | 12,31 | 997,5 | 0,9993 | 0,46 |
| 982,5 | 0,9843 | 11,87 | 998,0 | 0,9998 | 0,13 |
| 983,0 | 0,9848 | 11,44 |
|
|
|
10.13 XÁC ĐỊNH HÀM LƯỢNG METHANOL VÀ PROPAN-2-OL
Phương pháp
Tiến hành phương pháp sắc ký khí (Phụ lục 5.2), sử dụng các dung dịch sau:
Dung dịch chuẩn nội: Chuẩn bị một dung dịch chứa propan -1- ol (TT) 2,5 %.
Dung dịch thử: Thêm 2,0 ml dung dịch chuẩn nội vào bình định mức dung tích 50 ml có chứa dịch cất mẫu thử (dịch cất thu được từ phương pháp 3, chuyên luận xác định hàm lượng ethanol, Phụ lục 10.12), điều chỉnh hàm lượng ethanol tới 10 % (tt/tt) bằng cách pha loãng với nước hoặc với ethanol 90 % (TT) vừa đủ để đạt tới 50 ml dung dịch thử, tùy theo hàm lượng ethanol trong dịch cất cao hay thấp hơn 10,0 % (tt/tt).
Dung dịch đối chiếu: Chuẩn bị 50 ml dung dịch chứa 2,0 ml dung dịch chuẩn nội, 10 % (tt/tt) ethanol (TT), 0,05 % (tt/tt) propan-2-ol (TT) và một lượng methanol khan (TT) vừa đủ để có nồng độ tổng cộng của methanol trong dung dịch này là 0,05 % (tt/tt) kể cả lượng methanol chứa trong ethanol (TT).
Quá trình sắc ký
Có thể thực hiện bằng cách sử dụng cột thủy tinh đã nhồi hạt xốp copolymer ethylvinylbenzen divinyl- benzen (125 μm đến 150 μm) với nhiệt độ cột đặt và duy trì ở 130 °C, buồng tiêm ở 200 °C, detector 220 °C. Tiêm 1 μl mỗi dung dịch trên. Từ các sắc ký đồ thu được, tính hàm lượng methanol và propan-2-ol có trong sản phẩm gốc.
Độ nhạy của phương pháp
Với phương pháp này có thể phát hiện được hàm lượng methanol và propan-2-ol thấp hơn 0,025 % (tt/tt).
10.14 XÁC ĐỊNH DUNG MÔI TỒN DƯ
Quy trình mô tả trong phụ lục này được áp dụng để xác định dung môi tồn dư trong các trường hợp:
1. Định tính dung môi nhóm 1 và dung môi nhóm 2 tồn dư trong dược chất, tá dược, hay dược phẩm;
2. Thử giới hạn dung môi nhóm 1 và dung môi nhóm 2 khi chúng tồn tại trong dược chất, tá dược, hay dược phẩm;
3. Định lượng dung môi nhóm 2 khi lượng tồn dư lớn hơn 1000 phần triệu (0,1 %) hoặc định lượng dung môi nhóm 3 tồn dư khi có yêu cầu.
Các dung môi tồn dư nhóm 1, nhóm 2, nhóm 3 được liệt kê tại các bảng trong Phụ lục 10.14.1 Quy định đối với tạp chất là dung môi tồn dư.
Chuyên luận này giới thiệu 3 cách pha mẫu thử và các điều kiện kỹ thuật tiêm pha hơi các mẫu thử hóa hơi lên hệ thống sắc ký khí. Sử dụng hai hệ sắc ký, hệ sắc ký A thường được chọn trước, còn hệ sắc ký B thường được dùng để củng cố kết quả phát hiện. Việc chọn cách pha mẫu thử tùy thuộc vào độ tan của mẫu thử. Trong một số ít trường hợp cách pha mẫu tùy thuộc dung môi tồn dư cần kiểm tra. Các dung môi tồn dư sau đây không dễ dàng phát hiện được bằng các điều kiện tiêm pha hơi ghi trong chuyên luận này: formamid, 2-ethoxyethanol, 2-methoxyethanol, ethylen glycol, N-methylpyrrolidon va sulfolan. Phải áp dụng các qui trình khác thích hợp để kiểm tra sự tồn dư của các dung môi trên.
Khi dùng qui trình của chuyên luận này để định lượng các dung môi tồn dư, phải tiến hành thẩm định qui trình.
Tiến hành
Thực nghiệm bằng phương pháp sắc ký khí (Phụ lục 5.2) với kỹ thuật tiêm pha hơi tĩnh (static head- space injection).
Pha mẫu thử
Cách 1: Dùng để kiểm tra dung môi tồn dư trong các chất tan trong nước.
Dung dịch mẫu thử (1): Hòa tan 0,200 g mẫu thử trong nước, pha loãng với nước tới 20,0 ml.
Cách 2: Dùng để kiểm tra dung môi tồn dư trong các chất không tan trong nước.
Dung dịch mẫu thử (2): Hòa tan 0,200 g mẫu thử trong N,N-dimethylformamid (DMF) (TT), pha loãng tới 20,0 ml với cùng dung môi.
Cách 3: Dùng để kiểm tra N,N-dimethylacetamid và/ hoặc N,N- dimethylformamid khi biết rõ hoặc nghi ngờ có một hoặc cả hai dung môi này tồn dư trong mẫu thử.
Dung dịch mẫu thử (3): Hòa tan 0,200 g mẫu thử trong 1,3-dimethyl-2-imidazolidinon (DMI) và pha loãng đến 20,0 ml với cùng dung môi.
Khi không có cách pha mẫu nào nêu trên phù hợp với mẫu thử, thì cách pha mẫu thử và điều kiện tiêm pha hơi tĩnh áp dụng phải được chứng minh là phù hợp.
Dung dịch dung môi (a): Hòa tan 1,0 ml dung dịch dung môi tồn dư nhóm 1 chuẩn với 9 ml dimethyl sulfoxid và pha loãng với nước thành 100 ml. Pha loãng 1,0 ml dung dịch này với nước thành 10,0 ml.
Dung dịch dung môi (b): Hòa tan một lượng thích hợp dung môi tồn dư nhóm 2 trong dimethyl sulfoxid (TT). Pha loãng với nước thành 100,0 ml. Tiếp tục pha loãng để thu được dung dịch có nồng độ bằng 1/20 giới hạn quy định tại Bảng 10.14.1-2 trong Phụ lục 10.14.1 Quy định đối với tạp chất là dung môi tồn dư.
Dung dịch dung môi (c): Hòa tan 1,00 g dung môi hoặc các dung môi có trong mẫu thử với dimethyl sulfoxid (TT) hoặc nước (nếu thích hợp), và pha loãng với nước thành 100,0 ml. Tiếp tục pha loãng để thu được dung dịch có nồng độ bằng 1/20 giới hạn quy định tại Bảng 10.14.1-1 hoặc Bảng 10.14.1-2. trong Phụ lục 10.14.1 Qui định đối với tạp chất là dung môi tồn dư.
Dung dịch mẫu trắng: Chuẩn bị như cách pha dung dịch dung môi (c) nhưng không thêm dung môi cần xác định (để kiểm tra sự vắng mặt của các pic nhiễu).
Dung dịch thử: Lấy 5,0 ml dung dịch mẫu thử và 1,0 ml dung dịch mẫu trắng cho vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (a) (nhóm 1): Lấy 1,0 ml dung dịch dung môi (a) và 5,0 ml chất pha loãng thích hợp vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (a1) (nhóm 1): Lấy 1,0 ml dung dịch dung môi (a) và 5,0 ml dung dịch mẫu thử vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (b) (nhóm 2): Lấy 1,0 ml dung dịch dung môi (b) và 5,0 ml chất pha loãng thích hợp vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (c): Lấy 1,0 ml dung dịch dung môi (c) và 5,0 ml dung dịch mẫu thử vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (d): Lấy 1,0 ml dung dịch mẫu trắng và 5,0 ml chất pha loãng thích hợp vào một lọ đựng mẫu tiêm.
Đóng kín các lọ đựng mẫu tiêm nói trên bằng nút cao su có bao lớp polytetrafluoroethylen và giữ bởi một vòng chụp ngoài bằng nhôm. Lắc mạnh để có một dung dịch đồng nhất.
Các điều kiện tiêm pha hơi tĩnh có thể dùng:
| Thông số hoạt động | Cách pha mẫu | ||
|
| 1 | 2 | 3 |
| Nhiệt độ cân bằng (°C) | 80 | 105 | 80 |
| Thời gian cân bằng (min) | 60 | 45 | 45 |
| Nhiệt độ dòng chảy (°C) | 85 | 110 | 105 |
| Khí mang | Nitrogen hoặc heli dùng cho sắc ký khí ở áp suất thích hợp | ||
| Thời gian điều áp (s) | 30 | 30 | 30 |
| Thể tích tiêm (ml) | 1 | 1 | 1 |
Điều kiện sắc ký
Hệ sắc ký A
Cột mao quản thủy tinh hoặc cột có nòng rỗng, dài 30 m, đường kính trong 0,32 mm hoặc 0,53 mm được phủ bằng lớp các polymer liên kết mạng gồm polydimethylsiloxan 94 % và polycyanopropylphenylsiloxan 6 % (dày 1,8 μm hoặc 3,0 μm).
Khí mang: Nitrogen dùng cho sắc ký khí hoặc heli dùng cho sắc ký khí, tỷ lệ chia dòng 1: 5, tốc độ dòng khoảng 35 cm/s.
Detector ion hóa ngọn lửa [có thể dùng detector khối phổ hoặc để phát hiện dung môi tồn dư nhóm 1 ở dạng hợp chất clor hóa có thể dùng detector thu (bắt) điện tử].
Duy trì nhiệt độ cột ở 40 °C, trong 20 min, sau đó gia tăng nhiệt độ với tốc độ 10 °C/min cho đến khi đạt 240 °C, giữ ở 240 °C trong 20 min. Nhiệt độ buồng tiêm 140 °C, nhiệt độ detector 250 °C.
Hệ sắc ký B
Cột mao quản thủy tinh hoặc cột có nòng rỗng, dài 30 m có đường kính trong 0,32 mm hoặc 0,53 mm, được phủ macrogol 20 000 dày 0,25 μm.
Khí mang: Nitrogen dùng cho sắc ký khí hoặc heli dùng cho sắc ký khí, tỷ lệ chia dòng 1:5, tốc độ dòng khoảng 35 cm/s.
Detector ion hóa ngọn lửa [có thể dùng detector khối phổ hoặc để phát hiện dung môi tồn dư nhóm 1 ở dạng hợp chất clorid có thể dùng detector thu (bắt) điện tử].
Duy trì nhiệt độ cột ở 50 °C trong 20 min, sau đó gia tăng nhiệt độ với tốc độ 6 °C/min cho đến khi đạt 165 °C. Giữ ở 165 °C trong 20 min. Nhiệt độ buồng tiêm ở 140 °C, nhiệt độ detector ở 250 °C.
Phân tích trên hệ A
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a), ghi sắc ký đồ ở các điều kiện sao cho có thể xác định được tỷ lệ tín hiệu/nhiễu của 1,1,1-tricloroethan. Tỷ lệ tín hiệu/nhiễu của 1,1,1-tricloroethan không được nhỏ hơn 5; xem sắc ký đồ tại Hình 10.14-1.
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a1). Pic của các dung môi nhóm 1 thường được phát hiện. Tiêm 1 ml pha hơi của dung dịch đối chiếu (b), ghi sắc ký đồ ở các điều kiện sao cho hệ số phân giải giữa acetonitril và methylen clorid có thể xác định được. Hệ sắc ký A sẽ thích hợp nếu hệ số phân giải giữa acetonitril và methylen clorid không nhỏ hơn 1,0 và sắc ký đồ có dạng như Hình 10.14-2.
Tiêm 1 ml pha hơi của dung dịch thử lên cột phân tích của hệ A. Nếu sắc ký đồ thu được không có pic nào tương ứng với một trong các pic của những dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b) thì mẫu thử đạt yêu cầu. Nếu sắc ký đồ của mẫu thử có pic tương ứng với pic của một trong những dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b), thì phải tiếp tục phân tích trên hệ B.
Phân tích trên hệ B
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a) ghi sắc ký đồ ở các điều kiện sao cho có thể xác định được tỷ lệ tín hiệu/nhiễu của benzen. Tỷ lệ tín hiệu/nhiễu của benzen không được nhỏ hơn 5. Xem sắc ký đồ tại Hình 10.14-3.
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a1). Pic của các dung môi nhóm 1 thường được phát hiện. Tiêm 1 ml pha hơi của dung dịch đối chiếu (b), ghi sắc ký đồ ở các điều kiện sao cho hệ số phân giải giữa acetonitril và tricloroethylen có thể xác định được. Hệ sắc ký B sẽ thích hợp nếu hệ số phân giải giữa acetonitril và tricloroethylen không nhỏ hơn 1,0 và sắc ký đồ có dạng như Hình 10.14-4.
Tiêm 1 ml pha hơi của dung dịch thử. Nếu sắc ký đồ thu được không có pic nào tương ứng với một trong các pic của dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b) thì mẫu thử đạt yêu cầu. Nếu sắc ký đồ của mẫu thử có pic tương ứng với một trong những pic của dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b) và phù hợp với kết quả phân tích trên hệ A thì tiến hành như sau:
Tiêm 1 ml pha hơi của dung dịch đối chiếu (c) lên cột phân tích của hệ A hoặc B. Điều chỉnh độ nhạy của hệ thống sao cho chiều cao của pic dung môi tồn dư (hay của các dung môi tồn dư) không dưới 50 % của thang đo.
Tiêm 1 ml pha hơi của dung dịch đối chiếu (d) lên cột. Phải không có pic nhiễu nào được phát hiện. Tiêm 1,0 ml pha hơi của dung dịch thử và 1,0 ml pha hơi của dung dịch đối chiếu (c) lên cột. Tiêm lặp lại 2 lần nữa.
Diện tích pic trung bình của dung môi tồn dư trong sắc ký đồ cho bởi dung dịch thử không được lớn hơn một nửa (1/2) diện tích trung bình của pic tương ứng trong sắc ký đồ cho bởi dung dịch đối chiếu (c). Thử nghiệm chỉ có giá trị nếu độ lệch chuẩn tương đối của các hiệu số giữa ba cặp diện tích pic của chất phân tích thu được từ dung dịch đối chiếu (a) và dung dịch thử nhỏ hơn 15 %.
Khi hàm lượng dung môi tồn dư thuộc nhóm 2 hoặc nhóm 3 ở mức 0,1 % hoặc lớn hơn thì có thể tiến hành định lượng bằng phương pháp thêm chuẩn.
| 4. benzen | 14. 1,2-dicloroethan | 52. 1,1,1-tricloroethan |
| 10. carbon tetrachlorid | 15. 1,1-dicloroethylen |
|
Hình 10.14-1 - Sắc ký đồ các dung môi nhóm 1 phân tích trên hệ thống A theo qui trình 1
Detector ion hóa ngọn lửa
| 3. acetonitril | 17. dicloromethan | 49. pyridin | 61. tetralin |
| 11. cloroform | 23. 1,4-dioxan | 51. toluen | 62 methylcyclohexan |
| 13. cyclohexan | 29. hexan | 53. 1,1,2-tricloroethylen | 63. nitromethan |
| 30. 2-hexanon | 54. xylen (ortho, meta, para) | 64. 1,2-dimethoxyethan | |
| 16a. cis-1,2- dicloroethylen | 34. methanol | 58. clorobenzen | |
Hình 10.14-2 - Sắc ký đồ các dung môi nhóm 2 phân tích trên hệ thống A theo qui trình 1.
Detector ion hóa ngọn lửa
| 1. 1,1-dicloroethen | 3.carbon tetraclorid | 5. 1,2-dicloroethan |
| 2. 1,1,1-tricloroethan | 4.benzen |
|
Hình 10.14-3 - Sắc ký đồ các dung môi nhóm 1 phân tích trên hệ thống B theo qui trình 1.
Detector ion hóa ngọn lửa
| 1. methanol | 7. cloroform | 13. pyridin |
| 2. acetonitril | 8. cyclohexan | 14. toluen |
| 3. dicloromethan | 9. 1,2-dimethoxyethan | 15. 2-hexanon |
| 4. hexan | 10. 1,1,2-tricloroethen | 16. clorobenzen |
| 5. cis-1,2-dichloroethen | 11. methylcyclohexan | 17. xylen (ortho, meta, para) |
| 6. nitromethan | 12. 1,4-dioxan | 18. tetralin (tr= 28 min). |
Hình 10.14-4 - Sắc ký đồ các dung môi nhóm 2, phân tích trên hệ thống B theo qui trình 1.
Detector ion hóa ngọn lửa
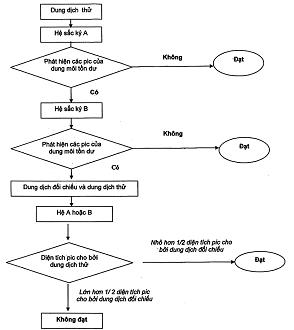
Hình 10.14-5 - Sơ đồ các bước tiến hành định tính và thử giới hạn các dung môi tồn dư
10.14.1 Quy định đối với tạp chất là dung môi tồn dư
(Theo tài liệu CPMP/ICH/283/95)
1. Mở đầu
Mục tiêu của Quy định này là đề ra lượng dung môi cho phép tồn dư trong dược phẩm, nhằm bảo đảm sự an toàn của người bệnh. Quy định khuyến cáo dùng các dung môi ít độc và đưa ra những giới hạn độc tính có thể chấp nhận được đối với một số dung môi.
Dung môi tồn dư trong dược phẩm là các chất hữu cơ bay hơi, được sử dụng hoặc sinh ra trong quá trình sản xuất các dược chất, tá dược hoặc trong quá trình bào chế các dược phẩm. Các dung môi này không loại bỏ được hoàn toàn trong quá trình sản xuất. Sự chọn lọc dung môi thích hợp dùng trong tổng hợp các dược chất có thể nâng cao sản lượng hoặc quyết định các đặc tính như dạng tinh thể, độ tinh khiết, tính hòa tan của sản phẩm. Vì vậy, đôi khi dung môi là một yếu tố quyết định trong quy trình tổng hợp. Quy định này không đề cập đến các dung môi dùng có cân nhắc kỹ lưỡng như một chất phụ gia, hoặc đến các solvat. Tuy nhiên hàm lượng dung môi trong các sản phẩm loại này phải được xác định và chứng minh hợp lý.
Vì các dung môi tồn dư không có tác dụng điều trị, các dung môi này phải được loại bỏ đến mức tối đa để đạt được các yêu cầu kỹ thuật của sản phẩm, việc thực hành tốt sản xuất (GMP) hoặc các yêu cầu chất lượng khác. Dược phẩm phải chứa một mức dung môi tồn dư không được cao hơn các dữ liệu an toàn. Phải tránh dùng một số dung môi có độc tính không thể chấp nhận được (dung môi Nhóm 1, Bảng 10.14.1-1), trừ khi lợi ích của việc sử dụng chúng được xác định chắc chắn. Một số dung môi có độc tính ít nguy hiểm hơn (dung môi Nhóm 2, Bảng 10.14.1-2) cũng cần phải dùng hạn chế, để bảo vệ người bệnh khỏi tác dụng độc hại. Tốt nhất phải dùng các dung môi ít độc (dung môi Nhóm 3, Bảng 10.14.1-3).
2. Phạm vi áp dụng
Các dung môi tồn dư trong dược chất, tá dược và dược phẩm thuộc phạm vi áp dụng của quy định này. Vì thế phải thực hiện phép thử tìm các dung môi tồn dư trong quá trình sản xuất hay tinh chế để kiểm soát sự hiện diện của chúng. Chỉ cần kiểm tra đối với các dung môi đã được sử dụng hay được sản sinh ra trong quá trình sản xuất hoặc tinh chế các dược chất, tá dược hoặc dược phẩm đó. Mặc dù các nhà sản xuất có thể chọn phương pháp xác định hàm lượng dung môi tồn dư trong sản phẩm, ta vẫn có thể tính hàm lượng đó, đi từ hàm lượng dung môi tồn dư trong các thành phần dùng để sản xuất ra sản phẩm đó. Nếu kết quả tính toán bằng hoặc thấp hơn giới hạn cho phép đã được khuyến cáo trong bản quy định này, thì không cần tiến hành thí nghiệm trên sản phẩm. Trái lại, nếu kết quả tính được vượt mức giới hạn cho phép, dược phẩm phải được thử nghiệm để biết chắc chắn lượng tồn dư dung môi đó có nằm trong phạm vi cho phép không. Sản phẩm cũng phải được thử nghiệm nếu có dùng dung môi trong quá trình sản xuất.
Quy trình này không áp dụng cho dược chất, tá dược hoặc sản phẩm mới đang trong quá trình nghiên cứu áp dụng lâm sàng và cũng không áp dụng cho các dược phẩm đang được lưu hành trên thị trường.
Quy định này áp dụng cho mọi dạng bào chế và mọi cách dùng thuốc. Trong một vài trường hợp, có thể cho phép giới hạn dung môi tồn dư cao hơn như khi dùng trong thời gian ngắn (30 ngày hay ít hơn), hoặc khi dùng dạng thuốc đắp. Việc chứng minh sự đúng đắn của giới hạn cao này phải dựa trên cơ sở của từng trường hợp một.
Xem Chú thích 2 (ở bên dưới) để biết thêm thông tin cơ bản có liên quan tới các dung môi tồn dư.
3. Các nguyên tắc cơ bản
Phân loại dung môi tồn dư theo mức độ nguy hiểm
Thuật ngữ “liều có thể dung nạp được cho mỗi ngày” (tolerable daily intake, TDI) được Chương trình quốc tế về an toàn hóa chất (IPCS) sử dụng và thuật ngữ “liều có thể dùng mỗi ngày” (acceptable daily intake ADI) được Tổ chức Y tế Thế giới, các Viện và các cơ quan quản lý sức khỏe quốc gia và quốc tế khác sử dụng để chỉ giới hạn hàm lượng các hóa chất độc có thể dùng mỗi ngày.
Thuật ngữ mới “liều phơi nhiễm được phép mỗi ngày” (permitted daily exposure, PDE) được nêu trong Quy định này là một lượng dung môi tồn dư trong dược phẩm có thể đưa vào cơ thể, để tránh nhầm với liều ADI của cùng dung môi.
Các dung môi tồn dư ghi trong Quy định này được liệt kê theo tên thông thường. Chúng được phân làm 3 nhóm, tùy thuộc khả năng gây độc đối với sức khỏe con người, như sau:
Nhóm 1: Các dung môi phải tránh sử dụng
Các chất gây ung thư cho người hay có khả năng gây ung thư cho người rõ rệt. Các chất gây nhiễm độc môi trường.
Nhóm 2: Các dung môi phải hạn chế sử dụng
Các chất gây ung thư trên động vật, không độc cho gen hoặc các tác nhân có thể gây độc không hồi phục như độc tính trên thần kinh hoặc gây quái thai. Các dung môi nghi có độc tính quan trọng, nhưng hồi phục được.
Nhóm 3: Các dung môi độc tính thấp
Các dung môi có độc tính thấp trên người: Không cần xác định liều gây tác hại cho sức khỏe.
Các dung môi Nhóm 3 có liều phơi nhiễm được phép mỗi ngày (PDE) bằng hoặc lớn hơn 50 mg/ngày.
Phương pháp xác định giới hạn phơi nhiễm
Phương pháp dùng để xác định liều PDE của các dung môi tồn dư được giới thiệu trong Chú thích 3. Tóm tắt các dữ liệu về độc tính dùng để thiết lập PDE được công bố trong Pharmeuropa Vol 1, No 1, Supplement April/1997.
Phương pháp xác định giới hạn dung môi Nhóm 2
Có hai cách có thể áp dụng để xác định giới hạn cho các dung môi Nhóm 2.
Cách 1:
Có thể dùng hàm lượng (nồng độ) giới hạn tính theo phần triệu (ppm) ghi trong Bảng 10.14.1-2. Các nồng độ này được tính theo công thức (1) với lượng chế phẩm dùng mỗi ngày cho là 10 g.
Nồng độ (phần triệu) = ![]() (1)
(1)
trong đó:
PDE là liều phơi nhiễm được phép mỗi ngày tính theo mg/ngày:
10 là số lấy làm lượng chế phẩm dùng trong mỗi ngày và tính theo g/ngày.
Giới hạn này áp dụng cho mọi dược chất, tá dược hoặc dược phẩm. Cách 1 có thể áp dụng nếu liều dùng hàng ngày không được biết hoặc không được quy định cụ thể. Khi mọi tá dược, dược chất có trong công thức bào chế đáp ứng với giới hạn cho bởi cách 1, các thành phần của chế phẩm có thể dùng theo bất kỳ tỷ lệ nào. Không cần tính toán gì thêm, nếu liều dùng mỗi ngày không quá 10 g. Chế phẩm có liều dùng lớn hơn 10 g mỗi ngày phải tính theo cách 2.
Cách 2:
Không nhất thiết mỗi thành phần của dược phẩm đều phải đáp ứng giới hạn đã đưa ra trong cách 1. Có thể dùng PDE tính theo mg/ngày ghi trong Bảng 10.14.1-2, cùng với liều tối đa mỗi ngày và công thức (1) để xác định hàm lượng dung môi tồn dư cho phép trong dược phẩm. Các giới hạn này được chấp nhận miễn là chứng minh được rằng dung môi tồn dư đã được giảm đến lượng thực tế tối thiểu. Các giới hạn này phải thực tế có liên quan đến độ chính xác của phép phân tích, điều kiện sản xuất, các thay đổi hợp lý của quá trình sản xuất và các giới hạn phải thực hiện các tiêu chuẩn công nghệ đương thời.
Có thể áp dụng cách 2 bằng cách cộng các lượng dung môi tồn dư trong mỗi thành phần của chế phẩm. Tổng lượng dung môi dùng trong ngày phải thấp hơn PDE.
Hãy xét một ví dụ áp dụng cách 1 và cách 2 cho dung môi acetonitril trong một dược phẩm. PDE của acetonitril là 4,1 mg/ngày. Như vậy, giới hạn tính theo cách 1 là 410 phần triệu (410 ppm). Lượng dùng tối đa mỗi ngày của dược phẩm là 5,0 g; dược phẩm chứa 2 tá dược. Thành phần dược phẩm và hàm lượng tối đa của acetonitril tồn dư thể hiện trong bảng sau đây:
| Thành phần | Lượng trong công thức bào chế | Hàm lượng acetonitril | Lượng phơi nhiễm mỗi ngày |
| Dược chất | 0,3 g | 800 × 10-6 | 0,24 mg |
| Tá dược 1 | 0,9 g | 400 × 10-6 | 0,36 mg |
| Tá dược 2 | 3,8 g | 800 × 10-6 | 3,04 mg |
| Dược phẩm | 5,0 g | 728 × 10-6 | 3,64 mg |
Tá dược 1 đáp ứng gới hạn tính theo cách 1.
Tá dược 2, dược chất và dược phẩm không đáp ứng giới hạn tính theo cách 1.
Tuy thế, dược phẩm tính theo cách 2 đáp ứng giới hạn 4,1 mg/ngày, và như vậy phù hợp với yêu cầu của quy định này.
Hãy xét một ví dụ khác, coi acetonitril là dung môi tồn dư. Lượng dùng tối đa mỗi ngày của một dược phẩm là 5,0 g, dược phẩm có chứa 2 tá dược. Thành phần dược phẩm và hàm lượng tối đa của acetonitril tồn dư thể hiện trong bảng sau:
| Thành phần | Lượng trong công thức chế | Hàm lượng acetonitril | Lượng phơi nhiễm mỗi ngày |
| Dược chất | 0,3 g | 800 × 10-6 | 0,24 mg |
| Tá dược 1 | 0,9 g | 2000 × 10-6 | 1,80 mg |
| Tá dược 2 | 3,8 g | 800 × 10-6 | 3,04 mg |
| Dược phẩm | 5,0 g | 1016 × 10-6 | 5,08 mg |
Trong ví dụ này, dược phẩm không đáp ứng giới hạn tính theo cách 1 và cả cách 2 bằng cách cộng. Nhà sản xuất phải thử nghiệm dược phẩm để xem quá trình bào chế có làm giảm mức độ tồn dư của acetonitril không. Nếu suốt quá trình bào chế, mức acetonitril không giảm tới giới hạn cho phép, khi đó nhà sản xuất phải tiến hành các bước làm giảm lượng acetonitril trong dược phẩm. Nếu tất cả các bước này không làm giảm được mức tồn dư của acetonitril, trong trường hợp đặc biệt, nhà sản xuất có thể cung cấp bản tóm tắt những cố gắng để giảm lượng dung môi tồn dư, nhằm đạt quy định này, và đưa ra lý lẽ phân tích nguy cơ/lợi ích khi dùng chế phẩm có chứa dung môi tồn dư ở mức cao này.
Qui trình phân tích dung môi tồn dư
Phương pháp phân tích dung môi tồn dư thông thường là dùng kỹ thuật sắc ký như sắc ký khí. Nếu thích hợp, dùng phương pháp xác định dung môi tồn dư mô tả trong dược điển. Ngoài ra, nhà sản xuất có thể tự do chọn lựa một quy trình phân tích có hiệu lực thích hợp nhất để áp dụng riêng. Nếu chỉ hiện diện dung môi Nhóm 3 thôi, có thể dùng một phương pháp không đặc hiệu như phương pháp Xác định mất khối lượng do làm khô (Phụ lục 9.6).
Thẩm định các phương pháp xác định dung môi tồn dư phải tuân thủ các quy định của ICH (Hội nghị quốc tế về hài hòa các yêu cầu kỹ thuật để đăng ký các thuốc dùng cho người) ghi trong “Văn bản thẩm định quy trình phân tích” (Text on validation of analytical procedures) và “Mở rộng văn bản thẩm định quy trình phân tích” (Extension of the ICH Text on validation of analytical procedures).
Báo cáo mức dung môi tồn dư
Nhà sản xuất dược phẩm cần nắm chắc thông tin về nồng độ dung môi tồn dư trong dược chất và tá dược để dược phẩm sản xuất ra đạt được mức của Quy định này.
Sau đây là các ví dụ về các thông tin mà nhà cung cấp tá dược hay dược chất có thể cung cấp cho nhà sản xuất dược phẩm. Nhà cũng cấp có thể chọn một trường hợp thích hợp trong số các trường hợp sau đây:
Chỉ có các dung môi Nhóm 3 hiện diện. Mất khối lượng do làm khô không quá 0,5 %.
Chỉ có các dung môi Nhóm 2 (X, Y...) hiện diện. Tất cả phải thấp hơn giới hạn tính theo cách 1 (ở đây, nhà cung cấp chỉ rõ tên các dung môi Nhóm 2: X, Y...).
Chỉ có các dung môi Nhóm 2 (X, Y...) và các dung môi Nhóm 3 hiện diện. Các dung môi Nhóm 2 tồn dư phải dưới mức giới hạn tính theo cách 1 và các dung môi Nhóm 3 tồn dư không quá 0,5 %.
Nếu chắc chắn hiện diện các dung môi Nhóm 1 thì phải định tính và định lượng chúng. Dung môi chắc chắn hiện diện là những dung môi được dùng trong giai đoạn cuối của quy trình sản xuất và các dung môi khác được dùng trước đó mà không được loại bỏ hoàn toàn bằng các biện pháp hữu hiệu.
Nếu dung môi Nhóm 2 hoặc Nhóm 3 hiện diện ở mức lớn hơn giới hạn cho phép tính theo Cách 1 (Nhóm 2) hay lớn hơn 0,5 % (Nhóm 3) thì chúng phải được định tính và định lượng.
4. Giới hạn dung môi tồn dư
Dung môi phải tránh sử dụng
Không được sử dụng các dung môi Nhóm 1 trong việc sản xuất dược chất, tá dược và dược phẩm. Chúng có độc tính không thể chấp nhận được, hoặc có tác hại đến môi trường. Khi buộc phải sử dụng chúng trong quy trình sản xuất một dược phẩm có tác dụng trị liệu vượt trội rõ rệt, phải tuân thủ giới hạn ghi trong Bảng 10.14.1-1; trừ trường hợp buộc phải chấp nhận giới hạn lớn hơn đã được giải thích chứng minh rõ. Dung môi 1,1,1-tricloroethan được xếp trong Bảng 10.14.1-1 vì là một tác nhân nguy hại cho môi trường. Giới hạn 1500 phần triệu dựa trên cơ sở thông tin về dữ liệu an toàn.
Dung môi phải hạn chế sử dụng
Các dung môi trong Bảng 10.14.1-2 phải được quy định mức giới hạn có trong dược phẩm vì độc tính vốn có của chúng. Các giá trị PDE được đưa ra dao động trong khoảng 0,1 mg/ngày. Hàm lượng tồn dư trong dược phẩm dao động trong khoảng 10 phần triệu.
Dung môi có độc tính thấp
Các dung môi Nhóm 3 (có trong Bảng 10.14.1-3) có thể coi như ít độc và có nguy cơ thấp đối với sức khỏe con người. Nhóm 3 bao gồm các dung môi không nguy hiểm đối với sức khỏe con người ở hàm lượng thường được chấp nhận trong dược phẩm. Tuy nhiên chưa có các nghiên cứu về độc tính trường diễn và về khả năng gây ung thư của nhiều dung môi thuộc nhóm này. Các dữ liệu hiện có cho thấy các dung môi này tỏ ra ít độc trong các nghiên cứu cấp diễn hoặc ngắn hạn và cho kết quả âm tính với những thử nghiệm độc tính trên gen. Do đó, coi như được phép dùng các dung môi này với lượng 50 mg mỗi ngày hoặc thấp hơn (tương ứng với hàm lượng 5000 × 10-6 hoặc 0,5 % tính theo cách 1) mà không cần phải thuyết minh, có thể dùng ở mức cao hơn, miễn là thực tế các mức đó có liên quan đến khả năng sản xuất và thực hành sản xuất tốt.
Dung môi chưa có đủ thông tin về độc tính
Các dung môi trong Bảng 10.14.1-4 có thể cũng được các nhà sản xuất dược chất, tá dược hoặc dược phẩm quan tâm. Tuy nhiên, chưa có các dữ liệu đầy đủ về độc tính của chúng để làm cơ sở cho việc xác định PDE. Khi sử dụng các dung môi này trong sản xuất, nhà sản xuất phải giải trình rõ ràng sự tồn dư của các dung môi này trong sản phẩm của mình.
Bảng 10.14.1-1 - Nhóm 1, các dung môi phải tránh sử dụng trong dược phẩm
| Dung môi | Nồng độ giới hạn (μg/g) | Độc tính |
| Benzen | 2 | Gây ung thư |
| Carbon tetraclorid | 4 | Độc và nguy hại cho môi trường |
| 1,2- Dicloroethan | 5 | Độc |
| 1,1- Dicloroethen | 8 | Độc |
| 1,1,1- Tricloroethan | 1500 | Nguy hại cho môi trường |
Bảng 10.14.1-2 - Nhóm 2, các dung môi phải hạn chế sử dụng trong dược phẩm
| Dung môi | Liều phơi nhiễm được phép mỗi ngày (PDE) (mg/ngày) | Nồng độ giới hạn (μg/g) |
| Acetonitril | 4,1 | 410 |
| Clorobenzen | 3,6 | 360 |
| Cloroform | 0,6 | 60 |
| Cyclohexan | 38,8 | 3880 |
| 1,2- Dicloroethen | 18,7 | 1870 |
| Dicloromethan | 6,0 | 600 |
| 1,2- Dimethoxyethan | 1,0 | 100 |
| N,N- Dimethylacetamid | 10,9 | 1090 |
| N,N- Dimethylformamid | 8,8 | 880 |
| 1.4- Dioxan | 3,8 | 380 |
| 2- Ethoxyethanol | 1,6 | 160 |
| Ethylenglycol | 6,2 | 620 |
| Formamid | 2,2 | 220 |
| Hexan | 2,9 | 290 |
| Methanol | 30,0 | 3000 |
| 2- Methoxyethanol | 0,5 | 50 |
| Methylbutylketon | 0,5 | 50 |
| Methylcyclohexan | 11,8 | 1180 |
| N- Methylpyrrolidon | 5,3 | 530 |
| Nitromethan | 0,5 | 50 |
| Pyridin | 2,0 | 200 |
| Sulfolan | 1,6 | 160 |
| Tetrahydrofuran | 7,2 | 720 |
| Tetralin | 1,0 | 100 |
| Toluen | 8,9 | 890 |
| 1,1,2- Tricloroethen | 0,8 | 80 |
| Xylen* | 21,7 | 2170 |
* Thường dùng hỗn hợp gồm m-xylen 60 %; p-xylen 14 %; o-xylen 9% và ethyl benzen 17 %.
Bảng 10.14.1-3: Nhóm 3, các dung môi phải được giới hạn vì GMP hoặc vì các yêu cầu chất lượng khác
| Acid acetic | Heptan |
| Aceton | Isobutyl acetat |
| Anisol | Isopropyl acetat |
| 1- Butanol | Methyl acetat |
| 2- Butanol | 3- Methyl-1-butanol |
| Butyl acetat | Methylethylketon |
| Tert - Butylmethyl ether | Methylisobutylketon |
| Cumen | 2- Methyl-1-propanol |
| Dimethylsulfoxid | Pentan |
| Ethanol | 1- Pentanol |
| Ethyl acetat | 1- Propanol |
| Ethyl ether | 2- Propanol |
| Ethyl format | Propyl acetat |
| Acid formic |
|
Bảng 10.14.1-4: Các dung môi chưa có đủ thông tin về độc tính
| 1,1- Diethoxypropan | Methylisopropylketon |
| 1,1- Dimethoxymethan | Methyltetrahydrofuran |
| 2,2- Dimethoxypropan | Petroleum |
| Isooctan | Acid tricloracetic |
| Isopropyl ether | Acid trifluoroacetic |
Chú thích 1: Thuật ngữ
Genotoxic carcinogens: Tác nhân gây ung thư do tác động lên gen hay chromosom.
LOEL: Viết tắt của từ “lowest-observed effect level” (mức phát hiện gây hại thấp nhất), liều thấp nhất dùng trong một thử nghiệm hay nhóm các thử nghiệm gây sự gia tăng có ý nghĩa sinh học về tần số hoặc về mức độ nghiêm trọng của bất kỳ các tác dụng nào đó trên người hay động vật bị phơi nhiễm.
Modifying factor: Hệ số hiệu chỉnh, là hệ số được chuyên gia độc chất học xác định và áp dụng cho các dữ liệu định lượng sinh học để tính các thông số an toàn cho người.
Neurotoxicity: Nhiễm độc thần kinh
NOEL: Viết tắt của từ “no-observed effect level” (mức không phát hiện gây hại), liều cao nhất của chất đã dùng mà không gây sự gia tăng có ý nghĩa sinh học về tần số hoặc về mức độ nghiêm trọng của bất kỳ các tác dụng nào đó trên người hay động vật bị phơi nhiễm.
PDE: viết tắt của từ “Permitted daily exposure”
Permitted daily exposure: Lượng cao nhất có thể dùng trong một ngày của dung môi tồn dư trong dược phẩm.
Reversible toxicity: Độc tính phục hồi được, tức là sẽ mất đi sau khi kết thúc phơi nhiễm.
Strongly suspected human carcinogen: Tác nhân nghi ngờ gây ung thư trên người, một chất không có dấu hiệu dịch tễ học là có khả năng gây ung thư nhưng lại cho kết quả dương tính về độc tính trên gen và các dấu hiệu rõ ràng về khả năng gây ung thư trên loài gặm nhấm.
Teratogenicity: Độc tính gây quái thai, sự cố gây dị tật về cấu trúc trong quá trình phát triển thai nhi khi sử dụng một chất trong thời kỳ mang thai.
Chú thích 2:
2.1 Quy chế bảo vệ môi trường đối với các dung môi hữu cơ bay hơi
Một số dung môi thường dùng trong sản xuất dược phẩm được liệt kê như các hóa chất độc trong các chuyên luận EHC (Environmental Health Criteria) và IRIS (Integrated Risk Information System). Trong các mục tiêu của các tổ chức như “Chương trình thế giới về an toàn hóa chất” (IPCS = International Programme on Chemical Safety), “Cơ quan bảo vệ môi trường Mỹ (USEPA_ = United States Environmental Protection Agency) và “Cơ quan quản lý thực phẩm và dược phẩm Mỹ (USFDA = United States Food and Drug Administration) có việc xác định các mức phơi nhiễm có thể chấp nhận được. Mục đích là bảo vệ sức khỏe con người và giữ gìn sự trong sạch vẹn toàn của môi trường, chống lại các tác dụng có thể có hại của các hóa chất do phơi nhiễm lâu dài tại môi trường. Các phương pháp dùng để xác định mức tối đa phơi nhiễm an toàn thường được căn cứ trên các thử nghiệm dài hạn. Khi không có các dữ liệu từ những thử nghiệm dài hạn, có thể dùng dữ liệu từ các thử nghiệm ngắn hạn với việc cải tiến cách tiếp cận như việc sử dụng các yếu tố hệ số an toàn lớn hơn. Cách tiếp cận nói ở trên đây chủ yếu liên quan tới các phơi nhiễm dài hạn hay suốt đời, đối với các cộng đồng dân cư, trong môi trường xung quanh như không khí, thức ăn, nước uống và các môi trường khác.
2.2. Dung môi tồn dư trong thuốc
Các giới hạn phơi nhiễm trong Quy định này được thiết lập căn cứ vào các phương pháp và các dữ liệu về độc tính mô tả trong các tiêu chuẩn EHC và trong các chuyên luận của EHC và IRIS. Tuy nhiên, một số giả định đặc biệt về dung môi tồn dư trong việc tổng hợp và bào chế các dược phẩm phải được tính đến khi thiết lập các giới hạn phơi nhiễm. Đó là:
1/ Những người bệnh (không phải toàn thể cộng đồng) phải sử dụng thuốc để phòng hoặc chữa bệnh.
2/ Việc giả định người bệnh bị phơi nhiễm suốt đời là không cần thiết đối với đa số các dược phẩm, song có thể thích hợp như một giả thiết nghiên cứu, nhằm giảm nguy cơ đến sức khỏe con người.
3/ Các dung môi tồn dư là những thành phần không thể tránh được trong sản xuất dược phẩm và còn thường là một phần của thuốc.
4/ Các dung môi tồn dư phải không được vượt quá các mức độ khuyến cáo, trừ những trường hợp đặc biệt.
5/ Các dữ liệu từ các nghiên cứu độc chất học dùng để xác định các mức chấp nhận được cho các dung môi tồn dư phải được thiết lập bằng cách sử dụng các quy trình thích hợp, chẳng hạn các quy trình do OECD mô tả và sử dụng cuốn sách đỏ của Tổ chức quản lý thực phẩm và dược phẩm của Mỹ (FDA Red book).
Chú thích 3: Phương pháp thiết lập các giới hạn phơi nhiễm
Phương pháp đánh giá nguy cơ của Gaylor - Kodell (Xem: Gaylor, D.W. và Kodell, R. L. - Thuật toán nội suy tuyến tính để đánh giá liều thấp của các chất độc. J. Environ Pathology, 4, 305, 1980) thích hợp với các dung môi gây ung thư thuộc Nhóm 1. Chỉ trong các trường hợp đã có các dữ liệu đáng tin cậy về tính gây ung thư thì phải áp dụng ngoại suy bằng cách dùng các mô hình toán học để thiết lập các giới hạn phơi nhiễm. Các giới hạn phơi nhiễm của các dung môi Nhóm 1 có thể được xác định với việc sử dụng một thông số an toàn lớn (từ 10 000 đến 100 000) đối với “mức không phát hiện gây hại” (NOEL). Việc phát hiện và định lượng các dung môi này phải được thực hiện bằng các kỹ thuật phân tích tiên tiến.
Các mức phơi nhiễm chấp thuận được của các dung môi Nhóm 2 ghi trong quy định này được thiết lập bằng cách tính các giá trị PDE theo các quy trình xác định giới hạn phơi nhiễm của các thuốc (xem PharmacopeiaI Forum 11-12/1989) và theo phương pháp đã được Chương trình thế giới về an toàn hóa chất (IPCS) thừa nhận nhằm đánh giá nguy cơ đối với sức khỏe con người do hóa chất (xem Environmental Health Criteria 170, WHO, 1994). Các phương pháp này tương tự như các phương pháp của Cơ quan bảo vệ môi trường Mỹ (USEPA) và của Cơ quan quản lý thực phẩm và dược phẩm Mỹ (USFDA) và các tổ chức khác nữa. Phương pháp được giới thiệu sơ lược ở đây nhằm cung cấp một hiểu biết tốt hơn nguồn gốc của các liều phơi nhiễm được phép mỗi ngày (PDE). Không nhất thiết phải thực hiện các phép tính toán này mà sử dụng ngay các giá trị PDE đã ghi trong các bảng trình bày ở Mục 4 của Quy định này.
Giới hạn PDE được tính từ “mức không phát hiện gây hại” (NOEL) hoặc từ “mức phát hiện gây hại thấp nhất” (LOEL) trong thử nghiệm trên súc vật như sau:
| PDE = | NOEL × thể trọng quy ước |
| F1 × F2 × F3 × F4 × F5 |
Trong đó:
Thể trọng quy ước là khối lượng cơ thể người trưởng thành, không phân biệt giới tính, được quy ước để tính ở đây là 50 kg;
F là các hệ số hiệu chỉnh.
PDE thường được tính từ NOEL. Khi không thiết lập được NOEL, có thể dùng LOEL. Các hệ số hiệu chỉnh đề ra ở đây để chuyển đổi các dữ liệu thử nghiệm trên súc vật sang cho người, cũng tương tự như các “hệ số không chắc chắn” (Uncertainty factors) dùng trong “Tiêu chuẩn trong sạch của môi trường” (EHC) (Xem Environmental Health Criteria 170, WHO, Geneva, 1994) và “hệ số hiệu chỉnh” (modifying factors) hay “hệ số an toàn” (safety factors) dùng trong PharmacopeiaI Forum. Giả định 100 % phơi nhiễm toàn thân được sử dụng trong tất cả các tính toán, bất kể dùng thuốc theo đường nào.
Các hệ số hiệu chỉnh
F1 là hệ số ngoại suy giữa các loài:
F1 = 2, khi ngoại suy từ thử nghiệm trên chó sang người;
F1 = 2,5, khi ngoại suy từ thử nghiệm trên thỏ sang người;
F1 = 3, khi ngoại suy từ thử nghiệm trên khỉ sang người;
F1 = 5, khi ngoại suy từ thử nghiệm trên chuột cống trắng (rat) sang người;
F1 = 10, khi ngoại suy từ thử nghiệm trên các loài động vật khác sang người;
F1 = 12, khi ngoại suy từ thử nghiệm trên chuột nhắt sang người.
F1 tính được khi so sánh tỷ lệ giữa diện tích bề mặt với khối lượng cơ thể của loài vật tham gia thử nghiệm và của người. Diện tích bề mặt (S) được tính như sau:
S = k.M 0,67
trong đó:
M là khối lượng cơ thể (khối lượng cơ thể của sinh vật tham gia thử nghiệm, dùng trong công thức trên, được liệt kê trong Bảng 10.14.2.5);
k = 10
F2 là hệ số chỉ sự khác biệt giữa các cá thể;
F2 = 10, thường dùng cho tất cả các dung môi hữu cơ và dùng thống nhất trong quy định này;
F3 là một hệ số không hằng định, dùng cho các thử nghiệm phơi nhiễm ngắn hạn;.
F3 = 1, cho các thử nghiệm kéo dài ít nhất bằng 1/2 đời sống của vật thí nghiệm (1 năm đối với loài gặm nhấm hoặc thỏ, 7 năm đối với chó, mèo hoặc khỉ);
F3 = 1, cho các thử nghiệm về sinh sản, kéo dài trong suốt thời gian hình thành các cơ quan phủ tạng;
F3 = 2, cho các thử nghiệm kéo dài 6 tháng, trên loài gặm nhấm hoặc 3,5 năm trên các con thú không thuộc loài gặm nhấm;
F3 = 5, cho các thử nghiệm kéo dài 3 tháng, trên loài gặm nhấm, hoặc 2 năm trên các con thú không thuộc loài gặm nhấm;
F3 = 10, cho các thử nghiệm ngắn hạn hơn.
Trong mọi trường hợp, khi thử nghiệm kéo dài trong khoảng thời gian nằm giữa 2 mốc thời gian quy định ở trên, thì ta cho F3 giá trị cao, ứng với mốc thời gian thử nghiệm ngắn. Ví dụ: F3 = 2 trong thử nghiệm kéo dài 9 tháng trên loài gặm nhấm (vì 9 tháng nằm trong mốc 1 năm và 6 tháng, do đó lấy F3 bằng F3 của mốc 6 tháng).
F4 là một hệ số áp dụng trong các trường hợp độc tính nghiêm trọng như: Gây ung thư không độc cho gen, độc tính thần kinh, độc tính gây quái thai. Trong các thử nghiệm về độc tính sinh sản, hệ số này được dùng như sau:
F4 = 1, khi gây độc trên bào thai cùng với gây độc trên mẹ;
F4 = 5, khi gây độc trên bào thai, nhưng không gây độc trên mẹ;
F4 = 5, khi gây quái thai cùng với gây độc trên mẹ;
F4 = 10, khi gây quái thai trên bào thai, nhưng không gây độc trên mẹ;
F5 là một hệ số không hằng định, áp dụng khi không thiết lập được “mức không phát hiện gây hại” (NOEL). Khi chỉ có thể xác định được “mức gây hại thấp nhất” (LOEL), có thể cho F5 giá trị cao, có thể tới F5 = 10, tùy thuộc vào mức độ nghiêm trọng của độc tính.
Khối lượng cơ thể (cân nặng) quy ước cho người trưởng thành, không phân biệt giới tính ở đây là 50kg. Khối lượng này tương đối thấp hơn khối lượng chuẩn mực (60 kg đến 70 kg) thường áp dụng trong các tính toán cùng loại (tính liều dùng thuốc) là nhằm mục đích an toàn.
Một số người bệnh có cân nặng nhẹ hơn 50 kg, những người bệnh này phải được xem xét điều chỉnh bằng cách dùng hệ số an toàn xác định PDE.
Nếu dung môi có trong một công thức dược phẩm chuyên dùng cho Nhi khoa, ta phải điều chỉnh thích hợp, vì khối lượng cơ thể của trẻ thấp.
Ví dụ áp dụng công thức tính PDE:
Hãy xét một thử nghiệm độc tính của acetonitril trên chuột nhắt, thử nghiệm này được tóm tắt trong Pharmaeuropa Vol 9, No1, Supplement April 1997 page S 24, “mức không phát hiện gây hại” (NOEL) là 50,7 mg/kg/ngày. PDE của acetonitril trong thử nghiệm này được tính như sau:
| PDE = | 50,7mg.kg-1.ngày-1 × 50kg | = 4,22 mg/ngày |
| 12 × 10 × 5 × 1 × 1 |
Trong đó
F1 = 12 là hệ số ngoại suy từ thử nghiệm trên chuột nhắt sang người;
F2 = 10 là hệ số khác biệt giữa các cá thể (thống nhất quy định là F2 = 10);
F3 = 5 khi thời gian thử nghiệm chỉ có 13 tuần (> 3 tháng);
F4 = 1 khi không thấy độc tính nghiêm trọng nào;
F5 = 1 khi “mức không phát hiện gây độc hại” (NOEL) được xác định.
Phương trình dùng cho khí lý tưởng PV = nRT được áp dụng để chuyển hàm lượng chất khí trong thử nghiệm dùng phương pháp xông (inhalation studies) từ đơn vị phần triệu thành đơn vị mg/l hay mg/m3.
Hãy xét thử nghiệm độc tính sinh sản trên chuột trắng bằng cách xông khí carbon tetraclorid (khối lượng phân tử 153,84) đã được tóm tắt trong Pharmaeuropa Vol 9. No1. Supplement April 1997, p.S9:
![]() Dùng công thức 1000 lít = 1 m3 để suy ra hàm lượng mg/m3
Dùng công thức 1000 lít = 1 m3 để suy ra hàm lượng mg/m3
Bảng 10.14.1-5. Các giá trị dùng tính toán trong tài liệu này
| Cân nặng của chuột cống trắng (Rat) | 425 g |
| Cân nặng của chuột cống trắng có thai (Pregnant rat) | 330 g |
| Cân nặng của chuột nhắt (Mouse) | 28 g |
| Cân nặng của chuột nhắt có thai (Pregnant mouse) | 30 g |
| Cân nặng của chuột lang (Guinea - pig) | 500 g |
| Cân nặng của khỉ Ấn Độ (Rhesus monkey) | 25 kg |
| Cân nặng của thỏ (có thai hoặc không có thai) | 4kg |
| Cân nặng của chó săn nhỏ (Beagle dog) | 11,5 kg |
| Dung lượng hô hấp của chuột cống trắng (Rat respiratory volume) | 290 l/ngày |
| Dung lượng hô hấp của chuột nhắt | 43 l/ngày |
| Dung lượng hô hấp của thỏ | 1440 l/ngày |
| Dung lượng hô hấp của chuột lang | 4301 l/ngày |
| Dung lượng hô hấp của người | 28800 l/ngày |
| Dung lượng hô hấp của chó | 9000 l/ngày |
| Dung lượng hô hấp của khỉ | 1150 l/ngày |
| Lượng nước chuột nhắt dùng | 5 ml/ngày |
| Lượng nước chuột trắng dùng | 30 ml/ngày |
| Lượng thức ăn chuột trắng dùng | 30 g/ngày |
10.15 XÁC ĐỊNH ETHYLEN OXYD VÀ DIOXAN TỒN DƯ
Thử nghiệm này xác định ethylen oxyd và dioxan tồn dư trong các chất tan trong nước hay trong dimethylacetamid. Đối với các chất không tan hoặc tan không hoàn toàn trong các dung môi trên, việc chuẩn bị dung dịch mẫu thử và các điều kiện sắc ký khí tiêm pha hơi được quy định trong chuyên luận riêng.
Xác định bằng phương pháp sắc ký khí (Phụ lục 5.2), kỹ thuật tiêm pha hơi (head-space gas chromatography).
Phương pháp 1: Dùng cho chất thử hòa tan hay trộn đều được trong nước.
Dung dịch thử: Cân 1,00 g (MT) chất thử vào một lọ nhỏ 10 ml (hoặc có thể tích thích hợp) và thêm 1,0 ml nước. Đóng nắp lọ và trộn để thu được dung dịch đồng nhất. Để yên ở 70 °C trong 45 min.
Dung dịch đối chiếu (a): Cân 1,00 g (MC) chất thử vào một lọ nhỏ như trên, thêm 0,50 ml dung dịch ethylen oxyd (TT3) và 0,50 ml dung dịch dioxan (TT1). Đóng nắp lọ và trộn đều để thu được một dung dịch đồng nhất. Để yên ở 70 °C trong 45 min:
Dung dịch đối chiếu (b): Trong lọ nhỏ thể tích 10 ml, thêm vào 0,50 ml dung dịch ethylen oxyd (TT3), 0,10 ml dung dịch acetaldehyd (TT) 0,001 % mới pha và 0,10 ml dung dịch dioxan (TT1). Đóng nắp lọ và trộn để thu được dung dịch đồng nhất. Để yên ở 70 °C trong 45 min.
Phương pháp 2: Dùng cho các chất thử hòa tan hay trộn đều được trong dimethylacetamid.
Dung dịch thử: Cần 1,00 g (MT) chất thử vào một lọ nhỏ 10 ml (hoặc có thể tích thích hợp) và thêm 1,0 ml dimethylacetamid (TT) và 0,2 ml nước. Đóng nắp lọ và trộn để thu được dung dịch đồng nhất. Để yên ở 90 °C trong 45 min
Dung dịch đối chiếu (a): Cân 1,00 g (MC) chất thử vào một lọ nhỏ như trên, thêm 1,0 ml dimethyl- acetamid (TT), thêm 0,10 ml dung dịch dioxan (TT) và 0,10 ml dung dịch ethylen oxyd (TT2). Đóng nắp lọ và trộn để thu được một dung dịch đồng nhất. Để yên ở 90 °C trong 45 min
Dung dịch đối chiếu (b): Trong lọ nhỏ thể tích 10 ml, thêm vào 0,10 ml dung dịch ethylen oxyd (TT2), 0,10 ml dung dịch acetaldehyd (TT) 0,01 % mới pha và 0,10 ml dung dịch dioxan (TT). Đóng nắp lọ và trộn đều để thu được dung dịch đồng nhất. Để yên ở 70 °C trong 45 min.
Quá trình sắc ký
Điều kiện tiêm pha hơi
Nhiệt độ cân bằng: 70 °C (90 °C đối với dung dịch trong dimethylacetamid).
Thời gian cân bằng: 45 min.
Nhiệt độ dòng chảy: 75 °C (150 °C đối với dung dịch trong dimethylacetamid).
Thời gian điều áp: 1 min.
Khí mang: Heli dùng cho sắc ký khí.
Thời gian tiêm: 12 s.
Điều kiện sắc ký
Cột thủy tinh hoặc thạch anh kích thước 30 m × 0,32 mm, mặt trong được phủ lớp polydimethylsiloxan dày 1,0 μm.
Khí mang: Heli (hoặc nitrogen) dùng cho sắc ký khí, tốc độ dòng khoảng 20 cm/s, tỷ lệ chia dòng 1:20.
Detector: ion hóa ngọn lửa.
Duy trì nhiệt độ cột ở 50 °C trong 5 min, sau đó tăng đến 180 °C với tốc độ 5 °C/min, sau đó lại tăng đến 230 °C với tốc độ 30 °C/min và giữ ở 230 °C trong 5 min. Nhiệt độ detector là 250 °C, nhiệt độ buồng tiêm là 150 °C.
Tiêm một lượng thích hợp pha hơi của dung dịch đối chiếu (b), chẳng hạn 1 ml. Điều chỉnh độ nhạy của hệ thống sao cho chiều cao các pic cho bởi ethylen oxyd và acetaldehyd trong sắc ký đồ đạt ít nhất 15 % thang đo. Thử nghiệm không có hiệu lực nếu hệ số phân giải giữa pic cho bởi acetaldehyd và pic cho bởi ethylen oxyd nhỏ hơn 2,0 và pic của dioxin và ethylene oxyd được phát hiện với tỷ lệ tín hiệu/nhiễu nhỏ hơn 5.
Tiêm lần lượt những lượng thích hợp pha hơi của dung dịch thử và dung dịch đối chiếu (a), chẳng hạn 1 ml (hoặc bằng lượng dung dịch đối chiếu (b) đã tiêm).
Tiêm lặp lại như trên thêm 2 lần nữa.
Diện tích trung bình của pic ethylen oxyd và dioxan trong sắc ký đồ cho bởi dung dịch thử không được lớn hơn một nửa (1/2) diện tích trung bình của pic tương ứng trong sắc ký đồ cho bởi dung dịch đối chiếu (a) (1 phần triệu ethylen oxyd và 50 phần triệu dioxan).
Kiểm tra độ tin cậy
Tính hiệu số của diện tích pic, theo từng cặp, cho bởi dung dịch thử và dung dịch đối chiếu (a), lần lượt ứng với ethylen oxyd và với dioxan. Thử nghiệm chỉ có hiệu lực nếu độ lệch chuẩn tương đối của ba giá trị khác biệt (3 hiệu số) ứng với ethylen oxyd không lớn hơn 15 % và độ lệch chuẩn tương đối của ba giá trị khác biệt (3 hiệu số) ứng với dioxan không lớn hơn 15 %.
Nếu lượng cân mẫu thử và chất đối chiếu nằm ngoài khoảng 0,995 g đến 1,005 g cần phải hiệu chỉnh kết quả cho thích hợp.
Hàm lượng ethylen oxyd tính theo phần triệu được tính bằng biểu thức:
AT × C / [(AC × MT) - (AT × MC)]
trong đó:
AT: diện tích pic tương ứng với ethylen oxyd trong sắc ký đồ cho bởi dung dịch thử;
AC: diện tích pic tương ứng với ethylen oxyd trong sắc ký đồ cho bởi dung dịch đối chiếu (a);
MT: khối lượng chất thử trong dung dịch thử tính theo gam;
MC: khối lượng chất thử trong dung dịch đối chiếu (a) tính theo gam;
C: khối lượng ethylen oxyd cho vào dung dịch đối chiếu (a) tính theo microgram.
Và hàm lượng dioxan tính theo phần triệu được tính bằng biểu thức:
DT × C/[(DC × MT) - (DT × MC)]
trong đó:
DT: diện tích pic tương ứng với dioxan trong sắc ký đồ cho bởi dung dịch thử;
DC: diện tích pic tương ứng với dioxan trong sắc ký đồ cho bởi dung dịch đối chiếu (a);
MT: khối lượng chất thử trong dung dịch thử tính theo gam;
MC: khối lượng chất thử trong dung dịch đối chiếu (a) tính theo gam;
C: khối lượng dioxan cho vào dung dịch đối chiếu (a) tính theo microgram.
10.16 ĐỊNH LƯỢNG N,N-DIMETHYLANILIN
Xác định bằng phương pháp sắc ký khí (Phụ lục 5.2).
Phương pháp 1
Dung dịch chuẩn nội: Hòa tan 50 mg N,N-diethylanilin (TT) trong 4 ml dung dịch acidhydrocloric 0,1 M (TT), pha loãng bằng nước tới 50 ml. Pha loãng 1 ml dung dịch thu được tới 100 ml bằng nước.
Dung dịch thử: Hòa tan 0,50 g chế phẩm trong 30,0 ml nước, thêm 1,0 ml dung dịch chuẩn nội. Điều chỉnh nhiệt độ của dung dịch tới 26 °C đến 28 °C. Thêm 1,0 ml dung dịch natri hydroxyd 40 % (TT). Lắc cho tan hoàn toàn. Thêm 2,0 ml trimethylpentan (TT). Lắc 2 min, để yên cho tách lớp, dùng lớp trên.
Dung dịch đối chiếu: Hòa tan 50,0 mg N,N-dimethylanilin (TT) trong 4,0 ml dung dịch acid hydrocloric 0,1 M (TT), pha loãng bằng nước tới 50,0 ml. Pha loãng 1 ml dung dịch thu được tới 100,0 ml bằng nước. Tiếp tục pha loãng 1,0 ml dung dịch thu được tới 30,0 ml bằng nước, thêm 1,0 ml dung dịch chuẩn nội, 1,0 ml dung dịch natri hydroxyd 40 % (TT), 2,0 ml trimethylpentan (TT). Lắc 2 min, để yên cho tách lớp, dùng lớp trên.
Điều kiện sắc ký
Cột mao quản bằng silica nung chảy, dài 25 m, đường kính trong 0,32 mm, phủ lớp polymer liên kết mạng polymethylphenylsiloxan dày 0,52 μm.
Khí mang: Heli dùng cho sắc ký khí, tỷ lệ chia dòng 1 : 20, áp suất đầu cột 5 kPa, lưu lượng 20 ml/min. Detector ion hóa ngọn lửa.
Bộ chia dòng (split-liner) là một cột dài khoảng 1 cm, nhồi diatomit dùng cho sắc ký khí tầm 10 % (kl/kl) polydimethylsiloxan.
Nhiệt độ: Duy trì nhiệt độ cột 150 °C trong 5 min, sau đó tăng đến 275 °C với tốc độ 20 °C/min, rồi giữ ở 275 °C trong 3 min. Nhiệt độ detector 300 °C, nhiệt độ buồng tiêm 220 °C.
Thể tích tiêm: 1 μl.
Thời gian lưu của N,N-dimethylanilin khoảng 3,6 min; của N,N-diethylanilin khoảng 5,0 min.
Phương pháp 2
Dung dịch chuẩn nội: Dung dịch naphtalen (TT) 0,005 % trong cyclohexan (TT).
Dung dịch thử: Hòa tan 1,00 g chế phẩm với 5 ml dung dịch natri hydroxyd 1 M (TT) trong ống thủy tinh có nút mài, thêm 1,0 ml dung dịch chuẩn nội. Đậy kín, lắc mạnh trong 1 min. Ly tâm nếu cần, dùng lớp trên.
Dung dịch đối chiếu: Thêm 2 ml acid hydrodoric đậm đặc (TT) và 20 ml nước vào 50,0 mg N,N-dimethylanilin (TT), lắc cho tan và pha loãng bằng nước tới 50,0 ml. Pha loãng 5,0 ml dung dịch này tới 250,0 ml bằng nước. Cho 1,0 ml dung dịch thu được vào ống thủy tinh có nút mài, thêm 5 ml dung dịch natri hydroxyd 1 M (TT) và 1,0 ml dung dịch chuẩn nội. Đậy kín, lắc mạnh trong 1 min, ly tâm nếu cần và dùng lớp trên.
Điều kiện sắc ký: Cột thủy tinh dài 2 m, đường kính trong 2 mm, được nhồi bằng diatomit silan hóa tẩm 3 % (kl/kl) polymethylphenylsiloxan.
Khí mang: Nitrogen dùng cho sắc ký khí, với lưu lượng 30 ml/min.
Detector ion hóa ngọn lửa.
Duy trì nhiệt độ cột ở 120 °C, nhiệt độ detector và buồng tiêm ở 150 °C.
Thể tích tiêm: 1 μl.
10.17 ĐỊNH LƯỢNG ACID 2-ETHYLHEXANOIC
Tiến hành phương pháp sắc ký khí (Phụ lục 5.2).
Dung dịch chuẩn nội: Hòa tan 100 mg acid 3-cyclohexyl propionic (TT) trong cyclohexan (TT) và pha loãng vừa đủ 100 ml với cùng dung môi.
Dung dịch thử: Thêm 4,0 ml dung dịch acid hydrocloric 33 % (tt/tt) vào 0,300 g mẫu thử. Lắc mạnh 2 lần, mỗi lần trong 1 min với 1,0 ml dung dịch chuẩn nội. Đợi tách lớp (nếu cần thiết, có thể ly tâm để tách lớp được tốt hơn), sử dụng dịch gộp của các lớp dịch chiết ở trên.
Dung dịch đối chiếu: Hòa tan 75,0 mg acid 2-ethylhexanoic (TT) trong dung dịch chuẩn nội và pha loãng vừa đủ 50,0 ml với dụng dịch chuẩn nội. Thêm 4,0 ml dung dịch acid hydrocloric 33 % (tt/tt) vào 1,0 ml dung dịch vừa pha. Lắc mạnh trong 1 min. Đợi tách lớp (nếu cần thiết, có thể ly tâm để tách lớp được tốt hơn). Gạn lấy lớp dịch chiết ở trên. Tiếp tục thêm 1,0 ml dung dịch chuẩn nội vào lớp dịch còn lại ở dưới và lắc mạnh trong 1 min. Đợi tách lớp (nếu cần thiết, có thể ly tâm để tách lớp được tốt hơn), sử dụng dịch gộp của các lớp dịch chiết ở trên.
Điều kiện sắc ký:
Cột silica nung chảy có nòng rỗng, chiều dài 10 m, đường kính trong 0,53 mm, phủ lớp pha tĩnh macrogol 20 000 2-nitroterephtalat (có bề dày 1,0 μm);
Khí mang: Heli dùng cho sắc ký, với tốc độ dòng 10 ml/min;
Detector ion hóa ngọn lửa (FID);
Chương trình nhiệt độ như sau:
|
| Thời gian (min) | Nhiêt độ (°C) | Tốc độ (°C/min) | Ghi chú |
| Cột | 0-2 | 40 | - | Đẳng nhiệt |
| 2-7,3 | 40 → 200 | 30 | Nhiệt độ tăng đều | |
| 7,3-10,3 | 200 | - | Đẳng nhiệt | |
| Buồng tiêm |
| 200 |
|
|
| Detector |
| 300 |
|
|
Thể tích tiêm: 1 μl.
Cách tiến hành:
Tiêm 1 μl dung dịch thử và 1 μl dung dịch đối chiếu.
Phép thử chỉ có giá trị khi độ phân giải giữa pic tương ứng với acid 2-ethylhexanoic (pic đầu tiên) và pic dung dịch chuẩn nội ít nhất là 2,0.
Hàm lượng phần trăm của acid 2-ethylhexanoic được tính theo công thức sau:
![]()
trong đó:
AT: diện tích pic tương ứng với acid 2-ethylhexanoic trên sắc ký đồ của dung dịch thử;
AC: diện tích pic tương ứng với acid 2-ethylhexanoic trên sắc ký đồ của dung dịch đối chiếu;
IT: diện tích pic tương ứng với chất chuẩn nội trên sắc ký đồ của dung dịch thử;
IC: diện tích pic tương ứng với chất chuẩn nội trên sắc ký đồ của dung dịch đối chiếu;
mT: lượng cân mẫu thử tính bằng gam;
mC: lượng cân acid 2-ethylhexanoic trong dung dịch đối chiếu, tính bằng gam.
10.18 XÁC ĐỊNH ACID ACETIC TRONG PEPTID TỔNG HỢP
Tiến hành bằng phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động:
Pha động A: Hòa tan 0,7 ml acid orthophosphoric (TT) trong vừa đủ 1000 ml nước; điều chỉnh pH tới 3,0 với dung dịch natri hydroxyd 40 % (TT).
Pha động B: Methanol dùng cho sắc ký lỏng (TT).
Dung dịch thử. Chuẩn bị như mô tả ở chuyên luận riêng.
Dung dịch đối chiếu: Pha dung dịch acid acetic 0,10 g/l từ acid acetic băng (TT), sử dụng hỗn hợp 5 thể tích pha động B và 95 thể tích pha động A làm dung môi.
Điều kiện sắc ký:
Cột thép không gỉ (250 mm × 4,6 mm), nhồi pha tĩnh C (5 μm).
Detector tử ngoại đặt ở bước sóng 210 nm.
Tốc độ dòng: 1,2 ml/min.
Thể tích tiêm: 10 μl.
Chương trình gradient dung môi như sau:
| Thời gian (min) | Pha động A (% tt/tt) | Pha động B (% tt/tt) |
| 0-5 | 95 | 5 |
| 5-10 | 95 → 50 | 5 → 50 |
| 10-20 | 50 | 50 |
| 20-22 | 50 → 95 | 50 → 5 |
| 22-30 | 95 | 5 |
Tiến hành sắc ký đối với dung dịch đối chiếu và dung dịch thử.
Trong các sắc ký đồ thu được, pic tương ứng với acid acetic có thời gian lưu khoảng 3 min đến 4 min. Đường nền có thể cao dần do hiện tượng rửa giải peptid.
Xác định hàm lượng acid acetic trong peptid dựa vào diện tích pic acid acetic tương ứng trong sắc ký đồ của dung dịch thử và dung dịch đối chiếu.
10.19 ĐỐT TRONG OXYGEN
Thiết bị
Nếu không có những qui định khác trong chuyên luận riêng, bình đốt là một bình nón bằng thủy tinh borosilicat dung tích ít nhất là 500 ml, nút thủy tinh mài có gắn một bộ phận mang mẫu thích hợp bằng platin hoặc bằng platin - iridi.
Phương pháp
Nghiền nhỏ mẫu thử. Đặt một lượng mẫu theo qui định vào giữa một mảnh giấy lọc không tro, kích thước 40 mm × 30 mm, có một dải nhỏ kích thước 10 mm × 30 mm. Nếu giấy lọc được qui định tẩm bằng lithi carbonat thì làm ẩm vùng giữa giấy lọc bằng dung dịch bão hòa lithi carbonat (TT) rồi sấy khô ở 100 °C đến 105 °C trước khi dùng. Gói mẫu thử vào trong giấy và cài vào bộ phận mang mẫu.
Thêm nước hoặc dung dịch hấp thụ như chỉ dẫn vào trong bình. Bơm khí oxygen vào bình qua một ống mà đầu cuối của nó đặt ở phía trên với khoảng cách vừa đủ so với mặt thoáng của chất lỏng hấp thụ. Làm ẩm cổ bình bằng nước. Nạp đầy khí oxygen vào bình. Đốt đầu tự do của dải giấy lọc hẹp bằng một dụng cụ thích hợp và cẩn thận đậy ngay nút bình lại. Giữ chắc nút. Khi bắt đầu cháy mạnh, lật ngược bình để tạo ra một lớp ngăn cách bằng nước nhưng cần chú ý ngăn không cho những sản phẩm cháy chưa hoàn toàn rơi xuống chất lỏng. Ngay sau khi đốt cháy xong, lắc bình thật mạnh để hòa tan hoàn toàn những sản phẩm đốt. Làm lạnh. Nếu không có những chỉ dẫn khác, sau khoảng 5 min, cẩn thận tháo nút ra. Rửa nút, dây, lưới platin và thành bình bằng nước rồi tiến hành tiếp như qui định trong chuyên luận riêng.
Đối với mẫu thử là chất lỏng, cho một lượng mẫu thử vào nang làm bằng methylcelulose có cỡ thích hợp đã có chứa sẵn khoảng 15 mg bột giấy lọc không tro. Đậy nắp nang, kẹp đầu cuối của dải giấy lọc hẹp vào giữa 2 phần thân và nắp. Buộc nang vào lưới platin.
Đối với mẫu thử là thuốc mỡ, cần phải bọc trong tờ giấy không thấm mỡ trước khi bọc trong giấy lọc.
Áp dụng cho những chế phẩm chứa iod
Đốt một lượng qui định mẫu thử bằng phương pháp đã mô tả ở trên, sử dụng chất lỏng hấp thụ là hỗn hợp 10 ml nước và 2 ml dung dịch natri hydroxyd 1 M (TT). Khi quá trình đốt đã hoàn thành, thêm vào bình một lượng thừa dung dịch brom trong acid acetic (TT) (khoảng 5 ml đến 10 ml) và để yên trong 2 min. Loại brom thừa bằng cách thêm acid formic (TT) (0,5 ml đến 1 ml). Rửa thành bình với nước cất. Đuổi hơi brom ở trên lớp chất lỏng bằng một luồng khí động. Thêm 1 g kali iodid (TT) và chuẩn độ bằng dung dịch natri thiosulfat 0,02 M (CĐ), dùng chỉ thị là dung dịch hồ tinh bột (TT) thêm vào lúc cuối của quá trình chuẩn độ.
1 ml dung dịch natri thiosulfat 0,02 M (CĐ) tương đương với 0,4230 mg iod.
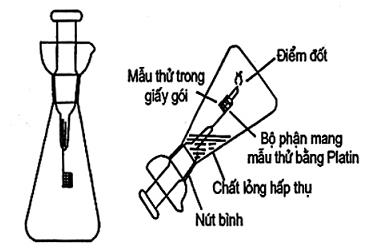
Hình 10.19 - Dụng cụ đốt trong oxygen
PHỤ LỤC 11
(Quy định)
11.1 GIỚI HẠN CHO PHÉP VỀ THỂ TÍCH CỦA CÁC DẠNG THUỐC LỎNG
Lượng hoạt chất có trong các dạng thuốc lỏng liên quan đến thể tích của chúng. Do thực tế sai số của các dụng cụ, trang thiết bị và việc thực hiện qui trình sản xuất mà mỗi dạng thuốc lỏng được phép có một khoảng chênh lệch nhất định về thể tích so với qui định ghi trên nhãn. Đó là giới hạn cho phép về thể tích.
Trừ các thuốc có qui định đặc biệt, các dạng thuốc lỏng có giới hạn cho phép về thể tích ghi trong Bảng 11.1.
Bảng 11.1 - Giới hạn cho phép chênh lệch (%) về thể tích của các thuốc dạng lỏng
| Loại thuốc | Thể tích ghi trên nhãn | Giới hạn cho phép | |
| Thuốc tiêm đơn liều: |
|
| |
| - Đo thể tích thuốc từng đơn vị chế phẩm. | Mọi thể tích | +10% | |
| - Trong trường hợp tiến hành đo thể tích thuốc từ nhiều đơn vị chế phẩm gộp lại với thuốc tiêm có thể tích ≤ 2 ml. | Tổng thể tích ghi trên nhãn của các đơn vị chế phẩm đem thử | +15% | |
| Thuốc tiêm đa liều | Mọi thể tích | Không dưới thể tích ghi trên nhãn | |
| Thuốc tiêm truyền | Tới 50 ml Trên 50 ml | +10 % +5% | |
| Thuốc dạng lỏng để uống (Dung dịch, Hỗn dịch, Nhũ dịch, Rượu, Sirô thuốc và Cao thuốc) | Đa liều | Mọi thể tích | Không dưới thể tích ghi trên nhãn |
| Đơn liều | Tới 20 ml Trên 20 ml đến 50 ml Trên 50 ml đến 150 ml Trên 150 ml | + 10% + 8% + 6% + 4% | |
| Thuốc nhỏ mắt, Thuốc nhỏ mũi, Thuốc nhỏ tai | Mọi thể tích | Không dưới thể tích ghi trên nhãn | |
| Thuốc dùng ngoài | Mọi thể tích | Không dưới thể tích ghi trên nhãn | |
CÁCH THỬ
Các dạng thuốc không thuộc dạng thuốc tiêm, thuốc tiêm truyền
Chế phẩm đa liều
Lấy ngẫu nhiên 5 đơn vị chế phẩm (ống, lọ...). Xác định thể tích từng đơn vị bằng bơm tiêm chuẩn hoặc ống đong chuẩn sạch, khô, có độ chính xác phù hợp. Thể tích mỗi đơn vị phải không dưới thể tích ghi trên nhãn. Nếu có một đơn vị không đạt phải tiến hành kiểm tra lần thứ hai giống như lần đầu. Chế phẩm đạt yêu cầu nếu trong lần thử này không có đơn vị nào có thể tích dưới thể tích ghi trên nhãn.
Chế phẩm đơn liều
Lấy ngẫu nhiên 10 đơn vị chế phẩm. Xác định thể tích từng đơn vị bằng bơm tiêm chuẩn hoặc ống đong chuẩn sạch, khô, có độ chính xác phù hợp. Thể tích mỗi đơn vị phải nằm trong khoảng từ thể tích ghi trên nhãn đến giới hạn cho phép (Bảng 11.1). Nếu có một đơn vị không đạt phải tiến hành kiểm tra lần thứ hai giống như lần đầu. Chế phẩm đạt yêu cầu nếu trong lần thử này không có đơn vị nào có thể tích nằm ngoài giới hạn cho phép.
Các dạng thuốc tiêm, thuốc tiêm truyền
Thuốc tiêm đơn liều
Thuốc tiêm có thể tích lớn hơn hoặc bằng 10 ml: Lấy 1 đơn vị chế phẩm để thử.
Thuốc tiêm có thể tích lớn hơn 3 ml và nhỏ hơn 10 ml: Lấy 3 đơn vị chế phẩm để thử.
Thuốc tiêm có thể tích nhỏ hơn hoặc bằng 3 ml: Lấy 5 đơn vị chế phẩm để thử.
Thuốc tiêm đem đo thể tích phải để cân bằng với nhiệt độ phòng và được phân tán đồng nhất. Dùng bơm tiêm khô, sạch, có dung tích không lớn hơn 3 lần so với thể tích cần đo, có gắn kim tiêm số 21 (21 gauge) và dài không quá 2,5 cm. Lấy toàn bộ thuốc của từng ống vào bơm tiêm, đẩy hết không khí trong bơm tiêm và kim tiêm ra ngoài. Chuyển lượng thuốc có trong bơm tiêm (để lại không bơm hết lượng thuốc còn trong kim) vào ống đong khô, sạch, có vạch chia phù hợp và được chuẩn hóa, ống đong có dung tích sao cho thể tích được đo chiếm tối thiểu 40 % thang đo của ống đong. Cũng có thể xác định thể tích (ml) thuốc tiêm bằng cách xác định khối lượng của lượng thuốc trong ống tiêm (g) rồi chia cho tỷ trọng (g/ml) của chế phẩm.
Đối với thuốc tiêm có thể tích nhỏ hơn hoặc bằng 2 ml, có thể lấy lượng thuốc từ một số lượng đơn vị chế phẩm vừa đủ và gộp lại để có được thể tích đáp ứng cho phép đo, sử dụng các bơm tiêm khô, riêng biệt cho mỗi ống. Đối với thuốc tiêm có thể tích lớn hơn hoặc bằng 10 ml, có thể xác định thể tích bằng cách chuyển thẳng lượng thuốc trong đơn vị chế phẩm vào ống đong hoặc cốc đã cân bì.
Chế phẩm dạng hỗn dịch hoặc nhũ tương phải được lắc đều trước khi rút ra để đo thể tích hoặc trước khi đo tỷ trọng. Chế phẩm nhớt hay ở dạng dầu có thể làm ấm theo hướng dẫn sử dụng ghi trên nhãn, nếu cần, và lắc kỹ ngay trước khi lấy ra để đo. Đo thể tích chế phẩm sau khi đã để nguội xuống 20°C đến 25°C.
Nếu tiến hành đo thể tích trên từng đơn vị chế phẩm, thể tích trong mỗi đơn vị phải nằm trong khoảng từ thể tích ghi trên nhãn đến giới hạn cho phép. Trong trường hợp đo mẫu gộp với thuốc tiêm có thể tích ≤ 2 ml, thể tích đo được phải nằm trong khoảng tổng thể tích ghi trên nhãn của các đơn vị chế phẩm lấy đem đo đến giới hạn cho phép.
Thuốc tiêm đa liều
Đối với thuốc tiêm đa liều mà trên nhãn có ghi số lượng liều và thể tích của từng liều, lấy một đơn vị chế phẩm, tiến hành lấy ra từng liều như với thuốc tiêm đơn liều, sử dụng các bơm tiêm riêng rẽ. Số lượng bơm tiêm dùng cho phép thử tương ứng với số liều ghi trên nhãn. Thể tích của mỗi liều không nhỏ hơn thể tích đơn vị liều ghi trên nhãn.
Thuốc tiêm truyền
Lấy 1 đơn vị chế phẩm. Chuyển toàn bộ lượng thuốc có trong đơn vị chế phẩm vào một ống đong khô, sạch có độ chính xác phù hợp và có dung tích sao cho thể tích được đo chiếm tối thiểu 40 % thang đo của ống đong. Thể tích đo được phải nằm trong khoảng từ thể tích ghi trên nhãn đến giới hạn cho phép.
11.2 PHÉP THỬ ĐỘ ĐỒNG ĐỀU HÀM LƯỢNG
Phép thử độ đồng đều hàm lượng của các chế phẩm đơn liều dựa trên cơ sở định lượng hàm lượng hoạt chất của từng đơn vị, để xác định mỗi hàm lượng riêng lẻ có nằm trong giới hạn cho phép so với hàm lượng trung bình hay không.
Phép thử này không áp dụng cho chế phẩm đa liều, thuốc truyền tĩnh mạch không phân liều, chế phẩm chứa vitamin, nguyên tố vi lượng và các trường hợp khác được phép miễn trừ.
Trừ khi có chỉ dẫn khác trong chuyên luận riêng, phép thử độ đồng đều hàm lượng được tiến hành trên 10 đơn vị riêng lẻ lấy ngẫu nhiên. Kết quả được đánh giá theo phương pháp sau:
Phương pháp 1
Áp dụng cho thuốc nang, thuốc bột không dùng pha tiêm, thuốc cốm, thuốc đạn, thuốc trứng
Chế phẩm đạt yêu cầu phép thử, nếu có không quá một đơn vị có hàm lượng nằm ngoài giới hạn từ 85 % đến 115 % và không có đơn vị nào có hàm lượng nằm ngoài giới hạn từ 75 % đến 125 % của hàm lượng trung bình.
Chế phẩm không đạt yêu cầu phép thử, nếu có quá ba đơn vị có hàm lượng nằm ngoài giới hạn từ 85 % đến 115 %, hoặc có một hay nhiều đơn vị có hàm lượng nằm ngoài giới hạn từ 75 % đến 125 % của hàm lượng trung bình.
Nếu hai hoặc ba đơn vị có hàm lượng nằm ngoài giới hạn từ 85 % đến 115 %, nhưng ở trong giới hạn từ 75 % đến 125 % của hàm lượng trung bình, thử lại trên 20 đơn vị khác lấy ngẫu nhiên. Chế phẩm đạt yêu cầu phép thử, nếu có không quá ba trong tổng số 30 đơn vị đem thử có hàm lượng nằm ngoài giới hạn từ 85 % đến 115 % và không có đơn vị nào có hàm lượng nằm ngoài giới hạn từ 75 % đến 125 % của hàm lượng trung bình.
Phương pháp 2
Áp dụng cho thuốc viên nén, thuốc bột pha tiêm, hỗn dịch để tiêm
Chế phẩm đạt yêu cầu phép thử, nếu hàm lượng của từng đơn vị nằm trong giới hạn từ 85 % đến 115 % của hàm lượng trung bình.
Chế phẩm không đặt yêu cầu phép thử, nếu có quá một đơn vị có hàm lượng nằm ngoài giới hạn từ 85 % đến 115 %, hoặc có một đơn vị có hàm lượng nằm ngoài giới hạn từ 75 % đến 125 % của hàm lượng trung bình.
Nếu có một đơn vị có hàm lượng nằm ngoài giới hạn từ 85 % đến 115 % của hàm lượng trung bình, thử lại trên 20 đơn vị khác lấy ngẫu nhiên. Chế phẩm đạt yêu cầu phép thử, nếu có không quá một trong tổng số 30 đơn vị đem thử có hàm lượng nằm ngoài giới hạn từ 85 % đến 115 % và không có đơn vị nào có hàm lượng nằm ngoài giới hạn từ 75 % đến 125 % của hàm lượng trung bình.
Phương pháp 3
Áp dụng cho thuốc dán (hấp thu qua da)
Chế phẩm đạt yêu cầu phép thử, nếu hàm lượng trung bình của 10 đơn vị nằm trong giới hạn từ 90 % đến 110 % hàm lượng ghi trên nhãn và hàm lượng của từng đơn vị phải nằm trong giới hạn từ 75 % đến 125 % của hàm lượng trung bình.
11.3 PHÉP THỬ ĐỘ ĐỒNG ĐỀU KHỐI LƯỢNG
Phép thử độ đồng đều khối lượng dùng để xác định độ đồng đều phân liều của chế phẩm, khi không có yêu cầu thử độ đồng đều hàm lượng.
Phương pháp thử
Phương pháp 1
Áp dụng cho thuốc viên nén, thuốc đạn, thuốc trứng, thuốc dán
Cân riêng biệt 20 đơn vị lấy ngẫu nhiên, tính khối lượng trung bình. Không được có quá hai đơn vị có khối lượng nằm ngoài giới hạn chênh lệch so với khối lượng trung bình quy định trong Bảng 11.3.1 và không được có đơn vị nào có khối lượng vượt gấp đôi giới hạn đó.
Phương pháp 2
Áp dụng cho thuốc nang, thuốc bột (đơn liều), thuốc cốm (không bao, đơn liều)
Cân khối lượng của một nang hay một gói (thuốc bột, thuốc cốm). Với viên nang cứng, tháo rời hai nửa vỏ nang, dùng bông lau sạch vỏ và cân khối lượng của vỏ. Với viên nang mềm, cắt mở nang, bóp hết thuốc ra, dùng ether hoặc dung môi hữu cơ thích hợp rửa vỏ nang, để khô tự nhiên cho đến khi hết mùi dung môi, cân khối lượng của vỏ nang. Với gói, cắt mở gói, lấy hết thuốc ra, dùng bông lau sạch bột thuốc bám ở mặt trong, cân khối lượng vỏ gói. Khối lượng thuốc trong nang hay gói là hiệu số giữa khối lượng nang thuốc hay gói và khối lượng vỏ nang hay vỏ gói. Tiến hành tương tự với 19 đơn vị khác lấy ngẫu nhiên. Tính khối lượng trung bình của thuốc trong nang hay gói. Kết quả được đánh giá dựa vào Bảng 11.3.1 giống như Phương pháp 1.
Phương pháp 3
Áp dụng cho thuốc bột để pha tiêm.
Loại bỏ hết nhãn, rửa sạch và làm khô bên ngoài. Loại bỏ hết các nút nếu có, cân ngay khối lượng cả vỏ và thuốc. Lấy hết thuốc ra, dùng bông lau sạch, nếu cần rửa với nước, sau đó với ethanol 96 % (TT), sấy ở 100 °C đến 105 °C trong 1 h. Nếu vỏ không chịu được nhiệt độ này, làm khô ở nhiệt độ thích hợp tới khối lượng không đổi, để nguội trong bình hút ẩm và cân. Hiệu số giữa hai lần cân là khối lượng của thuốc. Tiến hành tương tự với 19 đơn vị khác lấy ngẫu nhiên. Tính khối lượng trung bình của thuốc. Kết quả được đánh giá dựa vào Bảng 11.3.1 giống như Phương pháp 1.
Bảng 11.3.1 - Bảng quy định độ đồng đều khối lượng cho chế phẩm đơn liều
| Dạng bào chế | Khối lượng trung bình (KLTB) | % chênh lệch so với KLTB |
| Viên nén
| Nhỏ hơn hoặc bằng 80 mg | 10 |
| Viên bao phim | Lớn hơn 80 mg và nhỏ hơn 250 mg | 7,5 |
| Bằng hoặc lớn hơn 250 mg | 5 | |
| Thuốc nang: Thuốc bột (đơn liều); | Nhỏ hơn 300 mg | 10 |
| Thuốc cốm (không bao, đơn liều) | Bằng hoặc lớn hơn 300 mg | 7,5 |
| Thuốc bột để pha tiêm (đơn liều) | Lớn hơn 40 mg | 10 |
| Viên nén bao đường | Tất cả các loại | 10 |
| Thuốc đạn Thuốc trứng Cao dán | Tất cả các loại | 5 |
* Đối với thuốc bột để pha tiêm, khi khối lượng trung bình bằng hay nhỏ hơn 40 mg, chế phẩm không phải thử độ đồng đều khối lượng, nhưng phải thử độ đồng đều hàm lượng.
Phương pháp 4
Áp dụng cho thuốc bột (đa liều), thuốc cốm (đa liều), thuốc mỡ, cao xoa, cao động vật.
Cân khối lượng của một đơn vị đóng gói nhỏ nhất. Mở đồ chứa (gói, hộp, lọ...), lấy hết thuốc ra, cắt mở đồ chứa nếu cần để dễ dàng dùng bông lau sạch thuốc bám ở mặt trong, cân khối lượng của đồ chứa. Hiệu số giữa hai lần cân là khối lượng của thuốc. Tiến hành tương tự với bốn đơn vị khác lấy ngẫu nhiên. Tất cả các đơn vị phải có khối lượng nằm trong giới hạn chênh lệch so với khối lượng ghi trên nhãn quy định trong Bảng 11.3.2. Nếu có một đơn vị có khối lượng nằm ngoài giới hạn đó, tiến hành thử lại với năm đơn vị khác lấy ngẫu nhiên. Không được có quá một đơn vị trong tổng số 10 đơn vị đem thử có khối lượng nằm ngoài giới hạn qui định.
Bảng 11.3.2 - Bảng quy định độ đồng đều khối lượng cho chế phẩm đa liều
| Dạng bào chế | Khối lượng ghi trên nhãn (KLN) | % chênh lệch so với KLN |
| Thuốc bột (đa liều) | Nhỏ hơn hoặc bằng 0,50 g | 10 |
| Lớn hơn 0,50 g và bằng 1,50 g Lớn hơn 1,50 g và bằng 6,00 g Lớn hơn 6,00 g | 7 5 3 | |
| Thuốc cốm (đa liều) | Tất cả các loại | 5 |
| Thuốc kem, mỡ, bột nhão, gel Cao xoa | Nhỏ hơn hoặc bằng 10,0 g | 15 |
| Lớn hơn 10,0 g và bằng 20,0 g | 10 | |
| Lớn hơn 20,0 g và bằng 50,0 g | 8 | |
| Lớn hơn 50,0 | 5 | |
| Cao động vật | Nhỏ hơn hoặc bằng 100,0 g | 5 |
| Lớn hơn 100,0 g và bằng 200,0 g | 3 | |
| Lớn hơn 200,0 g | 2 |
11.4 PHÉP THỬ ĐỘ HÒA TAN CỦA DẠNG THUỐC RẮN PHÂN LIỀU
Phép thử này nhằm xác định sự phù hợp của chế phẩm đối với yêu cầu qui định về độ hòa tan dược chất của các dạng thuốc rắn phân liều dùng đường uống. Trong chuyên luận, một đơn vị được hiểu là 1 viên nén, 1 nang hoặc 1 lượng chế phẩm được chỉ rõ.
Thiết bị
Thiết bị kiểu giỏ quay (Thiết bị kiểu 1)
Hệ thống thiết bị này bao gồm những bộ phận sau:
Một bình hình trụ dung tích 1 lít có đáy hình bán cầu bằng thủy tinh hoặc bằng vật liệu trơ và trong suốt khác, miệng viền rộng có nắp đậy với những lỗ thích hợp (để cắm nhiệt kế....). Chiều cao của bình từ 160 mm đến 210 mm, đường kính trong từ 98 mm đến 106 mm. Vật liệu chế tạo bình phải không hấp phụ, không phản ứng hoặc tương tác với mẫu thử;
Một trục quay và một giỏ hình trụ như mô tả ở Hình 11.4.1 Các bộ phận trục và giỏ quay của thiết bị được chế tạo bằng thép không gỉ (SUS316) hoặc bằng một vật liệu trơ khác. Giỏ quay có thể được mạ một lớp vàng dày khoảng 2,5 μm. Đơn vị được đặt trong giỏ khô khi bắt đầu mỗi phép thử. Khoảng cách giữa đáy bình và đáy của giỏ luôn luôn giữ trong khoảng 25 mm ± 2 mm suốt quá trình thử. Trục quay được định vị sao cho trục của nó không bị lệch quá 2 mm so với trục xuyên tâm thẳng đứng của bình ở bất kỳ điểm nào;
Một động cơ có bộ phận điều tốc để duy trì tốc độ quay trong dung sai ± 4 % của tốc độ qui định. Động cơ được kết nối với một trục quay và khi vận hành thì quay nhẹ nhàng, không tạo nên sóng nước ảnh hưởng tới kết quả;
Bình được ngâm chìm một phần trong một bể cách thủy có kích thước phù hợp hoặc có thể được điều nhiệt bằng thiết bị thích hợp khác như áo nước. Bể cách thủy hay áo nước cho phép duy trì nhiệt độ bên trong bình môi trường hòa tan ở 37 °C ± 0,5 °C trong quá trình thử cũng như giữ cho môi trường trong bình chuyển động ổn định và nhẹ nhàng. Bất kỳ bộ phận nào của thiết bị, bao gồm cả bàn thí nghiệm hay nơi đặt thiết bị, đều không được có rung động, lắc lư đáng kể; chấp nhận những lay động rất nhẹ gây ra bởi bộ phận khuấy quay. Thiết bị được chế tạo sao cho có thể quan sát dễ dàng mẫu thử và bộ phận quay trong khi vận hành.
| 1. Lưới có mép được hàn: đường kính sợi lưới từ 0,25 đến 0,31 mm. Độ rộng mắt lưới từ 0,36 - 0,44mm |
| 2. Khi lắp giỏ quay vào trục quay và vận hành thì độ lệch tối đa tại "A" là 1,0 mm |
Hình 11.4.1 - Thiết bị thử độ hòa tan kiểu giỏ quay (kiểu 1)
(Kích thước tính bằng mm)
Thiết bị kiểu cánh khuấy (Thiết bị kiểu 2)
Thiết bị kiểu cánh khuấy giống như thiết bị kiểu 1 ngoại trừ có dùng một bộ phận khuấy, bao gồm một cánh khuấy và một trục mang nó. Trục quay được định vị sao cho trục của nó không bị lệch quá 2 mm so với trục xuyên tâm thẳng đứng của bình ở bất kỳ điểm nào. Trục này quay nhẹ nhàng, không tạo nên sóng nước ảnh hưởng tới kết quả. Đường thẳng đứng xuyên tâm của cánh khuấy cũng đi qua trục của trục quay và lắp sao cho đáy của cánh khuấy ngang bằng với đáy của trục. Cánh khuấy được mô tả trên Hình 11.4.2. Khoảng cách giữa đáy cánh khuấy và đáy bình luôn được duy trì ở 25 mm ± 2 mm trong quá trình vận hành phép thử. Trục quay và cánh khuấy được làm bằng kim loại hay vật liệu trơ thích hợp, được gắn kết thành một khối chắc chắn. Một kiểu thiết kế khác gồm 2 phần tháo lắp được cũng có thể được sử dụng miễn là 2 phần này gắn kết chắc chắn trong quá trình vận hành. Cánh khuấy và trục quay có thể được phủ một lớp mạ thích hợp để đảm bảo bề mặt hoàn toàn trơ. Đơn vị thử thường được đặt xuống đáy bình trước khi bắt đầu cho cánh khuấy quay, có thể dùng bộ phận giữ mẫu thử, làm bằng vật liệu trơ, được mô tả trên Hình 11.4.2a, để giữ cho mẫu thử không bị nổi lên. Những bộ phận giữ mẫu thử kiểu khác cũng có thể sử dụng miễn là được thẩm định đạt yêu cầu. Nếu trong chuyên luận có chỉ định dùng bộ phận giữ mẫu thử và không có qui định khác, thì dùng bộ phận giữ mẫu thử như trong Hình 11.4.2a.
Lưu ý:
1. Kích thước A và B không thay đổi quá 0,5 mm khi quay
2. Dung sai ± 1,0 mm trừ khi có quy định khác
Hình 11.4.2 - Thiết bị thử độ hòa tan kiểu cánh khuấy (kiểu 2)
(Kích thước tính bằng mm)
| A: Cặp giữ bằng sợi chịu acid B: Bộ phận đỡ bằng sợi chịu acid |
Hình 11.4.2a - Một kiểu bộ phận giữ mẫu thử
(Kích thước tính bằng mm)
Thiết bị kiểu buồng dòng chảy (Thiết bị kiểu 3)
Thiết bị này bao gồm 1 bình chứa và 1 bơm để bơm môi trường hòa tan; 1 buồng cho môi trường hòa tan chảy qua; 1 bể cách thủy để duy trì môi trường hòa tan ở 37 °C ± 0,5 °C. Dùng buồng dòng chảy có kích cỡ như qui định trong chuyên luận riêng.
Bơm đẩy môi trường hòa tan đi lên qua buồng dòng chảy. Sức đẩy của bơm ở trong khoảng từ 4 đến 16 ml/min với các tốc độ dòng tiêu chuẩn là 4, 8 và 16 ml/min. Nó phải chuyển tải được một dòng chảy hằng định (dao động ± 5 % tốc độ dòng qui định); giản đồ dòng phải ở dạng hình sin với tần số xung là 120 ± 10 xung/min. Loại bơm được khử xung cũng có thể được dùng. Qui trình thử độ hòa tan dùng buồng dòng chảy được qui định thông số đặc trưng về tốc độ dòng và tần số xung nếu có.
Buồng dòng chảy (xem Hình 11.4.3 và 11.4.4) được chế tạo bằng vật liệu trơ, trong suốt, lắp đặt thẳng đứng với hệ thống lọc (được qui định trong chuyên luận riêng) có khả năng ngăn ngừa những tiểu phân không hòa tan thoát ra từ mặt trên của buồng; đường kính buồng tiêu chuẩn là 12 mm và 22,6 mm; phần hình nón ngược phía dưới thường chứa đầy những hạt bi thủy tinh nhỏ có đường kính 1 mm, trong đó có 1 hạt đường kính 5 mm được định vị ở đáy để ngăn chất lỏng đi vào ống; một bộ phận giữ mẫu thử (xem Hình 11.4.3 và 11.4.4) được dùng để định vị các dạng phân liều đặc biệt. Buồng được nhúng chìm trong bể cách thủy, và được ổn nhiệt ở 37 °C ± 0,5 °C.
Thiết bị có 2 vòng khuyên để cặp giữ buồng dòng chảy với toàn hệ thống. Bơm được đặt tách riêng khỏi bộ phận hòa tan để giữ cho bộ phận này khỏi rung do bơm chạy. Vị trí của bơm không nên ở mức cao hơn bình chứa môi trường hòa tan. Các đoạn ống nối phải càng ngắn càng tốt. Dùng loại ống bằng vật liệu trơ thích hợp như polytetrafluoroethylen, có đường kính trong khoảng 1,6 mm và các đầu nối được viền nhẵn.
Hình 11.4.3 - Thiết bị thử độ hòa tan kiểu 3 loại buồng lớn
(Kích thước tính bằng mm)
Hình 11.4.4 - Thiết bị thử độ hòa tan kiểu 3 loại buồng nhỏ
(Kích thước tính bằng mm)
Đánh giá tính phù hợp của thiết bị
Việc xác định tính phù hợp của hệ thống thiết bị thử độ hòa tan phải bao gồm đáp ứng yêu cầu về kích thước và khoảng sai số được nêu ở trên. Những thông số quan trọng nhất của phép thử như nhiệt độ và thể tích môi trường hòa tan, tốc độ quay (Phương pháp giỏ quay và Phương pháp cánh khuấy) hay tốc độ dòng của môi trường hòa tan (Phương pháp buồng dòng chảy) cần phải theo dõi định kỳ trong quá trình thử.
Cần phải định kỳ hiệu chuẩn thiết bị thử độ hòa tan.
Phương pháp tiến hành
a. Phương pháp giỏ quay hoặc phương pháp cánh khuấy
Dạng thuốc giải phóng tức thời:
Cách tiến hành: Cho một thể tích xác định môi trường hòa tan (± 1 %) vào trong bình hòa tan, lắp ghép thiết bị, cân bằng nhiệt độ môi trường hòa tan ở 37 °C ± 0,5 °C rồi lấy nhiệt kế ra. Cho 1 đơn vị vào thiết bị, chú ý loại bọt khí khỏi bề mặt của mẫu thử. Cho vận hành thiết bị ngay ở tốc độ được qui định. Trong khoảng thời gian qui định, hay ở mỗi thời điểm qui định, lấy một phần mẫu môi trường hòa tan ở vùng giữa bề mặt môi trường hòa tan và mặt trên của giỏ quay hay mặt trên của cánh khuấy, cách thành bình ít nhất 10 mm để thử. Nếu phép thử qui định phải lấy mẫu nhiều lần, cần cấp bù một thể tích môi trường hòa tan mới ở 37 °C bằng thể tích dịch mẫu thử đã lấy đi, hoặc nếu không cần thiết phải cấp bù môi trường hòa tan thì tiến hành hiệu chỉnh sự thay đổi thể tích trong tính toán kết quả. Đậy nắp bình hòa tan trong suốt quá trình thử và kiểm tra nhiệt độ của môi trường hòa tan ở các thời điểm thích hợp.
Mẫu thử được lọc ngay sau khi lấy trừ khi đã chứng minh được việc lọc dịch thử là không cần thiết. Cần dùng màng lọc trơ, không hấp phụ dược chất hoặc không chứa những chất bị chiết ra gây trở ngại cho việc phân tích. Tiến hành xác định lượng dược chất được hòa tan theo phương pháp qui định. Lặp lại phép thử với những đơn vị thử khác.
Nếu dùng thiết bị tự động lấy mẫu hoặc thiết bị có thay đổi khác thì cần phải chứng tỏ rằng thiết bị này cho kết quả tương đương với kết quả thu được từ thiết bị chuẩn đã được mô tả trên đây.
Môi trường hòa tan: Sử dụng một loại môi trường hòa tan thích hợp. Đong, đo thể tích môi trường qui định trong điều kiện nhiệt độ từ 20 °C đến 25 °C. Nếu môi trường hòa tan là một dung dịch đệm, cần điều chỉnh pH sao cho ở trong khoảng pH qui định ± 0,05 đơn vị. Các chất khí hòa tan có thể tạo thành bọt khí làm thay đổi kết quả phép thử. Để tránh ảnh hưởng này, cần loại khí trước khi sử dụng.
Thời gian: Nếu qui định một thời điểm lấy mẫu kiểm tra, phép thử có thể kết thúc trong khoảng thời gian ngắn hơn nếu lượng dược chất hòa tan tối thiểu đã được đáp ứng. Mẫu thử được lấy ra chỉ ở những thời điểm qui định, thời điểm lấy mẫu được phép sai số ± 2 %.
Dạng thuốc giải phóng kéo dài:
Cách tiến hành: Tiến hành như mô tả trong mục Dạng thuốc giải phóng tức thời.
Môi trường hòa tan: Tiến hành như mô tả trong mục Dạng thuốc giải phóng tức thời.
Thời gian: Thường có 3 thời điểm lấy mẫu kiểm tra, được biểu thị bằng giờ.
Dạng thuốc giải phóng muộn:
Cách tiến hành: Theo phương pháp A hoặc phương pháp B dưới đây:
Phương pháp A:
Giai đoạn acid: Cho 750 ml acid hydrocloric 0,1 M vào bình hòa tan, lắp ráp thiết bị ổn định nhiệt độ môi trường hòa tan ở 37 °C ± 0,5 °C. Cho 1 đơn vị vào bình, đậy nắp bình rồi vận hành thiết bị với vận tốc được qui định trong chuyên luận riêng. Sau 2 h vận hành ở môi trường acid hydrocloric 0,1 M, lấy một một phần môi trường hòa tan để thử rồi tiến hành ngay Giai đoạn đệm tiếp theo. Xác định lượng dược chất được hòa tan bằng phương pháp thích hợp.
Giai đoạn đệm: Cần hoàn thành việc thêm dung dịch đệm và điều chỉnh pH trong thời gian không quá 5 min. Trong khi thiết bị vẫn đang vận hành ở vận tốc qui định, thêm vào bình hòa tan 250 ml dung dịch trinatri phosphat 0,20 M đã được ổn nhiệt ở 37 °C ± 0,5 °C. Điều chỉnh pH môi trường hòa tan đến 6,8 ± 0,05 nếu cần thiết, bằng acid hydrocloric 2 M (TT) hoặc bằng dung dịch natri hydroxyd 2 M (TT). Tiếp tục vận hành thiết bị 45 min nữa hoặc một khoảng thời gian theo quy định. Hết thời gian này, lấy một phần môi trường hòa tan để xác định lượng dược chất được hòa tan bằng phương pháp thích hợp.
Phương pháp B:
Giai đoạn acid: Cho 1000 ml acid hydrocloric 0,1 M vào bình hòa tan, lắp ráp thiết bị. Ổn định nhiệt độ môi trường hòa tan ở 37 °C ± 0,5 °C. Cho 1 đơn vị vào bình, đậy nắp bình rồi vận hành thiết bị với vận tốc được qui định trong chuyên luận riêng. Sau 2 h vận hành ở môi trường acid hydrocloric 0,1 M, lấy một phần môi trường hòa tan để thử rồi tiến hành ngay Giai đoạn đệm tiếp theo. Xác định lượng dược chất hòa tan bằng phương pháp thích hợp.
Giai đoạn đệm: Dùng môi trường là dung dịch đệm đã được ổn nhiệt trước ở 37 °C ± 0,5 °C. Rút hết dung dịch acid từ bình hòa tan đi rồi thêm 1000 ml đệm phosphat pH 6,8 được pha bằng cách trộn đều dung dịch acid hydrocloric 0,1 M với dung dịch trinatri phosphat 0,20 M theo tỷ lệ 3:1 và điều chỉnh pH môi trường hòa tan đến 6,8 ± 0,05 nếu cần thiết, bằng acid hydrocloric 2 M (TT) hoặc bằng dung dịch natri hydroxyd 2 M (TT). Cũng có thể thực hiện việc này bằng cách nhấc bình chứa acid ra khỏi máy, thay bằng một bình hòa tan khác chứa dung dịch đệm rồi chuyển đơn vị thử từ bình chứa acid sang bình dung dịch đệm. Tiếp tục vận hành thiết bị 45 min nữa hoặc một khoảng thời gian theo qui định. Hết thời gian này, lấy một lượng môi trường hòa tan để xác định lượng dược chất hòa tan theo phương pháp thích hợp.
Môi trường hòa tan: Tiến hành loại khí như mô tả trong mục Dạng thuốc giải phóng tức thời.
Thời gian: Các thời điểm lấy mẫu được phép sai số ± 2 %, trừ khi có qui định khác.
b. Phương pháp buồng dòng chảy
Dạng thuốc giải phóng tức thời:
Cách tiến hành: Cho các viên bi thủy tinh vào buồng thử như chỉ dẫn trong chuyên luận riêng. Đặt 1 đơn vị lên trên lớp viên bi, hoặc nếu có chỉ dẫn trong chuyên luận riêng thì đơn vị thử được đặt trên bộ phận giữ mẫu. Lắp đầu phễu lọc và cố định các bộ phận của thiết bị với nhau bằng kẹp giữ thích hợp. Bơm môi trường hòa tan đã được làm ấm đến (37 ± 0,5) °C đi qua đáy của buồng để có được tốc độ dòng theo quy định và được đo với độ chính xác 5 %. Thu các phần dịch hòa tan tại mỗi thời điểm qui định. Tiến hành định lượng như hướng dẫn. Lặp lại phép thử với những đơn vị khác.
Môi trường hòa tan: Tiến hành như chỉ dẫn trong mục Dạng thuốc giải phóng tức thời của Phương pháp giỏ quay và Phương pháp cánh khuấy.
Thời gian: Tiến hành như chỉ dẫn trong mục Dạng thuốc giải phóng tức thời của Phương pháp giỏ quay và Phương pháp cánh khuấy.
Dạng thuốc giải phóng kéo dài:
Cách tiến hành: Tiến hành như mô tả trong mục Dạng thuốc giải phóng tức thời của Phương pháp buồng dòng chảy.
Môi trường hòa tan: Tiến hành như mô tả trong mục Dạng thuốc giải phóng tức thời của Phương pháp buồng dòng chảy.
Thời gian: Thường có 3 thời điểm lấy mẫu, được biểu thị bằng giờ.
Dạng thuốc giải phóng muộn:
Cách tiến hành: Tiến hành như mô tả trong mục Dạng thuốc giải phóng tức thời của Phương pháp buồng dòng chảy.
Môi trường hòa tan: Tiến hành như mô tả trong mục Dạng thuốc giải phóng tức thời của Phương pháp buồng dòng chảy.
Thời gian: Các thời điểm lấy mẫu được phép sai số ± 2 %, trừ khi có qui định khác.
Đánh giá kết quả
a. Dạng thuốc giải phóng tức thời
Nếu không có chỉ dẫn gì khác, mẫu thử đạt tiêu chuẩn khi lượng dược chất hòa tan từ các đơn vị đem thử đáp ứng yêu cầu ghi trong Bảng 11.4.1. Phép thử phải trải qua bước tiếp theo nếu kết quả không đạt ở bước trước đó.
Giá trị Q, là lượng dược chất hòa tan theo qui định, được biểu thị bằng phần trăm so với lượng ghi trên nhãn; các giá trị 5 %, 15 % và 25 % cũng là phần trăm của lượng dược chất ghi trên nhãn.
Bảng 11.4.1 Tiêu chuẩn chấp nhận khi thử độ hòa tan đối với dạng thuốc giải phóng tức thời
| Bước | Số đơn vị thử | Tiêu chuẩn chấp nhận |
| S1 | 6 | Lượng dược chất hòa tan từ mỗi đơn vị không thấp hơn Q + 5 % |
| S2 | 6 | Lượng dược chất hòa tan trung bình của 12 đơn vị thử (S1 + S2) bằng hoặc lớn hơn Q và không có viên nào có lượng dược chất hòa tan thấp hơn Q - 15 % |
| S3 | 12 | Lượng dược chất hòa tan trung bình của 24 đơn vị thử (S1 + S2 + S3) bằng hoặc lớn hơn Q, không quá 2 đơn vị có lượng dược chất hòa tan thấp hơn Q - 15 % và không có đơn vị nào có lượng dược chất hòa tan thấp hơn Q - 25 % |
b. Dạng thuốc giải phóng kéo dài
Nếu không có chỉ dẫn gì khác, mẫu thử đạt tiêu chuẩn khi lượng dược chất hòa tan từ các đơn vị đem thử đáp ứng yêu cầu ghi trong Bảng 11.4.2. Phép thử phải trải qua bước tiếp theo nếu kết quả không đạt ở bước trước đó. Giới hạn lượng dược chất hòa tan ở từng thời điểm được biểu thị bằng phần trăm so với lượng ghi trên nhãn. Các giới hạn này gắn với mỗi giá trị Qi, là lượng dược chất được hòa tan ở mỗi một khoảng thời gian nhất định. Các giá trị 10 %, 20 % ghi trong Bảng 11.4.2 cũng là hàm lượng phần trăm của lượng dược chất ghi trên nhãn. Nếu có quy định từ 2 khoảng thời gian hay nhiều hơn, tiêu chuẩn chấp nhận được áp dụng cho từng khoảng thời gian.
Bảng 11.4.2 Tiêu chuẩn chấp nhận khi thử độ hòa tan đối với dạng thuốc giải phóng kéo dài
| Bước | Số đơn vị thử | Tiêu chuẩn chấp nhận |
| L1 | 6 | Không có đơn vị nào có lượng dược chất hòa tan nằm ngoài mỗi một trong các khoảng qui định và không có đơn vị nào có lượng dược chất hòa tan thấp hơn lượng dược chất hòa tan qui định tại thời điểm thử cuối cùng. |
| L2 | 6 | Lượng dược chất hòa tan trung bình của 12 đơn vị (L1 + L2) phải nằm trong mỗi khoảng qui định tương ứng và không được thấp hơn lượng dược chất hòa tan qui định ở thời điểm thử cuối cùng: không có đơn vị nào có lượng dược chất hòa tan vượt ra ngoài quá 10 % ở mỗi khoảng qui định tương ứng; không có đơn vị nào có lượng dược chất hòa tan thấp hơn quá 10 % lượng dược chất hòa tan qui định tại thời điểm thử cuối cùng. |
| L3 | 12 | Lượng dược chất hòa tan trung bình của 24 đơn vị (L1 + L2 + L3) phải nằm trong mỗi khoảng qui định tương ứng và không được thấp hơn lượng dược chất hòa tan qui định ở thời điểm cuối cùng; không quá 2 trong 24 đơn vị có lượng dược chất hòa tan vượt ra ngoài quá 10 % ở mỗi khoảng quy định tương ứng; không quá 2 trong 24 đơn vị có lượng dược chất hòa tan thấp hơn quá 10 % lượng dược chất hòa tan quy định tại thời điểm thử cuối cùng; không có một đơn vị nào có lượng dược chất hòa tan vượt ra ngoài quá 20 ở mỗi khoảng qui định tương ứng và thấp hơn quá 20 % lượng dược chất hòa tan qui định tại thời điểm thử cuối cùng. |
Dạng thuốc giải phóng muộn:
Giai đoạn acid: Nếu không có chỉ dẫn gì khác trong chuyên luận riêng, mẫu thử đạt yêu cầu ở giai đoạn này nếu lượng dược chất hòa tan tính bằng phần trăm so với lượng ghi trên nhãn của các đơn vị được thử đáp ứng yêu cầu ghi trong Bảng 11.4.3. Phép thử phải trải qua bước tiếp theo nếu kết quả không đạt ở bước trước đó.
Bảng 11.4.3 - Tiêu chuẩn chấp nhận khi thử độ hòa tan đối với dạng thuốc giải phóng muộn
| Bước | Số đơn vị thử | Tiêu chuẩn chấp nhận |
| A1 | 6 | Không có đơn vị nào có lượng dược chất hòa tan quá 10 % |
| A2 | 6 | Lượng dược chất hòa tan trung bình của 12 đơn vị thử (A1 + A2) không lớn hơn 10 % và không có viên nào có lượng dược chất hòa tan quá 25 % |
| A3 | 12 | Lượng dược chất hòa tan trung bình của 24 đơn vị thử (A1 + A2 + A3) không lớn hơn 10 %, không có đơn vị nào có lượng dược chất hòa tan quá 25 % |
Giai đoạn đệm: Nếu không có chỉ dẫn gì khác trong chuyên luận riêng, mẫu thử đạt yêu cầu khi lượng dược chất hòa tan từ các đơn vị đáp ứng yêu cầu ghi trong Bảng 11.4.4. Phép thử phải trải qua bước tiếp theo nếu kết quả không đạt ở bước trước đó. Giá trị Q trong Bảng 11.4.4 là 75 % lượng dược chất ghi trên nhãn được hòa tan, trừ phi có qui định khác trong chuyên luận riêng. Trị giá của Q là tổng lượng dược chất được hòa tan, biểu thị bằng phần trăm, của cả hai giai đoạn acid và đệm. Các giá trị 5 %, 15 % và 25 % trong Bảng 11.4.4 cũng là phần trăm của lượng dược chất ghi trên nhãn.
Bảng 11.4.4 - Tiêu chuẩn chấp nhận khi thử độ hòa tan đối với dạng thuốc giải phóng muộn
| Bước | Số đơn vị thử | Tiêu chuẩn chấp nhận |
| B1 | 6 | Không có đơn vị nào có lượng dược chất hòa tan thấp hơn Q + 5 % |
| B2 | 6 | Lượng dược chất hòa tan trung bình của 12 đơn vị thử (B1 + B2) bằng hoặc lớn hơn Q và không có viên nào có lượng dược chất hòa tan thấp hơn Q - 15 % |
| B3 | 12 | Lượng dược chất hòa tan trung bình của 24 đơn vị thử (B1 + B2+ B3) bằng hoặc lớn hơn Q, không quá 2 đơn vị có lượng dược chất hòa tan thấp hơn Q - 15 %, và không có đơn vị nào có lượng dược chất hòa tan thấp hơn Q - 25 % |
Một số quy định đối với phép thử độ hòa tan của viên nén và nang
Đối với viên nén hoặc nang giải phóng tức thời, nếu không có quy định trong chuyên luận riêng thì thời gian thử là 45 min. Lấy 6 đơn vị thử, lượng dược chất hòa tan từ mỗi đơn vị so với lượng ghi trên nhãn không được thấp hơn 70 %. Nếu có 1 đơn vị không đạt yêu cầu thì lặp lại phép thử với 6 đơn vị khác, lần này cả 6 đơn vị đều phải đạt.
Khi có quy định cho hai đơn vị thử hoặc nhiều hơn vào một bình thử, cũng tiến hành thử sáu lần như vậy. Trong mỗi lần thử, nếu không có chỉ dẫn khác trong chuyên luận riêng, lượng dược chất được hòa tan tính theo mỗi đơn vị không được ít hơn 70 % lượng dược chất ghi trên nhãn. Không được phép thử lại.
Trong trường hợp vỏ nang thuốc có ảnh hưởng đến kết quả phân tích, lấy ít nhất 6 nang thuốc, loại bỏ hết phần thuốc trong nang, hòa tan vỏ nang rỗng trong một thể tích môi trường hòa tan qui định. Tiến hành phân tích bằng phương pháp như đối với dung dịch mẫu thử được qui định trong chuyên luận riêng, sau đó tính hệ số hiệu chỉnh. Hệ số hiệu chỉnh này không được lớn hơn 25 % lượng ghi trên nhãn.
MỘT SỐ HƯỚNG DẪN CHO PHÉP THỬ ĐỘ HÒA TAN CỦA DẠNG THUỐC RẮN PHÂN LIỀU
Trong việc xác định độ hòa tan dược chất của các dạng thuốc rắn phân liều cần xác định rõ các điểm sau đây:
Loại thiết bị được sử dụng; trong trường hợp dùng thiết bị kiểu buồng dòng chảy, cần nói rõ loại buồng lớn hay buồng nhỏ;
Thành phần, thể tích và nhiệt độ của môi trường hòa tan;
Tốc độ quay hoặc tốc độ dòng của môi trường hòa tan;
Thời gian, phương pháp và lượng dung dịch hòa tan lấy để thử hoặc các điều kiện để theo dõi liên tục;
Phương pháp xác định lượng dược chất hòa tan trong mẫu thử;
Tiêu chuẩn chấp nhận.
Việc lựa chọn loại thiết bị hòa tan được dùng tùy thuộc vào các tính chất hóa lý của dạng bào chế thuốc. Khi một lượng lớn môi trường hòa tan được chỉ định sử dụng để đảm bảo điều kiện “sink”, hoặc khi sự thay đổi pH môi trường hòa tan trong quá trình thử là cần thiết, thiết bị kiểu buồng dòng chảy thường được ưu tiên sử dụng.
Các điều kiện thử
Việc dùng thiết bị kiểu giỏ quay hay thiết bị kiểu cánh khuấy, nói chung là dựa trên nguyên tắc vận hành ở điều kiện “sink”, có nghĩa là ở điều kiện đó, dược chất đã hòa tan vào dung dịch không có hiệu ứng đáng kể làm thay đổi độ hòa tan của phần dược chất còn lại. Thông thường, điều kiện “sink” có được khi thể tích môi trường hòa tan ít nhất bằng từ 3 đến 10 lần thể tích cần để bão hòa lượng hoạt chất.
Nói chung, nước thường được dùng làm dung môi để chuẩn bị môi trường hòa tan. Các thành phần của môi trường hòa tan được lựa chọn trên cơ sở đặc điểm hóa lý của dược chất và các tá dược sao cho các điều kiện môi trường này gần giống với điều kiện môi trường mà thuốc tiếp xúc sau khi uống. Điều cần đặc biệt quan tâm là pH và lực ion của môi trường hòa tan.
pH của môi trường hòa tan thường ở trong khoảng từ 1 đến 8. Trong những trường hợp cần thiết được biện giải thỏa đáng, có thể dùng môi trường hòa tan ở pH cao hơn. Đối với những giá trị pH thấp hơn ở vùng acid thì thường dùng acid hydrocloric 0,1 M. Một số môi trường hòa tan được khuyến cáo sử dụng được mô tả ở phần sau.
Nước được dùng làm môi trường hòa tan chỉ khi đã chứng minh được là sự thay đổi pH không ảnh hưởng đến đặc tính hòa tan của hoạt chất.
Trong những trường hợp đặc biệt, môi trường hòa tan có thể chứa enzym, chất hoạt động bề mặt và một số chất vô cơ hay hữu cơ khác. Để thử những chế phẩm chứa dược chất khó tan trong nước, có thể cần phải làm thay đổi môi trường hòa tan. Trong những trường hợp này, nên thêm vào môi trường hòa tan chất hoạt động bề mặt ở nồng độ thấp và tránh dùng các dung môi hữu cơ.
Không khí hòa tan trong môi trường hòa tan có thể ảnh hưởng tới kết quả của phép thử. Ảnh hưởng này càng rõ rệt đối với thiết bị buồng dòng chảy, vì thế, việc loại khí khỏi môi trường hòa tan là cần thiết nhằm tránh sự tạo thành bong bóng khí trong buồng dòng chảy. Phương pháp loại khí thích hợp như sau: Khuấy nhẹ và làm ấm môi trường lên khoảng 41 °C, lọc ngay dưới chân không, dùng màng lọc có kích thước lỗ lọc 0,45 μm hay bé hơn, đồng thời khuấy mạnh và tiếp tục khuấy trong chân không khoảng 5 min. Các kỹ thuật loại khí khác cũng có thể được sử dụng.
Khi dùng thiết bị kiểu giỏ quay hoặc kiểu cánh khuấy, thể tích môi trường hòa tan thường là 500 đến 1000 ml. Tốc độ khuấy thường được chọn là từ 50 r/min đến 100 r/min; tốc độ này không được vượt quá 150 r/min.
Đối với thiết bị buồng dòng chảy, tốc độ dòng của môi trường hòa tan thường đặt giữa 4 ml/min và 50 ml/min.
Môi trường hòa tan
Những môi trường sau có thể được sử dụng làm môi trường hòa tan.
Bảng 1 - Những ví dụ về môi trường hòa tan
| pH | Môi trường hòa tan |
| pH 1,0 | HCl |
| pH 1,2 | NaCl, HCl |
| pH 1,5 | NaCl, HCl |
| pH 4,5 | Đệm acetat hoặc đệm phosphat |
| pH 5,5 và 5,8 | Đệm acetat hoặc đệm phosphat |
| pH 6,8 | Đệm phosphat |
| pH 7,2 và 7,5 | Đệm phosphat |
Thành phần và cách pha chế các môi trường này được chỉ ra dưới đây:
Môi trường acid hydrocloric
Dung dịch acid hydrocloric 0,2 M,
Dung dịch natri clorid 0,2 M: Hòa tan 11,69 g natri clorid (TT) trong nước rồi pha loãng với nước đến đủ 1000,0 ml.
Để pha chế các môi trường có pH khác nhau, lấy 250 ml dung dịch natri clorid 0,2 M cho vào một bình định mức 1000 ml, thêm một thể tích acid hydrocloric 0,2 M như ghi trong Bảng 2 rồi thêm nước vừa đủ đến vạch.
Bảng 2 - Môi trường acid hydrocloric
| pH | 1,2 | 1,3 | 1,4 | 1,5 | 1,6 | 1,7 | 1,8 | 1,9 | 2,0 | 2,1 | 2,2 |
| HCl (ml) | 425,0 | 336,0 | 266,0 | 207,0 | 162,0 | 130,0 | 102,0 | 81,0 | 65,0 | 51,0 | 39,0 |
Cũng có thể thay thế natri clorid bằng kali clorid trong pha chế các môi trường acid hydrocloric.
Các dung dịch đệm acetat
Dung dịch acid acetic 2 M: Hòa trộn 120,0 g acid acetic băng (TT) với nước vừa đủ 1000,0 ml.
Dung dịch đệm acetat pH 4,5: Hòa tan 2,99 g natri acetat (TT) trong nước. Thêm 14,0 ml dung dịch acid acetic 2 M rồi pha loãng với nước vừa đủ 1000,0 ml.
Dung dịch đệm acetat pH 5,5: Hòa tan 5,98 g natri acetat (TT) trong nước. Thêm 3,0 ml dung dịch acid acetic 2 M rồi pha loãng với nước vừa đủ 1000,0 ml.
Dung dịch đệm acetat pH 4,8: Hòa tan 6,23 g natri acetat (TT) trong nước. Thêm 2,1 ml dung dịch acid acetic 2 M rồi pha loãng với nước vừa đủ 1000,0 ml.
Các dung dịch đệm phosphat
Để pha chế đệm phosphat với các pH khác nhau như trong Bảng 3, lấy 250 ml dung dịch kali dihydrophosphat 0,2 M (TT) cho vào một bình định mức 1000 ml, thêm một thể tích dung dịch natri hydroxyd 0,2 M (TT) ghi trong bảng rồi pha loãng với nước cho đủ 1000,0 ml.
Bảng 3 - Thành phần các đệm phosphat
| pH | 5,8 | 6,0 | 6,2 | 6,4 | 6,6 | 6,9 |
| NaOH (ml) | 18,0 | 28,0 | 40,5 | 58,0 | 82,0 | 112,0 |
| pH | 7,0 | 7,2 | 7,4 | 7,6 | 7,8 | 8,0 |
| NaOH (ml) | 145,5 | 173,5 | 195,5 | 212,0 | 222,5 | 230,5 |
Các dung dịch đệm phosphat khác
Dung dịch đệm phosphat pH 4,5: Hòa tan 13,61 g kali dihydrophosphat (TT) trong 750 ml nước. Điều chỉnh pH nếu cần bằng dung dịch natri hydroxyd 0,1 M (TT) hoặc bằng acid hydrocloric 0,1 M (TT). Pha loãng với nước đủ 1000,0 ml.
Dung dịch đệm phosphat pH 5,5 (TT).
Dung dịch đệm phosphat pH 6,8 (TT).
Đệm phosphat pH 7,2 (TT).
Dung dịch đệm phosphat 0,33 M pH 7,5 (TT).
Dịch ruột giả pH 6,8
Hòa lẫn 250 ml dung dịch chứa 6,8 g kali dihydrophosphat (TT), 77 ml dung dịch natri hydroxyd 0,2 M (TT) và 500 ml nước. Thêm 10 g bột tuyến tụy (TT), trộn đều và điều chỉnh pH nếu cần. Pha loãng với nước đủ 1000,0 ml.
Dịch dạ dày giả
Hòa tan 2,0 g natri clorid (TT) và 3,2 g bột pepsin (TT) trong nước. Thêm 80 ml acid hydrocloric 1 M (TT) rồi pha loãng với nước vừa đủ 1000 ml. Bột pepsin có thể không cần, nếu có yêu cầu.
Tăng dần pH
Đối với phép thử độ hòa tan với pH tăng dần, có thể áp dụng một trong các trình tự sau:
| Thời gian (h) | 0-1 | 1-2 | 2-3 | 3-4 | 4-5 | 5-6 | 6-7 | 7 | ||
| pH | 1,0 |
| ||||||||
| pH | 1,2 | 6,8 | ||||||||
| pH | 1,2 | 2,5 | 4,5 | 7,0 | 7,5 | |||||
| pH | 1,5 | 4,5 | 7,2 | |||||||
Để đạt được sự thay đổi pH, có thể thực hiện một trong các cách sau:
• Thay thế một dung dịch đệm bằng một dung dịch đệm khác (thay toàn bộ);
• Rút bỏ chỉ một nửa thể tích môi trường hòa tan mỗi lần (phương pháp thay thế một nửa) rồi bù vào bằng dung dịch đệm có pH cao hơn; ví dụ: pH khởi đầu là 1,2 và dung dịch đệm thay thế là đệm phosphat pH 7,5;
• Thêm vào dung dịch khởi đầu ở pH 1,5 một lượng bột hỗn hợp chứa tris(hydroxymethyl)aminomethan (TT) và natri acetat khan (TT) để thu được dung dịch pH 4,5 và một lượng lần thứ 2 để thu được dung dịch pH 7,2 như mô tả dưới đây:
Dung dịch acid hydrocloric pH 1,5: Hòa tan 2 g natri clorid (TT) trong nước (TT), thêm 31,6 ml dung dịch acid hydrocloric 1 M (TT) rồi pha loãng với nước vừa đủ 1000 ml;
Dung dịch đệm pH 4,5: Trộn lẫn 2,28 g tris(hydroxymethyl)aminomethan (TT) và 1,77 g natri acetat khan (TT). Hòa tan hỗn hợp này trong dung dịch acid hydrocloric pH 1,5 ở trên;
Dung dịch đệm pH 7,2: Trộn lẫn 2,28 g tris(hydroxymethyl)aminomethan (TT) và 1,77 g natri acetat khan (TT). Hòa tan hỗn hợp này trong dung dịch đệm pH 4,5 ở trên.
Thiết bị buồng dòng chảy có thể được dùng thử độ hòa tan trong môi trường thay đổi liên tục pH.
Đánh giá và thẩm định
Do bản chất của phép thử độ hòa tan, chất lượng thiết kế là yếu tố đánh giá quan trọng đối với thiết bị thử độ hòa tan in vitro. Bất kỳ một sai lệch nào như rung động hay lắc lư quá mức do phần cơ khí không hoàn hảo đều cần phải được loại trừ.
Việc đánh giá thiết bị thử độ hòa tan cần xem xét các kích thước và dung sai của thiết bị. Những thông số quan trọng nhất của phép thử như nhiệt độ và thể tích môi trường hòa tan, tốc độ quay hay tốc độ dòng, lượng mẫu thử cần lấy và qui trình thử cần phải định kỳ theo dõi đánh giá trong các khoảng thời gian sử dụng thiết bị.
Thiết bị thử độ hòa tan có thể được theo dõi đánh giá bằng cách thử một sản phẩm đối chiếu nhạy cảm với các điều kiện thủy động học. Những phép thử này có thể được thực hiện định kỳ hoặc liên tục để so sánh với các phòng kiểm nghiệm khác.
Trong quá trình thử, cần phải quan sát và kiểm tra một cách chặt chẽ. Điều này đặc biệt quan trọng khi cần biện giải bất kỳ một kết quả nào lọt ra ngoài khoảng mong đợi.
Việc kiểm định hệ thống thử độ hòa tan tự động đối với bộ phận lấy mẫu và bộ phận phân tích, hoặc việc pha chế môi trường hòa tan và vận hành phép thử, cần phải xem xét độ chính xác, độ đúng, và sự tránh nhiễm tạp do pha loãng, chuyển vận, làm sạch và các qui trình điều chế dung môi hay chuẩn bị mẫu.
Yêu cầu về độ hòa tan đối với các dạng thuốc uống
Yêu cầu về độ hòa tan được biểu thị bằng giá trị Q, là phần trăm lượng dược chất hòa tan so với lượng ghi trên nhãn trong khoảng thời gian qui định.
Dạng thuốc giải phóng tức thời (dạng quy ước):
Trong hầu hết các trường hợp, khi thử trong các điều kiện đã được khảo sát và chứng minh là phù hợp, tiêu chuẩn chấp nhận ở bước S1 là ít nhất 80 % lượng dược chất được giải phóng trong khoảng thời gian quy định, thông thường là 45 min hoặc ít hơn. Điều này tương ứng với giá trị Q bằng 75 %, đối chiếu với Bảng 11.4.1, ở bước S1, mỗi một giá trị của 6 đơn vị thử không được ít hơn Q + 5 % tức là không ít hơn 80 %.
Thông thường, tiêu chuẩn chấp nhận tại một thời điểm là đủ để chứng minh rằng phần lớn dược chất đã được giải phóng, tuy vậy trong một số trường hợp cần phải đánh giá thêm một thời điểm hoặc nhiều hơn để chứng minh chế phẩm đáp ứng yêu cầu về độ hòa tan.
Dạng thuốc giải phóng kéo dài:
Tiêu chuẩn độ hòa tan đối với dạng thuốc thuốc giải phóng kéo dài thường bao gồm từ 3 thời điểm thử trở lên. Thời điểm thử đầu tiên nhằm kiểm tra sự giải phóng dược chất nhanh không được định trước (sự “bùng” liều). Vì thế thông thường yêu cầu lượng dược chất được giải phóng tại thời điểm này nằm trong khoảng 20 % đến 30 %. Thời điểm thứ hai xác định cách thức hòa tan của chế phẩm và thường khoảng 50 % lượng dược chất được giải phóng. Thời điểm thử cuối cùng nhằm đảm bảo rằng sự giải phóng dược chất gần như hoàn tất, có nghĩa là trên 80 % lượng dược chất đã được giải phóng.
Dạng thuốc giải phóng muộn:
Dạng thuốc giải phóng muộn có thể giải phóng dược chất theo nhiều phân đoạn cách quãng hay giải phóng toàn bộ một lần tùy theo thiết kế công thức bào chế khi được thử trong môi trường hòa tan khác nhau, ví dụ trong các điều kiện pH tăng dần. Tiêu chuẩn độ hòa tan, vì thế, phải được xác định cho từng trường hợp một.
Những dạng thuốc kháng dịch vị yêu cầu ít nhất 2 thời điểm thử theo trình tự trước sau và 2 tiêu chuẩn khác nhau trong một phép thử song song. Trong phép thử theo trình tự, thời điểm thử thứ nhất có một giới hạn trên và được thử sau 1 h hoặc sau 2 h trong môi trường acid, còn điểm thử sau tại khoảng thời gian định trước trong một dung dịch đệm thích hợp (thường là pH 6,8).
Trong hầu hết các trường hợp, tiêu chuẩn chấp nhận ở bước B, là ít nhất 80 % lượng dược chất được giải phóng. Điều này tương ứng với giá trị Q bằng 75 %, đối chiếu với Bảng 11.4.4, ở bước B1, mỗi một giá trị của 6 đơn vị thử không được ít hơn Q + 5 % tức là không ít hơn 80 %.
11.5 PHÉP THỬ ĐỘ RÃ CỦA THUỐC ĐẠN VÀ THUỐC TRỨNG
Phép thử này xác định thuốc đạn và thuốc trứng có rã hoặc mềm đi hay không trong khoảng thời gian quy định, khi được đặt trong môi trường lỏng ở những điều kiện thử nghiệm chỉ định.
Thiết bị
a) Ống trong suốt bằng thủy tinh hay chất dẻo, cao 60 mm với đường kính bên trong 52 mm, thành dày thích hợp (Hình 11.5.1).
b) Bộ phận kim loại gồm hai đĩa kim loại không gỉ, mỗi đĩa có 39 lỗ tròn đường kính 4 mm và được phân bố như chỉ dẫn trong Hình 11.5.1. Đường kính của đĩa gần tương đương với đường kính bên trong của ống bao. Hai đĩa cách nhau khoảng 30 mm. Bộ phận kim loại được treo bằng ba móc kẹp cách đều nhau, gắn vào thành ngoài ống bao như chỉ dẫn trong Hình 11.5.1.
Khi thử độ rã của viên nén đặt âm đạo, bộ phận kim loại được gắn ở dưới với đầu móc kẹp hướng lên trên như mô tả trong Hình 11.5.2.
Cách thử
Thuốc đạn
Đặt một viên lên đĩa dưới của bộ phận kim loại, đưa bộ phận này vào ống bao và gắn chặt vào thành ống. Nếu không có chỉ dẫn khác, đặt thiết bị thử vào một bồn chứa ít nhất 4 lít nước ấm (36 °C đến 37 °C) có gắn dụng cụ khuấy chậm và giữ thiết bị thử ở vị trí thẳng đứng, ngập 90 mm so với mặt nước. Xoay ngược thiết bị thử 10 min một lần, tránh không để nhô lên khỏi mặt nước.
Lặp lại toàn bộ thử nghiệm với hai viên khác.
Thuốc được coi là rã, khi đáp ứng một trong những yêu cầu sau:
a) Tan hoàn toàn.
b) Phân tách ra các thành phần tạo thành, rồi tập trung trên bề mặt (các chất mỡ nóng chảy), chìm xuống đáy (bột không tan) hay hòa tan trong nước (các thành phần hòa tan), hoặc có thể phân tán theo một hay vài cách nêu trên.
c) Trở nên mềm, có thể biến dạng đáng kể, không nhất thiết bị phân tách hoàn toàn ra các thành phần tạo thành, nhưng không có nhân rắn chịu được sức ép của đũa thủy tinh.
Nếu không có quy định khác trong chuyên luận riêng, thời gian rã không quá 30 min đối với các thuốc đạn có tá dược thân mỡ và không quá 60 min đối với thuốc đạn tan trong nước.
Nang đặt trực tràng
Tiến hành như mô tả trong phần Thuốc đạn. Thuốc được coi là rã, khi vỏ gelatin vỡ ra, giải phóng các chất chứa bên trong.
Thời gian rã không được quá 30 min.
Thuốc trứng
Tiến hành và đánh giá như mô tả trong phần Thuốc đạn.
Nếu không có quy định khác trong chuyên luận riêng, thời gian rã không quá 60 min.
Nang đặt âm đạo
Tiến hành như mô tả trong phần Thuốc đạn và đánh giá như mô tả trong phần Nang đặt trực tràng.
Viên nén đặt âm đạo
Đặt thiết bị thử vào bồn có đường kính phù hợp, chứa nước ấm (36 °C đến 37 °C). Thêm dần nước ấm (36 °C đến 37 °C) cho đến khi mặt lỗ của đĩa kim loại vừa được phủ bằng một lớp nước. Đặt viên thuốc lên đĩa trên và đậy thiết bị bằng tấm kính để giữ các điều kiện ẩm thích hợp. Lặp lại toàn bộ thử nghiệm với hai viên khác.
Thuốc được coi là rã, khi đáp ứng một trong những yêu cầu sau:
a) Không còn cắn trên đĩa.
b) Nếu cắn vẫn còn, đấy chỉ là khối mềm hay rỗng xốp, không có nhân rắn chịu được sức ép của đũa thủy tinh.
Nếu không có quy định khác trong chuyên luận riêng, thời gian rã không quá 30 min.
Hình 11.5.1 - Thiết bị thử độ rã của thuốc đạn và thuốc trứng
(Kích thước tính bằng mm)
CHÚ DẪN: A - Viên nén đặt âm đạo; B - Tấm kính; C - Mặt nước.
Hình 11.5.2 - Thiết bị thử độ rã của viên nén đặt âm đạo
11.6 PHÉP THỬ ĐỘ RÃ CỦA VIÊN NÉN VÀ NANG
Phép thử này xác định viên nén hay nang có rã hay không trong khoảng thời gian quy định, khi được đặt trong môi trường lỏng ở những điều kiện thử nghiệm chỉ định.
Trong phép thử này, chế phẩm rã không có nghĩa là hòa tan hoàn toàn đơn vị chế phẩm hay thành phần hoạt chất.
Thuốc được coi là rã, khi đáp ứng một trong những yêu cầu sau:
a) Không còn cắn trên mặt lưới, trừ những mảnh vỏ bao không tan của viên nén hoặc vỏ nang trên mặt lưới; hoặc dính vào mặt dưới của đĩa, nếu sử dụng đĩa.
b) Nếu còn cắn, đấy là khối mềm không có nhân khô.
Sử dụng thiết bị A cho viên nén và nang cỡ bình thường (không dài quá 18 mm). Sử dụng thiết bị B cho viên nén và nang cỡ lớn.
Thiết bị A: Áp dụng cho viên nén và nang cỡ bình thường
Thiết bị
Thiết bị A thử độ rã viên nén và nang gồm:
Giá đỡ ống thử;
Cốc đáy bằng dung tích 1 lít, chiều cao (149 ± 11) mm, và đường kính trong (109 ± 9) mm để chứa môi trường thử;
Bộ phận điều nhiệt để làm nóng môi trường thử trong khoảng 35 °C đến 39 °C;
Bộ phận cơ chuyển động lên xuống cho giá đỡ ống thử ở tần số hằng định trong khoảng 29 r/min đến 32 r/min, với biên độ (55 ± 2) mm. Thể tích môi trường thử trong cốc phải đủ, để khi giá đỡ ống thử ở vị trí cao nhất, lưới kim loại phải ngập sâu ít nhất 15 mm so với bề mặt chất lỏng, còn khi ở vị trí thấp nhất, lưới kim loại phải cách đáy cốc ít nhất 25 mm và miệng các ống thử vẫn ở trên bề mặt chất lỏng. Thời gian để giá đỡ ống thử đi lên phải bằng với thời gian giá đỡ ống thử đi xuống và sự thay đổi của hướng chuyển động phải nhẹ nhàng không được đột ngột. Giá đỡ ống thử phải chuyển động thẳng đứng theo trục của nó, không được dao động ngang hoặc di chuyển ra khỏi trục thẳng đứng.
Giá đỡ ống thử: Giá đỡ ống thử bao gồm 6 ống trong suốt hở đầu, các ống dài (77,5 ± 2,5) mm, có đường kính trong (21,85 ± 1,15) mm và thành ống dày (1,9 ± 0,9) mm. Các ống được giữ ở vị trí thẳng đứng bởi hai đĩa, có đường kính (90 ± 2) mm và dày (6,75 ± 1,75) mm; có 6 lỗ, mỗi lỗ có đường kính (24 ± 2) mm, cách đều tâm dĩa và cách đều các lỗ còn lại. Gắn vào mặt dưới của đĩa bên dưới là một lưới kim loại, dệt hình vuông, có kích thước lỗ lưới (2,0 ± 0,2) mm và đường kính dây kim loại là (0,615 ± 0,045) mm. Các bộ phận được lắp ráp và được giữ chắc chắn bằng 3 ốc vít đi qua 2 đĩa. Giá đỡ ống thử được treo phù hợp vào bộ phận chuyển động tại một điểm trên trục của nó.
Thiết kế của giá đỡ ống thử có thể thay đổi, nhưng tiêu chuẩn của ống thủy tinh và lưới kim loại phải đúng quy định. Giỏ phải có kích thước như quy định ở Hình 11.6-1.
Đĩa: Chỉ được sử dụng đĩa nếu có chỉ dẫn hoặc được cho phép trong chuyên luận riêng hoặc chuyên luận chung tương ứng. Mỗi ống thử của giá đỡ ống thử có một đĩa hình trụ, đường kính (20,7 ± 0,15) mm và dày (9,5 ± 0,15) mm. Đĩa được làm bằng chất dẻo trong suốt có tỷ trọng riêng 1,18 đến 1,20. Đĩa có 5 lỗ song song theo trục đĩa và xuyên qua đĩa, 1 lỗ ở chính giữa, 4 lỗ còn lại nằm cách đều nhau trên vòng tròn bán kính 6 ± 0,2 mm tính từ tâm, các lỗ có đường kính (2 ± 0,1) mm. Có 4 rãnh hình thang, cân xứng, trên thành của đĩa, gần như vuông góc với bề mặt đĩa, bề mặt song song của nó trùng với hai mặt của đĩa và song song với trục nối 2 lỗ cạnh nhau, cách trục đĩa 6 mm. Cạnh đáy nhỏ của hình thang trên đáy của đĩa, có chiều dài (1,6 ± 0,1) mm và trung tâm của cạnh có độ sâu 1,5 đến 1,8 mm tính từ chu vi của đĩa. Cạnh đáy lớn của hình thang ở trên đỉnh của đĩa có chiều dài (9,4 ± 0,2) mm và trung tâm của cạnh có độ sâu (2,6 ± 0,1) mm tính từ chu vi của đĩa. Tất cả các mặt của đĩa phải phẳng.
Nếu sử dụng đĩa như chỉ dẫn, cho mỗi dĩa vào 1 ống và vận hành thiết bị. Đĩa phải đáp ứng kích thước như quy định ở Hình 11.6-1.
Việc sử dụng đĩa cải tiến có hệ thống phát hiện tự động có thể được sử dụng theo chỉ dẫn hoặc cho phép. Đĩa này phải đáp ứng yêu cầu về tỷ trọng và kích thước theo chuyên luận này.
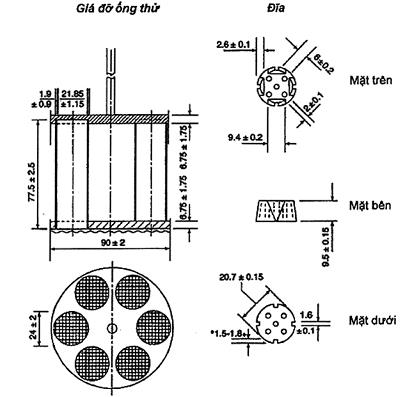
Hình 11.6.1 - Thiết bị thử độ rã của viên nén và nang cỡ bình thường
(Kích thước tính bằng mm)
Phương pháp thử
Nếu không có chỉ dẫn khác trong chuyên luận riêng, cho vào mỗi ống thử một viên nén hoặc nang. Nếu có chỉ dẫn trong chuyên luận chung tương ứng, cho một đĩa vào mỗi ống. Treo giá đỡ ống thử trong cốc có chứa môi trường theo chỉ dẫn được duy trì ở (37 ± 2) °C và vận hành thiết bị theo thời gian quy định. Lấy giá đỡ ống thử ra khỏi chất lỏng và quan sát chế phẩm thử. Mẫu thử đạt yêu cầu nếu tất cả 6 viên đều rã. Nếu có 1 đến 2 viên không rã, lặp lại phép thử với 12 viên khác. Mẫu thử đạt yêu cầu nếu không dưới 16 trong số 18 viên thử rã.
Thiết bị B: Áp dụng cho viên nén và nang cỡ lớn
Thiết bị
Thiết bị B có cấu tạo tương tự như thiết bị A, nhưng khác về cấu tạo của giá đỡ ống thử và đĩa.
Giá đỡ ống thử: Giá đỡ ống thử bao gồm 3 ống trong suốt hở đầu, các ống dài (77,5 ± 2,5) mm, có đường kính trong (33,0 ± 0,5) mm và thành ống dày (2,5 ± 0,5) mm. Các ống được giữ ở vị trí thẳng đứng bởi hai đĩa nhựa, có đường kính 97 mm và dày 7 mm; có 3 lỗ. Các lỗ cách đều tâm đĩa và cách đều các lỗ còn lại. Gắn vào mặt dưới của đĩa bên dưới là một lưới kim loại, dệt hình vuông, bằng dây kim loại có đường kính (0,63 ± 0,03) mm và có kích thước lỗ lưới (2,0 ± 0,2) mm. Hai đĩa được cố định cách nhau 77,5 mm bằng 3 thanh kim loại chốt thẳng đứng ở ngoại biên. Một thanh kim loại được gắn chặt vào tâm đĩa nhựa phía trên, sao cho giá đỡ ống thử lắp ráp được với bộ phận cơ có khả năng vận hành giá đỡ chuyển động lên xuống nhẹ nhàng ở tần số hằng định trong khoảng 29 đến 32 r/min, với biên độ (55 ± 2) mm.
Thiết kế của giá đỡ ống thử có thể thay, đổi, nhưng tiêu chuẩn của ống thủy tinh và lưới kim loại phải đúng quy định. Giỏ phải có kích thước như quy định ở Hình 11.6-2.
Đĩa: Chỉ được sử dụng đĩa nếu có chỉ dẫn hoặc được cho phép trong chuyên luận riêng hoặc chuyên luận chung tương ứng. Mỗi ống thử của giá đỡ ống thử có một đĩa hình trụ, đường kính (31,4 ± 0,13) mm và dày (15,3 ± 0,15) mm. Đĩa được làm bằng chất dẻo trong suốt có tỷ trọng riêng 1,18 đến 1,20. Đĩa có 7 lỗ song song theo trục đĩa và xuyên qua đĩa, các lỗ có đường kính (3,15 ± 0,1) mm, 1 lỗ ở chính giữa và 6 lỗ còn lại nằm cách đều nhau trên vòng tròn bán kính 4,2 mm tính từ tâm đĩa.
Nếu sử dụng đĩa như chỉ dẫn, cho mỗi đĩa vào 1 ống và vận hành thiết bị. Đĩa phải đáp ứng kích thước như quy định ở Hình 11.6-2.
Giá đỡ ống thử
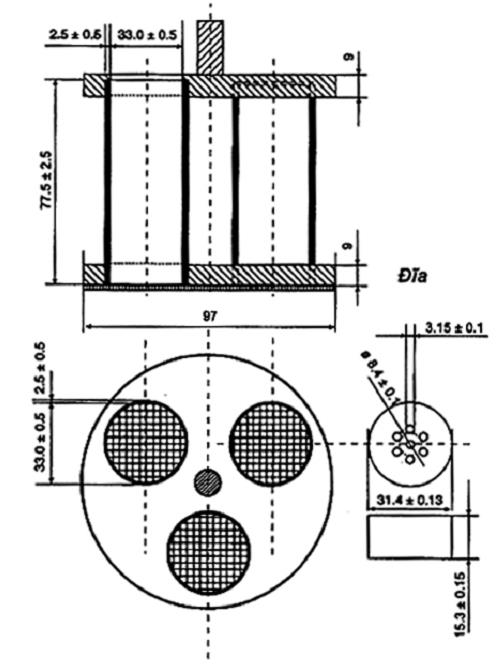
Hình 11.6.2 - Thiết bị thử độ rã của viên nén và nang cỡ lớn
(Kích thước tính bằng mm)
Phương pháp thử
Nếu không có chỉ dẫn khác trong chuyên luận riêng, tiến hành thử với 6 viên nén hoặc nang trên 2 giá đỡ ống thử lắp song song hoặc lặp lại phép thử trên 1 giá đỡ ống thử. Cho vào mỗi ống thử một viên nén hoặc nang. Nếu có chỉ dẫn trong chuyên luận chung tương ứng, cho một đĩa vào mỗi ống. Treo giá đỡ ống thử trong cốc có chứa môi trường theo chỉ dẫn được duy trì ở (37 ± 2) °C và vận hành thiết bị theo thời gian quy định. Lấy giá đỡ ống thử ra khỏi chất lỏng và quan sát chế phẩm thử. Mẫu thử đạt yêu cầu nếu tất cả 6 viên đều rã.
11.7 PHÉP THỬ ĐỘ RÃ CỦA VIÊN BAO TAN TRONG RUỘT
Thiết bị
Sử dụng thiết bị mô tả trong Phụ lục 11.6. Phép thử độ rã của viên nén và nang.
Phương pháp thử
Cho một viên vào mỗi ống thử, không dùng đĩa, treo giá đỡ ống thử trong cốc chứa dung dịch acid hydrocloric 0,1 N (TT), nếu không có chỉ dẫn khác trong chuyên luận riêng, vận hành thiết bị trong 120 min. Lấy giá đỡ ống thử ra khỏi chất lỏng. Trừ các mảnh vỏ bao, không một viên nào bị rã hay có dấu hiệu bị rạn nứt khiến hoạt chất bị hòa tan.
Thay chất lỏng trong cốc bằng đệm phosphat hỗn hợp pH 6,8 (TT), cho đĩa vào ống và vận hành thiết bị trong 60 min. Lấy giá đỡ ống thử ra khỏi chất lỏng. Nếu mẫu thử không đạt yêu cầu do viên bị dính vào đĩa, lặp lại phép thử với 6 viên khác và không dùng đĩa. Mẫu thử đạt yêu cầu nếu tất cả sáu viên đều rã.
11.8 XÁC ĐỊNH GIỚI HẠN TIỂU PHÂN
A. Xác định giới hạn tiểu phân không nhìn thấy bằng mắt thường
Tiểu phân trong thuốc tiêm và thuốc tiêm truyền là các hạt nhỏ không hòa tan, linh động, không phải là bọt khí, có nguồn gốc ngẫu nhiên từ bên ngoài.
Để xác định giới hạn tiểu phân có hai phương pháp mô tả dưới đây, Phương pháp 1 (dùng thiết bị đếm tiểu phân) và Phương pháp 2 (dùng kính hiển vi). Khi kiểm tra tiểu phân không nhìn thấy bằng mắt thường của thuốc tiêm và thuốc tiêm truyền, thường áp dụng Phương pháp 1. Tuy nhiên, đối với một số chế phẩm, phải áp dụng cả hai phương pháp để đánh giá.
Không phải tất cả các thuốc tiêm và thuốc tiêm truyền đều có thể kiểm tra được giới hạn tiểu phân không nhìn thấy bằng mắt thường bằng một trong hai phương pháp, hay cả hai phương pháp. Các chế phẩm không thật trong, hay có độ nhớt tăng cao, hoặc tạo bọt khí khi đổ vào thiết bị đếm tiểu phân, tiến hành thử theo Phương pháp 2. Nếu độ nhớt của chế phẩm quá cao, cản trở phương pháp kiểm tra, pha loãng với dung môi thích hợp để có thể tiến hành được thử nghiệm.
Kết quả kiểm tra một đơn vị riêng lẻ hay nhóm các đơn vị chế phẩm không có giá trị đương nhiên đối với các đơn vị không được kiểm tra. Bởi vậy, cần xây dựng kế hoạch lấy mẫu đủ số lượng thống kê, để từ kết quả kiểm tra có thể đánh giá được giới hạn tiểu phân của cả nhóm lớn các đơn vị chế phẩm.
Phương pháp 1: Dùng thiết bị đếm tiểu phân
Quy định chung
Tiến hành thử nghiệm trong các điều kiện hạn chế được nhiễm tiểu phân, tốt nhất là trong tủ lọc khí vô khuẩn.
Rửa thật cẩn thận dụng cụ thủy tinh và dụng cụ lọc (trừ màng lọc) bằng dung dịch tẩy rửa ấm, tráng lại bằng nước cho sạch hết chất tẩy. Ngay trước khi dùng, tráng lại các dụng cụ từ trên xuống dưới, bên ngoài, sau đó là bên trong với nước không có tiểu phân (TT).
Tránh tạo bọt khí trong chế phẩm đem thử, đặc biệt là khi rót vào dụng cụ để tiến hành đếm tiểu phân. Để kiểm tra môi trường có thích hợp cho thử nghiệm, dụng cụ thủy tinh có đủ sạch và nước sử dụng có hết tiểu phân hay không, tiến hành phép thử sau:
Kiểm tra giới hạn tiểu phân của 5 mẫu nước không có tiểu phân (TT), mỗi mẫu 5 ml, theo phương pháp mô tả ở dưới. Nếu trong cả 25 ml nước thử có trên 25 tiểu phân kích thước bằng 10 μm hay lớn hơn, phải tiến hành lại các bước chuẩn bị cho đến khi môi trường, dụng cụ thủy tinh và nước thích hợp cho thử nghiệm.
Dụng cụ
Sử dụng thiết bị thích hợp hoạt động theo nguyên tắc cản ánh sáng, cho phép tự động xác định kích thước tiểu phân và số lượng tiểu phân theo kích thước đã chọn.
Chuẩn hóa thiết bị bằng các hạt cầu chuẩn kích thước 10 μm đến 25 μm. Phân tán hạt chuẩn trong nước không có tiểu phân (TT). Tránh kết tụ tiểu phân trong quá trình phân tán.
Cách tiến hành
Trộn đều chế phẩm bằng cách lộn đi, lộn lại nhẹ nhàng dụng cụ chứa (đổ đựng) 20 lần liên tiếp. Nếu cần, tháo bỏ phần bảo vệ ngoài nắp đậy. Dùng tia nước không có tiểu phân (TT) rửa bề mặt nắp và cổ dụng cụ chứa (đồ đựng), mở nắp cẩn thận, tránh nhiễm tiểu phân từ bên ngoài. Loại bọt khí bằng cách để yên 2 min hoặc siêu âm.
Đối với thuốc tiêm và thuốc tiêm truyền có thể tích từ 25 ml trở lên, tiến hành thử với từng đơn vị. Đối với thuốc tiêm và thuốc tiêm truyền có thể tích nhỏ hơn 25 ml, gộp dung dịch của ít nhất 10 đơn vị trong một cốc sạch cho đủ 25 ml. Khi có chỉ dẫn đặc biệt, lấy dung dịch từ một số đơn vị thích hợp, pha loãng thành 25 ml với nước không có tiểu phân (TT) hoặc với dung môi thích hợp không có tiểu phân, khi nước không có tiểu phân (TT) không thích hợp.
Thuốc bột để pha tiêm được hòa tan trong nước không có tiểu phân (TT) hoặc dung môi thích hợp không có tiểu phân, khi nước không có tiểu phân (TT) không thích hợp.
Số mẫu thử phải đủ lượng thống kê. Đối với thuốc tiêm và thuốc tiêm truyền có thể tích từ 25 ml trở lên, số mẫu thử có thể nhỏ hơn 10, phụ thuộc vào kế hoạch lấy mẫu đã đề ra.
Lấy 4 mẫu thử, mỗi mẫu không dưới 5 ml, đưa vào máy đếm tiểu phân theo kích thước bằng 10 μm hay lớn hơn và bằng 25 μm hay lớn hơn.
Loại bỏ kết quả của mẫu đầu tiên. Tính số lượng tiểu phân trung bình của chế phẩm đã thử.
Đánh giá kết quả
Đối với thuốc tiêm và thuốc tiêm truyền có thể tích lớn hơn 100 ml: Chế phẩm đạt yêu cầu phép thử, nếu trung bình trong mỗi ml có không quá 25 tiểu phân kích thước bằng 10 μm hay lớn hơn và không quá 3 tiểu phân kích thước bằng hay lớn hơn 25 μm.
Đối với thuốc tiêm và thuốc tiêm truyền có thể tích nhỏ hơn hoặc bằng 100 ml: Chế phẩm đạt yêu cầu phép thử, nếu trung bình trong mỗi đơn vị đã kiểm tra có không quá 6000 tiểu phân kích thước bằng 10 μm hay lớn hơn và không quá 600 tiểu phân kích thước bằng 25 μm hay lớn hơn.
Nếu số lượng tiểu phân trung bình vượt quá giới hạn, kiểm tra chế phẩm bằng Phương pháp 2.
Phương pháp 2: Dùng kính hiển vi
Quy định chung
Tiến hành thử nghiệm trong các điều kiện hạn chế được nhiễm tiểu phân, tốt nhất là trong tủ lọc khí vô khuẩn.
Rửa thật cẩn thận dung cụ thủy tinh và dụng cụ lọc (trừ màng lọc) bằng dung dịch tẩy rửa ấm, tráng lại bằng nước cho sạch hết chất tẩy. Ngay trước khi dùng, tráng lại các dụng cụ từ trên xuống dưới, bên ngoài, sau đó là bên trong với nước không có tiểu phân (TT).
Để kiểm tra môi trường có thích hợp cho thử nghiệm, dụng cụ thủy tinh và màng lọc có đủ sạch, nước sử dụng có hết tiểu phân hay không, tiến hành phép thử sau:
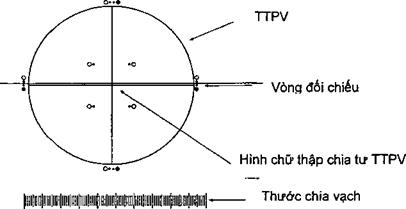
Hình 11.8.1 - Trắc vi thị kính
Kiểm tra giới hạn tiểu phân của 50 ml nước không có tiểu phân (TT) theo phương pháp mô tả ở dưới. Nếu có quá 20 tiểu phân kích thước bằng 10 μm hay lớn hơn hoặc có quá 5 tiểu phân kích thước bằng 25 μm hay lớn hơn trên màng lọc, phải tiến hành lại các bước chuẩn bị cho đến khi môi trường, dụng cụ thủy tinh, màng lọc và nước thích hợp cho thử nghiệm.
Dụng cụ
Sử dụng kính hiển vi thích hợp, bộ lọc để lưu giữ các tiểu phân và màng lọc để kiểm tra.
Kính hiển vi có độ phóng đại 100 lần ± 10 lần, được trang bị một trắc vi thị kính hiệu chỉnh bằng một trắc vi vật kính, một bàn soi có khả năng cố định và xoay quanh toàn bộ diện tích lọc của màng lọc, hai nguồn chiếu thích hợp cấp ánh sáng phản xạ bổ sung cho ánh sáng chếch.
Trắc vi thị kính (Hình 11.8.1) bao gồm một vòng tròn lớn có hình chữ thập chia tư, các vòng đối chiếu trong suốt hay màu đen đường kính 10 μm và 25 μm ở độ phóng đại 100 lần, một thước chia vạch 10 μm đã được kiểm định theo chuẩn quốc gia hoặc quốc tế với sai số tương đối cho phép ± 2 %. Vòng tròn lớn được gọi là thị trường phân vạch (TTPV).
Trong hai nguồn chiếu sáng, một nguồn cấp trường sáng phản xạ từ trong kính hiển vi và nguồn bên ngoài bổ trợ hội tụ, có khả năng điều chỉnh để rọi sáng khúc xạ chếch ở góc 10° đến 20°.
Bộ lọc để lưu giữ các tiểu phân bao gồm màng lọc thích hợp và đế lọc bằng thủy tinh hoặc vật liệu khác thích hợp, nối với máy hút chân không.
Màng lọc kích thước thích hợp màu đen hoặc xám sẫm, với lỗ xốp bằng 1,0 μm hoặc nhỏ hơn.
Cách tiến hành
Trộn đều chế phẩm bằng cách lộn đi, lộn lại nhẹ nhàng dụng cụ chứa 20 lần liên tiếp. Nếu cần, tháo bỏ phần bảo vệ ngoài nắp đậy. Dùng tia nước không có tiểu phân (TT) rửa bề mặt nắp và cổ bình chứa, mở nắp cẩn thận, tránh nhiễm tiểu phân từ bên ngoài.
Đối với thuốc tiêm và thuốc tiêm truyền có thể tích từ 25 ml trở lên, tiến hành thử với từng đơn vị. Đối với thuốc tiêm và thuốc tiêm truyền có thể tích nhỏ hơn 25 ml, gộp dung dịch của ít nhất 10 đơn vị trong một cốc sạch cho đủ 25 ml. Khi có chỉ dẫn đặc biệt, lấy dung dịch từ một số đơn vị thích hợp, pha loãng thành 25 ml với nước không có tiểu phân (TT) hoặc với dung môi thích hợp không có tiểu phân, khi nước không có tiểu phân (TT) không thích hợp.
Thuốc bột để pha tiêm được hòa tan trong nước không có tiểu phân (TT) hoặc dung môi thích hợp không có tiểu phân, khi nước không có tiểu phân (TT) không thích hợp.
Số mẫu thử phải đủ lượng thống kê. Đối với thuốc tiêm và thuốc tiêm truyền có thể tích từ 25 ml trở lên, số mẫu thử có thể nhỏ hơn 10, phụ thuộc vào kế hoạch lấy mẫu đã đề ra.
Dùng vài ml nước không có tiểu phân (TT) làm ẩm bên trong đế lọc đã gắn màng lọc. Chuyển toàn bộ dung dịch thử đã chuẩn bị, hay dung dịch trong một đơn vị, vào phễu lọc và hút chân không. Nếu cần, đổ từng lượng nhỏ vào phễu cho đến khi toàn bộ thể tích dung dịch thử được lọc. Sau lần đổ cuối cùng, bắt đầu dùng tia nước không có tiểu phân (TT) rửa thành bên trong đế lọc. Hút chân không cho tới khi bề mặt màng lọc không còn chất lỏng. Đặt màng lọc vào hộp lồng Petri và để khô tự nhiên bằng cách mở hé nắp hộp. Sau khi màng lọc khô, đặt hộp lồng Petri lên bàn soi của kính hiển vi và soi toàn bộ màng lọc dưới ánh sáng phản xạ từ nguồn chiếu. Đếm số tiểu phân bằng 10 μm hay lớn hơn và số tiểu phân bằng 25 μm hay lớn hơn. Hoặc, có thể đếm trên một phần của màng lọc, rồi tính số lượng trên cả màng lọc. Tính số lượng tiểu phân trung bình cho chế phẩm đem thử.
Quá trình phân loại tiểu phân được tiến hành bằng cách so sánh mỗi tiểu phân với các vòng đối chiếu 10 μm và 25 μm. Đường kính bên trong của các vòng đối chiếu trong suốt dùng để phân loại tiểu phân màu trắng và trong suốt, còn tiểu phân thẫm màu được phân loại bằng đường kính ngoài của các vòng đối chiếu màu đen.
Khi tiến hành kiểm tra theo Phương pháp 2, không phân loại hay liệt kê các chất vô định hình, nửa lỏng hoặc các chất không rõ ràng về hình thái khác, chúng có biểu hiện biến màu hay đổi màu trên màng lọc. Những chất này nhỏ, không biểu hiện rõ ràng, dạng sền sệt hay như có màng. Trong trường hợp này cần kiểm tra bổ sung bằng Phương pháp 1.
Đánh giá kết quả
Đối với thuốc tiêm và thuốc tiêm truyền có thể tích lớn hơn 100 ml: Chế phẩm đạt yêu cầu phép thử, nếu trung bình trong mỗi ml có không quá 12 tiểu phân kích thước bằng 10 μm hay lớn hơn và không quá 2 tiểu phân kích thước bằng 25 μm hay lớn hơn.
Đối với thuốc tiêm và thuốc tiêm truyền có thể tích nhỏ hơn hoặc bằng 100 ml: Chế phẩm đạt yêu cầu phép thử, nếu trung bình trong mỗi đơn vị đã kiểm tra có không quá 3000 tiểu phân kích thước bằng 10 μm hay lớn hơn và không quá 300 tiểu phân kích thước bằng 25 μm hay lớn hơn.
B. Xác định độ trong (Xác định tiểu phân nhìn thấy bằng mắt thường)
Tiểu phân trong thuốc tiêm và thuốc tiêm truyền là các hạt nhỏ không hòa tan, linh động, không phải là bọt khí, có nguồn gốc ngẫu nhiên từ bên ngoài.
Phép thử này là qui trình đơn giản để đánh giá độ trong của thuốc tiêm và thuốc tiêm truyền.
Dụng cụ
Thiết bị (Hình 11.8.2) là một bộ dụng cụ để soi bao gồm:
Bảng màu đen bề mặt mờ, kích thước thích hợp, gắn thẳng đứng.
Bảng màu trắng không loá (không bóng), kích thước thích hợp, gắn thẳng đứng bên cạnh bảng màu đen.
Hộp đèn có thể điều chỉnh với nguồn ánh sáng trắng được che chắn thích hợp và bộ khuếch tán ánh sáng thích hợp (như nguồn sáng bao gồm hai đèn huỳnh quang 13 W, mỗi ống dài 525 mm). Cường độ chiếu sáng tại vùng soi phải duy trì từ 2000 lux đến 3750 lux. Có thể phải sử dụng cường độ cao hơn đối với đồ chứa là thủy tinh màu hoặc plastic.
Cách tiến hành
Nếu không có chỉ dẫn khác trong chuyên luận riêng, lấy ngẫu nhiên 20 đơn vị. Loại bỏ mọi nhãn mác dán vào đồ chứa, rửa sạch và làm khô bên ngoài. Lắc nhẹ hay lộn đi, lộn lại chậm từng đơn vị, tránh không tạo thành bọt khí và quan sát khoảng 5 s trước bảng màu trắng. Tiến hành lặp lại trước bảng màu đen.
Đánh giá kết quả
Nếu có không quá một đơn vị có tiểu phân nhìn thấy bằng mắt thường, tiến hành kiểm tra lại với 20 đơn vị khác lấy ngẫu nhiên. Chế phẩm đạt yêu cầu phép thử, nếu có không quá một đơn vị trong số 40 đơn vị đem thử có tiểu phân nhìn thấy bằng mắt thường.
1. Bảng màu đen bề mặt mờ
2, 3. Bảng màu trắng không loá
4. Hộp đèn có thể điều chỉnh
Hình 11.8.2 - Dụng cụ để soi độ trong
PHỤ LỤC 12
(Quy định)
12.1 LẤY MẪU DƯỢC LIỆU
Lấy mẫu dược liệu là việc lựa chọn, thu thập các mẫu dược liệu cho việc kiểm tra chất lượng.
Mức độ đại diện của các mẫu dược liệu được lấy có ảnh hưởng trực tiếp đến độ chính xác và độ đúng của việc kiểm tra.
Các yêu cầu chung về việc lấy mẫu dược liệu như sau:
Kiểm tra trước khi lấy mẫu
Kiểm tra đối chiếu tên và nguồn gốc nguyên liệu;
Kiểm tra đặc điểm và hình dạng bao gói;
Kiểm tra sự nguyên vẹn, sạch sẽ, mức độ nhiễm mốc và tạp chất lạ của bao bì.
Các bao gói không bình thường cần được kiểm tra riêng một cách kỹ càng.
Ghi chép chi tiết kết quả kiểm tra.
Cách thức lấy mẫu
Cách thức lấy mẫu:
Tổng số bao gói dưới 5: Lấy mẫu từng bao gói.
Từ 5 đến dưới 100: Lấy mẫu 5 bao gói.
Từ 100 đến 1000: Lấy mẫu 5 % tổng số bao gói.
Trên 1000: Lấy 50 bao gói cộng thêm số bao gói bằng 1 % của tổng số bao gói vượt quá so với 1000 bao gói.
Dược liệu quý: Lấy mẫu từng bao gói, không kể số lượng các bao gói.
Dược liệu được lấy ở trên, giữa và cuối của mỗi bao gói bằng các phương tiện thích hợp (đối với bao gói lớn thì lấy sâu 10 cm dưới bề mặt của bao gói).
Đối với thuốc có kích thước lớn thì lấy mẫu đại diện thích hợp.
Khối lượng mẫu lấy:
Nếu lượng dược liệu dưới 5 kg thì số lượng mẫu được lấy không ít hơn 3 lần số lượng đem thử nghiệm. Nếu lượng dược liệu lớn hơn 5 kg thì số lượng mẫu lấy được xác định như sau:
Thuốc thông thường: 250 g đến 500 g;
Thuốc bột: 200 g;
Thuốc quý: 5 g đến 10 g (trừ khi có chỉ dẫn khác trong chuyên luận riêng).
Tạo mẫu đồng nhất
Mẫu sau khi lấy được trộn đều để có một mẫu đồng nhất dùng cho thử nghiệm. Nếu khối lượng mẫu đồng nhất lớn hơn vài lần so với mẫu thử nghiệm thì làm một mẫu trung bình.
Nếu dược liệu có kích thước nhỏ thì lấy một mẫu trung bình bằng phương pháp chia 4 như sau: San bằng mẫu thành hình vuông, chia mẫu theo 2 đường chéo thành 4 phần bằng nhau. Lấy 2 phần đối diện và trộn đều. Làm lại thao tác chia 4 cho đến khi thu được số lượng vừa đủ để làm mẫu thử và mẫu lưu.
Trong trường hợp các dược liệu có kích thước lớn thì lấy mẫu trung bình bằng phương pháp khác thích hợp.
Khối lượng của mẫu đồng nhất hoặc mẫu trung bình không ít hơn 3 lần số lượng của mẫu đem thử nghiệm. Lượng mẫu này được chia làm 3 phần, 1/3 dùng để phân tích, 1/3 để kiểm tra và số còn lại làm mẫu lưu giữ lại ít nhất 01 năm.
12.2 NHỮNG QUY ĐỊNH CHUNG VỀ KIỂM TRA CHẤT LƯỢNG DƯỢC LIỆU
Dược liệu trước khi đưa vào sử dụng đều phải đưa vào kiểm tra chất lượng và phải đạt yêu cầu chất lượng theo tiêu chuẩn đã đăng ký như tiêu chuẩn dược điển hoặc tiêu chuẩn cơ sở.
Kiểm tra chất lượng dược liệu bao gồm việc mô tả, định tính, các phép thử tinh khiết, xác định hàm lượng chất chiết được và định lượng hoạt chất trong dược liệu. Việc kiểm tra dược liệu được tiến hành theo quy định sau:
1. Lấy mẫu kiểm nghiệm (Phụ lục 12.1).
2. Sử dụng các mô tả của chuyên luận, một mẫu đối chiếu (dược liệu hay chất tinh khiết) thích hợp đã đạt yêu cầu chất lượng theo chuyên luận riêng để xác nhận kết quả kiểm nghiệm.
3. Nếu dược liệu được kiểm tra đã bị vụn nát thì dược liệu đó vẫn phải đáp ứng các yêu cầu chung, trừ yêu cầu về mô tả trong chuyên luận tương ứng.
4. Nếu dược liệu đòi hỏi phải được làm thành bột trước khi định tính, định lượng thì phải tán dược liệu đó thành bột, rây và trộn đều như được mô tả trong Phụ lục 3.5 và trong chuyên luận riêng.
5. Chỉ tiêu “Mô tả” bao gồm những mô tả về hình thái, kích thước, màu sắc, mùi vị, các đặc điểm của bề mặt, vết bẻ hay mặt cắt của dược liệu hoặc đặc điểm thể chất của dược liệu.
a. “Hình thái” là hình dạng của dược liệu khô. Thông thường dược liệu được quan sát mà không cần xử lý trước. Các loại dược liệu là lá hay hoa bị nhăn nheo, khô quăn có thể được làm ẩm, làm mềm và trải phẳng trước khi quan sát. Đối với một vài loại quả và hạt nếu cần có thể được làm mềm và loại bỏ vỏ hạt để kiểm tra đặc điểm bên trong.
b. “Kích thước” là chiều dài, đường kính và độ dày của dược liệu. Tiến hành đo trên một số mẫu. Cho phép một vài mẫu có giá trị hơi cao hơn hoặc thấp hơn giá trị đã xác định. Sử dụng thước đo chia vạch tới milimet. Đối với hạt hay vật có kích thước nhỏ, xếp 10 hạt gần nhau theo một hàng trên một tờ giấy có chia vạch tới milimet, đo và tính giá trị trung bình.
c. “Màu sắc” của dược liệu được quan sát bằng mắt thường ở ánh sáng ban ngày. Màu có thể được mô tả bằng các sắc độ như “hơi”, “đậm” hay “nhạt” (ví dụ màu hơi vàng, màu vàng đậm, màu vàng nhạt). Nếu màu được mô tả là màu phối hợp của hai màu thì màu chính là màu ghi trước (ví dụ trong màu nâu vàng thì màu nâu là màu chính).
d. Đặc điểm bên ngoài, bề mặt vết bẻ hay cắt ngang của dược liệu thường được quan sát trên dược liệu chưa sơ chế. Nếu quan sát thấy những đường vằn khác nhau trên mặt bẻ thì có thể cắt phẳng rồi quan sát.
e. “Mùi” của dược liệu được kiểm tra bằng cách ngửi trực tiếp hoặc sau khi bẻ gãy và vò nát. Cũng có thể ngửi sau khi làm ẩm dược liệu bằng nước nóng.
f. “Vị” của dược liệu được kiểm tra bằng cách nếm trực tiếp dược liệu hoặc nếm dịch chiết nước. Nên cẩn thận khi nếm những vị thuốc có độc.
6. Định tính là những phương pháp dùng để nhận biết dược liệu, bao gồm các kinh nghiệm truyền thống, phương pháp vi học và các phương pháp lý hóa.
a. Nhận biết dược liệu dựa theo kinh nghiệm bằng phương pháp đơn giản và truyền thống như sự chìm hay nổi trong nước, tiếng nổ, màu của ngọn lửa hay khói và mùi khi đốt cháy dược liệu v.v...
b. Định tính dược liệu bằng phương pháp vi học là việc quan sát đặc điểm của các tế bào, các mô của lát cắt, của bột hay (trong một vài trường hợp) của bề mặt dược liệu dưới kính hiển vi. (Phụ lục 12.18.)
c. Định tính lý học là việc xác định các chỉ số như độ tan, tỉ trọng, chiết xuất, năng suất quay cực v.v... của các dược liệu (Phụ lục 6).
d. Định tính hóa học là phép thử một vài thành phần trong dược liệu bằng các phản ứng hóa học. Phương pháp tiến hành được trình bày ở các chuyên luận dược liệu cụ thể.
e. Định tính sắc ký là việc sử dụng các phương pháp sắc ký như sắc ký lớp mỏng, sắc ký khí, sắc ký lỏng hiệu năng cao... để phát hiện một số thành phần có trong dược liệu; so sánh với chất chuẩn hay thành phần trong dược liệu chuẩn. Phương pháp tiến hành được trình bày ở Phụ lục 5 và các chuyên luận dược liệu cụ thể.
f. Định tính huỳnh quang là quan sát sự phát huỳnh quang của bề mặt hay mặt cắt dược liệu hoặc của dịch chiết dược liệu ở điều kiện thường hay sau khi cho tác dụng với acid, kiềm hay thuốc thử. Trừ khi có quy định riêng trong chuyên luận, mẫu thử được quan sát dưới ánh sáng tử ngoại ở bước sóng 365 nm, cách nguồn sáng khoảng 10 cm.
g. Trừ những quy định trong chuyên luận riêng, định tính vi thăng hoa thường được tiến hành như sau: Đặt một vòng kim loại đường kính khoảng 2 cm, cao khoảng 8 mm lên một tấm kim loại mỏng có kích thước hơi lớn hơn. Trải một lớp mỏng bột dược liệu trong vòng kim loại và đậy kín bằng một phiến kính bên trên đặt một miếng bông tẩm nước lạnh. Đặt tấm kim loại đã có dược liệu này lên một lưới amiant có 1 lỗ tròn đường kính khoảng 2 cm sao cho vòng kim loại có dược liệu nằm trên lỗ này. Đun nóng nhẹ phía dưới lỗ cho đến khi bột dược liệu bị cháy xém. Nhấc phiến kính ra và để nguội. Quan sát hình dạng và màu sắc của tinh thể chất được thăng hoa đọng lại trên phiến kính bằng kính hiển vi và/hoặc tiến hành phản ứng hóa học thích hợp đối với chất đã được thăng hoa.
7. Thử tinh khiết là cách kiểm tra độ tinh khiết của dược liệu. Tùy từng dược liệu mà thử tinh khiết có thể bao gồm một số hay tất cả các chỉ tiêu dưới đây:
a. Mất khối lượng do làm khô (Phụ lục 9.6 hay 12.13).
b. Tro toàn phần và tro không tan trong acid hydro-cloric (Phụ lục 9.7 và 9.8).
c. Các tạp chất hữu cơ, các bộ phận khác của dược liệu, các dược liệu bị biến màu, hư thối (Phụ lục 12.11).
d. Tỷ lệ vụn nát của dược liệu (Phụ lục 12.12).
e. Hàm lượng kim loại nặng (Phụ lục 9.4.11).
f. Dư lượng các chất bảo vệ thực vật (Phụ lục 12.17).
g. Xác định chất chiết được là xác định hàm lượng các chất trong dược liệu có thể chiết được bằng dung môi (nước, ethanol hay một dung môi khác) (Phụ lục 12.10).
8. Định lượng là việc xác định hàm lượng của một hay một số chất có trong dược liệu bằng phương pháp hóa học, lý học hoặc sinh học. Định lượng bao gồm cả việc xác định hàm lượng chất béo (Phụ lục 12.9), tinh dầu (Phụ lục 12.7) và xác định hoạt lực bằng các phép thử sinh học.
12.3 PHÉP THỬ XÁC ĐỊNH CHIẾT KIỆT ALCALOID
Các phép thử sau đây được dùng để xác định alcaloid đã được lấy kiệt hay chưa.
Các alcaloid được chiết bằng nước hoặc dung môi là ethanol - nước
Sau khi chiết ít nhất 3 lần, lấy 0,1 ml đến 0,2 ml của dịch chiết tiếp theo, acid hóa bằng dung dịch acid hydrocloric 2 M (TT), thêm 0,05 ml dung dịch kali tetraiodomercurat (TT) (thuốc thử Mayer) hoặc thêm 0,05 ml dung dịch kali iodobismuthat (TT) (thuốc thử Dragendorff) đối với các alcaloid thuộc họ Cà. Không được có tủa hay tạo dung dịch đục. Trong trường hợp dung môi chiết có độ cồn lớn hơn 25 % trở lên thì sau khi acid hóa, cần bay hơi hết dung môi (bằng cách đặt khay sứ chứa dịch chiết lên nồi cách thủy nóng) rồi mới thêm thuốc thử tạo tủa.
Các alcaloid được chiết bằng dung môi hữu cơ
Sau khi chiết ít nhất 3 lần, lấy 1 ml đến 2 ml dịch chiết tiếp theo, thêm 1 ml đến 2 ml dung dịch acid hydrocloric 0,1 M (TT), bay hơi hết dung môi hữu cơ, chuyển phần dịch lỏng còn lại vào một ống nghiệm và thêm 0,05 ml dung dịch kali tetraiodomercurat (TT) (thuốc thử Mayer) hoặc thêm 0,05 ml dung dịch kali iodobismuthat (TT) (thuốc thử Dragendorff) đối với các alcaloid thuộc họ Cà hoặc thêm 0,05 ml dung dịch iod-kali iodid (TT) (thuốc thử Bouchardat) đối với emetin. Không được thấy có tủa đục rõ tạo thành trong dung dịch.
12.4 ĐỊNH LƯỢNG ALDEHYD TRONG TINH DẦU
Cân chính xác khoảng 1g tinh dầu cần xác định aldehyd vào một ống thủy tinh có nút mài (kích thước khoảng 150 mm × 25 mm), thêm 5 ml toluen (TT) và 15 ml dung dịch hydroxylamin trong ethanol (TT), lắc mạnh và định lượng ngay với dung dịch kali hydroxyd 0,5 N trong ethanol 60 % (CĐ) cho tới khi màu đỏ chuyển sang màu vàng. Tiếp tục lắc và định lượng tới khi lớp dưới có màu vàng bền vững của chỉ thị sau khi đã lắc mạnh 2 min và để yên cho tách lớp; phản ứng xảy ra hoàn toàn trong khoảng 15 min. Kết quả chuẩn độ cho một giá trị tương đối về hàm lượng aldehyd trong mẫu.
Lặp lại quy trình định lượng như trên, dùng dung dịch thử sơ bộ ở trên thêm 0,5 ml dung dịch kali hydroxyd 0,5 N trong ethanol 60 % (CĐ) làm chuẩn màu cho điểm kết thúc định lượng. Tính hàm lượng aldehyd từ lần xác định thứ hai. Dùng đương lượng đã cho trong chuyên luận riêng.
12.5 ĐỊNH LƯỢNG CINEOL TRONG TINH DẦU
Cân 3,00 g mẫu thử vừa được làm khan bằng natri sulfat khan (TT) vào một ống nghiệm khô, thêm 2,10 g o-cresol (TT) đã chảy lỏng. Đặt ống nghiệm vào trong thiết bị xác định nhiệt độ đông đặc (Phụ lục 6.6) và làm lạnh, khuấy liên tục. Khi sự kết tinh bắt đầu, nhiệt độ hơi tăng lên một chút; ghi nhiệt độ cao nhất đạt được (t1).
Làm chảy hỗn hợp trên nồi cách thủy nhưng không được để nhiệt độ vượt quá 5 °C so với nhiệt độ t1. Đặt lại ống nghiệm vào trong thiết bị đã được duy trì ở nhiệt độ dưới t1 5°C.
Khi sự kết tinh lại xảy ra hoặc khi nhiệt độ của hỗn hợp hạ xuống 3 °C dưới t1, khuấy liên tục; ghi nhiệt độ cao nhất mà hỗn hợp đông lại (t2). Lặp lại quá trình này cho tới khi 2 giá trị cao nhất thu được (t2) chênh nhau không quá 0,2 °C. Nếu xảy ra quá trình chậm đông, thêm 1 tinh thể nhỏ của hỗn hợp gồm 3,00 g cineol (TT) và 2,10 g o-cresol (TT) đã chảy lỏng, để tạo sự kết tinh. Nếu t2 thấp hơn 27,4 °C, lặp lại thí nghiệm sau khi thêm 5,10 g hỗn hợp này.
Xác định phần trăm cineol (kl/kl) tương ứng với điểm đông (t2) từ Bảng 12.5.1 dưới đây và ta có các giá trị trung gian bằng phương pháp nội suy. Nếu có cho thêm 5,10 g hỗn hợp cineol và o-cresol, tính phần trăm (kl/kl) của cineol từ biểu thức:
2 × (A - 50)
trong đó:
A: giá trị tương ứng cho một điểm đông của t2 được lấy từ Bảng 12.5.1.
Bảng 12.5.1
| t2 | % cineol | t2 | % cineol |
| (°C) | (kl/kl) | (°C) | (kl/kl) |
| 24 | 45,5 | 40 | 67,0 |
| 25 | 47,0 | 41 | 68,5 |
| 26 | 48,5 | 42 | 70,0 |
| 27 | 49,5 | 43 | 72,5 |
| 28 | 50,5 | 44 | 74,0 |
| 29 | 52,0 | 45 | 76,0 |
| 30 | 53,5 | 46 | 78,0 |
| 31 | 54,5 | 47 | 80,0 |
| 32 | 56,0 | 48 | 82,0 |
| 33 | 57,0 | 49 | 84,0 |
| 34 | 58,5 | 50 | 86,0 |
| 35 | 60,0 | 51 | 88,5 |
| 36 | 61,0 | 52 | 91,0 |
| 37 | 62,5 | 53 | 93,5 |
| 38 | 63,5 | 54 | 96,0 |
| 39 | 65,0 | 55 | 99,0 |
12.6 ĐỊNH LƯỢNG TANINOID TRONG DƯỢC LIỆU
Có thể áp dụng 1 trong 2 phương pháp sau:
Phương pháp 1
Cân chính xác một khối lượng bột dược liệu đã rây qua rây số 355 (kích thước mắt rây 0,355 mm) và chứa khoảng 1 g taninoid, cho vào một bình nón, thêm 150 ml nước và đun trên cách thủy trong 30 min. Để nguội, chuyển hỗn hợp vào bình định mức 250 ml. Thêm nước vừa đủ tới vạch, để lắng, lọc phần dịch lỏng qua giấy lọc đường kính 125 mm, bỏ 50 ml dịch lọc đầu. Phần dịch lọc thu được dùng làm dung dịch thử.
Xác định chất chiết được trong nước toàn phần
Lấy chính xác 25 ml dung dịch thử đem bốc hơi đến khô, sấy cắn ở 105 °C trong 3 h. Để nguội trong bình hút ẩm chứa silicagel. Cân, được khối lượng T1 (g).
Xác định chất chiết được trong nước không liên kết với bột da
Lấy chính xác 100 ml dung dịch thử, thêm 6 g bột da khô. Lắc đều trong 15 min và lọc. Lấy chính xác 25 ml dịch lọc đem bốc hơi đến khô, sấy cắn ở 105 °C trong 3 h. Để nguội trong bình hút ẩm chứa silicagel. Cân, được khối lượng T2 (g).
Xác định bột da tan trong nước
Lấy chính xác 100 ml nước cất, thêm 6 g bột da khô. Lắc đều trong 15 min và lọc. Lấy chính xác 25 ml dịch lọc bốc hơi đến khô, sấy cắn ở 105 °C trong 3 h. Để nguội trong bình hút ẩm chứa silicagel. Cân, được khối lượng To (g).
Hàm lượng phần trăm (%) taninoid trong dược liệu được tính theo công thức:
![]()
trong đó:
a là khối lượng dược liệu (g).
Phương pháp 2
Tiến hành thử nghiệm trong điều kiện tránh ánh sáng.
Dung dịch chuẩn: Cân chính xác khoảng 50 mg acid galic chuẩn vào một bình định mức 100 ml màu nâu, thêm nước để hòa tan và vừa đủ đến vạch. Hút chính xác 5 ml dung dịch trên vào bình định mức 50 ml màu nâu, thêm nước vừa đủ, lắc đều (dung dịch có nồng độ acid galic khoảng 0,05 mg/ml).
Xây dựng đường chuẩn: Hút chính xác lần lượt 1,0 ml; 2,0 ml; 3,0 ml; 4,0 ml; 5,0 ml dung dịch chuẩn vào các bình định mức 25 ml riêng biệt màu nâu, thêm vào mỗi bình 1 ml thuốc thử phosphomolybdotungstic (TT), sau đó thêm lần lượt 11 ml, 10 ml, 9 ml, 8 ml, 7 ml nước vào các bình tương ứng, thêm dung dịch natri carbonat 29 % đến vạch, lắc đều. Đo độ hấp thụ của các dung dịch thu được ở 760 nm (Phụ lục 4.1), chuẩn bị song song một mẫu trắng. Xây dựng đường chuẩn với độ hấp thụ là trục tung và nồng độ dung dịch là trục hoành.
Dung dịch thử: Cân chính xác một lượng bột dược liệu (theo qui định của chuyên luận riêng) vào bình định mức 250 ml màu nâu, thêm 150 ml nước, để qua đêm, sau đó lắc siêu âm trong 10 min, để nguội, thêm nước vừa đủ, lắc đều, để lắng. Lọc, bỏ 50 ml dịch lọc đầu, hút chính xác 20 ml dịch lọc vào bình định mức 100 ml màu nâu, thêm nước vừa đủ đến vạch, lắc đều.
Tiến hành:
Hàm lượng phenol toàn phần: Hút chính xác 2 ml dung dịch thử vào bình định mức 25 ml màu nâu. Thêm 1 ml thuốc thử phosphomolybdotungstic (TT), trộn đều, thêm 10 ml nước, thêm dung dịch natri carbonat 29 % đến vạch, lắc đều, đo độ hấp thụ của dung dịch thu được như phương pháp trên và tính toán hàm lượng phenol theo acid galic trong dung dịch thử dựa trên đường chuẩn đã xây dựng.
Hàm lượng polyphenol không liên kết với casein: Hút chính xác 25 ml dung dịch thử vào bình nón nút mài 100 ml đã có 0,6 g casein, đậy kín. Đặt bình trong cách thủy ở nhiệt độ 30 °C trong 1 h, lắc đều, để nguội. Lọc, loại bỏ khoảng 5 ml dịch lọc đầu tiên, hút chính xác 2 ml dịch lọc sau cho vào bình định mức 25 ml màu nâu. Thêm 1 ml thuốc thử phosphomolybdotungstic (TT), trộn đều, thêm 10 ml nước và pha loãng bằng dung dịch natri carbonat 29 % đến vạch, lắc đều, đo độ hấp thụ của dung dịch thu được như phương pháp trên và tính toán hàm lượng phenol theo acid galic trong dung dịch dựa trên đường chuẩn đã xây dựng.
Hàm lượng taninoid tính theo acid galic trong dược liệu được tính theo công thức:
Taninoid toàn phần = (Hàm lượng phenol toàn phần) - (Hàm lượng polyphenol không liên kết với casein).
12.7 ĐỊNH LƯỢNG TINH DẦU TRONG DƯỢC LIỆU
Tinh dầu trong dược liệu được định lượng bằng cách cất kéo hơi nước trong dụng cụ cất như mô tả ở Hình 12.7. Dịch cất được hứng vào một ống chia độ, sử dụng xylen để giữ lại tinh dầu, pha nước được chảy tự động trở lại bình cất.
Dụng cụ
Dụng cụ định lượng tinh dầu bao gồm các bộ phận sau:
1. Một bình cầu thủy tinh đáy tròn có cổ ngắn với đường kính trong khoảng 29 mm.
2. Bộ phận ngưng cất (xem hình) nối kín được với bình cất, được làm từ thủy tinh có hệ số giãn nở thấp, bao gồm các bộ phận sau:
Khóa K’ có một lỗ thông khí, nhánh K có một lỗ đường kính khoảng 1 mm trùng khớp với lỗ thông khí, bề mặt cuối của nhánh K là thủy tinh mài có đường kính trong 10 mm.
Bầu hình quả lê J có thể tích 3 ml.
Ống JL chia vạch đến 0,01 ml.
Bầu tròn L có thể tích khoảng 2 ml.
M là một vòi 3 nhánh.
Điểm nối B cao hơn 20 mm so với vạch chia độ trên cùng.
3. Bộ phận đốt nóng có thể điều chỉnh được nhiệt độ.
4. Giá đỡ thẳng đứng với vòng đỡ nằm ngang có gắn vật liệu cách điện.
Tiến hành
Cho một thể tích dung môi cất theo quy định vào bình cất, thêm vài mảnh đá bọt và lắp bộ ngưng cất vào. Thêm nước qua phễu N tới mức B. Mở khóa K’, dùng pipet cho vào 1 ml xylen (TT) trừ khi có chỉ dẫn khác (tựa đầu pipet vào phía cuối của nhánh K). Đóng khóa K’ sao cho lỗ thông trùng khớp. Đun bình cho đến sôi, sau đó nếu không có chỉ dẫn gì khác thì điều chỉnh tốc độ cất sao cho cất được 2 ml/min đến 3 ml/min.
Xác định tốc độ cất như sau: Mở vòi 3 nhánh M để hạ mức dịch cất trong ống đến vạch (a) của bầu (J), khóa vòi M lại và xác định thời gian cần thiết để cất được đến vạch (b).
Mở vòi M và tiếp tục cất, điều chỉnh nhiệt độ đun để có tốc độ cất theo quy định. Cất trong 30 min. Ngừng cất, đọc thể tích xylen trong ống hứng chia độ khi nhiệt độ ống hứng trở về nhiệt độ phòng.
Cho vào bình cất một lượng mẫu theo quy định và tiếp tục cất với thời gian và tốc độ cất như quy định của chuyên luận riêng. Ngừng cất, đọc thể tích hỗn hợp tinh dầu và xylen trong ống hứng chia độ khi nhiệt độ ống hứng trở về nhiệt độ phòng. Thể tích đọc được lần này trừ đi thể tích xylen sẽ cho thể tích tinh dầu trong lượng mẫu định lượng. Tính toán kết quả thu được biểu thị theo ml tinh dầu có trong một kg mẫu thử hoặc theo phần trăm (số ml tinh dầu trong 100 g mẫu thử).
Hình 12.7 - Dụng cụ cất tinh dầu
(Kích thước tính bằng mm)
12.8 CÁC PHÉP THỬ CỦA TINH DẦU
Dầu béo và nhựa trong tinh dầu
Nhỏ 1 giọt tinh dầu lên giấy mỏng, để khô trong gió hay hơ nóng nhẹ cho bay hết mùi tinh dầu. Không được xuất hiện vết bóng mờ hoặc vết dầu mỡ.
Ester liên quan
Đun nóng 1 ml tinh dầu với 3 ml dung dịch kali hydroxyd 10 % trong ethanol 96 % (TT) mới pha trên cách thủy trong 2 min. Không được xuất hiện tinh thể trong vòng 30 min kể từ lúc bắt đầu để nguội.
Cắn sau khi bay hơi
Cắn sau khi bay hơi là phần trăm khối lượng của tinh dầu còn lại sau khi bay hơi trên cách thủy ở điều kiện như sau:
Thiết bị
Nồi cách thủy có nắp đậy với những lỗ có đường kính 70 mm.
Cốc cô bằng thủy tinh chịu nhiệt, trơ về mặt hóa học.
Bình hút ẩm.
Cách tiến hành
Cân 5,00 g tinh dầu (trừ khi có chỉ dẫn khác trong chuyên luận) vào một cốc cô đã được cân bì sau khi đun nóng trên cách thủy trong 1 h và làm nguội trong bình hút ẩm. Đặt cốc cô lên trên nồi cách thủy tại vị trí lỗ có đường kính 70 mm và giữ cho mực nước cách dưới mặt nồi cách thủy khoảng 50 mm trong suốt thời gian tiến hành (xem Hình 12.8). Đun sôi mạnh nước ở nồi cách thủy dưới áp suất thường. Cô tinh dầu trên nồi cách thủy sôi đến cạn trừ khi có chỉ dẫn khác trong chuyên luận riêng. Làm nguội cốc cô trong bình hút ẩm và cân khối lượng.
Hình 12.8. Dụng cụ xác định cắn tinh dầu sau khi bay hơi
(Kích thước tính bằng mm)
Độ tan trong ethanol
Hút chính xác 1,0 ml tinh dầu vào ống thủy tinh nút mài 25 ml hoặc 30 ml và đặt trong thiết bị ổn nhiệt duy trì ở nhiệt độ 20 °C ± 0,2 °C. Dùng buret có dung tích ít nhất 20 ml, thêm từng 0,1 ml ethanol có nồng độ như chỉ dẫn của chuyên luận cho đến khi tan thành dung dịch hoàn toàn, sau đó tiếp tục thêm từng 0,5 ml đến 20 ml, lắc mạnh thường xuyên. Đọc thể tích ethanol đã dùng để tạo thành dung dịch trong và nếu dung dịch bị đục hoặc có màu trắng sữa trước khi lượng ethanol thêm vào đạt 20 ml thì đọc thể tích tại thời điểm xuất hiện đục hoặc màu trắng sữa và đọc thể tích tại thời điểm hết đục hay hết màu trắng sữa.
Nếu dung dịch không trong, khi đã thêm 20 ml ethanol có nồng độ như chỉ dẫn của chuyên luận, lặp lại phép thử với ethanol có nồng độ cao hơn
Một tinh dầu được gọi là “Tan trong v hoặc hơn thể tích ethanol có nồng độ t” có nghĩa là khi dùng v thể tích ethanol để tạo thành dung dịch trong, dung dịch vẫn trong sau khi đã thêm ethanol cùng nồng độ đến 20 thể tích, so sánh với tinh dầu chưa pha loãng.
Một tinh dầu được gọi là “Tan trong v thể tích ethanol có nồng độ t, chuyển thành đục khi pha loãng” có nghĩa là khi dung dịch trong với v thể tích ethanol chuyển thành đục với v1 thể tích (v1 nhỏ hơn 20) và giữ nguyên tình trạng khi thêm từ từ ethanol cùng nồng độ đến 20 thể tích.
Một tinh dầu được gọi là “Tan trong v thể tích ethanol, chuyển thành đục giữa khoảng v1 và v2 thể tích ethanol” có nghĩa là khi dung dịch trong với v thể tích ethanol chuyển thành đục với v1 thể tích (v1 nhỏ hơn 20) và giữ nguyên tình trạng khi thêm từ từ ethanol cùng nồng độ đến v2 thể tích và chuyển thành dung dịch trong (v2 nhỏ hơn 20).
Một tinh dầu được gọi là “Tan thành dạng sữa” có nghĩa là khi dung dịch trong ethanol có màu hơi xanh nhẹ tương tự như màu của hỗn hợp chuẩn đục mới pha gồm 0,5 ml dung dịch bạc nitrat 1,7 % (kl/tt) trộn với 0,05 ml acid nitric (TT), thêm 50 ml dung dịch natri clorid 0,0012 % (TT), lắc đều và để yên trong chỗ tối 5 min.
Nước
Trộn 10 giọt tinh dầu với 1 ml carbon disulfid (TT), dung dịch phải trong.
12.9 DẦU BÉO
Tạp chất kiềm
Trộn 10 ml aceton (TT) và 0,3 ml nước trong ống nghiệm, thêm 0,05 ml dung dịch xanh bromophenol 0,04 % trong ethanol 96 % (TT), trung hòa dung dịch (nếu cần) bằng dung dịch acid hydrocloric 0,01 M (TT) hoặc dung dịch natri hydroxyd 0,01 M (TT). Thêm 10 ml dầu vào hỗn hợp trên, lắc và để lắng. Không quá 0,1 ml dung dịch acid hydrocloric 0,01 M (CĐ) cần thêm vào để làm chuyển màu lớp dung dịch phía trên sang màu vàng.
Định tính dầu béo bằng phương pháp sắc ký lớp mỏng
Tiến hành theo Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Chất hấp phụ là octadecylsilyl silica gel cho sắc ký lớp mỏng hiệu năng cao (HPTLC silica gel RP-18 là phù hợp).
Dung dịch thử: Hòa tan khoảng 20 mg (1 giọt) dầu trong 3 ml dicloromethan (TT), trừ khi có chỉ dẫn khác.
Dung dịch đối chiếu: Hòa tan khoảng 20 mg (1 giọt) dầu ngô trong 3 ml dicloromethan (TT).
Chấm riêng biệt lên bản mỏng 1 µl mỗi dung dịch trên. Triển khai sắc ký hai lần, mỗi lần 0,5 cm với dung môi khai triển là ether (TT). Triển khai sắc ký hai lần nữa, mỗi lần 8 cm với dung môi khai triển là hỗn hợp dicloromethan - acid acetic băng - aceton (20:40:50). Để khô bản mỏng ngoài không khí và phun dung dịch acid phosphomolybdic 10 % trong ethanol 96 % (TT). Sấy bản mỏng ở 120 °C trong khoảng 3 min và quan sát dưới ánh sáng tự nhiên, sắc ký đồ xuất hiện các vết đặc trưng như trên hình dưới đây.
| 1. Dầu lạc | 6. Dầu hạt cải |
| 2. Dầu vừng | 7. Dầu hạt lanh |
| 3. Dầu ngô | 8. Dầu oliu |
| 4. Dầu hạt cải | 9. Dầu hướng dương |
| (không có acid erucic) | 10. Dầu hạnh nhân |
| 5. Dầu đậu tương | 11. Dầu mầm lúa mì |
Hình 12.9: Sắc ký đồ định tính dầu béo
Phép thử các dầu tạp bằng phương pháp sắc ký lớp mỏng
Tiến hành theo Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Chất hấp phụ là kieselguhr G (TT). Làm ẩm bản mỏng khô bằng cách đặt trong bình có lớp dung môi dày 5 mm là hỗn hợp parafin lỏng - ether dầu hỏa (khoảng sôi 50 °C đến 70 °C) (10: 90). Để đến khi dung môi khai triển được ít nhất 12 cm, lấy bản mỏng ra và để khô ngoài không khí trong 5 min.
Pha động: Nước-acid acetic băng (1: 9).
Dung dịch thử: Đun hồi lưu 2 g dầu với 30 ml dung dịch kali hydroxyd 0,5 M trong ethanol (TT) trong 45 min, pha loãng với 50 ml nước, để nguội, chuyển sang bình gạn, lắc với ether ethylic (TT) 3 lần, mỗi lần 50 ml, loại bỏ lớp dịch chiết ether. Acid hóa lớp nước bằng acid hydrocloric (TT) rồi chiết bằng ether ethylic (TT) ba lần, mỗi lần 50 ml. Tập trung các dịch chiết ether, rửa bằng nước ba lần, mỗi lần 10 ml, làm khan dịch chiết ether bằng natri sulfat khan (TT) và lọc. Cô dịch chiết ether trên cách thủy, hòa tan 40 mg cắn trong 4 ml cloroform (TT). Các acid béo có thể thu được từ dung dịch đã xà phòng hóa trong quá trình xác định các chất không xà phòng hóa.
Dung dịch đối chiếu: Chuẩn bị giống như dung dịch thử nhưng dùng 2 g hỗn hợp gồm 19 thể tích dầu ngô và 1 thể tích dầu hạt cải thay cho mẫu thử.
Tiến hành: Chấm riêng biệt lên bản mỏng 3 µl mỗi dung dịch trên. Triển khai sắc ký đến khi dung môi đi được 8 cm. Lấy bản mỏng ra khỏi bình, làm khô bản mỏng ở 110 °C trong 10 min và trừ khi có chỉ dẫn khác trong chuyên luận, đặt bản mỏng vào bình bão hòa hơi iod. Sau một thời gian, xuất hiện các vết từ màu nâu đến nâu vàng. Lấy bản mỏng ra, để yên vài phút cho đến khi màu nâu trên nền bản mỏng biến mất. Phun lên bản mỏng dung dịch hồ tinh bột (TT). Các vết màu xanh xuất hiện, các vết này có thể chuyển thành màu nâu khi để khô và lại chuyển thành màu xanh sau khi phun nước. Trên sắc ký đồ của dung dịch thử luôn xuất hiện các vết với các giá trị Rf khoảng 0,5 (acid oleic) và khoảng 0,65 (acid linoleic), tương ứng với các vết trên sắc ký đồ của dung dịch đối chiếu. Trên sắc ký đồ của dung dịch thử có thể có vết với giá trị Rf khoảng 0,75 (acid linolenic) nhưng không được có vết có giá trị Rf khoảng 0,25 (acid erucic), tương ứng với vết xuất hiện trên sắc ký đồ của dung dịch đối chiếu.
Phép thử các dầu tạp bằng phương pháp sắc ký khí
Phép thử các dầu tạp được xác định bằng phương pháp sắc ký khí (Phụ lục 5.2), dựa trên các methyl ester của acid béo có trong dầu béo.
Phương pháp A
Phương pháp này không áp dụng với các dầu béo có chứa acid béo dạng glycerid với các nhóm epoxy-, hydroepoxy-, cyclopropyl hoặc nhóm cyclopropenyl, hoặc các dầu béo có chứa tỷ lệ lớn lượng acid béo có chuỗi carbon nhỏ hơn 8 hay dầu béo có chỉ số acid lớn hơn 2,0.
Dung dịch thử: Trừ các quy định trong chuyên luận riêng, làm khan dầu béo thử trước giai đoạn methyl hóa. Cân 1,0 g dầu vào bình đáy tròn 25 ml có cổ bình thủy tinh nối được với ống sinh hàn hồi lưu, cho vài viên đá bọt vào trong bình. Thêm 10 ml methanol khan (TT) và 0,2 ml dung dịch kali hydroxyd 6 % trong methanol. Lắp sinh hàn hồi lưu, cho khí nitơ chạy qua hỗn hợp với tốc độ khoảng 50 ml trong 1 min, lắc và đun sôi. Khi dung dịch trở nên trong suốt (thường sau khoảng 10 min), tiếp tục đun trong 5 min. Làm nguội bình dưới dòng nước và chuyển dung dịch sang bình gạn. Rửa bình bằng 5 ml heptan (TT) và chuyển dịch rửa vào bình gạn, lắc. Thêm 10 ml dung dịch natri clorid 20 % (TT) và lắc mạnh. Để yên cho tách lớp và chuyển lớp dung môi sang lọ có chứa natri sulfat khan (TT). Để lắng và lọc.
Dung dịch đối chiếu (a): Chuẩn bị 0,50 g hỗn hợp chuẩn với thành phần được mô tả ở một trong các bảng 12.9 như chỉ dẫn của chuyên luận riêng (nếu chuyên luận riêng không đề cập đến, sử dụng thành phần được mô tả trong Bảng 12.9.1). Hòa tan trong heptan (TT) và pha loãng đến 50,0 ml với cùng dung môi.
Dung dịch đối chiếu (b): Pha loãng 1 ml dung dịch đối chiếu (a) thành 10,0 ml bằng heptan (TT).
Dung dịch đối chiếu (c): Chuẩn bị 0,50 g hỗn hợp các methyl ester của acid béo(1) có thành phần tương ứng với các acid béo cần xác định của mẫu thử theo quy định của chuyên luận riêng. Hòa tan trong heptan (TT) và pha loãng đến 50,0 ml với cùng dung môi. Có thể sử dụng các hỗn hợp methyl ester bán sẵn.
Điều kiện sắc ký:
Cột:
Chất liệu: Silica nung chảy, thủy tinh hoặc thạch anh.
Kích cỡ: Dài 10 m đến 30 m, đường kính 0,2 mm đến 0,8 mm.
Pha tĩnh: Poly[(cyanopropyl)(methyl)][(phenyl)(methyl)] siloxan hoặc macrogol 20 000 (lớp phim dày 0,1 µm đến 0,5 µm) hoặc một số pha tĩnh thích hợp khác.
Khí mang: Khí heli dùng cho sắc ký hoặc khí hydrogen dùng cho sắc ký.
Tốc độ dòng: 1,3 ml/min (cho cột có đường kính 0,32 mm).
Tỷ lệ chia dòng: 1:100 hoặc ít hơn, tùy thuộc vào đường kính trong của cột sử dụng (1: 50 khi đường kính cột là 0,32 mm).
Nhiệt độ:
Cột: 160 °C đến 200 °C, tùy thuộc vào chiều dài và loại cột sử dụng (200 °C cho cột dài 30 m và bao bằng lớp macrogol 20 000). Nếu cần hoặc theo chỉ dẫn riêng, tăng nhiệt độ của cột với tốc độ 3 °C /min từ 170 °C đến 230 °C (cho cột dùng macrogol 20 000).
Buồng tiêm: 250 °C.
Detector 250 °C.
Phát hiện: Detector ion hóa ngọn lửa.
Thể tích tiêm: 1 µl.
Độ nhạy: Chiều cao của pic chính trong sắc ký đồ của dung dịch đối chiếu (a) từ 50 % đến 70 % toàn thang đo khi ghi sắc ký đồ.
Kiểm tra tính thích hợp của hệ thống sắc ký khi sử dụng hỗn hợp chuẩn trong các Bảng 12.9.1 hoặc 12.9.3
Độ phân giải: Không dưới 1,8 giữa pic methyl oleat và methyl stearat trong sắc ký đồ của dung dịch đối chiếu (a).
Tỷ lệ tín hiệu trên nhiễu: Không dưới 5 với pic methyl myristat trong sắc ký đồ của dung dịch đối chiếu (b).
Số đĩa lý thuyết: Không dưới 30000 tính theo pic methyl stearat trong sắc ký đồ của dung dịch đối chiếu (a).
Kiểm tra tính thích hợp của hệ thống sắc ký khi sử dụng hỗn hợp chuẩn trong Bảng 12.9.2
Độ phân giải: Không dưới 4,0 giữa pic methyl caprylat và methyl caprat trong sắc ký đồ của dung dịch đối chiếu (a).
Tỷ lệ tín hiệu trên nhiễu: Không dưới 5 với pic methyl caproat trong sắc ký đồ của dung dịch đối chiếu (b).
Số đĩa lý thuyết: Không dưới 15 000 tính theo pic methyl caprat trong sắc ký đồ của dung dịch đối chiếu (a).
Đánh giá sắc đồ
Tránh điều kiện thử nghiệm có chiều hướng làm che giấu pic (sự có mặt của các thành phần khác nhau rất ít về thời gian lưu, ví dụ như acid linolenic và acid arachidic).
Phân tích định tính
Xác định các pic trên sắc ký đồ của dung dịch đối chiếu (c), các pic có thể được xác định bằng đường chuẩn với sắc ký đồ của dung dịch đối chiếu (a) và thông tin ghi ở các Bảng 12.9.1, 12.9.2 và 12.9.3.
a) Sử dụng điều kiện đẳng nhiệt để tính logarit của sự biến đổi thời gian lưu như một hàm của số nguyên tử carbon trong acid béo; định tính các pic dựa trên đường thẳng lập được theo cách trên và giá trị "chiều dài chuỗi carbon tương ứng" của các pic khác nhau. Đường chuẩn của các acid béo no là một đường thẳng. Logarit sự biến đổi thời gian lưu của các acid béo không no nằm trên đường thẳng này và có vị trí tương ứng với giá trị không nguyên của số nguyên tử carbon được biết từ giá trị tương ứng với chiều dài chuỗi.
b) Sử dụng chương trình nhiệt độ tuyến tính để xác định thời gian lưu dựa trên số nguyên tử carbon của acid béo, định tính bằng cách so sánh với đường chuẩn của hỗn hợp chuẩn.
Phân tích định lượng
Thông thường, sử dụng phương pháp tính phần trăm mà trong đó tổng diện tích của tất cả các pic trên sắc ký đồ ngoại trừ dung môi được tính là 100 %. Hàm lượng của mỗi thành phần được tính dựa trên tỷ lệ của diện tích pic tương ứng so với tổng diện tích của tất cả các pic thu được từ mẫu đem thử. Loại bỏ các pic có diện tích nhỏ hơn 0,05 % so với tổng diện tích pic.
Trong một vài trường hợp như sự có mặt của các acid béo từ 12 nguyên tử carbon trở xuống, hệ số hiệu chỉnh có thể được sử dụng trong các chuyên luận riêng để chuyển đổi diện tích pic thành tỷ lệ phần trăm kl/kl.
Phương pháp B
Phương pháp này không áp dụng với các dầu béo có chứa acid béo dạng glycerid với các nhôm epoxy-, hydroepoxy-, cyclopropyl, nhóm cyclopropenyl, hoặc các dầu béo có chỉ số acid lớn hơn 2,0.
Dung dịch thử
Lấy 0,100 g chế phẩm cho vào ống ly tâm 10 ml có nắp xoáy. Hòa tan trong 1 ml heptan (TT) và 1 ml dimethyl carbonat (TT), đun nóng nhẹ (50 °C đến 60 °C) và lắc mạnh. Thêm 1 ml dung dịch natri (TT) 1,2 % trong methanol khan (TT) (được pha chế hết sức cẩn thận) khi dung dịch còn ấm, lắc mạnh trong 5 min. Thêm 3 ml nước và lắc mạnh trong 30 s. Ly tâm trong 15 min với tốc độ 1500 vòng/min. Tiêm 1 µl pha hữu cơ.
Dung dịch đối chiếu và đánh giá sắc đồ: Trừ khi có chỉ dẫn khác trong chuyên luận riêng, tiến hành như mô tả ở phương pháp A.
Cột:
Chất liệu: Silica nung chảy.
Kích cỡ: Dài 30 m, đường kính 0,25 mm.
Pha tĩnh: Macrogol 20 000 (lớp phim dày 0,25 µm).
Khí mang: Khí heli dùng cho sắc ký.
Tốc độ dòng: 0,9 ml/min
Tỷ lệ chia dòng: 1: 100.
Nhiệt độ:
|
| Thời gian | Nhiệt độ |
| Cột | 0 - 15 | 100 |
| 15 - 36 | 100 → 225 | |
| 36 - 61 | 225 | |
| Buồng tiêm |
| 250 |
| Detector |
| 250 |
Phát hiện: Detector ion hóa ngọn lửa.
Thể tích tiêm: 1 µI.
Phương pháp C
Phương pháp này không áp dụng với các dầu béo có chứa acid béo dạng glycerid với các nhóm epoxy-, hydroepoxy-, aldehyd, keton, cyclopropyl, nhóm cyclo-propenyl, và kết hợp với các đa acid béo không no hay các nhóm acetylenic do các nhóm này bị phá hủy một phần hay toàn bộ.
Dung dịch thử: Hòa tan 0,10 g mẫu thử trong 2 ml dung dịch natri hydroxyd 2 % trong methanol trong bình nón 25 ml và đun sôi hồi lưu trong 30 min. Thêm 2,0 ml dung dịch boron trifluorid (TT) qua ống sinh hàn và tiếp tục đun sôi trong 30 min. Thêm 4 ml heptan (TT) qua ống sinh hàn và tiếp tục đun sôi trong 5 min. Làm nguội, thêm 10 ml dung dịch natri clorid bão hòa (TT), lắc trong 15 s và thêm tiếp dung dịch natri clorid bão hòa (TT) cho đến khi pha dung môi trên dâng lên đến cổ bình. Lấy 2 ml pha dung môi trên, rửa bằng nước ba lần, mỗi lần 2 ml và làm khan bằng natrisulfat khan (TT).
Dung dịch đối chiếu và đánh giá sắc đồ: Trừ khi có chỉ dẫn khác trong chuyên luận riêng, tiến hành như mô tả ở phương pháp A.
Bảng 12.9.1 - Hỗn hợp chuẩn để lập đường chuẩn (2)
| Thành phần hỗn hợp | Tỷ lệ thành phần (phần trăm kl/kl) | ||
| Tương ứng với chiều dài chuỗi(3) | Đẳng nhiệt | Chương trình nhiệt độ tuyến tính | |
| Methyl laurat | 12,0 | 5 | 10 |
| Methyl myristat | 14,0 | 5 | 15 |
| Methyl palmitat | 16,0 | 10 | 15 |
| Methyl stearat | 18,0 | 20 | 20 |
| Methyl arachidat | 20,0 | 40 | 20 |
| Methyl oleat | 18,3 | 20 | 20 |
Bảng 12.9.2 - Hỗn hợp chuẩn để lập đường chuẩn(2)
| Thành phần hỗn hợp | Tỷ lệ thành phần (phần trăm kl/kl) | ||
| Tương ứng với chiều dài chuỗi(3) | Đẳng nhiệt | Chương trình nhiệt độ tuyến tính | |
| Methyl caproat | 6,0 | 5 | 10 |
| Methyl caprylat | 8,0 | 5 | 35 |
| Methyl caprat | 10,0 | 10 | 35 |
| Methyl laurat | 12,0 | 20 | 10 |
| Methyl myristat | 14,0 | 40 | 10 |
Bảng 12.9.3 - Hỗn hợp chuẩn để lập đường chuẩn(2)
| Thành phần hỗn hợp | Tỷ lệ thành phần (phần trăm kl/kl) | ||
| Tương ứng với chiều dài chuỗi(3) | Đẳng nhiệt | Chương trình nhiệt độ tuyến tính | |
| Methyl myristat | 14,0 | 5 | 15 |
| Methyl palmitat | 16,0 | 10 | 15 |
| Methyl stearat | 18,0 | 15 | 20 |
| Methyl arachidat | 20 | 20 | 15 |
| Methyl oleat | 18,3 | 20 | 15 |
| Methyl eicosenoat | 20,2 | 10 | 10 |
| Methyl behenat | 22,0 | 10 | 5 |
| Methyl lignocerat | 24,0 | 10 | 5 |
(1): Methyl ester của acid béo phải đạt quy định về chất chuẩn của Dược điển Việt Nam.
(2): Áp dụng cho hệ thống sắc ký khí với cột mao quản và buồng tiêm chia dòng, các thành phần có mạch dài nhất của hỗn hợp mẫu thử cần được thêm vào hỗn hợp chuẩn khi tiến hành phân tích định lượng sử dụng đường chuẩn.
(3): Giá trị được tính dựa trên đường chuẩn với cột macrogol 20 000.
12.10 XÁC ĐỊNH CÁC CHẤT CHIẾT ĐƯỢC TRONG DƯỢC LIỆU
Phương pháp xác định các chất chiết được bằng nước
Phương pháp chiết lạnh: Nếu không có chỉ dẫn đặc biệt trong chuyên luận riêng, cân chính xác khoảng 4.000 g bột dược liệu có cỡ bột nửa thô cho vào trong bình nón 250 ml đến 300 ml. Thêm chính xác 100,0 ml nước, đậy kín, ngâm lạnh, thỉnh thoảng lắc trong 6 h đầu, sau đó để yên 18 h. Lọc qua phễu lọc khô vào một bình hứng khô thích hợp. Lấy chính xác 20 ml dịch lọc cho vào một cốc thủy tinh đã cân bì trước, cô trong cách thủy đến cắn khô. Sấy cắn ở 105 °C trong 3 h, lấy ra để nguội trong bình hút ẩm 30 min, cân nhanh để xác định khối lượng cắn sau khi sấy, tính phần trăm lượng chất chiết được bằng nước theo dược liệu khô.
Phương pháp chiết nóng: Nếu không có chỉ dẫn đặc biệt trong chuyên luận riêng, cân chính xác khoảng 2,000 g đến 4,000 g bột dược liệu có cỡ bột nửa thô cho vào bình nón 100 ml hoặc 250 ml. Thêm chính xác 50,0 ml hoặc 100,0 ml nước, đậy kín, cân xác định khối lượng, để yên 1 h, sau đó đun sôi nhẹ dưới hồi lưu 1 h, để nguội, lấy bình nón ra, đậy kín, cân để xác định lại khối lượng, dùng nước để bổ sung phần khối lượng bị giảm, lọc qua phễu lọc khô vào một bình hứng khô thích hợp. Lấy chính xác 25 ml dịch lọc vào cốc thủy tinh đã cân bì trước, cô trong cách thủy đến cắn khô, cắn thu được sấy ở 105 °C trong 3 h, lấy ra để nguội trong bình hút ẩm 30 min, cân nhanh để xác định khối lượng cắn. Tính phần trăm lượng chất chiết được bằng nước theo dược liệu khô.
Phương pháp xác định các chất chiết được bằng ethanol hoặc methanol
Dùng các phương pháp tương tự như phương pháp xác định các chất chiết được bằng nước. Tùy theo chỉ dẫn trong chuyên luận riêng mà dùng ethanol hoặc methanol có nồng độ thích hợp để thay nước làm dung môi chiết. Với phương pháp chiết nóng thì nên đun trong cách thủy nếu dung môi chiết có độ sôi thấp.
12.11 XÁC ĐỊNH TẠP CHẤT LẪN TRONG DƯỢC LIỆU
Tạp chất lẫn trong dược liệu bao gồm tất cả các chất ngoài quy định của dược liệu đó như: Đất, đá, rơm rạ, cây cỏ khác, các bộ phận khác của cây không quy định làm dược liệu, xác côn trùng...
Cách xác định
Cân một lượng mẫu vừa đủ theo chỉ dẫn trong chuyên luận riêng hoặc trong ghi chú dưới đây, dàn mỏng trên tờ giấy, quan sát bằng mắt thường hoặc kính lúp, khi cần có thể dùng rây để phân tách tạp chất và dược liệu.
Cân phần tạp chất và tính phần trăm như sau:
![]()
trong đó:
a là khối lượng tạp chất tính bằng gam;
p là khối lượng mẫu thử tính bằng gam.
Ghi chú:
1. Trong một số trường hợp nếu tạp chất rất giống với thuốc, có thể phải làm các phản ứng định tính hóa học, phương pháp vật lý hoặc dùng kính hiển vi để phát hiện tạp chất. Tỷ lệ tạp chất được tính bao gồm cả tạp chất được phát hiện bằng phương pháp này.
2. Lượng mẫu lấy để thử nếu chuyên luận riêng không quy định thì lấy như sau:
Hạt và quả rất nhỏ (như hạt Mã đề): 10 g.
Hạt và quả nhỏ: 20 g.
Dược liệu thái thành lát: 50 g.
12.12 XÁC ĐỊNH TỶ LỆ VỤN NÁT CỦA DƯỢC LIỆU
Cân một lượng dược liệu nhất định (p gam) đã được loại tạp chất. Rây bằng rây có số quy định theo chuyên luận riêng hoặc chọn bằng tay những phần vụn đối với các bộ phận cây không thái nhỏ. Cân toàn bộ phần đã lọt qua rây và phần vụn đã chọn (a gam). Tính tỷ lệ vụn nát (X %) (từ kết quả trung bình của ba lần thực hiện) theo công thức:
![]()
Ghi chú:
Lượng dược liệu lấy để thử (tùy theo bản chất của dược liệu) từ 100 g đến 200 g.
Đối với dược liệu mỏng manh thì chỉ lắc nhẹ, tránh làm vụn nát thêm.
Phần bụi và bột vụn không phân biệt được bằng mắt thường được tính vào mục tạp chất.
12.13 XÁC ĐỊNH HÀM LƯỢNG NƯỚC BẰNG PHƯƠNG PHÁP CẤT VỚI DUNG MÔI
Dụng cụ
Dụng cụ xác định hàm lượng nước (Hình 12.13) gồm có bình cầu (A) nối qua ống (D) với ống hình trụ (B); ống này gắn với ống hứng chia độ (E) ở phía dưới và lắp với ống sinh hàn ngược (C) ở phía trên, ống hứng (E) được chia độ đến 0,1 ml để cho sai số của phép đọc thể tích không vượt quá 0,05 ml. Nguồn nhiệt thích hợp là bếp điện có biến trở hoặc đun cách dầu.
Cách tiến hành
Rửa sạch ống hứng và ống sinh hàn với nước rồi làm khô. Thêm 200 ml toluen (TT) và khoảng 2 ml nước vào bình cầu khô. Cất khoảng 2 h, để nguội trong 30 min rồi đọc thể tích nước cất được ở ống hứng (V1), chính xác đến 0,05 ml.
Thêm vào bình cầu một lượng mẫu thử đã cân chính xác tới 0,01 g có chứa khoảng 2 ml đến 3 ml nước. Thêm vài mảnh đá bọt. Đun nóng nhẹ trong 15 min; khi toluen bắt đầu sôi thì điều chỉnh nguồn cấp nhiệt để cất với tốc độ khoảng 2 giọt dịch cất trong 1 s. Khi đã cất được phần lớn nước sang ống hứng thì nâng tốc độ cất lên 4 giọt dịch cất trong 1 s. Tiếp tục cất cho đến khi mực nước cất được trong ống hứng không tăng lên nữa, dùng 5 ml đến 10 ml toluen (TT) rửa thành trong ống sinh hàn rồi cất thêm 5 min nữa. Sau đó, tách bộ cất khỏi nguồn cấp nhiệt, để cho ống hứng nguội đến nhiệt độ phòng. Nếu có những giọt nước còn đọng trên thành ống sinh hàn thì dùng 5 ml toluen (TT) để rửa kéo xuống. Khi lớp nước và lớp toluen đã được phân tách hoàn toàn, đọc thể tích nước trong ống hứng (V2). Tính tỷ lệ phần trăm nước trong mẫu thử theo công thức sau:
![]()
trong đó:
V1 là số ml nước cất được sau lần cất đầu;
V2 là số ml nước cất được sau hai lần cất;
m là số g mẫu đã cân đem thử.
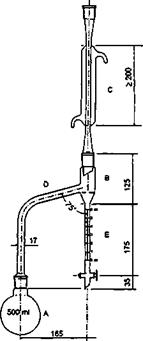
Hình 12.13 - Dụng cụ xác định hàm lượng nước bằng phương pháp cất với dung môi.
(Kích thước tính bằng mm)
12.15 CẮN KHÔ CỦA CÁC CHẤT CHIẾT ĐƯỢC TRONG DƯỢC LIỆU
Cho nhanh 2,00 g hoặc 2,0 ml mẫu thử vào một cốc đáy bằng có đường kính khoảng 50 mm và chiều cao khoảng 30 mm đã được sấy khô và xác định khối lượng. Cô đến khô cạn trên cách thủy và sấy ở 100 °C đến 105 °C trong 3 h. Lấy ra để nguội trong bình hút ẩm có chất hút ẩm phosphor pentoxyd hoặc silica gel và cân. Tính kết quả theo phần trăm khối lượng hoặc theo tỷ lệ gam trên lít.
12.16 MẤT KHỐI LƯỢNG DO LÀM KHÔ CỦA CÁC CHẤT CHIẾT ĐƯỢC TRONG DƯỢC LIỆU
Cân nhanh 0,50 g mẫu thử đã nghiền thành bột mịn vào một cốc đáy bằng có đường kính khoảng 50 mm và chiều cao khoảng 30 mm đã được sấy khô và xác định khối lượng, sấy ở 100 °C đến 105 °C trong 3 h. Lấy ra để nguội trong bình hút ẩm có chất hút ẩm phosphor pentoxyd hoặc silica gel và cân. Tính toán kết quả theo phần trăm khối lượng.
12.17 DƯ LƯỢNG HÓA CHẤT BẢO VỆ THỰC VẬT
Định nghĩa
Hóa chất bảo vệ thực vật là những chất hay hỗn hợp nhiều chất được sử dụng nhằm ngăn cản, tiêu diệt hoặc kiểm soát dịch hại đối với các loài cây hoặc động vật làm thuốc mà những chất này có thể gây tổn hại hoặc ảnh hưởng đến quá trình sản xuất, chế biến, bảo quản, vận chuyển hoặc buôn bán dược liệu. Khái niệm này bao gồm cả các chất điều hòa sinh trưởng thực vật, các chất làm rụng lá hoặc làm khô, hoặc bất cứ chất nào được sử dụng trước hoặc sau thu hoạch để bảo quản dược liệu khỏi bị hỏng trong quá trình bảo quản hoặc vận chuyển.
Giới hạn
Dược liệu được kiểm tra ít nhất phải đáp ứng yêu cầu về giới hạn ghi trong Bảng 12.17.1 (trừ những chỉ dẫn trong chuyên luận riêng).
Đối với hóa chất bảo vệ thực vật không liệt kê trong Bảng 12.17.1, giới hạn (mg/kg) của nó có thể được tính theo công thức sau:
![]()
trong đó:
ADI (mg/kg) là liều có thể đưa vào cơ thể mỗi ngày theo quy định của Tổ chức Lương thực Thế giới và Tổ chức Y tế thế giới (Codex Alimentarius, FAO - WHO);
M là thể trọng của người, bằng 60 kg;
MDD là liều dùng dược liệu hàng ngày, tính bằng kg.
Bảng 12.17.1
| Hợp chất | Giới hạn (mg/kg) |
| Alaclor | 0,02 |
| Tổng aldrin và dieldrin | 0,05 |
| Azinphos-methyl | 1,0 |
| Bromopropylat | 3,0 |
| Tổng cis - clordan, trans - clordan và oxyclordan | 0,05 |
| Clorfenvinphos | 0,5 |
| Cypermethrin và các đồng phân | 1,0 |
| Tổng o,p’ - DDT, p,p’ - DDT, p,p’ - DDE và p,p’ - TDE | 1,0 |
| Deltamethrin | 0,5 |
| Diazinon | 0,5 |
| Diclorvos | 1,0 |
| Dithiocarbamat (tính theo CS2) | 2,0 |
| Tổng α - Endosulfan, β - Endosulfan và Endosulfan sulfat | 3,0 |
| Endrin | 0,05 |
| Ethion | 2,0 |
| Fenitrothion | 0,5 |
| Fenvalerat | 1,5 |
| Fonofos | 0,05 |
| Tổng Heptaclor và Heptaclor epoxid | 0,05 |
| Hexaclorobenzen | 0,1 |
| Các đồng phân hexaclorocyclohexan, trừ đồng phân γ | 0,3 |
| Lindan (γ-Hexaclorocyclohexan) | 0,6 |
| Malathion | 1,0 |
| Methidathion | 0,2 |
| Parathion | 0,5 |
| Parathion - methyl | 0,2 |
| Permethrin | 1,0 |
| Phosalon | 0,1 |
| Piperonyl butoxid | 3,0 |
| Pirimiphos - methyl | 4,0 |
| Tổng pyrethrin | 3,0 |
| Tổng quintozen, pentacloroanalin và methyl pentaclorophenyl sulfid | 1,0 |
Lấy mẫu
Phương pháp:
Bao bì ít hơn 1 kg, trộn đều và lấy đủ lượng mẫu để phân tích. Bao bì từ 1 kg đến 5 kg, lấy những lượng tương đương và đủ để phân tích ở 3 vị trí: Phía trên, giữa và cuối bao bì. Trộn đều và lấy lượng mẫu đủ để phân tích. Bao bì lớn hơn 5 kg, lấy không ít hơn 250 g ở 3 vị trí: phía trên, giữa và cuối bao bì, trộn đều và lấy một lượng mẫu đủ để phân tích.
Số mẫu:
Nếu như số bao bì (n) ít hơn hoặc bằng 3, mỗi bao bì lấy một lượng mẫu theo phương pháp nêu trên. Nếu số bao bì lớn hơn 3, số mẫu cần lấy sẽ là ![]() , làm tròn số đến đơn vị số nguyên gần nhất.
, làm tròn số đến đơn vị số nguyên gần nhất.
Mẫu cần được phân tích ngay để tránh hóa chất bảo vệ thực vật có thể bị phân hủy. Nếu như không tiến hành phân tích ngay được, mẫu phải được bảo quản kín trong bao bì loại thích hợp dùng để đựng thực phẩm, ở nhiệt độ dưới 0 °C và tránh ánh sáng.
Thuốc thử
Tất cả các thuốc thử và dung môi không được nhiễm hóa chất bảo vệ thực vật. Nếu có thể, thường phải sử dụng các dung môi loại chất lượng đặc biệt, nếu không, các dung môi sử dụng cần phải mới được cất lại trong dụng cụ hoàn toàn bằng thủy tinh. Trong bất cứ trường hợp nào, cũng cần phải thực hiện kiểm tra mẫu trắng.
Dụng cụ
Rửa sạch dụng cụ, đặc biệt đối với dụng cụ thủy tinh để đảm bảo chúng sạch không có hóa chất bảo vệ thực vật, ví dụ ngâm trong dung dịch xà phòng không có phosphat ít nhất 16 h, rửa bằng nước cất nhiều lần đến sạch, tráng rửa bằng aceton và hexan hoặc heptan.
Phân tích định tính và định lượng hóa chất bảo vệ thực vật tồn dư
Quy trình phân tích áp dụng phải được kiểm định và chuẩn hóa theo quy chế hiện hành, đặc biệt, nó phải đáp ứng các yêu cầu dưới đây:
(a) Phương pháp lựa chọn, đặc biệt các bước xử lý làm sạch chất phân tích phải thích hợp đối với chất cần phân tích và không bị ảnh hưởng bởi các chất khác chiết cùng; giới hạn phát hiện và giới hạn định lượng được xác định đối với mỗi hóa chất bảo vệ thực vật và dựa trên nền của mẫu phân tích.
(b) Tỷ lệ thu hồi đối với mỗi hóa chất bảo vệ thực vật phải nằm trong khoảng 70 % đến 110 %.
(c) Độ lặp lại của phương pháp không được thấp hơn các giá trị ghi trong Bảng 12.17.2.
(d) Độ tái lặp của phương pháp không được thấp hơn các giá trị ghi trong Bảng 12.17.2.
(e) Nồng độ của các dung dịch thử, dung dịch đối chiếu và các thông số phân tích cài đặt trên máy phải tương ứng với đáp ứng tuyến tính của detector phân tích.
Bảng 12.17.2
| Nồng độ chất phân tích | Độ lặp lại | Độ tái lặp |
| 0,001 - 0,010 | 30 | 60 |
| > 0,010 - 0,100 | 20 | 60 |
| > 0,100 - 1,000 | 15 | 30 |
| > 1,000 | 10 | 20 |
Ghi chú:
Độ lặp lại (Repeatability): Độ chụm trong điều kiện các kết quả thử nghiệm độc lập với cùng một phương pháp, trên những mẫu giống hệt nhau, trong cùng phòng thí nghiệm, bởi cùng người thao tác, sử dụng cùng một thiết bị, trong khoảng thời gian ngắn.
Độ tái lặp (Reproducibility): Độ chụm trong điều kiện các kết quả thử nghiệm nhận được bởi cùng một phương pháp, trên các mẫu thử giống hệt nhau trong các phòng thí nghiệm khác nhau, với những người thao tác khác nhau, sử dụng thiết bị khác nhau.
Phương pháp phân tích dưới đây nhằm cung cấp thông tin để tham khảo
Xác định dư lượng hóa chất bảo vệ thực vật nhóm clor hữu cơ, nhóm phosphor hữu cơ và nhóm pyrethroid
Phương pháp dưới đây có thể được sử dụng. Tùy theo chất cần phân tích, nếu cần thiết có thể thay đổi quy trình phân tích mô tả dưới đây. Trong mọi trường hợp, đều có thể sử dụng bổ sung một loại cột khác có độ phân cực khác hoặc phương pháp phát hiện khác (ví dụ detector khối phổ) hoặc phương pháp phân tích khác (ví dụ phương pháp hóa miễn dịch) để khẳng định kết quả thu được.
1. Chiết
Thêm 100 ml aceton (TT) vào 10 g mẫu thử đã được làm thành bột thô và để yên 20 min. Thêm 1 ml dung dịch có chứa carbophenothion 1,8 µg/ml trong toluen (TT). Làm đồng nhất trong 3 min, sử dụng máy khuấy trộn tốc độ cao. Lọc và rửa cốc hai lần, mỗi lần với 25 ml aceton (TT). Gộp dịch chiết và dịch rửa, làm bay hơi dung môi ở nhiệt độ không quá 40 °C trong máy cất quay cho tới khi dung môi bị loại hầu như hoàn toàn. Thêm vào cắn vài mililít toluen (TT), tiếp tục làm bay hơi đến khi dung môi bị loại bỏ hầu như hoàn toàn. Hòa tan cắn trong 8 ml toluen (TT). Lọc qua màng lọc 45 µm, rửa bình, dụng cụ lọc và làm vừa đủ 10 ml bằng toluen (TT) (dung dịch A).
2. Làm sạch
2.1. Hóa chất bảo vệ thực vật nhóm clor hữu cơ, nhóm phosphor hữu cơ và nhóm pyrethroid
Tiến hành phương pháp sắc ký rây phân tử, Phụ lục 5.5.
Điều kiện sắc ký:
Cột thép không gỉ (0,30 m x 7,8 mm), pha tĩnh copolymer styren - divinylbenzen (5 µm) (TT), sử dụng toluen (TT) làm pha động với tốc độ dòng 1 ml/min.
Hiệu năng của cột: Tiêm 100 µl dung dịch có chứa đỏ methyl 0,05 % (TT) và oracet lam 2 R 0,05 % (TT) trong toluen (TT) vào hệ thống sắc ký. Cột chỉ thích hợp khi màu của dịch rửa giải thay đổi từ màu cam sang màu xanh da trời ở thể tích rửa giải 10,3 ml. Nếu cần chuẩn hóa cột, có thể sử dụng dung dịch pha trong toluen có chứa chất phân tích ở nồng độ thích hợp là chất có phân tử lượng thấp nhất (ví dụ diclorvos) và chất có phân tử lượng cao nhất (ví dụ deltamethrin). Xác định đoạn dung môi rửa giải có chứa cả hai chất.
Làm sạch dung dịch thử: Tiêm một thể tích xác định dung dịch A (100 µl đến 500 µI) vào hệ thống và tiến hành chạy sắc ký. Thu đoạn dung môi rửa giải đã xác định ở trên (dung dịch B). Các chất nhóm phosphor hữu cơ thường được rửa giải ở đoạn từ 8,8 ml đến 10,9 ml. Các chất nhóm clor hữu cơ và nhóm pyrethroid thường được rửa giải ở đoạn 8,5 ml đến 10,3 ml.
2.2. Hóa chất bảo vệ thực vật nhóm clor hữu cơ và nhóm pyrethroid
Cho một ít bông không có chất béo vào cột sắc ký (0,10 m x 5 mm), sau đó cho 0,5 g silica gel đã được xử lý vào cột. Xử lý silica gel như sau: Sấy silica gel dùng cho sắc ký trong tủ sấy ở 150 °C trong 4 h. Để nguội và thêm vào đó từng giọt một lượng nước tương ứng với 1,5 % khối lượng silica gel đem sấy. Lắc mạnh cho đến khi không còn các cục vón, tiếp tục lắc trên máy lắc cơ học trong 2 h. Luyện cột bằng 1,5 ml hexan (TT). Cột loại nhồi sẵn có chứa 500 mg silica gel loại thích hợp có thể được sử dụng, miễn là loại cột này đã được kiểm tra và chuẩn hóa trước.
Đuổi dung môi trong dung dịch B dưới luồng khí heli (TT) hoặc khí nitrogen không có oxy (TT) tới khi dung môi hầu như bị loại hoàn toàn, hòa tan cắn trong một thể tích toluen (TT) thích hợp (từ 200 µl đến 1 ml tùy theo thể tích dung dịch A đã tiêm ở giai đoạn thực hiện sắc ký rây phân tử).
Chuyển toàn bộ dung dịch lên cột và tiến hành sắc ký, sử dụng 1,8 ml toluen (TT) làm pha động. Thu hồi dịch rửa giải (dung dịch C).
3. Định lượng
3.1. Hóa chất bảo vệ thực vật nhóm phosphor hữu cơ
Tiến hành phân tích sắc ký khí (Phụ lục 5.2), sử dụng carbophenothion (TT) làm nội chuẩn. Nếu cần thiết, có thể sử dụng một chất nội chuẩn thứ hai để phân biệt khi có ảnh hưởng với pic của carbophenothion.
Dung dịch thử: Làm bay hơi dung dịch B dưới luồng khí heli (TT) gần tới khô hoàn toàn và hòa tan cắn trong 100 µl toluen (TT).
Dung dịch đối chiếu: Pha ít nhất 3 dung dịch có chứa các hóa chất bảo vệ thực vật cần xác định và carbo-phenothion ở nồng độ thích hợp để thiết lập đường chuẩn.
Điều kiện sắc ký:
Cột: Cột silica nung chảy (30 m x 0,32 mm) phủ lớp pha tĩnh poly(dimethyl)siloxan (TT) dày 0,25 µm. Khí mang hydrogen (TT), các khí mang khác như heli (TT) hoặc nitrogen (TT) cũng có thể sử dụng được, miễn là hệ thống sắc ký được chuẩn hóa một cách phù hợp.
Detector ion hóa ngọn lửa phosphor - nitrogen hoặc detector quang kế ngọn lửa.
Chương trình nhiệt độ buồng cột: Duy trì nhiệt độ 80 °C trong 1 min, tăng 30 °C/min tới 150 °C, duy trì trong 3 min, tăng 4 °C/min tới 280 °C và duy trì trong 1 min. Nhiệt độ cổng tiêm 250 °C, nhiệt độ detector 275 °C.
Thể tích tiêm: Tiêm một thể tích xác định các dung dịch, sắc ký đồ thu được với điều kiện mô tả trên sẽ cho kết quả thời gian lưu tương đối xấp xỉ các giá trị liệt kê ở Bảng 12.17.3.
Tính kết quả dựa vào diện tích pic và nồng độ của các dung dịch.
3.2. Hóa chất bảo vệ thực vật nhóm clor hữu cơ và pyrethroid
Tiến hành phân tích sắc ký khí (Phụ lục 5.2), sử dụng carbophenothion (TT) làm nội chuẩn. Nếu cần thiết, có thể sử dụng một chất nội chuẩn thứ hai để phân biệt khi có ảnh hưởng với pic của carbophenothion.
Dung dịch thử:
Làm bay hơi dung dịch C dưới luồng khí heli (TT) hoặc khí nitrogen không có oxy (TT) gần tới khô hoàn toàn và hòa tan cắn trong 500 µl toluen (TT).
Dung dịch đối chiếu:
Pha ít nhất 3 dung dịch có chứa các hóa chất bảo vệ thực vật cần xác định và carbophenothion ở nồng độ thích hợp để thiết lập đường chuẩn.
Điều kiện sắc ký:
Cột silica nung chảy (30 m x 0,32 mm) phủ lớp pha tĩnh poly(dimethyl)(diphenyl) siloxan (TT) dày 0,25 µm. Khí mang hydrogen (TT), các khí mang khác như heli (TT) hoặc nitrogen (TT) cũng có thể sử dụng được miễn là hệ thống sắc ký được chuẩn hóa một cách phù hợp.
Detector cộng kết điện tử.
Chương trình nhiệt độ buồng cột: Duy trì nhiệt độ 80 °C trong 1 min, tăng 30 °C/min tới 150 °C, duy trì trong 3 min, tăng 4 °C/min tới 280 °C và duy trì trong 1 min. Nhiệt độ cổng tiêm 250 °C, nhiệt độ detector 275 °C.
Thể tích tiêm: Tiêm một thể tích xác định các dung dịch, sắc ký đồ thu được với điều kiện mô tả trên sẽ cho kết quả thời gian lưu tương đối xấp xỉ các giá trị liệt kê ở Bảng 12.17.4.
Tính kết quả dựa vào diện tích pic và nồng độ các dung dịch.
Bảng -12.17.3
| STT | Hợp chất | Thời gian lưu tương đối |
| 1 | Diclorvos | 0,20 |
| 2 | Fonofos | 0,50 |
| 3 | Diazinon | 0,52 |
| 4 | Parathion - methyl | 0,59 |
| 5 | Clorpyrifos - methyl | 0,60 |
| 6 | Pirimiphos - methyl | 0,66 |
| 7 | Malathion | 0,67 |
| 8 | Parathion | 0,69 |
| 9 | Clorpyrifos | 0,70 |
| 10 | Methidathion | 0,78 |
| 11 | Ethion | 0,96 |
| 12 | Carbophenothion | 1,00 |
| 13 | Azinphos - methyl | 1,17 |
| 14 | Phosalon | 1,18 |
Bảng 12.17.4
| STT | Hợp chất | Thời gian lưu tương đối |
| 1 | α - Hexaclorocylohexan | 0,44 |
| 2 | Hexaclorbenzen | 0,45 |
| 3 | β - Hexaclorocylohexan | 0,49 |
| 4 | Lindan | 0,49 |
| 5 | δ - Hexaclorocylohexan | 0,54 |
| 6 | ε - Hexaclorocylohexan | 0,56 |
| 7 | Heptaclor | 0,61 |
| 8 | Aldrin | 0,68 |
| 9 | cis - Heptaclor epoxid | 0,76 |
| 10 | o,p’- DDE | 0,81 |
| 11 | α - Endosulfan | 0,82 |
| 12 | Dieldrin | 0,87 |
| 13 | p,p’- DDE | 0,87 |
| 14 | o,p’- DDD | 0,89 |
| 15 | Endrin | 0,91 |
| 16 | β - Endosulfan | 0,92 |
| 17 | o,p’- DDT | 0,95 |
| 18 | Carbophenothion | 1,00 |
| 19 | p,p’- DDT | 1,02 |
| 20 | cis - Permethrin | 1,29 |
| 21 | trans - Permethrin | 1,31 |
| 22 | Cypermethrin | 1,40 |
| 23 | Fenvalerat | 1,47; 1,49 |
| 24 | Deltamethrin | 1,54 |
12.18 ĐỊNH TÍNH DƯỢC LIỆU VÀ CÁC CHẾ PHẨM BẰNG KÍNH HIỂN VI
Định tính bằng kính hiển vi là phương pháp sử dụng kính hiển vi để nhận biết các đặc điểm của các mô, tế bào hoặc các chất chứa trong tế bào, ở các lát cắt, bột, các mô đã được làm rã ra, hoặc các tiêu bản bề mặt của dược liệu và các chế phẩm. Việc chọn mẫu đại diện để định tính và sự chuẩn bị tiêu bản phải phù hợp với những yêu cầu về định tính của mỗi loại mẫu cần kiểm tra. Các tiêu bản của các chế phẩm được chuẩn bị sau những xử lý thích hợp tùy theo các dạng chế phẩm khác nhau.
Tiêu bản dược liệu
Lát cắt ngang hoặc dọc
Chọn lấy một mẫu dược liệu thích hợp sao cho có đủ các đặc điểm thực vật như sau:
Thân và rễ nhỏ: Lấy một đoạn có đủ mặt cắt ngang.
Thân, rễ to và rễ củ: Lấy một phần có mặt cắt ngang hình quạt (có cấu tạo từ biểu bì đến tâm).
Vỏ thân: Lấy một phần cắt ngang hình chữ nhật (có cấu tạo từ bần đến tầng phát sinh libe-gỗ).
Lá: Lấy một đoạn gân giữa có dính mỗi bên một ít phiến lá.
Hoa: Bóc biểu bì hay cắt ngang từng bộ phận.
Quả và hạt nhỏ: Lấy nguyên cả quả và hạt.
Quả và hạt to: Lấy một phần, chọn vị trí cắt có đủ các đặc điểm.
Dùng lưỡi dao cạo, ống cắt vi phẫu cầm tay hay máy cắt vi phẫu cắt mẫu dược liệu thành từng lát mỏng 10 µm đến 20 µm. Cũng có thể kẹp mẫu trong parafin rắn, củ khoai lang, củ đậu v.v... để cắt. Trừ chỉ dẫn trong chuyên luận riêng, lát cắt có thể được soi ngay hoặc phải qua các bước xử lý sau đây trước khi đem soi kính:
Ngâm các lát cắt vào dung dịch cloramin T 5% (TT) đến khi lát cắt trắng ra thì rửa sạch cloramin bằng nước.
Ngâm lát cắt vào thuốc thử cloral hydrat (TT) khoảng 10 min rồi rửa sạch bằng nước cất.
Ngâm lát cắt trong dung dịch acid acetic 1 % (TT) trong khoảng 2 min rồi rửa thật kỹ lại bằng nước cất.
Ngâm lát cắt trong dung dịch lục iod (TT) hoặc xanh methylen (TT) trong khoảng từ 1 s đến 5 s, rửa nhanh bằng ethanol 60 % (TT) rồi rửa lại bằng nước cất.
Ngâm lát cắt trong dung dịch cairmn 40 (TT) tới khi thấy màu bắt rõ thì rửa bằng nước cất.
Tiêu bản bột
Lấy một lượng nhỏ bột (qua rây có kích thước mắt rây khoảng 250 µm), cho vào một giọt dung dịch soi (như nước, glycerin, cloral hydrat hay thuốc thử khác tùy thuộc vào mục đích soi) đã có sẵn trên lam kính, dùng kim mũi mác dàn đều cho bột thấm dung dịch, đậy lam kính, di nhẹ lam kính rồi quan sát dưới kính.
Tiêu bản bề mặt
Làm ẩm và làm mềm mẫu (nếu cần thiết), cắt lấy 2 mẩu nhỏ khoảng 4 mm2 (một mẩu để soi và một mẩu để đối chiếu) hoặc tước lấy một đoạn biểu bì, đặt lên một lam kính, thêm thuốc thử thích hợp rồi soi kính.
Tiêu bản mô đã làm rã
Nếu mẫu thử là mô mềm hoặc chỉ có ít, lẻ tẻ các mô gỗ thì có thể dùng phương pháp kali hydroxyd. Nếu mẫu thử cứng, có nhiều mô gỗ hoặc các mô gỗ tụ lại thành bó lớn thì dùng phương pháp acid cromic - nitric hay phương pháp kali clorat. Mẫu thử cần được cắt thành các đoạn dài 5 mm, đường kính 2 mm hoặc những mẫu nhỏ dày khoảng 1 mm trước khi làm rã.
a. Phương pháp kali hydroxyd: Cho mẫu thử vào một ống nghiệm, thêm dung dịch kali hydroxyd 5 % (TT) vừa đủ, đun nóng nhẹ (hoặc ngâm nóng trong cách thủy) đến khi ép bằng đũa thủy tinh thì mẫu bị vỡ ra. Gạn bỏ dung dịch kiềm, rửa sạch mẫu bằng nước rồi đặt mẫu lên lam kính. Dùng kim phẫu thuật xé mẫu ra rồi quan sát trong glycerin (TT).
b. Phương pháp acid cromic-nitric: Cho mẫu thử vào một ống nghiệm, thêm vừa đủ thuốc thử acid cromic - nitric (TT), ngâm đến khi ép bằng đũa thủy tinh thì mẫu bị vỡ ra. Gạn bỏ dung dịch acid, rửa bằng nước rồi làm tiếp như phương pháp a.
c. Phương pháp kali clorat: Cho mẫu thử vào trong ống nghiệm, thêm dung dịch acid nitric 50 % (TT) và một ít bột kali clorat (TT), đun nóng đến khi giảm sủi bọt. Thêm một lượng nhỏ kali clorat đúng lúc để duy trì sủi bọt nhẹ cho đến khi mẫu bị vỡ ra khi ép bằng đũa thủy tinh. Gạn bỏ dung dịch acid, rửa sạch mẫu bằng nước rồi làm tiếp như phương pháp a.
Tiêu bản phấn hoa và bào tử
Nghiền phấn hoa, bao phấn, hoa nhỏ hoặc túi bào tử (được làm mềm trong acid acetic băng (TT) bằng đũa thủy tinh và lọc vào một ống ly tâm, ly tâm rồi bỏ phần nước. Thêm vào cắn 1 ml đến 3 ml hỗn hợp mới pha của anhydrid acetic - acid sulfuric (9 : 1), đun nóng 2 min đến 3 min trên cách thủy, ly tâm. Rửa cắn bằng nước 2 lần, thêm 3 giọt đến 4 giọt dung dịch glycerin 50 % (TT) và dung dịch phenol 1 % (TT), làm tiêu bản trong fuchsin - glycerin - gelatin và soi. Có thể dùng cloral hydrat (TT) thay cho fuchsin - glycerin - gelatin để soi.
Tiêu bản của chế phẩm đông dược bao gồm cả thuốc bột
Để định tính các chế phẩm đông dược bao gồm cả thuốc bột, chuẩn bị tiêu bản như đã mô tả ở mục "Tiêu bản bột" ở phần 1. Lượng mẫu lấy như dưới đây được nghiền thành bột và lấy một lượng bột thích hợp, thêm từng giọt thuốc thử theo yêu cầu, khuấy kỹ để làm tách rời các tế bào hay tổ chức bị dính.
Thuốc bột hoặc bột của viên nang: Lấy một lượng bột thích hợp, nghiền nhỏ.
Viên nén: Lấy 2 đến 3 viên;
Hoàn hồ-nước, hoàn hồ, hoàn mật ong (loại bỏ lớp vỏ bao): Lấy vài viên.
Thuốc ngậm: Lấy 1 đến 2 viên.
Với hoàn mật ong có thể cắt thành lát mỏng và lấy một lượng mẫu thích hợp hoặc lấy một lượng cắn nhỏ sau khi loại bỏ mật ong và nước để làm tiêu bản.
Đo tế bào và các thành phần của tế bào
Để đo kích thước tế bào và các thành phần của tế bào dưới kính hiển vi, có thể dùng trắc vi thị kính. Trước hết, lắp trắc vi thị kính vào thị kính, sau đó định cỡ bằng bàn soi trắc vi kế. Để định cỡ, xoay thị kính và di chuyển bàn soi để cho các vạch trên 2 thang chia độ song song với những vạch "O" bên trải của chúng trùng nhau, sau đó tìm những vạch khác ở bên phải. Giá trị (µm) của một vạch trắc vi thị kính có thể được tính toán trên cơ sở những vạch chia của 2 thang chia độ giữa những dòng trùng nhau. Để đo mẫu, đem nhân số vạch đo được của trắc vi thị kính với giá trị (µm) của mỗi vạch. Nói chung, phép đo được thực hiện dưới một vật kính có độ phóng đại lớn, tuy nhiên, để đo những kích thước lớn hơn của sợi, lông v.v... thì nên dùng vật kính có độ phóng đại nhỏ. Ghi lại các giá trị cực đại và cực tiểu (µm). Cho phép có một vài giá trị cao hơn hay thấp hơn chút ít so với yêu cầu quy định trong chuyên luận dược điển.
Phát hiện màng tế bào
Màng tế bào hóa gỗ: Thêm 1 giọt đến 2 giọt thuốc thử phloroglucinol (TT), để yên một lúc, thêm 1 giọt acid hydrocloric (TT), vùng hóa gỗ xuất hiện màu đỏ tía.
Màng tế bào hóa bẩn hay hóa cutin: Thêm thuốc thử Sudan III (TT), để yên một lúc hay làm ấm nhẹ, xuất hiện màu đỏ cam hay màu đỏ.
Màng celulose: Thêm thuốc thử kẽm clorid-iod, hoặc lúc đầu thêm dung dịch iod 1 % (TT) để làm ẩm, để yên 1 min rồi thêm dung dịch acid sulfuric [33 ml acid sulfuric (TT) pha với nước cất vừa đủ 50 ml], sẽ xuất hiện màu xanh lam hay xanh tím.
Màng tế bào silic: Không có sự thay đổi gì khi thêm acid sulfuric (TT).
Phát hiện các thành phần của tế bào
Tinh bột: Thêm dung dịch iod (TT), sẽ xuất hiện màu xanh lam hay xanh tím.
Đặt mẫu trong dung dịch glycerin-acid acetic, quan sát dưới kính hiển vi phân cực thấy hiện tượng phân cực chéo của các hạt tinh bột, hiện tượng này mất đi ở những hạt tinh bột bị hóa hồ.
Hạt aleuron: Thêm dung dịch iod (TT), sẽ xuất hiện màu nâu hay màu vàng nâu.
Thêm dung dịch thủy ngân (II) nitrat (TT), xuất hiện màu đỏ gạch. Nếu dược liệu có dầu béo, phải loại dầu béo bằng ether (TT) hay ether dầu hỏa (TT) trước khi thử.
Dầu béo, tinh dầu hay nhựa:
Thêm dung dịch Sudan III (TT), sẽ xuất hiện màu đỏ da cam, màu đỏ hoặc đỏ tía.
Trong ethanol 90 % (TT), dầu béo không hòa tan (trừ dầu thầu dầu và dầu bông), trong khi đó tinh dầu thì hòa tan được.
Inulin: Thêm dung dịch 1-naphthol 10 % trong ethanol (TT), sau đó thêm acid sulfuric (TT), các tinh thể inulin chuyển sang màu đỏ tía và hòa tan nhanh.
Tinh thể calci oxalat:
Không hòa tan trong dung dịch acid acetic loãng (TT), tan trong dung dịch acid hydrocloric loãng (TT), không sủi bọt.
Hòa tan chậm trong dung dịch acid sulfuric 50 % (TT), nhưng sau khi để yên một lúc thì tách ra những tinh thể calci sulfat.
Calci carbonat: Hòa tan trong dung dịch acid hydro-cloric loãng (TT) và có sủi bọt.
Silic: Không hòa tan trong acid sulfuric (TT).
Các thuốc thử dùng cho phân tích vi thể
Dung dịch sắt (III) clorid: Hòa tan 1 g sắt (III) clorid (TT) trong nước và thêm nước vừa đủ 100 ml.
Thuốc thử cloral hydrat: Hòa tan 50 g cloral hydrat (TT) trong 15 ml nước và 10 ml glycerin (TT).
Dung dịch glycerin - acid acetic: Hòa lẫn các thể tích bằng nhau của glycerin (TT), acid acetic băng (TT) và nước.
Dung dịch Sudan III: Hòa tan 0,01 g Sudan III (TT) trong 5 ml ethanol 90 % (TT), thêm 5 ml glycerin (TT), lắc kỹ. Bảo quản trong lọ thủy tinh màu nâu và sử dụng trong thời gian 2 tháng.
Dung dịch đỏ rutheni: Cho một lượng vừa đủ đỏ rutheni (TT) vào 1 ml đến 2 ml dung dịch natri acetat 10 % (TT) để tạo thành màu đỏ vang. Dung dịch chỉ pha khi dùng.
Dung dịch phloroglucinol: Hòa tan 1 g phloroglucinol (TT) trong 100 ml ethanol 90 % (TT), lọc. Bảo quản trong lọ thủy tinh màu nâu, tránh ánh nắng.
Thuốc thử acid cromic - nitric:
Hòa tan 10 ml acid nitric (TT) trong 100 ml nước.
Hòa tan 10 g acid cromic (TT) trong 100 ml nước. Trộn lẫn các thể tích bằng nhau của 2 dung dịch trên trước khi dùng.
Fuchsin-glycerin-gelatin: Lấy 1,0 g gelatin (TT), thêm 6 ml nước, ngâm. Thêm 7 ml glycerin (TT), đun nóng và khuấy nhẹ để được hỗn hợp đồng nhất. Lọc qua gạc vào một hộp lồng, thêm một lượng vừa đủ fuchsin base [lấy 0,1 g fuchsin base (TT), thêm 600 ml ethanol (TT) và 80 ml dầu long não để hòa tan], trộn đều và để cho rắn lại.
Dung dịch 1 - naphthol: Lấy 10,5 ml dung dịch 1 - naphthol 15 % trong ethanol (TT), thêm chậm 6,5 ml acid sulfuric (TT), trộn đều, thêm 40,5 ml ethanol (TT) và 4 ml nước. Trộn đều.
Dung dịch thủy ngân (II) nitrat: Lấy 4,5 g thủy ngân (TT), thêm 3 ml acid nitric bốc khói (TT), trộn đều. Bảo quản trong lọ thủy tinh màu nâu, tránh ánh sáng.
Dung dịch kẽm clorid - iod: Lấy 8 g kali iodid (TT), thêm 8,5 ml nước để hòa tan. Sau đó thêm 2,5 g kẽm clorid khan (TT), hòa tan rồi thêm iod (TT) cho đến bão hòa. Bảo quản trong lọ thủy tinh màu nâu có nút mài.
Dung dịch lục iod: Hòa tan 0,3 g lục iod (TT) trong 100 ml nước, thêm 10 ml ethanol 90 % (TT), trộn đều.
Dung dịch carmin 40: Hòa tan 1 g carmin 40 (TT), 5 g kali sulfat (TT) và 0,5 g phenol (TT) trong 100 ml nước.
12.19 XÁC ĐỊNH CHỈ SỐ TRƯƠNG NỞ
Chỉ số trương nở là thể tích (ml) chiếm giữ của 1 g dược liệu, sau khi để trương nở trong nước hoặc một dung môi khác theo quy định trong 4 h, gồm tất cả các chất nhầy bám dính.
Cân chính xác khoảng 1 g dược liệu, để nguyên hoặc nghiền nhỏ ở mức độ thích hợp theo chỉ dẫn trong chuyên luận, cho vào 1 ống nghiệm thủy tinh 25 ml có nút mài, chiều cao 120 mm đến 130 mm, và chia độ 0,5 ml. Trừ những chỉ dẫn khác, làm ẩm dược liệu với 1,0 ml ethanol 96 % (TT), thêm 25 ml nước và đậy nút. Lắc kỹ 10 min một lần trong 1 h đầu, sau đó để yên 3 h. Sau khi bắt đầu thử nghiệm 1,5 h, loại bỏ những thể tích tự do của phần chất lỏng còn lại trong lớp dược liệu và những phần dược liệu nổi lên bề mặt của chất lỏng bằng cách quay ống nghiệm theo trục thẳng đứng.
Đo thể tích chiếm giữ của dược liệu bao gồm toàn bộ các chất nhầy bám dính. Mỗi mẫu tiến hành 3 lần thử nghiệm. Tính chỉ số trương nở từ kết quả trung bình của 3 lần thử nghiệm.
12.20 PHƯƠNG PHÁP CHẾ BIẾN ĐÔNG DƯỢC
Trong đông y, các vị thuốc được dùng để uống thường qua khâu chế biến như sau:
Sơ chế (Chế biến sơ bộ): Nhằm loại bỏ các bộ phận không dùng làm thuốc (Rễ con, lõi, gốc, hạch...) hoặc để ổn định dược liệu ngay từ đầu (phơi, sấy, xông diêm sinh...). Như vậy qua sơ chế, ta có nguyên liệu ban đầu được gọi là “thuốc sống”, tuy nhiên phải đáp ứng được các yêu cầu nhất định về chất lượng đã đề ra.
Phức chế (Chế biến hoàn chỉnh): Quá trình chế biến phức tạp hơn, nhằm giảm bớt độc tính, tác dụng không mong muốn hoặc thay đổi tính năng, tăng sự quy trình thuốc, nhiều khi còn ảnh hưởng đến cấu trúc của hoạt chất và tác dụng của vị thuốc đem chế. Như vậy qua phức chế ta thu được nguyên liệu mang ý nghĩa dược dụng và gọi là “thuốc chín”, đáp ứng được các yêu cầu trong điều trị.
Phương pháp dùng nước
Rửa: Nhằm làm sạch dược liệu, loại bỏ tạp chất hoặc có thể để khử mùi hôi tanh của một số dược liệu (dùng nước sắc của một số dược liệu có tinh dầu). Nước dùng để rửa phải đạt tiêu chuẩn nước sạch, thời gian rửa phụ thuộc vào tính chất của các dược liệu. Dược liệu có cấu tạo mỏng manh hoặc có thành phần dễ tan trong nước, hoặc có các chất nhầy cần rửa nhanh. Dược liệu có cấu trúc rắn chắc, có mùi vị khó chịu có thể rửa kỹ. Các dược liệu dễ bị nát thì không rửa.
Ngâm: Nhằm làm mềm dược liệu, giúp cho việc bào thái dễ dàng. Ngâm để loại muối bám vào dược liệu. Ngâm để giảm tính độc, ngâm để loại nhớt, ngứa. Nước để ngâm phải đạt tiêu chuẩn nước sạch, hoặc các dung dịch muối vô cơ như: Natri clorid, magnesi clorid, natri sulfat, calci hydroxyd, magnesi sulfat, phèn chua; hoặc nước sắc dược liệu như: Nước bồ kết, nước cam thảo, nước gạo, nước gừng... Thời gian ngâm phụ thuộc vào bản chất, kích thước dược liệu và nhiệt độ của mùa, mùa hè ngâm nhanh hơn mùa đông. Dụng cụ để ngâm dược liệu là thạp sành, bể xi măng, chậu nhựa, thùng nhựa, không dùng các dụng cụ bằng kim loại như tôn, sắt... Quá trình ngâm phải thường xuyên quấy đảo và thay nước nhiều lần.
Tẩm: Đem một phụ liệu nào đó tẩm vào dược liệu, rồi phơi khô hoặc phơi âm can đến khô.
Ủ: Nhằm làm mềm, giúp cho việc bào thái dễ dàng; để tạo dáng ngay sau thu hái; để tạo điều kiện lên men cho một số dược liệu khi chế biến, ủ để làm thơm dược liệu, ủ để phụ liệu chế ngấm vào dược liệu. Thời gian ủ phụ thuộc vào mục đích đối với từng loại dược liệu, nói chung nằm trong khoảng 30 min đến 2 h hoặc lâu hơn (12 h đến 24 h). Trong quá trình ủ, có thể dùng vải sạch hoặc bao tải sạch thấm ẩm đậy lên mặt dụng cụ đựng thuốc.
Thủy phi: Là phương pháp tán, nghiền dược liệu trong nước với mục đích thu được bột mịn, tránh nhiệt tạo ra khi tán làm hỏng thuốc, tránh làm dược liệu bị phân hủy, sinh chất độc khi nghiền tán trực tiếp. Cho dược liệu vào dụng cụ tán là sành, sứ..., không dùng dụng cụ bằng kim loại như sắt, đồng, tôn..., đổ nước ngập dược liệu, thường lượng nước gấp 10 lần lượng thuốc. Sau khi tán kỹ, hớt bỏ bụi rác bên trên, gạn lấy lớp nước bột huyền phù sang một dụng cụ khác, phần còn lại tiếp tục làm như thế nhiều lần. Gộp tất cả nước bột, để lắng, gạn bỏ lớp nước, lấy cắn đem phơi âm can hoặc sấy nhẹ đến khô. Dược liệu thường chế theo cách này như Thần sa, Chu sa, Thạch quyết minh...
Phương pháp dùng lửa (Hỏa chế)
Phương pháp sao
Dược liệu đem sao cần có sự phân chia đến kích thước thích hợp. Nếu là thuốc phiến thường có kích thước dài 3 cm đến 5 cm, dày 1 mm đến 3 mm, đồng thời phân ra to nhỏ khác nhau để khi sao đạt được sự đồng đều của thuốc và tránh cháy.
Sao không qua chất trung gian, không cho thêm phụ liệu (Thanh sao)
Vi sao: Nhằm diệt men, mốc hoặc làm khô và dậy mùi thơm đối với dược liệu có cấu tạo mỏng manh (hoa, lá...), hoặc dược liệu chứa các chất thơm, tinh dầu... Nhiệt độ sao nằm trong khoảng 50 °C đến 80 °C, sau khi làm nóng chảo, cho dược liệu vào, đảo đều đến khô hoặc có mùi thơm.
Sao vàng: Nhằm giảm bớt tính hàn, làm cho thuốc trở lên thơm, làm tăng quy kinh tỳ. Sau khi làm nóng chảo, cho dược liệu vào đảo đều, đến khi mặt ngoài của dược liệu chuyển màu vàng, nhiệt độ sao khoảng 100 °C đến 150 °C.
Sao vàng hạ thổ: Nhằm giảm bớt tính hàn của thuốc, đồng thời giảm bớt “hỏa độc” của thuốc, mặt khác làm giảm mùi khó chịu của thuốc. Sau khi sao thuốc tới vàng, nhân lúc còn nóng lấy ra đổ úp thuốc xuống nền đất đã quét sạch, đôi khi còn đào một hố sâu 10 cm, rộng 30 cm. Dụng cụ sao úp trên thuốc. Thời gian hạ thổ thường từ 10 min đến 15 min.
Sao vàng xém cạnh: Nhằm khử các mùi vị khó chịu của thuốc, tăng tác dụng tiêu thực của vị thuốc. Sau khi nóng chảo, cho dược liệu vào, đảo đều cho đến khi mặt ngoài xém cạnh là vừa, màu ruột thuốc vẫn giữ nguyên. Nhiệt độ sao vào khoảng 170 °C đến 200 °C. Thường tiến hành với các vị thuốc chua, chát hoặc tanh lợm như Binh lang, Thần khúc, Hòe giác....
Sao tồn tính (hắc sao): Nhằm tăng tác dụng tiêu thực, chỉ huyết của vị thuốc hoặc làm thay đổi tính năng của thuốc. Sau khi chảo nóng già, cho thuốc vào, đảo đều cho đến khi toàn bộ mặt ngoài bị đen đi, khi bỏ ra bên trong vẫn còn màu vàng. Nhiệt độ sao khoảng 200 °C.
Sao cháy (thán sao): Nhằm tăng tác dụng chỉ huyết của vị thuốc. Sau khi chảo nóng già, cho thuốc vào, đảo đều đến khi toàn bộ mặt ngoài của thuốc cháy đen (có mùi cháy) bên trong có màu nâu. So với sao tồn tính thì mức độ cháy cao hơn. Nhiệt độ sao khoảng 200 °C đến 240 °C. Kể cả sao tồn tính và sao cháy, trước khi đổ thuốc ra thường phun vào một ít nước lạnh để trừ hỏa độc.
Tẩm sao (Chích)
Sau khi tẩm với phụ liệu (nước sắc gừng, giấm...) rồi đem sao, nhằm tăng sự qui kinh, tăng tác dụng của vị thuốc.
Tẩm rượu sao: Dùng sức nóng, tính thăng đề của rượu để dẫn thuốc lên thượng tiêu (tác dụng thăng đề), để giảm bớt tính lạnh của vị thuốc, giảm bớt mùi vị khó chịu, tăng cường bổ can thận cho vị thuốc. Dùng rượu trắng có hàm lượng ethanol 35 % đến 40 %, với lượng 10 % đến 15 % (tt/kl). Sau khi tẩm, ủ thuốc 30 min đến 1 h, đem sao ở nhiệt độ sao vàng, khi có mùi thơm của rượu bốc lên là được.
Tẩm giấm sao (Thố chế): Nhằm tăng dẫn thuốc vào kinh can đởm, tăng cường tác dụng giảm đau, tiêu ứ huyết, giảm bớt mùi tanh của vị thuốc, làm xốp giòn vị thuốc giúp cho việc dễ tán, dễ chiết xuất. Tẩm dược liệu với 10 % đến 15 % giấm ăn (tt/kt), ủ 30 min đến 2 h, rồi sao vàng.
Tẩm nước muối sao (Diêm chế): Nhằm dẫn thuốc vào kinh thận và hạ tiêu. Tẩm dược liệu với 10 % đến 15 % dung dịch muối ăn 5 % (tt/kl). Sau khi tẩm đều, ủ 30 min đến 1 h, rồi sao vàng.
Tẩm mật sao (Mật chế): Dùng mật ong để chế thuốc. Nhằm tăng tác dụng dẫn thuốc vào kinh tỳ, tăng tác dụng bổ khí, nhuận phế và tăng tác dụng hoãn giải. Thường dùng mật ong dưới dạng mật luyện (mật ong đun sôi, lọc bỏ tạp, sáp...cô đến thể tích nhất định), thêm lượng mật được quy định trong từng chuyên luận vào dược liệu, thêm nước sạch (bằng khoảng 30 % lượng mật), tẩm đều vào dược liệu, ủ 1 h đến 6 h. Tiến hành sao nhỏ lửa, cho đến khi sờ không dính tay; dược liệu có màu vàng, mùi thơm mật, vị ngọt. Cũng có thể sao dược liệu tới khi bên ngoài có màu vàng, cho mật vào, đảo đều, tới khi không dính tay.
Tẩm nước gừng sao (Khương chế): Nhằm giảm bớt tính hàn, tăng sức phát tán, tăng khả năng tiêu thực, ôn trung. Tẩm dược liệu với 10 % đến 15 % dịch nước gừng tươi (tt/kl), ủ 30 min đến 1 h, đem sao vàng. Gừng già tươi 100 g, rửa sạch, giã nhỏ, thêm nước sạch, vắt lấy dịch đủ 100 ml.
Tẩm hoàng thổ sao: Nhằm tăng tác dụng quy tỳ vị, giảm bớt tính háo của thuốc. Lấy hoàng thổ (đất sét vàng), đất lòng bếp (Phục long can) hoặc đất lòng lò gạch, nghiền mịn, cứ 100 g đất thêm 1 lít nước đun sôi, quấy đều, gạn bỏ lớp trên, bỏ cặn, lấy lớp giữa. Cứ 1 kg dược liệu tẩm với 4 lít nước hoàng thổ, trộn đều, ủ 2 h đến 3 h. Sao vàng, chà sát rây bỏ hoàng thổ.
Tẩm sữa: Dùng sữa người (Nhũ nhân chế), sữa dê tươi (Dương nhũ chế) hoặc sữa bò tươi. Nhằm tăng tính dưỡng huyết, bổ âm và giảm bớt tính háo của thuốc. Dùng sữa tươi với tỷ lệ 10 % đến 15 % (tt/kl) để tẩm vào dược liệu, ủ 30 min. Vi sao tới thơm.
Tẩm nước vo gạo (Mễ cam chế): Nhằm giảm tính háo của thuốc. Dùng nước vo gạo đặc, mới vo, với tỷ lệ 10 % đến 15 % (tt/kl), tẩm đều, ủ 4 h đến 6 h, sao vàng.
Tẩm đồng tiện: Nhằm tăng tác dụng hoạt huyết, tư âm giáng hỏa. Dùng nước tiểu trẻ em khỏe mạnh, từ 5 đến 12 tuổi, nước tiểu mới đi, bỏ đoạn đầu đoạn cuối, với lượng từ 10 % đến 15 % (tt/kl). Sau khi tẩm trộn đều, ủ 30 min đến 1 h. Sao vàng.
Tẩm nước đậu đen, nước cam thảo: Nhằm giảm bớt tính chát của thuốc (tẩm với nước đậu đen), tăng khả năng giải độc (tẩm với Cam thảo). Lấy nước sắc đậu đen (100 g cho 1 lít dịch sắc), tẩm vào dược liệu với tỷ lệ 10 % đến 20 % (tt/kl). Cam thảo đem tán nhỏ, sắc lấy nước hoặc ngâm 24 h, lấy nước. Tẩm đều vào dược liệu, ủ cho ngấm khoảng 30 min. Sao khô.
Sao qua chất trung gian
Sao cám: Nhằm làm thơm thuốc, tăng quy kinh tỳ (Hoài sơn, Ý dĩ, Thần khúc, Bạch truật); giảm bớt tính háo của thuốc. Thường dùng cám gạo, với tỷ lệ giữa cám và dược liệu là 1/10 (kl/kl). Đầu tiên cho cám vào chảo đã đun nóng già, quấy đều, tới khi cám bốc khói trắng, cho dược liệu vào, đảo đều; sao đến khi bề mặt thuốc có màu vàng hoặc thâm; lấy ra, chà xát, rây bỏ cám.
Sao cách cát: Do nhiệt độ nâng cao, phá vỡ cấu trúc rắn của dược liệu, giúp cho việc bào chế (nghiền, sắc thuốc...) thuận lợi, đồng thời giảm mùi khó chịu, hoặc giảm bớt tính độc của dược liệu. Nhiệt độ sao nằm trong khoảng 250 °C đến 300 °C. Cát phải được làm sạch (đã qua rửa nhiều lần bằng cách cho nước sạch vào, quấy nhiều lần, gạn bỏ lớp nước trên, đổ ra phơi khô, sấy khô), đun nóng già, cho dược liệu vào đảo đều, đến lúc mặt ngoài phồng nứt; hoặc phồng thơm, lấy ra sàng bỏ cát.
Sao cách bột hoạt thạch hay văn cáp (vỏ hàu): Nhằm khử mùi hôi tanh khó chịu của thuốc nhất là các dược liệu có nguồn gốc động vật, đồng thời giúp cho dễ tán các dược liệu có thể chất dẻo dính. Đun nóng già bột trung gian, cho dược liệu vào đảo đều đến khi phồng giòn, xốp, sàng lấy dược liệu.
Nướng (Ổi): Nhằm tăng tính ấm và tăng khả năng dẫn thuốc vào tỳ vị. Thường dùng các phụ liệu có thể chất dẻo dính như bột gạo nếp (thêm nước làm áo bọc thuốc) hoặc giấy bản ẩm hoặc đất sét bọc thuốc, vùi vào tro nóng hay nướng trên bếp than.
Đốt
Nhằm làm sạch các lông, rễ phụ, sau khi phơi khô dược liệu, có thể đốt trực tiếp hoặc cho dược liệu lên các dàn sắt để đốt. Cũng có thể dùng nhiệt của một dụng cụ như dùi sắt đã hơ nóng đỏ, rồi đốt các lông.
Đốt rượu
Nhằm làm thơm vị thuốc, đốt cháy các lông có trên vị thuốc. Thường dùng ethanol 90 % để đốt.
Nung
Làm cho vị thuốc trở nên giòn xốp, có lợi cho việc nghiền tán bột; do ở nhiệt độ cao từ 300 °C đến 700 °C, có thể phá vỡ cấu trúc rắn chắc của thuốc, đôi khi có tác dụng giảm độc tính, hoặc biến tính của dược liệu. Thường tiến hành với các dược liệu có thể chất cứng rắn như Mẫu lệ, Long xỉ, Long cốt; hoặc tăng cường tính thu sáp của các vị thuốc như Phèn chua, Bằng sa...
Nung trực tiếp: Đặt dược liệu tiếp xúc trực tiếp trong lò (nhiệt độ 800 °C đến 1000 °C), nung cho đỏ đều, lấy ra để nguội.
Nung gián tiếp: Cho dược liệu vào nồi nung sạch, đậy nắp, trát kín bằng đất sét ẩm. Nhiệt độ nung thường trong khoảng 150 °C đến 200 °C. Có khi bọc dược liệu vào đất sét, rồi nung.
Sấy
Nhằm làm khô dược liệu, để chế biến, bảo quản trong quá trình phân phối, sử dụng. Tùy theo tính chất của dược liệu để điều chỉnh nhiệt độ sấy và phương pháp sấy cho thích hợp. Với các dược liệu chứa tinh dầu thì nhiệt độ sấy thường ở 50 °C, các dược liệu khác thường sấy ở 80 °C. Các phương pháp sấy thường được sử dụng như sấy khô trong tủ sấy ở áp suất thường, sấy trên bếp hoặc lò than, sấy bằng hồng ngoại....
Phương pháp kết hợp nước-lửa
Chưng
Nhằm thay đổi tính năng, giảm bớt tác dụng phụ giúp cho việc điều trị tốt hơn. Cho dược liệu vào dụng cụ chưng thích hợp như âu sành, inox..., có thể thêm phụ liệu (Sa nhân, Gừng...), đậy kín, đặt dụng cụ chưng này vào một dụng cụ lớn hơn, thêm nước vừa đủ để đun theo thời gian quy định cho mỗi dược liệu. Lấy dược liệu ra phơi hay sấy khô. Có trường hợp phải chưng, phơi nhiều lần (cửu chưng, cửu sái), cho đến lúc dược liệu mềm nhuận, đen bóng.
Đồ
Nhằm diệt men trong dược liệu để tránh thủy phân hoạt chất, để làm mềm cho dễ bào thái. Dược liệu được phân loại to nhỏ và xếp vào một dụng cụ thích hợp (chõ), loại to xếp dưới, loại nhỏ xếp trên. Nếu có phụ liệu thì xếp xen kẽ một lớp thuốc, một lớp phụ liệu, đậy chõ. Bắc chõ lên một nồi đáy, trát kín chỗ tiếp xúc bằng đất sét hay bột nhão của cám và lá khoai, rồi đồ trong khoảng thời gian quy định, đến lúc dược liệu chín mềm.
Nấu
Nhằm làm mềm dược liệu, tăng tác dụng của thuốc, giảm tác dụng không mong muốn, nhiều khi nấu với phụ liệu khác. Cho dược liệu vào dụng cụ nấu có dung tích thích hợp, loại to xếp dưới, loại nhỏ xếp trên để dược liệu chín đều. Đáy dụng cụ có thể lót vỉ để tránh tiếp xúc dược liệu với đáy nồi nấu. Đổ nước sạch hay dịch phụ liệu ngập dược liệu 5 cm đến 10 cm. Đun sôi và duy trì theo thời gian quy định. Trong quá trình đun có thể gạn nước và thêm nước mới nhiều lần. Khi dược liệu chín đều, lấy ra thái mỏng, phơi hoặc sấy khô tới độ ẩm quy định.
Tôi
Nhằm làm giòn dược liệu, giúp cho việc nghiền, tán dễ dàng, đồng thời khử các mùi khó chịu của thuốc. Dược liệu được rang cách cát hoặc nung tới khi đạt yêu cầu, khi đổ ra còn đang nóng thì nhúng ngập nhanh vào nước hay vào một dung dịch phụ liệu như như giấm hoặc nước Hoàng liên... Lấy ra phơi hoặc sấy khô.
Rán dầu
Nhằm làm giảm độc tính của dược liệu (đối với Mã tiền), làm cho dược liệu trở nên giòn, dễ tán nhỏ (đối với Xương báo). Thường rán bằng dầu lạc, dầu vừng. Đun sôi dầu, cho dược liệu vào rán cho đến khi có màu hơi vàng, dược liệu nổi lên, thể chất giòn, xốp. Vớt ra cho nhỏ hết dầu. Lượng dầu so với dược liệu thường có tỷ lệ khoảng 20 % đến 30 % (tt/kl).
PHỤ LỤC 13
(Quy định)
13.1 PHÉP THỬ HISTAMIN
Dùng chuột lang nặng 250 g đến 350 g. Để chuột nhịn đói trong 24 h, cho uống nước theo nhu cầu của chuột. Giết chuột đột ngột bằng một cú đánh mạnh vào đầu chuột.
Mổ bụng chuột, dùng kẹp, kẹp vào manh tràng, nâng lên và kéo ra phía trước, sẽ thấy hồi tràng nối vào manh tràng, cắt lấy một đoạn hồi tràng dài khoảng 2 cm, cho vào khay đựng dung dịch B (nếu làm chậm phải sục khí carbogen). Loại bỏ các chất còn trong đoạn ruột bằng cách dùng một pipet có dung dịch B lồng vào đầu đoạn ruột rồi để cho dung dịch B trong pipet chảy tự nhiên qua đoạn ruột, cần để pipet ở tư thế nghiêng sao cho mặt thoáng của dung dịch B không cao quá 2 cm so với đoạn ruột trong khay.
Thay dung dịch B mới vào khay. Buộc vào mỗi đầu của đoạn ruột một sợi chỉ mảnh và rạch một đường ngang nhỏ ở giữa đoạn ruột.
Cho đoạn ruột vào bình nuôi cơ quan cô lập có dung tích 10 ml đến 20 ml, chứa dung dịch B, và giữ ở nhiệt độ hằng định 34 °C đến 36 °C. Sục một hỗn hợp khí có 95 thể tích oxygen và 5 thể tích carbon dioxyd qua bình nuôi. Một đầu sợi chỉ buộc vào gần đáy bình nuôi, đầu kia nối với đầu ghi của một kimograph hoặc thiết bị thích hợp để ghi sự co bóp của ruột. Nếu dùng bút ghi trên giấy thì điều chỉnh sao cho biên độ có thể khuếch đại lên khoảng 20 lần. Sức căng của ruột nên vào khoảng 9,8 mN và điều chỉnh theo độ nhạy của ruột.
Thay dung dịch B trong bình nuôi cơ quan. Để yên 10 min. Tiếp tục thay dung dịch B 2 đến 3 lần như vậy nữa.
Thêm vào bình nuôi cơ quan, chính xác khoảng 0,2 ml đến 0,5 ml dung dịch histamin dihydroclorid có nồng độ nhất định để gây nên một đáp ứng gần tối đa có thể lặp lại được. Liều này gọi là liều "cao". Thay dung dịch B ở bình nuôi 3 lần trước mỗi lần thêm histamin. Nên thêm histamin vào những khoảng thời gian cách đều để ruột giãn hoàn toàn giữa các lần thêm (khoảng 2 min).
Tương tự, thêm những thể tích bằng nhau của dung dịch có nồng độ histamin thấp để cho đáp ứng có biên độ bằng khoảng một nửa biên độ của liều "cao" và có thể lặp lại được. Liều này gọi là liều "thấp". Tiếp tục thêm đều đặn các liều "cao" và liều "thấp" histamin như đã nêu ở trên và xen kẽ thêm cùng một thể tích dung dịch pha từ mẫu thử. Điều chỉnh độ pha loãng của dung dịch thử để cho sức co của ruột nhỏ hơn sức co của liều "cao".
Kiểm tra xem sức co của dung dịch thử có lặp lại không và xem đáp ứng với liều cao và liều thấp có như trước không.
Tính hoạt lực của chất thử ra microgam histamin base căn cứ vào độ pha loãng đã xác định ở trên. Lượng histamin xác định được không được vượt quá lượng ghi trong chuyên luận.
Nếu chất thử không gây co bóp ruột, pha một dung dịch khác và cho thêm một lượng histamin tối đa dung nạp được ghi trong chuyên luận và thử với dung dịch này. Theo dõi xem sự co cơ của dung dịch này có tương ứng với lượng histamin đã thêm không. Nếu dung dịch này không gây ra sự co cơ tương ứng hoặc sự co của dung dịch thử không lặp lại được, hoặc sau khi thử chất thử rồi, thử lại với histamin chuẩn liều "cao" và liều "thấp" mà đáp ứng có giảm đi thì kết quả thử là không có giá trị và phải thử chất hạ áp của thuốc theo chuyên luận "Phép thử các chất hạ áp, Phụ lục 13.3".
Dung dịch A:
| Natri cloriid | 160 g |
| Kali cloriid | 4,0 g |
| Calci clorid khan | 2,0 g |
| Magnesi clorid khan | 1,0 g |
| Dinatri hydrophosphat | 50 mg |
| Nước cất pha tiêm | vừa đủ 1000 ml |
Dung dịch B:
| Dung dịch A | 50 ml |
| Atropin sulfat | 0,5 mg |
| Natri hydrocarbonat | 1,0 g |
| D - glucose | 0,5 g |
| Nước cất pha tiêm | vừa đủ 1000 ml |
Dung dịch B chỉ pha ngay trước khi dùng và phải dùng trong vòng 24 h.
13.2 PHÉP THỬ NỘI ĐỘC TỐ VI KHUẨN
Phép thử nội độc tố vi khuẩn dùng để phát hiện hoặc định lượng nội độc tố của vi khuẩn gram âm có trong mẫu thử cần kiểm tra. Phương pháp sử dụng thuốc thử lysat là dịch phân giải tế bào dạng amip có trong máu một loài sam biển, Limulus polyphemus hoặc Tachy-pleus tridentatus.
Hiện nay có 3 phương pháp để thực hiện phép thử này: Phương pháp tạo gel dựa trên sự tạo thành gel khi cho thuốc thử vào dung dịch có chứa nội độc tố; phương pháp đo độ đục, dựa vào sự thay đổi độ đục của thuốc thử lysat khi tạo gel; phương pháp đo màu dựa trên sự thay đổi màu của phức hợp màu - peptid.
Tùy theo điều kiện và tính chất của mẫu thử, có thể áp dụng một trong các kỹ thuật thích hợp để thực hiện phép thử. Tuy nhiên, khi có nghi ngờ hoặc tranh chấp, kết luận cuối cùng sẽ dựa vào phương pháp tạo gel trừ khi có chỉ dẫn khác.
Phép thử được thực hiện trong điều kiện tránh nhiễm khuẩn.
Thiết bị và dụng cụ:
Tất cả dụng cụ dùng trong phép thử này đều phải không có chứa nội độc tố.
Với những dụng cụ thủy tinh hoặc bằng chất liệu chịu được nhiệt, loại chất gây sốt bằng cách sấy ở 250 °C trong ít nhất 30 min. Nếu dùng các dụng cụ là chất dẻo, như các đầu hút của micropipet hay các khay lỗ để làm phản ứng, chỉ dùng các loại có chất liệu không ảnh hưởng tới phép thử và không có chứa nội độc tố.
Nước để thử nội độc tố (nước BET): Là nước cất pha tiêm cho kết quả âm tính trong phép thử nội độc tố ở điều kiện quy định. Có thể dùng nước cất 3 lần hoặc một loại nước cất pha tiêm thu được bằng phương pháp thích hợp khác đạt yêu cầu trên.
Nếu không có chỉ dẫn gì khác, dùng nước BET làm dung môi để pha tất cả các dung dịch dùng trong phép thử.
Dung dịch acid hydrocloric 0,1 M BET và natri hydroxyd 0,1 M BET: Là dung dịch được pha từ acid hydrocloric đậm đặc hoặc natri hydroxyd rắn với nước BET trong một dụng cụ không có chứa nội độc tố. Các dung dịch này đạt yêu cầu nếu sau khi điều chỉnh đến pH 6,0 đến 8,0, cho kết quả âm tính trong điều kiện của phép thử.
Nội độc tố chuẩn và dung dịch chuẩn gốc: Nội độc tố chuẩn được sử dụng trong phép thử như một chất đối chiếu. Chuẩn được thiết lập và đánh giá theo nội độc tố chuẩn quốc tế. Hoạt lực của chuẩn được biểu thị theo đơn vị quốc tế quy định bởi Tổ chức Y tế Thế giới (International Unit = IU) hoặc đơn vị nội độc tố (Endotoxin Unit - EU). 1 IU tương đương với 1 EU.
Dung dịch chuẩn gốc nội độc tố được pha theo quy định đóng gói của từng nhà sản xuất. Trên nhãn và tờ hướng dẫn sử dụng kèm theo ghi rõ cách pha, điều kiện bảo quản và số đơn vị nội độc tố có trong dung dịch chuẩn gốc sau khi pha theo hướng dẫn.
Pha dung dịch chuẩn: Pha dung dịch chuẩn gốc với nước BET để được dãy chuẩn có nồng độ thích hợp. Trước khi pha, thường yêu cầu lắc mạnh dung dịch chuẩn gốc trong ít nhất 15 min, và mỗi lần pha loãng, trộn các dung dịch pha loãng ít nhất 30 s. Dùng các dung dịch chuẩn đã pha loãng càng sớm càng tốt để tránh mất hoạt lực.
Dung dịch thử:
Chuẩn bị dung dịch thử bằng cách hòa tan hoặc pha loãng mẫu thử với nước BET đến độ pha loãng thích hợp. Một số chất hoặc chế phẩm có thể cần phải hòa tan hoặc pha loãng trong dung dịch thân nước khác. pH của hỗn hợp phản ứng phải nằm trong khoảng quy định cho mỗi loại thuốc thử, thông thường trong khoảng 6,0 đến 8,0. Do vậy, nên điều chỉnh pH của dung dịch thử cho phù hợp bằng các dung dịch acid hydrocloric 0,1 M BET, natri hydroxyd 0,1 M BET hoặc một đệm thích hợp theo khuyến cáo của nhà sản xuất thuốc thử.
Phép thử không thích hợp với một số loại mẫu thử như các loại nhũ dịch, hỗn dịch... Một số chất có thể cho phản ứng dương tính hoặc âm tính giả với thuốc thử như các chế phẩm từ máu, polynucleotid; chế phẩm có chứa kim loại nặng, chất diện hoạt, nồng độ ion cao. Do vậy, cần phải khảo sát, tìm phương pháp xử lý và đánh giá trước khi áp dụng phép thử cho chế phẩm.
Độ pha loãng tối đa:
Độ pha loãng tối đa (MVD) là hệ số pha loãng lớn nhất cho phép để thu được dung dịch thử có chứa lượng nội độc tố có thể phát hiện được.
MVD được tính như sau:
| MVD = | Giới hạn nội độc tố x nồng độ dung dịch thử |
| λ |
trong đó:
Giới hạn nội độc tố (Endotoxin Limit = EL): Giới hạn nội độc tố thường được quy định trong các chuyên luận riêng. Chế phẩm đạt yêu cầu nếu lượng nội độc tố có trong chế phẩm thấp hơn giới hạn quy định. Giới hạn nội độc tố của các chế phẩm được tính trên cơ sở liều dùng cho người theo công thức sau:
EL = K/M
trong đó:
K là ngưỡng nội độc tố tính cho 1 kg thể trọng người có thể chịu đựng được mà không xảy ra phản ứng độc hại; Giá trị K được tham khảo trong Bảng 13.2.1 dưới đây;
M là liều tối đa của sản phẩm tính cho 1 kg thể trọng dùng dưới dạng đơn liều trong thời gian khoảng 1 h. Liều này được hiểu là lượng thuốc tối đa có thể được dùng trong vòng 1 h. Với các loại thuốc tiêm dưới da, tiêm bắp hoặc tiêm tĩnh mạch một lần sẽ được coi như dùng đơn liều. Với các dịch truyền tĩnh mạch trong thời gian dài, tính M theo lượng thuốc của liều có thể được truyền tối đa trong một giờ, dựa trên tốc độ truyền tiêu chuẩn nếu không có quy định riêng nào cho chế phẩm. Giá trị được dùng là liều tối đa theo khuyến cáo của nhà sản xuất, có thể mức liều đó vượt quá mức liều theo chỉ dẫn của thầy thuốc.
Nếu một sản phẩm có thể được dùng theo cả 2 đường tiêm trong màng cứng và các đường tiêm khác thì giới hạn nội độc tố cho sản phẩm được tính theo giá trị K của đường tiêm trong màng cứng vì có yêu cầu chặt chẽ hơn.
Bảng 13.2.1
| Loại sản phẩm | K (tính cho 1 kg thể trọng) |
| Các loại thuốc tiêm trong màng cứng | 0,2 |
| Các thuốc phóng xạ tiêm tĩnh mạch | 2,5 |
| Tất cả các thuốc tiêm trừ loại tiêm trong màng cứng hoặc thuốc phóng xạ | 5,0 |
Trên thực tế, các nhà sản xuất thường quy định giới hạn nội độc tố cho sản phẩm thấp hơn nhiều so với giới hạn tính theo ngưỡng chịu đựng của người bệnh, để khi dùng liều tối đa sản phẩm vẫn đảm bảo an toàn cho người bệnh.
Nồng độ của dung dịch thử:
Tính bằng mg/ml nếu giới hạn nội độc tố trong chuyên luận riêng quy định theo khối lượng (lU/mg).
Tính bằng đơn vị/ml nếu giới hạn nội độc tố trong chuyên luận riêng quy định theo đơn vị hoạt lực sinh học (IU/ đơn vị).
Tính bằng ml/ml nếu giới hạn nội độc tố trong chuyên luận riêng quy định theo thể tích (lU/ml).
λ là độ nhạy của thuốc thử được ghi trên nhãn (kỹ thuật tạo gel) hoặc nồng độ nội độc tố thấp nhất đã dùng để xây dựng đường cong chuẩn (phương pháp đo độ đục hoặc đo màu).
PHƯƠNG PHÁP TẠO GEL
Phương pháp tạo gel cho phép phát hiện hoặc xác định lượng nội độc tố dựa trên sự tạo gel của thuốc thử lysat với nội độc tố. Nồng độ nội độc tố yêu cầu để tạo gel với thuốc thử lysat trong điều kiện tiêu chuẩn là độ nhạy ghi trên nhãn của thuốc thử. Để đảm bảo độ chính xác và giá trị của phép thử, phải tiến hành phép thử kiểm tra để khẳng định độ nhạy ghi trên nhãn của thuốc thử lysat và kiểm tra các yếu tố ảnh hưởng theo hướng dẫn dưới đây.
Chuẩn bị thử
Kiểm tra độ nhạy của lysat
Độ nhạy của thuốc thử lysat ghi trên nhãn là nồng độ nội độc tố thấp nhất cần thiết để tạo gel với thuốc thử trong điều kiện xác định.
Phép thử kiểm tra độ nhạy được thực hiện mỗi khi có lô thuốc thử mới hoặc khi có sự thay đổi điều kiện thí nghiệm có thể ảnh hưởng tới kết quả của phép thử.
Tiến hành:
Chuẩn bị các dung dịch nội độc tố chuẩn với ít nhất 4 nồng độ tương đương 2 λ; λ; 1/2 λ và 1/4 λ bằng cách pha loãng dung dịch chuẩn gốc với nước BET, mỗi nồng độ cần 4 ống. Trong đó λ là độ nhạy ghi trên nhãn của lysat muốn kiểm tra. Pha thuốc thử lysat với nước BET theo hướng dẫn trên nhãn. Trộn một thể tích thuốc thử với đồng lượng dung dịch chuẩn (thường dùng 0,1 ml) cho mỗi ống. Ủ các ống nghiệm có chứa hỗn hợp phản ứng trong một khoảng thời gian quy định theo khuyến cáo của nhà sản xuất thuốc thử lysat [thường ở (37 ± 1) °C trong (60 ± 2) min], tránh bị rung động mạnh.
Đọc kết quả sau một khoảng thời gian quy định. Phản ứng dương tính nếu gel tạo thành không bị chảy ra khi nhẹ nhàng dốc ngược ống nghiệm (180 °). Phản ứng âm tính khi không tạo thành gel hoặc gel không đủ chắc, rất dễ bị rã khi dốc ngược ống nghiệm.
Phép thử không có giá trị nếu bất kỳ ống nào của dung dịch chuẩn nội độc tố có nồng độ thấp nhất (0,25 λ) cho phản ứng dương tính. Khi đó, cần kiểm tra kỹ lại các điều kiện thử nghiệm và lặp lại thí nghiệm.
“Điểm dừng” là ống cuối cùng có hệ số pha loãng lớn nhất trong dãy cho phản ứng dương tính.
Độ nhạy của thuốc thử được biểu thị là trung bình nhân của nồng độ điểm dừng (lU/ml), được tính như sau: Tính giá trị trung bình của các logarit nồng độ nội độc tố tại điểm dừng, sau đó lấy đối - logarit của giá trị trung bình.
Trung bình nhân nồng độ điểm dừng = anti log (Σe/f).
Trong đó:
Σe là tổng các logarit nồng độ điểm dừng của mỗi dãy đã thử;
f là số dãy chuẩn nội độc tố đã thử
Độ nhạy ghi trên nhãn của thuốc thử được khẳng định là đúng nếu trung bình nhân nồng độ điểm dừng lớn hơn hoặc bằng 0,5 λ và nhỏ hơn hoặc bằng 2,0 λ.
Kiểm tra yếu tố ảnh hưởng
Phép thử được tiến hành để xác định xem trong mẫu thử có chứa các yếu tố làm tăng hay ức chế phản ứng hay không. Khi có bất kỳ sự thay đổi điều kiện nào đó có thể ảnh hưởng đến kết quả thử nghiệm, cần thực hiện phép thử này.
Chuẩn bị các dung dịch A, B, C và D theo như Bảng 13.2.2. Các điều kiện thử nghiệm như nhiệt độ, thời gian ủ và đánh giá kết quả tương tự như trong phần Kiểm tra độ nhạy của lysat
Trung bình nhân nồng độ điểm dừng của dung dịch B và C được xác định theo hướng dẫn đã nêu trong phần Kiểm tra độ nhạy của lysat.
Phép thử có giá trị khi dung dịch A và D cho phản ứng âm tính và kết quả dung dịch C khẳng định đúng độ nhạy của thuốc thử.
Nếu độ nhạy của lysat tính theo dung dịch B lớn hơn 0,5 λ và nhỏ hơn 2,0 λ thì mẫu thử không chứa yếu tố ảnh hưởng và phép kiểm tra yếu tố ảnh hưởng đạt yêu cầu. Nếu không thỏa mãn các yêu cầu trên thì mẫu thử có chứa yếu tố ảnh hưởng tới phép thử.
Bảng 13.2.2 - Kiểm tra yếu tố ảnh hưởng
| Dung dịch | Nồng độ nội độc tố được thêm vào mỗi dung dịch | Dung môi | Hệ số pha loãng | Nồng độ nội độc tố sau khi pha loãng | Số ống nghiệm |
| A | 0/dung dịch thử | - | - | - | 4 |
| B | 2λ/dung dịch thử | dung dịch mẫu thử | 1 | 2 λ | 4 |
| 2 | 1 λ | 4 | |||
| 4 | 0,5 λ | 4 | |||
| 8 | 0,25 λ | 4 | |||
| C | 2λ/nước BET | Nước BET | 1 | 2 λ | 2 |
| 2 | 1 λ | 2 | |||
| 4 | 0,5 λ | 2 | |||
| 8 | 0,25 λ | 2 | |||
| D | 0/nước BET | - | - | - | 2 |
Trong đó:
Dung dịch A (dung dịch thử): Mẫu thử pha ở độ pha loãng ≤ MVD.
Dung dịch B (đối chứng dương tính có mẫu thử): Nội độc tố pha trong dung dịch thử để kiểm tra yếu tố ảnh hưởng.
Dung dịch C (khẳng định độ nhạy của lysat): Chuẩn nội độc tố pha trong nước BET với nồng độ khác nhau.
Dung dịch D (đối chứng âm tính): Nước BET.
Nếu mẫu thử có chứa yếu tố ảnh hưởng tới phép thử ở độ pha loãng nhỏ hơn MVD, làm lại phép kiểm tra yếu tố ảnh hưởng, dùng dung dịch thử có độ pha loãng lớn hơn nhưng không quá MVD.
Sử dụng thuốc thử có độ nhạy cao hơn sẽ cho phép mẫu thử được pha loãng nhiều hơn, khi đó có thể hạn chế được yếu tố ảnh hưởng. Ngoài ra, có thể xử lý để loại các yếu tố ảnh hưởng bằng phương pháp thích hợp như lọc, trung hòa, thẩm tách hoặc đun nóng. Để đảm bảo phương pháp xử lý đã chọn chắc chắn loại được yếu tố ảnh hưởng mà không làm mất nội độc tố, cần thực hiện phép thử Kiểm tra yếu tố ảnh hưởng với dung dịch thử sau khi xử lý và cho thêm chuẩn nội độc tố. Nếu phương pháp có hiệu quả, cần ghi vào tiêu chuẩn.
Phép thử giới hạn
Dựa trên sự tạo gel của thuốc thử lysat với nội độc tố, phương pháp này kiểm tra xem lượng nội độc tố có trong mẫu thử có lớn hơn giới hạn quy định hay không.
Tiến hành: Chuẩn bị dung dịch A, B, C và D theo Bảng 13.2.3, mỗi dung dịch 2 ống. Các điều kiện thử nghiệm như nhiệt độ, thời gian ủ và đánh giá kết quả tương tự như trong phần Kiểm tra độ nhạy của lysat.
Bảng 13.2.3
| Dung dịch | Nồng độ nội độc tố thêm vào cho mỗi dung dịch | Số ống nghiệm |
| A | 0/dung dịch thử | 2 |
| B | 2 λ/dung dịch thử | 2 |
| C | 2 λ/nước BET | 2 |
| D | 0/nước BET | 2 |
Trong đó:
Dung dịch A (dung dịch thử): Mẫu thử pha ở độ pha loãng ≤ MVD.
Dung dịch B (đối chứng dương tính có mẫu thử): Nội độc tố pha trong dung dịch thử.
Dung dịch C (đối chứng dương tính): Chuẩn nội độc tố pha trong nước BET.
Dung dịch D (đối chứng âm tính): Nước BET.
Đánh giá kết quả:
Phép thử có giá trị nếu cả 2 ống của dung dịch B và C đều cho kết quả dương tính và dung dịch D âm tính.
Mẫu thử đạt yêu cầu nếu kết quả âm tính ở cả hai ống nghiệm của dung dịch A.
Mẫu thử không đạt yêu cầu nếu kết quả dương tính trên cả hai ống nghiệm của dung dịch A khi độ pha loãng bằng MVD.
Nếu hai ống của dung dịch A cho kết quả khác nhau, một ống dương tính và một ống âm tính thì làm lại phép thử. Mẫu thử đạt yêu cầu nếu ở lần thử thứ hai cả hai ống đều cho kết quả âm tính.
Phép thử bán định lượng
Phép thử này xác định lượng nội độc tố có trong dung dịch mẫu thử bằng cách thực hiện phản ứng tiến dần tới điểm dừng trong quá trình tạo gel.
Tiến hành: Chuẩn bị dung dịch A, B, C và D theo Bảng 13.2.4, mỗi dung dịch 2 ống nghiệm.
Dung dịch thử dùng cho dung dịch A và B phải không có chứa yếu tố ảnh hưởng. Các điều kiện thử nghiệm như nhiệt độ, thời gian ủ và đánh giá kết quả tương tự như trong phần Kiểm tra độ nhạy của lysat.
Bảng 13.2.4
| Dung dịch | Nồng độ nội độc tố được thêm vào mỗi dung dịch | Dung môi | Hệ số pha loãng | Nồng độ nội độc tố sau khi pha loãng | Số ống nghiệm |
| A | 0/dung dịch mẫu thử | - | 1 | - | 2 |
| 2 | 2 | ||||
| 4 | 2 | ||||
| 8 | 2 | ||||
| B | 2λ/dung dịch mẫu thử | Dung dịch mẫu thử | 1 | 2 λ | 2 |
| C | 2λ/nước BET | Nước BET | 1 | 2 λ | 2 |
| 2 | 1 λ | 2 | |||
| 4 | 0,5 λ | 2 | |||
| 8 | 0,25 λ | 2 | |||
| D | 0/nước BET | - | - | - | 2 |
Trong đó:
Dung dịch A (dung dịch thử): Dung dịch thử có độ pha loãng ≤ MVD, không có yếu tố ảnh hưởng. Pha mẫu thử với nước BET theo các hệ số pha loãng 1/2, 1/4, 1/8... Có thể pha loãng tiếp để có kết quả phù hợp nhưng hệ số pha loãng dung dịch thử cuối cùng phải không quá MVD.
Dung dịch B (đối chứng dương tính có mẫu thử): Nội độc tố pha trong dung dịch A, để kiểm tra yếu tố ức chế sự tạo gel.
Dung dịch C (đối chứng dương tính): 2 dãy chuẩn nội độc tố pha trong nước BET
Dung dịch D (đối chứng âm tính): Dùng nước BET
Tính toán và đánh giá kết quả:
Phép thử có giá trị khi thỏa mãn 3 điều kiện sau:
a - Cả 2 ống của dung dịch D đều âm tính (đối chứng âm tính).
b - Cả 2 ống của dung dịch B đều dương tính (đối chứng dương tính có mẫu thử).
c - Trung bình nhân nồng độ điểm dừng của dung dịch C nằm trong khoảng 0,5 λ đến 2 λ.
Điểm dừng được xác định là ống nghiệm có hệ số pha loãng cao nhất trong dãy của dung dịch A cho phản ứng dương tính, và nồng độ nội độc tố ở điểm dừng của dung dịch A là tích số của độ pha loãng tại điểm đó nhân với hệ số λ.
Nồng độ nội độc tố trong dung dịch mẫu cần kiểm tra được xác định là trung bình nhân nồng độ điểm dừng của các dãy thử, tính trung bình nồng độ nội độc tố của 2 dãy (tương tự như phần Kiểm tra độ nhạy của lysat).
Nếu không có nồng độ nào của dung dịch A cho phản ứng dương tính, thì nồng độ nội độc tố của dung dịch A nhỏ hơn λ x hệ số pha loãng thấp nhất của dung dịch A.
Nếu tất cả các độ pha loãng đều cho phản ứng dương tính, nồng độ nội độc tố của dung dịch A là bằng hoặc lớn hơn tích của hệ số pha loãng lớn nhất của dung dịch A nhân với λ.
Có thể pha loãng tiếp để tìm được điểm dừng. Tính nồng độ nội độc tố của mẫu thử (EU/ml, EU/mg hoặc mEq hoặc EU/đơn vị), dựa trên nồng độ nội độc tố trung bình của dung dịch A. Mẫu thử đạt yêu cầu phép thử này nếu nồng độ nội độc tố có trong mẫu thử thấp hơn giới hạn nội độc tố quy định trong chuyên luận riêng.
PHƯƠNG PHÁP ĐO QUANG
Phương pháp đo độ đục
Phương pháp đo độ đục xác định lượng nội độc tố có trong dung dịch mẫu thử dựa trên đo mức độ thay đổi độ đục trong quá trình tạo gel của thuốc thử lysat. Hiện nay có 2 kỹ thuật được áp dụng: đo độ đục tại điểm dừng hoặc đo độ đục động học.
Đo độ đục tại điểm dừng dựa trên sự tương quan giữa nồng độ nội độc tố và độ đục của hỗn hợp phản ứng ở thời điểm xác định vào cuối của giai đoạn ủ.
Đo độ đục động học dựa trên sự tương quan giữa nồng độ nội độc tố và thời gian cần thiết để đạt tới độ đục định trước của hỗn hợp phản ứng hoặc tốc độ tăng độ đục của hỗn hợp.
Phép thử thường thực hiện ở (37 ± 1) °C, độ đục biểu thị bằng độ hấp thụ hay độ truyền qua.
Phương pháp đo màu
Phương pháp đo màu xác định nồng độ nội độc tố của các dung dịch mẫu thử dựa trên đo chất màu được giải phóng ra từ một cơ chất mang màu do phản ứng của nội độc tố với thuốc thử lysat.
Có 2 kỹ thuật đo: đo màu tại điểm dừng hoặc đo màu động học.
Đo màu tại điểm dừng dựa trên tương quan giữa nồng độ nội độc tố và đậm độ chất màu tạo thành của hỗn hợp phản ứng ở điểm cuối của giai đoạn ủ.
Đo màu động học dựa trên mối tương quan định lượng giữa nồng độ nội độc tố và thời gian cần thiết để hỗn hợp phản ứng đạt tới độ hấp thụ (hoặc độ truyền qua) định trước hoặc tốc độ tăng màu của hỗn hợp.
Phép thử thường thực hiện ở (37 ± 1) °C.
Chuẩn bị
Để đảm bảo độ đúng và giá trị của phương pháp đo màu hoặc đo độ đục, phải tiến hành phép thử Kiểm tra đường chuẩn và Kiểm tra yếu tố ảnh hưởng như sau:
Kiểm tra đường chuẩn: Phép thử được thực hiện với mỗi lô thuốc thử lysat mới. Dùng dung dịch nội độc tố chuẩn, pha ít nhất 3 nồng độ nội độc tố để tạo được đường chuẩn trong khoảng nồng độ nội độc tố ghi trong hướng dẫn của thuốc thử lysat đang sử dụng. Với mỗi nồng độ, phải dùng ít nhất 3 ống tùy theo điều kiện quy định cho thuốc thử lysat đang dùng (về tỷ lệ thể tích, thời gian ủ, nhiệt độ, pH...).*
Giá trị tuyệt đối của hệ số tương quan [r], phải lớn hơn hoặc bằng 0,980 cho khoảng nồng độ nội độc tố đã dùng.
Nếu phép thử không có giá trị, kiểm tra các điều kiện thử nghiệm và tiến hành lại như trên.
Phép thử Kiểm tra đường chuẩn phải được thực hiện mỗi khi có thay đổi một điều kiện nào đó có thể ảnh hưởng đến kết quả thử nghiệm.
Kiểm tra yếu tố ảnh hưởng: Chọn nồng độ nội độc tố vào khoảng giữa của đường chuẩn.
Chuẩn bị dung dịch A, B, C và D theo Bảng 13.2.5. Tiến hành phép thử với các dung dịch này sau khi đã chuẩn hóa các điều kiện theo quy định của loại thuốc thử lysat sử dụng (về thể tích, tỷ lệ thể tích dung dịch thử và thuốc thử lysat, thời gian ủ).
Phép thử Kiểm tra yếu tố ảnh hưởng phải được thực hiện mỗi khi có sự thay đổi có thể ảnh hưởng tới kết quả thử nghiệm.
Bảng 13.2.5
| Dung dịch | Nồng độ nội độc tố thêm vào mỗi dung dịch | Dung dịch được thêm nội độc tố | Số ống nghiệm hoặc lỗ phản ứng |
| A | 0 | Dung dịch mẫu | Không ít hơn 2 |
| B | Nồng độ ở khoảng giữa của đường cong chuẩn | Dung dịch mẫu | Không ít hơn 2 |
| C | Ít nhất 3 nồng độ | Nước BET | Mỗi nồng độ không ít hơn 2 |
| D | 0 | Nước BET | Không ít hơn 2 |
Trong đó:
Dung dịch A (dung dịch thử): Dung dịch mẫu pha loãng không quá MVD.
Dung dịch B: Thêm dung dịch chuẩn vào dung dịch mẫu thử để có nồng độ nội độc tố vào khoảng đoạn giữa của đường cong chuẩn.
Dung dịch C (đối chứng dương tính): Dung dịch chuẩn nội độc tố có nồng độ dùng trong thẩm định phương pháp như mô tả trong phần Kiểm tra đường chuẩn.
Dung dịch D (đối chứng âm tính): Dùng nước BET.
Phép thử chỉ có giá trị khi hệ số tương quan của đường chuẩn thu được từ dung dịch C lớn hơn hoặc bằng 0,980.
Dung dịch D phải cho kết quả không vượt quá giới hạn giá trị trắng theo quy định của thuốc thử lysat đã dùng, hoặc thấp hơn giới hạn nội độc tố phát hiện của lysat đã dùng.
Khả năng tìm lại được lượng nội độc tố chuẩn thêm vào dung dịch B bằng hiệu số nồng độ đã tìm thấy trong dung dịch B trừ đi nồng độ nội độc tố tìm được trong dung dịch A.
Nếu khả năng tìm lại lượng nội độc tố thêm vào nằm trong khoảng 50 % đến 200 %, dung dịch mẫu cần kiểm tra được coi là không có yếu tố ảnh hưởng.
Nếu nồng độ tìm lại được nằm ngoài khoảng quy định, dung dịch mẫu cần kiểm tra có chứa yếu tố ảnh hưởng. Khi đó, cần xử lý để loại yếu tố ảnh hưởng bằng các phương pháp thích hợp (như đã nêu trong phương pháp tạo gel). Sau đó, phải thực hiện đánh giá lại xem phương pháp xử lý có loại được yếu tố ảnh hưởng không.
Xác định lượng nội độc tố trong mẫu thử
Tiến hành: Chuẩn bị dung dịch A, B, C và D theo như Bảng 13.2.5, và theo qui trình như đã mô tả trong phần Kiểm tra yếu tố ảnh hưởng.
Tính nồng độ nội độc tố:
Tính nồng độ nội độc tố trong mỗi ống của dung dịch A theo đường chuẩn thu được từ dung dịch C. Phép thử không có giá trị trừ khi thỏa mãn các yêu cầu sau:
1. Giá trị tuyệt đối của hệ số tương quan của đường chuẩn theo dung dịch C lớn hơn hoặc bằng 0,980.
2. Lượng nội độc tố tìm lại được, tức là hiệu số nồng độ nội độc tố tìm lại được trong dung dịch B trừ đi nồng độ nội độc tố trong dung dịch A, phải nằm trong khoảng 50 % đến 200 %.
3. Kết quả của dung dịch D không vượt quá giới hạn giá trị trắng của thuốc thử lysat đã dùng, hoặc phải thấp hơn giới hạn nội độc tố phát hiện của lysat đã dùng.
Đánh giá kết quả:
Mẫu thử đạt phép thử nội độc tố vi khuẩn nếu nồng độ nội độc tố có trong mẫu thử tính từ nồng độ nội độc tố trung bình của các ống dung dịch A thấp hơn giới hạn nội độc tố quy định trong chuyên luận riêng.
* Nếu tỷ số giá trị logarit đáp ứng giữa nồng độ cao nhất so với nồng độ thấp nhất trong khoảng khảo sát lớn hơn 2, thì nên thêm một số nồng độ để thu được đường chuẩn có khoảng cách giữa các giá trị log phù hợp.
13.3 PHÉP THỬ CÁC CHẤT HẠ ÁP
Thử các chất hạ áp là phương pháp sinh học dựa vào tác dụng hạ huyết áp của thuốc đem thử trên mèo đã được gây mê so với tác dụng hạ áp của thuốc histamin chuẩn.
Chuẩn bị thí nghiệm
Động vật: Mèo khỏe mạnh, trưởng thành, đực hoặc cái (không mang thai), cân nặng từ 1,8 kg trở lên.
Nước muối heparin: Dung dịch chứa 50 đơn vị heparin trong 1 ml dung dịch natri clorid 0,9 % dùng để chống đông máu. Có thể dùng một dung dịch chống đông thích hợp thay thế.
Dung dịch histamin chuẩn: Dùng histamin dihydroclorid hoặc histamin monohydrophosphat bảo quản trong lọ kín, làm khô bằng silica gel 2 h trước khi cân. Hòa tan trong nước cất một lượng histamin đã được cân chính xác thành dung dịch 1 mg/ml histamin base. Ngay trước khi thử, pha loãng dung dịch histamin này (1 mg/ml) bằng dung dịch natri clorid 0,9 % đã tiệt trùng thành dung dịch có nồng độ 0,1 µg/ml tính theo histamin base.
Mẫu thử: Pha theo từng chuyên luận tương ứng trong dung dịch natri clorid 0,9 % đã tiệt khuẩn hoặc trong dung môi thích hợp, đến nồng độ cần thiết.
Tiến hành
Mèo được gây mê bằng cloralose hoặc một loại barbiturat thích hợp như phenobarbital v.v... để duy trì được huyết áp đồng đều trong suốt thời gian thí nghiệm, cố định động vật trên bàn mổ, cần làm nhẹ nhàng sao cho mèo không bị kích thích. Có biện pháp giữ để thân nhiệt mèo không giảm quá giới hạn sinh lý bằng đèn hoặc lò sưởi. Thông khí quản bằng một canun thủy tinh. Bộc lộ động mạch cảnh, lồng canun có chứa đầy nước muối heparin (hoặc dung dịch chống đông khác) vào động mạch cảnh. Nối canun với huyết áp kế thủy ngân hoặc một thiết bị ghi huyết áp khác đạt yêu cầu về độ nhạy. Bộc lộ tĩnh mạch đùi, luồn vào đó một kim tiêm đầu tù có chứa nước muối heparin, buộc cố định lại để tiêm mẫu thử hoặc histamin chuẩn.
Xác định độ nhạy của động vật với histamin bằng cách tiêm tĩnh mạch các liều 1 ml (0,1 µg) và 1,5 ml (0,15 µg) dung dịch histamin chuẩn cho mỗi kg thể trọng. Khoảng cách thời gian giữa các lần tiêm ít nhất là 1 min sau khi huyết áp đã trở lại mức bình thường. Bình thường một liều histamin 0,1 µg/kg thể trọng mèo làm huyết áp hạ khoảng 20 mmHg. Lặp lại liều thấp histamin chuẩn (1 ml/kg) ít nhất 2 lần. Động vật được dùng vào thí nghiệm khi sự giảm huyết áp là hằng định ở các liều 1 ml/kg và với liều 1,5 ml/ kg phải có độ hạ áp lớn hơn.
Sau khi đã đạt yêu cầu thử độ nhạy với histamin, tiêm tĩnh mạch cho mỗi kg mèo 1 ml dung dịch histamin chuẩn. Tiếp theo tiêm 2 liều liên tục của mẫu thử theo chỉ dẫn của từng chuyên luận và cuối cùng lại tiêm dung dịch histamin chuẩn với liều 1 ml/kg. Thời gian giữa các lần tiêm là hằng định như đã nêu ở trên. Nhắc lại chuỗi tiêm như trên một lần nữa và kết thúc bằng 1,5 ml dung dịch histamin chuẩn cho 1 kg mèo.
Đánh giá kết quả
a. Nếu độ hạ áp của dung dịch histamin chuẩn liều 1,5 ml/kg không lớn hơn liều 1 ml/kg thì thí nghiệm không có giá trị.
b. Chế phẩm đạt yêu cầu về thử các chất hạ áp nếu đạt cả 2 điều kiện sau:
Giá trị trung bình của các độ hạ áp khi tiêm mẫu thử không lớn hơn giá trị trung bình của các độ hạ áp khi tiêm dung dịch histamin chuẩn liều 1 ml/kg thể trọng mèo.
Mức hạ áp của mỗi lần tiêm mẫu thử đều không cao hơn mức hạ áp của dung dịch histamin chuẩn liều kết thúc 1,5 ml/kg.
c. Chế phẩm không đạt yêu cầu nếu một trong 2 điều kiện trên không đạt.
Chú ý: Động vật không được dùng để thử tác dụng hạ áp nữa nếu trong quá trình thử, đã có lần mẫu thử gây hạ áp lớn hơn mức hạ áp do liều kết thúc 1,5 ml dung dịch histamin chuẩn cho 1 kg mèo, hoặc giá trị hạ áp của dung dịch histamin chuẩn ở liều kết thúc 1,5 ml/kg nhỏ hơn giá trị trung bình của các liều 1 ml/kg đã tiêm trước đó.
13.4 PHÉP THỬ CHẤT GÂY SỐT
Phép thử được xác định bằng cách đo sự tăng thân nhiệt thỏ sau khi tiêm tĩnh mạch dung dịch vô khuẩn của chất cần kiểm tra.
Lựa chọn động vật thí nghiệm
Dùng thỏ trưởng thành, khỏe mạnh, cả 2 giống, cân nặng không dưới 1,5 kg, nuôi dưỡng bằng thức ăn không có chứa kháng sinh và thỏ không có dấu hiệu giảm cân trong quá trình thử nghiệm. Không dùng để thử nghiệm nếu:
a) Thỏ mới được dùng thử chất gây sốt có kết quả âm tính trong vòng 3 ngày trước đó, hoặc
b) Thỏ đã được dùng thử chất gây sốt có kết quả dương tính trong vòng 3 tuần.
Khu vực lưu giữ động vật
Thỏ được nuôi giữ riêng từng con trong khu vực yên tĩnh, có nhiệt độ ổn định. Cho thỏ nhịn ăn từ đêm trước khi thử cho đến khi thử xong, không cho uống nước trong quá trình thử. Tiến hành phép thử trong phòng yên tĩnh, không có tiếng ồn, nhiệt độ phòng chênh lệch không quá 3 °C so với khu vực nuôi giữ, hoặc thỏ phải được lưu giữ ở điều kiện phòng thí nghiệm trong khoảng ít nhất 18 h trước khi thử nghiệm.
Dụng cụ, bơm và kim tiêm: Tất cả các dụng cụ, bơm và kim tiêm phải được rửa sạch và tráng nước cất, sấy ở nhiệt độ 250 °C trong 30 min hoặc 200 °C trong 1 h.
Hộp, giá giữ thỏ: Các hộp, giá giữ thỏ cho trường hợp đo nhiệt độ bằng thiết bị điện được thiết kế để giữ ở cổ con vật nhưng không được chật quá, phần cơ thể còn lại được thoải mái để thỏ có thể ngồi ở tư thế bình thường. Không giữ thỏ bằng các loại kẹp hoặc giá có thể gây đau hoặc khó chịu cho con vật. Thỏ phải được cho vào hộp hoặc giá ít nhất 1 h trước khi thử và giữ ở đó trong suốt quá trình thử nghiệm.
Nhiệt kế: Nhiệt kế hoặc thiết bị điện dùng để ghi nhiệt độ có độ chính xác 0,1 °C và được đưa vào trực tràng của thỏ với độ sâu 5 cm. Độ sâu của nhiệt kế trong trực tràng phải giống nhau giữa các thỏ. Nếu dùng thiết bị điện, đầu đo nhiệt độ phải được đặt trong trực tràng trong suốt quá trình thử.
Thử nghiệm sơ bộ
Chỉ nên tiến hành thử sơ bộ với những thỏ lần đầu tiên được dùng để thử chất gây sốt.
Trong vòng 1 đến 3 ngày trước khi kiểm tra mẫu thử, tiêm tĩnh mạch tai 10 ml/kg thỏ dung dịch natri clorid 0,9 % không có chất gây sốt, đã làm ấm đến khoảng 38,5 °C trước khi tiêm. Ghi nhiệt độ thỏ, bắt đầu ít nhất 90 min trước khi tiêm và tiếp tục trong 3 h sau khi tiêm. Không dùng những thỏ có nhiệt độ thay đổi quá 0,6 °C vào thử nghiệm chính thức.
Thử nghiệm chính thức
Mỗi mẫu được thử trên một nhóm 3 thỏ.
Chuẩn bị và tiêm mẫu thử: Mẫu thử có thể được hòa tan trong một dung môi không có chất gây sốt, dung dịch natri clorid 0,9 % hoặc một chất lỏng được quy định trong chuyên luận riêng. Làm ấm dung dịch thử lên khoảng 38,5 °C trước khi tiêm. Tiêm chậm dung dịch thử vào tĩnh mạch tai thỏ trong khoảng thời gian không quá 4 min, trừ khi có chỉ dẫn gì khác trong chuyên luận riêng. Lượng mẫu thử được tiêm sẽ thay đổi tùy theo chế phẩm cần kiểm tra và được quy định trong chuyên luận riêng. Thể tích tiêm trong khoảng 0,5 ml/kg thể trọng thỏ đến 10 ml/kg thể trọng thỏ.
Theo dõi nhiệt độ và xác định đáp ứng:
Ghi nhiệt độ thỏ 30 min một lần, ít nhất 90 min trước khi tiêm và tiếp tục 3 h sau khi tiêm. Theo dõi nhiệt độ trước khi tiêm, không dùng vào thử nghiệm nếu:
Thỏ có chênh lệch nhiệt độ > 0,2 °C giữa 2 lần ghi liên tiếp, hoặc
Thỏ có nhiệt độ ban đầu cao hơn 39,8 °C hoặc thấp hơn 38 °C, hoặc
Nhiệt độ ban đầu của 3 thỏ trong cùng nhóm khác nhau quá 1 °C.
“Nhiệt độ ban đầu” của mỗi thỏ là trung bình của 2 giá trị nhiệt độ ghi được cách nhau 30 min, xác định trong khoảng 40 min ngay trước khi tiêm dung dịch thử. “Nhiệt độ tối đa” của mỗi thỏ là nhiệt độ cao nhất ghi được cho thỏ đó trong vòng 3 h sau khi tiêm. Chênh lệch giữa “nhiệt độ ban đầu” và “nhiệt độ tối đa” được gọi là đáp ứng. Khi chênh lệch là âm, kết quả được coi là đáp ứng bằng 0.
Đánh giá kết quả:
Đầu tiên thử trên một nhóm 3 thỏ, tùy thuộc vào kết quả thu được, thử thêm lần lượt từng nhóm 3 thỏ khác cho đến khi tổng cộng 4 nhóm, nếu cần. Nếu tổng đáp ứng của nhóm đầu tiên không vượt quá số ghi trong cột 2 của bảng dưới đây, thì mẫu thử đạt yêu cầu. Nếu tổng đáp ứng vượt quá số ghi trong cột 2 nhưng không vượt quá số ghi trong cột 3 thì lặp lại phép thử trên nhóm khác như đã nêu ở trên. Nếu tổng các đáp ứng lớn hơn số ghi trong cột 3 thì mẫu thử không đạt yêu cầu.
| Số thỏ | Mẫu thử đạt nếu tổng đáp ứng không vượt quá | Mẫu thử không đạt nếu tổng đáp ứng vượt quá |
| (1) | (2) | (3) |
| 3 | 1,15 °C | 2,65 °C |
| 6 | 2,80 °C | 4,30 °C |
| 9 | 4,45 °C | 5,95 °C |
| 12 | 6,60 °C | 6,60 °C |
Những thỏ có nhiệt độ tăng cao quá 1,2 °C thì loại và không bao giờ dùng lại cho phép thử chất gây sốt.
13.5 THỬ ĐỘC TÍNH BẤT THƯỜNG
Độc tính bất thường của thuốc được xác định bằng cách theo dõi số chuột chết trong thời gian 48 h sau khi cho chuột một liều thuốc qua đường dùng theo quy định.
Động vật thí nghiệm
Chuột nhắt trắng khỏe mạnh, cân nặng 18 g đến 22 g, chưa dùng vào thí nghiệm, nếu là chuột cái phải không có thai hoặc đang cho con bú, được chăn nuôi trong điều kiện bình thường
Chuẩn bị dung dịch thử
Nếu không có hướng dẫn gì khác, chuẩn bị dung dịch thử bằng cách hòa tan mẫu thử trong dung dịch natri clorid 0,9 % hoặc nước cất tiêm (nếu thử theo đường tiêm) hoặc phân tán đều mẫu thử trong nước cất (nếu thử theo đường uống) để thu được dung dịch hoặc hỗn dịch có chứa lượng chất cần thử phù hợp với mức liều quy định trong chuyên luận riêng.
Tiến hành thử
Dùng 5 chuột, cho mỗi con 0,5 ml dung dịch thử bằng một trong những đường dùng sau:
Tiêm tĩnh mạch: Tiêm vào tĩnh mạch đuôi của mỗi chuột với tốc độ hằng định trong 15 s đến 30 s.
Tiêm trong màng bụng: Tiêm qua lớp da bụng vào thẳng hốc bụng chuột.
Tiêm dưới da: Tiêm dưới da vào một chỗ ở lưng hoặc bụng.
Uống: Cho uống bằng một kim cong đầu tù hoặc một dụng cụ khác thích hợp, phải đảm bảo đưa thuốc vào trong thực quản hoặc dạ dày.
Đánh giá kết quả
Nếu không có hướng dẫn gì khác thì quan sát chuột 48 h sau khi dùng thuốc.
Nếu không có chuột nào chết thì mẫu thử đạt yêu cầu.
Nếu có chuột chết, làm lại thử nghiệm với 10 chuột khác, dùng chuột có thể trọng (19 ± 1) g. Sau 48 h, nếu không có chuột nào chết thì mẫu thử đạt yêu cầu.
13.6 THỬ GIỚI HẠN NHIỄM KHUẨN
XÁC ĐỊNH TỔNG SỐ VI SINH VẬT
Giới thiệu
Phép thử sau đây cho phép xác định số lượng khuẩn ưa nhiệt trung bình và nấm có thể phát triển được trong điều kiện hiếu khí.
Phép thử nhằm đánh giá chế phẩm thử có đáp ứng tiêu chuẩn chất lượng đã công bố về chỉ tiêu vi sinh hay không. Khi sử dụng phép thử cho mục đích này, cần tuân thủ theo chỉ dẫn dưới đây về số lượng mẫu đem thử, đánh giá kết quả.
Phép thử này không áp dụng được đối với chế phẩm thuốc có thành phần chính là vi sinh vật sống.
Có thể sử dụng qui trình kiểm tra vi sinh khác, bao gồm các phương pháp tự động nếu chứng minh được tính tương đương của chúng với phương pháp ghi trong dược điển.
Quy định chung
Tiến hành phép thử trong điều kiện đảm bảo tránh được sự tạp nhiễm vi sinh vật bên ngoài vào mẫu thử. Các biện pháp phòng tránh tạp nhiễm phải không ảnh hưởng đến bất cứ loài vi sinh vật nào có thể phát hiện được trong mẫu thử.
Nếu mẫu thử có chứa chất ức chế vi sinh vật thì cần phải loại bỏ hoặc trung hòa trước khi thử nghiệm. Khi sử dụng chất bất hoạt, cần chứng minh khả năng bất hoạt của chúng và đảm bảo chất bất hoạt đó không chứa độc tố với vi sinh vật.
Khi sử dụng chất diện hoạt trong quá trình chuẩn bị mẫu thử, phải đảm bảo chất diện hoạt đó không chứa độc tố với vi sinh vật và chất diện hoạt phải tương thích với chất bất hoạt khác cùng được sử dụng.
Phương pháp xác định tổng số vi sinh vật
Sử dụng phương pháp màng lọc hoặc phương pháp đĩa thạch. Phương pháp MPN (Most Probable Number) nói chung có độ chính xác thấp nhất trong số các phương pháp đếm vi sinh vật, tuy nhiên, với mẫu thử có số lượng vi sinh vật rất thấp thì đây lại là phương pháp phù hợp nhất.
Lựa chọn phương pháp thử dựa trên nhiều yếu tố như bản chất của mẫu thử và giới hạn vi sinh vật cho phép. Phương pháp được lựa chọn phải đảm bảo tiến hành trên lượng mẫu đủ để đánh giá được mẫu thử đáp ứng theo tiêu chuẩn, cần phải xác định sự phù hợp của phương pháp đã lựa chọn.
Kiểm tra chất lượng môi trường, sự phù hợp của phương pháp đếm, chứng âm tính
Yêu cầu chung
Phải chứng minh phương pháp thử có khả năng phát hiện vi sinh vật khi có mặt mẫu thử.
Phải chứng minh sự phù hợp của phương pháp khi có sự thay đổi trong quá trình tiến hành thử nghiệm hoặc có sự thay đổi về sản phẩm làm ảnh hưởng đến kết quả của phép thử.
Chuẩn bị chủng vi sinh vật
Có thể sử dụng hỗn dịch chủng vi sinh vật ổn định đã được tiêu chuẩn hóa hoặc chuẩn bị như trình bày dưới đây. Kỹ thuật lưu giữ chủng gốc phải đảm bảo vi sinh vật được cấy truyền không quá 5 lần từ giống gốc ban đầu. Cấy các chủng vi sinh vật và nấm riêng rẽ theo Bảng 13.6.1.
Sử dụng dung dịch đệm natri clorid-pepton pH 7,0 hoặc dung dịch đệm phosphat pH 7,2 để pha hỗn dịch chủng vi sinh vật. Để pha hỗn dịch A. niger, thêm polysorbat 80 (TT) với tỷ lệ 0,05 % vào dung dịch đệm. Hỗn dịch được sử dụng trong vòng 2 h, nếu bảo quản ở 2 °C đến 8 °C thì có thể sử dụng trong vòng 24 h. Ngoài ra, đối với A. niger và B. subtilis, thay vì chuẩn bị và pha loãng hỗn dịch chủng dạng sinh dưỡng thì có thể sử dụng hỗn dịch bào tử ổn định. Hỗn dịch bào tử được bảo quản ở 2 °C đến 8 °C trong khoảng thời gian đã được thẩm định.
Chứng âm tính
Để đánh giá điều kiện thử nghiệm, tiến hành mẫu chứng âm tính bằng cách sử dụng dung dịch pha loãng thay cho mẫu thử. Mẫu chứng âm tính phải không được có vi sinh vật phát triển. Mẫu chứng âm tính cũng có thể được tiến hành cùng lúc với mẫu thử như mô tả trong phần Cách tiến hành, cần phải xem xét hệ thống kỹ lưỡng khi mẫu chứng âm tính không đạt.
Kiểm tra chất lượng môi trường
Tiến hành kiểm tra chất lượng đối với mỗi lô môi trường pha chế sẵn hoặc mỗi lô môi trường được pha chế từ môi trường khô hay từ các thành phần riêng lẻ.
Cấy riêng rẽ vào môi trường thạch casein đậu tương hay môi trường lỏng casein đậu tương một lượng nhỏ vi sinh vật (không quá 100 CFU) theo chỉ dẫn trong Bảng 13.6.1. Cấy riêng rẽ vào môi trường thạch Sabouraud-dextrose một lượng nhỏ vi sinh vật (không quá 100 CFU) theo chỉ dẫn trong Bảng 13.6.1. Ủ môi trường theo điều kiện ghi trong Bảng 13.6.1.
Bảng 13.6.1- Chuẩn bị và sử dụng chủng vi sinh vật
| Vi sinh vật | Chuẩn bị chủng vi sinh vật | Kiểm tra chất lượng môi trường | Sự phù hợp của phương pháp đếm khi có mặt mẫu thử | ||
| Đếm tổng số vi sinh vật hiếu khí | Đếm tổng số nấm | Đếm tổng số vi sinh vật hiếu khí | Đếm tổng số nấm | ||
| Staphylococcus aureus ATCC 6538 NCIMB 9518 CIP 4.83 NBRC 13276 | Môi trường thạch casein đậu tương hoặc môi trường lỏng casein đậu tương 30 °C - 35 °C 18 h- 24 h | Môi trường thạch casein đậu tương và môi trường lỏng casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 3 ngày | - | Môi trường thạch casein đậu tương/MPN môi trường lỏng casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 3 ngày | - |
| Pseudomonas aeruginosa ATCC 9027 NCIMB 8626 CIP 82.118 NBRC 13275 | Môi trường thạch casein đậu tương hoặc môi trường lỏng casein đậu tương 30 °C - 35 °C 18 h - 24 h | Môi trường thạch casein đậu tương và môi trường lỏng casein đậu tương ≤ 100 CFU 30 °C -35 °C ≤ 3 ngày | - | Môi trường thạch casein đậu tương/MPN môi trường lỏng casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 3 ngày | - |
| Bacillus subtilis ATCC 6633 NCIMB 8054 CIP 52.62 NBRC 3134 | Môi trường thạch casein đậu tương hoặc môi trường lỏng casein đậu tương 30 °C - 35 °C 18 h - 24 h | Môi trường thạch casein đậu tương và môi trường lỏng casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 3 ngày | - | Môi trường thạch casein đậu tương/MPN môi trường lỏng casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 3 ngày | - |
| Candida albicans ATCC 10231 NCPF 3179 IP 48.72 NBRC 1594 | Môi trường thạch Sabouraud- dextrose hoặc môi trường lỏng Sabouraud- dextrose 20 °C - 25 °C 2 - 3 ngày | Môi trường thạch casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 5 ngày | Môi trường thạch Sabouraud- dextrose ≤ 100 CFU 20 °C - 25 °C ≤ 5 ngày | Môi trường thạch casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 5 ngày MPN: không áp dụng | Môi trường thạch Sabouraud- dextrose ≤ 100 CFU 20 °C - 25 °C ≤ 5 ngày |
| Aspergillus niger ATCC 16404 IMI 149007 IP 1431.83 NBRC 9455 | Môi trường thạch Sabouraud- dextrose hoặc môi trường thạch khoai tây dextrose 20 °C - 25 °C 5 - 7 ngày, hoặc đến khi thu được bào tử | Môi trường thạch casein đậu tương ≤ 100 CFU 30 °C - 35 °C ≤ 5 ngày | Môi trường thạch Sabouraud- dextrose ≤ 100 CFU 20 °C- 25 °C ≤ 5 ngày | Môi trường thạch casein đậu tương ≤ 100 CFU 30 °C -35 °C ≤ 5 ngày MPN: không áp dụng | Môi trường thạch Sabouraud- dextrose ≤ 100 CFU 20 °C - 25 °C ≤ 5 ngày |
Đối với môi trường thạch, số lượng vi sinh vật phát triển trên lô môi trường mới không được khác quá 2 lần so với số lượng tính được của hỗn dịch chủng đã được tiêu chuẩn hóa. Đối với chủng được pha chế khi dùng, sự phát triển của vi sinh vật trên lô môi trường mới sẽ được so sánh với sự phát triển của vi sinh vật trên lô môi trường đã đạt yêu cầu chất lượng.
Đối với môi trường lỏng, sự phát triển rõ ràng của vi sinh vật trên lô môi trường mới được so sánh với sự phát triển của vi sinh vật trên lô môi trường đã đạt yêu cầu chất lượng.
Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử
Chuẩn bị mẫu thử: Phương pháp chuẩn bị mẫu thử phụ thuộc vào tính chất vật lý của mẫu thử. Nếu không thể áp dụng cách nào trong các cách dưới đây, có thể sử dụng biện pháp khác để xử lý mẫu.
Mẫu thử tan trong nước: Hòa tan hoặc pha loãng (thường pha loãng 10 lần) mẫu thử trong dung dịch đệm natri clorid-pepton pH 7,0, dung dịch đệm phosphat pH 7,2 hoặc môi trường lỏng casein đậu tương. Điều chỉnh pH 6 đến 8, nếu cần. Có thể pha loãng tiếp nếu cần bằng cùng dung dịch pha loãng.
Mẫu thử có bản chất không phải chất béo, không tan trong nước: Pha loãng mẫu thử (thường pha loãng 10 lần) trong dung dịch đệm natri clorid-pepton pH 7,0, dung dịch đệm phosphat pH 7,2 hoặc môi trường lỏng casein đậu tương, có thể thêm chất diện hoạt như polysorbat 80 (TT) với tỷ lệ 1 g/l để tăng khả năng thấm nước của mẫu thử. Điều chỉnh pH 6 đến 8, nếu cần. Có thể pha loãng tiếp nếu cần bằng cùng dung dịch pha loãng.
Mẫu thử có bản chất là chất béo: Hòa mẫu thử trong isopropyl myristat (TT) đã được tiệt khuẩn trước bằng cách lọc qua màng lọc hoặc trộn mẫu thử với lượng nhỏ nhất có thể polysorbat 80 (TT) vô khuẩn hoặc một chất diện hoạt khác không có tính ức chế, làm nóng nếu cần ở nhiệt độ không quá 40 °C, trong một số ít trường hợp có thể sử dụng nhiệt độ không quá 45 °C. Trộn cẩn thận và nếu cần có thể duy trì trong bể ổn nhiệt. Thêm vừa đủ lượng dung dịch pha loãng đã được làm ấm sẵn để thu được độ pha loãng 10 lần. Duy trì nhiệt độ trong thời gian ngắn nhất và trộn cẩn thận để thu được nhũ dịch. Có thể tiếp tục pha loãng theo cấp số 10, trong đó dung dịch pha loãng được bổ sung polysorbat 80 (TT) hoặc chất diện hoạt không có tính ức chế khác với nồng độ thích hợp.
Dung dịch hoặc chất rắn dưới dạng aerosol: Chuyển mẫu thử lên màng lọc trong điều kiện vô khuẩn hoặc chuyển sang bình chứa vô khuẩn để tiếp tục lấy mẫu. Có thể sử dụng toàn bộ lượng mẫu hoặc một số liều nhất định từ các đơn vị đóng gói đem thử.
Thuốc dán: Loại bỏ lớp bảo vệ bên ngoài, và đặt miếng dán (mặt dính ngửa lên trên) vào bình vô khuẩn. Phủ lên bề mặt miếng dán một loại vật liệu xốp vô khuẩn, ví dụ tấm gạc vô khuẩn, để tránh cho các miếng dán dính vào nhau và cho vào miếng dán một lượng dung dịch pha loãng thích hợp có chứa chất bất hoạt như polysorbat 80 (TT) và/hoặc lecithin. Lắc kỹ ít nhất 30 min.
Cấy chủng và pha loãng:
Thêm vào mẫu thử đã được xử lý như mô tả ở phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử và thêm vào ống chứng (không có mẫu thử) một lượng hỗn dịch vi sinh vật có chứa không quá 100 CFU. Thể tích của hỗn dịch vi sinh vật không được vượt quá 1 % thể tích mẫu thử đã được pha loãng.
Để đánh giá độ phục hồi của vi sinh vật khi có mặt mẫu thử, tiến hành pha loãng mẫu thử ít nhất có thể. Nếu không thể pha loãng ít nhất do bản thân mẫu thử có tác dụng ức chế vi sinh vật hoặc mẫu thử khó tan trong nước, cần áp dụng các biện pháp phù hợp hơn. Nếu không thể loại bỏ được tác dụng ức chế vi sinh vật của mẫu thử, nên thêm hỗn dịch vi sinh vật vào mẫu thử sau khi đã trung hòa, pha loãng hoặc lọc mẫu thử.
Trung hòa/loại bỏ tác dụng của chất ức chế:
So sánh số lượng vi sinh vật hồi phục sau khi pha loãng như mô tả trong phần cấy chủng và pha loãng và ủ như quy định trong phần Hồi phục vi sinh vật khi có mặt mẫu thử với số lượng vi sinh vật hồi phục của ống chứng.
Nếu có sự ức chế vi sinh vật phát triển (số lượng vi sinh vật giảm quá 2 lần), thì phải thay đổi qui trình đếm số lượng vi sinh vật để khẳng định tính có giá trị của thử nghiệm. Sự thay đổi này có thể là: (1) tăng thể tích dung dịch pha loãng hoặc môi trường nuôi cấy, (2) phối hợp chất trung hòa đặc hiệu hoặc chất trung hòa phổ biến trong dung dịch pha loãng, (3) áp dụng phương pháp màng lọc, (4) phối hợp các biện pháp nêu trên.
Chất trung hòa:
Một số chất được dùng để trung hòa tác dụng của chất ức chế vi sinh vật (Bảng 13.6.2) được thêm vào dung dịch pha loãng hoặc môi trường nuôi cấy trước khi hấp tiệt khuẩn. Trước khi sử dụng, phải tiến hành một mẫu trắng có chất trung hòa, không có mẫu thử để khẳng định hoạt tính của chất trung hòa và khẳng định chất trung hòa không có độc tính đối với vi sinh vật.
Bảng 13.6.2 - Một số chất trung hòa thông dụng
| Chất ức chế | Chất/phương pháp trung hòa |
| Glutaraldehyd, dẫn chất thủy ngân | Natri hydrosulfit (Natri bisulfit) |
| Dẫn chất phenol, alcohol, các aldehyd, sorbat | Pha loãng |
| Các aldehyd | Glycin |
| Dẫn chất amoni bậc 4, parahydroxybenzoat (các paraben), bis-biguanid | Lecithin |
| Dẫn chất amoni bậc 4, iod, các paraben | Polysorbat |
| Dẫn chất thủy ngân | Thioglycolat |
| Dẫn chất thủy ngân, các halogen, các aldehyd | Thiosulfat |
| EDTA (edetat) | ion Mg2+ hoặc ion Ca2+ |
Nếu không tìm được phương pháp trung hòa phù hợp, có thể thấy việc không phân lập được vi sinh vật đã cấy vào mẫu thử cho thấy bản chất mẫu thử có tác dụng ức chế. Điều này cho thấy mẫu thử sẽ không có khả năng bị tạp nhiễm các vi sinh vật đã thử. Tuy nhiên, rất có thể mẫu thử chỉ ức chế một số vi sinh vật đã thử mà không ức chế vi sinh vật ngoài danh mục các vi sinh vật đã thử. Khi đó, tiến hành thử nghiệm với độ pha loãng cao nhất tương thích với sự phát triển vi sinh vật và phù hợp với giới hạn nhiễm khuẩn cho phép của mẫu thử.
Hồi phục vi sinh vật khi có mặt mẫu thử: Đối với mỗi vi sinh vật được liệt kê trong Bảng 13.6.1, tiến hành các thí nghiệm riêng rẽ. Chỉ đếm vi sinh vật được cho vào mẫu thử.
• Phương pháp màng lọc:
Sử dụng màng lọc có kích thước lỗ lọc không quá 0,45 µm. Bản chất màng lọc được lựa chọn sao cho khả năng lưu giữ vi sinh vật không bị ảnh hưởng bởi các thành phần của mẫu thử. Đối với mỗi loại vi sinh vật được ghi trong Bảng 13.6.1, sử dụng một màng lọc riêng.
Chuyển một lượng phù hợp mẫu thử (thường là lượng tương đương với 1 g hoặc 1 ml mẫu thử, hoặc lượng mẫu nhỏ hơn nếu dự đoán chứa nhiều vi sinh vật) đã được xử lý theo phần Chuẩn bị mẫu thử, Cấy chủng và pha loãng, Trung hòa/loại bỏ tác dụng của chất ức chế lên màng lọc, lọc ngay và rửa màng lọc với thể tích thích hợp của dung dịch pha loãng.
Để đếm tổng số vi sinh vật hiếu khí, chuyển màng lọc lên bề mặt đĩa môi trường thạch casein đậu tương. Để đếm tổng số nấm, chuyển màng lọc lên bề mặt đĩa môi trường thạch Sabouraud-dextrose. Ủ các đĩa môi trường theo điều kiện ghi ở Bảng 13.6.1. Sau đó đếm số lượng khuẩn lạc.
• Phương pháp đĩa thạch:
Với mỗi loại môi trường, tiến hành trên ít nhất 2 đĩa Petri và tính kết quả trung bình.
Phương pháp cấy trộn: Sử dụng đĩa Petri đường kính 9 cm, thêm vào mỗi đĩa 1 ml mẫu thử đã được xử lý theo phần Chuẩn bị mẫu thử, Cấy chủng và pha loãng, Trung hòa/loại bỏ tác dụng của chất ức chế và 15 ml đến 20 ml môi trường thạch casein đậu tương hoặc môi trường thạch Sabouraud- dextrose, cả hai môi trường có nhiệt độ không quá 45 °C. Nếu sử dụng dĩa Petri có đường kính lớn hơn, thể tích môi trường phải tăng lên cho phù hợp. Đối với mỗi loại vi sinh vật được ghi trong Bảng 13.6.1, sử dụng ít nhất 2 đĩa Petri. Ủ các đĩa môi trường theo điều kiện ghi ở Bảng 13.6.1. Tính trung bình số lượng khuẩn lạc đếm được đối với mỗi loại môi trường và từ đó tính ra số lượng vi sinh vật trong dịch cấy ban đầu.
Phương pháp cấy trải bề mặt: sử dụng đĩa Petri đường kính 9 cm, thêm vào mỗi đĩa 15 ml đến 20 ml môi trường thạch casein đậu tương hoặc môi trường thạch Sabouraud-dextrose ở nhiệt độ khoảng 45 °C và để môi trường đông. Nếu sử dụng đĩa Petri có đường kính lớn hơn, thể tích môi trường phải tăng lên cho phù hợp. Làm khô đĩa, ví dụ trong buồng thổi khí sạch (laminar air flow) hoặc tủ ấm. Đối với mỗi loại vi sinh vật được ghi trong Bảng 13.6.1, sử dụng ít nhất 2 đĩa Petri. Trải lên bề mặt đĩa môi trường thể tích không dưới 0,1 ml mẫu thử đã được xử lý theo phần Chuẩn bị mẫu thử, Cấy chủng và pha loãng, Trung hòa/loại bỏ tác dụng của chất ức chế. Ủ và đếm số lượng khuẩn lạc giống Mục Phương pháp cấy trộn.
• Phương pháp MPN (Most Probable Number):
Phương pháp MPN có độ chính xác và độ đúng kém hơn so với phương pháp màng lọc hoặc phương pháp đĩa thạch. Kết quả đếm tổng số nấm bằng phương pháp MPN càng cho sai số nhiều hơn. Chính vì vậy, phương pháp MPN chỉ được sử dụng khi không thể áp dụng phương pháp nào khác. Nếu sử dụng phương pháp MPN, cần tuân thủ các bước sau.
Chuẩn bị ít nhất 3 nồng độ pha loãng 10 lần của mẫu thử đã được xử lý như phần Chuẩn bị mẫu thử, Cấy chủng và pha loãng, Trung hòa/loại bỏ tác dụng của chất ức chế. Đối với mỗi độ pha loãng, cấy 1 g hoặc 1 ml vào 3 ống nghiệm có chứa 9 ml đến 10 ml môi trường lỏng casein đậu tương. Nếu cần, có thể thêm chất diện hoạt như polysorbat 80 (TT) hoặc các chất bất hoạt phù hợp khác vào môi trường. Như vậy, có tất cả 9 ống nghiệm cho 3 mức nồng độ pha loãng của mẫu thử.
Ủ tất cả các ống nghiệm trên ở 30 °C đến 35 °C trong không quá 3 ngày. Nếu không thể đọc kết quả chính xác do bản chất mẫu thử gây khó khăn cho việc quan sát sự phát triển của vi sinh vật thì cấy chuyển sang môi trường lỏng casein đậu tương hoặc môi trường thạch casein đậu tương, ủ tiếp ở nhiệt độ như trên trong 1 ngày đến 2 ngày, sau đó đọc kết quả. Xác định chỉ số MPN trong 1 g hoặc 1 ml mẫu thử theo Bảng 13.6.3.
Bảng 13.6.3 - Số lượng vi sinh vật lớn nhất có thể có trong chế phẩm
| Số ống nghiệm có sự phát triển của vi sinh vật | Số lượng vi sinh vật lớn nhất có thể có trong 1 g hoặc 1 ml | Độ tin cậy 95 % | ||
| Số g hoặc ml mẫu thử cho mỗi ống | ||||
| 0,1 | 0,01 | 0,001 | ||
| 0 | 0 | 0 | <3 | 0 - 9,4 |
| 0 | 0 | 1 | 3 | 0,1 - 9,5 |
| 0 | 1 | 0 | 3 | 0,1 - 10 |
| 0 | 1 | 1 | 6,1 | 1,2 - 17 |
| 0 | 2 | 0 | 6,2 | 1,2 - 17 |
| 0 | 3 | 0 | 9,4 | 3,5 - 35 |
| 1 | 0 | 0 | 3,6 | 0,2 - 17 |
| 1 | 0 | 1 | 7,2 | 1,2 - 17 |
| 1 | 0 | 2 | 11 | 4 - 35 |
| 1 | 1 | 0 | 7,4 | 1,3 - 20 |
| 1 | 1 | 1 | 11 | 4 - 35 |
| 1 | 2 | 0 | 11 | 4 - 35 |
| 1 | 2 | 1 | 15 | 5 - 38 |
| 1 | 3 | 0 | 16 | 5 - 38 |
| 2 | 0 | 0 | 9,2 | 1,5 - 35 |
| 2 | 0 | 1 | 14 | 4 - 35 |
| 2 | 0 | 2 | 20 | 5 - 38 |
| 2 | 1 | 0 | 15 | 4 - 38 |
| 2 | 1 | 1 | 20 | 5 - 38 |
| 2 | 1 | 2 | 27 | 9 - 94 |
| 2 | 2 | 0 | 21 | 5 - 40 |
| 2 | 2 | 1 | 28 | 9 - 94 |
| 2 | 2 | 2 | 35 | 9 - 94 |
| 2 | 3 | 0 | 29 | 9 - 94 |
| 2 | 3 | 1 | 36 | 9 - 94 |
| 3 | 0 | 0 | 23 | 5 - 94 |
| 3 | 0 | 1 | 38 | 9 - 104 |
| 3 | 0 | 2 | 64 | 16 - 181 |
| 3 | 1 | 0 | 43 | 9 - 181 |
| 3 | 1 | 1 | 75 | 17 - 199 |
| 3 | 1 | 2 | 120 | 30 - 360 |
| 3 | 1 | 3 | 160 | 30 - 380 |
| 3 | 2 | 0 | 93 | 18 - 360 |
| 3 | 2 | 1 | 150 | 30 - 380 |
| 3 | 2 | 2 | 210 | 30 - 400 |
| 3 | 2 | 3 | 290 | 90 - 990 |
| 3 | 3 | 0 | 240 | 40 - 990 |
| 3 | 3 | 1 | 460 | 90 - 1980 |
| 3 | 3 | 2 | 1100 | 200 - 4000 |
| 3 | 3 | 3 | > 1100 |
|
Kết quả và đánh giá
Khi đánh giá sự phù hợp của phương pháp màng lọc hoặc phương pháp đĩa thạch, chỉ số MPN phải không được khác quá 2 lần so với kết quả của ống chứng không chứa mẫu thử thu được theo phần Cấy chủng và pha loãng. Khi đánh giá sự phù hợp của phương pháp MPN, giá trị MPN phải nằm trong khoảng tin cậy 95 % của kết quả thu được của ống chứng.
Nếu các yêu cầu trên không đáp ứng đối với một hoặc nhiều loại vi sinh vật đem thử ở tất cả phương pháp thử thì lựa chọn phương pháp và điều kiện thử nào có đáp ứng gần nhất yêu cầu trên.
Cách tiến hành
Lượng mẫu thử dùng cho thử nghiệm
Trừ khi có chỉ dẫn riêng, lấy 10 g hoặc 10 ml mẫu thử để tiến hành thử nghiệm trong điều kiện đảm bảo như đã đề cập ở trên. Đối với thuốc khí dung chứa dung dịch hoặc chất rắn, tiến hành thử trên 10 đơn vị đóng gói. Đối với thuốc dán, tiến hành thử trên 10 miếng dán.
Lượng mẫu thử nghiệm có thể giảm xuống tùy theo lượng dược chất chính được đưa vào chế phẩm trong các trường hợp sau: Khối lượng dược chất chính không quá 1 mg trong 1 đơn vị (1 viên nén, 1 nang, 1 ống hoặc lọ thuốc tiêm...) hoặc khối lượng dược chất chính trong 1 g hoặc 1 ml mẫu thử (đối với dạng chế phẩm đa liều) nhỏ hơn 1 mg. Trong những trường hợp này, lượng mẫu thử nghiệm phải không được nhỏ hơn lượng mẫu có trong 10 đơn vị đóng gói hoặc 10 g hoặc 10 ml mẫu thử.
Đối với mẫu thử là nguyên liệu nhưng có khối lượng rất hạn chế hoặc cỡ lô quá nhỏ (dưới 1000 ml hoặc 1000 g), lượng mẫu thử nghiệm bằng 1 % cỡ lô, một số trường hợp lượng mẫu thử có thể nhỏ hơn và phải có quy định cụ thể.
Đối với mẫu thử mà tổng số đơn vị trong một lô dưới 200 (ví dụ mẫu thử nghiệm lâm sàng), có thể tiến hành thử nghiệm trên 2 đơn vị, trường hợp cỡ lô nhỏ hơn 100 đơn vị, có thể tiến hành thử nghiệm trên 1 đơn vị.
Lấy mẫu nguyên liệu hoặc mẫu thử một cách ngẫu nhiên. Để thu được đủ lượng mẫu theo yêu cầu, có thể phải trộn đều mẫu thử lấy từ các đơn vị đóng gói khác nhau.
Tiến hành
Phương pháp màng lọc: Sử dụng dụng cụ lọc được thiết kế phù hợp, cho phép có thể chuyển màng lọc vào môi trường nuôi cấy. Sử dụng phương pháp phù hợp theo mục Kiểm tra chất lượng môi trường, sự phù hợp của phương pháp đếm, chứng âm tính để chuẩn bị mẫu thử, và chuyển lượng mẫu thích hợp lên 2 màng lọc và tiến hành lọc ngay. Rửa màng lọc bằng qui trình phù hợp.
Để đếm tổng số vi sinh vật hiếu khí, chuyển một màng lọc lên bề mặt môi trường thạch casein đậu tương. Để đếm tổng số nấm, chuyển một màng lọc lên bề mặt môi trường thạch Sabouraud-dextrose. Ủ đĩa chứa môi trường thạch casein đậu tương ở 30 °C đến 35 °C trong 3 ngày đến 5 ngày và ủ đĩa chứa môi trường thạch Sabouraud-dextrose ở 20 °C đến 25 °C trong 5 ngày đến 7 ngày. Tính số CFU (Colony Forming Unit) trong 1 g hoặc 1 ml mẫu thử.
Khi thử nghiệm với thuốc dán, lọc riêng rẽ 10 % thể tích mẫu thử được chuẩn bị theo Phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử. trên 2 màng lọc vô khuẩn. Chuyển một màng lọc lên bề mặt môi trường thạch casein đậu tương để đếm tổng số vi sinh vật hiếu khí và chuyển màng lọc còn lại lên bề mặt môi trường thạch Sabouraud-dextrose để đếm tổng số nấm.
Phương pháp đĩa thạch:
• Phương pháp cấy trộn:
Sử dụng phương pháp phù hợp theo mục Kiểm tra chất lượng môi trường, sự phù hợp của phương pháp đếm, chứng âm tính để chuẩn bị mẫu thử. Tiến hành với ít nhất 2 đĩa Petri cho mỗi loại môi trường ở mỗi nồng độ pha loãng.
Ủ đĩa chứa môi trường thạch casein đậu tương ở 30 °C đến 35 °C trong 3 ngày đến 5 ngày và ủ đĩa chứa môi trường thạch Sabouraud-dextrose ở 20 °C đến 25 °C trong 5 ngày đến 7 ngày. Chọn các đĩa của một nồng độ pha loãng nhất định mà tại nồng độ pha loãng đó, số khuẩn lạc thu được là cao nhất và nhỏ hơn 250 đối với đĩa đếm tổng số vi sinh vật hiếu khí và nhỏ hơn 50 đối với đĩa đếm tổng số nấm. Từ đó tính giá trị trung bình đối với mỗi loại môi trường và tính số CFU trong 1 g hoặc 1 ml mẫu thử.
• Phương pháp cấy trải bề mặt:
Sử dụng phương pháp phù hợp theo mục Kiểm tra chất lượng môi trường, sự phù hợp của phương pháp đếm, chứng âm tính để chuẩn bị mẫu thử. Tiến hành với ít nhất 2 đĩa Petri cho mỗi loại môi trường ở mỗi nồng độ pha loãng. Điều kiện ủ và cách tính kết quả giống phương pháp cấy trộn.
Phương pháp MPN:
Sử dụng phương pháp phù hợp theo mục Kiểm tra chất lượng môi trường, sự phù hợp của phương pháp đếm, chứng âm tính để chuẩn bị mẫu thử. Ủ tất cả các ống nghiệm ở 30 °C đến 35 °C trong 3 ngày đến 5 ngày. Có thể cấy chuyển nếu cần theo qui trình phù hợp. Tại mỗi nồng độ pha loãng, ghi lại số lượng ống nghiệm có vi sinh vật phát triển. Xác định số lượng vi sinh vật trong 1 g hoặc 1 ml mẫu thử theo Bảng 13.6.3.
Đánh giá kết quả
Tổng số vi sinh vật hiếu khí được coi là số khuẩn lạc đếm được trên môi trường thạch casein đậu tương. Khuẩn lạc nấm phát hiện được trên môi trường này cũng được coi là một phần của tổng số vi sinh vật hiếu khí. Tổng số nấm được coi là số khuẩn lạc đếm được trên môi trường thạch Sabouraud- dextrose. Khi tổng số nấm vượt quá giới hạn chấp nhận của mẫu thử do vi khuẩn mọc trên môi trường này, có thể thêm kháng sinh vào môi trường thạch Sabouraud-dextrose (ví dụ 50 mg cloramphenicol vào 1 lít môi trường trước khi hấp tiệt khuẩn). Số lượng vi sinh vật đếm được bằng phương pháp MPN được coi là tổng số vi sinh vật hiếu khí. Nếu không thấy có khuẩn lạc mọc trên đĩa có độ pha loãng 1/10 thì kết luận chế phẩm có ít hơn 10 CFU trong 1 g hoặc 1 ml.
Dựa vào giới hạn nhiễm khuẩn cho phép, có thể đánh giá như sau:
101 CFU: Số lượng tối đa được chấp nhận = 20;
102 CFU: Số lượng tối đa được chấp nhận = 200;
103 CFU: Số lượng tối đa được chấp nhận = 2000 và cứ tiếp tục như vậy.
Các dung dịch và môi trường nuôi cấy được liệt kê trong mục Dung dịch pha loãng và môi trường.
XÁC ĐỊNH VI SINH VẬT GÂY BỆNH
Giới thiệu
Phép thử sau đây cho phép xác định sự không hiện diện hoặc hiện diện với số lượng hạn chế của một số vi sinh vật gây bệnh trong mẫu thử dưới những điều kiện đã quy định.
Phép thử nhằm đánh giá chế phẩm thử có đáp ứng tiêu chuẩn chất lượng đã công bố về chỉ tiêu vi sinh vật hay không. Khi sử dụng phép thử cho mục đích này, cần tuân thủ theo chỉ dẫn phía dưới về số lượng mẫu đem thử, đánh giá kết quả.
Có thể sử dụng qui trình xác định vi sinh vật khác, bao gồm cả các phương pháp tự động, khi chứng minh được tính tương đương của chúng với phương pháp ghi trong dược điển.
Quy định chung
Chuẩn bị mẫu thử theo mô tả ở phần Chuẩn bị mẫu thử.
Nếu mẫu thử có chứa chất ức chế vi sinh vật, cần loại bỏ hoặc trung hòa chất ức chế như mô tả trong phần Trung hòa/loại bỏ tác dụng của chất ức chế.
Khi sử dụng chất diện hoạt trong quá trình chuẩn bị mẫu thử, phải đảm bảo chất diện hoạt đó không chứa độc tố với vi sinh vật và chất diện hoạt phải tương thích với chất bất hoạt khác cùng được sử dụng như mô tả trong phần Trung hòa/loại bỏ tác dụng của chất ức chế.
Kiểm tra chất lượng môi trường, sự phù hợp của phương pháp, chứng âm tính
Phải chứng minh phương pháp thử có khả năng phát hiện vi sinh vật khi có mặt mẫu thử.
Phải chứng minh sự phù hợp của phương pháp khi có sự thay đổi trong quá trình tiến hành thí nghiệm hoặc có sự thay đổi về sản phẩm lảm ảnh hưởng đến kết quả của phép thử.
Chuẩn bị chủng vi sinh vật
Có thể sử dụng hỗn dịch chủng vi sinh vật ổn định đã được tiêu chuẩn hóa hoặc chuẩn bị như trình bày dưới đây. Kỹ thuật lưu giữ chủng gốc phải đảm bảo vi sinh vật được cấy truyền không quá 5 lần từ giống gốc ban đầu.
• Chủng vi sinh vật hiếu khí:
Cấy chủng vi sinh vật thử nghiệm riêng rẽ trên môi trường lỏng casein đậu tương hoặc môi trường thạch casein đậu tương và ủ ở 30 °C đến 35 °C trong 18 h đến 24 h. Cấy chủng Candida albicans riêng rẽ trên môi trường thạch Sabouraud-dextrose hoặc môi trường lỏng Sabouraud-dextrose và ủ ở 20 °C đến 25 °C trong 2 ngày đến 3 ngày.
- Staphylococcus aureus ATCC 6538, NCIMB 9518, CIP 4.83 hoặc NBRC 13276
- Pseudomonas aeruginosa ATCC 9027, NCIMB 8626, CIP 82.118 hoặc NBRC 13275
- Escherichia coli ATCC 8739, NCIMB 8545, CIP 53.126, hoặc NBRC 3972
- Salmonella enterica subsp. enterica serovar Typhimurium ATCC 14028 hoặc có thể thay thế bằng Salmonella enterica subsp. enterica serovar Abony NBRC 100797, NCTC 6017, hoặc CIP 80.39
- Candida albicans ATCC 10231, NCPF 3179, IP 48.72 hoặc NBRC 1594.
Sử dụng dung dịch đệm natri clorid-pepton pH 7,0 hoặc dung dịch đệm phosphat pH 7,2 để pha hỗn dịch chủng vi sinh vật. Hỗn dịch được sử dụng trong vòng 2 h, nếu bảo quản ở 2 °C đến 8 °C thì có thể sử dụng trong vòng 24 h.
• Clostridia:
Sử dụng chủng Clostridium sporogenes ATCC 11437 (NBRC 14293, NCIMB 12343, CIP 100651) hoặc ATCC 19404 (NCTC 532 hoặc CIP 79.3). Cấy chủng Clostridia trên môi trường tăng sinh cho Clostridia và ủ trong điều kiện kỵ khí ở 30 °C đến 35 °C trong 24 h đến 48 h. Ngoài ra, thay vì chuẩn bị chủng CI. sporogenes dạng sinh dưỡng như ở trên, có thể sử dụng hỗn dịch bào tử ổn định. Hỗn dịch bào tử được bảo quản ở 2 °C đến 8 °C trong khoảng thời gian đã được thẩm định.
Chứng âm tính
Để đánh giá điều kiện thử nghiệm, tiến hành mẫu chứng âm tính bằng cách sử dụng dung dịch pha loãng thay cho mẫu thử. Mẫu chứng âm tính phải không được có sự phát triển của vi sinh vật. Mẫu chứng âm tính cũng có thể được tiến hành như cùng lúc với mẫu thử như mô tả trong mục Cách tiến hành. Cần phải xem xét hệ thống kỹ lưỡng khi mẫu chứng âm tính không đạt.
Kiểm tra chất lượng môi trường
Tiến hành kiểm tra chất lượng đối với mỗi lô môi trường pha chế sẵn hoặc mỗi lô môi trường được pha chế từ môi trường khô hay từ các thành phần riêng lẻ.
Kiểm tra chất lượng môi trường theo chỉ dẫn trong Bảng 13.6.4.
Bảng 13.6.4 - Kiểm tra chất lượng môi trường
|
| Môi trường | Tính chất | Chủng thử nghiệm |
| Vi khuẩn Gram âm dung nạp mật | Môi trường lỏng tăng sinh Enterobacteria- Mossel | Kích thích sự phát triển | E. coli P. aeruginosa |
| Ức chế sự phát triển | S. aureus | ||
| Môi trường thạch muối mật tím đỏ | Kích thích sự phát triển và đặc hiệu | E. coli P. aeruginosa | |
| Escherichia coli | Môi trường lỏng MacConkey | Kích thích sự phát triển | E. coli |
| Ức chế sự phát triển | S. aureus | ||
| Môi trường thạch MacConkey | Kích thích sự phát triển và đặc hiệu | E. coli | |
| Salmonella | Môi trường lỏng tăng sinh Salmonella Rappaport Vassiliadis | Kích thích sự phát triển | Salmonella enterica subsp. enterica serovar Typhimurium hoặc Salmonella enterica subsp. enterica serovar Abony |
| Ức chế sự phát triển | S. aureus | ||
| Môi trường thạch Xylose Lysin Deoxycholat | Kích thích sự phát triển và đặc hiệu | Salmonella enterica subsp. enterica serovar Typhimurium hoặc Salmonella enterica subsp. enterica serovar Abony | |
| Pseudomonas aeruginosa | Môi trường thạch Cetrimid | Kích thích sự phát triển | P. aeruginosa |
| Ức chế sự phát triển | E. coli | ||
| Staphylococcus aureus | Môi trường thạch muối Mannitol | Kích thích sự phát triển và đặc hiệu | S. aureus |
| Ức chế sự phát triển | E. coli | ||
| Clostridia | Môi trường tăng sinh cho Clostridia | Kích thích sự phát triển | Cl. sporogenes |
| Môi trường thạch Columbia | Kích thích sự phát triển | Cl. sporogenes | |
| Candida albicans | Môi trường lỏng Sabouraud-dextrose | Kích thích sự phát triển | C. albicans |
| Môi trường thạch Sabouraud-dextrose | Kích thích sự phát triển và đặc hiệu | C. albicans |
Thử nghiệm kiểm tra khả năng kích thích sự phát triển vi sinh vật của môi trường lỏng:
Cấy vào môi trường lỏng một lượng nhỏ vi sinh vật phù hợp (không quá 100 CFU). Ủ ở nhiệt độ xác định trong thời gian không lâu hơn khoảng thời gian ngắn nhất mà phép thử quy định. Vi sinh vật phát triển tốt, rõ ràng trên lô môi trường mới khi so sánh với sự phát triển của vi sinh vật trên lô môi trường đã đạt yêu cầu chất lượng.
Thử nghiệm kiểm tra khả năng kích thích sự phát triển vi sinh vật của môi trường đặc:
Dùng kĩ thuật cấy trải bề mặt, cấy vào mỗi đĩa môi trường đặc một lượng nhỏ vi sinh vật phù hợp (không quá 100 CFU). Ủ ở nhiệt độ xác định trong thời gian không lâu hơn khoảng thời gian ngắn nhất mà phép thử quy định. So sánh sự phát triển của vi sinh vật trên lô môi trường mới với sự phát triển của vi sinh vật trên lô môi trường đã đạt yêu cầu chất lượng.
Thử nghiệm kiểm tra khả năng ức chế sự phát triển vi sinh vật của môi trường lỏng và đặc:
Cấy vào môi trường loại vi sinh vật phù hợp với lượng ít nhất là 100 CFU. Ủ ở nhiệt độ xác định trong thời gian không ít hơn khoảng thời gian dài nhất mà phép thử quy định. Vi sinh vật phải không phát triển trên môi trường.
Thử nghiệm kiểm tra tính đặc hiệu của môi trường:
Dùng kĩ thuật cấy trải bề mặt, cấy vào mỗi đĩa môi trường một lượng nhỏ vi sinh vật phù hợp (không quá 100 CFU). Ủ ở nhiệt độ xác định trong thời gian mà phép thử quy định. So sánh hình thái khuẩn lạc và phản ứng đặc hiệu của vi sinh vật trên lô môi trường mới với hình thái khuẩn lạc và phản ứng đặc hiệu của vi sinh vật trên lô môi trường đã đạt yêu cầu chất lượng.
Sự phù hợp của phương pháp thử
Đối với mỗi mẫu thử, tiến hành chuẩn bị mẫu thử như mô tả trong mục Cách tiến hành. Cấy chủng vi sinh vật chứng vào môi trường nuôi cấy tại thời điểm trộn mẫu thử vào môi trường. Cấy các chủng vi sinh vật một cách riêng rẽ. Số lượng vi sinh vật được cấy vào mẫu thử với lượng không quá 100 CFU.
Tiến hành thử nghiệm như mô tả trong mục Cách tiến hành và ủ trong thời gian ngắn nhất mà phép thử quy định.
Vi sinh vật gây bệnh được phát hiện bằng các phản ứng đặc hiệu được mô tả trong mục Cách tiến hành.
Nếu bản chất mẫu thử có tính ức chế vi sinh vật, cần thay đổi qui trình thử cho phù hợp (xem phần Trung hòa/loại bỏ tác dụng của chất ức chế).
Với mẫu thử có tính ức chế vi sinh vật nhưng phương pháp đã thử không thể trung hòa hoạt tính ức chế này được thì mẫu thử được coi là không bị nhiễm các vi sinh vật đã được thử nghiệm.
Cách tiến hành
Vi khuẩn Gram âm dung nạp mật
Chuẩn bị mẫu thử và tiền ủ:
Lấy không dưới 1 g hoặc 1 ml mẫu thử, pha loãng 10 lần như mô tả trong Phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử chỉ khác là dùng môi trường lỏng casein đậu tương thay cho dung dịch pha loãng, trộn đều và ủ ở 20 °C đến 25 °C trong một thời gian thích hợp để phục hồi lại vi khuẩn mà không làm tăng số lượng vi khuẩn (thường từ 2 h đến không quá 5 h).
Phát hiện vi khuẩn:
Trừ khi có chỉ dẫn riêng, chuyển một thể tích tương đương với 1 g hoặc 1 ml mẫu thử được chuẩn bị ở phần Chuẩn bị mẫu thử và tiền ủ vào 100 ml môi trường lỏng tăng sinh Enterobacteria-Mossel. Ủ ở 30 °C đến 35 °C trong 24 h đến 48 h. Cấy chuyển lên môi trường thạch muối mật tím đỏ. Ủ ở 30 °C đến 35 °C trong 18 h đến 24 h.
Mẫu thử không có Enterobacteria nếu không thấy khuẩn lạc mọc trên môi trường.
Đánh giá số lượng:
• Phân lập và cấy chuyển:
Cấy lượng mẫu thử đã được chuẩn bị theo phần Chuẩn bị mẫu thử và tiền ủ tương ứng chứa 0,1 g; 0,01 g và 0,001 g hoặc 0,1 ml; 0,01 ml và 0,001 ml vào thể tích thích hợp môi trường lỏng tăng sinh Enterobacteria-Mossel. Ủ ở 30 °C đến 35 °C trong 24 h đến 48 h. Cấy chuyển từ mỗi ống môi trường trên sang môi trường thạch muối mật tím đỏ. Ủ ở 30 °C đến 35 °C trong 18 h đến 24 h.
• Đánh giá kết quả:
Khuẩn lạc mọc trên môi trường chứng tỏ kết quả dương tính. Ghi lượng nhỏ nhất của chế phẩm mà ở đó cho kết quả dương tính và lượng lớn nhất của chế phẩm mà ở đó cho kết quả âm tính. Xác định số lượng vi khuẩn theo Bảng 13.6.5.
Bảng 13.6.5 - Đánh giá kết quả
| Kết quả đối với mỗi lượng sản phẩm | Số vi khuẩn có trong 1 gam hoặc 1 ml sản phẩm | ||
| 0,1 g hoặc 0,1 ml | 0,01 g hoặc 0,01 ml | 0,001 g hoặc 0,001 ml | |
| + | + | + | > 103 |
| + | + | - | < 103 và > 102 |
| + | - | - | < 102 và > 10 |
| - | - | - | < 10 |
Escherichia coli
Chuẩn bị mẫu thử và tiền ủ:
Lấy không dưới 1 g hoặc 1 ml, pha loãng 10 lần như mô tả trong phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử. Cấy 10 ml hoặc một lượng tương ứng với 1 g hoặc 1 ml mẫu thử vào một thể tích phù hợp môi trường lỏng casein đậu tương, trộn đều và ủ ở 30 °C đến 35 °C trong 18 h đến 24 h.
Phân lập và cấy chuyển:
Lắc bình môi trường lỏng casein đậu tương, cấy chuyển 1 ml môi trường lỏng casein đậu tương sang 100 ml môi trường lỏng MacConkey và ủ ở 42 °C đến 44 °C trong 24 h đến 48 h. Tiếp tục cấy chuyển sang đĩa môi trường thạch MacConkey và ủ ở 30 °C đến 35 °C trong 18 h đến 72 h.
Đánh giá kết quả:
Khuẩn lạc mọc trên môi trường cho thấy mẫu thử có thể có E. coli. Tiếp tục xác định bằng nhiều phản ứng sinh hóa khác.
Mẫu thử không có E. coli nếu không có khuẩn lạc mọc trên môi trường hoặc các phản ứng sinh hóa để xác định E. coli có kết quả âm tính.
Salmonella
Chuẩn bị mẫu thử và tiền ủ: Lấy lượng mẫu thử đã xử lý như mô tả trong Phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử tương ứng với 10 g hoặc 10 ml mẫu thử và cấy vào một thể tích phù hợp môi trường lỏng casein đậu tương, trộn đều và ủ ở 30 °C đến 35 °C trong 18 h đến 24 h.
Phân lập và cấy chuyển: Cấy chuyển 0,1 ml môi trường lỏng casein đậu tương sang 10 ml môi trường tăng sinh Salmonella Rappaport Vassiliadis và ủ ở 30 °C đến 35 °C trong 18 h đến 24 h. Tiếp tục cấy chuyển sang đĩa môi trường thạch xylose, lysin, deoxycholat và ủ ở 30 °C đến 35 °C trong 18 h đến 48 h.
Đánh giá kết quả:
Khuẩn lạc màu đỏ, có hoặc không có trung tâm màu đen mọc trên môi trường cho thấy mẫu thử có thể có Salmonella. Tiếp tục xác định bằng nhiều phản ứng sinh hóa khác.
Mẫu thử không có Salmonella nếu không có khuẩn lạc có đặc điểm mô tả ở trên mọc trên môi trường hoặc các phản ứng sinh hóa để xác định Salmonella có kết quả âm tính.
Pseudomonas aeruginosa
Chuẩn bị mẫu thử và tiền ủ: Lấy không dưới 1 g hoặc 1 ml mẫu thử, pha loãng 10 lần như mô tả trong phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử. Cấy 10 ml hoặc 1 lượng tương ứng với 1 g hoặc 1 ml mẫu thử vào một thể tích phù hợp môi trường lỏng casein đậu tương, trộn đều. Đối với thuốc dán, lọc một lượng mẫu thử tương đương với 1 miếng dán như mô tả trong phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử qua màng lọc vô khuẩn và cấy vào 100 ml môi trường lỏng casein đậu tương. Ủ ở 30 °C đến 35 °C trong 18 h đến 24 h.
Phân lập và cấy chuyển: Cấy chuyển sang đĩa môi trường thạch cetrimid và ủ ở 30 °C đến 35 °C trong 18 h đen 72 h.
Đánh giá kết quả:
Khuẩn lạc mọc trên môi trường cho thấy mẫu thử có thể có P. aeruginosa. Tiếp tục xác định bằng nhiều phản ứng sinh hóa khác.
Mẫu thử không có P. aeruginosa nếu không có khuẩn lạc mọc trên môi trường hoặc các phản ứng sinh hóa để xác định P. aeruginosa có kết quả âm tính.
Staphylococcus aureus
Chuẩn bị mẫu thử và tiền ủ:
Lấy không dưới 1 g hoặc 1 ml mẫu thử, pha loãng 10 lần như mô tả trong Phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử. Cấy 10 ml hoặc 1 lượng tương ứng với 1 g hoặc 1 ml mẫu thử vào một thể tích phù hợp môi trường lỏng casein đậu tương, trộn đều. Đối với thuốc dán, lọc một lượng mẫu thử tương đương với 1 miếng dán như mô tả trong Phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử qua màng lọc vô khuẩn và cấy vào 100 ml môi trường lỏng casein đậu tương. Ủ ở 30 °C đến 35 °C trong 18 h đến 24 h.
Phân lập và cấy chuyển:
Cấy chuyển sang đĩa môi trường thạch muối manitol và ủ ở 30 °C đến 35 °C trong 18 h đến 72 h.
Đánh giá kết quả:
Khuẩn lạc màu vàng hoặc trắng có vòng màu vàng bao quanh mọc trên môi trường cho thấy mẫu thử có thể có S. aureus. Tiếp tục xác định bằng nhiều phản ứng sinh hóa khác.
Mẫu thử không có S. aureus nếu không có khuẩn lạc có đặc điểm mô tả ở trên mọc trên môi trường hoặc các phản ứng sinh hóa để xác định S. aureus có kết quả âm tính.
Clostridia
Chuẩn bị mẫu thử và sốc nhiệt:
Chuẩn bị tối thiểu 20 ml mẫu thử được pha loãng 10 lần như mô tả trong phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử tương ứng với không dưới 2 g hoặc 2 ml mẫu thử. Chia mẫu thử thành 2 phần, mỗi phần ít nhất 10 ml. Một phần đun nóng tới 80 °C trong 10 min rồi làm lạnh ngay. Phần còn lại không được đun nóng.
Phân lập và cấy chuyển:
Cấy 10 ml hoặc lượng tương đương với 1 g hoặc 1 ml mẫu thử của cả 2 phần trên vào thể tích phù hợp môi trường tăng sinh cho Clostridia. Ủ cả hai mẫu ở điều kiện kỵ khí ở 30 °C đến 35 °C trong 48 h. Sau khi ủ, từ mỗi bình cấy chuyển sang môi trường thạch Columbia và tiếp tục ủ ở điều kiện kỵ khí ở 30 °C đến 35 °C trong 48 h đến 72 h.
Đánh giá kết quả:
Sự phát triển của trực khuẩn trong điều kiện kỵ khí (có hoặc không có bào tử) trên môi trường, phản ứng catalase âm tính cho thấy mẫu thử có thể có Clostridia. Tiếp tục xác định bằng nhiều phản ứng sinh hóa khác.
Mẫu thử không có Clostridia nếu không có khuẩn lạc có đặc điểm mô tả ở trên mọc trên môi trường hoặc các phản ứng sinh hóa để xác định Clostridia có kết quả âm tính.
Candida albicans
Chuẩn bị mẫu thử và tiền ủ:
Chuẩn bị mẫu thử như mô tả trong phần Chuẩn bị mẫu thử của mục Kiểm tra sự phù hợp của phương pháp đếm khi có mặt mẫu thử, cấy 10 ml hoặc một lượng tương ứng với 1 g hoặc 1 ml vào 100 ml môi trường lỏng Sabouraud-dextrose, trộn đều và ủ ở 30 °C đến 35 °C trong 3 ngày đến 5 ngày.
Phân lập và cấy chuyển:
Cấy chuyển sang môi trường thạch Sabouraud-dextrose và ủ ở 30 °C đến 35 °C trong 24 h đến 48 h.
Đánh giá kết quả:
Khuẩn lạc màu trắng mọc trên môi trường cho thấy mẫu thử có thể có C. albicans. Tiếp tục xác định bằng nhiều phản ứng sinh hóa khác.
Mẫu thử không có C. albicans nếu không có khuẩn lạc có đặc điểm mô tả ở trên mọc trên môi trường hoặc các phản ứng sinh hóa để xác định C. albicans có kết quả âm tính.
DUNG DỊCH PHA LOÃNG VÀ MÔI TRƯỜNG
Các dung dịch và môi trường nuôi cấy được chuẩn bị theo cách dưới đây. Có thể sử dụng môi trường khác khi chứng minh được sự phù hợp của chúng.
Dung dịch đệm gốc
Cân 34 g kali dihydrophosphat (TT) và cho vào bình định mức 1000 ml, thêm 500 ml nước tinh khiết, điều chỉnh pH 7,2 ± 0,2 bằng natri hydroxyd (TT). Thêm nước tinh khiết tới vạch, trộn đều. Phân chia vào các bình, tiệt khuẩn, bảo quản ở 2 °C đến 8 °C.
Dung dịch đệm phosphat pH 7,2
Pha loãng dung dịch đệm gốc với nước tinh khiết theo tỷ lệ 1: 800 (tt/tt) và tiệt khuẩn.
Dung dịch đệm natri clorid-pepton pH 7,0
| Kali dihydrophosphat | 3,6 g |
| Dinatri hydrophosphat dihydrat | 7,2 g tương ứng với 0,067 M phosphat |
| Natri clorid | 4,3 g |
| Pepton (từ thịt hoặc casein) | 1,0 g |
| Nước tinh khiết | 1000 ml |
Tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định
Môi trường lỏng casein đậu tương
| Casein thủy phân bởi pancreatin | 17,0 g |
| Bột đậu tương thủy phân bởi papain | 3,0 g |
| Natri clorid | 5,0 g 2,5 g |
| Dikali hydrophosphat | 2,5 g |
| Glucose monohydrat | 1000ml |
| Nước tinh khiết |
|
Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 7,3 ± 0,2 ở 25 °C. Tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường thạch Sabouraud-dextrose
| Dextrose | 40,0 g |
| Casein thủy phân bởi pancreatin | 5,0 g |
| Pepton từ mô động vật | 5,0 g |
| Thạch | 15,0 g |
| Nước tinh khiết | 1000 ml |
Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 5,6 ± 0,2 ở 25 °C. Tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường thạch khoai tây dextrose
| Dịch chiết từ khoai tây | 200,0 g |
| Dextrose | 20,0 g |
| Thạch | 15,0 g |
| Nước tinh khiết | 1000 ml |
Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 5,6 ± 0,2 ở 25 °C. Tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường Sabouraud-dextrose lỏng
| Dextrose | 20,0 g |
| Casein thủy phân bởi pancreatin | 5,0 g |
| Pepton từ mô động vật | 5,0 g |
| Nước tinh khiết | 1000 ml |
Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 5,6 ± 0,2 ở 25 °C. Tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường lỏng tăng sinh Enterobacteria-Mossel
| Genlatin thủy phân bởi pancreatin | 10,0 g |
| Glucose monohydrat | 5,0 g |
| Mật bò khô | 20,0 g |
| Kali dihydrophosphat | 2,0 g |
| Dinatri hydrophosphat dihydrat | 8,0 g |
| Xanh brilliant | 15 mg |
| Nước tinh khiết | 1000 ml |
Điều chỉnh pH sao cho sau khi tiệt khuẩn khuẩn, môi trường có pH 7,2 ± 0,2 ở 25 °C. Tiệt khuẩn bằng nhiệt ở 100 °C trong 30 min và làm lạnh ngay.
Môi trường thạch muối mật tím đỏ
| Cao nấm men | 3,0 g |
| Gelatin thủy phân bởi pancreatin | 7,0 g |
| Muối mật | 1,5 g |
| Natri clorid | 5,0 g |
| Glucose monohydrat | 10,0 g |
| Thạch | 15,0 g |
| Đỏ trung tính | 30 mg |
| Tím tinh thể | 2 mg |
| Nước tinh khiết | 1000 ml |
Điều chỉnh pH sao cho sau khi tiệt khuẩn, môi trường có pH 7,4 ± 0,2 ở 25 °C. Tiệt khuẩn bằng cách đun sôi, không được hấp trong nồi hấp.
Môi trường lỏng MacConkey
| Gelatin thủy phân bởi pancreatin Lactose monohydrat Mật bò khô Tía bromocresol Nước tinh khiết | 20,0 g 10,0 g 5,0 g 10 mg 1000 ml |
Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 7,3 ± 0,2 ở 25 °C. Tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường thạch MacConkey
| Gelatin thủy phân bởi pancreatin | 17,0 g |
| Pepton (thịt hoặc casein) | 3,0 g |
| Lactose monohydrat | 10,0 g |
| Natri clorid | 5,0 g |
| Muối mật | 1,5 g |
| Thạch | 13,5 g |
| Đỏ trung tính | 30,0 mg |
| Tím tinh thể | 1,0 mg |
| Nước tinh khiết | 1000 ml |
| Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 7,1 ± 0,2 ở 25 °C. Đun sôi 1 min, sau đó hấp tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định. Môi trường lỏng tăng sinh Salmonella Rappaport Vassiliadis | |
| Pepton đậu tương | 4,5 g |
| Magnesi clorid hexahydrat | 29,0 g |
| Natri clorid | 8,0 g |
| Dikali phosphat | 0,4 g |
| Kali dihydrophosphat | 0,6 g |
| Xanh malachit | 0,036 g |
| Nước tinh khiết | 1000 ml |
| Hòa tan, đun nóng nhẹ. Hấp tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định với nhiệt độ không quá 115 °C. Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 5,2 ± 0,2 ở 25 °C. Môi trường thạch xylose, lysin, desoxycholat | |
| Xylose | 3,5 g |
| L-Lysin | 5,0 g |
| Lactose monohydrat | 7,5 g |
| Sucrose | 7,5 g |
| Natri clorid | 5,0 g |
| Cao nấm men | 3,0 g |
| Đỏ phenol | 80 mg |
| Thạch | 13,5 g |
| Natri desoxycholat | 2,5 g |
| Natri thiosulfat | 6,8 g |
| Sắt amoni citrat | 0,8 g |
| Nước tinh khiết | 1000 ml |
Điều chỉnh pH sao cho sau khi tiệt khuẩn, môi trường có pH 7,4 ± 0,2 ở 25 °C. Đun sôi, để nguội đến 50 °C thì rót môi trường vào đĩa Petri. Không được hấp tiệt khuẩn trong nồi hấp.
Môi trường thạch cetrimid
| Gelatin thủy phân bởi pancreatin | 20,0 g |
| Magnesi clorid | 1,4 g |
| Dikali sulfat | 10,0 g |
| Cetrimid | 0,3 g |
| Thạch | 13,6 g |
| Nước tinh khiết | 1000 ml |
| Glycerol | 10 ml |
Đun sôi trong 1 min, khuấy đều. Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 7,2 ± 0,2 ở 25 °C. Tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường thạch muối manitol
| Casein thủy phân bởi pancreatin | 5,0 g |
| Pepton từ mô động vật | 5,0 g |
| Cao thịt bò | 1,0 g |
| D-Manitol | 10,0 g |
| Natri clorid | 75,0 g |
| Thạch | 15,0 g |
| Đỏ phenol | 0,025 g |
| Nước tinh khiết | 1000 ml |
Đun sôi trong 1 min, lắc đều. Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 7,4 ± 0,2 ở 25 °C. Hấp tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường tăng sinh cho Clostridia
| Cao thịt bò | 10,0 g |
| Pepton | 10,0 g |
| Cao nấm men | 3,0 g |
| Tinh bột dễ tan | 1,0 g |
| Glucose monohydrat | 5,0 g |
| Cystein hydroclorid | 0,5 g |
| Natri clorid | 5,0 g |
| Natri acetat | 3,0 g |
| Thạch | 0,5 g |
| Nước tinh khiết | 1000 ml |
Làm ẩm thạch, hòa tan các thành phần bằng cách đun nóng tới sôi, khuấy đều liên tục. Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 6,8 ± 0,2 ở 25 °C. Hấp tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định.
Môi trường thạch Columbia
| Casein thủy phân bởi pancreatin | 10,0 g |
| Pepton từ thịt | 5,0 g |
| Tim thủy phân bởi pancreatin | 3,0 g |
| Cao nấm men | 5,0 g |
| Tinh bột ngô | 1,0 g |
| Natri clorid | 5,0 g |
| Thạch | 10,0 g đến 15,0 g |
| Nước tinh khiết | 1000 ml |
Làm ẩm thạch, hòa tan bằng cách đun nóng tới sôi, khuấy đều liên tục. Điều chỉnh pH sao cho sau khi hấp tiệt khuẩn, môi trường có pH 7,3 ± 0,2 ở 25 °C. Hấp tiệt khuẩn trong nồi hấp theo quy trình đã được thẩm định. Để nguội tới 45 °C đến 50 °C, nếu cần, thêm 20 mg gentamycin base và rót vào đĩa Petri.
YÊU CẦU GIỚI HẠN NHIỄM KHUẨN
Nếu không có chỉ dẫn trong chuyên luận riêng thì giới hạn nhiễm khuẩn cho các loại thuốc không tiệt khuẩn trong quá trình sản xuất được ghi trong Bảng 13.6.6.
Bảng 13.6.6 - Yêu cầu giới hạn nhiễm khuẩn
| Loại chế phẩm | Tổng số vi sinh vật hiếu khí (CFU/g hoặc CFU/ml) | Tổng số nấm (CFU/g hoặc CFU/ml) | Vi sinh vật gây bệnh |
| Thuốc dùng điều trị bỏng và các vết loét sâu | Không có vi sinh vật trong 1 g (ml) | ||
| Nguyên liệu hóa dược | 103 | 102 | - |
| Thành phẩm hóa dược dùng để uống (dạng khô: viên nén, nang...) | 103 | 102 | Không có Escherichia coli trong 1 g (ml) |
| Thành phẩm hóa dược dùng để uống (dạng nước: siro, dung dịch...) | 102 | 101 | Không có Escherichia coli trong 1 g (ml) |
| Thuốc dùng theo đường trực tràng | 103 | 102 | - |
| Thuốc dùng theo đường niêm mạc miệng, lợi, răng, da, mũi, tai | 102 | 101 | Không có Staphylococcus aureus, Pseudomonas aeruginosa trong 1 g (ml) |
| Thuốc dùng theo đường âm đạo | 102 | 101 | Không có Staphylococcus aureus, Pseudomonas aeruginosa, Candida albicans trong 1 g (ml) |
| Thuốc dán (áp dụng cho 1 miếng dán) | 102 | 101 | Không có Staphylococcus aureus, Pseudomonas aeruginosa trong 1 miếng dán |
| Thuốc hít (bao gồm cả dạng khí dung) | 102 | 101 | Không có Staphylococcus aureus, Pseudomonas aeruginosa, vi khuẩn Gram âm dung nạp mật trong 1 g (ml). |
| Thuốc uống có nguồn gốc tự nhiên (động, thực vật, khoáng chất); cao thuốc, cồn thuốc dùng để sản xuất thuốc uống từ dược liệu | 104 | 102 | Không quá 102 CFU vi khuẩn Gram âm dung nạp mật trong 1 g (ml). Không có Salmonella trong 10 g (ml). Không có Escherichia coli, Staphylococcus aureus trong 1 g (ml). |
| Thuốc từ dược liệu (thuốc thang...) được xử lý bằng ethanol thấp độ hoặc nước nóng (không sôi) trước khi dùng | 105 | 104 | Không quá 104 CFU vi khuẩn Gram âm dung nạp mật trong 1 g (ml) Không có Salmonella trong 10 g (ml) Không có Escherichia coli trong 1 g (ml) |
| Thuốc từ dược liệu (thuốc thang, trà...) được xử lý bằng nước sôi trước khi dùng | 107 Số lượng tối đa được chấp nhận: 5 x 107 | 105 Số lượng tối đa được chấp nhận: 5 x 105 | Không quá 103 CFU Escherichia coli trong 1 g (ml) Không có Salmonella trong 10 g (ml) |
13.7 THỬ VÔ KHUẨN
Quy định chung
Phép thử này được áp dụng nhằm phát hiện sự có mặt của vi khuẩn, nấm trong các nguyên liệu, chế phẩm và dụng cụ mà theo Dược điển cần phải vô khuẩn. Tuy nhiên, kết quả âm tính chỉ có nghĩa là không phát hiện được vi sinh vật tạp nhiễm trong chế phẩm đã thử trong điều kiện của thử nghiệm.
Những biện pháp phòng tạp nhiễm vi sinh vật
Thử vô khuẩn phải được tiến hành trong điều kiện vô khuẩn. Để đạt được điều kiện này, khu vực thử nghiệm phải phù hợp với qui trình thử vô khuẩn được tiến hành. Các biện pháp phòng tránh tạp nhiễm phải đảm bảo không ảnh hưởng đến vi sinh vật có thể có trong mẫu thử. Khu vực thử vô khuẩn phải được đánh giá thường xuyên bằng phương pháp lấy mẫu tại khu vực làm việc hoặc bằng cách tiến hành mẫu đối chứng thích hợp.
Môi trường và nhiệt độ ủ
Có thể sử dụng môi trường được pha chế theo chỉ dẫn bên dưới hoặc sử dụng các môi trường thương mại có sẵn nếu chúng đáp ứng yêu cầu về khả năng dinh dưỡng (growth promotion test) theo mục Kiểm tra chất lượng môi trường. Môi trường thioglycolat lỏng được dùng để phát hiện vi khuẩn hiếu khí và kỵ khí. Môi trường lỏng casein đậu tương được dùng để phát hiện vi khuẩn hiếu khí và nấm.
Môi trường thioglycolat lỏng
| L - Cystin | 0,5 g |
| Thạch | 0,75 g |
| Natri clorid | 2,5 g |
| Glucose monohydrat/khan | 5,5/5,0 g |
| Cao nấm men (tan được trong nước) | 5,0 g |
| Casein thủy phân bởi pancreatin | 15,0 g |
| Natri thioglycolat | 0,5 g |
| hoặc acid thioglycolic | 0,3 ml |
| Dung dịch natri resazurin (dung dịch natri resazurin 1 g/lit mới pha) | 1,0 ml |
| Nước | 1000ml |
| pH sau khi tiệt khuẩn: 7,1 ± 0,2 |
|
Trộn L - Cystin, thạch, natri clorid, glucose, cao nấm men, casein thủy phân bởi pancreatin với nước và đun nóng đến tan hoàn toàn. Hòa tan natri thioglycolat hoặc acid thioglycolic vào dung dịch trên, thêm dung dịch natri hydroxyd 1M (nếu cần) để điều chỉnh pH sao cho môi trường sau khi tiệt khuẩn có pH 7,1 ± 0,2. Nếu cần phải lọc thì đun nóng lại dung dịch (tránh đun sôi) và lọc nóng qua giấy lọc đã thấm ướt. Thêm dung dịch resazurin, trộn đều và đóng môi trường vào ống (hoặc bình) thích hợp sao cho sau khi kết thúc quá trình ủ, không quá một nửa thể tích phía trên của môi trường trong ống (hoặc bình) bị chuyển màu chỉ thị do hấp thụ oxy. Hấp tiệt khuẩn theo qui trình đã được thẩm định. Bảo quản môi trường ở nhiệt độ 2 °C đến 25 °C trong bao bì vô khuẩn, kín khí. Nếu 1/3 thể tích phía trên của ống (hoặc bình) môi trường có màu hồng, môi trường không thích hợp để thử nghiệm. Có thể phục hồi lại môi trường bằng cách đun cách thủy cho mất màu rồi làm lạnh nhanh, chú ý tránh tạp nhiễm không khí không vô khuẩn vào môi trường và chỉ sử dụng môi trường đã phục hồi này một lần. Không sử dụng môi trường quá thời hạn sử dụng đã thẩm định.
Ủ môi trường thioglycolat lỏng ở 30 °C đến 35 °C.
Với các chế phẩm có chứa chất bảo quản nhóm thủy phân mà không áp dụng phương pháp màng lọc được, có thể ủ môi trường thioglycolat lỏng ở 20 °C đến 25 °C thay vì sử dụng môi trường lỏng casein đậu tương và trường hợp này môi trường thioglycolat lỏng phải đạt yêu cầu về khả năng dinh dưỡng theo mục Kiểm tra chất lượng môi trường.
Khi được quy định hoặc chứng minh rõ ràng, có thể sử dụng môi trường thioglycolat lỏng thay thế. Chuẩn bị hỗn hợp môi trường có thành phần giống như môi trường thioglycolat lỏng ở trên nhưng không có thành phần thạch và dung dịch natri resazurin, tiệt khuẩn giống như ở trên. Môi trường sau khi tiệt khuẩn có pH 7,1 ± 0,2. Đun nóng trong nồi cách thủy trước khi sử dụng và ủ ở 30 °C đến 35°C trong điều kiện kị khí.
Môi trường soybean casein lỏng
| Casein thủy phân bởi pancreatin | 17,0 g |
| Bột đậu tương thủy phân bởi papain | 3,0 g |
| Natri clorid | 5,0 g |
| Dikali hydrophosphat | 2,5 g |
| Glucose monohydrat/khan | 2,5/2,3 g |
| Nước | 1000 ml |
| pH sau khi tiệt khuẩn: 7,3 ± 0,2 |
|
Hòa tan tất cả các chất rắn trong nước, đun nóng nhẹ để cho tan hoàn toàn. Để nguội đến nhiệt độ phòng. Thêm dung dịch natri hydroxyd 1M (nếu cần) để điều chỉnh pH sao cho môi trường sau khi tiệt khuẩn có pH 7,3 ± 0,2. Lọc (nếu cần) để cho môi trường trong. Phân chia môi trường vào ống (hoặc bình) thích hợp và hấp tiệt khuẩn theo qui trình đã được thẩm định. Bảo quản môi trường ở nhiệt độ 2 °C đến 25 °C trong bao bì vô khuẩn, kín khí, trừ khi môi trường được sử dụng ngay. Không sử dụng môi trường quá thời hạn sử dụng đã thẩm định.
Ủ môi trường soybean casein lỏng ở 20 °C đến 25 °C.
Môi trường nuôi cấy phải đạt chất lượng theo mục Kiểm tra chất lượng môi trường. Có thể kiểm tra chất lượng môi trường trước hoặc song song với thử vô khuẩn mẫu thử.
Kiểm tra chất lượng môi trường
a) Độ vô khuẩn
Lấy ngẫu nhiên một vài ống (hoặc bình) môi trường mới sản xuất, đem ủ ở nhiệt độ 30 °C đến 35 °C trong 14 ngày đối với môi trường thioglycolat lỏng; ủ ở nhiệt độ 20 °C đến 25 °C trong 14 ngày đối với môi trường soybean casein lỏng. Các loại môi trường phải không được có vi khuẩn, nấm mọc.
b) Khả năng dinh dưỡng
Tiến hành kiểm tra khả năng dinh dưỡng đối với mỗi lô môi trường được pha chế sẵn (ready- prepared) hoặc mỗi lô môi trường được pha chế từ môi trường khô nhiều thành phần hoặc được pha chế từ các thành phần riêng lẻ. Sử dụng các chủng vi sinh vật phù hợp theo quy định trong Bảng 13.7.1
Bảng 13.7.1 - Chủng vi sinh vật phù hợp để kiểm tra khả năng dinh dưỡng và sự phù hợp của phương pháp
| Vi khuẩn hiếu khí |
|
| Staphyloccocus aureus | ATCC 6358, CIP 4.83, NCTC 10788, NCIMB 9518, NBRC 13276 |
| Bacillus subtilis | ATCC 6633, CIP 52.62, NCIMB 8054, NBRC 3134 |
| Pseudomonas aeruginosa | ATCC 9027, NCIMB 8626, CIP 82.118, NBRC 13275 |
| Vi khuẩn kị khí |
|
| Clostridium sporogenes | ATCC 19404, CIP 79.3, NCTC 532, ATCC 11437, NBRC 14293 |
| Nấm |
|
| Candida albicans | ATCC 10231, IP 48.72, NCPF 3179, NBRC 1594 |
| Aspergillus brasiliensis | ATCC 16404, IP 1431.83, IMI 149007, NBRC 9455 |
Cấy vào môi trường thioglycolat lỏng một lượng nhỏ (không quá 100 CPU) các vi sinh vật sau, chú ý cấy riêng rẽ vi sinh vật vào các ống (hoặc bình môi trường): Clostridium sporogenes, Pseudomonas aeruginosa, Staphyloccocus aureus, cấy vào môi trường soybean casein lỏng một lượng nhỏ (không quá 100 CFU) các vi sinh vật sau, chú ý cấy riêng rẽ vi sinh vật vào các ống (hoặc bình môi trường): Aspergillus brasiliensis, Bacillus subtilis, Candida albicans, Ủ trong không quá 3 ngày đối với vi khuẩn và không quá 5 ngày đối với nấm.
Cần đảm bảo chủng vi sinh vật đem cấy vào môi trường không được cấy truyền quá 5 lần từ chủng gốc ban đầu.
Môi trường đạt chất lượng nếu sau thời gian ủ, vi sinh vật đều mọc tốt.
Kiểm tra sự phù hợp của phương pháp
Tiến hành qui trình giống hệt với thử vô khuẩn mẫu thử, chỉ khác:
Phương pháp màng lọc: Sau khi chuyển mẫu thử lên màng lọc, thêm vào phần dung dịch pha loãng cuối cùng một lượng nhỏ (không quá 100 CFU) các vi sinh vật tương ứng.
Phương pháp cấy trực tiếp: Sau khi chuyển mẫu thử vào ống (hoặc bình) môi trường, thêm vào một lượng nhỏ (không quá 100 CFU) các vi sinh vật tương ứng.
Trong cả hai trường hợp trên, sử dụng vi sinh vật theo bảng 13.7.1. Đồng thời tiến hành kiểm tra khả năng dinh dưỡng của môi trường và các ống này được coi như mẫu đối chứng dương. Ủ tất cả các ống (hoặc bình) môi trường trong không quá 5 ngày.
Nếu sau thời gian ủ, sự phát triển của vi sinh vật trong ống môi trường chứa mẫu thử tốt, rõ ràng giống với sự phát triển của vi sinh vật trong ống đối chứng dương không chứa mẫu thử, điều đó có nghĩa mẫu thử không chứa chất ức chế vi sinh vật hoặc khả năng ức chế vi sinh vật của mẫu thử đã bị loại bỏ bởi điều kiện thí nghiệm. Lúc này có thể áp dụng qui trình thử vô khuẩn trên mẫu thử mà không cần bất cứ sự điều chỉnh nào.
Nếu sau thời gian ủ, sự phát triển của vi sinh vật trong ống môi trường chứa mẫu thử không rõ ràng như sự phát triển của vi sinh vật trong ống đối chứng dương không chứa mẫu thử, điều đó có nghĩa mẫu thử chứa chất ức chế vi sinh vật và khả năng ức chế vi sinh vật của mẫu thử không bị loại bỏ bởi điều kiện thí nghiệm. Lúc này cần thay đổi điều kiện thử nghiệm để loại bỏ tác dụng của chất ức chế và tiến hành kiểm tra sự phù hợp của phương pháp lại.
Tiến hành kiểm tra sự phù hợp của phương pháp khi:
- Tiến hành thử vô khuẩn trên một sản phẩm mới
- Có sự thay đổi về điều kiện thử vô khuẩn
Sự phù hợp của phương pháp có thể được tiến hành đồng thời với thử vô khuẩn mẫu thử.
Tiến hành thử vô khuẩn
Có thể tiến hành thử vô khuẩn mẫu thử bằng phương pháp màng lọc hoặc phương pháp cấy trực tiếp. Cần tiến hành các ống đối chứng âm tính. Phương pháp màng lọc ưu tiên được sử dụng, được lựa chọn khi bản chất mẫu thử cho phép có thể áp dụng phương pháp màng lọc được, ví dụ mẫu thử là dung dịch nước, dầu hay cồn, hoặc mẫu thử có thể hòa trộn hoặc hòa tan trong nước hoặc dung môi hữu cơ miễn là dung môi hữu cơ đó không ức chế vi sinh vật trong điều kiện của thử nghiệm.
Phương pháp màng lọc: Sử dụng màng lọc có kính thước lỗ lọc không quá 0,45 µm, kích thước lỗ lọc này đã được chứng minh là phù hợp trong việc lưu giữ vi sinh vật. Màng lọc celulose nitrat được dùng cho các chế phẩm dạng nước, dạng dầu và các dung dịch cồn thấp độ. Màng lọc celulose acetat được dùng cho các chế phẩm cồn cao độ. Có thể sử dụng loại màng lọc phù hợp cho các chế phẩm riêng biệt, ví dụ kháng sinh.
Kỹ thuật thử vô khuẩn mô tả dưới đây được áp dụng cho màng lọc có đường kính 50 mm. Nếu sử dụng màng lọc có đường kính lớn hơn, cần thay đổi thể tích dung dịch pha loãng và qui trình rửa cho phù hợp. Bộ dụng cụ lọc và màng lọc phải được tiệt khuẩn theo cách phù hợp. Bộ dụng cụ lọc phải được thiết kế sao cho có thể đưa mẫu thử và lọc mẫu thử trong điều kiện vô khuẩn, cho phép lấy màng lọc ra khỏi bộ lọc và cấy vào môi trường trong điều kiện vô khuẩn, hoặc cho phép có thể mang bộ dụng cụ đem ủ sau khi đã thêm môi trường vào bộ dụng cụ đó.
Dung dịch nước: chuyển một lượng nhỏ dung dịch pha loãng vô khuẩn thích hợp như dung dịch pepton thịt hoặc pepton casein 1 g/lit có pH 7,1 ± 0,2 lên màng lọc của bộ lọc vô khuẩn và lọc. Dung dịch pha loãng có thể chứa các các chất trung hòa và/hoặc các chất bất hoạt phù hợp trong trường hợp mẫu thử có chứa kháng sinh.
Chuyển mẫu thử lên màng lọc, nếu cần có thể pha loãng mẫu giống như đã tiến hành trong mục Kiểm tra sự phù hợp của phương pháp bằng dung dịch pha loãng phù hợp, chú ý lượng mẫu phải tuân thủ theo quy định trong bảng 13.7.2. Lọc ngay. Nếu mẫu thử có khả năng kháng khuẩn, rửa màng lọc không dưới 3 lần, thể tích dung dịch pha loãng của mỗi lần rửa phải giống như thể tích dung dịch pha loãng đã tiến hành trong mục Kiểm tra sự phù hợp của phương pháp. Chú ý không rửa quá 5 lần, mỗi lần 100 ml dung dịch pha loãng, ngay cả trong trường hợp kết quả kiểm tra sự phù hợp của phương pháp khi áp dụng qui trình rửa này không loại trừ được hoạt tính kháng khuẩn của mẫu thử. Chuyển toàn bộ màng lọc vào môi trường nuôi cấy hoặc cắt màng lọc trong điều kiện vô khuẩn thành 2 phần tương đương và cấy mỗi nửa màng lọc vào một loại môi trường nuôi cấy. Thể tích môi trường nuôi cấy phải giống như thể tích môi trường đã tiến hành trong mục Kiểm tra sự phù hợp của phương pháp. Ngoài ra, có thể thay thế bằng cách cho môi trường nuôi cấy vào bộ dụng cụ chứa màng lọc. Ủ môi trường trong không dưới 14 ngày.
Chất rắn có thể hòa tan được: Chú ý lượng mẫu cho vào mỗi loại môi trường phải tuân thủ theo quy định trong Bảng 13.7.2. Hòa tan mẫu thử trong dung môi pha loãng được cung cấp kèm theo mẫu thử, nước cất pha tiêm, nước muối sinh lý hay dung dịch pepton thịt hoặc pepton casein 1 g/l và tiếp tục tiến hành như mô tả đối với mẫu thử là dung dịch nước ở trên.
Mẫu thử dạng dầu/dung dịch dầu: Chú ý lượng mẫu cho vào mỗi loại môi trường phải tuân thủ theo quy định trong bảng 13.7.2. Chuyển trực tiếp mẫu thử (không cần pha loãng) lên màng lọc khô nếu mẫu thử là dung dịch dầu có độ nhớt thấp. Nếu mẫu thử là dung dịch dầu rất nhớt, nên pha loãng mẫu thử trong dung dịch pha loãng vô khuẩn thích hợp như isopropyl myristat, dung môi này đã được chứng minh là không có hoạt tính kháng khuẩn. Để cho dung dịch dầu tự chảy qua màng lọc rồi sau đó tiến hành lọc với áp lực lọc đều đặn. Rửa màng lọc không dưới 3 lần, mỗi lần 100 ml dung dịch pha loãng phù hợp như dung dịch pepton thịt hoặc pepton casein 1 g/lit có chứa chất diện hoạt phù hợp với nồng độ giống như nồng độ đã tiến hành trong mục Kiểm tra sự phù hợp của phương pháp, ví dụ có thể sử dụng polysorbat 80 ở nồng độ 10 g/l. Chuyển màng lọc vào trong môi trường hoặc ngược lại giống như đã mô tả đối với mẫu thử là dung dịch nước, ủ ở nhiệt độ và thời gian giống như trên.
Mẫu thử dạng mỡ, kem: Chú ý lượng mẫu cho vào mỗi loại môi trường phải tuân thủ theo quy định trong bảng 13.7.2. Nên pha loãng thuốc mỡ hoặc thuốc nhũ dịch kiểu pha nước-trong-dầu trong isopropyl myristat với tỷ lệ 1%, gia nhiệt dưới 40 °C nếu cần. Trong một số trường hợp hạn chế có thể gia nhiệt lên tới 44 °C. Tiến hành lọc nhanh và tiếp tục xử lí giống như mẫu thử dạng dầu/dung dịch dầu.
Phương pháp cấy trực tiếp
Chuyển trực tiếp lượng mẫu thử cho vào mỗi loại môi trường theo quy định trong Bảng 13.7.2. Chú ý thể tích mẫu thử không được vượt quá 10 % thể tích của môi trường, trừ khi có chỉ dẫn khác.
Nếu mẫu thử có hoạt tính kháng khuẩn, cần trung hòa hoạt tính này bằng chất trung hòa trước khi tiến hành thử vô khuẩn hoặc pha loãng mẫu thử trong một thể tích môi trường nuôi cấy đủ lớn. Khi thể tích mẫu thử lớn, cần phải sử dụng môi trường có nồng độ đặc hơn (cần phải tính đến mức độ pha loãng của mẫu thử khi xác định nồng độ đặc của môi trường nuôi cấy). Nếu có thể, nên chuyển trực tiếp môi trường đặc vào mẫu thử.
Mẫu thử dạng dung dịch dầu: Sử dụng môi trường có bổ sung chất diện hoạt phù hợp với nồng độ giống như nồng độ đã tiến hành trong mục Kiểm tra sự phù hợp của phương pháp, ví dụ có thể sử dụng polysorbat 80 ở nồng độ 10 g/l.
Mẫu thử dạng mỡ, kem: Pha loãng mẫu thử theo tỷ lệ 1:10 bằng cách nhũ hóa mẫu thử trong dung dịch pha loãng như dung dịch pepton thịt hoặc pepton casein 1 g/l có chứa chất diện hoạt. Chuyển mẫu thử đã pha loãng vào môi trường nuôi cấy không chứa chất diện hoạt.
Ủ môi trường đã cấy mẫu thử trong không dưới 14 ngày. Định kì quan sát sự phát triển của vi sinh vật trong quá trình ủ. Hàng ngày, lắc nhẹ nhàng môi trường nuôi cấy có chứa mẫu thử dạng dầu. Tuy nhiên, nếu sử dụng môi trường thioglycolat lỏng thì hạn chế lắc tối thiểu để duy trì điều kiện kị khí của môi trường này.
Mẫu thử là chỉ khâu phẫu thuật và chỉ khâu dùng trong thú y: Sử dụng lượng mẫu thử cho vào mỗi loại môi trường nuôi cấy theo quy định trong bảng 13.7.2. Mở bao bì đóng gói trong điều kiện vô khuẩn, cắt 3 đoạn của mỗi sợi chỉ khâu cho mỗi loại môi trường nuôi cấy. Tiến hành thử nghiệm trên 3 đoạn, mỗi đoạn 30 cm, cắt từ đoạn đầu, đoạn giữa và đoạn cuối của sợi chỉ khâu. Sử dụng các đoạn được cắt từ các bao gói mới mở. Chuyển từng đoạn vào môi trường nuôi cấy. Chú ý thể tích môi trường nuôi cấy phải đủ ngập mẫu thử (20 đến 150 ml).
Bảng 13.7.2 - Số lượng tối thiểu của đơn vị đóng gói cho vào mỗi loại môi trường
| Số lượng trong 1 đơn vị đóng gói | Số lượng tối thiểu cho vào môi trường |
| Chất lỏng: |
|
| - Dưới 1 ml | Toàn bộ đơn vị đóng gói |
| - 1 - 40 ml | 1/2 đơn vị đóng gói nhưng không dưới 1 ml |
| - Hơn 40 ml đến không quá 100 ml | 20 ml |
| - Hơn 100 ml | 10 % đơn vị đóng gói nhưng không dưới 20 ml |
| Chất lỏng kháng sinh | 1 ml |
| Chế phẩm không tan, chế phẩm dạng kem, mỡ được phân tán hoặc nhũ hóa | Lấy lượng tương ứng với ít nhất 200 mg thuốc |
| Chất rắn: |
|
| - Dưới 50 mg | Toàn bộ đơn vị đóng gói |
| - 50 đến dưới 300 mg | 1/2 đơn vị đóng gói nhưng không dưới 50 mg |
| - 300 mg đến 5 g | 150 mg |
| - Hơn 5 g | 500 mg |
| - Chỉ khâu phẫu thuật và chỉ khâu dùng trong thú y | 3 đoạn (mỗi đoạn dài 30 cm) |
| - Băng, bông, gạc phẫu thuật được đóng gói kín | 100 mg mỗi gối |
| - Chỉ khâu phẫu thuật và các dụng cụ khác được đóng gói dùng một lần | Toàn bộ đơn vị đóng gói |
| - Các dụng cụ y tế khác | Toàn bộ dụng cụ, cắt nhỏ dụng cụ hoặc tháo rời dụng cụ ra |
Bảng 13.7.3 - Số lượng tối thiểu đơn vị đóng gói của lô đem thử
| Số lượng đơn vị đóng gói của lô | Số lượng tối thiểu đơn vị đóng gói đem thử cho mỗi loại môi trường |
| Thuốc tiêm |
|
| - Không quá 100 đơn vị đóng gói | 10 % hoặc 4 đơn vị đóng gói, lấy số lớn hơn |
| - Trên 100 đến không quá 500 đơn vị đóng gói | 10 đơn vị đóng gói |
| - Trên 500 đơn vị đóng gói | 2 % hoặc 20 đơn vị đóng gói (đối với thuốc tiêm thể tích lớn: 10 đơn vị đóng gói), lấy số bé hơn |
| Thuốc nhỏ mắt và chế phẩm không tiêm |
|
| - Không quá 200 đơn vị đóng gói | 5 % hoặc 2 đơn vị đóng gói, lấy số lớn hơn |
| - Trên 200 đơn vị đóng gói | 10 đơn vị đóng gói |
| - Nếu chế phẩm được đóng gói dùng một lần, áp dụng quy định giống với thuốc tiêm |
|
| Chỉ khâu phẫu thuật và chỉ khâu dùng trong thú y | 2 % hoặc 5 đơn vị đóng gói, lấy số lớn hơn, tối đa 20 đơn vị đóng gói |
| Chế phẩm rắn |
|
| - Không quá 4 đơn vị đóng gói | Mỗi đơn vị đóng gói |
| - Trên 4 đến không quá 50 đơn vị đóng gói | 20 % hoặc 4 đơn vị đóng gói, lấy số lớn hơn |
| - Trên 50 đơn vị đóng gói | 2 % hoặc 10 đơn vị đóng gói, lấy số lớn hơn |
| - Nếu không rõ số lượng đơn vị đóng gói của lô, sử dụng số lượng tối đa theo quy định trong bảng - Nếu lượng mẫu trong một đơn vị đóng gói đủ cho 2 loại môi trường, số lượng đơn vị đóng gói đem thử trong bảng được hiểu là tính cho cả 2 loại môi trường | |
Quan sát và đánh giá kết quả
Định kì trong suốt quá trình ủ và khi kết luận, kiểm tra môi trường bằng mắt thường xem có sự phát triển của vi sinh vật hay không. Nếu bản chất của mẫu thử làm đục môi trường khiến khó quan sát bằng mắt thường sự phát triển của vi sinh vật thì sau 14 ngày ủ, cấy truyền một lượng nhỏ từ môi trường đó (mỗi ống không quá 1 ml) sang loạt môi trường mới cùng loại và tiếp tục ủ cả môi trường cũ và môi trường mới cấy truyền trong không dưới 4 ngày.
Nếu không quan sát thấy sự phát triển của vi sinh vật trong các môi trường, mẫu thử đạt chỉ tiêu vô khuẩn. Nếu quan sát thấy sự phát triển của vi sinh vật trong các môi trường, mẫu thử không đạt chỉ tiêu vô khuẩn, trừ trường hợp chỉ ra rõ ràng rằng thử nghiệm không có giá trị do các nguyên nhân không liên quan gì đến mẫu thử. Thử nghiệm được coi là không có giá trị khi:
a) Các số liệu về kiểm soát vi sinh vật của khu vực thử vô khuẩn không đạt;
b) Khi rà soát lại quá trình thử vô khuẩn phát hiện thấy có sai sót
c) Có sự phát triển của vi sinh vật trong ống đối chứng âm tính
d) Sau khi phân lập và định danh vi sinh vật thu được trong ống môi trường, sự phát triển của vi sinh vật này rõ ràng là do sai sót liên quan đến vật liệu hoặc kỹ thuật được áp dụng cho mẫu thử.
Nếu thử nghiệm không có giá trị, tiến hành thử vô khuẩn lại với số lượng mẫu giống như lần đầu.
Nếu không quan sát thấy sự phát triển của vi sinh vật trong các môi trường ở lần thử lặp lại, mẫu thử đạt chỉ tiêu vô khuẩn. Nếu quan sát thấy sự phát triển của vi sinh vật trong các môi trường ở lần thử lặp lại, mẫu thử không đạt chỉ tiêu vô khuẩn.
Áp dụng phép thử vô khuẩn vào thuốc tiêm, thuốc nhỏ mắt và các chế phẩm không tiêm có yêu cầu vô khuẩn
Khi sử dụng phương pháp màng lọc, lượng mẫu trong một đơn vị đóng gói ít nhất phải tuân thủ theo quy định trong Bảng 13.7.2, nếu có thể thì sử dụng toàn bộ lượng mẫu trong một đơn vị đóng gói, pha loãng nếu cần với khoảng 100 ml dung dịch pha loãng phù hợp như dung dịch pepton thịt hoặc pepton casein 1 g/l.
Khi sử dụng phương pháp cấy trực tiếp, lượng mẫu thử cho vào mỗi loại môi trường tuân theo quy định trong Bảng 13.7.2, trừ khi có chỉ dẫn khác. Nếu thể tích hoặc lượng mẫu thử trong một đơn vị đóng gói không đủ để tiến hành thử nghiệm, cần lấy mẫu thử từ 2 hoặc nhiều đơn vị đóng gói để cấy vào các môi trường khác nhau.
Số lượng tối thiểu đơn vị đóng gói đem thử
Số lượng tối thiểu đơn vị đóng gói đem thử được dựa trên cỡ lô của mẫu thử và được quy định tại Bảng 13.7.3.
13.8 XÁC ĐỊNH HIỆU QUẢ KHÁNG KHUẨN CỦA CHẤT BẢO QUẢN
Chất bảo quản là những chất được thêm vào các chế phẩm phân liều không vô trùng, nhất là các loại thuốc nước, để ngăn chặn sự phát triển của vi sinh vật có sẵn trong chế phẩm hay nhiễm vào chế phẩm trong quá trình sản xuất. Chất bảo quản cũng được thêm vào các chế phẩm vô trùng đa liều để ức chế sự phát triển của vi sinh vật nhiễm vào chế phẩm sau khi mở ra sử dụng.
Không được dùng chất bảo quản như một biện pháp thay thế cho thực hành tốt sản xuất thuốc, không được dùng chất bảo quản với mục đích duy nhất là làm giảm số lượng vi sinh vật trong chế phẩm không vô trùng hoặc để làm tăng hiệu quả của quá trình tiệt trùng đối với chế phẩm vô trùng đa liều.
Đa số các chất bảo quản là những hợp chất độc. Do đó, để bảo đảm an toàn, nồng độ chất bảo quản trong sản phẩm cuối phải thấp hơn mức có thể gây nguy hiểm cho người sử dụng.
Nồng độ chất bảo quản cần dùng có thể giảm đến mức tối thiểu nếu một hay nhiều hoạt chất trong công thức pha chế có khả năng kháng khuẩn và/hoặc kháng nấm.
Chất bảo quản và nồng độ sử dụng của nó phải ghi rõ trên bao bì của sản phẩm.
Hiệu quả bảo vệ sản phẩm của chất bảo quản phải được xác định ngay từ giai đoạn nghiên cứu và phát triển sản phẩm. Phương pháp xác định hiệu quả kháng khuẩn của chất bảo quản được trình bày dưới đây.
Nguyên tắc của phương pháp là cấy một hỗn dịch vi sinh vật chỉ thị thích hợp vào chế phẩm cần thử, tốt nhất là cấy trực tiếp vào bao bì đóng gói cuối cùng của sản phẩm, nếu có thể, ủ chế phẩm đã cấy vi sinh vật ở nhiệt độ thích hợp, sau đó lấy mẫu và đếm số lượng vi sinh vật sống còn lại trong sản phẩm sau mỗi khoảng thời gian ủ nhất định. Việc sử dụng chất bảo quản được xem là có hiệu quả nếu số lượng vi sinh vật giảm đáng kể hoặc không tăng theo thời gian.
Tiêu chuẩn để đánh giá hiệu quả kháng khuẩn của chất bảo quản tùy theo loại chế phẩm và đường dùng thuốc (xem Bảng 13.8.1).
Phân loại chế phẩm
Đối tượng của thử nghiệm xác định hiệu quả kháng khuẩn của chất bảo quản là các loại chế phẩm có thành phần chính là nước hay có sử dụng nước làm tá dược. Các chế phẩm này được chia thành 4 nhóm như sau:
Bảng 13.8.1
| Loại chế phẩm | Mô tả |
| 1 | Thuốc tiêm và thuốc dùng ngoài đường tiêu hóa bao gồm nhũ dịch, thuốc nhỏ tai, thuốc nhỏ mắt và thuốc nhỏ mũi vô trùng. |
| 2 | Thuốc nhỏ mũi không vô trùng, các chế phẩm dùng cho màng nhày, các chế phẩm dùng ngoài da, bao gồm cả các nhũ dịch. |
| 3 | Các loại thuốc uống (trừ thuốc kháng acid). |
| 4 | Thuốc uống kháng acid |
Vi sinh vật chỉ thị
Aspergillus niger ATCC 16404 (IP 1431.83, IMI 149 007).
Candida albicans ATCC 10231 (IP 48.72, NCPF 3179).
Pseudomonas aeruginosa ATCC 9027 (CIP 82.118, NCIMB 8626).
Staphylococcus aureus ATCC 6538 (CIP 4.83, NCTC 10788).
Môi trường nuôi cấy
Tất cả các môi trường sử dụng trong phụ lục này phải được kiểm tra khả năng dinh dưỡng, dùng các vi sinh vật chỉ thị nêu trên để thử, trước khi dùng.
Bảng 13.8.2 - Điều kiện nuôi cấy vi sinh vật
| Vi sinh vật chỉ thị | Môi trường nuôi cấy | Nhiệt độ ủ | Thời gian ủ |
| Pseudomonas aeruginosa | Môi trường lỏng casein đậu tương Môi trường thạch casein đậu tương | 30 °C đến 35 °C | 18 h đến 24 h |
| Staphylococcus aureus | Môi trường lỏng casein đậu tương Môi trường thạch casein đậu tương | 30 °C đến 35 °C | 18 h đến 24 h |
| Candida albicans | Môi trường Sabouraud lỏng Môi trường thạch Sabouraud | 20 °C đến 25 °C | 44 h đến 52 h |
| Aspergillus niger | Môi trường Sabouraud lỏng Môi trường thạch Sabouraud | 20 °C đến 25 °C | 6 ngày đến 10 ngày |
Chuẩn bị hỗn dịch vi sinh vật chỉ thị
Cấy riêng rẽ chủng vi sinh vật chỉ thị lên bề mặt môi trường rắn thích hợp, có thể sử dụng môi trường giới thiệu ở Bảng 13.8.2 hoặc môi trường khác có sẵn trên thị trường có cùng công dụng và có khả năng dinh dưỡng tương đương, sau đó ủ trong điều kiện như quy định ở Bảng 13.8.2. (xem Phụ lục 13.6. Thử giới hạn nhiễm khuẩn để biết thành phần của các môi trường dinh dưỡng).
Sau khi ủ, nếu cần, có thể tiếp tục cấy chuyển vi sinh vật thu được sang bề mặt môi trường dinh dưỡng mới, cho đến khi thu được vi sinh vật ở trong trạng thái phát triển tốt nhất. Tuy nhiên, số lần cấy chuyển không được quá 5 lần từ chủng gốc ban đầu.
Rửa bề mặt môi trường nuôi cấy các vi khuẩn và C. albicans bằng dung dịch natri clorid vô trùng 0,9 %, tập trung dịch rửa vào vật đựng thích hợp và bổ sung dung dịch natri clorid vô trùng 0,9 % để thu được hỗn dịch có khoảng 108 vi sinh vật sống (cfu) trong một ml. Để thu hoạch bào tử A. niger, cũng tiến hành như trên nhưng dùng dung dịch natri clorid vô trùng 0,9 % có chứa 0,05 % polysorbat 80, thêm dung dịch natri clorid vô trùng 0,9 % để được hỗn dịch có khoảng 108 cfu/ml.
Ngoài ra, có thể tăng sinh chủng gốc của mỗi vi sinh vật chỉ thị trong môi trường lỏng thích hợp, ví dụ môi trường giới thiệu ở Bảng 13.8.2, thu hoạch vi sinh vật chỉ thị bằng cách ly tâm, sau đó rửa và phân tán lại trong dung dịch natri clorid vô trùng 0,9 % để được hỗn dịch có khoảng 108 cfu/ml.
Đếm số lượng tế bào sống của mỗi hỗn dịch thu được bằng phương pháp đĩa thạch (xem Phụ lục 13.6 Thử giới hạn nhiễm khuẩn). Kết quả đếm (cfu/ml) được dùng để xác định lượng vi sinh vật đã cấy vào chế phẩm cần thử tại thời điểm bắt đầu thử nghiệm.
Bảo quản các hỗn dịch vi sinh vật chỉ thị trong tủ lạnh nếu không dùng ngay trong vòng 2 h. Các hỗn dịch vi khuẩn và C. albicans phải dùng trong vòng 24 h sau khi thu hoạch, hỗn dịch A. niger có thể bảo quản trong tủ lạnh trong 7 ngày.
Tiến hành
Cấy riêng rẽ từng hỗn dịch chủng vi sinh vật chỉ thị đã được chuẩn bị như trên trực tiếp vào 4 đơn vị đóng gói cuối cùng của chế phẩm cần thử, lắc kỹ để trộn đều. Thể tích hỗn dịch vi sinh vật cấy vào các đơn vị đóng gói được tính toán sao cho nồng độ vi sinh vật chỉ thị trong chế phẩm cần thử ngay sau khi cấy khoảng từ 105 cfu/ml đến 106 cfu/ml đối với chế phẩm loại 1, 2 và 3, khoảng 103 cfu/ml đến 104 cfu/ml đối với chế phẩm loại 4. Thể tích hỗn dịch đem cấy không được vượt quá 1 % thể tích chế phẩm cần thử, tốt nhất là từ 0,5 % đến 1 %. Nếu không thể cấy vi sinh vật chỉ thị trực tiếp vào đơn vị đóng gói cuối cùng của sản phẩm, chuyển một thể tích đủ lớn của chế phẩm cần thử, ít nhất 20 ml, vào 4 vật chứa vô trùng có thể tích thích hợp, có nắp kín và tiếp tục tiến hành như trên.
Ủ các vật chứa đã cấy vi sinh vật chỉ thị ở nhiệt độ 20 °C đến 25 °C, tránh ánh sáng. Lấy mẫu từ mỗi vật chứa lúc 0 h và sau những khoảng thời gian ủ nhất định như quy định ở Bảng 13.8.3, lượng mẫu cần lấy thường là 1 ml hoặc 1 g.
Ghi chép mọi thay đổi về mặt cảm quan của chế phẩm cần thử trong quá trình ủ. Xác định số lượng vi sinh vật tại mỗi thời điểm lấy mẫu bằng phương pháp đĩa thạch, dùng môi trường dinh dưỡng và nhiệt độ ủ như quy định ở Bảng 13.8.2, thời gian ủ từ 3 ngày đến 5 ngày đối các vi khuẩn và C. albicans, 3 ngày đến 7 ngày đối với A. niger.
Phải bảo đảm loại bỏ hoàn toàn hoạt tính kháng khuẩn và/hoặc kháng nấm của chế phẩm cần thử bằng phương pháp pha loãng, lọc, hay dùng chất trung hòa. Nếu sử dụng phương pháp pha loãng, điểm cần chú ý là độ chính xác của kết quả đếm sẽ giảm khi số lượng vi sinh vật khá nhỏ (nhỏ hơn 30 cfu/hộp đối với phương pháp hộp thạch, dùng hộp petri có đường kính từ 90 mm đến 100 mm). Nếu sử dụng chất trung hòa, phải có biện pháp kiểm tra thích hợp để bảo đảm nồng độ chất trung hòa đã dùng đủ để loại bỏ hoàn toàn hoạt tính kháng khuẩn của chế phẩm cần thử và để bảo đảm bản thân chất trung hòa không gây bất cứ ảnh hưởng bất lợi nào đến sự phát triển của vi sinh vật chỉ thị. Dùng nồng độ vi sinh vật chỉ thị, tính bằng cfu/ml, tại thời điểm bắt đầu thử nghiệm (Ro) và nồng độ vi sinh vật sống đếm được tại mỗi thời điểm lấy mẫu “t” (Rt), tính lượng giảm đi của mỗi vi sinh vật chỉ thị tại thời điểm đó (R):
Log Reduction + Log10Ro - Log10Rt
Nguyên tắc đánh giá hiệu quả kháng vi sinh vật của chất bảo quản
Hiệu quả kháng khuẩn của chất bảo quản hay hệ chất bảo quản được xem là đạt yêu cầu nếu kết quả thử nghiệm thỏa mãn các nguyên tắc đánh giá ở các Bảng 13.8.3. Số lượng vi sinh vật chỉ thị tại một thời điểm tăng không nhiều hơn 0,5 log10 cfu/ml so với kết quả đếm ở thời điểm gần kề trước đó thì được xem là “không tăng”.
Bảng 13.8.3. A - Chế phẩm loại 1
|
| Log Reduction | ||
| 7 ngày | 14 ngày | 28 ngày | |
| Vi khuẩn | ≥ 1,0 | ≥ 3,0 | Không tăng |
| Nấm men, nấm mốc | Không tăng | Không tăng | Không tăng |
Bảng 13.8.3. B - Chế phẩm loại 2
|
| Log Reduction | ||
| 7 ngày | 14 ngày | 28 ngày | |
| Vi khuẩn | - | ≥ 2,0 | Không tăng |
| Nấm men, nấm mốc | - | Không tăng | Không tăng |
Bảng 13.8.3.C - Chế phẩm loại 3
|
| Log Reduction | ||
| 7 ngày | 14 ngày | 28 ngày | |
| Vi khuẩn | - | ≥ 1,0 | Không tăng |
| Nấm men, nấm mốc | - | Không tăng | Không tăng |
Bảng 13.8.3.D - Chế phẩm loại 4
|
| Log Reduction | ||
| 7 ngày | 14 ngày | 28 ngày | |
| Vi khuẩn | - | Không tăng | Không tăng |
| Nấm men, nấm mốc | - | Không tăng | Không tăng |
13.9 XÁC ĐỊNH HOẠT LỰC THUỐC KHÁNG SINH BẰNG PHƯƠNG PHÁP THỬ VI SINH VẬT
Hoạt lực kháng sinh được xác định bằng so sánh khả năng ức chế sự phát triển vi sinh vật nhạy cảm của kháng sinh thử và kháng sinh chuẩn.
Kháng sinh chuẩn được sử dụng trong thử nghiệm phải được xác định hoạt tính một cách chính xác dựa trên chất chuẩn quốc tế.
Phép thử xác định hoạt lực phải được thiết kế sao cho có thể tính toán được tính có giá trị của mô hình toán học được dùng làm cơ sở cho phương trình hoạt lực. Nếu áp dụng mô hình đường thẳng song song, hai đường logarit liều-đáp ứng của chất chuẩn và chế phẩm thử phải song song với nhau và phải tuyến tính trong khoảng liều được dùng trong tính toán. Các điều kiện đó phải được kiểm định bởi các thử nghiệm tính có giá trị của phương pháp (validity test) ở xác suất đã cho, thường là 0,05. Có thể sử dụng các mô hình toán học khác, ví dụ mô hình tỷ số độ dốc nếu chứng minh được tính có giá trị của phương pháp.
Trừ trường hợp được chỉ dẫn trong chuyên luận riêng, giới hạn tin cậy (P = 0,95) của phép thử xác định hoạt lực phải không được dưới 95 % và không lớn hơn 105 % so với hoạt lực ước đoán.
Tiến hành định lượng theo phương pháp A hoặc B.
Phương pháp khuếch tán (A)
Làm lỏng môi trường phù hợp với điều kiện thử nghiệm và cấy một lượng chủng vi sinh vật nhạy cảm với kháng sinh thử vào môi trường ở nhiệt độ thích hợp, ví dụ 48 °C đến 50 °C đối với vi sinh vật dạng sinh dưỡng, sao cho có thể tạo ra được vòng vô khuẩn sắc nét, có đường kính phù hợp tại nồng độ kháng sinh sử dụng. Rót ngay môi trường đã cấy chủng vi sinh vật vào đĩa petri hoặc đĩa vuông với thể tích môi trường phù hợp để thu được lớp môi trường có bề dày đồng nhất từ 2 mm đến 5 mm. Ngoài ra, có thể dùng phương pháp tạo 2 lớp môi trường, trong đó chỉ có lớp môi trường phía trên là được cấy chủng vi sinh vật.
Bảo quản đĩa môi trường trên sao cho vi sinh vật không phát triển hoặc không bị chết trước khi đĩa môi trường được sử dụng và bề mặt của môi trường phải khô khi sử dụng.
Sử dụng dung môi và dung dịch đệm pha loãng theo Bảng 13.9.2 để chuẩn bị các dung dịch kháng sinh chuẩn và dung dịch kháng sinh thử có hoạt lực tương đương. Nhỏ các dung dịch này lên bề mặt của môi trường, ví dụ, vào các ống trụ vô khuẩn được làm bằng sứ, thép không gỉ hay vật liệu phù hợp khác, hoặc nhỏ vào các lỗ đã đục trong môi trường thạch. Phải nhỏ thể tích giống nhau vào các ống trụ hoặc vào các lỗ thạch. Ngoài ra, có thể sử dụng các khoanh giấy hấp phụ vô khuẩn có chất lượng phù hợp, đã thấm các dung dịch kháng sinh chuẩn và kháng sinh thử và đặt các khoanh giấy đó lên bề mặt của môi trường.
Để đánh giá tính có giá trị của phép thử, sử dụng ít nhất 3 nồng độ kháng sinh chuẩn và 3 nồng độ kháng sinh thử có hoạt lực tương đương nhau. Nên sử dụng các nồng độ kháng sinh theo cấp số nhân. Khi tính tuyến tính của hệ thống được khẳng định trên một số lượng đầy đủ các phép thử 3 nồng độ, thì đối với các phép thử thường ngày, có thể sử dụng phép thử 2 nồng độ. Tuy nhiên, trong trường hợp có tranh chấp, phải áp dụng phép thử 3 nồng độ.
Vị trí các dung dịch trong đĩa petri hoặc đĩa vuông được sắp xếp theo mô hình thống kê phù hợp, trường hợp sử dụng đĩa petri cỡ nhỏ không thể nhỏ quá 6 dung dịch thì phải sắp xếp các dung dịch kháng sinh chuẩn và thử một cách luân phiên để tránh các nồng độ lớn khuếch tán xen phủ nhau, ủ ở điều kiện nhiệt độ phù hợp trong 18 h. Thời gian khuếch tán trước khi ủ thường từ 1 h đến 4 h ở nhiệt độ phòng hoặc ở 4 °C để hạn chế tối đa sự ảnh hưởng của việc nhỏ các dung dịch kháng sinh ở các thời điểm khác nhau và cải thiện độ dốc của phương trình hồi qui.
Đo đường kính vòng vô khuẩn với độ chính xác tối thiểu là 0,1 mm hoặc đo diện tích vòng vô khuẩn với độ chính xác tương đương và tính toán hoạt lực bằng phương pháp thống kê thích hợp.
Với mỗi phép thử định lượng, sử dụng đủ số lần lặp lại của mỗi nồng độ để đảm bảo độ chính xác theo quy định, có thể phải tiến hành lặp lại phép thử nhiều lần, tập hợp kết quả theo phương pháp thống kê để đạt được độ chính xác theo quy định nhằm khẳng định chắc chắn hoạt lực của kháng sinh thử không thấp hơn giới hạn tối thiểu.
Phương pháp đo độ đục (B)
Cấy vào môi trường thích hợp hỗn dịch chủng vi sinh vật nhạy cảm với kháng sinh thử sao cho có thể gây ra sự ức chế đủ lớn tới sự phát triển của vi sinh vật. Cấy vào môi trường lượng vi sinh vật hợp lí nhằm thu được độ đục phù hợp, có thể đo được sau thời gian ủ là 4 h.
Môi trường sau khi đã cấy chủng phải được sử dụng ngay.
Sử dụng dung môi và dung dịch đệm theo bảng 13.9.3 để pha các dung dịch kháng sinh chuẩn và thử có hoạt lực tương đương.
Để đánh giá tính có giá trị của phép thử, sử dụng ít nhất 3 nồng độ kháng sinh chuẩn và 3 nồng độ kháng sinh thử có hoạt lực tương đương nhau. Nên sử dụng các nồng độ kháng sinh theo cấp số nhân. Để thu được sự tuyến tính theo qui định, cần lựa chọn 3 nồng độ liên tiếp từ một số lượng lớn các nồng độ, trong đó các nồng độ của dung dịch kháng sinh chuẩn và thử tương đương nhau.
Nhỏ một thể tích bằng nhau các dung dịch kháng sinh vào ống nghiệm, thêm môi trường đã cấy chủng với thể tích bằng nhau vào mỗi ống nghiệm (ví dụ 1 ml dung dịch kháng sinh và 9 ml môi trường). Với phép thử định lượng tyrothricin thêm 0,1 ml dung dịch kháng sinh vào 9,9 ml môi trường đã cấy chủng).
Song song chuẩn bị 2 ống nghiệm đối chứng không có kháng sinh, cả 2 ống này đều chứa môi trường đã cấy chủng, thêm ngay vào 1 ống 0,5 mL formaldehyd (TT). Các ống nghiệm này được dùng để chuẩn hóa thiết bị quang học dùng để đo sự phát triển của vi sinh vật.
Đặt các ống nghiệm theo phân bố ngẫu nhiên hoặc hình vuông Latin hoặc phân bố khối ngẫu nhiên trong bể điều nhiệt hoặc trong thiết bị phù hợp khác sao cho nhiệt độ của các ống nghiệm nhanh chóng đạt được nhiệt độ ủ theo quy định, duy trì các ống nghiệm ở nhiệt độ ủ trong 3 h đến 4 h, đảm bảo nhiệt độ và thời gian ủ đồng nhất giữa các ống nghiệm.
Sau thời gian ủ, làm ngừng sự phát triển của vi sinh vật bằng cách thêm 0,5 ml formaldehyd (TT) vào mỗi ống nghiệm hoặc bằng biện pháp xử lý nhiệt và đo độ đục bằng thiết bị quang học phù hợp cho kết quả có 3 chữ số có ý nghĩa. Ngoài ra, có thể đo độ đục của từng ống nghiệm sau các khoảng thời gian ủ hoàn toàn như nhau.
Tính toán hoạt lực dựa theo phương pháp thống kê phù hợp.
Sự tuyến tính giữa liều-đáp ứng (số liệu chuyển đổi hoặc không chuyển đổi) thường chỉ đạt được trên một khoảng rất hẹp. Khoảng này phải được dùng để tính toán hoạt lực và phải gồm ít nhất 3 nồng độ liên tiếp để cho phép có thể kiểm tra lại tính tuyến tính. Khi tính tuyến tính của cả hệ thống được khẳng định trên một số lượng đầy đủ các phép thử 3 nồng độ, thì đối với các phép thử thường ngày, có thể sử dụng phép thử 2 nồng độ, và phải được cơ quan có thẩm quyền chấp thuận. Tuy nhiên, trong trường hợp có tranh chấp, phải áp dụng phép thử 3 nồng độ.
Với mỗi phép thử định lượng, sử dụng đủ số lần lặp lại của mỗi nồng độ để đảm bảo độ chính xác theo quy định, có thể phải tiến hành lặp lại phép thử, tập hợp kết quả theo phương pháp thống kê để đạt được độ chính xác theo quy định nhằm khẳng định chắc chắn hoạt lực của kháng sinh thử không thấp hơn giới hạn tối thiểu.
Chủng vi sinh vật
Chuyên luận này giới thiệu một số chủng vi sinh vật và điều kiện sử dụng của chúng. Có thể sử dụng chủng vi sinh vật khác miễn là chủng đó nhạy cảm với kháng sinh thử, được sử dụng trong môi trường phù hợp, trong điều kiện nhiệt độ và pH phù hợp. Nồng độ của dung dịch kháng sinh phải được lựa chọn để đảm bảo có sự liên quan tuyến tính giữa logarit nồng độ với đáp ứng trong điều kiện thử nghiệm.
Chế tạo hỗn dịch cấy truyền
Bacillus cereus, Bacillus subtilis, Bacillus pumilus. Hỗn dịch bào tử được chuẩn bị như sau. Cấy chủng vi sinh vật lên bề mặt môi trường số 1 đã có sẵn mangan sulfat (TT) với nồng độ 0,001 g/l và ủ ở 35 °C đến 37°C trong 7 ngày. Dùng nước cất vô khuẩn để gặt vi khuẩn (chủ yếu dưới dạng bào tử). Làm nóng hỗn dịch bào tử ở 70 °C trong 30 min và pha loãng tới nồng độ thích hợp, thường là 107 đến 108 bào tử trong 1 ml. Hỗn dịch bào tử có thể sử dụng lâu dài nếu bảo quản ở nhiệt độ không quá 4 °C.
Ngoài ra, có thể chế tạo hỗn dịch bằng cách cấy chủng vi sinh vật vào môi trường số 3 và nuôi cấy ở 26 °C trong 4 ngày đến 6 ngày, sau đó, thêm lượng mangan sulfat (TT) phù hợp trong điều kiện vô khuẩn vào môi trường để thu được nồng độ 0,001 g/l, ủ thêm 48 h. Sử dụng kính hiển vi để kiểm tra xem bào tử có được hình thành với tỷ lệ đủ lớn trong hỗn dịch (khoảng 80 %) và li tâm. Hòa cắn trở lại trong nước cất vô khuẩn để thu được hỗn dịch có nồng độ 107 đến 108 bào tử trong 1 ml, làm nóng hỗn dịch ở 70 °C trong 30 min và bảo quản ở nhiệt độ không quá 4 °C.
Bordetella bronchiseptica. Nuôi cấy chủng vi sinh vật trong môi trường số 2 ở 35 °C đến 37 °C trong 16 h đến 18 h. Gặt chủng bằng nước cất vô khuẩn và pha loãng đến độ đục thích hợp.
Staphylococcus aureus, Klebsiella pneumoniae, Escherichia coli, Micrococus luteus, Staphylococcus epidermidis. Chuẩn bị chủng vi sinh vật giống như mô tả đối với chủng B. bronchiseptica, nhưng sử dụng môi trường số 1 và điều chỉnh độ đục hỗn dịch sao cho có sự tuyến tính tốt giữa liều-đáp ứng trong thử nghiệm bằng phương pháp đo độ đục, hoặc tạo ra được vòng vô khuẩn sắc nét, có đường kính phù hợp trong thử nghiệm bằng phương pháp khuếch tán.
Saccharomyces cerevisiae, Candida tropicalis. Nuôi cấy chủng vi sinh vật trong môi trường số 6 °C ở 30 °C đến 37 °C trong 24 h. Gặt chủng bằng dung dịch natri clorid 0,9% (TT) vô khuẩn. Pha loãng đến độ đục thích hợp bằng dung dịch natri clorid 0,9% (TT) vô khuẩn.
Dung dịch đệm
Dung dịch đệm số 1 đến 12 có pH từ 5,8 đến 8,0 được pha chế bằng cách: Lấy 50,0 ml dung dịch kali dihydrophosphat 0,2 M (TT), thêm dung dịch natri hydroxyd 0,2 M (TT) với lượng thể tích ghi ở Bảng 13.9.1. Thêm nước vừa đủ 200 ml.
Bảng 13.9.1- Thành phần dung dịch đệm
| Số dung dịch đệm | pH (± 0,1) | Dung dịch Natri hydroxyd 0,2 M (ml) |
| 1 | 5,8 | 3,72 |
| 2 | 6,0 | 5,70 |
| 3 | 6,2 | 8,60 |
| 4 | 6,4 | 12,60 |
| 5 | 6,6 | 17,80 |
| 6 | 6,8 | 23,65 |
| 7 | 7,0 | 29,63 |
| 8 | 7,2 | 35,00 |
| 9 | 7,4 | 39,50 |
| 10 | 7,6 | 42,80 |
| 11 | 7,8 | 45,20 |
| 12 | 8,0 | 46,80 |
Dung dịch đệm số 13 (pH 6,8 ± 0,1): Hòa tan 6,4 g kali dihydrophosphat (TT) và 18,9 g dinatri hydrophosphat (TT) trong 1000 ml nước.
Dung dịch đệm số 14 (pH 10,5 ± 0,1): Hòa tan 35,0 g dikali hydrophosphat (TT) trong 900 ml nước, thêm 20 ml dung dịch natri hydroxyd 1 M (TT), thêm nước vừa đủ 1000 ml.
Dung dịch đệm số 15 (pH 4,5 ± 0,1): Hòa tan 13,61 g kali dihydrophosphat (TT) trong 1000 ml nước.
Dung dịch đệm số 16 (pH 7,8 ± 0,1): Hòa tan 5,59 g dikali hydrophosphat (TT) và 0,41 g kali dihydrophosphat (TT) trong 1000 ml nước.
Dung dịch đệm số 17 (pH 8,0 ± 0,1): Hòa tan 16,73 g dikali hydrophosphat (TT) và 0,523 g kali dihydrophosphat (TT) trong 750 ml nước, điều chỉnh pH 8,0 bằng dung dịch natri hydroxyd 0,1 M (TT) hoặc dung dịch acid phosphoric 0,1 M (TT) nếu cần, thêm nước vừa đủ 1000 ml.
Dung dịch đệm số 18 (pH 6,0 ± 0,1): Hòa tan 8,63 g kali dihydrophosphat (TT) và 1,37 g dinatri hydrophosphat khan (TT) trong 750 ml nước, điều chỉnh pH 6,0 bằng dung dịch natri hydroxyd 0,1 M (TT) hoặc dung dịch acid phosphoric 0,1 M (TT) nếu cần, thêm nước vừa đủ 1000 ml.
Dung dịch đệm số 19 (pH 6,0 ± 0,1): Hòa tan 20,0 g dikali hydrophosphat (TT) và 80,0 g kali dihydrophosphat (TT) trong 1000 ml nước.
Dung dịch đệm số 20 (pH 5,6 ±0,1): Hòa tan 0,908 g kali dihydrophosphat (TT) trong vừa đủ 100 ml nước (dung dịch A). Hòa tan 1,161 g dikali hydrophosphat (TT) trong vừa đủ 100 ml nước (dung dịch B). Trộn 94,4 ml dung dịch A và 5,6 ml dung dịch B. Điều chỉnh pH 5,6 bằng dung dịch A hoặc B, nếu cần.
Môi trường
Có thể dùng các loại môi trường sau hoặc thay thế bằng môi trường khác tương đương.
Môi trường số 1
| Pepton | 6,0 g |
| Casein thủy phân bởi pancreatin | 4,0 g |
| Cao thịt bò | 1,5 g |
| Cao nấm men | 3,0 g |
| Glucose monohydrat | 1,0 g |
| Thạch | 15,0g |
| Nước | 1000 ml |
Môi trường số 2
| Casein thủy phân bởi pancreatin | 17,0 g |
| Bột đậu tương thủy phân bởi papain | 3,0 g |
| Natri clorid | 5,0 g |
| Dikali hydrophosphat | 2,5 g |
| Glucose monohydrat | 2,5 g |
| Thạch | 15,0 g |
| Polysorbat 80 | 10,0 g |
| Nước | 1000ml |
Hòa tan các thành phần (trừ polysorbat 80) trong nước, đun sôi, thêm polysorbat 80 ngay trước khi thêm nước vừa đủ.
Môi trường số 3
| Pepton | 6,0 g |
| Cao thịt bò | 1,5 g |
| Cao nấm men | 3,0 g |
| Natri clorid | 3,5 g |
| Glucose monohydrat | 1,0 g |
| Dikali hydrophosphat | 3,68 g |
| Kali dihydrophosphat | 1,32 g |
| Nước | 1000ml |
Môi trường số 4
| Cao tim | 1,5 g |
| Cao nấm men | 1,5 g |
| Pepton - casein | 5,0 g |
| Glucose monohydrat | 1,0 g |
| Natri clorid | 3,5 g |
| Dikali hydrophosphat | 3,68 g |
| Kali dihydrophosphat | 1,32 g |
| Kali nitrat | 2,0 g |
| Nước | 1000 ml |
Môi trường số 5
| Pepton | 5,0 g |
| Cao thịt | 3,0 g |
| Dikali hydrophosphat.12H2O | 26,9 g |
| Thạch | 10,0 g |
| Nước | 1000ml |
Sau khi tiệt khuẩn môi trường, thêm dikali hydrophosphat (TT) dưới dạng dung dịch vô khuẩn vào môi trường.
Môi trường số 6
| Pepton | 9,4 g |
| Cao nấm men | 4,7 g |
| Cao thịt bò | 2,4 g |
| Natri clorid | 10,0 g |
| Glucose monohydrat | 10,0 g |
| Thạch | 23,5 g |
| Nước | 1000 ml |
Môi trường số 7
| Glycerol | 10,0 g |
| Pepton | 10,0 g |
| Cao thịt | 10,0 g |
| Natri clorid | 3,0 g |
| Thạch | 15,0 g |
| Nước | 1000ml |
Môi trường số 8
| D-glucose | 10,0 g |
| Tryptone | 6,0 g |
| Cao nấm men | 2,0 g |
| Nước vừa đủ | 1000 ml |
| Môi trường số 9 |
|
| Pepton | 6,0 g |
| Casein thủy phân bởi pancreatin | 4,0 g |
| Cao nấm men | 3,0 g |
| Glucose monohydrat | 1,0 g |
| Thạch | 20,0 g |
| Nước | 1000 ml |
Môi trường số 10
| Pepton | 6,0 g |
| Cao thịt bò | 1,5 g |
| Cao nấm men | 6,0 g |
| Glucose | 1,0 g |
| Thạch | 15,0 - 20 g |
| Nước | 1000 ml |
Môi trường số 11
| Pepton | 5,0 g |
| Thạch | 15,0 g |
| Cao thịt bò | 3,0 g |
| Nước | 1000 ml |
Môi trường số 12
| Pepton | 10,0 g |
| Cao thịt | 3,0 g |
| Natri clorid | 30,0 g |
| Thạch | 20,0 g |
| Nước | 1000 ml |
Môi trường số 13
| Pepton | 6,0 g |
| Cao nấm men | 3,0 g |
| Cao thịt bò | 1,5 g |
| Thạch | 20,0 g |
| Nước | 1000 ml |
Môi trường số 14
| Pepton | 6,0 g |
| Cao nấm men | 3,0 g |
| Cao thịt bò | 1,5 g |
| Thạch | 15,0 g |
| Nước | 1000 ml |
Bảng 13.9.2 - Điều kiện định lượng kháng sinh bằng phương pháp khuếch tán
| Tên kháng sinh | Chất chuẩn | Dung môi ban đầu | Dung dịch đệm | Chủng vi sinh vật | Môi trường và pH (± 0,1) | Nhiệt độ ủ |
| Acetyl spiramycin | Acetyl spiramycin | Hòa tan acetyl spiramycin trong ethanol (TT) với tỷ lệ 5 mg trong 2 ml, sau đó thêm nước cất vừa đủ | Số 16 | Bacillus subtilis ATCC 6633 | Số 10 (pH 8,1) | 35 - 37 °C |
| Amphotericin B | Amphotericin B | Dimethyl sulfoxid (TT) | Số 14 | Saccharomyces cerevisiae ATCC 9763 IP 1432-83 | Số 6 (pH 6,1) | 35 - 37 °C |
| Bacitracin kẽm | Bacitracin kẽm | Dung dịch acid hydrocloric 0,01 N (TT) | Số 7 | Micrococcus luteus NCTC 7743 CIP 53.160 ATCC 10240 | Số 1 (pH 7,0) | 35 - 39 °C |
| Bleomycin sulfat | Bleomycin sulfat | Nước | Số 13 | Mycobacterium smegmatis ATCC 607 | Số 7 (pH 7,0) | 35 - 37 °C |
| Colistimethat natri | Colistimethat natri | Nước | Số 2 | Bordetella bronchiseptica NCTC 8344 CIP 53.157 ATCC 4617 Escherichia coli NCIB 8879 CIP 54.127 ATCC 10536 | Số 2 (pH 7,3) | 35 - 39 °C |
| Dihydrostreptomycin sulfat | Dihydrostrep-tomycin sulfat | Nước | Số 12 | Bacillus subtilis NCTC 8236 CIP 1.83 Bacillus subtilis NCTC 10400 CIP 52.62 ATCC 6633 | Số 1 (pH 7,9) | 30 - 37 °C |
| Doxycyclin hydroclorid | Doxycyclin hydroclorid | Dung dịch acid hydrocloric 0,01 N (TT) | Số 15 | Bacillus cereus ATCC 11778 | Số 9 (pH 6,0 - 7,0) | 35 - 37 °C |
| Erythromycin estolat Erythromycin ethyl sucinat Erythromycin stearat | Erythromycin | Methanol (TT) | Số 12 | Bacillus pumilus NCTC 8241 CIP 76.18 Bacillus subtilis NCTC 10400 CIP 52.62 ATCC 6633 | Số 1 (pH 7,9) | 30 - 37 °C |
| Framycetin sulfat | Framycetin sulfat | Nước | Số 12 | Bacillus subtilis NCTC 10400 CIP 52.62 ATCC 6633 Bacillus pumilus NCTC 8241 CIP 76.18 | Số 5 (pH 7,9) | 30 - 37 °C |
| Gentamycin sulfat | Gentamycin sulfat | Nước | Số 12 | Bacillus pumilus NCTC 8241 CIP 76.18 Staphylococcus epidermidis NICB 8853 CIP 68.21 ATCC 12228 | Số 1 (pH 7,9) | 35 - 39 °C |
| Josamycin | Josamicin | Hòa 30 mg trong 5 ml methanol (TT), thêm nước vừa đủ 100ml | Số 20 | Bacillus subtilis CIP 52.62 ATCC 6633 NCTC 10400 | Số 1 (pH 6,6) | 35 - 37 °C |
| Josamycin propiomat | Josamycin propiomat | Hòa 40 mg trong 20 ml methanol (TT), thêm dung dịch đệm số 20 vừa đủ 100 ml | Số 20 | Bacillus subtilis CIP 52.62 ATCC 6633 NCTC 10400 | Số 1 (pH 6,6) | 35 - 37 °C |
| Kanamycin monosulfat | Kanamycin monosulfat | Nước | Số 12 | Bacillus subtilis NCTC 10400 CIP 52.62 ATCC 6633 | Số 1 (pH 7,9) | 30 - 37 °C |
| Kanamycin acid sulfat |
|
|
| Staphylococcus aureus NCTC 7447 CIP 53.156 STCC 6538 P |
| 35 - 39 °C |
| Kitasamycin | Kitasamycin | Methanol (TT) | Số 17 | Bacillus subtilis ATCC 6633 | Số 11 (pH 7,9) | 30 - 37 °C |
| Lymecyclin | Lymecyclin |
| Số 1 | Bacillus pumilus NCTC 8241 CIP 76.18 | Số 1 (pH 6,6) | 37 - 39 °C |
| Neomycin sulfat | Neomycin sulfat | Nước | Số 12 | Bacillus pumilus NCTC 8241 | Số 5 (pH 7,9) | 30 - 37 °C |
| Số 17 | Bacillus pumilus NCTC 8241 CIP 76.18 | Số 1 (pH 8,0-8,1) | 35 - 39 °C | |||
| Số 12 | Bacillus subtilis NCTC 10400 CIP 52.62 ATCC 6633 | Số 5 (pH 7,9) | 30 - 37 °C | |||
| Netilmicin sulfat | Netilmicin sulfat | Nước | Số 12 | Staphylococcus aureus ATCC 6538 P CIP 53.156 | Số 1 (pH 7,9) | 32 - 35 °C |
| Nystatin | Nystatin | Dimethyl formamid (TT) | Số 19 | Saccharomyces cerevisiae ATCC 2601 | Số 6 (pH 6,0) | 29 - 31 °C |
| Oxytetracyclin dihydrat | Oxytetracyclin dihydrat | Dung dịch acid hydrocloric 0,01 N (TT) | Số 1 | Bacillus pumilus NCTC 8241 | Số 1 (pH 6,6) | 30 - 37 °C |
| Polymycin B sulfat | Polymycin B | Dung dịch đệm số 18 | Số 18 | Escherichia coli ATCC 8739 | Số 12 (pH 6,5 - 6,6) | 32 - 37 °C |
| Rifamycin natri | Rifamycin natri | Methanol (TT) | Số 7 | Micrococcus luteus NCTC 8340 CIP 53.45 ATCC 9341 | Số 1 (pH 6,6) | 35 - 39 °C |
| Spiramycin | Spiramycin | Methanol (TT) | Số 12 | Bacillus subtilis NCTC 10400 CIP 52.62 ATCC 6633 | Số 1 (pH 7,9) | 30 - 32 °C |
| Streptomycin sulfat | Streptomycin sulfat | Nước | Số 12 | Bacillus subtilis NCTC 8236 CIP 1.83 | Số 1 (pH 7,9) | 30 - 37 °C |
| Bacillus subtilis NCTC 10400 CIP 52.62 ATCC 6633 | 30 - 37 °C | |||||
| Teicoplanin | Teicoplanin | Dung dịch đệm số 2 | Số 2 | Bacillus subtilis NCTC 8236 CIP 52.62 ATCC 6633 | Số 11 (pH 7,8 - 8,0) | 35 - 37 °C |
| Tetracyclin hydroclorid | Tetracyclin hydroclorid | Dung dịch acid hydrocloric 0,01 N (TT) | Số 15 | Bacillus cereus ATCC 11778 | Số 13 (pH 6,0- 7.0) | 35 - 37 °C |
| Tobramycin | Tobramycin | Nước | Số 12 | Bacillus subtilis ATCC 6633 | Số 1 (pH 7,9) | 30 - 37 °C |
| Vancomycin | Vancomycin | Nước | Số 15 | Bacillus subtilis ATCC 6633 | Số 14 (pH 5,9) | 30 - 32 °C |
| Vancomycin hydroclorid | Vancomycin hydroclorid | Nước | Số 12 | Bacillus subtilis NCTC 8236 CIP 52.62 ATCC 6633 | Số 1 (pH 8,0) | 37 - 39 °C |
| Nước | Số 15 | Bacillus subtilis ATCC 6633 | Số 14 (pH 5,9) | 30 - 32 °C |
Bảng 13.9.3- Điều kiện định lượng kháng sinh bằng phương pháp đo độ đục
| Tên kháng sinh | Chất chuẩn | Dung môi ban đầu | Dung dịch đệm | Chủng vi sinh vật | Môi trường và pH (±0,1) | Nhiệt độ ủ |
| Colistimethat natri | Colistimethat natri | Nước | Số 7 | Escherichia coli NCIB 8666 CIP 2.83 ATCC 9637 | Số 3 (pH 7,0) | 35 - 37 °C |
| Dihydrostreptomycin sulfat | Dihydrostreptomy-cin sulfat | Nước | Số 12 | Klebsiella pneumoniae NCTC 7427 CIP 53.153 ATCC 10031 | Số 3 (pH 7,0) | 35 - 37 °C |
| Erythromycin estolat Erythromycin ethyl sucinat Erythromycin stearat | Erythromycin | Methanol (TT) | Số 12 | Klebsiella pneumoniae NCTC 7427 CIP 53.153 ATCC 10031 | Số 4 (pH 7,0) | 35 - 37 °C |
| Staphylococcus aureus NCTC 7447 CIP 53.156 ATCC 6538 P | Số 3 (pH 7,0) | 35-37 °C | ||||
| Framycetin sulfat | Framycetin sulfat | Nước | Số 12 | Staphylococcus aureus NCTC 7447 CIP 53.156 ATCC 6538 P | Số 3 (pH 7,0) | 35 - 37 °C |
| Gentamycin sulfat | Gentamycin sulfat | Nước | Số 7 | Staphylococcus aureus NCTC 7447 CIP 53.156 ATCC 6538 P | Số 3 (pH 7,0) | 35 - 37 °C |
| Gramicidin | Gramicidin | Methanol (TT) | Thêm vào dung dịch đệm số 7 polysorbat 80 (TT) với tỷ lệ 0,1 mg/ml | Enterococcus hirae CIP 58.55 ATCC 10541 Staphylococcus aureus ATCC 6538 P | Số 3 (pH 7,0) | 35 - 37 °C |
| Josamycin | Josamycin | Hòa 30 mg trong 5 ml methanol (TT), thêm nước vừa đủ 100 ml | Số 20 | Staphylococcus aureus CIP 53.156 ATCC 6538 P NCTC 7447 | Số 3 (pH 8,0) | 35 - 37 °C |
| Josamycin propionat | Josamycin propionat | Hòa 40 mg trong 20 ml methanol (TT), thêm dung dịch đệm số 20 vừa đủ 100 ml | Số 20 | Staphylococcus aureus CIP 53.156 ATCC 6538 P NCTC 7447 | Số 3 (pH 8,0) | 35 - 37 °C |
| Kanamycin monosulfat Kanamycin acid sulfat | Kanamycin monosulfat | Nước | Số 12 | Staphylococcus aureus NCTC 7447 CIP 53.156 ATCC 6538 P | Số 3 (pH 7,0) | 35 - 37 °C |
| Neomycin sulfat | Neomycin sulfat | Nước | Số 12 | Staphylococcus aureus NCTC 7447 CIP 53.156 ATCC 6538 P | Số 3 (pH 7,0) | 35 - 37 °C |
| Rifamycin natri | Rifamycin natri | Methanol (TT) | Số 7 | Escherichia coli NCIB 8879 CIP 54.127 ATCC 10536 | Số 3 (pH 7,0) | 35 - 37 °C |
| Spiramycin | Spiramycin | Methanol (TT) | Số 7 | Staphylococcus aureus NCTC 7447 CIP 53.156 ATCC 6538 P | Số 3 (pH 7,0) | 35 - 37 °C |
| Streptomycin sulfat | Streptomycin sulfat | Nước | Số 12 | Klebsiella pneumoniae NCTC 7427 CIP 53.153 ATCC 10031 | Số 3 (pH 7,0) | 35 - 37 °C |
| Tyrothricin | Gramycidin | Ethanol (TT) | Ethanol (TT) | Enterococcus hirae ATCC 10541 | Số 3 (pH 7,0) | 37 °C |
| Vancomycin hydroclorid | Vancomycin hydroclorid | Nước | Số 12 | Staphylococcus aureus CIP 53.156 ATCC 6538 P | Số 3 (pH 7,0) | 37 - 39 °C |
13.10 PHÂN TÍCH THỐNG KÊ KẾT QUẢ ĐỊNH LƯỢNG SINH HỌC
1. Mở đầu
Chuyên luận này hướng dẫn cách thiết kế thí nghiệm sinh học và phương pháp phân tích thống kê các kết quả thử nghiệm sinh học có trong Dược điển Việt Nam.
Có thể dùng kiểu thiết kế thí nghiệm và cách tính toán kết quả khác với phương pháp mô tả trong chuyên luận này, miễn là các phương pháp đó có độ tin cậy không kém phương pháp được mô tả ở đây.
Phương pháp sinh học được dùng để thử nghiệm các chất hay chế phẩm mà hoạt lực của chúng không thể xác định một cách chính xác bằng các phương pháp phân tích hóa - lý. Nguyên tắc của phương pháp là so sánh chế phẩm cần định lượng với một chế phẩm chuẩn để xác định lượng chế phẩm cần định lượng cho cùng tác động sinh học với lượng đã cho, tính bằng đơn vị, của chế phẩm chuẩn. Để đảm bảo độ chính xác của kết quả định lượng, các phép thử được tiến hành với chế phẩm chuẩn và chế phẩm cần định lượng phải thực hiện đồng thời và trong cùng điều kiện thí nghiệm.
Bất kỳ một định giá nào của thử nghiệm sinh học luôn luôn mắc phải sai số ngẫu nhiên do tính biến thiên vốn có của đáp ứng sinh học, do đó nếu có thể, khi tính kết quả của mỗi thử nghiệm phải tính sai số, kể cả khi dùng phương pháp chính thức. Vì vậy, trong chuyên luận này có hướng dẫn các phương pháp thiết kế thử nghiệm và cách tính sai số. Các phương pháp tính toán trình bày ở đây chỉ tính đến sai số ngẫu nhiên gây ra bởi các sinh vật thử và giả định rằng các sai số hệ thống, ví dụ sai số do cân, pha loãng,... là rất nhỏ và không ảnh hưởng đáng kể đến kết quả định lượng (do đó phải có biện pháp thích hợp để giảm sai số hệ thống đến mức có thể chấp nhận được). Trong mọi trường hợp, trước khi áp dụng một phương pháp thống kê bất kỳ, phải tiến hành thử sơ bộ một số lần thích hợp để bảo đảm chắc chắn tính khả dụng của phương pháp đó.
Độ chính xác của kết quả thử nghiệm sinh học được xác định bởi các giới hạn tin cậy, hoặc là khoảng tin cậy của hoạt lực. Giới hạn tin cậy ở xác suất 95 % thường được chọn trong các thử nghiệm sinh học, do đó cũng được chọn trong chuyên luận này. Khoảng tin cậy tính bằng các phương pháp toán thống kê giới thiệu ở đây có khả năng chứa hoạt lực thật của chế phẩm cần định lượng với xác suất 95 %. Một số chuyên luận Dược điển quy định giới hạn tin cậy phải không được vượt quá một ngưỡng nhất định, ví dụ phải nằm trong khoảng từ 95 % đến 105 % so với hoạt lực tính được, trong một số trường hợp cần phải lặp lại thử nghiệm hai hay nhiều lần để đạt được giới hạn tin cậy cho phép.
Chú giải các ký hiệu dùng trong chuyên luận này được trình bày trong bảng ở phần cuối của chuyên luận.
2. Ngẫu nhiên hóa
Việc phân bổ những xử lý (liều, nồng độ....) khác nhau của chế phẩm chuẩn và chế phẩm thử cho các đơn vị thí nghiệm (ví dụ động vật thí nghiệm, ống nghiệm,...) phải được thực hiện một cách hoàn toàn ngẫu nhiên. Bất kỳ một sự lựa chọn nào khác về những điều kiện thí nghiệm (ví dụ chọn lựa cân nặng, tuổi của động vật thí nghiệm, điều kiện môi trường thí nghiệm,...), cũng không được phép cố ý trong thiết kế thí nghiệm mà đều phải lựa chọn ngẫu nhiên. Chọn vị trí của chuồng nuôi động vật thí nghiệm trong phòng thí nghiệm, trình tự dùng các xử lý cũng phải được thực hiện một cách ngẫu nhiên.
Ngẫu nhiên hóa có thể thực hiện bằng cách ném xúc xắc, xào các lá bài có đánh số, dùng bảng số ngẫu nhiên hay các phần mềm vi tính thích hợp.
3. Các thử nghiệm dựa trên đáp ứng định lượng
3.1 Mô hình thống kê
3.1.1 Nguyên tắc chung
Hai mô hình thống kê thường dùng trong các thử nghiệm sinh học là mô hình đường thẳng song song và mô hình tỷ lệ độ dốc. Trong chuyên luận này chỉ giới thiệu mô hình đường thẳng song song. Để tìm hiểu thêm về mô hình còn lại, xin tham khảo Dược điển châu Âu IV hay các tài liệu thống kê khác.
Chỉ có thể áp dụng mô hình thống kê đường thẳng song song nếu thử nghiệm hội đủ các điều kiện sau:
1) Các xử lý khác nhau của chế phẩm chuẩn và chế phẩm cần định lượng đã được phân bổ ngẫu nhiên cho từng đơn vị thí nghiệm;
2) Các đáp ứng của mỗi xử lý tuân theo phân phối chuẩn;
3) Độ lệch chuẩn của các đáp ứng trong mỗi nhóm xử lý của cả chế phẩm chuẩn và chế phẩm cần định lượng không khác nhau có ý nghĩa.
Khi thử nghiệm được triển khai sử dụng, nhà phân tích phải xác định xem các số liệu thu thập được từ nhiều thử nghiệm có thỏa mãn các điều kiện lý thuyết sau không:
Điều kiện 1 có thể thỏa mãn nếu thực hiện đúng theo các hướng dẫn trình bày ở mục 2 (Ngẫu nhiên hóa).
Điều kiện 2 là một giả định mà trong thực tế hầu như đều được thỏa mãn. Những sai lệch nhỏ so với giả định này nói chung không gây ảnh hưởng lớn đến kết quả, nếu mỗi xử lý được tiến hành nhiều thí nghiệm lặp lại.
Điều kiện 3 có thể được kiểm tra bằng phép thử tính đồng nhất của các phương sai, ví dụ bằng phép kiểm Bartlett hoặc phép kiểm Hartley (xem các ví dụ trong mục 3.2.8).
Nếu điều kiện 2 và/hoặc điều kiện 3 không thỏa mãn, chuyển đổi đáp ứng y bằng ln(y), ![]() hay y2 có thể cho kết quả tốt.
hay y2 có thể cho kết quả tốt.
Phép chuyển đổi y thành ln(y) rất hữu ích trong trường hợp tính đồng nhất của các phương sai không thỏa mãn. Phép chuyển đổi này cũng giúp cải thiện tính chuẩn nếu phân phối bị lệch về bên phải.
Sự chuyển đổi y thành ![]() thường được dùng khi các quan sát tuân theo phân phối Poisson, ví dụ khi các quan sát thu được bằng phương pháp đếm.
thường được dùng khi các quan sát tuân theo phân phối Poisson, ví dụ khi các quan sát thu được bằng phương pháp đếm.
3.1.2 Các thử nghiệm thường nhật
Trong các thử nghiệm thường nhật, rất khó kiểm tra một cách hệ thống các điều kiện lý thuyết mô tả ở mục 3.1.1, bởi vì trong thực tế số quan sát trong một thử nghiệm thường nhỏ do đó ảnh hưởng đến độ nhạy của các phép kiểm thống kê. Tuy nhiên, trong những thử nghiệm cân xứng (là những thử nghiệm có số xử lý của chế phẩm chuẩn bằng với số xử lý của chế phẩm cần định lượng, ví dụ: thử nghiệm 2 + 2, 3 + 3,...) những sai lệch nhỏ so với tính chuẩn hay so với tính đồng nhất của phương sai không ảnh hưởng lớn đến kết quả thử nghiệm. Do đó, chỉ cần kiểm tra lại các điều kiện lý thuyết nói trên khi một loạt các thử nghiệm liên tiếp không thỏa mãn phép kiểm tính có giá trị (xem mục 3.2.4).
Ngoài 3 điều kiện lý thuyết đã nói trên, mô hình đường thẳng song song còn yêu cầu mỗi thử nghiệm phải thỏa mãn thêm 2 điều kiện sau:
4) Đường biểu diễn mối quan hệ giữa logarit liều và đáp ứng phải tuyến tính trong khoảng liều đã dùng.
5) Đường thẳng In(liều) - đáp ứng của chế phẩm cần định lượng phải song song với đường In(liều) - đáp ứng của chế phẩm chuẩn.
Chỉ có thể kiểm tra điều kiện 4 và 5 nếu thử nghiệm được tiến hành với ít nhất 3 nồng độ pha loãng của mỗi chế phẩm (định lượng 3 liều hay lớn hơn). Tuy nhiên, nếu tính tuyến tính của đường logarit liều - đáp ứng, trong một khoảng liều nhất định, đã được chứng minh bởi một số lượng đủ lớn các thử nghiệm có 3 liều trở lên, có thể tiến hành thử nghiệm chỉ với 2 liều của mỗi chế phẩm (thử nghiệm 2 liều) trong các thử nghiệm hàng ngày, dùng trong khoảng liều đã cho.
Trước khi tính hoạt lực và các giới hạn tin cậy, cần phải tiến hành phân tích phương sai để kiểm tra xem thử nghiệm có đáp ứng các điều kiện 4 và 5 hay không.
Các thử nghiệm dựa trên mô hình thống kê đường thẳng song song được trình bày trong mục 3.2.
Nếu có bất cứ điều kiện nào trong 5 điều kiện nói trên không thỏa mãn, các phương pháp tính toán giới thiệu ở đây sẽ không có giá trị. Khi đó, phải tiến hành rà soát lại các kỹ thuật thử nghiệm để tìm ra nguyên nhân.
Nếu các phép kiểm thống kê cho thấy bất kỳ một điều kiện nào trong 5 điều kiện nói trên không thỏa mãn, không được chuyển ngay sang một phép chuyển đổi khác, trừ khi có đủ bằng chứng rằng nguyên nhân gây ra hiện tượng đó không phải do ngẫu nhiên mà là kết quả của một sự thay đổi có hệ thống của các điều kiện thí nghiệm. Trước khi áp dụng một phép chuyển đổi mới vào các thử nghiệm thường nhật, phải lặp lại phép kiểm thống kê đã trình bày ở mục 3.1.1.
Để thu được một kết quả thử nghiệm đáng tin cậy, có thể phải tiến hành một vài thử nghiệm độc lập, sau đó phối hợp các kết quả thử nghiệm lại với nhau (xem mục 4 - Phối hợp các kết quả thử nghiệm). Nhằm mục đích kiểm soát chất lượng của các thử nghiệm hàng ngày, nên ghi các trị định giá về độ dốc hồi qui và sai số dư dưới dạng biểu đồ kiểm tra.
Nếu sai số dư lớn một cách bất thường, nguyên nhân đầu tiên phải nghĩ đến là có sai sót kỹ thuật nào đó. Nếu các khảo sát khẳng định có sai sót trong quá trình thử nghiệm, phải lặp lại thử nghiệm. Sai số dư cũng có thể rất lớn nếu trong dãy số liệu đó có một giá trị bất thường. Chỉ được loại bỏ giá trị nghi ngờ là bất thường nếu giá trị đó khác có ý nghĩa thống kê với các giá trị còn lại.
Một sai số dư nhỏ bất thường có thể xảy ra một lần hoặc một lúc nào đó làm cho các tỷ số F vượt quá giá trị tới hạn. Trong những trường hợp như vậy, có thể thay sai số dư định giá được của từng thử nghiệm riêng bằng một sai số dư trung bình dựa vào các số liệu trước đó trong biểu đồ kiểm tra.
3.1.3 Cách tính toán và những hạn chế
Dưới đây là 3 hạn chế áp đặt cho mỗi thử nghiệm nhằm làm đơn giản hóa việc tính toán và làm tăng độ chính xác của thử nghiệm:
a) Số nồng độ pha loãng của mỗi chế phẩm trong một thử nghiệm phải bằng nhau;
b) Tỷ lệ giữa các liều kế tiếp nhau phải là một hằng số đối với tất cả các xử lý trong một thử nghiệm, ví dụ: S2/S1 = S3/S2 =... = Z3/Z2;
c) Số các đơn vị thí nghiệm của mỗi xử lý phải bằng nhau.
Các công thức tính toán áp dụng cho một thiết kế thử nghiệm tuân thủ đúng 3 hạn chế nêu trên được giới thiệu ở mục 3.2. Đối với các thiết kế thử nghiệm không tuân theo các hạn chế đã nêu, các công thức tính toán sẽ rất phức tạp và không được giới thiệu trong chuyên luận này.
Trong mục 3.2.8 có trình bày một số ví dụ để minh họa về phương pháp tính thống kê. Có thể dùng các số liệu trong các ví dụ đó để kiểm tra các phần mềm dùng trong phân tích thống kê kết quả thử nghiệm sinh học.
3.2 Mô hình đường thẳng song song
3.2.1 Giới thiệu
Mô hình đường thẳng song song dựa trên quan hệ tuyến tính giữa đáp ứng Y và logarit X của liều D.
Y = a + bX
trong đó:
Y là đáp ứng mong đợi;
X là In (liều) = ln(D);
a và b là các hằng số.
Mô hình đường thẳng song song có thể được minh họa bởi đồ thị trong Hình 13.10.1. Trên đồ thị, trục hoành biểu diễn các logarit liều với nồng độ thấp ở bên trái và nồng độ cao ở bên phải, trục tung biễu diễn các đáp ứng đo được. Các đáp ứng riêng của mỗi xử lý được biểu thị bằng các chấm đen. Hai đường trên đồ thị biểu diễn mối quan hệ giữa logarit liều và đáp ứng tính được của chế phẩm chuẩn và chế phẩm cần định lượng.
Từ trục tung kẻ một đường thẳng bất kỳ song song với trục hoành, cắt hai đường In(liều) - đáp ứng tại hai điểm có hoành độ là lnzS và lnzU, đây là hai logarit nồng độ hoặc logarit liều của chế phẩm chuẩn và chế phẩm thử cho cùng đáp ứng sinh học.
Để cho thử nghiệm thỏa mãn, hoạt lực giả định của chế phẩm thử phải gần với hoạt lực thực. Dựa vào hoạt lực giả định này và hoạt lực ấn định của chế phẩm chuẩn, pha các độ pha loãng có hoạt lực tương đương sao cho hoạt lực ZU của chế phẩm thử U bằng hoạt lực ZS của chế phẩm chuẩn S.
Khoảng cách nằm ngang giữa 2 đường thẳng: ln(zS) - ln(zU) là hoạt lực thực của chế phẩm thử so với hoạt lực giả định của nó. Nếu đường thẳng của chế phẩm thử ở bên phải của chế phẩm chuẩn, hoạt lực giả định đã được định giá quá cao và việc tính toán sẽ cho hoạt lực định giá được thấp hơn hoạt lực giả định; cũng tương tự nếu đường thẳng của chế phẩm thử ở bên trái của chế phẩm chuẩn, hoạt lực giả định đã được định giá quá thấp và việc tính toán sẽ cho hoạt lực định giá được cao hơn hoạt lực giả định. Trong một thử nghiệm, nếu đoạn thẳng ln(zS) - ln(zU) càng nhỏ, hoạt lực giả định của chế phẩm cần thử nghiệm càng gần với hoạt lực thật của nó và kết quả thử nghiệm sẽ càng chính xác.
Hình 13.10.1 - Mô hình đường thẳng song song của một thử nghiệm 3 + 3.
Chú ý: Logarit tự nhiên In (hay loge) được sử dụng xuyên suốt trong chuyên luận này, do đó, antilogarit tương đương với ex. Tuy vậy, nếu muốn, hoàn toàn có thể dùng logarit cơ số 10 (hay log10) thay cho In, khi đó, antilogarit tương ứng với 10x.
3.2.2 Thiết kế thử nghiệm
Các thử nghiệm sinh học có thể được thiết kế theo một số cách khác nhau như trình bày dưới đây.
3.2.2.1 Thiết kế ngẫu nhiên hoàn toàn
Nếu toàn bộ các đơn vị thí nghiệm (động vật, ống nghiệm,...) tương đối đồng nhất, việc phân chia các đơn vị thí nghiệm cho các xử lý khác nhau phải được tiến hành một cách ngẫu nhiên, ví dụ dùng bảng hoán vị ngẫu nhiên.
Nếu chia các đơn vị thí nghiệm thành các phân nhóm, ví dụ theo vị trí vật lý hay theo ngày thí nghiệm, nên chia theo cách tính đồng nhất hơn là chỉ xét đến số lượng các đơn vị thí nghiệm, ta có thể tăng độ chính xác của thử nghiệm bằng cách áp dụng một hoặc nhiều hạn chế trong cách thiết kế thí nghiệm. Sự phân chia các đơn vị thí nghiệm theo các hạn chế đó cho phép loại bỏ những nguồn gây sai số không liên quan.
3.2.2.2 Thiết kế ngẫu nhiên theo khối
Trong kiểu thiết kế thí nghiệm này, có thể tách các nguồn biến thiên ra, ví dụ biến thiên do sự nhạy cảm giữa các lứa động vật thí nghiệm hoặc biến thiên giữa các hộp Petri trong định lượng vi sinh vật bằng phương pháp khuếch tán, khỏi sai số toàn phần. Thiết kế thí nghiệm ngẫu nhiên theo khối đòi hỏi mỗi khối (lứa động vật hoặc hộp Petri) phải có số xử lý bằng nhau và chỉ thích hợp nếu khối đủ lớn để có thể chứa tất cả các xử lý của thử nghiệm.
Bảng 13.10.6 trình bày công thức tính toán áp dụng cho các thử nghiệm thiết kế thí nghiệm ngẫu nhiên theo khối, trong đó mỗi xử lý chỉ xuất hiện một lần duy nhất trong mỗi khối (thiết kế thí nghiệm ngẫu nhiên theo khối không lặp). Ví dụ minh họa cho kiểu thiết kế thí nghiệm này được trình bày trong ví dụ 3.2.8.2.
3.2.2.3 Thiết kế hình vuông Latin
Kiểu thiết kế thí nghiệm này thích hợp với những thử nghiệm mà đáp ứng chịu ảnh hưởng của 2 nguồn biến thiên khác nhau, trong đó mỗi nguồn biến thiên có k mức hay k vị trí khác nhau. Ví dụ, trong định lượng kháng sinh bằng phương pháp khuếch tán, các xử lý có thể được sắp xếp thành k x k dãy trên một khay lớn, mỗi xử lý chỉ xuất hiện một lần duy nhất trong mỗi hàng và mỗi cột. Chỉ có thể áp dụng kiểu thiết kế thí nghiệm hình vuông Latin khi số hàng, số cột và số xử lý của mỗi chế phẩm đều phải bằng nhau.
Các đáp ứng được ghi vào một bảng hình vuông gọi là hình vuông Latin. Biến thiên do sự khác nhau về đáp ứng giữa k hàng và giữa k cột sẽ được tách khỏi biến thiên toàn phần, do đó làm giảm sai số của định lượng.
Bảng 13.10.6 trình bày công thức tính toán áp dụng cho các thử nghiệm hình vuông Latin, trong đó mỗi xử lý chỉ xuất hiện một lần duy nhất trong mỗi hàng và mỗi cột. Ví dụ minh họa cho kiểu thiết kế thí nghiệm này được trình bày trong ví dụ 3.2.8.3.
3.2.2.4 Thiết kế chéo
Kiểu thiết kế thí nghiệm này được áp dụng trong trường hợp thí nghiệm có thể chia thành các khối, nhưng mỗi khối chỉ có thể nhận hai xử lý, ví dụ một khối là một đơn vị thí nghiệm được thử hai lần vào hai thời điểm khác nhau. Mục đích của kiểu thiết kế chéo là làm tăng độ chính xác của thử nghiệm bằng cách loại trừ ảnh hưởng do sự khác nhau giữa các đơn vị thí nghiệm. Nếu thử nghiệm tiến hành với 2 liều của chế phẩm chuẩn và 2 liều của chế phẩm cần định lượng, ta có kiểu thí nghiệm chéo đôi.
Bảng 13.10.1- Cách sắp xếp các liều trong thiết kế thí nghiệm chéo
| Nhóm các đơn vị thí nghiệm | Giai đoạn I | Giai đoạn II |
| 1 | s1 | u2 |
| 2 | s2 | u1 |
| 3 | u1 | s2 |
| 4 | u2 | s1 |
Trong thiết kế chéo đôi, thử nghiệm được chia thành hai giai đoạn, mỗi giai đoạn tiến hành vào những thời điểm khác nhau. Các đơn vị thí nghiệm được chia thành bốn nhóm, mỗi nhóm nhận một trong bốn xử lý ở giai đoạn đầu của thí nghiệm. Đơn vị nhận một chế phẩm trong giai đoạn đầu sẽ nhận chế phẩm khác trong giai đoạn sau, và các đơn vị nhận liều thấp trong giai đoạn đầu sẽ nhận liều cao trong giai đoạn sau. Cách sắp xếp các liều cho ở Bảng 13.10.1. Ví dụ minh họa cho kiểu thiết kế thí nghiệm này được trình bày trong Ví dụ 3.2.8.4.
3.2.3 Phân tích phương sai
Mục này trình bày các công thức cần thiết để phân tích phương sai. Người đọc sẽ dễ hiểu hơn khi tham khảo thêm các ví dụ ở mục 3.2.8 và Bảng ghi chú các ký hiệu ở mục 6.
Các công thức trình bày ở đây có thể áp dụng cho các định lượng đối xứng trong đó một hay nhiều chế phẩm cần thử (U,.... Z) được so sánh với cùng một chế phẩm chuẩn S, với điều kiện thử nghiệm phải đáp ứng được các điều kiện sau: 1) tỷ lệ giữa các liều kế tiếp nhau của tất cả các chế phẩm phải là một hằng số, 2) số xử lý của mỗi chế phẩm và 3) số đơn vị thí nghiệm (số thử nghiệm lặp lại) trong mỗi xử lý phải bằng nhau (xem mục 3.1.3).
Ngoại trừ một vài khác biệt nhỏ trong cách tính sai số dư, phương pháp phân tích các số liệu của mỗi thử nghiệm sinh học về cơ bản là giống nhau đối với các kiểu thiết kế thí nghiệm ngẫu nhiên hoàn toàn, ngẫu nhiên theo khối và hình vuông Latin. Thiết kế thí nghiệm chéo có công thức tính hoàn toàn khác và do đó được trình bày trực tiếp trong Ví dụ 3.2.8.4.
Để chuẩn bị số liệu cho phân tích phương sai, đầu tiên phải tính tổng đáp ứng của mỗi liều, tổng đáp ứng và tương phản tuyến tính (linear contrast) của mỗi chế phẩm; đối với các định lượng có nhiều hơn hai liều của mỗi chế phẩm, cần tính thêm tương phản bậc hai (quadratic contrast). Các Bảng 13.10.2, 13.10.3 và 13.10.4 trình bày các công thức tính đó.
Bảng 13.10.2- Công thức tính toán cho các thử nghiệm hai liều đa chế phẩm
|
| Chế phẩm chuẩn S | Chế phẩm thử thứ nhất U | Chế phẩm thử thứ (h-1) Z |
| Tổng đáp ứng của liều thấp | S1 | U1 | Z1 |
| Tổng đáp ứng của liều cao | S2 | U2 | Z2 |
| Tổng đáp ứng của chế phẩm | S1 + S2 = S | U1 + U2 = U | Z1 + Z2 = Z |
| Tương phản tuyến tính | S2 - S1 = LS | U2 - U1 = LU | Z2 - Z1 = LZ |
Bảng 13.10.3 - Công thức tính toán cho các thử nghiệm ba liều đa chế phẩm
|
| Chế phẩm chuẩn S | Chế phẩm thử thứ nhất U | Chế phẩm thử thứ (h-1) Z |
| Tổng đáp ứng của liều thấp | S1 | U1 | Z1 |
| Tổng đáp ứng của liều trung gian | S2 | U2 | Z2 |
| Tổng đáp ứng của liều cao | S3 | U3 | Z3 |
| Tổng đáp ứng của chế phẩm | S1 + S2 + S3 = S | U1 + U2 + U3 = U | Z1 + Z2 + Z3 = Z |
| Tương phản tuyến tính | S3 - S1 = LS | U3 - U1 = LU | Z3 - Z1 = LZ |
| Tương phản bậc hai | S1 - 2S2 + S3 = QS | U1 - 2U2 + U3 = QU | Z1 - 2Z2 + Z3 = QZ |
Bảng 13.10.4 - Công thức tính toán cho các thử nghiệm bốn liều đa chế phẩm
|
| Chế phẩm chuẩn S | Chế phẩm thử thứ nhất U | Chế phẩm thử thứ (h-1) Z |
| Tổng đáp ứng của liều thấp nhất | S1 | U1 | Z1 |
| Tổng đáp ứng của liều thứ hai | S2 | U2 | Z2 |
| Tổng đáp ứng của liều thứ ba | S3 | U3 | Z3 |
| Tổng đáp ứng của liều cao nhất | S4 | U4 | Z4 |
| Tổng đáp ứng của chế phẩm | S1 + S2 + S3 + S4 = S | U1 + U2 + U3 + U4 = U | Z1 + Z2 + Z3 + Z4 = Z |
| Tương phản tuyến tính | 3S4 + S3 - S2 - 3S1 = LS | 3U4 + U3 - U2 - 3U1 = LU | 3Z4 + Z3 - Z2 - 3Z1 = LZ |
| Tương phản bậc hai | S1 - S2 - S3 + S4 = QS | U1 - U2 - U3 + U4 = QU | Z1 - Z2 - Z3 + Z4 = QZ |
| Tương phản bậc ba | 3S2 - S1 + S4 - 3S3 = JS | 3U2 - U1 + U4 - 3U3 = JU | 3Z2 - Z1 + Z4 - 3Z3 = JZ |
Trong bước kế tiếp, biến thiên toàn phần của thử nghiệm, còn gọi là sai số chung, được phân tích thành các biến thiên riêng phần như trình bày ở Bảng 13.10.5, các tổng các bình phương được tính từ các giá trị thu được ở các Bảng 13.10.2, 13.10.3 và 13.10.4.
Bảng 13.10.5- Phép thử tính có giá trị của thử nghiệm
| Nguồn biến thiên | Bậc tự do (f) | Tổng các bình phương | ||
| 2 liều/chế phẩm | 3 liều/chế phẩm | 4 liều/chế phẩm | ||
| Chế phẩm | h - 1 |
|
|
|
| Hồi qui tuyến tính | 1 |
|
|
|
| Tính không song song | h -1 |
|
|
|
| Tính không tuyến tính (Độ cong) | h (3 liều) 2h (4 liều) | - |
|
|
|
| ||||
Tính sai số dư (residual error) bằng cách lấy sai số toàn phần của thử nghiệm trừ đi các sai số riêng của các nguồn biến thiên khác nhau (Bảng 13.10.6), nguồn biến thiên có thể nhận dạng được tùy theo kiểu thiết kế thí nghiệm trong Bảng 13.10.6, ![]() là tổng bình phương của tất cả các đáp ứng đo được trong thử nghiệm, K là đại lượng để hiệu chỉnh:
là tổng bình phương của tất cả các đáp ứng đo được trong thử nghiệm, K là đại lượng để hiệu chỉnh:
![]()
Kết thúc phân tích phương sai bằng cách chia các tổng các bình phương tính được từ Bảng 13.10.5 cho bậc tự do tương ứng để được các bình phương trung bình.
Bình phương trung bình của sai số dư cũng được tính theo cách tương tự, dùng số liệu tương ứng ở Bảng 13.10.6.
Bình phương trung bình của mỗi biến số cần kiểm tra sẽ được biểu diễn dưới dạng một tỷ số với sai số dư s2, gọi là tỷ số F. Đánh giá mức ý nghĩa của các tỷ số F bằng cách so sánh F tính được (viết tắt là Fcal.)với giá trị F tới hạn (viết tắt là Fcrit.) tra từ Bảng 13.10.7 hay bằng cách dùng các phần mềm vi tính thích hợp. Nếu dùng bảng tra cứu, đọc Fcrit. từ Bảng 13.10.7 tại giao điểm giữa cột tương ứng với bậc tự do của bình phương trung bình của biến số cần kiểm tra (f1) và hàng tương ứng với bậc tự do của s2 (f2). Biến số cần kiểm tra được coi là có ý nghĩa nếu Fcal. > Fcrit. ở xác suất P = 0,05 hay rất có ý nghĩa nếu Fcal. > Fcrit. ở xác suất P = 0,01.
Bảng 13.10.6- Định giá của sai số dư
| Nguồn biến thiên | Bậc tự do (f) | Tổng các bình phương | ||
| Ngẫu nhiên hoàn toàn | Ngẫu nhiên theo khối | Hình vuông Latin | ||
| Giữa các xử lý | k-1 |
|
|
|
| Giữa các khối (hàng) | n -1 | - |
|
|
| Giữa các khối (cột) | n -1 | - | - |
|
| Sai số dư | Hiệu số | * | * | * |
| Sai số toàn phần | N -1 |
|
|
|
Lấy sai số toàn phần trừ đi các tổng các bình phương tương ứng của mỗi phương pháp thiết kế thí nghiệm.
3.2.4. Phép thử tính có giá trị của thử nghiệm
Kết quả thử nghiệm được coi là có giá trị thống kê (statistically valid) nếu:
1) Đại lượng hồi qui tuyến tính rất có ý nghĩa, Fcal. > Fcrit. ở xác suất P = 0,01 (P < 0,01), chứng tỏ độ dốc của đường In(liều) - đáp ứng rất khác 0.
2) Đại lượng không tuyến tính (non-linearity) phải không có ý nghĩa, Fcal. > Fcrit. ở xác suất 0,05 (P > 0,05), chứng tỏ thỏa mãn điều kiện 4, tức là các đường In(liều) - đáp ứng của chế phẩm chuẩn S và các chế phẩm thử (U,…,Z) thẳng.
3) Đại lượng không song song (non-parallelism) phải không có ý nghĩa, Fcal. > Fcrit. ở xác suất 0,05 (P > 0,05), chứng tỏ thử nghiệm đáp ứng điều kiện 5, hay nói cách khác các đường In(liều) - đáp ứng của các chế phẩm thử (U,...,Z) song song với đường In(liều) - đáp ứng của chế phẩm chuẩn S.
Nếu đại lượng không song song có ý nghĩa trong một thử nghiệm có h chế phẩm, kể cả chế phẩm chuẩn, một trong số các chế phẩm cần thử có thể có độ dốc của đường In (liều) - đáp ứng khác với các chế phẩm còn lại. Tính t’(phép kiểm Dunnett) của mỗi chế phẩm cần định lượng (U,...,Z) theo công thức:
|
|
| (3.2.4.-1) |
Đối với các thử nghiệm có bốn liều đơn chế phẩm, tính t' theo công thức:
|
|
|
|
So sánh mỗi t' tính được với giá trị tới hạn đọc từ Bảng 13.10.8, với f1 = h - 1, f2 là bậc tự do của s2. Nếu t’ có ý nghĩa thống kê với một chế phẩm bất kỳ, loại bỏ tất cả các số liệu liên quan đến chế phẩm đó và lặp lại từ đầu phép kiểm tính có giá trị với các chế phẩm còn lại.
Biến thiên do chế phẩm không được dùng trong phép thử tính có giá trị của thử nghiệm, tuy nhiên, trong những thử nghiệm mà sai số dư lớn một cách bất thường và tỷ số F của nguồn biến thiên do chế phẩm cao rất có ý nghĩa, hoạt lực giả định của chế phẩm cần định lượng rất khác với hoạt lực thật của nó, cần phải lặp lại thử nghiệm dùng hoạt lực tính được làm hoạt lực giả định.
Bảng 13.10.7- Giá trị tới hạn của tỷ số F
| f1 | |||||||||||
|
|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 20 | ∞ |
| f2 | 12 | 4,75 | 3,89 | 3,49 | 3,26 | 3,11 | 3,00 | 2,91 | 2,85 | 2,54 | 2,30 |
|
| 9,33 | 6,93 | 5,95 | 5,41 | 5,06 | 4,82 | 4,64 | 4,50 | 3,86 | 3,36 | |
| 15 | 4,54 | 3,68 | 3,29 | 3,06 | 2,90 | 2,79 | 2,71 | 2,64 | 2,33 | 2,07 | |
|
| 8,68 | 6,36 | 5,42 | 4,89 | 4,56 | 4,32 | 4,14 | 4,00 | 3,37 | 2,87 | |
| 20 | 4,35 | 3,49 | 3,10 | 2,87 | 2,71 | 2,60 | 2,51 | 2,45 | 2,12 | 1,84 | |
|
| 8,10 | 5,85 | 4,94 | 4,43 | 4,10 | 3,87 | 3,70 | 3,56 | 2,94 | 2,42 | |
| 25 | 4,24 | 3,39 | 2,99 | 2,76 | 2,60 | 2,49 | 2,40 | 2,34 | 2,01 | 1,71 | |
|
| 7,77 | 5,57 | 4,68 | 4,18 | 3,85 | 3,63 | 3,46 | 3,32 | 2,70 | 2,17 | |
| 30 | 4,17 | 3,32 | 2,92 | 2,69 | 2,53 | 2,42 | 2,33 | 2,27 | 1,93 | 1,62 | |
|
| 7,56 | 5,39 | 4,51 | 4,02 | 3,70 | 3,47 | 3,30 | 3,17 | 2,55 | 2,01 | |
| 40 | 4,08 | 3,23 | 2,84 | 2,61 | 2,45 | 2,34 | 2,25 | 2,18 | 1,84 | 1,51 | |
|
| 7,31 | 5,18 | 4,31 | 3,83 | 3,51 | 3,29 | 3,12 | 2,99 | 2,37 | 1,80 | |
| 60 | 4,00 | 3,15 | 2,76 | 2,53 | 2,37 | 2,25 | 2,17 | 2,10 | 1,75 | 1,39 | |
|
| 7,08 | 4,98 | 4,13 | 3,65 | 3,34 | 3,12 | 2,95 | 2,82 | 2,20 | 1,60 | |
| ∞ | 3,84 | 3,00 | 2,60 | 2,37 | 2,21 | 2,10 | 2,01 | 1,94 | 1,57 | 1,00 | |
|
|
|
|
|
|
|
|
|
|
|
|
|
| * Nếu F tính được > F tới hạn, biến số cần kiểm tra được coi là có ý nghĩa (dòng trên, p = 0,05), hay rất có ý nghĩa (dòng dưới, p = 0,01). f1 là bậc tự do của tử số, f2 là bậc tự do của mẫu số. | |||||||||||
Bảng 13.10.8- Giá trị tới hạn của t’
| f1 = (h - 1) = số chế phẩm cần định lượng | |||||||||
| f2 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| 5 | 2,57 | 3,03 | 3,29 | 3,48 | 3,62 | 3,73 | 3,82 | 3,90 | 3,97 |
| 6 | 2,45 | 2,86 | 3,10 | 3,26 | 3,39 | 3,49 | 3,57 | 3,64 | 3,71 |
| 7 | 2,36 | 2,75 | 2,97 | 3,12 | 3,24 | 3,33 | 3,41 | 3,47 | 3,53 |
| 8 | 2,31 | 2,67 | 2,88 | 3,02 | 3,13 | 3,22 | 3,29 | 3,35 | 3,41 |
| 9 | 2,26 | 2,61 | 2,81 | 2,95 | 3,05 | 3,14 | 3,20 | 3,26 | 3,32 |
| 10 | 2,23 | 2,57 | 2,76 | 2,89 | 2,99 | 3,07 | 3,14 | 3,19 | 3,24 |
| 11 | 2,20 | 2,53 | 2,72 | 2,84 | 2,94 | 3,02 | 3,08 | 3,14 | 3,19 |
| 12 | 2,18 | 2,50 | 2,68 | 2,81 | 2,90 | 2,98 | 3,04 | 3,09 | 3,14 |
| 13 | 2,16 | 2,48 | 2,65 | 2,78 | 2,87 | 2,94 | 3,00 | 3,06 | 3,10 |
| 14 | 2,14 | 2,46 | 2,63 | 2,75 | 2,84 | 2,91 | 2,97 | 3,02 | 3,07 |
| 15 | 2,13 | 2,44 | 2,61 | 2,73 | 2,82 | 2,89 | 2,95 | 3,00 | 3,04 |
| 16 | 2,12 | 2,42 | 2,59 | 2,71 | 2,80 | 2,87 | 2,92 | 2,97 | 3,02 |
| 17 | 2,11 | 2,41 | 2,58 | 2,69 | 2,78 | 2,85 | 2,90 | 2,95 | 3,00 |
| 18 | 2,10 | 2,40 | 2,56 | 2,68 | 2,76 | 2,83 | 2,89 | 2,94 | 2,98 |
| 19 | 2,09 | 2,39 | 2,55 | 2,66 | 2,75 | 2,81 | 2,87 | 2,92 | 2,96 |
| 20 | 2,09 | 2,38 | 2,54 | 2,65 | 2,73 | 2,80 | 2,86 | 2,90 | 2,95 |
| 24 | 2,06 | 2,35 | 2,51 | 2,61 | 2,70 | 2,76 | 2,81 | 2,86 | 2,90 |
| 30 | 2,04 | 2,32 | 2,47 | 2,58 | 2,66 | 2,72 | 2,77 | 2,82 | 2,86 |
| 40 | 2,02 | 2,29 | 2,44 | 2,54 | 2,62 | 2,68 | 2,73 | 2,77 | 2,81 |
| 60 | 2,00 | 2,27 | 2,41 | 2,51 | 2,58 | 2,64 | 2,69 | 2,73 | 2,77 |
| 120 | 1,98 | 2,24 | 2,38 | 2,47 | 2,55 | 2,60 | 2,65 | 2,69 | 2,73 |
| ∞ | 1,96 | 2,21 | 2,35 | 2,44 | 2,51 | 2,57 | 2,61 | 2,65 | 2,69 |
Nếu thử nghiệm thỏa mãn phép thử tính có giá trị, hay nói cách khác, thử nghiệm có giá trị thống kê, hoạt lực và các giới hạn tin cậy có thể được định giá bằng các phương pháp mô tả trong mục kế tiếp.
3.2.5 Định giá hoạt lực và các giới hạn tin cậy
Để định giá hoạt lực và các giới hạn tin cậy, trước hết phải tính đáp ứng trung bình của mỗi chế phẩm![]()
|
|
| (3.2.5.-1) |
Tiến hành tương tự như vậy với các chế phẩm khác.
Gọi l là khoảng cách giữa In các liều kế tiếp nhau của một chế phẩm bất kỳ (l là hằng số trong một thử nghiệm), ví dụ l = ln(S2) - ln(S1), độ dốc chung (b) của một thử nghiệm bao gồm h chế phẩm, mỗi chế phẩm có d liều khác nhau được tính theo công thức:
|
|
| (3.2.5.-2) |
Đối với các thử nghiệm bốn liều đơn chế phẩm, tính b theo công thức:
|
|
|
|
ln(tỷ lệ hoạt lực) của chế phẩm cần định lượng U được tính theo công thức:
|
|
| (3.2.5.-3) |
Gọi AU là hoạt lực giả định của chế phẩm U, logarit hoạt lực của U (MU) sẽ bằng:
| MU = M'U + ln (AU) |
| (3.2.5.-4) |
Hoạt lực tính được là ước lượng của hoạt lực thật của mỗi chế phẩm cần định lượng. Các giới hạn tin cậy của hoạt lực tính được là khoảng chứa hoạt lực thật của chế phẩm cần định lượng với xác suất 95 %. Logarit của các giới hạn tin cậy được tính theo công thức sau:
|
| (3.2.5.-5) | |
| với: |
| (3.2.5.-6) |
E là giá trị tính được từ Bảng 13.10.5, tính s2 bằng cách chia tổng các bình phương của biến số sai số dư trong Bảng 13.10.6 cho bậc tự do tương ứng của nó, t là giá trị đọc được từ Bảng 13.10.28 với xác suất P = 0,95 và f là bậc tự do của s2.
Trong những thử nghiệm đối xứng, công thức tính các giới hạn tin cậy có thể đơn giản hóa thành:
|
|
| (3.2.5.-7) | |
| với: |
|
|
|
Trong đó:
C là tiêu chí đánh giá ý nghĩa của đường hồi qui. Trong một thử nghiệm có độ dốc đạt yêu cầu, giá trị của C rất gần với 1. Nếu C < 0 hồi qui tuyến tính không có ý nghĩa.
Tính định giá hoạt lực của chế phẩm U (RU) và các giới hạn tin cậy của nó bằng cách lấy antilogarit các giá trị tính được từ các công thức 3.2.5.-4 và 3.2.5.-7.
|
| RU = anti ln (MU) | (3.2.5.-8) |
|
| (3.2.5.-9) | |
|
| (3.2.5.-10) | |
FLU là giới hạn tin cậy trên và FLL là giới hạn tin cậy dưới của hoạt lực tính được RU.
Nếu nồng độ dung dịch gốc của chế phẩm chuẩn và các chế phẩm thử không hoàn toàn bằng nhau, phải hiệu chỉnh hoạt lực tính được và các giới hạn tin cậy với một hệ số, gọi là hệ số hiệu chỉnh (xem Ví dụ 3.2.8.3). Hệ số hiệu chỉnh là tỷ số giữa nồng độ dung dịch gốc của chế phẩm chuẩn và nồng độ dung dịch gốc của mỗi chế phẩm cần định lượng.
3.2.6 Các giá trị bị mất
Trong một thử nghiệm đối xứng, một sự cố ngẫu nhiên có thể dẫn đến việc mất một hay nhiều giá trị đo, ví dụ động vật thí nghiệm chết, vòng vô khuẩn bị méo không đo được trong một thử nghiệm kháng sinh... Nếu sự cố xảy ra không liên quan đến thành phần của chế phẩm đã sử dụng, vẫn có thể tính được kết quả định lượng một cách chính xác, tuy nhiên khi đó các công thức tính toán sẽ trở nên rất phức tạp. Để có thể tiếp tục dùng các công thức tính toán đơn giản của thiết kế thí nghiệm đối xứng, có thể áp dụng một trong hai cách sau:
1) Nếu số đáp ứng của mỗi xử lý đủ lớn, giảm số đáp ứng trong các xử lý có số đáp ứng lớn hơn cho đến khi số đáp ứng của tất cả các xử lý bằng nhau. Nếu động vật thí nghiệm đã được phân bố một cách ngẫu nhiên cho mỗi xử lý, bỏ một hay vài đáp ứng, được chọn ngẫu nhiên, từ các xử lý có số đáp ứng lớn sẽ thu được thử nghiệm đối xứng. Đối với các thiết kế thí nghiệm ngẫu nhiên theo khối, cách đơn giản nhất là bỏ tất cả các đáp ứng của khối có giá trị bị mất. Ví dụ, chỉ giữ lại kết quả đo của các hộp Petri có đủ 6 vòng vô khuẩn trong thử nghiệm kháng sinh 3 + 3 bằng phương pháp khuếch tán, dùng hộp Petri.
2) Thay các giá trị bị mất bằng các giá trị tính được từ những giá trị còn lại. Công thức tính các giá trị mất được trình bày ở bên dưới, số bậc tự do của sai số toàn phần và sai số dư sẽ phải giảm đi một đơn vị cho mỗi giá trị bị mất. Cần nhớ rằng đây chỉ là phương pháp gần đúng, phương pháp chính xác luôn luôn cho kết quả đáng tin cậy hơn.
Nếu có nhiều quan sát bị mất, thay tất cả các giá trị bị mất, trừ một quan sát, bằng các giá trị ước lượng thô và dùng công thức thích hợp để tính giá trị của quan sát đó dựa trên các giá trị còn lại, kể cả các giá trị ước lượng thô. Thay quan sát bị mất bằng giá trị vừa tính được. Tiếp tục tính theo cách trên với giá trị ước đoán thô thứ nhất... Sau khi đã thay thế lần lượt tất cả các quan sát bị mất, lặp lại từ đầu chu trình tính toán, mỗi lần tính dùng một giá trị ước đoán hoặc giá trị tính được cho đến khi hai chu trình kế tiếp nhau cho cùng kết quả.
Kết quả thử nghiệm chỉ được chấp nhận nếu số các giá trị thay thế tương đối nhỏ (nhỏ hơn 5 %) so với tổng số đáp ứng của toàn bộ thử nghiệm, cần đặc biệt thận trọng trong trường hợp các giá trị bị mất có khuynh hướng tập trung trong một xử lý hay một khối.
Thiết kế ngẫu nhiên hoàn toàn
Trong kiểu thiết kế ngẫu nhiên hoàn toàn, thay giá trị bị mất bằng trung bình số học của các đáp ứng khác trong cùng xử lý.
Thiết kế ngẫu nhiên theo khối
Giá trị bị mất (y') được tính bằng công thức:
|
|
| (3.2.6.-1) |
Trong đó:
B’ là tổng đáp ứng của khối chứa giá trị bị mất, T' là tổng đáp ứng của xử lý chứa giá trị bị mất, G' là tổng tất cả các đáp ứng còn lại trong thử nghiệm, n và k theo thứ tự là số khối và số xử lý.
Để minh họa, giả sử giá trị của u1 trong khối thứ nhất (trong trường hợp này là hàng hay hộp Petri) ở ví dụ 3.2.8.2, Bảng 13.10.14 bị mất (u1 = 174). Với:
B' = 1.050
T' = 869
G' = 7.189
k = 6
n = 6
y’ = 173. Dùng giá trị tính được 173 thay cho đáp ứng của u1 trong hộp số 1 của Bảng 13.10.14 và tiếp tục tính toán như trình bày trong ví dụ 3.2.8.2 nhưng với bậc tự do của sai số toàn phần là 34, và bậc tự do của sai số dư là 24.
Thiết kế hình vuông Latin
Giá trị bị mất (y') được tính bằng công thức:
|
|
| (3.2.6.-2) |
Trong đó:
B’ và C’ là tổng các đáp ứng trong hàng và cột chứa giá trị bị mất. G' là tổng các đáp ứng còn lại trong thử nghiệm (Bảng 13.10.18), còn T' là tổng các đáp ứng của xử lý chứa giá trị bị mất (Bảng 13.10.19). Trong thiết kế hình vuông Latin k = n. Giả sử giá trị 161 của đáp ứng ở cột thứ 1 và hàng thứ 1 trong Bảng 13.10.18 và Bảng 13.10.19 (Ví dụ 3.2.8.3) bị mất, nó sẽ được thay thế bởi giá trị 150 tính được bằng công thức 3.2.6.-2 với:
B’ = 890
C’ = 876
T' = 791
G' = 6.175
k = 6
Các bậc tự do sẽ bị giảm xuống còn 19 đối với sai số dư và 34 đối với sai số toàn phần.
Thiết kế chéo
Nếu có giá trị bị mất trong kiểu thiết kế thí nghiệm chéo phải tham khảo các tài liệu thống kê vì công thức tính sẽ rất khác nhau tùy theo cách phối hợp các xử lý.
3.2.7 Các thử nghiệm đối xứng một phần
Nếu hoạt lực giả định của chế phẩm cần định lượng, là hoạt lực được dùng để tính lượng chế phẩm thử cần lấy khi pha các dung dịch của chế phẩm thử, khác xa với hoạt lực thật, nồng độ cao nhất hoặc nồng độ thấp nhất của chế phẩm thử có thể nằm lọt ra ngoài vùng tuyến tính của đường In(liều) - đáp ứng, do đó thử nghiệm sẽ không có giá trị thống kê do không thỏa mãn tính tuyến tính và/hoặc tính song song.
Có thể tính hoạt lực từ các số liệu còn lại, sau khi loại bỏ các đáp ứng của nồng độ cao nhất hoặc nồng độ thấp nhất của chế phẩm thử và dùng hoạt lực tính được làm hoạt lực giả định cho những lần thử nghiệm lặp lại.
Logarit tỷ lệ hoạt lực được tính bằng công thức:
|
|
| (3.2.7.-1) |
Công thức trên rất giống với phương trình 3.2.5.-3, tuy nhiên, In (tỷ lệ hoạt lực) sẽ bị trừ đi một đại lượng bằng l/2 nếu bỏ nồng độ thấp nhất, và cộng thêm l/2 nếu bỏ nồng độ cao nhất.
Tính các đáp ứng trung bình ![]() và
và ![]() theo cách tương tự như thử nghiệm cân xứng hoàn toàn (phương trình 3.2.5.-1), nhưng công thức tính độ dốc (b) có một số biến đổi tùy theo thiết kế thí nghiệm.
theo cách tương tự như thử nghiệm cân xứng hoàn toàn (phương trình 3.2.5.-1), nhưng công thức tính độ dốc (b) có một số biến đổi tùy theo thiết kế thí nghiệm.
Trong thử nghiệm hai liều đa chế phẩm, tính độ dốc bằng công thức:
|
|
| (3.2.7.-2) |
Chú ý: Trong công thức trên, tử số không bao gồm LU vì chế phẩm U chỉ còn một nồng độ.
Đối với thử nghiệm đơn chế phẩm:
|
|
| (3.2.7.-3) |
Trong thử nghiệm ba liều đa chế phẩm, tính các tương phản tuyến tính (LS ,…,LZ) theo công thức cho ở Bảng 13.10.3, riêng LU tính bằng công thức ở Bảng 13.10.2. Công thức tính độ dốc sẽ là:
|
|
| (3.2.7.-4) |
Đối với thử nghiệm đơn chế phẩm, công thức trên trở thành:
|
|
| (3.2.7.-5) |
3.2.8 Các ví dụ
Mục này trình bày một số ví dụ minh họa cách sử dụng các công thức tính toán liên quan đến mô hình đường thẳng song song.
Các ví dụ giới thiệu ở đây chỉ nhằm mục đích minh họa cho các phương pháp tính toán thống kê và không phải là phương pháp bắt buộc phải áp dụng nếu chuyên luận riêng cho phép dùng phương pháp khác.
Để có thể sử dụng các ví dụ trong chuyên luận này như nguồn số liệu tham khảo phục vụ cho việc kiểm tra các chương trình vi tính, thương mại hay tự biên soạn, dùng trong phân tích thống kê kết quả định lượng sinh học, các kết quả tính toán có thể chứa nhiều số thập phân hơn mức cần thiết. Có thể dùng phương pháp tính toán khác với phương pháp giới thiệu trong chuyên luận này, nhưng kết quả cuối cùng phải giống như các kết quả trong các ví dụ trình bày ở đây.
Ví dụ 3.2.8.1 Thử nghiệm hai liều đa chế phẩm với thiết kế ngẫu nhiên hoàn toàn
Thử nghiệm định lượng corticotrophin bằng phương pháp tiêm dưới da chuột cống trắng
Liều dùng của chế phẩm chuẩn là 0,25 và 1,0 đơn vị cho mỗi 100 g thể trọng chuột thí nghiệm. Hai chế phẩm thử đều có hoạt lực giả định là 1 đơn vị/mg và có liều dùng giống như chế phẩm chuẩn. Các đáp ứng riêng và đáp ứng trung bình của mỗi nhóm xử lý cho ở Bảng 13.10.9.
Bảng 13.10.9 - Đáp ứng metameter y - khối lượng acid ascorbic (mg) trên 100 g tuyến thượng thận
|
| Chế phẩm chuẩn S | Chế phẩm thử U | Chế phẩm thử Z | Tổng số | |||
| s1 | s2 | u1 | u2 | z1 | z2 | ||
|
| 300 | 289 | 310 | 230 | 250 | 236 |
|
|
| 310 | 221 | 290 | 210 | 268 | 213 |
|
|
| 330 | 267 | 360 | 280 | 273 | 283 |
|
|
| 290 | 236 | 341 | 261 | 240 | 269 |
|
|
| 364 | 250 | 321 | 241 | 307 | 251 |
|
|
| 328 | 231 | 370 | 290 | 270 | 294 |
|
|
| 390 | 229 | 303 | 223 | 317 | 223 |
|
|
| 360 | 269 | 334 | 254 | 312 | 250 |
|
|
| 342 | 233 | 295 | 216 | 320 | 216 |
|
|
| 306 | 259 | 315 | 235 | 265 | 265 |
|
| Trung bình | 332,0 | 248,4 | 323,9 | 244,0 | 282,2 | 250,0 |
|
| Phương sai (vari) | 1.026,7 | 483,8 | 725,0 | 718,7 | 854,6 | 784,7 |
|
| In (vari) | 6,9341 | 6,1817 | 6,5862 | 6,5774 | 6,7507 | 6,6653 |
|
Bảng 13.10.10- Các tổng đáp ứng và tương phản tuyến tính (xem công thức tính ở Bảng 13.10.2)
|
| Chế phẩm chuẩn S | Chế phẩm thư U | Chế phẩm thử Z | Tổng số | ||
| Liều thấp | S1 = 3.320 | U1 = 3.239 | Z1 = 2.822 |
| ||
| Liều cao | S1 = 2.484 | U2 = 2.440 | U2 = 2.500 |
| ||
| Tổng đáp ứng của chế phẩm | S = 5.804 | U = 5.679 | Z = 5.322 |
| ||
| Tương phản tuyến tính | Ls = -836 | Lu = -799 | Ls = -322 |
| ||
|
|
|
| ||||
|
|
|
| ||||
Tính các tổng các bình phương theo công thức trong các Bảng 13.10.5 và 13.10.6 dùng các số liệu tính được ở Bảng 13.10.10:
Chế phẩm = ![]() =
=![]()
Hồi qui tuyến tính = ![]() =
=![]() = 63.830,817 = E
= 63.830,817 = E
Không song song = ![]() =
=![]() = 8.218,233
= 8.218,233
Giữa các xử lý = ![]() =
=![]() = 78.305,68
= 78.305,68
Toàn phần = ![]() - K = 4.826.447 - 4.706.800,42 = 119.646,58
- K = 4.826.447 - 4.706.800,42 = 119.646,58
Sai số dư = Toàn phần - Giữa các xử lý = 119.646,58 - 78.305,68 = 41.340,90
Bảng 13.10.11- Phân tích phương sai
| Nguồn biến thiên | Bậc tự do | Tổng các bình phương | Bình phương trung bình | Tỷ số F | Xác suất |
| Chế phẩm | 2 | 6.256,6 | 3128,3 |
|
|
| Hồi qui tuyến tính | 1 | 63.830,8 | 63.830,8 | 83,4 | < 0,01 |
| Không song song | 2 | 8.218,2 | 4109,1 | 5,4 | < 0,05 |
| Giữa các xử lý | 5 | 78.305,7 |
|
|
|
| Sai số dư | 54 | 41.340,9 | 765,572 |
|
|
| Sai số toàn phần | 59 | 119.646,6 |
|
|
|
Phép thử tính có giá trị của thử nghiệm
Kết quả phân tích phương sai cho thấy, tỷ số F của biến số hồi qui Fcal.= 83,4 rất lớn so với F tới hạn ở xác suất
P = 0,01, f1 = 1 và f2 = 54: F(P=0,01;f1 = 1;f2=54) = 7,13(1), do đó biến số hồi qui rất có ý nghĩa.
Biến số không song song cũng có ý nghĩa, Fcal. = 5,4 > F(P= 0,05;f1 = 2;f2 = 54) =3,17. Khảo sát Bảng 13.10.10 cho thấy chế phẩm Z có tương phản tuyến tính LZ rất khác với LS và LU. Do đó, có khả năng độ dốc của đường In(liều) - đáp ứng của chế phẩm Z không phù hợp với các chế phẩm còn lại và là nguyên nhân làm cho tính có giá trị của thử nghiệm không thỏa mãn. Kiểm tra giả thuyết trên bằng phép kiểm Dunnett (t' được tính theo công thức 3.2.4 -1 ở mục 3.2.4):
Đối với chế phẩm U:
|
|
|
Đối với chế phẩm Z:
|
|
|
Đối với chế phẩm Z, ![]() = 2,94 > t’crit. = 2,27 đọc từ Bảng 13.10.8 với P = 0,05, f1 = 2 và f2 = 54, do đó đường In(liều) - đáp ứng của nó không song song với đường In(liều) - đáp ứng của chế phẩm chuẩn. Loại các số liệu liên quan đến chế phẩm Z và lặp lại phân tích chỉ với các số liệu của chế phẩm U và chế phẩm chuẩn, với
= 2,94 > t’crit. = 2,27 đọc từ Bảng 13.10.8 với P = 0,05, f1 = 2 và f2 = 54, do đó đường In(liều) - đáp ứng của nó không song song với đường In(liều) - đáp ứng của chế phẩm chuẩn. Loại các số liệu liên quan đến chế phẩm Z và lặp lại phân tích chỉ với các số liệu của chế phẩm U và chế phẩm chuẩn, với ![]() = 11.483 và
= 11.483 và ![]() = -1.635 (Bảng 13.10.10).
= -1.635 (Bảng 13.10.10).
|
|
| |
|
|
|
|
Bảng 13.10.12- Phân tích phương sai không có chế phẩm Z
| Nguồn biến thiên | Bậc tự do | Tổng các bình phương | Bình phương trung bình | Tỷ số F | Xác suất |
| Chế phẩm | 1 | 390,6 | 390,6 |
|
|
| Hồi qui tuyến tính | 1 | 66.830,6 | 66.830,6 | 90,5 | < 0,01 |
| Không song song | 1 | 34,2 | 34,2 | 0,05 | > 0,05 |
| Giữa các xử lý | 3 | 67.255,5 |
|
|
|
| Sai số dư | 36 | 26.587,3 | 738,54 |
|
|
| Sai số toàn phần | 39 | 93.842,8 |
|
|
|
Sau khi loại chế phẩm Z, kết quả phân tích phương sai cho thấy thử nghiệm thỏa mãn các yêu cầu về cả hồi qui và cả tính không song song.
Định giá hoạt lực và các giới hạn tin cậy
|
|
|
|
|
|
Tỷ lệ giữa các liều kế tiếp nhau bằng 1,0/0,25 = 4, do đó l = ln(4) = 1,3863, t = 2,03 với P = 0,95 và F = 36 (Bảng 13.10.28).
|
|
|
|
|
|
Hoạt lực giả định AU của chế phẩm U là 1 đơn vị/mg, do đó:
MU = M'U + ln (AU) = 0,1060 + 0 = 0,1060
Hoạt lực của chế phẩm U bằng antiln (MU) = antiln (0,1060) = 1,11 đơn vị/mg.
|
|
|
|
|
|
|
| |
Logarit các giới hạn tin cậy bằng:
|
|
|
Các giới hạn tin cậy là 0,82 và 1,51 đơn vị/mg.
Ví dụ 3.2.8.2 Thử nghiệm 3 liều đơn chế phẩm, với thiết kế thí nghiệm ngẫu nhiên theo khối không lặp thử nghiệm định lượng kháng sinh bằng phương pháp khuếch tán dùng hộp Petri
Các dung dịch thử của chế phẩm chuẩn s1, s2 và s3 có nồng độ lần lượt là 2, 4 và 8 lU/ml. Chế phẩm thử có hoạt lực giả định là 1.500 IU/ml và cũng được pha thành 3 dung dịch thử u1, u2 và u3 có nồng độ tương đương với nồng độ các dung dịch của chế phẩm chuẩn. Mỗi hộp Petri đều có 3 dung dịch của chế phẩm chuẩn và 3 dung dịch của chế phẩm thử, trong đó mỗi dung dịch thử chỉ xuất hiện một lần duy nhất trong mỗi hộp Petri (không lặp). Trật tự sắp xếp các dung dịch thử trong mỗi hộp được phân bố như trong Bảng 13.10.13.
Các đáp ứng riêng và đáp ứng trung bình của mỗi dung dịch thử cho ở Bảng 13.10.14.
Bảng 13.10.13- Cách bố trí các dung dịch thử trong các hộp
| Hộp số | Số vị trí trong hộp | |||||
| 1 | 2 | 3 | 4 | 5 | 6 | |
| 1 | u2 | u3 | s3 | s1 | u1 | s2 |
| 2 | s3 | u1 | s2 | u3 | s1 | u2 |
| 3 | u3 | u2 | s1 | s3 | s2 | u1 |
| 4 | s1 | s2 | u3 | u1 | u2 | s3 |
| 5 | u1 | s1 | u2 | s2 | s3 | u3 |
| 6 | s2 | s3 | u1 | u2 | u3 | s1 |
Các đáp ứng riêng và trị trung bình đáp ứng của mỗi nồng độ được cho ở Bảng 13.10.14.
Bảng 13.10.14- Đáp ứng y - đường kính vòng vô khuẩn (mm x 10)
| Hộp số | Chế phẩm chuẩn S | Chế phẩm thử U | Tổng khối | ||||
|
| s1 | s2 | s3 | u1 | u2 | u3 | |
| 1 | 176 | 205 | 235 | 174 | 202 | 232 | R1 = 1224 |
| 2 | 178 | 208 | 238 | 175 | 206 | 234 | R2 = 1239 |
| 3 | 178 | 207 | 237 | 177 | 203 | 236 | R3 = 1238 |
| 4 | 175 | 205 | 235 | 173 | 201 | 232 | R4 = 1221 |
| 5 | 176 | 206 | 235 | 174 | 204 | 231 | R5 = 1226 |
| 6 | 174 | 204 | 236 | 170 | 202 | 229 | R6 = 1215 |
| Tổng cột | 1057 | 1235 | 1416 | 1043 | 1218 | 1394 | 7363 |
| Trung bình | 176,2 | 205,8 | 236,0 | 173,8 | 203,0 | 232,3 |
|
| Phương sai | 2,6 | 2,2 | 1,6 | 5,4 | 3,2 | 5,9 |
|
Kiểm tra số liệu thu được bằng các phép thử tương tự như ở ví dụ 3.2.8.1 cho thấy thử nghiệm đáp ứng các điều kiện ở mục 3.1.1.
|
|
| |
|
|
|
|
Bảng 13.10.15- Các tổng đáp ứng và tương phản tuyến tính (xem công thức tính ở Bảng 13.10.3)
|
| Chế phẩm chuẩn S | Chế phẩm thử U | Tổng số |
| Liều thấp | S1 = 1.057 | U1 = 1.043 |
|
| Liều trung gian | S2 = 1.235 | U2 = 1.218 |
|
| Liều cao | S3 = 1.416 | U3 = 1.394 |
|
| Tổng đáp ứng của chế phẩm | S = 3.708 | U = 3.655 |
|
| Tương phản tuyến tính | Ls = 359 | Lu = 351 |
|
| Tương phản bậc hai | Qs = 3 | Qu = 1 |
|
Các tổng các bình phương được tính bằng các công thức trong Bảng 13.10.5 và 13.10.6 với các số liệu ở Bảng 13.10.15.
Chế phẩm = ![]() = 78,026
= 78,026
Hồi qui tuyến tính = ![]() = 21.004,167 = E
= 21.004,167 = E
Không song song = ![]() = 2,666
= 2,666
Độ cong = ![]() = 0,278
= 0,278
Giữa các xử lý = ![]() = 21.085,137
= 21.085,137
Giữa các khối = ![]() = 75,805
= 75,805
Toàn phần= ![]() - K = 21.188,97
- K = 21.188,97
Sai số dư = Toàn phần - Giữa các xử lý - Giữa các khối = 28,03
Bảng 13.10.16- Phân tích phương sai
| Nguồn biến thiên | Bậc tự do | Tổng các bình phương | Bình phương trung bình | Tỷ số F | Xác suất |
| Chế phẩm | 1 | 78,03 | 78,03 |
|
|
| Hồi qui tuyến tính | 1 | 21.004,17 | 21.004,17 | 18.737 | < 0,01 |
| Không song song | 1 | 2,67 | 2,67 | 2,4 | > 0,05 |
| Độ cong | 2 | 0,28 | 0,16 | 0,1 | > 0,05 |
| Giữa các xử lý | 5 | 21.085,14 |
|
|
|
| Giữa các khối (hộp Petri) | 5 | 75,80 | 15,16 | 13,5 | < 0,01 |
| Sai số dư | 25 | 28,03 | 1,121 |
|
|
| Sai số toàn phần | 35 | 21.188,97 |
|
|
|
Kết quả phân tích phương sai cho thấy sự khác nhau có ý nghĩa cao (P < 0,01) của kích thước đường kính vòng vô khuẩn giữa các hộp Petri. Nếu thí nghiệm thiết kế theo kiểu ngẫu nhiên hoàn toàn, biến thiên giữa các khối sẽ không được tách ra khỏi sai số toàn phần, do đó bình phương trung bình của sai số dư s2 sẽ lớn hơn, dẫn đến khoảng tin cậy sẽ rộng hơn. Đây là ưu điểm của thiết kế thí nghiệm ngẫu nhiên theo khối so với thiết kế thí nghiệm kiểu ngẫu nhiên hoàn toàn.
Phép thử tính có giá trị của thử nghiệm
Hồi qui tuyến tính rất có ý nghĩa (P < 0,01), các đại lượng không song song và độ cong không có ý nghĩa (P > 0,05), do đó thử nghiệm thỏa mãn cách tính hiệu lực.
Định giá hoạt lực và các giới hạn tin cậy
|
|
|
Tỷ lệ giữa các nồng độ kế tiếp bằng 2,0, do đó:
|
| I = ln(2)= 0,69315 |
|
|
|
|
|
|
|
|
|
|
| M = M' + ln(AU) = -0,0690 + ln(1.500) = 7,2442 |
|
Hoạt lực của chế phẩm U là R = antiln(M) = 1.400 IU/ml.
Với P = 0,95 và f = 25, t = 2,06 (Bảng 13.10.28), ta có:
|
|
|
|
|
|
|
| |
Các ln(giới hạn tin cậy) bằng:
![]() = ln(1.500)+1,0002x(- 0,0690)±
= ln(1.500)+1,0002x(- 0,0690)±![]() = 7,2442 ± 0,0160
= 7,2442 ± 0,0160
Các giới hạn tin cậy bằng 1.378 và 1.423 lU/ml.
Nếu phân tích bằng máy tính, các kết quả sẽ là:
| C = 1,000227 |
|
| |
|
| (C - 1)(CM'2 + 2H) = 0,00029128 |
| |
Khoảng tin cậy: 1.376,3 - 1.424,1 lU/ml.
Ví dụ 3.2.8.3 Thử nghiệm 3 liều đơn chế phẩm thiết kế hình vuông Latin không lặp
Thử nghiệm định lượng kháng sinh bằng phương pháp khuếch tán, dùng khay vuông
Chế phẩm chuẩn có hoạt lực biết trước 4.855 lU/mg. Chế phẩm thử có hoạt lực giả định là 5.600
lU/mg. Pha các dung dịch gốc bằng cách hòa tan 25,2 mg chế phẩm chuẩn và 21,4 mg chế phẩm thử trong lượng vừa đủ dung môi pha loãng để được 25 ml. Sau đó, pha loãng các dung dịch gốc đến nồng độ pha loãng 1/20 và tiếp tục pha loãng xa hơn với tỷ lệ pha loãng 1:1,5 để được các dung dịch thử cuối cùng của chuẩn và mẫu.
Các dung dịch thử của chế phẩm chuẩn và chế phẩm thử được thiết kế trên khay theo bố trí thí nghiệm hình vuông Latin như trong Bảng 13.10.17. Các đường kính vòng vô khuẩn của thử nghiệm được trình bày trong Bảng 13.10.18. Trung bình đường kính vòng vô khuẩn, phương sai (vari) và In(vari) của mỗi nhóm dung dịch thử trình bày trong Bảng 13.10.19.
Bảng 13.10.17- Cách bố trí các dung dịch thử trên khay
|
| 1 | 2 | 3 | 4 | 5 | 6 |
| 1 | s1 | u1 | u2 | s3 | s2 | u3 |
| 2 | u1 | u3 | s1 | s2 | u2 | s3 |
| 3 | u2 | s3 | s2 | s1 | u3 | u1 |
| 4 | s3 | s2 | u3 | u1 | s1 | u2 |
| 5 | s2 | u2 | s3 | u3 | u1 | s1 |
| 6 | u3 | s1 | u1 | u2 | s3 | s2 |
Phép thử tính đồng nhất của các phương sai
Đối với một nhóm gồm k phương sai, trong đó mỗi phương sai có f = (n - 1) bậc tự do, công thức tính sẽ như sau:
|
|
|
| Với k = 6, bậc tự do f = 5, công thức trên sẽ rút gọn thành: | |
|
|
|
| = |
|
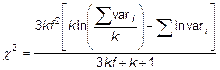
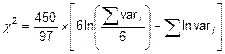
Giá trị tính được nhỏ hơn giá trị tới hạn 11,07 đọc từ Bảng 13.10.29 với P = 0,95 và f = (k- 1) = 5. Do đó, sự khác biệt giữa các phương sai của mỗi nhóm dung dịch thử không có ý nghĩa thống kê.
Bảng 13.10.18-Đáp ứng y - đường kính vòng vô khuẩn (mm x 10)
|
| 1 | 2 | 3 | 4 | 5 | 6 | Tổng hàng |
| 1 | 161 | 160 | 178 | 187 | 171 | 194 | R1 = 1.051 |
| 2 | 151 | 192 | 150 | 172 | 170 | 192 | R2 = 1.027 |
| 3 | 162 | 195 | 174 | 161 | 193 | 151 | R3 = 1.036 |
| 4 | 194 | 184 | 199 | 160 | 163 | 171 | R4 = 1.071 |
| 5 | 176 | 181 | 201 | 202 | 154 | 151 | R5 = 1.065 |
| 6 | 193 | 166 | 161 | 186 | 198 | 182 | R6 = 1.086 |
| Tổng cột | C1 = 1.037 | C2 = 1.078 | C3 = 1.063 | C4 = 1.068 | C5 = 1.049 | C6 = 1.041 |
|
Bảng 13.10.19- Trung bình đường kính vòng vô khuẩn và phương sai của mỗi nhóm dung dịch thử
| STT (hộp petri) | Chế phẩm chuẩn S | Chế phẩm thử U | Tổng số | |||||
| s1 | s2 | s3 | u1 | u2 | u3 | |||
| 1 | 161 | 171 | 187 | 160 | 178 | 194 |
| |
| 2 | 150 | 172 | 192 | 151 | 170 | 192 |
| |
| 3 | 161 | 174 | 195 | 151 | 162 | 193 |
| |
| 4 | 163 | 184 | 194 | 160 | 171 | 199 |
| |
| 5 | 151 | 176 | 201 | 154 | 181 | 202 |
| |
| 6 | 166 | 182 | 198 | 161 | 186 | 193 |
| |
| Tổng cột | 952 | 1059 | 1167 | 937 | 1048 | 1173 |
| |
| Trung bình | 158,67 | 176,50 | 194,50 | 156,17 | 174,67 | 195,50 |
| |
| Phương sai (vari) | 43,47 | 28,70 | 23,50 | 22,17 | 75,07 | 16,30 |
| |
| In (vari) | 3,772 | 3,357 | 3,157 | 3,099 | 4,318 | 2,791 |
| |
Bảng 13.10.20- Các tổng đáp ứng và tương phản tuyến tính (xem công thức tính ở Bảng 13.10.3)
|
| Chế phẩm chuẩn S | Chế phẩm thử U | Tổng số | ||
| Liều thấp | S1 = 952 | U1 = 937 |
| ||
| Liều trung gian | S2 = 1.059 | U2 = 1.048 |
| ||
| Liều cao | S3 = 1.167 | U3 = 1.173 |
| ||
| Tổng đáp ứng của chế phẩm | S = 3.178 | U = 3.158 |
| ||
| Tương phản tuyến tính | Ls = 215 | LU = 236 |
| ||
| Tương phản bậc hai | Qs = 1 | QU = 14 |
| ||
|
|
|
| |||
|
|
|
| |||
Các tổng các bình phương được tính bằng các công thức trong Bảng 13.10.5 và 13.10.6 với các số liệu ở Bảng 13.10.18 và Bảng 13.10.20.
Chế phẩm = ![]() =
= ![]() = 11,1111
= 11,1111
Hồi qui tuyến tính = ![]() = 8.475,0416 = E
= 8.475,0416 = E
Không song song = ![]() = 18,3750
= 18,3750
Độ cong (không tuyến tính) = ![]() = 5,4722
= 5,4722
Giữa các xử lý = ![]()
Giữa các hàng = ![]()
Giữa các cột = ![]()
Toàn phần= ![]() - K = 1.124.692 - 1.115.136 = 9.556
- K = 1.124.692 - 1.115.136 = 9.556
Sai số dư = Toàn phần - Giữa các xử lý - Giữa các hàng - Giữa các cột = 415,3333
Bảng 13.10.21- Phân tích phương sai
| Nguồn biến thiên | Bậc tự do | Tổng các bình phương | Bình phương trung bình | Tỷ số F | Xác suất |
| Chế phẩm | 1 | 11,1111 | 11,1111 |
|
|
| Hồi qui tuyến tính | 1 | 8.475,0416 | 8.475,0416 | 408,1 | < 0,01 |
| Không song song | 1 | 18,3750 | 18,3750 | 0,885 | > 0,05 |
| Độ cong | 2 | 5,4722 | 2,7361 | 0,132 | > 0,05 |
| Giữa các xử lý | 5 | 8510 |
|
|
|
| Giữa các hàng | 5 | 412 | 82,40 | 3.968 | < 0,05 |
| Giữa các cột | 5 | 218,6667 | 43,73 | 2,106 | > 0,05 |
| Sai số dư | 20 | 415,3333 | 20,7667 |
|
|
| Sai số toàn phần | 35 | 9.556 |
|
|
|
Phân tích phương sai cho thấy sự khác nhau có ý nghĩa (P < 0,05) giữa các hàng chứng tỏ thiết kế thí nghiệm hình vuông Latin cho độ chính xác cao hơn thiết kế thí nghiệm ngẫu nhiên hoàn toàn.
Phép thử tính có giá trị của thử nghiệm
Hồi qui tuyến tính rất có ý nghĩa (P < 0,01), tính không tuyến tính và tính không song song của các đường In(liều) - đáp ứng không có ý nghĩa (P > 0,05), do đó thử nghiệm thỏa mãn các phép thử tính có giá trị.
Định giá hoạt lực và các giới hạn tin cậy
Tỷ lệ giữa các nồng độ kế tiếp nhau bằng 1,5, do đó l = ln(1,5) = 0,405465. Với P = 0,95 và f = 20, t = 2,09 (Bảng 13.10.28).
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| M = M' + ln(AU) = -0,023974 + ln(5.600) = 8,606548 |
|
Hoạt lực của chế phẩm U là R = antiln(M) = 5.467,3 lU/mg.
|
|
|
|
|
|
|
|
Các ln(giới hạn tin cậy) được tính theo công thức:
![]() =
=
= ln(5.600) + 1,0108 x (- 0,023974) ± ![]() = 8,60629 ± 0,06878
= 8,60629 ± 0,06878
Các giới hạn tin cậy bằng 5.102,6 và 5.855,1 lU/mg ở P = 0,05
Vì các dung dịch gốc của chế phẩm chuẩn và chế phẩm thử có hoạt lực không hoàn toàn bằng nhau dựa trên hoạt lực giả định, cần hiệu chỉnh hoạt lực tính được và các giới hạn tin cậy bằng cách nhân với một hệ số hiệu chỉnh:
![]() 1,02091
1,02091
Sau khi hiệu chỉnh, hoạt lực tính được sẽ bằng 5.582 lU/mg với giới hạn tin cậy từ 5.209 lU/mg đến 5.977 lU/mg ở xác suất 95 %.
Ví dụ 3.2.8.4 Thiết kế chéo đôi
Thử nghiệm định lượng insulin bằng phương pháp tiêm dưới da thỏ
Các dung dịch của chế phẩm chuẩn có nồng độ 1 và 2 lU/ml. Hoạt lực giả định của chế phẩm thử là 40 lU/ml và được pha loãng thành các dung dịch có nồng độ tương đương với các dung dịch của chế phẩm chuẩn. Tiêm dưới da các thỏ thí nghiệm với liều 0,5 ml mỗi dung dịch thử theo thiết kế ở Bảng 13.10.22. Các kết quả thí nghiệm trình bày trong Bảng 13.10.23. Phương sai lớn cho thấy biến thiên lớn giữa các thỏ thí nghiệm và cần thiết phải dùng thiết kế thí nghiệm chéo.
Bảng 13.10.22- Cách sắp xếp các xử lý
| Ngày thí nghiệm | Nhóm thỏ | |||
| 1 | 2 | 3 | 4 | |
| Ngày I | s1 | s2 | u1 | u2 |
| Ngày II | u2 | u1 | s2 | s1 |
Phép thử tính đồng nhất của các phương sai bằng phép kiểm Hartley
Tính tỷ số F theo công thức:
![]()
Trong đó varmax. là phương sai lớn nhất và varmin. là phương sai nhỏ nhất trong k phương sai cần kiểm tra. F tính được (Fcal.= 5,3) nhỏ hơn F tới hạn (Fcrit. = 12,7) đọc từ Bảng 13.10.30 với P = 0,95, k = 8 và bậc tự do f = 7. Do đó, sự khác nhau giữa các phương sai của mỗi nhóm dung dịch thử không có ý nghĩa thống kê. Nếu dùng phép kiểm Bartlett với cùng số liệu, giá trị tính được sẽ là 6,4 so với giá trị tới hạn bằng 14,1.
Trong các thử nghiệm dùng thiết kế chéo, các tổng đáp ứng và tương phản tuyến tính được tính riêng rẽ cho mỗi giai đoạn thí nghiệm. Kết quả thu được trình bày trong Bảng 13.10.24.
Bảng 13.10.23-Đáp ứng y: glucose huyết (mg/100 ml) ở 1 và 2½ h
|
|
| Nhóm 1 |
|
| Nhóm 2 |
|
| Nhóm 3 |
|
| Nhóm 4 |
|
|
| s1 | u2 | Tổng số | s2 | u1 | Tổng số | u1 | s2 | Tổng số | u2 | s1 | Tổng số |
|
| 112 | 104 | 216 | 65 | 72 | 137 | 105 | 91 | 196 | 118 | 144 | 262 |
|
| 126 | 112 | 238 | 116 | 160 | 276 | 83 | 67 | 150 | 119 | 149 | 268 |
|
| 62 | 58 | 120 | 73 | 72 | 145 | 125 | 67 | 192 | 42 | 51 | 93 |
|
| 86 | 63 | 149 | 47 | 93 | 140 | 56 | 45 | 101 | 64 | 107 | 171 |
|
| 52 | 53 | 105 | 88 | 113 | 201 | 92 | 84 | 176 | 93 | 117 | 210 |
|
| 110 | 113 | 223 | 63 | 71 | 134 | 101 | 56 | 157 | 73 | 128 | 201 |
|
| 116 | 91 | 207 | 50 | 65 | 115 | 66 | 55 | 121 | 39 | 87 | 126 |
|
| 101 | 68 | 169 | 55 | 100 | 155 | 91 | 68 | 159 | 31 | 71 | 102 |
| Tổng cột | 765 | 662 |
| 557 | 746 |
| 719 | 533 |
| 579 | 854 | 5415 |
| Trung bình | 95,6 | 82,8 |
| 69,6 | 93,3 |
| 89,9 | 66,6 |
| 72,4 | 106,8 |
|
| Phương sai | 709,7 | 627,9 |
| 525,1 | 1.012,5 |
| 479,6 | 230,6 |
| 1.214,3 | 1.215,1 |
|
Bảng 13.10.24- Các tổng đáp ứng và tương phản tuyến tính
|
| Chế phẩm chuẩn S | Chế phẩm thử U | Tổng số |
| Ngày I Nồng độ thấp Nồng độ cao Tổng số |
S1I = 765 S2I = 557 SI = 1.322 |
U1I = 719 U2I = 579 UI = 1.298 | DI = 2.620 |
| Ngày II Nồng độ thấp Nồng độ cao Tổng số |
S1II = 854 S2II = 533 SII = 1.387 |
U1II = 746 U2II = 662 UII = 1.408 | DII = 2.795 |
| Tổng chế phẩm | S = 2.709 | U = 2.706 |
|
| Tương phản tuyến tính Ngày I Ngày II Tổng số | LSI = -208 LSII = - 321 LS = -529 | LUI = -140 LUII = - 84 LU = -224 | LI = -348 LII = -405
|
Phân tích phương sai trong các thử nghiệm thiết kế chéo phức tạp hơn các kiểu thiết kế thí nghiệm khác vì biến thiên do cấu phần tổng các bình phương, gây bởi tính song song không độc lập với biến thiên do cấu phần gây bởi sự khác nhau của thỏ. Tính song song của các đường hồi qui sẽ được kiểm tra bằng đại lượng bình phương trung bình của sai số dư thứ hai, được tính bằng cách lấy tổng các bình phương của cấu phần do sự khác nhau về thỏ (khối) trừ đi cấu phần không song song và hai cấu phần tương tác.
Ba cấu phần tương tác được đưa thêm vào phân tích phương sai là do sự lặp lại trong mỗi nhóm thỏ thí nghiệm, gồm:
Giữa các ngày x chế phẩm; Giữa các ngày x hồi qui; Giữa các ngày x tính song song.
Ba đại lượng này biểu thị khuynh hướng của 3 cấu phần (chế phẩm, hồi qui và song song) thay đổi từ ngày thí nghiệm này sang ngày thí nghiệm khác. Tính có giá trị của thử nghiệm phụ thuộc vào các tỷ số F tương ứng của ba đại lượng nói trên. Nếu F tính được rất có ý nghĩa, Fcal. > Fcrit. ở P = 0,01, cần phải rất thận trọng khi suy diễn các kết quả của thử nghiệm, và nếu có thể, phải lặp lại thử nghiệm.
|
|
|
|
|
|
|
|
Tính giá trị của tổng các bình phương của chế phẩm, hồi qui và không song song theo công thức trong Bảng 13.10.5 với số liệu trong Bảng 13.10.24.
Chú ý trong thử nghiệm này n = 16, là tổng số đáp ứng của mỗi xử lý qua hai ngày thí nghiệm.
Chế phẩm = ![]() =
= ![]() = 0,14
= 0,14
Hồi qui tuyến tính = ![]() = 8.859,52 = E
= 8.859,52 = E
Không song song = ![]() = 1.453,51
= 1.453,51
Giữa các khối (thỏ) = ![]() - 458.159,77 = 39.794,73
- 458.159,77 = 39.794,73
Trong đó Bi là các tổng đáp ứng của mỗi thỏ thí nghiệm trong Bảng 13.10.23.
Giữa các ngày = ![]() =
= ![]() -458.159,77 = 478,51
-458.159,77 = 478,51
Trong đó DI, và DII là tổng đáp ứng của mỗi ngày thí nghiệm.
Giữa các ngày x Chế phẩm =![]() =
= ![]()
Trong đó SI, SII, UI và UII là tổng đáp ứng của từng chế phẩm trong mỗi ngày.
Giữa các ngày x hồi qui = ![]()
Giữa các ngày x Không song song = ![]()
Trong đó LSI, LSll, LUI và LUII là tương phản tuyến tính của mỗi ngày thí nghiệm.
Toàn phàn = ![]() - K = 511.583 - 458.159,77 = 53.423,23
- K = 511.583 - 458.159,77 = 53.423,23
Sai số dư giữa các thỏ = Giữa các khối - Không song song - (Giữa các ngày x chế phẩm) - (Giữa các ngày x hồi qui) = 39.794,73 - 1.453,51 - 31,64 - 50,76 = 38.258,81
Sai số dư trong mỗi thỏ = Toàn phần - Giữa các khối - Giữa các ngày - Chế phẩm - Hồi qui - (Giữa các ngày x không song song) = 53.423,23 - 39.794,73 - 478,51 - 0,14 - 8.859,52 - 446,27 = 3.844,06
Phép thử tính có giá trị của thử nghiệm
Phân tích phương sai cho thấy thử nghiệm thỏa mãn phép thử tính có giá trị:
1) Hồi qui tuyến tính rất có ý nghĩa: tỷ số F của đại lượng hồi qui được tính dựa trên bình phương trung bình của sai số dư trong mỗi thỏ, Fcal. = 64,5 > Fcrit. = 7,63 với P = 0,01, f1 = 1 và f2 = 28.
2) Tính không song song của đường hồi qui: phép thử tính không song song trong thiết kế chéo dựa trên bình phương trung bình của sai số dư giữa các thỏ, Fcal. = 1,06 < F(P=0 05, f1 = 1, f2 = 28) = 4,19, chứng tỏ các đường In(liều) - đáp ứng của chuẩn và mẫu song song với nhau.
3) Ba cấu phần tương tác đều không có ý nghĩa, các tỷ số F tính được lần lượt là 0,02, 0,04 và 3,25 và đều nhỏ hơn Fcrit. = 4,19.
Bảng 13.10.25- Phân tích phương sai
| Nguồn biến thiên | Bậc tự do | Tổng các bình phương | Bình phương trung bình | Tỷ số F | Xác suất |
| Không song song | 1 | 1.453,5 | 1.453,5 | 1,06 | > 0,05 |
| Giữa các ngày x chế phẩm | 1 | 31,6 | 31,6 | 0,02 | > 0,05 |
| Giữa các ngày x hồi qui | 1 | 50,8 | 50,8 | 0,04 | > 0,05 |
| Sai số dư giữa các thỏ | 28 | 38.258,8 | 1.366,4 |
|
|
| Giữa các khối (thỏ) | 31 | 39.794,7 | 1.283,7 |
|
|
| Chế phẩm | 1 | 0,1 | 0,1 | 0,00 | > 0,05 |
| Hồi qui | 1 | 8.859,5 | 8.859,5 | 64,5 | < 0,01 |
| Giữa các ngày | 1 | 478,5 | 478,5 | 3,48 | > 0,05 |
| Giữa các ngày x không song song | 1 | 446,3 | 446,3 | 3,25 | > 0,05 |
| Sai số dư trong mỗi thỏ | 28 | 3.844,1 | 137,3 |
|
|
| Toàn phần | 63 | 53.423,2 | 847,9878472 |
|
|
Định giá hoạt lực và cốc giới hạn tin cậy
Tỷ lệ pha loãng bằng 2, do đó l = ln(2) = 0,69315. Với P = 0,95 và f = 28, t = 2,05 (Bảng 13.10.28).
|
|
|
|
|
|
|
|
|
|
|
|
|
| M = M' + ln(AU) = 0,00276 + ln(40) = 3,6916 |
|
Hoạt lực R = antiln(M) = antiln(0,00276) = 40,1 lU/ml.
|
|
|
|
|
|
|
|
|
|
| |
Logarit các giới hạn tin cậy bằng:
![]() = 3,6918 ± 0,1830
= 3,6918 ± 0,1830
Khoảng tin cậy của hoạt lực từ 33,4 đến 48,2 IU/ml.
4. Phối hợp các kết quả thử nghiệm
4.1 Mở đầu
Để đáp ứng các yêu cầu của Dược điển, ví dụ yêu cầu về độ chính xác, có thể phải lặp lại hai hay nhiều thử nghiệm độc lập và phối hợp các kết quả thử nghiệm để được kết quả cuối cùng chính xác và đáng tin cậy hơn.
Hai thử nghiệm được coi là độc lập với nhau nếu quá trình thực hiện của mỗi thử nghiệm không gây bất cứ ảnh hưởng nào đến kết quả của thử nghiệm khác. Điều đó có nghĩa là các sai số ngẫu nhiên của các yếu tố chủ yếu ảnh hưởng đến kết quả thử nghiệm (ví dụ: sự pha loãng chế phẩm chuẩn và chế phẩm thử, độ nhạy của chỉ thị sinh học) của một thử nghiệm phải độc lập với các sai số ngẫu nhiên tương ứng của thử nghiệm kia. Các thử nghiệm thực hiện vào những ngày kế tiếp nhau dùng cùng dung dịch gốc của chế phẩm chuẩn không phải là những thử nghiệm độc lập.
Có nhiều phương pháp phối hợp kết quả của các thử nghiệm độc lập, tuy nhiên, dưới đây chỉ trình bày ba phương pháp gần đúng do tính đơn giản và dễ áp dụng của chúng.
Trước khi phối hợp, các hoạt lực phải được hiệu chỉnh với hoạt lực giả định của mỗi chế phẩm thử (xem Ví dụ 3.2.8.3) và phải được biểu diễn dưới dạng logarit.
4.2 Phối hợp cân chỉnh (weighted combination) các kết quả thử nghiệm
Phương pháp này có thể áp dụng nếu thỏa mãn các điều kiện sau:
1) Các thử nghiệm phải độc lập với nhau;
2) Đại lượng C của mỗi thử nghiệm phải nhỏ hơn 1,1 (xem công thức 3.2.5-6 để biết công thức tính và ý nghĩa của C);
3) Bậc tự do của sai số dư của mỗi thử nghiệm không được nhỏ hơn 6, tốt nhất là lớn hơn 15;
4) Sự khác nhau giữa các định giá hoạt lực riêng phải không có ý nghĩa thống kê (xem mục 4.2.2).
Nếu các điều kiện trên không thỏa mãn, phương pháp này không được áp dụng. Khi đó dùng phương pháp giới thiệu ở mục 4.3 để tính hoạt lực trung bình và lấy kết quả tính được làm hoạt lực giả định cho lần thử nghiệm kế tiếp.
4.2.1 Tính các hệ số cân chỉnh W
Giả sử các kết quả đo của mỗi n' thử nghiệm đã được phân tích để được n' giá trị logarit hoạt lực M và các giới hạn tin cậy tương ứng. Đối với mỗi thử nghiệm, tính theo logarit khoảng tin cậy L bằng cách lấy giới hạn tin cậy trên trừ đi giới hạn tin cậy dưới. Tính hệ số cân chỉnh W cho mỗi giá trị của M theo phương trình 4.2.1.-1, với t có cùng giá trị như khi tính các giới hạn tin cậy.
|
|
| (4.2.1.-1) |
4.2.2 Tính đồng nhất của các hoạt lực
Để kiểm tra tính đồng nhất của một dãy các logarit hoạt lực, thành lập biểu thức có phân phối xấp xỉ phân phối c2 như sau:
|
|
| (4.2.2.-1) |
| với |
|
|
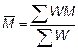
Nếu ![]() tính được nhỏ hơn giá trị tương ứng với bậc tự do f = (n' - 1) tra từ Bảng 13.10.29, sự khác nhau giữa các hoạt lực không có ý nghĩa thống kê và, do đó, có thể tiếp tục tính hoạt lực trung bình và các giới hạn tin cậy theo các công thức ở mục 4.2.3.
tính được nhỏ hơn giá trị tương ứng với bậc tự do f = (n' - 1) tra từ Bảng 13.10.29, sự khác nhau giữa các hoạt lực không có ý nghĩa thống kê và, do đó, có thể tiếp tục tính hoạt lực trung bình và các giới hạn tin cậy theo các công thức ở mục 4.2.3.
Nếu ![]() tính được lớn hơn giá trị tới hạn tương ứng đọc từ Bảng 13.10.29, các hoạt lực tính được không đồng nhất và không thể sử dụng các công thức ở mục 4.2.3, thay vào đó, có thể dùng các công thức ở mục 4.2.4.
tính được lớn hơn giá trị tới hạn tương ứng đọc từ Bảng 13.10.29, các hoạt lực tính được không đồng nhất và không thể sử dụng các công thức ở mục 4.2.3, thay vào đó, có thể dùng các công thức ở mục 4.2.4.
4.2.3 Tính hoạt lực trung bình có cân chỉnh (weighted mean) và các giới hạn tin cậy
Logarit hoạt lực trung bình có cân chỉnh được tính theo công thức:
|
|
| (4.2.3.-1) |
Độ lệch chuẩn của logarit hoạt lực trung bình là căn bậc hai của nghịch đảo của tổng các hệ số cân chỉnh W:
|
|
| (4.2.3.-2) |
và các logarit giới hạn tin cậy của hoạt lực trung bình được tính theo phương trình sau:
|
|
| (4.2.3.-3) |
Trong đó t là giá trị tới hạn đọc được từ Bảng 13.10.29 với bậc tự do bằng tổng số bậc tự do của các sai số dư của mỗi thử nghiệm riêng.
4.2.4 Hoạt lực trung bình có cân chỉnh và các giới hạn tin cậy dựa trên biến thiên trong mỗi thử nghiệm và giữa các thử nghiệm
Khi phối hợp các kết quả của một số thử nghiệm lặp lại, giá trị ![]() có thể có ý nghĩa. Biến thiên của các hoạt lực có thể phân tích thành hai cấu phần, gồm:
có thể có ý nghĩa. Biến thiên của các hoạt lực có thể phân tích thành hai cấu phần, gồm:
1) Biến thiên trong mỗi thử nghiệm ![]()
2) Biến thiên giữa các thử nghiệm ![]()
với ![]() là trung bình chưa cân chỉnh (trung bình số học) của các M. Biến thiên đầu thay đổi từ thử nghiệm này đến thử nghiệm khác, trong khi biến thiên sau là chung cho tất cả các M.
là trung bình chưa cân chỉnh (trung bình số học) của các M. Biến thiên đầu thay đổi từ thử nghiệm này đến thử nghiệm khác, trong khi biến thiên sau là chung cho tất cả các M.
Đối với mỗi M, tính hệ số cân chỉnh theo công thức:
|
|
|
|
Dùng W' thay thế cho W trong các công thức ở mục 4.2.3, với t = 2.
4.3 Phối hợp phi cân chỉnh (unweighted combination) các kết quả thử nghiệm
Cách phối hợp các kết quả thử nghiệm đơn giản nhất là tính trung bình số học của n’ logarit hoạt lực M và sau đó tính độ lệch chuẩn của nó theo công thức:
|
|
| (4.3.-1) |
Các logarit giới hạn tin cậy bằng:
|
|
| (4.3.-2) |
Trong đó t có (n’ - 1) bậc tự do. Vì số các thử nghiệm cần phối hợp thường nhỏ nên giá trị của t khá lớn.
4.4 Ví dụ
Bảng 13.10.26 liệt kê các hoạt lực cùng các giới hạn tin cậy của 6 thử nghiệm độc lập của cùng một chế phẩm và bậc tự do của sai số dư tương ứng. Tất cả các thử nghiệm đều đáp ứng điều kiện 1, 2 và 3 trong mục 4.2. Các hệ số cân chỉnh W được tính theo công thức 4.2.1.-1 ở mục 4.2.
Phép thử tính đồng nhất của các hoạt lực
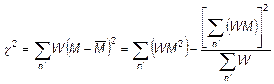 = 2.123.323,8792 -
= 2.123.323,8792 - ![]() = 4,42
= 4,42
![]() tính được 4,42 nhỏ hơn giá trị tới hạn 11,07 đọc từ Bảng 13.10.29 với bậc tự do f = 5. Do đó, sự khác nhau giữa các hoạt lực không có ý nghĩa thống kê, và do đó đạt tất cả các điều kiện.
tính được 4,42 nhỏ hơn giá trị tới hạn 11,07 đọc từ Bảng 13.10.29 với bậc tự do f = 5. Do đó, sự khác nhau giữa các hoạt lực không có ý nghĩa thống kê, và do đó đạt tất cả các điều kiện.
Bảng 13.10.26 - Hoạt lực và các giới hạn tin cậy của sáu thử nghiệm độc lập
| Lần thử nghiệm | Hoat lực R (IU/lọ) | Các giới hạn tin cậy (lU/lọ) | Bậc tự do DF | |
| Giới hạn dưới | Giới hạn trên | |||
| 1 | 18.367 | 17.755 | 19.002 | 20 |
| 2 | 18.003 | 17.415 | 18.610 | 20 |
| 3 | 18.064 | 17.319 | 18.838 | 20 |
| 4 | 17.832 | 17.253 | 18.429 | 20 |
| 5 | 18.635 | 17.959 | 19.339 | 20 |
| 6 | 18.269 | 17.722 | 18.834 | 20 |
| Tổng số |
|
|
| 120 |
Bảng - 13.10.27
Với f = 20, P = 0,05, bảng 13.10.28 cho t = 2,09. Tính W = 4t2/L2 theo công thức 4.2.1.-1
|
| M=ln(R) | L* | W | WM | WM2 |
|
| 9,8183 | 0,0679 | 3.777,8301 | 37.091,9101 | 364.179,9033 |
|
| 9,7983 | 0,0664 | 3.951,6825 | 38.719,7456 | 379.387,4392 |
|
| 9,8017 | 0,0841 | 2.462,5579 | 24.137,1950 | 236.584,9721 |
|
| 9,7887 | 0,0659 | 4.003,1116 | 39.185,4586 | 383.576,6531 |
|
| 9,8328 | 0,0740 | 3.175,7400 | 31.226,4061 | 307.042,9060 |
|
| 9,8130 | 0,0609 | 4.699,6807 | 46.117,7833 | 452.552,0054 |
| Tồng số | 58,8528 |
| 22.070,6028 | 216.478,4988 | 2.123.323,8792 |
* L = ln(giới hạn tin cậy trên) - ln(giới hạn tin cậy dưới)
Hoạt lực trung bình và các giới hạn tin cậy
Hoạt lực trung bình và các giới hạn tin cậy của nó được tính bằng các công thức ở mục 4.2.3 và các số liệu trong Bảng 13.10.26 và 13.10.27.
|
|
|
|
|
|
|
|
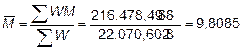
Với số bậc tự do f = 120, P = 0,95, t = 1,98.
|
|
|
|
Bằng cách lấy antilogarit các giá trị tính được, hoạt lực trung bình bằng 18.187 lU/lọ với khoảng tin cậy ở xác suất 95 % từ 17.946 đến 18.431 lU/lọ.
5. Các Bảng tra cứu
Bảng 13.10.28 - Bảng giá trị tới hạn của phân phối t
|
| P |
|
| P |
|
| f | 0,05 | 0,01 | f | 0,05 | 0,01 |
| 1 | 12,71 | 63,66 | 18 | 2,10 | 2,88 |
| 2 | 4,30 | 9,92 | 19 | 2,09 | 2,86 |
| 3 | 3,18 | 5,84 | 20 | 2,09 | 2,85 |
| 4 | 2,78 | 4,60 | 21 | 2,08 | 2,83 |
| 5 | 2,57 | 4,03 | 22 | 2,07 | 2,82 |
| 6 | 2,45 | 3,71 | 23 | 2,07 | 2,81 |
| 7 | 2,36 | 3,50 | 24 | 2,06 | 2,80 |
| 8 | 2,31 | 3,36 | 25 | 2,06 | 2,79 |
| 9 | 2,26 | 3,25 | 26 | 2,06 | 2,78 |
| 10 | 2,23 | 3,17 | 27 | 2,05 | 2,77 |
| 11 | 2,20 | 3,11 | 28-29 | 2,05 | 2,76 |
| 12 | 2,18 | 3,05 | 30 | 2,04 | 2,75 |
| 13 | 2,16 | 3,01 | 40-43 | 2,02 | 2,7 |
| 14 | 2,14 | 2,98 | 57-63 | 2,00 | 2,66 |
| 15 | 2,13 | 2,95 | 102-126 | 1,98 | 2,26 |
| 16 | 2,12 | 2,92 | 600-∞ | 1,96 | 2,58 |
| 17 | 2,11 | 2,90 |
|
|
|
Nếu giá trị quan sát lớn hơn giá trị trong bảng, nó được coi là có ý nghĩa (P = 0,05) hay rất có ý nghĩa (P = 0,01).
Bảng 13.10.29 - Bảng giá trị tới hạn của ![]()
|
| P |
|
| P |
|
| f | 0,05 | 0,01 | f | 0,05 | 0,01 |
| 1 | 3,84 | 6,63 | 11 | 19,68 | 24,73 |
| 2 | 5,99 | 9,21 | 12 | 21,03 | 26,22 |
| 3 | 7,81 | 11,34 | 13 | 22,36 | 27,69 |
| 4 | 9,49 | 13,28 | 14 | 23,68 | 29,14 |
| 5 | 11,07 | 15,09 | 15 | 25,00 | 30,58 |
| 6 | 12,59 | 16,81 | 16 | 26,30 | 32,00 |
| 7 | 14,07 | 18,48 | 20 | 31,41 | 37,57 |
| 8 | 15,51 | 20,09 | 25 | 37,65 | 44,31 |
| 9 | 16,92 | 21,67 | 30 | 43,77 | 50,89 |
| 10 | 18,31 | 23,21 | 40 | 55,76 | 63,69 |
| Nếu giá trị quan sát lớn hơn giá trị trong bảng, nó được coi là có ý nghĩa (P = 0,05) hay rất có ý nghĩa (P = 0,01). | |||||
Bảng 13.10.30 Giá trị tới hạn của tỷ số F = varmax/varmin
k là số phương sai trong nhóm xử lý, mỗi phương sai có f bậc tự do (Hartley's test).
|
| k | 4 | 6 | 8 | 9 | 10 | 12 |
| f |
| ||||||
| 4 | 20,6 | 29,5 | 37,5 | 41,1 | 44,6 | 51,4 | |
|
| 49 | 69 | 89 | 97 | 106 | 120 | |
| 5 | 13,7 | 18,7 | 22,9 | 24,7 | 26,5 | 29,9 | |
|
| 28 | 38 | 46 | 50 | 54 | 60 | |
| 6 | 10,4 | 13,7 | 16,3 | 17,5 | 18,6 | 20,7 | |
|
| 19,1 | 25 | 30 | 32 | 34 | 37 | |
| 7 | 8,44 | 10,8 | 12,7 | 13,5 | 14,3 | 15,8 | |
|
| 14,5 | 18,4 | 22 | 23 | 24 | 27 | |
| 8 | 7,18 | 9,03 | 10,5 | 11,1 | 11,7 | 12,7 | |
|
| 11,7 | 14,5 | 16,9 | 17,9 | 18,9 | 21 | |
| 9 | 6,31 | 7,80 | 8,95 | 9,45 | 9,91 | 10,7 | |
|
| 9.9 | 12,1 | 13,9 | 14,7 | 15,3 | 16,6 | |
| 10 | 5,67 | 6,92 | 7,87 | 8,28 | 8,66 | 9,34 | |
|
| 8,6 | 10,4 | 11,8 | 12,4 | 12,9 | 13,9 | |
Nếu F tính được lớn hơn giá trị trong bảng, biến số cần kiểm tra được coi là có ý nghĩa (dòng trên, P = 0,05), hay rất có ý nghĩa (dòng dưới, P = 0,01).
6. Bảng chú giải các ký hiệu
| Ký hiệu | Định nghĩa |
| b | Độ dốc hồi qui tuyến tính của đường thẳng liều - đáp ứng hay In(liều) - đáp ứng của tất cả các chế phẩm trong một thử nghiệm |
| d | Số mức liều của mỗi chế phẩm trong một thử nghiệm đối xứng |
| e | Cơ số của logarit tự nhiên, e = 2,718281828.... |
| f | Số bậc tự do |
| h | Số chế phẩm trong một thử nghiệm, kể cả chế phẩm chuẩn |
| k | Số xử lý trong một thử nghiệm, k = dh |
| n | Số thí nghiệm lặp lại hoặc số đơn vị thí nghiệm của mỗi xử lý |
| n' | Số các định giá hoạt lực riêng |
| s2 | Trị định giá của phương sai được tính từ bình phương trung bình của sai số dư. |
| s | Trị định giá của Độ lệch chuẩn, (= |
| s1,s2,s3 | Nồng độ liều thấp, trung gian và cao của chế phẩm chuẩn S. Trong một thử nghiệm chỉ có 2 nồng độ của mỗi chế phẩm s2 đại diện cho nồng độ cao |
| t | Thống kê t của student (Bảng 13.10.28) |
| t’ | Thống kê t’ của Dunnett (Bảng 13.10.8) |
| u1...z3 | Nồng độ (liều) của các chế phẩm thử U... Z |
| y | Đáp ứng riêng hay đáp ứng riêng đã được chuyển đổi |
| y' | Đáp ứng tính được dùng để thay thế cho giá trị bị mất |
|
| Đáp ứng trung bình của chế phẩm chuẩn và các chế phẩm thử |
| AU...AZ | Hoạt lực giả định của các chế phẩm thử U... Z, là hoạt lực được dùng để tính lượng chế phẩm cần lấy khi pha các dung dịch thử |
| B' | Tổng đáp ứng của mỗi đơn vị thí nghiệm (1 đến 2n) trong thiết kế chéo đôi |
| S’ | Tổng đáp ứng của khối hay hàng có chứa giá trị bị mất |
| C | Đại lượng biểu thị ý nghĩa của hồi qui, dùng trong tính toán các giới hạn tin cậy (phương trình 3.2.5-5). Trong một số tài liệu thống kê kết quả thử nghiệm sinh học, ý nghĩa của hồi qui được biểu thị theo g:
|
| C1...Cn | Tổng đáp ứng của mỗi cột (1 đến n) trong thiết kế hình vuông Latin |
| C’ | Tổng đáp ứng của cột không kể một giá trị bị mất trong thiết kế hình vuông Latin |
| D | liều |
| DI,DII | Tổng đáp ứng của giai đoạn I và giai đoạn II trong thiết kế chéo đôi |
| E | Tổng các bình phương của đại lượng hồi qui (Bảng 13.10.5) |
| F | Tỳ số của hai trị định giá độc lập của phương sai theo phân phối F (Bảng 13.10.7) |
| G' | Tổng đáp ứng trong một thử nghiệm không kể giá trị bị mất |
| I | Khoảng cách giữa các In(liều) kế tiếp nhau, I = ln(s2/s1) = ln(s2) - ln(s1) |
| K | Đại lượng được dùng để tính tổng các bình phương trong phân tích phương sai
|
| L | Độ rộng của logarit giới hạn tin cậy, L = ln(giới hạn tin cậy trên) - ln(giới hạn tin cậy dưới) |
| Ls...Lz | Tương phản tuyến tính của chế phẩm chuẩn và các chế phẩm thử (Bảng 13.10.2, 13.10 3 và 13.10.4) |
| M | Trị định giá của logarit hoạt lực, trong thử nghiệm đa chế phẩm, M được dùng kèm với một ký tự (U...Z) ghi ở dưới, ví dụ MU, để chỉ rõ một chế phẩm riêng (M = InR). |
|
| Trung bình của một vài trị định giá độc lập của M |
| M’ | Logarit tỷ lệ hoạt lực hay trị định giá của logarit hoạt lực trước khi hiệu chỉnh với hoạt lực giả định (M’ = InR’) |
| N | Tổng số đáp ứng trong một thử nghiệm |
| NS, NU | Tổng số đáp ứng của chế phẩm S và U |
| P | Xác suất |
| Qs…Qz | Tương phản bậc hai của chế phẩm chuẩn và các chế phẩm thử |
| R | Hoạt lực định giá được dùng với một ký tự (U....Z) ghi ở dưới, ví dụ RU để chỉ rõ một chế phẩm thử riêng trong một thử nghiệm đa chế phẩm (R = antilnM) |
| R' | Định giá hoạt lực hoặc tỷ số hoạt lực trước khi hiệu chỉnh bởi hoạt lực giả định (R’ = antilnM’). |
| R1...Rn | Tổng đáp ứng của mỗi hàng (1 đến n) trong thiết kế hình vuông Latin, hay mỗi khối trong thiết kế ngẫu nhiên theo khối. |
| S | Chế phẩm chuẩn |
| S | Tổng đáp ứng của chế phẩm chuẩn |
| S1, S2, S3 | Tổng đáp ứng của liều (nồng độ) thấp, trung gian và cao của chế phẩm chuẩn S. Trong một thử nghiệm chỉ có 2 mức liều, S2 là tổng đáp ứng của liều cao |
| T' | Tổng đáp ứng trong một xử lý không kể giá trị bị mất |
| U...Z | Tổng đáp ứng của các chế phẩm thử U...Z |
| U...Z | Các chế phẩm cần thử |
| U1, U2, U3 | Tổng đáp ứng của liều thấp, trung gian và cao của chế phẩm U, trong một định lượng chỉ có 2 nồng độ của mỗi chế phẩm, U2 đại diện cho nồng độ cao |
| W | Hệ số cân chỉnh dùng trong phối hợp một số định giá độc lập (phương trình 4.2.1.-1) |
| X | In(liều) |
PHỤ LỤC 14. HƯỚNG DẪN ĐÁNH GIÁ SINH KHẢ DỤNG VÀ TƯƠNG ĐƯƠNG SINH HỌC IN VIVO THUỐC GENERIC
Thuốc phát minh là thuốc được cấp phép lưu hành đầu tiên, trên cơ sở đã có đầy đủ các số liệu về chất lượng, an toàn và hiệu quả. Thuốc generic là một thuốc thành phẩm nhằm thay thế một thuốc phát minh được sản xuất không có giấy phép nhượng quyền của công ty phát minh và được đưa ra thị trường sau khi bằng phát minh và các bản quyền đã hết hạn. Theo quy định trong đăng ký thuốc của Việt Nam cũng như của nhiều nước khác, đối với các thuốc generic có tác dụng toàn thân mà bản thân dược chất và dạng bào chế không chứng minh được sinh khả dụng tương tự như thuốc phát minh hoặc thuốc đối chứng (đối với các trường hợp không xác định được thuốc phát minh) thì phải có các số liệu chứng minh thuốc generic có tương đương sinh học so với thuốc phát minh hoặc thuốc đối chứng. Hiện nay, Bộ Y tế ban hành thông tư (được cập nhật định kỳ) quy định danh sách các thuốc generic phải nộp hồ sơ chứng minh tương đương sinh học khi nộp hồ sơ đăng ký sản xuất, lưu hành.
Trong đánh giá Tương Đương Sinh Học (TĐSH) in vivo thuốc generic thì khái niệm sinh khả dụng được định nghĩa là tốc độ và mức độ hấp thu dược chất từ chế phẩm thuốc vào hệ tuần hoàn chung. Các đại lượng được sử dụng để biểu thị tốc độ và mức độ hấp thu dược chất vào hệ tuần hoàn chung được ứng dụng trong nghiên cứu TĐSH bao gồm: Cmax (nồng độ đỉnh); Tmax (thời gian từ khi bắt đầu dùng thuốc đến khi đạt đến nồng độ đỉnh) và AUC (diện tích dưới đường cong nồng độ - thời gian). Hai chế phẩm thuốc được coi là tương đương sinh học nếu sinh khả dụng của chúng khác nhau không đáng kể khi dùng cùng mức liều trong cùng điều kiện thử nghiệm.
Hướng dẫn này đưa ra một số nguyên tắc cơ bản trong đánh giá tương đương sinh học để thu được kết quả tin cậy.
Những yêu cầu cơ bản đối với phương pháp phân tích xác định nồng độ dược chất và/hoặc chất chuyển hóa có tác dụng trong mẫu sinh học
Các phương pháp sắc ký, như sắc ký khí, sắc ký lỏng hiệu năng cao sử dụng các detector khác nhau như quang phổ tử ngoại - khả kiến, huỳnh quang, phổ khối thường được sử dụng trong nghiên cứu đánh giá tương đương sinh học để xác định nồng độ thuốc (dược chất và/hoặc chất chuyển hóa-chất cần phân tích) trong các mẫu sinh học (thường là các mẫu máu, huyết thanh hay nước tiểu) của người tình nguyện tham gia nghiên cứu. Những phương pháp này có tính đặc hiệu cao; có khả năng tách và định lượng trên cùng một hệ thống và cùng thời điểm. Trong một số trường hợp, có thể sử dụng phương pháp phân tích sinh hóa và sinh học nếu phương pháp có đủ độ đúng, độ chính xác, độ đặc hiệu để xác định được nồng độ thuốc trong các mẫu sinh học.
Do quá trình phân tích mẫu sinh học bị ảnh hưởng bởi nhiều yếu tố như lượng mẫu ít, nồng độ thấp, lẫn nhiều tạp chất là các chất nội sinh (lipid, protein, chất nội sinh, chất chuyển hóa...) và sự khác nhau giữa các cá thể, nên phương pháp phân tích phải được thiết lập và thẩm định để đảm bảo độ tin cậy.
1. Tính đặc hiệu: Phải chứng minh được rằng chất xác định được là dược chất hay chất chuyển hóa có tác dụng. Sự phân tích mẫu không bị ảnh hưởng bởi các chất nội sinh và chất chuyển hóa có liên quan. Báo cáo kết quả phải bao gồm cả sắc ký đồ của mẫu trắng (dịch sinh học), mẫu chất chuẩn pha trong dịch sinh học và mẫu thử thu được sau khi dùng thuốc. Trong nghiên cứu tính đặc hiệu, phải thực hiện phương pháp thêm chất chuẩn để loại bỏ sai số do nền mẫu. Khi xác định nồng độ chất phân tích trong mẫu sinh học bằng phương pháp sắc ký lỏng - khối phổ cần phải có các nghiên cứu ảnh hưởng của nền mẫu.
2. Đường chuẩn và khoảng tuyến tính: Mối quan hệ giữa đáp ứng với nồng độ của chất phân tích phải được đánh giá bằng phương trình toán học phù hợp. Phương trình này phải được xác định bằng các phương pháp phân tích hồi quy phù hợp (như phương pháp bình phương nhỏ nhất). Khoảng tuyến tính là khoảng nồng độ từ thấp nhất đến cao nhất của đường chuẩn có mối quan hệ tuyến tính giữa đáp ứng (xác định bằng các thiết bị phân tích phù hợp) và nồng độ dược chất có trong mẫu. Trong khoảng này, phép phân tích phải thỏa mãn các yêu cầu về độ đúng và độ chính xác theo qui định. Đường chuẩn phải có ít nhất 6 nồng độ của chất chuẩn pha trong cùng một mẫu dịch sinh học (không bao gồm mẫu điểm 0 - mẫu có nồng độ hoạt chất cần phân tích bằng 0). Đường chuẩn hay khoảng tuyến tính phải bao gồm toàn bộ khoảng nồng độ của các mẫu cần phân tích. Không xác định nồng độ chất cần phân tích có trong mẫu thử bằng phương pháp ngoại suy.
3. Độ đúng và độ chính xác: Độ đúng và độ chính xác được xác định cùng lúc bằng cách sử dụng tối thiểu 3 nồng độ của mẫu chuẩn kiểm tra, 1 nồng độ gần với giá trị nhỏ nhất của đường chuẩn (LLOQ); 1 nồng độ gần với điểm giới hạn trên của đường chuẩn và 1 nồng độ ở gần điểm giữa. Mỗi nồng độ phải được xác định trên ít nhất 5 mẫu.
Độ chính xác có thể được biểu thị bằng hệ số biến thiên (CV) trong ngày và giữa các ngày, được xác định trên mẫu chuẩn kiểm tra. Nói chung, CV không nên vượt quá 15 %, riêng điểm gần giới hạn định lượng dưới cho phép không vượt quá 20 %.
Độ đúng được biểu thị là khả năng tiến tới gần nồng độ thực nhất của chất phân tích trong mẫu sinh học được xác định bằng phương pháp đặc biệt. Điều đó có thể được biểu thị bằng khả năng tìm lại tương đối, và phải nằm trong khoảng 85 % đến 115 %, nhưng có thể chấp nhận 80 % đến 120 % đối với điểm gần giới hạn định lượng dưới.
4. Giới hạn định lượng dưới: Giới hạn định lượng là nồng độ thấp nhất của đường chuẩn có thể xác định được với độ đúng và độ chính xác cho phép. Giới hạn định lượng dưới ít nhất phải thỏa mãn khả năng phân tích nồng độ của mẫu thử lấy ở thời điểm bằng 3 đến 5 lần thời gian bán thải hoặc bằng 1/20 đến 1/10 giá trị Cmax của chất phân tích.
5. Độ ổn định của mẫu thử: Độ ổn định của mẫu sinh học có chứa chất phân tích cần được khảo sát khi bảo quản ở nhiệt độ phòng, đông lạnh trong khoảng thời gian khác nhau để xác định điều kiện bảo quản và thời gian bảo quản mẫu sau khi lấy.
6. Tỷ lệ thu hồi: Khả năng tìm lại sau khi chiết được đánh giá với ít nhất 3 nồng độ: Cao, trung bình và thấp của đường chuẩn. Giới hạn của chỉ tiêu này có thể tham khảo những tài liệu liên quan.
7. Mẫu chuẩn kiểm tra (QC): Mẫu chuẩn kiểm tra được dùng để so sánh khi định lượng là những mẫu tự tạo biết trước nồng độ, chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng.
8. Phân tích mẫu thử: Định lượng các mẫu thử bằng phương pháp phân tích đã được thẩm định. Mỗi mẫu thử có thể phân tích một lần hoặc lặp lại nếu cần. Toàn bộ các mẫu thử của mỗi người tình nguyện phải được phân tích trong cùng một điều kiện. Một lô mẫu phân tích (các mẫu phân tích trong cùng một điều kiện - trong thời gian một buổi hoặc một ngày) bao gồm toàn bộ các mẫu thử của một hoặc một số người tình nguyện; các mẫu của đường chuẩn/ khoảng tuyến tính và các mẫu QC - với tối thiểu 3 mức nồng độ tương ứng với khoảng nồng độ thấp, trung bình và cao của đường chuẩn/khoảng tuyến tính và tương ứng với mỗi mức nồng độ phải có tối thiểu hai mẫu. Số lượng các mẫu QC của một lô phân tích không được ít hơn 5 % tổng số mẫu thử và và cũng không được ít hơn 6 mẫu. Nồng độ chất cần phân tích trong mỗi mẫu thử và các mẫu QC được xác định dựa trên kết quả phân tích các mẫu của đường chuẩn trong lô phân tích. Chỉ chấp nhận kết quả của một lô mẫu phân tích khi: các mẫu đường chuẩn/khoảng tuyến tính đạt yêu cầu về độ đúng, chính xác và độ lệch giữa kết quả xác định nồng độ chất cần phân tích trong các mẫu QC phải sai khác không quá ± 15 % so với giá trị nồng độ lý thuyết tương ứng.Tiêu chuẩn chấp nhận kết quả lô phân tích được quy định bởi các hướng dẫn cụ thể của Cơ quan quản lý.
Quy định chung đối với một nghiên cứu đánh giá tương đương sinh học
Lựa chọn đối tượng tham gia nghiên cứu
Tiêu chuẩn: Đối tượng tham gia nghiên cứu phải tự nguyện tham gia vào nghiên cứu và được lựa chọn theo những tiêu chuẩn phù hợp nhất định. Trong nghiên cứu tương đương sinh học, đa số trường hợp thường chọn đối tượng tham gia nghiên cứu là nam giới khỏe mạnh. Một số trường hợp cần thiết chỉ sử dụng đối tượng là phụ nữ, khi đó cần giải thích rõ. Với thuốc dùng cho trẻ em, vẫn nên chọn người lớn để thử nghiệm. Các tiêu chuẩn cụ thể của đối tượng tham gia nghiên cứu như sau:
a. Giới tính: Nam hoặc nữ.
b. Tuổi: Thông thường từ 18 tuổi đến 55 tuổi. Trong cùng một nhóm nghiên cứu, giữa các cá thể, khác nhau không quá 10 tuổi.
c. Cân nặng, chiều cao cơ thể: Chỉ số khối lượng cơ thể (Body mass index - BMI) được tính bằng thương số giữa cân nặng cơ thể (đơn vị tính kg) và bình phương chiều cao cơ thể (đơn vị tính m2) của đối tượng tham gia nghiên cứu và phải nằm trong giới hạn trung bình từ 18,0 đến 25,0. Cân nặng cơ thể của các cá thể trong cùng nhóm nghiên cứu nên gần giống nhau.
d. Người tình nguyện phải có đủ năng lực hành vi dân sự, khỏe mạnh, không có tiền sử về bệnh tim mạch, bệnh gan, bệnh đường tiêu hóa, bệnh chuyển hóa không bình thường hoặc bệnh về hệ thần kinh.
e. Không có tiền sử về dị ứng và hạ huyết áp tư thế.
f. Không dùng các đối tượng nghiện ma túy, nghiện rượu, nghiện thuốc lá.
g. Trong quá trình thử nghiệm không được sử dụng thuốc lá, rượu và những thức uống khác có chứa cafein.
h. Không dùng thuốc có chứa dược chất cùng với thuốc nghiên cứu trong vòng 2 tuần trước và trong quá trình thử nghiệm.
i. Chấp thuận tự nguyện ký vào bản cam kết tự nguyện tham gia nghiên cứu.
Đối tượng tham gia nghiên cứu phải được kiểm tra sức khoẻ bao gồm các chỉ tiêu: khám lâm sàng tổng quát, huyết áp, nhịp tim, chức năng gan, thận, phổi và công thức máu. Các kết quả xét nghiệm phải trong giới hạn bình thường. Khi thử những thuốc đặc biệt, có thể phải kiểm tra thêm một số chỉ tiêu khác, ví dụ như đường huyết khi thử nghiệm những thuốc có ảnh hưởng đến đường huyết hay điện tim đối với các thuốc ảnh hưởng đến hoạt động của tim... Nếu đối tượng là phụ nữ thì phải tiến hành làm các xét nghiệm và đảm bảo không mang thai trong quá trình tham gia nghiên cứu.
Số lượng: Số lượng người tình nguyện phải đủ theo các yêu cầu, quy định liên quan của Cơ quan quản lý và được tính theo nguyên tắc dùng với số lượng nhỏ nhất có thể được để đảm bảo yếu tố đạo đức trong các nghiên cứu y sinh học nhưng vẫn đảm bảo yêu cầu, mức độ tin cậy thống kê của phép thử. Trong nghiên cứu tương đương sinh học, số lượng người tình nguyện tham gia nghiên cứu được tính trên các dữ liệu: mức độ tin cậy thống kê của phép thử (thường α = 0,1); khoảng giới hạn tương đương sinh học yêu cầu (thường là 80 % -125 %); mức độ dao động các thông số dược động học trên đối tượng tham gia nghiên cứu và tỷ lệ trung bình thông số dược động học giữa thuốc thử và thuốc đối chứng. Với mỗi nghiên cứu tương đương sinh học, cần xác định cụ thể và nêu rõ số lượng người tình nguyện trong đề cương nghiên cứu tương đương sinh học trình Hội đồng đạo đức trong nghiên cứu Y sinh học phê duyệt theo quy định, số lượng người tình nguyện tham gia nghiên cứu có thể thay đổi tùy từng thuốc, từng nghiên cứu và các quy định liên quan nhưng không được ít hơn 12 người.
Thuốc nghiên cứu
Thuốc đối chứng: Thuốc (chế phẩm) đối chứng dùng trong nghiên cứu tương đương sinh học phải là sản phẩm đã được cấp phép sản xuất, lưu hành dựa trên các số liệu an toàn, hiệu quả điều trị của sản phẩm và được lựa chọn theo các nguyên tắc với thứ tự ưu tiên lần lượt sau:
- Thuốc phát minh đang lưu hành tại Việt Nam;
- Thuộc danh mục thuốc đối chứng của tổ chức Y tế thế giới;
- Thuốc phát minh đang được lưu hành tại một trong các nước thành viên của Hội nghị hòa hợp quốc tế và các nước liên kết (gồm các nước thuộc EU, Mỹ, Nhật Bản, Canada, Australia, Thụy Sỹ);
- Trong trường hợp không xác định được thuốc phát minh, lựa chọn thuốc đối chứng là thuốc đã được cấp phép và đang được lưu hành tại một trong các nước thành viên của Hội nghị hòa hợp quốc tế và các nước liên kết hoặc thuốc đã được đánh giá chất lượng bởi Tổ chức Y tế Thế giới.
Quy định cụ thể thuốc đối chứng dùng trong một nghiên cứu tương đương sinh học do Bộ Y tế ban hành. Thuốc đối chứng được lựa chọn phải đạt tiêu chuẩn chất lượng và phải có nguồn gốc, xuất xứ rõ ràng mới được sử dụng trong nghiên cứu tương đương sinh học. Trong nghiên cứu tương đương sinh học nên lựa chọn thuốc đối chứng có dạng bào chế phù hợp với dạng bào chế của thuốc thử.
Thuốc thử: Thuốc (chế phẩm) thử trong nghiên cứu tương đương sinh học phải được nghiên cứu, sản xuất theo tiêu chuẩn thực hành tốt sản xuất thuốc (GMP) và các quy định liên quan của Cơ quan quản lý với cỡ lô phù hợp. Chỉ được sử dụng lô thuốc thử được sản xuất với cỡ lô đủ lớn, có đầy đủ hồ sơ chế biến và đạt các yêu cầu chất lượng theo tiêu chuẩn đồng thời có hàm lượng dược chất so với hàm lượng ghi trên nhãn sai khác không quá 5 % so với lô thuốc đối chứng trong nghiên cứu tương đương sinh học.
Thiết kế và phê duyệt đề cương nghiên cứu
Theo quy định, tất cả các nghiên cứu tương đương sinh học đều phải xây dựng đề cương nghiên cứu. Đề cương nghiên cứu trình bày rõ mô hình/ thiết kế nghiên cứu; phương pháp, nội dung nghiên cứu; phương pháp thống kê/ đánh giá kết quả nghiên cứu... Đề cương nghiên cứu đều phải được Hội đồng đạo đức trong nghiên cứu Y sinh học phê duyệt chấp thuận trước khi tiến hành triển khai nghiên cứu.
Lựa chọn mô hình nghiên cứu: Kiểu nghiên cứu phổ biến nhất trong nghiên cứu tương đương sinh học, so sánh sinh khả dụng giữa một thuốc thử và một thuốc đối chứng là nghiên cứu đơn liều, chéo 2 x 2 (2 thuốc, 2 giai đoạn, 2 trình tự thử thuốc). Để hạn chế ảnh hưởng của sự khác biệt giữa các cá thể và giai đoạn thử nghiệm, các đối tượng nghiên cứu được chia ngẫu nhiên thành 2 nhóm. Một nhóm sẽ được sử dụng thuốc thử trước và thuốc chứng sau; ngược lại, nhóm kia sử dụng thuốc chứng trước và thuốc thử sau. Giữa 2 giai đoạn thử, cần có một khoảng thời gian để đảm bảo cho thuốc của lần thử trước loại hết ra khỏi cơ thể (thường từ 1 đến 2 tuần, gấp khoảng 5 - 7 lần thời gian bán thải t1/2 của thuốc). Đối với các thuốc có thời gian bán thải kéo dài, có thể áp dụng thiết kế nghiên cứu song song. Trong nghiên cứu song song, đối tượng tham gia thử nghiệm được chia thành 2 nhóm, một nhóm uống thuốc thử và nhóm còn lại uống thuốc đối chứng. Ngoài mô hình nghiên cứu kể trên, trong một số trường hợp có thể áp dụng các mô hình nghiên cứu khác như: nghiên cứu đa liều (xác định các thông số dược động học của thuốc ở trạng thái ổn định - sau khi dùng thuốc từ 5 đến 7 liều); nghiên cứu lặp lại (sử dụng thuốc thử và/hoặc thuốc đối chứng lặp lại). Nghiên cứu đa liều có thể được áp dụng trong nghiên cứu tương đương sinh học khi:
a. Mức độ hấp thu dược chất từ dạng thuốc tương tự nhau, nhưng khác nhau về tốc độ hấp thu.
b. Sinh khả dụng giữa các cá thể khác nhau nhiều.
c. Sau khi uống liều đơn, nồng độ của thuốc dưới dạng tiền chất của thuốc hoặc chất chuyển hóa thấp, không thể xác định được bởi phương pháp phân tích thích hợp.
d. Thuốc giải phóng có kiểm soát, tác dụng kéo dài.
Đối với các nghiên cứu so sánh sinh khả dụng/tương đương sinh học của 2 thuốc thử so với 1 thuốc đối chứng (trường hợp thuốc thử có nhiều hàm lượng) hoặc nghiên cứu ảnh hưởng của thức ăn/ đồ uống tới sinh khả dụng của thuốc thử dạng rắn dùng đường uống đồng thời đánh giá tương đương sinh học của thuốc thử so với thuốc đối chứng, người ta thường sử dụng thiết kế nghiên cứu chéo 3 x 3 (3 thuốc, 3 giai đoạn, 3 hoặc 6 trình tự/phác đồ sử dụng thuốc - tùy theo mô hình nghiên cứu). Tương tự như thiết kế nghiên cứu chéo 2 x 2, giữa mỗi giai đoạn cần có thời gian nghỉ để đảm bảo cho thuốc sử dụng ở giai đoạn trước được thải trừ hết khỏi cơ thể. Thời gian nghỉ (thời gian thải trừ thuốc) giữa mỗi giai đoạn thường gấp khoảng 5 - 7 lần thời gian bán thải của thuốc. Tất cả các nghiên cứu tương đương sinh học có thiết kế nghiên cứu khác với mô hình nghiên cứu phổ biến đều phải nêu rõ lý do giải thích cho thiết kế nghiên cứu và thiết kế nghiên cứu phải được trình bày rõ ràng, chi tiết trong đề cương nghiên cứu trình Hội đồng đạo đức trong nghiên cứu Y sinh học phê duyệt.
Xác định liều thử: Liều thử trong nghiên cứu tương đương sinh học phải giống với liều dùng trong lâm sàng và nên sử dụng mức liều thấp nhất có thể được mà vẫn xác định được chính xác các thông số dược động học trên đối tượng tham gia nghiên cứu khi sử dụng mức liều đó. Trong nghiên cứu tương đương sinh học tốt nhất là liều thử của thuốc thử phải giống với liều thử của thuốc chứng. Trong trường hợp phải sử dụng liều khác nhau, cần nêu rõ lý do và phải hiệu chỉnh các giá trị dược động học thu được một cách phù hợp theo tỷ lệ mức liều dùng.
Thời điểm lấy mẫu: Sau khi sử dụng thuốc, tiến hành lấy các mẫu sinh học (thường là mẫu máu) của mỗi người tình nguyện tại những thời điểm xác định để xác định nồng độ thuốc/ dược chất có trong các mẫu. Tiến hành xác định các thông số dược động học dựa trên kết quả xác định nồng độ thuốc/dược chất có trong các mẫu. Do vậy, thiết kế thời điểm lấy mẫu trong nghiên cứu tương đương sinh học rất quan trọng để thu được kết quả nghiên cứu tin cậy. Một đường cong nồng độ thuốc trong máu người tình nguyện theo thời gian hoàn thiện phải bao gồm cả các pha: hấp thu, phân bố và thải trừ của thuốc.
(1) Nghiên cứu đơn liều: cần phải lấy một điểm trước khi dùng thuốc (mẫu trắng, thời điểm 0) và ít nhất 11 điểm khác sau khi sử dụng thuốc bao gồm nên có ít nhất 3 - 4 điểm lấy mẫu trước khi đạt tới đỉnh của đường cong nồng độ - thời gian (pha hấp thu), khoảng 3 điểm lấy mẫu xung quanh giá trị đỉnh và không ít hơn 4 - 6 điểm sau giá trị đỉnh (pha thải trừ). Tổng số điểm lấy mẫu của đường cong nồng độ - thời gian không ít hơn 12. Thời gian lấy mẫu nên kéo dài tới khoảng 3 đến 5 lần thời gian bán thải của dược chất hoặc khi nồng độ dược chất trong mẫu máu bằng khoảng 1/10 đến 1/20 giá trị nồng độ đỉnh. Đối với các thuốc có thời gian bán thải quá dài, có thể kết thúc lấy mẫu ở thời điểm sau khi uống thuốc 72 h.
(2) Nghiên cứu đa liều: Sau khi người tình nguyện đã sử dụng được một số liều trong tổng số liều cần phải sử dụng trong nghiên cứu (thường là từ liều thứ 3 - 5 trong tổng số 5 - 7 liều dùng), trong một khoảng thời gian bằng 7 lần thời gian bán thải của thuốc cần phải lấy 3 mẫu tại 3 thời điểm (dự đoán có nồng độ cực tiểu) liên tiếp, xác định nồng độ thuốc có trong mỗi mẫu để đảm bảo rằng nồng độ thuốc trong máu người tình nguyện đã đạt đến trạng thái ổn định. Nên lấy mẫu máu vào cùng một thời điểm (thường là buổi sáng) của những ngày khác nhau (phù hợp nhất là lấy mẫu ngay trước khi sử dụng thuốc liều kế tiếp) để có thể so sánh được và hạn chế những ảnh hưởng của thời gian tới dược động học. Sau khi nồng độ thuốc đạt đến trạng thái ổn định, tiến hành cho người tình nguyện sử dụng liều cuối trong tổng số liều dùng của một giai đoạn nghiên cứu và tiến hành lấy các mẫu máu của người tình nguyện trong khoảng thời gian dùng liều cuối theo các thời điểm xác định. Đường cong nồng độ thuốc trong máu theo thời gian của người tình nguyện ở trạng thái ổn định (AUCss0-t) tương tự như đường cong nồng độ - thời gian khi sử dụng đơn liều (AUC0-t) bao gồm các thời điểm ở pha hấp thu, các điểm xung quanh giá trị đỉnh của đường cong và các thời điểm ở pha thải trừ. Khoảng thời gian từ điểm 0 (sử dụng liều cuối) đến điểm lấy mẫu cuối cùng trong suốt khoảng liều ở trạng thái ổn định bằng t chính là khoảng thời gian giữa các lần dùng thuốc.
Các mẫu máu (huyết tương, huyết thanh hay máu toàn phần) phải bảo quản đông lạnh ngay sau khi lấy để chờ phân tích. Khi không thể xác định được nồng độ thuốc trong huyết tương, có thể thực hiện trên một mẫu sinh học khác như nước tiểu, nhưng chất xác định được và phương pháp phân tích phải phù hợp đảm bảo kết quả xác định nồng độ thuốc trong các mẫu đúng, chính xác và tin cậy được.
Tiến hành nghiên cứu
Nghiên cứu đơn liều: Người tình nguyện nhịn ăn qua đêm (ít nhất là 10 h trước khi uống thuốc). Sáng hôm sau, mỗi người sẽ được cho uống một liều thuốc thử hoặc thuốc đối chứng với 240 ml nước ấm. Người tình nguyện không được uống nước trong vòng 1h trước và sau khi uống thuốc, trừ lượng nước sử dụng khi uống thuốc. Đối với các nghiên cứu ảnh hưởng của thức ăn tới sinh khả dụng của thuốc hoặc nghiên cứu tương đương sinh học ở tình trạng no, sau khi nhịn ăn qua đêm, người tình nguyện ăn một bữa ăn tiêu chuẩn và uống thuốc với 240 ml nước ấm sau khi ăn 30 min. Bữa ăn tiêu chuẩn trước khi uống thuốc của người tình nguyện thường là bữa ăn giàu năng lượng; nhiều chất béo, tuân thủ theo đúng các quy định liên quan và phải được quy định cụ thể trong đề cương nghiên cứu. Nếu không có yêu cầu gì đặc biệt, người tình nguyện dùng bữa ăn tiêu chuẩn 4 h sau khi uống thuốc. Khẩu phần ăn được quy định giống nhau cho tất cả người tình nguyện và cho các giai đoạn của mỗi nghiên cứu. Tiến hành lấy mẫu máu tĩnh mạch theo thời điểm lấy mẫu đã thiết kế.
Nghiên cứu đa liều: Với các nghiên cứu có nhịp dùng thuốc là 24 h (dùng thuốc 1 lần/ngày), nên uống thuốc vào mỗi buổi sáng sau khi đã nhịn ăn ít nhất 10 h, sau đó không ăn gì trong vòng 2 h đến 4 h sau khi uống thuốc. Với các nghiên cứu có chế độ dùng thuốc 2 lần/ ngày, liều thuốc đầu tiên nên cho người tình nguyện uống vào buổi sáng sau khi đã nhịn đói ít nhất 10 h, sau đó không ăn gì trong vòng 2 h đến 4 h; liều thứ 2 được uống trước hoặc sau bữa ăn 2 h và tiếp tục nhịn ăn trong vòng 2 h sau khi uống thuốc. Mỗi liều được uống với 240 ml nước ấm. Nói chung, người tình nguyện có thể uống nước sau khi uống thuốc 1 h đến 2 h. Khi thuốc đối chứng là chế phẩm qui ước, nên thử theo mức liều và cách dùng thông thường đã sử dụng trên lâm sàng, nhưng nên chọn mức liều tương đương với liều của thuốc thử dạng giải phóng có kiểm soát hoặc kéo dài.
Theo quy định, các mẫu sinh học (máu, huyết tương, huyết thanh) sau khi lấy sẽ được bảo quản đông lạnh ngay để chờ phân tích. Sau khi uống thuốc, người tình nguyện nên tránh các vận động phải gắng sức. Quá trình lấy các mẫu máu của người tình nguyện phải được thực hiện tại cơ sở y tế có đủ nhân viên y tế (bác sỹ, điều dưỡng); đủ điều kiện chăm sóc sức khỏe và xử lý các biến cố bất lợi, các tai biến y tế nếu xảy ra. Các biến cố bất lợi, các tác dụng phụ xảy ra cần phải xử trí và điều trị kịp thời. Trường hợp xảy ra biến cố bất lợi nghiêm trọng, cần phải ngừng ngay nghiên cứu và báo cáo các bên liên quan theo đúng quy định.
Phân tích dược động học: Lập các bảng và hình để biểu thị các dữ liệu nồng độ thuốc trong huyết tương vào những thời điểm lấy mẫu khác nhau của từng cá thể, giá trị trung bình và độ lệch chuẩn. Sau đó tính các thông số dược động học tương đối của mỗi cá thể, giá trị trung bình và độ lệch chuẩn cho mỗi thông số.
Nghiên cứu đơn liều: Những thông số dược động học chính cần phải xác định trong nghiên cứu tương đương sinh học với thiết kế nghiên cứu đơn liều bao gồm: nồng độ đỉnh trong máu (Cmax), diện tích dưới đường cong nồng độ - thời gian (AUC), thời gian bán thải của thuốc (t1/2) và thời điểm đạt tới nồng độ đỉnh trong máu (Tmax). Giá trị Cmax và Tmax biểu thị bằng số liệu thu được trực tiếp từ thí nghiệm và không phải tính toán. Giá trị AUC0-tn (diện tích dưới đường cong nồng độ - thời gian từ thời điểm 0 đến thời điểm t) được tính theo phương pháp hình thang, với tn là thời điểm lấy mẫu cuối cùng có thể định lượng được. Giá trị AUC0-∞ (diện tích dưới đường cong nồng độ - thời gian từ thời điểm 0 đến vô cùng) được tính toán theo công thức:
|
| AUC0-∞ = AUC0-tn + Ctn/λz |
|
Trong đó: Ctn là nồng độ của thuốc tại thời điểm lấy mẫu cuối cùng có thể xác định được bằng phương pháp phân tích thuốc trong dịch sinh học được sử dụng trong nghiên cứu; λz là hằng số tốc độ thải trừ. Giá trị t1/2 có thể được tính bằng công thức:
|
| T1/2 = 0,693/λz |
|
Trong đó: λz là hằng số tốc độ thải trừ được tính từ độ dốc của đoạn tuyến tính trên đường biểu diễn logarit nồng độ - thời gian.
Thông thường, một nghiên cứu tương đương sinh học có thời điểm lấy mẫu phù hợp thì tỷ số:
| (AUC0-tn - AUC0-∞ ) x 100% ≥ 80 |
|
Nghiên cứu đa liều: Sau khi uống đa liều (thông thường từ 5 - 7 liều) với khoảng cách thời gian giữa các liều dùng như nhau, thì thuốc có thể đạt tới trạng thái ổn định. Tiến hành lấy mẫu máu sau khi đạt đến trạng thái liều ổn định, phân tích xác định nồng độ thuốc trong các mẫu máu và tính giá trị các thông số dược động học cơ bản của thuốc trạng thái ổn định như nồng Cmax, Tmax, AUCss0-t hay giá trị nồng độ trung bình ở trạng thái cân bằng (Cav), khoảng dao động nồng độ thuốc trong máu (DF)... Các giá trị Cssmax, Tssmax; AUCss0-t ở trạng thái cân bằng được tính toán bằng phương pháp tương tự tương ứng như thử nghiệm đơn liều. Nồng độ thuốc trung bình ở trạng thái cân bằng (Cav) có thể được tính như sau:
|
| Cav = (AUCss0-t)/ t |
|
Trong đó: AUCss0-t là diện tích dưới đường cong nồng độ thuốc - thời gian từ thời điểm 0 đến thời điểm t trong suốt khoảng liều ở trạng thái ổn định và t là khoảng thời gian giữa các lần dùng thuốc.
Khoảng dao động của nồng độ thuốc trong máu có thể được tính theo biểu thức:
|
| DF = 100% x (Cssmax-Cmin)/Cav |
|
Trong đó: Cssmax là nồng độ đỉnh, thu được từ các số liệu thực, sau khi dùng liều cuối cùng ở trạng thái ổn định và Cmin là nồng độ cực tiểu ở thời điểm cuối trong khoảng thời gian dùng liều cuối cùng ở trạng thái cân bằng. Nếu thuốc chứng cũng là thuốc giải phóng chậm hay thuốc tác dụng kéo dài, DF/t của thuốc thử phải không lớn hơn 143 % giá trị của thuốc chứng.
Ngoài các thông số dược động học cơ bản trong nghiên cứu tương đương sinh học kề trên, tùy theo mục đích cũng như các mô hình nghiên cứu cụ thể còn có thể xác định thêm các thông số dược động học khác như giá trị t1/2 hấp thu; thời gian lưu trú trung bình (mean retention/residence time - MRT); diện tích dưới đường cong nồng độ tích lũy - thời gian (area under the moment curve - AUMC)...
Tính toán sinh khả dụng
Nghiên cứu đơn liều: Sinh khả dụng F được tính toán lần lượt bằng cách sử dụng AUC0-tn và AUC0-∞ của mỗi cá thể, đồng thời tính giá trị trung bình và độ lệch chuẩn. Khi liều của thuốc thử (T) giống với liều của thuốc đối chứng (R):
|
| F = (AUC0-tn)T/ (AUC0-tn)R x 100% F = (AUC0-∞)T/ (AUC0-∞)R x 100% |
|
Khi liều thuốc thử khác với liều thuốc đối chứng và chất được phân tích đặc trưng cho dược động học tuyến tính, chỉ số F có thể thay đổi phụ thuộc vào liều và được thể hiện dưới đây:
|
| F = [(AUC0-tn)T x DR/(AUC0-tn)R x DT] x 100% F = [(AUC0-∞)T x DR/(AUC0-∞)R x DT] x 100% |
|
Trong đó: DR và DT là liều uống của thuốc chứng và thuốc thử.
Phân tích các chất chuyển hóa: Một vài thuốc là dạng tiền chất của thuốc không thể định lượng được trong máu vì dạng tiền chất của thuốc chuyển hóa rất nhanh trong cơ thể. Sinh khả dụng của những loại thuốc này có thể được đánh giá qua đáp ứng thích hợp của chất chuyển hóa có hoạt tính.
|
| F = [(AUC0-tn)mT x DR/(AUC0-tn) mR x DT] x 100% F = [(AUC0-∞)mT x DR/(AUC0-∞)mR x DT] x 100% |
|
Trong đó: m là ký hiệu cho chất chuyển hóa.
Đánh giá kết quả chủ yếu dựa vào số liệu AUC0-tn và AUC0-∞ được dùng như một số liệu để tham khảo. Nghiên cứu đa liều: Sinh khả dụng F được tính toán dựa trên số liệu AUCss0-t của mỗi cá thể. Khi liều của thuốc thử (T) giống với liều của thuốc đối chứng (R), sinh khả dụng ở trạng thái ổn định được xác định bằng công thức:
|
| F= (AUCss0-t)T/ (AUCss0-t)R x 100% |
|
Trong đó: AUCss0-t là diện tích dưới đường cong nồng độ thuốc - thời gian từ thời điểm 0 đến thời điểm t trong suốt khoảng liều ở trạng thái ổn định.
Đánh giá tương đương sinh học (phân tích thống kê các số liệu dược động học): Phân tích thống kê và đánh giá tương đương sinh học nên tập trung vào các thông số dược động học chính (AUC, Cmax và Tmax). Các giá trị Cmax và AUC phải chuyển đổi sang giá trị thang logarit. Phân tích phương sai (ANOVA) xác định ảnh hưởng của các yếu tố như trình tự/phác đồ sử dụng thuốc, cá thể, thuốc, giai đoạn thử thuốc... tới kết quả của nghiên cứu tương đương sinh học bằng phương pháp phân tích thống kê phù hợp tương ứng với mô hình nghiên cứu đã sử dụng (nghiên cứu song song, chéo 2 x 2; chéo 3 x 3 hay nghiên cứu lặp lại...). Từ kết quả phân tích phương sai, xác định khoảng tin cậy 90 % cho tỷ lệ InCmax và khoảng tin cậy 90 % cho tỷ lệ InAUC giữa thuốc thử và thuốc chứng tương ứng của mỗi người tình nguyện theo phương pháp thống kê phù hợp (two one-sided test với độ tin cậy α = 0,1). So sánh giá trị Tmax của thuốc thử và thuốc đối chứng (số liệu không chuyển sang thang logarit) bằng một trong các phương pháp thống kê phi tham số Wilcoxon signed rank test, Friedman test, Kruskal- Wallis test, ANCOVA hoặc một phương pháp thống kê phi tham số khác phù hợp với mô hình nghiên cứu đã sử dụng.
Nếu giới hạn khoảng tin cậy 90% của các tỷ số Cmax, AUCo-tn, AUC0-∞ (nghiên cứu đơn liều) hoặc Cssmax , AUCss0-t (nghiên cứu đa liều) tương ứng giữa thuốc thử và thuốc đối chứng nằm trong khoảng 80.0 % đến 125,0 % và giá trị Tmax của thuốc thử và thuốc đối chứng khác nhau không có ý nghĩa thống kê thì thuốc thử tương đương sinh học in vivo so với thuốc đối chứng.
Chấp nhận hai thuốc có giới hạn khoảng tin cậy 90 % tương ứng của các tỷ số Cmax và AUC nằm trong khoảng 80,0 % đến 125,0 % và Tmax khác nhau có ý nghĩa thống kê tương đương sinh học in vivo nếu sự khác nhau về tốc độ hấp thu (giá trị Tmax) của thuốc thử so với thuốc đối chứng là có chủ đích và chủ đích này được trình bày trên nhãn của sản phẩm theo đúng các quy định liên quan hoặc cung cấp thêm được bằng chứng lâm sàng phù hợp cho thấy sự khác nhau về tốc độ hấp thu của thuốc thử không ảnh hưởng đến an toàn và hiệu quả điều trị của thuốc trên lâm sàng.
Đối với một số thuốc/dược chất có khoảng điều trị hẹp, khoảng chấp nhận đối với khoảng tin cậy 90 % của tỷ số Cmax và/hoặc AUC có thể phải thu nhỏ hơn (chỉ từ 90 % đến 110 %) so với khoảng chấp nhận thông thường từ 80 % đến 125 %. Cơ quan quản lý sẽ có quy định cụ thể trong trường hợp này. Ngoài ra, đối với các thuốc/dược chất không thuộc nhóm có khoảng điều trị hẹp, có mức độ dao động các thông số dược động học trên đối tượng sử dụng lớn, có thể xem xét mở rộng khoảng chấp nhận đối với khoảng tin cậy 90 % của tỷ số Cmax rộng hơn so với khoảng chấp nhận thông thường 80 % -125 %, có thể mở rộng tới 75 % - 133 % hoặc 70 % -143 %. Cơ quan quản lý sẽ xem xét từng trường hợp và quy định cụ thể cho các trường hợp thay đổi khoảng giới hạn chấp nhận đối với khoảng tin cậy 90 % của tỷ số Cmax.
Đối với nghiên cứu sinh khả dụng/tương đương sinh học của một thuốc thử dạng bào chế tác dụng kéo dài (phóng thích hoạt chất kéo dài) so với một thuốc đối chứng dạng bào chế quy ước, mức độ hấp thụ của 2 thuốc trong nghiên cứu là tương đương sinh học với nhau nếu giá trị AUC đáp ứng được những yêu cầu của tương đương sinh học (khoảng tin cậy 90 % của tỷ số InAUC nằm trong giới hạn từ 80,0 % đến 125,0 %) và thuốc thử có khả năng giải phóng dược chất có kiểm soát hoặc kéo dài so với thuốc đối chứng (giá trị Cmax giảm và Tmax kéo dài hơn thuốc đối chứng; giá trị DF/t của thuốc thử phải lớn hơn 143 % giá trị của thuốc chứng).
PHỤ LỤC 15
(Quy định)
15.1 XÁC ĐỊNH ĐỘ SỐNG CỦA VẮC XIN BCG
Kiểm tra độ sống
Các vật liệu
Dung dịch Sauton loãng 1/4 được sử dụng để pha loãng vắc xin BCG.
Môi trường nuôi cấy: Lowenstein - Jensen.
Vắc xin BCG mẫu thử và mẫu chuẩn.
Hoàn nguyên và pha loãng vắc xin
Vắc xin BCG được xác định độ sống theo phương pháp sau:
Hoàn nguyên ít nhất 5 mg vắc xin BCG vào 5 ml dung dịch Sauton loãng 1/4 vô khuẩn để tạo hỗn dịch cơ bản chứa 1 mg vắc xin BCG/1 ml. Sau khi hoàn nguyên, hỗn dịch này phải được giữ ở 4 °C trong thời gian tối thiểu 15 min, sau đó pha theo công thức sau:
1) 1 ml hỗn dịch cơ bản + 99 ml dung dịch Sauton loãng 1/4 = 1/100 (I).
2) 1 ml hỗn dịch pha loãng I + 99 ml dung dịch Sauton loãng 1/4 = 1/10 000 (II).
3) 4 ml hỗn dịch pha loãng II + 4 ml dung dịch Sauton loãng 1/4 = 1/20 000 (III).
4) 2 ml hỗn dịch pha loãng II + 6 ml dung dịch Sauton loãng 1/4 = 1/40 000 (IV).
5) 2 ml hỗn dịch pha loãng II + 14 ml dung dịch Sauton loãng 1/4 = 1/80 000 (V).
6) 2 ml hỗn dịch pha loãng II + 30 ml dung dịch Sauton loãng 1/4 = /160 000 (VI).
Bảng 1
| Điều kiện xác định | Công thức tính |
| 1. | 1. |
| 2. | 2. |
| 3. | 3. |
| 4. | 4. |
Trong đó:
![]() là số khuẩn lạc trung bình mọc trên các ống môi trường Lowenstein - Jensen khi cấy huyền dịch BCG ở độ pha thứ 1;
là số khuẩn lạc trung bình mọc trên các ống môi trường Lowenstein - Jensen khi cấy huyền dịch BCG ở độ pha thứ 1;
![]() là số khuẩn lạc trung bình mọc trên các ống môi trường Lowenstein - Jensen khi cấy huyền dịch BCG ở độ pha thứ 2;
là số khuẩn lạc trung bình mọc trên các ống môi trường Lowenstein - Jensen khi cấy huyền dịch BCG ở độ pha thứ 2;
![]() là số khuẩn lạc trung bình mọc trên các ống môi trường Lowenstein - Jensen khi cấy huyền dịch BCG ở độ pha thứ 3;
là số khuẩn lạc trung bình mọc trên các ống môi trường Lowenstein - Jensen khi cấy huyền dịch BCG ở độ pha thứ 3;
d là độ pha loãng huyền dịch vắc xin BCG;
v là thể tích hỗn dịch cấy trên một ống môi trường Lowenstein - Jensen;
Hằng số ![]() tương đương với so lượng tối ưu khuẩn lạc có thể đếm được trên một ống môi trường Lowenstein-Jensen.
tương đương với so lượng tối ưu khuẩn lạc có thể đếm được trên một ống môi trường Lowenstein-Jensen.
Nuôi cấy
Lắc đều hỗn dịch của mỗi độ pha trong 20 s, cấy vào môi trường Lowenstein - Jensen 0,1 ml/ống theo trình tự sau:
2 độ pha đầu: Mỗi độ pha loãng cấy lên 5 ống môi trường.
Độ pha thứ 3 cấy lên 10 ống môi trường.
Đối với vắc xin tươi (trước khi đông khô): Cấy lên các ống môi trường Lowenstein - Jensen huyền dịch vắc xin BCG từ 3 độ pha loãng cuối IV, V, VI.
Đối với vắc xin BCG đông khô giữ ở 4 °C: Cấy huyền dịch vắc xin BCG từ 3 độ pha loãng III, IV, V.
Đối với vắc xin BCG đông khô ủ ở 37 °C: Cấy huyền dịch vắc xin BCG từ 3 độ pha loãng II, III, IV.
Các ống môi trường đã được cấy huyền dịch vắc xin BCG ở các độ pha khác nhau như trên sẽ được láng đều, để khô bề mặt và ủ ở 37 °C trong 28 ngày.
Đếm khuẩn lạc
Các ống môi trường được cấy huyền dịch BCG và ủ ở 37 °C sẽ được đưa ra đếm số khuẩn lạc mọc trên từng ống sau 21 ngày và 28 ngày.
Tính kết quả
Số đơn vị sống trong 1 ml vắc xin được tính theo 1 trong 4 công thức ở Bảng 1:
Hoặc kết quả độ sống của mỗi loạt vắc xin BCG có thể được tính theo chương trình phần mềm Whoprogram /BCG/Biofarma.
Tại nhà sản xuất, tiến hành kiểm tra độ sống ở tất cả các loạt vắc xin thành phẩm (bao gồm cả độ sống trước đông khô, sau đông khô và sau khi ủ ổn định nhiệt).
Những loạt vắc xin BCG có độ sống không đảm bảo với giới hạn cho phép ở lần kiểm tra thứ nhất phải nhắc lại thử nghiệm:
Nếu thử nghiệm độ sống lần 1 không đạt yêu cầu nhưng thử nghiệm có giá trị (valid test), phải nhắc lại thử nghiệm với số lượng mẫu và số ống môi trường Lowenstein-Jensen gấp đôi so với lần 1.
Nếu thử nghiệm độ sống lần 1 không đạt yêu cầu nhưng thử nghiệm không có giá trị (invalid test) thì chỉ cần nhắc lại thử nghiệm với số lượng mẫu và số ống môi trường bằng lần 1.
Nếu lần thử nghiệm nhắc lại, độ sống vẫn không đạt thì phải hủy bỏ loạt vắc xin BCG đó.
Kiểm tra độ sống của vắc xin BCG phải tiến hành song song với vắc xin BCG mẫu chuẩn để kiểm tra chất lượng môi trường dùng cho thử nghiệm độ sống. Độ sống của vắc xin BCG mẫu chuẩn trong mỗi thử nghiệm phải ổn định và nằm trong giới hạn giá trị trung bình ± 2SD.
Tiêu chuẩn chấp thuận
Đối với vắc xin BCG phòng bệnh lao, giới hạn độ sống cho phép là từ 1 triệu đến 6 triệu đơn vị sống trong 1 mg vắc xin (1x106 đvs/mg đến 6x106 đvs/mg vắc xin BCG).
Kiểm tra tính ổn định nhiệt
Số đơn vị sống của vắc xin BCG sau khi ủ ở 37 °C trong 28 ngày phải đạt ít nhất là 20 % so với mẫu ủ ở 4 °C trong 28 ngày.
Các vật liệu
Dung dịch Sauton loãng 1/4 được sử dụng để pha loãng vắc xin BCG.
Môi trường nuôi cấy: Lowenstein-Jensen.
Vắc xin BCG mẫu thử và mẫu chuẩn.
Trước khi kiểm tra tính ổn định nhiệt, vắc xin BCG đông khô mẫu thử phải được ủ ở các nhiệt độ khác nhau trong 28 ngày như dưới đây:
Dạng 1 mg/ống: Ủ 10 ống ở 4 °C trong 28 ngày và 10 ống ở 37 °C trong 28 ngày.
Dạng 0,5 mg/ống: Ủ 20 ống ở 4 °C trong 28 ngày và 20 ống ở 37 °C trong 28 ngày.
Tiến hành
Kiểm tra tính ổn định nhiệt của vắc xin BCG bằng cách đếm độ sống của loạt vắc xin này sau khi ủ ở 4 °C và 37 °C trong 28 ngày. Quy trình đếm độ sống được thực hiện như ở phần Kiểm tra độ sống. Tính tỷ lệ % số đơn vị sống BCG mọc ở 37 °C so với ở 4 °C, theo công thức sau:
Tỷ lệ % ổn định nhiệt = ![]()
trong đó:
A: số ĐVS/mg vắc xin BCG ở 37 °C trong 28 ngày;
B: số ĐVS/mg vắc xin BCG ở 4 °C trong 28 ngày.
ĐVS: đơn vị sống.
Những loạt vắc xin BCG có tỷ lệ % ổn định nhiệt không đạt yêu cầu ở lần kiểm tra thứ nhất phải nhắc lại thử nghiệm:
Nếu thử nghiệm tỷ lệ % ổn định nhiệt lần 1 không đạt yêu cầu nhưng thử nghiệm có giá trị, phải nhắc lại thử nghiệm với số lượng mẫu và số ống môi trường Lowenstein-Jensen gấp đôi so với lần 1.
Nếu thử nghiệm tỷ lệ % ổn định nhiệt lần 1 không đạt yêu cầu nhưng thử nghiệm không có giá trị thì chỉ cần nhắc lại thử nghiệm với số lượng mẫu và số ống môi trường bằng lần 1.
Nếu lần thử nghiệm nhắc lại, tỷ lệ % ổn định nhiệt vẫn không đạt thì phải hủy bỏ loạt vắc xin BCG đó. Kiểm tra tỷ lệ % ổn định nhiệt của vắc xin BCG phải tiến hành song song với vắc xin BCG mẫu chuẩn để kiểm tra chất lượng môi trường dùng cho thử nghiệm độ sống. Độ sống của vắc xin BCG mẫu chuẩn trong mỗi thử nghiệm phải ổn định và nằm trong giới hạn giá trị trung bình ± 2SD.
Tiêu chuẩn chấp thuận
Tỷ lệ % số đơn vị sống BCG mọc ở 37 °C so với ở 4 °C phải không được thấp hơn 20 %.
15.2 XÁC ĐỊNH ĐỘ CHÂN KHÔNG CỦA VẮC XIN BCG
Vắc xin BCG được đông khô và hàn kín trong điều kiện chân không (0,001 mmHg) và phải được kiểm tra độ chân không bằng thiết bị Tesla vacuum.
Kiểm tra độ chân không trong các ống vắc xin BCG đông khô không sớm hơn 2 tháng sau khi hàn ống. Khi kiểm tra chân không, được phép dùng những ống vắc xin phát ra ánh sáng màu tím nhạt đến trắng xanh và loại bỏ những ống vắc xin không phát ra ánh sáng hoặc phát ra ánh sáng màu đỏ hình tên lửa. Trước khi phân phát, vắc xin phải được kiểm tra lại về độ chân không (do đơn vị sản xuất kiểm tra). Nếu phát hiện thấy quá 5 % số ống không còn chân không thì lô vắc xin này phải lưu lại thêm 1 tháng nữa, sau đó kiểm tra chân không lại. Nếu vẫn có hơn 1 % số ống không đạt chân không thì lô vắc xin đó phải bỏ đi. Những số liệu kiểm tra này phải ghi vào hồ sơ của lô vắc xin.
Tiêu chuẩn chấp thuận:
Loạt vắc xin BCG đông khô được coi là đạt độ chân không khi có không ít hơn 95 % tổng số ống đạt tiêu chuẩn về độ chân không.
15.3 XÁC ĐỊNH ĐỘ PHÂN TÁN CỦA VẮC XIN BCG
Nguyên lý
Vắc xin BCG đông khô sau khi được hoàn nguyên với nước pha thuốc tiêm để kiểm tra độ phân tán của vắc xin này.
Nếu vắc xin có độ phân tán kém, tạo cụm nhiều sẽ có nguy cơ gây sưng hạch hoặc sưng hạch mủ cho trẻ được tiêm vắc xin.
Tiến hành
Vắc xin BCG đông khô được hoàn nguyên trong nước muối sinh lý (pha từ nước để pha thuốc tiêm) để có đậm độ 1 mg/ml.
Sử dụng nước muối sinh lý dùng để hoàn nguyên vắc xin BCG làm dung dịch mẫu trắng để chuẩn máy đo. Huyền dịch vắc xin được lắc kỹ trước khi đo mật độ quang (OD).
Đo mật độ quang của hỗn dịch vắc xin ở 2 bước sóng: 434 nm và 630 nm
Tính kết quả
Sau khi đo được OD của huyền dịch vắc xin BCG ở 2 bước sóng nêu trên, tính kết quả theo công thức:
Độ phân tán = ![]()
trong đó:
OD1 là mật độ quang của hỗn dịch vi khuẩn đo được ở bước sóng 434 nm (lọc màu xanh lam);
OD2 là mật độ quang của hỗn dịch vi khuẩn đo được ở bước sóng 630 nm (lọc màu đỏ).
Ở công thức này mẫu số (![]() -
- ![]() ) luôn là một hằng số và bằng 0,1618.
) luôn là một hằng số và bằng 0,1618.
Nếu loạt vắc xin BCG đông khô không đạt yêu cầu thì phải nhắc lại thử nghiệm:
- Nếu thử nghiệm lần 1 có giá trị (valid test) thì cần phải nhắc lại thử nghiệm với số lượng mẫu thử nghiệm gấp đôi so với thử nghiệm lần 1.
- Nếu thử nghiệm lần 1 không có giá trị (invalid test) thì chỉ cần nhắc lại thử nghiệm với số lượng mẫu thử nghiệm bằng thử nghiệm lần 1.
Kết quả của lần thử nghiệm nhắc lại nếu không đạt yêu cầu thì loạt vắc xin BCG đông khô đó coi như không đạt yêu cầu về độ phân tán và phải hủy bỏ; nếu đạt yêu cầu thì loạt vắc xin BCG đông khô đó coi như đạt yêu cầu về độ phân tán.
Tiêu chuẩn chấp thuận: Độ phân tán của vắc xin BCG cho phép là ≥ 0,9.
15.4 XÁC ĐỊNH TÍNH AN TOÀN VẮC XIN DTwP HẤP PHỤ
An toàn đặc hiệu
Tính an toàn đặc hiệu của vắc xin bạch hầu - uốn ván - ho gà toàn tế bào được kiểm tra trên mẫu bán thành phẩm cuối cùng hay vắc xin thành phẩm khi cần thiết và được tiến hành như sau:
An toàn đặc hiệu đối với thành phần bạch hầu và uốn ván
Chọn 5 chuột lang khoẻ mạnh, cùng giới (nếu khác giới phải nhốt riêng lồng), có trọng lượng 250 g đến 350 g, chưa sử dụng vào bất cứ thử nghiệm nào trước đó; tiêm dưới da cho mỗi chuột lang ít nhất 5 liều đơn vắc xin cho người (liều vắc xin như ghi trên nhãn). Theo dõi chuột hàng ngày và cân trọng lượng chuột hàng tuần trong 3 tuần (đối với thành phần uốn ván) và 6 tuần (đối với thành phần bạch hầu).
Nếu có chuột chết phải mổ để kiểm tra phủ tạng về dấu hiệu nhiễm độc bạch hầu (tuyến thượng thận đỏ).
Nếu có hơn một chuột lang chết trong thử nghiệm lần 1, phải nhắc lại thử nghiệm; nếu lần thử nghiệm nhắc lại không có chuột nào có biểu hiện nhiễm độc do độc tố bạch hầu hay uốn ván và có ít nhất 2/3 số chuột thử nghiệm sống sót sau khoảng thời gian theo dõi như quy định (3 tuần đối với vắc xin uốn ván và 6 tuần đối với vắc xin bạch hầu) thì loạt vắc xin DTwP đó đạt yêu cầu về an toàn đặc hiệu đối với thành phần bạch hầu và uốn ván.
- Nếu có hơn một chuột lang bị chết vì những nguyên nhân không đặc hiệu và thử nghiệm không có giá trị (invalid test), phải nhắc lại thử nghiệm với số lượng động vật thí nghiệm và mẫu thử bằng lần 1; nếu vẫn có hơn một chuột lang chết ở lần thử nghiệm thứ 2 thì loạt vắc xin không đạt về an toàn đặc hiệu đối với thành phần bạch hầu và uốn ván, phải hủy bỏ.
- Nếu thử nghiệm lần 1 không đạt yêu cầu nhưng thử nghiệm có giá trị (valid test), phải nhắc lại thử nghiệm với số lượng mẫu thử và số lượng chuột lang gấp đôi. Nếu thử nghiệm nhắc lại vẫn có quá 1/3 số chuột chết do bất kỳ lý do nào thì loạt vắc xin DTwP này sẽ phải hủy bỏ.
Tiêu chuẩn chấp thuận
Vắc xin đạt yêu cầu nếu không có chuột lang nào có dấu hiệu liệt do uốn ván hoặc triệu chứng nhiễm độc bạch hầu và ít nhất 80 % chuột sống sót, khoẻ mạnh và lên cân trong thời gian 6 tuần (đối với thành phần bạch hầu) hoặc 3 tuần (đối với thành phần uốn ván).
An toàn đặc hiệu đối với thành phần ho gà toàn tế bào (wP)
Dùng 20 chuột nhắt trắng có trọng lượng 14 g đến 16 g, cùng giới (nếu có cả 2 giới, cần phân chia đều trong các nhóm) cho mỗi mẫu vắc xin thử và cho nhóm chứng.
Chuột được ăn uống đầy đủ trước khi tiêm 1 h và trong suốt thời gian thí nghiệm. Tổng trọng lượng của từng nhóm chuột được xác định ngay trước lúc tiêm. Mỗi chuột được tiêm vào ổ bụng 0,5 ml dung dịch chứa tối thiểu nửa liều đơn vắc xin cho người. Nhóm chứng được tiêm 0,5 ml nước muối sinh lý (tốt nhất chứa cùng hàm lượng chất bảo quản như có trong dung dịch tiêm cho nhóm thí nghiệm). Tổng trọng lượng các nhóm chuột được xác định lại vào 72 h và 7 ngày sau tiêm.
Tiêu chuẩn chấp thuận:
Loạt vắc xin đạt yêu cầu nếu đạt cả 3 tiêu chuẩn sau:
- Sau 72 h tổng trọng lượng chuột của mỗi nhóm không ít hơn trước tiêm.
- Sau 7 ngày trọng lượng trung bình mỗi chuột không ít hơn 60 % so với nhóm chứng.
- Chuột chết không quá 5 %.
Nếu thử nghiệm lần 1 không đạt một trong 3 tiêu chuẩn nêu trên phải nhắc lại thử nghiệm:
- Nếu thử nghiệm lần 1 không có giá trị (invalid test): Chỉ cần nhắc lại thử nghiệm với số lượng chuột và số lượng mẫu bằng lần 1.
- Nếu thử nghiệm lần 1 có giá trị (valid test): Phải nhắc lại thử nghiệm với số lượng chuột nhắt trắng và số lượng mẫu gấp đôi so với lần 1.
Kết quả của lần kiểm tra nhắc lại, nếu đạt yêu cầu 3 tiêu chuẩn nêu trên và giá trị trung bình của 2 lần thử nghiệm (về % tăng trọng chuột của nhóm vắc xin thử nghiệm so với nhóm chứng sau 7 ngày theo dõi) đạt yêu cầu thì loạt vắc xin đó mới được coi là đạt yêu cầu về an toàn đặc hiệu ho gà. Nếu ở lần kiểm tra nhắc lại không đạt yêu cầu thì loạt vắc xin đó không đạt yêu cầu an toàn đặc hiệu thành phần ho gà và phải bị hủy bỏ.
An toàn không đặc hiệu (Độc tính bất thường)
Thử nghiệm được tiến hành trên chuột nhắt trắng và chuột lang khoẻ mạnh không tiêm bất cứ gì trước khi thử nghiệm. Tiêm vào ổ bụng cho 5 chuột nhắt trắng có trọng lượng 17 g/con đến 22 g/con, mỗi con một liều tiêm cho người; 2 chuột lang có trọng lượng 250 g/con đến 350 g/con, mỗi con một liều tiêm cho người nhưng không quá 1 ml/con.
Theo dõi tình trạng sức khoẻ và cân trọng lượng chuột hàng ngày.
Tiêu chuẩn chấp thuận:
Vắc xin được coi là không có độc tính bất thường nếu tất cả các động vật thí nghiệm sống khoẻ mạnh và lên cân trong thời gian ít nhất 7 ngày và không có dấu hiệu nhiễm độc.
Nếu thử nghiệm lần 1 không đạt yêu cầu nêu trên, phải nhắc lại thử nghiệm. Thử nghiệm nhắc lại nếu đạt yêu cầu thì loạt vắc xin đó được coi là đạt an toàn không đặc hiệu; nếu không đạt yêu cầu thì loạt vắc xin đó bị coi là không đạt an toàn không đặc hiệu và phải huỷ bỏ.
Đối với thử nghiệm nhắc lại:
- Nếu thử nghiệm lần 1 không đạt yêu cầu nhưng thử nghiệm có giá trị: phải nhắc lại thử nghiệm với số lượng mẫu và số lượng chuột gấp đôi lần 1.
- Nếu thử nghiệm lần 1 không đạt yêu cầu nhưng thử nghiệm không có giá trị: chỉ cần nhắc lại thử nghiệm với số lượng mẫu và số lượng chuột bằng lần 1.
15.5 XÁC ĐỊNH ĐẬM ĐỘ VI KHUẨN HO GÀ
Nguyên tắc
Thực tế một hỗn dịch vi khuẩn ho gà có đậm độ 10 tỷ vi khuẩn/ml tương ứng với 10 IOU (đơn vị độ đục quốc tế) khi nhìn bằng mắt thường.
Có thể sử dụng bộ so độ đục chuẩn quốc tế của WHO để so bằng mắt thường hoặc chuẩn trên máy quang phổ kế khi xác định đậm độ của hỗn dịch B. pertussis 18323 trong thử nghiệm công hiệu vắc xin ho gà.
Cách tiến hành
Trong quá trình sản xuất vắc xin ho gà, mỗi lần gặt sinh khối cần xác định đậm độ vi khuẩn của nước cốt ho gà. Độ đục nước cốt ho gà và nước cốt ho gà cô đặc được xác định bằng cách so sánh với ống mẫu chuẩn quốc tế hoặc quốc gia và quan sát bằng mắt thường hoặc đo trên máy quang phổ kế (không được muộn hơn hai tuần sau khi gặt và trước khi huyền dịch vi khuẩn được đưa vào bất kỳ quy trình pha chế nào tiếp theo).
Pha loãng nước cốt trong nước muối sinh lý cho đến khi soi bằng mắt thường thấy tương đương với ống chuẩn 10 IOU.
Tính độ đục của mẫu thử bằng cách nhân với số lần pha loãng.
Có thể đo độ đục của huyền dịch vi khuẩn ho gà trên máy đo quang phổ ở bước sóng 560 nm. Đậm độ nước cốt ho gà được sử dụng làm cơ sở để tính toán khi pha chế vắc xin bán thành phẩm cuối cùng.
Thành phần ho gà trong vắc xin DTP hỗn hợp không vượt quá 20 đơn vị độ đục quốc tế (IOU) trong một liều đơn vắc xin cho người. Tỷ lệ thành phần các chủng B. Pertussis 18323 không được thay đổi khi hỗn hợp vắc xin, phải đăng ký rõ trong hồ sơ và được sự chấp thuận của cơ quan Kiểm định quốc gia.
15.6 XÁC ĐỊNH HÀM LƯỢNG TWEEN 20 TRONG VẮC XIN VÀ SINH PHẨM
Nguyên lý
Dựa vào phản ứng của polyethoxylat trong thành phần của Tween 20 với amoni cobaltothiocyanat tạo thành hợp chất màu xanh lơ tan trong dicloromethan.
Tiến hành
Mẫu thử được điều chỉnh bằng nước cất để có nồng độ protein khoảng 100 pg/ml. Hút 1 ml mẫu thử vào ống nghiệm, thêm 5 ml ethanol 95 % (TT), ly tâm 3500 r/min trong 10 min ở 20 °C. Chuyển phần nước sang ống nghiệm khác, rửa tủa bằng 1 ml ethanol 95 % (TT), chuyển nốt nước rửa tủa vào ống nghiệm. Đun cách thủy phần nước thu được ở 80 °C cho đến khi còn lại khoảng 0,5 ml; thêm 1 ml nước cất vào ống nghiệm.
Hút dung dịch Tween 20 chuẩn 1 mg/ml vào các ống nghiệm lần lượt theo thể tích 10 µl; 25 µl; 50 µl; 75 µl; 100 µl, thêm nước cất vừa đủ 1 ml.
Mẫu trắng là 1 ml nước cất.
Thêm 2 ml dicloromethan (TT) vào mỗi ống nghiệm đựng mẫu trắng, mẫu chuẩn, mẫu thử. Thêm tiếp 3 ml amoni cobaltothiocyanat, lắc kỹ, để yên ở nhiệt độ phòng trong 90 min. Hút bỏ phần nước nổi bằng máy hút chân không, đo mật độ quang (Phụ lục 4.1) của lớp dicloromethan màu xanh lơ phía dưới ở bước sóng 620 nm.
Dựng đường chuẩn, từ đó tính ra hàm lượng Tween 20 trong mẫu thử.
Đơn vị tính: µg/100 µg protein.
Cách pha các dung dịch:
Dung dịch amoni cobaltothiocyanat. Hòa tan 6,0 g cobalt nitrat (TT) và 40 g amoni thiocyanat (TT) trong nước cất, thêm nước cất vừa đủ 200 ml, lọc qua giấy lọc.
Dung dịch Tween 20 chuẩn 10 mg/ml: Cân 1 g Tween 20 vào bình định mức 100 ml, thêm nước cất vừa đủ, lắc đều. Pha loãng bằng nước cất 10 lần trước khi dùng.
Tiêu chuẩn chấp thuận
Tùy từng loại vắc xin và sinh phẩm.
Hàm lượng Tween 20 có trong bán thành phẩm viêm gan B không lớn hơn 50 µg/100 µg protein.
15.7 KIỂM TRA VÔ TRÙNG VẮC XIN/SINH PHẨM
Việc kiểm tra vô trùng vắc xin/sinh phẩm được thực hiện trong điều kiện vô trùng. Kiểm soát môi trường thử nghiệm và sử dụng mẫu đối chứng âm tính thích hợp để kiểm tra và đảm bảo điều kiện vô trùng.
Môi trường nuôi cấy
Chuẩn bị môi trường nuôi cấy
Hai môi trường sử dụng để kiểm tra vô trùng vắc xin/sinh phẩm là môi trường lỏng thioglycolat (Fluid Thioglycollate Medium: FTM) và casein đậu tương (Soybean-casein digest medium: SCDM). FTM dự định dùng để phát hiện vi khuẩn kỵ khí nhưng cũng có thể xác định được vi khuẩn hiếu khí. SCDM dùng để phát hiện vi khuẩn hiếu khí và nấm. Khi vắc xin/sinh phẩm có chứa chất bảo quản là merthiolat, có thể dùng FTM thay thế cho SCDM trong phương pháp cấy trực tiếp khi môi trường này đạt yêu cầu trong thử nghiệm kiểm tra tính tăng sinh (Growth promotion test). Các môi trường tương đương khác cũng có thể được sử dụng nếu chúng đạt được yêu cầu về tính tăng sinh và được cơ quan Kiểm định Quốc gia chấp thuận.
Công thức và cách pha hai môi trường trên như sau:
Môi trường lỏng thioglycolat:
| L - Cystin | 0,50 g |
| Natri clorid | 2,50 g |
| Glucose monohydrat (C6H12O6.H2O) | 5,50 g |
| Thạch bột có độ ẩm nhỏ hơn 15 % | 0,75 g |
| Cao nấm men (tan trong nước) | 5,00 g |
| Casein thuỷ phân bằng pancreatin (men tụy) | 15,00 g |
| Nước | 1000 mL |
| Natri thioglycolat | 0,50 g |
| hoặc acid thioglycolic | 0,3 mL |
| Dung dịch natri resazurin (1 g/L) mới pha | 1 mL |
Trộn lần lượt sáu thành phần đầu như thứ tự ở trên trong nước nóng cho đến khi tan hoàn toàn. Cho thêm natri thioglycolat (hoặc acid thioglycolic), chuyên sang một bình chứa thích hợp và thêm nước vừa đủ, làm tan hoàn toàn trong một nồi cách thủy. Chỉnh pH của dung dịch (nếu cần) bằng dung dịch natri hydroxyd 1 N (TT) sao cho pH sau tiệt trùng (pH cuối cùng) là 7,0 đến 7,2. Nếu cần thiết phải lọc, đun nóng dung dịch (tránh đun sôi) rồi lọc qua giấy lọc ướt khi dung dịch còn đang nóng. Thêm dung dịch natri resazurin, trộn đều. Chia môi trường vào các chai/ống chứa thích hợp. Hấp tiệt trùng môi trường trong lò đã được thẩm định ở 121 °C trong 18-20 min. Vặn chặt nút các chai/ống môi trường và bảo quản ở nhiệt độ 20 °C đến 30 °C.
Nếu quá 1/3 trên của chai/ống môi trường chuyển thành màu hồng thì không nên sử dụng nữa. Có thể sử dụng lại một lần nữa bằng cách đun nóng môi trường trong bể ổn nhiệt đến khi màu hồng biến mất rồi làm lạnh nhanh. Không dùng môi trường quá thời hạn sử dụng đã được thẩm định.
Môi trường lỏng casein đậu tương:
| Casein thủy phân bởi pancreatin | 17,0 g |
| Bột đậu tương thủy phân bởi papain | 3,0 g |
| Natri clorid | 5,0 g |
| Dikali hydrophosphat | 2,5 g |
| Glucose monohydrat | 2,5 g |
| Nước tinh khiết | 1000 ml |
Hòa tan tất cả các thành phần vào nước, đun nóng nhẹ cho tan hoàn toàn, để nguội đến nhiệt độ phòng. Chỉnh pH của dung dịch (nếu cần) bằng dung dịch natri hydroxyd 1M (TT) hoặc dung dịch acid hydrocloric 1 N (TT) sao cho pH sau tiệt trùng (pH cuối cùng) là 7,1 đến 7,5. Lọc trong nếu cần. Phân chia môi trường vào các chai/ống. Hấp tiệt trùng môi trường trong lò đã được thẩm định ở 121 °C trong 18-20 min. Bảo quản môi trường ở nhiệt độ phòng (20 °C đến 30 °C).
Không dùng môi trường quá thời hạn sử dụng đã được thẩm định.
Kiểm tra chất lượng môi trường
Việc kiểm tra chất lượng mỗi loạt môi trường được thực hiện trước hoặc song song với kiểm tra vô trùng vắc xin/sinh phẩm.
Tất cả các loạt môi trường dùng trong thử nghiệm vô trùng vắc xin/sinh phẩm cần được kiểm tra về hai tính chất: tính vô trùng và khả năng cung cấp chất dinh dưỡng cho sự phát triển của vi khuẩn và nấm (tính tăng sinh).
Kiểm tra tính vô trùng
Lấy 2 % số chai/ống của mỗi loạt môi trường nhưng ít nhất là 5 chai/ống. Ủ FTM ở nhiệt độ 30°C đến 35 °C ít nhất 14 ngày. Ủ SCDM (hoặc FTM khi dùng thay thế) ở nhiệt độ 20 °C đến 25 °C ít nhất 14 ngày.
Môi trường đạt yêu cầu về vô trùng nếu sau nuôi cấy các chai/ống môi trường trong, không có lắng cặn ở đáy; các chai/ống FTM không mất chỉ thị màu.
Kiểm tra tính tăng sinh
Nguyên tắc: đánh giá khả năng phát triển của các chủng thử thách trong môi trường bằng cách cho vào mỗi chai/ống môi trường không quá 100 CFU chủng thử thách.
Bảng 1- Chủng thích hợp cho kiểm tra chất lượng môi trường
| Loại vi sinh vật | Tên | Ký hiệu |
| Vi khuẩn hiếu khí | Staphylococcus aureus | ATCC 6538, CIP 4.83, NCTC 10788, NCIMB 9518, NBRC 13276 |
| Bacillus subtilis | ATCC 6633, CIP 52.62, NCIMB 8054, NBRC 3134 | |
| Pseudomonas aeruginosa | ATCC 9027, NCIMB 8626, CIP 82.118, NBRC 13275 | |
| Vi khuẩn kị khí | Clostridium sporogenes | ATCC 19404, CIP 79.3, NCTC 532, ATCC 11437, NBRC 14293 |
| Nấm | Candida albicans | ATCC 10231, IP 48.72, NCPF 3179, NBRC 1594 |
| Aspergillus brasiliensis (Aspergillus niger) | ATCC 16404, IP 1431.83, IMI 149007, NBRC 9455. |
Có thể thay thế Pseudomonas aeruginosa bằng Kocuria rhizophila (Micrococcus luteus) ATCC 9341. Trong trường hợp không cần vi khuẩn tạo nha bào thì có thể thay thế Clostridium sporogenes bằng Bacteroides vulgatus (ATCC 8482).
Các chủng này không được cấy chuyển quá 5 lần kể từ chủng gốc.
Chủng dùng kiểm tra FTM gồm Clostridium sporogenes, Pseudomonas aeruginosa, Staphylococcus aureus. Chủng dùng kiểm tra TSB gồm Aspergillus brasiliensis, Bacillus subtilis, Candida albicans. Các chủng thích hợp khác cũng có thể sử dụng nếu được cơ quan Kiểm định Quốc gia chấp thuận.
Môi trường đạt yêu cầu về tính tăng sinh nếu các chủng vi khuẩn trong các chai/ống môi trường mọc trong vòng 3 ngày, các chủng nấm trong các chai/ống môi trường mọc trong vòng 5 ngày.
Mẫu kiểm tra
Số lượng vắc xin/sinh phẩm cần cho 1 lần kiểm tra vô trùng tùy thuộc vào số lượng thành phẩm mỗi loạt và dạng đóng gói của vắc xin/sinh phẩm, cần lấy mẫu đại diện cho quá trình đóng ống lúc đầu và cuối.
Bảng 2 - Số lượng mẫu cần cho thử nghiệm vô trùng
| Số lượng thành phẩm (Lọ/ống) | Số lượng mẫu cần lấy (Lọ/ống) | |
| Lượng vắc xin/sinh phẩm trong mỗi lọ ≥ 2 ml | Lượng vắc xin/sinh phẩm trong mỗi lọ < 2 ml | |
| <100 | 10% nhưng không ít hơn 4 | 20% nhưng không ít hơn 8 |
| ≥ 100 và < 500 | Ít nhất 10 | Ít nhất 20 |
| ≥ 500 | Ít nhất 20 | Ít nhất 40 |
Vắc xin/sinh phẩm được chia đều để cấy vào mỗi loại môi trường.
Phương pháp thực hiện
Tiến hành theo phương pháp màng lọc hoặc phương pháp cấy trực tiếp.
Phương pháp màng lọc
Sử dụng phương pháp màng lọc khi bản chất của vắc xin/sinh phẩm cho phép: các dung dịch có thể qua được màng lọc; các chế phẩm chứa cồn, dầu, các chế phẩm có thể trộn hoặc tan trong dung môi là nước hoặc dầu (các dung môi không chứa chất ức chế vi khuẩn/nấm).
Bộ lọc và màng lọc phải được tiệt trùng trước khi sử dụng.
Sử dụng màng có kích thước lỗ lọc nhỏ hơn hoặc bằng 0,45 μm, đường kính khoảng 47 mm. Màng cellulose nitrat dùng cho các dung dịch có dung môi là nước, dầu và các dung dịch có độ cồn thấp. Màng cellulose acetat dùng cho các dung dịch có độ cồn cao.
Thấm ướt màng lọc bằng dung dịch vô trùng thích hợp, chẳng hạn dung dịch pepton nồng độ 1 g/L, pH 7,1 ± 0,2. Cho mẫu thử trực tiếp lên màng lọc, hút mẫu qua màng lọc bằng một bơm chân không.
Nếu vắc xin/sinh phẩm có đặc tính ức chế sự phát triển của vi khuẩn/nấm, rửa màng lọc ít nhất ba lần bằng dung dịch rửa. Loại dung dịch rửa và thể tích mỗi lần rửa được xác định trong thử nghiệm độ phù hợp của phương pháp. Không rửa màng lọc quá 5 lần, mỗi lần 100 mL kể cả khi thử nghiệm độ phù hợp của phương pháp chỉ ra rằng như thế là chưa đủ để loại bỏ ảnh hưởng của chất ức chế.
Cho môi trường vào bình chứa màng lọc của bộ lọc hoặc cắt màng lọc thành hai phần bằng nhau rồi cho mỗi phần vào một chai môi trường nuôi cấy.
Các động tác cho mẫu thử lên màng lọc, cắt màng lọc và chuyển vào môi trường nuôi cấy phải được thực hiện trong điều kiện vô trùng để tránh lây nhiễm từ bên ngoài.
Phương pháp cấy trực tiếp
Bảng 3 - Thể tích vắc xin/sinh phẩm cần lấy từ mỗi lọ để cấy vào mỗi loại môi trường
| Thể tích có trong mỗi lọ | Thể tích tối thiểu cho mỗi loại môi trường |
| < 1 mL | Toàn bộ vắc xin/sinh phẩm có trong mỗi lọ |
| Từ 1 mL đến 20 mL | 1 mL |
| > 20 mL và < 100 mL | 5 mL |
| > 100 mL | 10 % vắc xin/sinh phẩm có trong mỗi lọ nhưng không ít hơn 20 mL |
Cấy trực tiếp vắc xin/sinh phẩm cần kiểm tra vào môi trường sao cho thể tích mẫu thử không quá 10 % thể tích môi trường (trừ trường hợp đặc biệt).
Nếu vắc xin/sinh phẩm có tính chất ức chế sự phát triển của vi khuẩn/nấm, phải loại bỏ được đặc tính ức chế này bằng cách trung hòa với một chất trung hòa thích hợp hoặc hòa loãng trong thể tích môi trường đủ lớn khi thực hiện thử nghiệm vô trùng vắc xin/sinh phẩm. Nên xác định thể tích môi trường đủ lớn để loại bỏ được ảnh hưởng của chất ức chế. Mỗi lần xác định, thể tích này được sử dụng cho những lần thử nghiệm sau đó, trừ khi có thay đổi trong thành phần của vắc xin/sinh phẩm.
Đối với các vắc xin/sinh phẩm mà chính mẫu thử làm đục môi trường hoặc lắng cặn ở đáy ống môi trường tới mức không xác định được có hay không có sự phát triển của vi khuẩn/nấm; sau 3 đến 7 ngày nuôi cấy, cấy chuyển ít nhất 1 mL của mỗi ống môi trường đó sang ống môi trường mới tương ứng.
Ủ môi trường và theo dõi
Ủ FTM ở nhiệt độ 30 °C đến 35 °C ít nhất 14 ngày. Ủ SCDM (hoặc FTM khi dùng thay thế) ở nhiệt độ 20 °C đến 25 °C ít nhất 14 ngày. Các ống môi trường cấy chuyển ủ trong thời gian ít nhất 7 ngày. Theo dõi các ống môi trường vào những khoảng thời gian thích hợp và vào ngày cuối cùng để phát hiện sự phát triển của vi khuẩn/nấm trong các ống môi trường.
Đánh giá kết quả
Nếu không có sự phát triển của vi khuẩn/nấm trong các ống môi trường cấy vắc xin/sinh phẩm, mẫu kiểm tra đạt yêu cầu về vô trùng.
Nếu có sự phát triển của vi khuẩn/nấm trong các ống môi trường cấy vắc xin/sinh phẩm, mẫu kiểm tra không đạt yêu cầu về vô trùng trừ khi có thể chứng minh thử nghiệm đã thực hiện không có giá trị bằng cách thực hiện lại thử nghiệm hoặc bằng phương pháp khác.
Thử nghiệm không có giá trị khi xảy ra một trong các tình huống sau:
• Kết quả giám sát vi sinh môi trường trong khi thực hiện thử nghiệm không đạt yêu cầu;
• Sau khi xem xét lại qui trình thực hiện thử nghiệm phát hiện ra sai sót;
• Có vi khuẩn hoặc nấm mọc trong các chứng âm;
• Chủng phân lập từ thử nghiệm được xác định chắc chắn là do nguyên vật liệu hoặc kỹ thuật trong quá trình thực hiện thử nghiệm.
Thử nghiệm độ phù hợp của phương pháp
Thực hiện thử nghiệm độ phù hợp của phương pháp khi:
• Kiểm tra vô trùng cho sản phẩm mới;
• Khi có thay đổi trong điều kiện thực hiện thử nghiệm vô trùng vắc xin/sinh phẩm.
Thử nghiệm này nên được thực hiện để chứng tỏ thử nghiệm vô trùng có khả năng phát hiện được vi khuẩn/nấm có trong mẫu thử, nhất là khi vắc xin/sinh phẩm có khả năng ức chế sự phát triển của vi khuẩn/nấm.
Thực hiện thử nghiệm giống như thực hiện thử nghiệm vô trùng vắc xin/sinh phẩm, chỉ khác ở những điểm sau:
• Phương pháp màng lọc: Sau khi đã cho vắc xin/sinh phẩm qua màng lọc, cho thêm không quá 100 CFU chủng thử thách vào dung dịch rửa màng lọc lần cuối cùng.
• Phương pháp cấy trực tiếp: Sau khi đã cấy vắc xin/sinh phẩm vào môi trường, cho thêm không quá 100 CFU chủng thử thách vào mỗi ống môi trường.
Sử dụng cùng chủng thử thách với thử nghiệm kiểm tra tính tăng sinh. Thực hiện thử nghiệm kiểm tra tính tăng sinh làm chứng dương.
Ủ tất cả các ống môi trường đã cấy vắc xin/sinh phẩm không quá 5 ngày.
Nếu sau 5 ngày nuôi cấy, có dấu hiệu mọc của vi khuẩn/nấm trong các ống môi trường chứa vắc xin/sinh phẩm và không yếu hơn mức độ mọc trong các ống chứng dương thì vắc xin/sinh phẩm không chứa chất ức chế sự phát triển của vi khuẩn/nấm hoặc ảnh hưởng của chất ức chế đã được loại bỏ. Thử nghiệm vô trùng vắc xin/sinh phẩm có thể được thực hiện mà không cần phải thay đổi.
Nếu sau 5 ngày nuôi cấy, không có dấu hiệu mọc của vi khuẩn/nấm trong các ống môi trường cấy vắc xin/sinh phẩm hoặc yếu hơn mức độ mọc trong các ống chứng dương thì vắc xin/sinh phẩm có chứa chất ức chế làm ảnh hưởng đến sự phát triển của vi khuẩn/nấm. Thay đổi điều kiện thực hiện thử nghiệm vô trùng vắc xin/sinh phẩm và nhắc lại thử nghiệm độ phù hợp của phương pháp.
15.8 MÔI TRƯỜNG DÙNG ĐỂ PHÁT HIỆN VI KHUẨN HIẾU KHÍ, KỴ KHÍ VÀ NẤM
Quy định chung
Môi trường nuôi cấy dùng để kiểm tra vô trùng phải do cơ quan Kiểm định Quốc gia quy định. Môi trường này phải bảo đảm sự phát triển tốt cho phần lớn các loại vi sinh vật hiếu khí, kị khí thường có trong không khí của cơ sở sản xuất.
Để kiểm tra tính vô trùng của các sinh phẩm phải dùng môi trường thioglycolat dạng nước. Thành phần của môi trường được mô tả trong phần tiếp theo. Mỗi loạt môi trường thioglycolat mới được điều chế phải được kiểm tra về tính vô trùng, khả năng làm cho vi sinh vật phát triển (dinh dưỡng) và tính trung hòa.
Để kiểm tra tính vô trùng của các chế phẩm không có chất bảo quản thimerosal, môi trường phải được dùng trong vòng 3 tuần lễ kể từ ngày pha chế. Môi trường sau khi sản xuất phải bảo quản ở nhiệt độ phòng và tránh ánh sáng.
Để kiểm tra sinh phẩm có chứa thimerosal có thể dùng môi trường thioglycolat pha mới, môi trường phải được dùng trong vòng 7 ngày đến 10 ngày, kể từ ngày sản xuất.
Thời hạn sử dụng cụ thể của môi trường thioglycolat được xác định bằng cách kiểm tra tính chất trung hòa của môi trường sau khi pha chế.
Thành phần và cách pha chế môi trường Thioglycolat lỏng
| L - Cystin | 0,50 g |
| Natri clorid | 2,50 g |
| Glucose monohydrat (C6H12O6.H2O) | 5,50 g |
| Thạch (agar) | 0,75 g |
| Chiết xuất men (tan trong nước) 5,00 g | 5,00 g |
| Casein thủy phân bằng men tụy | 15,00 g |
| Nước cất | 1000,0 mL |
| Natri thioglycolat | 0,50 g |
Natri resazurin (dung dịch 0,01 % mới pha) 1,00 ml pH cuối cùng 7,0 đến 7,2 (sau khi hấp ở 121 °C từ 18 min đến 20 min)
Cách pha chế:
Cho lần lượt 6 thành phần đầu vào cốc. Khuấy trong 1 ít nước nóng, cho phần nước còn lại, sau đó đun cách thủy cốc đựng môi trường, chú ý để cho L-cystin được tan hoàn toàn. Cho natri thioglycolat, tiếp đó cho dung dịch natri hydroxyd 1 N vừa đủ sao cho khi hấp pH phải là 7,0 đến 7,2. Đun nóng lại, không được đun sôi, nếu cần, lọc qua giấy lọc ướt, sau đó cho thêm natri resazurin. Phân chia vào ống nghiệm thích hợp tùy theo yêu cầu. Hấp ở 121 °C trong thời gian từ 18 min đến 20 min. Khi lấy từ lò hấp ướt ra, làm lạnh ngay dưới vòi nước lạnh cho tới 25 °C. Bảo quản ở nhiệt độ 2 °C - 25 °C, tránh ánh sáng. Nếu quá 1/3 phần trên ống môi trường đổi thành màu hồng, không nên dùng hoặc nếu dùng phải hấp hơi nước lại một lần nữa cho đến khi màu hồng biến mất rồi làm lạnh nhanh. Môi trường bảo quản quá 3 tuần không dùng được.
Có thể dùng môi trường bột của các hãng thương mại để pha chế môi trường này, vừa đảm bảo chất lượng vừa pha chế rất đơn giản.
Yêu cầu về chất lượng của môi trường nuôi cấy
Môi trường thioglycolat sau khi điều chế phải đạt các yêu cầu sau:
Tính vô trùng: Phải vô trùng (Phụ lục 15.7).
Tính chất phát triển: Phải bảo đảm cho chủng Bacillus subtilis ATCC 6633, Micrococcus luteus ATCC 9341, Clostridium sporogenes ATCC 11437 và Candida albicans ATCC 10231 phát triển tốt với liều gây nhiễm là 10 đến 100 CFU/0,1 ml chủng/ống môi trường.
15.9 KIỂM TRA ĐỘC TÍNH ĐẶC HIỆU (AN TOÀN ĐẶC HIỆU) TRONG VẮC XIN BCG ĐÔNG KHÔ
Thử nghiệm “Kiểm tra độc tính đặc hiệu (an toàn đặc hiệu) trong vắc xin BCG đông khô” dùng để phát hiện độc tính đặc hiệu của vắc xin BCG đông khô do Mycobacteria gây ra. Thử nghiệm này được áp dụng đối với mẫu bán thành phẩm hoặc mẫu thành phẩm vắc xin BCG.
Dùng ít nhất 6 chuột lang cùng giới (nếu là chuột cái phải không được có thai) có phản ứng âm tính với tuberculin, cân nặng mỗi chuột khoảng 250 g đến 400 g.
Tiêm dưới da cho mỗi chuột một liều vắc xin tương đương với ít nhất 50 liều tiêm trong da cho người. Sau khi tiêm vắc xin, phải theo dõi chuột lang ít nhất là 6 tuần. Cuối thời kỳ theo dõi, tất cả chuột lang đều phải được mổ, kiểm tra đại thể các phủ tạng xem có các dấu hiệu của bệnh lao tiến triển không. Mặt khác, trong thời kỳ theo dõi nếu có chuột nào chết cũng phải mổ kiểm tra như trên. Nếu sau 6 tuần theo dõi các chuột đều khoẻ mạnh, tăng cân; không có biểu hiện bệnh lao tiến triển và ít nhất có 2/3 số chuột sống sót cho đến hết thời gian theo dõi thì loạt vắc xin BCG đó đạt yêu cầu về an toàn đặc hiệu. Trong vòng 42 ngày theo dõi, nếu có hơn 1/3 số chuột lang thử nghiệm chết nhưng xác định không phải chết do lao thì thử nghiệm được lặp lại. Nếu:
Lần thử nghiệm thứ nhất không đạt yêu cầu và không có giá trị (invalid test): Thử nghiệm nhắc lại chỉ cần tiến hành với ít nhất 6 chuột lang khác.
Lần thử nghiệm thứ nhất không đạt yêu cầu và có giá trị (valid test): thử nghiệm phải được nhắc lại với số lượng mẫu và số lượng chuột gấp đôi.
Nếu trong lần thử nghiệm thứ 2 vẫn có hơn 1/3 số chuột lang chết (kể cả được xác định chuột chết không phải do lao) thì loạt vắc xin BCG đó coi như không an toàn và phải xem xét lại nguồn cung cấp chuột, hoặc/ và kiểm tra quy trình sản xuất với sự xác nhận của cơ quan Kiểm định Quốc gia.
Nếu trong lần thử nghiệm nhắc lại các chuột đều khỏe mạnh, tăng cân, không có biểu hiện bệnh lao tiến triển và ít nhất có 2/3 số chuột sống sót cho đến hết thời gian theo dõi thì loạt vắc xin BCG đó đạt yêu cầu về tính an toàn đặc hiệu.
Nếu phát hiện thấy chuột có biểu hiện của bệnh lao tiến triển thì loạt vắc xin đó phải hủy bỏ và phải đình chỉ sản xuất các loạt vắc xin tiếp theo. Toàn bộ vắc xin trong kho phải giữ lại để tiến hành thanh tra và tìm ra nguyên nhân. Việc sản xuất chỉ được tiếp tục khi có sự chấp thuận của cơ quan Kiểm định Quốc gia.
15.10 THỬ NGHIỆM NHẬN DẠNG HUYẾT THANH MIỄN DỊCH
Mục đích của thử nghiệm nhận dạng huyết thanh miễn dịch là nhằm khẳng định huyết thanh thử nghiệm chỉ chứa protein từ loài động vật đăng kí trong sản xuất.
Có thể tiến hành theo 1 trong 2 kỹ thuật sau:
Kỹ thuật khuếch tán miễn dịch (Ouchterlony)
Nguyên lý
Kỹ thuật này dựa trên nguyên lý khuếch tán kép tự do của kháng nguyên và kháng thể từ các giếng riêng biệt được tạo trong gel agarose 1 % vào môi trường và tạo thành cung tủa do phản ứng đặc hiệu của chúng.
Vật liệu và thiết bị.
Phiến kính.
Agarose 1 % trong đệm phosphat pH 7,4.
Hộp ẩm.
Huyết thanh miễn dịch thử nghiệm.
Huyết thanh kháng loài.
Dung dịch nhuộm: Coomasle blue 0,025 %.
Dung dịch tẩy màu: Methanol - acetic acid - nước cất (4 : 1 : 5).
Tiến hành
Đổ gel agarose 1 % lên phiến kính.
Dùng dụng cụ đục giếng loại có đường kính 3 mm tạo 7 giếng trên phiến kính (1 giếng ở giữa, 6 giếng cách đều xung quanh). Nhỏ huyết thanh kháng loài vào các giếng xung quanh và huyết thanh thử nghiệm vào giếng ở giữa.
Đặt phiến kính vào hộp ẩm từ 12 h đến 48 h.
Nhuộm gel với dung dịch nhuộm coomasie blue 0,025 %. Tẩy màu bằng dung dịch tẩy màu, sau đó rửa bằng nước cất.
Để khô phiến kính ở nhiệt độ phòng.
Kỹ thuật điện di miễn dịch
Nguyên lý
Dưới tác động của điện trường và trong môi trường gel agarose, kháng nguyên tích điện âm từ giếng ở phía cực âm và kháng thể tích điện dương từ giếng ở phía cực dương sẽ di chuyển ngược chiều nhau, khi gặp nhau sẽ hình thành đường tủa có thể nhìn thấy được.
Vật liệu và thiết bị.
Phiến kính.
Thạch 3 % trong nước cất.
Agarose 1,5 % trong đệm barbital pH 8,4.
Huyết thanh miễn dịch thử nghiệm.
Huyết thanh kháng loài.
Máy điện di, nguồn điện.
Dung dịch nhuộm, rửa, tẩy màu....
Tiến hành
Đổ thạch nền 3 % lên phiến kính.
Đổ agarose 1,5 % lên trên thạch nền.
Sau khi agarose đông, đục 2 giếng có đường kính 3 mm, khoảng cách giữa 2 giếng là 4 mm đến 5 mm. Nhô huyết thanh thử nghiệm và huyết thanh kháng loài vào mỗi giếng.
Đặt phiến kính vào máy điện di.
Tiến hành điện di ở 10 V/cm từ 10 min đến 60 min tùy thử nghiệm.
Ngừng điện di, quan sát kết quả.
Để dễ quan sát đường tủa, cần loại protein không tủa bằng cách ngâm phiến kính trong đệm PBS. Phủ giấy lọc Whatman lên phiến kính và sấy ở 37 °C đến khô.
Nhuộm xanh với Coomasie blue trong 20 min.
Tẩy màu, rửa với nước cất, để khô.
Nhận định kết quả
Huyết thanh miễn dịch thử nghiệm được nhận dạng đúng khi xuất hiện đường tủa giữa mẫu thử nghiệm và huyết thanh kháng loài tương ứng.
15.11 XÁC ĐỊNH AN TOÀN CHUNG CỦA VẮC XIN VÀ SINH PHẨM
Xác định độc tính bất thường trong vắc xin - sinh phẩm được tiến hành trên chuột nhắt và chuột lang. Triệu chứng nhiễm độc trên chuột có thể biểu hiện như sau:
Thay đổi diện mạo bên ngoài, xù lông.
Trạng thái bất thường, giảm hoạt động.
Chuột giảm cân.
Chuột chết do nhiễm độc.
Trên chuột lang
Mỗi thử nghiệm dùng 2 chuột lang, cân nặng mỗi chuột từ 250 g đến 350 g, chưa dùng cho thí nghiệm nào trước đó, khỏe mạnh, tăng trọng bình thường trong thời gian cách ly 3 ngày đến 7 ngày.
Tiêm ổ bụng cho mỗi chuột 1 liều tiêm cho người nhưng không quá 5 ml; trừ một số vắc xin, sinh phẩm đặc biệt sẽ theo chuyên luận riêng. Liều cho người được trình bày trên nhãn sản phẩm.
Cân trọng lượng từng con trước khi tiêm và cân ít nhất 1 lần vào cuối giai đoạn theo dõi. Chuột phải được quan sát kỹ 2 h đầu sau tiêm và hàng ngày trong suốt 7 ngày để phát hiện dấu hiệu lâm sàng ốm do nhiễm độc.
Thử nghiệm đạt yêu cầu nếu toàn bộ chuột thí nghiệm khoẻ mạnh, tăng trọng và không có biểu hiện bệnh lý hoặc nhiễm độc.
Nếu có nhiều hơn 1 chuột chết thì thử nghiệm không đạt yêu cầu (nếu thử nghiệm đó đã được xác định là có giá trị).
Nếu có 1 chuột chết hoặc chỉ ra dấu hiệu ốm do nhiễm độc trong lần thử đầu tiên (thử nghiệm có giá trị), cần làm lại thử nghiệm với số lượng chuột gấp đôi và lượng mẫu thử gấp đôi. Thử nghiệm đạt yêu cầu nếu trong lần thử nghiệm thứ 2 không có chuột nào chết hoặc chỉ ra dấu hiệu ốm do nhiễm độc. Nếu thử nghiệm lần đầu không có giá trị thì thử nghiệm lặp lại với số lượng mẫu và số lượng động vật thí nghiệm bằng lần đầu.
Trên chuột nhắt
Mỗi thử nghiệm dùng 5 chuột nhắt Swiss, cân nặng mỗi chuột từ 17 g đến 22 g, chưa dùng thí nghiệm nào trước đó, khỏe mạnh, tăng trọng bình thường trong thời gian cách ly 3 ngày.
Tiêm ổ bụng 1 liều tiêm cho người nhưng không quá 1 ml, trừ một số vắc xin sinh phẩm đặc biệt sẽ theo chuyên luận riêng. Liều cho người được trình bày trên nhãn sản phẩm.
Cân trọng lượng từng con trước khi tiêm và cân ít nhất 1 lần vào cuối giai đoạn theo dõi. Chuột phải được quan sát kỹ 2 h đầu sau tiêm và hàng ngày trong suốt 7 ngày để phát hiện dấu hiệu lâm sàng ốm do nhiễm độc.
Thử nghiệm đạt yêu cầu nếu toàn bộ chuột thí nghiệm khỏe mạnh, tăng trọng bình thường và không có biểu hiện bệnh lý hoặc nhiễm độc.
Nếu có nhiều hơn 1 chuột chết thì thử nghiệm không đạt yêu cầu (nếu thử nghiệm là có giá trị).
Nếu có 1 chuột chết hoặc chỉ ra dấu hiệu ốm do nhiễm độc trong lần thử đầu tiên (thử nghiệm có giá trị) cần làm lại thử nghiệm với số lượng chuột gấp đôi và lượng mẫu thử gấp đôi. Thử nghiệm đạt yêu cầu nếu trong lần thử nghiệm thứ 2 không có chuột nào chết hoặc chỉ ra dấu hiệu ốm do nhiễm độc. Nếu thử nghiệm lần đầu không có giá trị thì thử nghiệm lặp lại với số lượng mẫu và số lượng động vật thí nghiệm bằng lần đầu.
15.12 XÁC ĐỊNH CHẤT GÂY SỐT TRONG VẮC XIN VÀ SINH PHẨM
Thử nghiệm chất gây sốt là phương pháp sinh học dùng để đánh giá tính chất gây sốt của vắc xin, sinh phẩm dựa trên sự tăng thân nhiệt của thỏ trước và sau khi tiêm vào tĩnh mạch dung dịch mẫu thử.
Lựa chọn động vật thí nghiệm
Thỏ khoẻ mạnh, trưởng thành, đực hoặc cái (nhưng không được mang thai), cân nặng không ít hơn 1,5 kg, được nuôi dưỡng bằng thức ăn không chứa chất kháng sinh và không bị tụt cân trong suốt 1 tuần trước thử nghiệm. Trong thời gian 3 ngày trước khi thí nghiệm, không sử dụng thỏ này cho những thí nghiệm tương tự khác. Cũng có thể sử dụng những thỏ trước đó 3 tuần đã làm thử nghiệm chất gây sốt nhưng kết quả lần thử trước là âm tính.
Phòng thí nghiệm
Thử nghiệm được tiến hành trong phòng yên tĩnh, tránh mọi tiếng động và ánh sáng làm ảnh hưởng đến kết quả thí nghiệm và nhiệt độ phòng không chênh lệch 3 °C so với nơi ở cũ.
Thỏ được nuôi riêng trong phòng yên tĩnh có nhiệt độ 20 °C đến 25 °C, cho ăn thức ăn tổng hợp không có chất kháng sinh. Trước ngày tiêm, để thỏ nhịn ăn qua đêm (nhưng được uống nước) cho đến khi thí nghiệm kết thúc. Không cho uống nước trong thời gian đang làm thí nghiệm.
Thiết bị, dụng cụ
Nhiệt kế: Để đo nhiệt độ của thỏ có thể dùng nhiệt kế hoặc thiết bị điện có độ chính xác 0,1 °C. Khi đo nhiệt độ, nhiệt kế phải đặt sâu trong trực tràng thỏ ít nhất 5 cm, phải để tối thiểu 5 min. Nếu dùng thiết bị điện, phải đặt yên trong trực tràng ít nhất 90 min trước khi tiêm và giữ nguyên vị trí đó suốt thời gian thí nghiệm.
Dụng cụ thủy tinh, bơm tiêm, kim tiêm: Tất cả phải được rửa sạch bằng nước để pha thuốc tiêm (WFI) và sấy khô 250 °C trong 30 min hoặc 200 °C trong 60 min, nếu dùng đồ nhựa phải không chứa chất gây sốt và phải vô trùng.
Lồng thỏ chuyên dụng:
Trước khi bắt đầu, đặt thỏ trong lồng chuyên dụng trong tư thế thỏa mái không ít hơn 1 h và duy trì ở đó trong suốt thời gian làm thí nghiệm.
Thử nghiệm thăm dò (preliminary test):
1 ngày đến 3 ngày trước khi tiến hành tiêm vắc xin, sinh phẩm cần kiểm tra sơ bộ về tính nhậy cảm cho các thỏ thí nghiệm bằng cách tiêm vào tĩnh mạch 10 ml/kg trọng lượng dung dịch nước muối sinh lý 0,9 % không chứa chất gây sốt đã được làm ấm lên khoảng 38,5 °C. Ghi nhiệt độ trước tiêm và tiếp tục trong vòng 3 h sau khi tiêm dung dịch nước muối sinh lý. Nếu thỏ nào có nhiệt độ dao động so với nhiệt độ ban đầu > 0,6 °C sẽ bị loại, không dùng đưa vào thử nghiệm chính.
Thử nghiệm chính (main test): Đối với mẫu vắc xin, sinh phẩm.
Thỏ thí nghiệm là thỏ đạt các tiêu chuẩn trên, được để trong lồng chuyên dụng ít nhất 60 min để ổn định. Sau đó tiến hành đo nhiệt độ thỏ 2 lần mỗi lần cách nhau 30 min (nếu đo tay) hoặc theo chương trình cài đặt (nếu đo bằng thiết bị). Nhiệt độ ban đầu của thỏ là giá trị trung bình của các lần đo này. Tiêu chuẩn đưa vào thí nghiệm là những thỏ có nhiệt độ chênh lệch giữa các lần đo không quá 0,2 °C và nhiệt độ ban đầu nằm trong khoảng 38 °C đến 39,8 °C. Mỗi mẫu thử được tiêm cho một nhóm thỏ 3 con, nhiệt độ chênh lệch trong nhóm không quá 1 °C.
Dung dịch dùng để tiêm thỏ là vắc xin, sinh phẩm nếu ở dạng nước hoặc được hoàn nguyên bằng dung dịch nước hồi chỉnh đi kèm nếu ở dạng đông khô. Trừ một số vắc xin, sinh phẩm đặc biệt sẽ theo chuyên luận riêng (có thể được pha loãng bằng dung dịch nước muối sinh lý không chứa chất gây sốt). Tiêm chậm dung dịch vào tĩnh mạch vành tai của mỗi thỏ trong khoảng thời gian không kéo dài quá 4 min. Thể tích được tiêm cho mỗi thỏ tương ứng với 1 ml/kg cân nặng. Nên làm ấm dung dịch đến 38,5 °C trước khi tiêm. Đo nhiệt độ thỏ ở những khoảng thời gian tùy thuộc vào thiết bị sử dụng (nếu đo tay cứ 1 h đo 1 lần; trường hợp dùng thiết bị, nhiệt độ được đo tự động theo chương trình cài đặt) trong khoảng thời gian 3 h sau khi tiêm. Nhiệt độ tối đa là nhiệt độ cao nhất đo được trong khoảng thời gian này.
Đánh giá kết quả
Đáp ứng của mỗi thỏ là hiệu số của nhiệt độ tối đa sau khi tiêm mẫu thử và nhiệt độ ban đầu.
Đáp ứng của thỏ bằng 0 nếu nhiệt độ tối đa sau khi tiêm bằng hoặc thấp hơn nhiệt độ ban đầu.
Mẫu thử được coi là đạt yêu cầu về tiêu chuẩn chất gây sốt nếu nhiệt độ chênh lệch của mỗi thỏ ≤ 0,6 °C và tổng nhiệt độ chênh lệch của 3 thỏ ≤ 1,3 °C. Nếu > 2,4 °C coi như không đạt yêu cầu.
Nếu tổng nhiệt độ chênh lệch của 3 thỏ nằm trong khoảng > 1,3 °C đến 2,4 °C thì thử nghiệm cần phải tiến hành thêm trên 3 thỏ khác. Mẫu thử đạt yêu cầu về tiêu chuẩn chất gây sốt khi tổng nhiệt độ chênh lệch (cộng dồn) của cả 6 thỏ ≤ 3 °C. Nếu > 4,1 °C coi như không đạt yêu cầu.
Nếu tổng nhiệt độ chênh lệch (cộng dồn) của 6 thỏ nằm trong khoảng > 3 °C đến 4,1 °C thì cần phải làm thêm lần cuối trên 3 thỏ khác. Mẫu thử được coi là đạt yêu cầu về tiêu chuẩn chất gây sốt khi tổng nhiệt độ chênh lệch của 9 thỏ (cộng dồn) phải ≤ 4,9 °C. Nếu > 4,9 °C coi như không đạt yêu cầu và phải hủy vắc xin hay sinh phẩm thử nghiệm.
15.14 PHƯƠNG PHÁP LẤY MẪU VÀ LƯU MẪU
Tiêu chuẩn này quy định phương pháp lấy mẫu cho các loại sinh phẩm sau đây:
• Các loại vắc xin làm từ vi khuẩn hoặc virus, dùng để phòng bệnh cho người.
• Các dung dịch dùng để hồi chỉnh các sinh phẩm dạng đông khô.
• Các sinh phẩm điều trị: SAT, SAD,....
PHƯƠNG PHÁP LẤY MẪU
Nguyên tắc
Phải lấy mẫu ở công đoạn sản xuất, với số lượng đủ tính đại diện.
Các mẫu phải lấy trong điều kiện vô khuẩn bằng những dụng cụ đã vô khuẩn.
Cách lấy mẫu
Mẫu được lấy trong các giai đoạn sau:
Lấy mẫu bán thành phẩm cuối cùng
Trước khi lấy mẫu phải lắc kỹ chai hoặc bình chứa bán thành phẩm cuối cùng. Lấy ít nhất 10 ml cấy vào trong 2 loại môi trường thử.
Lấy mẫu trong quá trình đóng ống
Để kiểm tra tính vô khuẩn trong quá trình đóng ống phải lấy mẫu ít nhất tại thời điểm đầu, giữa và cuối quá trình đóng ống. Tổng số mẫu lấy tùy thuộc số lượng thành phẩm (xem Bảng 15.14).
Đối với quá trình đóng ống mà sau đó sinh phẩm phải đông khô thì sau giai đoạn đông khô cũng phải lấy mẫu để kiểm tra vô khuẩn.
Lấy mẫu thành phẩm
Mẫu thành phẩm cần kiểm tra phải đảm bảo đại diện cho cả loạt thành phẩm. Mẫu lấy kiểm tra phải bao gồm cả mẫu của giai đoạn đầu, giai đoạn giữa và cuối của quá trình đóng ống. Nếu loạt bán thành phẩm cùng trong bình chứa (tank), nhưng được đóng làm nhiều lần, thì mẫu lấy của mỗi lần (gọi là một loạt đóng ống) phải đại diện cho toàn bộ quá trình đóng ống của lần đó và được kiểm tra riêng về mặt vô trùng, số lượng mẫu cần lấy để kiểm tra do cơ quan Kiểm định Quốc gia quy định.
Số lượng mẫu lấy
Mỗi giai đoạn phải lấy số mẫu phù hợp để kiểm định.
Công thức lấy mẫu để kiểm tra theo quy định của Tổ chức Y tế Thế giới là 0,4 ![]() ; trong đó N là số lượng ống, lọ của loạt thành phẩm đó.
; trong đó N là số lượng ống, lọ của loạt thành phẩm đó.
Đối với loạt thành phẩm có ít hơn 100 ống thì phải lấy 10 % số ổng để kiểm tra.
Cần lưu ý: Riêng với thử nghiệm vô trùng, số lượng mẫu lấy ở mỗi giai đoạn phải đủ cho ít nhất 3 lần thử nghiệm phòng trường hợp phải nhắc lại thử nghiệm.
Lưu mẫu
Mỗi loạt sinh phẩm thành phẩm đều phải giữ mẫu lưu. Mẫu lưu này được giữ cho đến khi vắc xin và sinh phẩm hết hạn sử dụng. Phải ghi nhãn cẩn thận số loạt và tên sinh phẩm. Các cơ sở sản xuất cần phải lưu một lượng mẫu đủ để có thể tiến hành kiểm tra vô khuẩn lại khi cần thiết.
Số lần kiểm định
Thông thường, số lượng mẫu lấy phải bảo đảm cho các thử nghiệm kiểm định của 3 cấp như sau: Kiểm định sản xuất (kiểm định cấp I); Kiểm định địa phương (kiểm định cấp II) và Kiểm định Quốc gia (kiểm định cấp III).
Bảng 15.14- Số lượng mẫu được lấy cho thử nghiệm vô khuẩn vắc xin, sinh phẩm
| Số lượng thành phẩm /loạt | Thể tích được đóng trong 1 ống hoặc lọ vắc xin, sinh phẩm | |||||||||||||||||
| 0,5 ml | 1 ml đến 1,5 ml | ≥ 2 ml | ||||||||||||||||
| Tổng số | SX | KĐĐP | Tổng số | SX | KĐĐP | Tổng số | SX | KĐĐP | ||||||||||
| M | Ô | M | Ô | M | Ô | M | Ô | M | Ô | M | Ô | M | Ô | M | Ô | M | Ô | |
| Từ 501 đến 1000 | 16 | 16 | 2 | 8 | 2 | 8 | 6 | 12 | 3 | 6 | 3 | 6 | 12 | 12 | 6 | 6 | 6 | 6 |
| Từ 1001 đến 2000 | 24 | 24 | 3 | 12 | 3 | 12 | 8 | 16 | 4 | 8 | 4 | 8 | 18 | 18 | 9 | 9 | 9 | 9 |
| Từ 2001 đến 3000 | 24 | 24 | 3 | 12 | 3 | 12 | 10 | 20 | 4 | 10 | 5 | 10 | 22 | 22 | 11 | 11 | 11 | 11 |
| Từ 3001 đến 4000 | 24 | 24 | 3 | 12 | 3 | 12 | 12 | 24 | 6 | 12 | 6 | 12 | 26 | 26 | 13 | 13 | 13 | 13 |
| Từ 4001 đến 5000 | 32 | 32 | 4 | 16 | 4 | 16 | 14 | 28 | 7 | 14 | 7 | 14 | 28 | 28 | 14 | 14 | 14 | 14 |
| Từ 5001 đến 6000 | 32 | 32 | 4 | 16 | 4 | 16 | 16 | 32 | 8 | 16 | 8 | 16 | 32 | 32 | 16 | 16 | 16 | 16 |
| Từ 6001 đến 7000 | 32 | 32 | 4 | 16 | 4 | 16 | 16 | 32 | 8 | 16 | 8 | 16 | 34 | 34 | 17 | 17 | 17 | 17 |
| Từ 7001 đến 8000 | 40 | 40 | 5 | 20 | 5 | 20 | 18 | 36 | 9 | 18 | 9 | 18 | 36 | 36 | 18 | 18 | 18 | 18 |
| Từ 8001 đến 9000 | 40 | 40 | 5 | 20 | 5 | 20 | 20 | 40 | 10 | 20 | 10 | 20 | 38 | 38 | 19 | 19 | 19 | 19 |
| Từ 9001 đến 10000 | 40 | 40 | 5 | 20 | 5 | 20 | 20 | 40 | 10 | 20 | 10 | 20 | 40 | 40 | 20 | 20 | 20 | 20 |
| > 10.000 | 40 | 40 | 5 | 20 | 5 | 20 | 20 | 40 | 10 | 20 | 10 | 20 | 40 | 40 | 20 | 20 | 20 | 20 |
Ghi chú:
M: Số mẫu được lấy để kiểm định.
Ô: Số lượng lọ vắc xin, sinh phẩm lấy để kiểm tra vô khuẩn.
SX: Kiểm định sản xuất (cấp I).
KĐĐP: Kiểm định địa phương (cấp II).
Số lượng mẫu lấy để kiểm tra vô khuẩn của sản phẩm tối thiểu phải đủ cho kiểm định sản xuất, kiểm định địa phương và lưu mẫu. Nếu Kiểm định Quốc gia cần kiểm tra lại thử nghiệm vô khuẩn thì số lượng mẫu thử bằng số lượng mẫu mà kiểm định địa phương đã làm.
Trường hợp cần hủy một loạt sinh phẩm do không đạt tiêu chuẩn lý học (đục, tủa) thì toàn bộ số ống đó phải chuyển qua kiểm định địa phương để tìm nguyên nhân.
Số lượng thành phẩm cần lấy để kiểm tra vô khuẩn sẽ tùy thuộc vào tổng số ống của mỗi loạt và thể tích sinh phẩm có trong 1 ống hoặc lọ (Bảng 15.14).
Xử lý mẫu
Mẫu kiểm định phải bảo quản theo những quy định phù hợp cho từng loại sinh phẩm và phải được tiến hành kiểm định ngay. Nếu chưa kiểm định được, nhất thiết phải bảo quản ở nhiệt độ quy định.
Các mẫu kiểm định phải có thông tin như sau:
|
| Tên sản phẩm; Ngày sản xuất; Số loạt; Số lượng sản phẩm; Yêu cầu kiểm định; Số lượng mẫu gửi kiểm định; Ngày lấy mẫu; Đơn vị gửi mẫu; Hạn dùng; Dạng đóng gói; Nhà sản xuất; Điều kiện bảo quản; Số đăng kí. |
PHƯƠNG PHÁP LƯU MẪU
Tại cơ quan kiểm định quốc gia chỉ lưu mẫu sinh phẩm ở dạng thành phẩm cuối cùng, mẫu lưu phải được bảo quản ít nhất đến khi hết hạn sử dụng.
Mẫu lưu do phòng kiểm định cấp 2 giữ tại các cơ sở sản xuất. Các mẫu kiểm định cấp 2 được lấy cùng thời điểm với mẫu gửi cho cơ quan Kiểm định Quốc gia. Mẫu gửi Kiểm định Quốc gia cần bảo quản đúng nhiệt độ quy định trong suốt quá trình vận chuyển (trong trường hợp đơn vị sản xuất ở cách xa cơ quan kiểm định).
Các mẫu lưu của từng sinh phẩm phải được đóng gói cẩn thận, gắn xi (hoặc có băng bảo đảm) ngoài bao bì phải ghi rõ các thông tin sau:
|
| Tên sinh phẩm; Số loạt; Số lượng lưu mẫu; Ngày lưu mẫu; Nhiệt độ bảo quản trong thời gian lưu mẫu; Thời gian lưu mẫu tùy thuộc vào từng loại sinh phẩm và hạn dùng của sinh phẩm đó. |
15.15 XÁC ĐỊNH HIỆU GIÁ HUYẾT THANH KHÁNG ĐỘC TỐ BẠCH HẦU
Hiệu giá huyết thanh kháng độc tố bạch hầu được xác định bằng cách so sánh khả năng trung hòa một lượng cố định độc tố bạch hầu của kháng độc tố bạch hầu thử nghiệm và kháng độc tố bạch hầu chuẩn thông qua thử nghiệm trung hòa độc tố trên chuột lang hoặc trong da thỏ.
Thử nghiệm trung hòa độc tố bạch hầu trên chuột lang
Xác định liều độc tố bạch hầu thử nghiệm (L+)
L+ là lượng độc tố bạch hầu nhỏ nhất còn lại sau khi trung hòa với 1 đơn vị quốc tế (IU) của kháng độc tố bạch hầu chuẩn đủ để gây chết 1 chuột lang có trọng lượng đã xác định trong thời gian từ 4 ngày đến 6 ngày.
Pha kháng độc tố bạch hầu chuẩn với nước muối sinh lý (0,85 %) để có dung dịch kháng độc tố chứa 3,0 lU/ml. Pha độc tố bạch hầu với dung dịch Jensen pepton để có dung dịch độc tố chứa 3 Lf/ml đến 4 Lf/ml.
Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống: 1,0 ml dung dịch kháng độc tố bạch hầu chuẩn chứa 3.0 lU/ml, một thể tích thay đổi dung dịch độc tố bạch hầu (ví dụ 0,6 ml; 0,7 ml; 0,8 ml; 0,9 ml; 1,0 ml;... tùy theo độc tính của độc tố bạch hầu) và nước muối sinh lý để vừa đủ 6,0 ml trong mỗi ống nghiệm. Lắc đều các ống, để yên ở 37 °C trong 60 min, tránh ánh sáng.
Tiêm 2,0 ml vào dưới đa đùi cho mỗi chuột lang cân nặng từ 250 g đến 300 g, dùng 2 chuột cho mỗi độ pha.
Theo dõi và ghi chép số chuột bị chết trong thời gian từ 4 ngày đến 6 ngày.
Cách xác định: Hỗn dịch nào có lượng độc tố bạch hầu nhỏ nhất sau khi tiêm gây chết 100 % chuột trong vòng 4 ngày đến 6 ngày sẽ có chứa lượng độc tố 3 L+/6ml hay 1 L+// 2ml (một liều tiêm/1 chuột lang).
Xác định hiệu giá huyết thanh kháng độc tố bạch hầu
Là xác định số đơn vị quốc tế kháng độc tố bạch hầu có trong 1,0 ml của huyết thanh kháng độc tố bạch hầu thử nghiệm.
Pha huyết thanh kháng độc tố bạch hầu chuẩn với nước muối sinh lý để có dung dịch chứa 3,0 đơn vị quốc tế /ml.
Pha độc tố bạch hầu với dung dịch Jensen pepton để có dung dịch độc tố chứa 3L+ trong một thể tích đã xác định.
Pha huyết thanh kháng độc tố bạch hầu thử nghiệm với nước muối sinh lý để có các độ pha 1/300; 1/400; 1/500; 1/600; 1/700; vv... tùy theo hiệu giá có thể có được của kháng độc tố bạch hầu thử nghiệm. Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống: Một lượng dung dịch độc tố bạch hầu chứa 3L+; 3.0 ml dung dịch kháng độc tố bạch hầu thử nghiệm ở mỗi độ pha loãng và nước muối sinh lý để vừa đủ 6.0 ml trong mỗi ống nghiệm.
Pha mẫu chứng: Cho vào 3 ống nghiệm lần lượt: Một lượng dung dịch độc tố bạch hầu có chứa 3 L+, một trong 3 lượng 0,9 ml; 1,0 ml; 1,1 ml dung dịch kháng độc tố bạch hầu chuẩn chứa 3,0 lU/ml và sau đó một lượng nước muối sinh lý để vừa đủ 6,0 ml trong mỗi ống nghiệm.
Lắc đều các ống, để yên ở 37 °C trong 60 min, tránh ánh sáng.
Tiêm 2,0 ml vào dưới da đùi cho mỗi chuột nhắt, dùng 2 chuột cho mỗi độ pha.
Theo dõi và ghi chép số chuột bị chết trong thời gian từ 4 ngày đến 6 ngày.
Cách tính hiệu giá
Thử nghiệm chỉ có giá trị khi mẫu chứng có chứa 0,9 ml và 1,0 ml dung dịch kháng độc tố bạch hầu chuẩn gây chết 100 % chuột trong vòng 4 ngày đến 6 ngày và mẫu chứng có chứa 1,1 ml dung dịch kháng độc tố bạch hầu chuẩn không được gây chết chuột trong thời gian theo dõi.
Dung dịch nào có chứa lượng kháng độc tố bạch hầu thử nghiệm lớn nhất không bảo vệ được chuột trong thời gian 4 ngày đến 6 ngày giống như mẫu chứng có chứa 1,0 ml dung dịch kháng độc tố bạch hầu chuẩn sẽ có chứa 1,0 lU/ml.
Hiệu giá của huyết thanh kháng độc tố bạch hầu được tính như sau:
Hiệu giá (lU/ml) = 1,0 x N trong đó:
N: số lần pha loãng kháng độc tố bạch hầu thử nghiệm;
1,0: số IU có trong dung dịch kháng độc tố bạch hầu chuẩn dùng để trung hòa 1L+ trong thử nghiệm.
Thử nghiệm trung hòa độc tố bạch hầu trong da thỏ
Xác định liều độc tố bạch hầu thử nghiệm (L+/30)
L+/30 là lượng độc tố bạch hầu nhỏ nhất còn lại sau khi trung hòa với 1/30 đơn vị quốc tế (IU) của kháng độc tố bạch hầu chuẩn đủ để gây phản ứng Shick trên da thỏ trong 48 h.
Pha kháng độc tố bạch hầu chuẩn với nước muối sinh lý để có dung dịch kháng độc tố chứa 1,0 lU/ml. Pha độc tố bạch hầu với dung dịch Jensen pepton để có dung dịch độc tố chứa khoảng 1,0 Lf/ml.
Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống: 1,0 ml dung dịch kháng độc tố bạch hầu chuẩn chứa 1,0 lU/ml, một thể tích thay đổi dung dịch độc tố bạch hầu (ví dụ 0,2 ml; 0,3 ml; 0,4 ml; 0,5 ml; 0,6 ml;... tùy theo độc tính của độc tố bạch hầu) và nước muối sinh lý để vừa đủ 3,0 ml trong mỗi ống nghiệm.
Lắc đều các ống nghiệm và để yên ở 37 °C trong 60 min, tránh ánh sáng.
Tiêm 0,1 ml trong da thỏ, loại có cân nặng từ 2,5 kg/con đến 3,0 kg/con.
Theo dõi thỏ thử nghiệm và đo đường kính quầng đỏ trong vòng 48 h.
Cách xác định: Độ pha nào của độc tố bạch hầu tạo phản ứng Shick với đường kính từ 12 mm đến 15 mm sẽ có chứa 30 L+/30 trong 3,0 ml hay L+/30 trong 0,1 ml.
Xác định hiệu giá huyết thanh kháng độc tố bạch hầu
Pha huyết thanh kháng độc tố bạch hầu chuẩn với nước muối sinh lý để có dung dịch chứa 1,0 đơn vị quốc tế /ml.
Pha độc tố bạch hầu với dung dịch Jensen pepton để có dung dịch độc tố chứa 30 L+/30 trong một thể tích đã xác định.
Pha huyết thanh kháng độc tố bạch hầu thử nghiệm với nước muối sinh lý để có các độ pha 1/300; 1/400; 1/500; 1/600; 1/700;... tùy theo hiệu giá có thể có được của kháng độc tố bạch hầu thử nghiệm. Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống. Một lượng dung dịch độc tố bạch hầu chứa 30 L+/30 đã xác định; 1,0 ml dung dịch kháng độc tố bạch hầu thử nghiệm ở mỗi độ pha loãng và nước muối sinh lý để vừa đủ 3,0 ml trong mỗi ống nghiệm.
Pha mẫu chứng: Cho vào 3 ống nghiệm lần lượt: Một lượng dung dịch độc tố bạch hầu chứa 30 L+/30, một trong 3 lượng 0,9 ml; 1,0 ml; 1,1 ml dung dịch kháng độc tố bạch hầu chuẩn chứa 1,0 lU/ml và sau đó một lượng nước muối sinh lý để vừa đủ 3,0 ml trong mỗi ống nghiệm.
Lắc đều các ống, để yên ở 37 °C trong 60 min, tránh ánh sáng.
Tiêm 0,1 ml trong da thỏ, loại có cân nặng từ 2,50 kg/con đến 3,0 kg/con, dùng 3 thỏ cho mỗi thử nghiệm. Theo dõi thỏ thử nghiệm và đo đường kính quầng đỏ trong vòng 48 h.
Cách tính hiệu giá
Thử nghiệm chỉ có giá trị khi mẫu chứng chứa 1,0 ml dung dịch kháng độc tố bạch hầu chuẩn tạo phản ứng Shick với đường kính từ 12 mm đến 15 mm. Hỗn dịch nào chứa lượng kháng độc tố bạch hầu thử nghiệm lớn nhất tạo phản ứng Shick với đường kính 12 mm đến 15 mm tương đương với mẫu chứng chứa 1,0 ml kháng độc tố bạch hầu chuẩn sẽ có chứa 1 lU/ml.
Hiệu giá của huyết thanh kháng độc tố bạch hầu thử nghiệm được tính như sau:
Hiệu giá (lU/ml) = 1,0 x N
trong đó:
N: số lần pha loãng kháng độc tố bạch hầu thử nghiệm;
1,0: số IU có trong dung dịch kháng độc tố bạch hầu chuẩn dùng để trung hòa 30 L+/30 trong thử nghiệm
15.16 XÁC ĐỊNH HIỆU GIÁ HUYẾT THANH KHÁNG ĐỘC TỐ UỐN VÁN
Hiệu giá huyết thanh kháng độc tố uốn ván được xác định bằng cách so sánh mức độ bảo vệ giữa liều kháng độc tố uốn ván thử nghiệm và kháng độc tố uốn ván chuẩn đối với chuột nhắt trắng có cân nặng từ 16 g đến 18 g sau khi đã trung hòa một lượng cố định độc tố uốn ván.
Xác định liều độc tố uốn ván thử nghiệm (L+/10)
L+/10 là lượng độc tố uốn ván nhỏ nhất còn lại sau khi trung hòa với 0,1 đơn vị quốc tế (IU) của kháng độc tố uốn ván chuẩn đủ để gây chết 1 chuột có trọng lượng đã xác định trong thời gian 96 h.
Pha kháng độc tố uốn ván chuẩn với nước muối sinh lý để có dung dịch kháng độc tố uốn ván chứa 0,5 lU/ml.
Pha độc tố uốn ván với dung dịch Jensen pepton để có dung dịch độc tố uốn ván 3 Lf/ml đến 5 Lf/ml.
Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống: 1,0 ml dung dịch kháng độc tố uốn ván chuẩn chứa 0,5 lU/ml, một thể tích thay đổi dung dịch độc tố uốn ván (ví dụ 0,1 ml; 0,2 ml; 0,3 ml; 0,4 ml; 0,5 ml... tùy theo độc tính của độc tố uốn ván) và nước muối sinh lý để vừa đủ 2,5 ml trong mỗi ống nghiệm.
Lắc đều các ống nghiệm và để yên ở 37 °C trong 45 min, tránh ánh sáng.
Tiêm 0,5 ml dưới da lưng cho mỗi chuột nhắt, dùng 4 chuột cho mỗi độ pha.
Theo dõi và ghi chép số chuột bị chết trong 96 h.
Cách xác định: Hỗn dịch nào có lượng độc tố thấp nhất sau khi tiêm gây chết chuột trong vòng 96 h sẽ có chứa lượng độc tố 5 L+/10 trong 2,5 ml hay 1 L+/10 trong 0,5 ml (một liều tiêm/1 chuột).
Xác định hiệu giá huyết thanh kháng độc tố uốn ván
Hiệu giá huyết thanh kháng độc tố uốn ván là số đơn vị quốc tế (IU) kháng độc tố uốn ván có trong 1,0 ml huyết thanh thử nghiệm.
Pha huyết thanh kháng độc tố uốn ván chuẩn với nước muối sinh lý để có dung dịch chứa 0,5 IU /ml.
Pha độc tố uốn ván với dung dịch Jensen pepton để có dung dịch độc tố uốn ván chứa 5 L+10 trong 1 thể tích xác định.
Pha huyết thanh kháng độc tố uốn ván thử nghiệm với nước muối sinh lý để có các độ pha 1/2600; 1/2800; 1/3000; 1/3200; 1/3400;... tùy theo hiệu giá có thể có được của kháng độc tố uốn ván thử nghiệm.
Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống: 1 thể tích dung dịch độc tố uốn ván chứa 5L+10; 1,0 ml dung dịch kháng độc tố uốn ván thử nghiệm ở mỗi độ pha loãng và nước muối sinh lý để vừa đủ 2,5 ml trong mỗi ống nghiệm.
Pha mẫu chứng: Cho vào 3 ống nghiệm lần lượt: Một thể tích dung dịch độc tố uốn ván chứa 5 L+10, một trong 3 lượng 0,9 ml; 1,0 ml; 1,1 ml dung dịch kháng độc tố uốn ván chuẩn chứa 0,5 lU/ml và sau đó một lượng nước muối sinh lý để vừa đủ 2,5 ml trong mỗi ống nghiệm.
Lắc đều các ống nghiệm và để yên ở 37 °C trong 45 min, tránh ánh sáng.
Tiêm 0,5 ml dưới da lưng cho mỗi chuột nhắt, dùng 4 chuột cho mỗi độ pha.
Theo dõi và ghi chép số chuột bị chết trong 96 h.
Cách tính hiệu giá
Thử nghiệm chỉ có giá trị khi mẫu chứng có chứa 0,9 ml và 1,0 ml dung dịch kháng độc tố uốn ván chuẩn gây chết 100 % chuột trong vòng 96 h và mẫu có chứa 1,1 ml dung dịch kháng độc tố uốn ván chuẩn không được gây chết chuột trong thời gian theo dõi.
Dung dịch nào có chứa lượng kháng độc tố uốn ván thử nghiệm lớn nhất không bảo vệ được chuột trong thời gian 96 h giống như mẫu chứng có chứa 1,0 ml dung dịch kháng độc tố uốn ván chuẩn sẽ có chứa 0,5 lU/ml.
Hiệu giá của huyết thanh kháng độc tố uốn ván được tính như sau:
Hiệu giá (lU/ml) = 0,5 x N
trong đó:
N: số lần pha loãng kháng độc tố uốn ván thử nghiệm;
0,5: số IU có trong 1 ml dung dịch kháng độc tố uốn ván chuẩn dùng trong thử nghiệm.
15.17 XÁC ĐỊNH HIỆU GIÁ HUYẾT THANH KHÁNG DẠI
Hiệu giá huyết thanh kháng dại được xác định dựa trên nguyên lý của phản ứng trung hòa in vivo của một liều cố định virus dại thử thách với các độ pha loãng khác nhau của huyết thanh kháng dại.
Xác định hiệu giá của virus thử thách
Chủng virus dại thử thách được dùng trong thử nghiệm xác định hiệu giá của huyết thanh kháng dại là chủng CVS (Challenge virus strain), được giữ ở dạng hỗn dịch 20 % não chuột, bảo quản ở nhiệt độ -70 °C.
Làm tan băng nhanh ống đựng chủng virus dại thử thách dưới vòi nước chảy.
Pha loãng bậc 10 với dung dịch huyết thanh ngựa thường 2 % (đã được bất hoạt 30 min ở 56 °C) để có nồng độ từ 2 x 10-2, 2 x 10-3.... đến 2 x 10-9.
Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống: 0,5 ml hỗn dịch virus của mỗi độ pha loãng, từ 2 x 10-4 đến 2 x 10-9 và 0,5 ml dung dịch huyết thanh ngựa thường 2 %.
Lắc đều các ống, để yên ở 37 °C trong 90 min.
Tiêm 0,03 ml vào não của mỗi chuột nhắt, dùng 6 chuột cho mỗi độ pha, cân nặng mỗi chuột từ 14 g đến 16 g. Các ống chủng được giữ trong nước đá trong suốt quá trình tiêm.
Theo dõi và ghi chép số chuột bị chết từ ngày thứ 6 đến ngày thứ 20 sau khi tiêm.
Tính LD50 theo Spearman - Kaber.
Xác định hiệu giá huyết thanh kháng dại
Pha loãng huyết thanh kháng dại chuẩn quốc tế, huyết thanh kháng dại thử nghiệm và trung hòa với virus thử thách:
Pha huyết thanh kháng dại chuẩn quốc tế với nước muối sinh lý để có dung dịch huyết thanh kháng dại chuẩn chứa 10 đơn vị quốc tế (IU)/ml.
Pha huyết thanh kháng dại thử nghiệm với nước muối sinh lý để có dung dịch huyết thanh kháng dại thử nghiệm chứa khoảng 10 IU/ml.
Pha loãng bậc 5 hai dung dịch trên với dung dịch huyết thanh ngựa thường 2 % để có các độ pha 1/5; 1/25; 1/125; 1/625 và 1/3125.
Pha hỗn dịch virus thử thách đã biết hiệu giá để có 200 LD50/0,03 ml.
Trong một dãy các ống nghiệm, lần lượt cho vào mỗi ống: 0,5 ml huyết thanh kháng dại ở mỗi độ pha loãng và 0,5 ml hỗn dịch virus thử thách có chứa 200 LD50/0,03 ml.
Lắc đều các ống nghiệm và để yên ở 37 °C trong 90 min.
Giữ các ống trong nước đá trong suốt quá trình tiêm.
Tiêm 0,03 ml vào não của mỗi chuột nhắt, dùng 6 chuột có cân nặng từ 14 g đến 16 g cho mỗi độ pha của kháng huyết thanh.
Theo dõi và ghi chép số chuột bị chết từ ngày thứ 6 đến ngày thứ 20 sau khi tiêm.
Xác định số LD50 của chủng virus dại trong thử nghiệm (nhóm chứng):
Từ hỗn dịch virus pha để trung hòa có chứa 200 LD50/0,03 ml đã nêu ở trên, được xem là 10° để từ đó pha loãng bậc 10 thành 3 độ pha 10-1; 10-2 và 10-3.
Trong một dãy ống nghiệm, lần lượt cho vào mỗi ống: 0,5 ml hỗn dịch virus từ mỗi độ pha và 0,5 ml dung dịch huyết thanh ngựa thường 2 %.
Lắc đều các ống nghiệm và để yên ở 37 °C trong 90 min.
Giữ các ống trong nước đá.
Tiêm 0,03 ml vào não của mỗi chuột nhắt, dùng 6 chuột có trọng lượng 14 g đến 16 g cho mỗi độ pha của hỗn dịch virus dại.
Theo dõi và ghi chép số chuột bị chết từ ngày thứ 6 đến ngày thứ 20 sau khi tiêm.
Tính LD50 theo bảng Spearman - Kaber.
Tính kết quả
Thử nghiệm chỉ có giá trị khi số LD50 để trung hòa huyết thanh kháng dại là 100 LD50 đến 300 LD50 trong 0,03 ml.
Tính ED50 của huyết thanh kháng dại theo phương pháp Spearman - Kaber.
Hiệu giá của huyết thanh kháng dại được tính ra đơn vị quốc tế trong 1 ml và bằng:
Anti log (ED50 huyết thanh chuẩn quốc tế: ED50 huyết thanh thử nghiệm) x 2 x N trong đó:
2: số IU có trong 1 ml huyết thanh kháng dại chuẩn quốc tế dùng trong thử nghiệm;
N: số lần pha loãng huyết thanh kháng dại thử nghiệm.
15.18 XÁC ĐỊNH HÀM LƯỢNG NITƠ TOÀN PHẦN CỦA VẮC XIN VÀ SINH PHẨM BẰNG THUỐC THỬ NESSLER
Nguyên lý
Phương pháp dựa vào tính chất của thuốc thử Nessler cho phản ứng màu với ion amoni (NH4+) được tạo thành sau khi vô cơ hóa các chất protein.
Phương pháp tiến hành
a. Chuẩn bị mẫu thử
Cho 0,2 ml chế phẩm và 0,2 ml acid sulfuric (TT) vào một ống thủy tinh và đem đốt trên bếp cát. Để tăng tốc độ phản ứng, thỉnh thoảng cho vài giọt nước oxy già đậm đặc (TT) vào ống đốt sau khi làm nguội. Khi chế phẩm đã được vô cơ hóa hoàn toàn, dung dịch mất màu, cho nước cất vừa đủ b ml thu được dung dịch A.
Lấy 1 ml dung dịch A vào ống nghiệm, thêm 8,5 ml nước cất và 0,5 ml thuốc thử Nessler, được mẫu thử.
b. Chuẩn bị mẫu trắng: 9,5 ml nước cất và 0,5 ml thuốc thử Nessler (TT).
c. Dựng đường chuẩn
Hút dung dịch amoni sulfat chuẩn có hàm lượng nitơ 0,05 mg/ml vào các ống nghiệm theo thứ tự sau: 0,1 ml; 0,2 ml; 0,3 ml; 0,4 ml; 0,5 ml; 0,6 ml và 0,7 ml. Cho nước cất vừa đủ 9,5 ml và 0,5 ml thuốc thử Nessler (TT).
Lắc đều các ống nghiệm.
Đo quang phổ ở bước sóng 420 nm.
Vẽ đường cong chuẩn.
Lượng nitơ toàn phần trong chế phẩm được tính theo công thức sau:
|
|
|
|
trong đó:
a: lượng Nitơ tìm được trên đường chuẩn (mg);
b: thể tích dung dịch A (ml);
6,25: hệ số chuyển đổi nitơ thành protein;
100: chuyển đổi thành phần trăm;
1,0: lượng mẫu thử sau pha loãng đem so màu (ml);
0,2: lượng mẫu thử đem vô cơ hóa (ml);
1000: Hệ số chuyển đổi g thành mg.
Dung dịch amoni sulfat chuẩn có hàm lượng nitơ 0,05 mg/ml: Cân 0,2375 g amoni sulfat (đã được sấy khô trong bình hút ẩm bằng silica gel đến trọng lượng không đổi), hòa tan vào vừa đủ 500 ml nước cất. Dung dịch vừa pha có hàm lượng nitơ là 0,1 mg/ml, trước khi dùng pha loãng 2 lần bằng nước cất.
Tiêu chuẩn cho phép
Hàm lượng nitrơ toàn phần trong vắc xin và sinh phẩm tùy theo yêu cầu đối với từng loại mẫu thử (xem trong phần các vắc xin và sinh phẩm cụ thể).
15.19 THỬ NGHIỆM NHẬN DẠNG THÀNH PHẦN BẠCH HẦU - UỐN VÁN - HO GÀ TRONG VẮC XIN DTwP HẤP PHỤ
Các thành phần bạch hầu, uốn ván, ho gà toàn tế bào trong vắc xin DTwP hấp phụ được tiến hành kiểm tra nhận dạng theo các bước như sau:
Giải hấp phụ
Vắc xin Bạch hầu - Uốn ván - Ho gà (toàn tế bào) hấp phụ được tách gel bằng cách cho thêm natri citrat với nồng độ 5 % ủ ở 37 °C trong 48 h. Sau đó ly tâm 2000 r/min trong 15 min. Hoặc ly tâm vắc xin ở 3000 r/min. Cho dung dịch EDTA-natri phosphat vào phần kết tủa sau ly tâm với nồng độ 20 %. Đánh đều tạo dung dịch đồng nhất. Ủ ở 37 °C trong 18 h. Ly tâm 3000 r/min trong 10 min. Nước nổi được dùng làm dung dịch kiểm tra. Nước nổi được dùng để nhận dạng thành phần bạch hầu và uốn ván bằng phản ứng lên bông, cặn ly tâm dùng để nhận dạng thành phần ho gà có trong vắc xin bằng phản ứng ngưng kết trên phiến kính với các huyết thanh kháng ho gà đặc hiệu.
Nhận dạng thành phần bạch hầu và uốn ván
Phương pháp lên bông
Phương pháp tiến hành (phản ứng lên bông).
Dùng 5 ống nghiệm thủy tinh có đường kính 10 mm, đánh số thứ tự từ 1 đến 5 để làm thử nghiệm kiểm tra sự có mặt của thành phần bạch hầu.
Dùng 5 ống nghiệm thủy tinh khác có đường kính 10 mm, đánh số thứ tự từ 6 đến 10 để làm thử nghiệm kiểm tra sự có mặt của thành phần uốn ván.
Huyết thanh chuẩn kháng độc tố bạch hầu hoặc uốn ván được pha loãng đến nồng độ 20 lU/ml.
Cho vào dãy ống nghiệm từ 1 đến 5 lượng huyết thanh chuẩn kháng độc tố bạch hầu tăng dần đều trong khoảng tương ứng với lượng kháng nguyên giải độc tố bạch hầu có trong nước nổi vắc xin. Tương tự, cho vào dãy ống nghiệm từ 6 đến 10 lượng huyết thanh chuẩn kháng độc tố uốn ván tăng dần đều trong khoảng tương ứng với lượng kháng nguyên giải độc tố uốn ván có trong nước nổi vắc xin. Thêm nước muối sinh lý vào các ống cho đủ 1 ml.
Cho vào mỗi ống nghiệm 1 ml nước nổi.
Đậy các ống nghiệm bằng giấy parafin. Lắc đều và đặt vào nồi cách thủy ở nhiệt độ 45 °C. Quan sát liên tục dưới ánh đèn, ghi nhận thời gian của ống nghiệm đầu tiên xuất hiện sự lên bông và khoảng thời gian lên bông (Kf).
Đánh giá kết quả
Hàm lượng Lf/ml của thành phần bạch hầu hoặc uốn ván tính theo công thức;
|
|
|
|
trong đó:
Vc là thể tích huyết thanh chuẩn kháng độc tố ở ống đầu tiên có sự lên bông (ml).
a là số đơn vị huyết thanh chuẩn kháng độc tố (lU/ml).
b là số ml nước nổi.
Hiện tượng lên bông xảy ra chỉ chứng tỏ trong mẫu thử nghiệm có chứa giải độc tố bạch hầu hoặc uốn ván chứ không phản ánh công hiệu của giải độc tố.
Ví dụ
Lượng huyết thanh chuẩn kháng độc tố bạch hầu (20 lU/ml) và lượng huyết thanh chuẩn kháng độc tố uốn ván (20 IU/ml) được cho vào các ống như sơ đồ sau:
| Ống số | Thể tích huyết thanh (ml) | Ống số | Thể tích (ml) |
| 1 | 0,3 | 6 | 0,16 |
| 2 | 0,4 | 7 | 0,18 |
| 3 | 0,5 | 8 | 0,20 |
| 4 | 0,6 | 9 | 0,22 |
| 5 | 0,7 | 10 | 0,24 |
Ống số 3 lên bông đầu tiên tức hàm lượng Lf/ml của thành phần bạch hầu có trong 1 ml vắc xin thử là: (0,5 ml huyết thanh x 20 IU / ml)/1 ml = 10 Lf/ ml
Tương tự, nếu thấy ống số 8 lên bông đầu tiên có nghĩa là hàm lượng Lf/ml của thành phần uốn ván có trong 1 ml vắc xin thử là 4 Lf/ ml.
Hiện tượng lên bông xảy ra ở ví dụ này chứng tỏ trong mẫu thử nghiệm có chứa giải độc tố bạch hầu hoặc uốn ván chứ không phản ánh công hiệu của giải độc tố.
Phương pháp khuyếch tán miễn dịch kép trên thạch - Ouchterlony
Chuẩn bị các dung dịch
Dung dịch 1: Dung dịch đệm Helena buffer
Cân 18 g đệm Helena Buffer, hòa tan trong nước cất 3 lần, pha loãng vừa đủ 1500 ml với cùng dung môi.
Dung dịch 2: Dung dịch EDTA 0,15 M, được pha như sau: Cân 2,8 g EDTA, hòa tan trong 40 ml nước cất, sau đó điều chỉnh pH về 8,0± 0,3 bằng dung dịch natri hydroxyd (TT), thêm vừa đủ 50 ml dung dịch bằng nước cất. Sử dụng trong vòng 1 tháng kể từ ngày pha.
Dung dịch 3: Dung dịch dinatri hydro phosphat 0,25 M, được pha như sau: Cân 4,45 g dinatri hydro phosphat (TT), hòa tan hoàn toàn trong 80 ml nước cất, thêm nước cất vừa đủ 100 ml. Sử dụng trong vòng 1 tháng kể từ ngày pha.
Dung dịch 4: Hỗn hợp bao gồm 0,1 ml dung dịch EDTA và 4,9 ml dung dịch dinatri phosphat.
Dung dịch 5: Pha loãng dung dịch kháng huyết thanh uốn ván (NIBSC, 66/21, 1400 lU/ml) trong 14 ml nước cất pha tiêm để tạo thành 100 lU/ống, lấy ra mỗi tuýp ly tâm 500 μl, sau đó bảo quản ở nhiệt độ dưới -20 °C.
Dung dịch 6: Pha loãng giải độc tố uốn ván chuẩn (NIBSC, TEFT, 1000 Lf/ml) trong 10 ml nước cất pha tiêm để tạo thành 100 lU/ống, lấy ra mỗi túyp ly tâm 500 µl, sau đó bảo quản ở nhiệt độ dưới -20 °C.
Giải hấp phụ
Ly tâm 5 ml vắc xin cần kiểm tra ở 3000 r/min trong 10 min.
Lấy 1 ml Dung dịch 4 cho vào phần kết tủa sau khi ly tâm, trộn đều (bằng voltex) để tạo được dung dịch đồng nhất. Để dung dịch thu được trong tủ ấm ở (37±1) °C ít nhất 18 h. Tiếp tục ly tâm dung dịch ở 3000 r/min trong 10 min. Chuyển phần dịch nổi sang 1 ống sạch làm dung dịch kiểm tra.
Tiến hành
Làm tan hoàn toàn (đến khi dung dịch trong suốt) dung dịch agarose 1 % trong Dung dịch 1 bằng lò vi sóng hoặc đun cách thủy, để nguội đến (56 ± 1) °C.
Dùng pipet 25 ml chuyển 18 ml dung dịch agarose sang bản phim gelbond đã được cố định bằng khuôn thạch.
Để mẫu cố định trong 15 min (có thể đặt vào tủ lạnh nếu cần).
Đục bản gel thành các giếng có đường kính 4 mm, như chỉ dẫn ở hình sau:

Nhỏ 10 µl antitoxin/antiserum Dung dịch 5 vào giếng 5; 6; 7; 8.
Nhỏ 20 µl Dung dịch kiểm tra (mẫu thử) vào giếng 1; 2; 3 và 10 µl vào giếng 9; 10; 11.
Nhỏ 10µl Dung dịch 6 (chứng dương) vào giếng 4, 10µl chứng âm (dd 7) vào giếng 12.
Đặt bản gel vào buồng ẩm bão hòa hơi nước trong 48 h.
Làm khô hoàn toàn bản gel (cho đến khi bản gel trong suốt) bằng giấy thấm nước.
Nhuộm màu:
Ngâm bản gel trong dung dịch Coomassie stain solution (1X) 10 min, trong quá trình ngâm lắc nhẹ bản gel bằng máy lắc ngang.
Tẩy màu:
Chuyển bản gel vào bình chứa dung dịch Destain solution Coomassie R- 250 (1X). Lắc nhẹ bản gel trong 5 min.
Rửa lại bản gel bằng nước cất.
Để khô, đọc kết quả.
Đánh giá kết quả
Đọc kết quả:
Quan sát đường tủa màu xanh của kháng thể uốn ván chuẩn với giải độc tố uốn ván trong vắc xin tại vị trí Dung dịch kiểm tra (trong khoảng giữa giếng 1 và 9 với giếng 5, khoảng giữa giếng 2 và 10 với giếng 6, khoảng giữa giếng 3 và 11 với giếng 7).
Tiêu chuẩn đánh giá:
• Thử nghiệm có giá trị khi tuân thủ đúng quy trình (các yếu tố, điều kiện của quá trình thực hiện là đảm bảo không ảnh hưởng đến kết quả thí nghiệm).
• Tại chứng dương, xuất hiện đường tủa của kháng thể uốn ván chuẩn với giải độc tố uốn ván chuẩn.
• Tại chứng âm, không xuất hiện đường tủa.
• Thử nghiệm được coi là có giá trị khi thỏa mãn cả 3 tiêu chuẩn trên. Nếu không thỏa mãn 1 trong 3 tiêu chuẩn, thử nghiệm được coi là không có giá trị.
Đánh giá:
Vắc xin được coi là đạt tiêu chuẩn khi tại vị trí kiểm tra dung dịch (trong khoảng giữa giếng 1 và 9 với giếng 5, khoảng giữa giếng 2 và 10 với giếng 6, khoảng giữa giếng 3 và 11 với giếng 7) xuất hiện đường tủa màu xanh của kháng thể uốn ván chuẩn với giải độc tố uốn ván trong vắc xin.
Nếu kết quả không đạt mà thử nghiệm không có giá trị, lặp lại thử nghiệm như lần đầu.
Nếu kết quả không đạt mà thử nghiệm có giá trị thì lặp lại thử nghiệm với số lượng mẫu gấp đôi lần đầu. Ở lần lặp lại, vắc xin được coi là đạt tiêu chuẩn khi tại vị trí kiểm tra dung dịch (trong khoảng giữa giếng 1 và 9 với giếng 5, khoảng giữa giếng 2 và 10 với giếng 6, khoảng giữa giếng 3 và 11 với giếng 7) xuất hiện đường tủa màu xanh của kháng thể uốn ván chuẩn với giải độc tố uốn ván trong vắc xin. Vắc xin được coi là không đạt yêu cầu khi không có đường kết tủa ở các vị trí trên.
Nhận dạng thành phần ho gà trong vắc xin DTwP
Cặn của vắc xin DTP hấp phụ sau khi ly tâm được ngưng kết trên phiến kính với kháng huyết thanh ho gà đặc hiệu týp 1, 2, 3.
Hiện tượng ngưng kết xảy ra chứng tỏ trong mẫu thử nghiệm có chứa vi khuẩn ho gà.
3. Tiêu chuẩn chấp thuận
Có hiện tượng lên bông khi làm phản ứng với huyết thanh chuẩn kháng độc tố bạch hầu, uốn ván và có phản ứng ngưng kết với huyết thanh chuẩn ho gà đặc hiệu typ 1, 2, 3.
Hàm lượng Lf/ml của giải độc tố bạch hầu ≤ 30 Lf/ml.
Hàm lượng Lf/ml của giải độc tố uốn ván ≤ 10 Lf/ml.
15.20 XÁC ĐỊNH ĐỘC TỐ THẦN KINH TỒN DƯ TRONG VẮC XIN BẠI LIỆT UỐNG
Khỉ dùng để thử nghiệm có cân nặng không dưới 1,5 kg. Dùng khỉ loài Macaca hoặc Cercopithecus. Hỗn dịch bán thành phẩm đã lọc phải thử song song với chế phẩm chuẩn bằng cách tiêm vào sừng trước tủy sống khỉ tại đoạn thắt lưng. Trước khi tiêm phải lấy máu khỉ và kiểm tra để chứng tỏ huyết thanh của chúng không chứa kháng thể trung hòa đối với từng týp virus bại liệt. Những tiêu bản tổ chức học khi đã đọc xong cần phải được lưu lại trong thời gian 10 năm để làm bằng chứng.
Số lượng khỉ
Vắc xin thử và mẫu chuẩn đồng týp cần thử nghiệm song song với số lượng khỉ bằng nhau. Phân chia ngẫu nhiên nhóm khỉ sẽ được tiêm vắc xin thử hoặc mẫu chuẩn. Phải đạt được ít nhất 11 khỉ dương tính cho vắc xin thử và ít nhất 11 khỉ dương tính cho mẫu chuẩn đối với týp 1 và týp 2 (khỉ dương tính là khỉ có những tổn thương thần kinh đặc hiệu do virus bại liệt ở hệ thần kinh trung ương nhưng không bị liệt). Đối với týp 3 phải có ít nhất 18 khỉ dương tính cho vắc xin thử và 18 khỉ dương tính cho mẫu chuẩn. Có thể tiêm một lúc nhiều loạt vắc xin bán thành phẩm mà chỉ cần 1 mẫu chuẩn đồng týp.
Để có thể có được 11 và 18 khỉ dương tính thường là tiêm 12 và 20 khỉ tương ứng.
Gây mê khỉ bằng ketamin hydroclorid hoặc những thuốc gây mê phù hợp khác.
Hàm lượng virus của vắc xin thử và mẫu chuẩn trong liều tiêm
Hàm lượng virus của vắc xin thử và mẫu chuẩn đồng týp tiêm cho khỉ phải càng giống nhau càng tốt và nằm trong khoảng 105,5 đến 106,5 CCID50/0,1 ml. Chỉ tiêm một đậm độ.
Theo dõi khỉ sau tiêm
Theo dõi khỉ sau tiêm từ 17 ngày đến 22 ngày để phát hiện triệu chứng nghi do virus bại liệt hoặc các virus khác. Những khỉ sống qua 24 h và chết trước 11 ngày sau tiêm cần phải giải phẫu để xác định xem có phải chết do virus bại liệt hay không.
Tất cả những khỉ chết do những nguyên nhân không phải virus bại liệt thì không tính vào kết quả nhưng vẫn phải ghi vào phiếu theo dõi.
Những khỉ liệt nặng hoặc hấp hối và tất cả những khỉ sống qua giai đoạn theo dõi đều phải giải phẫu đại thể và vi thể.
Số lượng các tiêu bản phải đánh giá
* Phải làm tiêu bản từng con khỉ để kiểm tra tổ chức học ít nhất các vùng sau:
Tủy sống: Vùng phình lưng, vùng phình cổ.
Phần trên và phần dưới của hành tủy.
Não giữa (mesocephalon).
Cầu não, tiểu não.
Đồi thị.
Vùng vận động của vỏ não.
Tiêu bản cắt mỏng 8 µm đến 15 µm và nhuộm gailo-cyanin hoặc nhuộm Nissl.
* Số tiêu bản tối thiểu phải kiểm tra như sau:
12 tiêu bản đại diện cho vùng phình lưng.
10 tiêu bản đại diện cho vùng phình cổ.
2 tiêu bản vùng hành tủy.
1 tiêu bản vùng cầu não và tiểu não.
1 tiêu bản não giữa.
Đồi thị phải và trái, mỗi bên 1 tiêu bản.
Vỏ não bán cầu phải và trái mỗi bên 1 tiêu bản.
Cho điểm hoạt tính virus trên tiêu bản
Dùng phương pháp cho điềm theo mức độ tổn thương để đánh giá hoạt tính của virus trong từng nửa tiêu bản. Kiểu tổn thương như là thâm nhiễm tế bào hoặc phá hủy tế bào thần kinh đều quan trọng, các tổn thương được cho điểm như sau:
Điểm 1 - Chỉ có thâm nhiễm tế bào (điểm này không đủ để tính là khỉ dương tính).
Điểm 2 - Thâm nhiễm tế bào và có hủy hoại nơron tối thiểu.
Điểm 3 - Thâm nhiễm tế bào với hủy hoại nhiều nơron.
Điểm 4 - Hủy hoại nhiều nơron có hoặc không có thâm nhiễm tế bào.
Ghi điểm và báo cáo theo biểu mẫu chuẩn.
Khỉ có tổn thương nơron nhưng không có vết tiêm vẫn được coi là khỉ (+).
Khỉ có vết tiêm trên tiêu bản nhưng không có tổn thương nơron là khỉ (-).
Tiêu bản có tổn thương do tiêm nhưng không có tổn thương virus đặc hiệu thì không tính điểm.
Đọc và cho điểm từng nửa tiêu bản (NTB) của vùng thắt lưng (L); cổ (C); não (B).
Điểm tổn thương của từng con khỉ dương tính được tính như sau:
|
|
| Tổng số điểm L | + | Tổng số điểm C | + | Tổng số điểm B |
| LS | = | Tổng số NTB | Tổng số NTB | Tổng số NTB | ||
| 3 | ||||||
Đánh giá thử nghiệm độc tính thần kinh
Dựa trên hoạt tính ở vùng phình lưng và mức độ lan truyền lên vùng phình cổ và não, so sánh hoạt tính giữa vắc xin thử và mẫu chuẩn.
Việc xuất xưởng hoặc hủy bỏ vắc xin dựa trên điểm tổng thể của toàn bộ súc vật thử nghiệm. Những khỉ có hoạt tính cao khác thường hoặc ở vùng thắt lưng hoặc ở mức độ lan truyền đều phải tính đến trong đánh giá cuối cùng.
Bán thành phẩm đã lọc đạt tiêu chuẩn nếu số khỉ dương tính đạt yêu cầu và không có khác biệt có ý nghĩa thống kê về lâm sàng cũng như tổ chức học giữa vắc xin thử và mẫu chuẩn.
15.21 XÁC ĐỊNH CÔNG HIỆU CỦA VẮC XIN BẠI LIỆT UỐNG
Xác định công hiệu (hiệu giá) của vắc xin bại liệt uống gồm 2 nội dung:
Xác định hiệu giá virus trong vắc xin bán thành phẩm (đơn týp).
Xác định hiệu giá virus trong vắc xin thành phẩm (tam liên).
Xác định hiệu giá virus trong vắc xin bán thành phẩm (đơn týp)
Nguyên tắc
Công hiệu của vắc xin phòng bại liệt uống được xác định bởi liều gây hủy hoại 50 % tế bào cảm thụ Hep- 2 Cincinnati (CCID50) trong phương pháp vi lượng.
Tiến hành
Pha loãng bậc 10 mẫu chuẩn và vắc xin cần thử từ 10-1 đến 10-8 bằng môi trường duy trì tế bào.
Cho 0,1 ml các đậm độ virus vào mỗi giếng của phiến nhựa, mỗi đậm độ dùng từ 8 giếng đến 10 giếng. Cho 0,1 ml hỗn dịch tế bào chứa 1.104 tế bào Hep-2 vào mỗi giếng.
Đậy nắp phiến nhựa và cho các phiến nhựa vào tủ 5% CO2, nhiệt độ nuôi cấy là 36 °C trong 7 ngày.
Theo dõi
Theo dõi sự hủy hoại của tế bào từ ngày thứ 3 đến ngày thứ 7.
Tính kết quả
Đọc kết quả cuối cùng vào ngày thứ 7 và xử lý số liệu theo phương pháp thống kê Reed-Muench hoặc Karber.
Thí nghiệm có giá trị nếu hiệu giá của vắc xin mẫu chuẩn sai số ± 0,5 so với hiệu giá đã biết (tính theo log10 hiệu giá).
Xác định hiệu giá của vắc xin bại liệt uống trong thành phẩm tam liên
Nguyên tắc
Vắc xin bại liệt uống tam liên bao gồm 3 týp virus bại liệt sống giảm độc, do đó cần xác định hiệu giá của từng týp thành phần (týp 1, týp 2, týp 3) và hiệu giá tổng cả 3 týp (týp 1 + týp 2 + týp 3). Muốn chuẩn độ hiệu giá của từng týp phải dùng kháng huyết thanh (KHT) đặc hiệu để trung hòa 2 týp còn lại. Hiệu giá của vắc xin Sabin được tính bằng lượng virus Sabin gây hủy hoại 50 % tế bào cảm thụ trong 1 liều dùng cho người (số CCID50/0,1 ml) (CCID50 Cell Culture Infective Dose).
Các bước tiến hành
Pha loãng bậc 0,5 Iog10 vắc xin mẫu chuẩn và vắc xin thử bằng môi trường duy trì.
Pha hỗn dịch kháng huyết thanh số 1 chứa kháng huyết thanh týp 2 và týp 3.
Pha hỗn dịch kháng huyết thanh số 2 chứa kháng huyết thanh týp 1 và týp 3.
Pha hỗn dịch kháng huyết thanh số 3 chứa kháng huyết thanh týp 1 và týp 2.
Cho 0,05 ml/giếng hỗn dịch kháng huyết thanh số 1 vào phiến số 1 (xác định hiệu giá týp 1).
Cho 0,05 ml/giếng hỗn dịch kháng huyết thanh số 2 vào phiến số 2 (xác định hiệu giá týp 2).
Cho 0,05 ml/giếng hỗn dịch kháng huyết thanh số 3 vào phiến số 3 (xác định hiệu giá týp 3).
Cho 0,05 ml/giếng môi trường duy trì vào tất cả các giếng của phiến số 4 (xác định hiệu giá tổng).
Cho 0,05 ml của từng độ pha loãng vắc xin vào các dãy tương ứng của cả 4 phiến, mỗi độ pha loãng gây nhiễm 8 giếng. Bắt đầu gây nhiễm từ độ pha loãng cao nhất.
Thêm 0,05 ml môi trường duy trì vào các giếng chứng huyết thanh (SC - serum control) và chứng tế bào (CC- cell control).
Đậy nắp phiến cho vào tủ ấm 5% CO2. Trong trường hợp để tủ ấm thường thì gói kín trong giấy bạc hoặc trong túi nilông.
Ủ ở nhiệt độ 36 °C trong vòng 3 h để virus và kháng huyết thanh đặc hiệu kết hợp với nhau.
Chuẩn bị hỗn dịch tế bào Hep-2 nồng độ 1 - 2 x 105/ ml rồi cho 0,1 ml hỗn dịch này vào toàn bộ các giếng của cả 4 phiến. Nồng độ này thường đảm bảo tế bào sẽ mọc thành 1 lớp trên phiến trong vòng 2 ngày đến 3 ngày.
Đậy nắp phiến rồi cho vào tủ ấm 5% CO2. Trong trường hợp để ở tủ ấm thường thì dán kín bằng màng dán phiến (microtest film).
Ủ ở 36 °C trong 7 ngày.
Theo dõi và tính kết quả
Theo dõi sự hủy hoại của tế bào và tính kết quả như đã trình bày ở phần trên. Tế bào ở các giếng chứng huyết thanh và chứng tế bào phải phát triển tốt.
Vắc xin thành phẩm coi như đạt yêu cầu nếu:
Týp 1 có ít nhất 106,0 CCID50/0,1ml
Týp 2 có ít nhất 105,0 CCID50/0,1ml
Týp 3 có ít nhất 105,0 CCID50/0,1ml
Toàn bộ qui trình xác định công hiệu của vắc xin bại liệt uống phải được tiến hành trong điều kiện vô khuẩn
15.22 XÁC ĐỊNH CÔNG HIỆU THÀNH PHẦN UỐN VÁN TRONG VẮC XIN HẤP PHỤ CHỨA GIẢI ĐỘC TỐ UỐN VÁN
Công hiệu của thành phần uốn ván trong vắc xin DTwP được xác định bằng phương pháp so sánh liều hữu hiệu 50 % (ED50) của vắc xin chuẩn và vắc xin thử. Kết quả được tính bằng phần mềm Probit analysis (phần mềm WHO program).
MIỄN DỊCH
Pha vắc xin
Sử dụng ít nhất 3 đến 4 độ pha loãng bậc 2 nhưng không quá 5 độ pha cho cả vắc xin mẫu chuẩn và vắc xin mẫu thử trong dung dịch đệm sinh lý (dung dịch natri clorid 0,85 %).
Các độ pha vắc xin được chọn trên nguyên tắc ED50 không nằm lệch ngoài khoảng giữa độ pha loãng cao nhất (liều miễn dịch thấp nhất) và độ pha loãng thấp nhất (liều miễn dịch cao nhất), có nghĩa là chọn sao cho ở độ pha loãng có thể bảo vệ được 50 % động vật thí nghiệm rơi vào giữa đường cong. Tại mỗi phòng thí nghiệm có thể căn cứ vào các số liệu trước đó và chủng loại chuột sử dụng trong thử nghiệm để chọn độ pha phù hợp. Nếu không có các số liệu trước đó có thể tham khảo cách pha dưới đây.
Đối với vắc xin mẫu chuẩn, có thể chọn 3 độ pha bậc 2, trong đó độ pha loãng thấp nhất (độ pha đầu tiên) chứa 20 lU/ml. Các độ pha tiếp theo là 10 lU/ml và 5 lU/ml.
Đối với vắc xin thử, tùy theo hàm lượng kháng nguyên giải độc tố uốn ván chứa trong vắc xin và bản chất của loại vắc xin (hấp phụ, có hay không có thành phần ho gà) có thể chọn 1 trong 2 dải pha loãng bậc 2 với độ pha loãng đầu tiên 1/10 đến 1/15 (vắc xin TT, TD, Td) hay 1/25 đến 1/40 (vắc xin DTP, DTP-Hib - Heb...). Các độ pha tiếp theo theo bậc 2 tính từ độ pha đầu tiên.
Tiêm miễn dịch
Mỗi độ pha của vắc xin mẫu chuẩn và vắc xin mẫu thử được tiêm dưới da ít nhất cho 16 chuột nhắt trắng khỏe mạnh, 4 đến 5 tuần tuổi, có cân nặng mỗi chuột từ 14 g đến 20 g nhưng không được chênh lệch quá 4 g, liều tiêm mỗi con 0,5 ml. Dùng chuột cùng giới tính; nếu không cùng giới tính phải chia đều số chuột đực, cái vào các nhóm và nhốt riêng.
Sau tiêm miễn dịch, chăm sóc và theo dõi chuột trong 28 ngày và tiến hành tiêm thử thách.
TIÊM THỬ THÁCH
Xác định liều độc tố thử thách
Chỉ sử dụng độc tố có ít nhất 10.000 LD50/Lf. Độc tố được pha loãng ngay trước khi sử dụng để có các độ pha như: 0,0004 Lf/ ml; 0,0002 Lf/ ml; 0,0001 Lf/ ml và 0,00005 Lf/ ml. Mỗi độ pha tiêm cho một nhóm gồm 5 con chuột nhắt trắng, mỗi chuột có trọng lượng 16 g ± 0,3 g.
Theo dõi chuột hàng ngày trong 96 h, ghi lại số chuột còn sống trong mỗi độ pha.
Độ pha loãng độc tố gây chết 50 % số chuột vào ngày thứ 4 (sau 96 h) được tính là 1 LD50.
Tiêm thử thách
Dựa vào kết quả LD50 được xác định ở trên để độ pha dung dịch độc tố thử thách tương ứng với 100 LD50/ml trong dung dịch Jensen vô khuẩn có 1 % pepton.
Sau 28 ngày toàn bộ chuột đã miễn dịch bởi vắc xin mẫu chuẩn và mẫu thử được tiêm dưới da 0,5 ml với liều độc tố thử thách chứa 100 LD50/ml (50 LD50/0,5 ml).
Nhóm chuột chứng: Từ dung dịch độc tố thử thách pha thành 3 độ pha 2 LD50, 1 LD50, 0,5 LD50. Mỗi độ pha tiêm cho 5 chuột nhắt trắng có trọng lượng 16 g ± 0,3 g.
Theo dõi chuột
Theo dõi chuột thử thách hàng ngày trong 5 ngày. Ghi lại số chuột sống, chết, liệt ở mỗi độ pha miễn dịch của cả vắc xin mẫu thử và mẫu chuẩn.
Triệu chứng liệt uốn ván:
Liệt cứng hoặc co giật kích động (sau thử thách 2 ngày).
Chuột đi nhón cao chân và chân sau liệt co cứng tiếp đến 2 chân trước. Khi nắm vào chuột cảm thấy rất cứng do các cơ bị co cứng, cổ cong vẹo về phía lưng. Khi kích thích bằng tiếng động như quẹt bút trên nắp thanh lồng chuột, những chuột này sẽ phản ứng bằng cách co cứng cơ hoặc co giật.
TÍNH KẾT QUẢ
Dựa vào số chuột sống sót trong mỗi độ pha, công hiệu được xác định bằng phương pháp Probit analysis (Phần mềm WHO program).
Thử nghiệm có giá trị tin cậy nếu:
• Liều độc tố thử thách chứng minh được chứa từ 50 đến 200 LD50.
• Ở độ pha loãng thấp nhất (liều miễn dịch cao nhất) bảo vệ được trên 50 % động vật thí nghiệm.
• Ở độ pha loãng cao nhất (liều miễn dịch thấp nhất) bảo vệ được ít hơn 50 % động vật thí nghiệm. Đồ thị biểu diễn sự tương quan giữa liều miễn dịch và đáp ứng trên chuột của cả vắc xin chuẩn và vắc xin thử phải song song và tuyến tính.
Tiêu chuẩn chấp thuận
Tiêu chuẩn công hiệu của thành phần uốn ván trong vắc xin tùy theo tiêu chuẩn qui định của Tổ chức y tế thế giới cho loại vắc xin đó. Trong đó giới hạn tin cậy, công hiệu của vắc xin phải nằm trong khoảng từ 50 đến 200 % so với công hiệu quy định và giới hạn dưới phải đạt tối thiểu bằng tiêu chuẩn công hiệu quy định của vắc xin đó.
15.23 XÁC ĐỊNH CÔNG HIỆU THÀNH PHẦN BẠCH HẦU TRONG VẮC XIN HẤP PHỤ CHỨA GIẢI ĐỘC TỐ BẠCH HẦU
Xác định công hiệu của giải độc tố bạch hầu hoặc thành phần bạch hầu trong vắc xin phối hợp, hấp phụ được xác định bằng 1 trong 2 phương pháp: Thử thách trên chuột lang hoặc chuẩn độ huyết thanh chuột nhắt trên tế bào Vero.
PHƯƠNG PHÁP THỬ THÁCH TRÊN CHUỘT LANG
Miễn dịch cho chuột
Sử dụng ít nhất 3 độ pha loãng bậc 2 (nhưng không quá 5 độ pha) cho cả vắc xin mẫu chuẩn và vắc xin thử. Mỗi độ pha dùng ít nhất 16 chuột lang khoẻ mạnh, 3 đến 5 tuần tuổi, chưa sử dụng cho bất kỳ mục đích nào trước đó. Cân nặng mỗi chuột từ 250 g đến 350 g. Có thể sử dụng chuột khác giới nhưng phải phân chia đều cho các độ pha vắc xin và nhốt riêng lồng.
Các vắc xin được pha bằng nước muối sinh lý vô khuẩn và lựa chọn các độ pha sao cho ED50 trung bình không nằm ngoài các độ pha loãng của vắc xin. Mỗi chuột trong các nhóm miễn dịch được tiêm dưới da 1 ml huyền dịch của độ pha vắc xin tương ứng. Thời gian miễn dịch là 28 ngày.
Chọn ra 3 nhóm chuột đối chứng, mỗi nhóm 4 con đến 6 con, không tiêm gì và nuôi song song với các nhóm chuột miễn dịch bởi vắc xin.
Ví dụ pha vắc xin mẫu chuẩn và vắc xin mẫu thử như sau:
Vắc xin bạch hầu mẫu chuẩn chứa 180 lU/ống được hoàn nguyên trong 18 ml nước muối sinh lý (NMSL), pha thành ít nhất 3 độ pha loãng bậc 2 để có các độ pha tương ứng với 10 IU/ ml; 5 IU/ ml và 2,5 IU/ ml như sau:
A (1/18) = 1 ống vắc xin mẫu chuẩn +18 ml
B (1/36) = 9 ml A + 9 ml NMSL
C (1/72) = 9 ml B + 9ml NMSL
Vắc xin mẫu thử cũng được pha thành ít nhất 3 độ pha loãng bậc 2 bằng nước muối sinh lý vô khuẩn như sau:
A (1/ 20) = 1 ml vắc xin mẫu thử + 19 ml NMSL
B (1/40) = 9 ml A + 9 ml NMSL
C (1/80) = 9 ml B + 9 ml NMSL
Thử thách
Độc tố bạch hầu dùng cho thử thách phải có ít nhất 10.000 LD50/Lf. Sau 4 tuần, toàn bộ chuột lang đã tiêm miễn dịch được tiêm thử thách dưới da 1 ml độc tố bạch hầu chứa 100 LD50/ml.
Pha loãng độc tố bạch hầu để có dung dịch chứa 1 LD50, dùng tiêm cho nhóm chuột đối chứng, mỗi con 1 ml/dưới da.
Theo dõi chuột hàng ngày trong 5 ngày, ghi lại số chuột sống và chết trong mỗi độ pha miễn dịch và các nhóm chuột đối chứng.
Tính kết quả
Căn cứ vào số chuột sống sót trong mỗi độ pha vắc xin vào ngày thứ 5 sau khi thử thách, công hiệu vắc xin được xác định bằng phương pháp Probit analysis.
Vắc xin đạt yêu cầu về công hiệu bạch hầu nếu có không ít hơn 30 IU trong một liều đơn vắc xin cho người với điều kiện 95 % khoảng tin cậy của công hiệu tìm được nằm trong giới hạn từ 50 % đến 200 %, hoặc khi giới hạn cận dưới của 95 % khoảng tin cậy của công hiệu ≥ 30 IU trong một liều đơn vắc xin cho người.
Thử nghiệm có giá trị khi
Liều thử thách chứng minh chứa xấp xỉ 50 - 200 LD50/ml.
Giá trị ED50 trung bình của vắc xin mẫu chuẩn và vắc xin thử nằm trong khoảng giữa liều miễn dịch thấp nhất và cao nhất.
Thử nghiệm cần đáp ứng được tiêu chuẩn về tính tuyến tính và tính song song giữa liều miễn dịch và sự đáp ứng của chuột giữa vắc xin chuẩn và mẫu thử.
PHƯƠNG PHÁP CHUẨN ĐỘ HUYẾT THANH CHUỘT NHẮT TRÊN TẾ BÀO VERO
Miễn dịch cho chuột
Mỗi vắc xin mẫu chuẩn và vắc xin mẫu thử được tiến hành 4 độ pha loãng bậc 2. Các vắc xin được pha loãng bởi nước muối sinh lý vô khuẩn. Lựa chọn các độ pha vắc xin căn cứ vào hàm lượng giải độc tố bạch hầu có trong từng loại vắc xin.
Mỗi độ pha dùng ít nhất 8 chuột nhắt trắng khỏe mạnh, chưa sử dụng cho bất kỳ mục đích nào trước đó. Trọng lượng chuột 12 g/con đến 14 g/con, 3 đến 4 tuần tuổi, cùng giới (nếu khác giới phải phân đều cho các độ pha khác nhau và nhốt riêng lồng).
Mỗi chuột trong từng nhóm được tiêm dưới da 0,5 ml huyền dịch vắc xin tương ứng với từng độ pha của vắc xin mẫu thử và mẫu chuẩn. Thời gian miễn dịch là 5 tuần.
Ví dụ: Pha vắc xin mẫu chuẩn và thử như sau:
Vắc xin bạch hầu mẫu chuẩn chứa 688 IU được hoàn nguyên trong 8 ml nước muối sinh lý để có hỗn dịch (*) chứa 88 IU/ ml. Tiếp tục pha loãng bậc 2 để có 4 độ pha A - B - C - D như sau:
A (1/2) = 8 ml (*) + 8 ml NMSL
B (1/4) = 8 ml A + 8 ml NMSL
C (1/8) = 8 ml B + 8 ml NMSL
D (1/16) = 8 ml C + 8 ml NMSL
Ví dụ vắc xin thử pha như sau:
A (1/5) = 4 ml vắc xin + 16 ml NMSL
B (1/10) = 8 ml A + 8 ml NMSL
C (1/20) = 8 ml B + 8 ml NMSL
D (1/40) = 8 ml C + 8 ml NMSL
Lấy máu và thu thập huyết thanh chuột miễn dịch
Sau khi tiêm miễn dịch 5 tuần, mỗi chuột được lấy máu riêng rẽ (lấy máu tim bằng bơm tiêm loại 1 ml vô trùng) và tách lấy huyết thanh miễn dịch. Huyết thanh được bất hoạt ở 56 °C trong 30 min. Sau đó có thể giữ các mẫu huyết thanh chuột miễn dịch ở nhiệt độ -20 °C cho đến khi chuẩn độ huyết thanh trên tế bào.
Chuẩn độ kháng thể kháng bạch hầu trên tế bào Vero
Chuẩn bị tế bào Vero
Chuẩn bị môi trường M199:
Môi trường M199 bột khô hòa tan trong nước cất 2 lần. Điều chỉnh đến pH 7,0 - 7,2 bằng dung dịch natri bicarbonat. Môi trường nuôi cấy tế bào có chứa penicilin và streptomycin và 5 % đến 10 % huyết thanh bào thai bê.
Nuôi tế bào Vero:
Tế bào Vero giữ trong nitrogen lỏng được lấy ra và làm tan băng nhanh dưới vòi nước. Ly tâm loại bỏ phần nước. Cặn được hòa trong môi trường M199 và nuôi trong chai nhựa 25 cm2 ủ ở 37 °C. Sau 4 đến 6 ngày, khi tế bào mọc kín thành một lớp, dùng dung dịch trypsin 0,25 % tách tế bào và cấy chuyển qua chai nuôi tế bào 75 cm2. Tiếp tục ủ ở 37 °C trong 4 ngày đến 6 ngày. Khi tế bào mọc kín thành một lớp, dùng dung dịch trypsin 0,25 % tách tế bào. Pha thành hỗn dịch tế bào có đậm độ là 2,5 x 105 tế bào/ml dùng để chuẩn độ.
Tìm liều độc tố bạch hầu ức chế tế bào Lm/10 000
Liều độc tố Lm/10 000 là liều độc tố tối thiểu sau khi trung hòa với 0,0001 IU kháng độc tố bạch hầu còn khả năng ức chế sự phát triển của tế bào Vero.
Xác định liều Lm/10 000 của độc tố bạch hầu bằng phương pháp sau đây:
Độc tố bạch hầu chuẩn được pha loãng thành nhiều nồng độ khác nhau trong môi trường M199 để có nồng độ khoảng 0,2 Lf/ml.
Pha loãng độc tố bạch hầu 0,2 Lf/ml trên phiến chuẩn độ từ cột 1 đến 11.
Thêm huyết thanh chuẩn kháng bạch hầu có nồng độ 0,002 lU/ml vào các giếng trên.
Sau khi ủ phiến 1 h ở nhiệt độ phòng, thêm vào tất cả các giếng dung dịch tế bào vero có nồng độ 2.5 x 105 tế bào/ml.
Lắc nhẹ và dán kín phiến bằng giấy dán phiến.
Ủ ở 37 °C trong 5 ngày đến 6 ngày.
Đọc kết quả và tính liều Lm/10 000 của độc tố bạch hầu.
Chuẩn độ kháng thể kháng bạch hầu trên tế bào Vero
Tiến hành
Mỗi phiến dùng để chuẩn độ 8 mẫu huyết thanh của 1 độ pha vắc xin.
Cho 50 μl môi trường M199 vào tất cả các giếng của phiến (trừ giếng A11 và A12). Riêng giếng G11( G12, H11, H12 cho 100 μl.
Lấy 8 mẫu huyết thanh chuột đã tiêm miễn dịch của mỗi độ pha vắc xin cho lần lượt vào các giếng từ A1 đến H1, mỗi giếng 50 μl. Pha loãng bậc 2 từ dãy số 1 đến dãy số 10.
Huyết thanh chuẩn kháng bạch hầu có 10 IU/ml pha thành 0,008 IU/ ml (**).
Cho 50 μl huyết thanh (**) vào các giếng A11, A12 và B11, B12. Từ B11 và B12 pha loãng bậc 2 xuống C11, C12 và D11, D12; sau khi pha loãng và trộn đều ở các giếng D11, D12 thì bỏ đi 50 μl ở mỗi giếng trong 2 giếng này.
Pha độc tố chuẩn để có liều Lm/10 000, cho 50 μl độc tố vào tất cả các giếng trừ giếng G11, G12, H11, H12 (chứng tế bào).
Lắc phiến và ủ 1 h ở nhiệt độ phòng. Cho thêm vào các giếng 50 μl hỗn dịch tế bào có đậm độ 2,5 x 105 tế bào/ml.
Dán kín phiến bằng màng dán phiến và để ở 37 °C trong 5 ngày đến 6 ngày. Đọc kết quả.
Cách đọc và tính kết quả
Có thể đọc kết quả bằng cách trực tiếp soi các giếng chuẩn độ dưới kính hiển vi phản pha hoặc dựa vào việc đổi màu để phân biệt giếng “âm tính" và “dương tính”. Sự thay đổi màu từ đỏ sang vàng được coi như không có sự ức chế trao đổi chất do các độc tố đã bị trung hòa bởi các kháng thể kháng độc tố. Hiệu giá kháng thể được tính theo điểm là giếng cuối cùng vẫn còn tế bào mọc (môi trường có màu vàng).
Căn cứ vào số giếng “dương tính", tính kết quả theo phần mềm Parallel line (phần mềm WHO program).
Thử nghiệm có giá trị khi
Giếng A11 - D11 và A12 - D12 cho thấy rõ liều Lm/10 000 được pha đúng: Sau 6 ngày không xảy ra sự ức chế trao đổi chất ở giếng A11 và 12 và B11 và 12 (màu vàng). Ngược lại có xảy ra sự ức chế trao đổi chất ở giếng C11 và 12 và D11 và 12 (màu đỏ).
Sự ức chế trao đổi chất sẽ xảy ra ở giếng E11 và 12 và F11 và 12 (màu đỏ) là những giếng chứng độc tố (không có kháng huyết thanh để trung hòa độc tố nên độc tố đã gây hủy hoại tế bào).
Ở các giếng G11 và 12 và H11 và 12 các tế bào phát triển bình thường và môi trường có màu vàng, đó là những giếng chứng tế bào (không có độc tố và kháng huyết thanh bạch hầu).
Thử nghiệm phải đạt được các tiêu chuẩn đặt ra trong thử nghiệm Parallel line nghĩa là phải tạo được đường thẳng và đường song song trong mối tương quan giữa liều miễn dịch và đáp ứng.
Tộ số Yg của vắc xin chuẩn (là số các giếng dương tính trung bình ở mỗi độ pha vắc xin) sẽ phải nằm trong giá trị trung bình tích lũy ± 2SD của tất cả các thí nghiệm tiến hành trước đó.
Tiêu chuẩn chấp thuận
Vắc xin đạt yêu cầu về công hiệu bạch hầu nếu có không ít hơn 30 IU trong một liều đơn vắc xin cho người với điều kiện 95 % khoảng tin cậy của công hiệu tìm được nằm trong giới hạn từ 50 % đến 200 %, trừ khi giới hạn cận dưới của 95 % khoảng tin cậy của công hiệu ≥ 30 IU trong liều đơn vắc xin cho người.
Nếu kết quả không đạt yêu cầu nhưng thử nghiệm có giá trị (valid test) thì phải nhắc lại thử nghiệm trên mẫu huyết thanh miễn dịch còn lưu và làm thử nghiệm kép. Kết quả sẽ là giá trị trung bình của 2 thử nghiệm nhắc lại. Nếu thử nghiệm nhắc lại đạt yêu cầu thì loạt vắc xin được coi là đạt công hiệu thành phần bạch hầu. Nếu thử nghiệm nhắc lại không đạt yêu cầu, loạt vắc xin bị coi là không đạt công hiệu thành phần bạch hầu và phải hủy bỏ.
Nếu kết quả không đạt yêu cầu nhưng thử nghiệm không có giá trị (invalid test) thì chỉ cần nhắc lại thử nghiệm với số lượng mẫu và số lượng chuột bằng lần 1 (trình tự thử nghiệm lặp lại giống lần 1). Nếu ở lần thử nghiệm nhắc lại vẫn không đạt yêu cầu thì loạt vắc xin bị coi là không đạt yêu cầu về công hiệu thành phần bạch hầu và phải hủy bỏ.
15.24 XÁC ĐỊNH CÔNG HIỆU THÀNH PHẦN HO GÀ TOÀN TẾ BÀO TRONG VẮC XIN PHỐI HỢP, HẤP PHỤ
Công hiệu của vắc xin ho gà trong vắc xin được xác định bằng cách so sánh liều hữu hiệu 50 % (ED50) của vắc xin mẫu chuẩn so với vắc xin mẫu thử. Kết quả được tính bằng phương pháp Probit analysis (phần mềm WHO program).
Pha vắc xin
Sử dụng ít nhất 3 độ pha loãng bậc 5 nhưng không quá 5 độ pha cho cả vắc xin chuẩn và vắc xin thử. Pha vắc xin trong một loại dung dịch đệm sinh lý (dung dịch natri clorid 0,85 %). Các độ pha vắc xin được chọn dựa vào các số liệu trước đó và tùy thuộc vào chủng loại chuột thí nghiệm nhưng phải dựa trên nguyên tắc ED50 không được lệch ngoài khoảng giữa độ pha loãng cao nhất và độ pha loãng thấp nhất.
Nếu không có sẵn số liệu trước đó có thể pha 3 độ pha bậc 5 của vắc xin chuẩn và vắc xin thử như ví dụ dưới đây:
Vắc xin mẫu chuẩn
| A = 1/30 | = | 1 ống mẫu chuẩn | + 30 ml | NMSL |
| B =1/150 | = | 5 ml A | + 20 ml | NMSL |
| C = 1/750 | = | 5 ml B | + 20 ml | NMSL |
| Vắc xin DTP mẫu thử |
|
|
| |
| A = 1/5 | = | 5 ml vắc xin mẫu thử | + 20 ml | NMSL |
| B = 1/ 25 | = | 5 ml dung dịch A | + 20 ml | NMSL |
| C = 1/125 | = | 5 ml dung dịch B | + 20 ml | NMSL |
Tiêm miễn dịch
Chọn chuột nhắt trắng có cân nặng từ 10 g đến 18 g, nhưng các chuột trong một thử nghiệm không được chênh lệch trọng lượng quá 4 g. Dùng chuột cùng giới, nếu khác giới phải chia đều số chuột đực, cái vào các nhóm và nhốt riêng.
Chuột được tiêm miễn dịch sau khi đã được ăn uống đầy đủ 1 h. Tiêm ổ bụng 0,5 ml/1 chuột. Mỗi độ pha vắc xin tiêm cho một nhóm chuột, số lượng chuột trong mỗi nhóm tùy theo kết quả thẩm định thử nghiệm nhưng không được ít hơn 20 chuột. Tiêm miễn dịch theo thứ tự từ độ pha loãng nhất (từ độ pha C đến A) cho cả loạt vắc xin mẫu thử và mẫu chuẩn.
Bốn nhóm chuột đối chứng: Mỗi nhóm 10 con, được nuôi song song với nhóm chuột miễn dịch dùng để kiểm chứng độc lực của chủng thử thách.
Trong thời gian miễn dịch (14 ngày đến 17 ngày), chuột phải khỏe và tăng trọng bình thường, ít nhất 94 % số chuột đã được miễn dịch ở mỗi độ pha của cả vắc xin mẫu chuẩn và vắc xin thử nghiệm phải được sống sót và khỏe mạnh cho đến ngày thử thách.
Tiêm thử thách
Sau 14 ngày đến 17 ngày miễn dịch, chuột được thử thách với chủng B. pertussis 18323. Chủng thử thách có thể dùng chủng tươi hoặc chủng ở dạng huyền dịch đông băng (10 tỷ vi khuẩn ho gà) được bảo quản trong nitơ lỏng. Chủng thử thách được pha trong dung dịch casein 1 %. LD50 của chủng thử thách phải nằm trong khoảng 100 đến 1000 vi khuẩn.
Cách pha dung dịch casein 1 %
Hòa tan 10 g bột casein trong 1000 ml nước cất 2 lần, cho thêm 2,3 g natri clorid. Điều chỉnh đến pH 7 - 7,2 bằng dung dịch natri hydroxyd hoặc acid hydrocloric loãng. Hấp 121 °C trong 30 min.
Dung dịch casein 1 % phải được kiểm tra an toàn bằng cách tiêm vào não cho 5 chuột nhắt, mỗi con 0,02 ml. Theo dõi 3 ngày, toàn bộ chuột thí nghiệm phải sống. Nếu có 1 chuột chết phải pha lại dung dịch.
Cách pha hỗn dịch vi khuẩn để kiểm tra liều LD50 và để thử thách
Chủng B. pertussis 18323 được cấy truyền đời 2 hoặc đời 3 trên môi trường Bordet - Gengou có chứa 15 % máu cừu hoặc ngựa, huyền dịch vi khuẩn ho gà gặt từ nuôi cấy 20 h đến 22 h được dùng để pha thành hỗn dịch thử thách hoặc sử dụng huyền dịch vi khuẩn ho gà đậm độ 10 tỷ vi khuẩn/ml đã bảo quản trong nitrogen lỏng để pha dung dịch thử thách. Trong khi pha cũng như trong khi tiêm thử thách, hỗn dịch vi khuẩn ho gà phải giữ trong đá lạnh. Thời gian bắt đầu pha đến khi tiêm xong hỗn dịch vi khuẩn thử thách không được quá 150 min. Xác định đậm độ vi khuẩn ho gà bằng mắt thường khi so với ống độ đục chuẩn quốc tế 10 IOU hoặc đo OD bằng máy quang phổ kế để tính số vi khuẩn ho gà trong hỗn dịch gặt. Pha loãng huyền dịch vi khuẩn ho gà bằng casein 1 % để có nồng độ là 10 x 109 vi khuẩn/ml (*). Nếu sử dụng chủng ho gà được bảo quản trong nitơ lỏng thì phải làm tan băng nhanh trước khi thử thách. Các độ pha loãng của chủng ho gà được pha theo sơ đồ dưới đây trong dung dịch casein 1 %.
Ví dụ: Sơ đồ các độ pha loãng của chủng ho gà trong dung dịch casein 1 %
Chủng B. pertussis 18323 giữ trong nitrogen lỏng là 10 x 109/ml (*) = A0
| A1 | = | 1/2 | = | 5 x 109/ ml | = | 1 ml A0 | + | 1 ml đệm Casein 1 % |
| A2 | = | 1/4 | = | 5 x 109/2 ml | = | 1 ml A1 | + | 1 ml đệm Casein 1 % |
| A3 | = | 1/40 | = | 5 x 106/ 0,02 ml | = | 1 ml A2 | + | 9 ml đệm Casein 1 % |
| A4 | = | 1/400 | = | 5 x 105/ 0,02 ml | = | 1 ml A3 | + | 9 ml đệm Casein 1 % |
| A5 | = | 1/2000 | “ | 100 000/0,02 ml | = | 3 ml A4 | + | 12 ml đệm Casein 1 % |
| A6 | = | 1/20000 | = | 10 000/0,02 ml | = | 1 ml A5 | + | 9 ml đệm Casein 1 % |
| A7 | = | 1/100000 | = | 2000/0,02 ml | = | 1 ml A6 | + | 4 ml đệm Casein 1 % |
| A8 | = | 1/500000 | = | 400/0,02 ml | = | 1 ml A7 | + | 4 ml đệm Casein 1 % |
| A9 | = | 1/2500000 | - | 80/0,02 ml | = | 1 ml A8 | + | 4 ml đệm Casein 1 % |
Tiêm chuột
Sau 14 ngày đến 17 ngày, toàn bộ chuột đã miễn dịch bởi vắc xin mẫu chuẩn và mẫu thử được tiêm thử thách vào não, mỗi con nhận 0,02 ml hỗn dịch chứa 100 000 vi khuẩn (độ pha A5) bằng một bơm tiêm bán tự động Hamilton.
Nhóm chuột đối chứng, mỗi nhóm 10 con được tiêm với các độ pha từ A6 đến A9 tương ứng với hỗn dịch chứa 10 000 vi khuẩn; 2000 vi khuẩn; 400 vi khuẩn và 80 vi khuẩn.
Độ pha hỗn dịch chứa 80 vi khuẩn sẽ được cấy ít nhất trên 2 đĩa thạch Bordet Gengou sau khi pha xong hỗn dịch và sau khi kết thúc tiêm thử thách. Ủ đĩa thạch ở 35 °C trong 3 ngày đến 4 ngày, đếm số khuẩn lạc mọc. Thông thường số khuẩn lạc đạt khoảng 30 %.
Lưu ý:
• Trong quá trình pha và tiêm thử thách, dung dịch chủng phải luôn giữ trong đá lạnh.
• Thời gian tính từ khi bắt đầu pha chủng đến khi kết thúc thử nghiệm không được quá 2,5 h.
• Tất cả vật liệu chứa chủng ho gà đều phải tiệt trùng bằng nồi hấp 121 °C trong 15 min đến 20 min hoặc luộc sôi trong 30 min trước khi rửa dụng cụ.
• Những chuột chết trong 3 ngày đầu sau tiêm thử thách được coi là chuột chết không mang tính đặc hiệu của ho gà. Do đó chỉ những chuột chết từ ngày thứ 4 đến ngày 14 mới được tính kết quả.
• Ngày thứ 14 sau thử thách: Tất cả những chuột chết, hay có dấu hiệu liệt rõ và sưng phồng tấy đầu đều được tính là chuột chết.
Tính kết quả
Dựa vào số chuột sống sót trong mỗi nhóm chứng, dùng phương pháp Reed Muench để tính LD50 của chủng thử thách. Xem ví dụ dưới đây:
Ví dụ:
| Số Vi khuẩn | Số Chuột | Số Sống | Số Chết | Số tích lũy | Tổng số | % chết | |
| Sống | Chết |
| |||||
| 10 000 | 10 | 1 | 9 | 1 | 22 | 23 | 95 |
| 2 000 (B) | 10 | 3 | 7 | 4 | 13 | 17 | 76 (b) |
| 400 (A) | 10 | 6 | 4 | 10 | 6 | 16 | 37 (a) |
| 80 | 10 | 8 | 2 | 18 | 2 | 20 | 10 |
Theo ví dụ trên, LD50 nằm trong khoảng giữa 2 liều 2000 và 400 vi khuẩn, số % chuột chết tương ứng với liều sát trên 50 % là 76 %, số % chuột chết tương ứng liều sát dưới 50 % là 37 %
Tính LD50 theo công thức sau:
|
|
|
|
trong đó:
A là liều gây chết vừa sát dưới 50 % (trong ví dụ trên là 400);
A là % chết tương ứng với A (trong ví dụ trên là 37 %);
B là liều gây chết vừa sát trên 50 % (trong ví dụ trên là 2000);
b là % chết tương ứng với B (trong ví dụ trên là 76 %);
f là hệ số pha loãng (trong thử nghiệm này là pha loãng bậc 5).
Thay bằng con số cụ thể là:
|
|
|
|
LD50 = 680 vi khuẩn
Dùng phần mềm Probit analysis của WHO program để tính công hiệu của vắc xin ho gà.
Thử nghiệm được coi là có giá trị khi:
ED50 của vắc xin chuẩn và ED50 vắc xin thử nghiệm nằm giữa độ pha loãng nhất và độ pha đặc nhất của các vắc xin đó.
Trong suốt thời gian miễn dịch cho đến ngày thử thách số chuột trong mỗi độ pha không được chết quá 6 %.
Liều thử thách phải chứa 100 đến 1000 LD50, như vậy liều LD50 phải chứa 100 đến 1000 vi khuẩn.
Thử nghiệm phải đạt được tiêu chuẩn của chương trình Probit Analysis. Yêu cầu thể hiện bằng sự tương quan giữa liều miễn dịch và đáp ứng trên chuột (tính tuyến tính) và tương quan giữa vắc xin chuẩn và vắc xin thử (tính song song).
Tiêu chuẩn chấp thuận
Vắc xin đạt yêu cầu nếu công hiệu không ít hơn 4 IU trong một liều đơn vắc xin cho người và giới hạn dưới của 95 % khoảng tin cậy của công hiệu tìm được không ít hơn 2 IU trong một liều đơn vắc xin cho người.
Nếu thử nghiệm lần 1 có giá trị (valid) và đạt tiêu chuẩn chấp thuận thì vắc xin đó coi như đạt yêu cầu về công hiệu. Nhưng nếu không đạt tiêu chuẩn chấp thuận thì thử nghiệm đó phải được làm nhắc lại. Công hiệu vắc xin đạt tiêu chuẩn chấp thuận khi kết quả trung bình nhân của các lần thử nghiệm có giá trị không thấp hơn tiêu chuẩn chấp thuận cho phép.
Nếu lần 1 không đạt yêu cầu và thử nghiệm không có giá trị (invalid) thì phải làm lại thử nghiệm. Kết quả lần thử nghiệm đầu sẽ không được dùng để xem xét. Nếu kết quả lần thử nghiệm nhắc lại có giá trị và đạt tiêu chuẩn chấp thuận thì loạt vắc xin đó coi như đạt yêu cầu về công hiệu. Nếu không đạt yêu cầu thì phải làm lại như ở trên.
15.25 XÁC ĐỊNH HÀM LƯỢNG FORMALDEHYD TỒN DƯ TRONG VẮC XIN VÀ SINH PHẨM
Nguyên lý
Có hai phương pháp xác định hàm lượng formaldehyd tồn dư trong vắc xin và sinh phẩm:
• Phương pháp dựa vào phản ứng tạo chất màu chinol dưới tác dụng của thuốc thử fuchsin và aldehyd hòa tan trong nước.
• Định lượng formaldehyd bằng cách so màu của 3,5-diacetyl-1,4-dihydrolutidin ở bước sóng 415 nm. Phức hợp tạo thành có màu vàng chanh do phản ứng của formaldehyd tự do trong mẫu thử với acetylaceton và amoni trong môi trường acid nhẹ.
Phương pháp tiến hành
*Phương pháp 1
Lấy 1 ml mẫu thử, cho nước cất vừa đủ 5 ml, thêm vào 1 ml thuốc thử fuchsin acid, trộn đều; đậy ống nghiệm bằng nút cao su và đợi 1 h. Đo mật độ quang học của dung dịch bằng kính lọc màu tím than (bước sóng 400 nm ± 5 nm).
Tiến hành làm song song trên mẫu trắng theo trình tự như trên nhưng thay 1 ml mẫu thử bằng 1 ml nước cất. Kết quả xác định được bằng cách so màu của mẫu vắc xin thử với dung dịch chuẩn có mật độ quang học gần nhau nhất.
Dựng đường chuẩn
Lấy 1 ml formaldehyd chuẩn 0,4 % thêm nước cất vừa đủ 100 ml vào (dung dịch 0,004 %).
Lấy 0,5 ml, 1,0 ml, 2,0 ml, 3,0 ml, 4,0 ml, 5,0 ml dung dịch chuẩn formaldehyd 0,004 % cho vào các ống nghiệm và cho nước cất vừa đủ 5,0 ml, thêm 1,0 ml dung dịch thuốc thử. Đợi 1 h sau đó tiến hành phân tích như đã mô tả ở trên.
Cách pha dung dịch formaldehyd chuẩn 0,4 %: Trước khi pha tiến hành xác định sơ bộ hàm lượng formaldehyd có trong dung dịch formaldehyd (dung dịch phải chứa không ít hơn 37 % formaldehyd). Cân 1 g dung dịch formaldehyd 40 % (cân chính xác, trong trường hợp dung dịch chứa ít hơn 40 % thì phải tính toán lại cho thích hợp) cho vào một bình định mức 100 ml, cho nước cất vừa đủ tới vạch (dung dịch 0,4 %). Trước khi dùng pha loãng 100 lần.
Cách pha thuốc thử fuchsin: Hòa tan 2 g fuchsin kiềm vào 120 ml nước cất đã đun sôi, để nguội. Hòa 2 g natri sulfit (TT) vào 20 ml nước cất và trộn vào dung dịch trên, thêm 2 ml acid hydrocloric (TT). Cho nước cất vừa đủ 200 ml và chờ ít nhất 1 h mới đem sử dụng. Dung dịch này phải luôn luôn mới.
*Phương pháp 2
Lấy khoảng 3 ml đến 4 ml mẫu thử ly tâm 4000 r/ min trong 10 min, lấy phần nước (nếu mẫu thử trong thì không cần ly tâm). Pha loãng mẫu thử nếu cần thiết.
Lấy chính xác 3 ml mẫu thử cho vào ống nghiệm.
Dựng đường chuẩn: Pha loãng 10 lần dung dịch formaldehyd chuẩn 0,04 %, từ đó pha các dung dịch formaldehyd chuẩn 0,0002 %; 0,0004 %; 0,0008 %; 0,0012 %.
Lấy chính xác 3 ml các dung dịch chuẩn đã pha loãng vào các ống nghiệm.
Mẫu trắng dùng 3 ml nước cất.
Thêm 3 ml dung dịch acetylaceton vào mỗi ống nghiệm đựng mẫu trắng, mẫu chuẩn và mẫu thử.
Lắc đều, đun cách thuỷ 100 °C trong 15 min.
Để nguội ở nhiệt độ phòng.
Tiến hành đo mật độ quang ở bước sóng 415 nm.
Cách tính kết quả:
|
| X = na |
|
trong đó:
X: hàm lượng formaldehyd có trong mẫu thử (%);
a: hàm lượng formaldehyd có trong mẫu thử tìm được trên đường chuẩn (%);
n: độ pha loãng mẫu.
Cách pha dung dịch formaldehyd chuẩn 0,04 %: Cân 311 mg hexamethylentetramin hòa tan trong vừa đủ 1000 ml nước cất, dung dịch có chứa 0,04 % formaldehyd, bảo quản dung dịch ở nhiệt độ phòng dùng trong 6 tháng.
Cách pha dung dịch acetylaceton: Cân 150 g amoni acetat (TT), hòa tan trong khoảng 500 ml nước cất. Thêm 3 ml acid acetic đậm đặc, 2 ml acetylaceton vào, khuấy đều, thêm nước cất vừa đủ 1000 ml. Bảo quản dung dịch ở 2 °C đến 8 °C, dùng trong 2 tháng.
Tiêu chuẩn chấp thuận
Mỗi loại vắc xin, sinh phẩm có tiêu chuẩn khác nhau. Các tiêu chuẩn này dựa theo quy định của WHO cho từng loại vắc xin và sinh phẩm. Tuy nhiên, lượng formaldehyd tồn dư trong vắc xin, sinh phẩm không được vượt quá 0,02 %.
15.26 XÁC ĐỊNH HÀM LƯỢNG NATRI CLORID VỚI SỰ CÓ MẶT CỦA PROTEIN BẰNG PHƯƠNG PHÁP ĐỊNH LƯỢNG GIÁN TIẾP (PHƯƠNG PHÁP CHARPENTIER-VOLHARD)
Nguyên lý
Phương pháp dựa trên phản ứng của ion Cl- với một lượng bạc nitrat thừa.
Bạc nitrat thừa sẽ được chuẩn độ bằng dung dịch amoni sulfocyanid với chỉ thị màu phèn sắt amoni.
Protein trong mẫu thử được oxy hóa bằng kali per-manganat trong môi trường acid trước khi tiến hành định lượng natri clorid.
Phương pháp tiến hành
Hút chính xác một thể tích (V ml) mẫu thử tương ứng với khoảng 0,2 mg hoạt chất cho vào một bình nón dung tích 50 ml.
Thêm 5 ml dung dịch bạc nitrat 0,01 N.
Thêm 1 ml acid nitric đậm đậc (TT).
Thận trọng đun trên bếp cho đến khi sôi.
Thêm từng giọt dung dịch bão hòa kali permanganat cho đến khi có màu tím.
Tiếp tục đun sôi trong 5 min.
Thêm một ít đường glucose bột để khử màu.
Trên đáy bình xuất hiện tủa trắng như bông.
Làm nguội.
Thêm 0,5 ml dung dịch phèn sắt amoni bão hòa.
Chuẩn độ bạc bằng dung dịch amoni sulfocyanid 0,01 N (CĐ) cho đến khi có màu hồng.
Song song tiến hành làm mẫu trắng (thay mẫu thử là nước cất).
Lượng natri clorid có trong mẫu thử tính bằng mg % theo công thức:
|
|
|
|
trong đó:
A: thể tích dung dịch amoni sulfocyanid 0,01 N (CĐ) dùng để chuẩn độ mẫu trắng (ml);
B: thể tích dung dịch amoni sulfocyanid 0,01 N (CĐ) dùng để chuẩn độ mẫu thử (ml);
K: hệ số điều chỉnh của dung dịch amoni suffo-cyanid 0,01 N;
0,585: lượng natri clorid tương ứng với 1 ml dung dịch amoni sulfocyanid 0,01 N (mg);
V: số ml dung dịch mẫu thử đem kiểm tra;
100: chuyển thành %.
15.27 XÁC ĐỊNH HÀM LƯỢNG NHÔM Al+++ TRONG VẮC XIN VÀ SINH PHẨM
Phương pháp 1
Nguyên lý:
Phương pháp dựa trên phản ứng tạo thành phức chất của một lượng thừa dung dịch muối natri của acid ethylen diamin tetra acetic (EDTA) với ion nhôm (Al+++) và sau đó chuẩn độ lượng thừa EDTA bằng dung dịch đồng sulfat 0,01 M.
Phương pháp tiến hành:
Lấy 1 ml acid sulfuric đậm đặc (TT), 6 giọt acid nitric đậm đặc (TT) và 3 ml mẫu thử cho vào bình nón đun nóng cho đến khi thấy khói trắng dày đặc thoát ra.
Tiếp tục đun nóng và cho thêm vài giọt acid nitric đậm đặc (TT) cho đến khi dung dịch trong bình mất màu. Để nguội bình.
Thận trọng thêm 10 ml nước cất. Nếu dung dịch còn đục, đun tiếp cho đến khi trong.
Thêm dung dịch natri hydroxyd 50 % vào bình cho đến khi dung dịch có màu đỏ vàng [dùng dung dịch da cam methyl (TT) làm chất chỉ thị].
Nếu dung dịch đục, hòa tan tủa bằng cách thêm dung dịch acid sulfuric 1 M, cho thêm chính xác vào bình 25 ml dung dịch dinatri diethylen diamin tetraacetat 0,01 M (CĐ) và 10 ml dung dịch đệm acetat pH 4,4 [có chứa 68 g natri acetat (TT); 38,5 g amoni acetat (TT); 125 ml acid acetic (TT) và nước vừa đủ 500 ml], Đun sôi nhẹ trong 3 min.
Thêm 1 ml dung dịch chỉ thị màu pyridylazonaphtol 0,1 % trong ethanol 95 %. Khi còn nóng chuẩn độ ngay dung dịch dinatri diethylen diamin tetraacetat thừa bằng dung dịch đồng sulfat 0,01 M (CĐ) cho đến khi xuất hiện màu nâu tía, ta được A ml dung dịch đồng sulfat 0,01 M. Song song tiến hành làm mẫu trắng (thay mẫu thử bằng nước cất) được B ml dung dịch đồng sulfat 0,01 M.
1,0 ml dung dịch dinatri diethylen diamin tetraacetat 0,01 M (CĐ) tương đương với 0,2698 mg Al+++. Lượng nhôm x (Al+++ mg/ml) trong mẫu thử được tính theo công thức sau:
| X | = | B - A | x | 0,2689 |
| Thể tích mẫu thử |
Phương pháp 2
Nguyên lý:
Hàm lượng nhôm (Al+++) có trong mẫu thử được xác định bằng cách so màu do phản ứng của nhôm với stilbazo. Nhôm hydroxyd có trong mẫu thử dưới tác dụng của acid nitric và nhiệt độ chuyển thành Al+++.
Phương pháp tiến hành:
Trước khi kiểm tra, mẫu thử được lắc đều thành hỗn dịch đồng nhất. Thêm 0,2 ml acid nitric đậm đặc vào 1 ml mẫu thử. Đun sôi cách thủy trong khoảng 1 h. Pha loãng bằng nước cất mẫu thử đến nồng độ thích hợp (thường pha loãng 25 và 50 lần).
Lấy 1ml mẫu thử vào bình nón, thêm nước cất vừa đủ 5 ml. Thêm 10 ml dung dịch đệm acetat (pH = 5,55 - 5,75), 5 ml dung dịch stilbazo, 5 ml nước cất. Để ở nhiệt độ phòng 20 min, đo quang ở bước sóng 510 nm.
Song song tiến hành dựng đường chuẩn với các dung dịch nhôm chuẩn 2, 4, 6, 8, 10 microgam/ml, tương đương với 1 ml, 2 ml, 3 ml, 4 ml, 5 ml nhôm chuẩn 2 microgam/ml. Dựa vào đường chuẩn tính ra hàm lượng nhôm có trong mẫu thử.
Song song tiến hành làm mẫu trắng: Thay mẫu thử bằng 1ml nước cất.
Cách pha dung dịch nhôm chuẩn 0,1 %: Cân chính xác 0,895 g nhôm clorid (AlCl3.6H2O) hòa tan vào 100 ml nước cất 2 lần. Trước khi dùng pha loãng dung dịch nhôm chuẩn 0,1 % 500 lần bằng nước cất, thu được dung dịch nhôm chuẩn 2 microgam/ml.
Cách pha dung dịch stilbazo 0,035 %: Hòa tan 0,035 g stilbazo trong vừa đủ 100 ml nước cất. Lọc qua giấy lọc. Dung dịch bảo quản ở nhiệt độ 2 °C đến 8 °C sử dụng được trong 1 tuần.
Cách pha dung dịch đệm acetat (pH = 5,55 - 5,75):
Dung dịch A: Hòa tan 69,05 g natri acetat (TT) vào 500 ml nước cất.
Dung dịch B: Hòa 5,72 ml acid acetic (TT) trong vừa đủ 100 ml nước cất.
Trộn dung dịch A vào dung dịch B theo tỷ lệ 9 : 1. Đo pH.
Tiêu chuẩn chấp thuận
Hàm lượng Al+++ trong vắc xin không được vượt quá 1,25 mg Al+++/1 liều tiêm cho người.
15.28 XÁC ĐỊNH HÀM LƯỢNG PHENOL TRONG VẮC XIN VÀ SINH PHẨM
Phương pháp 1
Nguyên lý
Phương pháp dựa vào phản ứng tạo màu của thuốc thử Folin với phenol.
Phương pháp tiến hành
Vắc xin mẫu thử được ly tâm với vận tốc 3000 r/min trong 15 min. Bỏ cặn, lấy 1 ml phần nước ở phía trên cho vào bình định mức 100 ml và cho nước cất vừa đủ 100 ml, lắc đều.
Lấy 0,5 ml dung dịch trên, thêm 9 ml nước cất, 0,5 ml thuốc thử Folin (TT) pha loãng 2 lần, 2 ml dung dịch natri carbonat 20 % (TT). Lắc đều. Đun cách thủy cho sôi trong 1 min. Làm nguội ở nhiệt độ phòng.
Song song tiến hành xây dựng đường chuẩn:
Cách dựng đường chuẩn:
Hút dung dịch phenol chuẩn 1: 1000 (TT) vào các ống nghiệm với lượng như sau: 0,1 ml; 0,2 ml; 0,3 ml; 0,4 ml; 0,5 ml; 0,6 ml, 0,7 ml; 0,8 ml; 0,9 ml; 1,0 ml. Thêm vào các ống nghiệm trên mỗi ống một lượng nước cất vừa đủ 9,5 ml; 0,5 ml thuốc thử Folin pha loãng 2 lần; 2 ml dung dịch natri carbonat 20 % (TT). Lắc đều. Đun cách thủy cho sôi trong 1 min. Làm nguội ở nhiệt độ phòng.
Song song tiến hành làm mẫu trắng (dùng nước cất thay cho mẫu vắc xin thử nghiệm).
Đem so màu trên máy quang phổ với bước sóng 310 nm (cả với mẫu thử và mẫu chuẩn).
Cách tính kết quả:
|
|
|
|
trong đó:
X: hàm lượng phenol có trong mẫu thử (%);
a x 2: hàm lượng phenol có trong 1 ml mẫu đã pha loãng tìm được trên đường chuẩn;
100: độ pha loãng của mẫu thử.
Tiêu chuẩn chấp thuận
Mỗi loại vắc xin, sinh phẩm có tiêu chuẩn khác nhau về hàm lượng phenol (dựa vào tiêu chuẩn của WHO hoặc tiêu chuẩn Việt Nam cho vắc xin hay sinh phẩm đó). Hàm lượng phenol trong vắc xin thành phẩm nói chung không được vượt quá 0,12 g/lít. Trừ những quy định cụ thể khác. Theo nguyên tắc chung, hàm lượng phenol trong vắc xin thành phẩm không được thấp hơn so với hàm lượng cho thấy có khả năng kháng khuẩn và không được cao hơn 115 % hàm lượng đăng ký trên nhãn.
Pha dung dịch phenol chuẩn:
Cân chính xác 1 g phenol (TT) hòa tan trong vừa đủ 100 ml nước cất. Trước khi dùng pha loãng 1000 lần bằng nước cất.
Pha dung dịch natri carbonat 20 %:
Cân 20 g natri carbonat (TT) hòa tan trong vừa đủ 100 ml nước cất. Lọc vào chai đựng.
Phương pháp 2
Mẫu thử được trộn đều để làm thử nghiệm. Pha loãng mẫu thử bằng nước cất (nếu cần thiết) để có nồng độ phenol khoảng 15 μg/ml.
Chuẩn bị mẫu phenol chuẩn theo các nồng độ 5 μg; 10 μg; 15 μg; 20 μg và 30 μg /ml.
Lấy 5 ml các dung dịch mẫu thử và mẫu chuẩn vào ống nghiệm, thêm 5 ml dung dịch đệm borat pH 9,0; 5 ml dung dịch aminopyrazolon và 5 ml dung dịch kali fericyanid. Lắc đều, để yên 10 min, đo quang phổ (Phụ lục 4.1) ở bước sóng 546 nm. Dựa vào đường chuẩn để tính ra hàm lượng phenol có trong mẫu thử.
Cách pha dung dịch đệm borat pH 9,0:
Dung dịch A: Hòa tan 6,18 g acid boric (TT) trong vừa đủ 1000 ml dung dịch kali clorid 0,1 M.
Dung dịch B: Dung dịch natri hydroxyd 0,1 M (TT).
Trộn 1000 ml dung dịch A với 420 ml dung dịch B thu được dung dịch đệm pH 9,0.
Cách pha dung dịch aminopyrazolon:
Dung dịch 4-aminophenazon 1 % trong dung dịch đệm borat pH 9,0.
15.29 XÁC ĐỊNH HÀM LƯỢNG THIMEROSAL TRONG VẮC XIN VÀ SINH PHẨM
Có nhiều phương pháp để xác định hàm lượng thimerosal trong vắc xin và sinh phẩm. Tùy theo khả năng hiện có về thiết bị và hóa chất tại cơ sở để chọn 1 trong 2 phương pháp thử nghiệm sau đây: Phương pháp hóa lý và phương pháp vi sinh vật.
Phương pháp hóa lý
Nguyên lý:
Dựa vào phản ứng giải phóng ion thủy ngân tự do từ thimerosal, kết hợp với dithizon thành dithizon- thủy ngân, xác định bằng cách đo quang.
Cách tiến hành:
Làm song song 2 mẫu (mẫu vắc xin thử và mẫu chưng nước cất), lấy 0,2 ml mỗi mẫu cho vào từng bình nút mài riêng biệt có dung tích 100 ml.
Cho vào mỗi bình 1,2 ml acid sulfuric (TT) đã pha loãng 2 lần bằng nước cất 2 lần tính theo thể tích và đun nhẹ bình số 1 (chứa mẫu vắc xin thử) trong cách thủy cho đến khi tan hết. Thêm vào mỗi bình 5 ml dung dịch kali permanganat 5% (TT), lắc đều và để yên trong 1 h.
Loại bỏ kali permanganat thừa bằng cách thêm 1,5 ml dung dịch hydroxylamin clorid 10% (TT), thêm 35 ml nước cất 2 lần, 5 ml dung dịch acid acetic 6 M (TT) và trộn đều. Dùng buret thêm chính xác vào mỗi bình 10 ml dung dịch dithizon 0,001% và lắc 30 s.
Chuyển toàn bộ dung dịch thử của từng bình vào mỗi bình lắng gạn riêng biệt, tách lớp cloroform qua một lớp bông đã hấp và sấy khô vào các ống nghiệm. Đo quang phổ ở bước sóng 620 nm.
Hàm lượng thimerosal (X,%) có trong vắc xin thử được tính theo công thức:
![]()
trong đó:
a là lượng thủy ngân (μg) xác định được theo đường chuẩn;
0,2 là số ml dung dịch vắc xin thử;
49,55 là hàm lượng thủy ngân có trong 100 phần của thimerosal;
100 là 100 phần thimerosal;
100 là đổi ra phần trăm;
1 000 000 là hệ số chuyển đổi từ μg sang g.
Cách dựng đường chuẩn:
Cân chính xác 0,5 g thủy ngân, hòa vào 15 ml acid nitric đậm đặc (TT) và thêm nước cất vừa đủ 500 ml, được dung dịch thủy ngân có hàm lượng 1 mg/ml (DD*). Xác định hàm lượng thủy ngân như sau: Lấy 10 ml DD*, cho thêm 10 giọt dung dịch phèn sắt amoni 10% và chuẩn độ từ từ với dung dịch amoni sulfocyanid 0,01 N (CĐ) cho đến khi xuất hiện màu da cam.
1 ml dung dịch amoni sulfocyanid 0,01 N (CĐ) tương đương với 0,001003 g thủy ngân.
Lấy 5 bình nút mài có dung tích 100 ml để tiến hành song song 5 thí nghiệm với 5 mẫu: 0,4 ml, 0,6 ml, 0,8 ml, 1,0 ml, 1,2 ml dung dịch thủy ngân chuẩn trên. Thêm vào mỗi bình 1,2 ml acid sulfuric (TT) đã pha loãng 2 lần bằng nước cất 2 lần và 30 ml nước cất 2 lần. Nếu không có nước cất 2 lần thì sử dụng nước xử lý dithizon như sau: Cho vào mỗi bình gạn 10 ml dung dịch dithizon (TT) (0,01 g dithizon/1 lít nước), sau đó tách phần cloroform ra. Nếu màu của dithizon thay đổi thì làm đi làm lại vài lần cho đến khi màu của dithizon không thay đổi. Thêm 5 ml dung dịch natri clorid 0,9% (TT) và 5 ml dung dịch acid acetic 6 M (TT) và tiến hành tiếp tục như đối với mẫu kiểm tra ghi ở phần trên. Các kết quả thu được cho phép lập một đường chuẩn về thủy ngân.
Cách pha dung dịch dithizon 0,001%:
Cân chính xác 10 mg dithizon (TT) hòa tan vào cloroform (TT) và thêm vừa đủ 100 ml với cùng dung môi.
Bảo quản dung dịch ở chỗ tối 4 °C đến 8 °C trong vòng 1 tháng. Trước khi dùng pha loãng 10 lần.
Phương pháp vi sinh vật
Nguyên lý:
Phương pháp thực hiện dựa trên sự hình thành vòng ức chế kháng khuẩn được tạo bởi dung dịch thimerosal chuẩn và thimerosal có trong mẫu thử; so sánh đường kính vòng ức chế kháng khuẩn.
Cách tiến hành:
Pha thạch thường, đun nóng chảy hoàn toàn và đổ thạch dày 4 mm lên một phiến kính hình chữ nhật phẳng. Để thạch nguội hoàn toàn, đục lỗ thạch (đường kính 8 mm) sao cho khoảng cách giữa các lỗ thạch ít nhất là 30 mm. Cấy chủng Staphylococcus aureus trên thạch nghiêng, ủ ở 37 °C trong 18 h đến 24 h. Gặt và pha vi khuẩn bằng nước muối sinh lý vô khuẩn thành huyền dịch 109 vi khuẩn/ml. Láng đều huyền dịch vi khuẩn (0,5 ml/đĩa thạch) trên đĩa thạch đã đục lỗ. Nhỏ vào mỗi lỗ 100 μl dung dịch thimerosal chuẩn chứa 1 g thimerosal/1 lít ở các độ pha loãng 1/2,5, 1/5, 1/10, 1/20, 1/40, 1/80, 1/160. Ủ ở 37 °C trong 24 h.
Đọc kết quả:
Đo đường kính các vùng ức chế vi khuẩn mọc của các đậm độ thimerosal chuẩn, xây dựng đường chuẩn. Dựa vào đường chuẩn và đường kính vùng ức chế của vắc xin mẫu thử để tính ra hàm lượng thimerosal có trong vắc xin mẫu thử.
Tiêu chuẩn chấp thuận
Hàm lượng thimerosal trong mỗi vắc xin và sinh phẩm có thể khác nhau tùy theo yêu cầu cụ thể, nhưng không được thấp hơn hàm lượng cho thấy có khả năng kháng khuẩn và không cao hơn 115% hàm lượng thimerosal ghi trên nhãn.
Yêu cầu chung cho hàm lượng thimerosal trong các vắc xin và sinh phẩm có sử dụng thimerosal làm chất bảo quản đều không được vượt quá 0,02%.
15.31 XÁC ĐỊNH HIỆU LỰC VẮC XIN DẠI THEO PHƯƠNG PHÁP NIH
Nguyên lý
Thử nghiệm NIH (National Institute of Health - USA) được Tổ chức Y tế Thế giới chính thức công nhận để kiểm tra công hiệu vắc xin dại bất hoạt. Công hiệu vắc xin dại được xác định bằng cách so sánh liều hữu hiệu bảo vệ chuột chống lại liều gây chết của virus dại với một vắc xin dại mẫu chuẩn đã biết trước công hiệu (lU/ml). Thử nghiệm được tiến hành song song 2 vắc xin (mẫu thử và mẫu chuẩn), sau 14 ngày thử thách với cùng 1 liều chủng virus thử thách CVS.
Phương pháp tiến hành
Pha vắc xin và gây miễn dịch:
Pha vắc xin mẫu chuẩn bằng nước vô khuẩn để tiêm (hoặc PBS pH 7,2 - 7,4) để có được độ pha có chứa khoảng 2 lU/ml. Phân chia vào các ống một lượng vắc xin đủ dùng cho một lần pha tiêm miễn dịch. Bảo quản ở -20 °C.
Pha vắc xin thử: Nếu là vắc xin đông khô phải hoàn nguyên bằng nước hồi chỉnh cho tới nồng độ liều tiêm cho người.
Dung dịch vắc xin chuẩn và dung dịch vắc xin thử đã hoàn nguyên tiếp tục được pha loãng thành ít nhất 3 độ pha trong nước muối sinh lý (hoặc PBS). Các độ pha được chọn sao cho có ED50 trung bình không nằm ngoài các độ pha loãng của vắc xin mẫu thử và vắc xin mẫu chuẩn, thường sử dụng độ pha loãng bậc 5 là 1/5,1/25, 1/125, 1/625 để pha tiêm miễn dịch.
Gây miễn dịch: Mỗi độ pha loãng vắc xin được gây miễn dịch cho ít nhất 16 chuột nhắt trắng, 4 đến 5 tuần tuổi, 11 g đến 15 g. Mỗi chuột tiêm 0,5 ml vào phức mạc. Tiêm 2 mũi vào ngày 0 và ngày 7. Nhóm chuột đối chứng gồm 40 con được nuôi song song trong cùng điều kiện với nhóm chuột miễn dịch. Chuột đối chứng được dùng để chuẩn độ hiệu giá chủng virus thử thách, mỗi độ pha loãng tiêm 10 chuột.
Pha chủng CVS và thử thách.
Nhóm chuột miễn dịch được tiêm thử thách với liều trong khoảng từ 10 đến 100 LD50. Thường dùng liều thử thách có chứa 25 đến 50 LD50.
Ví dụ: sau 5 lần chuẩn độ hiệu giá chủng CVS là 10-6,81. Như vậy để có được liều 25 LD50 phải dùng độ pha đậm đặc hơn 1,4 log tức là độ pha 10-5,4.
Cách pha cụ thể:
Dùng huyết thanh ngựa 2% pha hỗn dịch CVS 20% để có được hỗn dịch 10% (hỗn dịch A). Sau đó pha tiếp:
|
| Độ pha loãng | Ký hiệu |
| 0,3 ml A + 2,7 ml huyết thanh 2% | 10-2 | B |
| 0,3 ml B + 2,7 ml huyết thanh 2% | 10-3 | C |
| 0,3 ml C + 2,7 ml huyết thanh 2% | 10-4 | D |
| 1 ml D + 24 ml huyết thanh 2% | 10-5,4 | E |
Lấy độ pha E tiêm cho tất cả nhóm chuột miễn dịch, mỗi chuột tiêm 0,03 ml vào não.
Để chuẩn độ hiệu giá chủng CVS thử thách thực tế trong thử nghiệm, tiếp tục pha loãng bậc 10 từ hỗn dịch E (10-5,4) để có các độ pha 10-6,4, 10-7,4, 10-8,4. Mỗi độ pha chủng CVS tiêm vào não cho 10 chuột, mỗi chuột 0,03 ml.
Theo dõi và đọc kết quả:
Sau khi tiêm thử thách, theo dõi các nhóm chuột trong vòng 14 ngày. Những chuột liệt hoặc chết sau ngày thứ 5 mới được tính vào kết quả. Những chuột có dấu hiệu co giật, liệt, rối loạn vận động được coi như chết vì bệnh dại. Cần ghi chép kết quả hàng ngày. Đọc và tính kết quả cuối cùng vào ngày thứ 14 sau tiêm thử thách.
Tính kết quả:
Hiệu giá chủng thử thách CVS tính theo phương pháp Reed-Muench.
Công hiệu (ED50) tính theo chương trình Probit analysis của WHO.
Tiêu chuẩn chấp thuận: Vắc xin dại phải đạt công hiệu ≥ 2,5 lU/liều.
15.32 XÁC ĐỊNH HÀM LƯỢNG NITƠ PROTEIN CỦA VẮC XIN VÀ SINH PHẨM BẰNG THUỐC THỬ NESSLER
Nguyên lý
Phương pháp dựa vào tính chất của thuốc thử Nessler cho phản ứng màu với ion amoni (NH4+) được tạo thành sau khi vô cơ hóa các protein.
Phương pháp tiến hành
a. Chuẩn bị mẫu thử
Cho 1 ml chế phẩm và 1 ml dung dịch acid tricloracetic 20% (TT) vào một ống ly tâm 10 ml, để ở nhiệt độ 4 °C đến 8 °C trong 10 h đến 12 h. Sau đó ly tâm 3500 r/min trong 1 h, loại bỏ phần nước. Rửa tủa bằng 2 ml dung dịch acid tricloracetic 5%, tiếp tục ly tâm 3500 r/min trong 1 h, loại bỏ phần nước. Thêm vào cặn 0,2 ml acid sulfuric đậm đặc (TT) đem vô cơ hóa trên bếp cát cho đến khi dung dịch mất màu. Để tăng tốc độ phản ứng thỉnh thoảng cho vài giọt dung dịch oxy già đậm đặc vào ống đốt sau khi làm nguội. Khi chế phẩm đã được vô cơ hóa hoàn toàn, dung dịch mất màu, thêm nước cất 2 lần vừa đủ b ml, thu được dung dịch A.
Lấy 1 ml dung dịch A vào ống nghiệm, thêm 8,5 ml nước cất 2 lần và 0,5 ml thuốc thử Nessler (TT), được mẫu thử.
b. Chuẩn bị mẫu trắng gồm: 9,5 ml nước cất 2 lần và 0,5 ml thuốc thử Nessler (TT).
c. Dựng đường chuẩn:
Hút dung dịch amoni sulfat chuẩn có hàm lượng 0,05 mg nitơ/ml vào các ống nghiệm theo thứ tự sau: 0,1 ml; 0,2 ml; 0,3 ml; 0,4 ml; 0,5 ml; 0,6 ml; 0,7 ml. Thêm nước cất 2 lần vừa đủ 9,5 ml và 0,5 ml thuốc thử Nessler (TT).
Lắc đều các ống nghiệm.
Đo mật độ quang (Phụ lục 4.1) ở bước sóng 420 nm.
Vẽ đồ thị đường chuẩn.
Lượng nitơ protein trong chế phẩm được tính theo công thức sau:
![]()
trong đó:
a: lượng nitơ tìm được trên đường chuẩn (mg);
b: thể tích dung dịch A (ml);
1,0: thể tích mẫu sau pha loãng đem so màu (ml);
1,0: thể tích mẫu đem tủa rồi vô cơ hóa (ml);
6,25: hệ số chuyển đổi protein từ nitơ.
Cách pha dung dịch amoni sulfat chuẩn có hàm lượng nitơ 0,05 mg/ml:
Cân 0,2375 g amoni sulfat (đã được sấy khô trong bình hút ẩm bằng silica gel đến trọng lượng không đổi), hòa tan vừa đủ 500 ml bằng nước cất 2 lần. Dung dịch vừa pha có hàm lượng niơ 0,1 mg/ml, trước khi dùng pha loãng gấp đôi bằng nước cất 2 lần.
Tiêu chuẩn cho phép
Hàm lượng nitơ protein trong vắc xin và sinh phẩm tùy theo yêu cầu đối với từng loại mẫu thử.
15.33 XÁC ĐỊNH pH CỦA VẮC XIN VÀ SINH PHẨM
Nguyên lý
Trị số pH của một dung dịch vắc xin hay sinh phẩm được xác định bằng cách đo thế hiệu giữa điện cực chỉ thị nhạy cảm với ion hydrogen (thường là điện cực thủy tinh) và một điện cực so sánh (ví dụ điện cực calomel bão hòa). pH là một giá trị biểu thị quy ước nồng độ ion hydrogen của một dung dịch. pH của một dung dịch mẫu thử liên quan với pH của dung dịch mẫu chuẩn theo biểu thức sau:
![]()
trong đó:
E là điện thế, tính bằng von, của pin chứa dung dịch mẫu thử;
Es là điện thế, tính bằng von, của pin chứa dung dịch mẫu chuẩn (đã biết);
pHs là pH của dung dịch mẫu chuẩn;
k là hệ số có giá trị thay đổi theo nhiệt độ được ghi ở bảng dưới đây:
| Nhiệt độ | k |
| 15 °C | 0,0572 |
| 20 °C | 0,0582 |
| 25 °C | 0,0592 |
| 30 °C | 0,0601 |
| 35 °C | 0,0611 |
Phương pháp tiến hành
pH của một dung dịch vắc xin, sinh phẩm được đo bằng máy đo pH (pH meter). Máy đo pH là một điện thế kế có trở kháng đầu vào gấp ít nhất 100 lần trở kháng của các điện cực sử dụng và thường được phân độ theo đơn vị pH và có độ nhạy để phát hiện được những thay đổi cỡ 0,05 đơn vị pH hoặc ít nhất 0,003 V. Các điện cực thủy tinh là phù hợp và các kiểu máy đo pH, kể cả máy đo pH hiện số, đều phải đáp ứng yêu cầu trên. Vận hành máy đo pH tùy theo loại máy đo pH và tùy theo hướng dẫn của nhà sản xuất. Tất cả các phép đo đều cần phải tiến hành trong cùng một điều kiện nhiệt độ khoảng từ 20 °C đến 25 °C, trừ trường hợp có quy định khác trong chuyên luận riêng.
Trước khi đo pH của mẫu thử, cần chuẩn định máy đo pH bằng dung dịch pH chuẩn do nhà sản xuất cung cấp (tối thiểu sử dụng 2 dung dịch pH chuẩn mà pH của dung dịch mẫu thử phải nằm trong khoảng đó). Ví dụ: dung dịch mẫu thử cần đo pH thường nằm trong khoảng pH 4 - 7, cần sử dụng 2 dung dịch pH chuẩn là pH 4 và pH 7 để chuẩn định máy đo pH; hoặc nếu dung dịch mẫu thử cần đo pH thường nằm trong khoảng pH 7 -10, cần sử dụng 2 dung dịch pH chuẩn là pH 7 và pH 10 để chuẩn định máy đo pH trước khi đo pH của dung dịch mẫu thử.
Tráng đầu điện cực pH bằng nước cất 2 lần, dùng giấy thấm mềm chuyên dụng để thấm khô đầu điện cực và tiến hành đo pH của dung dịch mẫu thử. Số lượng mẫu thử tối thiểu dùng để kiểm tra pH là 15 ml đến 20 ml và nên để ở nhiệt độ phòng trước khi đo. Lắc đều và rót mẫu thử vào cốc đựng mẫu, nhúng ngập đầu điện cực vào cốc mẫu thử và để cho chỉ số pH hiện trên màn hình ổn định thì xác định pH của mẫu thử. Sau khi đo pH của mẫu thử xong, cần tráng đầu điện cực bằng nước cất 2 lần, dùng giấy thấm mềm chuyên dụng để thấm khô đầu điện cực và nhúng ngập đầu điện cực của máy đo pH vào dung dịch bảo quản điện cực, ngắt điện của máy đo pH.
Tiêu chuẩn chấp thuận
Tiêu chuẩn pH chấp thuận tùy theo yêu cầu đối với từng loại mẫu thử.
15.34 XÁC ĐỊNH HÀM LƯỢNG PROTEIN TOÀN PHẦN TRONG VẮC XIN VÀ SINH PHẨM
Protein có trong vắc xin và sinh phẩm được xác định theo nhiều phương pháp khác nhau, tùy theo từng loại vắc xin và sinh phẩm.
Phương pháp Lowry
Nguyên lý:
Protein có trong mẫu vắc xin thử bị tủa với acid tricloracetic nóng. Xác định lượng tủa để tính ra lượng protein có trong vắc xin, sinh phẩm.
Phương pháp tiến hành:
Pha loãng mẫu thử (nếu cần thiết) để chỉnh hàm lượng protein của mẫu thử trong khoảng 20 μg/ml đến 120 μg/ml. Thêm 1 ml dung dịch acid tricloracetic 10% vào một ống nghiệm có chứa 1 ml mẫu thử. Đun nóng ở 80 °C trong nồi cách thủy trong 15 min. Làm nguội ở nhiệt độ phòng.
Ly tâm ở tốc độ 3500 r/min trong 30 min, loại bỏ phần dung dịch nước.
Thêm vào phần lắng cặn 2 ml dung dịch acid tricloracetic 5%. Lắc kỹ trên máy lắc và ly tâm lại một lần nữa ở tốc độ 3500 r/min trong 30 min, loại bỏ phần dung dịch nước.
Thêm 2,5 ml dung dịch Alkalin vào mỗi ống nghiệm nêu trên và ủ ở nhiệt độ phòng (thời gian ủ tùy từng loại mẫu thử) để hòa tan phần lắng cặn.
Thêm 2,5 ml nước cất và 0,5 ml thuốc thử Folin (pha loãng 1/2), ủ ở 37 °C trong 30 min. Ly tâm với tốc độ 3500 r/min đến 6000 r/min trong 30 min (với các chế phẩm chứa chất hấp phụ nhôm). Đo mật độ quang học ở bước sóng 750 nm trên quang phổ kế.
Song song tiến hành thử nghiệm với mẫu trắng: thay vào 1 ml mẫu thử là 1 ml nước cất.
Dựng đường chuẩn: Dựng đường chuẩn với dung dịch protein chuẩn (dung dịch albumin chuẩn ở các nồng độ 25 μg/ml; 50 μg/ml; 100 μg/ml; 150 μg/ml). Dựa vào hàm lượng protein tìm được trên đường chuẩn tính ra hàm lượng protein có trong mẫu thử.
Cách pha dung dịch Alkalin (pha ngay trước khi dùng):
Dung dịch đồng sulfat pentahydrat 2%: Hòa tan 2 g đồng sulfat pentahydrat vào vừa đủ 100 ml nước cất.
Dung dịch natri tartrat 4%: Hòa tan 4 g natri tartrat trong vừa đủ 100 ml nước cất.
Trộn lẫn 2 dung dịch trên được dung dịch A.
Hòa tan 0,8 g natri hydroxyd và 4 g natri carbonat trong vừa đủ 100 ml nước cất được dung dịch B.
Trộn 50 ml dung dịch B và 1 ml dung dịch A được dung dịch Alkalin.
Đo độ hấp thụ thực của protein ở bước sóng 280 nm
Nguyên lý:
Protein trong dung dịch hấp thụ tia tử ngoại ở bước sóng 280 nm, do sự có mặt các amino acid dãy thơm, chủ yếu là tyrosin và tryptophan trong cấu trúc protein.
Phương pháp tiến hành:
Dung dịch thử: Pha loãng mẫu thử đến hàm lượng protein trong khoảng 0,2 mg/ml đến 2 mg/ml, bằng dung dịch đệm thích hợp.
Dung dịch chuẩn: Pha loãng mẫu chuẩn bằng cùng dung dịch đệm với mẫu thử và có hàm lượng protein tương ứng với mẫu thử.
Giữ dung dịch thử và chuẩn ở nhiệt độ phòng. Đo độ hấp thụ các dung dịch này bằng cóng thạch anh ở bước sóng 280 nm, dùng nước cất làm mẫu trắng.
Hàm lượng protein trong mẫu thử (Cu) được-tính bởi công thức:
CU = CS (AU/AS)
trong đó:
CS: hàm lượng protein trong dung dịch chuẩn;
AU: độ hấp thụ của mẫu thử;
AS: độ hấp thụ của dung dịch chuẩn.
Phương pháp Bradford
Nguyên lý:
Dựa trên sự kết hợp giữa thuốc nhuộm Coomasie với protein trong môi trường acid.
Phương pháp tiến hành:
Pha loãng mẫu thử bằng nước cất đến hàm lượng thích hợp trong khoảng đường chuẩn.
Chuẩn bị dung dịch chuẩn BSA (bovin serum albumin) có hàm lượng trong khoảng 0,1 mg/ml đến 1 mg/ml.
Dùng nước cất làm mẫu trắng.
Thêm 2,5 ml thuốc nhuộm Coomasie 20% vào 0,05 ml mẫu trắng, mẫu chuẩn và mẫu thử, lắc đều. Để yên 10 min ở nhiệt độ phòng. Đo mật độ quang (Phụ lục 4.1) ở bước sóng 595 nm (không dùng cóng thạch anh do sẽ bị thuốc nhuộm gắn vào).
Hàm lượng protein trong mẫu thử được tính toán dựa vào đường chuẩn.
Phương pháp đồng/bicinchoninic acid
Nguyên lý:
Dựa vào sự khử của ion Cu2+ thành Cu1+ do protein, dùng acid bicinchoninic (BCA) để xác định Cu1+.
Phương pháp tiến hành:
Pha loãng mẫu thử bằng nước cất đến hàm lượng thích hợp trong khoảng đường chuẩn.
Pha dung dịch chuẩn bằng nước cất (không ít hơn 5 nồng độ) có hàm lượng trong khoảng 10 μg/ml đến 1200 μg/ml.
Dùng nước cất làm mẫu trắng.
Thêm 2 ml thuốc thử đồng-BCA vào 0,1 ml mẫu trắng, mẫu chuẩn và mẫu thử, lắc đều. Ủ trong nồi cách thủy 37 °C trong 30 min. Làm nguội ở nhiệt độ phòng trong 60 min, đo mật độ quang (Phụ lục 4.1) ở bước sóng 562 nm, dùng cóng thạch anh.
Dựa vào đường chuẩn tính ra hàm lượng protein có trong mẫu thử.
Pha thuốc thử BCA: Hòa tan 10 g dinatri bicin-choninat, 20 g natri carbonat monohydrat, 1,6 g natri tatrat, 4 g natri hydroxyd và 9,5 g natri hydrogen carbonat vào nước cất. Điều chỉnh pH 11,25 nếu cần bằng dung dịch natri hydroxyd hoặc dung dịch natri hydrogen carbonat. Thêm nước cất vừa đủ 1000 ml, lắc đều.
Pha thuốc thử đồng - BCA: Trộn 1 ml dung dịch đồng sulfat 40 g/l với 50 ml thuốc thử BCA.
Phương pháp biuret
Nguyên lý:
Dựa vào sự tương tác của ion Cu2+ với protein trong dung dịch alkalin, sản phẩm thu được có độ hấp thụ cực đại ở bước sóng 545 nm.
Phương pháp tiến hành:
Pha loãng mẫu thử bằng dung dịch nước muối sinh lý đến hàm lượng thích hợp trong khoảng đường chuẩn.
Pha dung dịch chuẩn bằng dung dịch nước muối sinh lý (không ít hơn 3 nồng độ) có hàm lượng trong khoảng từ 0,5 mg/ml đến 10 mg/ml.
Dùng dung dịch nước muối sinh lý làm mẫu trắng.
Lấy một thể tích dung dịch mẫu thử, thêm đồng lượng dung dịch natri hydroxyd 60 g/l, lắc đều. Ngay lập tức cho thuốc thử biuret với lượng bằng 0,4 lần thể tích dung dịch mẫu thử và lắc nhanh. Để yên ở nhiệt độ phòng 15 min. Trong vòng 90 min sau khi cho thuốc thử biuret, đo mật độ quang (Phụ lục 4.1) của mẫu thử ở bước sóng 545 nm.
Song song tiến hành với mẫu trắng và mẫu chuẩn.
Dựa vào đường chuẩn tính ra hàm lượng protein có trong mẫu thử.
Pha thuốc thử biuret: Hòa tan 3,46 g đồng sulfat (TT) trong 10 ml nước cất nóng, để nguội (dung dịch A). Hòa tan 34,6 g natri citrat (TT) và 20 g natri carbonat khan (TT) vào 80 ml nước cất nóng, để nguội (dung dịch B). Trộn dung dịch A và B, thêm nước cất vừa đủ 200 ml. Dùng trong 6 tháng, không dùng nếu thấy vẩn đục hoặc có tủa.
Phương pháp quang phổ huỳnh quang
Nguyên lý:
Dựa vào dẫn xuất của protein với o-phthaldehyd do chất này phản ứng với các amin chính của protein (N- terminal amino acid và nhóm ε-amino của lysin tồn dư).
Phương pháp tiến hành:
Pha loãng mẫu thử bằng nước muối sinh lý (TT) đến hàm lượng thích hợp trong khoảng đường chuẩn. Điều chỉnh pH từ 8 đến 10,5 trước khi thêm thuốc thử phthaldehyd.
Pha dung dịch chuẩn bằng nước muối sinh lý (TT) (không ít hơn 5 nồng độ) có hàm lượng trong khoảng từ 10 μg/ml đến 200 μg/ml. Điều chỉnh pH từ 8 đến 10,5 trước khi thêm thuốc thử phthaldehyd. Dùng nước muối sinh lý (TT) làm mẫu trắng.
Trộn 10 μl các dung dịch mẫu thử, mẫu chuẩn, mẫu trắng với 0,1 ml thuốc thử phthaldehyd, để yên ở nhiệt độ phòng trong 15 min. Thêm 3 ml dung dịch natri hydroxyd 0,5 M và lắc đều. Đo cường độ huỳnh quang các dung dịch mẫu chuẩn và mẫu thử ở bước sóng kích thích 340 nm và bước sóng phát xạ giữa 440 nm và 445 nm.
Dựa vào đường chuẩn tính ra hàm lượng protein có trong mẫu thử.
Pha dung dịch đệm borat: Hòa tan 61,83 g acid boric trong nước cất và điều chỉnh pH 10,4 bằng dung dịch kali hydroxyd, thêm nước cất vừa đủ 1000 ml, lắc đều.
Pha dung dịch gốc phthaldehyd: Hòa tan 1,20 g o-phthaldehyd trong 1,5 ml methanol (TT), thêm 100 ml dung dịch đệm borat (TT), lắc đều. Thêm 0,6 ml dung dịch macrogol 23 lauryl ether và lắc đều. Bảo quản ở nhiệt độ phòng sử dụng trong 3 tuần.
Pha thuốc thử phthaldehyd: Thêm 15 μl 2-mercaptoethanol vào 5 ml dung dịch gốc phthaldehyd. Chuẩn bị dung dịch này trước khi dùng 30 min. Sử dụng trong vòng 24 h.
Tiêu chuẩn chấp thuận
Theo quy định trong chuyên luận riêng.
15.35 XÁC ĐỊNH ĐỘ ẨM TỒN DƯ TRONG VẮC XIN ĐÔNG KHÔ
Nguyên lý
Sử dụng phương pháp Karl Fischer để xác định độ ẩm tồn dư trong vắc xin đông khô theo nguyên lý chung là dựa trên phản ứng toàn lượng của nước với lưu huỳnh dioxyd và iod trong sự có mặt của alcohol và một chất base hữu cơ thích hợp.
Hiện nay có nhiều thiết bị để đo độ ẩm tồn dư trong các vắc xin, sinh phẩm đông khô, tuy nhiên nguyên tắc của các thiết bị này đều phải cấu tạo sao cho thao tác thuận tiện và tránh ẩm.
Phương pháp tiến hành
Tùy theo thiết bị của các hãng khác nhau nên thao tác theo hướng dẫn vận hành của hãng đó.
Phương pháp phổ biến hiện nay để đo độ ẩm tồn dư của các vắc xin và sinh phẩm đông khô là phương pháp Karl Fischer. Vận hành máy đo độ ẩm tồn dư theo hướng dẫn sử dụng. Rót dung dịch chuẩn anod và cathod vào khoang tương ứng đến đúng mức quy định, cần lưu ý qua một thời gian nhất định, methanol trong dung dịch cathod có thể thấm qua màng trao đổi ion bằng cách thẩm thấu. Để giảm bớt tình trạng này không nên rót hóa chất đầy quá mức quy định.
Bật máy và cài đặt phương pháp lựa chọn. Nếu máy đo độ ẩm tồn dư không hoạt động trong vòng 3 tuần thì vẫn nên bật máy vài giờ trong khoảng thời gian này dù không tiến hành kiểm tra mẫu gì để nạp pin và đảm bảo tất cả các thông tin của chương trình vẫn được duy trì. Cần đảm bảo trước khi nạp mẫu thử tỷ lệ% độ ẩm trên màn hình phải ổn định và ở mức độ thấp để đủ đạt được kết quả theo yêu cầu.
Cân chính xác khối lượng tổng của cả mẫu thử và vật chứa mẫu.
Nhập khối lượng tổng của cả mẫu thử và vật chứa mẫu vào máy, sau đó nhập khối lượng của vật chứa mẫu. Đưa mẫu vào khoang bên ngoài qua đường bơm mẫu (nếu vận hành trong điều kiện độ ẩm cao và sáng thì nên đóng kín nắp khoang chứa mẫu thử khi vận hành).
Khi thực hiện xong thử nghiệm, máy in sẽ tự động in ra kết quả độ ẩm tồn dư của mẫu thử.
Tiêu chuẩn chấp thuận
Độ ẩm tồn dư trong vắc xin và sinh phẩm đông khô không được vượt quá 3% (≤ 3%), trừ khi có quy định trong chuyên luận riêng.
15.36 PHÁT HIỆN MYCOPLASMA BẰNG PHƯƠNG PHÁP NUÔI CẤY
Nguyên vật liệu
Mẫu thử nghiệm: Nước nổi nuôi tế bào, hỗn dịch virus, vắc xin bán thành phẩm và thành phẩm.
Môi trường nuôi cấy: Môi trường lỏng 1: LM1 (pH 7,6 đến 7,8; môi trường có màu đỏ) và môi trường lỏng 2: LM2 (pH 7,2; môi trường có màu vàng cam), dưới dạng 100 ml/chai hoặc 10 ml/ống.
Môi trường rửa lọc: Môi trường PPLO lỏng.
Môi trường đặc: Môi trường thạch PPLO.
Màng lọc 0,1 μm, xy lanh 10 ml và dụng cụ cần thiết đủ cho tiến hành thử nghiệm.
Các bước tiến hành
Chuẩn bị môi trường
Pha môi trường LM1 (pH 7,6 đến 7,8): 500 ml.
Nước cất pha tiêm: 375 ml.
Môi trường PPLO lỏng: 8 g.
Đỏ phenol 0,4%: 2,5 ml.
Hấp ở 121 °C trong 15 min, sau đó thêm 75 ml huyết thanh ngựa, 50 ml Yeast Extract 25%, 6 ml glucose 25% và 1,25 ml penicilin G. Trộn đều, ra chai 100 ml và ống thử 10 ml.
Pha môi trường LM2 (pH 7,0 đến 7,2): 500 ml.
Nước để pha thuốc tiêm: 375 ml.
Môi trường PPLO lỏng: 8 g.
L-Arginin: 1,5 g.
Đỏ phenol 0,4%: 2,5 ml.
Hấp ở 121 °C trong 15 min sau đó thêm 75 ml huyết thanh ngựa, 50 ml Yeast Extract 25%, và 1,25 ml penicilin G, 1 ml acid hydrocloric. Trộn đều, ra chai 100 ml và ống thử 10 ml.
Pha môi trường thạch PPLO: 500 ml.
Thạch PPLO: 13,5 g.
Nước để pha thuốc tiêm: 375 ml.
Hấp tại 121 °C trong 15 min sau đó thêm 75 ml huyết thanh ngựa, 50 ml Yeast Extract 25% và 1,25 ml penicilin G. Trộn đều, sau đó đổ thạch ra phiến 6 giếng: 8 ml/giếng, bảo quản 37 °C trước khi thử độ nhạy 1 ngày.
Pha môi trường rửa màng lọc:
Môi trường PPLO lỏng: 2,1 g.
Nước để pha thuốc tiêm: 100 ml.
Hấp ở 121 °C trong 15 min.
Các bước tiến hành
• Lọc mẫu: Sử dụng xylanh 10 ml hút 6 ml mẫu thử vào giá lọc 0,1 μm và bơm mẫu qua lọc cho đến hết.
• Rửa lọc: Sử dụng 30 ml môi trường rửa lọc và xylanh 10 ml, bỏ kim, sau đó bơm từ từ cho đến khi môi trường rửa qua hết lọc.
• Tháo lọc, lấy giấy lọc cho vào dĩa petri vô trùng, cắt đôi giấy lọc, cho mỗi nửa giấy lọc vào mỗi chai môi trường LM1 và LM2 đã chuẩn bị sẵn.
• Chai môi trường sau gây nhiễm được nuôi ở 37 °C trong 28 ngày.
• Cấy truyền trên môi trường lỏng: sau gây nhiễm 14 ngày, lấy mẫu từ chai môi trường nuôi cấy chuyển sang ống môi trường nuôi cấy cùng loại: 0,2 ml/ống thử, 3 ống thử mỗi loại môi trường và đặt tại 37 °C trong 14 ngày.
• Cấy truyền trên môi trường đặc: Thạch được chuẩn bị trên phiến 6 giếng, gây nhiễm 10 μl từ ống thử nghi ngờ lên chính giữa mặt thạch, dán kín phiến và nuôi tại 37 °C trong 10 ngày.
Đọc kết quả
• Đọc kết quả lần gây nhiễm đầu tiên: Ngày thứ 28.
• Đọc kết quả lần cấy truyền: Ngày thứ 14 trên môi trường lỏng và ngày thứ 10 trên môi trường thạch;
• Kết quả dương tính khi màu môi trường LM1 chuyển từ màu đỏ sang màu vàng cam; màu môi trường LM2 từ màu vàng cam sang màu đỏ ánh tím và có khuẩn lạc giống hình trứng ốp lếp trên môi trường thạch.
• Kết quả âm tính khi màu môi trường của 2 loại môi trường lỏng không thay đổi giống như ống thử đối chứng và không có khuẩn lạc mọc trên môi trường thạch.
15.37 XÁC ĐỊNH HÀM LƯỢNG Vi POLYSACHARID CỦA VẮC XIN THƯƠNG HÀN Vi POLYSACHARID
Nguyên lý
Sử dụng phản ứng điện di miễn dịch rocket để xác định hàm lượng Vi polysacharid trong vắc xin thương hàn Vi.
Phương pháp tiến hành
Pha dung dịch đệm và thạch agarose
• Pha dung dịch đệm barbital 1/2.
• Rót dung dịch đệm barbital 1/2 vào bể điện di.
• Pha thạch agarose (0,5 g thạch agarose hòa vào 40 ml dung dịch đệm barbital 1/2), đun cách thủy hoặc đun trên máy khuấy từ gia nhiệt cho đến khi thạch tan chảy hoàn toàn.
• Để thạch nguội đến khoảng 56 °C, cho 1 ml huyết thanh thỏ kháng Vi vào và lắc nhẹ tròn đều chai thạch sao cho huyết thanh thỏ kháng Vi hòa tan đều vào thạch nhưng không bị tạo bọt.
Tạo bản gel và đục lỗ thạch
• Đổ thạch trên khuôn, chờ thạch nguội hoàn toàn.
• Đục lỗ thạch bằng bộ đục lỗ chuyên dụng sao cho các giếng trên bản gel phải cách mép bản gel ít nhất 2 cm và cách nhau ít nhất 1 cm. Số lượng giếng trên bản gel phải được tính toán sao cho có đủ 5 giếng cho các hàm lượng kháng nguyên Vi chuẩn, 2 giếng (làm kép) cho mỗi mẫu thử.
Pha mẫu kháng nguyên Vi chuẩn
• Pha loãng kháng nguyên Vi chuẩn bằng dung dịch PBS ở các độ pha loãng như sau: 100 μg: 50 μg: 25 μg: 12,5 μg; 6,25 μg Vị polysacharid /ml.
• Lần lượt nhỏ mẫu chuẩn ở các độ pha loãng như trên vào từng giếng trên bản gel (5 μl/giếng); cho vào 2 giếng tiếp theo mỗi giếng 5 μl vắc xin mẫu thử.
Chạy điện di miễn dịch Rocket
• Đặt bản gel vào máy điện di theo hướng mẫu đi từ cực âm sang cực dương.
• Bật nguồn, cài đặt các thông số thích hợp về hiệu điện thế, cường độ dòng điện và thời gian chạy điện di của mẫu.
• Dùng giấy lọc Whatman làm cầu nối từ bể dung dịch điện di tới bản gel ở cả 2 đầu bản gel.
• Bấm nút "Run" để chạy điện di.
Rửa bản gel
• Sau khi chạy điện di xong, tắt nguồn, lấy bản gel ra khỏi máy điện di, cho vào hộp nhựa trong có nắp kín.
• Cho nước muối sinh lý ngập bản gel và ngâm, lắc nhẹ bằng máy lắc trong 3 h (thay nước muối sinh lý 1 h/lần).
• Hút hết nước muối sinh lý ra và thay bằng nước cất. Ngâm và lắc hộp nước cất có chứa bản gel liên tục. Sau khi ngâm được 1 h, thay nước cất 30 min/lần.
Ủ bản gel
• Đổ toàn bộ nước cất ra khỏi hộp.
• Phủ một lớp giấy lọc lên mặt bản gel để tránh cho bản gel không bị khô không đồng đều.
• Ủ bản gel qua đêm trong tủ ấm 37 °C.
Nhuộm và tẩy bản gel
• Bóc bỏ lớp giấy lọc trên mặt bản gel.
• Nhuộm bản gel bằng dung dịch nhuộm màu trong 15 min. Trong quá trình nhuộm, lắc đều hộp dung dịch nhuộm màu có chứa bản gel bằng máy lắc.
• Hút bỏ toàn bộ dung dịch nhuộm màu và bơm dung dịch tẩy màu vào hộp chứa bản gel; ngâm, lắc liên tục cho đến khi nhìn rõ những cột tên lửa trên bản gel. Nếu bản gel vẫn còn xanh đậm, có thể thay dung dịch tẩy màu mới nhằm rút ngắn thời gian tẩy màu của bản gel.
Đọc và tính kết quả
• Bản gel được đưa ra khỏi hộp nhựa và đặt trên phiến kính trong.
• Dùng thước đo chuyên dụng để đo chiều cao các cột tên lửa trên bản gel, từ các giếng kháng nguyên chuẩn và mẫu thử.
• Dựa vào chiều cao các cột tên lửa, xây dựng được đường chuẩn và tính kết quả hàm lượng kháng nguyên Vi từ mẫu thử bằng chương trình Excell.
Đánh giá kết quả
• Loạt vắc xin thương hàn Vi polysacharid được coi là đạt yêu cầu về chỉ số hàm lượng Vi polysacharid khi thử nghiệm có giá trị (valid) và đạt 35 μg đến 65 μg polysacharid/1,0 ml vắc xin.
• Nếu thử nghiệm xác định hàm lượng Vi polysacharid không có giá trị (invalid) vì một lý do nào đó thì phải nhắc lại thử nghiệm với số lượng mẫu bằng lượng mẫu của lần kiểm tra thứ nhất.
• Nếu thử nghiệm xác định hàm lượng Vi polysacharid có giá trị (valid) nhưng không đạt yêu cầu về hàm lượng như quy định thì nhắc lại thử nghiệm với số lượng mẫu gấp đôi. Ở lần kiểm tra thứ hai nếu hàm lượng Vi polysacharid đạt yêu cầu (nằm trong khoảng cho phép của tiêu chuẩn chấp thuận thì loạt vắc xin đó được coi là đạt yêu cầu về hàm lượng Vi polysacharid. Nếu kết quả hàm lượng Vi polysacharid không đạt yêu cầu thì loạt vắc xin đó phải hủy bỏ, không được phép xuất xưởng đưa vào sử dụng.
Tiêu chuẩn chấp thuận
(25 μg ± 30%) Vi polysacharid/ liều đơn vắc xin dùng cho người (0,5 ml).
15.38 XÁC ĐỊNH HÀM LƯỢNG POLYSACHARID TRONG VẮC XIN VÀ SINH PHẨM
Nguyên lý
Polysacharid có trong mẫu thử được chuyển thành đường đơn, sau đó phản ứng với anthron tạo thành hợp chất màu. Đo hợp chất màu này ở bước sóng 620 nm, từ đó xác định hàm lượng polysacharid trong mẫu thử.
Tiến hành
Pha loãng dung dịch glucose chuẩn 500 μg/ml bằng nước cất thành các nồng độ 2,5 μg/ml; 5,0 μg/ml; 7,5 μg/ml; 10,0 μg/ml; 12,5 μg/ml. Mẫu thử được điều chỉnh bằng nước cất để có nồng độ protein khoảng 200 μg/ml.
Hút 1 ml mẫu thử và mỗi dung dịch glucose chuẩn vừa pha loãng trên vào ống nghiệm, thêm 4 ml dung dịch anthron 0,2%; lắc đều, đun cách thủy 100 °C trong 15 min. Làm nguội nhanh trong nước đá và để ở nhiệt độ phòng trong 60 min.
Đo mật độ quang (Phụ lục 4.1) ở bước sóng 620 nm, dựng đường chuẩn, từ đó tính ra hàm lượng polysacharid trong mẫu thử.
Đơn vị tính: μg/100 μg protein.
Cách pha các dung dịch:
• Dung dịch glucose chuẩn 500 μg/ml: Hòa tan 500 mg glucose bằng nước cất trong bình định mức 100 ml, thêm nước cất vừa đủ. Trước khi dùng, pha loãng 10 lần bằng nước cất.
• Dung dịch anthron 0,2%: Cân 0,2 g anthron chuyển vào bình định mức 100 ml, thêm acid sulfuric 95% vào vừa đủ 100 ml.
Bảo quản dung dịch ở 2 °C đến 10 °C trong chai thủy tinh màu, sử dụng trong vòng 2 tuần sau khi pha.
Tiêu chuẩn chấp thuận
Tùy từng loại vắc xin và sinh phẩm. Đối với bán thành phẩm viêm gan B, hàm lượng polysacharid không lớn hơn 10 μg/100 μg protein.
15.39 XÁC ĐỊNH ĐỘ TINH KHIẾT KHÁNG NGUYÊN HbsAg
Thử nghiệm xác định độ tinh khiết kháng nguyên HBsAg được thực hiện trong giai đoạn bán thành phẩm của vắc xin viêm gan B.
Độ tinh khiết kháng nguyên HBsAg được xác định bằng phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Dung dịch đệm PBS pH 6,8.
Cách pha dung dịch đệm PBS pH 6,8: Cân 3,48 g natri phosphat monobasic, 9,75 g natri phosphat dibasic, 23,38 g natri clorid và 0,1 g natri azid, hòa tan trong vừa đủ 1 lít nước khử ion. Điều chỉnh pH bằng 6,8. Lọc qua màng lọc 0,45 μm.
Điều kiện sắc ký:
Cột TKgel G3000SW, Nhật Bản.
Nhiệt độ phòng
Tốc độ dòng: 0,6 ml/min.
Detector quang phổ tử ngoại đặt ở bước sóng 280 nm.
Thể tích tiêm: 50 μl.
Lọc mẫu thử qua màng lọc 0,45 μm trước khi cho mẫu vào cột.
Tiến hành sắc ký với dung dịch thử.
Tiêu chuẩn chấp thuận: Tỷ lệ diện tích pic HBsAg so với tổng số các pic không nhỏ hơn 95%.
15.40 XÁC ĐỊNH HÀM LƯỢNG LIPID TRONG VẮC XIN VÀ SINH PHẨM
Nguyên lý
Dựa trên phản ứng của lipid với vanilin trong môi trường acid sulfuric và acid phosphoric tạo thành hợp chất màu. Đo hợp chất màu này ở bước sóng 540 nm, từ đó xác định hàm lượng lipid trong mẫu thử.
Tiến hành
Mẫu thử được điều chỉnh bằng nước cất để có nồng độ protein khoảng 200 μg/ml.
Hút dung dịch dầu ô liu (olive oil) chuẩn 500 μg/ml vào các ống nghiệm lần lượt 0,1 ml; 0,2 ml; 0,3 ml; 0,4 ml; thêm nước cất vừa đủ 1 ml.
Hút 1 ml mẫu thử vào ống nghiệm, 1 ml nước cất vào mẫu trắng. Thêm 3 ml acid sulfuric 95% vào mỗi ống nghiệm, lắc mạnh bằng máy lắc. Đun cách thủy 100 °C trong 10 min, làm lạnh bằng nước đá trong 5 min. Thêm 13 ml dung dịch phosphovanilin (TT) vào mỗi ống nghiệm, lắc kỹ. Đun cách thủy 37 °C ± 2 °C trong 30 min, làm lạnh, để yên ở nhiệt độ phòng 30 min.
Đo mật độ quang (Phụ lục 4.1) ở bước sóng 540 nm, dựng đường chuẩn, từ đó tính ra hàm lượng lipid trong mẫu thử.
Đơn vị tính: μg/100 μg protein.
Cách pha các dung dịch
• Dung dịch dầu ô liu (olive oil) chuẩn 5000 μg/ml: Cân 0,25 g olive oil vào bình định mức 50 ml, thêm ethanol (TT) vừa đủ, lắc kỹ. Bảo quản dung dịch ở 4 °C đến 7 °C, dùng trong 1 tháng. Trước khi dùng, pha loãng 10 lần bằng nước cất.
• Dung dịch vanilin 0,6%: Hòa tan 6,0 g vanilin trong 6 ml đến 7 ml ethanol (TT), chuyển vào bình định mức 1 lít, thêm nước cất đến định mức.
• Dung dịch phosphovanilin: Cho 350 ml dung dịch vanilin 0,6% và 50 ml nước cất vào cốc thủy tinh, khuấy đều. Thêm 600 ml acid phosphoric 85% (TT) trong khi đang khuấy từ.
Tiêu chuẩn chấp thuận: Tùy từng loại vắc xin và sinh phẩm.
Đối với bán thành phẩm viêm gan B, hàm lượng lipid không lớn hơn 100 μg/100 μg protein.
15.41 XÁC ĐỊNH HÀM LƯỢNG CESI TRONG VẮC XIN VÀ SINH PHẨM
Nguyên lý
Cesi tồn dư trong quá trình tinh khiết HBsAg được xác định bằng phương pháp sắc ký trao đổi ion.
Tiến hành
Mẫu chuẩn: Hút 1 ml nước khử ion vào 3 ống nghiệm, thêm lần lượt 0,2 ml; 0,4 ml; 0,6 ml dung dịch cesi chuẩn 0,001%.
Mẫu thử: Hút 1 ml mẫu đã pha loãng 5 lần bằng nước khử ion vào ống nghiệm, thêm 0,4 ml dung dịch chuẩn. Lọc các dung dịch mẫu chuẩn và mẫu thử qua màng lọc 0,22 μm.
Điều kiện vận hành máy sắc ký ion: Cột lonPac Cs-12, detector đo độ dẫn điện 5 mcS đến 10 mcS, tốc độ dòng 1,0 ml/min, thể tích mẫu thử: 100 μl, nhiệt độ phòng.
Dựa vào diện tích pic để tính ra hàm lượng cesi trong mẫu thử.
Đơn vị tính: μg/20 μg protein.
Cách pha các dung dịch
Dung dịch acid hydrocloric 0,04 M (dung dịch rửa giải): Lọc 1,5 lít nước khử ion qua màng lọc 0,45 μm, hút 3,3 ml acid hydrocloric đậm đặc (TT) pha trong vừa đủ 1 lít nước khử ion vừa lọc trên.
Dung dịch tetrabutyl amoni hydroxyd (TBAOH) (dung dịch tái sinh): Hút 66,67 ml TBAOH vào ống đong 2 lít, thêm nước khử ion vừa đủ 2 lít, khuấy đều.
Dung dịch cesi chuẩn 0,01%: Cân chính xác khoảng 10 mg (a) cesi clorid, chuyển vào bình định mức 100 ml, thêm nước khử ion vừa đủ, lắc đều.
Pha loãng 10 lần dung dịch cesi chuẩn 0,01% bằng nước khử ion trước khi dùng. Hàm lượng cesi thực có trong dung dịch (X) được tính bằng công thức:
![]()
trong đó:
132,9 là phân tử lượng của Cs;
168,4 là phân tử lượng của CsCI;
10 là hệ số pha loãng dung dịch chuẩn;
1000 chuyển từ mg sang μg.
Tiêu chuẩn chấp thuận
Không lớn hơn 5 μg/20 μg protein.
PHỤ LỤC 16
(Quy định)
16.1 CÁC PHƯƠNG PHÁP TIỆT KHUẨN
Một sản phẩm được coi là vô khuẩn nếu nó hoàn toàn không chứa bất kỳ vi sinh vật sống nào. Phương pháp tiệt khuẩn là phương pháp làm cho một sản phẩm trở thành vô khuẩn. Do không thể đảm bảo độ vô khuẩn của sản phẩm bằng cách thử vô khuẩn, nên độ vô khuẩn của sản phẩm chỉ có thể đảm bảo bằng cách áp dụng một quy trình sản xuất đã được thẩm định thích hợp. Đối với mỗi quy trình tiệt khuẩn đã chọn phải tiến hành khảo sát để đảm bảo hiệu quả tiệt khuẩn và tính toàn vẹn của sản phẩm, bao gồm cả vật chứa và bao gói bên ngoài, trước khi áp dụng vào thực tế. Trong quá trình sản xuất phải tuân thủ đúng quy trình tiệt khuẩn đã thẩm định. Nếu có thay đổi lớn trong quy trình tiệt khuẩn, bao gồm cả sự thay đổi kích cỡ lô, phải tái thẩm định quy trình. Trong khi xây dựng quy trình sản xuất thuốc vô khuẩn phải tuân theo các nguyên tắc thực hành tốt sản xuất GMP, cụ thể là:
Sử dụng công nhân lành nghề đã trải qua quá trình huấn luyện thích hợp;
Thiết kế nhà xưởng phù hợp;
Thiết kế máy móc, thiết bị sản xuất sao cho dễ vệ sinh và tiệt khuẩn;
Áp dụng các biện pháp phòng ngừa thích hợp nhằm làm giảm số lượng vi sinh vật trong sản phẩm trước khi tiệt khuẩn;
Thẩm định quy trình sản xuất cho tất cả các công đoạn trong dây chuyền sản xuất có nguy cơ nhiễm khuẩn;
Thiết lập và thực thi chương trình giám sát chất lượng môi trường sản xuất và các quy trình kiểm tra trong quá trình sản xuất.
Các phương pháp mô tả dưới đây chủ yếu dùng để diệt hoặc loại bỏ vi khuẩn, nấm men và nấm mốc ra khỏi sản phẩm cần tiệt khuẩn. Đối với các sản phẩm sinh học có nguồn gốc từ người hay động vật, trong quá trình thẩm định phải chứng minh khả năng loại bỏ hay làm bất hoạt virus của quy trình tiệt khuẩn.
Nếu có thể, nên ưu tiên chọn phương pháp tiệt khuẩn thành phẩm. Nếu không thể tiệt khuẩn thành phẩm được, có thể loại vi khuẩn bằng phương pháp lọc qua màng lọc giữ lại vi khuẩn hay phương pháp sản xuất vô khuẩn. Trong mọi trường hợp, vật chứa và nút đậy phải có khả năng duy trì được tính vô khuẩn của sản phẩm cho đến khi hết hạn dùng.
Mức bảo đảm vô khuẩn
Sự khử khuẩn bằng tác nhân vật lý và hóa học tuân theo quy luật số mũ, do đó luôn luôn có khả năng còn vi sinh vật sống tồn tại trong sản phẩm sau khi tiệt khuẩn với một xác suất thống kê nhất định. Mức bảo đảm vô khuẩn SAL (sterility assurance level) của một quy trình tiệt khuẩn đã cho là xác suất tồn tại một đơn vị đóng gói không vô khuẩn trong toàn bộ lô sản phẩm sau khi tiệt khuẩn bằng quy trình đó.
Ví dụ:
SAL = 10-6 có nghĩa là có khả năng có không nhiều hơn một vi sinh vật sống trong 1 x 106 đơn vị đóng gói của sản phẩm đã tiệt khuẩn. Đối với một quy trình tiệt khuẩn đã cho, SAL phụ thuộc số lượng vi sinh vật có trong sản phẩm trước khi tiệt khuẩn, sức đề kháng của nó với tác nhân tiệt khuẩn và môi trường trong đó vi sinh vật tồn tại trong quá trình xử lý. Khi có thể, sử dụng mức bảo đảm vô khuẩn SAL để đánh giá hiệu quả tiệt khuẩn của các phương pháp mô tả trong chuyên luận này. SAL của mỗi quy trình tiệt khuẩn được thiết lập trong khi thẩm định quy trình.
Phương pháp và điều kiện tiệt khuẩn
Tiến hành tiệt khuẩn theo một trong các phương pháp giới thiệu dưới đây. Có thể cải tiến hoặc kết hợp các phương pháp đã giới thiệu, tuy nhiên phải tiến hành thẩm định phương pháp đã cải tiến để bảo đảm hiệu quả tiệt khuẩn của phương pháp cũng như để đảm bảo tính toàn vẹn của sản phẩm cần tiệt khuẩn, bao gồm cả bao bì sản phẩm. Đối với mọi phương pháp tiệt khuẩn, phải theo dõi các thông số vận hành quan trọng trong suốt quá trình tiệt khuẩn của mỗi lô sản phẩm nhằm đảm bảo các thông số đo được phù hợp với các thông số kỹ thuật đã thiết lập trước đó trong quá trình thẩm định phương pháp, kể cả trường hợp tiệt khuẩn trong điều kiện chuẩn.
Phương pháp tiệt khuẩn thành phẩm
Đối với phương pháp tiệt khuẩn thành phẩm, điểm quan trọng cần phải chú ý là sự phân bố không đồng đều của tác nhân tiệt khuẩn trong buồng tiệt khuẩn. Phải xác định được vị trí trong buồng tiệt khuẩn mà tác nhân tiệt khuẩn khó đến nhất (ví dụ vị trí có nhiệt độ thấp nhất trong nồi hấp) để bố trí sắp xếp sản phẩm có kiểu dáng và kích thước bao bì khác nhau.
Tiệt khuẩn bằng nhiệt ẩm
Phương pháp tiệt khuẩn bằng nhiệt ẩm được tiến hành trong một thiết bị chuyên dùng gọi là nồi hấp, tác nhân tiệt khuẩn là hơi nước bão hòa dưới áp suất cao. Đây là phương pháp ưu tiên chọn lựa, khi có thể, nhất là đối với các dung dịch nước. Điều kiện chuẩn của phương pháp tiệt khuẩn bằng nhiệt ẩm là đun nóng ở 121 °C trong ít nhất 15 min đối với các thành phẩm là dung dịch nước. Có thể tiến hành tiệt khuẩn ở điều kiện nhiệt độ và thời gian khác với điều kiện chuẩn nếu chứng minh được hiệu quả của chế độ tiệt khuẩn đã chọn. Phải khống chế số lượng vi sinh vật trong sản phẩm trước khi tiệt khuẩn bằng cách áp dụng quy trình sản xuất và các biện pháp phòng ngừa thích hợp để đạt được mức bảo đảm vô khuẩn SAL là 10-6 hay nhỏ hơn.
Trong quá trình tiệt khuẩn, phải theo dõi và ghi chép lại nhiệt độ và áp suất bên trong nồi hấp. Nhiệt độ thường được theo dõi bằng các đầu dò nhiệt đặt ở phần nguội nhất của nồi hấp. Thông số vận hành của mỗi chu trình tiệt khuẩn được ghi lại dưới dạng biểu đồ nhiệt độ - thời gian, hay bằng một cách thích hợp khác.
Khi đánh giá hiệu quả của quá trình tiệt khuẩn bằng phương pháp sinh học, dùng chỉ thị sinh học thích hợp theo Phụ lục 16.2 Chỉ thị sinh học dùng cho tiệt khuẩn.
Tiệt khuẩn bằng nhiệt khô
Tiệt khuẩn bằng nhiệt khô được thực hiện trong tủ sấy có gắn thiết bị đối lưu không khí để đảm bảo sự phân phối nhiệt đồng đều trong toàn bộ khoang tiệt khuẩn. Điều kiện chuẩn của phương pháp tiệt khuẩn bằng nhiệt khô là sấy ở nhiệt độ tối thiểu 160 °C trong ít nhất 2 h. Có thể tiến hành tiệt khuẩn ở điều kiện nhiệt độ và thời gian khác với điều kiện chuẩn nếu chứng minh được hiệu quả của chế độ tiệt khuẩn đã chọn. Phải khống chế số lượng vi sinh vật trong sản phẩm trước khi tiệt khuẩn bằng cách áp dụng quy trình sản xuất và các biện pháp phòng ngừa thích hợp để đạt được mức bảo đảm vô khuẩn SAL là 10-6 hay nhỏ hơn.
Trong quá trình tiệt khuẩn phải tiến hành theo dõi nhiệt độ bên trong tủ sấy. Nhiệt độ thường được đo bằng các đầu dò nhiệt đặt ở phần nguội nhất của tủ sấy. Phải ghi chép và lưu trữ thông số nhiệt độ của mỗi chu trình tiệt khuẩn.
Khi đánh giá hiệu quả của quá trình tiệt khuẩn bằng phương pháp sinh học, dùng chỉ thị sinh học thích hợp theo Phụ lục 16.2 Chỉ thị sinh học dùng cho tiệt khuẩn.
Tiệt khuẩn và khử chất gây sốt các dụng cụ thủy tinh thường tiến hành ở nhiệt độ cao hơn 220 °C. Trong trường hợp này, có thể thay chỉ thị sinh học bằng nội độc tố vi khuẩn bền nhiệt. Để kiểm tra quá trình tiệt khuẩn, cấy một hay nhiều vật liệu cần tiệt khuẩn với ít nhất 1000 đơn vị nội độc tố vi khuẩn. Lượng nội độc tố còn lại sau khi sấy tiệt khuẩn phải không được nhiều hơn 1/1000 so với lượng nội độc tố ban đầu (xem Phụ lục 13.2. Phép thử nội độc tố vi khuẩn).
Tiệt khuẩn bằng bức xạ ion hóa
Phương pháp này được tiến hành bằng cách cho sản phẩm trong bao bì cuối tiếp xúc với bức xạ ion hóa. Hai nguồn bức xạ ion hóa thường dùng là: tia γ phát ra từ nguồn phóng xạ thích hợp (ví dụ đồng vị phóng xạ Cobalt 60), và chùm electron năng lượng cao được gia tốc bởi máy gia tốc electron.
Đối với phương pháp tiệt khuẩn bằng bức xạ ion hóa, liều hấp thu chuẩn là 25 kGy. Có thể tiến hành tiệt khuẩn với liều hấp thu khác với liều hấp thu chuẩn nếu chứng minh được hiệu quả của chế độ tiệt khuẩn đã chọn. Phải khống chế số lượng vi sinh vật trong sản phẩm trước khi tiệt khuẩn bằng cách áp dụng quy trình sản xuất và các biện pháp phòng ngừa thích hợp để đạt được mức bảo đảm vô khuẩn SAL là 10-6 hay nhỏ hơn.
Trong quá trình tiệt khuẩn phải định kỳ đo bức xạ hấp thu bởi sản phẩm cần tiệt khuẩn bằng thiết bị đo liều thích hợp. Thiết bị đo liều phải được hiệu chỉnh với nguồn bức xạ chuẩn ít nhất mỗi năm một lần. Khi đánh giá hiệu quả của quá trình tiệt khuẩn bằng phương pháp sinh học, dùng chỉ thị sinh học thích hợp theo Phụ lục 16.2. Chỉ thị sinh học dùng cho tiệt khuẩn.
Tiệt khuẩn bằng chất khí
Chất khí thường dùng trong phương pháp này là ethylen oxyd. Đây là loại khí dễ cháy, có khả năng gây đột biến và có thể để lại vết chất độc trong vật liệu đem tiệt khuẩn, do đó chỉ nên chọn phương pháp này khi không thể chọn phương pháp nào khác. Phải có biện pháp thích hợp để bảo đảm sự thẩm thấu của khí và hơi ẩm vào bên trong vật liệu cần tiệt khuẩn cũng như làm giảm lượng ethylen oxyd hoặc sản phẩm phân hủy của nó trong sản phẩm đã tiệt khuẩn xuống dưới mức có thể gây nguy hiểm khi sử dụng sản phẩm.
Khi có thể, nên theo dõi và ghi chép nồng độ khí, độ ẩm tương đối, nhiệt độ và thời gian tiệt khuẩn của mỗi lô tiệt khuẩn.
Đánh giá hiệu quả của mỗi lô tiệt khuẩn bằng chỉ thị sinh học thích hợp (xem Phụ lục 16.2 Chỉ thị sinh học dùng cho tiệt khuẩn).
Tiệt khuẩn bằng phương pháp lọc
Phương pháp lọc thường được dùng để tiệt khuẩn các dung dịch kém bền nhiệt, không thể tiệt khuẩn trong bao bì cuối. Nguyên tắc của phương pháp là dẫn dung dịch cần tiệt khuẩn qua vật liệu lọc có khả năng giữ vi khuẩn. Vật liệu lọc thường dùng là các màng lọc có kích thước lỗ lọc 0,22 μm hay nhỏ hơn, có khả năng giữ lại 100% vi khuẩn Pseudomonas diminuta (ATCC 19146, NCIMB 11091, CIP 103020) trong điều kiện thí nghiệm thích hợp. Kiểm tra khả năng giữ khuẩn của màng lọc bằng cách lọc qua màng lọc một hỗn dịch vi khuẩn Pseudomonas diminuta trong môi trường nuôi cấy tryptone soya broth, hay môi trường dinh dưỡng tương đương, có nồng độ thích hợp sao cho mỗi cm2 bề mặt màng lọc có ít nhất 107 cfu và tốc độ lọc lớn hơn 30 Psi. Ủ dịch lọc ở 32 °C trong điều kiện hiếu khí, phải không được có vi khuẩn phát triển.
Quy trình sản xuất và môi trường sản xuất các sản phẩm tiệt khuẩn bằng phương pháp lọc phải được thiết kế thích hợp để giảm thiểu tối đa sự nhiễm khuẩn. Phải thường xuyên kiểm tra chất lượng môi trường sản xuất bằng một chương trình giám sát chất lượng môi trường đã được thẩm định. Các máy móc, thiết bị, vật chứa và nắp đậy phải được tiệt khuẩn trước khi sử dụng bằng phương pháp thích hợp. Vị trí lọc tiệt khuẩn nên bố trí càng gần nơi đóng ống càng tốt và thời điểm tiệt khuẩn nên bố trí càng gần thời điểm đóng ống càng tốt. Tất cả các thao tác sau khi lọc tiệt khuẩn phải thực hiện trong điều kiện vô khuẩn.
Cần có biện pháp thích hợp để tránh sự hấp thu hoạt chất trên màng lọc, cũng như để tránh sự thôi tạp chất từ màng lọc vào dịch lọc.
Trước khi lọc phải kiểm tra để đảm bảo màng lọc không bị rách trong quá trình lắp ráp và tiệt khuẩn hệ thống lọc, sau khi lọc xong cũng phải kiểm tra tính toàn vẹn của màng lọc bằng phương pháp thích hợp, ví dụ phương pháp đo áp suất điểm sủi bọt (bubble-point test) hay phương pháp đo chênh áp suất trước và sau khi qua màng lọc (pressure hold test).
Vì hiệu quả của quá trình lọc tiệt khuẩn phụ thuộc vào số lượng vi sinh vật trong sản phẩm trước khi lọc nên nếu không thể làm giảm số lượng vi sinh vật bằng phương pháp khác, nên lọc trước dung dịch cần tiệt khuẩn qua tiền lọc có khả năng giữ vi khuẩn.
Sản xuất vô khuẩn
Trong quá trình sản xuất vô khuẩn, sản phẩm chờ đóng gói, vật chứa rỗng và nắp đậy được tiệt khuẩn riêng rẽ sau đó kết hợp với nhau trong điều kiện vô khuẩn để thu được sản phẩm cuối. Bởi vì sản phẩm đã đóng gói không được tiệt khuẩn bổ sung bằng bất kỳ phương pháp nào khác nên quá trình sản xuất phải tiến hành trong điều kiện môi trường có chất lượng rất cao. Trước khi phối hợp, mỗi thành phần của sản phẩm cuối phải được tiệt khuẩn riêng bằng phương pháp thích hợp, ví dụ bằng nhiệt khô với vật chứa bằng thủy tinh, bằng nhiệt ẩm với nắp đậy bằng nhựa và bằng phương pháp lọc đối với các dung dịch. Để duy trì tính vô khuẩn của mỗi thành phần trong quá trình sản xuất phải chú ý đến các yếu tố sau:
Môi trường sản xuất;
Con người;
Các bề mặt tiếp xúc trực tiếp với sản phẩm hở;
Quy trình tiệt khuẩn các vật liệu bao gói và quy trình vận chuyển chúng vào khu vực đóng gói;
Thời gian lưu trữ tối đa của sản phẩm chờ đóng gói trước khi đóng vào bao bì cuối.
Để đảm bảo tính vô khuẩn của thành phẩm, cả quy trình tiệt khuẩn và quy trình đóng gói đều phải được thẩm định bằng phương pháp thích hợp trước khi áp dụng trong thực tế. Thẩm định quy trình đóng gói phải bao gồm thử nghiệm dùng môi trường dinh dưỡng nuôi cấy vi sinh vật thay cho sản phẩm thật (media fill test). Phải lấy mẫu để kiểm tra độ vô khuẩn tất cả các lô của bất kỳ sản phẩm nào được tiệt khuẩn bằng phương pháp lọc và/hoặc sản xuất vô khuẩn trước khi xuất xưởng (Xem Phụ lục 13.7 Thử vô khuẩn để biết thêm chi tiết).
16.2 CHỈ THỊ SINH HỌC DÙNG CHO TIỆT KHUẨN
Chỉ thị sinh học dùng cho tiệt khuẩn là những chế phẩm sinh học đã tiêu chuẩn hóa, được sản xuất từ các vi sinh vật chọn lọc, dùng để đánh giá hiệu quả của các quy trình tiệt khuẩn. Chỉ thị sinh học thường được sản xuất bằng cách cấy một lượng bào tử vi sinh vật chỉ thị lên vật mang trơ, ví dụ băng giấy lọc, bản mỏng thủy tinh hay ống plastic, sau đó đóng gói vật mang đã cấy khuẩn vào bao bì thích hợp nhằm bảo vệ sản phẩm tránh bị biến chất và tạp nhiễm. Vật liệu dùng làm bao gói phải bền, không bị phân hủy trong quá trình tiệt khuẩn, nhưng phải cho tác nhân tiệt khuẩn thấm vào bên trong để tiếp xúc với vi khuẩn. Hỗn dịch bào tử vi khuẩn đóng trong ống thủy tinh kín cũng có thể được dùng làm chỉ thị sinh học.
Đối với các chế phẩm lỏng, có thể cấy trực tiếp bào tử vi khuẩn chỉ thị vào một số đơn vị đóng gói đại diện của sản phẩm cần tiệt khuẩn hay vào chất lỏng có thành phần gần giống sản phẩm, nếu không thể cấy trực tiếp vào sản phẩm thật. Trong trường hợp này, phải có biện pháp kiểm tra thích hợp để đảm bảo bản thân chế phẩm hay sản phẩm giả đều không có khả năng ức chế vi khuẩn chỉ thị.
Các thông tin bắt buộc phải cung cấp kèm theo mỗi chỉ thị sinh học bao gồm: tên loài vi khuẩn dùng làm vi sinh vật đối chiếu, số định danh loài của bảo tàng giống gốc, số lượng bào tử sống trên mỗi vật mang, hạn dùng và trị số D. Trị số D là tham số tiệt khuẩn (khoảng thời gian hoặc liều hấp thụ) cần để làm giảm lượng bào tử sống xuống còn 10% so với lượng ban đầu. Chỉ thị sinh học có thể gồm hai hay nhiều loài vi khuẩn trên một vật mang, nhưng không được lẫn tạp khuẩn. Ngoài ra, trên nhãn chỉ thị sinh học phải cung cấp thông tin về môi trường nuôi cấy và điều kiện ủ.
Để kiểm tra một quy trình tiệt khuẩn, đặt chỉ thị sinh học tại vị trí được giả định, hoặc đã được xác định trước bằng phương pháp vật lý thích hợp khi có thể, là nơi mà tác nhân tiệt khuẩn khó luân chuyển đến nhất trong buồng tiệt khuẩn. Sau khi cho tiếp xúc với tác nhân tiệt khuẩn, chuyển vật mang vào trong môi trường dinh dưỡng thích hợp trong điều kiện vô khuẩn và đem ủ. Đối với chỉ thị sinh học đóng trong ống thủy tinh kín có chứa sẵn môi trường dinh dưỡng thì đem ủ ngay.
Các vi khuẩn chỉ thị được chọn lựa theo nguyên tắc:
a) Sức đề kháng của vi khuẩn chỉ thị đối với phương pháp tiệt khuẩn đã cho phải lớn hơn sức đề kháng của tất cả các vi khuẩn gây bệnh và các vi khuẩn có khả năng nhiễm vào sản phẩm cần tiệt khuẩn;
b) Không gây bệnh;
c) Dễ nuôi cấy.
Sau khi ủ, nếu quan sát thấy vi khuẩn chỉ thị phát triển, chứng tỏ quy trình tiệt khuẩn đã sử dụng không đạt yêu cầu.
1. Tiệt khuẩn bằng nhiệt ẩm
Chỉ thị sinh học được dùng để thẩm định quy trình tiệt khuẩn bằng nhiệt ẩm là bào tử của vi khuẩn Bacillus stearothermophilus (ví dụ: ATCC 7953, NCTC 10007, NCIMB 8157, hay CIP 52.81). Số lượng bào tử sống trên mỗi vật mang phải từ 5 x 105 trở lên. Trị số D ở 121 °C ± 1 °C (ký hiệu D121) phải lớn hơn 1,5 min. Phải kiểm tra để bảo đảm ở chế độ tiệt khuẩn ở 121 °C ± 1 °C trong 6 min chỉ thị sinh học vẫn còn vi khuẩn sống, nhưng bị diệt hoàn toàn ở chế độ tiệt khuẩn 121 °C ± 1 °C trong 15 min.
2. Tiệt khuẩn bằng nhiệt khô
Chỉ thị sinh học được dùng để thẩm định quy trình tiệt khuẩn bằng nhiệt khô là bào tử của vi khuẩn Bacillus subtilis (ví dụ: var. niger ATCC 9372, NCIMB 8058, CIP 77.18). Số lượng bào tử sống trên mỗi vật mang không được ít hơn 1 x 105 và trị số D ở 160 °C phải từ 1 min đến 3 min. Trong trường hợp tiệt khuẩn ở nhiệt độ cao hơn 220 °C, ví dụ tiệt khuẩn và khử chất gây sốt các dụng cụ thủy tinh, có thể dùng nội độc tố vi khuẩn bền nhiệt thay cho bào tử vi khuẩn (xem Phụ lục 16.1. Các phương pháp tiệt khuẩn để biết thêm chi tiết).
3. Tiệt khuẩn bằng bức xạ ion hóa
Chỉ thị sinh học có thể được sử dụng để kiểm tra mỗi lô tiệt khuẩn trong điều kiện sản xuất bình thường như một biện pháp bổ sung để thẩm định phương pháp tiệt khuẩn bằng bức xạ ion hóa. Thường dùng bào tử của vi khuẩn Bacillus pumilus (ví dụ: ATCC 27.142, NCTC 10327, NCIMB 10692, CIP 77.25). Số lượng bào tử sống trên mỗi vật mang không được ít hơn 1 x107. Trị số D không được nhỏ hơn 1,9 kGy. Phải kiểm tra để bảo đảm không còn vi khuẩn sống sau khi cho chỉ thị sinh học tiếp xúc với bức xạ ion hóa ở liều hấp thu 25 kGy (liều hấp thu tối thiểu).
4. Tiệt khuẩn bằng chất khí
Chỉ thị sinh học được sử dụng trong tất cả các quy trình tiệt khuẩn bằng chất khí, cả trong thẩm định hiệu quả tiệt khuẩn của quy trình lẫn trong điều kiện vận hành bình thường. Với tác nhân tiệt khuẩn là ethylen oxyd, thường dùng bào tử của vi khuẩn Bacillus subtilis (ví dụ: var. niger ATCC 9372, NCIMB 8058, CIP 77.18). Số lượng bào tử sống trên mỗi vật mang không được ít hơn 5 x 105. Trị số D phải lớn hơn 2,5 min khi tiến hành tiệt khuẩn ở 54 °C, độ ẩm tương đối là 60% và nồng độ ethylen oxyd trong khí mang là 600 mg/l. Phải kiểm tra để bảo đảm không còn vi khuẩn sống sau khi xử lý chỉ thị sinh học trong 60 min ở điều kiện nêu trên, nhưng phải còn vi khuẩn sống khi xử lý trong 15 min ở 30 °C (30 °C, độ ẩm tương đối là 60%, 600 mg/l). Để kiểm tra khả năng sức đề kháng của bào tử với tác nhân tiệt khuẩn khi thiếu độ ẩm, cho chỉ thị sinh học tiếp xúc với ethylen oxyd nồng độ 600 mg/l ở nhiệt độ 54 °C trong 60 min không có độ ẩm, phải còn vi khuẩn sống.
PHỤ LỤC 17
(Quy định)
ĐỒ ĐỰNG CẤP 1 DÙNG CHO CHẾ PHẨM DƯỢC
Đồ đựng dùng cho chế phẩm dược hay bao bì đựng thuốc là phần gắn bó hữu cơ với cấu trúc của dạng bào chế vì chúng giữ thuốc ở liều lượng mong muốn và đảm bảo cho thuốc ổn định chất lượng ở nhiều chỉ tiêu liên quan. Mặt trong của đồ đựng thuốc (bao bì cấp 1) do tiếp xúc trực tiếp với thuốc nên phải được chọn lựa cẩn thận đề khi dùng không ảnh hưởng tới bản chất, độ ổn định của thuốc trong quá trình chứa đựng, vận chuyển, bảo quản dù chỉ là thời gian ngắn. Việc làm kín như hàn ống thủy tinh hay nút, nắp và xi sáp kín cũng là một phần của đồ đựng cấp 1. Những yêu cầu khái quát về đồ đựng đã được ghi trong mục những quy định chung. Chuyên luận này cung cấp những thông tin cần thiết và cung cấp những tiêu chuẩn cần đáp ứng đối với những nguyên liệu chính như thủy tinh và chất dẻo dùng để chế tạo bao bì đựng thuốc cấp 1.
17.1 ĐỒ ĐỰNG BẰNG THỦY TINH DÙNG CHO CHẾ PHẨM DƯỢC
Đồ đựng bằng thủy tinh dùng cho chế phẩm dược là vật dụng mà mặt trong của chúng tiếp xúc trực tiếp với thuốc, nên tùy vào bản chất của thuốc mà chọn loại thủy tinh thích hợp để chế tạo đồ đựng. Trước khi đóng thuốc, đồ đựng phải được xử lý đạt độ sạch theo yêu cầu của từng dạng thuốc như giới hạn hạt bụi và vi sinh vật...
Đồ đựng thủy tinh có thể là đồng chất như ống tiêm, nhưng với chai lọ thì phải có nắp, nút thích hợp bằng thủy tinh hoặc vật liệu khác, như cao su, chất dẻo... Nhóm đồ dựng chế tạo từ thủy tinh có nhiều loại như: Chai, lọ, ống, bơm tiêm...
Chai, lọ đựng thuốc
Chai, lọ đựng thuốc là loại đồ đựng có thành tương đối dày và được đóng kín bằng một loại nắp, nút và các phụ tùng thích hợp. Nút có thể là thủy tinh, cao su hoặc chất dẻo. Những chất đựng ở trong chai, lọ có thể được lấy ra dùng một lần (bao bì đơn liều) hoặc nhiều lần (đồ đựng đa liều). Chai, lọ có thể in, khắc vạch đánh dấu thể tích. Riêng với chai đựng thuốc tiêm truyền và các chế phẩm tương tự phải khắc vạch đánh dấu thể tích từ 2 hướng lên xuống theo chiều cao của chai.
Chai, lọ đựng thuốc tiêm phải dùng loại thủy tinh trung tính phù hợp, trong suốt, không màu hoặc trường hợp thủy tinh có xử lý đặc biệt có thể làm thủy tinh hơi mờ nhưng vẫn phải giữ mức trong suốt đủ để quan sát thuốc đựng bên trong.
Chai, lọ bằng thủy tinh để đựng thuốc đặc biệt như khí dung ở áp suất cao phải được chế tạo đặc biệt để chịu áp lực cao của khí đẩy và an toàn như độ dày hoặc bao bọc chai, lọ bằng nhựa.
Chai, lọ đựng máu và những chế phẩm của máu
Là những đồ đựng hình trụ, có thành dày thích hợp đáp ứng yêu cầu về độ bền trong điều kiện sử dụng, có dung tích khác nhau và là những thủy tinh trung tính, trong và không màu.
Ống tiêm rỗng
Là ống có thành mỏng, đầu và miệng ống có thể thuôn nhỏ hoặc cổ bồng miệng loe, miệng ống để hở và được hàn kín sau khi đã đóng đủ thuốc.
Thuốc ở trong ống thường chỉ lấy ra dùng một lần.
Ống đựng thuốc tiêm phải dùng loại thủy tinh trung tính phù hợp.
Cũng có thể sản xuất ống tiêm rỗng đạt độ sạch theo yêu cầu và hàn kín. Khi đóng thuốc, ống được đưa vào máy tự động đốt cắt làm hở miệng ống, đóng thuốc và hàn kín.
Bơm tiêm đựng thuốc bằng thủy tinh
Bơm tiêm bằng thủy tinh để đựng thuốc có thể gặp trong thực tế, theo đó thuốc được đóng trong bơm tiêm và cung cấp đi liền với kim tiêm và phụ tùng khác. Đó là loại đồ đựng đặc biệt dùng cho một số loại thuốc tiêm phân liều, thường ở dạng lỏng và được đóng gói kín, vô trùng và dùng một lần.
Chất lượng của thủy tinh
Thủy tinh là muối silicat như natri silicat, calci silicat... chế tạo bằng cách nấu chảy hỗn hợp của silic oxyd (SiO2) và các chất phụ gia. Thủy tinh thường trong suốt, không màu. Thủy tinh màu là có thêm một lượng nhỏ oxyd kim loại mà sự lựa chọn tùy thuộc vào sự hấp thụ quang phổ người ta mong muốn.
Thủy tinh trung tính chứa một lượng đáng kể bor oxyd, nhôm oxyd thay cho một phần oxyd kim loại kiềm. Do thành phần khác biệt của chúng nên thủy tinh trung tính có độ bền với nhiệt cao và độ bền với nước rất cao.
Thủy tinh kiềm chứa oxyd kim loại kiềm, chủ yếu là natri oxyd và oxyd kim loại kiềm thổ, chủ yếu là calci oxyd. Với những thành phần như vậy nên thủy tinh kiềm có độ bền với nước vừa phải.
Độ bền hóa học của đồ đựng thủy tinh đối với thuốc được biểu thị bằng độ bền với nước trong những điều kiện quy định của thử nghiệm. Khi tiếp xúc với nước, thủy tinh có thể giải phóng, hòa tan những chất gốc kim loại từ cấu trúc phân tử của thủy tinh nhất là ion natri và ion kim loại kiềm, kiềm thổ tạo ra dịch có tính kiềm ít hoặc nhiều. Định lượng độ kiềm này có thể đánh giá được độ bền với nước của thủy tinh, qua đó phân loại và chọn loại đồ đựng thủy tinh phù hợp với dạng thuốc. Khi thử nghiệm bằng cách cho nước tiếp xúc với mặt trong của đồ đựng thủy tinh trong điều kiện quy định, sau đó định lượng độ kiềm của dịch, kết quả này gọi là độ kiềm hay độ bền bề mặt của đồ đựng thủy tinh. Khi thử nghiệm bằng cách đập vỡ bao bì thủy tinh rồi lấy lượng chính xác, tán bột và cho tiếp xúc với nước trong điều kiện quy định, sau đó định lượng độ kiềm của dịch thu được, kết quả này gọi là độ kiềm hay độ bền tổng cộng của vật liệu làm đồ đựng thủy tinh.
Chuyên luận này quy định đánh giá độ bền bề mặt với nước của đồ đựng thủy tinh và được tiến hành bằng chuẩn độ kiềm giải phóng ra trong điều kiện xác định. Tùy theo độ bền bề mặt với nước mà đồ đựng thủy tinh được phân loại theo các cấp như sau:
Đồ đựng thủy tinh cấp I: Là thủy tinh trung tính có độ bền với nước cao do thành phần hóa học của thủy tinh.
Đồ đựng thủy tinh cấp II: Thường là thủy tinh kiềm đã xử lý bề mặt thích hợp nên có độ bền với nước khá cao.
Đồ đựng thủy tinh cấp III: Thường là thủy tinh kiềm, chỉ có độ bền với nước vừa phải.
Đồ đựng thủy tinh cấp IV: Thường là thủy tinh kiềm, có độ bền với nước ở mức thấp.
Nhà sản xuất thuốc phải chịu trách nhiệm chọn những đồ đựng thích hợp cho từng loại thuốc khác nhau.
Đồ đựng thủy tinh cấp I nói chung thích hợp với tất cả những chế phẩm tiêm, máu và những sản phẩm của máu.
Đồ đựng thủy tinh cấp II nói chung thích hợp cho những chế phẩm có tính acid hay trung tính dùng để tiêm.
Đồ đựng thủy tinh cấp III nói chung thích hợp cho những chế phẩm không có nước hay thuốc trong dung môi dầu dùng để tiêm, những bột dùng để tiêm và những chế phẩm dùng ngoài đường tiêm.
Đồ dùng thủy tinh cấp IV hay gọi là thủy tinh thường, chỉ dùng cho những chế phẩm không dùng để tiêm.
Có thể dùng thủy tinh màu hay không màu cho những chế phẩm không dùng để tiêm.
Các thuốc tiêm thường được đóng trong đồ đựng thủy tinh không màu.
Đồ đựng thủy tinh màu cũng có thể được dùng cho những thuốc tiêm chứa hoạt chất nhạy cảm với ánh sáng. Những đồ đựng thủy tinh cho chế phẩm để tiêm dù ở dạng lỏng hoặc bột đều phải đảm bảo cho việc quan sát kiểm tra được tính chất cảm quan của thuốc đựng ở bên trong.
Những đồ đựng thủy tinh cho chế phẩm tiêm không được đem dùng lại, trừ đồ đựng thủy tinh cấp I, thường là chai, lọ nhưng phải có những quy định cụ thể cho vấn đề này. Những đồ đựng thủy tinh để đựng máu và những sản phẩm từ máu thì không được dùng lại.
Mặt trong của đồ đựng thủy tinh để đóng thuốc có thể được xử lý đặc biệt để cải thiện độ bền đối với nước hoặc tạo ra tính không thấm nước... nhưng phải chứng minh tính an toàn của các biện pháp này. Mặt ngoài cũng có thể được xử lý để làm giảm độ ma sát và tăng khả năng chống sự mài mòn, nhưng không để nhiễm bẩn mặt trong.
Khi những đồ đựng thuốc bằng thủy tinh có những bộ phận không phải thủy tinh thì chỉ áp dụng những thử nghiệm đối với phần thủy tinh của đồ đựng.
ĐỘ BỀN ĐỐI VỚI NƯỚC CỦA MẶT TRONG ĐỒ ĐỰNG BẰNG THỦY TINH
Phép thử này đánh giá độ bền bề mặt trong của đồ đựng bằng thủy tinh đối với nước và chỉ áp dụng cho đồ đựng mới, chưa qua sử dụng.
Số lượng mẫu thử nghiệm và thể tích dung dịch cần lấy để định lượng phụ thuộc vào dung tích của đồ đựng, theo chỉ dẫn ở Bảng 17.1.1.
Bảng 17.1.1 - Số lượng mẫu và thể tích dung dịch cần lấy để định lượng
| Dung tích quy định của đồ đựng (ml) | Số lượng mẫu | Thế tích dung dịch để định lượng (ml) |
| Đến 3 | Ít nhất 30 | 25,0 |
| 5 hoặc ít hơn | Ít nhất 10 | 50,0 |
| 6 đến 30 | Ít nhất 5 | 50,0 |
| Trên 30 | Ít nhất 3 | 100,0 |
Phương pháp thử
Rửa sạch mẫu ngay trước khi thử nghiệm bằng cách súc với nước cất không có carbon dioxyd (TT) ít nhất 3 lần. Đổ hết nước rửa ra và đóng nước cất không có carbon dioxyd (TT) vào trong đồ dựng với lượng quy định. Với chai, lọ thì đậy kín miệng bằng một đĩa thủy tinh trung tính hoặc giấy nhôm mà trước đó đã rửa sạch bằng nước cất không có carbon dioxyd (TT). Với ống thì hàn kín bằng đèn gas. Đặt đồ đựng đã đóng đầy nước cất vào khay của nồi hấp, các khay không được chạm vào nước trong nồi và nước của nồi hấp phải là nước cất. Đậy chặt nắp nồi hấp. Điều chỉnh nhiệt độ của nồi như sau: Để ở 100 °C trong 10 min cho khí trong nồi thoát ra hết qua van xả.
Nâng nhiệt độ từ 100 °C lên 121 °C trong 20 min.
Duy trì nhiệt độ 121 °C ± 1 °C trong 60 min.
Hạ nhiệt độ từ 121 °C xuống 100 °C trong khoảng 40 min, thông hơi để tránh chân không.
Lấy mẫu thử ra khỏi nồi hấp theo các thao tác chung và làm nguội nhanh mẫu bằng nước thường. Thời gian tiến hành định lượng trong vòng 60 min sau khi lấy ra khỏi nồi hấp. Lấy dung dịch ra khỏi đồ đựng và trộn lẫn với nhau. Cho vào một bình nón một lượng dung dịch đã chỉ dẫn ở Bảng 17.1.1. Cho vào bình nón thứ 2 một lượng nước cất không có carbon dioxyd (TT) tương đương lượng dung dịch ở bình 1 (mẫu trắng). Thêm vào mỗi bình 0,05 ml dung dịch đỏ methyl (TT) tính cho mỗi 25 ml dung dịch thử. Định lượng bằng dung dịch acid hydrocloric 0,01 M (CĐ) cho tới khi chuyển sang màu tương đương với màu của mẫu trắng. Số ml dung dịch acid hydrocloric 0,01 M (CĐ) dùng để trung hòa lượng kiềm do đồ đựng thủy tinh nhả vào nước là hiệu số giữa kết quả của mẫu thử và mẫu trắng và được gọi là độ bền đối với nước của mặt trong đồ đựng, được quy định theo 100 ml dung dịch sau khi hấp và đem định lượng. Giới hạn quy định cho thử độ bền đối với nước của đồ đựng thủy tinh đựng thuốc không được vượt quy định ở Bảng 17.1.2.
Bảng 17.1.2 - Giới hạn quy định cho thử độ bền đối với nước của đồ đựng thủy tinh.
| Dung tích của đồ đựng (ml) | Thế tích dung dịch acid hydrocloric 0,01 M tính cho 100 ml dung dịch thử (ml) | |
| Thủy tinh cấp I hoặc cấp II | Thủy tinh cấp III | |
| Đến 1 | 2,0 | 20,0 |
| Trên 1 đến 2 | 1,8 | 17,6 |
| Trên 2 đến 5 | 1,3 | 13,2 |
| Trên 5 đến 10 | 1,0 | 10,2 |
| Trên 10 đến 20 | 0,80 | 8,1 |
| Trên 20 đến 50 | 0,60 | 6,1 |
| Trên 50 đến 100 | 0,50 | 4,8 |
| Trên 100 đến 200 | 0,40 | 3,8 |
| Trên 200 đến 500 | 0,30 | 2,9 |
| Trên 500 | 0,20 | 2,2 |
GHI CHÚ: Lượng nước đóng đủ vào đồ đựng thủy tinh khi thử nghiệm như sau:
Với chai, lọ: Đóng đến 90% dung tích tràn đầy.
Với ống: Đóng đến vai của ống hay tương đương với dung tích của thuốc sẽ đóng vào ống theo quy định.
Phân biệt thủy tinh cấp I và cấp II
Số lượng mẫu thử nghiệm và thể tích dung dịch cần lấy để định lượng theo chỉ dẫn ở Bảng 17.1.1.
Phương pháp thử
Súc sạch đồ đựng 2 lần với nước, sau đó đóng đầy dung dịch acid hydrofluoric 4% (tt/tt) và để yên 10 min ở nhiệt độ phòng rồi đổ đi, súc lại đồ đựng bằng nước ít nhất 5 lần.
Tiến hành ngay thử độ bền đối với nước của mặt trong đồ đựng bằng thủy tinh. Kết quả thu được đem so sánh theo Bảng 17.1.2. Mẫu thủy tinh đã xử lý với acid hydrofluoric 4% (tt/tt) đạt cấp độ:
Thủy tinh cấp I: Nếu thể tích dung dịch acid hydrocloric 0,01 M (CĐ) định lượng không vượt quá quy định cho thủy tinh cấp I hoặc cấp II ghi tại Bảng 17.1.2.
Thủy tinh cấp II: Nếu thể tích dung dịch acid hydro-cloric 0,01 M (CĐ) định lượng vượt quá quy định cho thủy tinh cấp I hoặc cấp II, nhưng không vượt quá dung dịch quy định cho thủy tinh cấp III, ghi tại Bảng 17.1.2.
Giới hạn arsen
Áp dụng cho đồ đựng thủy tinh đựng thuốc tiêm dung môi là nước.
Những ống thủy tinh phải thử nghiệm như sau: Tiến hành thử trên những ống đã được rửa 5 lần với nước vừa mới cất. Chuẩn bị một dung dịch thử như trong thử độ bền đối với nước với một số ống thích hợp để tạo ra 50 ml dịch thử.
Dùng ống hút để lấy 10 ml dung dịch thử cho vào bình nút mài, thêm 10 ml acid nitric (TT) và làm bay hơi cho tới khô trên cách thủy. Làm khô cắn trong tủ sấy ở 130 °C trong 30 min. Để nguội, thêm vào cắn 10,0 ml dung dịch hydrazin molybdat lắc để hòa tan và đun 20 min trên cách thủy có ống sinh hàn ngược. Để nguội tới nhiệt độ phòng. Xác định độ hấp thụ của dung dịch ở bước sóng cực đại khoảng 840 nm (Phụ lục 4.1) và dùng mẫu trắng là 10,0 ml dung dịch hydrazin molybdat.
Độ hấp thụ của dung dịch thử không được vựợt quá độ hấp thụ của dung dịch chuẩn được chuẩn bị trong cùng điều kiện bằng cách dùng 0,1 ml dung dịch arsen mẫu 10 phần triệu As (TT) thay cho dung dịch thử.
Dung dịch hydrazin molybdat: Hòa tan 0,1 g amoni molybdat (TT) vào 10 ml nước có chứa 1,5 ml acid sulfuric (TT). Pha loãng thành 90 ml với nước, thêm 1 ml dung dịch hydrazin sulfat 0,15% (TT) và thêm nước vừa đủ 100 ml.
17.2 ĐỒ ĐỰNG BẰNG KIM LOẠI CHO THUỐC MỠ TRA MẮT
Ống bằng kim loại có thể gấp được dùng đóng thuốc mỡ tra mắt phải đáp ứng thử nghiệm sau đây về tiểu phân kim loại:
Lấy ngẫu nhiên 50 ống trong một lô ống cần kiểm tra. Rửa sạch từng ống bằng máy rung hay máy thổi. Đun chảy một lượng tá dược thuốc mỡ tra mắt thích hợp và đóng đầy vào từng ống. Gấp kín đáy ống bằng 2 nếp gấp và để qua đêm ở nhiệt độ 15 °C đến 20 °C.
Dùng một hệ thống lọc chân không có lắp phễu lọc hình trụ bằng thép không gỉ hoặc bằng sứ, đáy phễu phẳng có đục nhiều lỗ thủng nhỏ, có thể đun nóng được, đường kính trong của phễu đặt vừa giấy lọc kích thước 4,25 cm. Cho vào phễu miếng giấy lọc có lỗ xốp thích hợp. Đun nóng phễu lên nhiệt độ cao hơn nhiệt độ nóng chảy của tá dược.
Mở nắp ống tá dược đã để qua đêm, dốc ngược ống, bóp đều từ phía đầu kín của ống cho tá dược đi qua đầu ống đã mở vào phễu đã đun nóng, thời gian bóp hết ống tá dược không dưới 20 s, hút chân không để lọc. Bóp và lọc hết toàn bộ 50 ống. Khi đã lọc hết tá dược chảy lỏng, rửa thành phễu và giấy lọc liên tiếp ba lần, mỗi lần với 30 ml cloroform. Để cho giấy lọc khô, kẹp giấy lọc giữa 2 phiến kính để quan sát.
Dùng một kính phóng đại có thước đo được chia ô vuông có cạnh 1 mm, mỗi cạnh lại được chia nhỏ thành 0,2 mm để quan sát giấy lọc dưới ánh sáng chiếu xiên góc. Đếm số tiểu phân quan sát được trong thị trường và ghi lại số tiểu phân kim loại dài từ 1 mm trở lên, số tiểu phân kim loại dài từ 0,5 mm đến dưới 1 mm và số tiểu phân kim loại dài từ 0,2 mm đến dưới 0,5 mm.
Thực hiện tiếp 2 lần nữa việc quan sát giấy lọc ở 2 vị trí khác, sao cho ánh sáng chiếu tới từ các hướng khác nhau. Tính số trung bình của những tiểu phân kim loại đếm được ứng với 3 khoảng giới hạn kích thước nêu trên có trong 3 thị trường đã quan sát.
Mỗi tiểu phân kim loại phát hiện được trên giấy lọc ứng với một số điểm cụ thể như sau:
Tiểu phân từ 1 mm trở lên 50 điểm
Tiểu phân từ 0,5 mm đến dưới 1 mm 10 điểm
Tiểu phân từ 0,2 mm đến dưới 0,5 mm 2 điểm
Tiểu phân dưới 0,2 mm 0 điểm
Cộng toàn bộ số điểm để đánh giá (không tính điểm đối với các tiểu phân dưới 0,2 mm)
Lô ống đạt yêu cầu nếu tổng số điểm dưới 100 điểm. Lô ống không đạt yêu cầu nếu tổng số điểm trên 150 điểm. Trường hợp tổng số điểm là từ 100 điểm đến 150 điểm, thì thử lại với 50 ống khác và lô thử đạt yêu cầu nếu tổng số điểm trong 2 lần thử ít hơn 150 điểm.
17.3 ĐỒ ĐỰNG VÀ NÚT BẰNG CHẤT DẺO
Chất dẻo hay nhựa dẻo là các hợp chất cao phân tử thiên nhiên hoặc tổng hợp. Đồ đựng bằng chất dẻo dùng cho chế phẩm dược là những vật dụng được chế tạo theo khuôn mẫu phù hợp để đựng thuốc và mặt trong của chúng tiếp xúc trực tiếp với thuốc. Nếu đồ đựng là chai, lọ, ống hoặc loại tương tự thì thường phải có nút đi kèm. Nút để đậy kín đồ đựng là một phần của đồ đựng, đồng thời phải có biện pháp thích hợp như xi sáp, hàn... để khi đóng nút đồ đựng phải có độ kín đạt yêu cầu.
Ở phạm vi rộng hơn, đồ đựng chế tạo bằng chất dẻo còn có những loại khác không cần có nút để làm kín, như túi, ống hàn kín bằng nhiệt...
Nguyên liệu chất dẻo dùng chế tạo đồ đựng thuốc có thể là một hay phối chế từ nhiều polymer và có thể thêm một số chất. Những chất thêm vào có thể là chất chống oxy hóa, chất ổn định, chất làm dẻo, chất làm bóng, chất mầu.
Đồ đựng và nút làm bằng chất dẻo có thể dùng để đựng nhiều dạng thuốc theo đường dùng khác nhau:
Đựng thuốc tiêm như chai, túi, ống.
Đựng thuốc nhỏ mắt, thuốc tra mắt như lọ, ống.
Đựng thuốc uống và thuốc dùng ngoài như chai, lọ, hoặc vài loại đặc biệt khác.
Các chất dẻo để chế tạo đồ đựng thuốc thường dùng như polyethylen (loại tỷ trọng thấp hoặc cao) ký hiệu là: PE, polypropylen ký hiệu là PP, polyvinyl clorid ký hiệu là PVC, ethylen vinyl acetat copolymer, polyethylen terphtalat...
Đồ đựng và nút làm bằng chất dẻo có nhiều ưu điểm như nhẹ, bền, rẻ tiền... nhưng cũng có những nhược điểm như có thể thấm hơi nước, thấm khí từ môi trường, chống tia cực tím không cao, nhả chất phụ gia có thể gây độc cho người sử dụng, làm ô nhiễm môi trường.
Yêu cầu chất lượng chung
Đồ đựng và nút làm bằng chất dẻo phải có chất lượng riêng biệt cho đồ đựng thuốc tiêm, đồ đựng thuốc nhỏ mắt, thuốc tra mắt và đồ đựng thuốc uống và thuốc dùng ngoài hay đồ đựng cho các dạng thuốc ngoài đường tiêm.
Những nguyên liệu làm đồ đựng không được có thành phần có thể chiết ra một lượng chất làm giảm hoạt lực và giảm tính ổn định hoặc làm tăng độc tính của thuốc. Những chất giảm tĩnh điện, những chất giải phóng khuôn chỉ có thể dùng cho đồ đựng thuốc dùng ngoài khi được phép. Những nguyên liệu làm chất phụ gia nào được phép dùng phải mô tả đặc tính và được ghi trong Dược điển; những chất phụ gia khác cũng có thể được dùng nếu được cơ quan có thẩm quyền cho phép trong từng trường hợp.
Để chọn một đồ đựng bằng chất dẻo thích hợp và có thể đánh giá được khả năng rủi ro thì cần phải biết đầy đủ về công thức sản xuất chất dẻo đó, bao gồm tất cả những nguyên liệu cho thêm vào trong quá trình sản xuất đồ đựng. Đồ đựng bằng chất dẻo được lựa chọn cho bất kỳ một chế phẩm đặc biệt nào cũng phải đảm bảo các yêu cầu sau:
• Khi đựng thuốc, chất dẻo không được hấp thụ hoạt chất thuốc lên bề mặt và không được để cho thuốc thấm vào trong chất dẻo.
• Chất dẻo không được tạo ra một lượng chất đủ để làm ảnh hưởng đến sự bền vững của thuốc đựng ở trong hoặc tạo ra khả năng gây độc.
• Để kiểm tra sự tương hợp của đồ đựng và chất đựng ở trong, đảm bảo không có sự thay đổi có hại đến chất lượng chế phẩm thì phải thực hiện nhiều phép thử khác nhau như: Kiểm tra không có sự thay đổi về tính chất lý học; xác định chất bị mất và chất được thêm do sự thấm hút, phát hiện sự thay đổi pH; đánh giá về những thay đổi gây ra bởi ánh sáng; những thử nghiệm hóa học và những thử nghiệm sinh học cần thiết.
• Những đồ đựng sản xuất hàng hoạt phải phù hợp với mẫu vật về mọi phương diện. Chúng phải đảm bảo không có thay đổi về thành phần, về phương pháp sản xuất và quan trọng nhất là không dùng nguyên liệu phế loại. Những mẫu lấy từ nơi sản xuất phải được kiểm tra để đảm bảo phù hợp với vật mẫu.
Quy trình thử nghiệm hóa học, sinh học mô tả dưới đây được áp dụng cho đồ dựng bằng chất dẻo dùng cho chế phẩm dược. Phải thấy rằng những thử nghiệm này chưa đủ để xác định độ an toàn hoặc sự thích hợp của đồ đựng bằng chất dẻo mà cần thiết phải xem xét kết quả những thử nghiệm kết hợp với thông tin ở trên. Nếu có rủi ro, nhà sản xuất phải xem xét lại tiêu chuẩn của đồ đựng và thành phần của chất dẻo hoặc chất lượng của thành phần bị hư hoặc quy trình sản xuất và chế biến bị thay đổi.
17.3.1 ĐỒ ĐỰNG BẰNG CHẤT DẺO DÙNG CHO NHỮNG CHẾ PHẨM KHÔNG PHẢI THUỐC TIÊM
Những thử nghiệm chung
Độ kín
Đóng đầy 10 bình với nước, đậy bình bằng những nút thích hợp, lộn ngược bình và giữ ở nhiệt độ phòng trong 24 h. Bất cứ bình nào cũng không có hiện tượng rò rỉ.
Độ gấp uốn
Phép thử này áp dụng cho những đồ đựng có thể bóp để lấy những chất đựng ở trong ra. Khi bóp ống phải lấy ra ít nhất 90% thể tích hay khối lượng chứa danh định với tốc độ chảy quy định ở nhiệt độ phòng.
Những thử nghiệm áp dụng cho đồ đựng thuốc lỏng để uống
Gồm 2 thử nghiệm là độ trong của nước chiết và cắn không bay hơi.
Độ trong của nước chiết
Chọn những phần không có nhãn, không có vết in và không dát mỏng từ những đồ đựng thích hợp theo cách lấy tự nhiên để cho đủ diện tích tổng cộng của mẫu yêu cầu và phải tính diện tích của cả hai mặt. Cắt những phần này thành những miếng hẹp và dài, để không có mảnh nào có diện tích tổng cộng lớn hơn 20 cm2. Rửa những mảnh này cho hết những chất ở bên ngoài bằng cách lắc chúng ít nhất 2 lần riêng biệt với nước, mỗi lần 30 s, sau đó để ráo hết nước.
Chọn những phần đã cắt và đã rửa của mẫu thử, với diện tích bề mặt tổng cộng là 1250 cm2, cho vào một bình (vừa mới được làm sạch với hỗn hợp acid cromic (TT) và rửa nhiều lần với nước và thêm 250 ml nước. Đậy bình bằng một cốc và hấp ở 121 °C trong 30 min. Làm một mẫu trắng để so sánh, dùng 250 ml nước. Để nguội rồi quan sát nước chiết. Nước chiết phải không màu, không đục hơn mẫu trắng.
Cắn không bay hơi
Bốc hơi 100 ml nước chiết từ phép thử Độ trong của nước chiết tới khô, sấy ở 105 °C tới khối lượng không đổi. Cắn không được nhiều hơn 12,5 mg.
Độ thấm hơi nước
Độ thấm hơi nước hay độ ngấm hơi nước qua bao bì có ảnh hưởng rất lớn đến các thuốc cần mức độ chống ẩm cao như thuốc nang, thuốc bột và những thuốc nhạy cảm khác... Do vậy tùy yêu cầu có thể thử nghiệm đặc tính này cho đồ đựng bằng nhựa như chỉ dẫn ở mục: Đồ đựng bằng chất dẻo cho chế phẩm tiêm (Phụ lục 17.3.2).
17.3.2 ĐỒ ĐỰNG BẰNG CHẤT DẺO DÙNG CHO CHẾ PHẨM THUỐC TIÊM
Yêu cầu chung
Nguyên liệu
Chỉ những chất dẻo tinh khiết, không màu, không mùi, mới được dùng làm nguyên liệu để chế tạo đồ đựng thuốc tiêm. Đồ đựng thuốc tiêm có thể được chế tạo từ một hay nhiều polymer như polyethylen, polypropylen, polyvinyl clorid và có thể thêm các chất phụ gia để chống oxy hóa, làm trơn, hóa dẻo, ổn định nhưng không được dùng các chất để tạo màu.
Đặc tính
Đồ đựng phải đủ trong để kiểm tra được bằng mắt thường thuốc chứa bên trong. Đồ đựng đã đóng thuốc phải chịu được phương pháp tiệt khuẩn bằng nhiệt hoặc các phương pháp tiệt khuẩn khác. Sau khi tiệt khuẩn, đồ đựng không được có dấu hiệu bị co lại, méo mó, biến màu, mất độ trong, rạn nứt, chảy dính hoặc bất kỳ sự hư hỏng nào khác. Đồ đựng phải không cho vi sinh vật thâm nhập vào thuốc sau khi đã hàn kín. Đồ đựng có thể là túi hoặc chai có kích thước, hình dáng thích hợp cho việc sử dụng (có thể thêm các nút gắn, dây treo khi tiêm truyền).
Thử nghiệm về tính chất của đồ đựng
Thử độ kín, độ gấp uốn
Phải đáp ứng những thử nghiệm trong chuyên luận Đồ đựng chất dẻo cho những chế phẩm không phải thuốc tiêm (Phụ lục 17.3.1).
Độ trong của đồ đựng
Hỗn dịch chuẩn: Hòa tan 1,0 g hydrazin sulfat (TT) trong một ít nước, thêm nước vừa đủ 100 ml, để yên trong 6 h. Thêm 25,0 ml dung dịch hexamin 10% (TT) vào 25 ml dung dịch này, trộn kỹ và để yên trong 24 h. Hỗn dịch thu được bền trong khoảng 2 tháng. Pha loãng 15 ml hỗn dịch này với nước vừa đủ 1000 ml. Hỗn dịch chuẩn chỉ dùng trong vòng 24 h.
Pha loãng hỗn dịch chuẩn để thu được hỗn dịch có độ hấp thụ ở bước sóng khoảng 640 nm là 0,37 đến 0,43 (pha loãng khoảng 16 lần). Cho một thể tích hỗn dịch thu được bằng dung tích quy định vào 5 đồ đựng. Độ đục của hỗn dịch khi nhìn qua đồ đựng phải đục hơn so với nước cất đựng trong đồ đựng tương ứng.
Độ ngấm hơi nước
Cho nước vào 5 đồ đựng theo dung tích quy định và hàn kín bằng một miếng polyethylen, nhôm mỏng hay một cách khác thích hợp. Cân chính xác mỗi đồ đựng và để yên (không phủ gì ở trên) trong 14 ngày ở độ ẩm tương đối 60% ± 5% và ở nhiệt độ trong khoảng 20 °C và 25 °C. Cân lại các đồ đựng. Khối lượng giảm đi không được vượt quá 0,2%.
Những đồ đựng bằng chất dẻo polyvinyl clorid (PVC) dùng cho thuốc tiêm (tiêm truyền tĩnh mạch) phải đạt thêm những phép thử sau:
Chất di(2-ethylhexyl) phthalat chiết được: Không quá 0,010% (kl/tt).
Dùng một ống cấp, kim hoặc bộ phận nối thích hợp. Cho vào đồ đựng một thể tích dung môi chiết bằng khoảng một nửa dung tích. Hút hết không khí ra và hàn kín ống cấp. Đặt đồ đựng theo hướng nằm ngang vào một nồi cách thủy và giữ nhiệt độ ở 36 °C đến 38 °C trong 60 min ± 1 min, không lắc. Lấy đồ đựng ra khỏi nồi cách thủy, đảo ngược đồ đựng cẩn thận 10 lần và chuyển dịch chứa ở trong sang một bình thủy tinh. Đo ngay độ hấp thụ ở cực đại khoảng 272 nm (Phụ lục 4.1). Tính phần trăm của di(2-ethylhexyl) phthalat theo đường chuẩn.
Dung môi chiết: Ethanol đã pha loãng để có tỷ trọng tương đối từ 0,9373 đến 0,9378 và làm ẩm tới 37 °C trong một bình có nút kín.
Đường chuẩn di(2-ethylhexyl) phthalat: Pha 5 dung dịch chuẩn có chứa 0,020%; 0,010%: 0,0050%; 0,0020%; 0,0010% (kl/tt) di(2-ethylhexyl) phthalat trong dung môi chiết. Đo độ hấp thụ trong cùng điều kiện.
Thử nghiệm về chất liệu của đồ đựng
Dùng phần đồ đựng không có nhãn, không có vết in hoặc không bị dát mỏng hoặc hạt chất dẻo trong trường hợp đồ đựng được chế tạo đồng thời với quá trình đóng thuốc và hàn kín.
Bari
Làm ẩm 2 g mẫu thử với acid hydrocloric (TT) và nung trong một chén bạch kim. Hòa tan tro trong 10 ml dung dịch acid hydrocloric 1 M (TT), lọc và thêm 1 ml dung dịch acid sulfuric 1M (TT) vào dịch lọc. Độ đục không lớn hơn hỗn dịch đục chuẩn thu được bằng cách cho 1ml dung dịch acid sulfuric 1M (TT) vào một hỗn hợp 10 ml dung dịch bari chuẩn (10 phần triệu Ba) và 10 ml dung dịch acid hydrocloric 1M (TT).
Kim loại nặng
Cho 2,5 g mẫu thử vào một bình đáy tròn, cổ dài, thêm 20 ml acid sulfuric (TT) và đốt thành than trong khoảng 10 min. Thêm từng giọt nước oxy già (100 thể tích) vào dung dịch nóng cho tới khi hết màu, đun nóng sau mỗi lần thêm cho tới khi có khói trắng bay lên. Để nguội, dùng 10 ml nước cất để chuyển hết cắn từ bình sang đĩa bạch kim và bốc hơi đến khô. Hòa tan cắn vào 10 ml dung dịch acid hydrocloric 1 M (TT). Lọc nếu cần, thêm nước để được 25 ml (dung dịch A).
Thêm 1,2 ml dung dịch thioacetamid (TT) vào hỗn hợp gồm 10 ml dung dịch A và 2 ml đệm acetat pH 3,5 (TT), lắc trộn đều ngay và để yên trong 2 min. Nếu dung dịch tạo thành có màu vàng, màu phải không được đậm hơn màu vàng thu được bằng cách dùng 10 ml dung dịch cadmi mẫu 10 phần triệu Cd (TT) thay cho dung dịch A. Nếu là màu nâu phải không được đậm hơn màu nâu thu được bằng cách dùng một hỗn hợp 5 ml dung dịch chì mẫu 10 phần triệu Pb (TT) và 5 ml nước thay cho dung dịch A.
Thiếc
Thêm 5 ml dung dịch acid sulfuric 20% (TT), 1 ml dung dịch natri dodecyl sulfat 1% (kl/tt) và 1 ml thuốc thử kẽm dithiol (TT) vào 10 ml dung dịch A trong phép thử kim loại nặng. Đun nóng trong nồi cách thủy đúng 1 min. Làm nguội và để yên trong 30 min. Nếu dung dịch tạo thành có màu đỏ thì không được đậm hơn màu đỏ thu được bằng cách dùng 10 ml dung dịch thiếc mẫu 5 phần triệu Sn (TT) thay cho dung dịch A.
Kẽm
Lấy 1 ml dung dịch A trong phép thử kim loại nặng, thêm nước cất vừa đủ 100 ml. Lấy 10 ml dung dịch thu được, thêm 5 ml dung dịch đệm acetat pH 4,4 (TT) và 1 ml dung dịch natri thiosulfat 0,1 M (TT), 5 ml dung dịch dithizon 0,001% trong cloroform (TT). Lắc và để yên 2 min. Màu tím trong lớp cloroform không được đậm hơn màu thu được bằng cách dùng hỗn hợp 2ml dung dịch kẽm mẫu 10 phần triệu Zn (TT) và 8 ml nước thay cho dung dịch thử. Tiến hành một mẫu trắng kiểm tra, dùng 10 ml nước thay cho 10 ml dung dịch thử. Thử nghiệm không có giá trị nếu lớp cloroform thu được trong mẫu trắng có màu xanh lục.
Cắn nung
Lấy 5 g đồ đựng đã cắt nhỏ cho vào một chén nung thích hợp đã cân bì. Nung ở 800 °C ± 25 °C tới khối lượng không đổi. Để chén nung cho nguội trong bình hút ẩm sau mỗi lần nung, lượng cắn không được quá 0,1%.
Thử nghiệm trên dịch chiết
Thử nghiệm hóa lý
Những thử nghiệm sau đây thực hiện trên dịch chiết từ đồ đựng là chất dẻo, lượng chất dẻo tính theo diện tích bề mặt (cả 2 mặt) và chiết ở nhiệt độ quy định. Mẫu chất dẻo đồng nhất về chất liệu được cắt thành những miếng dài khoảng 5 cm và rộng khoảng 0,3 cm. Chuyển mẫu đã chia nhỏ vào một bình thủy tinh hình trụ dung tích 250 ml có nút mài, thêm khoảng 150 ml nước. Lắc khoảng 30 s, gạn bỏ nước và rửa lại một lần nữa. Cho vào bình chiết thích hợp một lượng mẫu đã chuẩn bị có diện tích bề mặt khoảng 1200 cm2 nếu bề dầy của mẫu là 0,5 mm hoặc mỏng hơn, hoặc 600 cm2 nếu bề dày lớn hơn 0,5 mm. Thêm 200 ml nước và chiết nóng trong nồi cách thủy ở 70 °C trong 24 h hoặc trong nồi hấp ở 120 °C trong 30 min. Làm nguội nhưng không dưới 20 °C. Lấy 20,0 ml dịch chiết vào một bình thích hợp để dùng cho phép thử dung lượng đệm. Gạn ngay dịch chiết còn lại vào một bình sạch và đậy kín để thử các phép thử dưới đây. Dùng nước để làm mẫu trắng trong những phép thử sau:
Độ trong và màu sắc: Dịch chiết phải trong (Phụ lục 9.2) và không màu (Phụ lục 9.3).
Độ hấp thụ ánh sáng: Lọc dịch chiết nếu cần và dịch lọc có độ hấp thụ ánh sáng không quá 0,08 ở 220 nm đến 240 nm và không quá 0,05 ở 240 nm đến 360 nm (Phụ lục 4.1). Dùng nước để làm mẫu trắng.
pH: Cứ mỗi 20 ml dịch chiết và mẫu trắng cho thêm 1 ml dung dịch kali clorid 0,1% (TT), xác định pH của dung dịch (Phụ lục 6.2). Hiệu số pH của hai dung dịch không được lớn hơn 1,5.
Chất không bay hơi: Cho 50,0 ml dịch chiết vào một chén nung thích hợp đã cân bì và được làm sạch bằng acid. Bốc hơi trên cách thủy cho tới khô và sấy cắn ở 105 °C trong 1 h. Song song làm mẫu trắng. Hiệu số giữa cắn của dịch chiết và mẫu trắng không được vượt quá 15 mg.
Ghi chú: Nếu là cắn dầu thì kiểm tra lại quá trình bay hơi, giai đoạn làm khô, và giảm nhiệt nếu dầu có xu hướng bám vào thành của chén nung.
Cắn nung (Không cần phải làm thử nghiệm này nếu kết quả thử nghiệm cắn không bay hơi không vượt quá 5 mg): Tiến hành với chất không bay hơi thu được từ mẫu thử và mẫu trắng, nhưng cho thêm cùng một lượng acid sulfuric (TT) vào mỗi chén nung. Hiệu số giữa cắn nung của mẫu thử và mẫu trắng không vượt quá 5 mg.
Kim loại nặng
Dịch chiết có thể lọc nếu cần, lấy 20,0 ml cho vào 1 trong 2 ống Nessler, điều chỉnh pH tới khoảng giữa 3,0 và 4,0 với dung dịch acid acetic 1 M (TT) hay dung dịch amoniac 5 M (TT), pha loãng với nước để thành 35 ml và trộn đều.
Cho vào ống Nessler kia 2,0 ml dung dịch chì mẫu 1 phần triệu Pb (TT) và 20 ml nước. Điều chỉnh pH vào khoảng giữa 3,0 và 4,0 với dung dịch acid acetic 1 M (TT) hay dung dịch amoniac 5 M (TT). Pha loãng với nước để thành 35 ml và trộn đều.
Cho vào mỗi ống 10 ml dung dịch dihydrosulfid (TT) vừa mới pha, thêm nước thành 50 ml và trộn đều. Trong 10 min, nếu dịch chiết có màu nâu thì không được đậm hơn màu trong ống chuẩn.
Dung lượng đệm: Lẩy 20 ml dịch chiết đem chuẩn độ với dung dịch acid hydrocloric 0,01 M (CĐ) hay dung dịch natri hydroxyd 0,01 M (CĐ) tới pH 7,0 xác định bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2). Song song chuẩn độ với 20 ml mẫu trắng. Nếu mẫu thử và mẫu trắng dùng cùng loại dung dịch chuẩn độ thì hiệu của hai thể tích đã dùng ở trên không vượt quá 10,0 ml, nếu dùng hai dung dịch chuẩn độ khác loại cho mẫu thử hay mẫu trắng thì tổng số thể tích không lớn hơn 10,0 ml.
Những chất bị oxy hóa: Cho 20,0 ml dịch chiết vào một bình thủy tinh nút mài, thêm 20,0 ml dung dịch kali permanganat 0,002 M (CĐ) và 1 ml dung dịch acid sulfuric 10% (kl/kl), đun sôi 3 min. Để nguội, thêm 0,1 g kali iodid (TT), lắc trộn đều và để yên 10 min trong tối. Chuẩn độ bằng dung dịch natri thiosulfat 0,01 M (CĐ). Dùng 0,25 ml dung dịch hồ tinh bột (TT) cho vào khoảng cuối chuẩn độ làm chỉ thị màu. Song song làm mẫu trắng, hiệu số giữa hai lần định lượng không khác nhau quá 1,0 ml.
Thử nghiệm sinh học
Những thử nghiệm sau đây được xây dựng để đánh giá đáp ứng sinh học của động vật đối với những vật liệu là chất dẻo hay polymer khác khi tiêm các chất chiết từ mẫu thử nghiệm.
Lượng mẫu thử được lấy theo diện tích bề mặt đem chiết. Khi diện tích bề mặt không thể xác định được thì dùng 0,2 g mẫu thử cho mỗi ml dịch chiết. Cần thận trọng khi chuẩn bị các mẫu thử để tiêm, tránh nhiễm khuẩn và các chất lạ.
Thử nghiệm áp dụng cho các loại chất dẻo và các polymer đúng với điều kiện sử dụng. Khi các đồ đựng phải rửa hoặc tiệt khuẩn trước khi dùng thì thử nghiệm cũng phải tiến hành trên mẫu đồ đựng đã được xử lý với cùng quy trình rửa và tiệt khuẩn.
Mẫu thử là dịch chiết từ mẫu đồ đựng cần thử. Mẫu trắng là dung môi dùng để chiết được xử lý trong cùng điều kiện và quy trình như mẫu thử. Mẫu đối chiếu âm tính là mẫu không cho phản ứng trong cùng điều kiện thử.
a) Thử nghiệm toàn thân
Thử nghiệm dùng để đánh giá đáp ứng toàn thân của chuột nhắt sau khi tiêm dịch chiết từ mẫu cần thử. Động vật thí nghiệm: Chuột nhắt trắng khỏe mạnh, cùng nguồn gốc, cân nặng 18 g đến 22 g, chưa dùng vào thí nghiệm nào trước đó. Chuột được cho ăn và uống nước bình thường.
Dụng cụ
Nồi hấp có khả năng duy trì nhiệt độ ở 121 °C ± 2 °C, có thêm hệ thống nước làm lạnh để làm nguội bình đựng mẫu thử đến khoảng 20 °C ngay sau khi hấp.
Tủ sấy có khả năng duy trì nhiệt độ ở 50 °C ± 2 °C hoặc 70 °C ± 2 °C.
Dùng ống nghiệm hoặc ống nuôi cấy có nắp xoáy là thủy tinh trung tính (thủy tinh loại I) để chiết mẫu.
Chuẩn bị dụng cụ
Các dụng cụ thủy tinh cần tráng bằng hỗn hợp acid cromic hoặc acid nitric nóng, súc rửa kỹ với nước, tráng lại bằng nước cất. Rửa các dụng cụ để cắt lần lượt với aceton và dicloromethan trước khi dùng để cắt mẫu. Tất cả các dụng cụ khác phải được rửa với chất tẩy rửa thích hợp rồi tráng kỹ bằng nước cất. Làm khô và tiệt khuẩn các dụng cụ bằng phương pháp thích hợp.
Dung môi chiết: Việc lựa chọn dung môi chiết, trong sổ các dung môi chiết dưới đây, cần đại diện cho thành phần dung môi có trong chế phẩm sẽ tiếp xúc trực tiếp với đồ đựng chất dẻo đem thử nghiệm. Dung dịch tiêm natri clorid 0,9% (vô khuẩn và không có chất gây sốt).
Ethanol 5% (TT) trong dung dịch tiêm natri clorid 0,9%.
Polyethylen glycol 400.
Dầu thực vật: dùng dầu vừng, dầu lạc hoặc dầu hạt bông mới tinh chế và phải đạt thêm các yêu cầu sau: Dùng 3 thỏ thí nghiệm theo quy định phép thử tiêm trong da. Tiêm trong da 0,2 ml vào mỗi điểm, tiêm 10 điểm trên mỗi thỏ. Quan sát chỗ tiêm 24 h, 48 h và 72 h sau khi tiêm. Không được có điểm nào có phản ứng dưới dạng vết đỏ hay phồng có đường kính lớn hơn 0,5 cm.
Chuẩn bị mẫu thử
Chọn và cắt mẫu thử thành những mảnh nhỏ theo kích thước hướng dẫn trong Bảng 17.3.2.1. Loại bỏ những vật lạ như xơ vải và những mảnh vụn quá nhỏ bằng cách cho mẫu đã cắt vào ống đong thủy tinh loại I, dung tích 100 ml, có nút mài, thêm 70 ml nước để pha thuốc tiêm. Lắc trong khoảng 30 s, gạn bỏ hết nước, làm lại một lần nữa. Với phần mẫu dùng chiết bằng dung môi là dầu thực vật, cần sấy khô mẫu thử ở nhiệt độ không quá 50 °C.
Bảng 17.3.2.1
| Dạng chất dẻo | Bề dày | Lượng mẫu cho mỗi 20 ml dung môi chiết | Chia nhỏ thành |
| Màng mỏng hay tờ | < 0,5 mm | Tương đương diện tích bề mặt 120 cm2 (cả hai mặt) | Những dải khoảng 5 cm x 0,3 cm |
| Từ 0,5 mm đến 1 mm | Tương đương diện tích bề mặt 60 cm2 (cả hai mặt) | ||
| Ống | < 0,5 mm | Chiều dài (cm) = 120 (cm2): [Chu vi đường tròn trong + chu vi đường tròn ngoài của ống] (cm) | Những phần khoảng 5 cm x 0,3 cm |
| Từ 0,5 mm đến 1 mm | Chiều dài (cm) = 60 (cm2): [Chu vi đường tròn trong + chu vi đường tròn ngoài của ống] (cm) | ||
| Phiến mỏng, ống hoặc đã tạo khuôn | > 1 mm | Tương đương với tổng diện tích bề mặt 60 cm2 (toàn bộ bề mặt tiếp xúc) | Những miếng khoảng 5 cm x 0,3 cm |
Ghi chú: Không lau mẫu bằng khăn khô/ẩm hoặc rửa bằng dung môi hữu cơ.
Chuẩn bị dịch chiết: Cho lượng mẫu thử đã xử lý đúng yêu cầu vào dụng cụ chiết, thêm 20 ml dung môi chiết. Chuẩn bị như vậy với từng loại dung môi chiết theo yêu cầu phép thử. Song song chuẩn bị một mẫu trắng dùng 20 ml cho mỗi loại dung môi và xử lý trong cùng điều kiện. Chiết bằng cách hấp trong nồi hấp ở 120 °C trong 60 min hoặc trong tủ ấm ở 70 °C trong 24 h hoặc ở 50 °C trong 72 h, tùy thuộc vào loại chất dẻo đem thử.
Làm nguội ngay đến nhiệt độ phòng nhưng không dưới 20 °C, lắc mạnh trong vài phút và gạn ngay mỗi dịch chiết vào một bình khô, vô khuẩn. Bảo quản các dịch chiết ở nhiệt độ từ 20 °C đến 30 °C nhưng không dùng để thử nếu để quá 24 h.
Ghi chú: Điều kiện chiết không được làm thay đổi trạng thái vật lý của mẫu thử, như làm chảy hoặc làm nóng chảy mẫu thử, dẫn tới làm giảm diện tích bề mặt mẫu thử. Có thể cho phép các mảnh dính nhẹ vào nhau. Nên cho từng mảnh nhỏ đã rửa sạch vào dung môi chiết. Nếu dùng các ống nuôi cấy để chiết với dầu thực vật trong nồi hấp thì phải dán kín mép quanh nút với băng dính tốt để ngăn hơi nước không vào trong dịch chiết.
Tiến hành
Lắc mạnh dịch chiết trước mỗi liều tiêm để đảm bảo chất chiết được phân bố đều. Chú ý không lấy các tiểu phân nhìn thấy được để tiêm tĩnh mạch.
Dùng 5 chuột cho một nhóm, mỗi nhóm thử với một dịch chiết của mẫu thử hoặc mẫu trắng. Liều tiêm và đường tiêm theo hướng dẫn ở Bảng 17.3.2.2. Riêng dịch chiết với dung môi polyethylene glycol và mẫu trắng tương ứng cần pha loãng với 4,1 thể tích dung dịch tiêm natri clorid 0,9% để được dung dịch có nồng độ khoảng 200 mg polyethylene glycol trong 1 ml.
Bảng 17.3.2.2
| Dịch chiết hay mẫu trắng | Liều cho 1 kg | Đường tiêm | Tốc độ tiêm |
| Dung dịch tiêm natri clorid 0,9% | 50 ml | Tĩnh mạch | 100 |
| Dung dịch 5% (tt/tt) của ethanol trong dung dịch tiêm natri clorid 0,9% | 50 ml | Tĩnh mạch | 100 |
| Polyethylenglycol 400 | 10g | Trong màng bụng |
|
| Dầu thực vật | 50 ml | Trong màng bụng |
|
Quan sát tất cả các chuột ngay sau khi tiêm, sau 4 h và ít nhất vào các thời điểm 24 h, 48 h và 72 h sau khi tiêm. Nếu trong thời gian theo dõi không có chuột nào của nhóm tiêm mẫu thử có phản ứng sinh học lớn hơn đáng kể so với nhóm tiêm mẫu trắng thì mẫu thử đạt yêu cầu. Nếu có 2 chuột hoặc nhiều hơn bị chết hoặc có 2 chuột hoặc nhiều hơn có biểu hiện bất thường như co giật, suy nhược hoặc có 3 chuột trở lên bị giảm cân (> 2 g) thì mẫu thử không đạt yêu cầu.
Nếu có một chuột bị chết hoặc có dấu hiệu phản ứng sinh học thì thử lại trên 10 chuột khác. Với lần thử lại, mẫu thử đạt yêu cầu nếu tất cả 10 chuột đều sống và không có biểu hiện sinh học khác nhau đáng kể so với nhóm chứng.
b) Thử nghiệm tiêm trong da
Thử nghiệm này được xây dựng để đánh giá phản ứng tại chỗ của thỏ với dịch chiết mẫu thử sau khi tiêm vào trong da.
Động vật thí nghiệm: Chọn thỏ trắng, da mỏng, có thể cắt lông thật ngắn và da không bị tổn thương hoặc bị kích ứng do cọ xát. Trong thời gian theo dõi, tránh tiếp xúc vào những chỗ tiêm trừ khi cần phân biệt giữa nốt phù nề với vết dầu đọng.
Ghi chú: Những thỏ trước đó đã dùng cho các phép thử không liên quan như thử chất gây sốt hoặc thỏ đã nghỉ một thời gian sau lần thử trước, có thể dùng vào phép thử này nếu da sạch và không bị tổn thương.
Tiến hành: Trong ngày thí nghiệm, cắt lông trên phần lưng thỏ, về cả hai bên sống lưng, một khoảng đủ rộng để thử. Tránh gây kích ứng và làm tổn thương da. Dùng máy hút để loại hết lông rơi ra, khi cần có thể lau nhẹ da bằng ethanol loãng và để khô trước khi tiêm. Mỗi loại dịch chiết cần thử trên 2 thỏ, một bên tiêm dịch chiết và một bên tiêm mẫu trắng theo hướng dẫn trong Bảng 17.3.2.3. Để tránh lãng phí, trên cùng một thỏ có thể tiêm một vài loại dịch chiết hoặc dịch chiết của vài mẫu khác nếu kết quả không ảnh hưởng lẫn nhau. Lắc mạnh mỗi dịch chiết trước khi lấy để tiêm. Mẫu dịch chiết với polyethylen glycol 400 và mẫu trắng tương ứng, cần pha loãng với dung dịch tiêm natri clorid 0,9% để có nồng độ polyethylen glycol khoảng 120 mg trong 1 ml.
Bảng 17.3.2.3
| Dịch chiết hay mẫu trắng | Sổ vị trí (cho mỗi thỏ) | Liều cho mỗi vị trí (μl) |
| Mẫu thử | 5 | 200 |
| Mẫu trắng | 5 | 200 |
Quan sát các chỗ tiêm để phát hiện các phản ứng của mô như ban đỏ, phù nề hoặc hoại tử. Có thể lau nhẹ bằng ethanol loãng để dễ quan sát và đánh giá kết quả. Quan sát tất cả các thỏ thí nghiệm tại các thời điểm 24 h, 48 h và 72 h sau khi tiêm. Chấm điểm tất cả các chỗ tiêm của mẫu trắng và mẫu thử theo thang điểm quy định trong Bảng 17.3.2.4.
Bảng 17.3.2.4
| Ban đỏ và tạo thành vẩy | Điểm | Tạo thành phù nề * | Điểm |
| Không có ban đỏ | 0 | Không có phù nề | 0 |
| Ban đỏ rất nhẹ (vừa đủ nhận thấy) | 1 | Phù nề rất nhẹ (vừa đủ nhận thấy) | 1 |
| Ban đỏ nhận thấy rõ | 2 | Phù nề nhẹ (viền phù nề nổi rõ) | 2 |
| Ban đỏ vừa phải tới đậm | 3 | Phù nề vừa phải (phù nề cao khoảng 1 mm) | 3 |
| Ban đỏ đậm (sặc đỏ) tới tạo thành vẩy nhẹ | 4 | Phù nề nặng (phù nề cao trên 1 mm và lan rộng hơn khoảng tiếp xúc) | 4 |
* Không kể những vết nề (cơ học) không phải viêm.
Trong thời gian theo dõi, nếu cần có thể cắt lại lông thỏ để dễ quan sát. Tính điểm ban đỏ và phù nề trung bình cho mẫu trắng và mẫu thử ở mỗi lần chấm điểm (24 h, 48 h và 72 h) cho mỗi thỏ. Sau 72 h, cộng tất cả các điểm ban đỏ và phù nề cho riêng mẫu thử và mẫu trắng. Chia tổng số điểm của từng mẫu cho 12 (2 thỏ x 3 lần ghi điểm x 2 lần ghi loại điểm) để xác định điểm trung bình chung với mỗi mẫu thử và mẫu trắng tương ứng. Phép thử đạt yêu cầu nếu hiệu số giữa điểm trung bình của mẫu thử và mẫu trắng tương ứng nhỏ hơn hoặc bằng 1. Nếu ở bất kỳ lần ghi điểm nào đó, điểm phản ứng trung bình của mẫu thử nghi ngờ cao hơn mẫu trắng, làm lại thêm trên 3 thỏ khác. Phép thử đạt yêu cầu nếu điểm trung bình giữa mẫu thử và mẫu trắng khác nhau nhỏ hơn hoặc bằng 1,0.
c) Thử nghiệm chất gây sốt
Thử nghiệm này được quy định cho các đồ đựng bằng chất dẻo dùng cho thuốc tiêm truyền. Dùng dung môi chiết là dung dịch tiêm natri clorid 0,9% không có chất gây sốt. Chuẩn bị mẫu thử và dịch chiết như hướng dẫn trong phép thử tiêm toàn thân. Các dụng cụ chiết phải đảm bảo không có chất gây sốt. Tiến hành theo thử nghiệm chất gây sốt (Phụ lục 13.4), tiêm 10 ml dịch chiết cho 1 kg thỏ.
17.3.3 ĐỒ ĐỰNG BẰNG CHẤT DẺO DÙNG CHO CHẾ PHẨM NHỎ MẮT
Đồ đựng bằng chất dẻo cho các chế phẩm nhỏ mắt có thể được chế tạo từ các polymer có khối lượng phân tử đồng nhất trong một khoảng nhất định. Có thể dùng polypropylen hoặc đồng polymer của propylen với không quá 25% ethylen hoặc hỗn hợp polypropylen và không quá 25% polyethylen và thường có thêm các chất hóa dẻo, ổn định, chống oxy hóa, tạo màu, làm trơn.
Để lựa chọn đồ đựng bằng chất dẻo phù hợp với chế phẩm nhỏ mắt cần phải tiến hành các thử nghiệm về thành phần của chất dẻo, quy trình xử lý đồ đựng, môi trường tiếp xúc, khả năng hấp thụ và tính thấm của các chất thêm vào, điều kiện bảo quản... có thể ảnh hưởng đến chế phẩm.
Đồ đựng bằng chất dẻo cho chế phẩm thuốc nhỏ mắt phải đáp ứng những thử nghiệm sau:
Thử độ kín, độ gấp uốn, độ trong của dịch chiết, cắn không bay hơi
Phải đáp ứng những thử nghiệm quy định cho đồ đựng bằng chất dẻo dùng cho chế phẩm không phải thuốc tiêm (Phụ lục 17.3.1).
Thử nghiệm tiêm toàn thân, tiêm trong da
Phải đáp ứng những thử nghiệm quy định cho đồ đựng bằng chất dẻo dùng cho chế phẩm thuốc tiêm (Phụ lục 17.3.2).
Thử độ kích ứng mắt
Thử nghiệm này để đánh giá đáp ứng khi nhỏ dịch chiết của mẫu thử vào mắt thỏ.
Dung môi chiết:
(a) Dung dịch tiêm natri clorid 0,9%.
(b) Dầu thực vật.
Chuẩn bị mẫu thử và dịch chiết: Tiến hành giống như chuẩn bị dịch chiết để thử nghiệm tiêm toàn thân.
Động vật thí nghiệm:
Chọn những thỏ trắng khỏe mạnh, trước đó chưa dùng để thử kích ứng mắt. Nhà chăn nuôi phải không có mùn cưa, vỏ bào hay những vật liệu khác có thể làm kích ứng mắt thỏ. Kiểm tra cả hai mắt thỏ trước khi thử và chỉ dùng những thỏ mà mắt không bị kích ứng để thí nghiệm.
Thử sự thích ứng của mắt thỏ: Dùng một thỏ, nhỏ vào một mắt 100 μl mẫu trắng đã chuẩn bị trong thử nghiệm tiêm toàn thân và nhỏ vào mắt kia 100 μl nước cất vô khuẩn để tiêm. Mắt thỏ được coi là thích ứng nếu không nhận thấy sự khác nhau có ý nghĩa giữa hai mắt.
Quy trình thử: Dùng 3 thỏ trắng cho mỗi dịch chiết đem thử. Nhốt thỏ ở nơi chắc chắn nhưng cần nhẹ nhàng và yên tĩnh. Kéo nhẹ và hạ mí mắt dưới xa con ngươi để làm thành một hốc lõm và nhỏ khoảng 100 μl nước cất vô khuẩn để tiêm, giữ mi mắt khoảng 30 s. Làm tương tự để nhỏ vào mắt kia 100 μl dịch chiết mẫu thử đã chuẩn bị ở thử nghiệm tiêm toàn thân. Quan sát mắt thỏ vào các thời điểm 24 h, 48 h và 72 h sau khi nhỏ. Phép thử đạt yêu cầu nếu dịch chiết của mẫu thử chứng tỏ không có kích ứng đáng kể trong quá trình theo dõi so với mẫu trắng và mắt thỏ thích ứng với thử nghiệm. Nếu có hiện tượng kích ứng với mắt nhỏ nước cất vô khuẩn để tiêm [hoặc mắt thỏ không thích ứng với thử nghiệm] thì làm lại thí nghiệm với 3 thỏ khác. Trong lần thử lại, tất cả các thỏ phải đạt yêu cầu thử nghiệm.
17.4 DỤNG CỤ TIÊM TRUYỀN ĐÃ TIỆT KHUẨN (Bộ dây truyền dịch)
Dụng cụ tiêm truyền (Bộ dây truyền dịch) dùng để dẫn các chế phẩm như thuốc tiêm thể tích lớn, máu và các chế phẩm từ máu qua đường tĩnh mạch hoặc đường khác thích hợp trong điều trị và dinh dưỡng.
Cấu tạo
Bộ dây truyền dịch có phần chính là một ống dẫn hình trụ bằng chất dẻo gắn chặt với các bộ phận khác gồm: kim chọc nút chai, bầu đếm giọt, màng lọc máu, khóa (bộ phận điều chỉnh lưu lượng chảy), kim tiêm, màng lọc không khí. Y cụ này được sản xuất với các kích cỡ khác nhau theo yêu cầu của trị liệu.
Vật liệu và thiết kế sản xuất
Vật liệu dùng để chế tạo ra bộ dây truyền dịch thường gồm nhựa dẻo như polyvinyl clorid (PVC), ethylenvinyl acetat (EVA) và kim loại không gỉ. Những vật liệu này phải là loại dùng cho y tế. Vật liệu dùng làm ống dẫn, bầu đếm giọt phải là loại trong suốt. Việc chọn vật liệu cho sản phẩm phải không ảnh hưởng tới chất lượng của dịch được dẫn truyền, đặc biệt không tác động đến các thành phần của máu, cũng như đảm bảo an toàn trong khi dùng.
Bộ dây truyền dịch phải vô khuẩn và không có chất gây sốt; chỉ dùng một lần, không được tiệt trùng để dùng lại.
Tiêu chuẩn kỹ thuật chung
Bộ dây truyền dịch đã tiệt khuẩn, đóng gói đúng quy định phải đáp ứng các phép thử sau:
Chuẩn bị dung dịch thử nghiệm (dung dịch T): Thiết lập một hệ thống quay vòng khép kín gồm 3 bộ dây truyền dịch (được nối liền với nhau) và một bình thủy tinh borosilicat dung tích 300 ml. Lắp một bộ phận cấp và điều chỉnh nhiệt vào bình. Rót 250 ml nước để pha thuốc tiêm vào bình và điều nhiệt ở 37 °C ± 1 °C. Cho nước lưu thông trong hệ thống qua các bộ dây theo chiều sẽ được dùng để truyền dịch làm thành một vòng khép kín (nước từ bình - qua dây - trở lại bình) với tốc độ 1000 ml/h trong 2 h. Trong quá trình chiết rửa có thể dùng một bơm kiểu nhu động thích hợp để đẩy nước lưu thông dễ dàng, nhưng không ảnh hưởng tới kết quả các thử nghiệm sau đó. Thu toàn bộ dịch và để nguội (dán nhãn dung dịch T).
Độ trong và màu sắc dung dịch
Dung dịch T phải trong suốt (Phụ lục 9.2) và không màu (Phụ lục 9.3, phương pháp 2).
Độ hấp thụ ánh sáng
Đo độ hấp thụ ánh sáng của dung dịch T trong khoảng từ 230 nm đến 360 nm (Phụ lục 4.1).
Độ hấp thụ đọc được là không quá 0,3 tại bất kỳ bước sóng nào trong khoảng từ 230 nm đến 250 nm, và không quá 0,15 tại bất kỳ bước sóng nào trong khoảng từ 251 nm đến 360 nm.
Giới hạn acid - kiềm
Lấy 25 ml dung dịch T, thêm 0,15 ml dung dịch chứa 0,1% xanh bromothymol (TT), 0,02% đỏ methyl (TT) và 0,2% phenolphtalein (TT) trong ethanol 96% (TT). Màu của dung dịch phải chuyển sang xanh khi thêm không quá 0,5 ml dung dịch natri hydroxyd 0,01 M (CĐ).
Lấy 25 ml dung dịch T, thêm 0,2 ml dung dịch da cam methyl (TT). Dung dịch phải bắt đầu chuyển màu khi thêm không quá 0,5 ml dung dịch acid hydrocloric 0,01 M (CĐ).
Cắn không bay hơi
Bốc hơi 50 ml dung dịch T đến khô trên cách thủy và sấy đến khối lượng không đổi ở 100 °C đến 105 °C. Song song làm mẫu trắng với 50 ml nước để pha thuốc tiêm. Lượng cắn thu được của mẫu thử không lớn hơn quá 1,5 mg so với lượng cắn thu được của mẫu trắng.
Chất khử
Phép thử này được tiến hành trong vòng 4 h từ khi chuẩn bị dung dịch T. Lấy 20 ml dung dịch T, thêm 1 ml dung dịch acid sulfuric 1 M (TT), 20 ml dung dịch kali permanganat 0,002 M (CĐ). Đun sôi 3 min, sau đó làm nguội nhanh. Thêm 1 g kali iodid (TT) chuẩn độ bằng dung dịch natri thiosulfat 0,01 M (CĐ), dùng 0,25 ml dung dịch hồ tinh bột (TT) làm chỉ thị. Song song làm mẫu trắng với 20 ml nước để pha thuốc tiêm. Lượng dung dịch natri thiosulfat 0,01 M (CĐ) dùng cho mẫu thử không lớn hơn quá 2,0 ml so với lượng dùng cho màu trắng.
Tiểu phân lạ
Đóng đầy dung dịch natri lauryl sulfat 0,01% (kl/tt) (TT) đã được lọc trước qua phễu thủy tinh xốp với đường kính lỗ lọc khoảng 10 μm đến 16 μm và làm nóng đến 37 °C vào bộ dây truyền dịch qua đường vào thông thường. Thu dung dịch từ đường ra của bộ dây và quan sát. Dung dịch phải trong suốt và thực tế không có các tiểu phân hoặc sợi khi quan sát bằng mắt thường (coi các tiểu phân hoặc sợi với đường kính bằng hoặc lớn hơn 50 μm là có thể quan sát được bằng mắt thường).
Tốc độ dòng chảy
Đặt đầu vào của một bộ dây truyền dịch ở độ cao 1 m. Cho 50 ml một dung dịch có độ nhớt bằng 3 mPa.s (dung dịch polyethylen glycol 4000 3,3% (kl/tt) của trong nước ở 20 °C là thích hợp) chảy qua bộ dây với khóa để ở trạng thái mở hoàn toàn. Thời gian chảy hết lượng dung dịch thử nghiệm phải không quá 90 s.
Độ bền áp lực
Bịt kín một đầu của bộ dây truyền dịch, kể cả ống thông khí (nếu có). Mở các khóa. Gắn đầu kia của bộ dây với đầu ống chứa khí nén, ống có gắn sẵn thiết bị điều áp. Nhúng chìm một bộ dây vào một thùng nước ở 20 °C đến 23 °C. Nén khí vào dây với áp suất 100 kPa trong 1 min. Không được có bọt khí thoát ra từ bộ dây.
Độ trong suốt của dây truyền dịch
Pha hỗn dịch chuẩn: Trộn đều 25 ml dung dịch hydrazin sulfat 1% trong nước (đã pha sau 4 h đến 6 h) với một dung dịch có chứa 2,5 g hexamin (TT) trong 25,0 ml nước và để yên trong 24 h. Hỗn dịch này phải được bảo quản trong bình thủy tinh có bề mặt trong thật nhẵn, có thể ổn định trong vòng 2 tháng. Trước khi dùng phải lắc đều sau đó lấy 15 ml hỗn dịch cho vào bình định mức, pha loãng với nước cất tới vừa đủ 1000 ml, thu được hỗn dịch chuẩn.
Pha loãng hỗn dịch chuẩn 8 lần để thử các bộ dây truyền dịch có đường kính ngoài của ống dẫn nhỏ hơn 5 mm; pha loãng hỗn dịch chuẩn 16 lần để thử các bộ dây truyền dịch có đường kính ngoài của ống dẫn bằng hoặc lớn hơn 5 mm.
Cho hỗn dịch chuẩn đã pha loãng phù hợp chạy qua bộ dây cần thử. Độ đục và các bọt khí của hỗn dịch chuẩn đã pha loãng khi chảy qua bộ dây phải được nhận biết rõ bằng mắt thường khi so sánh với bộ dây khác cùng lô đã đóng đầy nước cất.
Thử vô khuẩn
Tiến hành phép thử vô khuẩn (Phụ lục 13.7) với các chỉ dẫn thêm sau:
Nếu bộ dây truyền dịch quy định chỉ có mặt trong là vô khuẩn: Cho 50 ml dung môi A (được chuẩn bị theo chỉ dẫn ở Phụ lục Thử vô khuẩn 13.7), chảy qua bộ dây truyền dịch và thu lấy toàn bộ lượng dung môi này để thử. Tiến hành thử theo phương pháp màng lọc. Nếu bộ dây truyền dịch quy định cả mặt trong và mặt ngoài đều vô khuẩn: Mở đồ bao gói bằng các dụng cụ tiệt khuẩn. Nếu dùng phương pháp màng lọc để thử: Đặt bộ dây vào một đồ đựng vô khuẩn thích hợp có chứa sẵn một lượng dung môi A đủ để làm ngập bộ dây và ngâm rửa một bộ dây trong 10 min. Nếu dùng phương pháp cấy trực tiếp để thử: Đặt cả bộ dây vào trong một đồ đựng vô khuẩn thích hợp có chứa sẵn một lượng môi trường đủ để ngâm chìm toàn bộ dây đem thử. Bộ dây truyền dịch phải vô khuẩn.
Chất gây sốt
Nối kết liên tiếp 5 bộ dây truyền dịch với nhau. Cho chảy qua hệ thống này 250 ml dung dịch natri clorid 0,9% vô khuẩn, không có chất gây sốt với tốc độ chảy không quá 10 ml/min. Hứng dịch rửa vào một đồ đựng không có chất gây sốt, dung dịch thu được để tiến hành thử chất gây sốt với liều 10 ml/kg thể trọng thỏ (Phụ lục. 13.4).
Bộ dây truyền dịch không được chứa chất gây sốt.
Ethylen oxyd
Nếu trên nhãn có ghi là bộ dây truyền dịch được tiệt khuẩn bằng ethylen oxyd thì dư lượng chất này không vượt quá 0,001% (kl/kl).
Tiến hành theo phương pháp sắc ký khí (Phụ lục 5.2).
Dung dịch thử. Lấy bộ dây truyền dịch ra khỏi đồ bao gói và cân. Cắt dây thành từng đoạn dài không quá 1 cm và bỏ vào một lọ thủy tinh dung tích 250 ml đến 500 ml đã có sẵn 150 ml dimethylacetamid (TT). Nút lọ lại bằng một nút thích hợp và cột nút thật chắc chắn. Đặt lọ ở 69 °C đến 71 °C trong 16 h. Dùng khí nóng thu được từ lọ để tiêm vào cột.
Dung dịch chuẩn gốc: Tiến hành trong tủ hút như sau: Chuyển 50 ml dimethylacetamid (TT) vào một lọ thủy tinh 50 ml, nút lọ lại và cột nút chắc chắn. Cân khối lượng lọ (chính xác đến 0,1 mg). Làm đầy một tiêm bơm (bằng polyethylen hoặc polypropylen) có dung tích 50 ml với khí ethylen oxyd (TT). Giữ cho khí tiếp xúc với mặt trong của bơm tiêm khoảng 3 min rồi đẩy hết khí ra khỏi bơm tiêm. Làm đầy lại bơm tiêm với 50 ml khí ethylen oxyd (TT) khác. Lắp một kim tiêm (loại dùng tiêm dưới da) vào bơm tiêm, đẩy bớt khí ra khỏi bơm tiêm đến khi thể tích khí trong bơm tiêm còn 25 ml. Tiêm từ từ lượng khí trong bơm tiêm này vào lọ đã chuẩn bị trên. Lắc nhẹ nhàng, tránh không cho kim tiêm tiếp xúc với dimethylacetamid trong lọ. Cân lại khối lượng lọ. Sự tăng khối lượng lọ chính là lượng khí ethylen oxyd đã được hòa tan. Từ sự tăng khối lượng này (khoảng 45 mg đến 60 mg), tính toán nồng độ chính xác của dung dịch khí ethylen oxyd trong dimethylacetamid (khoảng 1g/lít).
Pha các dung dịch chuẩn từ 1 đến 7: Chuẩn bị 7 lọ (cùng loại như đã dùng để chuẩn bị dung dịch thử), đánh số từ 1 đến 7, trong mỗi lọ đã có sẵn 150 ml dimethylacetamid (TT). Lần lượt cho vào các lọ, theo thứ tự từ 1 đến 7, các thể tích chính xác dung dịch chuẩn gốc sau: 0 ml; 0,05 ml; 0,10 ml; 0,20 ml; 0,50 ml; 1,00 ml và 2,00 ml (tương ứng với khoảng 0 μg; 50 pig; 100 μg; 200 μg; 500 μg; 1000 μg và 2000 μg ethylen oxyd). Đậy nút các lọ và cột nút vào cổ lọ cho thật chắc chắn. Đặt các lọ ở 69 °C đến 71 °C trong 16 h. Dùng khí nóng thu được từ các lọ để tiêm vào cột.
Điều kiện sắc ký
Cột thép không gỉ (1,5 m x 6,4 mm) nhồi diatomit silan hóa được tẩm 30% (kl/kl) polyethylen glycol 1500.
Khí mang: Heli với tốc độ dòng 20 ml/min.
Detector: lon hóa ngọn lửa.
Nhiệt độ vận hành: Cột ở 40 °C; buồng tiêm ở 100 °C; dectector ở 150 °C.
Cách tiến hành:
Xây dựng đường cong chuẩn: Lần lượt tiêm riêng biệt 1 ml khí nóng thu được từ các lọ đựng dung dịch chuẩn từ 1 đến 7 vào cột sắc ký và xây dựng đường cong chuẩn từ các chiều cao của các pic đáp ứng thu được và lượng ethylen oxyd có trong mỗi lọ tương ứng.
Tiêm 1 ml khí nóng thu được từ lọ đựng dung dịch thử. Dựa vào chiều cao của pic đáp ứng thu được trong sắc ký đồ của dung dịch thử và căn cứ vào đường cong chuẩn đã được xây dựng ở trên, tính ra lượng ethylen oxyd có trong mẫu thử.
Cần kiểm tra để đảm bảo rằng không có sự ảnh hưởng của các pic khác lên pic của ethylen oxyd bằng một trong hai cách sau: 1) Tiến hành sắc ký một dung dịch được chuẩn bị như dung dịch thử quy định ở trên, nhưng thay một bộ dây truyền dịch cần thử bằng một bộ dây không được tiệt khuẩn; 2) Tiến hành quy trình sắc ký theo quy định ở trên, nhưng dùng cột thép không gỉ (3 m x 3,2 mm) nhồi diatomit silan hóa được tầm 20% (khối lượng trên khối lượng) triscyanoethoxypropan, duy trì ở nhiệt độ cột 60 °C.
Đóng gói, bảo quản
Mỗi bộ dây truyền dịch được đóng trong bao bì kín thích hợp, duy trì được trạng thái vô khuẩn. Dán nhãn đáp ứng quy cách và ghi rõ kỹ thuật vô khuẩn trong sản xuất.
Bảo quản nơi khô ráo, mát và tránh va chạm.
17.5 NÚT CAO SU DÙNG CHO CHAI ĐỰNG THUỐC TIÊM VÀ THUỐC TIÊM TRUYỀN
Nút cao su dùng cho chai, lọ đựng các chế phẩm thuốc tiêm nước, bột, hay bột đông khô, được làm từ các nguyên liệu thu được bằng cách lưu hóa các chất hữu cơ cao phân tử có tính đàn hồi, có nguồn gốc thiên nhiên hay tổng hợp, với các chất phụ gia thích hợp (chất lưu hóa, chất tăng tốc độ lưu hóa, chất ổn định, chất màu...).
Nút cao su được dùng cho nhiều chế phẩm thuốc tiêm, thuốc tiêm truyền khác nhau, đòi hỏi nút có những tính chất khác nhau. Việc lựa chọn nút cao su cho một chế phẩm thuốc cần đảm bảo để trong quá trình tiếp xúc trực tiếp với thuốc, nút cao su không hấp phụ các thành phần của thuốc, cũng như không nhả tạp chất vào thuốc, với mức có thể làm biến đổi chất lượng thuốc hoặc gây độc hại. Nút cao su phải không cho các chất thấm qua vào thuốc làm ảnh hưởng đến chất lượng thuốc, có độ đàn hồi tốt, đảm bảo nút chặt khít miệng chai, lọ, cho phép kim tiêm xuyên qua mà không rơi ra các mảnh vụn, khi rút kim ra lỗ thủng phải tự bít kín ngay để không gây ô nhiễm thuốc, nút phải ổn định trong suốt thời gian bảo quản và sử dụng. Nút cao su phải không gây tượng kỵ với thuốc chứa bên trong.
Nút cao su có thể được phân thành 2 loại:
• Loại I: Đáp ứng được các yêu cầu nghiêm ngặt nhất và được ưa dùng.
• Loại II: Là loại có các tính chất cơ học phù hợp cho các trường hợp sử dụng đặc biệt (ví dụ như loại dùng cho nhiều liều, chọc chai nhiều lần), do cấu tạo thành phần hóa học mà nút loại II không thể đáp ứng được các yêu cầu nghiêm ngặt như loại I.
Tính chất
Nút cao su là loại nút đàn hồi, trong mờ hoặc đục mờ, màu phụ thuộc vào thành phần các chất có trong nút. Nút phải đồng nhất và không có các vật lạ ngẫu nhiên rơi vào thuốc như sợi, tiểu phân lạ hay mảnh vụn cao su. Nút cao su phải không tan trong tetrahydrofuran nhưng có thể trương nở.
Độ bền của nút khi đâm kim
Đối với phép thử độ bền của nút khi đâm kim, khả năng tự bịt kín lỗ đâm kim, nút được xử lý như xử lý nút ở phần chuẩn bị dịch chiết, sau đó làm khô nút.
Đối với nút thiết kế đâm qua bằng kim tiêm dưới da, tiến hành thử như sau: Lấy 12 chai đã rửa sạch, sấy khô. Cho vào mỗi chai một thể tích nước bằng dung tích danh định bớt đi 4 ml. Đậy chai bằng nút cần kiểm tra. Đóng nắp nhôm bên ngoài và để yên trong 16 h. Dùng kim tiêm dưới da đã được bôi trơn có mũi kim nhọn vát một góc 12° ± 2°, đường kính ngoài 0,8 mm, dài 40 mm, gắn vào một bơm tiêm sạch. Đâm kim xuyên qua nút chai, bơm vào chai 1 ml nước và hút ra 1 ml không khí. Tiến hành 4 lần trên mỗi nút chai, mỗi lần đâm ở một vị trí khác nhau. Mỗi kim chỉ sử dụng cho một nút. Nếu kim bị cùn phải thay kim mới. Lọc nước ở trong chai qua màng có lỗ lọc khoảng 0,45 μm. Đếm các mảnh cao su có thể nhìn thấy bằng mắt thường đã rơi ra.Tổng số mảnh cao su đếm được ở 12 chai không được quá 5.
Độ bền của nút khi tiệt khuẩn
Cho nước vào chai sạch với thể tích bằng khoảng 2/3 dung tích, đậy bằng nút cần kiểm tra, đóng nắp nhôm bên ngoài. Sau khi hấp ở 121 °C ± 2 °C trong 20 min, nút không được biến dạng hay chảy dính.
Độ kín của nút
Cho nước theo đúng dung tích vào 5 chai. Đậy chai bằng nút cần thử. Đóng nắp nhôm bên ngoài. Hấp ở 121 °C ± 2 °C trong 30 min. Nhúng chìm các chai vào dung dịch xanh methylen 0,1% (TT) trong 1 h ở áp suất 650 mmHg. Khi áp suất trở lại bình thường, dung dịch không được thấm vào bên trong chai.
Khả năng tự bịt kín lỗ đâm kim
Phép thử này chỉ áp dụng với nút cao su dùng cho chế phẩm thuốc tiêm đóng nhiều liều.
Cho nước theo đúng thể tích danh định vào 10 chai. Đậy chai bằng nút cần thử. Đóng nắp nhôm bên ngoài. Dùng kim tiêm có đường kính ngoài 0,8 mm, dài 40 mm, đâm xuyên qua nút, tiến hành 10 lần trên mỗi nút chai, mỗi lần đâm ở một vị trí khác nhau. Mỗi nút dùng một kim tiêm mới. Xếp chai theo chiều thẳng đứng, chìm trong dung dịch xanh methylen 0,1% (TT). Giảm áp suất bên ngoài tới 27 kPa trong 10 min. Trả lại áp suất khí quyển nhưng vẫn để chai chìm trong đó 30 min. Rửa sạch bên ngoài chai. Không có chai nào có vết dung dịch màu.
Giới hạn sulfid dễ bay hơi
Cho nút (có thể cắt nếu cần) với diện tích bề mặt 20 cm2± 2 cm2 vào bình nón dung tích 100 ml và thêm 50 ml dung dịch acid citric 2%. Đặt giấy tẩm chì acetat lên trên miệng bình và giữ giấy bằng cách úp một cốc thủy tinh lên miệng bình. Hấp ở 121 °C ± 2 °C trong 30 min, màu đen hiện lên không được đậm hơn màu của mẫu chuẩn (gồm 0,154 mg natri sulfid (TT) và 50 ml dung dịch acid citric (TT) 2%), thực hiện giống như mẫu thử trong cùng thời gian.
Các phép thử đối với dịch chiết
Chuẩn bị dịch chiết (mẫu thử):
Lấy một số nút cao su tương ứng với diện tích bề mặt 100 cm2 cho vào bình thủy tinh, thêm nước cất ngập nút, đun sôi 5 min rồi đem rửa lại nút 5 lần bằng nước cất nguội.
Cho các nút đã rửa sạch vào bình thủy tinh trung tính (thủy tinh loại I) có miệng rộng, thêm 200 ml nước cất và cân bình. Đậy bình bằng một cốc thủy tinh trung tính hoặc giấy nhôm đã rửa sạch. Cho vào nồi hấp và nâng nhiệt độ đến 121 °C ± 2 °C trong thời gian từ 20 min đến 30 min và duy trì ở nhiệt độ này trong 30 min. Để nguội đến nhiệt độ phòng trong khoảng 30 min. Thêm nước đến bằng khối lượng ban đầu. Lắc đều và gạn ngay lấy phần dung dịch bên trên (dung dịch A). Lắc dung dịch A trước khi thực hiện mỗi phép thử.
Mẫu trắng:
Cho 200 ml nước cất vào một bình thủy tinh trung tính (thủy tinh loại I) và thực hiện các bước tiếp theo trong cùng điều kiện như khi chuẩn bị mẫu thử.
Độ đục của dịch chiết từ nút
Dung dịch A không được đục hơn hỗn dịch đối chiếu II (với nút loại I) và hỗn dịch đối chiếu III (với nút loại II) (Phụ lục 9.2).
Màu sắc của dịch chiết từ nút
Dung dịch A không được có màu đậm hơn màu của dung dịch VL5 (Phụ lục 9.3).
Giới hạn acid - kiềm
Cho 20 ml dung dịch A vào bình nón, thêm 0,1 ml dung dịch xanh bromothymol (TT). Dung dịch phải chuyển sang màu xanh khi thêm không quá 0,3 ml dung dịch natri hydroxyd 0,01M (CĐ) hoặc dung dịch phải chuyển sang màu vàng khi thêm không quá 0,8 ml dung dịch acid hydrocloric 0,01 M (CĐ).
Giới hạn kim loại nặng
Không được quá 2 phần triệu (Phụ lục 9.4.8).
Lấy 12 ml dung dịch A cho vào ống nghiệm, thêm 2 ml dung dịch đệm acetat pH 3,5 (TT), lắc đều. Thêm 1,2 ml dung dịch thioacetamid (TT). Để yên trong 2 min. Màu tạo thành không được đậm hơn mẫu chuẩn làm trong cùng điều kiện, thay 12 ml dung dịch A bằng 10 ml dung dịch chì chuẩn 2 phần triệu Pb (TT) và 2 ml dung dịch A.
Giới hạn chất khử
Lấy 20 ml dung dịch A (mới được điều chế trong vòng 4 h), thêm 1 ml dung dịch acid sulfuric 1M (CĐ) và 20 ml dung dịch kali permanganat 0,002 M (CĐ). Đun sôi 3 min. Làm nguội. Thêm 1 g kali iodid (TT) và chuẩn độ ngay bằng dung dịch natri thiosulfat 0,01 M (CĐ), chỉ thị hồ tinh bột.
Tiến hành song song với 20 ml mẫu trắng.
Thể tích dung dịch natri thiosulfat 0,01 M (CĐ) sử dụng cho mẫu thử so với mẫu trắng không được vượt quá 3 ml (với nút loại I) và 7 ml (với nút loại II).
Giới hạn cắn khô
Làm bay hơi trên cách thủy 50 ml dung dịch A đến khô, sấy cắn ở 100 °C đến 105 °C đến khối lượng không đổi. Lượng cắn khô không được quá 2 mg (với nút loại I) và 4 mg (với nút loại II).
Giới hạn amoni
Kiềm hóa 5 ml dung dịch A bằng dung dịch natri hydoxyd 2M (CĐ), pha loãng bằng nước cất đến 15 ml, thêm 0,3 ml thuốc thử Nessler (TT), để yên 30 s. Màu tạo thành không được đậm hơn mẫu chuẩn. Mẫu chuẩn gồm 10 ml dung dịch amoni mẫu 1 phần triệu NH4 (TT), một thể tích dung dịch natri hydoxyd 2M (TT) bằng thể tích đã cho vào mẫu thử, thêm nước đến 15 ml và 0,3 ml thuốc thử Nessler (TT).
Độ hấp thụ ánh sáng
Lấy dung dịch A (mới được điều chế trong vòng 5 h) lọc qua màng có lỗ lọc 0,45 μm, bỏ đi vài ml dịch lọc đầu. Đo độ hấp thụ (Phụ lục 4.1) của dịch lọc trong dải có bước sóng từ 220 nm đến 360 nm, dùng nước cất làm mẫu trắng để hiệu chuẩn. Độ hấp thụ ánh sáng không được vượt quá 0,2 (loại I) và 0,4 (loại II).
Giới hạn kẽm hòa tan
Không được quá 5 μg kẽm trong 1 ml dung dịch A.
Xác định bằng phương pháp quang phổ hấp thụ nguyên tử (Phụ lục 4.4).
Dung dịch thử. Lấy 10 ml dung dịch A cho vào bình định mức 100 ml, thêm dung dịch acid hydrocloric 0,1M (TT), đến vạch, lắc đều.
Dung dịch đối chiếu: Chuẩn bị dung dịch đối chiếu bằng cách dùng dung dịch kẽm mẫu 10 phần triệu Zn (TT) pha loãng với dung dịch acid hydrocloric 0,1M (TT).
Bước sóng: 213,9 nm.
Nguồn sáng: Đèn cathod rỗng kẽm.
Ngọn lửa: Không khí - acetylen.
17.6 BƠM TIÊM VÔ KHUẨN BẰNG CHẤT DẺO SỬ DỤNG MỘT LẦN
Bơm tiêm vô khuẩn bằng chất dẻo sử dụng một lần là dụng cụ y tế sử dụng được ngay để đưa thuốc tiêm vào cơ thể. Loại bơm tiêm này đáp ứng vô khuẩn và không có chất gây sốt, chỉ sử dụng một lần không được tái tiệt khuẩn hoặc dùng lại. Bơm tiêm gồm có một ống bơm tiêm hình trụ và một pít tông, giữa ống bơm tiêm và pít tông có thể có một vòng đàn hồi để làm kín khít, kim tiêm có thể gắn vào ống bơm tiêm và không tháo rời được. Mỗi bơm tiêm được bao gói bảo quản riêng để đảm bảo vô khuẩn, ống bơm tiêm phải trong để có thể đọc được dễ dàng lượng thuốc cần dùng và có thể kiểm tra bằng mắt thường bọt khí và các phần tử lạ.
Chất dẻo và nguyên liệu có tính đàn hồi làm ống bơm tiêm và pít tông phải đáp ứng các yêu cầu kỹ thuật thích hợp hoặc các yêu cầu của cơ quan y tế có thẩm quyền. Nguyên liệu thông dụng nhất là polypropylen và polyethylen. Loại bơm tiêm này phải đáp ứng các tiêu chuẩn hiện hành về kích cỡ và tính năng.
Có thể tráng dầu silicon (Phụ lục 17.7) vào mặt trong của ống bơm tiêm để giúp vận hành bơm tiêm được nhẹ nhàng nhưng không được thừa dư làm ảnh hưởng tới chất lượng thuốc tiêm khi sử dụng. Mực, hồ dán, chất kết dính dùng để đánh dấu trên bơm tiêm hoặc trên đồ bao gói và tại những nơi cần thiết khi lắp ráp và đóng gói bơm tiêm, không được để ngấm qua các thành ống.
Dung dịch S: Pha chế dung dịch S một cách cẩn thận không để bị nhiễm bẩn bởi các tiểu phân lạ. Lấy một số lượng bơm tiêm đủ để tạo 50 ml dung dịch. Cho nước để pha thuốc tiêm vào các bơm tiêm đến dung tích quy định và để ở nhiệt độ 37 °C trong 24 h. Gộp các dung dịch lấy ra từ bơm tiêm vào trong một bình thủy tinh borosilicat thích hợp.
Độ trong và màu sắc dung dịch: Dung dịch S phải trong (Phụ lục 9.2) và không màu (Phụ lục 9.3 Phương pháp 2) và không được có các tiểu phân rắn lạ.
Giới hạn acid-kiềm: Lấy 20 ml dung dịch S, thêm 0,1 ml dung dịch xanh bromothymol (TT). Dung dịch phải chuyển màu khi thêm không quá 0,3 ml dung dịch natrihydroxyd 0,01 M (CĐ) hoặc dung dịch acid hydrocloric 0,01 M (CĐ).
Độ hấp thụ ánh sáng: Độ hấp thụ ánh sáng của dung dịch S (Phụ lục 4.1) không được quá 0,4 trong khoảng bước sóng từ 220 nm đến 360 nm.
Ethylen oxid: Nếu nhãn ghi rõ có dùng ethylen oxid để tiệt khuẩn thì lượng ethylen oxyd không được lớn hơn 10 phần triệu.
Xác định bằng phương pháp sắc ký khí (Phụ lục 5.2).
Điều kiện sắc ký.
Cột thép không gỉ (1,5 m x 6,4 mm) được nhồi diatomit silan hóa và tẩm 30% (kl/kl) macrogol 1500.
Khí mang: Heli dùng cho sắc ký.
Tốc độ dòng: 20 ml/min.
Detector ion hóa ngọn lửa.
Duy trì nhiệt độ vận hành cột ở 40 °C, buồng tiêm ở 100 °C, detector ở 150 °C.
Kiểm tra đảm bảo không có các pic khác ảnh hưởng lên pic của ethylen oxyd bằng một trong hai cách sau: 1) Tiến hành sắc ký sử dụng bơm tiêm không được tiệt khuẩn hoặc; 2) Tiến hành quy trình sắc ký theo quy định như trên nhưng dùng cột thép không gỉ (3 m x 3,2 mm) được nhồi diatomit silan hóa được tẩm 20% (kl/kl) triscyanoethoxypropan (TT). Duy trì nhiệt độ cội ở 60 °C.
Dung dịch ethylen oxyd: Tiến hành trong tủ hút. Lấy 50,0 ml dimethylacetamid (TT) vào một lọ thủy tinh dung tích 50 ml, nút lọ lại và đóng nút chắc chắn. Cân khối lượng lọ (chính xác đến 0,1 mg). Nạp đầy khí ethylen oxyd (TT) vào một bơm tiêm (bằng polyethylen hoặc polypropylen) có dung tích 50 ml. Giữ cho khi tiếp xúc với mặt trong của bơm tiêm khoảng 3 min, rồi đẩy hết khí ra khỏi bơm tiêm và lại nạp đầy 50 ml khí ethylen oxyd (TT) vào bơm tiêm. Lắp một kim tiêm (loại tiêm dưới da) vào bơm tiêm và đẩy bớt khí ra khỏi bơm tiêm đến khi thể tích khí trong bơm tiêm còn 25 ml. Tiêm từ từ lượng khí trong bơm tiêm này vào lọ đã chuẩn bị trên. Lắc nhẹ nhàng, tránh không cho kim tiếp xúc với dimethylacetamid trong lọ. Cân lại khối lượng lọ. Khối lượng tăng khoảng 45 mg đến 60 mg. Dùng khối lượng tăng này để tính toán nồng độ chính xác của dung dịch khí ethylen oxyd trong dimethylacetamid (khoảng 1g/lít).
Xây dựng đường cong chuẩn: Chuẩn bị 7 lọ thủy tinh (cùng loại như đã dùng để chuẩn bị dung dịch thử), trong mỗi lọ đã có sẵn 150 ml dimethylacetamid (TT). Lần lượt cho vào các lọ 0 ml; 0,05 ml; 0,10 ml; 0,20 ml; 0,50 ml; 1,00 ml và 2,00 ml dung dịch ethylen oxid tương ứng với khoảng 0 μg; 50 μg; 100 μg; 200 μg; 500 μg; 1000 μg và 2000 μg ethylen oxyd. Đậy nút các lọ và đóng nút chắc chắn. Đặt các lọ này vào tủ sấy ở nhiệt độ 70 °C ± 1 °C trong 16 h. Tiêm 1 ml khí nóng của từng lọ vào cột và vẽ đường cong chuẩn từ chiều cao của các pic và khối lượng ethylen oxyd có trong mỗi lọ.
Cách tiến hành: Lấy bơm tiêm ra khỏi đồ bao gói và cân bơm tiêm. Cắt bơm tiêm thành từng mảnh có kích thước cạnh không quá 1 cm và cho vào trong một lọ thủy tinh dung tích 250 ml đến 500 ml đã có sẵn 150 ml dimethylacetamid (TT). Nút lọ bằng một nút thích hợp và đóng nút chắc chắn. Đặt lọ vào tủ sấy ở nhiệt độ 70 °C ± 1 °C trong 16 h. Lấy 1 ml khí nóng từ lọ và tiêm vào cột. Tính khối lượng ethylen oxyd trong lọ dựa vào đường cong chuẩn và chiều cao của pic thu được.
Dầu Silicon
Tính diện tích bề mặt trong (cm2) của một bơm tiêm theo biểu thức:
![]()
trong đó:
v là dung tích quy định của bơm tiêm (cm3);
h là chiều cao của phần chia vạch khắc (cm).
Lấy một lượng bơm tiêm đủ để có diện tích bề mặt trong khoảng từ 100 cm2 đến 200 cm2. Hút vào mỗi bơm tiêm một lượng methylen clorid (TT) bằng một nửa dung tích quy định và hút tiếp không khí vào cho tới dung tích quy định. Rửa bề mặt trong tương ứng dung tích quy định với dung môi này bằng cách đảo ngược bơm tiêm 10 lần liên tiếp, dùng ngón tay có bọc màng chất dẻo (trơ với methylen clorid) để bịt kín kim tiêm. Đẩy các dịch chiết trong bơm tiêm vào một đĩa đã cân bì và tiến hành chiết như vậy với các bơm tiêm còn lại. Bốc hơi dịch chiết đến khô trên cách thủy. Sấy khô ở nhiệt độ 100 °C đến 105 °C trong 1 h. Khối lượng cắn không được quá 0,25 mg/cm2 diện tích bề mặt trong.
Định tính cắn bằng phương pháp quang phổ hấp thụ hồng ngoại (Phụ lục 4.2). Cắn có dải hấp thụ đặc trưng của dầu silicon ở 805 cm-1, 1020 cm-1, 1095 cm-1, 1260 cm-1 và 2960 cm-1.
Chất khử: Lấy 20,0 ml dung dịch S, thêm 2 ml acid sulfuric (TT) và 20,0 ml dung dịch kalipermanganat 0,002 M (CĐ). Đun sôi 3 min. Sau đó làm nguội ngay. Thêm 1 g kali iodid (TT) và chuẩn độ ngay bằng dung dịch natri thiosulfat 0,01 M (CĐ), dùng 0,25 ml dung dịch hồ tinh bột (TT) làm chỉ thị. Song song làm mẫu trắng với 20,0 ml nước để pha thuốc tiêm (TT). Hiệu số giữa hai thể tích dung dịch chuẩn độ đã tiêu thụ không được lớn hơn 3,0 ml.
Độ trong suốt: Đóng đầy nước vào một bơm tiêm (mẫu trắng) và đóng đầy hỗn dịch chuẩn đục gốc (Phụ lục 9.2) đã pha loãng 10 lần vào một bơm tiêm khác. Dùng hỗn dịch chuẩn đục gốc đã để yên ở 20 °C ± 2 °C trong 24 h. Quan sát bằng mắt thường trong ánh sáng khuếch tán trên nền đen. Độ đục của hỗn dịch phải được nhận biết rõ khi so sánh với mẫu trắng.
Thử vô khuẩn (Phụ lục 13.7): Bơm tiêm quy định vô khuẩn phải đáp ứng phép thử vô khuẩn tiến hành như sau: Sử dụng kỹ thuật vô khuẩn, mở đồ bao gói, tháo rời các bộ phận và đặt mỗi thứ vào một bình chứa thích hợp có chứa sẵn một lượng môi trường cấy thích hợp đủ để ngâm chìm hoàn toàn bộ phận đó. Bơm tiêm quy định chỉ mặt trong là vô khuẩn phải đáp ứng phép thử vô khuẩn tiến hành như sau: Dùng 50 ml môi trường cấy cho mỗi bơm tiêm để thử. Sử dụng kỹ thuật vô khuẩn, tháo bộ phận bảo vệ kim tiêm ra và ngâm chìm kim tiêm trong môi trường cấy. Rửa xịt bơm tiêm 5 lần bằng cách kéo pít tông tới mức cao nhất có thể được.
Chất gây sốt (Phụ lục 13.4): Bơm tiêm có dung tích quy định từ 15 ml trở lên phải đáp ứng phép thử chất gây sốt. Dùng ít nhất 3 bơm tiêm để thử. Cho dung dịch natri clorid 0,9% (TT) không có chất gây sốt vào bơm tiêm tới dung tích quy định và giữ ở nhiệt độ 37 °C trong 2 h. Gộp toàn bộ các dung dịch này trong điều kiện vô khuẩn vào một bình chứa không có chất gây sốt và tiến hành thử nghiệm ngay. Dùng dung dịch này để thử chất gây sốt với liều 10 ml/kg cân nặng thỏ.
Ghi nhãn
Nhãn trên bao bì ghi rõ:
• Số lô;
• Mô tả bơm tiêm;
• Bơm tiêm này chỉ được sử dụng một lần.
• Nhãn trên bao bì ngoài ghi rõ:
• Phương pháp tiệt khuẩn;
• Bơm tiêm này vô khuẩn hoặc chỉ mặt trong bơm tiêm là vô khuẩn;
• Tên, địa chỉ và logo nhà sản xuất;
• Không được sử dụng bơm tiêm này nếu đồ bao gói bị hỏng hoặc bộ phận bảo vệ vô khuẩn không khít.
17.7 SILICON
1. Dầu silicon dùng bôi trơn
Dầu Silicon dùng bôi trơn là một poly (dimethylsiloxan) được tạo thành bằng cách thủy phân và đa trùng ngưng của dicloromethylsilan và clorotrimethylsilan. Dầu silicon dùng bôi trơn có nhiều loại khác nhau, mỗi loại có chỉ số độ nhớt quy định đặt sau tên sản phẩm.
Dầu Silicon dùng bôi trơn có độ polymer hóa (n = 400 đến 1200) tương đương độ nhớt động học của chúng trong khoảng từ 1000 mm2.s-1 đến 30000 mm2.s-1.
Tính chất
Chất lỏng không màu, trong, có độ nhớt khác nhau. Thực tế không tan trong nước và trong methanol, trộn lẫn được với ethyl acetat, methyl ethylceton và toluen, rất khó tan trong ethanol khan.
Định tính
A. Độ nhớt động học ở 25 °C (Xem phép thử Độ nhớt).
B. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hồng ngoại đối chiếu của dầu silicon dùng bôi trơn chuẩn. Không tính đến miền phổ từ 850 cm-1 đến 750 cm-1 vì ở dải phổ này có sự khác biệt nhỏ tùy theo độ polymer hóa.
C. Đốt nóng 0,5 g chế phẩm trong một ống nghiệm trên ngọn lửa nhỏ đến khi bắt đầu có khói trắng xuất hiện. Úp ngược ống nghiệm này lên một ống nghiệm thứ hai có chứa 1 ml dung dịch muối natri của acid cromotropic (TT) 0,1% trong acid sulphuric (TT) sao cho khói tiếp xúc với dung dịch. Lắc ống nghiệm thứ hai trong khoảng 10 s và đun nóng trên cách thủy trong 5 min. Dung dịch có màu tím.
D. Trong một chén platin, chuẩn bị như phép thử tro sulfat (Phụ lục 9.9); dùng 50 mg chế phẩm, cắn thu được là bột màu trắng và cho phản ứng của silicat (Phụ lục 8.1).
Độ acid
Lấy 2,0 g chế phẩm, thêm 25 ml hỗn hợp đồng thể tích ethanol (TT) và ether (TT) đã được trung hòa trước với 0,2 ml dung dịch xanh bromothymol (TT) làm chỉ thị và lắc. Lượng dung dịch natri hydroxyd 0,01 M (CĐ) dùng để chuyền màu của dung dịch thành màu xanh lam không được quá 0,15 ml.
Độ nhớt
Xác định độ nhớt lực học ở 25 °C (Phụ lục 6.3). Tính độ nhớt động học, lấy tỷ trọng tương đối là 0,97. Độ nhớt động học không được nhỏ hơn 95% và không lớn hơn 105% độ nhớt quy định trên nhãn.
Dầu khoáng
Lấy 2 ml chế phẩm vào ống nghiệm và quan sát dưới ánh sáng tử ngoại ở 365 nm. Huỳnh quang của chế phẩm không được đậm hơn huỳnh quang của 2 ml dung dịch có chứa 0,1 phần triệu quinin sulfat trong dung dịch acid sulphuric 0,005 M được quan sát trong cùng điều kiện.
Hợp phần phenylat
Chỉ số khúc xạ (Phụ lục 6.1) không được lớn hơn 1,410.
Kim loại nặng
Không được quá 5 phần triệu.
Hỗn hợp dung môi: Dung dịch amoniac 2 M - dung dịch hydroxylamin hydroclorid 0,2% (1:9)
Dung dịch thử: Trộn 1,0 g chế phẩm với dicloromethan (TT) và pha loãng thành 20 ml với cùng dung môi. Thêm 1,0 ml dung dịch dithizon 0,002% trong dicloromethan (TT) mới pha, 0,5 ml nước và 0,5 ml hỗn hợp dung môi. Lắc kỹ ngay trong 1 min.
Dung dịch đối chiếu: Song song chuẩn bị dung dịch đối chiếu như sau: Lấy 20 ml dicloromethan (TT) thêm 1 ml dung dịch dithizon 0,002% trong dicloromethan (TT) mới pha, 0,5 ml dung dịch chì mẫu 10 phần triệu Pb và 0,5 ml hỗn hợp dung môi. Lắc kỹ ngay trong 1 min.
Màu đỏ của dung dịch thử không được đậm hơn màu đỏ của dung dịch đối chiếu.
Chất bay hơi
Không được quá 2,0%.
Lấy 2,00 g chế phẩm vào một đĩa có đường kính 60 mm và sâu 10 mm, sấy ở nhiệt độ 150 °C trong 24 h.
Nhãn
Theo quy định hiện hành và cần có thông tin sau:
Độ nhớt quy định bằng con số đặt ở sau tên của sản phẩm.
Chế phẩm được sử dụng làm chất bôi trơn.
2. Cao su Silicon dùng làm nút chai và ống dẫn
Cao su Silicon đáp ứng các yêu cầu dưới đây thì phù hợp để sản xuất nút chai và ống dẫn.
Cao su Silicon được điều chế bằng phản ứng liên kết chéo một polysiloxan mạch thẳng cấu tạo chủ yếu từ những nhóm dimethylsiloxy cùng với một số ít nhóm methylvinylsiloxy; các đầu mạch được khóa bằng các nhóm trimethylsiloxy hoặc dimethylvinylsiloxy.
Công thức tổng quát của polysiloxan như sau:
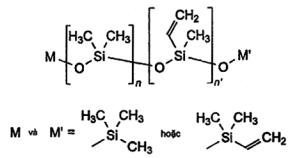
Phản ứng liên kết chéo được thực hiện ở nhiệt độ nóng với:
- 2,4-diclorobenzoyl peroxyd đối với các sản phẩm được chế tạo bằng phương pháp đùn;
- 2,4-diclorobenzoyl peroxyd, hoặc dicumyl peroxyd, hoặc OO-(1,1-dimethylethyl) O-isopropyl monoperoxycarbonat, hoặc 2,5-bis[(1,1-dimethylethyl)dioxy]-2,5-dimethylhexan cho các sản phẩm được chế tạo bằng phương pháp đúc;
hoặc,
- bằng phản ứng hydrosilyl hóa polysiloxan có nhóm SiH với xúc tác platin.
Trong tất cả các trường hợp, chất phụ gia thích hợp được dùng là silic dioxyd và đôi khi là một lượng nhỏ phụ gia cơ silic (α,ω-dihydroxypolydimethylsiloxan).
Tính chất
Vật liệu trong suốt hay trong mờ, thực tế không tan trong các dung môi hữu cơ; một số dung môi như cyclohexan, hexah và dicloromethan làm cho vật liệu phồng nở nhưng hồi phục được.
Định tính
A. Phổ hồng ngoại thu được bằng phương pháp phản xạ nhiều lần đối với chất rắn (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hồng ngoại đối chiếu của của silicon đàn hồi chuẩn.
B. Đun nóng 1,0 g chế phẩm trong ống nghiệm trên ngọn lửa nhỏ cho đến khi khói trắng xuất hiện. Lật ngược ống nghiệm lên trên một ống nghiệm khác chứa 1 ml dung dịch muối natri của acid cromotropic (TT) 0,1% trong acid sulfuric (TT) sao cho khói trắng tiếp xúc với thuốc thử. Lắc ống nghiệm chứa thuốc thử trong 10 s rồi đun nóng trên cách thủy trong 5 min. Dung dịch trong ống nghiệm chuyển thành màu tím.
C. 50 mg cắn còn lại sau khi nung phải cho phản ứng của silicat (Phụ lục 8.1).
Chuẩn bị dung dịch S
Nếu cần thiết, đem cắt nguyên liệu cần kiểm tra thành những mảnh nhỏ có kích thước mỗi cạnh không quá 1 cm.
Lấy 25 g mẫu thử cho vào một bình thủy tinh borosilicat cổ mài. Thêm 500 ml nước rồi đun sôi hồi lưu trong 5 h. Để nguội, gạn lấy dung dịch.
Độ trong của dung dịch
Dung dịch S phải trong (Phụ lục 9.2).
Giới hạn acid - kiềm
Lấy 100 ml dung dịch S, thêm 0,15 ml dung dịch xanh bromothymol (TT). Khi thêm không quá 2,5 ml dung dịch natri hydroxyd 0,01 N (CĐ), dung dịch chuyển sang màu xanh lam. Lấy 100 ml dung dịch S khác, thêm 0,2 ml dung dịch da cam methyl (TT). Khi thêm không quá 1,0 ml acid hydrocloric 0,01 N (CĐ), dung dịch phải bắt đầu chuyển từ vàng sang da cam.
Tỷ trọng tương đối
Từ 1,05 đến 1,25. Xác định bằng phương pháp dùng lọ đo tỷ trọng pycnomet (Phụ lục 6.5), như sau: Cân chính xác lọ pycnomet rỗng (M1), đổ đầy ethanol (TT) vào lọ pycnomet, giữ ở 20 °C rồi cân lại (M4). Đổ ethanol đi, làm khô pycnomet. Cho chế phẩm đã được cắt nhỏ vào pycnomet đến khoảng 1/3 - 1/2 thể tích. Đậy nút rồi cân lại (M2). Thêm ethanol (TT) đến vạch, lau khô mặt ngoài lọ rồi lại cân (M3). Chú ý không được để bọt khí còn lại giữa lớp ethanol và mẫu thử.
Tính kết quả theo công thức sau:
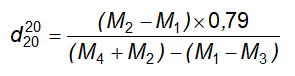
Chất khử
Thêm 1 ml acid sulfuric loãng (TT) và 20 ml dung dịch kali permanganat 0,002 M (CĐ) vào 20 ml dung dịch S. Để yên 15 min. Thêm 1 g kali iodid (TT) rồi chuẩn độ ngay bằng dung dịch natri thiosulfat 0,01 N (CĐ), dùng 0,25 ml dung dịch hồ tinh bột (TT) làm chỉ thị. Tiến hành làm một mẫu trắng với 20 ml nước cất thay cho 20 ml dung dịch S. Chênh lệch thể tích dung dịch chuẩn độ của mẫu thử và mẫu trắng không được quá 1,0 ml.
Chất tan trong hexan
Không được quá 3%.
Bốc hơi trên cách thủy 25 ml dung dịch thu được trong phép thử Hợp phần phenylat trong một cốc thủy tinh rồi sấy khô ở 100 °C đến 105 °C trong 1 h. Khối lượng cắn không được quá 15 mg.
Hợp phần phenylat
Cho 2,0 g mẫu thử vào một bình thủy tinh borosilicat cổ mài, thêm 100 ml hexan (TT). Đun sôi hồi lưu trong 4 h. Làm nguội, lọc nhanh dung dịch qua phễu thủy tinh xốp, số độ xốp 16 (Phụ lục 3.4). Gộp dịch lọc vào một bình kín, đậy nút để tránh bay hơi. Đo độ hấp thụ tử ngoại của dung dịch ở dải sóng từ 250 nm đến 340 nm (Phụ lục 4.1); độ hấp thụ đo được không được quá 0,4.
Dầu khoáng
Lấy 2 g mẫu thử cho vào một bình nón dung tích 100 ml có chứa 30 ml hỗn hợp 5 thể tích amoniac (TT) và 95 thể tích pyridin (TT). Để yên 2 h, thỉnh thoảng lắc. Gạn lấy dung dịch pyridin đem quan sát ở ánh sáng tử ngoại có bước sóng 365 nm. Huỳnh quang của dung dịch thu được không được đậm hơn huỳnh quang của dung dịch mẫu đối chiếu có chứa 1 phần triệu quinin sulfat (TT) trong acid sulfuric 0,01 N (CĐ) khi quan sát trong cùng điều kiện.
Chất dễ bay hơi
Không được quá 0,5% đối với cao su silicon được điều chế với peroxyd.
Không được quá 2,0% đối với cao su silicon khi điều chế có dùng platin làm xúc tác.
Cân 10,0 g chế phẩm đã được làm khô trước trong bình hút ẩm có calci clorid khan (TT) trong 48 h. Sấy ở 200 °C trong 4 h, để nguội trong bình hút ẩm rồi cân lại.
Silicon đàn hồi được điều chế với peroxyd phải đáp ứng các phép thử bổ sung như sau:
Peroxyd tồn dư
Không được quá 0,08% tính theo diclorobenzoyl peroxyd
Lấy 5 g mẫu thử cho vào một bình thủy tinh borosilicat, thêm 150 ml dicloromethan (TT) rồi đậy nút bình. Lắc bình bằng máy lắc cơ học trong 16 h. Lọc nhanh và thu dịch lọc vào một bình cổ mài. Đuổi không khí trong bình bằng khí nitrogen không có oxy (TT), thêm 1 ml dung dịch natri iodid 20% (TT) trong acid acetic khan (TT), đậy nút bình, lắc mạnh rồi để yên trong 30 min tránh ánh sáng. Thêm 50 ml nước cất rồi chuẩn độ ngay bằng dung dịch natri thiosulfat 0,01 N (CĐ), dùng 0,25 ml dung dịch hồ tinh bột (TT) làm chỉ thị. Tiến hành song song một mẫu trắng. Chênh lệch thể tích dung dịch chuẩn độ của mẫu thử và mẫu trắng không được lớn hơn 2,0 ml.
Cao su silicon được điều chế với xúc tác platin cần phải đáp ứng phép thử bổ sung sau:
Platin
Không được quá 30 phần triệu.
Nung 1,0 g mẫu thử trong một chén nung bằng thạch anh, nâng nhiệt độ từ từ cho đến khi thu được cắn màu trắng. Chuyển cắn vào một chén nung graphit. Thêm vào chén nung thạch anh 10 ml hỗn hợp mới pha của 1 thể tích acid nitric (TT) và 3 thể tích acid hydrocloric (TT), đun nóng trên cách thủy trong 1 đến 2 min rồi chuyển hỗn hợp vào chén nung graphit. Thêm 5 mg kali clorid (TT) và 5 ml acid hydrofluoric (TT) rồi làm bay hơi đến khô trên cách thủy. Lại thêm 5 ml acid hydrofluoric (TT) rồi làm bay hơi đến khô; lặp lại quá trình này thêm 2 lần nữa. Hòa tan cắn trong 5 ml acid hydrocloric 1 M (TT) có làm ấm trên cách thủy. Để nguội, thêm dung dịch thu được vào 1 ml dung dịch thiếc (II) clorid (TT) 25% trong acid hydrocloric 1 M (TT), rửa chén nung graphit bằng vài mililít dung dịch acid hydrocloric 1 M (TT) rồi pha loãng đến đủ 10,0 ml bằng dung dịch acid trên. Chuẩn bị đồng thời một dung dịch so sánh như sau: thêm 1,0 ml dung dịch platin mẫu 30 phần triệu Pt (TT) vào 1 ml dung dịch thiếc (II) clorid (TT) 25% trong dung dịch acid hydrocloric 1 M (TT) rồi pha loãng đến đủ 10,0 ml bằng dung dịch acid hydrocloric 1 M. Màu của dung dịch thử không được đậm hơn màu của dung dịch so sánh.
Ghi nhãn
Theo quy định hiện hành và cần ghi rõ phương pháp điều chế dùng peroxyd hay dùng xúc tác platin.
PHỤ LỤC 18
BẢNG NGUYÊN TỬ LƯỢNG CÁC NGUYÊN TỐ
Theo tài liệu của Liên đoàn quốc tế về hóa học thuần túy và ứng dụng xuất bản năm 2001 (Pure Appl. Chem. 2003, 75, 1107).
| Tên nguyên tố | Ký hiệu | Nguyên tử số | Nguyên tử lượng |
| Argon | Ar | 18 | 39,948 |
| Arsen | As | 33 | 74,9216 |
| Bạc (Argentum) | Ag | 47 | 107,8682 |
| Bari | Ba | 56 | 137,327 |
| Beryli | Be | 4 | 9,0122 |
| Bismuth | Bi | 83 | 208,9804 |
| Bor | B | 5 | 10,811 |
| Brom | Br | 35 | 79,904 |
| Cadmi | Cd | 48 | 112,411 |
| Calci | Ca | 20 | 40,078 |
| Carbon | C | 6 | 12,0107 |
| Ceri | Ce | 58 | 140,116 |
| Cesi | Cs | 55 | 132,9054 |
| Chì (Plumbum) | Pb | 82 | 207,2 |
| Clor | Cl | 17 | 35,453 |
| Cobalt | Co | 27 | 58,9332 |
| Crom | Cr | 24 | 51,9961 |
| Đồng (Cuprum) | Cu | 29 | 63,546 |
| Dysprosi | Dy | 66 | 162,5 |
| Erbi | Er | 68 | 167,259 |
| Europi | Eu | 63 | 151,964 |
| Fluor | F | 9 | 18,9984 |
| Gadolini | Gd | 64 | 157,25 |
| Gali | Ga | 31 | 69,723 |
| Germani | Ge | 32 | 72,64 |
| Hafni | Hf | 72 | 178,49 |
| Heli | He | 2 | 4,0026 |
| Holmi | Ho | 67 | 163,9303 |
| Hydrogen | H | 1 | 1,0079 |
| Indi | In | 49 | 114,818 |
| lod | I | 53 | 126,9045 |
| Iridi | Ir | 77 | 192,217 |
| Kali | K | 19 | 39,0983 |
| Kẽm (Zincum) | Zn | 30 | 65,409 |
| Krypton | Kr | 36 | 83,798 |
| Lanthan | La | 57 | 138,9055 |
| Lithi | Li | 3 | 6,941 |
| Luteti | Lu | 71 | 174,967 |
| Lưu huỳnh (Sulfur) | S | 16 | 32,065 |
| Mangan | Mn | 25 | 54,9380 |
| Mangnesi | Mg | 12 | 24,305 |
| Molybden | Mo | 42 | 95,94 |
| Natri | Na | 11 | 22,9898 |
| Neodymi | Nd | 60 | 144,24 |
| Neon | Ne | 10 | 20,1797 |
| Nhôm (Aluminium) | AI | 13 | 26,9815 |
| Nickel (Niccolum) | Ni | 28 | 58,6934 |
| Niobi | Nb | 41 | 92,9064 |
| Nitrogen | N | 7 | 14,0067 |
| Osmi | Os | 76 | 190,23 |
| Oxygen | O | 8 | 15,9994 |
| Paladi | Pd | 46 | 106,42 |
| Phosphor | P | 15 | 30,9738 |
| Platin | Pt | 78 | 195,078 |
| Prasodymi | Pr | 59 | 140,9077 |
| Rheni | Re | 75 | 186,207 |
| Rhodi | Rh | 45 | 102,9055 |
| Rubidi | Rb | 37 | 85,4678 |
| Rutheni | Ru | 44 | 101,07 |
| Samari | Sm | 62 | 150,36 |
| Sắt (Iron) | Fe | 26 | 55,845 |
| Scandi | Sc | 21 | 44,9559 |
| Selen | Se | 34 | 78,96 |
| Silic (Silicium) | Si | 14 | 28,0855 |
| Stibi (Antimony) | Sb | 51 | 121,760 |
| Stronti | Sr | 38 | 87,62 |
| Tantal | Ta | 73 | 180,9479 |
| Techneti | Tc | 43 | (98) |
| Telur | Te | 52 | 127,6 |
| Terbi | Tb | 65 | 158,9253 |
| Thali | Tl | 81 | 204,3833 |
| Thiếc (Stannium) | Sn | 50 | 118,7 |
| Thori | Th | 90 | 232,0381 |
| Thuli | Tm | 69 | 168,9342 |
| Thủy ngân (Hydragyrum) | Hg | 80 | 200,59 |
| Titan | Ti | 22 | 47,867 |
| Uran | U | 92 | 238,0289 |
| Vanadi | V | 23 | 50,9415 |
| Vàng (Aurum) | Au | 79 | 196,9665 |
| Wolfram | W | 74 | 183,84 |
| Xenon | Xe | 54 | 131,293 |
| Yterbi | Yb | 70 | 173,04 |
| Ytri | Y | 39 | 88,9059 |
| Zirconi | Zr | 40 | 91,224 |
|
FILE ĐƯỢC ĐÍNH KÈM THEO VĂN BẢN
|
(1) Trong bảng 13.10.7 không có f2 = 54. Tuy nhiên F(P=0,01;f1 = 1;f2=40) = 7,31 nên ở đây Fcrit. = F(P=0,01;f1 = 1;f2=54) phải < 7,31, còn Fcal = 83,4, do đó Fcal. > Fcrit nên biến số hồi qui rất có ý nghĩa. Nếu có bảng tra cứu chi tiết hơn, có f2 = 54 thì Fcrit = 7,13. Trị số này có thể tìm được bằng phép nội suy của bảng 13.10.7 hoặc cũng có thể tính được bằng hàm FINV(p,f1,f2) của chương trình bảng tính Excel
Bạn chưa Đăng nhập thành viên.
Đây là tiện ích dành cho tài khoản thành viên. Vui lòng Đăng nhập để xem chi tiết. Nếu chưa có tài khoản, vui lòng Đăng ký tại đây!
