- Tổng quan
- Nội dung
- VB gốc
- Tiếng Anh
- Hiệu lực
- VB liên quan
- Lược đồ
-
Nội dung hợp nhất
Tính năng này chỉ có tại LuatVietnam.vn. Nội dung hợp nhất tổng hợp lại tất cả các quy định còn hiệu lực của văn bản gốc và các văn bản sửa đổi, bổ sung, đính chính... trên một trang. Việc hợp nhất văn bản gốc và những văn bản, Thông tư, Nghị định hướng dẫn khác không làm thay đổi thứ tự điều khoản, nội dung.
Khách hàng chỉ cần xem Nội dung hợp nhất là có thể nắm bắt toàn bộ quy định hiện hành đang áp dụng, cho dù văn bản gốc đã qua nhiều lần chỉnh sửa, bổ sung.
- Tải về
Quyết định 1832/QĐ-BYT 2022 tài liệu "Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học"
| Cơ quan ban hành: | Bộ Y tế |
Số công báo:
Số công báo là mã số ấn phẩm được đăng chính thức trên ấn phẩm thông tin của Nhà nước. Mã số này do Chính phủ thống nhất quản lý.
|
Đang cập nhật |
| Số hiệu: | 1832/QĐ-BYT | Ngày đăng công báo: | Đang cập nhật |
| Loại văn bản: | Quyết định | Người ký: | Nguyễn Trường Sơn |
|
Ngày ban hành:
Ngày ban hành là ngày, tháng, năm văn bản được thông qua hoặc ký ban hành.
|
01/07/2022 |
Ngày hết hiệu lực:
Ngày hết hiệu lực là ngày, tháng, năm văn bản chính thức không còn hiệu lực (áp dụng).
|
Đang cập nhật |
|
Áp dụng:
Ngày áp dụng là ngày, tháng, năm văn bản chính thức có hiệu lực (áp dụng).
|
Đã biết
|
Tình trạng hiệu lực:
Cho biết trạng thái hiệu lực của văn bản đang tra cứu: Chưa áp dụng, Còn hiệu lực, Hết hiệu lực, Hết hiệu lực 1 phần; Đã sửa đổi, Đính chính hay Không còn phù hợp,...
|
Đã biết
|
| Lĩnh vực: | Y tế-Sức khỏe |
TÓM TẮT QUYẾT ĐỊNH 1832/QĐ-BYT
Ngày 01/7/2022, Bộ Y tế đã ra Quyết định 1832/QĐ-BYT về việc ban hành tài liệu chuyên môn "Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học".
Theo đó, thiếu máu là tình trạng giảm hemoglobin (HGB) trong máu của người bệnh so với người cùng giới, cùng lứa tuổi và cùng điều kiện sống, gây ra các biểu hiện thiếu oxy ở các mô và tổ chức của cơ thể. Mức độ giảm hemoglobin trong máu xuống 5% so với giá trị tham chiếu (theo tuổi, giới, điều kiện sống) có giá trị chẩn đoán xác định tình trạng thiếu máu.
Bên cạnh đó, dựa vào lâm sàng thiếu máu được xác định là dấu hiệu thiếu oxy các mô; tùy mức độ thiếu máu và đáp ứng của cơ thể, như: Mệt mỏi, hoa mắt, chóng mһt, giảm tập trung, chán ăn; cảm giác tức ngực, khó thở nhất là khi gắng sức hoặc đi lại nhiều; cảm giác hồi hộp, đánh trống ngực; Da xanh, niêm mạc nhợt; móng tay khô, dễ gãy; tóc khô, dễ rụng; mất kinh ở nữ.
Ngoài ra, 03 nguyên tắc điều trị thiếu máu bao gồm: Xác định và điều trị theo nguyên nhân; phối hợp điều trị nguyên nhân và truyền bù khối hồng cầu; Chỉ định truyền chế phẩm khối hồng cầu dựa vào huyết sắc tố và lâm sàng; Duy trì lượng huyết sắc tố tối thiểu từ 80 g/L (những trường hợp có bệnh lý tim, phổi mạn tính nên duy trì từ 90 g/L).
Quyết định có hiệu lực từ ngày ký.
Xem chi tiết Quyết định 1832/QĐ-BYT có hiệu lực kể từ ngày 01/07/2022
Tải Quyết định 1832/QĐ-BYT
 Quyết định 1832/QĐ-BYT PDF (Bản có dấu đỏ)
Quyết định 1832/QĐ-BYT PDF (Bản có dấu đỏ) Quyết định 1832/QĐ-BYT DOC (Bản Word)
Quyết định 1832/QĐ-BYT DOC (Bản Word)|
BỘ Y TẾ |
CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
QUYẾT ĐỊNH
VỀ VIỆC BAN HÀNH TÀI LIỆU CHUYÊN MÔN “HƯỚNG DẪN CHẨN ĐOÁN VÀ ĐIỀU TRỊ MỘT SỐ BỆNH LÝ HUYẾT HỌC”
---------------------
BỘ TRƯỞNG BỘ Y TẾ
Căn cứ Luật Khám bệnh, chữa bệnh năm 2009;
Căn cứ Nghị định số 75/2017/NĐ-CP ngày 20 tháng 6 năm 2017 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Theo đề nghị của Cục trưởng Cục Quản lý khám, chữa bệnh.
QUYẾT ĐỊNH:
Điều 1. Ban hành kèm theo Quyết định này tài liệu chuyên môn “Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học”.
Điều 2. Tài liệu chuyên môn “Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học” được áp dụng tại các cơ sở khám bệnh, chữa bệnh trong cả nước.
Điều 3. Quyết định này có hiệu lực kể từ ngày ký, ban hành và thay thế Quyết định số 1494/QĐ-BYT ngày 22/04/2015 của Bộ trưởng Bộ Y tế về việc ban hành tài liệu chuyên môn “Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học”.
Điều 4. Các ông, bà: Chánh Văn phòng Bộ, Chánh thanh tra Bộ, Tổng Cục trưởng, Cục trưởng và Vụ trưởng các Tổng cục, Cục, Vụ thuộc Bộ Y tế, Giám đốc Sở Y tế các tỉnh, thành phố trực thuộc trung ương, Giám đốc các Bệnh viện trực thuộc Bộ Y tế, Thủ trưởng Y tế các ngành chịu trách nhiệm thi hành Quyết định này./.
|
Nơi nhận: |
KT. BỘ TRƯỞNG |
HƯỚNG DẪN
CHẨN ĐOÁN VÀ ĐIỀU TRỊ MỘT SỐ BỆNH LÝ HUYẾT HỌC
(Ban hành kèm theo Quyết định số 1832/QĐ-BYT ngày 01 tháng 7 năm 2022 của Bộ trưởng Bộ Y tế)
CHỈ ĐẠO BIÊN SOẠN
PGS.TS. Nguyễn Trường Sơn
CHỦ BIÊN
PGS.TS. Lương Ngọc Khuê
ĐỒNG CHỦ BIÊN
TS. Bạch Quốc Khánh
THAM GIA BIÊN SOẠN VÀ THẨM ĐỊNH
ThS. Nguyễn Vũ Bảo Anh
TS. Vũ Đức Bình
BSCKII. Võ Thị Thanh Bình
TS. Nguyễn Hữu Chiến
TS. Dương Quốc Chính
ThS. Trần Thị Mỹ Dung
BSCKII. Phù Chí Dũng
TS. Vương Ánh Dương
BSCKII. Phạm Tuấn Dương
TS. Nguyễn Ngọc Dũng
TS. Nguyễn Thị Thu Hà
PGS.TS Lê Xuân Hải
BSCKII. Phan Quang Hòa
TS. Hoàng Thị Hồng
ThS. Vũ Thị Bích Hường
TS. Nguyễn Trọng Khoa
TS. Nguyễn Bá Khanh
TS. Bạch Quốc Khánh
BSCKII. Mai Lan
ThS. Nguyễn Thị Mai
TS. Nguyễn Thị Mai
TS. Huỳnh Văn Mẫn
TS. Trần Kiều My
TS. Hoàng Thị Thanh Nga
ThS. Nguyễn Quốc Nhật
PGS.TS Huỳnh Nghĩa
PGS.TS Nguyễn Thị Minh Phương
PGS.TS Vũ Minh Phương
BSCKII. Nguyễn Thị Lan Phương
TS. Trần Ngọc Quế
PGS.TS Nguyễn Hà Thanh
BSCKII. Nguyễn Thị Thảo
ThS. Trần Thu Thủy
BSCKII. Tôn Thất Minh Trí
BSCKII. Võ Thị Thanh Trúc
PGS.TS Nguyễn Quang Tùng
TS. Trần Thanh Tùng
GS.TS. Phạm Quang Vinh
ThS. Phạm Hải Yến
Thư ký biên soạn
TS. Nguyễn Hữu Chiến
ThS. Đoàn Văn Chính
ThS. Trương Lê Vân Ngọc
LỜI GIỚI THIỆU
Năm 2015 Bộ Y tế đã ký ban hành Quyết định số 1494/QĐ-BYT ban hành Tài liệu chuyên môn “Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học”. Từ đó, tài liệu này đã trở thành tài liệu chuyên môn hướng dẫn thực hành lâm sàng hữu ích đối với các bác sĩ trong công tác chẩn đoán và điều trị các bệnh máu. Đồng thời, đây cũng là tài liệu tham khảo quan trọng trong giảng dạy, đào tạo cho các cán bộ y tế không chỉ ở lĩnh vực Huyết học mà còn nhiều lĩnh vực Y học khác, đáp ứng hoạt động khám bệnh, chữa bệnh tại các bệnh viện trong cả nước. Nhờ đó nhiều người bệnh tại các cơ sở khám chữa bệnh khác nhau đã được chẩn đoán và điều trị theo các phác đồ thống nhất. Điều này mang lại nhiều lợi ích cho người bệnh, cho việc trao đổi thông tin và phối hợp giữa các cơ sở Y tế trong cả nước.
Tuy nhiên, những năm qua nhờ sự phát triển, tiến bộ của khoa học kỹ thuật nói chung và nhất là lĩnh vực y sinh học, nhiều tiêu chuẩn chẩn đoán, tiên lượng được áp dụng, nhiều phương pháp, kỹ thuật mới, thuốc mới đã được nghiên cứu giúp lựa chọn phương pháp điều trị hiệu quả. Để cập nhật những tiến bộ mới cho công tác chẩn đoán, điều trị mang lại lợi ích cho người bệnh ở Việt Nam, chuẩn hóa các hướng dẫn chuyên môn, dưới sự chỉ đạo của Bộ Y tế, các chuyên gia đầu ngành là các giáo sư, bác sĩ trong lĩnh vực Huyết học - Truyền máu của cả nước đã biên soạn lại tài liệu “Hướng dẫn chẩn đoán và điều trị một số bệnh lý Huyết học”. Tài liệu cập nhật này gồm 49 bài, chia làm 3 chương: Chương I: Bệnh máu tổng hợp gồm 19 bài, Chương II: Bệnh máu ác tính gồm 19 bài và Chương III: Truyền máu, chỉ định xét nghiệm, thủ thuật có 11 bài.
Thay mặt Ban biên soạn, xin trân trọng cảm ơn Lãnh đạo Bộ Y tế, cảm ơn các thành viên trong Ban biên soạn và các chuyên gia đã rất cố gắng, dành nhiều trí tuệ, kinh nghiệm và thời gian quý báu để biên soạn, sửa đổi, bổ sung, góp ý hoàn thiện cuốn tài liệu này. Chúng tôi xin trân trọng giới thiệu cùng bạn đọc Tài liệu chuyên môn “Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học”. Trong quá trình biên soạn rất khó tránh khỏi thiếu sót, chúng tôi rất mong nhận được các ý kiến đóng góp của độc giả để nội dung cuốn sách ngày càng được hoàn chỉnh hơn.
Xin trân trọng cảm ơn.
Hà Nội, năm 2022
Thay mặt các tác giả
PGS.TS. LƯƠNG NGỌC KHUÊ, TS. BẠCH QUỐC KHÁNH
DANH MỤC CHỮ VIẾT TẮT
ACT (Activated Clotting Time): Thời gian máu đông hoạt hóa
ALIP (Abnormal localization of immature precursors): Khu trú bất thường của các tế bào đầu dòng chưa trưởng thành
ALL (Acute lymphoblastic leukemia): Lơ xê mi lympho cấp (bệnh bạch cầu cấp dòng lympho)
AML (Acute myelogenous leukemia): Lơ xê mi tủy cấp (bệnh bạch cầu cấp dòng tủy)
ANA (Anticorps anti-nucleaires): Kháng thể kháng nhân
APL (acute promyelocytic leukemia): Lơ xê mi cấp thể tiền tủy bào
APS (Anti-phospholipid syndrome): Hội chứng Anti phospholipid
APTT (Activated Partial Thromboplastin Time): Thời gian thromboplastin một phần hoạt hóa
ATG: Anti-Thymocyte Globuline
CLL (Chronic Lymphocytic Leukemia): Lơ xê mi lympho mạn (bệnh bạch cầu mạn dòng lympho)
CML (Chronic myeloid leukemia): Lơ xê mi tủy mạn (bệnh bạch cầu mạn dòng tủy)
CMML (Chronic Myelo-Monocytic Leukemia): Lơ xê mi tủy - mono mạn (bệnh bạch cầu mạn dòng tủy - mono)
CMV: Cytomegalovirus
CR (Complete remission): Lui bệnh hoàn toàn
CT (Closure time): Thời gian tạo nút cầm máu tiểu cầu
DIC (Disseminated Intravascular Coagulation): Đông máu rải rác trong lòng mạch
ET (Essential thrombocythemia): Tăng tiểu cầu tiên phát
FISH (Fluorescence In Situ Hybridization):Kỹ thuật lai huỳnh quang tại chỗ
GP: Glycoprotein
GVHD (Graft-versus-host disease): Bệnh ghép chống chủ
Hb (Hemoglobin): Huyết sắc tố
HC: Hồng cầu
HCL (Hairy Cell Leukemia): Lơ xê mi tế bào tóc
HIT (Heparin Induced Thrombocytopenia): Giảm tiểu cầu do heparin
HLA (Human leukocyte antigen): kháng nguyên bạch cầu người
HLH (Hemophagocytic lymphohistiocytosis):Hội chứng thực bào tế bào máu
HUS (Hemolytic Uremic Syndrome): Hội chứng tan máu tăng ure huyết
ITP (Immune thrombocytopenic purpura): Ban xuất huyết giảm tiểu cầu miễn dịch
IT (Intrathecal): Nội tủy
LA (Lupus Anticoagulant): Chất kháng đông lupus
MCH (Mean corpuscular hemoglobin): Lượng huyết sắc tố trung bình hồng cầu
MCHC (Mean corpuscular hemoglobin concentration): Nồng độ huyết sắt tố trung bình của hồng cầu
MCV (Mean corpuscular volume): Thể tích trung bình khối hồng cầu
MDS (Myelodysplastic Syndrome): Hội chứng rối loạn sinh tủy
MM (Multiple Myeloma): Đa u tủy xương
MPDs (Myeloproliferative diseases): Các bệnh tăng sinh tủy
MPNs (Myeloproliferative neoplasms): Các bệnh tăng sinh tủy ác tính
MRD (Minimal residual disease): Tồn dư tối thiểu của bệnh
NST: Nhiễm sắc thể
PC: Protein C
PCC (Prothombin Complex Concentrate): Phức hợp prothrombin cô đặc
PCL (Plasma cell leukemia): Lơ xê mi tế bào dòng plasmo
PCR (Polymerase Chain Reaction): Phản ứng tổng hợp chuỗi
PFA (Platelet Funtion Analyzer): Đánh giá tổng quát chức năng tiểu cầu
PMF (Primary myelofibrosis): Xơ tủy nguyên phát
PNH (Paroxysmal noctural hemoglobinuria): Đái huyết sắc tố kịch phát ban đêm
PS: Protein S
PT (Prothrombin Time): Thời gian prothrombin
PV (Polycythemia vera): Đa hồng cầu nguyên phát
RDW (Red cell distribution width): Dải phân bố kích thước hồng cầu
TEG (ThromboElastography): Đàn hồi đồ cục máu
TM: Tĩnh mạch
TMC: Tĩnh mạch chậm
TMDD: Thiếu máu dai dẳng
TT (ThrombinTime): Thời gian Thrombin
TTP (Thrombotic Thrombocytopenic Purpura): Ban xuất huyết giảm tiểu cầu huyết khối
vWD (von Willebrand Disease): Bệnh von Willebrand
WHO (World Health Organization): Tổ chức y tế thế giới
MỘT SỐ KHÁI NIỆM SỬ DỤNG TRONG CÁC BÀI VIẾT
1. Điều trị tấn công: Là điều trị cảm ứng để đạt lui bệnh.
2. Điều trị củng cố: Là điều trị nhằm đạt tình trạng lui bệnh ổn định.
3. Điều trị duy trì: Là điều trị nhằm kéo dài ổn định tình trạng lui bệnh tránh tái phát.
Ghi chú:
- Các phác đồ điều trị dựa trên cơ sở những thuốc đang có giấy phép lưu hành (visa) tại Việt Nam. Trường hợp thuốc được cấp visa mới/ lại sẽ được cập nhật phác đồ điều trị theo quy định của Bộ Y tế.
- Khi dụng sử dụng thuốc nhóm Corticoid cần phối hợp với các thuốc bảo vệ niêm mạc dạ dày (khi có triệu chứng).
MỤC LỤC
CHƯƠNG 1: BỆNH MÁU TỔNG HỢP
1. THIẾU MÁU: XẾP LOẠI, CHẨN ĐOÁN VÀ ĐIỀU TRỊ
2. THIẾU MÁU THIẾU SẮT
3. BỆNH HUYẾT SẮC TỐ (Thalassemia và Huyết sắc tố bất thường)
4. SUY TỦY XƯƠNG
5. ĐÁI HUYẾT SẮC TỐ KỊCH PHÁT BAN ĐÊM
6. THIẾU MÁU TAN MÁU TỰ MIỄN
7. HỘI CHỨNG EVANS
8. GIẢM TIỂU CẦU MIỄN DỊCH NGUYÊN PHÁT
9. RỐI LOẠN CHỨC NĂNG TIỂU CẦU
10. ĐÔNG MÁU RẢI RÁC TRONG LÒNG MẠCH
11. HỘI CHỨNG ANTIPHOSPHOLIPID
12. HEMOPHILIA MẮC PHẢI
13. BỆNH VON WILLEBRAND (Von-Willebrand Disease - VWD)
14. CÁC RỐI LOẠN CHẢY MÁU BẨM SINH HIẾM GẶP
15. HỘI CHỨNG BAN XUẤT HUYẾT GIẢM TIỂU CẦU HUYẾT KHỐI - TAN MÁU URE TĂNG (Thrombotic Thrombocytopenic Purpura - Hemolytic Uremic Syndrome: TTP- HUS)
16. HỘI CHỨNG THỰC BÀO TẾ BÀO MÁU
17. CHẨN ĐOÁN VÀ XỬ TRÍ BIẾN CHỨNG SỐT NHIỄM TRÙNG Ở BỆNH NHÂN GIẢM BẠCH CẦU HẠT
18. HỒI SỨC HUYẾT HỌC
19. DỰ PHÒNG VÀ ĐIỀU TRỊ NHIỄM NẤM XÂM NHẬP Ở BỆNH NHÂN HUYẾT HỌC
CHƯƠNG II. BỆNH MÁU ÁC TÍNH
20. LƠ XÊ MI CẤP (Bệnh bạch cầu cấp)
21. LƠ XÊ MI LYMPHO CẤP Ở TRẺ EM
22. LƠ XÊ MI TỦY CẤP TRẺ EM
23. LƠ XÊ MI TỦY MẠN (Bệnh bạch cầu mạn dòng tủy)
24. LƠ XÊ MI TỦY MONO MẠN (Bệnh bạch cầu mạn dòng tủy-mono)
25. ĐA HỒNG CẦU NGUYÊN PHÁT
26. TĂNG TIỂU CẦU TIÊN PHÁT
27. XƠ TỦY NGUYÊN PHÁT
28. HỘI CHỨNG RỐI LOẠN SINH TỦY
29. U LYMPHO HODGKIN
30. U LYMPHO KHÔNG HODGKIN
31. U LYMPHO HODGKIN (TRẺ EM)
32. U LYMPHO KHÔNG HODGKIN (TRẺ EM)
33. LƠ XÊ MI LYMPHO MẠN (Bệnh bạch cầu mạn dòng lympho)
34. LƠ XÊ MI TẾ BÀO TÓC
35. LƠ XÊ MI TẾ BÀO PLASMO
36. BỆNH WALDENSTRÖM
37. ĐA U TUỶ XƯƠNG
38. U PLASMO ĐƠN ĐỘC
CHƯƠNG III. TRUYỀN MÁU, CHỈ ĐỊNH XÉT NGHIỆM, THỦ THUẬT
39. CHỈ ĐỊNH VÀ SỬ DỤNG MÁU, CHẾ PHẨM MÁU TRONG LÂM SÀNG
40. CHỈ ĐỊNH GẠN TÁCH THÀNH PHẦN MÁU TRONG ĐIỀU TRỊ
41. XỬ TRÍ TAI BIẾN TRUYỀN MÁU
42. GHÉP TẾ BÀO GỐC TỰ THÂN ĐIỀU TRỊ BỆNH MÁU
43. GHÉP TẾ BÀO GỐC ĐỒNG LOÀI ĐIỀU TRỊ BỆNH MÁU
44. HƯỚNG DẪN CHỈ ĐỊNH VÀ ĐÁNH GIÁ MỘT SỐ XÉT NGHIỆM TẾ BÀO - MÔ BỆNH HỌC CƠ QUAN TẠO MÁU
45. CHỈ ĐỊNH VÀ ĐÁNH GIÁ KẾT QUẢ MỘT SỐ XÉT NGHIỆM ĐÔNG CẦM MÁU
46. CHỈ ĐỊNH MỘT SỐ XÉT NGHIỆM MIỄN DỊCH HUYẾT HỌC
47. CHỈ ĐỊNH XÉT NGHIỆM PHÁT HIỆN ĐỘT BIẾN GEN TRONG CÁC BỆNH MÁU DI TRUYỀN
48. CHỈ ĐỊNH XÉT NGHIỆM DI TRUYỀN TẾ BÀO VÀ SINH HỌC PHÂN TỬ TRONG CHẨN ĐOÁN VÀ THEO DÕI ĐIỀU TRỊ BỆNH MÁU ÁC TÍNH
49. HƯỚNG DẪN CHỈ ĐỊNH XÉT NGHIỆM HUYẾT THANH HỌC NHÓM MÁU
1. THIẾU MÁU: XẾP LOẠI, CHẨN ĐOÁN VÀ ĐIỀU TRỊ
1. ĐẠI CƯƠNG
- Thiếu máu là tình trạng giảm hemoglobin (HGB) trong máu của người bệnh so với người cùng giới, cùng lứa tuổi và cùng điều kiện sống, gây ra các biểu hiện thiếu oxy ở các mô và tổ chức của cơ thể.
- Mức độ giảm hemoglobin trong máu xuống 5% so với giá trị tham chiếu (theo tuổi, giới, điều kiện sống) có giá trị chẩn đoán xác định tình trạng thiếu máu.
2. CĂN CỨ XẾP LOẠI THIẾU MÁU
Thiếu máu có thể được xếp loại dựa vào mức độ, diễn biến, nguyên nhân và đặc điểm hồng cầu. Mỗi cách xếp loại có ý nghĩa và ứng dụng khác nhau trong việc tiếp cận chẩn đoán và tìm nguyên nhân gây thiếu máu.
2.1. Một số cách xếp loại thiếu máu
a. Theo mức độ: Chủ yếu dựa vào nồng độ huyết sắc tố. Cách xếp loại này giúp ra quyết định truyền máu, nhất là đối với các trường hợp thiếu máu mạn tính.
- Thiếu máu nhẹ: Huyết sắc tố từ 90 đến 120 g/L.
- Thiếu máu vừa: Huyết sắc tố từ 60 đến dưới 90 g/L.
- Thiếu máu nặng: Huyết sắc tố từ 30 đến dưới 60 g/L.
- Thiếu máu rất nặng: Huyết sắc tố dưới 30 g/L.
b. Theo diễn biến (cấp và mạn): Giúp tiếp cận nguyên nhân và thái độ xử trí.
- Trường hợp thiếu máu do mất máu cấp tính, do điều chỉnh đáp ứng sớm của cơ thể, giá trị hematocrit sẽ phản ánh khá trung thành thể tích máu bị mất nên thường được sử dụng trong cấp cứu ngoại khoa để ước lượng thể tích máu cần bù do mất đi.
- Trường hợp thiếu máu hoặc mất máu mạn tính, mức độ thiếu máu dựa chủ yếu vào nồng độ huyết sắc tố.
c. Theo nguyên nhân
- Mất máu: Do chảy máu (xuất huyết tiêu hóa, trĩ, kinh nguyệt, đái máu…).
- Tan máu: Do tăng phá hủy hồng cầu vì nguyên nhân tại hồng cầu hoặc nguyên nhân khác (tan máu bẩm sinh hoặc miễn dịch, sốt rét...).
- Giảm hoặc rối loạn sinh máu: Do tủy xương giảm sinh hoặc rối loạn sinh các tế bào máu (suy tủy xương, rối loạn sinh tủy, bệnh máu ác tính, ung thư di căn…) hoặc do thiếu yếu tố tạo máu (erythropoietin, acid amin, acid folic và vitamin B12; thiếu sắt…).
d. Theo đặc điểm hồng cầu: Là cách xếp loại thường được sử dụng để giúp tiếp cận và chẩn đoán nguyên nhân gây thiếu máu.
2.2. Một số chỉ số dùng để xếp loại thiếu máu
a. Thể tích trung bình hồng cầu (MCV- Mean corpuscular volume): Phản ảnh kích thước hồng cầu, nói lên thiếu máu hồng cầu to, thiếu máu hồng cầu nhỏ hay hồng cầu bình thường. Giá trị bình thường MCV là 80-100 fl (10-15 lít).
b. Nồng độ huyết sắc tố trung bình hồng cầu (MCHC- Mean corpuscular hemoglobin concentration): Là lượng huyết sắc số có trong 1 lít hồng cầu; bình thường: 320-360 g/L. Dựa vào MCHC xếp loại thiếu máu bình sắc hay nhược sắc (MCHC < 320 g/l).
c. Dải phân bố kích thước hồng cầu (RDW- Red cell distribution width): Phản ánh sự đồng đều về kích thước giữa các hồng cầu; Bình thường là 11-14%. Nếu RDW > 14: Kích thước của các hồng cầu không đồng đều.
d. Chỉ số hồng cầu lưới: Phản ánh khả năng tăng sinh hồng cầu của tủy xương khi thiếu máu. Các chỉ số thường dùng là tỷ lệ % và số lượng tuyệt đối hồng cầu lưới; bình thường là 0,5-1%, tương đương 20 đến 40 G/L máu toàn phần.
Hồng cầu lưới giảm phản ánh tình trạng tủy đáp ứng kém; hồng cầu lưới tăng ≥ 2% nói lên: Thiếu máu có hồi phục.
3. CHẨN ĐOÁN HIẾU MÁU
|
Tiếp cận trường hợp thiếu máu (1) Xác định thiếu máu và các triệu chứng liên quan (2) Xác định mức độ thiếu máu và định hướng nguyên nhân (3) Tìm nguyên nhân gây thiếu máu. |
3.1. Chẩn đoán xác định thiếu máu: Dựa vào lâm sàng và xét nghiệm HGB.
a. Lâm sàng: Là dấu hiệu thiếu oxy các mô; tùy mức độ thiếu máu và đáp ứng của cơ thể, như:
- Mệt mỏi, hoa mắt, chóng mặt, giảm tập trung, chán ăn; cảm giác tức ngực, khó thở nhất là khi gắng sức hoặc đi lại nhiều; cảm giác hồi hộp, đánh trống ngực.
- Da xanh, niêm mạc nhợt; móng tay khô, dễ gãy; tóc khô, dễ rụng; mất kinh ở nữ.
b. Xét nghiệm: Chẩn đoán xác định thiếu máu khi nồng độ huyết sắc tố giảm trên 5% so với giá trị tham chiếu.
3.2. Xác định mức độ thiếu máu
Xác định mức độ thiếu máu dựa vào nồng độ huyết sắc với thiếu máu mạn; dựa vào lâm sàng, lượng máu mất và hematocrit với thiếu máu cấp.
3.3. Tìm nguyên nhân thiếu máu:
- Thu thập các triệu chứng và yếu tố liên quan:
+ Yếu tố dịch tễ (tuổi, giới, nghề nghiệp…); tiền sử bệnh, sử dụng thuốc và gia đình;
+ Khám lâm sàng phải đầy đủ và cẩn thận để phát hiện các biểu hiện kèm theo như: Sốt, nhiễm khuẩn, vàng da; khám hệ thống gan, lách và hạch ngoại vi.
- Các xét nghiệm hóa sinh thường quy, test Coombs, định lượng enzym: G6PD, pyruvate kinase, điện di huyết sắc tố và sức bền hồng cầu; dự trữ sắt, acid folic, vitamin B12, erythropoietin; tốc độ máu lắng, định lượng CRP, fibrinogen; Kháng thể kháng nhân, kháng thể kháng chuỗi kép DNA; ký sinh trùng sốt rét, giun móc.
- Xét nghiệm tủy đồ đánh giá giảm sinh tủy hay bệnh lý khác của tủy xương: Thiếu máu nguyên hồng cầu khổng lồ, lơ xê mi cấp hay mạn, rối loạn sinh tủy…
- Tìm nguyên nhân mất máu: Soi dạ dày, soi đại-trực tràng…
- Dựa vào chỉ số hồng cầu để định hướng nguyên nhân gây thiếu máu, cụ thể:
+ Dựa vào thể tích trung bình hồng cầu (bảng 1).
Bảng 1. Định hướng nguyên nhân thiếu máu dựa vào kích thước hồng cầu
|
Hồng cầu nhỏ (MCV < 80fl) |
Hồng cầu bình thường (MCV: 80-100fl) |
Hồng cầu to (MCV > 100fl) |
|
- Thiếu sắt - Thalassemia - Bệnh huyết sắc tố E - Thiếu máu do viêm mạn tính |
- Mất máu - Bệnh thận - Thiếu máu do viêm mạn tính - Bệnh hồng cầu hình liềm - Bệnh gan mạn tính - Rối loạn sinh tủy - Suy tủy xương |
- Thiếu a.folic, B12 - Bệnh gan, rượu - Suy tủy xương - Điều trị hóa chất, thuốc kháng virus - Tan máu tự miễn - Rối loạn sinh tủy |
+ Dựa vào các chỉ số hồng cầu lưới để đánh giá đáp ứng bù trừ của tủy xương trước tình trạng thiếu máu:
a). Chỉ số hồng cầu lưới tăng: Tìm các nguyên nhân ngoài tủy như tan máu hoặc mất máu mạn tính, tan máu bẩm sinh (do huyết sắc tố hoặc do màng hồng cầu…);
b). Chỉ số hồng cầu lưới giảm: Có thể tủy xương không đáp ứng bù đủ do tổn thương tại tủy hoặc do thiếu hụt các yếu tố cần thiết để tạo máu (erythropoietin, acid folic, vitamin B12…).
4. ĐIỀU TRỊ THIẾU MÁU
4.1. Nguyên tắc điều trị thiếu máu
- Xác định và điều trị theo nguyên nhân; phối hợp điều trị nguyên nhân và truyền bù khối hồng cầu.
- Chỉ định truyền chế phẩm khối hồng cầu dựa vào huyết sắc tố và lâm sàng.
- Duy trì lượng huyết sắc tố tối thiểu từ 80 g/L (những trường hợp có bệnh lý tim, phổi mạn tính nên duy trì từ 90 g/L).
4.2. Điều trị một số nguyên nhân thiếu máu cụ thể
4.2.1. Thiếu máu thiếu yếu tố tạo máu
a. Thiếu máu do thiếu sắt: (Xin xem thêm bài thiếu máu thiếu sắt)
- Tìm và điều trị nguyên nhân gây thiếu sắt: U xơ tử cung, trĩ, giun móc…
- Chế độ ăn giàu sắt: Rau xanh, bí ngô, nho, chuối, thịt nạc, thịt bò, lòng đỏ trứng.
- Bổ sung sắt theo đường uống (liều trung bình 100-200 mg/ngày) hoặc tĩnh mạch:
+ Dạng viên: Sắt (II) sulfat phối hợp với acid folic (50mg sắt/ viên), uống 2-4 viên/ngày, trong 3-6 tháng;
+ Dạng dung dịch uống: Trong 3-6 tháng;
+ Sắt sucrose: 20mg/ml, pha truyền tĩnh mạch với tỷ lệ 1ml thuốc với 20ml dung dịch NaCl 0,9%, tổng lượng sắt tùy thuộc vào mức độ thiếu hụt và nồng độ hemoglobin, truyền 2-3 lần/tuần, sau đó chuyển sang bổ sung bằng chế phẩm đường uống.
- Xét nghiệm tế bào máu ngoại vi định kỳ 1-2 tuần/lần cho đến khi nồng độ huyết sắc tố đạt 100 g/L, sau đó kiểm tra định kỳ hàng tháng đến hết phác đồ.
b. Thiếu máu do thiếu acid folic và/hoặc vitamin B12
- Tìm và điều trị nguyên nhân: Tổn thương gan do rượu, cắt đoạn dạ dày…
- Chế độ ăn: Rau xanh, đậu đỗ, các loại nấm, chuối, dưa hấu, thịt, cá, gan, trứng.
- Bổ sung chế phẩm acid folic: 5mg hàng ngày trong vòng 4-6 tháng; vitamin B12: 1mg tiêm bắp 3 lần/tuần, tổng liều 10mg; sau đó duy trì 1mg/tháng.
- Xét nghiệm tế bào máu ngoại vi định kỳ hàng tháng để theo dõi và đánh giá kết quả điều trị.
4.2.2. Thiếu máu do tan máu tự miễn
(Xin xem thêm bài Tan máu tự miễn)
a. Điều trị ức chế miễn dịch bằng corticoid
- Methylprednisolone liều thông thường:
+ Liều 1-2mg/kg/ngày trong 2-4 tuần. Ở người già, liều corticoid khởi đầu có thể thấp hơn (0,6mg/kg/ngày). Duy trì liều điều trị đến khi huyết sắc tố đạt từ 80-100 g/L, sau đó giảm từ 5 đến 10 mg/tuần cho đến khi đạt liều duy trì;
+ Liều duy trì 10mg/ngày có thể kéo dài 3-6 tháng, uống hàng ngày hoặc cách ngày;
+ Đánh giá đáp ứng tốt khi nồng độ huyết sắc tố và tỷ lệ hồng cầu lưới tăng dần...
- Methylprednisolone liều cao: Chỉ định tương tự với phác đồ methylprednisolone thông thường với ưu điểm hạn chế được các biến chứng do sử dụng thuốc lâu dài mà vẫn đạt tác dụng ức chế miễn dịch tốt. Cụ thể:
+ 1.000 mg/ngày (tương đương 20 mg/kg), trong 1-3 ngày;
+ Pha với dung dịch đẳng trương, truyền trong khoảng 90 phút;
+ Lưu ý biến chứng tăng kali bằng cách theo dõi điện tim liên tục 24 giờ;
+ Giảm xuống liều từ 3-5mg/kg/ngày trong 3-5 ngày tiếp theo;
+ Sau đó duy trì liều 1-2mg/kg/ngày đến khi đạt kết quả điều trị.
b. Điều trị gamma globulin tĩnh mạch
- Chỉ định: Cơn tan máu rầm rộ hoặc khi không đáp ứng hoặc chống chỉ định dùng methylprednisolone.
- Liều dùng: 400 mg/kg/ngày x 5 ngày hoặc 1.000 mg/ngày x 2 ngày, nhắc lại sau mỗi chu kỳ 21 ngày.
c. Cắt lách
- Chỉ định khi không đáp ứng hoặc phụ thuộc corticoid với liều trên 20mg/ ngày và/ hoặc không có điều kiện sử dụng gamma globulin tĩnh mạch.
- Phải cân nhắc với những người bệnh dưới 25 tuổi vì nguy cơ nhiễm khuẩn nặng sau cắt lách.
d. Điều trị hóa chất và các thuốc ức chế miễn dịch
Chỉ định với những trường hợp tái phát sau cắt lách và những phương pháp điều trị khác. Có thể dùng một trong các phác đồ sau:
- Cyclophosphamide liều 2 mg/kg/ngày, duy trì 3-6 tháng. Kiểm tra lâm sàng và xét nghiệm tế bào máu, hóa sinh định kỳ hàng tháng.
- Hoặc azathioprine liều 1,5mg/kg/ngày kéo dài khoảng 3 tháng để đạt và duy trì tác dụng ức chế miễn dịch có hiệu quả. Kiểm tra định kỳ hàng tháng.
- Hoặc cyclosporin A liều ban đầu 5mg/kg/ngày, chia 2 lần, sau đó giảm xuống 3 mg/kg/ngày kết hợp với methylprednisolone 5mg/ngày. Điều trị kéo dài từ 3-6 tháng rồi đánh giá lại. Kiểm tra định kỳ hàng tháng.
- Hoặc mycophenolate mofetil liều 500-2000 mg/ngày trong 3-6 tháng cũng có tác dụng trong một số trường hợp tan máu tự miễn liên quan đến Hội chứng rối loạn sinh tủy.
e. Một số phương pháp điều trị khác
- Kháng thể kháng CD20 (rituximab):
+ Chỉ định chủ yếu trong trường hợp thất bại khi sử dụng các phác đồ trên;
+ Liều dùng 375mg/m2 da/tuần, trong 2-4 tuần. Theo dõi và kiểm tra định kỳ hàng tháng.
- Trao đổi huyết tương: Ít chỉ định, trừ khi hiệu giá tự kháng thể quá cao. Thường trao đổi khoảng 60% thể tích huyết tương của người bệnh.
- Ngoài ra còn có thể sử dụng danazol, tia xạ vùng lách hoặc ghép tế bào gốc tạo máu… Tuy nhiên, hiệu quả còn thấp và phức tạp, nhiều biến chứng.
4.2.3. Thiếu máu do tủy giảm sinh: (Xin xem thêm bài Suy tủy xương).
a. Điều trị hỗ trợ: Truyền khối hồng cầu, khối tiểu cầu…
b. Ghép tế bào gốc tạo máu đồng loại: Nguồn tế bào gốc từ người cho trong gia đình (máu ngoại vi, dịch tủy xương hoặc máu dây rốn) hoặc người cho không liên quan đến huyết thống.
c. Điều trị ức chế miễn dịch
- ATG (anti-thymocyte-globuline): 40 mg/ngày/4 ngày, thường kết hợp với cyclosporin A, liều 6-12mg/kg trong liên tục 6 tháng và methylprednisolone, liều 1 mg/kg trong 2-4 tuần đầu.
- Ngoài ra, có thể sử dụng thêm:
+ G-CSF (filgrastim) 300mcg/ ngày có thể giúp tăng số lượng bạch cầu tạm thời, tránh nguy cơ nhiễm khuẩn nặng.
+ Androgen (testosterone).
4.2.4. Thiếu máu do bệnh lý hồng cầu bẩm sinh
- Truyền khối hồng cầu, duy trì huyết sắc tố khoảng 90 g/L. Tốt nhất là truyền khối hồng cầu hòa hợp nhóm tối đa (phù hợp phenotype).
- Thải trừ sắt định kỳ, đặc biệt khi nồng độ ferritin vượt quá 1.000 ng/ml.
- Cắt lách: Sẽ làm giảm bớt yêu cầu truyền máu trong trường hợp α-thalassemia.
- Ghép tế bào gốc tạo máu đồng loài: Sử dụng nguồn tế bào gốc từ máu dây rốn, máu ngoại vi hoặc tủy xương của người cho hòa hợp HLA.
4.2.5. Thiếu máu do mất máu mạn
- Tìm nguyên nhân để điều trị: Cầm máu và điều trị ổ loét dạ dày-tá tràng, cắt u xơ tử cung, tẩy giun móc…
- Bù sắt và các yếu tố cần thiết cho tạo máu (xem phần trên).
TÀI LIỆU THAM KHẢO
1. Phùng Xuân Bình (2011), “Sinh lý máu”, Sinh lý học, Nhà xuất bản Y học.
2. Phạm Quang Vinh (2012), “Thiếu máu: phân loại và điều trị thiếu máu”, Bệnh học nội khoa. Nhà xuất bản Y học.
3. Đỗ Trung Phấn (2006), Bài giảng Huyết học-Truyền máu sau đại học, Nhà xuất bản Y học.
4. Nguyễn Quang Tùng (2013), “Các thông số tế bào máu ngoại vi”, Huyết học-Truyền máu cơ bản (Tài liệu đào tạo cử nhân kỹ thuật y học), Nhà xuất bản Y học.
5. Turgeon M.L (2005), “Erythrocytes”, in the Clinical Hematology: Theory and Procedures, 4th edition, p. 71-190.
6. Hoffman R. et al, (2009), “Red blood cells”, in the Hematology: Basic Principles and Practice, 5th edition.
7. Greer J.P. et al (2004), “Disorders of Red Cells”, in the Wintrobe’s Clinical Hematology, 2nd edition.
2. THIẾU MÁU THIẾU SẮT
1. ĐẠI CƯƠNG
- Thiếu máu thiếu sắt là tình trạng thiếu máu xảy ra do cơ thể không đủ sắt đáp ứng nhu cầu tạo hồng cầu.
- Thiếu máu thiếu sắt là tình trạng bệnh lý rất phổ biến trên thế giới, gặp ở mọi vùng miền, tuy nhiên gặp tỷ lệ cao nhiều ở các nước nghèo. Bệnh có thể gặp ở mọi lứa tuổi và ở cả hai giới nhưng phụ nữ độ tuổi sinh đẻ và trẻ em chiếm tỷ lệ cao hơn.
2. NGUYÊN NHÂN THIẾU SẮT
2.1. Không cung cấp đủ nhu cầu sắt
Do tăng nhu cầu sử dụng sắt của cơ thể như trẻ dậy thì, phụ nữ thời kỳ có thai, kinh nguyệt, cho con bú…; Do chế độ ăn không có đủ lượng sắt; Do cơ thể bị giảm hấp thu sắt như bị viêm loét dạ dày, cắt đoạn dạ dày,ruột…
2.2. Mất sắt do mất máu mạn tính
- Chảy máu đường tiêu hóa, tiết niệu. Mất máu do kinh nguyệt nhiều, do phẫu thuật, chấn thương,…
- Tan máu trong lòng mạch: Bệnh đái huyết sắc tố kịch phát ban đêm.
2.3. Rối loạn chuyển hóa sắt bẩm sinh (Hypotransferrinemia):
Cơ thể không tổng hợp được transferrin vận chuyển sắt. Bệnh rất hiếm gặp.
3. TRIỆU CHỨNG
3.1. Triệu chứng lâm sàng: Diễn biến từ từ qua 3 giai đoạn:
- Giai đoạn 1: Chỉ giảm sắt dự trữ, người bệnh chưa bị thiếu máu, thường có một số triệu chứng của nguyên nhân gây thiếu sắt.
- Giai đoạn 2: Đã cạn sắt dự trữ và giảm sắt vận chuyển, người bệnh chưa có biểu hiện rõ tình trạng thiếu máu, có triệu chứng của nguyên nhân gây thiếu sắt; bắt đầu có triệu chứng của thiếu sắt như: Mất tập trung, mệt mỏi.
- Giai đoạn 3: Thể hiện cả triệu chứng của thiếu máu và thiếu sắt. Tùy theo mức độ thiếu máu mà các triệu chứng của hội chứng thiếu máu sẽ khác nhau, mức độ nặng có thể choáng, ngất.
3.2. Xét nghiệm
- Tổng phân tích tế bào máu: Thiếu máu hồng cầu nhỏ nhược sắc, hồng cầu lưới giảm; số lượng bạch cầu và tiểu cầu trong giới hạn bình thường.
- Sinh hóa máu: Sắt huyết thanh giảm, ferritin giảm, transferrin tăng; khả năng gắn sắt toàn thể tăng; độ bão hòa transferrin giảm. Cần làm các xét nghiệm sinh hóa máu như sau: Bộ bilan sắt để khẳng định nếu bệnh nhân chưa rõ thiếu sắt (transferin, độ bão hòa transferin, transferin receptor, khả năng gắn sắt toàn thể); Tầm soát chức năng chung để phát hiện bệnh kèm theo: Glucose, chức năng thận, chức năng gan, điện giải đồ, bộ mỡ máu, LDH, chức năng tuyến giáp, định lượng acid folic và vitamin B12; haptoglobin...
- Xét nghiệm đông máu: Fibrinogen, PT, APTT, TT, D-Dimer để tầm soát đông cầm máu của cơ thể; Các xét nghiệm chức năng tiểu cầu: Ngưng tập tiểu cầu với các chất; xét nghiệm Von-Willerrand; định lượng yếu tố đông máu... nếu bệnh nhân có tình trạng chảy máu bất thường (rong kinh, xuất huyết tiêu hóa...).
- Các xét nghiệm khi có truyền máu và chế phẩm: Xét nghiệm vi sinh (HBV, HCV, HIV); xét nghiệm nhóm máu; sàng lọc và định danh kháng thể bất thường;
- Một số xét nghiệm tìm nguyên nhân: Soi dạ dày, soi đại tràng, siêu âm ổ bụng, tìm ký sinh trùng đường ruột (trứng giun móc trong phân); CD55, CD59 (chẩn đoán bệnh đái huyết sắc tố kịch phát ban đêm); coombs trực tiếp, coomb gián tiếp; kháng thể kháng nhân, kháng thể kháng ds-DNA; tủy đồ, sinh thiết tủy xương... để tìm các nguyên nhân thiếu máu khác kèm theo.
- Các xét nghiệm khác tầm soát tình trạng bệnh lý kèm theo: Tổng phân tích nước tiểu, tế bào nước tiểu, siêu âm bụng, X-quang phổi...
4. CHẨN ĐOÁN
4.1. Chẩn đoán xác định:
Dựa vào lâm sàng, xét nghiệm:
- Tổng phân tích tế bào máu: Thiếu máu hồng cầu nhỏ nhược sắc.
- Sinh hóa máu: Ferritin huyết thanh < 30ng/mL và hoặc độ bão hòa transferrin < 30%.
4.2. Chẩn đoán nguyên nhân:
Dựa vào lâm sàng, các xét nghiệm để chẩn đoán nguyên nhân thiếu máu thiếu sắt là do giảm cung cấp sắt hay mất sắt do mất máu hoặc do các nguyên nhân phối hợp.
4.3. Chẩn đoán phân biệt
a. Thalassemia:
- Lâm sàng: Biểu hiện thiếu máu từ nhỏ, thường có vàng da, lách to, có thể có tiền sử gia đình.
- Xét nghiệm: Ferritin thường tăng; transferrin bình thường hoặc giảm; độ bão hòa transferrin tăng; khả năng gắn sắt toàn thể bình thường; bilirubin gián tiếp thường tăng; điện di hay sắc kí Hb phát hiện bất thường thành phần Hb.
b. Thiếu máu trong viêm mạn tính:
- Lâm sàng: Tình trạng viêm mạn tính như viêm đa khớp dạng thấp, lao, lupus…
- Xét nghiệm: Sắt huyết thanh giảm, ferritin tăng, transferrin bình thường, độ bão hòa transferrin bình thường hoặc giảm, khả năng gắn sắt toàn thể tăng, tốc độ máu lắng tăng; protein phản ứng (CRP) tăng.
c. Thiếu máu trong suy dinh dưỡng:
- Lâm sàng: Tình trạng gầy, yếu. Có nguyên nhân dẫn đến suy dinh dưỡng như đói ăn, nhịn ăn,… trong thời gian dài.
- Xét nghiệm: Thiếu máu hồng cầu nhỏ nhược sắc, protein huyết thanh giảm.
5. ĐIỀU TRỊ
5.1. Nguyên tắc điều trị
- Hạn chế truyền máu, chỉ truyền máu trong trường hợp thiếu máu nặng, mất bù.
- Bổ sung các dạng chế phẩm sắt bằng truyền tĩnh mạch hoặc dung dịch uống, viên nén, khuyến khích sử dụng thuốc bổ sung sắt dạng uống; chỉ định sử dụng sắt đường truyền tĩnh mạch trong các trường hợp:
+ Thiếu máu thiếu sắt nặng, rất nặng;
+ Cơ thể không hấp thu được sắt uống: Cắt đoạn ruột, dạ dày, bệnh bẩm sinh;
+ Thiếu máu trong khi bệnh mạn tính hoặc viêm nhiễm đang tiến triển.
- Giai đoạn sớm khi mới thiếu sắt chưa thiếu máu: Bổ sung sắt qua thức ăn và uống các chế phẩm chứa sắt.
- Thời gian bổ sung sắt: Kéo dài, nên tiếp tục bổ sung sắt thêm ba tháng sau khi lượng huyết sắc tố trở đã về bình thường.
- Phối hợp với điều trị nguyên nhân: Cần tìm được nguyên nhân gây thiếu sắt để điều trị đồng thời với điều trị thiếu máu thiếu sắt.
5.2. Các chế phẩm thuốc bổ sung sắt
a. Dạng uống:
- Ferrous sulfate; ferrous gluconate; ferrous fumarate.
- Liều lượng: 2mg sắt/kg/ngày.
- Thời gian dùng thuốc: 6 tháng đến 12 tháng.
Nên bổ sung thêm vitamin C hoặc uống thêm nước cam, chanh để tăng khả năng hấp thu sắt.
Lưu ý: Thuốc hấp thu tốt nhất khi uống vào lúc đói, tuy nhiên nếu bị kích ứng dạ dày thì có thể uống trong lúc ăn; phân có màu đen, táo (không phải do xuất huyết tiêu hóa).
b. Dạng truyền tĩnh mạch:
- Iron sucrose; ferric gluconate; ferric carboxymaltose; Iron isomaltoside-1000; Ferumoxytol; Iron dextran (low molecular weight form)
- Cách tính liều lượng thuốc bổ sung sắt dạng tiêm:
Tổng liều (mg) = P (kg) x (Hb đích (g/L) - Hb thực (g/L)) x 0,24 + 500 mg
+ P: Trọng lượng cơ thể (kg);
+ Hb: Nồng độ huyết sắc tố (g/L).
5.3. Điều trị nguyên nhân
Phải chẩn đoán và điều trị nguyên nhân gây thiếu sắt một cách triệt để, tránh gây thiếu sắt tái phát.
6. CÁC XÉT NGHIỆM THEO DÕI ĐIỀU TRỊ
- Tổng phân tích tế bào máu, hồng cầu lưới.
- Sinh hóa máu: Sắt; ferritin; bộ bilan sắt; xét nghiệm chức năng gan; chức năng thận, điện giải đồ... để theo dõi đáp ứng thuốc và kiểm tra các tác dụng phụ khi điều trị thuốc.
- Các xét nghiệm theo dõi điều trị nguyên nhân gây thiếu máu của bệnh nhân: Chức năng tuyến giáp, xét nghiệm đông cầm máu, nội soi dạ dày, nội soi đại tràng, xét nghiệm phân...
7. PHÒNG BỆNH
- Bổ sung sắt trong suốt thời kỳ mang thai.
- Không nên uống trà, cà phê ngay sau ăn.
- Nên nuôi trẻ bằng sữa mẹ hoặc sữa bổ sung sắt dành cho trẻ trong năm đầu đời, vì sắt trong sữa mẹ được hấp thu hơn sữa bột.
TÀI LIỆU THAM KHẢO
1. Nguyễn Công Khanh (2004), Phân loại và chẩn đoán thiếu máu, Thiếu máu thiếu sắt, Thalassemia, Suy tuỷ xương, Huyết học lâm sàng Nhi khoa, Nhà xuất bản Y học Hà Nội, trang 33-35, 63-78, 132-146, 165-171.
2. Nguyễn Nghiêm Luật (2006), Chuyển hoá sắt và rối loạn chuyển hoá sắt, Bài giảng hoá sinh sau đại học.
3. Nguyễn Xuân Ninh (2004), Bệnh thiếu máu do thiếu sắt và biện pháp phòng chống, Một số chuyên đề Huyết học truyền máu tập I, nhà xuất bản Y học Hà Nội, trang 250 - 262.
4. Thái Quý, Nguyễn Hà Thanh (2006), Chuyển hoá sắt và thiếu máu thiếu sắt, Bài giảng huyết học truyền máu, nhà xuất bản Y học, Hà Nội, trang 208 - 213.
5. Beard J, Connor J (2003), Iron deficiency alter brain development and functioning, American society for nutritional sciencies journal 133, p. 1468 - 72.
6. Ernest B (2005), Disorder of iron metabolism. William Hematology 7th edition, p.511-37
7. Tomas Ganz. Iron deficency and overload. William Hematology 9th edition. p. 650 - 672
8. James SW & Micheal RM (2005), Hem biosynthesis and its disorder. Hematology, 4th edition, p. 449 - 451.
9. Rockville (2006). Screening for iron deficiency anemia-including iron supplementation for children and pregnant women: ecommendation statement. Agency for Healthcare Research and Quality, U.S. Preventive Services Task Force.
10. Guenter Weiss, M.D., and Lawrence T. Goodnough, M.D. (2005) Anemia of Chronic Disease, The new England journal of medicine 352:10
11. Anthony Lopez, Patrice Cacoub, Iain C Macdougall, Laurent Peyrin-Biroulet (2016). Iron deficiency anaemia. Thelancet. Vol 387, p 907 - 914.
3. BỆNH HUYẾT SẮC TỐ (Thalassemia và Huyết sắc tố bất thường)
1. ĐẠI CƯƠNG
Huyết sắc tố (Hemoglobin - Hb) là thành phần cơ bản của hồng cầu, được cấu tạo bởi hai loại chuỗi globin là alpha globin và không alpha globin. Ở người trưởng thành bình thường, Hb là sự kết hợp của 2 chuỗi α globin và 2 chuỗi β globin.
Bệnh do đột biến gen tổng hợp chuỗi globin dẫn đến giảm hoặc mất tổng hợp chuỗi globin được gọi là Thalassemia, thiếu hoặc không có chuỗi α globin gọi là Alpha thalassemia, thiếu hoặc không có chuỗi β globin gọi là Beta thalassemia.
Bệnh cũng có thể do gen globin đột biến để tạo ra một chuỗi globin có cấu trúc khác gọi là bệnh huyết sắc tố bất thường, trong đó HbE và Hb Constant Spring (HbCs) rất phổ biến ở Việt Nam. Thalassemia và bệnh huyết sắc tố bất thường có thể phối hợp với nhau như Beta thalassemia /HbE, Alpha thalassemia/HbCs (HbH HbCs). Gen quy định tổng hợp chuỗi α globin nằm trên nhiễm sắc thể (NST) 16, mỗi NST có gen α1 globin và gen α2 globin. Gen quy định tổng hợp chuỗi không αglobin nằm trên NST 11, mỗi NST có một cụm gen gồm gen β, δ và γ globin.
2. CHẨN ĐOÁN
2.1. Lâm sàng
Thalassemia là bệnh mạn tính. Các hội chứng và triệu chứng thường gặp sau:
- Hội chứng thiếu máu mạn.
- Hội chứng hoàng đảm.
- Lách to, gan to.
- Tăng sinh tạo máu phản ứng: Phì đại các xương dẹt làm biến dạng đầu, mặt như trán dô, mũi tẹt, bướu chẩm…
- Quá tải sắt: Da xạm đen, khô; tổn thương cơ quan như suy gan, suy nội tiết, suy tim… Thời gian xuất hiện: Mức độ rất nặng, gây phù thai, thường ở 3 tháng cuối của thai kỳ; Mức độ nặng, trẻ có biểu hiện thiếu máu sớm khi được vài tháng tuổi; Mức độ trung bình biểu hiện khi trẻ được 4-6 tuổi; Mức độ nhẹ, triệu chứng thiếu máu rất kín đáo, có thể được phát hiện khi có các bệnh lý khác kèm theo.
2.2. Cận lâm sàng
- Tổng phân tích tế bào máu ngoại vi: Thiếu máu, hồng cầu nhỏ, nhược sắc, kích thước không đều, hình thái đa dạng như hình bia bắn, hình giọt nước, có thể có hồng cầu non ra máu ngoại vi; hồng cầu lưới tăng.
- Sức bền thẩm thấu hồng cầu (Osmotic Fragility - OF): Tăng.
- Test DCIP (dichlorophenolindophenol): Dương tính khi có huyết sắc tố E.
- Xác định thành phần huyết sắc tố: Có Hb bất thường hoặc thay đổi tỷ lệ thành phần Hb.
- Xác định đột biến gen globin: Phát hiện các đột biến trên gen globin.
- Sinh hóa máu:
+ Bilirubin toàn phần tăng, bilirubin gián tiếp tăng;
+ Biểu hiện tình trạng thừa sắt: Sắt huyết thanh tăng, ferritin tăng, độ bão hòa transferin tăng,...;
+ Có thể thay đổi chỉ số hormon (biểu hiện của suy tuyến nội tiết): Tuyến yên (LH, GH, ACTH, FSH,...), tuyến sinh dục (FSH, Estradiol, progesterol, prolactin, testosterol, Gn-RH, IGFI, IGFBP-3…), tuyến giáp (T3, T4, FT3, FT4, TSH), tuyến cận giáp (PTH, calcitonin), tụy nội tiết (Insulin, peptid C, Fructosamin, HbA1C);
+ Có thể thay đổi các chỉ số: Glucose, GOT, GPT, photphatase kiềm, GGT, Ure, creatinin, acid uric, Protein, Albumin, Globulin, LDH, Canxi, Phosphate, Magne, Vitamin B12, Acid folic.
- Những xét nghiệm để giúp phòng ngừa và phát hiện biến chứng:
+ Định nhóm máu hệ ABO, Rh (D, C, c, E,e), MNS (Mia); định nhóm Kid (Jka, Jkb), Duffy (Fya, Fyb), P1Pk, M,N,S,s, Lea, Leb... trước khi truyền máu lần đầu;
+ Test Coomb trực tiếp, gián tiếp;
+ Sàng lọc kháng thể bất thường trước mỗi lần truyền máu;
+ Định danh kháng thể bất thường (khi sàng lọc kháng thể bất thường dương tính);
+ Xác định hiệu giá kháng thể miễn dịch (nếu có điều kiện thực hiện được);
+ Sàng lọc virus lây qua đường truyền máu: HBsAg (cho bệnh nhân truyền máu lần đầu, hoặc có tiền sử truyền máu mà HBsAg (-) cách đó trên 30 ngày); anti-HCV, anti HIV (cho bệnh nhân truyền máu lần đầu, hoặc có tiền sử truyền máu mà kết quả trước đó anti- HCV, anti HIV (-) trên 90 ngày);
+ Đếm bản coppy HCV (nếu HCV dương tính), HBV (nếu HBsAg dương tính);
+ Đông máu: Bộ xét nghiệm đông máu huyết tương (Fibrinogen, PT, APTT, TT);
+ Xét nghiệm một số yếu tố đông máu: Protein C, Protein S, AT III, yếu tố II, V, VII, X, D-dimer, Rotem… (tùy theo tình trạng lâm sàng của người bệnh mà có những chỉ định cụ thể);
+ Siêu âm ổ bụng: Gan, lách, mật...;
+ Siêu âm hạch nếu có các bất thường nghi ngờ trên lâm sàng;
+ Siêu âm tinh hoàn, buồng trứng, tử cung;
+ Siêu âm mạch trong các trường hợp nghi ngờ có huyết khối, tắc mạch hoặc các biểu hiện lâm sàng bất thường khác
+ Chụp cộng hưởng từ gan, tim để đánh giá mức độ quá tải sắt tại gan và tim (1 lần/năm);
+ X-Quang xương: Đặc biệt các xương dẹt (xương sọ, xương sườn, khung chậu, cột sống thắt lưng…), xương bàn tay;
+ Đo mật độ xương trung ương (cổ xương đùi, đốt sống thắt lưng…);
+ Nội soi đường tiêu hóa trong các trường hợp nghi ngờ có tổn thương;
+ Điện tâm đồ;
+ Siêu âm tim;
+ Chụp cộng hưởng từ hạt nhân: Đánh giá biến chứng sinh máu ngoài tủy tại các vị trí nghi ngờ có chèn ép
2.3. Chẩn đoán xác định
a. Chẩn đoán xác định bệnh:
- Triệu chứng lâm sàng: Hội chứng thiếu máu, hội chứng hoàng đảm, gan, lách to.
- Xét nghiệm: Thiếu máu nhược sắc hồng cầu nhỏ, bất thường thành phần huyết sắc tố và /hoặc có đột biến gen globin.
b. Chẩn đoán người mang gen:
- Lâm sàng: Không có triệu chứng; có thể quan hệ huyết thống với người bệnh.
- Xét nghiệm:
+ Tổng phân tích tế bào máu ngoại vi: Hb giảm nhẹ hoặc bình thường, hồng cầu nhỏ MCV giảm nhược sắc MCH giảm) kích thước không đều (RDW > 14%);
+ Sức bền thẩm thấu hồng cầu: Tăng;
+ Test DCIP: Dương tính trong bệnh huyết sắc tố E;
+ Xác định thành phần huyết sắc tố: Có Hb bất thường hoặc thay đổi thành phần Hb;
+ Xác định đột biến gen globin: Phát hiện các đột biến, kiểu đột biến trên gen globin;
+ Sinh hóa máu: Sắt, ferritin huyết thanh bình thường hoặc tăng.
2.4. Chẩn đoán thể bệnh
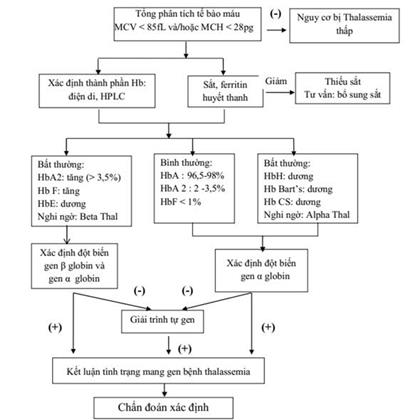
a. Alpha thalassemia
- Xét nghiệm thành phần huyết sắc tố có Hb Bart’s và/hoặc HbH.
- Và/ hoặc xét nghiệm DNA: Có đột biến gen alpha globin như: --SEA; --THAI; α3.7; α4.2;…
b. Beta thalassemia
- Xét nghiệm thành phần huyết sắc tố có HbA2 tăng và/ hoặc HbF tăng.
- Và/hoặc xét nghiệm DNA: Có đột biến gen beta globin như: Cd17; Cd 41/42; Cd71/72; Cd95; IVS1-1; IVS1-5; IVS2- 654;...
c. Bệnh huyết sắc tố bất thường
- Bệnh huyết sắc tố bất thường (chuỗi α globin): Xét nghiệm thành phần huyết sắc tố có HbCs hay Hb Quong Sze (HbQs), Hb Pakse’,… Và/hoặc xét nghiệm DNA: Có đột biến điểm trên gen α globin như: codon 142 (T>C); Codon 125 (T>C), codon 142 (A>T), codon 125 (T>C),...
- Bệnh huyết sắc tố bất thường (chuỗi β globin): Xét nghiệm thành phần huyết sắc tố có HbS (hồng cầu hình liềm) hay HbE, HbC và/ hoặc xét nghiệm DNA: Có đột biến trên gen β globin như: Đột biến codon 26 (G>A) tạo HbE; codon 6 (A>T) tạo HbS; codon 6 (G>A) tạo HbC…
- Phối hợp thalassemia với bệnh huyết sắc tố bất thường: Khi xét nghiệm có cả đặc điểm của thalassemia và bệnh huyết sắc tố bất thường. Ở Việt Nam thường gặp là beta thalasemia phối hợp với huyết sắc tố E và alpha-thalasemia phối hợp với huyết sắc tố HbCs.
2.5. Chẩn đoán mức độ bệnh
a. Alpha-thalassemia
- Mức độ rất nặng: Mất cả 4 gen alpha globin (gọi là bệnh Hb Bart’s).
+ Lâm sàng: phù thai, 100% chết trước hoặc ngay sau sinh; biểu hiện phù thai, vàng da, thiếu máu, gan to, rau thai to và mủn. Thường có biểu hiện ở 3 tháng cuối của thai kỳ. Mẹ có thể bị ngộ độc thai nghén khi mang thai;
+ Thành phần huyết sắc tố có Hb Bart’s chiếm chủ yếu, xét nghiệm DNA có đột biến mất 4 gen αglobin như đồng hợp tử SEA, THAI, FIL… (kiểu gen α0/α0).
- Mức độ trung bình - nhẹ: Mất 3 gen alpha globin (bệnh Hb H).
+ Thiếu máu nhẹ hoặc vừa; bệnh nặng hơn khi có điều kiện thuận lợi như sốt, có thai, bệnh lý khác,...;
+ Hb giảm nhẹ hoặc vừa, hồng cầu nhỏ nhược sắc, có thể thấy thể Heinz; Thành phần huyết sắc tố có HbH; xét nghiệm DNA có các đột biến làm mất 3 gen alpha globin như: --SEA/ -α3.7; --SEA/ -α4.2 hoặc thể phối hợp bệnh huyết sắc tố --SEA/-αCs… (kiểu gen α0/α+).
- Mức độ nhẹ/ người mang gen: Tổn thương 2 gen alpha globin.
+ Không có biểu hiện lâm sàng;
+ Hb bình thường; hồng cầu nhỏ, nhược sắc; Thành phần huyết sắc tố trong giới hạn bình thường hoặc tỷ lệ HbA2 trong giới hạn thấp (lúc mới sinh có thể thấy Hb Bart’s chiếm 2 - 5%); Xét nghiệm DNA có các đột biến làm mất 2 gen alpha globin như: --SEA / αα hoặc -α3.7/-α3.7 hoặc -α 4.2/-α4.2, hoặc ααHbCs/ -α3.7 ,… (kiểu gen α0/α+, α+/α+).
- Thể ẩn/ người mang gen: Mất 1 gen α globin.
+ Không có biểu hiện lâm sàng;
+ Các chỉ số hồng cầu có thể trong giới hạn bình thường hoặc hồng cầu nhỏ nhược sắc; Thành phần huyết sắc tố trong giới hạn bình thường (lúc mới sinh có thể thấy Hb Bart’s 1-2%); Xét nghiệm DNA có các đột biến làm mất 1 gen alpha như: -α3.7 /αα; -α4.2/αα…(kiểu gen α+/α).
b. Beta-thalassemia
- Mức độ nặng:
+ Có đủ các hội chứng thiếu máu, hội chứng hoàng đảm, gan to, lách to. Triệu chứng thiếu máu xuất hiện trước 2 tuổi. Lách thường to độ III, IV;
+ Hb thường dưới 70g/L; thành phần huyết sắc tố có HbF > 50%; HbA giảm hay không có xét nghiệm DNA có đột biến kiểu: β0/β0; β0/β+.
- Mức độ trung bình:
+ Triệu chứng xuất hiện khi trẻ trên 2 tuổi; lách to độ II, III;
+ Hb khoảng 70 - 100g/L; Thành phần huyết sắc tố có HbF từ 10-50%; xét nghiệm DNA có đột biến kiểu: β0/β+; β+/β+; β+/(α β0).
- Mức độ nhẹ/ người mang gen:
+ Không có biểu hiện lâm sàng;
+ Hb giảm nhẹ hoặc bình thường; HC nhỏ, nhược sắc; Thành phần huyết sắc tố có HbA2 tăng (> 3,5%) và hoặc HbF tăng; Kiểu gen β0/β, β+/β+.
c. Chẩn đoán mức độ phụ thuộc truyền máu
- Thalassemia phụ thuộc truyền máu (Transfusion Dependent Thalassemia - TDT): Bệnh nhân cần phải truyền máu định kỳ, nếu không được truyền máu định kỳ bệnh nhân sẽ có nhiều biến chứng và giảm tuổi thọ. Nhóm này bao gồm β-thalassemia nặng, β-thalassemia/HbE nặng, β-thalassemia nặng (HbBart’s nếu còn sống), một số β-thalassemia và α-thalassemia trung bình.
- Thalassemia không phụ thuộc truyền máu (Non Transfusion Dependent Thalassemia - NTDT): Bệnh nhân không phải truyền máu định kỳ để duy trì sự sống, tuy nhiên họ có thể phải truyền máu trong những điều kiện cụ thể. Nhóm này bao gồm β- thalassemia trung bình và nhẹ, β-thalassemia/HbE trung bình và nhẹ, α-thalassemia trung bình và nhẹ.
Bảng 2. Tiêu chuẩn phân loại theo truyền máu
|
Đặc điểm |
Phụ thuộc truyền máu |
Không phụ thuộc truyền máu |
|
Tuổi xuất hiện triệu chứng |
< 2 |
> 2 |
|
Hb cơ bản (g/L) |
< 70 |
70-100 |
|
Gan/lách to |
To nhiều |
To vừa - nhiều |
|
Chậm phát triển thể chất/ chậm dậy thì |
(+++) / (++++) |
(-) đến(++) |
|
Thiếu máu ảnh hưởng đến cuộc sống hàng ngày |
Có |
Không |
|
Biến dạng xương |
Có |
Không hoặc nhẹ |
|
Kiểu tổn thương gen |
Nặng Ví dụ: β0/β0 |
Nhẹ Ví dụ: β+/β+ |
|
Phối hợp tổn thương gen làm bệnh nhẹ hơn |
Không |
Có Ví dụ: β+/(α0β0) |
|
Phối hợp tổn thương làm bệnh nặng hơn |
Có Ví dụ: β+/(ααα β0) |
Không |
2.6. Chẩn đoán biến chứng
a. Quá tải sắt:
- Sinh hóa máu: Dựa vào chỉ số Ferritin huyết thanh để đánh giá chung tình trạng quá tải sắt.
|
Ferritin (ng/ml) |
Mức độ qui tải sắt |
|
< 300 |
Bình thường |
|
300 - 1000 |
Nhẹ |
|
1000 - 2500 |
Trung bình |
|
≥ 2500 |
Nặng |
- Chụp cộng hưởng từ hạt nhân gan (MRI): Đánh giá tình trạng nhiễm sắt tại gan.
|
Sắt trong gan (LIC) (mg sắt/g gan khô) |
Mức độ qui tải sắt |
|
< 2 |
Bình thường |
|
2 - 7 |
Nhẹ |
|
7 - 15 |
Trung bình |
|
≥ 15 |
Nặng |
(LIC: Liver Iron concentration - Nồng độ sắt trong gan)
- Chụp cộng hưởng từ hạt nhân tim (MRI): Đánh giá tình trạng nhiễm sắt tại tim.
|
MRI T2* (ms) |
Mức độ qui tải sắt |
|
> 20 |
Bình thường |
|
15 - 20 |
Nhẹ |
|
10 - 15 |
Trung bình |
|
< 10 |
Nặng |
(T2*: Là chỉ số đánh giá tình trạng nhiễm sắt tại tim qua kỹ thuật chụp cộng hưởng từ; đơn vị tính là ms (mili second - mili giây).
b. Suy tuyến nội tiết: Dựa vào lâm sàng và xét nghiệm.
- Đái tháo đường: Cần theo dõi định kỳ 6 tháng/lần.
Với người bệnh đã bị đái tháo đường thì cần xét nghiệm định kỳ hàng tháng để đánh giá hiệu quả điều trị.
- Tuyến yên: Cần theo dõi định kỳ 6 tháng/lần.
- Tuyến sinh dục: Cần theo dõi định kỳ hàng năm, bắt đầu khi trẻ được 13 tuổi đối với nữ và 14 tuổi đối với nam.
- Tuyến giáp, tuyến cận giáp: Cần theo dõi định kỳ hàng năm, bắt đầu khi người bệnh được 15 tuổi.
c. Tổn thương gan: Dựa vào lâm sàng và xét nghiệm.
- Xét nghiệm chức năng gan (định kỳ mỗi lần vào viện hoặc tối thiểu 3 tháng/lần).
- HBsAg, anti- HCV theo quy định về chỉ định xét nghiệm.
d. Tổn thương xương: Dựa vào lâm sàng và xét nghiệm.
- Chụp XQ xương: Đặc biệt xương dẹt (xương sọ, xương sườn, khung chậu, cột sống thắt lưng), xương bàn tay (đối với trẻ dưới 12 tuổi để đánh giá tốc độ trưởng thành).
- Đo mật độ xương trung ương: Cổ xương đùi, đốt sống thắt lưng.
Cần theo dõi và đánh giá lại định kỳ hàng năm.
e. Tổn thương tim mạch: Dựa vào lâm sàng và xét nghiệm.
- Đo điện tâm đồ, siêu âm tim.
Cần theo dõi và đánh giá lại định kỳ hàng năm, đối với trường hợp đã có biến chứng thì phải khám và làm xét nghiệm theo chỉ định.
f. Rối loạn đông cầm máu: Dựa vào lâm sàng và xét nghiệm.
Khi có biểu hiện lâm sàng nghi ngờ có rối loạn đông cầm máu, đặc biệt là bệnh nhân đã cắt lách cần lưu ý tình trạng tắc mạch.
2.7. Chẩn đoán phân biệt
a. Thiếu máu thiếu sắt: Cần phân biệt với thalassemia mức độ nhẹ.
- Tiền sử bản thân và gia đình không bị thiếu máu mạn tính.
- Lâm sàng: Có nguyên nhân (mất máu, giảm cung cấp sắt, tăng nhu cầu sắt…).
- Xét nghiệm:
+ Sinh hóa máu: Sắt giảm, ferritin giảm, transferrin tăng, khả năng gắn sắt toàn thể tăng, độ bão hòa transferrin giảm; bilirubin bình thường;
+ Thành phần huyết sắc tố: Bình thường.
b. Thiếu máu tan máu tự miễn:
- Tiền sử bản thân và gia đình không bị thiếu máu mạn tính.
- Lâm sàng: Bệnh diễn biến cấp tính, không bị biến dạng xương đầu, mặt.
- Xét nghiệm:
+ Tổng phân tích tế bào máu: Thiếu máu hồng cầu bình thường hoặc hồng cầu to;
+ Thành phần huyết sắc tố: Bình thường;
+ Nghiệm pháp Coomb trực tiếp, gián tiếp: Dương tính.
Lưu ý: Trong thalassemia, những người truyền máu nhiều lần cũng có thể có kết quả Coombs gián tiếp dương tính (do sinh kháng thể bất thường).
3. ĐIỀU TRỊ
3.1. Điều trị cơ bản:
a. Truyền khối hồng cầu:
- Bắt đầu truyền khi:
+ Hb < 70g/L trong hai lần xét nghiệm cách nhau trên 2 tuần;
+ Hb > 70g/L - 90g/L nhưng có các biến chứng như: Chậm phát triển, lách to nhiều, biến dạng xương, sinh máu ngoài tủy.
- Với bệnh nhân mức độ nặng nên duy trì huyết sắc tố trước truyền khoảng 90 g/L.
- Khoảng cách các đợt truyền máu: Mức độ nặng 2 - 5 tuần/đợt; mức độ trung bình 1 - 3 tháng/đợt (để hạn chế tình trạng huyết sắc tố giảm xuống quá thấp).
Lưu ý khi truyền khối hồng cầu:
- Xét nghiệm kháng nguyên hồng cầu ngoài hệ ABO để truyền máu hòa hợp kháng nguyên hệ hồng cầu (xem mục 2.2).
- Xét nghiệm sàng lọc kháng thể bất thường trước mỗi đợt truyền máu; Nếu người bệnh có kháng thể bất thường dương tính, cần thực hiện việc chọn máu phù hợp, tốt nhất là hòa hợp phenotype.
- Nên truyền khối hồng cầu lọc bạch cầu hoặc nghèo bạch cầu.
- Nên truyền khối cầu tươi (mới điều chế trong vòng 2 tuần).
b. Thải sắt
Tiêu chuẩn bắt đầu dùng thuốc thải sắt: Khi có một hoặc nhiều tiêu chuẩn sau:
- Người bệnh đã nhận ≥ 10 đơn vị khối hồng cầu.
- Ferritin huyết thanh ≥ 800ng/ml hoặc Ferritin huyết thanh ≥ 500ng/ml và người bệnh có nguy cơ tăng tích lũy sắt nhanh, như truyền máu định kỳ hàng tháng.
- Xét nghiệm MRI gan - tim có bằng chứng quá tải sắt (LIC ≥ 5mg/g).
- Tuổi bệnh nhân: Tùy theo từng loại thuốc thải sắt.
Tiêu chuẩn ngừng điều trị thải sắt: Khi ferritin < 300 ng/mL hoặc LIC < 3mg/g.
Các thuốc thải sắt:
(1) Deferrioxamin (tên khác là deferoxamine):
+ Liều lượng: Trẻ em 20 - 40 mg/kg/ngày; Người lớn: 40 - 60 mg/kg/ngày;
+ Cách dùng: Tiêm dưới da hoặc truyền tĩnh mạch liên tục 8 - 12 giờ/ngày, 5-6 ngày/tuần. Trường hợp quá tải sắt nặng thì dùng liên tục cả tuần.
Thận trọng:
+ Không nên dùng cho trẻ dưới 3 tuổi (do có nguy cơ làm chậm phát triển hệ xương), người bệnh đang có biểu hiện nhiễm trùng, người bệnh đang bị viêm gan cấp hoặc suy gan;
+ Với phụ nữ có thai, có thể sử dụng thuốc trong 3 tháng giữa và cuối chu kỳ thai, không dùng trong 3 tháng đầu.
(2) Deferipron: Sử dụng thuốc này khi thuốc Deferrioxamin không hiệu quả.
+ Liều lượng: Tối đa 75mg/kg/ngày;
+ Cách dùng: Uống, chia 3 lần/ngày;
+ Thận trọng: Không dùng cho phụ nữ có thai, không dùng cho trẻ dưới 6 tuổi. Nên cân nhắc khi sử dụng cho trẻ dưới 10 tuổi (do chưa có dữ liệu nghiên cứu về ảnh hưởng của thuốc);
+ Tác dụng phụ: Có thể gặp các triệu chứng như đau khớp, giảm bạch cầu hạt trung tính, nôn, thay đổi vị giác, chán ăn, tổn thương gan…
Kết hợp 2 thuốc thải sắt Deferrioxamin và Deferipron:
+ Chỉ định: Khi dùng liệu pháp 1 thuốc không hiệu quả hoặc khi người bệnh có tình trạng nhiễm sắt nặng hoặc đã có biến chứng tim mạch do quá tải sắt.
(3) Deferasirox: Nếu có thể thì nên lựa chọn điều trị ngay từ đầu.
+ Liều lượng trung bình:
● Dạng bào chế viên nén phân tán: 20mg - 30mg/kg/ngày;
● Dạng bào chế viên nén bao phim: 14mg - 21 mg/kg/ngày.
+ Cách dùng: Uống 1 lần/ngày, trước ăn 30 phút;
+ Thận trọng: Không dùng cho phụ nữ có thai, người bệnh bị suy thận; không nên sử dụng cho trẻ dưới 2 tuổi (vì chưa đủ cơ sở dữ liệu về độ an toàn);
+ Tác dụng phụ: Có thể gặp các triệu chứng đau bụng, buồn nôn, nôn, tiêu chảy, co thắt (thường kéo dài < 1 tuần), phát ban, tăng creatinin.
3.2. Ghép tế bào gốc đồng loài
- Là phương pháp điều trị triệt để, có hiệu quả cao.
- Chỉ định ghép tế bào gốc: Thalassemia mức độ nặng, dưới 16 tuổi, chưa có quá tải sắt mức độ nặng và có người cho tế bào gốc phù hợp HLA.
3.3. Điều trị hỗ trợ
a. Cắt lách:
- Nguyên tắc: Không khuyến khích việc cắt lách, cố gắng trì hoãn càng lâu càng tốt vì sau cắt lách người bệnh có nguy cơ huyết khối, nhiễm trùng.
- Chỉ cân nhắc cắt lách trong các trường hợp sau:
+ Khi người bệnh tăng nhu cầu truyền máu > 200 ml/kg/năm để giữ Hb đạt > 90g/L sau truyền (không kèm các nguyên nhân khác có thể làm giảm Hb);
+ Tăng tình trạng quá sắt (mặc dù người bệnh vẫn đang thải sắt theo phác đồ);
+ Lách quá to gây cản trở sinh hoạt hàng ngày hoặc gây đau cho người bệnh;
+ Giảm bạch cầu hoặc tiểu cầu do cường lách.
- Lưu ý:
+ Không nên cắt lách cho trẻ dưới 5 tuổi;
+ Người bệnh nên được tiêm phòng các vắc xin phòng viêm phổi (Pneumococcal), viêm màng não mủ (Meningococcal), não mô cầu, cúm (hemophilus influenza typ B) trước khi cắt lách ít nhất 3 tuần. Nên tiêm nhắc lại vaccine phòng viêm phổi, viêm não sau mỗi 5 năm;
+ Người bệnh có nguy cơ nhiễm trùng cao nhất trong thời gian 1 - 4 năm ngay sau cắt lách;
+ Người bệnh có nguy cơ huyết khối cao nhất trong thời gian 2 tuần đầu sau cắt lách và trong suốt 6 tháng sau cắt lách.
b. Thuốc tăng tạo HbF (α2/γ2)
- Mục đích: Thuốc làm tăng tổng hợp chuỗi γ-globin để kết hợp với chuỗi α-globin dư thừa tạo thành HbF, hiệu quả là giảm bớt lượng chuỗi α-globin dư thừa trong Beta thalassemia, làm tăng chất lượng hồng cầu.
- Thuốc tăng tạo HbF:
+ Hydroxyurea: 10-20mg/kg/ngày;
+ Erythropoietin 10.000 UI/tuần.
- Chỉ định: Beta Thalassemia mức độ trung bình.
- Đánh giá: Hiệu quả tốt khi Hb tăng thêm 10g/L sau 6 tháng.
3.4. Điều trị biến chứng: Là một phần rất quan trọng để nâng cao tuổi thọ và chất lượng cuộc sống của người bệnh.
- Hiện nay, hầu hết người bệnh thalassemia đều có từ một đến nhiều biến chứng, phổ biến là suy các tuyến nội tiết, viêm gan (C, B), suy gan, tổn thương xương, suy tim,… Vì vậy, người bệnh cần được kiểm tra định kỳ, toàn diện để phát hiện sớm các biến chứng và được điều trị kịp thời, đầy đủ theo đúng phác đồ.
- Điều trị suy tuyến nội tiết bằng liệu pháp bổ sung hormon.
- Điều trị suy gan, viêm gan do nhiễm HBV, HCV theo đúng phác đồ đặc hiệu.
- Điều trị biến chứng xương khớp bằng thuốc bổ sung canxi, vitamin D, bisphosphonate (theo phác đồ điều trị chống loãng xương).
- Điều trị suy tim, tăng áp lực động mạch phổi, rối loạn nhịp… theo phác đồ đặc hiệu.
- Điều trị rối loạn đông máu tùy theo từng giai doạn và diễn biến cụ thể mà sử dụng phác đồ phù hợp.
3.5. Chế độ ăn uống
Tránh quá tải sắt bằng cách không uống các thuốc có chứa sắt, hạn chế ăn các thức ăn có nhiều sắt. Nên có chế độ ăn cân bằng giàu dinh dưỡng, nhiều rau quả tươi để bổ sung acid folic, bổ sung canxi, kẽm và vitamin D, vitamin E.
4. PHÒNG BỆNH
- Người bệnh thalassemia cần được khám và điều trị định kỳ để hạn chế các biến chứng của bệnh.
- Người bệnh và người mang gen bệnh thalassemia cần được tư vấn để không sinh ra con bị bệnh Thalassemia.
- Bộ Y tế đã có quy định chi tiết phác đồ sàng lọc trước sinh, xin tham khảo Quyết định số 1807/QĐ-BYT ngày 21/4/2020.
Phụ lục
- MCV (mean corpuscular volume): Thể tích trung bình hồng cầu.
- MCH (mean corpuscular hemoglobin): Lượng huyết sắc tố trung bình hồng cầu.
- Thành phần huyết sắc tố ở người trưởng thành bình thường:
+ HbA1 (α2β2): 96-98%;
+ HbA2 (α2δ2): 0,5- 3,2%;
+ HbF (α2γ2): < 0,5%.
TÀI LIỆU THAM KHẢO
1. Phạm Quang Vinh (2006), “Cấu trúc chức năng Tổng hợp huyết sắc tố”, ”Bệnh huyết sắc tố”, Bài giảng huyết học truyền máu, Nhà xuất bản Y học.
2. Nguyễn Công Khanh (2004) ”Bệnh Hemoglobin”, Huyết học lâm sàng nhi khoa, Nhà xuất bản Y học.
3. Androulla Eleftheriou, About Thalassemia, TIF, 2005
4. David J.Weatherall (2006), Disorders of globin synthesis: the Thalassemia, William Heamatology 7th edition, p. 633-66.
5. Guideline for the clinical management of Thalassemia (2008), Thalassemia International Ferderation, 2nd edition.
6. Guideline for the management of non transfusion dependent Thalassemia (2013), Thalassemia International Ferderation.
7. Emergency management of Thalassemia (2012), Thalassemia International Ferderation.
8. Fucharoen G (2004), A simplified screening strategy for Thalassemia and hemoglobin E in rural communities in South - East Asia, Bull World Health Organ, 82(5):364-72.
9. Kanokwan Sanchaisuriya, A Reliable Screening Protocol for Thalassemia and Hemoglobinopathies in Pregnancy, Am J Clin Pathol 2005;123:113-118.
10. Management of hemoglobin disorder (2007), Report of joint WHO - TIF meeting
11. Prevention of Thalassemia and other haemoglobin disorders, Volume 1 (2013), Thalassemia International Ferderation, 2nd edition.
12. Prevention of Thalassemia and other haemoglobin disorders, Volume 2 (2012), Thalassemia International Ferderation, 2nd edition.
13. Standards for the Clinical Care of Children and Adults with Thalassaemia in the UK (2008), United Kingdom Thalassaemia Society.
4. SUY TỦY XƯƠNG
1. ĐẠI CƯƠNG
Suy tủy xương là bệnh lý của tế bào gốc tạo máu với đặc điểm là giảm ba dòng tế bào máu ngoại vi do sự giảm sinh tế bào máu của tủy xương. Nguyên nhân gây bệnh gồm có 2 nhóm nguyên nhân chính là do bẩm sinh và mắc phải. Suy tủy xương mắc phải thường gặp do những nguyên nhân sau:
- Hóa chất: Benzene là hóa chất hàng đầu gây suy tủy và một số bệnh ung thư. Thuốc trừ sâu, diệt cỏ như DDT (Chorophenothanediclorodiphenyltricloroethane) và thuốc nổ TNT cũng có khả năng gây bệnh suy tủy.
- Virus: Có liên quan với bệnh suy tủy xương gồm: B19 parvovirus thường gây bất sản dòng hồng cầu. Epstein-Bar virus là virus gây nhiễm trùng tăng bạch cầu đơn nhưng cũng gây suy tủy. Các virus viêm gan cũng có thể gây suy tủy như HBV, HCV và các virus non-A, non-B, non-C.
- Tia xạ.
- Do thuốc: Có nhiều loại thuốc gây suy tủy xương nhưng tỉ lệ gây bệnh cao nhất là Chloramphenicol (có thể tới 1/20.000 người sử dụng), tiếp đến là quinacrin với tỉ lệ mắc là 28/1 triệu người sử dụng. Một số thuốc kháng viêm không steroid cũng có thể gây suy tủy xương với tần suất thấp hơn.
- Thai nghén: Một số trường hợp xuất hiện bệnh khi mang thai và có thể hồi phục sau khi sinh.
- Không rõ căn nguyên: Trên thực tế, có khoảng trên 90% các trường hợp suy tủy xương là chưa tìm được nguyên nhân.
- Suy tuỷ xương có thể phối hợp đồng thời hoặc tiến triển thành các bệnh lý huyết học khác: PNH, MDS, AML.
2. CHẨN ĐOÁN
2.1. Lâm sàng
- Hội chứng thiếu máu: Da xanh, niêm mạc nhợt, mệt mỏi, chóng mặt, đau đầu, khó thở, khó tập trung, nhịp tim nhanh…
- Hội chứng xuất huyết: Xuất huyết đa hình thái, chủ yếu dưới da và niêm mạc, nhưng cũng có thể xuất huyết nội tạng.
- Hội chứng nhiễm trùng: Sốt, viêm loét miệng, nhiễm trùng da, mô mềm và các dấu hiệu nhiễm trùng cơ quan khác như hô hấp, tiết niệu…
2.2. Cận lâm sàng
a. Tế bào máu ngoại vi: Biểu hiện giảm 3 dòng ngoại vi, cụ thể:
- Giảm hồng cầu, huyết sắc tố.
- Hồng cầu bình sắc, kích thước bình thường.
- Giảm bạch cầu, chủ yếu giảm bạch cầu hạt trung tính, tăng tỉ lệ tế bào lympho.
- Giảm số lượng tiểu cầu.
- Giảm số lượng hồng cầu lưới.
b. Xét nghiệm tủy xương
- Trên xét nghiệm tủy đồ: Số lượng tế bào tủy giảm, giảm tế bào dòng hồng cầu, bạch cầu và mẫu tiểu cầu; tăng tỉ lệ tế bào lympho; không có tế bào ác tính.
- Xét nghiệm mô bệnh học tủy xương: Rất nghèo tế bào sinh máu, tủy sinh máu chủ yếu bị mỡ hóa ít xơ hóa, rải rác còn một số vùng có tế bào nhưng chủ yếu là lympho; không có tế bào lạ hoặc tế bào ác tính.
- Tế bào CD34 trong tủy giảm; tăng T CD8, giảm tỷ lệ CD4/CD8.
- Cấy cụm tế bào tủy có hiện tượng giảm tạo cụm.
- Định lượng TNF và INF trong dịch tủy tăng.
- Công thức nhiễm sắc thể, đứt gãy nhiễm sắc thể.
c. Các xét nghiệm định hướng nguyên nhân gây bệnh
- CMV (IgM, IgG), EBV (IgM, IgG), HBsAg, Anti HCV, HIV: Loại trừ nguyên nhân suy tủy xương do virus.
- Kháng thể kháng nhân (ANA), kháng thể kháng dsDNA: Loại trừ do bệnh hệ thống và tự miễn.
d. Xét nghiệm khác
- CD55/CD59 hồng cầu và bạch cầu.
- HLA để xem mức độ hoà hợp xét ghép tế bào gốc (những bệnh nhân có kế hoạch ghép tế bào gốc đồng loài).
2.3. Chẩn đoán xác định: Kết hợp tiêu chuẩn ở máu ngoại vi và sinh thiết tủy xương:
- Máu ngoại vi có hai trong 3 tiêu chuẩn sau:
+ Hemoglobin < 100g/L;
+ Tiểu cầu < 50G/L;
+ Bạch cầu trung tính < 1,5G/L.
- Mật độ tế bào tủy trên sinh thiết còn dưới 25%.
2.4. Chẩn đoán mức độ bệnh
a. Suy tủy xương thể nặng
- Mật độ tế bào tủy còn < 25% trên sinh thiết tủy xương, và
- Có hai 2 trong 3 tiêu chuẩn:
+ Bạch cầu trung tính < 0,5G/L;
+ Tiểu cầu < 20G/L;
+ Hồng cầu lưới < 20G/L.
b. Suy tủy xương thể rất nặng: Tiêu chuẩn như suy tủy xương thể nặng nhưng bạch cầu trung tính < 0,2G/L.
c. Thể trung bình: Không có đủ tiêu chuẩn của hai thể trên.
Lưu ý: Khái niệm giảm sinh tủy (Hypoplasia) được đánh giá chủ yếu qua mật độ tế bào tủy trên tiêu bản sinh thiết khi còn 25-50% nhưng dưới 30% so với người cùng giới và lứa cùng tuổi và ít có ý nghĩa tiên lượng về mặt lâm sàng.
2.5. Chẩn đoán phân biệt
a. Rối loạn sinh tủy
- Trên tủy đồ thấy số lượng tế bào tủy bình thường hoặc tăng, có rối loạn hình thái của các dòng tế bào máu, có thể gặp một tỉ lệ blast.
- Trên sinh thiết tủy có thể gặp sự khu trú bất thường của các tế bào đầu dòng - ALIPs (Abnormal Localization of Immature Precursors).
- Xét nghiệm nhiễm sắc thể có thể phát hiện bất thường.
b. Lơ xê mi cấp
- Lâm sàng cũng có hội chứng thiếu máu, xuất huyết, nhiễm trùng.
- Máu ngoại vi: Phần lớn là có gặp một số tế bào blast ác tính.
- Sự khác biệt trên lâm sàng là có thể có gan, lách, hạch to.
- Xét nghiệm tủy đồ thấy có tế bào blast ác tính (với tỉ lệ ≥ 20%).
c. Đái huyết sắc tố kịch phát ban đêm
- Lâm sàng có thiếu máu, có thể có xuất huyết.
- Có thể giảm 2 hoặc 3 dòng ngoại vi.
- Có thể giảm các tế bào máu trong tủy.
- Khác biệt với suy tủy xương ở chỗ người bệnh thường có biểu hiện đái ra huyết sắc tố ban đêm và tan máu: Như tăng hồng cầu lưới máu ngoại vi, tăng bilirubin gián tiếp.
- Xét nghiệm CD55, CD59 trên màng hồng cầu và bạch cầu hạt (bằng kỹ thuật Flowcytometry) bị giảm là dấu hiệu đặc trưng để chẩn đoán bệnh.
3. ĐIỀU TRỊ
Bao gồm cả điều trị đặc hiệu và điều trị hỗ trợ.
3.1. Sơ đồ điều trị
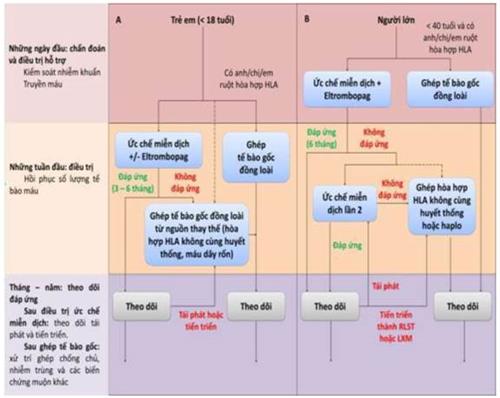
3.2. Điều trị đặc hiệu
3.2.1. Ghép tế bào gốc tạo máu: Là phương pháp điều trị hiệu quả nhất hiện tại.
- Chỉ định:
+ Tuổi ≤ 55;
+ Có nguồn tế bào gốc phù hợp: Lựa chọn hàng một là nguồn tế bào gốc từ anh chị em ruột hòa hợp HLA hoàn toàn; Các lựa chọn khác (máu dây rốn, haplo, hoặc máu dây rốn kết hợp với haplo).
+ Người bệnh suy tủy xương nặng hoặc rất nặng.
- Những biến chứng liên quan đến ghép:
+ Các biến chứng sớm: Viêm loét niêm mạc, viêm tắc tĩnh mạch trên gan, nhiễm trùng, xuất huyết, tổn thương tạng: Viêm bàng quang chảy máu, độc cơ tim…, bệnh ghép chống chủ, hội chứng mọc mảnh ghép, thải ghép, mảnh ghép mọc kém, CMV tái hoạt động...;
+ Các biến chứng muộn: Ung thư thứ phát, bệnh ghép chống chủ, nhiễm trùng, suy chức năng các tuyến nội tiết...
3.2.2. Các phương pháp điều trị đặc hiệu không phải ghép tế bào gốc đồng loài:
- Bệnh nhân không điều trị bằng phương pháp ghép tế bào gốc đồng loài, tùy thuộc vào khả năng cung cấp thuốc tại những cơ sở điều trị cũng như điều kiện của người bệnh có thể sử dụng các phác đồ dựa trên:
+ Các thuốc ức chế miễn dịch như ATG (Anti thymocyte globulin) có nguồn gốc từ thỏ hoặc ngựa, Cyclosporin A, …;
+ Các thuốc kích thích sinh tế bào máu như Androgen, Eltrombopag, G-CSF…
3.2.2.1. Các phác đồ ức chế miễn dịch:
a. ATG (anti thymocyte globulin) có nguồn gốc từ thỏ hoặc ngựa kết hợp cyclosporin: ATG ngựa kết hợp Cyclosporin A là phác đồ ức chế miễn dịch có hiệu quả cao nhất. Có thể sử dụng ATG thỏ để thay thế.
- ATG ngựa (Horse antithymocyte globulin hay viết tắt hATG) 40mg/kg/ngày trong 4 ngày.
Hoặc ATG thỏ (Rabbit antithymocyte globulin): Chú ý có 2 loại
+ Viết tắt rATG: 3,5 - 15mg/kg/ngày trong 5 ngày;
- Cyclosporin A 10mg/kg/ngày chia 2 cách 12 giờ. Duy trì nồng độ thuốc từ 200-400ng/ml trong 6 tháng đầu, sau đó giảm dần liều duy trì thêm tối thiểu 6 tháng tới 12 tháng, hoặc có thể kéo dài tuỳ mức độ đáp ứng và phụ thuộc liều CSA.
- Có thể sử dụng methylprednisolone 1mg/kg/ngày trong 14 ngày đầu, giảm dần liều và ngừng thuốc trong 2 tuần tiếp theo để dự phòng phản ứng huyết thanh với ATG.
- Lưu ý:
+ Các tác dụng phụ có thể có của cyclosporin A là: Tăng huyết áp, run, rậm lông, hạ magiê máu, tăng men gan, suy thận, đau nhức xương, co giật… (điều trị khi có biểu hiện hoặc dự phòng các tác dụng phụ);
+ Xét nghiệm theo dõi: Tế bào máu hàng ngày; đường máu, kali, calci, magiê máu, chức năng thận, enzym gan 1-2 lần/tuần; nồng độ cyclosporin A 1 lần/ tuần, trừ trường hợp đặc biệt có tác dụng phụ gây độc gan, thận cần định lượng theo diễn biến để chỉnh liều thuốc.
b. Cyclosporin A đơn trị: Có thể sử dụng cho các bệnh nhân có thể trạng yếu, bệnh kết hợp, suy giảm chức năng các cơ quan như gan, thận, tim mạch…
3.2.2.2. Sử dụng các chất kích thích sinh máu
a. Eltrombopag: Là chất chủ vận thụ thể của Thrombopoietin (TPO). Được chỉ định dùng kết hợp với ATG và cyclosporin A trong điều trị hàng một hoặc ở các bệnh nhân tái phát/ không đáp ứng với các phương pháp điều trị trước đó (không nên chỉ định ở những bệnh nhân có tổn thương nhiễm sắc thể số 7).
- Liều khởi đầu 25-50mg/ngày nếu sau 2 tuần không đáp ứng có thể tăng thêm 50mg.
- Liều tối đa: 150mg/ngày (người lớn, trẻ > 12 tuổi), 75mg/ngày (6-11 tuổi), 2,5mg/kg (2-5 tuổi). Tuy nhiên, với người châu Á, liều có thể giảm đi 1/2 so với liều chuẩn theo khuyến cáo của Viện sức khỏa Hoa Kỳ.
- Đánh giá đáp ứng điều trị khi: Hemoglobin > 100g/L, số lượng tiểu cầu > 50 G/L, bạch cầu trung tính > 1 G/L.
b. G-CSF: Được dùng kết hợp với các phác đồ *ức chế miễn dịch để rút ngắn thời gian giảm bạch cầu hạt và có giá trị dự đoán sớm nhóm bệnh nhân không đáp ứng với điều trị hàng một.
c. Androgen: Có tác dụng làm tăng lượng huyết sắc tố (thường có hiệu quả trên người bệnh thể trung bình).
3.3. Điều trị hỗ trợ
a. Truyền máu:
- Truyền khối hồng cầu khi huyết sắc tố dưới 80g/L, duy trì ở mức 90-100g/L.
- Truyền khối tiểu cầu khi tiểu cầu < 10G/L hoặc có tình trạng xuất huyết trên lâm sàng; tốt nhất là truyền tiểu cầu từ 1 người cho.
- Nếu có kế hoạch ghép tế bào gốc thì cần truyền chế phẩm máu được chiếu xạ, lọc bạch cầu và xét nghiệm CMV IgM âm tính.
b. Dự phòng và điều trị nhiễm trùng
- Người bệnh nằm phòng sạch, cách ly với những người bệnh nhiễm trùng khác.
- Dự phòng kháng sinh (nhóm quinolon, colistin, neomycin,… chống nấm đường uống (Itraconazole, voriconazole hoặc posaconazole,…).
- Khi có biểu hiện nhiễm trùng người bệnh cần được sử dụng kháng sinh phổ rộng sớm đồng thời với việc phân lập vi khuẩn hoặc nấm từ những bệnh phẩm nghi ngờ.
- Truyền khối bạch cầu hạt trong trường hợp nhiễm trùng quá nặng, đe doạ đến tính mạng bệnh nhân.
c. Các điều trị hỗ trợ khác
- Thải sắt khi Ferritin > 800 ng/ml (tham khảo thêm bài Bệnh huyết sắc tố). Có thể sử dụng các loại thuốc thải sắt khác nhau, nhưng thường dùng là desferrioxamine với liều 20-60mg/kg/ngày hoặc deferiprone 75mg/kg/ngày.
- Điều trị các biến chứng và tác dụng phụ của các thuốc điều trị như hạ áp, giảm đường máu, bổ sung canxi, magne, kali…
4. THEO DÕI ĐIỀU TRỊ
4.1. Lâm sàng:
- Tình trạng thiếu máu, xuất huyết và dấu hiệu nhiễm trùng.
4.2. Cận lâm sàng:
- Tổng phân tích tế bào máu, hồng cầu lưới; Sinh hóa máu: Acid uric, ure, creatinin, billirubin, GOT/GPT, điện giải đồ, magie, sắt, ferritin, định lượng nồng độ thuốc (cyclosporin A,...) 1-2 lần/ tuần với trường hợp điều trị nội trú và hàng tháng với điều trị ngoại trú.
- Đánh giá rối loạn đường huyết và calci máu sau mỗi 3 tháng sau điều trị.
- Bộ mỡ máu: Đánh giá trong vòng 3 tháng sau điều trị. Kiểm tra định kì mỗi 3 tháng trong trường hợp bệnh nhân có tiền sử rồi loạn mỡ máu hoặc kiểm tra sau 6 tháng với trường hợp không rối loạn.
- Huyết tuỷ đồ, sinh thiết tuỷ xương, công thức nhiễm sắc thể: Đánh giá lại sau 1 năm/lần, trừ trường hợp bệnh nghi ngờ tái phát hoặc chuyển thể.
- Xét nghiệm CD55/CD59: Mỗi 6 tháng đối với các trường hợp trước đó âm tính, 3 tháng với các trường hợp trước đó dương tính.
- Xét nghiệm vi sinh: EBV, CMV trong trường hợp nghi ngờ virus tái hoạt động; xét nghiệm lại HBV, HCV, HIV theo khuyến cáo trong trường hợp bệnh nhân có tiền sử truyền máu nhiều lần.
4.3. Đáp ứng điều trị sau điều trị
a. Sau ghép tế bào gốc: Theo hướng dẫn ghép tế bào gốc ở bệnh Suy tuỷ xương.
b. Sau điều trị ức chế miễn dịch ATG
|
|
Suy tủy xương mức độ nặng |
Suy tủy xương khác |
|
CR (đáp ứng hoàn toàn) |
Số lượng Tiểu cầu > 150 G/L. Số lượng BCHTT > 1,5 G/L. Lượng Hb bình thường |
Số lượng Tiểu cầu > 150 G/L. Số lượng BCHTT > 1,5 G/L. Lượng Hb bình thường |
|
PR (đáp ứng một phần) |
Không phụ thuộc truyền máu |
Không phụ thuộc truyền máu Cải thiện các dòng tế bào |
|
Không đáp ứng |
Bệnh nhân có đầy đủ tiêu chuẩn của suy tuỷ xương mức độ nặng |
Không đáp ứng các tiêu chí trên |
- Trường hợp sau điều trị hATG được 3-6 tháng: Nếu tình trạng bệnh nhân chưa đáp ứng cần xem xét điều trị miễn dịch lần 2 bằng rATG hoặc có kế hoạch ghép tế bào gốc khác (ghép không cùng huyết thông không phù hợp HLA, máu dây rốn, haplotype, hoặc kết hợp các nguồn ghép,…) càng sớm càng tốt.
4. TIÊN LƯỢNG
- Người bệnh suy tủy xương thể rất nặng với bạch cầu trung tính giảm dưới 0,2G/L đáp ứng rất kém với các thuốc ức chế miễn dịch và có tiên lượng rất xấu. Người bệnh suy tủy thể nặng có tỉ lệ tử vong 25% trong vòng 4 tháng đầu và 50% trong vòng 1 năm nếu không ghép tế bào gốc.
- Người bệnh được ghép tế bào gốc có tỉ lệ đáp ứng chung khoảng 70%.
- Người bệnh điều trị ATG kết hợp CSA thì cải thiện được 70%; nhưng sau 10 năm thì có khoảng 40% tiến triển thành đái huyết sắc tố kịch phát ban đêm, rối loạn sinh tủy hoặc lơ xê mi cấp.
- Người bệnh chỉ điều trị triệu chứng khi không đáp ứng với các phương pháp điều trị đặc hiệu có tỉ lệ tử vong khoảng 80% trong vòng 2 năm.
TÀI LIỆU THAM KHẢO
1. Nguyễn Hà Thanh, suy tủy xương, bài giảng Huyết học - Truyền máu sau đại học (2019), nhà xuất bản y học: 224-234.
2. Brodsky.R.A (2009), Acquired Aplastic Anemia, Wintrobe’s Clinical Hematology1, pp 1180-1192.
3. DeZern A.E and Brodsky R.A. Clinical management of aplastic anemia. Rev Hematol. 2011 April; 4(2): 221-230.
4. George.B.S, Marshall A.L (2006), Aplastic anemia, William Hematology, 7, pp. 419-430.
5. Guidelines for the diagnosis and management of acquired aplastic anaemia, British Journal of Haematology, 2003, 123, 782-801.
6. MacMillan ML, Walters MC, Gluckman E. Transplant outcomes in bone marrow failure syndromes and hemoglobinopathies. Semin Hematol. Jan 2010;47(1):37-45.
7. Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. Aug 4 2011;365(5):4308.
8. Young N.S and Maciejewski J.P (2005). Aplastic anemia. Hematology Basic principle and practice,4, pp 381-407.
9. Novartis. Eltrombopag tablets for oral use. Prescribing Information. March 2017.
10. Scheinberg P. and Young N.S. (2012). How I treat acquired aplastic anemia. Blood, 120(6), 1185-1196.
11. Peslak S.A., Olson T., and Babushok D.V. (2017). Diagnosis and Treatment of Aplastic Anemia. Current Treatment Options in Oncology, 18(12).
12. For the EBMT SAA Working Party, Ecsedi M., Lengline É., et al. (2019). Use of eltrombopag in aplastic anemia in Europe. Annals of Hematology, 98(6), 1341-1350.
13. Scheinberg P. (2018). Activity of eltrombopag in severe aplastic anemia. 2(21), 9.
14. Scheinberg P., Nunez O., Weinstein B., et al. (2011). Horse versus Rabbit Antithymocyte Globulin in Acquired Aplastic Anemia. New England Journal of Medicine, 365(5), 430-438.
15. Killick S.B., Bown N., Cavenagh J., et al. (2016). Guidelines for the diagnosis and management of adult aplastic anaemia. British Journal of Haematology, 172(2), 187-207.
16. Lee S.-E., Yahng S.-A., Cho B.-S., et al. (2016). Impact of pretransplant red cell transfusion on outcome after allogeneic stem cell transplantation in adult patients with severe aplastic anemia. Bone Marrow Transplantation, 51(10), 1323-1329.
17. Hayakawa, J., Kanda, J., Akahoshi, et al. (2017). Meta-analysis of treatment with rabbit and horse antithymocyte globulin for aplastic anemia. International Journal of Hematology, 105(5), 578-586.
5. ĐÁI HUYẾT SẮC TỐ KỊCH PHÁT BAN ĐÊM
1. ĐẠI CƯƠNG
- Đái huyết sắc tố kịch phát ban đêm (Paroxysmal noctural hemoglobinuria - PNH) là bệnh lý do phá hủy hồng cầu và giải phóng huyết sắc tố vào nước tiểu tại thời điểm ban đêm. Bệnh được coi là một tiền lơ xê mi (preleukemia).
- Là bệnh rối loạn đơn dòng mắc phải của tế bào gốc tuỷ xương, do đột biến gen PIG-A (phosphatidylinositol glycan class A) gây gián đoạn sinh tổng hợp GPI (glycosylphosphatidylinositol), làm thiếu hụt tất cả GPI protein gắn kết trên màng tế bào, đặc biệt là protein điều hoà bổ thể CD59 (chất ức chế màng phản ứng ly giải) và CD55 (yếu tố thúc đẩy tiêu huỷ), dẫn đến tăng nhạy cảm với bổ thể của tế bào, tan máu trong lòng mạch, thúc đẩy các yếu tố viêm và hệ thống thải hemoglobin.
- Hay gặp ở các nước châu Á, chủ yếu ở độ tuổi từ 30-59.
2. CHẨN ĐOÁN
2.1. Lâm sàng
a. Tan máu trong lòng mạch
- Thường biểu hiện tan máu mạn tính trong lúc ngủ nên nước tiểu sẫm màu vào buổi sáng do đái huyết sắc tố. Tuy nhiên, chỉ có 25% trường hợp có huyết sắc tố.
- Biểu hiện tan máu tăng khi có nhiễm trùng hoặc do một số yếu tố khác kích hoạt bổ thể làm tăng tan máu gây cơn tan máu.
b. Huyết khối
- Là nguyên nhân chính gây tử vong. Huyết khối xuất hiện chủ yếu ở các trường hợp tế bào dòng bạch cầu hạt thiếu hụt > 50% CD55 và CD59; thường gặp huyết khối tĩnh mạch ổ bụng, tĩnh mạch não và nhồi máu phổi có thể đe dọa tính mạng người bệnh; ít gặp hơn là huyết khối tĩnh mạch sâu ở chi, huyết khối tĩnh mạch mào tinh hoàn và xoang hang gây căng cứng dương vật; tắc tĩnh mạch dưới da giống phát ban.
c. Giảm chức năng tuỷ xương
- Biểu hiện suy tuỷ xương (50-60% bệnh nhân) hoặc thiếu máu dai dẳng trong rối loạn sinh tủy (15-20%).
- Bệnh PNH có thể có phối hợp suy tuỷ xương (STX); thường gặp ở người trẻ châu Á; biểu hiện: Thiếu máu, hồng cầu lưới giảm, có thể kèm theo bạch cầu và tiểu cầu giảm.
- Người bệnh suy tuỷ xương sau điều trị thuốc ức chế miễn dịch có thể tiến triển thành đái huyết sắc tố kịch phát ban đêm.
- Bệnh PNH có biểu hiện tắc mạch gặp khoảng 30% người bệnh châu Âu, nhưng dưới 15% ở người bệnh châu Á. Ngược lại, PNH liên quan tổn thương tuỷ xương như suy tuỷ hay gặp ở châu Á.
d. Các triệu chứng khác
- Thiếu máu, mệt mỏi do nhiều nguyên nhân.
- Bệnh thận mạn tính là hậu quả tan máu trong lòng mạch thời gian dài gây tắc vi mạch thận, lắng đọng sắt.
- Có thể biểu hiện tăng áp động mạch phổi mức độ nhẹ đến trung bình.
- Rối loạn co bóp thực quản, rối loạn cương dương.
- Hiếm gặp: Gan và lách to.
2.2. Xét nghiệm
a. Đếm tế bào dòng chảy (Flow cytometry)
- Cho thấy có thiếu hụt CD55 và CD59 (các protein GPI-A) ở các hồng cầu ngoại vi, đó là căn cứ để chia ra các thể bệnh.
- Có thể thấy có thiếu hụt CD55 và CD59 ở các bạch cầu hạt và/hoặc các bạch cầu mono. Tỷ lệ quần thể thiếu hụt CD55 và CD59 liên quan thuận với nguy cơ tắc mạch (nguy cơ tăng cao gấp 8 lần ở nhóm thiếu hụt > 50% so với nhóm bệnh nhân thiếu hụt < 50%).
- Ngoài ra, có thể phát hiện sự thiếu hụt của các GPI-A khác như CD16, CD24 ở bạch cầu hạt và CD14, CD24 ở bạch cầu mono.
b. Đếm tế bào dòng chảy độ phân giải cao (High resolution Flow cytometry) FLAER (fluorescein-tagged proaerolysin)
Có thể phát hiện được quần thể rất nhỏ tế bào đái huyết sắc tố kịch phát ban đêm với tỷ lệ khoảng 0,003%. Đặc biệt, trong trường hợp đái huyết sắc tố kịch phát ban đêm kết hợp với suy tủy xương hoặc rối loạn sinh tủy có số lượng bạch cầu thấp.
c. Xét nghiệm tủy xương
- Chỉ định trong trường hợp: Giảm các dòng tế bào máu nặng.
- Trường hợp STX-PNH: Tủy nghèo tế bào, nhưng dòng hồng cầu có thể tăng sinh tương đối.
- Xác định di truyền tế bào: Có khoảng 25% bệnh nhân PNH có bất thường.
2.3. Khuyến cáo sàng lọc người bệnh mắc đái huyết sắc tố kịch phát ban đêm
- Người bệnh có biểu hiện đái huyết sắc tố.
- Người bệnh có biểu hiện tan máu trong lòng mạch, tăng LDH nhưng xét nghiệm Coombs âm tính; xét nghiệm sắt và ferritin có thể bình thường hoặc tăng.
- Người bệnh có huyết khối tĩnh mạch ở vị trí bất thường kèm bằng chứng tan máu trong lòng mạch:
+ Hội chứng Budd-Chiari;
+ Huyết khối tĩnh mạch (tĩnh mạch mạc treo, tĩnh mạch cửa, tĩnh mạch não, tĩnh mạch ngoại vi...).
- Người bệnh suy tủy xương: Khi bắt đầu chẩn đoán và hàng năm dù không có biểu hiện tan máu.
- Người bệnh rối loạn sinh tủy thể thiếu máu dai dẳng, giảm các tế bào máu.
- Người bệnh có biểu hiện nuốt khó hoặc đau bụng kèm theo có biểu hiện của tan máu trong lòng mạch.
2.4. Các xét nghiệm trong chẩn đoán và xử trí PNH
Những trường hợp có biểu hiện lâm sàng của tan máu (Đái huyết sắc tố kịch phát ban đêm cổ điển và đái huyết sắc tố kịch phát ban đêm kết hợp với suy tủy xương):
- Tại thời điểm chẩn đoán, cần xét nghiệm CD55 và CD59, cả dòng hồng cầu và bạch cầu hạt.
- Sau khi đã được chẩn đoán, cần xét nghiệm CD55 và CD59 hai tháng/lần trong 2 năm, sau đó mỗi năm một lần nếu người bệnh ổn định.
- Nếu có bằng chứng của lâm sàng tiến triển, cần tiến hành thêm những xét nghiệm khác.
2.5. Chẩn đoán xác định
- Hồng cầu và bạch cầu có thiếu hụt CD55 và CD59.
- Tổng phân tích tế bào máu: Thấy giảm hồng cầu; có thể giảm bạch cầu, tiểu cầu và hồng cầu lưới.
- LDH, bilirubin huyết thanh tăng.
- Tuỷ đồ, sinh thiết tuỷ xương: Có thể có hình ảnh giảm sinh hoặc rối loạn sinh tủy.
- Phân tích di truyền tế bào: Có thể có bất thường trong rối loạn sinh tủy.
3. XẾP LOẠI
Bảng 3: xếp loại đái huyết sắc tố kịch phát ban đêm của nhóm nghiên cứu thế giới như sau:
|
Xếp loại |
PNH cổ điển |
PNH kết hợp tổn thương tủy xương |
PNH không có biểu hiện lâm sàng |
|
Mức độ tan máu trong lòng mạch |
Nước tiểu đỏ rõ (đái huyết sắc tố đại thể thường xuyên và kéo dài) |
Nhẹ đến trung bình (đái huyết sắc tố đại thể thường xuyên và kéo dài) |
Không có bằng chứng lâm sàng hay xét nghiệm hóa sinh có tan máu trong lòng mạch. |
|
Tỷ lệ tan máu trong lòng mạch |
Nước tiểu đỏ (đái máu đại thể, tăng cao LDH) |
Mức độ nhẹ (các xét nghiệm tan máu thường biểu hiện bất thường tối thiểu) |
Không có biểu hiện lâm sàng hay bằng chứng trên trên xét nghiệm của tan máu trong lòng mạch. |
|
Tuỷ xương |
Hồng cầu tuỷ xương tăng sinh với hình thái bình thường hoặc gần bình thường (hiếm gặp bất thường nhiễm sắc thể) |
Có kết hợp tổn thương tuỷ xương (Suy tuỷ xương hoặc rối loạn sinh tuỷ) |
Có kết hợp tổn thương tuỷ xương (Suy tuỷ xương hoặc rối loạn sinh tuỷ) |
|
Đếm tế bào dòng chảy (Flow cytometry: FC) |
Tỷ lệ dòng bạch cầu hạt thiếu hụt CD55 và CD59 > 50% |
Tỷ lệ dòng bạch cầu hạt thiếu hụt CD55 và CD59 < 50%. |
Tỷ lệ dòng bạch cầu hạt thiếu hụt CD55 và CD59 rất thấp < 1%, phải xét nghiệm bằng FC độ phân tích cao. |
Ghi chú: Để xếp loại thể bệnh PNH cần xét nghiệm tế bào dòng chảy dòng bạch cầu, không phải hồng cầu do có thể sai số vì một số hồng cầu thiếu hụt protein GPI-A đã bị phá hủy bởi bổ thể hoặc truyền máu ảnh hưởng tới đánh giá quần thể hồng cầu.
4. ĐIỀU TRỊ
4.1. Phương hướng điều trị
Hiện nay, chiến lược điều trị cho PNH có sự thay đổi duy nhất là điều trị ức chế bổ thể và ghép tế bào gốc đồng loài.. Ghép tế bào gốc tạo máu đồng loài được lựa chọn nếu nhóm thuốc bổ thể không có sẵn và cho bệnh nhân PNH có suy tủy xương nặng. Điều trị ức chế miễn dịch cho bệnh nhân PNH có suy tủy xương để cải thiện tổn thương tuỷ xương.
a. Đái huyết sắc tố niệu kịch phát ban đêm cổ điển
- Ghép tế bào gốc tạo máu đồng loài được đánh giá là phương pháp có hiệu quả cao nhất giúp khỏi được bệnh.
b. Đái huyết sắc tố niệu kịch phát ban đêm không có biểu hiện lâm sàng
Không có điều trị đặc hiệu cho PNH; tập trung vào điều trị ức chế miễn dịch nhằm cải thiện các tế bào máu.
c. Đái huyết sắc tố kịch phát ban đêm có suy tuỷ xương
- Hướng điều trị gồm ghép tế bào gốc đồng loài hoặc cyclophosphamid liều cao hoặc điều trị ATG kết hợp cyclosporin A.
- Nếu người bệnh đái huyết sắc tố kịch phát ban đêm chưa đủ tiêu chuẩn xếp nhóm suy tủy xương nặng thì theo dõi hoặc điều trị thuốc ức chế miễn dịch.
4.2. Điều trị cụ thể
4.2.1. Điều trị thiếu máu
- Trước khi bắt đầu điều trị cần phải đánh giá để có hướng điều trị: Mức độ thiếu máu do tan máu hay do giảm chức năng tủy xương. Có thiếu sắt do đái huyết sắc tố hay không.
- Điều trị thiếu máu: Truyền khối hồng cầu, bổ sung sắt trong trường hợp sắt giảm.
4.2.2. Điều trị tan máu: trong điều kiện Việt Nam
- Methylprednisolone: Có thể điều trị 1 thời gian ngắn (1 tuần) với liều thấp (10-20mg methylprednisolone) các trường hợp tan máu cấp.
d. Điều trị hỗ trợ:
- Truyền hồng cầu khối mà không cần hồng cầu rửa.
- Bổ sung acid folic 1-2mg/ ngày uống và vitamin B12 do tăng sinh dòng hồng cầu trong tuỷ xương.
- Bổ sung sắt uống khi thiếu sắt do mất sắt mạn tính do đái máu.
- Phòng và điều trị nhiễm trùng sớm bằng kháng sinh vì nhiễm trùng có thể gây cơn tan máu: Có thể dự phòng bằng kháng sinh đường uống hàng ngày.
4.2.3. Dự phòng huyết khối và điều trị huyết khối
a. Dự phòng huyết khối
- Chỉ định dự phòng: Trường hợp thiếu hụt CD55 và CD59 ở bạch cầu hạt > 50%; cần dự phòng bằng warfarin, duy trì INR (international normalized ratio): 2,0-3,0.
- Chống chỉ định dự phòng chống đông cho các trường hợp sau:
+ Bệnh diễn biến kéo dài mà chưa có biểu hiện huyết khối lần nào;
+ Người bệnh có nguy cơ chảy máu liên quan đến suy tuỷ.
b. Điều trị huyết khối
- Tắc mạch cấp: Điều trị chống đông bằng heparin.
- Hội chứng Budd-Chiari có huyết khối: Điều trị chống đông và can thiệp mạch.
- Những người bệnh có tiền sử huyết khối phải điều trị chống đông duy trì kéo dài.
Lưu ý: Trong trường hợp giảm tiểu cầu vẫn có thể phải điều trị chống đông, nếu cần sẽ truyền tiểu cầu hỗ trợ.
- Người bệnh thất bại với wafarin liều chuẩn, khi đó lựa chọn wafarin liều cao (duy trì INR từ 3,0-4,0) hoặc tiêm dưới da heparin trọng lượng phân tử thấp.
4.2.4. Điều trị trường hợp có tổn thương tủy xương
Những trường hợp có suy tủy xương hoặc rối loạn sinh tủy có thể điều trị thuốc ức chế miễn dịch.
4.2.5. Ghép tế bào gốc tạo máu đồng loài
a. Chỉ định
- Suy tuỷ xương kết hợp đái huyết sắc tố kịch phát ban đêm.
- Đái huyết sắc tố niệu kịch phát ban đêm có các biến chứng chính:
+ Huyết khối nguy hiểm đe dọa tính mạng, tái diễn nhiều lần;
+ Thiếu máu tan máu phụ thuộc truyền máu;
+ Chuyển thể như MDS hoặc lơ xê mi.
b. Một số vấn đề liên quan đến ghép
- Khuyến cáo phác đồ điều kiện hóa: Cyclophosphamid phối hợp ATG đối với người bệnh Đái huyết sắc tố kịch phát ban đêm/ Suy tuỷ xương. Thể đái huyết sắc tố kịch phát ban đêm cổ điển, nên sử dụng phác đồ diệt tuỷ mạnh hơn.
- Ghép nửa hoà hợp không diệt tuỷ sử dụng cyclophosphamide sau ghép để giảm biến chứng GVHD hoặc kết hợp ghép nửa hoà hợp và máu dây rốn không cùng huyết thống... là lựa chọn nếu bệnh nhân không có người hiến phù hợp HLA.
- Không có các biến chứng đặc hiệu đái huyết sắc tố kịch phát ban đêm liên quan đến ghép. Biến chứng ghép chống chủ (Graft-versus-host disease: GVHD): GVHD cấp mức độ nặng xảy ra ở 1/3 người bệnh ghép; tỷ lệ GVHD mạn khoảng 35%.
- Tỷ lệ đáp ứng nếu ghép từ anh chị em ruột phù hợp HLA khoảng 50-60%.
5. THEO DÕI
5.1. Lâm sàng: Tình trạng thiếu máu, xuất huyết, tan máu, tắc mạch.
5.2. Cận lâm sàng:
- Tổng phân tích tế bào máu, sinh hoá máu (glucose, chức năng gan, thận...), đông cầm máu trong mỗi lần khám, điều trị.
- Xét nghiệm CD55/CD59:
+ Đối với người bệnh có bằng chứng thiếu hụt CD55/CD59 đánh giá 6 tháng/lần trong vòng 2 năm hoặc khi có triệu chứng lâm sàng tiến triển, sau đó đánh giá 1 năm/lần nếu bệnh ổn định.
+ Đối với người bệnh điều trị Eculizumab: Đánh giá 1 năm/ lần hoặc khi lâm sàng thay đổi;
+ Đối với người bệnh sau ghép tế bào gốc: 3 tháng/ lần cho đến khi âm tính, sau đó giảm dần hàng năm, hoặc làm lại khi nghi ngờ tái phát.
TÀI LIỆU THAM KHẢO
1. Bessler M, Hiken J. 2008. The pathophysiology of paroxysmal nocturnal haemoglobinuria. Hematology, 104-110.
2. Hillmen P. 2008. The Role of Complement Inhibition in PNH. Hematology,116-123.
3. Hillmen P. 2011. Chapter 11: Paroxysmal Nocturmal hemoglobinuria. Postgraduate Haematology, sixth edition (Victor Hoffbrand, Daniel Catovsky, Edwảd GD Tuddenham, Anthony Green).
4. Neal S. Young, 2009. Paroxysmal Nocturnal Haemoglobinuria and myelodysplastic syndrome: Clonal expansion of PIG-A mutant hematopoietic cells in bone marrow failure. Haematologica, 94(1): 3-7.
5. Rosse WF, Schirier SL, Landaw SA, 2013. Diagnosis and treatment of paroxysmal nocturnal hemoglobinuria. Uptodate.
6. Parker CJ, 2010. Chapter 40: Paroxysmal Nocturmal hemoglobinuria. Williams Hematology eighth edition (Marshall, Thomas J.Kipps, Uri Seligsohn, Kenneth Kaushansky, Josef T. Prachal).
7. Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2003;102:3587-3591
8. Hill A, Kelly RJ, and Hillmen P, 2013. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood, 121(25), 4985-4996.
9. Lucio Luzzatto. Recent advances in the pathogenesis and treatment of paroxysmal nocturnal hemoglobinuria. F1000 Research 2016, 5(F1000 Faculty Rev): 209. Last updated: 25 Dec 2016.
10. Anita Hill, Amy E. DeZern, Taroh Kinoshitaand Robert A. Brodsky. Paroxysmal nocturnal haemoglobinuria. Nature Reviews (2017). Vo. 3, Article No. 17028.
11. Morag Griffin, Judith Marshand Anita Hill. Concurrent treatment of aplastic anemia/paroxysmal nocturnal hemoglobinuria syndrome with immunosuppressive therapy and eculizumab: a UK experience. Haematologica 2018; 103-346.haem
12. Salvatrice Mancuso et alls. Paroxysmal nocturnal hemoglobinuria: When delay in diagnosis and long therapy occurs. Hematology Reports 2018; 10:7523.
13. Dimitrios C. Mastellos, Edimara S. Reis, Despina Yancopoulou, Antonio M. Risitanoand John D. Lambris. Expanding complement therapeutics for the treatment of paroxysmal nocturnal hemoglobinuria. Seminars in Hematology 2018.
6. THIẾU MÁU TAN MÁU TỰ MIỄN
1. ĐẠI CƯƠNG
Thiếu máu tan máu tự miễn là bệnh thiếu máu do đời sống của hồng cầu bị rút ngắn bởi sự xuất hiện tự kháng thể chống hồng cầu. Các kháng thể này có thể hoạt động ở 37ºC (kháng thể nóng) hoặc dưới 37ºC (kháng thể lạnh) và cũng có khi hoạt động ở cả hai mức nhiệt độ (kháng thể hỗn hợp). Tan máu tự miễn do kháng thể nóng chiếm đến trên 80-90% các trường hợp. Hồng cầu bị hủy do bị thực bào ở hệ thống liên võng nội mô hoặc do sự hoạt hóa bổ thể làm ly giải hồng cầu.
2. CHẨN ĐOÁN
2.1. Lâm sàng
- Hội chứng thiếu máu: Thường xuất hiện nhanh có thể kèm theo sốt.
- Hội chứng hoàng đảm: Xuất hiện đồng thời với hội chứng thiếu máu.
- Gan, lách có thể to.
2.2. Cận lâm sàng
a. Máu ngoại vi
- Hồng cầu: Số lượng giảm, kích thước bình thường hoặc to; thiếu máu càng nặng thì kích thước hồng cầu có xu hướng càng to.
- Lượng huyết sắc tố và hematocrit giảm.
- Hồng cầu lưới tăng.
b. Sinh hóa
- Bilirubin tăng, chủ yếu tăng bilirubin gián tiếp.
- LDH tăng, haptoglobin giảm.
c. Xét nghiệm tủy đồ, sinh thiết tủy xương
Tủy, sinh giàu tế bào, dòng hồng cầu tăng sinh mạnh, hồng cầu lưới tủy tăng. Dòng bạch cầu hạt và mẫu tiểu cầu phát triển bình thường.
d. Xét nghiệm huyết thanh học
- Xét nghiệm Coombs trực tiếp dương tính với bản chất kháng thể là IgG, IgM, C3d, IgA, hoặc IgD.
- Xét nghiệm Coombs gián tiếp có thể dương tính (nếu dương tính nên sàng lọc định danh kháng thể bất thường).
- Cần định nhóm kháng nguyên ngoài hệ ABO để truyền máu hòa hợp phenotype.
- Sàng lọc kháng thể bất thường, định danh kháng thể bất thường.
- Xét nghiệm chọn máu để có đơn vị máu truyền phù hợp.
e. Các xét nghiệm bệnh lý tự miễn
Cần phải sàng lọc để phát hiện bệnh phối hợp:
- Kháng thể kháng nhân, kháng thể kháng ds-DNA.
- Kháng đông nội sinh, kháng thể kháng phospholipid (anti beta2- glycoprotein IgM, IgG; anti cardiolipin IgM, IgG), Lupus Anticoagulation test.
- Kháng thể kháng tiểu cầu, kháng thể kháng bạch cầu trung tính...
3. CHẨN ĐOÁN PHÂN BIỆT
3.1. Bệnh hồng cầu hình cầu bẩm sinh
- Lâm sàng cũng có hội chứng thiếu máu, hoàng đảm, nhưng thường biểu hiện mạn tính.
- Xét nghiệm tế bào máu thấy hồng cầu mất vùng sáng trung tâm, sức bền hồng cầu thường giảm; xét nghiệm Coombs trực tiếp âm tính.
3.2. Đái huyết sắc tố kích phát ban đêm
- Lâm sàng cũng có thiếu máu, hoàng đảm nhưng cơn tan máu thường về đêm, có từng đợt.
- Xét nghiệm cũng có tăng bilirubin gián tiếp, hồng cầu lưới tăng nhưng xét nghiệm Coombs trực tiếp âm tính; CD55, CD59 trên màng hồng cầu có thiếu hụt.
3.3. Tan máu trong bệnh hệ thống
- Có hội chứng thiếu máu và hội chứng hoàng đảm.
- Xét nghiệm Coombs trực tiếp có thể dương tính.
- Người bệnh thường có những tổn thương các cơ quan phối hợp như da, thận, khớp, tim…
- Kháng thể kháng nhân và dsDNA dương tính là xét nghiệm khẳng định bệnh. Khi nghi ngờ có thể làm thêm ANA 8 profile (anti dsDNA, anti RNP, anti Sm, anti SS-A/Ro, anti SS-B/La, anti Scl-70, anti CENP-B, anti Jo 1).
3.4. Tan máu trong bệnh U lympho
- Biểu hiện tan máu tự miễn, xét nghiệm Coombs trực tiếp dương tính. Tuy nhiên, người bệnh có thể nổi hạch (hay u), vã mồ hôi đêm, sút cân; xét nghiệm hạch hay u ngoài hạch phát hiện bệnh. Tình trạng này hay gặp nên với bệnh thiếu máu tan máu tự miễn, bao giờ cũng phải loại trừ u lympho đặc biệt là người bệnh lớn tuổi.
- Trường hợp này cần làm thêm các xét nghiệm để chẩn đoán và tiên lượng bệnh u lympho như nhuộm HE, nhuộm hóa mô miễn dịch sinh thiết hạch/ tủy xương; Xét nghiệm các IgA, IgG, IgM, IgE, Beta2 microglobulin, LDH; CT-scanner để tìm các tổn thương hạch trong nội tạng...
4. ĐIỀU TRỊ
4.1. Methylprednisolone
- Liều dùng: 1 - 2mg/kg/ngày. Khi có đáp ứng (huyết sắc tố > 80g/L) thì giảm liều dần (30% liều/ tuần).
- Trường hợp cơn tan máu rầm rộ, nguy cơ đe doạ tính mạng có thể dùng methylprednisolone liều cao (bolus):
+ Liều 1g/ngày trong 3 ngày; sau đó:
+ 3-4mg/kg/ngày trong 3-5 ngày;
+ 1-2mg/kg/ngày; khi có đáp ứng thì giảm dần liều và duy trì.
- Liều duy trì cần xác định riêng cho từng cá thể do đáp ứng thuốc khác nhau ở mỗi người bệnh nhằm đạt được sự cân bằng giữa hiệu quả và tác dụng phụ của thuốc.
- Có thể ngừng thuốc khi huyết sắc tố của người bệnh về bình thường với liều duy trì ở mức thấp (khoảng 4mg/ngày hoặc < 0,1mg/kg) trong 1 năm mà không có tái phát.
Lưu ý tác dụng phụ của thuốc, có thể dùng thuốc hỗ trợ: Chống loét dạ dày, hạ huyết áp, can xi, giảm đường huyết…
- Sau 3 tuần điều trị nếu không đáp ứng có thể kết hợp với các thuốc ức chế miễn dịch khác.
4.2. Các thuốc ức chế miễn dịch
- Azathioprine
+ Chỉ định: Bệnh không đáp ứng với điều trị corticoid;
+ Liều dùng: 50-100mg/ngày trong 4 tháng.
- Cyclophosphamid
+ Chỉ định: Bệnh không đáp ứng với điều trị corticoid;
+ Liều dùng: 50-200mg/ngày trong 3-6 tháng.
- Cyclosporin A: Liều dùng: 50-200mg/ngày trong 3-6 tháng.
- Vincristin: 1mg/tuần tối thiểu 3 tuần.
- Mycophenolate mofetil:
+ Liều dùng: 500mg - 2.000mg/ngày trong 1-3 tháng;
+ Lưu ý tác dụng giảm bạch cầu hoặc ức chế tủy xương của nhóm thuốc này, lúc đó nên dừng thuốc hoặc giảm liều. Dùng thuốc kích bạch cầu nếu bạch cầu giảm < 1 G/L.
4.3. Gamma globulin
- Chỉ định trong trường hợp cấp cứu: Cơn tan máu rầm rộ, đáp ứng kém với truyền máu và corticoid.
- Liều dùng: Tổng liều là 2g/kg (0,4g/kg/ngày x 5 ngàyhoặc 1g/kg/ngày x 2 ngày).
4.4. Cắt lách
Chỉ định trong trường hợp:
- Điều trị 3-6 tháng bằng corticoid và các thuốc ức chế miễn dịch thất bại hoặc phụ thuộc liều cao hay tác dụng phụ nặng nề của corticoid.
- Không có các bệnh lý nội khoa khác.
- Người bệnh tự nguyện.
Lưu ý: Sau cắt lách nếu không có đáp ứng thì tiếp tục sử dụng corticoid và các thuốc ức chế miễn dịch ban đầu.
4.5. Rituximab
- Là kháng thể đơn dòng chống CD20 của tế bào B lympho, làm giảm sản xuất kháng thể chống hồng cầu.
- Chỉ định khi các phương pháp điều trị ức chế miễn dịch và cắt lách không có hiệu quả.
- Liều dùng: 375mg/m2/tuần x 4 tuần (4 đợt điều trị).
4.6. Điều trị hỗ trợ
a. Truyền máu
- Nên truyền máu có lựa chọn đơn vị máu phù hợp nhất.
- Tốt nhất là truyền máu có hòa hợp thêm các nhóm máu ngoài hệ ABO (truyền hồng cầu phenotype).
- Nên truyền chậm và theo dõi sát các dấu hiệu lâm sàng.
b. Trao đổi huyết tương: Nhằm loại bỏ tối đa lượng kháng thể lưu hành trong máu, làm giảm mức độ tan máu trên lâm sàng.
c. Điều trị và dự phòng các biến chứng của thuốc và bệnh.
- Hạ huyết áp, giảm đường máu, bổ sung canxi, kali, các thuốc bảo vệ dạ dày…
- Lọc máu ngoài thận trong trường hợp có suy thận cấp.
d. Một số biện pháp điều trị khác như:
- Cắt tuyến ức, thuốc ức chế chuyển hóa Purine có tác dụng trên một số các trường hợp diễn biến dai dẳng.
- Khi bệnh nhân không đáp ứng với các phương pháp điều trị có thể sử dụng Cyclophosphamid liều 50mg/kg x 4 ngày có kèm theo kích bạch cầu.
4.7. Theo dõi trong qui trình điều trị
a. Lâm sàng: Đánh giá mức độ thiếu máu, màu sắc và số lượng nước tiểu, huyết áp, những biểu hiện ở dạ dày…
b. Cận lâm sàng:
- Xét nghiệm tế bào máu ngoại vi, hồng cầu lưới 2-3 lần/ tuần;
- Các chỉ số đường huyết, điện giải, canxi, bilirubin, men gan, chức năng thận 1-2 lần/tuần;
- Xét nghiệm Coombs: 1 lần/ 1-2 tuần.
c. Các xét nghiệm cơ bản khác
- Xét nghiệm đông máu: Fibrinogen, PT, rAPTT, TT, D-Dimer, rượu để tầm soát các tình trạng rối loạn đông máu đi kèm trong bệnh cảnh bệnh rối loạn tự miễn.
- Xét nghiệm nước tiểu: Tổng phân tích nước tiểu, tế bào nước tiểu để đánh giá tình trạng toàn thân, chức năng thận.
- Xét nghiệm chẩn đoán hình ảnh: Siêu âm bụng, X-quang phổi để chẩn đoán tình trạng toàn thân.
- Xét nghiệm nhóm máu, sàng lọc kháng thể bất thường, định danh kháng thể bất thường, xét nghiệm vi sinh (HCV, HBV, HIV) khi bệnh nhân có truyền máu và chế phẩm máu.
5. TIÊN LƯỢNG
Tỉ lệ đáp ứng chung với corticoid là khoảng 80%. Với những trường hợp phải cắt lách thì tỉ lệ đáp ứng từ 38-82%. Khi sử dụng rituximab tỉ lệ đáp ứng có thể đạt tới 80% các trường hợp. Tỉ lệ sống tại thời điểm 10 năm khoảng 73% với nguyên nhân tử vong chính là nhiễm trùng và tắc mạch phổi. Nếu bệnh phối hợp với hội chứng anti-phospholipid hoặc giảm tiểu cầu tự miễn (hội chứng Evans) thì tỉ lệ tử vong sẽ cao hơn.
TÀI LIỆU THAM KHẢO
1. Nguyễn Thị Minh An (2006), Thiếu máu tan máu miễn dịch (2006), Bài giảng Huyết học - Truyền máu Sau đại học, Tr 198-208.
2 Lechner.K and Jager.U (2010 ), How I treat autoimmune hemolytic anemias in adults, Blood 116: 1831-1838.
3. Friedberg R.C, Johari V.P (2009) Autoimmune Hemolytic Anemia, Wintrobe’s Clinical Hematology, pp.966-977.
4. Pacman.C.H (2006), Hemolytic Anemia from Immune Injury, William Hematology, 7, pp. 729-750.
7. HỘI CHỨNG EVANS
1. ĐẠI CƯƠNG
Hội chứng Evans là bệnh lý tan máu tự miễn kết hợp đồng thời với giảm tiểu cầu miễn dịch; trong một số trường hợp có thể kết hợp với giảm bạch cầu hạt trung tính.
2. CHẨN ĐOÁN
2.1. Lâm sàng
- Hội chứng thiếu máu: Thường xuất hiện nhanh, người bệnh có thể có sốt.
- Hội chứng hoàng đảm: Xuất hiện đồng thời với hội chứng thiếu máu.
- Hội chứng xuất huyết: Xuất huyết dưới da và niêm mạc nhưng cũng có thể xuất huyết nội tạng.
- Gan lách có thể to.
2.2. Cận lâm sàng
a. Tế bào máu ngoại vi: Hồng cầu, huyết sắc tố và hematocrit giảm; tiểu cầu giảm; hồng cầu lưới tăng.
b. Xét nghiệm tủy đồ, sinh thiết tủy xương: Tủy giàu tế bào, dòng hồng cầu tăng sinh, tăng hồng cầu lưới; mật độ mẫu tiểu cầu bình thường hoặc tăng (chủ yếu tuổi có hạt chưa sinh tiểu cầu); dòng bạch cầu hạt phát triển bình thường.
c. Xét nghiệm sinh hóa: Bilirubin tăng, chủ yếu tăng bilirubin gián tiếp; haptoglobin giảm, LDH tăng.
d. Xét nghiệm huyết thanh học: Xét nghiệm Coombs trực tiếp dương tính với bản chất kháng thể là IgG, IgM, IgA, IgD hoặc C3d; xét nghiệm Coombs gián tiếp có thể dương; xét nghiệm định nhóm kháng nguyên ngoài hệ ABO để tìm đơn vị máu phù hợp khi truyền máu (sàng lọc và định danh kháng thể bất thường).
e. Xét nghiệm miễn dịch:
- Kháng thể kháng tiểu cầu, kháng thể kháng bạch cầu trung tính.
- Kháng thể kháng dsDNA và kháng thể kháng nhân âm tính, nếu nghi ngờ có thể làm thêm ANA 8 profile (anti dsDNA, anti RNP, anti Sm, anti SS-A/Ro, anti SS-B/La, anti Scl-70, anti CENP-B, anti Jo 1).
- Kháng đông nội sinh, kháng thể kháng phospholipid (anti beta2- glycoprotein IgM, IgG; anti cardiolipin IgM, IgG), Lupus Anticoagulation test.
2.3. Chẩn đoán phân biệt
a. Bệnh lupus ban đỏ hệ thống:
- Lâm sàng: Có thể thiếu máu, xuất huyết; nhưng có tổn thương đặc trưng của bệnh lupus ban đỏ như ban ở da, đau khớp, rụng tóc, tràn dịch các màng, tổn thương thận…
- Xét nghiệm: Máu ngoại vi có thể giảm 1, 2 hoặc cả 3 dòng, nhưng có kháng thể kháng nhân và kháng thể kháng dsDNA.
b. Ban xuất huyết giảm tiểu cầu huyết khối hoặc hội chứng tan máu có tăng urê huyết (TTP - HUS):
- Lâm sàng: Có thể thiếu máu, xuất huyết, nhưng thường vàng da; có thể sốt, có biểu hiện thần kinh như li bì, lú lẫn; biểu hiện ở thận như thiểu niệu hoặc vô niệu.
- Xét nghiệm: Máu ngoại vi có giảm hồng cầu, tiểu cầu, có gặp mảnh vỡ hồng cầu; hoạt tính enzym ADAMTS 13 giảm.
- Trao đổi huyết tương trong điều trị sẽ làm giảm nhanh những triệu chứng về thần kinh.
3. ĐIỀU TRỊ
3.1. Methylprednisolon
- Liều dùng: 1-2mg/kg/ngày. Khi có đáp ứng (huyết sắc tố > 80g/L và tiểu cầu > 50G/L) thì giảm liều dần (30% liều/ tuần).
- Trường hợp cơn tan máu rầm rộ hoặc xuất huyết nhiều nơi, nguy cơ đe doạ tính mạng có thể dùng methylprednisolone liều cao (bolus):
+ Liều 1g/ngày trong 3 ngày;
+ Sau đó dùng liều 3-4mg/kg/ngày trong 3-5 ngày;
+ Sau đó dùng liều 1-2mg/kg/ngày. Khi có đáp ứng thì giảm dần liều và duy trì;
+ Liều duy trì cần xác định riêng cho từng cá thể do tính chất đáp ứng với thuốc khác nhau ở mỗi người bệnh nhằm đạt được sự cân bằng giữa hiệu quả và tác dụng phụ của thuốc.
- Có thể ngừng thuốc khi huyết sắc tố và tiểu cầu của người bệnh trở về bình thường với liều duy trì ở mức thấp (khoảng 4-5mg/ ngày) trong vòng 1 năm mà không có tái phát.
- Lưu ý tác dụng phụ của thuốc như: Tăng huyết áp, đái tháo đường, biểu hiện Cushing, loãng xương, loét dạ dày... Do vậy, cần phát hiện sớm các dấu hiệu này để điều chỉnh liều methylprednisolone và dùng thuốc hỗ trợ kịp thời, phù hợp.
- Sau 3 tuần điều trị mà người bệnh không đáp ứng có thể kết hợp với các thuốc ức chế miễn dịch khác.
3.2. Các thuốc ức chế miễn dịch
a. Azathioprine:
- Chỉ định: Bệnh không đáp ứng với điều trị corticoid.
- Liều dùng: 50-100mg/ngày, có thể dùng trong 3- 4 tháng.
- Khi có đáp ứng thì giảm liều, nếu bạch cầu trung tính giảm < 1,5G/L thì ngừng thuốc.
b. Cyclophosphamid:
- Chỉ định: Bệnh không đáp ứng với điều trị corticoid.
- Liều dùng: 50-100mg/ngày, có thể dùng trong 3-4 tháng.
- Khi có đáp ứng thì giảm liều, nếu bạch cầu trung tính giảm < 1,5G/L thì ngừng thuốc.
c. Cyclosporin A:
- Liều dùng: 50-200mg/ngày trong 3-6 tháng.
- Duy trì nồng độ thuốc trong máu từ 200-400ng/dl.
- Giảm liều dần khi có đáp ứng.
Lưu ý điều trị một số tác dụng phụ của thuốc.
d. Vincristin:
Liều dùng 1mg/tuần tối thiểu 3 tuần.
e. Mycophenolate mofetil:
Liều dùng: 500mg-2.000mg/ngày trong 1-3 tháng.
Lưu ý: Tác dụng giảm bạch cầu hoặc ức chế tủy xương của nhóm thuốc này, lúc đó nên dừng thuốc hoặc giảm liều.
3.3. Gamma globulin
- Chỉ định trong trường hợp cấp cứu: Cơn tan máu rầm rộ hoặc xuất huyết nhiều vị trí, người bệnh đáp ứng kém với truyền máu, truyền tiểu cầu và methylprednisolone.
- Liều dùng: Tổng liều là 2g/kg (0,4g/kg/ngày x 5 ngàyhoặc 1g/kg/ngày x 2 ngày).
3.4. Cắt lách: Chỉ định với những trường hợp:
- Điều trị 3-6 tháng bằng corticoid và các thuốc ức chế miễn dịch thất bại hoặc phụ thuộc liều cao corticoid.
- Không có các bệnh lý nội khoa khác.
- Người bệnh tự nguyện.
- Tủy xương sinh máu tốt
Lưu ý: Sau cắt lách nếu không có đáp ứng thì tiếp tục sử dụng corticoid và các thuốc ức chế miễn dịch ban đầu.
3.5. Rituximab
- Là kháng thể đơn dòng chống CD20 của tế bào B lympho, làm giảm sản xuất kháng thể chống hồng cầu.
- Chỉ định khi các phương pháp điều trị ức chế miễn dịch và cắt lách không có hiệu quả.
- Liều dùng: 375mg/m2/tuần x 4 tuần (4 đợt điều trị).
3.6. Điều trị hỗ trợ
a. Truyền máu:
- Nên truyền máu có lựa chọn đơn vị máu phù hợp nhất.
- Tốt nhất là truyền máu có hòa hợp thêm các nhóm máu ngoài hệ ABO.
- Nên truyền chậm và theo dõi sát các dấu hiệu lâm sàng.
b. Truyền khối tiểu cầu:
- Được chỉ định trong trường hợp có xuất huyết trên lâm sàng hoặc tiểu cầu < 10G/L.
- Nên truyền khối tiểu cầu gạn tách từ một người cho. Với khối tiểu cầu từ nhiều người cho (khối tiểu cầu pool) nên truyền số lượng lớn ngay từ đầu, liều lượng có thể tới 6-8 đơn vị/ngày.
c. Trao đổi huyết tương: Nhằm loại bỏ tối đa lượng kháng thể lưu hành trong máu, làm giảm mức độ tan máu và xuất huyết trên lâm sàng.
d. Điều trị và dự phòng các biến chứng của thuốc: Hạ huyết áp, giảm đường máu, bổ sung canxi, kali, các thuốc bảo vệ dạ dày…
3.7. Một số phương pháp điều trị khic
- Ghép tế bào gốc tạo máu.
4. DIỄN BIẾN VÀ TIÊN LƯỢNG
Với điều trị bằng Methylprednisolone là liệu pháp duy nhất thì tỉ lệ đáp ứng đạt được 82% các trường hợp. Cắt lách đạt tỉ lệ đáp ứng khoảng 78% nhưng sau 5 năm thì tỉ lệ đáp ứng chỉ còn 52%. Khi điều trị bằng rituximab có thể đạt được 80% số người bệnh có đáp ứng. Trong những người bệnh có chỉ định sử dụng gamma globulin thì 60% người bệnh là có đáp ứng. Sau 10 năm tỉ lệ tử vong khoảng 20% các trường hợp, nguyên nhân chính chủ yếu là nhiễm khuẩn và các bệnh lý tim mạch kèm theo.
5. THEO DÕI ĐIỀU TRỊ
a. Lâm sàng: Mức độ thiếu máu, màu sắc và số lượng nước tiểu, tình trạng xuất huyết, số đo huyết áp, những biểu hiện ở dạ dày…
b. Cận lâm sàng: Tế bào máu ngoại vi, hồng cầu lưới 2-3 lần/tuần. Các chỉ số đường huyết, điện giải, canxi, bilirubin, men gan, chức năng thận 1-2 lần/tuần. Xét nghiệm Coombs 1 lần/1-2 tuần.
c. Các xét nghiệm cơ bản khác
- Xét nghiệm đông máu: Fibrinogen, PT, rAPTT, TT, D-Dimer, rượu để tầm soát các tình trạng rối loạn đông máu đi kèm.
- Xét nghiệm nước tiểu: Tổng phân tích nước tiểu, tế bào nước tiểu để đánh giá tình trạng xuất huyết.
- Xét nghiệm chẩn đoán hình ảnh: Siêu âm bụng, X-quang phổi để chẩn đoán tình trạng toàn thân.
- Xét nghiệm nhóm máu hệ ABO, vi sinh (HBsAg, HCV Ab, HCV Ab) khi bệnh nhân có truyền máu và chế phẩm máu.
TÀI LIỆU THAM KHẢO
1. Costallat G. L, Appenzeller S, Costallat L. T (2011). Evans syndrome and Systemic Lupus Erythematosus: Clinical presentation and outcome. Joint Bone Spine.
2. Kashif M, Qureshi A, Adil S. N, Khurshid M. (2010), Successful use of rituximab in Evans syndrome and refractory immune thrombocytopenic purpura. J Pak Med Assoc, pp: 64-5.
3. Mathew P, Chen G, Wang W. (1997), Evans syndrome: results of a national survey. J Pediatr Hematol Oncol, 19(5), pp: 433-7.
4. Michel M, Chanet V, Dechartres A, Morin A. S, Piette J. P. (2009), The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood, 114, pp: 3167-3172.
5. Raetz E, Beatty P. G, Adams R. H. (1997), Treatment of severe Evans syndrome with an allogeneic cord blood transplant. Bone Marrow Transplant, 20(5), pp: 427-9.
6. Teachey D. T, Manno C. S, Axsom K. M, et al. (2005), Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS). Blood, 105(6), pp: 2443-8.
8. GIẢM TIỂU CẦU MIỄN DỊCH NGUYÊN PHÁT
1. ĐẠI CƯƠNG
1.1. Khái niệm
Giảm tiểu cầu miễn dịch nguyên phát (GTCMDNP) được xác định khi giảm số lượng tiểu cầu đơn độc, số lượng tiểu cầu ở máu ngoại vi < 100 (G/L) và không có nguyên nhân hoặc bệnh lý nào khác gây ra tình trạng giảm tiểu cầu.
1.2. Danh pháp
- Theo sự đồng thuận của Nhóm Thực Hành Quốc Tế năm 2009 (International Working Group - IWG) đề xuất áp dụng tên gọi “Immune ThrombocytoPenia” (viết tắt là ITP - Giảm tiểu cầu miễn dịch (nguyên phát)) thay cho “Idiopathic Thrombocytopenic Purpura” (Ban xuất huyết giảm tiểu cầu vô căn) và “Immune Thrombocytopenic Purpura” (Ban xuất huyết giảm tiểu cầu miễn dịch).
- Tên gọi mới nhằm nhấn mạnh đến cơ chế miễn dịch và tình trạng giảm tiểu cầu nguyên phát của bệnh. Hiện nay, ITP đã được xác định và không còn là “idiopathic - vô căn” (trong tên gọi Idiopathic Thrombocytopenic Purpura) hoặc “purpura - ban xuất huyết” (trong tên gọi Immune Thrombocytopenic Purpura - Vì theo các nghiên cứu có khoảng 30% bệnh nhân chỉ có giảm tiểu cầu mà không có triệu chứng xuất huyết.
2. TRIỆU CHỨNG LÂM SÀNG VÀ XÉT NGHIỆM
2.1. Triệu chứng lâm sàng: Có thể gặp các triệu chứng và hội chứng sau:
- Hội chứng xuất huyết: Người bệnh thường có xuất huyết tự nhiên dưới da đa hình thái (dạng chấm, nốt, mảng hoặc đám xuất huyết), đa lứa tuổi (màu sắc nốt xuất huyết thay đổi theo thời gian: Đỏ, tím, xanh, vàng sau đó mất đi không để lại dấu vết) hoặc người bệnh có chảy máu chân răng, chảy máu mũi, nôn ra máu, đi ngoài phân đen, kinh nguyệt kéo dài, đi tiểu ra máu… Khoảng 20-30% bệnh nhân người lớn mới được chẩn đoán không có biểu hiện xuất huyết; ở trẻ em tỷ lệ này khoảng 10%. Triệu chứng xuất huyết nên được phân chia mức độ nặng (theo WHO) hoặc NCI-CTCAE (bảng 4).
- Hội chứng thiếu máu: Có thể gặp và mức độ thiếu máu tương xứng với mức độ xuất huyết.
- Hội chứng nhiễm trùng: Bệnh nhân ITP có thể nhiễm trùng thứ phát hoặc tăng nguy cơ nhiễm trùng do điều trị ức chế miễn dịch hoặc cắt lách.
- Gan, lách, hạch ngoại vi không to.
Bảng 4. Mức độ xuất huyết theo WHO và NCI – CTCAE
Ghi chú: NCI - CTCAE (National Cancer Institute Common Terminology Criteria for Adverse Events).
2.2. Xét nghiệm
- Tổng phân tích tế bào máu ngoại vi:
+ Số lượng tiểu cầu giảm < 100 G/L;
+ Số lượng hồng cầu, lượng huyết sắc tố, số lượng bạch cầu và công thức bạch cầu thường trong giới hạn bình thường;
+ Số lượng hồng cầu và lượng huyết sắc tố có thể giảm nếu có xuất huyết nhiều và mức độ giảm tương xứng với mức độ xuất huyết.
- Tủy đồ:
+ Mật độ tế bào tủy bình thường hoặc tăng;
+ Số lượng mẫu tiểu cầu thường tăng sinh, một số bình thường;
+ Dòng hồng cầu và bạch cầu hạt phát triển bình thường;
+ Không gặp tế bào ác tính.
- Xét nghiệm cầm - đông máu:
+ Thời gian máu chảy/PFA: Kéo dài;
+ Co cục máu: Cục máu không co hoặc co không hoàn toàn;
+ Các xét nghiệm PT, APTT, TT, fibrinogen: Bình thường;
+ Kháng thể kháng phospholipid (anti beta2-glycoprotein IgM, IgG; anti cardiolipin IgM, IgG): Có thể gặp ở 40% trường hợp ITP;
+ Xét nghiệm TEG/ ROTEM thường có hình ảnh giảm đông.
- Kháng thể đặc hiệu kháng GPIIb-IIIa (hoặc GPIb) trên bề mặt tiểu cầu: Dương tính gặp trong khoảng 60 - 80% trường hợp ITP mạn hoặc ITP dai dẳng.
- Các xét nghiệm khác:
+ Bilan xét nghiệm virus (HbsAg, HCV, HIV, CMV, Epstein Barr, parovius…): Âm tính;
+ Bilan xét nghiệm bệnh miễn dịch: nghiệm pháp Coombs, ANA, anti dsDNA, lupus ban đỏ hệ thống…: Âm tính. Xét nghiệm ANA dương tính có thể tiên lượng ITP mạn tính ở trẻ em;
+ Xét nghiệm chức năng tuyến giáp (FT3, FT4, TSH): Bình thường;
+ Định lượng các immunoglobulin (IgA, IgG, IgM): Bình thường;
+ Xét nghiệm tìm vi khuẩn HP: Test thở, xét nghiệm phân tìm kháng nguyên: thường âm tính.
+ Xét nghiệm kháng thể kháng tiểu cầu: Dương tính ở 60-70% bệnh nhân.
3. CHẨN ĐOÁN
3.1. Chẩn đoán xác định
Theo Đồng thuận của Nhóm Thực Hành Quốc Tế (International Working Group - IWG) năm 2009 về bệnh Giảm tiểu cầu miễn dịch nguyên phát xác định: “Giảm tiểu cầu miễn dịch nguyên phát - ITP được xác định khi: Giảm số lượng tiểu cầu đơn độc, số lượng tiểu cầu ở máu ngoại vi < 100 G/L và không có nguyên nhân hoặc bệnh lý nào khác gây ra tình trạng giảm tiểu cầu”.
Như vậy, chẩn đoán GTCMDNP là chẩn đoán loại trừ dựa trên: Triệu chứng lâm sàng và xét nghiệm (đã nêu ở phần 2) và lưu ý loại trừ các nguyên nhân như:
- Tiền sử hoặc nguy cơ cao mắc các bệnh liên quan đến giảm tiểu cầu:
+ Nhiễm HBV, HCV, HIVhoặc các nhiễm trùng khác...;
+ Rối loạn miễn dịch/ tự miễn khác: Lupus ban đỏ hệ thống, hội chứng tăng sinh lympho...
- Tiền sử nghiện rượu hoặc xơ gan.
- Tiền sử tiêm vắcxin và sử dụng thuốc trong thời gian 2 tuần...
3.2. Chẩn đoán phân biệt: Cần chẩn đoán phân biệt giảm tiểu cầu miễn dịch nguyên phát với một số bệnh:
3.2.1. Giảm tiểu cầu di truyền: Thường có tiền sử gia đình, giảm số lượng tiểu cầu kèm theo thay đổi thể tích trung bình tiểu cầu, các xét nghiệm đánh giá chức năng tiểu cầu bị rối loạn… (ví dụ: Tiểu cầu kích thước to trong hội chứng Bernard - Soulier; tiểu cầu kích thước rất nhỏ trong hội chứng Wiskott - Aldrich…).
3.2.2. Do giảm sinh tiểu cầu: Suy tủy xương, lơ xê mi cấp, rối loạn sinh tủy, xơ tủy, ung thư di căn tủy xương…
3.2.3. Giảm giảm tiểu cầu trong các bệnh có cơ chế miễn dịch khác: Lupus ban đỏ hệ thống, hội chứng TTP/ HUS, hội chứng anti phospholipid, hội chứng Evans, bệnh suy giảm miễn dịch, HIT,…
3.2.4. Giảm tiểu cầu khác: Giảm tiểu cầu ở người nghiện rượu, xơ gan, đông máu rải rác trong lòng mạch, tác dụng phụ hóa trị liệu, tác dụng phụ của thuốc, nhiễm CMV, thủy đậu, nhiễm virus viêm gan B/C, HIV, tiêm chủng, nhiễm HP, sau ghép…
3.2.5. Phá hủy/giảm tiểu cầu thông qua kháng thể đồng loài: Giảm tiểu cầu do kháng thể đồng loài ở trẻ sơ sinh, giảm tiểu cầu do kháng thể đồng loài sau truyền tiểu cầu...
3.3. Chẩn đoán giai đoạn bệnh
- GTCMDNP mới (newly diagnosis): 3 tháng đầu sau chẩn đoán.
- GTCMDNP dai dẳng: Bệnh kéo dài từ 3 đến 12 tháng sau chẩn đoán.
- GTCMDNP mạn tính: Bệnh kéo dài hơn 12 tháng sau chẩn đoán.
- GTCMDNP kháng trị: Bao gồm tất cả các tiêu chuẩn sau:
+ Kéo dài hơn ba tháng;
+ Không đáp ứng hoặc mất đáp ứng với cắt lách và Rituximab;
+ Số lượng tiểu cầu < 50 G/L.
- GTCMDNP phụ thuộc Corticoid: người bệnh cần tiếp tục điều trị prednisone > 5mg/ngày (hoặc lượng corticoid tương đương) hoặc các liệu trình corticoid lặp lại để duy trì số lượng tiểu cầu > 30 G/l và/ hoặc để không có xuất huyết.
4. ĐIỀU TRỊ
4.1. Nguyên tắc điều trị
- Điều trị cá thể hóa dựa trên nguy cơ xuất huyết (bảng 4) và mức độ xuất huyết (theo WHO - bảng 4).
- Duy trì số lượng tiểu cầu ở mức an toàn với độc tính thuốc thấp nhất.
- Bệnh nhân có SLTC ≤ 20 G/l: Điều trị nội trú.
- Bệnh nhân có SLTC ≤ 30 G/l và không có triệu chứng xuất huyết hoặc xuất huyết mức độ nhẹ (mức độ I và II - theo WHO) trở lên: Có chỉ định điều trị.
- Bệnh nhân có SLTC ≥ 30 G/l và không có triệu chứng xuất huyết hoặc xuất huyết mức độ nhẹ (mức độ I và II - theo WHO): Theo dõi tiếp.
- Theo dõi độc tính và tác dụng phụ của thuốc trong điều trị là rất quan trọng, đặc biệt là điều trị corticoid kéo dài.
Bảng 5. Một số yếu tố tiên lượng và đánh giá nguy cơ
|
Yếu tố tiên lượng tự giới hạn bệnh |
Yếu tố nguy cơ diễn biến mạn tính |
Yếu tố nguy cơ xuất huyết nặng |
|
- Trẻ em, người trẻ tuổi - Tiền sử nhiễm trùng trước đó - Biểu hiện đột ngột - Triệu chứng ban đầu có xuất huyết |
- Người lớn, đặc biệt >60 tuổi - Không có tiền sử nhiễm trùng hoặc các rối loạn khác - Khởi phát từ từ - Triệu chứng ban đầu: xuất huyết ít hoặc phát hiện giảm tiểu cầu tình cờ và không có xuất huyết |
- SLTC < 20-30G/l và có nhiều đám xuất huyết - Xuất huyết niêm mạc - Tiền sử xuất huyết nặng trước đó, xuất huyết đường tiết niệu - Không đáp ứng corticoid, sốt, nhiễm trùng hoặc >60 tuổi |
Bảng 6. Một số tiêu chuẩn đánh giá đáp ứng điều trị
|
Mức độ đip ứng |
Tiêu chuẩn |
|
Đáp ứng hoàn toàn (CR) |
SLTC ≥ 100 G/L và không xuất huyết (dựa vào kết quả giữa 2 lần xét nghiệm, cách nhau trên 7 ngày). |
|
Đáp ứng (R) |
SLTC ≥ 30 G/L và tăng hơn 2 lần so với SLTC ban đầu, không có xuất huyết (dựa vào kết quả giữa 2 lần xét nghiệm, cách nhau trên 7 ngày). |
|
Không đáp ứng (NR) |
SLTC < 30 G/L hoặc tăng ít hơn 2 lần SLTC ban đầu hoặc có xuất huyết (dựa vào kết quả giữa 2 lần xét nghiệm, cách nhau trên 1 ngày). |
|
Mất đáp ứng sau khi đạt đáp ứng hoàn toàn (CR) |
SLTC < 100 G/L hoặc có xuất huyết (dựa vào kết quả giữa 2 lần xét nghiệm, cách nhau trên 1 ngày). |
|
Mất đáp ứng sau khi đạt đáp ứng (R) |
SLTC < 30 G/L hoặc tăng ít hơn 2 lần trị số ban đầu hoặc có xuất huyết (dựa vào kết quả giữa 2 lần xét nghiệm, cách nhau hơn 1 ngày). |
|
Thời gian đáp ứng |
từ khi đạt được đáp ứng hoàn toàn (CR) hoặc đáp ứng (R) đến khi mất đáp ứng. |
4.2. Điều trị cấp cứu
- Chỉ định: SLTC < 10 G/L và/hoặc bệnh nhân có tình trạng xuất huyết nghiêm trọng đe dọa tính mạng (Xuất huyết não, xuất huyết tiêu hóa, niệu - sinh dục…) (mức độ III, IV - theo WHO) hoặc cần nâng cao SLTC để chuẩn bị phẫu thuật.
- Có thể sử dụng một trong các phác đồ sau:
+ IG: 1g/kg/ngày x 2 ngày (truyền tĩnh mạch);
+ Methylprednisolone liều cao: 1g/ngày x 3 ngày (truyền tĩnh mạch trong 1 giờ);
+ Truyền khối tiểu cầu gạn tách (liều gấp 2-3 lần) kết hợp với IVIG (50-100 µg/ kg, tiêm tĩnh mạch trong 3-5 phút);
+ Anti D: 50 - 75 µg/ kg cân nặng; áp dụng cho bệnh nhân Rh (D) dương, chưa cắt lách;
+ Yếu tố VII hoạt hóa tái tổ hợp: Liều 90µg/kg/lần cách nhau mỗi 3h đến khi ngừng chảy máu hoặc liều duy nhất 270 µg/kg (nếu thất bại với tất cả các phương pháp trên và bệnh nhân cần cầm máu khẩn cấp).
Sau khi áp dụng các phương pháp điều trị cấp cứu trên: Người bệnh sẽ được theo dõi SLTC hàng ngày. Nếu SLTC > 20 G/l hoặc tình trạng xuất huyết nghiêm trọng đe dọa tính mạng đã được kiểm soát, bệnh nhân nên được tiếp tục điều trị theo phác đồ điều trị thường quy (Xem “điều trị thường quy”).
4.3. Điều trị thường quy ở người lớn
4.3.1. Điều trị “hàng một”: Corticosteroid
- Chỉ định: Bệnh nhân mới chẩn đoán, SLTC < 30G/l, không có triệu chứng xuất huyết hoặc xuất huyết nhẹ (mức độ 0 - II), chọn một trong các phương án dưới đây:
+ Methylprednisolone: 0,5 - 2,0 mg/kg/ngày, uống hoặc tiêm TM x1-2 tuần, tối đa 3 tuần. Sau khi có đáp ứng, giảm liều mỗi 10mg/ tuần đến liều 0,5 mg/kg/ngày, sau đó giảm liều mỗi 5mg/tuần; hoặc
+ Methylprednisolone: 1-2 mg/kg cân nặng/ngày, tiêm TM, 1-5 ngày (tối đa 1000mg). Sau khi có đáp ứng, giảm liều mỗi 10mg/ tuần đến liều 0,5 mg/kg/ngày, sau đó giảm liều mỗi 5mg/tuần; hoặc
+ Dexamethasone 40 mg/ngày x 4 ngày/chu kỳ x 4-6 chu kỳ cách nhau mỗi 14 - 28 ngày (truyền TM).
4.3.2. Điều trị “hàng thứ hai”
Chỉ định: Khi bệnh nhân không đáp ứng với điều trị corticoid hoặc phụ thuộc corticoid > 3 tháng.
4.3.2.1. Thuốc ức chế miễn dịch khác: Kết hợp corticoid với một trong các thuốc sau:
a. Mycophenolate Mofetil:
- Liều dùng: 0,5- 2g/ngày trong 4 - 8 tuần (uống).
- Khi số lượng tiểu cầu về bình thường: Giảm liều dần và điều trị duy trì 0,5g/ ngày trong 3 tháng, sau đó có thể dừng thuốc và tiếp tục theo dõi.
b. Cyscloporin A:
- Liều dùng: 5-10 mg/kg/ngày, chia 2 lần cách nhau 12 tiếng.
- Định lượng nồng độ thuốc duy trì khoảng 200 ng/ml.
- Sau 6-8 tháng giảm dần liều thuốc và theo dõi.
- Ngừng thuốc nếu sau 3 tháng không đáp ứng
c. Azathioprin:
- Liều: 100-200mg/ ngày trong vòng ít nhất 4 tháng.
- Lưu ý: Nhóm thuốc này có tác dụng phụ gây giảm bạch cầu.
d. Cyclophosphamide:
- Liều dùng: 50-200mg/ ngày trong 4 - 8 tuần (uống).
- Khi số lượng tiểu cầu về bình thường: giảm liều dần và điều trị duy trì 50mg/ ngày trong 3 tháng, sau đó có thể dừng thuốc và tiếp tục theo dõi.
e) Vincristin:
Liều dùng: Vincristin 1-2 mg/ngày, truyền tĩnh mạch mỗi tuần 1 lần, ít nhất 3 tuần.
4.3.2.2. Cắt lách
- Chỉ định: Thất bại hoặc phụ thuộc Corticosteroid, ITP mạn tính.
- Tiêm phòng vacxin trước cắt lách ít nhất 4 tuần (Streptococcus pneumoniae, Haemophilus influenzae type B, Neisseria meningitides) và kháng sinh phòng ngừa nhiễm trùng.
- Tỉ lệ đáp ứng khoảng 80%, tỉ lệ đáp ứng sau 5 năm khoảng 60 - 70%. Cắt lách theo phương pháp nội soi có tỉ lệ tử vong do biến chứng là 0,2 %. Sau cắt lách không cần điều trị nếu số lượng TC > 30 G/L và không xuất huyết. Sau cắt lách có thể điều trị phối hợp Corticosteroid liều thấp (0,2mg/kg/ngày) để giữ TC > 30 G/L và không xuất huyết.
4.3.2.3. Thuốc chủ vận thụ thể kích thích tạo tiểu cầu (Thrombopoietin receptor agonist) (Eltrombopag và Romiplostim)
Chỉ định: Bệnh nhân GTCMDNP mạn tính và thất bại với ít nhất một điều trị khác (Corticosteroid, IVIG, cắt lách) hoặc bệnh nhân chống chỉ định cắt lách.
a. Eltrombopag:
- Liều dùng: Khởi đầu 50 mg/ngày (uống).
- Duy trì: Điều chỉnh liều để đạt SLTC ≥ 50 G/L, không vượt quá 75mg/ngày.
+ Khi 200 < SLTC < 400 G/L: Giảm liều còn 25mg/ngày, sau 2 tuần đánh giá lại;
+ Khi SLTC > 400 G/L: Ngừng thuốc, theo dõi SLTC 2 lần mỗi tuần. Sau 2 tuần nếu vẫn duy trì > 400 G/L, ngừng thuốc luôn;
+ Nếu SLTC giảm lại < 150 G/L, bắt đầu điều trị lại với liều 25mg/ngày.
- Ngừng điều trị nếu không đáp ứng với liều 75 mg /ngày x 4 tuần.
- Tác dụng phụ: Hội chứng giả cúm, nhức đầu, buồn nôn, đau cơ, tăng men gan, suy gan cấp, huyết khối, trầm cảm…
- Điều chỉnh liều: 25mg/ngày nếu có tăng men gan và dừng thuốc khi men gan tăng gấp 3 trị số bình thường liên tục trên 4 tuần.
- Theo dõi: Tổng phân tích tế bào máu ngoại vi hàng tuần và ALT, AST, billirubin mỗi 2 tuần ở giai đoạn điều chỉnh liều, mỗi tháng khi liều dùng ổn định. Lặp lại mỗi tuần nếu có bất thường.
Lưu ý: Có thể gây nhiễm độc gan cấp nếu dùng chung interferon và ribarivin ở bệnh nhân nhiễm HCV.
b. Romiplostim:
- Liều dùng: Liều khởi đầu 1µg/kg/tiêm dưới da mỗi tuần, điều chỉnh liều tăng dần mỗi 1 µg/ tuần để đạt mục tiêu SLTC > 50 G/l, tối đa 10 µg/kg.
- Kiểm tra SLTC hàng tuần, sau đó mỗi 4 tuần/ lần.
- Nếu bệnh nhân có xuất huyết nặng có thể bắt đầu với liều 3-5 µg/ tuần.
- Xem xét dừng thuốc khi điều trị 4 tuần không đáp ứng hoặc SLTC > 400 G/l.
- Có thể điều trị ở bệnh nhân giảm chức năng gan/thận, không điều trị ở phụ nữ có thai.
4.3.2.4. Anti CD 20 (Rituximab):
- Chỉ định: Bệnh nhân điều trị thất bại/tái phát sau Corticoid và có chống chỉ định cắt lách hoặc không đồng ý cắt lách, thất bại sau cắt lách.
- Liều: 375mg/m2 da mỗi tuần x 4 tuần. Đáp ứng trong 4 - 8 tuần. Tỉ lệ đáp ứng là 60% và đáp ứng hoàn toàn là 40 %. Có thể phải lặp lại mỗi năm nếu tái phát.
4.4. Điều trị GTCMDNP trong một số trường hợp khác
4.4.1. Điều trị GTCMDNP ở phụ nữ có thai
- Chỉ định điều trị khi:
+ Khi có xuất huyết trên lâm sàng;
+ Trước bất kỳ thủ thuật xâm lấn/ phẫu thuật;
+ SLTC < 20 - 30 G/l ở 3 tháng đầu hoặc 3 tháng giữa thai kỳ;
+ SLTC< 50 G/l ở 3 tháng cuối thai kỳ. Phẫu thuật lấy thai cần SLTC > 50 G/l, gây tê tủy sống cần SLTC > 80G/l.
- Không điều trị các thuốc (corticoid, UCMD...) trong 12 tuần đầu của thai kỳ.
- Sau 12 tuần có thể điều trị Corticoid với liều Methylprednisolone 1mg/kg/ngày.
- Nếu điều trị corticoid không đáp ứng, cân nhắc điều trị IVIG(1g/kg/ngày x 2 ngày (truyền tĩnh mạch) hoặc anti D (50 - 75 µg/ kg cân nặng; áp dụng cho bệnh nhân Rh (D) dương, chưa cắt lách).
- Chống chỉ định với các loại thuốc ức chế miễn dịch khác.
- Khi có chỉ định phẫu thuật hoặc chuyển dạ, SLTC phù hợp để an toàn cho chuyển dạ là > 50G/l, nếu gây tê tủy sống cần SLTC > 80G/l. Nếu chưa đạt được SLTC như trên thì kết hợp truyền KTC trước và sau chuyển dạ hoặc phẫu thuật.
4.4.2. Điều trị GTCMDNP ở trẻ em:
4.4.2.1. Điều trị “hàng một”:
a. Corticosteroid:
- Chỉ định: Bệnh nhân mới chẩn đoán, SLTC < 30G/l, không có triệu chứng xuất huyết hoặc xuất huyết nhẹ (mức độ 0 - II), chọn một trong các phương án dưới đây:
+ Methylprednisolone: 2-4 mg/kg/ngày (tối đa 120 mg/ngày) x 5-7 ngày; hoặc
+ Methylprednisolone: 500 mg/m2 da/ ngày x 10 - 20 ngày, sau đó giảm liều trong vòng 1 - 2 tuần và ngừng thuốc; hoặc
b. Globulin miễn dịch (Truyền tĩnh mạch Immunoglobulin-IVIG)
- Chỉ định:
+ Trẻ sơ sinh và dưới 2 tuổi;
+ Trường hợp chảy máu cấp tính, đe dọa tính mạng;
+ Trường hợp cần tăng nhanh số lượng tiểu cầu hoặc chuẩn bị phẫu thuật.
- Liều dùng:
+ Thể cấp tính: 0,4g/kg/ngày x 5 ngày hoặc 1g/kg/ngày x 2 ngày (tổng liều 2g/kg cân nặng), truyền tĩnh mạch trong 3-6h;
+ Thể mạn tính: 1g/kg/ngày x 2 ngày, sau đó điều trị xen kẽ methylprednisolone và IVIG (0,4-1g/kg) phụ thuộc đáp ứng của người bệnh (có thể nhắc lại IVIG 10 ngày/lần nếu người bệnh đáp ứng kém hoặc tác dụng phụ của corticoid nhiều).
4.4.2.2. Điều trị “hàng thứ hai”:
a. Cắt lách
- Chỉ định:
+ Chỉ nên chỉ định cắt lách ở trẻ em > 5 tuổi, đã được chẩn đoán > 2 năm và điều trị nội khoa không đáp ứng; hoặc
+ Có nguy cơ chảy máu nặng, đe dọa tính mạng và không đáp ứng với điều trị nội khoa;
+ Thể mạn tính, có xuất huyết trên lâm sàng và số lượng tiểu cầu luôn < 30G/L, không đáp ứng với điều trị nội khoa.
- Tiêm phòng vacxin trước cắt lách ít nhất 4 tuần (Streptococcus pneumoniae, Haemophilus influenzae type B, Neisseria meningitides).
- Trường hợp có SLTC < 50G/L, đặc biệt <20G/L cần điều trị corticoid và/ hoặc IVIg trước phẫu thuật để nâng cao SLTC, giảm tối đa nguy cơ chảy máu trong và sau phẫu thuật.
- Truyền khối tiểu cầu gạn tách trước và trong phẫu thuật để giảm nguy cơ chảy máu.
- Điều trị methylprednisolone trước, trong và sau khi cắt lách (để đề phòng ức chế thượng thận sau khi cắt lách).
- Uống kháng sinh dự phòng sau khi cắt lách (penecillin V, erythromycin… trong 6 tháng đến 1 năm).
b. Anti-(Rh) D
- Chỉ định:
+ Trường hợp không đáp ứng với methylprednisolone;
+ Tình trạng xuất huyết nặng, đe dọa tính mạng hoặc có chỉ định phẫu thuật cấp cứu.
- Liều đơn độc được khuyến nghị: 50 - 100 μg/ kg, tiêm tĩnh mạch trong 3-5 phút.
Ghi chú: Anti- (Rh) D không được chỉ định cho người bệnh Rh (-) và hiệu quả rất thấp trên người bệnh đã cắt lách.
c. Các thuốc ức chế miễn dịch khác: Ít được sử dụng. Chỉ lựa chọn khi có chảy máu nặng, đe dọa tính mạng, nguy cơ tử vong và biến chứng cao. Liều dùng: Tương tự như ở người lớn.
d. Thuốc chủ vận thụ thể kích thích tạo tiểu cầu (Thrombopoietin receptor agonist) (Eltrombopag)
- Chỉ định: Điều trị giảm tiểu cầu nguyên phát mạn tính ở bệnh nhân trên 1 tuổi kháng trị với các điều trị khác (Corticoid, IVIG).
- Liều khởi đầu: 25mg, một lần mỗi ngày.
- Cách dùng thuốc: Uống ít nhất 2 giờ trước hoặc 4 giờ sau khi dùng các sản phẩm khác như thuốc kháng acid, các thuốc có thành phần Canxi và sản phẩm từ sữa. Theo dõi điều chỉnh thuốc để duy trì số lượng tiểu cầu > 50 G/l.
4.4.3. Bệnh nhân GTCMDNP có nhiễm virus viêm gan C, HIV
- Bệnh nhân nhiễm HCV: Cân nhắc tiếp tục điều trị antivirus nếu không có chống chỉ định, theo dõi sát SLTC. Thuốc điều trị giảm tiểu cầu thích hợp là IVIG.
- Bệnh nhân nhiễm HIV: Tiếp tục điều trị antivirus cho đến khi có biến chứng xuất huyết nặng. Điều trị giảm tiểu cầu thích hợp là IVIG và anti D. Nếu thất bại, cân nhắc cắt lách.
4.4.4. Bệnh nhân GTCMDNP có nhiễm khuẩn HP
- Một số nghiên cứu trên thế giới cho thấy hiệu quả tăng tiểu cầu đối với nhóm bệnh nhân diệt vi khuẩn HP thành công.
- Trường hợp SLTC > 30 G/L: Chỉ điều trị diệt HP.
- Trường hợp SLTC < 30 G/L: Điều trị GTCMDNP và điều trị diệt HP.
- Phác đồ điều trị diệt HP 3 thuốc: Một thuốc ức chế bơm proton (PPI) (ví dụ Omeprazol 20mg: 1 viênx2 lần/ngày), Amoxicillin (1gx2 lần/ngày) và Clarithromycin (500mg x 2 lần/ngày) trong 2 tuần (Grade 2B).
4.5. Điều trị hỗ trợ
4.5.1. Truyền khối tiểu cầu:
- Được chỉ định khi bệnh nhân có xuất huyết và/ hoặc SLTC < 20G/L.
- Các loại chế phẩm tiểu cầu theo thứ tự ưu tiên: KTC gạn tách, KTC pool lọc bạch cầu, KTC túi đơn.
4.5.2. Tranexamic acid:
- Chỉ định: Xuất huyết mức độ nặng (độ III - IV theo WHO).
- Có thể dùng cả đường uống và tiêm.
- Liều dùng: 500-1000 mg/ lần x 2 đến 4 lần/ ngày, tối đa 4.000 mg/ngày.
- Trường hợp nặng: Truyền tĩnh mạch với liều 0,1g/kg/30 phút đầu sau đó truyền liên tục 0,5-1g/h đến khi ngừng xuất huyết.
- Chống chỉ định: Người bệnh đi tiểu ra máu.
4.5.3. Các điều trị khác:
- Bổ sung Canxi và vitamin D phòng tránh loãng xương.
- Thuốc kháng tiết acid dạ dày nếu bệnh nhân có triệu chứng ở dạ dày.
- Trao đổi huyết tương: Thường áp dụng trong các trường hợp xuất huyết nặng, diễn biến cấp tính, có thể kèm theo các bệnh lý khác như viêm gan, tan máu miễn dịch...
5. THEO DÕI ĐIỀU TRỊ
- Theo dõi huyết áp, tình trạng toàn thân hàng ngày.
- Xét nghiệm:
+ Tổng phân tích tế bào máu ngoại vi: 2 ngày/ lần;
+ Sinh hóa máu: Glucose, chức năng gan, chức năng thận; bộ mỡ máu; điện giải... 2 lần/ tuần; bilan viêm CRP, pro-calcitonin khi lâm sàng có dấu hiệu nhiễm trùng;
+ Đông máu: Fibrinogen, PT, APTT, TT, D-Dmer 1 lần/ tuần;
+ Các xét nghiệm bệnh tự miễn: coombs trực tiếp, coombs gián tiếp; kháng thể kháng nhân, kháng thể kháng ds-DNA, kháng đông nội sinh; kháng đông ngoại sinh; LA test; kháng thể kháng Phospholipid (anti beta2- glycoprotein IgM, IgG; anti cardiolipin IgM, IgG) khi bệnh nhân có các triệu chứng gợi ý chuyển thể bệnh, bệnh tiến triển, xuất hiện bệnh kèm theo;
+ Xét nghiệm đánh giá chức năng tim (điện tâm đồ, siêu âm tim), chức năng phổi; xét nghiệm tủy xương khi bệnh nhân cần điều trị phẫu thuật cắt lách;
+ Các xét nghiệm khác phụ thuộc tình trạng bệnh lý kèm theo: Glucose, HbA1C; tổng phân tích nước tiểu, tế bào nước tiểu, siêu âm bụng, X-quang phổi...;
+ Các xét nghiệm khi có truyền máu và chế phẩm: xét nghiệm vi sinh (HBV, HCV, HIV); xét nghiệm nhóm máu; sàng lọc và định danh kháng thể bất thường;
+ Xét nghiệm kháng thể kháng tiểu cầu bằng phương pháp flowcytometry khi có truyền tiểu cầu không hiệu lực.
TÀI LIỆU THAM KHẢO
1. Shosaku Nomura (2016), Advances in Diagnosis and Treatments for Immune Thrombocytopenia, Clinical Medicine Insights: Blood Disorders Vol 9, p.15-22.
2. Michele P. L.,Terry B. G. et al (2017), Clinical updates in adult immune thrombocytopenia, Blood, Vol 129 (21), p. 2829-2835.
3. Lambert MP, Gernsheimer TB (2017), Clinical updates in adult immune thrombocytopenia, Blood, Vol 129, p. 2829-2835.
4. Neunert C., Lim W. et al (2017), The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia, Blood, Vol 117, p.4190-4207.
5. Provan D., Stasi R., et al (2010), International consensus report on the investigation and management of primary immune thrombocytopenia. Blood:115 - 168.
6. Rodeghiro F., Stasi R. et al (2009). Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood: 113: 2386-93.
7. British Committee for Standards in Haematology General Haematology. Task Force (2003), Br J Haematol. 120:574-596.
8. Douglas B. C., James B. B. et al (2005). How I treat idiopathic thrombocytopenic purpura (ITP). Blood: 106:2244-2251.
9. Yunfeng Cheng, Raymond S.M. Wong et al (2003). Initial Treatment of Immune Thrombocytopenic Purpura with High-Dose Dexamethasone.The new England Journal of medicine. August 28, 349 (9), 278 - 289.
10. Marshall A. Lichman et all (2016), Primary immune thrombocytopenia, Williams Hematology, 9th Edition, 1999- 2024.
9. RỐI LOẠN CHỨC NĂNG TIỂU CẦU
1. ĐẠI CƯƠNG
- Rối loạn chức năng tiểu cầu là bệnh lý do bất thường các glycoprotein (GP), hạt, enzym hoặc receptor của tiểu cầu... dẫn đến bất thường các chức năng dính, ngưng tập, giải phóng và hoạt tính tiền đông máu của tiểu cầu.
- Bệnh có thể do di truyền hoặc mắc phải, triệu chứng thường gặp là xuất huyết dưới da, xuất huyết niêm mạc (chảy máu mũi, chảy máu chân răng, rong kinh).
2. XẾP LOẠI
Tùy theo nguyên nhân, chia thành các nhóm như sau:
2.1. Rối loạn chức năng tiểu cầu bẩm sinh: Có các nhóm nguyên nhân chính:
a. Do bất thường glycoprotein (GP)
- Bất thường GP Ib (CD42b,c) /IX (CD42a)/V làm cho tiểu cầu không gắn được với yếu tố von - Willebrand do đó tiểu cầu không dính vào lớp collagen dưới nội mạc (Hội chứng Bernard - Soulier).
- Bất thường GP IIb/IIIa (CD41/CD61) làm cho tiểu cầu không gắn được với các chất kích tập (fibrinogen) dẫn đến tiểu cầu không ngưng tập được với nhau (bệnh suy nhược tiểu cầu Glanzmann).
- Bất thường GPIb (CD42b,c) gây bệnh von-Willebrand thể tiểu cầu.
- Bất thường Glycoprotein IV (CD36).
- Bất thường Glycoprotein VI.
b. Do bất thường tương tác khung - màng tế bào: Hội chứng Wiskott - Aldrich.
c. Do bất thường hạt tiểu cầu
- Thiếu hụt kho dự trữ δ.
- Hội chứng tiểu cầu xám (thiếu hạt α).
- Thiếu hụt kho dự trữ α, δ.
- Bệnh rối loạn chức năng tiểu cầu Quebec (yếu tố V Quebec).
d. Do bất thường hoạt tính tiền đông máu của tiểu cầu (hội chứng Scott).
đ. Do bất thường receptor hoạt hóa, dẫn truyền tín hiệu và hiện tượng tiết của tiểu cầu.
- Bất thường receptor dẫn truyền tín hiệu hoạt hóa đặc hiệu (agonist - specific signal transduction).
- Bất thường protein gắn guanosis - triphosphat (GTP).
- Bất thường chuyển hóa acid arachidonic và sản xuất thromboxan.
- Bất thường phopholipase C, Gαq, huy động và đáp ứng calci.
e. Bất thường cấu trúc khung tế bào tiểu cầu.
f. Bất thường yếu tố phiên mã dẫn đến bất thường chức năng tiểu cầu.
- RUNX1 (bất thường chức năng tiểu cầu gia đình với khuynh hướng chuyển thành lơ xê mi cấp dòng tủy).
- GATA -1.
- FLI1 (tiểu cầu dị hình lưỡng hình với hạt alpha khổng lồ và giảm tiểu cầu/ hội chứng Paris - Trousseau/Jacobsen).
- GFI1B.
2.2. Rối loạn chức năng tiểu cầu mắc phải thứ phát
a. Một số nhóm bệnh chính:
- Rối loạn chức năng tiểu cầu trong các bệnh lý Huyết học: Hội chứng tăng sinh tủy mạn tính, lơ xê mi cấp và mạn, hội chứng rối loạn sinh tủy, đái huyết sắc tố kịch phát ban đêm…
- Rối loạn chức năng tiểu cầu trong bệnh lý không phải huyết học: Tăng ure máu...
- Rối loạn chức năng tiểu cầu do thuốc: Aspirin, dipyridamole, thuốc kháng viêm non - steroid, ticlopidine, clopidogrel, abciximab, eptifibatide, tirofiban…
b. Nguyên nhân: Do chế độ dinh dưỡng.
- Chế độ ăn kiêng với dầu cá giàu ω-3 dẫn đến giảm acid arachidonic tiểu cầu.
- Các đồ ăn/thực phẩm có hành/chiết xuất hành dẫn đến ức chế tổng hợp acid arachidonic tiểu cầu.
- Các đồ ăn/thực phẩm có tỏi ức chế gắn tiểu cầu với fibrinogen dẫn đến giảm NTTC.
- Các đồ ăn/thực phẩm có thì là/nghệ dẫn đến ức chế ngưng tập tiểu cầu và sinh tổng hợp eicosanoid…
3. CHẨN ĐOÁN
- Rối loạn chức năng tiểu cầu là một nhóm bệnh lý phức tạp. Biểu hiện, mức độ của triệu chứng lâm sàng và xét nghiệm phụ thuộc mức độ giảm hoặc bất thường chức năng tiểu cầu thường.
- Nhóm bệnh lý này có một số triệu chứng lâm sàng và xét nghiệm chung. Ngoài ra, còn có các triệu chứng riêng tùy theo bệnh/ hội chứng cụ thể. Chẩn đoán bệnh lý rối loạn chức năng tiểu cầu dựa trên các đặc điểm chung và riêng sau:
3.1. Đặc điểm chung
a. Triệu chứng lâm sàng
Triệu chứng thường gặp là xuất huyết ở dưới da và niêm mạc như: Chảy máu chân răng, chảy máu mũi, xuất huyết tiêu hóa, tiểu đỏ, kinh nguyệt kéo dài, xuất huyết não, chảy máu sau nhổ răng, sau phẫu thuật… Hiếm gặp xuất huyết tiêu hóa hoặc chảy máu khớp.
b. Xét nghiệm
- Số lượng và hình thái tiểu cầu: Thường bình thường.
- Thời gian máu chảy: Thường kéo dài.
- Co cục máu đông: Co không hoàn toàn hoặc không co.
- PFA (platelet function analysis): Kéo dài.
- Ngưng tập tiểu cầu với các chất kích tập (ADP, Thrombin..): Giảm hoặc không ngưng tập.
3.2. Đặc điểm của một số bệnh/ hội chứng rối loạn chức năng tiểu cầu.
Bảng 7. Đặc điểm một số bệnh/hội chứng rối loạn chức năng tiểu cầu bẩm sinh
|
Bệnh/ hội chứng |
Đặc điểm di truyền |
Cơ chế bệnh sinh |
Triệu chứng lâm sàng |
Thay đổi xét nghiệm |
|
Hội chứng Bernard - Soulier |
Di truyền lặn, NST thường, do tổn thương 1 trong 3 gen mã hóa phức hợp GPIb/IX/ V của tiểu cầu. |
Thiếu GP Ib/IX làm cho tiểu cầu không gắn được với yếu tố von - Willebrand (và một số chất như thrombin, P- selectin, yếu tố XI, XII…, và bất thường hoạt tính tiền đông) dẫn đến tiểu cầu không dính được với collagen (lớp dưới nội mô). |
Chảy máu cam, chảy máu chân răng, xuất huyết dưới da, xuất huyết tiêu hóa, tiểu đỏ, kinh nguyệt kéo dài, xuất huyết não. |
- Số lượng tiểu cầu giảm. - Tiểu cầu kích thước lớn (tiểu cầu khổng lồ). - Thể tích khối tiểu cầu (PCT): Bình thường. - Thời gian máu chảy kéo dài - Cục máu: Co không hoàn toàn hoặc không co. - PFA: Kéo dài. - Ngưng tập tiểu cầu với: + Ristocetin: Không ngưng tập. + ADP, adrenalin, collagen: Bình thường (có thể tăng). + Thrombin: liều thấp: giảm; liều cao: bình thường. - Định lượng yếu tố von Willerbrand: Bình thường - Định lượng GPIb /IX /V tiểu cầu: Giảm hoặc không có. |
|
Suy nhược tiểu cầu Glanzmann |
Di truyền lặn (NTS số 17). |
Thiếu/bất thường chức năng GP IIb/IIIa làm cho tiểu cầu không gắn được với các chất kích tập (fibrinogen,..) dẫn đến tiểu cầu không ngưng tập với nhau được. |
Chảy máu mũi, chân răng; xuất huyết dưới da, xuất huyết tiêu hóa, tiểu đỏ, kinh nguyệt kéo dài, xuất huyết não, chảy máu sau phẫu thuật. |
- Số lượng và hình thái tiểu cầu: Bình thường. - Thời gian máu chảy kéo dài. - PFA: Kéo dài. - Ngưng tập tiểu cầu với: + ADP, epinephrine, thrombin, collagen, thromboxane A2:Giảm nặng hoặc không ngưng tập. + Ristocetin 0,5 mg/ml: Không ngưng tập + Ristocetin 1,5mg/ml: Bình thường - Định lượng GP IIb/IIIa: Giảm. - Định lượng CD41/CD61: giảm |
|
Bệnh von- Willebrand thể tiểu cầu. |
Di truyền trội, NST thường. |
Bất thường GPIb …bất thường liên kết với vWF multimer dẫn đến cạn kiệt vWF multimer trong huyết tương và giảm đời sống tiểu cầu. |
Bệnh nhân thường có xuất huyết niêm mạc từ nhẹ đến trung bình (chảy máu mũi, chảy máu chân răng, xuất huyết dưới da…) |
- Số lượng tiểu cầu: có thể giảm nhẹ - Kích thước tiểu cầu: Có thể lớn. - Thời gian máu chảy kéo dài. - Yếu tố von - Willerbrand huyết tương: Giảm. - Ngưng tập tiểu cầu với ristocetin (nồng độ thấp) và botrocetin: Tăng. |
|
Hội chứng Wiskott - Aldrich |
Di truyền lặn trên NST X |
Bất thường GP Sialophorin của tiểu cầu. |
- Nhiễm trùng tái phát. - Eczema. - Lách to (hiếm gặp). - Có thể có bệnh ác tính kèm theo (u lympho, leukemia, …) |
- Số lượng tiểu cầu: Có thể giảm. - Kích thước tiểu cầu: Nhỏ. - Thời gian máu chảy kéo dài. - Bất thường miễn dịch do bất thường lympho T |
|
Hội chứng tiểu cầu Quebec |
Di truyền trội NST thường |
Thiếu hụt multimerin và thrombospon- din tiểu cầu dẫn đến plasmin tăng thoái giáng các protein trong hạt α |
Chảy máu kéo dài sau chấn thương. |
- Giảm số lượng tiểu cầu. - Thời gian máu chảy có thể bình thường hoặc kéo dài. - Ngưng tập tiểu cầu: + Epinephrin: giảm + Các chất kích tập khác: bình thường hoặc giảm - Định lượng yếu tố V tiểu cầu giảm. - Định lượng yếu tố V huyết tương bình thường. - Các xét nghiệm đánh giá hệ thống tiêu sợi huyết (Fibrinogen, D-dimer, plasminogen, plasmin - α2 antiplasmin, u - PA): bình thường. |
|
Thiếu hụt kho dự trữ δ |
Di truyền lặn trên NST thường |
Thiếu hạt đặc (hạt δ). |
Chảy máu mũi, chảy máu chân răng, xuất huyết dưới da. |
- Số lượng và hình thái tiểu cầu: Bình thường. - Thời gian máu chảy: Có thể kéo dài. - PFA: kéo dài. - Ngưng tập tiểu cầu với: + ADP, epinephrine: sóng ngưng tập 1 bình thường, sóng ngưng tập 2 giảm. + Collagen: giảm hoặc không ngưng tập. + Thrombin: Không ngưng tập. |
|
Hội chứng tiểu cầu xám |
Di truyền lặn trên NST thường (có thể di truyền trội NST thường hoặc NST X). |
Thiếu hạt α. |
- Chảy máu mũi, chảy máu chân răng, xuất huyết dưới da. - Lách to (có thể). |
- Số lượng tiểu cầu thường giảm. - Thời gian máu chảy kéo dài. - Tiểu cầu kích thước lớn, hình bầu dục, nhạt màu. - Co cục máu: Co không hoàn toàn hoặc không co. - PFA: Kéo dài. - Ngưng tập tiểu cầu với: + Collagen, thrombin: Giảm +ADP, adrenalin: Bình thường hoặc giảm nhẹ. - Tủy xương có thể tăng sinh xơ (liên quan đến tăng sinh máu ngoài tủy). |
|
Rối loạn hoạt tính tiền đông máu của tiểu cầu. |
Hiếm gặp, di truyền NST thường. |
Bất thường khả năng hoạt hóa của tiểu cầu để kích hoạt phản ứng đông máu và bất thường giải phóng các vi hạt, vi túi của tiểu cầu. |
Chảy máu sau nhổ răng, sau phẫu thuật; phụ nữ rong kinh, chảy máu kéo dài sau sinh đẻ. |
- Thời gian máu chảy: Bình thường. - Thời gian prothombin: kéo dài. - NTTC: bình thường. - Định lượng yếu tố 3 tiểu cầu: Giảm. - Điều trị: truyền KTC, phức hợp prothrombin hoạt hóa cô đặc (lưu ý nguy cơ huyết khối). |
Bảng 8. Đặc điểm một số bệnh/ hội chứng rối loạn chức năng tiểu cầu mắc phải
|
Bệnh/ hội chứng |
Cơ chế bệnh sinh |
Thay đổi xét nghiệm |
Điều trị |
|
|
Bệnh lý Huyết học: - Hội chứng tăng sinh tủy mạn tính. - Lơ xê mi cấp, mạn. - Hội chứng rối loạn sinh tủy. - Đái huyết sắc tố kịch phát ban đêm… |
- Bất thường kích thước tiểu cầu - Bất thường receptor màng tiểu cầu - Bất thường phóng thích, chuyển hóa acid arachidonic. - Bất thường tiết Epinephrin/ADP/Collagen dẫn đến giảm NTTC - Rối loạn hoạt tính tiền đông tiểu cầu. - Biểu hiện các marker bất thường trên các hạt - Thiếu hụt kho dự trữ mắc phải. - Rối loạn sinh tổng hợp mẫu tiểu cầu. |
- Rối loạn xét nghiệm đặc hiệu: tùy theo bệnh chính. - Thời gian máu chảy, PFA: Kéo dài. - Co cục máu đông: Co không hoàn toàn hoặc không co. - Ngưng tập tiểu cầu với các chất kích tập: Có thể bình thường, giảm hoặc không ngưng tập. |
Điều trị bệnh chính và điều trị triệu chứng bằng truyền khối tiểu cầu đậm đặc (nếu bệnh nhân có SLTC giảm nặng và/hoặc nguy cơ xuất huyết nặng). |
|
|
Tăng ure máu. |
- Bất thường khả năng dính, ngưng tập và chế tiết của tiểu cầu. |
- Tăng ure máu. - Thời gian máu chảy, PFA: Kéo dài. - Co cục máu đông: Co không hoàn toàn hoặc không co. - Ngưng tập tiểu cầu với các chất kích tập: Có thể bình thường, giảm hoặc không ngưng tập. |
- Điều trị bệnh chính. - Lọc thận nhân tạo. - Truyền khối tiểu cầu đậm đặc. - DDAVP 0.3 mcg/kg truyền TM 15-30’ có thể lặp lại sau mỗi 12 hoặc 24h. |
|
|
Rối loạn chức năng tiểu cầu do thuốc |
Aspirin |
Ức chế không đảo ngược men cyclo - oxygenase (acetyl hóa cyclo - oxygenase) làm giảm tổng hợp Thromboxan A2. |
- Số lượng tiểu cầu bình thường. - Thời gian máu chảy, PFA: Kéo dài. - Ngưng tập tiểu cầu với các chất kích tập: Có thể bình thường, giảm hoặc không ngưng tập. |
Tạm dừng thuốc nếu có nguy cơ chảy máu cao. |
|
Dipyridamole |
Khóa kênh dẫn của Adenosin. |
- Số lượng tiểu cầu bình thường. - Thời gian máu chảy, PFA: Kéo dài. - Ngưng tập tiểu cầu với các chất kích tập: Có thể bình thường, giảm hoặc không ngưng tập. |
|
|
|
Thuốc kháng viêm không steroid |
Ức chế men cyclooxygenase thuận nghịch. |
Ngưng tập tiểu cầu: Giảm hoặc không ngưng tập (về bình thường khi hết tác dụng của thuốc). |
|
|
|
Ticlopidine, Clopidogrel: |
Kháng receptor của ADP trên màng tiểu cầu |
Ngưng tập tiểu cầu với các chất kích tập: Giảm hoặc không ngưng tập. |
|
|
|
Abciximab, Eptifibatide, Tirofiban |
Ức chế GP IIb/IIIa tiểu cầu. |
Thời gian máu chảy kéo dài; Ngưng tập tiểu cầu với các chất kích tập: Giảm hoặc không ngưng tập. |
|
|
|
|
Ticagrelor, cangrelor, elinogrel |
Kháng receptor nonthienopydine P2Y12 thuận nghịch |
Thời gian máu chảy kéo dài; Ngưng tập tiểu cầu với các chất kích tập: Giảm hoặc không ngưng tập. |
|
|
Nitroprusside |
Tăng cGMP của tiểu cầu |
Thời gian máu chảy kéo dài; Ngưng tập tiểu cầu với các chất kích tập: Giảm hoặc không ngưng tập. |
|
|
|
Nitroglycerin Propanolol |
Giảm hạt đặc hiệu |
Ngưng tập tiểu cầu với các chất kích tập giảm hoặc không ngưng tập. |
|
|
|
Ức chế kênh Calci (verapamil, nifedipine, diltrazem) |
Ức chế ngưng tập tiểu cầu |
Ngưng tập tiểu cầu với các chất kích tập giảm hoặc không ngưng tập. |
|
|
|
Dextran |
Hấp phụ lên màng tiểu cầu dẫn đến rối loạn ngưng tập và phản ứng tiết, rối loạn hoạt tính tiền đông của tiểu cầu. |
Thời gian máu chảy kéo dài |
|
|
|
Heparin |
Ức chế hoạt hóa của tiểucầu với thrombin, liều cao gây bất thường chức năng yếu tố von Willebrand tiểu cầu. |
Ngưng tập tiểu cầu với các chất thrombin, ristocetin giảm hoặc không ngưng tập. |
|
|
|
|
Penicillin |
Ức chế không thuận nghịch receptor ADP/Epinephrin, bất thường tương tác giữa yếu tố von Willebrand - màng tiểu cầu, dẫn đến giảm phản ứng dính, giảm tiết, giảm hoạt hóa, giảm NTTC. |
Thời gian máu chảy kéo dài. Ngưng tập tiểu cầu với các chất giảm hoặc không ngưng tập. |
|
|
Tim phổi nhân tạo |
- Giảm NTTC, giảm ngưng kết TC với ristocetin, giảm hạt dự trữ α,δ. - Hình thành các mảnh tiểu cầu và tăng các sản phẩm thoái giáng fibrin (PDFs) trong quá trình chạy tim phổi nhân tạo gây rối loạn chức năng tiểu cầu. |
|
- Tráng các bề mặt nhân tạo của thiết bị tim phổi nhân tạo bằng heparin. - Sử dụng bơm trung tâm tốt hơn bơm “cuộn”. - Có thể sử dụng yếu tố VIIa cho bệnh nhân sau phẫu thuật mà không đáp ứng được bằng các điều trị RLĐM thông thường. |
|
Chú thích: GP (Glycoprotein).
4. ĐIỀU TRỊ
- Không có phương pháp điều trị đặc hiệu, chủ yếu là truyền khối tiểu cầu nếu chảy máu nhiều.
- Đối với rối loạn chức năng tiểu cầu thứ phát: Loại bỏ nguyên nhân gây rối loạn chức năng tiểu cầu, điều trị bệnh chính (Suy thận, tăng ure máu: Lọc thận nhân tạo…).
- Điều trị triệu chứng:
+ Cầm máu tại chỗ cho người bệnh chảy máu mũi bằng cách chèn ép tại chỗ, ngẩng cao đầu, nhét mèche; thắt hoặc làm tắc động mạch phía trong hàm trên;
+ Thuốc chống tiêu sợi huyết: Thường được chỉ định cho bênh nhân chảy máu chân răng, chảy máu sau nhổ răng. Tranexamic acid 0,5-1,0 g/lần x 3-4 lần/ ngày (tiêm tĩnh mạch) hoặc dung dịch tranexamic acid HANK 5% x 10ml/ lần x 3 - 4 lần/ ngày (xúc miệng) đến khi cầm máu;
+ Người bệnh sau truyền khối tiểu cầu có thể xuất hiện kháng thể kháng tiểu cầu (Ví dụ: Kháng thể kháng phức hợp GP IIb/IIIa, GPIb/IX/V… của tiểu cầu), đang có xuất huyết nặng có thể xem xét để chỉ định điều trị bằng yếu tố VII hoạt hóa (liều dùng: 90 µ/kg x 3 liều, cách nhau 3 giờ; hoặc liều duy nhất 270 µ/kg; tiêm nhắc lại liều 90 µ/kg sau 6 giờ nếu vẫn chảy máu);
+ Bổ sung sắt ở người bệnh thiếu máu thiếu sắt do chảy máu quá nhiều hoặc chảy máu kéo dài.
- Các biện pháp khác:
+ Loại bỏ các nguyên nhân có khả năng gây chảy máu;
+ Không sử dụng các thuốc có ảnh hưởng đến chức năng tiểu cầu như: Aspirin, các thuốc chống viêm không steroid, heparin;
+ Sử dụng các vật liệu cầm máu tại chỗ:
● Gelfoam (tẩm acid tranexamic + thrombin);
● Keo fibrin (chứa fibrinogen + thrombin; có thể kèm chất chống tiêu sợi huyết
hoặc các thành phần tương tự).
5. CÁC XÉT NGHIỆM THEO DÕI
- Tổng phân tích tế bào máu: Đánh giá mức độ thiếu máu, làm khi bệnh nhân vào viện, hoặc định kì mỗi tháng, hoặc sau điều trị thiếu máu.
- Sinh hóa: Glucose, chức năng gan thận, sắt và ferritin.
- HBV, HCV, HIV: Kiểm tra định kì theo quy định để theo dõi nguy cơ lây nhiễm các bệnh truyền qua đường truyền máu (những trường hợp truyền máu và chế phẩm máu).
- Kháng thể kháng tiểu cầu: Khi đáp ứng kém trên lâm sàng, nghi ngờ có kháng thể kháng tiểu cầu.
- Rotem (Extem, Fibtem): Đánh giá hiệu quả truyền khối tiểu cầu (xét nghiệm trước và sau truyền khối tiểu cầu).
TÀI LIỆU THAM KHẢO
1. Nguyễn Ngọc Minh (2007), “Bệnh lý rối loạn chức năng tiểu cầu bẩm sinh”, Bài giảng Huyết học - Truyền máu sau Đại học, NXB Y học, tr. 483 - 504.
2. Joel S. Bennett (2005), “Hereditary disorders of platelet function”, Hematology Basic principles and practice, 4th, p: 2327 - 2345.
3. José A. López and Perumal Thiagarajan (2005), “Accuired disorders of platelet function”, Hematology Basic principles and practice, 4th, p: 2347 - 2367.
4. Marshall A. Lichman et all (2006), “Hereditary qualitative platelet disorders: overview”, Williams Hematology, 7th Edition, pp: 4326 - 4421.
5. Marshall A. Lichman et all (2016), “Acquired qualitative platelet disorders: overview”, Williams Hematology, 9th Edition, pp: 4422 - 4482.
6. Thomas J. K., Diane J. N (2009), “Qualitative disorders of platelet function”, Wintrobe’s Clinical Hematology, 12th, pp. 1361 - 1378.
10. ĐÔNG MÁU RẢI RÁC TRONG LÒNG MẠCH
(Disseminated Intravascular Coagulation: DIC)
1. ĐẠI CƯƠNG
Khái niệm: Đông máu rải rác trong lòng mạch (Disseminated Intravascular Coagulation: DIC) là một hội chứng đặc trưng bởi sự hoạt hóa đông máu quá mức, mất tính khu trú, bắt nguồn từ nhiều nguyên nhân, gây lắng đọng fibrin, hình thành huyết khối vi mạch ở nhiều cơ quan trong cơ thể dẫn tới tình trạng chảy máu mất kiểm soát và huyết khối.
2. NGUYÊN NHÂN
DIC do rất nhiều nguyên nhân gây nên, vì vậy hội chứng này có thể gặp trong nhiều tình trạng bệnh lý, nhiều chuyên khoa. Tuy nhiên, thường gặp tỷ lệ cao ở những tình trạng bệnh lý sau:
- Nhiễm trùng: Đặc biệt là các nhiễm trùng nặng (hệ hô hấp, đường tiết niệu, đường mật...).
- Bệnh lý ác tính hệ tạo máu:
+ Bạch cầu cấp thể tiền tủy bào hay gặp nhất;
+ U lympho ác tính và các bệnh lý ác tính cơ quan tạo máu khác.
- Ung thư tạng đặc (thường liên quan đến tình trạng di căn).
- Tổn thương tổ chức, mô: Chấn thương, bỏng, tiêu cơ vân, sau phẫu thuật...
- Rối loạn liên quan đến sản khoa: Tiền sản giật, hội chứng HELLP, rau bong non...
- Bệnh lý liên quan đến mạch máu:
+ Phình tách động mạch chủ bụng và ngực;
+ Khối u liên quan đến thành mạch máu;
+ Viêm mạch máu...
- Tổn thương gan: Suy gan cấp, viêm gan cấp, xơ gan.
- Viêm tụy cấp.
- Sốc.
- Độc tố và phản ứng miễn dịch:
+ Tan máu, truyền nhầm nhóm máu;
+ Rắn độc cắn;
+ Thải ghép.
- Hạ thân nhiệt...
3. TRIỆU CHỨNG
Để chẩn đoán DIC, cần dựa vào các triệu chứng lâm sàng (bao gồm cả bệnh lý nền, triệu chứng của DIC) và kết quả xét nghiệm dưới đây:
3.1. Triệu chứng lâm sàng
a. Triệu chứng của bệnh chính gây DIC
b. Triệu chứng của DIC
- Trên nền bệnh chính có thể gây DIC, người bệnh có thể có sốc do bệnh chính hoặc do DIC.
- Xuất huyết: Xuất huyết dưới da, niêm mạc, chảy máu nội tạng, chảy máu nơi tiêm truyền, chảy máu bất thường ở vết mổ.
- Huyết khối: Thường gặp hoại tử đầu chi, tắc vi mạch.
- Có thể người bệnh đồng thời có cả xuất huyết và huyết khối.
- Suy đa phủ tạng: Nguyên nhân thường do huyết khối gây tắc vi mạch ở các tạng.
3.2. Thay đổi xét nghiệm
Các xét nghiệm thay đổi ở bệnh nhân DIC phản ánh tình trạng hoạt hóa, tiêu thụ các yếu tố đông máu, mất cân bằng trong quá trình tiêu sợi huyết.
3.2.1. Xét nghiệm phản ánh tình trạng tiêu thụ các yếu tố tham gia vào quá trình đông máu
- Số lượng tiểu cầu (SLTC): Số lượng tiểu cầu thường giảm trong DIC, sự thay đổi tiểu cầu theo thời gian có ý nghĩa trong chẩn đoán và theo dõi tiến triển của DIC. Tuy nhiên, xét nghiệm SLTC có độ nhạy cao nhưng độ đặc hiệu thấp đặc biệt khi bệnh lý nền là bệnh lý liên quan đến hệ tạo máu như Lơ xê mi, suy tủy xương...
- PT/APTT: Kéo dài trong khoảng 50 - 60% bệnh nhân DIC, liên quan đến tình trạng tiêu thụ yếu tố đông máu, suy chức năng gan, mất yếu tố đông máu do mất máu nặng...
Tuy nhiên, gần 50% bệnh nhân chẩn đoán DIC có xét nghiệm PT, APTT bình thường, thậm chí ngắn do sự lưu hành của các yếu tố đông máu hoạt hóa như thrombin, Xa, thúc đẩy sự hình thành fibrin. Do vậy, xét nghiệm PT, APTT bình thường cũng không thể loại trừ DIC, cần lặp lại xét nghiệm này để theo dõi.
- Fibrinogen: Fibrinogen giảm trong DIC liên quan đến tiêu thụ yếu tố đông máu và hoạt động của hệ thống tiêu sợi huyết. Trong những trường hợp DIC liên quan đến tình trạng viêm, nồng độ fibrinogen thường trong giới hạn bình thường, thậm chí có thể tăng. Những bệnh lý nền như Lơ xê mi, phình tách động mạch, rối loạn liên quan sản khoa, mất máu nặng thì dấu hiệu giảm fibrinogen khá rõ rệt, nhanh chóng.
- Nồng độ một số yếu tố đông máu (V, VII, VIII, X): Giảm.
3.2.2. Sản phẩm thoái giáng của fibrin/fibrinogen (FDP) và D-Dimer
- FDP (Fibrin/fibrinogen degradation): Là sản phẩm của quá trình phân cắt fibrin và fibrinogen bởi plasmin.
- D-dimer là sản phẩm của sự thoái giáng fibrin dưới tác dụng của plasmin, được tạo ra sau khi đã hình thành cục máu đông bền vững.
- D-Dimer và FDPs thường tăng trong hầu hết các trường hợp DIC, là một tiêu chuẩn trong chẩn đoán DIC hiện nay. Tuy nhiên, cả D-dimer và FDP đều có độ nhạy thấp, tăng trong các trường hợp như huyết khối, nhiễm trùng, có thai.
3.2.3. Dấu ấn phân tử của sự hoạt hóa đông máu
Các xét nghiệm Fibrin monomer hòa tan, TAT (thrombin - antithrombin), Prothrombin F1+2 là những xét nghiệm phản ánh tình trạng hoạt hóa đông máu.
- Xét nghiệm Fibrin monomer hòa tan (sFM): Là sản phẩm của quá trình phân cắt fibrinogen dưới tác dụng của thrombin, hình thành ở giai đoạn hoạt hóa đông máu, trước khi cục đông được hình thành, phản ánh tình trạng tăng đông. Nồng độ sFM tăng từ 3 - 5 ngày trước khi cục đông hình thành và giảm xuống sau 3 ngày xuất hiện cục đông.
- Xét nghiệm đánh giá sự hình thành thrombin: Vì thrombin lưu hành trong huyết tương sẽ nhanh chóng kết hợp với antithrombin, do vậy không thể định lượng trực tiếp thrombin trong huyết tương. Các xét nghiệm như TAT, Prothrombin F1+2 đánh giá gián tiếp sự hình thành thrombin.
- Xét nghiệm TAT (Thrombin - Antithrombin): Nồng độ TAT tăng trong các trường hợp tăng hoạt hóa đông máu, hình thành thrombin. Xét nghiệm phát hiện phức hệ TAT gián tiếp phản ánh tình trạng hình thành thrombin.
- Phức hệ Prothrombin F1+2: Prothrombin F1+2 là peptide được giải phóng ra từ prothrombin trong quá trình hình thành thrombin. Định lượng Prothrombin F1+2 gián tiếp đánh giá sự hình thành thrombin.
3.2.4. Hệ thống các chất kháng đông sinh lý
Các chất kháng đông sinh lý giúp điều hòa quá trình đông máu, hạn chế quá trình đông máu lan rộng. Các chất này gồm có Antithrombin, protein S, protein C, thrombomodulin, sự thay đổi hệ thống các chất kháng đông sinh lý này làm quá trình đông máu mất tính khu trú. Sự giảm nồng độ các chất kháng đông sinh lý đặc biệt được đề cập đến trong DIC liên quan đến tình trạng nhiễm trùng, dẫn đến hình thành những cục đông nhỏ, hậu quả là suy chức năng các cơ quan.
3.2.5. Xét nghiệm liên quan đến hoạt động của hệ thống tiêu sợi huyết
- PIC: Plasmin -α2- plasmin inhibitor complex: Là xét nghiệm đánh giá gián tiếp sự hình thành plasmin. Trong một số nghiên cứu các tác giả nhận thấy rằng ở bệnh nhân DIC thường có PIC tăng, điều đó phản ánh có sự tăng hoạt động của hệ thống tiêu sợi huyết, đặc biệt ở nhóm DIC thể ưu thế tiêu sợi huyết.
- PAI - 1: Tế bào nội mạc hoạt hóa tăng tổng hợp PAI.
- Alpha2 antiplasmin: Tăng.
- Plasminogen: Giảm.
- Nghiệm pháp Von-Kaulla: Có thể dương tính (khi tiêu sợi huyết thứ phát nổi trội) hoặc âm tính.
- ROTEM, TEG: Hình ảnh giảm đông, có thể có hình con quay gặp trong một số trường hợp DIC có tiêu sợi huyết thứ phát tối cấp.
3.2.6. Xét nghiệm khác
- Có mảnh hồng cầu trên tiêu bản máu ngoại vi do hiện tượng tan máu vi mạch.
- Có thể gặp giảm huyết sắc tố nếu người bệnh có chảy máu, thay đổi các kết quả xét nghiệm sinh hóa nếu có suy thận, suy gan.
4. CHẨN ĐOÁN
4.1. Chẩn đoán xác định: Hiện tại ở Việt Nam, chẩn đoán DIC được áp dụng theo tiêu chuẩn do Hiệp hội cầm máu và tắc mạch quốc tế (ISTH) đề xuất; Cụ thể:
- Trên lâm sàng có một bệnh lý nền có thể gây DIC.
- Xét nghiệm: Đánh giá qua thang điểm:
|
Chỉ số |
Điểm |
|
Số lượng tiểu cầu >100G/L 50 - 100G/L < 50G/L |
0 1 2 |
|
Thời gian Prothrombin PTs > PT chứng dưới 3s PTs > PT chứng 3 - 6s PTs > PT chứng > 6s |
0 1 2 |
|
Fibrinogen Fibrinogen > 1g/l Fibrinogen < 1g/l |
0 1 |
|
Các chất liên quan đến fibrin (FDP, D-dimer, FM) - Không tăng - Tăng vừa - Tăng cao |
0 2 3 |
|
Tổng điểm: ≥ 5: DIC thực sự, lặp lại xét nghiệm hàng ngày; < 5: Nghi ngờ DIC tiềm tàng, lặp lại XN sau 1 - 2 ngày. Lưu ý tính động học của các xét nghiệm |
|
4.2. Chẩn đoán phân biệt
Phân biệt DIC với một số hội chứng bệnh lý thiếu máu tan máu vi mạch khác:
Bảng 9. Phân biệt DIC và một số hội chứng khác.
|
|
TTP |
HUS |
HELLP |
DIC |
|
Tổn thương thần kinh trung ương |
+++ |
+/- |
+/- |
+/- |
|
Suy thận |
+/- |
+++ |
+ |
+/- |
|
Sốt |
+/- |
-/+ |
- |
- |
|
Suy gan |
+/- |
+/- |
+++ |
+/- |
|
Tăng huyết áp |
-/+ |
+/- |
+++ |
- |
|
Tan máu |
+++ |
++ |
+ |
+ |
|
Giảm tiểu cầu |
+++ |
++ |
++ |
+++ |
|
Bất thường XN đông máu |
- |
- |
+/- |
+++ |
Ghi chú:
- TTP (Thrombotic Thrombocytopenic Purpura): Ban xuất huyết giảm tiểu cầu huyết khối.
- HUS (Hemolytic Uremic Syndrome): Hội chứng tan máu tăng ure.
- HELLP (Hemolysis Elevated Liver enzymes Low Platelet count): Tan máu tăng men gan giảm tiểu cầu.
5. ĐIỀU TRỊ
Lưu ý cần làm đầy đủ các xét nghiệm tế bào máu ngoại vi, đông máu (cơ bản, chuyên sâu), các xét nghiệm tìm nguyên nhân (như ổ ung thư, nhiễm khuẩn, chẩn đoán hình ảnh…) để vừa điều trị nguyên nhân vừa điều trị DIC. Cần theo dõi xét nghiệm đông máu và hiệu quả chống đông liên tục tùy theo loại thuốc sử dụng dựa trên các khuyến cáo quốc tế.
5.1. Điều trị bệnh chính gây DIC
Điều trị tích cực bệnh chính để loại bỏ nguyên nhân gây DIC đóng vai trò quan trọng trong điều trị hội chứng này.
5.2. Điều trị DIC
a. Điều trị giảm đông, chống chảy máu bằng chế phẩm máu: Đóng vai trò quyết định trong cứu sống người bệnh, nhất là những trường hợp chảy máu nặng.
- Chỉ định: Lâm sàng có xuất huyết, hoặc kết quả xét nghiệm giảm đông nặng, nguy cơ xuất huyết cao.
- Loại chế phẩm và liều lượng:
+ Truyền khối tiểu cầu khi lâm sàng có chảy máu và số lượng tiểu cầu < 50G/L hoặc không chảy máu nhưng số lượng tiểu cầu < 20G/L; Liều lượng: Duy trì số lượng tiểu cầu > 50 G/L;
+ Truyền huyết tương tươi đông lạnh khi có chảy máu và PT % giảm < 50% hoặc không chảy máu nhưng PT% < 30%; liều lượng: 15 - 20ml/kg/24h, ngừng truyền khi PT ≥ 70%. Trường hợp chống chỉ định truyền huyết tương tươi đông lạnh do quá tải tuần hoàn, thay thế bằng phức hợp prothrombin cô đặc;
+ Truyền tủa lạnh yếu tố VIII (cryoprecipitate) khi fibrinogen < 1,5g/l; ước tính truyền 3g fibrinogen sẽ nâng được nồng độ fibrinogen lên 1g/L;
+ Truyền khối hồng cầu khi Hb < 80 g/L hoặc người bệnh tiếp tục chảy máu.
b. Điều trị thuốc chống đông
Sử dụng heparin với liều điều trị trong trường hợp DIC mà lâm sàng có huyết khối. Có thể sử dụng heparin không phân đoạn (UFH) hoặc heparin trọng lượng phân tử thấp (LMWH).
- Heparin trọng lượng phân tử thấp (LMWH):
+ Liều lượng: 50 - 100 UI anti Xa/kg/12giờ, tiêm dưới da;
+ Theo dõi bằng xét nghiệm anti Xa.
- Heparin tiêu chuẩn (heparin standard):
+ Liều lượng: 300-500 UI/6 giờ, truyền tĩnh mạch;
+ Theo dõi: Xét nghiệm APTT: Duy trì ở mức APTT kéo dài gấp 1,5 đến 2.5 lần so với chứng (tiến hành xét nghiệm 4 - 6 giờ /lần) hoặc xét nghiệm anti Xa.
Điều chỉnh liều dựa vào xét nghiệm anti Xa khi điều trị LMWH và APTT hoặc anti Xa khi điều trị UFH; Diễn biến của DIC, cụ thể: Kết quả nồng độ D- Dimer, nồng độ fibrin monomer hòa tan, nghiệm pháp rượu, kết hợp triệu chứng xuất huyết, huyết khối trên lâm sàng.
- Dự phòng huyết khối bằng heparin (UFH hoặc LMWH) với liều dự phòng được khuyến cáo cho những trường hợp DIC không có chảy máu nghiêm trọng hoặc có thể kiểm soát được nguy cơ chảy máu.
- Chú ý: Khi điều trị heparin, cần đánh giá nồng độ antithrombin để có điều chỉnh phù hợp.
c. Điều trị thuốc chống tiêu sợi huyết: Acid tranexamic.
- Chỉ định: Lâm sàng chảy máu nặng đe dọa tính mạng người bệnh và kết quả xét nghiệm thể hiện tình trạng tiêu sợi huyết thứ phát nổi trội: Fibrinogen giảm nặng, D-Dimer tăng rất cao, nghiệm pháp Von-Kaulla dương tính, hình ảnh ROTEM, TEG có biểu hiện tiêu sợi huyết.
- Liều lượng: Tranexamic acid 1g/8h.
- Ngừng điều trị khi fibrinogen ≥ 2g/l.
Luôn cân nhắc giữa nguy cơ chảy máu và huyết khối của bệnh nhân khi dùng thuốc.
5.3. Các biện pháp hỗ trợ khác
- Điều trị suy phủ tạng (nếu có).
- Khôi phục thể tích tuần hoàn.
- Duy trì thăng bằng kiềm toan.
- Bổ sung folate.
6. THEO DÕI ĐIỀU TRỊ
DIC là hội chứng tiến triển nhanh, biểu hiện lâm sàng của DIC rất phức tạp: Từ hầu như không có biểu hiện lâm sàng ở giai đoạn còn bù (non - overt DIC) tới biểu hiện lâm sàng nặng nề: Chảy máu, suy đa phủ tạng do huyết khối tắc mạch ở giai đoạn DIC mất bù (overt DIC). Vì vậy, để chẩn đoán sớm cũng như đánh giá tiến triển, điều chỉnh thuốc, chế phẩm kịp thời, hàng ngày cần tiến hành các xét nghiệm sau đây:
- Tổng phân tích tế bào máu ngoại vi (chú ý diễn biến động học số lượng tiểu cầu và huyết sắc tố).
- Đông máu cơ bản: PT, APTT, TT và fibrinogen.
- Định lượng D- Dimer.
- Định lượng fibrin monomer hòa tan (FM) hoặc nghiệm pháp rượu.
- Nghiệm pháp Von-Kaulla.
- Định lượng antithrombin.
- ROTEM hoặc TEG.
- Kiểm tra nồng độ anti Xa khi điều trị heparin trọng lượng phân tử thấp để chỉnh liều. Đối với những trường hợp nặng: Chảy máu đe doạ tính mạng, tiêu sợi huyết nặng phải điều trị thuốc chống tiêu sợi huyết. Cần tiến hành kiểm tra các xét nghiệm nêu trên 6 giờ 1 lần để điều chỉnh chế phẩm, thuốc kịp thời.
7. TIÊN LƯỢNG
DIC là tình trạng bệnh lý có thể gặp ở nhiều chuyên khoa. Cụm từ “cái chết đang đến” được dùng để phản ánh mức độ nặng của DIC. Việc chẩn đoán và điều trị sớm DIC giúp cải thiện tiên lượng của bệnh nhân. Tiên lượng bệnh nhân DIC phụ thuộc vào bệnh lý nền DIC, mức độ rối loạn đông máu và việc can thiệp xử trí.
Tiếp cận chẩn đoán và điều trị DIC
TÀI LIỆU THAM KHẢO
1. Nguyễn Anh Trí (2011), Thông báo của hội nghị khoa học toàn quốc về Hemophilia và Đông máu ứng dụng lần thứ VI, Y học thực hành (791) số 11/2011, tr.67-68.
2. British Committee for Standards in Haematology (2009). Guidelines for the Diagnosis and Management of Disseminated Intravascular Coagulation.
3. Hideo Wada, Esteban C. Gabazza et al (2003). Comparison of Diagnostic Criteria for DisseminatedIntravascular Coagulation (DIC): Diagnostic Criteria ofthe International Society of Thrombosis and Hemostasis(ISTH) and of the Japanese Ministry of Health and Welfare for Overt DIC. American Journal of Hematology 2003; 74:17-22.
4. Jong Hwa Lee, Jae Woo Song and Kyung Soon Song (2007). Diagnosis of Overt Disseminate Intravascular Coagulation: A comparative study using criteria from the ISTH versus the Korean Society on Thrombosis and Hemostasis. Yonsei Med J 2007; 48(4): 595-600.
5. Mike Laffan, Richard Manning (2011), Investigation of haemostasis, Practical Hematology, Eleventh Edition ,Churchill Living Stone, p. 392-446
6. Raj S. Kasthuri and Nigel S. Key (2010), Disseminated intravascular coagulation and other microangiopathies, Practical Hemostasis and Thrombosis, Second Edition, A John Wiley & Sons Publication, p.123-135.
7. Marcel Levi and Marie Scully (2018), How I treat disseminated intravascular coagulation, Blood, 131: 845-854
11. HỘI CHỨNG ANTIPHOSPHOLIPID
1. ĐẠI CƯƠNG
Hội chứng Antiphospholipid (Antiphospholipid syndrome - APS) là một hội chứng có biểu hiện lâm sàng chủ yếu là tắc mạch hoặc sảy thai tái diễn, với sự xuất hiện của kháng thể chống phospholipid hoặc chống lại các quyết định kháng nguyên protein gắn phospholipid.
2. CHẨN ĐOÁN
2.1. Lâm sàng
Biểu hiện rất đa dạng, ở nhiều cơ quan đó là tắc mạch, thiếu máu cục bộ hoặc nhồi máu và các biến chứng thai nghén. Các biểu hiện này có thể xuất hiện độc lập hoặc đồng thời với bệnh chính (trường hợp hội chứng Antiphospholipid thứ phát).
|
Các biểu hiện lâm sàng liên quan tới hội chứng Antiphospholipid
|
2.2. Xét nghiệm:
Chỉ định xét nghiệm tìm kháng thể kháng phospholipid (anti-phospholipid antibodies = kháng thể aPL) bao gồm: Kháng thể kháng Cardiolipin (anti-Cardiopilin = aCL), kháng thể kháng β2-Glycoprotein I (anti - β2Glycoprotein I = aβ2GI), kháng thể kháng phosphatidylcholin, kháng thể kháng phosphatidylethanolamine, kháng thể kháng phosphotidylserin) và kháng đông Lupus (LA - Lupus Anticoagulant) trong những trường hợp sau:
- Tắc tĩnh mạch và/ hoặc động mạch ở người dưới 50 tuổi.
- Tắc mạch ở những vị trí không thường gặp hoặc liên quan đến bệnh tự miễn.
- APTT kéo dài không rõ nguyên nhân.
- Phụ nữ có biến chứng thai nghén không giải thích được.
- Lupus ban đỏ hệ thống, bệnh tự miễn: Nếu âm tính, làm lại định kỳ (có thể xuất hiện kháng thể trong tương lai).
2.3. Chẩn đoán xác định
Theo tiêu chuẩn Sydney 2006 của ISTH
a. Tiêu chuẩn lâm sàng
- Bằng chứng của huyết khối: Huyết khối tĩnh mạch hoặc động mạch hoặc mao mạch, khẳng định bằng chẩn đoán hình ảnh hoặc mô bệnh học và không có biểu hiện của viêm thành mạch.
- Biến chứng thai sản (một trong các biểu hiện sau):
+ Một hoặc nhiều lần thai chết lưu sau 10 tuần không giải thích được mà hình thái phôi thai bình thường;
+ Một hoặc nhiều lần đẻ non trước 34 tuần mà nguyên nhân là do sản giật, tiền sản giật nặng hoặc kém phát triển bánh rau;
+ Ba hoặc hơn ba lần sảy thai tự phát trước 10 tuần mà không có bất thường hormon của thai phụ hay bất thường nhiễm sắc thể của bố, mẹ.
b. Tiêu chuẩn xét nghiệm
- LA test dương tính.
- Kháng thể kháng cardiolipin dương tính.
- Kháng thể kháng β2-Glycoprotein I dương tính.
- Kháng thể kháng phosphatidylcholin dương tính.
- Kháng thể kháng phosphatidylethanolamine dương tính.
- Kháng thể kháng phosphotidylserin dương tính.
Các xét nghiệm được coi là có ý nghĩa khi dương tính ít nhất 2 lần, cách nhau ít nhất 12 tuần.
Chẩn đoán xác định hội chứng Antiphospholipid đòi hỏi ít nhất một tiêu chuẩn lâm sàng và một tiêu chuẩn xét nghiệm.
3. XẾP LOẠI
3.1. APS tiên phát: Hội chứng Antiphospholipid không tìm được nguyên nhân hay bệnh lý phối hợp.
3.2. APS thứ phát: Hội chứng Antiphospholipid sau hoặc kèm theo:
- Lupus ban đỏ hệ thống.
- Các bệnh tự miễn và tổ chức liên kết khác.
- Các bệnh cảm ứng do thuốc: Procainamid, hydralazin, quinidin, pherothiazin, penicillin…
3.3. APS toàn phát (thê thảm) (CAPS-Catastrophic APS): Diễn biến cấp tính, và toàn phát, tổn thương nhiều cơ quan, thiếu máu cục bộ, bít tắc mạch máu nhỏ lan tỏa; chẩn đoán dựa trên các tiêu chuẩn:
- Tắc mạch từ 3 tổ chức/ cơ quan trở lên trong đó tắc vi mạch ở ít nhất 1 cơ quan.
- Các biểu hiện xảy ra đồng thời hoặc không quá một tuần.
- Xét nghiệm có kháng thể aPL.
4. CHẨN ĐOÁN PHÂN BIỆT
4.1. Phân biệt với các bệnh lý có thể sinh kháng thể aPL như: Nhiễm trùng: giang mai, bệnh Lyme, nhiễm HIV…; hoặc sau sử dụng một số thuốc. Phân biệt dựa trên tiền sử bệnh, triệu chứng lâm sàng, xét nghiệm xác định bệnh (xét nghiệm TPHA tìm giang mai, xét nghiệm HIV…), làm lại xét nghiệm tìm kháng thể aPL sau mỗi 6 tuần.
4.2. Phân biệt với hemophilia (APTT kéo dài), đặc biệt là hemophilia A mắc phải (có kháng yếu tố VIII): Phân biệt bằng định lượng yếu tố đông máu đường nội sinh, xét nghiệm định lượng hoạt tính kháng yếu tố VIII.
4.3. APS toàn phát (thê thảm): Phân biệt với đông máu nội mạch rải rác (DIC), xuất huyết giảm tiểu cầu huyết khối (TTP).
5. ĐIỀU TRỊ
5.1. Điều trị và dự phòng huyết khối
Sử dụng heparin tiêu chuẩn hoặc heparin trọng lượng phân tử thấp, sau đó chuyển sang kháng vitamin K (Wafarin) hoặc aspirin (70 - 100 mg/ngày) tùy thuộc vào từng bệnh nhân, dùng lâu dài. INR thông thường duy trì ở mức 2-3.
Liều điều trị tắc mạch:
- Heparin tiêu chuẩn (5.000-10.000 U/12h tiêm dưới da hoặc tiêm tĩnh mạch, khởi đầu 5.000U, sau đó truyền tĩnh mạch liên tục 1.000-2.000 U/giờ. Duy trì rAPTT 1,5 - 2,0.
- Heparin trọng lượng phân tử thấp (Low Molecular Weigh Heparin = LMWH): 1,5 mg/kg/ngày hoặc 1mg/kg x 2 lần/ngày, tiêm dưới da. Theo dõi bằng anti Xa (duy trì anti Xa 0,5-1 U/ml).
- Đối với những trường hợp vẫn xuất hiện tắc mạch khi đang điều trị dự phòng bằng kháng vitamin K thì cần điều chỉnh:
+ Tăng liều kháng vitamin K, duy trì INR 3- 4; hoặc
+ Phối hợp với Aspirin liều thấp 70-100 mg/ngày; hoặc
+ Đổi sang LWMH 20 - 40mg/ngày tiêm dưới da 1 lần/ngày (liều dự phòng).
- Có thể cân nhắc điều trị thuốc hạ mỡ máu (nhóm statin) ở những người bệnh có mỡ máu cao, xơ vữa mạch.
- Không có chỉ định điều trị dự phòng huyết khối nếu người bệnh không có biểu hiện tắc mạch hay tiền sử sảy thai trước đó. Theo dõi dấu hiệu tắc mạch trên lâm sàng và theo dõi xét nghiệm kháng thể 3 tháng/ lần.
5.2. Điều trị APS có biến chứng thai nghén
- Có kháng thể aPL, không có tiền sử tắc mạch và sảy thai, có hoặc không bệnh tự miễn kết hợp: Theo dõi chặt chẽ, chưa cần dùng thuốc. Nếu có hiệu giá kháng thể aPL cao, tiền sử gia đình có bệnh tự miễn, đau nửa đầu… có thể cân nhắc aspirin liều thấp.
- Người có tiền sử sảy thai ≥ 2 lần: Trao đổi với người bệnh về lợi ích và nguy cơ của việc điều trị.
+ Chưa có thai nhưng có kế hoạch có thai trong thời gian gần: Dự phòng aspirin liều thấp, dùng LWMH (liều dự phòng) ngay khi xác nhận có thai.
+ Đã có thai: Điều trị dự phòng bằng LWMH, phối hợp aspirin 70-100 mg/ngày, ngừng trước sinh 24 giờ và dùng lại sau khi sinh 4-6 giờ, thời gian dùng thuốc sau sinh kéo dài tối thiểu 6-8 tuần.
- Người bệnh tiền sử sảy thai > 3 lần, có hoặc không có tiền sử huyết khối và SLE, hoặc cả hai:
+ Đang điều trị dự phòng bằng kháng vitamin K: Ngừng thuốc kháng vitamin K;
+ Điều trị dự phòng huyết khối bằng LWMH, phối hợp aspirin 70-100 mg/ngày, ngừng trước sinh 24 giờ và dùng lại sau khi sinh 4-6 giờ, thời gian dùng thuốc sau sinh kéo dài tối thiểu 6-8 tuần.
- Theo dõi thai sản: Theo dõi trên siêu âm sự phát triển thai và dịch ối mỗi 4 tuần và theo dõi trên siêu âm doppler lưu lượng máu tử cung - rau lúc 20-24 tuần.
5.3. Điều trị hội chứng antiphospholipid toàn phát
Cần điều trị sớm, phối hợp nhiều biện pháp với mục đích loại bớt kháng thể và điều trị tắc mạch.
- Methylprednisolone liều cao: 1.000mg/ngày x 3 ngày, sau đó duy trì 1-2 mg/kg/ngày.
- Chống đông: Heparin.
- IVIG: 1g/kg/ngày x 1-2 ngày hoặc 0,4g/kg/ngày x 5 ngày.
- Trao đổi huyết tương.
- Có thể cân nhắc sử dụng thuốc tiêu sợi huyết: Streptokinase, urokinase, t-PA…
- Cyclophosphamid (2-3 mg/kg/ngày): CAPS liên quan đến Lupus ban đỏ hệ thống.
- Có thể cân nhắc sử dụng Rituximab.
5.4. Điều trị các bệnh lí đi kèm:
- Corticoid: Chỉ sử dụng khi có APS toàn phát, giảm tiểu cầu nặng, tan máu…
- Thuốc ức chế miễn dịch như Azathioprin, Cyclophosphamid, Mycophenolate mofetil…: Chỉ dùng để điều trị SLE, giảm tiểu cầu, tan máu, suy thận APL, không dùng đối với APS nguyên phát…
- Hydroxycholoquine: 200 - 400 mg/ngày (đối với SLE và APS thứ phát).
5.5. Theo dõi điều trị
- Theo dõi các dấu hiệu tắc mạch trên lâm sàng và chẩn đoán hình ảnh.
- Theo dõi xét nghiệm 3 tháng (12 tuần)/lần: Kháng thể LA, kháng thể kháng Cardiolipin, kháng thể kháng β2-Glycoprotein I.
- Theo dõi điều trị thuốc:
+ Sử dụng warfarin, theo dõi bằng chỉ số INR trong xét nghiệm PT;
+ Sử dụng heparin tiêu chuẩn, theo dõi bằng xét nghiệm APTT;
+ Sử dụng LMWH, theo dõi bằng xét nghiệm anti Xa.
- Theo dõi tác dụng phụ của thuốc trong quá trình điều trị.
TÀI LIỆU THAM KHẢO
1. Nguyễn Anh Trí (2011), “Hội chứng antiphospholipid”, Nhà xuất bản Y học.
2. Claudio Galarza Maldonado, Maria R Kourlovitch, Priscila Andrade - Sanchez et all, 2013, “Treating obstetric antiphospholipid syndrome”, Int. J. Clin. Rheumatol, 8 (3), 407-414.
3. David Garcia, Doruk Erkan, 2018, “Diagnosis and Management of the Antiphospholipid syndrom”, N Engl Journal of Med, 378; 2010 - 21.
4. David Kreeling, Ian mackie, Garry W. Moore, 2012, “Guideline on the investigation and management of antiphospholipid syndrome”, British Journal of Haematology, 157, 47 - 58.
5. Jacob H. Rand and Lucia Wolgast, 2016, “The Antiphospholipid Syndrome”, Williams Hematology, 9th edition, 2233 - 2252.
6. Jose A. Gomez - Puerta, Ricard Cervera, 2014, “Diagnosis and Classification of the antiphospholipid syndrome”, Journal of Autoimmunity 48-49, 20 - 25.
7. Miyakis S, Lockshin MD, Atsumi T, et al, 2006, “International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome”, J Thromb Haemost,4:p295-306.
12. HEMOPHILIA MẮC PHẢI
1. ĐẠI CƯƠNG
Hemophilia mắc phải là bệnh chảy máu bất thường do giảm yếu tố đông máu VIII/IX do tự kháng thể gây nên. Kháng thể kháng VIII thường có bản chất là IgG, gây chảy máu nặng nề trên lâm sàng. Mặc dù là một bệnh hiếm gặp nhưng việc điều trị hemophilia mắc phải gặp nhiều khó khăn do những chảy máu nguy hiểm đến tính mạng, khó kiểm soát và chi phí điều trị lớn.
2. CHẨN ĐOÁN
2.1 Chẩn đoán xác định
2.1.1 Triệu chứng lâm sàng
- Gặp ở cả nam và nữ, thường ở người lớn tuổi, có các bệnh ung thư, tự miễn kèm theo hoặc có tiền sử dùng thuốc, hoặc phụ nữ sau đẻ.
- Biểu hiện thường là xuất huyết dưới da, mô mềm, triệu chứng thường rầm rộ; có thể chảy máu vị trí khác như đái máu, xuất huyết tiêu hóa, chảy máu sau đẻ, sau phẫu thuật; ít gặp chảy máu khớp.
- Không có tiền sử chảy máu trước đó, gia đình không có người dễ chảy máu.
2.1.2 Xét nghiệm
a. Hemophilia A mắc phải
- APTT kéo dài.
- Kháng đông nội sinh dương tính (phụ thuộc thời gian và nhiệt độ).
- Yếu tố VIII < 40%.
- Có chất ức chế yếu tố VIII (nồng độ thường tính bằng đơn vị Bethesda - BU).
- PT, TT, fibrinogen, số lượng tiểu cầu bình thường.
b. Hemophilia B mắc phải
- APTT kéo dài.
- Kháng đông nội sinh dương tính (không phụ thuộc thời gian và nhiệt độ).
- Yếu tố IX < 40%.
- Có chất ức chế yếu tố IX (nồng độ thường tính bằng đơn vị Bethesda - BU).
- PT, TT, fibrinogen, số lượng tiểu cầu bình thường.
2.2. Chẩn đoán phân biệt
2.2.1 Với Hemophilia bẩm sinh
- Thường gặp ở nam giới, có tiền sử chảy máu bất thường tái phát nhiều lần, gia đình họ mẹ có người nam giới bị chảy máu bất thường.
- Hay chảy máu ở khớp, cơ; ít chảy máu ở mô mềm.
- Xét nghiệm: APTT kéo dài, yếu tố VIII/IX giảm, có thể có kháng đông nội sinh và chất ức chế VIII/IX nếu đã được điều trị.
2.2.2 Có chất chống đông lupus
- Lâm sàng: Thường ở người bệnh có bệnh tự miễn kèm theo như Lupus ban đỏ hệ thống; có biểu hiện tắc mạch (ở người trẻ), sảy thai liên tiếp; hoặc chảy máu bất thường.
- Xét nghiệm:
+ APTT kéo dài;
+ Kháng đông nội sinh không phụ thuộc thời gian và nhiệt độ: Dương tính;
+ Có các kháng thể kháng phospholipid dương tính như: LA, anti - cardiolipin, anti-β2 glycoprotein…
2.3. Chẩn đoán nguyên nhân
Làm các xét nghiệm chẩn đoán nguyên nhân gây bệnh như kháng thể kháng nhân, kháng thể kháng ds-DNA, marker ung thư, siêu âm, CT ổ bụng, lồng ngực…
3. ĐIỀU TRỊ
3.1. Điều trị khi có chảy máu cấp
3.1.1. Khi nồng độ chất ức chế ≤ 5BU
a. Đối với chảy máu nhẹ: Như xuất huyết dưới da hoặc tụ máu cơ nhưng không gây ảnh hưởng đến chức năng thì thường chỉ cần dùng:
- Tranexamic acid liều 25 mg/ kg cân nặng 3 - 4 lần/ ngày uống hoặc tiêm tĩnh mạch 10 mg/kg cân nặng 3-4 lần/ ngày.
- Desmopressin (nếu Hemophilia A mắc phải): 0,3mcg/kg - tiêm dưới da hoặc truyền tĩnh mạch. Có thể nhắc lại mỗi 8 - 12 giờ. Chú ý cân bằng nước điện giải và theo dõi nồng độ natri máu nếu dùng nhắc lại.
b. Đối với chảy máu trung bình và nặng: Nâng nồng độ yếu tố VIII/IX
- Hemophilia A mắc phải:
+ Bằng yếu tố VIII cô đặc: 20ui/kg với mỗi đơn vị Bethesda của chất ức chế + 40 ui/kg/ngày hoặc 100-200ui/kg/ngày, tiêm tĩnh mạch.
+ Desmopressin: 0,3mcg/kg, tiêm dưới da hoặc truyền tĩnh mạch.
- Hemophilia B mắc phải:
+ Yếu tố IX cô đặc 100-200ui/kg/ngày, tiêm tĩnh mạch 1-2 liều.
+ Theo dõi đáp ứng lâm sàng và nồng độ yếu tố VIII/IX. Nếu lâm sàng đáp ứng tốt, nồng độ yếu tố VIII/IX tăng thì liều dùng tiếp theo 30-50UI/kg 1 lần/ngày với Hemophilia B mắc phải và 2 lần/ngày với Hemophilia A mắc phải. Nếu lâm sàng không đáp ứng và nồng độ yếu tố VIII/IX không tăng thì chuyển sang dùng các chất bắc cầu.
3.1.2. Khi nồng độ chất ức chế > 5 BU hoặc khi không đáp ứng với yếu tố VIII/IX cô đặc
Có thể dùng các chất bắc cầu để điều trị chảy máu bao gồm phức hợp prothrombin hoạt hóa cô đặc (Activated Prothrombin Complex Concentrate - APCC) hoặc yếu tố VIIa (chỉ dùng 1 trong 2 loại chế phẩm).
+ APCC: 50-100 ui/kg/ngày nhắc lại mỗi 8-12 giờ đến khi ngừng chảy máu, tổng liều không quá 200ui/kg/ngày; hoặc
+ VIIa: 90 - 120 mcg/kg, nhắc lại mỗi 2-3 giờ đến khi ngừng chảy máu; hoặc dùng liều duy nhất 270 mcg/kg, nếu còn chảy máu thì nhắc lại sau 4 - 6 giờ liều 90 mcg/kg.
Kết hợp với Tranexamic acid liều 15-25 mg/kg x 2-3 lần/ngày, không dùng kết hợp với APCC do nguy cơ huyết khối.
3.2. Điều trị chất ức chế
Điều trị ức chế miễn dịch càng sớm càng tốt sau khi có chẩn đoán.
- Nếu yếu tố VIII ≥ 1% và ức chế VIII ≤ 20BU/ml:
+ Hàng 1: Methylprednisolon hoặc Prednisolon 1mg/kg/ngày dùng đơn độc 3-4 tuần.
+ Hàng 2: Nếu không đáp ứng: Kết hợp với cyclophosphamid 1,5 - 2mg/kg/ngày uống tối đa 6 tuần; hoặc Mycophenolate mofetil 1g/ngày trong tuần đầu tiên, sau đó 2g/ngày; hoặc Rituximab 375mg/m2 da/tuần tối đa 4 tuần. Có thể thay cyclophosphamide bằng azathioprin cho những phụ nữ trong tuổi sinh đẻ.
- Nếu yếu tố VIII < 1% và ức chế VIII > 20BU/ml:
+ Hàng 1: Corticoid kết hợp với cyclophosphamid hoặc rituximab trong 3-4 tuần;
+ Hàng 2: Trường hợp không đáp ứng, thêm cyclophosphamid hoặc rituximab nếu chưa dùng.
- Trao đổi huyết tương: Dùng kết hợp với điều trị yếu tố VIII liều cao
3.3. Điều trị các bệnh kèm theo
Vì hemophilia mắc phải hay gặp ở người lớn tuổi, có các bệnh kèm theo nên cần chú ý điều trị các bệnh kèm theo cũng như lưu ý các biến chứng do dùng thuốc như đái tháo đường, tăng huyết áp, nhiễm trùng…
3.4. Theo dõi người bệnh
- Vì tỉ lệ tái phát cao nên cần theo dõi xét nghiệm APTT và nồng độ yếu tố VIII/IX ít nhất mỗi tháng 1 lần trong vòng 6 tháng đầu; 2-3 tháng/ lần trong vòng 12 tháng và mỗi 6 tháng trong vòng 2 năm kể từ khi đáp ứng hoàn toàn với điều trị.
- Đối với các người bệnh có tiền sử hemophilia mắc phải cần kiểm tra kĩ đông cầm máu trước các thủ thuật có can thiệp hoặc phẫu thuật.
4. TIÊN LƯỢNG
Tiên lượng nặng, tỉ lệ tử vong 6-22%. Người bệnh nên được điều trị tại cơ sở chuyên khoa.
5. XÉT NGHIỆM THEO DÕI
- Tổng phân tích tế bào máu ngoại vi: Đánh giá tình trạng thiếu máu, mất máu do chảy máu; giảm bạch cầu và tiểu cầu khi điều trị ức chế miễn dịch. Cần làm khi bệnh nhân vào viện, kiểm tra định kỳ hàng tuần hoặc 2 lần/ tuần nếu điều trị ức chế miễn dịch hoặc làm bất cứ khi nào cần để theo dõi tình trạng mất máu cấp tùy thuộc vào bệnh cảnh lâm sàng.
- Sinh hóa: Chức năng gan thận, protein, albumin, glucose, điện giải đồ, canxi, canxi ion hóa khi vào viện và hàng tuần khi điều trị ức chế miễn dịch và corticoid.
- Đông máu: APTT, định lượng VIII, định lượng ức chế VIII hàng tuần để đánh giá hiệu quả điều trị ức chế miễn dịch, hoặc bất cứ khi nào nếu cần kiểm tra để đánh giá hiệu quả cầm máu.
- Vi sinh: Cấy vết thương, cấy máu tìm vi khuẩn, nấm nếu có tình trạng nhiễm trùng, vết thương hở…
TÀI LIỆU THAM KHẢO
1. Massimo Franchini and Giuseppe Lippi, “Acquired factor VIII inhibitors”, Blood, 2008, 112, 220-225
2. Paul Giangrande, “Acquired Hemophilia”, 11/2012, No 38, World Federation of Hemophilia
3. Lilian Tengborn, Jan Astermark, Jørgen Ingerslev, Anne Mäkipernaa , Geir E. Tjønnfjord , Páll T. Önundarson, 2009, Acquired Hemophilia - Nordisk guideline for diagnosis and treatment.
4. Peter W Collins, Elizabeth Chalmers, Daniel Hart, Ian Jennings, Ri Liesner, Savita Rangarajan, Kate Talks, Michael Williams and Charles R. M. Hay, 2013, Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO, British Journal of Haematology, 2013, 162, 758-773.
5. Nemes, Fabio Pellegrini, Lilian Tengborn and Paul Knoebl, Francesco Baudo, Peter Collins, Angela Huth-Kühne, Hervé Lévesque, Pascual Marco, László, 2012, Management of bleeding in acquired Hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry, Blood, 5 July 2012, vol 120, number 1, 39-46.
6. Knöbl, Paul. “Prevention and Management of Bleeding Episodes in Patients with Acquired Hemophilia A.” Drugs vol. 78,18 (2018): 1861-1872. doi:10.1007/s40265-018-1027-y
7. Andreas Tiede, Peter Collins, Paul Knoebl, Jerome Teitel, Craig Kessler, Midori Shima, Giovanni Di Minno, Roseline d’Oiron, Peter Salaj, Victor Jiménez-Yuste, Angela Huth- Kühne, Paul Giangrande. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica 2020;105(7):1791-1801; https://doi.org/10.3324/haematol.2019.230771.
13. BỆNH VON WILLEBRAND (Von-Willebrand Disease - VWD)
1. ĐẠI CƯƠNG
Yếu tố von Willebrand (von Willebrand factor-vWF) là glycoprotein kết dính có trên bề mặt tiểu cầu, tế bào nội mạc và trong huyết tương với 3 chức năng chính:
- Làm trung gian cho tiểu cầu dính vào vị trí nội mạc bị tổn thương qua việc gắn với GpIb/IX của tiểu cầu vào collagen của lớp dưới nội mạc.
- Kích thích tiểu cầu ngưng tập qua việc gắn receptor GpIIb/IIIa.
- Gắn và bảo vệ yếu tố VIII khỏi sự phá hủy của protein C trong tuần hoàn.
Bệnh von Willebrand (von Willebrand disease - vWD) là bệnh do bất thường về số lượng họăc chất lượng vWF dẫn tới suy yếu tương tác giữa tiểu cầu với thành mạch, gây ra khiếm khuyết trong giai đoạn cầm máu ban đầu. Bệnh di truyền nhiễm sắc thể thường, gặp ở cả nam và nữ và đặc trưng bởi biểu hiện chảy máu bất thường ở da và niêm mạc.
2. CHẨN ĐOÁN
2.1. Triệu chứng lâm sàng
- Triệu chứng nổi bật là chảy máu niêm mạc: Chảy máu mũi, chảy máu răng miệng, xuất huyết tiêu hóa, rong kinh; xuất huyết dưới da hoặc chảy máu sau can thiệp, chấn thương, phẫu thuật; tuy nhiên cũng có khi bị chảy máu cơ, khớp. Triệu chứng có thể biểu hiện từ nhẹ đến nặng tùy từng thể. Nên sử dụng công cụ sàng lọc tiêu chuẩn tìnhh trạng chảy máu (như BAT của ISTH 2010 (xem phụ lục I)) để cân nhắc có cần làm xét nghiệm hay không Đối với nam giới, trẻ em hoặc người liên quan trực hệ với bệnh nhân von Willebrand, ngay cả khi sàng lọc BAT không rõ triệu chứng chảy máu, nếu vẫn nghi ngờ bị bệnh vẫn cần làm xét nghiệm kiểm tra.
- Có thể có thiếu máu do chảy máu nhiều và kéo dài.
- Tiền sử gia đình có người biểu hiện chảy máu bất thường, triệu chứng chảy máu của các thành viên trong gia đình có thể không giống nhau.
2.2. Xét nghiệm
Để chẩn đoán bệnh von Willebrand cần một phức hợp xét nghiệm, bên cạnh đó do nồng độ yếu tố von Willebrand và yếu tố VIII phụ thuộc nhiều vào các hoạt động thể lực, stress, vòng kinh nên các xét nghiệm cần được làm ít nhất 2 lần ở 2 thời điểm khác nhau.
- Thời gian máu chảy: kéo dài;
- Co cục máu đông: không co hoặc co không hoàn toàn;
- Số lượng tiểu cầu: bình thường, có thể giảm (trong typ 2B); ngưng tập tiểu cầu với ristocetin: giảm; thời gian có nút tiểu cầu (closure time - PFA): kéo dài;
- APTT: kéo dài;
- PT, TT, fibrinogen: bình thường
- Yếu tố VIII: giảm hoặc bình thường;
- Giảm yếu tố von Willebrand kháng nguyên (vWF:Ag) và yếu tố von Willebrand hoạt tính (vWF:Act); giảm hoạt tính vWF phụ thuộc tiểu cầu (vWF:GPIbM, vWF: GPIbR), giảm khả năng gắn của yếu tố von Willebrand với collagen (vWF:CBA); định lượng multimer vWF: bình thường hoặc bất thường.
3. XẾP LOẠI vWD
Có nhiều đột biến trên gen có tác động đến cấu trúc và chức năng của yếu tố von Willerbrand. Căn cứ vào sự bất thường về số lượng và chất lượng yếu tố von Willebrand, năm 2021, Hội Huyết học Mỹ (Asscociation of Hematology - ASH), Hội Đông máu và Tắc mạch Quốc tế (International Society of Thrombosis and Hemostasis - ISTH), Quỹ Hemophilia Mỹ (National Hemophilia Foundation - NHF) và Liên đoàn Hemophilia Thế giới (World Federation of Hemophilia - WFH) nhóm họp và thống nhất xếp loại bệnh von Willerbrand như sau (bảng 1):
Bảng 1. Xếp loại bệnh von Willebrand theo ASH ISTH NHF WFH 2021
|
Type |
Đặc điểm |
|
1 |
Giảm số lượng VWF với tỷ lệ được bảo toàn giữa VWF:Ag, VWF:Act và FVIII; phân phối multimer bình thường. |
|
1C |
Giảm số lượng VWF với tỷ lệ được bảo toàn giữa VWF:Ag, VWF:Act và FVIII; tăng VWF polypeptid so với VWF:Ag |
|
2A |
Giảm hoạt tính vWF phụ thuộc tiểu cầu với sự thiếu hụt đặc hiệu các multimer trọng lượng phân tử cao |
|
2B |
Tăng liên kết với GPIbα, thường dẫn đến giảm tiểu cầu |
|
2M |
Giảm hoạt tính vWF phụ thuộc tiểu cầu nhưng không thiếu hụt các multimer trọng lượng phân tử cao |
|
2N |
Giảm gắn kết của yếu tố VIII với VWF |
|
3 |
Thiếu hụt hoàn toàn hoặc gần hoàn toàn VWF |
|
Von Willebrand typ tiểu cầu |
Khiếm khuyết chức năng của GPIbα tiểu cầu, dẫn đến liên kết quá mức giữa tiểu cầu và VWF gây ra giảm tiểu cầu và mất multimer trọng lượng phân tử cao |
|
Hội chứng von Willebrand mắc phải |
Giảm VWF và đặc biệt mất các multimer trọng lượng phân tử cao do tác động của lực cắt cơ học (ví dụ, hẹp eo động mạch chủ dẫn đến hội chứng Heyde), hấp phụ trên các khối u (ví dụ, bệnh Waldenstrom hoặc khối u Wilms), hoặc sự hình thành chất ức chế tự miễn dịch |
Chú thích:
VWF: yếu tố von Willebrand
vWF:Ag: kháng nguyên yếu tố von Willebrand
vWF:Act: hoạt tính yếu tố von Willebrand, còn gọi là đồng yếu tố ristocetin
(vWF:Rco)
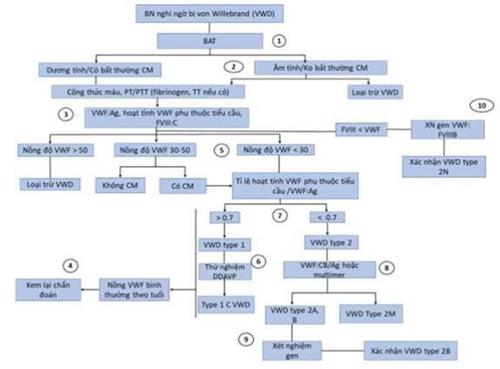
Sơ đồ 1: Sơ đồ chẩn đoán VWD theo ASH ISTH NHF WFH 2021
Chú thích:
CM: Chảy máu
vWD: bệnh von Willebrand
vWF: yếu tố von Willebrand
BAT: Bleeding asessment tool - công cụ đánh giá chảy máu của ISTH 2010
FVIIIB: Khả năng gắn yếu tố VIII
VWF:CB: Khả năng gắn collagen
FVIII:C: Hoạt tính yếu tố VIII
DDAVP: desmopressin
4. CHẨN ĐOÁN PHÂN BIỆT
4.1. Với bệnh Hemophilia A: Chảy máu tái phát nhiều lần và có yếu tố VIII giảm:
- Chủ yếu gặp ở nam giới;
- Hay chảy máu ở cơ, khớp;
- Chỉ có yếu tố VIII giảm, vWF bình thường, các xét nghiệm thăm dò giai đoạn cầm máu kì đầu như thời gian máu chảy, co cục máu đông, ngưng tập tiểu cầu với ristocetin, thời gian có nút tiểu cầu (PFA): Bình thường.
4.2. Với các trường hợp bệnh von Willebrand mắc phải
Giảm số lượng hoặc chức năng yếu tố von Willebrand thứ phát do bệnh tự miễn, ung thư, tăng sinh lympho hoặc đa u tủy xương, bệnh lí tim mạch…
- APTT kéo dài, vWF và VIII giảm, ngưng tập tiểu cầu với ristocetin giảm.
- Không có tiền sử cá nhân và gia đình về chảy máu lâu cầm.
5. ĐIỀU TRỊ
Người bệnh cần được đăng kí và quản lý tại các bệnh viện đa khoa, các cơ sở huyết học hoặc trung tâm điều trị các rối loạn chảy máu để được cấp thẻ người bệnh có các thông tin liên quan đến chẩn đoán, được phối hợp chăm sóc bởi các chuyên khoa có liên quan như: Huyết học, Răng hàm mặt, Sản phụ khoa, Ngoại khoa…
5.1. Nâng nồng độ vWF
a. DDAVP (Desmopressin) có tác dụng giải phóng vWF nội sinh nhưng chỉ có tác dụng trong vòng 2- 3 liều, sử dụng tốt cho việc điều trị chảy máu và sau can thiệp nhỏ như nhổ răng, phẫu thuật nhỏ cho các người bệnh type 1, 2A, 2M và 2N. Không dùng DDAVP cho người bệnh type 2B do có thể làm trầm trọng thêm việc giảm tiểu cầu; không dùng cho người bệnh type 3 do ít tác dụng.
- Liều: 0,3mcg/kg tiêm dưới da hoặc pha trong 100 ml nước muối sinh lí truyền trong vòng 15 phút (tác dụng đạt đỉnh sau 1 giờ); hoặc dùng dạng xịt mũi liều 300 mcg/xịt. Có thể nhắc lại sau 12-24 giờ.
- Tác dụng phụ: Hạ Natri máu. Theo dõi điện giải đồ hàng ngày, hạn chế nước đưa vào.
Lưu ý: Đáp ứng với DDAVP của mỗi người bệnh khác nhau, vì vậy cần làm test thử bằng cách định lượng nồng độ VIII và VWF trước và sau khi truyền, nếu tăng lên thì tiếp tục dùng và cần định lượng yếu tố hàng ngày. Nếu không tăng hoặc hết tăng thì không dùng nữa.
b. Tủa lạnh: Giàu vWF và yếu tố VIII. Liều lượng: 1-2 đơn vị tủa lạnh/ngày, 1 lần/ngày.
Nhược điểm: Không dự đoán được nồng độ vWF nâng lên và có thể có nguy cơ lây nhiễm các bệnh truyền qua đường máu.
c. Yếu tố VIII cô đặc có vWF: Có một số yếu tố VIII cô đặc có nguồn gốc huyết tương có chứa vWF.
- Chỉ định khi có chảy máu trung bình, nặng hoặc cần phẫu thuật.
- Tương tự như yếu tố VIII, 1 đơn vị vWF:Act/kg cân nặng có thể nâng nồng độ vWF:Act bệnh nhân lên được 2%.
- Liều 15 đơn vị (ui) FVIII/kg/ngày cho chảy máu trung bình và 30 đơn vị FVIII/kg cho chảy máu nặng, nhắc lại mỗi 24 giờ.
- Đối với những trường hợp phẫu thuật lớn: Cần duy trì cả nồng độ yếu tố VIII và vWF: Act ≥ 50% ít nhất 3 ngày sau mổ hoặc kéo dài lâu hơn tuỳ vào tình trạng lâm sàng. Nồng độ cần đạt, thời gian duy trì tuỳ thuộc vào từng cá nhân dựa vào loại phẫu thuật, tiền sử chảy máu, lượng yếu tố VIII và vWF có sẵn. Những trường hợp bệnh nhân có nguy cơ huyết khối cao thì tránh nâng nồng độ lên quá cao (> 150%) kết hợp với tranexamic acid.
d. VWF cô đặc: Nguồn gốc huyết tương hoặc tái tổ hợp
Liều thông thường 40-80 ui vWF:Act/kg, có thể nhắc lại 40-50 ui/kg sau 8-24h tuỳ thuộc vào tình trạng chảy máu trên lâm sàng.
Lưu ý:
- Yếu tố VIII tái tổ hợp không chứa vWF, vì vậy không dùng được cho người bệnh von Willebrand.
- Trong một số trường hợp nồng độ VIII cơ sở của người bệnh thấp thì cần cân nhắc bổ sung thêm yếu tố VIII sau khi dùng vWF tinh khiết.
- Cần theo dõi nồng độ yếu tố VIII và vWF hàng ngày để điều chỉnh liều.
5.2. Điều trị hỗ trợ
- Tranexamic acid:
+ Chỉ định: Chảy máu niêm mạc như chảy máu răng miệng, rong kinh...;
+ Chống chỉ định: Đái máu;
+ Liều lượng: 15-25 mg/kg mỗi 6-8 giờ, uống hoặc tiêm tĩnh mạch chậm;
+ Dung dịch tranexamic acid 5% dùng để ngậm, súc họng trong trường hợp chảy máu lợi hoặc sau nhổ răng.
- Kiểm soát rong kinh bằng thuốc tránh thai đường uống chứa estrogen hoặc progesterone.
5.3. Điều trị biến chứng
- Thiếu máu: Bổ sung sắt, truyền khối hồng cầu trường hợp thiếu do mất máu.
- Điều trị các bệnh lây qua đường truyền máu.
- Điều trị các biến chứng khác nếu có.
5.4. Quản lý và theo dõi bệnh nhân von Willebrand
Giống như bệnh nhân mắc các rối loạn chảy máu di truyền khác, người bệnh vWD cần được quản lê, tư vấn, điều trị tốt để có thể sống chung với bệnh. Việc quản lý bao gồm:
- Lập hồ sơ quản lý.
- Cấp thẻ người bệnh bao gồm các thông tin về chẩn đoán, mức độ bệnh, tình trạng có chất ức chế yếu tố đông máu.
- Tư vấn về bệnh và tư vấn di truyền.
- Điều trị chảy máu và hạn chế biến chứng chảy máu: Sử dụng chế phẩm máu và các thuốc, biện pháp hỗ trợ. Đối với vWD typs 3 có biểu hiện chảy máu nhiều cần bổ sung định kỳ yếu tố đông máu để hạn chế biến chứng do chảy máu gây ra, liều thông thường 40-60 ui/kg cân nặng x 2 lần/tuần.
- Phối hợp với các chuyên ngành có liên quan trong chăm sóc toàn diện: Răng hàm mặt, ngoại khoa, sản khoa…
- Kiểm tra định kỳ mỗi 1-3 tháng và điều trị nếu có các bệnh nhiễm qua đường truyền máu và các kháng thể kháng yếu tố đông máu.
5.5. Xét nghiệm theo dõi điều trị
- Tổng phân tích tế bào máu ngoại vi: Đánh giá mức độ thiếu máu, làm khi vào viện và định kì hàng tháng, hoặc khi lâm sàng nghi ngờ mất máu, thiếu máu, theo dõi điều trị thiếu máu.
- Đông máu cơ bản (Fibrinogen, PT, APTT, TT), định lượng yếu tố VIII, vWF:Ag, vWF:Act, vWF phụ thuộc tiểu cầu… hàng ngày để đánh giá hiệu quả của điều trị và điều chỉnh liều yếu tố đông máu hoặc khi nghi ngờ có kháng thể chống yếu tố đông máu.
- Sinh hóa: Chức năng gan, thận, sắt, ferritin, protein, albumin và các chỉ số khác tuỳ thuộc vào tình trạng của người bệnh
- Vi sinh: Kiểm tra HBV, HCV, HIV theo quy định để theo dõi nguy cơ lây nhiễm các bệnh qua đường truyền máu; cấy máu, cấy vết thương tìm vi khuẩn, nấm khi nghi ngờ có nhiễm trùng máu, nhiễm trùng vết thương…
- Chẩn đoán hình ảnh (siêu âm, X-quang): Đánh giá tình trạng chảy máu, tắc mạch và các bệnh lý kèm theo.
6. BỆNH VON WILLEBRAND MẮC PHẢI
Bệnh Von Willebrand mắc phải là bệnh giảm số lượng hoặc chất lượng yếu tố von Willebrand thứ phát do bệnh tự miễn, tăng sinh lympho, tăng sinh tủy, ung thư, tim mạch hoặc các bệnh khác. Đây là bệnh rất hiếm gặp; yếu tố von Willebrand bị bất thường do 1 trong những nguyên nhân sau:
- Kháng thể kháng yếu tố von Willebrand.
- Hấp thụ trên bề mặt tiểu cầu hoặc tế bào biến hình (transformed cell).
- Tăng phân giải protein.
6.1. Chẩn đoán
Để chẩn đoán bệnh von Willebrand mắc phải không có một xét nghiệm riêng biệt nào đủ để loại trừ hoặc khẳng định bệnh.
- Các xét nghiệm thể hiện như bệnh von Willebrand bẩm sinh.
- Không có tiền sử chảy máu bất thường trước đó cũng như tiền sử gia đình có người chảy máu bất thường.
- Có các bệnh lí kèm theo như ung thư, tự miễn, tăng sinh lympho, tăng sinh tủy…
- Có tăng sinh bất thường gamma globulin.
- Có kháng thể kháng yếu tố von Willebrand.
6.2. Điều trị
- Điều trị chảy máu giống như trong bệnh von Willebrand.
- IgIV trong các trường hợp bệnh von Willebrand mắc phải trên nền bệnh MGUS hoặc tăng IgG.
- Gạn huyết tương nếu tăng sinh gamma globulin.
- Kết hợp điều trị các bệnh lí kèm theo.
TÀI LIỆU THAM KHẢO
1. Andreas Tiede, Jacob H. Rand,Ulrich Budde,Arnold Ganser, andAugusto B. Federici, 2011, How I treat the acquired von Willebrand syndrome, Blood, vol. 117 no. 25 6777-6785.
2. David Lillicrap, Paula James, (2009), von Willebrand disease: An introduction for the primary care physician, World Federation of Hemophilia. Treatment of Hemophilia, N0 47.
3. Edith Fressinaud and Dominique Meyer, “von Willebrand diseae: biological diagnosis”, Textbook of Hemophilia, Blackwell Publishing 2005, 272-278.
4. Francesca Stufano, Luciano Baronciani, Flora Peyvandi, 2017, Diagnosis of von Willebrand disease phenotypic characterization, World Federation of Hemophilia, Treatment of Hemophilia, No55.
5. Jill Johnsen and David Ginsburg, (2016), von Willebrand disease, Williams Hematology, 9th edition, Mc Graw-Hill Education, 2163- 2182.
6. J . E . Sadler, U. Budde, J . C. J . Eikenboom, E . J. Favaloro, F. G. H. Hill, L. Holmberg, J. Ingerslev, C. A. Lee, D. Lillicrap, P. M. Mannucci, C. Mazurier, D. Meyer, W. L. Nichols, M. Nishino, I. R. Peake, F . Rodeghiero, R. Schneppenheim, Z. M. RuggeriI, A. Srivastava, R. R. Montgomery and A. B . Federici, The Working party on Von Willebrand disease classification, (2006), Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor, Journal of Thrombosis and Haemostasis, Volume 4, Issue 10, 2103-2114.
7. Laffan MA, Lester W, O'Donnell JS, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014;167(4):453-465. doi:10.1111/bjh.13064
8. Paula D. James, Nathan T. Connell, Barbara Ameer, Jorge Di Paola, Jeroen Eikenboom, Nicolas Giraud, Sandra Haberichter, Vicki Jacobs-Pratt, Barbara Konkle, Claire McLintock, Simon McRae, Robert R. Montgomery, James S. O’Donnell, Nikole Scappe, Robert Sidonio, Veronica H. Flood, Nedaa Husainat, Mohamad A. Kalot, Reem A. Mustafa; ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv 2021; 5 (1): 280-300. doi: https://doi.org/10.1182/bloodadvances.2020003265
9. Nathan T. Connell, Veronica H. Flood, Romina Brignardello-Petersen, Rezan Abdul- Kadir, Alice Arapshian, Susie Couper, Jean M. Grow, Peter Kouides, Michael Laffan, Michelle Lavin, Frank W. G. Leebeek, Sarah H. O’Brien, Margareth C. Ozelo, Alberto Tosetto, Angela C. Weyand, Paula D. James, Mohamad A. Kalot, Nedaa Husainat, Reem A. Mustafa; ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv 2021; 5 (1): 301-325. doi: https://doi.org/10.1182/bloodadvances.2020003264
10. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, Neunert C, Lillicrap D; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010 Sep;8(9):2063-5. doi: 10.1111/j.1538-7836.2010.03975.x. PMID: 20626619.
11. Elbatarny M, Mollah S, Grabell J, Bae S, Deforest M, Tuttle A, Hopman W, Clark DS, Mauer AC, Bowman M, Riddel J, Christopherson PA, Montgomery RR; Zimmerman Program Investigators, Rand ML, Coller B, James PD. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014 Nov;20(6):831-5. doi: 10.1111/hae.12503. Epub 2014 Sep 6. PMID: 25196510; PMCID: PMC4251588.
Phụ lục:
THANG ĐIỂM ĐANH GIÁ TÌNH TRẠNG CHẢY MÁU (Bleeding Asessment Tool -BAT)THEO ISTH 2010
|
Đặc điểm |
0 |
1 |
2 |
3 |
4 |
|
Chảy máu cam |
không có hoặc không đáng kể |
Kéo dài trên 10’ hoặc 5 lần/năm |
Chỉ tư vấn * |
Dùng thuốc chống tiêu sợ huyết hoặc các biện pháp cầm máu cơ học |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
|
Xuất huyết dưới da |
không có hoặc không đáng kể |
>5 vết bầm hoặc bầm tim > 1cm |
Chỉ tư vấn * |
Lớn |
có xuất huyết tự phát cần truyền máu |
|
Chảy máu từ vết thương nhỏ |
không có hoặc không đáng kể |
Kéo dài trên 10’ hoặc 5 lần/năm |
Chỉ tư vấn* |
Phẫu thuật để cầm máu |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
|
Tiểu máu |
không có hoặc không đáng kể |
Tiếu máu đại thể ở hiện tại |
Chỉ tư vấn* |
Phẫu thuật để cầm máu hoặc bổ xung sắt. |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
|
Xuất huyết tiêu hóa |
không có |
Có nhưng không phải điều trị gì (không liên quan bệnh lý dạ dày, tăng huyết áp, dị dạng mạch, trĩ.) |
Chỉ tư vấn* |
Phẫu thuật để cầm máu hoặc dùng thuốc chống tiêu sợi huyết |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
|
Chảy máu chân răng, khoang miệng |
không có hoặc không đáng kể |
Hiện tại |
Chỉ tư vấn* |
Phẫu thuật để cầm máu hoặc dùng thuốc chống tiêu sợi huyết |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
|
Chảy máu sau can thiệp nha khoa |
không có hoặc không đáng kể |
nhỏ hơn 25% số lần nhổ răng hoặc thủ thuật có chảy máu |
lớn hơn 25% số lần nhổ răng hoặc thủ thuật có chảy máu. |
Phải khâu phục hồi hoặc nhét bông cầm máu. |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
|
Chảy máu sau phẫu thuật hoặc can thiệp |
không có hoặc không đáng kể |
nhỏ hơn 25% số lần phẫu thuật có chảy máu |
lớn hơn 25% số lần phẫu thuật có chảy máu |
Phẫu thuật để cầm máu hoặc dùng thuốc chống tiêu sợi huyết |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
|
Rong kinh |
không có hoặc không đáng kể |
Chỉ tư vấn* hoặc thay băng vệ sinh thường xuyên hơn 2h/lần hoặc có máu cục. |
Phải nghỉ học/làm > 2 lần/năm hoặc phải dùng thuốc chống tiêu fibrin hoặc dùng thuôc nội tiết, bổ sung sắt. |
Phải dùng thuốc chồng tiêu fibrin kết hợp thuốc nội tiết hoặc kinh huyệt kéo dài diễn ra >12 tháng. |
Rong kinh cấp tính cần nhập viện và điều trị khẩn cấp hoặc - Yêu cầu truyền máu, Điều trị thay thế, Desmopressin, hoặc - Yêu cầu nong & cắt bỏ hoặc cắt bỏ nội mạc tử cung hoặc cắt tử cung) |
|
Xuất huyết sau sinh |
không có hoặc không đáng kể |
Chỉ tư vấn* hoặc đã dùng ocytocin hoặc gây lo lắng > 6 tuần |
Bổ xung sắt hoặc dùng thuốc chống tiêu fibrin |
Yêu cầu truyền máu, Điều trị thay thế, Desmopressin hoặc yêu cầu kiểm tra dưới gây mê/ sử dụng bóng/ chèn tử cung. |
bất kì tình trang yêu cầu phẫu thuật, can thiệp thủ thuật xâm lấn nào |
|
Tụ máu cơ tự phát |
không có |
sau chấn thương và không cần điều trị |
tự phát và không cần điều trị |
tự phát hoặc sau chấn thương yêu cầu điều trị thay thế hoặc dùng desmopressin |
Tự phát hoặc sau chấn thương cần can thiệp phẫu thuật hoặc truyền máu |
|
Xuất huyết khác |
không có |
sau chấn thương và không cần điều trị |
tự phát và không cần điều trị |
tự phát hoặc sau chấn thương yêu cầu điều trị thay thế hoặc dùng desmopressin |
Tự phát hoặc sau chấn thương cần can thiệp phẫu thuật hoặc truyền máu |
|
Chảy máu thần kinh TW tự phát |
không có |
- |
- |
chấn thương dưới màng cứng cần can thiệp |
chấn thương nội sọ cần can thiệp |
|
Chảy máu khác # |
không có |
Có nhưng tự khỏi |
Chỉ tư vấn * |
Phẫu thuật cầm máu hoặc dùng thuốc chống tiêu fibrin. |
Truyền máu hoặc điều trị thay thế (sử dụng các thành phần máu cầm máu và rFVIIa) hoặc desmopressin |
# Chảy máu khác gồm: chảy máu cuống rốn, tụ máu giữa da đầu và hộp sọ ở trẻ sơ sinh, tụ máu ở má do bú trong khi bú mẹ /bú bình, xuất huyết kết mạc hoặc chảy máu quá nhiều sau cắt bao quy đầu hoặc lấy máu tĩnh mạch. Sự hiện diện của các triệu chứng trên khi còn nhỏ đòi hỏi phải điều tra chi tiết độc lập với tổng điểm.
* Chỉ tư vấn: bệnh nhân được đánh giá y tế và được giới thiệu đến bác sĩ chuyên khoa hoặc được đề nghị kiểm tra xét nghiệm chi tiết hơn.
Các tiêu chuẩn xác định các yếu tố trên
- Chảy máu mũi có ý nghĩa: Bất kì chảy máu mũi nào, đặc biệt xuất hiện sau tuổi dậy thì gây ra sự quan tâm của bệnh nhân được coi là có ý nghĩa, quan trọng. Không nên coi là có ý nghĩa khi nó kéo dài dưới 10 phút, tần xuất nhỏ hơn 5 lần/ năm, có liên quan đến nhiễm trùng hô hấp trên hoặc các nguyên nhân xác định khác.
- Xuất huyết dưới da có ý nghĩa : khi xuất hiện trên 5 lần/ năm, kích thước lớn hơn 1 cm khu vực tiếp xúc, được mô tả chi tiết bởi bệnh nhân hoặc người nhà bệnh nhân, hoặc xuất huyết xảy ra tự nhiên.
- Chảy máu vết thương nhỏ được coi là đáng kể khi phải yêu cầu thay băng thường xuyên; Chảy máu không đáng kể từ các vết thương bao gồm những vết thương có thời gian chảy máu <10 phút và các tổn thương thường yêu cầu khâu ở các đối tượng bình thường (ví dụ: dưới cằm). Triệu chứng biểu hiện trên nhiều lần cũng nên được coi là quan trọng.
- Chảy máu khoang miệng: Chảy máu chân răng nên được coi là đáng kể khi nó gây ra chảy máu kéo dài trên 10 phút. Chảy máu chân răng hoặc chảy máu sau gẫy răng nên được coi là đáng kể khi nó cần có sự hỗ trợ hoặc giám sát của bác sĩ, hoặc kéo dài ít nhất 10 phút (chảy máu liên quan đến nhổ răng được xem xét riêng). Chảy máu xảy ra sau khi cắn môi, má và lưỡi nên được coi là đáng kể khi nó kéo dài ít nhất 10 phút hoặc gây ra sưng lưỡi hoặc miệng.
- Xuất huyết tiêu hóa: Bất kỳ xuất huyết tiêu hóa nào không giải thích được bởi sự hiện diện của một bệnh cụ thể nên được coi là đáng kể.
- Tiểu máu: Chỉ có tiểu máu đại thể (từ nước tiểu màu đỏ đến màu hồng nhạt) không được giải thích bởi sự hiện diện của một bệnh tiết niệu cụ thể nên được coi là đáng kể.
- Nhổ răng: Bất kỳ chảy máu mới nào xảy ra sau khi nhổ răng yêu cầu chăm sóc y tế hoặc chảy máu kéo dài khi nhổ răng nên được coi là đáng kể.
- Chảy máu sau phẫu thuật: Bất kỳ chảy máu nào được đánh giá bởi bác sĩ phẫu thuật là kéo dài bất thường, đó là gây ra sự chậm trễ trong xuất viện, hoặc yêu cầu một số điều trị hỗ trợ được coi là đáng kể.
- Rong kinh: Bất kỳ chảy máu nào cản trở các hoạt động hàng ngày như làm việc, việc nhà, tập thể dục hoặc các hoạt động xã hội trong hầu hết các kỳ kinh nguyệt nên được coi là đáng kể. Tiêu chí cho chảy máu đáng kể có thể bao gồm bất kỳ điều nào sau đây: thay băng vệ sinh nhiều hơn thường xuyên hơn 2 giờ/ lần; chảy máu kinh nguyệt kéo dài 7 ngày trở lên; và sự hiện diện của các cục máu đông > 1 cm kết hợp với cường kinh.
- Chảy máu sau sinh. Chảy máu âm đạo hoặc chảy máu tử cung (lochia) kéo dài hơn 6 tuần. Bất kỳ chảy máu trong thời gian ít hơn được đánh giá bởi bác sĩ sản khoa là nặng bất thường hoặc kéo dài, gây ra sự chậm trễ trong việc xuất viện, đòi hỏi một số hỗ trợ điều trị, đòi hỏi phải thay băng vệ sinh thường xuyên hơn mỗi 2 giờ hoặc nguyên nhân thiếu máu tiến triển cũng được coi là đáng kể.
- Tụ máu cơ, khớp tự phát: Bất kỳ chảy máu cơ/khớp tự phát (không liên quan đến chấn thương) được coi là đáng kể.
- Chảy máu thần kinh trung ương. Bât kì xuất huyết thần kinh trung ương nào có triệu chứng lâm sàng được coi là đáng kể.
- Các triệu chứng chảy máu khác: Khi những triệu chứng chảy máu này xảy ra trong giai đoạn sơ sinh được tính 1 điểm. Các triệu chứng này khi được báo cáo bởi bệnh nhân hoặc người nhà nên luôn luôn nhắc nhở cần có xét nghiệm kiểm tra chi tiết.
Chỉ số bình thường:
- Nam giới trưởng thành: 0-3, từ 4 trở lên là bất thường
- Nữ giới trưởng thành: 0-5, từ 5 trở lên là bất thường
- Trẻ em cả nam và nữ: 0-2, từ 3 trở lên là bất thường
TÀI LIỆU THAM KHẢO
1. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, Neunert C, Lillicrap D; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010 Sep;8(9):2063-5. doi: 10.1111/j.1538- 7836.2010.03975.x. PMID: 20626619.
2. Elbatarny M, Mollah S, Grabell J, Bae S, Deforest M, Tuttle A, Hopman W, Clark DS, Mauer AC, Bowman M, Riddel J, Christopherson PA, Montgomery RR; Zimmerman Program Investigators, Rand ML, Coller B, James PD. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014 Nov;20(6):831-5. doi: 10.1111/hae.12503. Epub 2014 Sep 6. PMID: 25196510; PMCID: PMC4251588.
14. CÁC RỐI LOẠN CHẢY MÁU BẨM SINH HIẾM GẶP
Các rối loạn chảy máu hiếm gặp chiếm khoảng 3-5% bệnh nhân rối loạn đông máu, bao gồm thiếu các yếu tố đông máu di truyền không phải hemophilia như thiếu yếu tố II, yếu tố V, thiếu yếu tố V và VIII kết hợp, thiếu yếu tố VII, X, XI, fibrinogen. Biểu hiện chảy máu của các bệnh lý này thường đa dạng, đa số liên quan đến việc giảm hoạt tính của các yếu tố đông máu.
1. BẤT THƯỜNG FIBRINOGEN BẨM SINH
Fibrinogen là một glycoprotein có trọng lượng phân tử lớn, có nhiều chức năng: tạo cục máu đông, ngưng tập tiểu cầu, gắn thrombin... Fibrinogen được tổng hợp ở gan, do gen nằm trên nhiễm sắc thể số 4 quy định. Bất thường fibrinogen có thể là:
- Không có fibrinogen trong máu (afibrinogenemia).
- Giảm nồng độ fibrinogen với cấu trúc bình thường (hypofibrinogenemia).
- Bất thường cấu trúc fibrinogen (dysfibrinogemia).
Trên thực tế rất khó phân biệt giữa giảm và bất thường fibrinogen.
1.1. Chẩn đoán
a. Biểu hiện lâm sàng
Triệu chứng nổi bật là chảy máu, xuất hiện tự nhiên hoặc sau chấn thương, biểu hiện từ nặng đến nhẹ tùy thuộc vào mức độ thiếu hụt:
- Triệu chứng sớm nhất là chảy máu rốn ở trẻ sơ sinh.
- Hay gặp xuất huyết dưới da và niêm mạc; chảy máu cơ, khớp ít gặp hơn.
- Chảy máu kéo dài sau đứt tay, sau can thiệp, phẫu thuật.
- Xuất huyết nội tạng: Chảy máu não, xuất huyết tiêu hóa, trong ổ bụng…
- Ở phụ nữ: Kinh nguyệt số lượng nhiều, rong kinh, chảy máu sau đẻ, sảy thai 3 tháng đầu, chảy máu sau mãn kinh. Phụ nữ bị bất thường fibrinogen có nguy cơ sảy thai cao hơn người bình thường, nguyên nhân là do vai trò của fibrinogen trong việc làm tổ của thai.
Ở người bệnh có bất thường fibrinogen có thể gặp tắc động mạch và tĩnh mạch - cơ chế còn chưa rõ.
b. Xét nghiệm
- Định lượng fibrinogen: Fibrinogen < 1,5 G/L hoặc không có fibrinogen.
- PT, APTT, TT, thời gian reptilase: Kéo dài (trong đó nhạy nhất là TT).
- Thời gian máu chảy: Kéo dài.
- Co cục máu đông: Không co hoặc co không hoàn toàn.
- Bất thường chức năng fibrinogen: Không tương ứng giữa nồng độ fibrinogen đo bằng phương pháp miễn dịch và phương pháp protein đông máu.
- Cần loại trừ nguyên nhân gây ra giảm fibrinogen mắc phải như DIC, xơ gan...
- Đối với người bệnh có biểu hiện tắc mạch cần loại trừ các nguyên nhân khác gây tắc mạch như định lượng antithrombin, protein C, protein S...
1.2. Điều trị
Dựa vào đặc điểm lâm sàng của người bệnh và tiền sử gia đình để lựa chọn điều trị.
- Đối với trường hợp chảy máu và với người bệnh cần phẫu thuật thì cần điều trị thay thế fibrinogen bằng các chế phẩm như: Tủa lạnh, huyết tương tươi, huyết tương tươi đông lạnh, fibrinogen cô đặc.
- Liều fibrinogen cô đặc: 50-100 mg/kg cân nặng trong ngày đầu, sau đó 20mg/kg trong những ngày sau. Thông thường nồng độ fibrinogen trong máu đạt 1G/L là đủ để cầm chảy máu. Chú ý: Thời gian bán hủy của fibrinogen truyền vào là 3-5 ngày.
- Có thể sử dụng thuốc ức chế tiêu fibrin (Tranexamic acid), đặc biệt khi cần nhổ răng, liều 15-25mg/kg/ngày. Cần cân nhắc ở các người bệnh có nguy cơ cao tắc mạch như có thai, phẫu thuật, bất động lâu hoặc người bệnh có tiền sử tắc mạch.
- Đối với phụ nữ có thai cần điều trị dự phòng bằng bổ sung fibrinogen càng sớm càng tốt để tránh sảy thai, nồng độ cần đạt là 1G/L; điều trị trong và sau đẻ, duy trì nồng độ fibrinogen > 1G/L để tránh chảy máu.
- Điều trị huyết khối nếu có.
2. THIẾU HỤT YẾU TỐ II
Yếu tố II (prothrombin) là yếu tố phụ thuộc vitamin K, được tổng hợp ở gan. Gen mã hóa yếu tố II nằm trên nhiễm sắc thể số 11. Thiếu hụt yếu tố II hiếm gặp, khoảng 1/2.000.000 người. Đây là bệnh di truyền lặn, có thể là giảm nồng độ nhưng chất lượng bình thường (typ1), hoặc bất thường prothrombin (giảm hoạt tính nhưng kháng nguyên bình thường- typ 2).
2.1. Chẩn đoán
a. Biểu hiện lâm sàng
- Chảy máu khớp và cơ: Hay gặp nhất.
- Có thể có xuất huyết dưới da và niêm mạc.
- Chảy máu sau phẫu thuật, chấn thương.
- Chảy máu rốn ở trẻ sơ sinh, thường gặp ở thể nặng.
- Xuất huyết não...
b. Xét nghiệm
- Cả PT và APTT đều kéo dài.
- Yếu tố II giảm.
- Các yếu tố đông máu khác bình thường.
2.2. Điều trị
Điều trị chảy máu bằng chế phẩm có chứa yếu tố II. Hiện chưa có yếu tố II cô đặc tinh khiết trên thị trường. Các chế phẩm có thể chọn lựa:
- Huyết tương tươi, huyết tương tươi đông lạnh liều 15-20ml/kg cân nặng.
- Phối hợp với tranexamic acid, liều 15-25 mg/kg cân nặng.
3. THIẾU HỤT YẾU TỐ V
Yếu tố V do tế bào gan và mẫu tiểu cầu chế tiết ra. Đa số yếu tố V có mặt trong huyết tương nhưng có khoảng 20% yếu tố V có mặt trong hạt α của tiểu cầu. Yếu tố V kém bền vững trong huyết tương lưu trữ. Gen mã hóa yếu tố V nằm trên nhiễm sắc thể số 1, bệnh di truyền lặn. Thiếu hụt yếu tố V hiếm gặp với tỉ lệ 1:1 triệu người.
3.1. Chẩn đoán
a. Lâm sàng
- Xuất huyết dưới da.
- Chảy máu niêm mạc, đặc biệt là chảy máu mũi.
- Chảy máu khớp và cơ (ít gặp hơn Hemophilia).
- Có thể có xuất huyết não.
b. Xét nghiệm
- Cả PT và APTT đều kéo dài.
- Kháng đông nội sinh: Âm tính.
- Định lượng yếu tố V giảm: Đây là tiêu chuẩn vàng để chẩn đoán.
- Cần định lượng yếu tố VIII để loại trừ trường hợp thiếu hụt V và VIII phối hợp.
3.2. Điều trị
- Hiện chưa có chế phẩm yếu tố V cô đặc.
- Huyết tương tươi đông lạnh: Liều 15-20 ml/kg cân nặng.
- Chú ý cân nhắc truyền khối tiểu cầu trong một số trường hợp nếu có ngưng tập tiểu cầu giảm.
- Phối hợp với tranexamic acid, liều 15-25 mg/kg cân nặng.
4. THIẾU HỤT YẾU TỐ V VÀ VIII PHỐI HỢP
Là rối loạn gây ra do bất thường vận chuyển yếu tố V và VIII từ lưới nội bào đến thể Golgi để tiết ra ngoài. Đây là bệnh di truyền lặn nhiễm sắc thể thường.
4.1. Chẩn đoán
a. Lâm sàng
- Thường chảy máu sau phẫu thuật hoặc sau can thiệp như nhổ răng.
- Hiếm khi chảy máu tự phát.
- Ở phụ nữ có thể có rong kinh và chảy máu sau đẻ.
b. Xét nghiệm
- Cả PT và APTT đều kéo dài, APTT thường kéo dài hơn.
- Nồng độ yếu tố V và VIII thường từ 5- 20%.
- Kháng đông nội sinh, ngoại sinh âm tính.
- Fibrinogen bình thường.
- Số lượng tiểu cầu bình thường.
4.2. Điều trị
- Huyết tương tươi đông lạnh có thể nâng cả nồng độ yếu tố V và VIII lên, liều 15 - 20 ml/kg cân nặng.
- Yếu tố VIII cô đặc.
5. THIẾU HỤT YẾU TỐ VII
Thiếu hụt yếu tố VII có tỉ lệ mắc bệnh 1/500.000, không phụ thuộc vào địa lí và chủng tộc. Thiếu hụt yếu tố VII trong cộng đồng có thể cao hơn do có cả người có triệu chứng và người không có triệu chứng. Gen mã hóa yếu tố VII nằm trên cánh dài nhiễm sắc thể 13, bệnh di truyền lặn.
Cần phân biệt với giảm yếu tố VII do nguyên nhân mắc phải: Ăn kiêng, tuổi già, béo phì... Hơn nữa nguồn sinh phẩm thromboplastin để chẩn đoán có ảnh hưởng lớn đến kết quả xét nghiệm.
5.1. Chẩn đoán
a. Biểu hiện lâm sàng
Thường chảy máu ở những cơ quan mà việc đông máu phụ thuộc nhiều vào con đường đông máu ngoại sinh như: Não, ruột, tử cung, nhau thai, phổi, tim. Giữa nồng độ yếu tố VII và biểu hiện chảy máu không liên quan với nhau.
- Xuất huyết dưới da và niêm mạc: Chảy máu mũi, rong kinh, chảy máu sau đẻ.
- Xuất huyết nội tạng: Chảy máu phổi, xuất huyết não…
- Chảy máu khớp.
- Một số người bệnh có biểu hiện tắc mạch, cơ chế chưa rõ.
b. Xét nghiệm
- PT kéo dài trong khi APTT, TT, fibrinogen, số lượng tiểu cầu bình thường.
- Định lượng yếu tố VII giảm.
Lưu ý: Không nên bảo quản lạnh mẫu máu trước khi làm xét nghiệm vì có thể gây hoạt hóa lạnh yếu tố VII và làm tăng nồng độ yếu tố VII hơn so với nồng độ thực có của người bệnh.
5.2. Điều trị
- Yếu tố VII: Liều 30-40mcg/kg cân nặng, liều duy nhất hoặc nhắc lại 2-3 lần mỗi 3-4 giờ.
- Yếu tố VIIa: Liều 15-30mcg/kg cân nặng, nhắc lại mỗi 2-4 giờ, đến khi ngừng chảy máu.
- Phức hợp prothrombin cô đặc: 30ui/kg liều tấn công sau đó 10-20ui/kg cân nặng mỗi 6-24 giờ đến khi ngừng chảy máu.
- Huyết tương: Liều 15ml/kg cân nặng.
6. THIẾU HỤT YẾU TỐ XI
Gen mã hóa yếu tố XI nằm trên NST số 4, bệnh di truyền lặn. Xu hướng chảy máu của người bệnh thiếu yếu tố XI có thể phụ thuộc vào nồng độ yếu tố đông máu khác như yếu tố VIII và yếu tố von Willebrand.
6.1. Chẩn đoán
a. Lâm sàng
- Thường gặp chảy máu kéo dài sau phẫu thuật, đặc biệt tại các vùng có nguy cơ tiêu fibrin cao như miệng, mũi và đường sinh dục - tiết niệu.
- Hiếm khi chảy máu tự phát.
- Chảy máu khớp: Ít gặp.
- Ở phụ nữ: Rong kinh, chảy máu sau đẻ.
b. Xét nghiệm
- APTT kéo dài (phụ thuộc nhiều vào sinh phẩm).
- Kháng đông nội sinh âm tính.
- Yếu tố XI giảm.
Lưu ý: Vì giới hạn thấp của nồng độ yếu tố XI là 60-70%, cho nên đôi khi xét nghiệm APTT không đủ nhạy để phát hiện thiếu hụt yếu tố XI. Trong trường hợp nghi ngờ thì cần làm định lượng yếu tố XI ngay cả khi APTT bình thường.
6.2. Điều trị
- Việc điều trị người bệnh thiếu yếu tố XI không đơn giản bởi vì xu hướng chảy máu là rất khác nhau ở các người bệnh. Hơn nữa đôi khi việc điều trị có liên quan đến nguy cơ tắc mạch, đặc biệt là ở những người bệnh lớn tuổi.
- Với một số tiểu phẫu như nhổ răng, sinh thiết da: Thường không cần điều trị thay thế.
- Với các phẫu thuật khác: Nồng độ yếu tố XI cần đạt là 30% trong 5 ngày cho tiểu phẫu và 45% trong ít nhất 10 ngày với đại phẫu.
- Nguồn yếu tố XI: Huyết tương, huyết tương tươi đông lạnh (15-20ml/kg cân nặng/ngày), yếu tố XI cô đặc.
Lưu ý: Cần theo dõi nồng độ yếu tố XI hàng ngày để điều chỉnh liều, tránh nguy cơ tắc mạch. Thời gian bán hủy của yếu tố XI là 46-52giờ.
- Thuốc ức chế tiêu fibrin: acid tranexamic 15-25mg/kg cân nặng. Không dùng đồng thời với yếu tố XI cô đặc do nguy cơ gây tắc mạch.
7. THIẾU HỤT YẾU TỐ X
Yếu tố X là một protease có vai trò quan trọng nhất trong sự hoạt hóa prothrombin. Gen mã hóa yếu tố X nằm trên NST số 13 gần gen yếu tố VII vì vậy chúng có liên quan với nhau. Yếu tố X được tổng hợp ở gan. Tỉ lệ mắc thiếu yếu tố X khoảng 1:1 triệu người dân.
7.1. Chẩn đoán
a. Lâm sàng
Giảm nặng yếu tố X có biểu hiện chảy máu nặng giống bệnh Hemophilia:
- Xuất huyết dưới da và niêm mạc: chảy máu chân răng, chảy máu mũi, rong kinh ở phụ nữ.
- Chảy máu khớp, cơ.
- Xuất huyết não…
b. Xét nghiệm
- Cả PT và APTT kéo dài.
- Kháng đông nội sinh và ngoại sinh âm tính.
- Yếu tố X giảm.
- Các yếu tố đông máu khác bình thường.
7.2. Điều trị
- Mục tiêu: Nâng nồng độ yếu tố X lên trên 20%.
- Huyết tương, huyết tương tươi đông lạnh: 15-20ml/kg cân nặng.
- 1 đơn vị yếu tố X/kg có thể nâng được nồng độ yếu tố X lên khoảng 1,5%. Thời gian bán hủy của yếu tố X là 60 giờ.
- Người bệnh có chảy máu tái phát khớp có chỉ định điều trị dự phòng 1-2 lần/tuần.
8. THIẾU HỤT YẾU TỐ XIII
Thiếu yếu tố XIII hiếm gặp, khoảng 1 người mắc bệnh/ 1 triệu người. Người dị hợp tử gen yếu tố XIII thường không có biểu hiện lâm sàng. Bệnh di truyền lặn, nhiễm sắc thể thường. Thời gian bán hủy của yếu tố XIII là 7-12 ngày.
8.1. Chẩn đoán
a. Lâm sàng: Biểu hiện chảy máu rất đa dạng tùy thuộc vào tổn thương phân tử.
- Người bệnh có thể có biểu hiện chảy máu rốn kéo dài và có nguy cơ xuất huyết não ngay trong thời kì sơ sinh.
- Xuất huyết dưới da, chảy máu cơ, chảy máu khớp.
- Phụ nữ có thai có thể bị sảy thai vì vậy cần được điều trị dự phòng.
b. Xét nghiệm
- Tất cả các xét nghiệm sàng lọc đông máu đều bình thường.
- Cục máu đông dễ tan trong môi trường acid acetic 2%.
- Định lượng yếu tố XIII giảm.
8.2. Điều trị
- Bổ sung yếu tố XIII khi có chảy máu.
- Nguồn yếu tố XIII: tủa lạnh, huyết tương tươi đông lạnh.
- Vì sự khó khăn trong định lượng yếu tố XIII và do bệnh hiếm gặp nên chưa có khuyến cáo chung về nồng độ yếu tố XIII cần đạt khi phẫu thuật.
- Do nguy cơ cao bị chảy máu não nên cần điều trị dự phòng cho người bệnh mỗi 4-6 tuần.
9. THIẾU HỤT CÁC YẾU TỐ ĐÔNG MÁU PHỤ THUỘC VITAMIN K (Các yếu tố II, VII, IX và X)
Bình thường, các yếu tố II, VIII, IX và X được tổng hợp ở dạng tiền chất và chưa có hoạt tính đông máu. Với sự có mặt của vitamin K trong vai trò chất xúc tác, các tiền chất này được chuyển thành các chất có hoạt tính đông máu bởi sự chuyển acid glutamic gần acid amin cuối cùng thành Gama-carboxyglutamyl. Quá trình chuyển hóa này xảy ra ở gan, do gen nằm trên nhiễm sắc thể thường, di truyền lặn quy định. Tổn thương gen này dẫn tới giảm các yếu tố II, VII, IX, X.
9.1. Chẩn đoán
a. Lâm sàng
- Nặng: Thường biểu hiện ngay trong thời kì sơ sinh như chảy máu rốn, xuất huyết não.
- Nhẹ hơn: Chảy máu niêm mạc, chảy máu sau mổ…
- Bệnh có thể ảnh hưởng đến cả các yếu tố phụ thuộc vitamin K khác như protein C, protein S, protein GIa tử cung… và sự cốt hóa xương. Trẻ bị thiếu hụt nặng có thể có biểu hiện lâm sàng tương tự nhiễm độc warfarin bào thai như thiểu sản mũi, thiểu sản đầu xa ngón chân tay, rỗ đầu xương, điếc nhẹ do giảm dẫn truyền.
b. Xét nghiệm
- Cả PT và APTT đều kéo dài.
- Giảm nồng độ II, VII, IX, X, PC, PS; mức độ nặng: < 5%.
- Cần loại trừ các nguyên nhân thiếu hụt vitamin K mắc phải và tiếp xúc với các thuốc kháng vitamin K.
9.2. Điều trị
- Bổ sung vitamin K đường uống hoặc đường tiêm, liều 5- 15mg/ngày.
- Trường hợp nặng: Truyền huyết tương, huyết tương tươi đông lạnh, phức hợp prothrombin cô đặc.
10. VẤN ĐỀ QUẢN LÝ NGƯỜI BỆNH BỊ RỐI LOẠN CHẢY MÁU BẨM SINH DI TRUYỀN
Giống như Hemophilia, các rối loạn chảy máu bẩm sinh di truyền hiện nay đều chưa chữa khỏi được, người bệnh cần được quản lý, tư vấn, điều trị tốt để có thể sống chung với bệnh. Việc quản lý bao gồm:
- Lập hồ sơ quản lý.
- Cấp thẻ người bệnh bao gồm các thông tin về chẩn đoán, mức độ bệnh, tình trạng có chất ức chế yếu tố đông máu.
- Tư vấn về bệnh và tư vấn di truyền.
- Điều trị chảy máu và dự phòng chảy máu: Sử dụng chế phẩm máu và các thuốc, biện pháp hỗ trợ.
- Phối hợp với các chuyên ngành có liên quan trong chăm sóc toàn diện: Răng hàm mặt, phục hồi chức năng, ngoại khoa, sản khoa…
- Kiểm tra định kì mỗi 1-3 tháng và điều trị nếu có các bệnh nhiễm qua đường truyền máu và các kháng thể kháng yếu tố đông máu.
TÀI LIỆU THAM KHẢO
1. Flora Peyvandi and Marzia Menegatti, 2016, Inherited Deficiencies of Coagulation Factors II, V, V+VIII, VII, X, XI, and XIII, Williams Hematology 9th edition, 2133 - 2150.
2. Kenneth D. Friedman, George M. Rodgers, (2009), Inherited Coagulation disorders, Wintrobe’s Clinical Hematology, Vol 2, Lippincot Williams and Wilkins, 1379 - 1424.
3. Marguerite Neerman - Arber and Philippe de Moerloose, 2016, Hereditary Fibrinogen Anormalities, Williams Hematology 9th edition, 2151 - 2161.
4. Mumford, A.D., Ackroyd, S., Alikhan, R., Bowles, L., Chowdary, P., Grainger, J., Mainwaring, J., Mathias, M., O'Connell, N. and (2014), Guideline for the diagnosis and management of the rare coagulation disorders. Br J Haematol, 167: 304-326. doi:10.1111/bjh.13058
5. Paula HB Bolton-Maggs, (2006), The rare coagulation disorder, Treatment of Hemophilia N39, World Federation of Hemophilia.
6. Roberta Palla, Flora Peyvandi, Amy D. Shapiro; Rare bleeding disorders: diagnosis and treatment. Blood 2015; 125 (13): 2052-2061. doi: https://doi.org/10.1182/blood-2014-08-532820
15. HỘI CHỨNG BAN XUẤT HUYẾT GIẢM TIỂU CẦU HUYẾT KHỐI - TAN MÁU URE TĂNG (Thrombotic Thrombocytopenic Purpura - Hemolytic Uremic Syndrome: TTP-HUS)
1. ĐẠI CƯƠNG
Hội chứng ban xuất huyết giảm tiểu cầu huyết khối (TTP) và Hội chứng tan máu ure tăng (HUS) là hai trong số những bệnh lý huyết khối vi mạch (thrombotic microangiopathy: TMAs), nhóm các rối loạn do sự hình thành huyết khối vi mạch, dẫn đến giảm tiểu cầu, tan máu vi mạch và suy chức năng cơ quan. TTP và HUS có thể có tính di truyền hoặc mắc phải.
2. CƠ CHẾ BỆNH SINH
2.1. TTP
Việc thiếu hụt yếu tố ADAMTS13 (di truyền hay mắc phải) sẽ dẫn đến việc dư thừa von Willebrand, yếu tố có vai trò quan trọng trong ngưng tập tiểu cầu, từ đó dẫn đến vi huyết khối tại các vi mạch có thể làm suy các cơ quan nhanh chóng.
2.2. HUS
Cơ chế bệnh sinh của HUS có thể liên quan đến độc tố của vi khuẩn như E.coli, Salmonella, vi khuẩn Gram âm (hay gặp ở trẻ em) hoặc có thể do rối loạn quá trình điều hòa bổ thể ở thể không điển hình (aHUS).
3. TRIỆU CHỨNG VÀ CHẨN ĐOÁN TTP
Trường hợp TTP điển hình, dựa vào ngũ chứng: Thiếu máu tan máu vi mạch, giảm tiểu cầu, rối loạn thần kinh, sốt và suy thận.
Tuy nhiên, trên thực tế ít khi gặp đầu đủ cả ngũ chứng; trong khi đó, nếu chờ đợi sẽ chậm trễ trong điều trị. Vì vậy, nên nghĩ đến TTP khi có biểu hiện của tan máu huyết khối vi mạch và giảm tiểu cầu trên lâm sàng và xét nghiệm mà không xác định được nguyên nhân.
3.1. Lâm sàng
Biểu hiện lâm sàng của TTP khá đa dạng.
- Giai đoạn sớm, các triệu chứng không đặc hiệu: Sốt, mệt mỏi, đau đầu, buồn nôn, nôn, tiêu chảy…
- Khi bệnh tiến triển, các triệu chứng biểu hiện tình trạng thiếu máu tan máu, giảm tiểu cầu, thiếu máu cục bộ:
+ Thiếu máu, xuất huyết;
+ Rối loạn thần kinh: Thiếu máu cục bộ, nhức đầu, lú lẫn, hôn mê;
+ Đau ngực, hiếm gặp nhồi máu cơ tim;
+ Suy thận.
3.2. Xét nghiệm
- Giảm số lượng tiểu cầu (thường < 30G/l).
- Bằng chứng của tan máu vi mạch không do miễn dịch:
+ Giảm huyết sắc tố, hematocrit;
+ Giảm haptoglobin;
+ Có mảnh hồng cầu trên tiêu bản máu ngoại vi: Theo khuyến cáo của hội đồng tiêu chuẩn quốc tế về huyết học (ICSH), cần tính tỷ lệ mảnh vỡ hồng cầu khi quan sát tối thiểu 1.00 hồng cầu trên tiêu bản máu ngoại vi. Tỷ lệ mảnh vỡ hồng cầu > 1% có giá trị gợi ý TMA; trên 4% trong chẩn đoán TMA liên quan đến ghép tế bào gốc tạo máu do trong quá trình điều trị (hóa trị, xạ trị) cũng có thể xuất hiện mảnh vỡ hồng cầu;
+ Tăng bilirubin toàn phần và gián tiếp; LDH tăng;
+ Phản ứng Coombs trực tiếp âm tính.
- Các xét nghiệm đông máu:
+ APTT, PT, fibrinogen: Bình thường;
+ Tăng D-Dimer và/ hoặc FDP.
- Tăng ure, creatinine.
- Giảm hoạt tính ADAMTS13 (A Disintegrin And Metalloproteinase with a Thrombospondin type 1 motif, member 13):
+ Đây là xét nghiệm rất có giá trị trong chẩn đoán TTP, nồng độ ADAMTS13 huyết tương < 10% có độ đặc hiệu cao trong chẩn đoán TTP;
+ ADAMTS13 có thể giảm nhẹ trong nhiều bệnh lý khác nhau, các bệnh lý tan máu vi mạch khác. Nghiên cứu gần đây chỉ ra rằng ADAMTS13 < 20 % có độ nhạy và độ đặc hiệu cao trong chẩn đoán TTP.
- Kháng thể kháng ADAMTS13: Dương tính.
- Xét nghiệm kháng thể tự miễn: ANA, LA.
Phác đồ xét nghiệm
|
Vòng đầu |
|
|
Xét nghiệm |
Kết quả |
|
Số lượng tiểu cầu |
Giảm |
|
Huyết sắc tố |
Giảm |
|
Haptoglobin |
Giảm |
|
Tiêu bản máu ngoại vi |
Có mảnh vỡ hồng cầu |
|
Hồng cầu lưới |
Tăng |
|
Nước tiểu |
Protein niệu |
|
Bilirubin, creatinin, LDH |
Tăng |
|
Coombs trực tiếp |
Âm tính |
|
APTT, PT, Fibrinogen |
Bình thường |
|
D-Dimer/FDP/FM |
Tăng |
|
Vòng 2 |
|
|
Hoạt tính ADAMTS-13 |
Giảm |
|
Kháng thể kháng ADAMTS13 |
Dương tính (TTP mắc phải). |
3.3. Chẩn đoán xác định
- Ít nhất 3/5 triệu chứng và ADAMTS 13 < 10%.
3.4. Chẩn đoán phân biệt
3.4.1. Phân biệt với đông máu rải rác trong lòng mạch (DIC)
- Về lâm sàng, khó phân biệt 2 hội chứng này, nhất là khi DIC có suy đa phủ tạng.
- Bệnh nhân DIC có thể xuất hiện TMA cùng với các bệnh lý nền như chảy máu, nhiễm trùng, ung thư…
- Do tình trạng tiêu thụ yếu tố đông máu trong DIC, bệnh nhân thường có PT, APTT kéo dài, fibrinogen giảm, D-dimer tăng cao.
- Các xét nghiệm PT, APTT, fibrinogen thường trong giới hạn bình thường ở người bệnh TTP- HUS còn trong DIC lại kéo dài bất thường, mặc dù D- Dimer đều tăng trong cả 2 hội chứng này.
3.4.2. Phân biệt với Hội chứng tan máu tăng men gan và giảm tiểu cầu (HELLP)
Thường xảy ra ở thai phụ có nhiễm độc thai nghén và thường xảy ra ở giai đoạn 3 tháng cuối của thai kỳ, trong khi TTP có thể xảy ra ở mọi thời điểm trong quá trình mang thai. Mặc dù cả hai tình trạng bệnh lý đều có giảm số lượng tiểu cầu, bệnh lý huyết khối vi mạch, tuy nhiên trong TTP thường gặp chủ yếu là biểu hiện rối loạn thần kinh, trong khi đó HELLP thường có biểu hiện chính là suy gan, men gan tăng cao.
|
Biểu hiện |
TTP |
HUS |
HELLP |
DIC |
|
Rối loạn thần kinh |
+++ |
+/- |
+/- |
+/- |
|
Suy thận |
+/- |
+++ |
+ |
+/- |
|
Sốt |
+/- |
-/+ |
- |
+/- |
|
Tan máu |
+++ |
++ |
++ |
+++ |
|
Giảm tiểu cầu |
+++ |
++ |
++ |
+++ |
|
Bất thường XN đông máu |
- |
- |
+/- |
+++ |
|
Suy gan |
+/- |
+/- |
+++ |
+/- |
3.4.3. Các trường hợp TMA do thuốc
- TMA có thể xảy ra trong những tuần đầu tiên khi bắt đầu điều trị Ticlodipine, bệnh nhân thường giảm nặng ADAMTS13 do kháng thể kháng ADAMTS13, đáp ứng với trao đổi huyết tương.
- Điều trị Clopidogrel TMA thường xuất hiện trong tuần đầu tiên, không liên quan đến giảm ADAMTS 13 và kháng thể kháng ADAMTS13.
- Một số thuốc khác như quinine, cyclosporine, tacrolimus, estrogen…
- Một số thuốc như mitomycin, gemcitabine gây TMA, nồng độ ADAMTS13 thường bình thường hoặc giảm nhẹ.
3.4.4. TMA sau ghép
TMA xảy ra ở bệnh nhân ghép tạng, ghép tế bào gốc tạo máu có thể do nhiễm trùng, ghép chống chủ, thải ghép, thuốc.
3.4.5. TMA liên quan đến ung thư
- TMA có thể xảy ra ở bệnh nhân ung thư dạ dày, vú, phổi, tuyến tiền liệt... và thường xảy ra ở bệnh nhân di căn. Cơ chế do sự tắc nghẽn vi tuần hoàn bởi tế bào ung thư, huyết khối vi mạch, DIC, hóa trị liệu.
- Phần lớn bệnh nhân có ADAMTS13 bình thường hoặc giảm nhẹ.
3.4.6. TMA liên quan đến tăng huyết áp ác tính
Bệnh nhân tăng huyết áp ác tính có thể có tan máu vi mạch, giảm tiểu cầu, suy thận. Ngược lại bệnh nhân TMA có thể tăng huyết áp, đặc biệt ở bệnh nhân có tổn thương thận. Kiểm soát huyết áp ổn định sẽ cải thiện tình trạng tan máu vi mạch.
3.5. Xếp loại
3.5.1. TTP di truyền
- Hội chứng Upshaw - Schulman (USS): Là thể bệnh khá hiếm gặp, biểu hiện bệnh lý tan máu vi mạch, giảm tiểu cầu, tái phát nhiều lần, tỷ lệ tử vong rất cao. Xét nghiệm cho thấy ADAMTS13 giảm nặng, phân tích DNA có đột biến gen mã hóa tổng hợp ADAMTS13 nằm trên nhiễm sắc thể 9q34.
- Biểu hiện lâm sàng tương tự TTP mắc phải nhưng thường gặp ở trẻ em với các biểu hiện vàng da, tan máu sơ sinh, không có bất đồng về nhóm máu hệ ABO cũng như hệ Rh và những hệ nhóm máu khác; các biểu hiện bệnh lý này thường tái phát nhiều lần trong thời niên thiếu của trẻ.
b. TTP mắc phải
Thiếu hụt men ADAMTS13 thường là hậu quả của một tình trạng bệnh lý hoặc do thuốc và nhiều khi không xác định được nguyên nhân:
- Mắc phải xác định được nguyên nhân:
+ Do thuốc: Thuốc tránh thai, ticlopidine, cyclosporin…;
+ Sau truyền tuỷ, ghép tạng;
+ Thai nghén: Sau đẻ và nhiễm độc thai nghén nặng.
- Mắc phải: Không xác định được nguyên nhân: Bao gồm TTP kinh điển ở người lớn và TTP - HUS không liên quan độc tố của E.coli 0157: H7 ở trẻ em.
3.6. Điều trị
Lưu ý: Trước khi điều trị cần làm đầy đủ các xét nghiệm đánh giá như phần phác đồ xét nghiệm và các xét nghiệm tìm nguyên nhân, như: chụp CT, MRI sọ não, chức năng gan, thận, các xét nghiệm tìm bệnh lý tự miễn, xét nghiệm tìm ổ ung thư, xét nghiệm gen (nếu có)… Làm các xét nghiệm về tế bào máu, đông máu, sinh hóa và ADAMTS 13 trong quá trình điều trị để theo dõi và đánh giá.
3.6.1. Điều trị TTP thứ phát
Điều trị bệnh chính và kết hợp với các phương pháp điều trị như:
a. Trao đổi huyết tương
- Mục đích: Loại kháng thể (trong trường hợp TTP) và cung cấp ADAMTS13.
- Thường sử dụng huyết tươi đông lạnh hoặc huyết tương đã tách tủa (cryosupernatant) và phải bắt đầu ngay khi có các biểu hiện: Giảm tiểu cầu, thiếu máu huyết tán vi quản và không tìm được nguyên nhân khác gây nên những bất thường này. Trao đổi huyết tương sớm giúp giảm tỷ lệ tử vong của TTP từ 90% xuống còn khoảng 10-30%.
- Liều lượng: 40 - 60 ml/kg cân nặng (1,0 - 1,5 thể tích huyết tương), tiến hành ngày 1 lần, kéo dài tối thiểu thêm 2 ngày sau khi số lượng tiểu cầu và LDH về bình thường.
- Nếu sau 7 ngày không đáp ứng: Tăng tần số lên 2 lần/ngày.
b. Truyền huyết tương tươi
- Chỉ định: Trường hợp không thể tiến hành trao đổi huyết tương được hoặc thời gian chờ để trao đổi huyết tương ≥ 12 giờ.
- Liều lượng: 20 - 40ml/kg cân nặng/ 24 giờ, cần lưu ý tình trạng quá tải.
c. Truyền khối hồng cầu: Khi thiếu máu nặng.
d. Truyền tiểu cầu: Chỉ truyền tiểu cầu khi chảy máu nặng đe dọa tính mạng người bệnh.
e. Thuốc kết hợp:
- Methylprednisolone: Kết hợp với trao đổi huyết tương, thường sử dụng methylprednisolone với liều 2mg/kg cân nặng/ 24 giờ bằng đường truyền tĩnh mạch, trong 3 ngày liền. Trường hợp không đáp ứng, có thể tăng methylprednisolone: 1g/24 giờ.
- Acid folic: Tất cả người bệnh TTP thứ phát đều được điều trị acid folic với liều lượng: 3-5mg/24 giờ và bằng đường uống.
- Thuốc kháng tiểu cầu: Aspirin liều thấp có thể sử dụng khi SLTC > 50G/L.
- Thuốc chống đông: Người bệnh có nguy cơ huyết khối, sử dụng heparin trọng lượng phân tử thấp khi SLTC > 50G/L.
f. Những trường hợp TTP dai dẳng (Số lượng tiểu cầu < 150G/L, LDH không về bình thường sau 7 ngày trao đổi huyết tương):
(1) Tiến hành trao đổi huyết tương với liều gấp đôi: 80ml/kg cân nặng /lần hoặc 40ml/kg cân nặng 2 lần mỗi ngày và trao đổi huyết tương bằng huyết tương đã tách tủa (cryosupernatant);
(2) Gammaglobulin: Liều 2g/kg cân nặng/ 24 giờ;
(3) Vincristin 1mg/ 24 giờ, tuần 2 lần trong 4 tuần.
g. Những trường hợp TTP tái phát: Có thể kết hợp corticoid với cắt lách hoặc dùng rituximab.
h. Những trường hợp không đáp ứng với các biện pháp điều trị thông thường: Có thể xem xét để chỉ định điều trị bằng rituximab với liều 375mg/ lần/ tuần x 4 tuần.
i. Theo dõi, đánh giá tiến triển qua các chỉ số
- Tình trạng tổn thương thần kinh.
- Lượng huyết sắc tố.
- Số lượng tiểu cầu.
- Nồng độ LDH, ure, creatinin máu.
3.6.2. Điều trị TTP di truyền
- Không tiến hành trao đổi huyết tương, chỉ truyền huyết tương tươi; Điều trị dự phòng bằng truyền huyết tương 3-4 tuần/ lần.
- Đối với người lớn, phác đồ điều trị TTP và HUS tương tự, vì vậy việc phân biệt rõ ràng 2 hội chứng này nhiều khi không nhất thiết phải đặt ra.
4. TRIỆU CHỨNG VÀ CHẨN ĐOÁN HUS
4.1. Triệu chứng lâm sàng:
HUS đặc trưng bởi 3 triệu chứng: Huyết khối vi mạch, giảm tiểu cầu và tổn thương thận. Triệu chứng sẽ phụ thuộc vào nguyên nhân gây bệnh.
Theo cách phân chia cổ điển thì HUS bao gồm 2 thể:
- Thể HUS điển hình (D+ HUS) hay còn gọi là thể liên quan tiêu chảy: HUS có nhiễm trùng E.Coli, có tiêu chảy; có thể gặp ở bất kỳ lứa tuổi nào nhưng thường gặp nhất ở trẻ em dưới 10 tuổi.
- Thể HUS không điển hình (D-HUS) hay còn gọi là thể không liên quan tiêu chảy: HUS không có nhiễm trùng E.Coli, không tiêu chảy; thể này thường ít gặp hơn thể D+HUS.
- Hiện nay, HUS được chia thành 2 thể bệnh chính là STEC-HUS (90%) do độc tố của vi khuẩn (Shiga-toxin (Stx) producing Escherichia Coli-STEC) và aHUS-atypical HUS (10%) với những trường hợp không liên quan đến E Coli, là hậu quả của sự rối loạn điều hòa hệ thống bổ thể do đột biến gen hoặc sự xuất hiện của các tự kháng thể, và bệnh thường khởi phát do những tác nhân như nhiễm trùng, ung thư, vắc xin, bệnh tự miễn…
Các triệu chứng thường thấy như sau:
- Thường gặp ở trẻ em.
- Tiêu chảy, thường phân có máu.
- Đau bụng, quặn và đầy hơi.
- Nôn.
- Sốt.
Tất cả các trường hợp HUS bất kể nguyên nhân gì đều có thể có tổn thương mạch máu dẫn đến tan máu không do tự miễn và tổn thương thận do vậy sẽ có các triệu chứng tùy theo mức độ bệnh: Thiếu máu, tiểu sẫm, phù, tăng huyết áp.
4.2. Xét nghiệm:
Thể hiện tình trạng tan máu vi mạch, tổn thương thận và giảm tiểu cầu:
- Hb thường < 10g/l.
- Tăng hồng cầu lưới, có mảnh vỡ hồng cầu trên tiêu bản máu ngoại vi.
- Giảm haptoglobin.
- Tăng LDH.
- TC < 150G/l, thường gặp < 40G/l.
- Tăng ure, creatinin.
- Có hồng cầu và protein trong nước tiểu.
- Nồng độ ADAMTS 13 > 10%.
- Xét nghiệm nguyên nhân:
+ STEC HUS: Xét nghiệm thấy bằng chứng E. Coli sinh độc tố Shiga;
+ a HUS: C3, C4, yếu tố H bổ thể, kháng thể kháng yếu tố H, B, I; Xét nghiệm đột biến gen.
4.3. Chẩn đoán
4.3.1. Chẩn đoán xác định
- Xét nghiệm ADAMTS 13 hoạt tính > 10%.
- STEC-HUS: Dựa vào lâm sàng, có tiền sử tiêu chảy khoảng 2 tuần trước đó, thiếu máu tan máu không do miễn dịch, suy thận. STEC-HUS: Xét nghiệm thấy độc tố vi khuẩn hoặc nuôi cấy có vi khuẩn trong phân. Có thể dựa vào sinh thiết thận để chẩn đoán xác định.
- a HUS: Chẩn đoán sau khi loại trừ TTP, STEC-HUS và TMA thứ phát khác.
4.3.2. Chẩn đoán phân biệt
- DIC: Có thay đổi các chỉ số đông máu huyết tương, trong khi HUS không thấy có sự thay đổi này.
- TTP: ADAMTS13 hoạt tính < 10%, trong khi với HUS tỷ lệ này vẫn > 10%.
4.4. Điều trị
- Các xét nghiệm theo dõi điều trị tương tự TTP.
- Với STEC-HUS: Điều trị chủ yếu là điều trị hỗ trợ bao gồm đảm bảo cân bằng nước, điện giải, kiểm soát huyết áp, lọc thận tùy theo mức độ suy thận. Việc sử dụng kháng sinh, Eculizumab và PEX, truyền huyết tương, ức chế miễn dịch còn nhiều tranh cãi, thường không được khuyến cáo với STEC-HUS.
- Với a HUS:
+ Khuyến cáo hàng đầu trong điều trị aHUS là thuốc ức chế hoạt hóa và sản xuất bổ thể C5a, C5b do gắn với C5;
+ Trao đổi huyết tương liều 60-75ml/kg hoặc truyền huyết tương liều 10-20 ml/kg với bệnh nhân nghi ngờ aHUS khi không có Eculizumab;
+ Đảm bảo cân bằng nước, điện giải thông qua lượng dịch, loại dịch cần truyền.
+ Kiểm soát huyết áp bằng thuốc hạ huyết áp và lượng dịch đưa vào;
+ Lọc thận: Tuỳ theo mức độ suy của thận;
+ Ghép thận;
+ Ức chế miễn dịch như corticoid, cyclophosphamid phối hợp trao đổi huyết tương được sử dụng trong trường hợp aHUS có kháng thể chống yếu tố H bổ thể (Complement factor H).
5. TIÊN LƯỢNG
Tỷ lệ tử vong của TTP-HUS rất cao (95%) nếu không được điều trị; chẩn đoán sớm và điều trị kịp thời, nhất là sau khi phương pháp trao đổi huyết tương được áp dụng rộng rãi, 80 - 90% người bệnh được cứu sống.
TÀI LIỆU THAM KHẢO
1. Nguyễn Ngọc Minh, (2007), Ban xuất huyết giảm tiểu cầu huyết khối - hội chứng tăng ure máu huyết tán, Bài giảng Huyết học Truyền máu sau Đại học; NXB Y học Hà nội; Tr: 505-509.
2. BCSH Secretary, British Society for Haematology, (2003), Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias, British Journal of Haematology, Volume 120, Issue 4, pp: 556-573.
3. Mortimer Poncz, (2009), Antibody- Mediated thrombotic disorders idiopathic thrombotic thrombocytopenic purpura and heparin - induced thrombocytopenia: overview, William Hematology seventh edition, pp: 4877-4897
4. Sanchez-Corral P, Melgosa M, (2010), Advances in understanding the aetiology of atypical Haemolytic Uraemic Syndrome, Br J Haematol 2010, 150, pp: 529-542.
5. Taylor CM, Machin S, Wigmore SJ, Goodship TH, (2010), Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol. 2010, 148:37-47.
6. Marie Scully, Piers Blombery, (2014), Management of thrombotic thrombocytopenic purpura: current perspectives. Journal of blood medicine 2014: 15 - 23.
7. M .Saha et al, (2017), Thrombotic thrombocytopenic purpura: pathogenesis, diagnosis and potential novel therapeutics, Journal of thrombosis and haemostasis. 2017: 1 - 12.
16. HỘI CHỨNG THỰC BÀO TẾ BÀO MÁU
(Hemophagocytic lymphohistiocytosis-HLH)
1. ĐẠI CƯƠNG
Hội chứng thực bào máu (Hemophagocytic syndromes) là bệnh cảnh do tăng hoạt hóa quá mức các tế bào thực bào, dẫn đến tiêu hủy tế bào máu bình thường. Bệnh có nguồn gốc là tăng sinh lympho-mô bào. Phân lớn bệnh là do bẩm sinh, tuy nhiên có thể do mắc phải. Bệnh thường gặp ở trẻ em, tỷ lệ tử vong có thể lên tới 60%.
2. NGUYÊN NHÂN
- HLH được chia làm 2 thể: Nguyên phát (tính gia đình-Familial HLH-FHLH) và thứ phát, hay mắc phải (Secondary HLH-sHLH, hay acquired HLH).
- Hội chứng thực bào máu có tính chất gia đình (Familial HLH-FHLH) là một bệnh di truyền hay bệnh do đột biến gen gây suy giảm tế bào NK và tế bào T gây độc, giảm sản xuất các cytokine. Tỷ lệ phát hiện được các loại đột biến gen là 20-40%. Có thể gặp di truyền gen lặn, đột biến gen perforin nằm trên nhiễm sắc thể 9,10 gây suy giảm hiện tượng chết theo chương trình. Bệnh có thể được khởi động sau một tình trạng nhiễm khuẩn.
- Hội chứng thực bào máu thứ phát (Secondary HLH-sHLH) là hậu quả của sự hoạt hóa quá mức hệ thống đại thực bào-mono do nhiều nguyên nhân khác nhau như: Rối loạn miễn dịch (gặp trong nhiễm vi khuẩn, virus, ký sinh trùng), các loại ung thư (lơ xê mi cấp, u lympho hay các ung thư tạng đặc…). Hội chứng thực bào thứ phát có thể thoái triển tự nhiên nhưng cũng có thể tiến triển nặng hơn và dẫn đến tử vong.
3. TRIỆU CHỨNG
3.1. Lâm sàng
- Triệu chứng chủ yếu là sốt kéo dài.
- Gan to, lách to chiếm > 90% các trường hợp.
- Triệu chứng thần kinh trung ương thường thấy ở các trường hợp nặng, biểu hiện chủ yếu là hội chứng màng não.
3.2. Cận lâm sàng
a. Xét nghiệm tế bào và mô bệnh học máu, tạo máu
- Tế bào máu ngoại vi và hồng cầu lưới: Giảm các dòng tế bào máu.
- Tủy đồ và mô bệnh học tủy xương: Phát hiện hình ảnh thực bào tế bào máu, trong đó các đại thực bào hoạt hóa đang thực bào hồng cầu, bạch cầu, tiểu cầu và các tế bào khác.
- Sinh thiết hạch, lách hoặc gan phát hiện hình ảnh thực bào tế bào máu trong trường hợp xét nghiệm tủy xương không xác định rõ.
b. Xét nghiệm đông máu: Fibrinogen, APTT, PT, D-dimer.
c. Sinh hóa: Tăng triglycerid, ferritin, thay đổi nồng độ các immunoglobulin. Các xét nghiệm cần tầm soát khác: Glucose, chức năng gan, chức năng thận, điện giải, bộ mỡ máu, LDH; Bilan viêm (khi lâm sàng có dấu hiệu nhiễm trùng): CRP, pro-calcitonin; Các marker ung thư tầm soát bệnh ác tính.
d. Xét nghiệm chẩn đoán hình ảnh
- Siêu âm bụng: Phát hiện gan, lách và hạch to.
- X quang ngực: Phát hiện thâm nhiễm phổi.
- MRI sọ não: Đánh giá mức độ tổn thương thần kinh trung ương khi có triệu chứng gợi ý trên lâm sàng.
- CT-scanner ngực bụng: Tìm các bệnh lý ác tính như u lympho, ung thư các tạng, tình trạng di căn.
- Nội soi dạ dày, đại tràng tìm khối u đường ruột.
đ. Xét nghiệm dịch não tủy: Đánh giá tế bào, nồng độ protein, glucose để phát hiện thâm nhiễm thần kinh trung ương.
e. Xét nghiệm miễn dịch (thường sử dụng kỹ thuật đếm tế bào dòng chảy - Flow Cytometry).
- Phát hiện giảm hoặc mất hoạt tính của tế bào NK.
- Định lượng nồng độ receptor interleukin-2 hòa tan (sCD25).
f. Xét nghiệm chẩn đoán mức độ phân tử
- Phát hiện các đột biến gen liên quan đến protein perforin để chẩn đoán xác định HLH nguyên phát: PFR1, UNC13D, STX11, STXBP2.
- Phát hiện các đột biến gen của những nhóm bệnh cần chẩn đoán phân biệt: LYST (hội chứng Chesdiak-Higashi); RAB27A (hội chứng Griscelli).
g. Xét nghiệm tìm nguyên nhân
- Tìm các nguyên nhân gây sốt: Bệnh tự miễn khác, kháng thể kháng nhân, kháng thể kháng dsADN, cấy máu, CRP.
- Các xét nghiệm thăm dz nhiễm virus, vi khuẩn và ký sinh trùng: CMV, EBV, HIV, HSV, HHV6+8, rubella, variellae virus, parvovirus, adenovirus, leishmaniasis, brucellosis, lao, mycoplasma, giang mai, định lượng DNA virus CMV, EBV
h. Định type HLA người bệnh và gia đình người bệnh: Khi cần ghép tế bào gốc.
k. Xét nghiệm nước tiểu: Tế bào, sinh hóa nước tiểu, cấy nước tiểu để tìm nguyên nhân nhiễm trùng.
4. CHẨN ĐOÁN
4.1. Chẩn đoán xác định
Chẩn đoán xác định (theo tiêu chuẩn chẩn đoán Hội chứng thực bào máu-2004) khi có 1 trong 2 nhóm tiêu chuẩn sau:
a. Nhóm tiêu chuẩn về sinh học phân tử
- Người bệnh được chẩn đoán Hội chứng thực bào máu nguyên phát khi có tổn thương một trong các gen đặc hiệu: PFR1 (vị trí: 10q21-22), UNC13D (vị trí: 17q25), STX11 (vị trí: 6q24), STXBP2.
b. Nhóm tiêu chuẩn về lâm sàng và xét nghiệm
Nếu không có tiêu chuẩn về sinh học phân tử, người bệnh được chẩn đoán Hội chứng thực bào máu khi có 5/8 tiêu chuẩn sau:
Tiêu chuẩn về lâm sàng:
- Sốt kéo dài.
- Lách to.
Tiêu chuẩn về xét nghiệm:
- Giảm ít nhất 2 trong 3 dòng tế bào máu:
+ Hemoglobin < 90 g/L (với trẻ nhỏ < 4 tuần tuổi thì hemoglobin < 100 G/L);
+ Số lượng tiểu cầu < 100 G/L;
+ Số lượng bạch cầu hạt trung tính < 1 G/L.
- Tăng triglycerid máu ≥ 3,0 mmol/l (tương đương ≥ 265 mg/dl) và/hoặc giảm fibrinogen < 1,5 g/L.
- Tăng ferritin ≥ 500 ng/ml.
- Tế bào và mô bệnh học: Hình ảnh đại thực bào đang thực bào tế bào máu trong tủy xương, lách hoặc hạch. Không kèm theo các tình trạng ác tính khác trong trường hợp hội chứng thực bào nguyên phát. Nếu không có thì không loại trừ và làm lại tủy đồ sau 1-2 tuần.
- Tế bào NK giảm hoặc mất hoạt tính.
- CD25 hòa tan (IL-2 receptor) ≥ 2400 U/ml.
4.2. Chẩn đoán phân biệt
Cần phân biệt với một số bệnh có biểu hiện tương tự hội chứng thực bào đó là:
- Bệnh mô bào Langerhans (Langerhans cell histiocytosis): Sinh thiết tổ chức có hình ảnh tế bào Langerhans với những hạt hồng trong bào tương, CD 1 (+), protein S100 bộc lộ trong bào tương…
- Hội chứng tăng sinh lympho liên quan nhiễm sắc thể X (X-linked lympho- proliferative syndrome-XLP): Giảm sinh tủy, tăng sinh lympho tủy, giảm sản xuất CD 27, có tổn thương Xq25.
- Hội chứng Chesdiak-Higashi: Liên quan đến tình trạng nhiễm khuẩn Staphylococus và Streptococcus, giảm bạch cầu trung tính, mất sắc tố, tổn thương thần kinh, rối loạn đông máu và có gen LYST: Vị trí 1q42.1-q42.2.
- Hội chứng Griscelli: Suy giảm miễn dịch và có gen RAB27A vị trí 15q21.
5. ĐIỀU TRỊ
5.1. Nguyên tắc chung
- Đây là một bệnh máu ác tính tiến triển nhanh, nguy cơ tử vong cao nên cần điều trị nghiêm ngặt theo phác đồ.
- Cần đánh giá toàn diện người bệnh trước điều trị.
- Chẩn đoán xác định Hội chứng thực bào nguyên phát hay thứ phát để có hướng điều trị phù hợp.
- Đối với Hội chứng thực bào máu thứ phát, cần ưu tiên điều trị nguyên nhân trước hoặc song song với điều trị theo phác đồ Hội chứng thực bào máu 2004 tùy thuộc vào nguyên nhân cụ thể (xem sơ đồ 2).
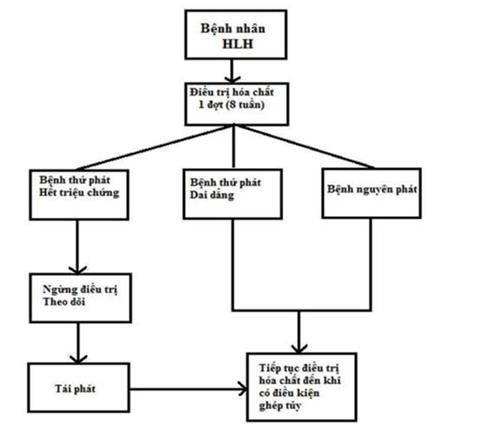
Sơ đồ 2. Tóm tắt hướng dẫn điều trị (theo HLH 2004).
5.2. Điều trị bệnh chính
a. Điều trị hóa chất tấn công 8 tuần
- Hóa trị liệu toàn thân bao gồm 3 loại hóa chất:
+ Etoposide;
+ Dexamethasone;
+ Cyclosporin A.
Liều thuốc cụ thể:
|
|
Etoposide |
Dexamethasone |
Cyclosporin A |
|
Tuần 1 |
150 mg/m2 (TM) 2 lần/tuần |
10 mg/m2/ngày Đường uống hoặc TM |
Khởi đầu 6 mg/kg/ngày, uống chia 2 lần, sau 1 tuần xét nghiệm chỉnh liều để đảm bảo nồng độ thuốc trong máu 200 µg/L |
|
Tuần 2 |
150 mg/m2 (TM) 2 lần/tuần |
10 mg/m2/ngày Đường uống hoặc TM |
|
|
Tuần 3 |
150 mg/m2 (TM) 1 lần/tuần |
5 mg/m2/ngày Đường uống hoặc TM |
|
|
Tuần 4 |
150 mg/m2 (TM) 1 lần/tuần |
5 mg/m2/ngày Đường uống hoặc TM |
|
|
Tuần 5 |
150 mg/m2 (TM) 1 lần/tuần |
2,5 mg/m2/ngày Đường uống hoặc TM |
|
|
Tuần 6 |
150 mg/m2 (TM) 1 lần/tuần |
2,5 mg/m2/ngày Đường uống hoặc TM |
|
|
Tuần 7 |
150 mg/m2 (TM) 1 lần/tuần |
1,25 mg/m2/ngày Đường uống hoặc TM |
|
|
Tuần 8 |
150 mg/m2 (TM) 1 lần/tuần |
Giảm liều và dừng |
- Tiêm tủy sống:
+ Chỉ định nếu sau 2 tuần điều trị, các bất thường về thần kinh vẫn tiến triển hoặc không cải thiện;
+ Tiêm tủy sống: 2 lần trong 1 đợt điều trị 4 tuần. Mỗi lần tiêm cần phối hợp:
● < 1 tuổi: Methotrexate: 6mg, predisolon: 4mg = 3,2 mg methylprednisolon;
● 1-2 tuổi: Methotrexate: 8mg, predisolon: 6mg = 4,8 mg methylprednisolon;
● 2-3 tuổi: Methotrexate: 10mg, predisolon: 8mg= 6,4 mg methylprednisolon;
● > 3 tuổi: Methotrexate: 12mg, prednisolon: 10mg= 8mg methylprednisolon.
- Các hướng dẫn bổ sung:
+ Cyclosporin A: Gây độc cho thận, cần theo dõi chức năng thận để có điều chỉnh thích hợp, giảm liều theo mức lọc cầu thận;
+ Etoposide: Gây ức chế tủy, dừng thuốc hoặc giảm liều khi số lượng bạch cầu hạt trung tính < 0,5 G/L;
+ Dexamethasone: Gây ức chế miễn dịch, tổn thương dạ dày, cần bổ sung thuốc bảo vệ dạ dày và kháng sinh chống nấm, kháng virus dự phòng.
b. Điều trị củng cố:
- Áp dụng tùy theo các mức đáp ứng điều trị của người bệnh (thường bắt đầu từ tuần 9 và kéo dài không quá tuần 40):
+ Đối với người bệnh Hội chứng thực bào máu nguyên phát, có tổn thương gen đặc hiệu, chuyển sang điều trị củng cố và tiến hành ghép tế bào gốc đồng loài càng sớm càng tốt;
+ Đối với người bệnh Hội chứng thực bào máu thứ phát, đạt tiêu chuẩn đáp ứng sau 8 tuần điều trị tấn công, có thể dừng điều trị và theo dõi;
+ Đối với người bệnh Hội chứng thực bào máu thứ phát, bệnh dai dẳng sau 8 tuần điều trị tấn công hoặc tái phát sau khi hết triệu chứng, tiếp tục điều trị củng cố và ghép tế bào gốc đồng loài càng sớm càng tốt.
- Liều thuốc cụ thể như sau:
+ Etoposide: 150 mg/m2 tĩnh mạch, 2 tuần/ 1 lần;
+ Dexamethasone 10 mg/m2 trong vòng 3 ngày, 2 tuần/ 1 đợt;
+ Cyclosporin A: Liều điều chỉnh tùy từng người bệnh để đảm bảo nồng độ thuốc trong máu đạt 200 µG/L.
c. Ghép tế bào gốc
- Ưu tiên lựa chọn ghép tế bào gốc đồng loài hòa hợp HLA.
- Trong trường hợp không tìm được người cho hòa hợp HLA, có thể lựa chọn ghép tế bào gốc hòa hợp một nửa (haplotype) từ người cho anh chị em ruột.
d. Người bệnh không có điều kiện ghép tế bào gốc
Điều trị duy trì tối đa 40 tuần. Có thể lựa chọn 1 trong 4 cách dưới đây, sau đó giảm liều và dừng nếu như bệnh không có biểu hiện tái hoạt động:
- Tiếp tục áp dụng phác đồ như điều trị củng cố (tuần 9 đến tuần 40).
- Điều trị luân phiên truyền etoposide và dexamethasone từ 2-4 tuần và tiếp tục điều trị cyclosporin A như trên. Sau đó, người bệnh có thể được điều trị bằng etoposide và dexamethasone liều như trên nhưng 2 tuần/ 1 lần.
- Điều trị bằng cyclosporin A và dexamethasone, với liều như trên và luân phiên nhau từ tuần 9-40.
- Chỉ điều trị bằng cyclosporin A hoặc dexamethasone đến tuần 40.
đ. Điều trị cứu vãn (salvage therapy)
- Trong một số trường hợp đặc biệt, các biện pháp điều trị trên không có tác dụng hoặc đem lại nhiều biến chứng nặng phải ngừng thuốc, có thể sử dụng đến một số loại thuốc nhóm kháng thể đơn dòng đối kháng dòng lympho như ATG, kháng interferon gamma.
5.3. Điều trị hỗ trợ
- Điều trị hỗ trợ ban đầu:
+ Điều trị nhiễm trùng bằng kháng sinh và điều trị giảm bạch cầu (có thể dùng thuốc kích thích tăng sinh dòng bạch cầu hạt (G-CSF) khi số lượng bạch cầu trung tính < 0,5 G/L);
+ Điều trị kháng sinh phổ rộng phù hợp (cho đến khi có kết quả kháng sinh đồ);
+ Có thể dự phòng bằng cotrimoxazole (liều 30 mg/kg, 2-3 lần/ tuần);
+ Thuốc chống nấm đường uống;
+ Liệu pháp chống virus đối với những người bệnh bị nhiễm virus;
+ Gammaglobulin tĩnh mạch (0,5 g/kg, tĩnh mạch) mỗi 4 tuần (áp dụng trong điều trị tấn công và điều trị củng cố).
- Thiếu máu: Cần truyền khối hồng cầu để duy trì hemoglobin > 90 g/L.
- Giảm tiểu cầu: Chỉ định truyền khối tiểu cầu khi số lượng tiểu cầu < 20 G/L hoặc xuất huyết nặng đe dọa tính mạng người bệnh.
- Điều trị các rối loạn đông máu nếu có.
- Các hỗ trợ khác: Truyền dịch, thải độc, hạ acid uric, bảo vệ tế bào gan.
6. ĐÁNH GIÁ ĐÁP ỨNG
- Đáp ứng (đạt được tất cả tiêu chuẩn sau 2 tuần hoặc 4 tuần điều trị tấn công): Hết sốt, lách nhỏ lại, số lượng tiểu cầu ≥ 100 G/L, fibrinogen ≥ 1,5 G/L, giảm 25% nồng độ ferritin.
- Lui bệnh (đạt được tất cả tiêu chuẩn sau 8 tuần điều trị tấn công): Hết sốt, gan không sờ thấy, Hb ≥ 90 g/L; số lượng tiểu cầu ≥ 100 G/L; số lượng bạch cầu trung tính ≥ 0,5G/L; triglycerid < 3 mmol/l; ferritin < 500 ng/ml; dịch não tủy bình thường; giảm nồng độ CD25 hòa tan. Theo dõi xét nghiệm CD25 mỗi 4 tuần.
- Bệnh dai dẳng: Nếu không đạt được các tiêu chuẩn lui bệnh.
- Bệnh tái phát (sau khi đã đạt lui bệnh, xuất hiện lại ≥ 3 tiêu chuẩn sau): Sốt; lách to; tiểu cầu < 100 G/L; triglycerid ≥ 3 mmol/l; fibrinogen < 1,5 G/L; có hiện tượng thực bào tế bào máu; tăng nồng độ ferritin trở lại; CD25 hòa tan ≥ 2.400 U/ml. Tổn thương thần kinh trung ương dù đơn độc cũng đủ để chẩn đoán tái phát.
7. THEO DÕI ĐIỀU TRỊ
- Tổng phân tích tế bào máu và hồng cầu lưới: 2 lần/ tuần khi điều trị nội trú.
- Tủy đồ và sinh thiết tủy xương khi bệnh nhân không lui bệnh, tái phát, kiểm tra điều trị.
- Xét nghiệm đông máu: Fibrinogen, APTT, PT, D-dimer.
- Xét nghiệm sinh hóa: Glucose, acid uric, chức năng gan, chức năng thận, điện giải, bộ mỡ máu, LDH hàng tuần; Bilan viêm (CRP, pro-calcitonin) khi lâm sàng có dấu hiệu nhiễm trùng.
- Xét nghiệm chẩn đoán hình ảnh: Siêu âm ổ bụng, X-quang phổi, siêu âm hạch, CT ngực bụng nếu bệnh nhân có triệu chứng mới gợi ý.
- Xét nghiệm nước tiểu: Tổng phân tích nước tiểu, tế bào nước tiểu.
- Xét nghiệm vi sinh (HBsAg, HCV Ab, HIV Ab): Với bệnh nhân có truyền máu và chế phẩm máu.
- Xét nghiệm huyết thanh học nhóm máu: Với bệnh nhân có truyền máu.
- Xét nghiệm CD25: Theo dõi hàng tháng.
8. TIÊN LƯỢNG
- Nếu không được điều trị, người bệnh hội chứng thực bào nguyên phát chỉ có thể sống được khoảng 2 tháng, thường tử vong trong tình trạng nhiễm khuẩn nặng và suy đa phủ tạng.
- Áp dụng theo protocol Hội chứng thực bào máu 2004 (trình bày trên) có tỷ lệ sống trên 2 năm khoảng 55-60%. Tuy nhiên, có 20-40% người bệnh hội chứng thực bào thứ phát có thể tử vong ngay trong các đợt điều trị tấn công do nhiều nguyên nhân, mà chủ yếu là nhiễm khuẩn.
- Tỷ lệ người bệnh sống > 3 năm sau ghép tế bào gốc đồng loài khoảng 60%.
TÀI LIỆU THAM KHẢO
1. Stepp SE, Dufourcq-Lagelouse R, Le DF et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999; 286(5446):1957-1959.
2. Henter JI, Horne A, Arico M et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48(2):124-131.
3. Schneider EM, Lorenz I, Muller-Rosenberger M, Steinbach G, Kron M, Janka-Schaub GE. Hemophagocytic lymphohistiocytosis is associated with deficiencies of cellular cytolysis but normal expression of transcripts relevant to killer-cell-induced apoptosis. Blood 2002; 100(8): 2891-2898.
4. Chairman: Jan-Inger Henter, MD, PhD, Stockholm, Sweden. Treatment protocol of the second international HLH study 2004. Hemophagocytic Lymphohistiocytosis Study Group. January 2004
5. Michael B. Jordan, Carl E. Allen, Sheila Weitzman, Alexandra H. Filipovich and Kenneth L. McClain. How we treat hemophagocytic lymphohistocytosis. Prepublished online August 9, 2011; doi: 10.1182/blood-2011-03-278127.
17. CHẨN ĐOÁN VÀ XỬ TRÍ BIẾN CHỨNG SỐT NHIỄM TRÙNG Ở BỆNH NHÂN GIẢM BẠCH CẦU HẠT
1. ĐẠI CƯƠNG
1.1. Định nghĩa
Sốt giảm bạch cầu được định nghĩa là có nhiệt độ cơ thể đo 1 lần ở miệng > 38,3ºC hoặc nhiệt độ > 38ºC kéo dài trên 1 giờ, kèm theo có số lượng bạch cầu hạt < 0,5G/l hoặc có nguy cơ sẽ hạ xuống dưới mức 0,5G/l.
1.2. Dịch tễ và lâm sàng
Sốt giảm bạch cầu hạt là một trong các biến chứng nguy hiểm nhất của điều trị hóa chất trong ung thư nói chung và bệnh máu ác tính nói riêng. Tỷ lệ tử vong chung là khoảng
5% ở các bệnh ung thư mô đặc, 11 % ở các bệnh nhân ung thư hệ tạo máu. Tiên lượng xấu đối với các trường hợp kết quả cấy máu có vi khuẩn, tỷ lệ tử vong là 18% với vi khuẩn gram âm, 5% với vi khuẩn gram dương. Tỷ lệ tử vong cũng khác nhau tùy thuộc vào bảng điểm tiên lượng MASCC, dưới 3% nếu điểm MASCC ≥ 21; 36% nếu MASCC < 15. Bệnh nhân lớn tuổi sốt giảm bạch cầu hạt sau điều trị hóa chất có tỷ lệ tử vong cao hơn và các biến chứng nặng nề hơn.
Tỷ lệ dương tính đối với cấy máu cũng phụ thuộc nhiều yếu tố như có sử dụng kháng sinh dự phòng, có đặt catheter tĩnh mạch trung tâm. Ở bệnh nhân ung thư hệ tạo máu, nếu có sử dụng kháng sinh dự phòng thì tỷ lệ dương tính khoảng 17%, còn không sử dụng kháng sinh dự phòng thì tỷ lệ khoảng 31%.
2. CHẨN ĐOÁN
Để chẩn đoán được các trường hợp sốt giảm bạch cầu hạt, cần thực hiện đầy đủ các bước thăm khám để đưa ra kết luận:
a. Bệnh nhân đang có catheter hay không.
b. Triệu chứng nhiễm trùng:
- Hệ hô hấp.
- Hệ tiêu hóa.
- Da.
- Hệ tiết niệu sinh dục, tầng sinh môn.
- Tai mũi họng.
- Hệ thần kinh trung ương.
c. Tiền sử cấy vi khuẩn dương tính trước đây.
d. Thăm khám thường quy:
- Xét nghiệm máu đánh giá chức năng tủy xương, thận, gan.
- Đông máu cơ bản.
- Protein C phản ứng.
- Cấy máu ít nhất 2 lần, kể cả cấy từ catheter.
- Cấy nước tiểu, tổng phân tích nước tiểu.
- Cấy và soi đờm.
- Cấy và soi phân.
- Cấy tổ chức da.
- Chụp phổi.
đ. Các xét nghiệm chuyên sâu hơn:
- Chụp CT ngực độ phân giải cao.
- Xét nghiệm dịch rửa phế quản.
Sau khi thăm khám cần tiến hành đánh giá phân loại bệnh nhân dựa theo bảng điểm MASCC (Bảng 14).
Bảng 14. Bảng điểm MASCC
|
Đặc điểm |
Điểm |
|
Không có triệu chứng hoặc triệu chứng nhẹ |
5 |
|
Triệu chứng ở mức vừa |
3 |
|
Triệu chứng nặng |
0 |
|
Không tụt huyết áp (HA tâm thu > 90 mmHg) |
5 |
|
Không có COPD |
4 |
|
U đặc hoặc u lympho không có nhiễm nấm từ trước |
4 |
|
Không mất nước |
3 |
|
Đang là bệnh nhân ngoại trú (khi có sốt) |
3 |
|
Tuổi < 60 |
2 |
Nhóm nguy cơ thấp: MASCC ≥ 21 điểm
Nhóm nguy cơ cao: MASCC < 21 điểm Tối đa 26 điểm.
MASCC: Multinational Association for Supportive Care in Cancer
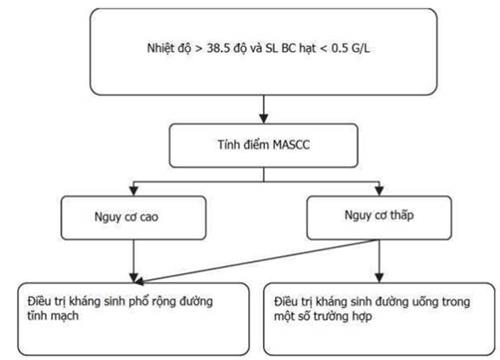
Sơ đồ 3. Đánh giá và xử trí bệnh nhân sốt giảm bạch cầu hạt
3. ĐIỀU TRỊ
3.1. Bệnh nhân nguy cơ thấp:
- Gồm các bệnh nhân có số lượng bạch cầu hạt giảm dưới 0,5G/l dưới 7 ngày; có huyết động ổn định, không mắc Lơ xê mi cấp hoặc không có suy tạng, không viêm phổi, không đặt catheter, không nhiễm khuẩn mô mềm thì có thể sử dụng kháng sinh đường uống.
- Kháng sinh khởi đầu được khuyến cáo: Ciprofloxacin 500-750mg uống 2 lần/ ngày hoặc levofloxacin 750mg uống 1 lần/ ngày kết hợp với amoxicillin-clavulanic acid 625mg uống 3 lần/ ngày (nếu trước đó bệnh nhân đã dùng quinolone dự phòng thì phải thay bằng phác đồ kháng sinh tĩnh mạch như điều trị bệnh nhân nguy cơ cao).
- Bệnh nhân nguy cơ thấp nhưng có các yếu tố nguy cơ kể trên thì dùng kháng sinh đường tĩnh mạch giống như bệnh nhân nguy cơ cao.
- Bệnh nhân hết sốt sau 48h có thể chuyển dần từ kháng sinh đường tĩnh mạch sang Quinolone đường uống.
3.2. Bệnh nhân nguy cơ cao: Gồm những bệnh nhân giảm bạch cầu hạt dưới 0,5G/l kéo dài trên 7 ngày.
a. Lựa chọn kháng sinh
- Tùy thuộc vào các loại vi khuẩn thường gặp và tình trạng kháng kháng sinh tại mỗi cơ sở y tế hoặc mỗi khu vực để đưa ra lựa chọn phù hợp nhất, đặc biệt cần quan tâm đến MRSA (tụ cầu vàng kháng Methicilin) và các loại vi khuẩn gram âm kháng thuốc.
- Có thể lựa chọn Cephalosporin thế hệ 3-4 (hoặc Carbapenem), dùng đơn độc hoặc kết hợp với một nhóm kháng sinh khác.
- Nếu có nguy cơ nhiễm khuẩn huyết cao hoặc giảm bạch cầu hạt lâu dài thì lựa chọn ưu thế là kết hợp Beta lactam với Aminoglycoside.
b. Lựa chọn thay thế và các chỉ định đặc biệt
- Bệnh nhân có đặt catheter: Nếu nghi ngờ nhiễm khuẩn liên quan đến catheter, cần cấy máu ngoại vi và cấy chân catheter, tính thời gian cấy dương tính chênh lệch giữa cấy catheter và cấy máu. Nếu thời gian chênh lệch > 2h thì nhiều khả năng nhiễm khuẩn huyết do catheter.
+ Nếu nghi nhiễm khuẩn do catheter, chưa có kết quả cấy và tình trạng bệnh nhân vẫn ổn định thì không cần rút catheter. Ưu tiên sử dụng Vancomycin hoặc thay thế bằng Teicoplanin;
+ Nếu có kết quả cấy dương tính với tụ cầu, bệnh nhân vẫn ổn định, có thể vẫn lưu catheter;
+ Rút catheter khi có nhiễm khuẩn quanh chân catheter, tạo thành ổ, nhiễm khuẩn huyết dai dẳng dù đã điều trị phù hợp, hoặc nhiễm nấm, đặc biệt là Candida.
- Viêm phổi: Nếu có chẩn đoán viêm phổi dựa trên lâm sàng và kết quả hình ảnh, cần thêm một loại kháng sinh chống các vi khuẩn không điển hình (Legionella và Mycoplasma) như nhóm Macrolide phối hợp với Beta lactam.
Bệnh nhân có thở nhanh, giảm SpO2 cần nghi ngờ có nhiễm Pneumocystic Jerovecii, nên điều trị bằng Co-trimoxazole liều cao.
- Nhiễm trùng da: Thêm Vancomycin để điều trị các trường hợp nghi tụ cầu vàng.
- Nhiễm trùng trong ổ bụng hoặc khung chậu: Thêm Metronidazole.
- Tiêu chảy: Thêm Metronidazole, cần chú ý Clostridium difficile.
- Nhiễm nấm Candida: Bệnh nhân có nguy cơ cao nhiễm Candida với các biểu hiện:
+ Giảm bạch cầu hạt lâu dài;
+ Mắc các bệnh lý ác tính huyết học với điều trị hóa chất diệt tủy;
+ Cấy máu để có bằng chứng nhưng thường cho kết quả lâu;
+ Điều trị theo kinh nghiệm khi thất bại với kháng sinh diệt khuẩn bình thường sau 3-7 ngày điều trị;
+ Lựa chọn kháng sinh theo kinh nghiệm đầu tay là Amphotericin B gắn lipid hoặc nhóm Echinocandin như Caspofungin, đặc biệt là các trường hợp đã sử dụng nhóm Azole trước đó (như Fluconazole);
+ Fluconazole lựa chọn đầu tay cho các trường hợp nguy cơ thấp đối với nhiễm Aspergillus xâm nhập và chưa từng sử dụng nhóm Azole dự phòng;
+ Sử dụng kháng sinh chống nấm ít nhất 14 ngày đến khi hết giảm bạch cầu hạt.
- Nhiễm nấm phổi xâm nhập: Hay gặp ở các bệnh nhân Lơ xê mi cấp hoặc ghép tủy đồng loại vì thường có giảm bạch cầu hạt kéo dài:
+ Thường xuyên đánh giá mức độ đáp ứng với kháng sinh;
+ Nếu nghi ngờ nhiễm Aspergillus xâm nhập, cần chụp CT-scaner độ phân giải cao, với hình ảnh liềm hơi điển hình, hoặc xét nghiệm dịch phế quản;
+ Lựa chọn kháng sinh Voriconazole hoặc Amphotericin B gắn lipid.
- Nhiễm trùng nghi do virus: Bổ sung Aciclovir. Nếu nghi ngờ CMV thì dùng thêm Ganciclovir.
- Nghi viêm não, màng não: Chọc dịch não tủy, điều trị viêm màng não do vi khuẩn bằng kháng sinh đồ, ban đầu khi chưa có kháng sinh đồ có thể kết hợp Ceftazidim với Ampicilin (để bao phủ cả Listeria), hoặc Meropenem liều 6 g/ngày. Viêm màng não do virus sử dụng Aciclovir liều cao.
- Nhiễm trùng do vi khuẩn gram âm kháng thuốc: Kết hợp meropenem + colistin hoặc phác đồ 2 kháng sinh nhóm carbapenem (meropenem + ertapenem).
4. ĐÁNH GIÁ ĐÁP ỨNG
Cần đánh giá hàng ngày, tùy theo thực tế và mức độ nặng của bệnh nhân, nhất là các bệnh nhân hồi sức có thể theo dõi mỗi 2-4h.
4.1. Các chỉ tiêu đánh giá bao gồm:
- Diễn biến sốt.
- Xét nghiệm tế bào máu ngoại vi.
- Chức năng thận.
4.2. Nếu bệnh nhân hết sốt và BC hạt ≥ 0,5 G/L trong 48h, xử trí như sau:
- Nhóm nguy cơ thấp, không tìm ra nguyên nhân: Chuyển kháng sinh đường uống.
- Nhóm nguy cơ cao, không tìm ra nguyên nhân: Có thể dừng Aminoglycosid, giữ lại nhóm kháng sinh đang phối hợp còn lại.
- Có nguyên nhân: Tiếp tục điều trị theo kháng sinh đồ.
4.3. Nếu bệnh nhân vẫn sốt sau 48h:
- Lâm sàng ổn định: Tiếp tục theo kháng sinh ban đầu.
- Lâm sàng không ổn định: Điều chỉnh kháng sinh, có thể thêm nhóm Glycopeptide (Vancomycin, Teicoplanin), hoặc phối hợp Carbapenem với Glycopeptide.
4.4. Thời gian điều trị:
- Nếu SLBC hạt ≥ 0,5 G/L, bệnh nhân hết triệu chứng, hết sốt trong 72h, cấy máu âm tính, có thể dừng kháng sinh.
- Nếu số lượng bạch cầu hạt < 0,5 G/L, hết sốt trong 5-7 ngày, không có biến chứng gì thêm, có thể dừng kháng sinh, ngoại trừ các trường hợp nguy cơ cao và Lơ xê mi cấp (thường duy trì kháng sinh ít nhất 10 ngày hoặc cho đến khi bạch cầu hạt ≥ 0,5 G/L).
- Nếu vẫn sốt dù số lượng bạch cầu hạt đã hồi phục, cần hội chẩn các chuyên gia về vi sinh và cân nhắc sử dụng thuốc chống nấm.
Phụ lục: Liều kháng sinh tham khảo
|
Nhóm KS |
Tên thuốc |
Đường dùng |
Liều |
|
|
Trẻ em |
Người lớn |
|||
|
Cephalosporin |
Ceftazidim Cefoperazone Ceftriaxone Cefepime |
Tĩnh mạch |
25-100 mg/kg/ngày |
2-4 g/ngày, chia 2 lần (liều tối đa 12 g/24h) |
|
Carbapenem |
Imipenem |
Tĩnh mạch |
25mg/kg mỗi 8-12h |
0,5-1g mỗi 8h (tổng liều 2-3 g/24h, không quá 50mg/kg/24h hoặc 4g/24h) |
|
Meropenem |
Tĩnh mạch |
10-20 mg/kg mỗi 8h |
0,5-1g mỗi 8h, tối đa đến 2g mỗi 8h |
|
|
Quinolone |
Ciprofloxacin |
Uống |
7,5-15mg/kg/ngày chia 2 lần |
500 mg/ngày (uống 1 lần hoặc chia 2 lần) |
|
Tĩnh mạch |
5-10 mg/kg/ngày |
400 mg/ngày (chia 2 lần) |
||
|
Levofloxacin |
Uống/TM |
|
500mg/ngày |
|
|
Moxifloxacin |
Uống/TM |
|
400mg/ngày |
|
|
Glycopeptide |
Vancomycin |
Tĩnh mạch |
50 mg/kg/6h |
0,5-1g/12h |
|
Teicoplanin |
Tĩnh mạch |
6-10 mg/kg/ngày |
400mg/ngày (tối đa 12mg/kg/ngày) |
|
|
Beta lactam + kháng Betalactamase |
Amoxicilin + A.Clavulanic |
Uống |
40-50 mg/kg/ngày |
1-2 g/ngày |
|
Ampicilin + Sulbactam |
Uống/TM |
150mg/kg/ngày |
1,5g-tối đa 12g/ngày |
|
|
Tazobactam + piperacillin |
Tĩnh mạch |
240-300 mg piperacilin/kg/ngày chia 3-4 lần |
4,5g mỗi 6-8 giờ |
|
|
Kháng penicillinase |
Methicillin |
Tĩnh mạch |
50-100 mg/kg/ngày |
1-2g/6h (Tối đa 12g) |
|
Macrolide |
Clarythromycin |
Uống |
7,5 -15 mg/kg/12h |
0,5-1g/ngày |
|
Azithromycin |
Uống |
10 mg/kg/ngày |
1-2 g/ngày |
|
|
Aminoside |
Amikacin |
Tĩnh mạch |
15 mg/kg/ngày |
1g/ngày |
|
Tobramycin |
Tĩnh mạch |
2 mg/kg/8h |
2 mg/kg/8h |
|
|
Gentamycin |
Tĩnh mạch |
2 mg/kg/8h |
2mg/kg/8h |
|
|
Polymycin B |
Colistin |
Tĩnh mạch |
75.000-150.000 UI/kg/ngày chia 2 lần |
Liều nạp 9.000.000 UI Duy trì 4.500.000 UI mỗi 12 giờ |
|
Cotrimoxazole |
Trimethoprim + Sulfamethoxazole |
Uống |
48 mg/kg/ngày |
480 mg x 4 viên/ngày (chia 2) |
|
Imidazole |
Metronidazole |
Uống/TM |
10-15 mg/kg/ngày |
0,5-1 g/ngày |
|
Triazole |
Fluconazole |
Uống/TM |
3-12 mg/kg/ngày |
150-400 mg/ngày |
|
Itraconazole |
Uống/TM |
|
200-400 mg/ngày |
|
|
Voriconazole |
Uống/TM |
5-7mg/kg/12h |
4-6 mg/kg/12h |
|
|
Posaconazole |
Uống/TM |
|
400mg/12h |
|
|
Amphotericin |
Amphotericin deoxicholate |
Tĩnh mạch |
|
Test: 1mg/20-30phút Liều đầu 0,25-0,5mg/kg/ngày Duy trì: 0,25-1 mg/kg/ngày |
|
Amphotericin B gắn lipid |
Tĩnh mạch |
3-5 mg/kg/ngày |
3-5mg/kg/ngày (tối đa 40mg/kg) |
|
|
Echinocandin |
Caspofungin |
Tĩnh mạch |
|
50-70mg/ngày |
|
Micafungin |
Tĩnh mạch |
|
100-150 mg/ngày |
|
|
Kháng virus |
Acyclovir |
Uống |
|
1- 4g/ngày |
|
Tĩnh mạch |
10-20 mg/kg/8h |
5-10 mg/kg/8h |
||
|
Ganciclovir |
Uống |
|
3g/ngày |
|
|
Tĩnh mạch |
|
5-6mg/kg/ngày |
||
|
Foscarnet |
tĩnh mạch |
|
90mg/kg x 2 lần/ngày |
|
TÀI LIỆU THAM KHẢO
1. J. de Naurois1, I. Novitzky-Basso2, M. J. Gill3, F. Marti Marti1, M. H. Cullen1 & F. Roila (2010). Management of febrile neutropenia: ESMO Clinical Practice Guidelines, Annals of Oncology 21 (Supplement 5): v252-v256.
2. Alison G. Freifeld,1 Eric J. Bow, Kent A. Sepkowitz, Michael J. Boeckh, James I. Ito, Craig A. Mullen, Issam I. Raad, Kenneth V. Rolston, Jo-Anne H. Young, and John R. Wingard (2010). Clinical Practice Guideline for the Use of Antimicrobial Agents in Neutropenic Patients with Cancer: 2010 Update by the Infectious Diseases Society of America. Clinical Infectious Diseases 2011;52(4):e56-e93.
3. Mary Denshaw-Burke, Thomas E.Herchline (2013).Neutropenic Fever Empiric Therapy. www.Emedicine.Medscape.com/article/2012185.
4. Kern WV, Marchetti O, Drgona L, Akan H, Aoun M, Akova M, de Bock R, Paesmans M, Viscoli C, Calandra T (2013). Oral antibiotics for fever in low-risk neutropenic patients with cancer: a double-blind, randomized, multicenter trial comparing single daily moxifloxacin with twice daily ciprofloxacin plus amoxicillin/clavulanic acid combination therapy--EORTC infectious diseases group trial XV. J Clin Oncol.31(9):1149-56.Epub 2013 Jan 28.
18. HỒI SỨC HUYẾT HỌC
1. CHẨN ĐOÁN VÀ ĐIỀU TRỊ MỘT SỐ TÌNH TRẠNG CẤP CỨU GẶP TRONG CÁC BỆNH LÝ HUYẾT HỌC
1.1. Chèn ép tủy sống và thâm nhiễm thần kinh trung ương
Do tế bào ác tính (thường trong lơ xê mi) thâm nhiễm thần kinh trung ương hoặc do u (u lympho, u plasmo) chèn ép tủy.
1.1.1. Chẩn đoán
a. Triệu chứng lâm sàng
Đau đầu: Đau liên tục, tăng dần, có thể kèm theo nhìn mờ, buồn nôn. Đau lưng: khu trú hoặc lan dọc theo một bên, đau tăng lên khi ho hoặc vận động. Rối loạn cảm giác: Cảm giác bỏng rát, tăng cảm giác da, tê bì. Rối loạn vận động: Phản xạ gân xương lúc đầu tăng, giai đoạn sau thường giảm. Liệt một hoặc nhiều dây thần kinh sọ. Tùy vị trí tủy bị chèn mà có các biểu hiện tổn thương thần kinh theo khu vực đặc thù
b. Cận lâm sàng
Chụp MRI: Có thể thấy hình ảnh khối u chèn ép, hoặc hình ảnh phù não. Chọc dịch não tủy: Thâm nhiễm các tế bào ác tính, protein trong dịch não tủy tăng, phản ứng Pandy (+).
1.1.2. Điều trị
- Dexamethasone liều 10 mg, sau đó chuyển sang 4 mg tiêm tĩnh mạch cứ 6 giờ một lần, giảm dần liều trong 2-3 tuần tiếp theo.
- Tiêm hóa chất nội tủy: Methotrexate 12 mg + cytarabine 50 mg + dexamethasone 8 mg, cách ngày tiêm một lần đến khi xét nghiệm dịch não tủy không còn tế bào ác tính.
- Phẫu thuật mở cung sau đốt sống giải phóng chèn ép đối với tủy.
- Tia xạ trực tiếp vào khối u.
1.2. Hội chứng tiêu khối u
Hội chứng tiêu khối u là hậu quả của tình trạng hủy tế bào quá mức, dẫn tới giải phóng đột ngột các ion và các chất chuyển hóa trung gian trong tế bào vào tuần hoàn.
1.2.1. Chẩn đoán
a. Triệu chứng lâm sàng
- Rối loạn cảm giác, mệt, loạn nhịp tim.
- Cơn tetani, dấu hiệu Chvostek và Trousseau, mệt mỏi, co rút cơ, co thắt phế quản, co giật, có thể trụy tim.
- Mệt mỏi, buồn nôn, nôn, chán ăn, kích thích thần kinh cơ, kém tập trung, ngứa, xuất huyết dưới da. Có thể có biểu hiện viêm màng ngoài tim, quá tải tuần hoàn, khó thở.
- Đau khớp và đau quặn thận.
b. Xét nghiệm
- Tăng cao creatinin và axit uric trong máu.
- Rối loạn điện giải: Kali và phosphat máu tăng, giảm nồng độ calci máu.
- Tăng cao nồng độ LDH máu.
1.2.2. Điều trị
- Theo dõi 1-3 lần /ngày về: Urea máu, creatinine, acid uric, kali, calci, phosphat và LDH nếu nguy cơ cao. Theo dõi trước, trong và ít nhất 3 ngày sau kết thúc hóa trị liệu.
- Bù dịch: Bù dịch đường tĩnh mạch, bắt đầu 24-48 giờ trước hóa trị liệu và tiếp tục đến 48-72 giờ sau kết thúc hóa trị liệu. Lượng dịch khoảng 4-5 L/ngày (3 L/m2/ngày).
- Lợi tiểu: Có thể sử dụng Furosemid uống, với liều 40-80 mg/ngày, bảo đảm lượng nước tiểu ít nhất 3 L/ngày.
- Kiềm hóa nước tiểu: Dung dịch natri bicarbonate 1,4% truyền tĩnh mạch. Theo dõi các chỉ số pH niệu, bicarbonate huyết thanh, acid uric máu.
- Allopurinol liều 300-600 mg/ngày để dự phòng (nếu nồng độ axit uric máu < 475 µmol/l) và 600-900 mg/ngày để điều trị hội chứng tiêu khối u. Dùng Rasburicase khi điều trị allopurinol không tác dụng với liều dùng: 0,15-0,2 mg/kg/ngày tiêm tĩnh mạch (nếu nồng độ axit uric máu > 475 µmol/l), dùng tối đa 7 ngày.
- Điều trị giảm kali máu: Chế đố ăn giảm kali. Truyền dịch, dùng thuốc lợi tiểu và bổ sung kali đường uống. Dùng calcium gluconate đường tĩnh mạch để bảo vệ tim khi nồng độ kali trên 6,5 mmol/l hoặc có biểu hiện rối loạn điện tim đồ do tăng kali máu.
- Lọc máu cấp cứu: Khi có tình trạng tăng kali máu hoặc tăng phospho máu dai dẳng không đáp ứng với điều trị, quá tải tuần hoàn, tăng urea máu, giảm calci máu có biểu hiện lâm sàng và tăng acid uric > 595 µmol/l mặc dù đã dùng nhiều liều rasburicase.
1.3. Tình trạng tắc mạch do tăng bạch cầu
1.3.1. Chẩn đoán
- Triệu chứng lâm sàng: Tùy vào vị trí mạch tắc mà có các triệu chứng:
+ Biểu hiện thần kinh từ mức độ nhẹ (đau đầu, chóng mặt); đến nặng (mê sảng, phù gai thị, chảy máu võng mạc, xuất huyết nội sọ);
+ Biểu hiện tại phổi bao gồm khó thở tăng dần, rối loạn nhịp thở, giảm nồng độ oxygen máu. Chụp X quang thấy các vùng mờ lan toả cả 2 phế trường;
+ Nhồi máu lách hoặc tắc tĩnh mạch dương vật...
- Xét nghiệm: Số lượng bạch cầu tăng rất cao, thường trên 100 G/l.
1.3.2. Điều trị
Gạn bạch cầu cấp cứu bằng máy tách thành phần máu tự động. Truyền dịch đường tĩnh mạch 3 L/m2/ngày. Hydroxyurea uống 50-100 mg/kg/ngày.
1.4. Tình trạng tắc mạch do tăng tiểu cầu
1.4.1. Chẩn đoán
- Triệu chứng lâm sàng: Tê bì, đau đầu ngón tay, ngón chân, đau đầu, ù tai, rối loạn ý thức; biểu hiện tắc tĩnh mạch hoặc động mạch.
- Xét nghiệm: Số lượng tiểu cầu tăng cao. Siêu âm Doppler có thể thấy huyết khối.
1.4.2. Điều trị
Gạn tiểu cầu khi số lượng tiểu cầu > 1.000 G/l. Aspirin liều thấp 75-100 mg/ngày, tuy nhiên hạn chế dùng khi người bệnh có biến chứng xuất huyết. Hydroxyurea uống 50-100 mg/kg/ngày.
1.5. Hội chứng tăng độ quinh máu toàn phần do tăng số lượng hồng cầu
1.5.1. Chẩn đoán
- Triệu chứng lâm sàng: Đau đầu, nhìn mờ, chóng mặt, mất thị lực hoặc thính lực đột ngột (thường là 1 bên). Soi đáy mắt phát hiện ứ máu tĩnh mạch võng mạc, xuất huyết võng mạc, phù gai thị.
- Xét nghiệm: Tăng độ quánh máu toàn phần (bình thường là 4,4-6,3 mPas/giây).
1.5.2. Điều trị
Rút máu điều trị. Ban đầu cách ngày rút máu 1 lần. Sau đó giảm dần số lần rút máu để duy trì hematocrit < 0,45 L/L. Dùng thuốc giảm tế bào như hydroxyurea với liều khởi đầu 10-30 mg/kg/ngày để bổ sung hiệu quả điều trị của rút máu trong điều trị đa hồng cầu nguyên phát.
1.6. Hội chứng tăng độ quánh huyết tương
1.6.1. Chẩn đoán
- Gây ra bởi sự tăng tiết quá mức globulin miễn dịch đơn dòng (bệnh đa u tủy xương) hoặc IgM đơn dòng (bệnh Waldenström macroglobulinemia) trong huyết tương.
- Triệu chứng lâm sàng: Đau đầu, nhìn mờ, chóng mặt, mất thị lực hoặc thính lực đột ngột (thường là 1 bên), chảy máu mũi, chảy máu chân răng. Soi đáy mắt có thể gặp hiện tượng ứ máu trong các tĩnh mạch võng mạc, xuất huyết võng mạc, phù gai thị.
- Xét nghiệm: Tăng độ quánh huyết tương (bình thường 1,6-1,8 mPas/giây), độ quánh huyết tương càng tăng cao thì mức độ triệu chứng càng nặng; protein máu tăng cao; hình ảnh hồng cầu chuỗi tiền.
1.6.2. Điều trị
Trao đổi huyết tương càng sớm càng tốt, đặc biệt ở những người bệnh có triệu chứng lâm sàng; điều trị bệnh lý chính gây ra tình trạng tăng độ quánh huyết tương.
1.7. Viêm loét niêm mạc đường tiêu hóa và tiêu chảy
1.7.1. Chẩn đoán
- Triệu chứng lâm sàng: Tổn thương viêm, loét, chảy máu niêm mạc miệng và/ hoặc ống tiêu hóa; đi ngoài phân lỏng nhiều lần; buồn nôn.
- Xét nghiệm: Nội soi và sinh thiết niêm mạc ống tiêu hóa giúp chẩn đoán phân biệt với các nguyên nhân khác (bệnh ghép chống chủ, nhiễm trùng).
1.7.2. Điều trị
Nhịn ăn hoàn toàn, nuôi dưỡng tĩnh mạch. Morphin 10-20 mg/ngày nếu tiêu chảy quá nhiều, điều chỉnh liều morphin theo đáp ứng lâm sàng. Súc miệng dung dịch Natri gualenate hydrate + lidocain giúp giảm tình trạng viêm và đỡ đau cho người bệnh. Điều trị thuốc giảm tiết acid và thuốc bọc niêm mạc nếu loét dạ dày.
1.8. Bệnh ghép chống chủ cấp
Xin xem thêm trong bài “Ghép tế bào gốc đồng loài”.
Bệnh ghép chống chủ (graft versus host disease - GVHD) là một biến chứng thường gặp sau ghép tế bào gốc tạo máu đồng loài, hoặc sau khi truyền chế phẩm máu không được chiếu xạ cho người bệnh bị suy giảm miễn dịch. Bệnh ghép chống chủ được chia thành bệnh ghép chống chủ cấp (acute graft versus host disease - aGVHD) và bệnh ghép chống chủ mạn (chronic graft versus host disease - cGVHD).
1.8.1. Chẩn đoán
- Chẩn đoán aGVHD dựa vào các triệu chứng lâm sàng ở da, gan và ống tiêu hóa. Phân loại mức độ tổn thương dựa trên hệ thống tính điểm của Glucksberg.
- Biểu hiện ở da thường gặp nhất trong aGVHD, chiếm trên 75% người bệnh. Biểu hiện sớm ở lòng bàn tay và bàn chân với các nốt sẩn nhỏ, đỏ và ngứa. Tổn thương nặng biểu hiện bằng xơ cứng da hoặc tróc vảy.
- Biểu hiện ở ống tiêu hóa bao gồm buồn nôn, nôn, đau bụng, rối loạn tiêu hoá, đi ngoài phân lỏng như nước. Nội soi ống tiêu hóa thấy niêm mạc phù nề hoặc tạo thành mảng cứng. Sinh thiết tổn thương giúp chẩn đoán phân biệt aGVHD với các bệnh lý khác.
- Biểu hiện ở gan ít gặp, chiếm 20% trong số các người bệnh có aGVHD; biểu hiện vàng da, gan to và tăng men gan.
1.8.2. Điều trị
- Methylprednisolone liều tấn công 2 mg/kg/ngày trong ít nhất một đến hai tuần, giảm liều chậm dưới 10% mỗi tuần.
- Các trường hợp kháng điều trị corticoid thường có tiên lượng xấu, cần điều trị phối hợp corticoid với một số thuốc như là CSA, tacrolimus, MMF, ATG, infliximab.
1.9. Hội chứng đông máu rải rác trong lòng mạch
- Chi tiết xin xem trong bài Hội chứng đông máu rải rác trong lòng mạch (DIC).
- Chẩn đoán DIC theo tiêu chuẩn của Hội đông máu tắc mạch quốc tế, có tính đến đặc thù của DIC gặp trong lơ xê mi cấp và theo dõi xét nghiệm có tính động học. Các xét nghiệm cơ bản bao gồm: đếm số lượng tiểu cầu, định lượng fibrinogen, APTT, PT, D-dimer, ROTEM;
- Nguyên tắc điều trị DIC: Điều trị bệnh lý là nguyên nhân gây DIC (ví dụ lơ xê mi cấp thể tiền tủy bào); Điều trị thay thế bằng chế phẩm máu (khối tiểu cầu, huyết tương tươi đông lạnh, tủa lạnh); Điều trị heparin tiêu chuẩn hoặc LMWH.
2. ĐIỀU TRỊ HỖ TRỢ NÂNG CAO THỂ TRẠNG, BẢO VỆ GAN THẬN VÀ ĐƯỜNG TIÊU HÓA
2.1. Điều trị hỗ trợ nâng cao thể trạng
Bảo đảm cân bằng và đủ chất dinh dưỡng như chất đạm, chất béo, tinh bột. Thức ăn cho người bệnh suy giảm miễn dịch cần vô trùng tối đa bằng cách ăn thức ăn mềm, nấu chín, hoa quả rửa sạch và gọt vỏ, nhúng qua nước sôi nếu cần. Nếu người bệnh tổn thương niêm mạc đường tiêu hóa không nuốt được hoặc không hấp thu được, thì cần nuôi dưỡng đường tĩnh mạch.
2.2. Điều trị hỗ trợ bảo vệ gan và thận
Bù dịch bằng cách uống nhiều nước hoặc bù dịch đường tĩnh mạch.
2.3. Điều trị hỗ trợ bảo vệ niêm mạc đường tiêu hóa
Dự phòng loét dạ dày do stress bằng thuốc bọc niêm mạc và thuốc giảm tiết acid. Vệ sinh miệng họng bằng cách đánh răng sạch, súc miệng bằng dung dịch sát khuẩn, điều trị kháng sinh và thuốc chống viêm giảm đau dạng gel. Bổ sung vitamin C. Dùng thuốc bọc niêm mạc, điều trị kháng sinh đường ruột nếu có nhiễm trùng kèm theo. Nếu tổn thương viêm loét nặng cần chỉ định nuôi dưỡng đường tĩnh mạch.
3. ĐIỀU TRỊ HỖ TRỢ CHỐNG ĐỘC TÍNH CỦA THUỐC HÓA CHẤT
3.1. Tác dụng phụ huyết học
Thuốc hóa chất là gây tình trạng suy tủy sau điều trị, làm người bệnh thiếu máu, xuất huyết do giảm tiểu cầu và nhiễm trùng cơ hội do giảm bạch cầu hạt; lưu ý phòng và dùng chế phẩm máu nếu cần.
3.2. Tác dụng phụ không phải huyết học của một số loại hóa chất
Sử dụng mesna phòng ngừa độc tính của Cyclophosphamid và ifosfamid liều cao (viêm bàng quang chảy máu). Sử dụng calcifolinat (acid folinic) phòng ngừa độc tính của methotrexat liều cao. Các thuốc anthracyclin gây độc cơ tim, ví dụ: Daunorubicin có thể gây độc cơ tim khi tổng liều vượt quá 550 mg/m2. ATRA có thể gây hội chứng ATRA; điều trị bằng dexamethasone 10 mg mỗi 12 giờ đường tĩnh mạch trong ít nhất 3 ngày.
4. ĐIỀU TRỊ TRIỆU CHỨNG ĐAU
Điều trị giảm đau mang tính cá thể hóa rất cao, việc sử dụng thuốc tùy thuộc vào đáp ứng của từng người bệnh. Các biện pháp chính bao gồm: Điều trị bằng thuốc, gây tê, phẫu thuật thần kinh, điều trị tâm lý.
4.1. Cơn đau mức độ nhẹ hoặc vừa phải:
Dùng các thuốc sau: Aspirin và Paracetamol với liều lên đến 1000 mg/4 giờ. Paracetamol phối hợp codein (30 mg codein + 500 mg Paracetamol). Tramadol: Mạnh gấp 2 lần codein, viên 50 mg ít gây táo bón. Thuốc kháng viêm không chứa Steroid: Ibuprofen 200-400 mg x 3 lần/ngày; diclofenac 100-150 mg x 3 lần/ngày.
4.2. Cơn đau nặng, dữ dội
- Sử dụng thuốc viên morphin sulfat phóng thích có kiểm soát. Liều khởi đầu cho người lớn là 30-60 mg/chia 2 lần/ ngày uống hoặc bơm qua sonde dạ dày.
- Nếu sử dụng thuốc opioid đường uống không còn tác dụng, phải chỉ định Morphin đường tiêm, dùng liều nhỏ 5-10 mg tiêm dưới da 4 giờ/ lần.
- Fentanyl: Thấm qua da nên có thể dùng dưới dạng dán, cung cấp 1 lượng thuốc chậm qua da kéo dài đến 3 ngày.
5. ĐIỀU TRỊ GIẢM NHẸ MỘT SỐ TRIỆU CHỨNG ĐƯỜNG TIÊU HÓA
5.1. Nôn và buồn nôn
Các thuốc chống nôn thông dụng bao gồm: Metoclopramide liều 10 mg x 1-3 lần/ngày; hoặc ondansetron (thuốc kháng thụ thể 5-HT3 chọn lọc) liều 4 mg x 2 lần/ngày; thường dùng điều trị nôn do hóa trị liệu.
5.2. Táo bón
- Để hỗ trợ nhu động ruột nên yêu cầu người bệnh vận động nhẹ, tăng dần tùy theo thể trạng, giữ vệ sinh sau khi đi ngoài, ăn thức ăn có nhiều rau quả.
- Sử dụng thuốc:
+ Thuốc làm tăng khối lượng phân, làm tăng kích thích đường ruột (ví dụ: Thuốc Polynu bào chế từ dược liệu);
+ Thuốc làm mềm phân:
● Bisacodyl: Là thuốc nhuận tràng tiếp xúc, kích thích các chất từ niêm mạc ruột.
Liều dùng 5-15 mg/ngày có ở dạng viên hoặc viên đặt hậu môn;
● Dầu paraffin 10-20 ml (dùng buổi tối);
● Thuốc kích thích hoạt động cơ trơn, chẳng hạn glycerol.
5.3. Chướng bụng
Điều trị tình trạng chướng hơi, khó tiêu, cổ trướng tùy theo nguyên nhân. Dùng thuốc lợi tiểu, chẳng hạn furosemide 40-120 mg/ngày. Truyền albumin người giúp tăng nồng độ protein máu, giảm cổ trướng.
5.4. Khô miệng
Đánh răng, súc miệng với nước súc miệng có soda hay bicarbonate. Điều trị nhiễm nấm Candida vùng miệng bằng kháng sinh chống nấm, chà rửa lưỡi bẩn bằng dung dịch sát khuẩn. Bôi môi bằng kem vaselin hay dầu thực vật.
6. ĐIỀU TRỊ KHÁNG SINH CHO NGƯỜI BỆNH GIẢM BẠCH CẦU HẠT
Chi tiết xin xem trong bài Chẩn đoán và xử trí biến chứng sốt nhiễm trùng ở người bệnh giảm bạch cầu hạt.
- Sốt giảm bạch cầu được định nghĩa là khi nhiệt độ cơ thể > 38,3ºC hoặc có 2 lần đo nhiệt độ > 38ºC, kèm theo có số lượng bạch cầu hạt < 0,5 G/l hoặc có nguy cơ sẽ hạ xuống dưới mức 0,5 G/l.
- Giảm bạch cầu hạt nhưng chưa có sốt cần điều trị kháng sinh dự phòng bằng kháng sinh nhóm ciprofloxacin, acyclovir và kháng sinh chống nấm nhóm azole. Sốt do giảm bạch cầu hạt điều trị bằng kháng sinh phổ rộng đường tĩnh mạch theo kinh nghiệm, nhóm cephalosporin thế hệ 3-4 hoặc carbapenem. Điều chỉnh kháng sinh dựa theo đáp ứng điều trị và kết quả xác định tác nhân nhiễm khuẩn.
7. ĐIỀU TRỊ HỖ TRỢ BẰNG TRUYỀN MÁU
- Chi tiết xin đọc trong bài chỉ định sử dụng máu và chế phẩm máu.
- Các chế phẩm máu thường dùng bao gồm: Khối hồng cầu; Khối tiểu cầu; Huyết tương tươi đông lạnh; Tủa lạnh; Chế phẩm máu chiếu xạ. Ngoài ra còn có biện pháp truyền máu phenotype, tức là truyền chế phẩm máu hòa hợp không chỉ hệ nhóm máu ABO và Rh mà còn phù hợp với các hệ nhóm máu khác.
TÀI LIỆU THAM KHẢO
1. Bộ Y tế, Thông tư 26 “Hướng dẫn hoạt động truyền máu”, 2013.
2. Freifeld AG, Bow EJ, Sepkowitz KA, Boeckh MJ, Ito JI, Mullen CA, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 Update by the Infectious Diseases Society of America. Clin Infect Dis. Feb 15 2011; 52(4):427-31.
3. Jagasia MH, Arrowsmith ER. Complications of hematopoietic neoplasms. In: Wintrobe MM, Greer JP, Foerster J, et al. Wintrobe's Clinical Hematology. Vol II. 11th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2003:1919-44.
4. Mughal TI, Ejaz AA, Foringer JR, Coiffier B. An integrated clinical approach for the identification, prevention, and treatment of tumor lysis syndrome. Cancer Treat Rev. Apr 2010; 36(2): 164-76.
19. DỰ PHÒNG VÀ ĐIỀU TRỊ NHIỄM NẤM XÂM NHẬP Ở BỆNH NHÂN HUYẾT HỌC
1. ĐẠI CƯƠNG
Nhiễm nấm xâm nhập (invasive fungal infection) là tình trạng nhiễm nấm máu hoặc nhiễm nấm sâu lan tỏa ở nội tạng (gan, lách …). Đây là một bệnh nhiễm trùng cơ hội thường gặp ở bệnh nhân suy giảm miễn dịch (giảm bạch cầu hạt trung tính, sau ghép tế bào gốc tạo máu, sử dụng corticoid kéo dài…). Tỷ lệ tử vong ước tính của bệnh nhân nhiễm Candida máu khoảng 40%, nhiễm Aspergillus phổi từ 50 - 75%.
2. NGUYÊN NHÂN GÂY BỆNH
Candida: Tỷ lệ nhiễm nấm xâm nhập do Candida ở bệnh nhân bệnh máu ác tính không được điều trị dự phòng khoảng 8 - 24%. Candida có thể tìm thấy trên da và niêm mạc của người bình thường; khi mất tính toàn vẹn của da và niêm mạc (chủ yếu là tổn thương niêm mạc đường tiêu hóa do hóa trị) sẽ dẫn đến sự xâm nhập của Candida vào mô tạng và máu. Một đường xâm nhập phổ biến khác của Candida là qua catheter tĩnh mạch trung tâm. Biểu hiện thường gặp nhất của nhiễm Candida xâm nhập là nhiễm Candida máu, ít gặp hơn là các trường hợp nhiễm nấm sâu ở nội tạng.
Aspergillus: Tỷ lệ nhiễm nấm xâm nhập do Aspergillus ở bệnh nhân lơ xê mi tủy cấp khoảng 2 - 28%. Đường xâm nhập chủ yếu của Aspergillus là qua đường không khí và gây tổn thương ở xoang, đường hô hấp. Biểu hiện hay gặp nhất của nhiễm Aspergillus xâm nhập là viêm phổi.
Các loại nấm hiếm gặp khác: Mucormycosis, Fusarium spp và Scedosporium spp.
3. YẾU TỐ NGUY CƠ
Bảng 15. Yếu tố nguy cơ nhiễm nấm
|
Nguy cơ thấp |
- Ghép tự thân; - Lơ xê mi lympho cấp trẻ em (trừ viêm phổi do Pneumocystic carinii); - U lympho. |
|
Nguy cơ trung bình |
- BCTT 0,1-0,5 G/l < 3 tuần; - BC lympho < 0,5 G/l + kháng sinh; - Lớn tuổi; - Có sonde tĩnh mạch trung tâm. |
|
Nguy cơ trung bình cao |
- BCTT 0,1-0,5 G/l > 3-5 tuần; - Lơ xê mi tủy cấp; - Tia xạ toàn thân; - Ghép đồng loài với người cho hòa hợp. |
|
Nguy cơ cao |
- BCTT < 0,1 G/l > 3 tuần; - BCTT < 0,5 G/l > 5 tuần; - Tổn thương niêm mạc miệng và đường tiêu hóa do hóa trị; - Sử dụng kháng sinh phổ rộng dài ngày; - Sử dụng thuốc ức chế miễn dịch dài ngày; - Bệnh nhân có đặt catheter tĩnh mạch trung tâm (CVC, PICC line, Port-a-cath); - Bệnh nhân lơ xê mi cấp hoặc rối loạn sinh tủy giai đoạn muộn, bệnh tái phát/ kháng thuốc; - Bệnh nhân lơ xê mi tủy cấp trên 65 tuổi; - Bệnh nhân ghép đồng loài không cùng huyết thống - Bệnh nhân có nhiều bệnh lý mạn tính kèm theo: đái tháo đường, bệnh phổi tắc nghẽn mạn tính, nhiễm virus đường hô hấp, toàn trạng theo ECOG kém; - Suy giảm chức năng thực bào do hội chứng rối loạn sinh tủy; - Tình trạng kê sinh của nấm Aspergillus ở đường hô hấp từ trước; ký sinh của nấm Candida ở nhiều vị trí trong cơ thể; - Phòng điều trị không có HEPA filter, gần khu vực đang sửa chữa cải tạo phòng ốc; - Bệnh nhân được cấy ghép các dụng cụ nhân tạo: stent, sonde... |
4. CHẨN ĐOÁN
4.1. Nhiễm Candida xâm nhập
Có 2 hình thức nhiễm Candida xâm nhập, đó là nhiễm Candida máu và nhiễm Candida lan tỏa ở nội tạng (gan, lách, thận, hệ thần kinh trung ương, mắt, nội tâm mạc…).
a. Triệu chứng lâm sàng của nhiễm Candida máu rất đa dạng, gồm:
- Sốt, nhất là ở bệnh nhân đã được dùng kháng sinh mạnh phổ rộng.
- Tổn thương da dạng ban hoặc nốt đỏ.
- Tổn thương ở mắt: Viêm màng bồ đào và võng mạc.
- Tổn thương cơ dạng áp xe hiếm gặp.
- Nặng hơn có thể sốc nhiễm trùng và suy đa tạng.
b. Triệu chứng xét nghiệm của nhiễm Candida xâm nhập gồm:
- Cấy máu dương tính với Candida, tuy nhiên chỉ 50% trường hợp nhiễm Candida máu cho kết quả dương tính.
- Sinh thiết tổ chức tổn thương thấy Candida.
- Phát hiện beta-D-glucan, một thành phần của vách tế bào nấm, tuy vậy, không đặc hiệu cho Candida do beta-D-glucan có mặt ở nhiều loại nấm khác nhau. Do đó, xét nghiệm này được khuyến cáo thực hiện cùng với cấy máu và sinh thiết tổ chức tổn thương để tăng giá trị chẩn đoán.
- Xét nghiệm T2Candida phát hiện và khuếch đại ADN của nấm Candaida từ bệnh phẩm máu bằng kỹ thuật cộng hưởng từ. Xét nghiệm có thể cho kết quả sau 3 - 5 giờ, sớm hơn rất nhiều so với kỹ thuật nuôi cấy.
- Xét nghiệm PCR phát hiện ADN của nấm Candida. Ưu điểm của kỹ thuật này là nhạy hơn trong trường hợp bệnh nhân nhiễm Candida lan tỏa ở tạng.
4.2. Nhiễm Aspergillus xâm nhập
Nhiễm Aspergillus xâm nhập biểu hiện chủ yếu ở phổi và xoang, xảy ra sau khi hít phải bào tử của nấm. Ít gặp hơn là những trường hợp xâm nhập qua đường tiêu hóa hoặc trực tiếp qua da do tiếp xúc.
a. Triệu chứng lâm sàng của nhiễm Aspergillus phổi:
- Sốt.
- Đau ngực.
- Khó thở.
- Ho, có thể ho ra máu.
Tuy nhiên, ngay cả khi không có những triệu chứng điển hình cũng không loại trừ được nhiễm Aspergillus ở bệnh nhân nguy cơ cao, do bệnh nhân giảm bạch cầu hạt trung tính thường chỉ có sốt mà không có triệu chứng khu trú tại phổi.
b. Triệu chứng xét nghiệm của nhiễm Aspergillus phổi:
- Chụp cắt lớp vi tính phổi: Một hoặc nhiều nốt tổn thương nhu mô phổi có hoặc không kèm theo hình ảnh hang hóa; tổn thương xâm nhập cây phế quản ngoại vi (dấu hiệu tree-in-bud); tổn thương lan tỏa dạng kính mờ (dấu hiệu halo).
- Xét nghiệm phát hiện galactomannan huyết thanh: Đây là một thành phần chính của vách tế bào nấm Aspergillus.
- Nhuộm soi và cấy đờm hoặc dịch rửa phế quản thấy nấm Aspergillus.
- Sinh thiết tổn thương phổi dưới hướng dẫn của chụp cắt lớp vi tính: thấy hình ảnh nấm sợi xâm nhập mô phế quản gần các mạch máu, kèm theo hình ảnh hoại tử.
- Xét nghiệm ADN của nấm Aspergillus bằng kỹ thuật PCR.
4.3. Chẩn đoán mức độ
- Nhiễm nấm xâm nhập có bằng chứng (proven): Tìm thấy thành phần tế bào nấm cùng với bằng chứng tổn thương tổ chức hoặc nuôi cấy phát hiện nấm từ bệnh phẩm ở những vị trí vô trùng (ví dụ như phổi hoặc da).
- Nhiễm nấm xâm nhập nhiều khả năng (probable): Có triệu chứng lâm sàng phù hợp với nhiễm nấm xâm nhập và phát hiện được nấm gây bệnh bằng phương pháp nuôi cấy hoặc marker sinh học (beta-D-glucan, galactomannan).
- Có thể nhiễm nấm xâm nhập (possible): Chỉ có triệu chứng lâm sàng và yếu tố nguy cơ nhiễm nấm xâm nhập mà không có bằng chứng về vi nấm học.
5. ĐIỀU TRỊ DỰ PHÒNG Ở BỆNH NHÂN NGUY CƠ CAO
Bảng 16. Khuyến cáo dự phòng nhiễm nấm xâm nhập trên bệnh nhân huyết học nguy cơ trung bình và cao (theo Mạng ung thư Quốc gia Hoa Kỳ - 2018)
|
Bệnh lý- Nguy cơ |
Thuốc chống nấm dự phòng |
Thời gian điều trị dự phòng |
|
Lơ xê mi lympho cấp |
Fluconazol, itraconazol hoặc Micafungin, Amphotericin B |
Đến khi hết giai đoạn giảm bạch cầu hạt trung tính |
|
Hội chứng rối loạn sinh tủy Lơ xê mi tủy cấp |
Posaconazol (khuyến cáo mạnh) Voriconazol, Fluconazol, itraconazol, Micafungin hoặc Amphotericin B |
|
|
Ghép tế bào gốc tự thân có tình trạng viêm niêm mạc |
Fluconazol, hoặc Micafungin (cả 2 đều được khuyến cáo mạnh) itraconazole |
|
|
Ghép tế bào gốc tự thân không có tình trạng viêm niêm mạc |
Cân nhắc không cần dự phòng (khuyến cáo trung bình) |
|
|
Ghép tế bào gốc đồng loài (giai đoạn giảm bạch cầu hạt trung tính) |
Fluconazol, hoặc Micafungin (cả 2 đều được khuyến cáo mạnh) Voriconazol, Posaconazol, hoặc Amphotericin B (khuyến cáo trung bình) itraconazole |
Trong giai đoạn giảm bạch cầu hạt trung tính |
|
Ghép tế bào gốc đồng loài có bệnh ghép chống chủ |
Posaconazol (khuyến cáo mạnh) Voriconazol, Echinocandin, hoặc Amphotericin B (tất cả được khuyến cáo trung bình) itraconazole |
Đến khi hết triệu chứng của bệnh ghép chống chủ |
Bảng 17. Liều thuốc chống nấm dự phòng
|
Thuốc |
Liều dự phòng |
|
Posaconazole, dung dịch uống |
200 mg uống 3 lần/ngày |
|
Posaconazole, viên |
300 mg uống 1 lần/ngày (2 lần vào ngày đầu) |
|
Amphotericin B dạng lipid, khí dung |
12,5 mg khí dung 2 lần/tuần |
|
Amphotericin B dạng lipid, truyền tĩnh mạch |
50 mg mỗi 48 giờ hoặc 5 mg/kg 2 lần/tuần |
|
Caspofungin |
50 mg/ngày, truyền tĩnh mạch |
|
Micafungin |
50-100 mg/ngày, truyền tĩnh mạch |
|
Fluconazole |
400 mg uống 1 lần/ngày |
|
Itraconazole, viên nang |
200 mg uống 2 lần/ngày |
|
Voriconazole |
200 mg uống 2 lần/ngày |
6. ĐIỀU TRỊ BỆNH NHÂN NHIỄM NẤM XÂM NHẬP
Nguyên tắc điều trị:
- Cân nhắc một số yếu tố khi lựa chọn thuốc kháng nấm: tình trạng bệnh nhân, tác nhân gây bệnh, thuốc sẵn có, vị trí nhiễm trùng.
- Khởi đầu điều trị bằng đường tiêm truyền tĩnh mạch và chuyển sang dùng đường uống sau khi bệnh đáp ứng tốt, chức năng đường tiêu hóa còn nguyên vẹn.
- Với những tác nhân gây bệnh kháng nhiều loại thuốc kháng nấm, lựa chọn thuốc dựa trên kháng sinh đồ.
- Kết hợp điều trị thuốc kháng nấm song song với nâng đỡ hệ thống miễn dịch, bao gồm: Giảm liều corticoid (nếu đang sử dụng), cân nhắc sử dụng G-CSF và truyền bạch cầu hạt nếu dự tính bạch cầu hạt không hồi phục trong 3-4 ngày tiếp theo.
- Rút sớm catheter tĩnh mạch trung tâm nếu có thể.
- Cấy máu cách ngày nếu bệnh nhân nhiễm nấm máu.
6.1. Điều trị theo kinh nghiệm
a. Thời điểm điều trị: Nếu sau 72 giờ, ở bệnh nhân giảm bạch cầu hạt < 0,5 G/l đang điều trị kháng sinh phổ rộng thích hợp vẫn tồn tại sốt > 38ºC hoặc sốt lại sau 3-7 ngày thì nên bắt đầu điều trị kháng nấm theo kinh nghiệm ngay.
b. Thuốc lựa chọn:
- Caspofungin 70 mg/ngày 1, sau đó 50 mg/ngày truyền tĩnh mạch.
- Amphotericin B dạng lipid 3-5 mg/kg/ngày truyền tĩnh mạch.
- Amphotericin B dạng desoxycholate 0,5-1 mg/kg/ngày truyền tĩnh mạch.
- Voriconazole 6 mg/kg/12 giờ trong ngày 1, sau đó 3 mg/kg/12 giờ truyền tĩnh mạch hoặc 200 mg/12 giờ uống.
- Itraconazole 250 mg/ngày truyền tĩnh mạch.
c. Thời gian điều trị:
- Ít nhất 14 ngày.
- Trong giai đoạn giảm bạch cầu hạt.
6.2. Điều trị nhiễm Candida xâm nhập
a. Lựa chọn hàng 1:
- Echinocandin (caspofungin: Liều nạp 70mg, sau đó 50mg/ngày; micafungin 100mg/ ngày; anidulafungin: Liều nạp 200mg, sau đó 100mg/ngày).
- Amphotericin B dạng lipid 3-5mg/kg/ngày.
- Amphotericin B dạng desoxycholate 0,5-1 mg/kg/ngày (nếu không có khả năng dùng echinocandin hoặc amphotericin B dạng lipid).
b. Lựa chọn thay thế:
- Fluconazole liều nạp 800mg (12mg/kg), sau đó 400mg (6mg/kg)/ ngày cho những bệnh nhân có tình trạng lâm sàng không quá nặng và chưa dùng azole trước đó.
- Voriconazole truyền tĩnh mạch 400mg (6mg/kg) 2 lần/ ngày x 2 ngày đầu, sau đó 200-300mg (3-4 mg/kg) 2 lần/ ngày.
c. Khi tình trạng lâm sàng ổn định, có thể chuyển sang dùng thuốc đường uống:
- Fluconazole uống 400mg (6 mg/kg)/ngày ở bệnh nhân lâm sàng ổn định, cấy máu âm tính, kháng nấm đồ nhạy với fluconazole.
- Voriconazole uống 200-300mg (3-4 mg/kg) x 2 lần/ngày ở bệnh nhân lâm sàng ổn định, cấy máu âm tính, kháng nấm đồ nhạy với voriconazole.
d. Thời gian điều trị: Tối thiểu 2 tuần sau khi cấy máu âm tính, hết giai đoạn giảm bạch cầu hạt và không còn triệu chứng do nhiễm candida máu.
6.3. Điều trị nhiễm Aspergillus xâm nhập
a. Lựa chọn hàng 1:
- Voriconazole truyền tĩnh mạch 400mg (6mg/kg) 2 lần/ngày x 2 ngày đầu, sau đó 200-300mg (3-4 mg/kg) 2 lần/ ngày.
- Amphotericin B dạng lipid 3-5 mg/kg/ ngày.
- Amphotericin B dạng desoxycholate 0,6-0,7 mg/kg/ngày (nếu không có khả năng dùng voriconazole hoặc amphotericin B dạng lipid).
b. Khi tình trạng lâm sàng ổn định, có thể chuyển sang dùng thuốc đường uống:
- Voriconazole uống 200-300mg (3-4 mg/kg) x 2 lần/ngày.
c. Điều trị cứu vãn:
- Posaconazole dung dịch uống 200mg x 3 lần/ngày; hoặc truyền tĩnh mạch 300mg x 2 lần trong ngày đầu, sau đó 300mg/ ngày.
- Itraconazole uống 200 mg x 2 lần/ngày.
- Phối hợp Voriconazole + Echinocandin.
d. Thời gian điều trị:
- Tối thiểu 6-12 tuần, phụ thuộc vào mức độ suy giảm miễn dịch, vị trí nhiễm nấm, đáp ứng với điều trị.
- Tiếp tục điều trị dự phòng nếu bệnh nhân vẫn duy trì thuốc ức chế miễn dịch.
TÀI LIỆU THAM KHẢO
1. NCCN clinical practice guidelines in oncology: prevention and treatment of cancer- related infections. Version 1.2018.
2. Marcio Nucci, Elias Anaissie (2014). How we treat invasive fungal diseases in patients with acute leukemia: the importance of an individualized approach. Blood: 3858-3869.
3. Sibylle C. Mellinghoff, Jens Panse et al (2018). Primary prophylaxis of invasive fungal infections in patients with haematological malignancies: 2017 update of the recommendations of the Infectious Diseases Working Party (AGIHO) of the German Society for Haematology and Medical Oncology (DGHO). Annals of Hematology 97:197-207.
4. Ben De Pauw, Thomas J. Walsh et al (2008). Revised Definitions of Invasive Fungal Disease from the European Organization for Research and Treatment of Cancer/Invasive. Clin Infect Dis: 46(12): 1813-1821.
5. Thomas F. Patterson, George R. Thompson et al (2016). Practice Guidelines for the Diagnosis and management of Aspergillosis: 2016 Update by the Infectious diseases Society of America. Clin Infect Dis: 63(4): e1-60.
6. Peter G. Pappas, Carol A. Kauffman et al (2016). Clinical Practice Guideline for the management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin Infect Dis: 62(4): e1-50.
7. Frederic Tissot, Samir Agrawal et al (2017). ECIL-6 guidelines for the treatment of invasive candidiasis, aspergillosis and mucormycosis in leukemia and hematopoietic stem cell transplant patients. Hematologica, vol 102(3): 433-444.
CHƯƠNG II. BỆNH MÁU ÁC TÍNH
20. LƠ XÊ MI CẤP (Bệnh bạch cầu cấp)
1. ĐẠI CƯƠNG
Lơ xê mi (LXM) cấp là một nhóm bệnh máu ác tính, có đặc trưng là sự tăng sinh một loại tế bào non - ác tính (tế bào blast), nguồn gốc tại tuỷ xương. Có nhiều yếu tố nguy cơ làm tăng tỷ lệ mắc lơ xê mi cấp, như: Tia xạ; hoá chất; virus HTLV1, HTLV2; yếu tố di truyền; lơ xê mi cấp thứ phát sau hội chứng rối loạn sinh tủy (MDS), các bệnh tăng sinh tủy ác tính (MPNs), sau dùng thuốc hóa chất.
2. CHẨN ĐOÁN
2.1. Triệu chứng lâm sàng
- Hội chứng thiếu máu.
- Hội chứng xuất huyết: Thường do giảm tiểu cầu, xuất huyết tự nhiên, hay gặp ở da - niêm mạc, nặng hơn có thể gặp xuất huyết nội tạng. Đông máu rải rác trong lòng mạch (DIC), đặc biệt hay gặp trong lơ xê mi tiền tủy bào cấp.
- Hội chứng nhiễm trùng: Sốt, viêm loét miệng họng, viêm phổi, nhiễm trùng da...
- Hội chứng thâm nhiễm: Gan, lách, hạch to, phì đại lợi, thâm nhiễm da, thâm nhiễm thần kinh trung ương...
- Có thể gặp triệu chứng tắc mạch do tăng bạch cầu.
- Biểu hiện toàn thân do bệnh lý ác tính: Mệt mỏi, gầy sút, suy sụp nhanh.
2.2. Triệu chứng xét nghiệm
a. Xét nghiệm tế bào máu ngoại vi
- Thiếu máu bình sắc, hồng cầu kích thước bình thường, hồng cầu lưới giảm.
- Bạch cầu thường tăng, nhưng có thể bình thường hoặc giảm, gặp một tỷ lệ tế bào non (tế bào blast) ác tính.
- Số lượng tiểu cầu giảm.
b. Xét nghiệm tủy xương
- Tuỷ đồ là xét nghiệm quyết định chẩn đoán, cho thấy các tế bào blast chiếm tỷ lệ ≥ 20% các tế bào có nhân trong tủy; các dòng hồng cầu, bạch cầu hạt và mẫu tiểu cầu bị lấn át bởi tế bào blast.
- Sinh thiết tuỷ xương được chỉ định trong trường hợp chọc hút tuỷ không chẩn đoán xác định được do tủy nghèo tế bào thấy ổ tế bào blast.
c. Xét nghiệm miễn dịch phát hiện dấu ấn của tế bào non - ác tính
- Nhuộm hóa học tế bào sử dụng dịch hút tủy xương: Peroxidase, sudan đen, PAS.
- Phân tích biểu hiện kháng nguyên tế bào blast (CD) bằng kỹ thuật đếm tế bào dòng chảy (flow cytometry).
d. Xét nghiệm nhiễm sắc thể và gen có thể gặp một số bất thường:
- Phân tích công thức NST tủy xương.
- Phân tích các đột biến gen.
- Với LXM tủy cấp cần làm các đột biến sau: c-KIT, FLT3-ITD, FLT3-TKD, NPM1, CEBPA, IDH1, IDH2, ASXL1, TP53, RUNX1-RUNXT1, CBFB-MYH11, MLLT3-KMT2A, GATA2…
- Với LXM lympho cấp (acute lymphoblastic leukemia - ALL): NST Ph - t(9;22) và/hoặc gen bcr-abl, t(4;11), t(1;19), t(12;21), t(11;19)… hoặc tái tổ hợp gen MLL, tái sắp xếp gen KMT2A, đột biến ETV6-RUNX1...
e. Xét nghiệm hóa sinh
Chức năng gan thận, điện giải đồ, calci, phospho, LDH, aicd uric để phát hiện hội chứng tiêu khối u.
f. Xét nghiệm các virus
Viêm gan B, viêm gan C, HIV, CMV.
g. Chụp cắt lớp vi tính hoặc cộng hưởng từ sọ não: Nếu có triệu chứng thần kinh.
h. Xét nghiệm dịch não tủy: Để phát hiện thâm nhiễm thần kinh trung ương (đặc biệt trong LXM lympho cấp).
i. Điện tâm đồ và siêu âm tim: Đánh giá chức năng tâm thu thất trái (đặc biệt với với các bệnh nhân có tiền sử bệnh tim mạch hoặc sẽ điều trị bằng nhóm thuốc anthracycline hoặc tia xạ vùng ngực).
j. Xét nghiệm HLA: Cho bệnh nhân để tìm nguồn tế bào gốc phù hợp cho ghép tế bào gốc đồng loài.
2.3. Tiêu chuẩn chẩn đoán
2.3.1. Chẩn đoán xác định
- Dựa vào triệu chứng lâm sàng điển hình của bệnh.
- Xét nghiệm tuỷ đồ thấy tế bào blast ≥ 20% tế bào có nhân trong tuỷ.
2.3.2. Chẩn đoán thể bệnh và xếp loại lơ xê mi cấp
Chẩn đoán thể bệnh lơ xê mi cấp dựa vào các bảng xếp loại của WHO và FAB.
2.3.2.1. Xếp loại lơ xê mi tủy cấp (Bạch cầu cấp dòng tủy)
- Xếp loại lơ xê mi tủy cấp theo FAB 1986 có bổ sung.
- Kết hợp miễn dịch chia thành 8 thể, từ M0 đến M7 (bảng 18).
Bảng 18. Xếp loại LXM tủy cấp theo FAB kết hợp dấu ấn miễn dịch
|
Thể bệnh |
Đặc điểm hình thái, hóa học tế bào |
Dấu ấn miễn dịch |
|
M0 |
Tế bào non chưa biệt hóa ≥ 90% các tế bào có nhân không thuộc dòng hồng cầu, không có thể Auer, <3% MPO+ |
CD34+ |
|
M1 |
Tế bào non chưa trưởng thành dòng tủy ≥ 90% các tế bào có nhân không thuộc dòng hồng cầu, hiếm gặp thể Auer, > 3% MPO+ |
HLA-DR, CD13, CD33, CD15, CD11± |
|
M2 |
Tế bào non đầu dòng tủy < 90% các tế bào có nhân không thuộc dòng hồng cầu, nhiều thể Auer |
HLA-DR, CD13, CD33, CD15, CD11± |
|
M3 |
Lơ xê mi tiền tủy bào cấp Dưới nhóm: M3v |
CD33, CD13, CD15, CD11 |
|
M4 |
Lơ xê mi tủy-mono cấp Dưới nhóm: M4eo |
HLA-DR, CD34±, CD33, CD15±, CD14, CD64, CD11 |
|
M5 |
Lơ xê mi mono cấp |
HLA-DR, CD34±, CD33, CD15±, CD14, CD64, CD11 |
|
M6 |
> 80% là các tiền thân hồng cầu ≥ 30% là các nguyên tiền hồng cầu tỷ lệ blast dòng tủy < 20% |
Glycophorin A |
|
M7 |
≥ 50% blast thuộc dòng mẫu tiểu cầu |
HLA-DR, CD61, CD42, CD34±, CD33± |
Xếp loại lơ xê mi tủy cấp theo Tổ chức Y tế thế giới (WHO) 2016
a. LXM tủy cấp có kèm theo bất thường vật chất di truyền tái diễn
- LXM tủy cấp với t(8;21)(q22;q22); gen RUNX1-RUNX1T1.
- LXM tủy cấp với inv(16)(p13.1;q22) hoặc t(16;16)(p13.1;q22); gen CBFβ/MYH11.
- LXM cấp thể tiền tủy bào với gen PML/RARα.
- LXM tủy cấp với t(9;11)(p22;q23); gen MLLT3/KMT2A.
- LXM tủy cấp với t(6;9)(p23;q34); gen DEK/NUP214.
- LXM tủy cấp với inv(3)(q21;q26.2) hoặc t(3;3)(q21.3;q26.2); gen GATA2, MECOM.
- LXM tủy cấp (dòng mẫu tiểu cầu) với t(1;22)(p13;q13); gen RBM15-MKL1.
- Thể để xuất bổ sung LXM tủy cấp với BCR-ABL1.
- LXM tủy cấp có biến đổi gen NPM1.
- LXM tủy cấp có biến đổi gen CEBPA.
- Thể đề xuất bổ sung LXM tủy cấpvới đột biến RUNX1.
b. LXM tủy cấp có liên quan với hội chứng rối loạn sinh tủy.
c. LXM tủy cấp có liên quan đến điều trị.
d. LXM tủy cấp không xếp loại được theo các cách khác
- LXM tủy cấp có sự biệt hóa tối thiểu.
- LXM tủy cấp không có sự trưởng thành.
- LXM cấp dòng tuỷ có sự trưởng thành.
- LXM cấp tủy-mono.
- LXM cấp mono/nguyên bào mono.
- LXM cấp dòng hồng cầu.
- LXM cấp dòng mẫu tiểu cầu.
- LXM cấp dòng bạch cầu hạt ưa base.
- Tăng tế bào tủy cấp kèm theo xơ tủy.
e. Sarcoma tủy.
f. Tăng sinh dòng tủy có liên quan đến hội chứng Down
- Sinh tủy bất thường thoáng qua (transient abnormal myelopoiesis - TAM).
- LXM tủy cấp liên quan đến hội chứng Down.
g. Tân sản tế bào tua non dạng tương bào.
h. LXM cấp hỗn hợp dòng
- LXM cấp tế bào không biệt hóa.
- LXM cấp kiểu hình hỗn hợp (mixed phenotype acute leukemia - MPAL) với t(9;22)(q34.1;q11.2); gen BCR-ABL1.
- MPAL dòng tế bào B/tủy, không phân loại được theo cách khác.
- MPAL dòng tế bào T/tủy, không phân loại được theo cách khác.
2.3.2.2. Xếp loại LXM lympho cấp (Bạch cầu cấp dòng lympho).
a. Xếp loại LXM lympho cấp theo FAB: chia 3 thể
Bảng 19. Xếp loại LXM lympho cấp theo FAB 1986
|
Thể bệnh theo FAB |
Đặc điểm hình thái tế bào |
|
- ALL thể L1 |
Các tế bào có kích thước nhỏ, đồng đều |
|
- ALL thể L2 |
Các tế bào có kích thước lớn; to nhỏ không đồng đều |
|
- ALL thể L3 |
Các tế bào có kích thước lớn, nhiều hốc trong nguyên sinh chất |
b. Xếp loại LXM lympho cấp theo WHO 2016
Đặc điểm dấu ấn miễn dịch theo dòng lympho B và T
Bảng 20. Xếp loại LXM lympho cấp (Acute lymphoblastic leukemia-ALL) theo đặc trưng dấu ấn miễn dịch
|
Thể bệnh |
Đặc điểm dấu ấn miễn dịch |
|
|
Dòng lympho B |
ALL tế bào B sớm (pro-B) |
CD 19 (+), CD79a (+), CD22 (+), TdT (+), CD10 (-), sIg (-) |
|
ALL tế bào tiền B (pre-B) |
CD 19 (+), CD79a (+), CD22 (+), cIg (+), CD10 (+) [CD10 còn gọi là kháng nguyên B phổ biến (Common B ALL antigen - CALLA)] |
|
|
ALL tế bào B trưởng thành (mature B/Burkitt) |
TdT (-), sIg (+) |
|
|
Dòng lympho T |
ALL tế bào T sớm (pro-T) |
cCD3 (+), CD7 (+), TdT (+) CD1a (-), CD2 (-), sCD3 (-), CD4 (-), CD8 (-), CD34 (+/-) |
|
ALL tế bào tiền T (pre-T) |
cCD3 (+), CD7 (+), CD2 (+), TdT (+) CD1a (-), CD4 (-), CD8 (-), CD34 (+/-) |
|
|
ALL tế bào T vỏ (cortical T- ALL) |
cCD3 (+),CD7 (+), CD1a (+), CD2 (+), CD4 (+), CD8 (+), CD34 (-) |
|
|
ALL tế bào T tủy (medullary T-ALL) |
sCD3 (+), cCD3 (+),CD7 (+), CD1a (-), CD2 (+), CD4 (+) hoặc CD8 (+), CD34 (-) |
|
Chú thích: cIG: immunoglobulin bào tương (cytoplasmic); sIg: immunoglobulin bề mặt (surface).
- Các thể bệnh LXM lympho cấp theo WHO 2016
(1) LXM cấp/u lympho ác tính dòng tế bào lympho B:
+ LXM cấp/u lympho ác tính dòng tế bào lympho B không phân loại được theo cách khác.
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với các biến đổi di truyền tái diễn:
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với t(9;22)(q34;q11.2); BCR/ABL1;
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với t(v;11q23); KMT2A tái tổ hợp;
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với t(12;21)(p13;q22); ETV6/RUNX1;
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với biến đổi thêm bội;
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với biến đổi thiểu bội;
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với t(5;14)(q31;q32): IL3-IGH;
+ LXM cấp/u lympho ác tính dòng tế bào lympho B với t(1;19)(q23;p13.3): TCF3-PBX1;
+ Thể đề xuất bổ sung LXM cấp/u lympho ác tính dòng tế bào lympho B biểu hiện tương tự BCR-ABL (BCR-ABL like);
+ Thể đề xuất bổ sung LXM cấp/u lympho ác tính dòng tế bào lympho B với iAMP21.
(2) LXM cấp/u lympho ác tính dòng tế bào lympho T:
+ Thể đề xuất bổ sung: LXM cấp/u lympho ác tính dòng tế bào tiền thân T sớm (early T-cell precursor).
(3) Thể đề xuất bổ sung: LXM cấp/u lympho ác tính dòng tế bào lympho NK.
2.3.3. Chẩn đoán phân biệt
Lơ xê mi cấp cần được chẩn đoán phân biệt với phản ứng giả lơ xê mi gặp trong nhiễm trùng, ung thư di căn tủy xương, hội chứng rối loạn sinh tủy (MDS), các bệnh tăng sinh tủy ác tính (MPNs)… Phân biệt bằng kết hợp các triệu chứng lâm sàng, xét nghiệm tiêu chuẩn chẩn đoán lơ xê mi nêu trên.
3. ĐIỀU TRỊ LƠ XÊ MI CẤP
- Điều trị Lơ xê mi cấp là một phương pháp điều trị chuyên khoa sâu. Do đó, việc điều trị chỉ có thể được thực hiện ở các cơ sở chuyên ngành huyết học, do bác sĩ được đào tạo chuyên ngành huyết học và có kinh nghiệm điều trị hóa chất/ ghép tế bào gốc tạo máu thực hiện.
- Các cơ sở y tế không chuyên khoa huyết học chủ yếu thực hiện việc phát hiện, chẩn đoán bệnh, điều trị ban đầu trước khi chuyển tuyến chuyên khoa, cũng như theo dõi người bệnh ngoại trú giữa các đợt điều trị hóa chất và sau khi ghép tế bào gốc tạo máu.
- Do vậy, trong bài này chúng tôi chỉ mô tả nguyên tắc điều trị, nguyên tắc theo dõi điều trị và một số phác đồ điều trị thường dùng để tham khảo.
3.1. Điều trị lơ xê mi tủy cấp (Acute myeloblastic leukemia-AML) không phải thể tiền tủy bào (APL)
3.1.1. Phân nhóm tiên lượng theo bất thường NST và gen
|
Nhóm tiên lượng |
Bất thường NST và gen |
|
Tốt |
t(8;21)(q22;q22.1); RUNX1-RUNX1T1 inv(16)(p13.1q22) hoặc t(16 ;16)(p13.1;q22); CBFB-MYH11 Đột biến kép CEBPA Đột biến NPM1 không kèm FLT3-ITD hoặc có FLT3-ITD mức thấp |
|
Trung bình |
Đột biến NPM1 kèm FLT3-ITD mức cao Không có đột biến NPM1 và FLT3-ITD hoặc FLT3-ITD mức thấp (không kèm các tổn thương di truyền xấu khác) t(9;11)(p21.3;q23.3); MLLT3-KMT2A Các bất thường di truyền tế bào không thuộc nhóm xấu hoặc tốt |
|
Xấu |
t(6;9)(p23;q34.1); DEK-NUP214 t(v;11q23.3); tái sắp xếp KMT2A t(9;22)(q34.1;q11.2) ; BCR-ABL1 inv(3)(q21.3q26.2) hoặc t(3;3)(q21.3 ;q26.2); GATA2, MECOM(EVI1) -5 hoặc del(5q); -7 hoặc -17/bất thường 17p Đa tổn thương NST; có NST đơn (monosomies) Đột biến FLT3-ITD mức cao không kèm đột biến NPM1 Đột biến RUNX1 Đột biến ASXL1 Đột biến TP53 |
3.1.2. Phác đồ hóa trị liệu tiêu chuẩn
a. Đối với người bệnh dưới 60 tuổi
Phác đồ hóa trị liệu tiêu chuẩn bao gồm: Phác đồ tấn công (điều trị cảm ứng) “3+7”, củng cố bằng cytarabin liều cao (Hi-DAC) 4 đợt. Cụ thể như sau:
- Phác đồ “3+7”.
- Phác đồ cytarabin liều cao (Hi-DAC).
- Lơ xê mi mono cấp hoặc tủy-mono hoặc tủy cấp có số lượng bạch cầu lúc chẩn đoán > 50 G/L cần được điều trị dự phòng thâm nhiễm thần kinh trung ương.
b. Đối với người bệnh trên 60 tuổi
Tùy theo thể trạng người bệnh, có thể sử dụng cytarabin liều thấp (20mg/m2 da tiêm dưới da trong 10 ngày), hoặc phác đồ “3+7” giảm số ngày điều trị (“2+5”), hoặc Decitabine 20mg/m2 da/ngày x 5 ngày, hoặc Azacitidine 75mg/m2 da tiêm tĩnh mạch hoặc dưới da trong 7 ngày.
3.1.3. Điều trị LXM tủy cấp tái phát, kháng thuốc
Với LXM tủy cấp tái phát hoặc kháng thuốc, có thể sử dụng các phác đồ hóa trị liệu liều cao như phác đồ ADE, MEC, HAM, FLAG ± Daunorubicin hoặc Idarubicin hoặc Mitoxantron, Hi-DAC ± Daunorubicin hoặc Mitoxantron; nên tiến tới ghép đồng loài nếu đủ điều kiện.
3.1.4. Chỉ định ghép tế bào gốc tạo máu đồng loài (chi tiết xin xem bài Ghép tế bào gốc tạo máu)
- AML nguy cơ thấp nên được điều trị hóa chất. Ghép tế bào gốc tạo máu đồng loài được chỉ định sau khi tái phát và đã điều trị đạt lui bệnh hoàn toàn lần 2.
- AML nguy cơ hoặc cao ít khi đáp ứng tốt với hóa trị liệu vì thế nên chỉ định ghép tế bào gốc tạo máu đồng loài sau lui bệnh hoàn toàn lần 1.
3.2. Điều trị Lơ xê mi cấp thể tiền tủy bào (APL)
a. Phác đồ CALGB 9710
- Điều trị tấn công:
+ Người lớn: ATRA (45 mg/m2 da/ngày đến khi lui bệnh hoàn toàn: Tối đa 90 ngày); Trẻ em: ATRA 25mg/m2 da/ngày chia hai lần;
+ Daunorubicin 50 mg/m2 da/ngày, truyền tĩnh mạch ngày 3-6;
+ Ara-C 200 mg/m2 da/ngày, truyền tĩnh mạch ngày 3-9.
- Điều trị củng cố: 2-3 đợt:
+ ATRA: Người lớn 45 mg/m2 da/ngày; trẻ em: 25mg/m2 da/ngày (uống ngày 1-7);
+ Daunorubicin 50 mg/m2 da/ngày, truyền tĩnh mạch ngày 3-5.
- Điều trị duy trì: Bằng ATRA 45 mg/m2 da đường uống hàng ngày trong 15 ngày mỗi 3 tháng, mercaptopurine 60 mg/m2 da 1 lần hàng ngày và methotrexate 20 mg/m2 da 1 lần hàng tuần trong 2 năm.
- Đối với Lơ xê mi cấp tiền tủy bào tái phát, lựa chọn điều trị bằng Arsenic trioxide (ATO) với liều: ATO 0,15 mg/kg/ngày đến khi lui bệnh hoàn toàn trong tủy xương, tối đa 60 liều, trung bình 35 liều. Điều trị củng cố bằng ATO với liều như trên, 25 liều trong vòng 5 tuần.
- Đối với người bệnh trên 60 tuổi: Điều trị ATRA đơn độc hoặc ATO.
b. Phác đồ theo NCCN Guidelines Version 3.2020
- Nhóm APL nguy cơ thấp (BC máu ngoại vi ≤ 10G/l):
+ Phác đồ tấn công:
● ATRA: 45 mg/m2/ngày, uống chia 2 lần, liên tục đến khi có cải thiện lâm sàng;
● ATO: 0,15 mg/kg/ngày, truyền tĩnh mạch ngày 1 lần x 4 tuần.
Sau 28 ngày làm lại xét nghiệm tủy, đánh giá lui bệnh trước khi chuyển sang điều trị phác đồ củng cố.
+ Phác đồ củng cố:
● ATRA: 45 mg/m2/ngày, uống chia 2 lần, mỗi đợt uống trong 2 tuần, cách 4 tuần/đợt x 7 đợt;
● ATO: 0,15 mg/kg/ngày x 5 ngày/tuần x 4 tuần, cách 8 tuần/đợt x 4 đợt.
- Nhóm APL nguy cơ cao (BC máu ngoại vi > 10G/l):
+ Phác đồ tấn công:
● ATRA: 45 mg/m2/ngày, uống chia 2 lần, liên tục đến khi có cải thiện lâm sàng;
● Daunorubicin: 50 mg/m2 da/ngày, truyền tĩnh mạch ngày 3-6;
● Cytarabine: 200 mg/m2 da/ngày, truyền tĩnh mạch ngày 3-9.
Sau 28 ngày làm lại xét nghiệm tủy, đánh giá lui bệnh trước khi chuyển sang điều trị phác đồ củng cố.
+ Phác đồ củng cố:
● ATO: 0,15 mg/kg/ngày x 5 ngày/tuần x 5 tuần liên tục/1 đợt, cách 7 tuần/ đợt x 2 đợt.
+ Sau đó điều trị tiếp 2 đợt:
● Daunorubicin: 50 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-3;
● ATRA: 45 mg/m2/ngày, uống chia 2 lần, liên tục ngày 1-7.
Điều trị duy trì: Sau kết thúc điều trị củng cố xét nghiệm lại tủy, đánh giá lui bệnh, xét nghiệm gen PML/RARα định kỳ, theo dõi khả năng tái phát cho đến tối thiểu là 2 năm.
- Chuyển sang phác đồ điều trị duy trì trong 2 năm cho cả 2 nhóm nguy cơ, sau khi kết thúc điều trị củng cố bằng các thuốc:
+ ATRA: 45 mg/m2/ngày, uống chia 2 lần, mỗi đợt uống trong 2 tuần, cách 3 tháng/đợt;
+ 6-MP: 50mg/m2/ngày, uống hàng ngày;
+ Methotrexate: 15mg/m2/lần, uống tuần 1 lần.
- Điều trị APL tái phát:
+ Với nhóm bệnh nhân có thời gian tái phát sớm (< 6 tháng), phác đồ đã điều trị trước đó là ATO+ATRA: Dùng phác đồ hóa chất giống như APL nhóm nguy cơ cao điều trị lần đầu, kết hợp tiêm hóa chất nội tủy. Xét ghép tế bào gốc tự thân nếu đạt lui bệnh, gen PML/RARα âm tính, ghép tế bào gốc đồng loài nếu gen PML/RARα dương tính. Nếu không ghép được thì tiếp tục điều trị phác đồ ATO x 6 đợt;
+ Với nhóm bệnh nhân tái phát điều trị trước đó chưa có ATO, hoặc có thời gian tái phát muộn (≥ 6 tháng), phác đồ đã điều trị trước đó đã có ATO: Dùng phác đồ hóa chất ATRA 45 mg/m2 da/ngày ± ATO 0,15 mg/kg/ngày ± Daunorubicin 60mg/m2 da/ngày x 3 ngày, liên tục đến khi tủy xương hồi phục, kết hợp tiêm hóa chất nội tủy. Nếu đạt lui bệnh xét ghép tế bào gốc tự thân khi xét nghiệm gen PML/RARα âm tính, ghép tế bào gốc đồng loài khi gen PML/RARα dương tính;
+ Nếu APL tái phát điều trị hóa chất không đạt lui bệnh, xét khả năng ghép tế bào gốc đồng loài.
Lưu ý:
+ Điều chỉnh rối loạn đông máu;
+ Theo dõi và xử trí hội chứng biệt hóa;
+ Theo dõi độc tính của ATO trên tim mạch, thận (creatinine) và rối loạn điện giải (Ca, K, Cl, Mg);
+ Điện tim được làm trước điều trị tấn công, đánh giá khoảng QTc, làm lại hàng tuần để theo dõi. Trong các đợt sau làm điện tim trước mỗi đợt điều trị.
3.3. Điều trị Lơ xê mi lympho cấp ở người lớn
3.3.1. Nguyên tắc điều trị
- Dựa trên các tiêu chí: Lâm sàng, miễn dịch, tế bào di truyền, NCCN guideline chia thành các nhóm điều trị sau:
+ LXM lympho cấp ở bệnh nhân thanh thiếu niên và người trẻ tuổi (adolescent and young adult - AYA) có NST Ph: tuổi 15-39;
+ LXM lympho cấp ở bệnh nhân thanh thiếu niên và người trẻ tuổi (adolescent and young adult - AYA) không có NST Ph: tuổi 15-39;
+ LXM lympho cấp ở bệnh nhân người lớn (adult) có NST Ph: tuổi 40-64;
+ LXM lympho cấp ở bệnh nhân người lớn (adult) không có NST Ph: tuổi 40-64.
- Liệu trình điều trị gồm: Tấn công (điều trị cảm ứng), củng cố, dự phòng thâm nhiễm hệ thần kinh trung ương, điều trị duy trì. Bên cạnh các phác đồ mô tả dưới đây, có thể sử dụng một số phác đồ như GRAALL 2005, phác đồ Hyper-CVAD phối hợp với L-asparaginase. Với ALL có NST Ph(+) hóa trị liệu phối hợp với các thuốc ức chế hoạt tính tyrosine kinase (TKIs) và phối hợp với rituximab cho ALL có CD20(+).
3.3.2. Phác đồ điều trị
a. Điều trị LXM lympho cấp người lớn (adult)
Phác đồ CALGB 8811 (bao gồm điều trị tấn công, củng cố, duy trì, dự phòng thâm nhiễm thần kinh trung ương).
Đợt I: Điều trị tấn công (4 tuần):
- Cyclophosphamide 1.200 mg/m2 (800 mg/m2 nếu bệnh nhân trên 60 tuổi) đường tĩnh mạch; ngày 1.
- Daunorubicin 45 mg/m2/ngày (30 mg/m2/ ngày nếu bệnh nhân trên 60 tuổi) đường tĩnh mạch; ngày 1-3.
- Vincristine 2 mg đường tĩnh mạch; ngày 1, 8, 15 và 22.
- Methylprednisone 60 mg/m2/ngày đường uống; ngày 1-21 (ngày 1-7 nếu bệnh nhân trên 60 tuổi).
- L-asparaginase 6.000 UI/m2 tiêm dưới da; ngày 5, 8, 11, 15, 18 và 22.
Đợt II: Tăng cường sớm (4 tuần/chu kỳ, Tổng cộng 2 chu kỳ):
- Methotrexate 15 mg tiêm tủy sống; ngày 1;
- Cyclophosphamide 1.000 mg/m2 đường tĩnh mạch; ngày 1.
- 6-mercaptopurine 60 mg/m2/ngày đường uống; ngày 1-14.
- Cytarabine 75 mg/m2/ngày tiêm dưới da; ngày 1-4 và 8-11.
- Vincristine 2 mg đường tĩnh mạch; ngày 15 và 22.
- L-asparaginase 6.000 IU/m2 tiêm dưới da; ngày 15, 18, 22 và 25.
Đợt III: Dự phòng thâm nhiễm thần kinh trung ương và duy trì giữa kỳ (12 tuần):
- Tia xạ não 2.400 cGy ngày 1-12.
- Methotrexate 15 mg tiêm tủy sống; ngày 1, 8, 15, 22 và 29.
- 6-mercaptopurine 60 mg/m2/ngày đường uống; ngày 1-70.
- Methotrexate 20 mg/m2 đường uống; ngày 36, 43, 50, 57 và 64.
Đợt IV: Tăng cường muộn (8 tuần):
- Daunorubicin 30 mg/m2 đường tĩnh mạch; ngày 1, 8 và 15.
- Vincristine 2 mg đường tĩnh mạch; ngày 1, 8 và 15.
- Dexamethasone 10 mg/m2/ngày đường uống; ngày 1-14.
- Cyclophosphamide 1.000 mg/m2 đường tĩnh mạch; ngày 29.
- 6-mercaptopurine 60 mg/m2/ngày đường uống; ngày 29-42.
- Cytarabine 75 mg/m2/ngày tiêm dưới da; ngày 29-32 và 36-39.
Đợt V: Duy trì kéo dài (đến 24 tháng tính từ thời điểm chẩn đoán), lặp lại mỗi 4 tuần:
- Vincristine 2 mg đường tĩnh mạch; ngày 1.
- Methylprednisone 60 mg/m2/ngày đường uống; ngày 1-5.
- Methotrexate 20 mg/m2 đường uống; ngày 1, 8, 15 và 22.
- 6-mercaptopurine 80 mg/m2/ngày đường uống; ngày 1-28.
Phác đồ hóa trị liệu liều cao phân liều (hyperfractionated CVAD)
- Thường được sử dụng với LXM lympho cấp tái phát hoặc các thể đáp ứng kém với hoá trị liệu liều tiêu chuẩn (mature B-ALL, Burkit leukemia). Nguyên tắc chung là sử dụng liệu trình điều trị quay vòng nhiều đợt, mỗi đợt ngắn ngày nhưng hoá chất sử dụng liều rất cao. Phác đồ thường dùng là Hyper-CVAD, bao gồm 8 đợt điều trị liều cao phân liều, không điều trị duy trì;
- Phác đồ Hyper-CVAD: Gồm 8 đợt điều trị, chia thành 2 liệu trình A và B, điều trị xen kẽ.
b. Điều trị bệnh nhân LXM lympho cấp ở bệnh nhân thanh thiếu niên và người trẻ tuổi (AYA)
Phác đồ GRAALL 2005
Giai đoạn test corticoid:
Thực hiện từ ngày 1-7, tối đa là ngày thứ 10.
- Methylprednisone: liều 60 mg/m2 đường uống hoặc methylprednisolone liều 48mg/m2 đường tĩnh mạch từ ngày 1-7 (hoặc đến ngày thứ 10).
Sau đó bệnh nhân được chia thành 2 nhóm để lựa chọn phác đồ điều trị là nhóm có NST Ph(+) và nhóm có NST Ph(-).
Nhóm bệnh nhân có NST Ph(-) điều trị như sau:
(1) Giai đoạn điều trị cảm ứng (điều trị tấn công)
- Methylprednisone liều 60 mg/m2 đường uống (hoặc methylprednisolone liều 48 mg/m2 đường tĩnh mạch) ngày 1-14.
- Daunorubicin liều 50 mg/m2 đường tĩnh mạch ngày 1, 2, 3, 15, 16.
- Vincristine liều 2 mg đường tĩnh mạch ngày 1, 8, 15, 22.
- Cyclophosphamide liều 750 mg/m2 đường tĩnh mạch (truyền trong 3 giờ) ngày 1, 15.
- L-Asparaginase liều 6.000 UI/m2 đường tĩnh mạch ngày 8, 10, 12, 20, 22, 24, 26, 28.
- Tiêm tủy sống: Methotrexate 15 mg, cytarabine 40 mg, methylprednisolone 40 mg ngày 1, 8.
- G-CSF 5µg/kg/ngày bắt đầu từ ngày 18 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
Trong trường hợp chưa đạt lui bệnh hoàn toàn sau điều trị tấn công thì cần điều trị hóa chất bổ sung (điều trị cứu vãn) bằng các thuốc sau:
- Mitoxantron liều 12 mg/m2 tiêm dưới da từ ngày 1-3.
- Cytarabin liều 1g/m2/12 giờ đường tĩnh mạch (truyền trong 3 giờ) ngày 1-4.
- G-CSF liều 300 UI tiêm dưới da từ ngày 9 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
(2) Giai đoạn điều trị củng cố 1 và 2
* Đợt (block) cytarabin: (Block 1 và 4)
- Cytarabin 2.000 mg/m2/12 giờ đường tĩnh mạch (truyền trong 2 giờ) ngày 1,2.
- Dexamethasone 10 mg/12 giờ đường uống ngày 1,2.
- L-Asparaginase 10.000 UI/m2 đường tĩnh mạch ngày 3.
- G-CSF 300 UI tiêm dưới da ngày 9-13.
* Block MTX: (Block 2 và 5)
- Vincristine liều 2 mg đường tĩnh mạch ngày 15.
- Methotrexate (MTX) liều 3.000 mg/m2 đường tĩnh mạch (truyền 24 giờ) ngày 15 dùng với thuốc giải độc Calci folinat/Acid folinic.
- 6-Mercartopurine liều 60 mg/m2 đường uống ngày 15-21.
- L-Asparaginase 10.000 UI/m2 đường tĩnh mạch ngày 16.
- G-CSF liều 300 UI tiêm dưới da ngày 23-27.
* Block CPM: (Block 3 và 6)
- Methotrexate liều 25 mg/m2 đường tĩnh mạch ngày 29.
- Cyclophosphamide liều 500 mg/m2 đường tĩnh mạch (truyền trong 3 giờ) ngày 29, 30.
- Etoposide liều 75mg/m2 đường tĩnh mạch ngày 29, 30.
- Tiêm tủy sống ngày 29.
- G-CSF liều 300 UI tiêm dưới da ngày 31 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
(3) Giai đoạn điều trị tăng cường muộn
Đối với các bệnh nhân không qua điều trị cứu vãn:
- Methylprednisone liều 60 mg/m2 đường uống (hoặc methylprednisolone liều 48 mg/m2 đường tĩnh mạch) ngày 1-14.
- Daunorubicin liều 30 mg/m2 đường tĩnh mạch ngày 1, 2, 3, 15, 16.
- Vincristine liều 2 mg đường tĩnh mạch ngày 1, 8, 15, 22.
- Cyclophosphamide liều 750 mg/m² đường tĩnh mạch ngày 1, 15.
- L-Asparaginase liều 6.000 UI/m² đường tĩnh mạch ngày 10, 12, 20, 22, 24, 28.
- G-CSF liều 300 UI tiêm dưới da từ ngày 8 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
- Tiêm tủy sống ngày 1, 8.
Đối với các bệnh nhân được điều trị cứu vãn:
- Mitoxantrone liều 12 mg/m2 đường tĩnh mạch ngày 1, 2, 3.
- Cytarabin 2.000 mg/m2/12 giờ đường tĩnh mạch (truyền trong 2 giờ) ngày 1, 2, 3, 4.
- G-CSF liều 300 UI tiêm dưới da từ ngày 9 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
- Tiêm tủy sống ngày 8, 15.
(4) Giai đoạn điều trị củng cố 3
- Block Ara-C: như trên.
- Block MTX: như trên.
- Block CPM: như trên.
(5) Giai đoạn xạ trị
- Liều chiếu xạ: 18 Gy x 10 (5 lần/1 tuần) với tổng liều là 180 Gy.
- 6-Mercaptopurine liều 60mg/m2/ngày đường uống trong suốt thời gian xạ trị.
Nếu bệnh nhân có biểu hiện thâm nhiễm LXM vào tinh hoàn, cần cân nhắc xạ trị khu trú.
(6) Giai đoạn điều trị duy trì
- Vincristine liều 2 mg đường tĩnh mạch ngày 1.
- Methylprednisone liều 40 mg/m2/ngày đường uống trong 7 ngày.
- 6-Mercaptopurine liều 60 mg/m2/ngày đường uống.
- Methotrexate 25 mg/m2/tuần đường uống (vào tuần 2, 3, 4).
Những bệnh nhân thuộc vào nhóm nguy cơ cao được chỉ định ghép tế bào gốc đồng loài sau giai đoạn điều trị củng cố nếu có người cho phù hợp HLA và độ tuổi < 55.
Nhóm bệnh nhân có NST Ph(+) điều trị như sau:
(1) Giai đoạn điều trị cảm ứng (điều trị tấn công)
- Doxorubicin liều 50 mg/m2 truyền tĩnh mạch trong 24 giờ ngày 4.
- Cyclophosphamide liều 300 mg/m2/12 giờ, truyền tĩnh mạch ngày 1, 2, 3.
- Vincristine liều 2 mg đường tĩnh mạch ngày 4, 11.
- Dexamethasone liều 40 mg đường tĩnh mạch ngày 1-4, 11-14.
- Imatinib liều 800 mg đường uống chia làm 2 lần.
- G-CSF 5 liều µg/kg/ngày bắt đầu từ ngày 18 phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
- Tiêm tủy sống: Methotrexate 15mg, cytarabine 40mg, methylprednisolon 40mg ngày 1, 8, 15.
(2) Giai đoạn điều trị sau tấn công
- Methotrexate liều 1.000mg/m2 truyền tĩnh mạch trong 24 giờ dùng với thuốc giải độc Calcifolinat.
- Cytarabin liều 3.000 mg/m2/12 giờ đường tĩnh mạch (trong 2 giờ).
- Imatinib liều 800 mg đường uống chia làm 2 lần sáng và tối.
- Tiêm tủy sống.
- G-CSF liều 300 UI tiêm dưới da từ ngày 6 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
(3) Giai đoạn điều trị củng cố
Các đột điều trị củng cố được thực hiện xen kẽ, bao gồm:
Đợt 3/5/7: (giống điều trị cảm ứng)
- Vincristine liều 2 mg đường tĩnh mạch ngày 4, 11.
- Doxorubicine liều 50mg/m2 đường tĩnh mạch ngày 4.
- Cyclophosphomide liều 300mg/m2/12 giờ đường tĩnh mạch ngày (truyền trong 3 giờ) ngày 1, 2, 3.
- Dexamethasone liều 40mg đường uống ngày 1-4, 8-11.
- Imatinib mesylate liều 600mg đường uống chia 2 lần sáng và tối ngày 1-14.
- Tiêm tủy sống ngày 1.
- G-CSF liều 300 UI tiêm dưới da từ ngày 15 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
Đợt 4/6/8: (giống điều trị sau tấn công)
- Methotrexate liều 1g/m2 đường tĩnh mạch dùng với thuốc giải độc Lederfoline ngày 1.
- Cytarabin liều 3.000mg/m2/12 giờ đường tĩnh mạch ngày 2, 3.
- Imatinib liều 600 mg đường uống chia làm 2 lần sáng và tối ngày 1-14.
- Tiêm tủy sống ngày 1.
- G-CSF liều 300 UI tiêm dưới da từ ngày 9 cho đến khi phục hồi số lượng bạch cầu hạt trung tính > 1 G/l.
(4) Giai đoạn điều trị duy trì
|
Thời gian Thuốc |
Tháng 1-5 |
Tháng 6 |
Tháng 7-11 |
Tháng 12 |
|
Vincristine |
Ngày 1 hàng tháng |
Lặp lại đợt 3/5/7 |
Ngày 1 hàng tháng |
Lặp lại đợt 4/6/8 |
|
Prednisone |
Ngày 1-5 hàng tháng |
Ngày 1 - ngày 5 hàng tháng |
||
|
Imatinib |
Đường uống chia làm 2 lần hàng ngày |
|||
- Vincristine liều 2mg đường tĩnh mạch vào ngày đầu hàng tháng.
- Methylprednisone liều 200mg/ngày trong 5 ngày từ ngày 1-5 hàng tháng.
- Imatinib liều 600mg đường uống chia làm 2 lần.
c. Điều trị LXM lympho cấp có NST Ph(+) bằng thuốc nhắm đích
Hiện nay, các thuốc TKI như imatinib được sử dụng điều trị LXM lympho cấp có NST Ph(+). Các thuốc này thường được phối hợp với đa hóa trị liệu để tăng thêm hiệu quả điều trị. Chẳng hạn, có thể phối hợp imatinib (liều 600 mg/ngày đường uống) với phác đồ hóa trị liệu Hyper-CVAD.
3.4. Điều trị hỗ trợ (xin xem chi tiết trong bài Hồi sức huyết học)
- Chống thiếu máu, xuất huyết bằng các chế phẩm máu.
- Dự phòng và điều trị nhiễm trùng bằng kháng sinh và yếu tố kích thích sinh máu.
- Phòng ngừa hội chứng tiêu khối u bằng allopurinol, truyền dịch, lợi niệu, kiềm hoá nước tiểu.
- Gạn bạch cầu khi số lượng bạch cầu quá cao, nguy cơ tắc mạch.
3.5. Theo dõi đáp ứng điều trị
- Xét nghiệm tủy đồ: Được thực hiện 3-4 tuần sau điều trị để đánh giá mức độ đáp ứng và có thể thực hiện 2 tuần sau điều trị để đánh giá đáp ứng ban đầu, làm cơ sở lựa chọn phác đồ tấn công bổ sung nếu không lui bệnh.
- Tiêu chuẩn lui bệnh về huyết học: Bằng xét nghiệm tủy đồ (3-4 tuần sau điều trị) theo tiêu chuẩn của Viện Ung thư quốc gia Hoa Kỳ (1990):
+ Lui bệnh hoàn toàn: Lâm sàng ổn định, số lượng bạch cầu trung tính > 1,5G/L, Hematocrit > 0,3 l/l, số lượng tiểu cầu > 100G/L, không còn tế bào blast ở máu ngoại vi, tỷ lệ tế bào blast trong tủy xương < 5% (trên nền tủy sinh máu bình thường);
+ Lui bệnh không hoàn toàn: Tỷ lệ tế bào blast ở tủy xương từ 5-20%;
+ Không lui bệnh: Tỷ lệ tế bào blast ở tủy xương > 20%.
- Phát hiện tồn dư tối thiểu của bệnh: Kỹ thuật đếm tế bào dòng chảy đa màu (ngưỡng phát hiện < 1 x 10-4), kỹ thuật PCR định lượng (ngưỡng phát hiện < 1 x 10-5/6).
- Xét nghiệm đánh giá lại gen bệnh sau 2-3 chu kỳ hoá chất hoặc trước khi ghép tế bào gốc tạo máu.
- Bệnh nhân tái phát cần làm lại xét nghiệm chẩn đoán như bệnh nhân mới.
3.6. Các xét nghiệm đinh giá và theo dõi điều trị
- Tổng phân tích tế bào máu ngoại vi, được làm ít nhất 2 ngày 1 lần.
- Sinh hóa máu: Chức năng gan, thận, điện giải, acid uric, photpho, LDH..., ít nhất 1 tuần/lần.
- Bộ xét nghiệm đông máu, D-Dimer, rotem…, ít nhất 1 tuần/lần.
- Các xét nghiệm vi sinh: virus viêm gan, HIV… làm sàng lọc trước khi điều trị, lặp lại theo quy định. Các xét nghiệm đánh giá nhiễm trùng: cấy vi khuẩn, vi nấm, CRP, PCT, CMV, EBV, kháng sinh đồ…, nếu lâm sàng có dấu hiệu nhiễm trùng.
- Huyết thanh học nhóm máu, làm lúc chẩn đoán hoặc khi cần truyền máu.
- Xét nghiệm tế bào và sinh hóa nước tiểu, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Chẩn đoán hình ảnh, thăm dò chức năng, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Xét nghiệm Huyết tủy đồ và có thể cả STTX làm lúc chẩn đoán và làm lại sau các đợt điều trị; Gen đột biến lơ xê mi cấp, Nhiễm sắc thể được làm lúc chẩn đoán và làm lại để đánh giá sau mỗi 2 đợt điều trị hóa chất.
- Xét nghiệm Flowcytometry được làm lúc chẩn đoán và làm lại sau mỗi đợt điều trị hóa chất để đánh giá bệnh tồn lưu tối thiểu.
- Một số xét nghiệm khác được chỉ định tùy theo bệnh nền và bệnh mắc kèm của người bệnh.
PHỤ LỤC
|
Phác đồ |
Cách dùng |
|
|
3 + 7 |
- Daunorubicin 60 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-3; hoặc Idarubicin 12mg/m2/ngày x 3 ngày; - Cytarabin 100 - 200 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-7 |
|
|
Hi-DAC |
Cytarabin 3.000 mg/m2 da/12 giờ x 2 lần/ngày, truyền tĩnh mạch ngày 1, 3, 5 |
|
|
ADE |
- Daunorubicin 45 - 60 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-3 - Etoposide 100 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-3 - Cytarabin 100 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-7 |
|
|
MEC |
- Mitoxantron 8 mg/m2 da/ngày, truyền tĩnh mạch trong 30 phút,ngày 1-5 - Etoposide 100 mg/m2 da/ngày, truyền tĩnh mạch trong 60 phút, ngày 1-5 - Cytarabin 1.000 mg/m2 da/ngày, truyền tĩnh mạch trong 3 giờ, ngày 1-5 |
|
|
HAM |
- Cytarabin 3.000 mg/m2 da/12 giờ x 2 lần/ngày, truyền tĩnh mạch trong 3 giờ, ngày 1-3 - Mitoxantron 10 mg/m2 da/ngày, truyền tĩnh mạch trong 1 giờ, ngày 3-5 |
|
|
FLAG |
- Fludarabin 30 mg/m2 da/ngày, truyền tĩnh mạch 30 phút ngày1-5 - Cytarabin 2.000 mg/m2 da/ngày, truyền tĩnh mạch 3 giờ, bắt đầu sau khi truyền xong fludarabin 3,5 giờ, ngày 1-5 - Filgrastim 5 mcg/kg tiêm dưới da ngày 0-5 |
|
|
Hyper CVAD xen kẽ course A và B mỗi course trong 28 ngày |
Course A |
- Cyclophosphamide 300mg/m2 truyền tĩnh mạch trong 3 giờ, 12 giờ một lần, ngày 1-3 - Methotrexate 12 mg tiêm tủy sống ngày 2 - Doxorubicin 50 mg/m2 truyền tĩnh mạch liên tục 24 giờ, ngày 4 - Vincristine 2 mg truyền tĩnh mạch ngày thứ 4, 11 - Dexamethasone 40 mg/ngày truyền tĩnh mạch hoặc uống ngày 1-4 và 11-14 - Cytarabine 100 mg tiêm tủy sống ngày 7 |
|
Course B |
- Methotrexat: 1.000mg/m2 truyền tĩnh mạch liên tục 24 giờ ngày 1; - Cytarabine 3.000mg/m2 truyền tĩnh mạch trong 2 giờ, 12 giờ một lần, ngày 2, 3 |
|
TÀI LIỆU THAM KHẢO
1. Arber D, Orazi A, Hasserjian R, et al. (2016). The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20), pp. 2391-2405.
2. Kantarjian H et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemie. Cancer 2004; 101:2788.
3. National Comprehensive Cancer Network (NCCN) (2016).Clinical Practice Guidelines in oncology: Acute Myelogenous Leukemia. Vesion 2.2016.
4. Powell BL et al. Effect of consolidation with arsenic trioxide (As2O3) on event-free survival (EFS) and overall survival (OS) among patients with newly diagnosed acute promyelocytic leukemia (APL): North American Intergroup Protocol C9710. 2007 ASCO annual meeting. Abstract 2.
5. Rowe JM et al. ECOG, MRC/NCRI Adult leukemia working party. Induction therapy for adults with acute lymphoblastic leukemia: result of more than 1500 patients from the International ALL trial: MRC UKALL XII/ECOG E2993. Blood 2005; 106:3760.
6. Thomas DA et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt’s-type lymphoma or acute lymphoblastic leukemia. Cancer 2006; 106:156921. LƠ XÊ MI LYMPHO CẤP Ở TRẺ EM
(Bệnh bạch cầu cấp dòng lympho ở trẻ em)
1. ĐẠI CƯƠNG
Lơ xê mi lympho cấp (bệnh bạch cầu cấp dòng lympho) là bệnh lý tăng sinh ác tính các tế bào dòng lympho của hệ thống tạo máu. Bệnh chiếm tỉ lệ khoảng 75% bệnh máu ác tính và khoảng 25-30% ung thư ở trẻ em. Tần suất bệnh khoảng 3-4 trường hợp/ 100.000 em bé da trắng và khoảng 2.500-3.000 trường hợp phát hiện mới/ năm tại Hoa Kỳ. Bệnh thường gặp ở nhóm từ 2-5 tuổi. Hầu hết các trường hợp không do di truyền mà do thay đổi di truyền somatic. Cho đến nay, với sự tiến bộ của khoa học trong việc chẩn đoán sớm, xếp loại nguy cơ theo đặc điểm tế bào - sinh học phân tử và hóa trị liệu, chăm sóc hồi sức huyết học đã giúp cho bệnh lơ xê mi lympho cấp có khả năng được chữa khỏi.
2. CHẨN ĐOÁN BỆNH
2.1. Triệu chứng lâm sàng
- Triệu chứng toàn thân:
+ Mệt mỏi, gầy sút cân, chán ăn;
+ Sốt: Thường gặp sốt thất thường, kém đáp ứng với điều trị kháng sinh;
+ Hội chứng thiếu máu từ nhẹ đến nặng;
+ Hội chứng xuất huyết;
+ Hội chứng nhiễm trùng.
- Triệu chứng thâm nhiễm:
+ Hạch to, hạch to khu trú hoặc toàn thân;
+ Lách to;
+ Gan to;
+ Đau xương: Biểu hiện sớm như đi khập khiễng, không chịu đi bộ, đau khớp, đau các xương dài;
+ Thần kinh trung ương: Đau đầu, nôn và buồn nôn, li bì hoặc kích thích, cứng gáy, phù gai thị, liệt thần kinh sọ;
+ Hệ sinh dục-tiết niệu: Ít gặp, tinh hoàn to, đau;
+ Đường tiêu hóa: Ít gặp, có thể gặp thâm nhiễm đại tràng gây hội chứng như thương hàn, hoặc hoại tử và chảy máu thành ruột như viêm ruột.
2.2. Triệu chứng xét nghiệm
- Huyết đồ:
+ Bạch cầu: Số lượng tăng, bình thường hoặc giảm, có thể có bạch cầu non ra máu ngoại vi (nguyên bào lympho);
- Huyết sắc tố: Thường giảm, mức độ từ nhẹ đến nặng.
- Tiểu cầu: Thường giảm hoặc có thể bình thường.
- Tủy đồ: Tỷ lệ tế bào non ác tính trong tủy ≥ 20%. Mẫu tiểu cầu giảm.
+ Hóa học tế bào: PAS (+), Soudan - black (-), Peroxydase (-).
- Miễn dịch tế bào:
+ Dấu ấn tế bào non: CD34 (+), HLA-DR (+), TdT (+);
+ Dòng lympho B (85%):
● Pro B-ALL: HLA-DR+, TdT+, CD19+ (5%);
● Common ALL: HLA-DR+, TdT+, CD19+, CD10+ (65%);
● Pre B-ALL: HLA-DR+, TdT+, CD19+, CD10±, cytoplasmic IgM+ (15%).
+ Dòng Lympho T (15%):
● Pre T-ALL: TdT+, Cytoplasmic CD3+, CD7+ (2%);
● T cell-ALL: TdT+, Cytoplasmic CD3+, CD1a/2/3+, CD5+ (13%).
- Di truyền tế bào và sinh học phân tử:
+ Bất thường về NST:
● Giảm bội (hypodiploid) < 46NST;
● 46 NST với cấu trúc bất thường (pseudodiploid);
● Trên lưỡng bội 47-50 NST (hyperdiploid);
● > 50 NST (hyper-hyperdiploid).
+ Các đột biến gen:
● Gen kết hợp TEL-AML1 tạo bởi chuyển đoạn t(12;21) (p13q22). t(12;21), 22% của Pre-B ALL;
● Gen kết hợp BCR-ABL tạo bởi chuyển đoạn t(9;22) (q34q11). t(9;22), 3% ALL trẻ em;
● Tái sắp xếp gen MLL tại vị trí 11q23 ảnh hưởng 80% của trẻ nhũ nhi, 3% của ALL trẻ lớn;
● Các chuyển đoạn gen của lơ xê mi lympho B cấp liên quan gen MYC trên NST 8q24. 80% B-ALL có t(8;14) (q24;q32);
● > 50% trường hợp Lơ xê mi lympho T cấp có đột biến hoạt động liên quan đến gen NOTCH1.
2.3. Chẩn đoán xác định
Chẩn đoán Lơ xê mi lympho cấp dựa trên các tiêu chuẩn lâm sàng, hình thái học, nhuộm hoá học tế bào, phân loại miễn dịch và các biến đổi di truyền đặc trưng của dòng lympho. Chẩn đoán xác định khi nguyên bào lympho chiếm trên 20% tế bào có nhân trong tuỷ, mang dấu ấn miễn dịch đặc trưng dòng lympho.
3. ĐIỀU TRỊ
Các phác đồ điều trị được căn cứ vào các nhóm nguy cơ. Thường sử dụng các phác đồ đa hoá trị liệu như: FRALLE 2000, NOPHO, BFM, UK, CCG 1961, CCG1991, COG, COALL, Study VII… trong phác đồ có các thuốc, hoá chất như prednison, dexamethasone, vincristine, methotrexate, anthracycline, cytarabine, L-asparaginase, cyclophosphamide, mercaptopurine,…, kết hợp tiêm tuỷ sống và xạ trị. Phác đồ thường được sử dụng hiện nay là phác đồ FRALLE 2000. Dưới đây là phác đồ điều trị lơ xê mi lympho cấp trẻ em mới chẩn đoán theo FRALLE 2000:
3.1. Phân nhóm nguy cơ theo phác đồ FRALLE 2000
3.1.1. Phân nhóm nguy cơ tổng quát
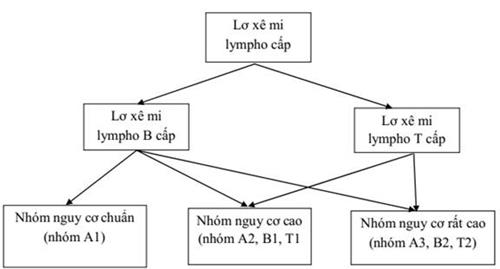
Sơ đồ 4. Phân nhóm nguy cơ tổng quát ở bệnh nhân lơ xê mi lympho cấp trẻ em
Ghi chú: Các nhóm cụ thể căn cứ vào kết quả điều trị tấn công
3.1.2. Tiêu chuẩn phân nhóm nguy cơ phác đồ FRALLE 2000
a. Nhóm A: Nguy cơ chuẩn (Standard risk)
- Lơ xê mi lympho B cấp, 1-10 tuổi số lượng bạch cầu ≤ 50G/L và có đủ các yếu tố sau:
+ Không có thâm nhiễm hệ thần kinh trung ương;
+ Không có t(9;22), t(4;11) hoặc bộ nhiễm sắc thể < 44;
+ BCR-ABL hay MLL-AF4 âm tính;
+ Không hiện diện sự tái sắp xếp gen MLL phát hiện bằng kỹ thuật Southern blot hay FISH cho trường hợp CD10 (+) yếu;
+ CD10 (+);
+ Không có trisomy 21;
+ Không điều trị corticoid trước đó;
+ Không phải ALL thể L3.
- Nhóm A sẽ được chia ra 3 phân nhóm A1, A2, A3 ở ngày 21 dựa vào sự đánh giá tế bào blast trong tủy vào ngày 21 (bất kể sự nhạy cảm với corticoid vào ngày 8 hay không):
+ Blast < 5% (týp M1): Nhóm A1;
+ Blast 6-25% (týp M2): Nhóm A2;
+ Blast > 25% (týp M3): Nhóm A3.
- Người bệnh có MRD (+) (≥ 10-2) vào ngày 35-42 sẽ được chuyển sang nhóm A3 bất kể lúc đầu thuộc nhóm nào.
b. Nhóm B: Nguy cơ cao (high risk)
- Lơ xê mi lympho B cấp de novo và có từ một trong những tiêu chuẩn sau:
+ Tuổi > 10;
+ Có thâm nhiễm hệ thần kinh trung ương;
+ Bạch cầu > 50G/L;
+ Có t(9;22), t(4;11) hoặc bộ NST < 44;
+ BCR-ABL hay MLL-AF4 dương tính;
+ Hiện diện sự tái sắp xếp gen MLL phát hiện bằng kỹ thuật Southern blot hay FISH cho trường hợp CD10 (+) yếu.
- Xếp nhóm B1 và B2 vào ngày 21 của điều trị tấn công dựa vào sự nhạy cảm với corticoid vào ngày 8 và nhạy cảm hóa trị vào ngày 21.
|
Nhóm B1 (Tất cả các tiêu chuẩn sau) |
Nhóm B2 (Chỉ có từ 1 trong các tiêu chuẩn) |
|
- Không có thể thiểu bội < 44, hoặc t(4;11) hoăc t(9 ;22) - Không có gen MLL-AF4 hay BCR-ABL - Nhạy cảm corticoid ngày 8 - Nhạy cảm hóa trị ngày 21 |
- Có thể thiểu bội < 44, hoặc t(4;11) hoặc t(9 ;22) - Có gen MLL-AF4 hay BCR-ABL - Kháng corticoid ngày 8 - Kháng hóa trị ngày 21 - MRD (+) (≥ 10-2) vào ngày 35-42 |
c. Nhóm T: Lơ xê mi lympho T cấp. Chia nhóm T1 và T2 dựa vào tiêu chuẩn:
|
Nhóm T1 (Khi có đủ cả 3 tiêu chuẩn) |
Nhóm T2 (Có từ 1 trong 3 tiêu chuẩn) |
|
- Nhạy cảm corticoid ngày 8 - Nhạy cảm hóa trị ngày 21 - MRD ngày 35 < 10-2 |
- Kháng corticoid ngày 8 - Kháng hóa trị ngày 21 - MRD ngày 35 > 10-2 |
Lưu ý:
- Tiêu chuẩn nhạy cảm corticoid trên huyết đồ ngày 8:
+ Nhạy cảm corticoid ngày 8: Tế bào blast trong máu ngoại vi ngày 8: < 1 G/L;
+ Kháng corticoid ngày 8: Tế bào blast trong máu ngoại vi ngày 8: ≥ 1 G/L.
- Tiêu chuẩn nhạy cảm hóa trị ngày 21:
+ Nhạy với hóa trị ngày 21: Tủy đồ ngày 21 có blast ≤ 5%;
+ Kháng hóa trị ngày 21: Tủy đồ ngày 21 có blast > 5%.
3.2. Điều trị Lơ xê mi lympho cấp theo FRALLE 2000
Phác đồ điều trị thường được sử dụng là phác đồ FRALLE 2000 và được phân chia điều trị 5 giai đoạn khác nhau, có tính liên tục, bắt buộc phải tuân thủ chính xác và chặt chẽ; mỗi giai đoạn chuyển đổi đều được đánh giá và có tiêu chuẩn để bắt đầu sử dụng thuốc, các giai đoạn điều trị bao gồm:
+ Giai đoạn cảm ứng;
+ Điều trị tấn công;
+ Điều trị củng cố;
+ Điều trị tăng cường 1;
+ Điều trị trung gian;
+ Điều trị tăng cường 2;
+ Điều trị duy trì.
Lưu ý:
- Trong trường hợp bệnh nhi được chẩn đoán lơ xê mi lympho cấp có nhiễm sắc thể Philadelphia (Ph+) hoặc có gen BCR-ABL1 có thể điều trị bằng phác đồ hóa chất kết hợp với thuốc ức chế Tyrosine kinase thế hệ 1 hoặc các thuốc thế hệ tiếp theo như Imatinib liều 340mg/m2 từ khi bắt đầu điều trị…
- Phác đồ CAALL-F01 năm 2017 cải tiến từ phác đồ FRALLE, thay thế L-asparaginase bằng chế phẩm thế hệ tiếp theo.
3.2.1. Phác đồ FRALLE 2000 điều trị lơ xê mi lympho B cấp
3.2.1.1. Phác đồ điều trị lơ xê mi lympho B cấp - nhóm A
a. Sơ đồ điều trị tổng quát
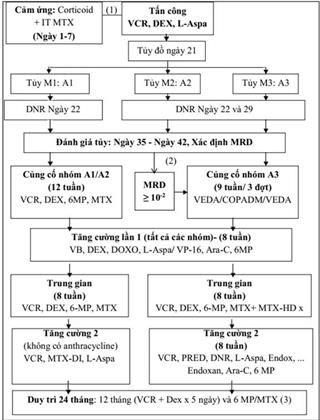
(1) Làm tiêu bản máu ngoại vi và xác định số lượng tế bào non (blasts) vào ngày 8;
(2) Đánh giá MRD tủy vào ngày 35 hoặc ngày 42. Nếu MRD ≥ 10-2 thì chuyển sang điều trị theo nhóm A3 cho đến hết phác đồ.
(3) Đối với nhóm A2/A3 và các bệnh nhân có MRD ≥ 10-2 sau điều trị tăng cường 2 : Trong giai đoạn điều trị duy trì, dexa được thay thế bằng methylprednisone x 7 ngày.
b. Phác đồ điều trị chi tiết
b.1. Giai đoạn cảm ứng
- Methylprednisone: 60mg/m2/ngày chia 2 lần (uống hay truyền tĩnh mạch) từ ngày 1 đến ngày 7. Nếu dùng đường tĩnh mạch: Sử dụng methylprednisolone cùng liều như trên).
b.2. Giai đoạn tấn công: Từ ngày 8, bắt đầu ngay sau giai đoạn cảm ứng
- Dexamethasone: 6 mg/m2/ngày (chia 3 lần uống/ tĩnh mạch): Ngày 8-28.
- Vincristine: 1,5 mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch, không quá 2 mg) các ngày: 8, 15, 22, 29.
- Daunorubicin: 40 mg/m2 (truyền tĩnh mạch trong 60 phút) ngày 22/29.
- L-asparaginase: 6.000 UI/m2 (tiêm bắp/ tiêm tĩnh mạch trong 60 phút) từ ngày 10, 12, 14,16, 18, 20, 23, 25, 27.
- Tiêm tủy sống (IT): Ngày 1 (MTX), ngày 15 (MTX, AraC, depomedrol) theo tuổi người bệnh.
Lưu ý:
- Tiêm tủy sống chỉ với methotrexate vào ngày 1 (không có methylprednisone) với điều kiện tiểu cầu > 100G/L.
- Làm huyết đồ để đánh giá sự nhạy cảm với methylprednisone vào ngày 8.
- Sự kháng corticoid sẽ được đánh giá trong mỗi nhóm A1, A2, A3.
- Thay methylprednisone bằng dexamethasone bắt đầu từ ngày 8.
- Tủy đồ ngày 21 là yếu tố quyết định số liều điều trị Daunorubicine:
+ Tủy M1: Dùng Daunorubicine vào ngày 22, sau đó điều trị theo nhóm A1;
+ Tủy M2, M3: Chỉ định Daunorubicine vào ngày 22 và ngày 29. Sau khi đạt lui bệnh hoàn toàn:
● Nếu tủy M2 vào ngày 21: Điều trị theo nhóm A2;
● Nếu tủy M3 vào ngày 21: Điều trị theo nhóm A3.
- Làm tủy đồ + MRD vào cuối giai đoạn tấn công (ngày 35-42), chậm nhất là ngày 42 để đánh giá lui bệnh. Người bệnh có MRD (+) vào ngày 35-42 (≥10-2) sẽ tiếp tục điều trị củng cố theo nhóm A3 (bắt đầu VEDA 1 ngay khi có thể). Kiểm tra MRD tủy trước khi bắt đầu giai đoạn VEDA.
b.3. Giai đoạn củng cố
- Nhóm A1/A2
+ Mercaptopurine: 75 mg/m2/ngày (uống), ngày 1 đến 77;
+ Vincristine: 1,5 mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 1, 8, 29, 36, 57, 64 (không quá 2mg);
+ Dexamethasone: 6 mg/m2/ngày (uống, 3 lần/ngày), ngày 1-5, ngày 29-33, ngày 57-61;
+ Methotrexate: 25 mg/m2/ lần (uống), ngày 8, 15, 22, 36, 43, 64, 71, 78;
+ Tiêm tủy sống: Methotrexat ngày 1, 29, 57 (tùy theo tuổi người bệnh).
Trong lúc chờ kết quả MRD, người bệnh sẽ được điều trị theo giai đoạn củng cố của nhóm A1.
+ Nếu MRD (+): Giai đoạn củng cố sẽ không được kết thúc, người bệnh sẽ được điều trị theo giai đoạn củng cố của nhóm A3 (bắt đầu từ giai đoạn VEDA 1) ngay khi có thể. Kiểm tra MRD một cách hệ thống ngay trước khi bắt đầu VEDA. Sau đó người bệnh sẽ được điều trị như nhóm A3. Nếu MRD lần 2 vẫn cao (> 10-2): Hội chẩn lại.
+ Nếu MRD (-): Người bệnh được tiếp tục cho đến hết giai đoạn củng cố của nhóm A1. Sau đó, người bệnh sẽ được điều trị như nhóm A1.
- Nhóm A3: (3 đợt liên tiếp: VEDA/COPADM2000/VEDA)
Đợt 1: VEDA 1
Bắt đầu ngay khi bạch cầu trung tính > 1G/L và TC > 100G/L.
+ Dexamethasone: 20mg/m2/ngày (chia 3 lần, uống hoặc tĩnh mạch) ngày 1-5;
+ Vincristine: 1,5mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch) ngày 1 (không quá 2mg);
+ Cytarabine: 2g/m2/ mỗi lần x 2 lần/ngày 1, 2. Truyền tĩnh mạch/ 3giờ. Tổng cộng 8g/m2;
+ VP-16: 150mg/m2/ngày (truyền tĩnh mạch/1 giờ), ngày 3, 4, 5;
+ Tiêm tủy sống 3 thuốc: MTX + Depomedrol + AraC ngày 5.
G-C SF: 150 μg/m2/ngày (= 5 μg/kg/ ngày) tiêm dưới da, bắt đầu từ ngày 7. Tiếp tục cho đến khi bạch cầu trung tính > 1G/L trong 3 ngày.
Lưu ý:
+ Người bệnh nhóm A1 và A2 có MRD (+) vào ngày 35-42, được đưa vào nhóm A3, phải được kiểm tra MRD trước khi bắt đầu block COPADM;
+ Nếu MRD lần 2 vẫn cao (> 10-2): Hội chẩn lại.
3 đợt VEDA1, COPADM, VEDA 2: Điều trị trong vòng 9 tuần.
Đợt 2: COPADM2000
Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L.
+ Methylprednisone: 60mg/m2/ngày, ngày 1-5;
+ Vincristine: 1,5mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 1 (không quá 2mg);
+ Methotrexate: 5.000mg/m2/ngày (truyền tĩnh mạch/ 24 giờ), ngày 1;
(Thuốc giải Acid folininic từ giờ thứ 36 của MTX)
+ Cyclophosphamide: 500mg/m2/lần x 2lần/ ngày (truyền tĩnh mạch/60phút), ngày 2, 3 (tổng cộng 2g/m2);
+ Doxorubicin: 40mg/m2/ngày (truyền tĩnh mạch/1 giờ), ngày 2;
+ Tiêm tủy sống 3 thuốc: MTX + methylprednisolone + cytarabin, ngày 1.
Đợt 3: VEDA 2 (giống đợt 1)
Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L vào các ngày 1-7.
b.4. Giai đoạn tăng cường 1
- Nhóm A1/ A2 và A3
Đợt I: Bắt đầu ngay khi bạch cầu trung tính > 1 G/L và tiểu cầu > 100 G/L
+ Dexamethasone: 10 mg/m2/ngày (uống); ngày 1-15 sau đó giảm liều đến ngày 21;
+ Vincristine: 1,5mg/m2 (không quá 2mg) tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch, ngày 1, 8, 15;
+ Doxorubicine: 25 mg/m2 (truyền tĩnh mạch trong 60 phút); ngày 1, 8, 15;
+ L-asparaginase: 6.000 UI/m2 (tiêm bắp/ truyền tĩnh mạch trong 60 phút); ngày 2, 4, 6, 9, 11, 13;
+ Tiêm tủy sống 3 thuốc: Ngày 1, 29 tùy theo tuổi người bệnh.
Đợt II: Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L
+ Mercaptopurine: 75mg/m2/ngày (uống), ngày 29-49;
+ Etoposide: 150mg/m2/ngày (truyền tĩnh mạch trong 60 phút); ngày 29, 36, 43;
+ Cytarabine: 30mg/m2 x 2 lần/ngày (tiêm dưới da); ngày 29, 30, 36, 37, 43, 44;
+ Tiêm tủy sống 3 thuốc: Ngày 29, tùy theo tuổi người bệnh.
Lưu ý:
+ Hóa trị ngày 36, 43 được tiếp tục bất kể phân tích huyết học và không có vấn đề về lâm sàng;
+ Siêu âm tim, điện tim trước mỗi mũi doxorubicin.
b.5. Giai đoạn trung gian
- Nhóm A1/ A2:
Bắt đầu khi bạch cầu trung tính + monocyte > 1G/L và tiểu cầu > 100G/L
+ Vincristine: 1,5 mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 1, 29 (không quá 2 mg);
+ Dexamethasone: 6 mg/m2/ ngày (uống, chia 3 lần/ngày), ngày 1-5, ngày 29-33;
+ Mercaptopurine: 75 mg/m2/ngày (uống), từ ngày 1-56;
+ Methotrexate: 25 mg/m2/lần (uống), ngày 8, 15, 22, 36, 43, 50;
+ Tiêm tủy sống: Ngày 1, 29.
Lưu ý:
+ Trường hợp lâm sàng ổn định, giai đoạn này được tiếp tục (mà không có sự thay đổi) khi bạch cầu trung tính > 0,5G/L và tiểu cầu > 50G/L.
- Nhóm A3: Gồm 2 đợt liên tiếp nhau:
ĐỢT 1: Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L
+ Vincristine:1,5mg/m2/lần (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 1, 15 (không quá 2mg);
+ Dexamethasone: 6mg/m2/ngày (chia 3 lần, uống), ngày 1-5;
+ Mercaptopurine: 50mg/m2/ngày (uống), ngày 1-28;
+ Methotrexate: 5.000mg/m2/ngày (truyền tĩnh mạch/ 24 giờ) ngày 1, 15;
(Thuốc giải Acid folinic bắt đầu từ giờ thứ 36 của MTX)
+ Methotrexat: 25mg/m2/lần (uống) ngày 8, 22;
+ Tiêm tủy sống 3 thuốc: Vào giờ 24 của MTX ngày 2, 16.
ĐỢT 2: Bắt đầu ngay khi bạch cầu trung tính > 1G/L và TC > 100G/L
+ Vincristine: 1,5mg/m2/lần (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 29 (không quá 2mg);
+ Dexamethasone: 6mg/m2/ngày (chia 3 lần, uống), ngày 29-33;
+ Mercaptopurine: 50mg/m2/ngày (uống), ngày 29-49;
+ Methotrexate: 5.000mg/m2/ngày (truyền tĩnh mạch/ 24 giờ), ngày 29;
(Thuốc giải Acid folinic bắt đầu từ giờ thứ 36 của MTX)
+ Methotrexat: 25mg/m2/lần (uống), ngày 36, 43, 50;
+ Tiêm tủy sống 3 thuốc: Vào giờ 24 của MTX ngày 30.
Lưu ý:
+ Liều mercaptopurine ở đây chỉ 50mg/m2/ngày vì giai đoạn này có MTX liều cao;
+ Ngày 15: Bắt đầu khi bạch cầu trung tính > 0,5G/L và tiểu cầu > 50G/L.
b.6. Giai đoạn tăng cường 2
- Nhóm A1/A2: Bắt đầu từ ngày 57 của giai đoạn trung gian khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L
+ Vincristine: 1,5mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 1, 10, 20, 30 (không quá 2mg);
+ Methotrexate: 100 mg/m2 (truyền tĩnh mạch/ 15phút), ngày 1, 10, 20, 30;
+ L-Asparaginase: 20.000 UI/m2 (truyền tĩnh mạch/ 60phút), ngày 2, 11, 21, 31;
+ Tiêm tủy sống 3 thuốc: Ngày 1.
Lưu ý:
+ Không dùng acid folinic sau MTX liều trung bình;
+ Trường hợp không có vấn đề về lâm sàng, giai đoạn này được tiếp tục (mà không có sự thay đổi) khi bạch cầu trung tính > 0,5G/L và tiểu cầu > 50G/L;
+ Hướng dẫn dùng L-asparaginase liều cao:
● Ngày 1, 10, 20, 30: Dùng 1/10 tổng liều truyền tĩnh mạch 60 phút;
● Ngày 2, 11, 21, 31: Dùng 9/10 tổng liều truyền tĩnh mạch ≥ 2 giờ.
+ Kiểm tra MRD sau kết thúc điều trị tăng cường 2.
- Nhóm A3: Gồm 2 đợt liên tiếp nhau:
ĐỢT 1: Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L
+ Methylprednisone: 40mg/m2/ngày (chia 3 lần, uống), ngày 1-15. Giảm liều từ ngày 15 và ngừng vào ngày 21.
+ Vincristine: 1,5mg/m2/ngày (tĩnh mạch chậm hoặc truyền tĩnh mạch) ngày 1, 8, 15 (không quá 2mg);
+ L-asparaginase: 6.000 UI/m2/lần (tĩnh mạch chậm/ 60 phút), tổng cộng 6 mũi, ngày 3, 5, 7, 9, 11, 13;
+ Daunorubicine: 30mg/m2/lần (tĩnh mạch chậm/ 60 phút), ngày 1, 8, 15;
+ Tiêm tủy sống 3 thuốc: Ngày 1.
ĐỢT 2: Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L
+ Mercaptopurine: 50 mg/m2/ngày (uống), ngày 29-49;
+ Cyclophosphamide: 1g/m2/ngày (tĩnh mạch chậm/ 60phút), ngày 29;
+ Cytarabine: 30mg/m2/lần x 2 lần/ngày (tiêm dưới da), ngày 29-30, ngày 36-37, ngày 43-44;
+ Tiêm tủy sống 3 thuốc: Ngày 29.
Điều trị hỗ trợ
+ Dịch truyền: 2.000 ml/m2/ngày;
+ Sử dụng mesna: Tổng liều bằng 2 lần tổng liều cyclophosphamide, truyền tĩnh mạch trước cyclophosphamide 30phút, sau cyclophosphamide 4 giờ và 8 giờ.
Lưu ý:
+ Hóa trị ngày 36, 43 được tiếp tục bất kể số lượng tế bào máu, với điều kiện không có vấn đề về lâm sàng;
+ Kiểm tra MRD sau kết thúc điều trị tăng cường 2;
b.7. Giai đoạn duy trì
- Nhóm A1/A2 có MRD âm tính (< 10-2):
+ Bắt đầu từ ngày 40 của giai đoạn tăng cường II, khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L;
+ Thời gian điều trị duy trì kéo dài trong 24 tháng (nam và nữ), trong đó có 12 tháng tái tấn công (RI) với vincristine và dexamethasone trong năm đầu tiên.
Bao gồm:
+ Vincristine: 1,5mg/m2/ngày (tĩnh mạch chậm hoặc truyền tĩnh mạch). Vincristine vào ngày 1 mỗi tháng, trong 12 tháng đầu (không quá 2mg);
+ Dexamethasone: 6mg/m2/ngày (uống), ngày 1-5 mỗi tháng, trong 12 tháng đầu;
+ Methotrexate: 25mg/m2/tuần (uống), ngày 8, 15, 22 mỗi tháng;
+ Mercaptopurine: 75mg/m2/ngày (uống), ngày 1-28;
+ Tiêm tủy sống 3 thuốc: Mỗi 3 tháng bắt đầu từ các tháng 1, 4, 7, 10, 13, 16, 19 và 22 (tổng cộng 8 mũi).
- Nhóm A3 và A1/A2 có MRD dương tính (≥ 10-2):
+ Bắt đầu ngày 57 sau giai đoạn tăng cường II, điều trị tương tự nhóm A1 nhưng thay dexa bằng corticoid 40 mg/m2 ngày 1-7;
+ Tổng cộng có 6 mũi IT (duy trì tháng 3, 6, 9, 12, 15, 18).
3.2.1.2. Phác đồ điều trị lơ xê mi lympho B cấp - nhóm B
a. Sơ đồ điều trị tổng quát: FRALLE 2000-B: NHÓM B
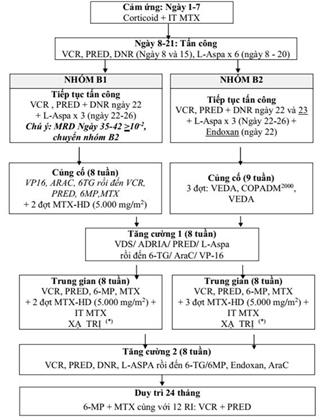
(*) Những người bệnh thuộc nhóm B2 nhưng nhỏ hơn 4 tuổi: Không xạ trị dự phòng trên hệ thần kinh trung ương.
b. Phác đồ điều trị chi tiết
b.1. Giai đoạn cảm ứng:
- Methylprednisone: 60m/ngày chia 2 lần (uống hay truyền tĩnh mạch), ngày 1-7.
- Nếu dùng đường tĩnh mạch: Sử dụng methylprednisolone (cùng liều như trên).
b.2. Giai đoạn tấn công (B1/ B2): Bắt đầu từ ngày 8, ngay sau điều trị cảm ứng:
- Methylprednisone: 40mg/m2, ngày 8-28 (uống hoặc tĩnh mạch), chia 3 lần từ ngày 29: Giảm liều dần, ngừng vào ngày 35.
- Vincristine: 1,5mg/m2, ngày 8, 15, 22, 29. Truyền tĩnh mạch chậm hoặc truyền tĩnh mạch (không quá 2mg).
- L-Asparaginase: 6.000 UI/m2, ngày 10, 12, 14, 16. Tiêm bắp hoặc truyền tĩnh mạch chậm trong 1 giờ ngày 18, 20, 23, 25, 27.
- Daunorubicine: 40mg/m2, ngày 8, 15, 22 (B1, B2). Với nhóm B2, bệnh nhân được điều trị thêm Daunorubicin vào ngày 23. Truyền tĩnh mạch chậm trong 1 giờ.
- Cyclophosphamide: 1.000 mg/m2, + ngày 22 (Riêng nhóm B2) truyền tĩnh mạch chậm 30 phút.
- Tiêm tủy sống 3 thuốc theo tuổi, ngày 8, 15 (với MTX, DEPO, cytarabin). Ngày 22 nếu người bệnh có tổn thương thần kinh trung ương lúc chẩn đoán.
Lưu ý:
- IT chỉ với methotrexate vào ngày 1 (không có corticoid) với điều kiện tiểu cầu > 100G/L. IT mũi 1 nên được thực hiện sớm khi có thể (không chậm quá ngày 4 của điều trị cảm ứng bằng corticoid) trừ trường hợp chống chỉ định (bạch cầu cao có triệu chứng, u trung thất to...).
- Trường hợp BC tăng cao (> 500G/L): Có thể bổ sung vincristine trước ngày 8.
- Vincristine có thể chỉ định liền ở những người bệnh có hội chứng tắc mạch lúc chẩn đoán, hoặc bổ sung nhanh (trước ngày 3 của điều trị cảm ứng bằng corticoid) khi tình trạng lâm sàng diễn tiến xấu đi hoặc tăng nhanh số lượng bạch cầu (khi đã dùng corticoid).
- Xét nghiệm huyết đồ để đánh giá sự nhạy cảm với corticoid vào ngày 8.
- Sự kháng corticoid sẽ được đánh giá để phân nhóm B1, B2.
- Tủy đồ (ngày 21): Đánh giá nhạy cảm với hóa trị liệu ban đầu.
- Người bệnh có tổn thương thần kinh trung ương lúc chẩn đoán: Bổ sung thêm 1 mũi IT vào ngày 22.
- Từ ngày 22, dựa vào kết quả huyết + tủy đồ → người bệnh được chia làm 2 nhóm B1 và B2.
- Làm tủy đồ, MRD và FISH, PCR, nhiễm sắc thể (nếu có bất thường lúc chẩn đoán) vào ngày 35-42 để đánh giá lui bệnh.
- Người bệnh nhóm B1 có MRD dương (vào ngày 35-42) sẽ được tiếp tục điều trị giai đoạn củng cố theo nhóm B2. Người bệnh này sẽ ghép tế bào gốc khi đạt lui bệnh hoàn toàn lần 1 (CR1) nếu có người cho phù hợp HLA.
b.3. Giai đoạn củng cố
b.3.1. Nhóm B1: Gồm 2 đợt
ĐỢT 1: Bắt đầu khi bạch cầu trung tính + monocyte > 1G/L và tiểu cầu > 100G/L
- Mercaptopurine: 50 mg/m2/ngày: 60mg/m2/ngày (uống), ngày 1-21.
- Etoposide: 150mg/m2 (truyền tĩnh mạch trên 1 giờ), ngày 1, 8, 15.
- Cytarabine: 30mg/m2/ngày (tiêm dưới da), ngày 1, 2, 8, 9, 15, 16 (tổng cộng 12 mũi).
- Tiêm tủy sống 3 thuốc ngày 1, người bệnh có tổn thương thần kinh trung ương lúc chẩn đoán + ngày 15.
ĐỢT 2: Bắt đầu khi bạch cầu trung tính > 0,5G/L và tiểu cầu > 50G/L
- Vincristine: 1,5 mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 29 (không quá 2mg).
- Methylprednisone: 40 mg/m2/ngày (uống, chia 3 lần/ngày), ngày 29-35.
- Mercaptopurine: 50 mg/m2/ngày (uống), ngày 29-49.
- Methotrexate: 25 mg/m2/lần (uống), ngày 36, hoặc
5.000mg/m2/ngày (truyền tĩnh mạch 24 giờ), ngày 9, 43.
- Tiêm tủy sống 3 thuốc: Ngay sau khi kết thúc MTX-HD (giờ 24), ngày 30, 44.
Lưu ý:
- Thuốc ngày 8, 15, 43 được tiếp tục bất kể số lượng tế bào máu (nếu không có bất thường về lâm sàng, sinh hóa).
- Đối với người bệnh có tổn thương thần kinh trung ương lúc chẩn đoán: Bổ sung thêm 1 mũi tiêm tủy sống vào ngày 15.
- Nếu kết quả MRD dương (vào ngày 35-42): Người bệnh được tiếp tục điều trị giai đoạn củng cố của nhóm B2 ngay khi có thể và sẽ làm lại MRD lần 2 và ghép tế bào gốc khi đạt CR1 nếu có người cho phù hợp.
- Nếu sử dụng MTX liều cao: Dùng acid folinic bắt đầu từ giờ thứ 36 (xem phụ lục).
- Ngừng co-trimoxazol 3 ngày trước và 5 ngày sau khi dùng MTX liều cao (xem phụ lục).
- Ngừng hóa trị và nghỉ từ ngày 50-57.
b.3.2. Nhóm B2
- 3 đợt liên tiếp: VEDA/COPADM2000/VEDA.
- Giống như củng cố nhóm A3.
b.4. Giai đoạn tăng cường 1 (giống nhau cho B1 và B2): Gồm 2 đợt:
ĐỢT 1: Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L.
- Dexamethasone: 10mg/m2, ngày 1-14, chia 3 lần, uống, tĩnh mạch.
Ngày 15: Giảm liều dần và ngừng ở ngày 21.
- Vincristine 1,5mg/m2/ngày (tối đa 2mg), tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch, ngày 1, 8, 15
- L-Asparaginase: 6.000UI/m2, ngày 3, 5, 7, 9, 11, 13 truyền tĩnh mạch trong 60 phút.
- Doxorubicin: 25mg/m2, ngày 1, 8, 15. Truyền tĩnh mạch trong 60 phút.
- Tiêm tủy sống 3 thuốc ngày 1 (ngày 15).
ĐỢT 2: Bắt đầu ngay khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L.
- Mercaptopurine: 50 mg/m2/ngày: 60mg/m2, ngày 29-49. Uống 1 lần, sáng, đói.
- Etoposide: 150mg/m2, ngày 29, 36, 43. Truyền tĩnh mạch trong 60 phút.
- Cytarabine: 30mg/m2 x 2, ngày 29-30, ngày 36-37, ngày 43-44. Tiêm dưới da (tổng cộng 12 mũi).
- Tiêm tủy sống 3 thuốc, ngày 29.
Lưu ý:
- Thuốc ngày 8, 15, 36, 43 được tiếp tục bất kể số lượng tế bào máu (và không có vấn đề gì về lâm sàng, sinh hóa).
- Ngừng hóa trị và nghỉ từ ngày 50-57.
b.5. Giai đoạn trung gian
b.5.1. Nhóm B1
Bắt đầu ngay khi bạch cầu trung tính và monno > 1G/L và tiểu cầu > 100G/L
- Vincristine: 1,5mg/m2, ngày 1, 15, 29, 43. Tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch (tối đa 2mg).
- Methylprednisone: 40mg/m2, ngày 1-7; ngày 29-35. Chia 3 lần, uống.
- Mercaptopurine: 50mg/m2, ngày 1-49. Uống, sáng, đói.
- Methotrexate: 25mg/m2, ngày 8, 15, 22, 36, 43. Uống 1 lần, sáng, đói.
5.000mg/m2, ngày 1, 29. Truyền tĩnh mạch 24 giờ.
- Tiêm tủy sống 3 thuốc ngày 1, 29 ngay sau khi kết thúc MTX-HD (giờ 24).
Lưu ý:
- Đối với nhóm B1 có tổn thương thần kinh trung ương lúc chẩn đoán:
+ Bổ sung một mũi tiêm tủy sống (*) vào ngày 15 và không thực hiện hóa trị liệu ngày 43;
+ Xạ trị cho những người bệnh này vào khoảng ngày 40-55. Tổng liều xạ trị là 24Gy cho đến C2 cho trẻ > 4 tuổi, và liều này giảm xuống 18Gy cho trẻ < 4 tuổi.
- Sử dụng MTX liều cao: Dùng acid folinic vào giờ thứ 36 (xem phụ lục).
- Ngừng co-trimoxazol 3 ngày trước và 5 ngày sau khi dùng MTX liều cao (xem phụ lục).
b.5.2. Nhóm B2
Bắt đầu ngay khi bạch cầu trung tính và mono > 1 G/L và tiểu cầu > 100 G/L
- Vincristine: 1,5mg/m2/lần (tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch) ngày 1, 15, 29 (không quá 2mg). Bổ sung thêm ngày 43 (nếu không có xạ trị TKTW).
- Methylprednisone: 40mg/m2/ngày (chia 3 lần, uống), ngày 1-7, ngày 29-35.
- Mercaptopurine: 50mg/m2/ngày (uống), ngày 1-49.
- Methotrexat: 5.000mg/m2/ngày (truyền tĩnh mạch/24 giờ), ngày 1, 15, 29, 43.
- Methotrexat: 25 mg/m2/lần (uống), ngày 8, 22, 36.
- Tiêm tủy sống 3 thuốc (vào giờ 24 sau MTX), ngày 2, 16, 30, 43.
Lưu ý:
- Acid folinic (thuốc giải MTX) bắt đầu từ giờ thứ 36 của MTX.
- Đối với trẻ > 4 tuổi: Chỉ định xạ trị dự phòng thần kinh trung ương từ ngày 40-55 và không có hóa trị vào ngày 43 (gồm MTX và IT).
- Đối với trẻ < 4 tuổi: Không xạ trị thần kinh trung ương.
- Chiếu xạ dự phòng thần kinh trung ương: Được thực hiện cho tất cả người bệnh trẻ em trên 4 tuổi từ ngày 40-55 với liều 18Gy cho đến C2. Trẻ dưới 4 tuổi sẽ không được xạ trị và được bổ sung thêm hóa trị liệu với MTX + IT vào ngày 43.
- Chiếu xạ điều trị thần kinh trung ương: Dành cho người bệnh có tổn thương thần kinh trung ương lúc chẩn đoán (CNS+) vào khoảng ngày 40-55. Liều xạ trị là 24Gy cho đến C2 cho trẻ > 4 tuổi, và liều này giảm xuống 18Gy cho trẻ < 4 tuổi.
- Sử dụng MTX liều cao: Dùng acid folinique vào giờ thứ 36 (xem phụ lục).
- Ngừng co-trimoxazol 3 ngày trước và 5 ngày sau khi dùng MTX liều cao (xem phụ lục).
- Ngừng hóa trị liệu và nghỉ từ ngày 50-57.
b.6. Giai đoạn tăng cường 2 (chung cả B1 và B2)
- Liệu trình như tăng cường 2 của nhóm A3.
b.7. Giai đoạn duy trì
- Bắt đầu từ ngày 57 của giai đoạn tăng cường II, khi bạch cầu trung tính > 1G/L và tiểu cầu > 100G/L.
- Thời gian điều trị duy trì kéo dài trong 24 tháng (nam lẫn nữ), trong đó có 12 tháng tái tấn công (RI) với vincristine và methylprednisone trong năm đầu tiên.
Bao gồm:
- Vincristine: 1,5mg/m2/ngày (tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 1 mỗi tháng, trong 12 tháng đầu (không quá 2mg).
- Methylprednisone: 40mg/m2/ngày (uống), ngày 1-7 mỗi tháng, trong 12 tháng đầu.
- Methotrexate: 25mg/m2/tuần (uống), ngày 8, 15, 22 mỗi tháng (ngừng vào tuần có Vincristine).
- Mercaptopurine: 75mg/m2/ngày (uống): Liên tục.
- Tiêm tủy sống 3 thuốc mỗi 3 tháng vào ngày 1 của tái tấn công, bắt đầu từ RI tháng thứ 3 (RI tháng 3, 6, 9, 12,15,18), tổng cộng 6 mũi.
Lưu ý:
- Điều kiện duy trì mỗi tháng khi bạch cầu trung tính > 0,5G/L, tiểu cầu > 100G/L.
- Người bệnh đã xạ trị thì không được tiêm tủy sống nữa.
- Tiếp tục phòng ngừa Pneumocystic carinii trong giai đoạn duy trì với bactrim 25mg/kg x 1 liều, 3 ngày mỗi tuần.
3.2.2. Phác đồ điều trị lơ xê mi lymho T cấp (FRALLE 2000 T)
3.2.2.1. Sơ đồ điều trị tổng quát

3.2.2.2. Phác đồ điều trị chi tiết
a. Giai đoạn cảm ứng:
- Methylprednisone: 60mg/m2/ngày chia 2 lần (uống hay tĩnh mạch), ngày 1-7.
Nếu dùng đường tĩnh mạch: Sử dụng methylprednisolone cùng liều như trên.
b. Giai đoạn tấn công: Bắt đầu từ ngày 8, ngay sau điều trị cảm ứng
- Methylprednisone: 40mg/m2, ngày 8-28, uống hoặc tĩnh mạch, chia 3 lần.
Từ ngày 29: Giảm liều dần và ngừng vào ngày 35.
- Vincristine: 1,5mg/m2, ngày 8, 15, 22, 29. Tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch (không quá 2mg).
- Daunorubicine: 40mg/m2, ngày 8, 9, 10, 15. Truyền tĩnh mạch trong 60 phút.
- L-Asparaginase: 6.000UI/m2, ngày 10, 12, 14, 16, 18, 20, 23, 25, 27. Tiêm bắp hoặc truyền tĩnh mạch trong 1 giờ.
- Cyclophosphamide: 1.000 mg/m2, ngày 8. Truyền tĩnh mạch trong 30 phút.
- Tiêm tủy sống 3 thuốc theo tuổi, ngày 8, 15 (với MTX, DEPO, cytarabin). Ngày 22 nếu người bệnh có tổn thương thần kinh trung ương lúc chẩn đoán.
Lưu ý:
- Người bệnh có tổn thương thần kinh trung ương lúc chẩn đoán: Bổ sung thêm 1 mũi tiêm tủy sống vào ngày 22.
- Huyết đồ (ngày 8): Đánh giá nhạy cảm với methylprednisone.
+ Nhạy với methylprednisone: Khi tế bào blast ở máu ngoại vi (ngày 8): < 1G/L;
+ Kháng với methylprednisone: Khi tế bào blast ở máu ngoại vi (ngày 8): ≥ 1G/L;
- Tủy đồ (ngày 21): Đánh giá nhạy cảm với hóa trị liệu ban đầu.
+ Nhạy hóa trị liệu: Khi tế bào blast trong tủy vào ngày 21: ≤ 5%;
+ Kháng hóa trị liệu: Khi tế bào blast trong tủy vào ngày 21: > 5%.
Từ ngày 22 theo kết quả huyết + tủy đồ người bệnh được phân thành 2 nhóm T1 và T2
- Xét nghiệm tủy đồ, MRD và FISH, PCR, Karyotype (nếu có bất thường lúc chẩn đoán) vào ngày 35-42 để đánh giá lui bệnh và theo dõi sau khi đạt lui bệnh.
- Người bệnh có MRD dương (≥ 10-2) vào ngày 35-42 sẽ được tiếp tục điều trị giai đoạn củng cố theo nhóm T2 khi đủ điều kiện cho đến hết phác đồ. Những người bệnh này sẽ ghép tế bào gốc khi đạt CR1 nếu có người cho phù hợp HLA.
c. Giai đoạn củng cố
c1. Nhóm T1
ĐỢT 1: Bắt đầu khi bạch cầu đoạn trung tính và monocyte > 1G/L và tiểu cầu > 100G/L
- Mercaptopurine 50 mg/m2/ngày (uống), ngày 1-21.
- Cyclophosphamide: 1.000mg/m2 (truyền tĩnh mạch trên 1 giờ), ngày 1, 15.
- Cytarabine: 30mg/m2/12 giờ (tiêm dưới da), ngày 1-2, ngày 8-9, ngày 15-16 (tổng cộng 12 mũi).
- Tiêm tủy sống 3 thuốc vào ngày 1, 15.
ĐỢT 2: Bắt đầu khi bạch cầu đoạn trung tính > 0,5G/L và tiểu cầu > 50G/L
- Vincristine: 1,5mg/m2 (tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 29, 43 (không quá 2mg/ngày).
- Methylprednisone: 40 mg/m2/ngày (uống, chia 3 lần/ngày), ngày 29-35.
- Mercaptopurine: 50 mg/m2/ngày (uống), ngày 29-49.
- Methotrexate: 25 mg/m2/lần (uống), ngày 36.
- Methotrexate: 5.000mg/m2/ngày (truyền tĩnh mạch 24 giờ), ngày 29, 43.
- Tiêm tủy sống 3 thuốc vào giờ 24 của Methotrexat liều cao (ngày 30, 44).
Lưu ý:
- Thuốc ngày 8, 15, 43 được tiếp tục bất kể số lượng tế bào máu (nếu không có bất thường về lâm sàng, sinh hóa).
- Nếu kết quả MRD vào ngày 35-42 dương: Người bệnh được tiếp tục điều trị giai đoạn củng cố nhóm T2 ngay khi có thể và sẽ làm lại MRD lần hai. Nếu MRD lần hai dương, người bệnh bắt buộc phải ghép tế bào gốc khi đạt CR1 nếu có người cho phù hợp.
- Nếu sử dụng MTX liều cao: Dùng acid folinique vào giờ thứ 36 (xem phụ lục).
- Ngừng Co-trimoxazol 3 ngày trước và 5 ngày sau khi dùng MTX liều cao (xem phụ lục).
- Ngừng hóa trị và nghỉ từ ngày 50-57.
c2. Nhóm T2 (3 đợt liên tiếp: VEDA/COPADM2000/VEDA)
Liệu trình như củng cố nhóm A3.
d. Giai đoạn tăng cường 1 (chung cả nhóm T1 và T2)
Liệu trình như tăng cường 1 của B1 và B2.
e. Giai đoạn trung gian
e1. Nhóm T1: Giống như nhóm B1. Xạ trị dự phòng (từ ngày 40-55) cho các bênh nhân trên 4 tuổi có BC lúc nhập viện ≥ 100G/L (liều xạ giống như nhóm B2). Không có hóa trị liệu ngày 43 (VCR, MTX, IT).
e2. Nhóm T2: Như nhóm B2.
g. Giai đoạn tăng cường 2: Chung cả nhym T1 và T2
Liệu trình như tăng cường 2 của B1 và B2.
f. Giai đoạn duy trì: Chung cho cả nhóm T1 và T2
Bắt đầu từ ngày 57 của giai đoạn tăng cường II, khi bạch cầu đoạn trung tính > 1G/L và tiểu cầu > 100G/L.
Thời gian điều trị duy trì kéo dài trong 18 tháng (nam lẫn nữ), trong đó có 12 tháng tái tấn công (RI) với vincristine và dexamethasone trong năm đầu tiên.
Bao gồm:
- Vincristin: 1,5mg/m2/ngày (tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch), ngày 1 mỗi tháng, trong 12 tháng đầu (không quá 2mg/ngày).
- Methylprednisone: 40mg/m2/ngày (uống), ngày 1-7 mỗi tháng, trong 12 tháng đầu.
- Methotrexate: 25mg/m2/tuần (uống), ngày 8, 15, 22 mỗi tháng (ngừng vào tuần có Vincistine).
- Mercaptopurine: 75mg/m2/ngày (uống): Liên tục.
- Tiêm tủy sống 3 thuốc mỗi 3 tháng vào ngày 1 của tái tấn công, bắt đầu từ RI tháng thứ 3 (RI tháng 3, 6, 9). Tổng cộng 3 mũi.
Lưu ý:
- Tái tấn công mỗi 4 tuần. Điều kiện là bạch cầu trung tính > 0,5G/L, tiểu cầu > 100G/L.
- Tiếp tục phòng ngừa Pneumocystic carinii trong giai đoạn duy trì với Bactrim 25 mg/kg x 1 liều, 3 ngày mỗi tuần. Trường hợp người bệnh không chịu đựng được giai đoạn duy trì, ngừng Bactrim và chuyển sang Pentacarinat phun khí dung mỗi tháng.
3.3. Các tiêu chuẩn đánh giá mức độ đip ứng đối với điều trị đặc hiệu theo NCCN 2016:
3.3.1. Tiêu chuẩn đánh giá lui bệnh
- Tiêu chuẩn lui bệnh hoàn toàn về mặt huyết học (CR):
+ Không phát hiện tế bào ác tính trong máu ngoại vi hay biểu hiện thâm nhiễm ngoài tủy (hết các triệu chứng gan to, lách, hạch to, thâm nhiễm thần kinh trung ương);
+ Phục hồi các dòng tế bào sinh máu trong tuỷ; Tỷ lệ blast < 5% tổng số tế bào có nhân trong tủy;
+ Số lượng tuyệt đối bạch cầu đoạn máu ngoại vi > 1 G/l;
+ Số lượng tiểu cầu > 100 G/l;
+ Không tái phát trong vòng 4 tuần.
- Lui bệnh không hoàn toàn về mặt huyết học (CRi): Có đầy đủ tiêu chuẩn lui bệnh hoàn toàn ngoại trừ:
+ Số lượng bạch cầu hạt máu ngoại vi < 1 G/l;
+ Số lượng tiểu cầu < 100 G/l;
+ Không lui bệnh về mặt huyết học (NR): Không đạt được các tiêu chuẩn CR khi kết thúc điều trị cảm ứng.
3.3.2. Tiêu chuẩn tái phát
- Tái phát tuỷ xương đơn độc: Sau khi đạt lui bệnh hoàn toàn, bệnh nhi có biểu hiện thiếu máu, xuất huyết dưới da, sốt, hạch to trở lại..., xuất hiện tế bào blast trong máu ngoại vi, tuỷ xương có ≥ 25% tế bào blast.
- Tái phát tuỷ xương kết hợp: Tái phát tuỷ xương + có ít nhất 1 vị trí tái phát ngoài tuỷ xương.
- Tái phát TKTW: Có hoặc không có đau đầu, buồn nôn, dịch não tuỷ có > 5 bạch cầu/mm3 và có tế bào blast, hoặc có u nội sọ, tổn thương các dây thần kinh sọ, tổn thương võng mạc không giải thích được nguyên nhân.
- Tái phát ngoài tủy khác: Có hiện diện của nguyên bào lympho ở thận hay tinh hoàn...
3.4. Bilan để theo dõi và đánh giá trong suốt qui trình điều trị và khi kết thúc phác đồ điều trị.
Tổng hợp các bilan cần chú ý trong phác đồ:
3.4.1. Bilan trước điều trị: Các xét nghiệm cần làm tại thời điểm chẩn đoán:
- Huyết tủy đồ: Hình thái và hóa học tế bào.
- Dấu ấn miễn dịch.
- Di truyền và sinh học phân tử.
- Sinh thiết tủy xương: Có thể dược chỉ định trong trường hợp tủy nghèo tế bào, tỷ lệ blast < 20% tế bào có nhân trong tủy.
- Sinh hóa máu: Chức năng gan, thận, acid uric, LDH.
- Đông máu cơ bản:
+ Fibrinogen, PT, APTT, TT;
+ Nghiệm pháp rượu;
+ D-dimer.
- Các virus: HBV, HCV, HIV, EBV, CMV.
- Định nhóm máu hồng cầu: ABO, Rh(D).
- Điện tâm đồ và siêu âm tim.
- Chẩn đoán hình ảnh:
+ X-quang tim phổi;
+ Siêu âm ổ bụng, siêu âm tim.
- Xét nghiệm dịch não tủy.
- Xét nghiệm HLA:
+ Nhóm 1: HLA-A; HLA-B; HLA-C;
+ Nhóm 2: HLA-DR; HLA-DQ.
Lưu ý: Bệnh nhân cần được khám răng hàm mặt, tai mũi họng trước khi điều trị.
3.4.2. Bilan trong quá trình điều trị
- Huyết đồ ngày 8: Xác định tình trạng nhạy cảm corticoid.
- Tủy đồ ngày 21: Xác định tình trạng nhạy cảm hóa trị liệu.
- Tủy đồ + MRD vào ngày 35-42: Xác định tình trạng lui bệnh và đánh giá MRD lần 1.
- Tủy đồ + MRD vào ngày 21/29 giai đoạn củng cố: Đánh giá MRD lần 2, dành cho các người bệnh có MRD (+) vào ngày 35-42. Xét nghiệm MRD có thể được đánh giá vào mỗi giai đoạn tiếp theo nếu người bệnh có MRD (+) trước đó và được đánh giá lại trước khi người bệnh điều trị duy trì.
- Tủy đồ trước mỗi một giai đoạn điều trị (tấn công, củng cố, tăng cường 1, trung gian, tăng cường 2).
- Xét nghiệm chẩn đoán hình ảnh và thăm dz chức năng: XQ tim phổi, siêu âm ổ bụng, siêu âm tim và CT-scanner, MRI nếu có nghi ngờ thâm nhiễm trước mỗi giai đoạn điều trị (tấn công, củng cố, tăng cường 1, trung gian, tăng cường 2).
- Bilan theo dõi trong mỗi đợt điều trị: Xét nghiệm sinh hóa máu, tổng phân tích tế bào máu ngoại vi, đông máu huyết tương, xét nghiệm dịch não tủy trong quá trình điều trị. Trong trường hợp bệnh nhân điều trị Methotrexat liều cao, cần theo dõi xét nghiệm định lượng Methotrexat trong máu, nước tiểu, theo dõi pH nước tiểu để đánh giá nguy cơ độc tố của Methotrexat liều cao.
3.4.3. Bilan kết thúc điều trị
- Huyết tủy đồ, đánh giá MRD, đánh giá chức năng cơ quan khi bệnh nhân kết thúc điều trị.
- Khi bệnh ở giai đoạn ổn định, theo dõi xét nghiệm tổng phân tích tế bào máu ngoại vi, sinh hóa máu, đông máu huyết tương hàng tháng, làm Huyết tủy đồ nếu nghi ngờ tái phát.
3.5. Điều trị tái phát
- Điều trị hóa chất phác đồ tái phát: Có thể lựa chọn một trong các phác đồ BFM- REZ 85, COPRALL 2005, FLAG, FLAG-Daunorubicin, FLAG-IDA, Mito-FLAG, sau đó ghép tủy đồng loài (chỉ định ghép xin xem trong bài ghép tế bào gốc tạo máu).
- Tái phát thần kinh trung ương hay tái phát tinh hoàn đơn thuần: Sử dụng phác đồ O2P2 (Mỹ).
Phác đồ cụ thể:
3.5.1. Phác đồ ALL- REZ-BFM 85 có cải tiến: Bao gồm 9 đợt (cảm ứng + luân phiên R1, R2, mỗi đợt 4 lần). Mỗi đợt điều trị cách nhau 21 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
a. Cảm ứng P:
- Methylprednisone 100 mg/m2, uống hoặc tiêm, truyền TM ngày 1-7 và 15-21.
- Vincristine 1,5 mg/m2 tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch ngày 1, 8, 15, 22.
- Methotrexate 1 g/m2 truyền tĩnh mạch trong 36h, ngày 1.
- Leucovorin 15 mg/m2, tiêm tĩnh mạch chậm giờ 48 và 54 sau khi bắt đầu truyền Methotrexat (Có thể tiêm tăng số liều trong trường hợp bệnh nhân có nguy cơ loét hoặc có loét niêm mạc).
- Cytarabine liều cao 3g/m2/12h truyền tĩnh mạch ngày 15, 16.
- L-aspaginase 10.000UI/m2 tiêm bắp ngày 2,3 và ngày 17, 18.
b. R1:
- Methylprednisone 100 mg/m2, uống hoặc tiêm, truyền tĩnh mạch ngày 1-5.
- Vincristine 1,5 mg/m2 tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch ngày 1.
- Methotrexate 1 g/m2 truyền tĩnh mạch trong 36h, bắt đầu từ ngày 1.
- Leucovorin 15 mg/m2, tiêm tĩnh mạch chậm giờ 48 và 54 sau khi bắt đầu truyền Methotrexat (Có thể tiêm tăng số liều trong trường bệnh nhân có nguy cơ loét hoặc có loét niêm mạc).
- Cytarabine 300/m2 truyền tĩnh mạch ngày 5.
- Teniposid (Etoposide) 165 mg/m2 truyền tĩnh mạch trong 1h, ngày 5.
- L-aspaginase 10.000UI/m2 tiêm bắp ngày 6-8.
- Mercaptopurin 100 mg/m2 uống ngày 1-5.
- Tiêm tủy sống Methotrexate ngày 1 (Theo lứa tuổi).
c. R2:
- Dexamethasone 20 mg/m2, uống hoặc tiêm, truyền tĩnh mạch ngày 1-5.
- Vincristine 1,5 mg/m2 tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch ngày 1.
- Methotrexate 1 g/m2 truyền tĩnh mạch trong 36h, ngày 1.
- Leucovorin 15 mg/m2, tiêm tĩnh mạch chậm giờ 48 và 54 sau khi bắt đầu truyền Methotrexat (Có thể tiêm tăng số liều trong trường bệnh nhân có nguy cơ loét hoặc có loét niêm mạc).
- Daunorubicin 50 mg/m2 truyền tĩnh mạch ngày 5.
- Ifosfamid 400 mg/m2 truyền tĩnh mạch, ngày 1-5.
- Mercaptopurin 100 mg/ m2/ngày uống ngày 1-5.
- Tiêm tủy sống Methotrexate ngày 1 (Theo lứa tuổi).
Điều trị luân phiên R1 và R2 mỗi loại 4 đợt
Điều trị duy trì: Bắt đầu khi hoàn thành tất cả cic đợt R1 và R2:
- Mercaptopurin 50 mg/m2 uống hàng ngày trong 2 năm.
- Methotrexate 50 mg/m2 hàng tuần tĩnh mạch hoặc uống trong 2 năm.
- Tiêm tủy sống 3 loại (Methotrexate, cytarabin, hydrocortison hoặc dexamethasone) (theo lứa tuổi) 6 tuần/lần trong 2 năm.
3.5.2. Phác đồ Cooprall 2005:
Phác đồ Cooprall bao gồm nhiều đợt hóa chất với các đợt điều trị phụ thuộc vào vị trí và thời gian tái phát.
Phân nhóm điều trị:
Nhóm S1
Bao gồm tất cả trường hợp tái phát ngoài tủy đơn độc (kể cả ở hệ thần kinh TW) xảy ra ở thời điểm ≥ 6 tháng sau khi ngưng điều trị lần 1, bất kể là kiểu hình lympho T hay không phải lympho T.
Nhóm S2
- Tất cả các Lơ xê mi lympho B cấp tái phát tại tủy xương vào thời điểm ≥ 6 tháng sau khi ngưng điều trị lần 1.
- Tất cả các Lơ xê mi lympho B cấp tái phát nhiều nơi (kết hợp) với thời gian từ lúc chẩn đoán ban đầu đến lúc tái phát ≥ 18 tháng.
- Tái phát ngoài tủy đơn độc, sớm xảy ra trong quá trình điều trị hoặc < 6 tháng sau khi ngưng điều trị này.
Nhóm S3-S4
- Tất cả Lơ xê mi lympho B cấp tái phát tủy đơn độc lần thứ 1: Xảy ra vào thời điểm:
+ Dưới 18 tháng kể từ lúc chẩn đoán ban đầu: (S4);
+ ≥ 18 tháng kể từ lúc chẩn đoán ban đầu và < 6 tháng sau khi ngưng điều trị: (S3).
- Tất cả Lơ xê mi lympho B cấp tái phát tủy kết hợp lần thứ 1: Xảy ra vào thời điểm < 18 tháng kể từ sau chẩn đoán ban đầu (S4).
- Tất cả Lơ xê mi lympho T cấp hay U lympho không Hodgkin tế bào T tái phát tủy hay kết hợp bất kể thời gian đạt CR1 (S4).
- Tất cả Lơ xê mi lympho cấp với NST Ph(+) tái phát hoặc Lơ xê mi lympho cấp có tái sắp xếp gen MLL tái phát (S4).
Phác đồ điều trị theo phân nhóm:
- Với nhóm S1: Điều trị cảm ứng Block F1 và Block F2 sau đó luân phiên R2 và R1 mỗi loại 3 đợt, sau đó duy trì 12 tháng.
- Với nhóm S2: Điều trị cảm ứng Block F1 và Block F2 sau đó luân phiên R2 và R1 mỗi loại 3 đợt, sau đó ghép tế bào gốc tạo máu hoặc 4 đợt sau đó duy trì 2 năm nếu không ghép tế bào gốc.
- Với nhóm S3-4: Điều trị cảm ứng Block F1 và Block F2 sau đó luân phiên R2 và R1 mỗi loại 2 đợt sau đó ghép tế bào gốc.
Mỗi đợt điều trị cách nhau 21 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
Phác đồ điều trị cụ thể:
- Cảm ứng: Lần lượt với Block F1 và Block F2.
a. Block F1:
- Dexamethasone 20 mg/m2, uống hoặc tiêm, truyền TM ngày 1-5.
- Vincristine 1,5 mg/m2 tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch ngày 1, 6.
- Methotrexate 1 g/m2 truyền tĩnh mạch trong 36h, ngày 1.
- Leucovorin 15 mg/m2, tiêm tĩnh mạch chậm giờ 48 và 54 sau khi bắt đầu truyền Methotrexat (Có thể tiêm tăng số liều trong trường bệnh nhân có nguy cơ loét hoặc có loét niêm mạc).
- L-aspaginase 6.000UI/m2 tiêm bắp x 6 mũi từ ngày 4
- Tiêm tủy sống 3 loại (Methotrexate, cytarabin, prednisone) (theo lứa tuổi) ngày 2 (có thể thêm ngày 7 nếu có nguy cơ cao thâm nhiễm thần kinh trung ương).
b. Block F2:
- Dexamethasone 20 mg/m2, uống hoặc tiêm, truyền tĩnh mạch ngày 1-5.
- Vincristine 1,5 mg/m2 tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch ngày 1.
- Cytarabine 3g/m2/12h ngày 1, 2.
- L-aspaginase 6.000UI/m2 tiêm bắp x 6 mũi từ ngày 4.
- Tiêm tủy sống 3 loại (Methotrexate, cytarabin, prednisone) (theo lứa tuổi) ngày 5.
c. Block R2:
- Dexamethasone 20 mg/m2, uống hoặc tiêm, truyền tĩnh mạch ngày 1-5, sau đó giảm xuống ½ liều ngày 6.
- Vincristine 1,5 mg/m2) tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch ngày 1.
- Methotrexate 1 g/m2 truyền tĩnh mạch trong 36h, ngày 1.
- Leucovorin 15 mg/m2, tiêm tĩnh mạch chậm giờ 48 và 54 sau khi bắt đầu truyền Methotrexat (Có thể tiêm tăng số liều trong trường bệnh nhân có nguy cơ loét hoặc có loét niêm mạc).
- Daunorubicin 35 mg/m2 truyền tĩnh mạch ngày 5.
- Ifosfamid 400 mg/m2 truyền tĩnh mạch, ngày 1-5.
- Mercaptopurin 100 mg/m2 uống ngày 1-5.
- L-aspaginase 6.000UI/m2 tiêm bắp x 6 mũi từ ngày 6.
- Tiêm tủy sống 3 loại (Methotrexate, cytarabin, prednisone) (theo lứa tuổi) ngày 2 (có thể thêm ngày 6 nếu có nguy cơ cao thâm nhiễm thần kinh trung ương).
d. Block R1:
- Dexamethasone 20 mg/m2, uống hoặc tiêm, truyền TM ngày 1-5, sau đó giảm xuống ½ liều ngày 6.
- Vincristine 1,5 mg/m2 tiêm tĩnh mạch chậm hoặc truyền tĩnh mạch ngày 1, 6.
- Methotrexate 1 g/m2 truyền tĩnh mạch trong 36h, ngày 1.
- Leucovorin 15 mg/m2, tiêm tĩnh mạch chậm giờ 48 và 54 sau khi bắt đầu truyền Methotrexat (Có thể tiêm tăng số liều trong trường bệnh nhân có nguy cơ loét hoặc có loét niêm mạc).
- Cytarabine liều cao 2g/m2/12h truyền tĩnh mạch ngày 5.
- Mercatopurine 100 mg/m2 uống ngày 1-5.
- L-aspaginase 6.000UI/m2 tiêm bắp x 6 mũi từ ngày 6.
Tiêm tủy sống 3 loại (Methotrexate, cytarabin, prednisone) (theo lứa tuổi) ngày 2.
3.5.3. Phác đồ FLAG-IDA:
- Fludarabin 25-30 mg/m2 da truyền tĩnh mạch ngày 1-5.
- Cytarabin 2.000 mg/m2 da truyền tĩnh mạch ngày 1-5.
- G-CSF 5 mcg/kg cân nặng/ngày tiêm dưới da từ ngày 0 đến khi phục hồi bạch cầu hạt trung tính (> 1,5G/L).
- Idarubicin 10 mg/m2 da/ngày tiêm tĩnh mạch ngày 1-3.
Điều trị 1-2 đợt. Mỗi đợt điều trị cách nhau 28 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
3.5.4. Phác đồ FLAG-Daunorubicin
- Fludarabin 25-30 mg/m2 da truyền tĩnh mạch ngày 1-5.
- Cytarabin 2.000 mg/m2 da truyền tĩnh mạch ngày 1-5.
- G-CSF 5 mcg/kg cân nặng/ngày tiêm dưới da từ ngày 0 đến khi phục hồi bạch cầu hạt trung tính (> 1,5G/L).
- Daunorubicin 40-60 mg/m2 da/ngày tiêm tĩnh mạch ngày 1-3 hoặc ngày 1-3-5.
Điều trị 1-2 đợt. Mỗi đợt điều trị cách nhau 28 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
3.5.5. Phác đồ Mito-FLAG
- Fludarabin 25-30 mg/m2 da truyền tĩnh mạch ngày 1-5.
- Cytarabin 2.000 mg/m2 da truyền tĩnh mạch ngày 1-5.
- G-CSF 5 mcg/kg cân nặng/ngày tiêm dưới da từ ngày 0 đến khi phục hồi bạch cầu hạt trung tính (> 1,5G/L).
- Mitoxantrone 10 mg/m2 da/ngày tiêm tĩnh mạch ngày 1.
Điều trị 1-2 đợt. Mỗi đợt điều trị cách nhau 28 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
3.5.6. Một số phác đồ khác:
- Phác đồ O2P2 cho bệnh nhân tái phát thần kinh trung ương đơn độc hoặc tái phát tinh hoàn đơn độc: bao gồm 106 tuần điều trị với các thuốc cơ bản như Prednisone, dexamethasone, cyclophosphamide, etoposide, daunorubicine, methotrexat, vincristin, L-Aspaginase, Mercatopurine, kết hợp tiêm tủy sống và xạ trị.
3.6. Cập nhật hướng điều trị mới:
Điều trị nhắm đích kết hợp điều trị hóa đích bao gồm:
- Liệu pháp phân tử:
+ Ức chế tyrosine kinase ABL1 (imatinib) cho bệnh nhân Ph+ hoặc Ph-like B-ALL hoặc T-ALL với NUP-214- ABL1;
+ Ức chế JAK (ruxolitinib) cho Ph-like hoặc ETP hoặc JAK-dependent ALL;
+ Bortezomib cho B/T-ALL tái phát;
+ Ức chế Notch (LY3039478) cho T-ALL.
- Liệu pháp kháng thể miễn dịch: anti-CD19, anti-CD22.
- Liệu phip tế bào: CD19 CAR T cells, CD22 CAR T cells.
TÀI LIỆU THAM KHẢO
1. Campana D. Status of minimal residual disease testing in childhood haematological malignances. Br J Haematol. 2008 Aug 15; [Epub ahead of print] PMID: 18710378.
2. Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008; 111: 5477-5485.
3. Gaynon PS, Angiolillo AL, Carroll WL, et al. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children’s Oncology Group Report. Leukemia. 2010; 24:285-297.
4. Moricke A, Zimmermann M, Reiter A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010; 24: 265-284.
5. Pui C, Carroll W, Meshinchi S, Arceci RJ. Biology, risk stratification and therapy of pediatric acute leukemia: an update. J Clin Oncol. 2010 In press.
6. Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009; 360:2730-2741.
7. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006; 354:166-178.
8. Tallen G, Ratei R, Mann G, et al. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010; 28:2339-2347.
9. FRALLE 2000-B/T. Version amendée mars 2003.
10. Cooprall version 2005
11. https://www.researchgate.net/publication/20974508_BFM_Group_Treatment_Results_in_Relapsed_Childhood_Acute_Lymphoblastic_Leukemia
12. NCCN (2016). Pediatric acute lymphoblastic leukemia. Version 2.2016. NCCN clinical practice guidelines in oncology. https://www.nccn.org/professionals/physician_gls/pdf/ped_all.pdf.
13. NCCN (2020). Pediatric acute lymphoblastic leukemia. Version 2.2020. NCCN clinical practice guidelines in oncology
22. LƠ XÊ MI TUỶ CẤP TRẺ EM
(Bệnh bạch cầu cấp dòng tủy trẻ em)
1. ĐẠI CƯƠNG
Lơ xê mi (LXM) tuỷ cấp là một nhóm bệnh máu ác tính. Đặc trưng của bệnh là sự tăng sinh một loại tế bào non ác tính (tế bào blast) có nguồn gốc dòng tủy tại tuỷ xương.
Ở trẻ em, LXM tủy cấp hiếm gặp hơn LXM lympho cấp, chiếm khoảng 18% các LXM ở trẻ em, tỷ lệ gặp cao nhất ở trẻ dưới 1 tuổi.
2. NGUYÊN NHÂN
Nguyên nhân gây LXM tủy cấp trẻ em chưa được sáng tỏ. Tuy nhiên, các yếu tố được cho là nguy cơ gây LXM tủy cấp ở trẻ bao gồm: Tia xạ, hóa chất, yếu tố di truyền (hội chứng Down, thiếu máu Fanconi…), cách sống của bố mẹ (nghiện rượu, thuốc lá…).
3. CHẨN ĐOÁN
3.1. Triệu chứng lâm sàng
- Hội chứng thiếu máu.
- Hội chứng xuất huyết.
- Có thể gặp tình trạng đông máu rải rác trong lòng mạch (DIC), đặc biệt rất hay gặp trên người bệnh LXM cấp thể tiền tủy bào.
- Hội chứng nhiễm trùng: Sốt, viêm loét miệng họng, viêm phổi, nhiễm trùng da...
- Hội chứng thâm nhiễm: Gan to, lách to, hạch to, phì đại lợi, thâm nhiễm da, đau xương, thâm nhiễm thần kinh trung ương với các dấu hiệu thần kinh khu trú...
- Trong trường hợp số lượng bạch cầu tăng cao có thể gặp triệu chứng tắc mạch do tăng bạch cầu: Tắc mạch não, tắc mạch phổi, tắc mạch dương vật…
- Biểu hiện toàn thân do bệnh lý ác tính: Mệt mỏi, gầy sút, suy sụp nhanh.
3.2. Triệu chứng xét nghiệm
3.2.1. Xét nghiệm tế bào máu ngoại vi và tủy xương
a. Xét nghiệm tế bào máu ngoại vi: Thường có các biểu hiện sau:
- Thiếu máu bình sắc, hồng cầu kích thước bình thường, hồng cầu lưới giảm.
- Số lượng bạch cầu thường tăng, nhưng có thể bình thường hoặc giảm; gặp một tỷ lệ tế bào non (tế bào blast) - ác tính.
- Số lượng tiểu cầu giảm.
b. Xét nghiệm tủy xương:
- Xét nghiệm tủy đồ: Số lượng tế bào tủy thường tăng, tế bào blast tăng sinh, hình thái dòng tủy, chiếm tỷ lệ ≥ 20% các tế bào có nhân trong tủy. Các dòng hồng cầu, bạch cầu hạt và mẫu tiểu cầu bị lấn át bởi tế bào blast.
- Nhuộm hoá học tế bào cho phép chẩn đoán thể bệnh LXM cấp theo bảng xếp loại FAB:
+ Thường dương tính với MPO, Soudan - Black, âm tính với PAS;
+ Kỹ thuật nhuộm esterase không đặc hiệu được sử dụng trong chẩn đoán LXM cấp dòng mono vì các tế bào thuộc dòng này cho phản ứng dương tính mạnh và bị ức chế bởi NaF, trong khi tế bào dòng bạch cầu hạt tuy cũng cho phản ứng dương tính nhưng không bị ức chế bởi NaF.
- Sinh thiết tuỷ xương được chỉ định trong trường hợp chọc hút tủy không chẩn đoán xác định được do tủy nghèo tế bào.
3.2.2. Đặc điểm dấu ấn miễn dịch và di truyền
a. Dấu ấn miễn dịch đặc trưng trong LXM tủy cấp (AML)
- Tế bào non chưa biệt hóa: CD34, CD117 (c-kit): tế bào dòng tủy/tế bào gốc.
- Dòng bạch cầu hạt: CD13, CD15, CD 33, cytoplasmic MPO, HLA-DR (dương tính trong đa số thể AML, âm tính trong APL).
- Dòng mono: CD13, CD15, CD33, CD14.
- Dòng mẫu tiểu cầu: CD41 (Platelet glycoprotein IIb/IIIa complex), CD61 (Platelet glycoprotein IIIa).
- Dòng hồng cầu: Glycophorin A.
b. Bất thường di truyền và ý nghĩa tiên lượng trong LXM tủy cấp trẻ em
- Nguy cơ thấp: Bất thường NST t(8;21) tạo đột biến gen AML1/ETO (15%), t(15;17) tạo đột biến gen PML/RARα, hoặc inv(16) tạo đột biến gen CBFβ/MYH11 (10%), đột biến gen liên quan đến sắp xếp lại gen MLL (20%) (một số thuộc nhóm tiên lượng xấu); đột biến NPM1, đột biến CEFBA (hiếm gặp hơn ở người lớn).
- Nguy cơ trung bình: Công thức NST bình thường, bất thường NST +8, -Y, +6; t(1;11)(q21;q23) tạo gen MLL-AF3.
- Nguy cơ cao: Bất thường NST -7, -5, tổn thương đồng thời nhiều NST (≥ 3), hoặc đột biến gen FLT3(FLT3-ITD), t(9;11)(p12;q23) tạo gen MLL-AF4, t(6;11)(q27;q23) tạo gen MLL-AF10.
3.2.3. Tiêu chuẩn chẩn đoán
Chẩn đoán xác định LXM tuỷ cấp dựa vào biểu hiện lâm sàng và xét nghiệm, cụ thể là:
- Dựa vào triệu chứng lâm sàng điển hình của bệnh.
- Dựa vào triệu chứng cận lâm sàng: Xét nghiệm tuỷ đồ thấy tế bào blast ≥ 20% tế bào có nhân trong tuỷ mang đặc tính của dòng tủy (Hoá học tế bào dương tính với MPO, Soudan black).
- Mang dấu ấn miễn dịch đặc trưng dòng tủy (CD 13, CD33, CD 117, CD15, CD 64, CD14…).
3.3. Xếp loại LXM tủy cấp trẻ em: Tương tự xếp loại người lớn (tham khảo bài Lơ xê mi cấp):
- Xếp loại LXM tủy cấp theo FAB 1986 có bổ sung.
- Xếp loại theo WHO 2016.
4. ĐIỀU TRỊ
4.1. Điều trị hóa chất
4.1.1. Điều trị LXM tuỷ cấp trẻ em (trừ thể tiền tủy bào)
Hiện nay, có nhiều phác đồ điều trị LXM tủy cấp trẻ em, dựa trên các hóa chất cơ bản như: Daunorubicin (Idarubicin), Cytarabine, Etoposide, Mitoxantrone, Fludarabine… trải qua các giai đoạn: tấn công, củng cố, duy trì. Nhiều thuốc điều trị mới cũng đang được thử nghiệm điều trị trên LXM tủy cấp trẻ em, đặc biệt trên nhóm bệnh nhi nguy cơ cao, tái phát/ kháng thuốc. Một số phác đồ đang được áp dụng:
a. Phác đồ MASPORE 2006
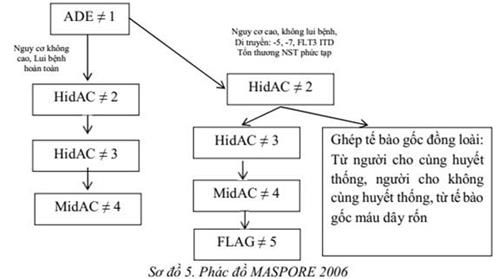
Phác đồ Cụ thể:
- Cảm ứng ADE # 1
+ Truyền tĩnh mạch Cytarabine 100mg/m2/12 giờ tiêm tĩnh mạch bolus cho 20 liều (10 ngày);
+ Truyền tĩnh mạch Daunorubicin 50mg/m2/liều mỗi ngày vào ngày 1, 3 và 5 truyền 20 giờ nếu có đường truyền tĩnh mạch trung tâm, hoặc đường bolus tiêm tĩnh mạch nếu không có đường trung tâm;
+ Truyền tĩnh mạch Etoposide 100 mg/m2/liều trong 250 ml NaCl 0,9% trong vòng 1 giờ ngày 1-5;
+ Tiêm tủy sống (IT): Ngày 1.
- HidAC # 2
+ Truyền tĩnh mạch cytarabine 3.000mg/m2/12 giờ với 6 liều (3 ngày);
+ Tiêm tủy sống (IT): Ngày 1.
- HidAC # 3
+ Truyền tĩnh mạch cytarabine 3.000mg/m2/12 giờ truyền với 6 liều (3 ngày);
+ Tiêm tủy sống (IT): Ngày 1.
- MiDAC # 4
+ Truyền tĩnh mạch Mitoxantrone 10 mg/m2/ngày trong 250ml NaCl 0,9% ngày 1-5 (tổng cộng 5 liều);
+ Truyền tĩnh mạch cytarabine 1.000 mg/m2/12 giờ truyền vào ngày 1-3 (tổng cộng 6 liều).
- Củng cố FLAG # 5: Cho bệnh nhân có nguy cơ cao mà không cần ghép tế bào gốc:
+ GCSF 5µg/kg/ngày tiêm dưới da từ ngày 1 đến khi số lượng tuyệt đối bạch cầu đoạn trung tính > 1G/L trong 3 ngày liên tiếp;
+ Truyền tĩnh mạch Fludarabine 25mg/m2/ngày trong 100ml NaCl 0,9% ngày 1-5;
+ Truyền tĩnh mạch Cytarabine 2.000mg/m2/ngày trong 250ml NaCl 0,9% trong 5 ngày (bắt đầu 4 giờ sau khi bắt đầu truyền fludarabine).
 Lưu ý: Liều tiêm hóa chất nội tủy theo tuổi:
Lưu ý: Liều tiêm hóa chất nội tủy theo tuổi:
|
|
MTX |
Hydrocortisone |
Cytarabine |
Thể tích |
|
< 1 tuổi |
5 mg |
5 mg |
15 mg |
3 ml |
|
1-2 tuổi |
7,5 mg |
7,5 mg |
20 mg |
4ml |
|
2-3 tuổi |
10 mg |
10 mg |
25 mg |
5ml |
|
>3 tuổi |
12,5 mg |
12,5 mg |
30 mg |
6ml |
b. Phác đồ BFM-83
- Điều trị cảm ứng:
+ Cytarabine 100mg/m2/ngày, truyền tĩnh mạch liên tục, ngày 1, 2;
+ Daunorubicin 60mg/m2, truyền tĩnh mạch trong 30 phút, ngày 3-5;
+ Cytarabine 100mg/m2/12h, truyền tĩnh mạch, ngày 3-8;
+ Etoposid 150mg/m2, truyền tĩnh mạch 60 phút, ngày 6-8;
+ Methotrexat (12,5 mg) tiêm tủy sống, ngày 1.
- Củng cố:
+ Methylprednison 40mg/m2, tiêm tĩnh mạch trong 28 ngày, giảm liều bằng 1/2, 1/4, 1/8 liều ban đầu;
+ Vincristine 1,5mg/m2, truyền tĩnh mạch hàng tuần x 4 tuần đầu (tối đa 2mg/m2);
+ Adriamycin 30mg/m2, truyền tĩnh mạch hàng tuần x 4 tuần, vào thứ hai;
+ Cytarabine 75mg/m2, truyền tĩnh mạch hàng ngày, 4 ngày một tuần, bắt đầu ngày thứ 3 đến thứ 6, tổng cộng 16 liều;
+ Mercaptopurine 50mg/m2, uống hàng ngày trong 28 ngày đầu;
+ Cyclophosphamid 500mg/m2, TMx2, liều đầu ngày 35,liều 2 ngày 63;
+ Cytarabine 75mg/m2/ngày, TM bắt đầu ngày 35, 4 ngày một tuần, tổng cộng 16 liều;
+ Mercaptopurine 50mg/m2, uống hàng ngày, từ ngày 35-63.
- Duy trì: 12 chu kỳ, mỗi chu kỳ bao gồm 8 tuần:
+ Doxorubicin 25 mg/m2 ngày 1 trong 12 tháng đầu;
+ Cytarabine 40 mg/m2 ngày 1-4 và ngày 29-32;
+ Mercaptopurine 40 mg/m2, uống hàng ngày, từ ngày 1-56.
c. Phác đồ hóa trị liệu tiêu chuẩn: Điều trị như LXM tủy cấp người lớn, bao gồm: Phác đồ tấn công (điều trị cảm ứng) “3+7”, ADE, củng cố bằng cytarabin liều cao (HDAC) 2 đợt. Cụ thể như sau:
- Phác đồ "3+7":
+ Daunorubicin 45-60 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-3; hoặc Idarubicin 12mg/m2/ngày x 3 ngày;
+ Ara-C 100-200 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-7.
- Phác đồ ADE
+ Truyền tĩnh mạch Cytarabine 100 mg/m2/ liều 12 giờ 20 liều (10 ngày);
+ Truyền tĩnh mạch Daunorubicin 50mg/m2/ liều mỗi ngày vào ngày 1, 3 và 5;
+ Truyền tĩnh mạch Etoposide 100 mg/m2/ liều truyền trong vòng 1 giờ ngày 1-5.
- Phác đồ cytarabin liều cao:
+ Ara-C 3.000 mg/m2 da/12 giờ x 2 lần/ngày, truyền tĩnh mạch ngày 1, 3, 5.
Các đợt điều trị cách nhau 28 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
- Cần lưu ý rằng LXM cấp dòng mono hoặc tủy-mono hoặc dòng tủy có SLBC lúc chẩn đoán > 50G/L có tỷ lệ thâm nhiễm thần kinh trung ương đáng kể nên cần được điều trị dự phòng thâm nhiễm thần kinh trung ương như đối với LXM cấp dòng lympho.
4.1.2. Điều trị LXM cấp thể tiền tủy bào
a. Phân nhóm nguy cơ
Nhóm nghiên cứu Italian Group for Adult Hematologic Diseases (GINEMA) và Spanish PETHEMA phân nhóm nguy cơ đối với bệnh nhân APL theo số lượng bạch cầu và số lượng tiểu cầu:
- Nguy cơ thấp SLBC < 10G/L và SLTC > 40G/L.
- Nguy cơ trung bình SLBC < 10G/L và SLTC < 40G/L.
- Nguy cơ cao SLBC > 10G/L và SLTC < 40G/L.
b.Phác đồ điều trị cụ thể
Các phác đồ điều trị thường phối hợp điều trị nhắm đích với ATRA (all trans retinoic acid) và hóa trị liệu, bao gồm các đợt tấn công, củng cố và duy trì.
- Phác đồ IC - APL 2006:
+ ATRA 25mg/m2 da/ngày chia hai lần đến khi lui bệnh hoàn toàn: tối đa 90 ngày);
+ Daunorubicin 60 mg/m2 da/ngày, truyền tĩnh mạch ngày 2-4-6-8;
+ Dexamethasone 2,5mg/m2/12h x 15 ngày.
- Điều trị củng cố: Điều trị theo nhóm nguy cơ, bao gồm 3 đợt
* Nhóm nguy cơ thấp:
Đợt 1:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Daunorubicin 25 mg/m2 da/ngày, truyền tĩnh mạch ngày 1,2,3,4.
Đợt 2:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Mitoxantrone 10 mg/m2 da/ngày, truyền tĩnh mạch ngày 1,2,3.
Đợt 3:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Daunorubicin 60 mg/m2 da/ngày, truyền tĩnh mạch ngày 1.
* Nhóm nguy cơ trung bình:
Đợt 1:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Daunorubicin 35 mg/m2 da/ngày, truyền tĩnh mạch ngày 1,2,3,4.
Đợt 2:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Mitoxantrone 10 mg/m2 da/ngày, truyền tĩnh mạch ngày 1,2,3.
Đợt 3:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Daunorubicin 60 mg/m2 da/ngày, truyền tĩnh mạch ngày 1,2.
* Nhóm nguy cơ cao:
Đợt 1:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Daunorubicin 25 mg/m2 da/ngày, truyền tĩnh mạch ngày 1,2,3,4;
+ Ara-C 1.000mg/m2/ngày, truyền tĩnh mạch ngày 1,2,3,4.
Đợt 2:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Mitoxantrone 10 mg/m2 da/ngày, truyền tĩnh mạch ngày 1,2,3,4,5.
Đợt 3:
+ ATRA 25 mg/m2 da/ngày x 15 ngày, đường uống;
+ Daunorubicin 60 mg/m2 da/ngày, truyền tĩnh mạch ngày 1;
+ Ara-C 150 mg/m2 da/8h, truyền tĩnh mạch ngày 1,2,3,4.
Mỗi đợt điều trị cách nhau 14-21 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
- Điều trị duy trì bằng ATRA 25 mg/m2 da đường uống hàng ngày trong 15 ngày mỗi 3 tháng, 6-MP 50 mg/m2 da 1 lần hàng ngày và methotrexate 15 mg/m2 da 1 lần hàng tuần trong 2 năm.
- Hội chứng ATRA có thể xảy ra trong 21 ngày đầu của điều trị với các triệu chứng như: sốt, hạ huyết áp, giữ nước, suy hô hấp, viêm niêm mạc, dịch màng phổi, màng tim, các đám mờ trên X-quang phổi, suy thận cấp, suy chức năng gan, thường đi kèm với tăng bạch cầu. Điều trị bằng dexamethasone 10 mg mỗi 12 giờ đường tĩnh mạch trong ít nhất 3 ngày. Có thể tạm thời dừng thuốc ATRA cho đến khi hội chứng ATRA giảm hoặc hết.
- Đối với LXM cấp tiền tủy bào tái phát, lựa chọn điều trị bằng Arsenic trioxide (ATO) với liều: ATO 0,15 mg/kg/ngày đến khi lui bệnh hoàn toàn trong tủy xương, tối đa 60 liều, trung bình 35 liều. Điều trị củng cố bằng ATO với liều như trên, 25 liều trong vòng 5 tuần. Chỉ định ghép tế bào gốc cho bệnh nhân có PML/RARA (+) sau điều trị ATO.
- Hiện nay, nhiều phác đồ trên thế giới đã cho phép điều trị phối hợp ATRA và ATO như điều trị hàng 1 cho bệnh nhân APL ở người lớn và trẻ em.
4.1.3. Điều trị LXM tuỷ cấp có hội chứng Down
Sử dụng phác đồ MASPORE 2006 cho bệnh nhân có hội chứng Down với hóa trị liệu giảm cường độ liều qua 4 giai đoạn:
- Cảm ứng ADE # 1
+ Truyền tĩnh mạch Cytarabine 100 mg/m2 da/12h, truyền tĩnh mạch bolus cho 14 liều (7 ngày);
+ Truyền tĩnh mạch Daunorubicin 50mg/m2 da/liều mỗi ngày vào ngày 1 và ngày 3 truyền 20 giờ nếu có đường trung tâm, hoặc đường bolus truyền tĩnh mạch nếu không có đường trung tâm;
+ Truyền tĩnh mạch Etoposide 100 mg/m2 da/liều trong 250ml NaCl 0,9% trong vòng 1 giờ ngày 1-3;
+ Tiêm tủy sống (IT): Ngày 1.
- HidAC # 2
+ Truyền tĩnh mạch cytarabine 1.000mg/m2 da/12h với 6 liều (3 ngày);
+ Tiêm tủy sống (IT): Ngày 1.
- HidAC # 3
+ Truyền tĩnh mạch cytarabine 1.000mg/m2 da/12h truyền với 6 liều (3 ngày);
+ Tiêm tủy sống (IT): Ngày 1.
- MiDAC # 4 (nếu không đạt lui bệnh hoàn toàn sau điều trị tấn công ADE)
+ Truyền tĩnh mạch Mitoxantrone 10 mg/m2/ngày trong 250ml NaCl 0,9% ngày 1-3 (tổng cộng 5 liều);
+ Truyền tĩnh mạch cytarabine 1.000 mg/m2 da/12h truyền vào ngày 1-3 (tổng cộng 6 liều).
4.2. Ghép tế bào gốc
Chỉ định ghép tế bào gốc tạo máu cho LXM tủy cấp trẻ em trong các trường hợp:
- Nhóm nguy cơ cao: Sau khi đạt Lui bệnh hoàn toàn lần 1.
- Bệnh nhi thất bại với điều trị hóa chất tấn công.
- Bệnh nhi tái phát: Sau khi đạt lui bệnh lần 2.
4.3. Điều trị nhắm đích và một số thuốc điều trị mới
- Hóa chất thế hệ mới: Một số hoá chất thế hệ mới đang được nghiên cứu áp dụng điều trị nhằm giảm độc tính của hoá chất trên bệnh nhân. Một số đang được nghiên cứu trên bệnh nhân nhi có hiệu quả là:
+ Các thuốc nucleoside analogs mới: Clofarabine.
+ Thuốc ức chế Proteazome: Bortezomib.
- Điều trị nhắm đích:
+ Thuốc ức chế FLT3: Sorafenib và một số thuốc đang trong giai đoạn thử nghiệm;
+ Thuốc ức chế IDH: IDH 1, IDH 2;
+ Liệu pháp tế bào CAR-T cell.
4.4. Điều trị LXM tuỷ cấp trẻ em tái phát, kháng thuốc
Với LXM tủy cấp tái phát hoặc kháng thuốc, có thể sử dụng các phác đồ hóa trị liệu liều cao như phác đồ ADE, FLAG-IDA, Mito-FLAG, CLAG, HAM, Cytarabin + Mitoxantron; hoặc phác đồ Cytarabin liều cao; nên tiến tới ghép đồng loại nếu đủ điều kiện. Một số nghiên cứu sử dụng thuốc nucleoside analogs mới như Clofarabine kết hợp cytarabine hoặc các thuốc điều trị nhắm đích (thuốc ức chế FLT3, kháng thể đơn dòng…) trên bệnh nhi LXM cấp dòng tủy tái phát/kháng thuốc cũng đang được nghiên cứu.
Điều trị hóa chất: Với 1 số hóa chất tác dụng mạnh, có thể sử dụng 1 số phác đồ như điều trị AML người lớn:
- Phác đồ FLAG-IDA:
+ Fludarabin 25-30 mg/m2 da, truyền tĩnh mạch ngày 1-5;
+ Cytarabin 2.000 mg/m2 da, truyền tĩnh mạch ngày 1-5;
+ G-CSF 5 mcg//kg cân nặng/ngày, tiêm dưới da từ ngày 0 đến khi phục hồi bạch cầu hạt trung tính (> 1,5G/L);
+ Idarubicin 10 mg/m2 da/ngày, truyền tĩnh mạch ngày 1-3.
Điều trị 1-2 đợt. Mỗi đợt điều trị cách nhau 28 ngày hoặc 1 tuần sau khi tủy xương hồi phục sau đợt điều trị trước đó (số lượng bạch cầu hạt > 1,5G/L; số lượng tiểu cầu > 100G/L).
- Phác đồ FLAG-Daunorubicin:
+ Fludarabin 25-30 mg/m2 da, truyền tĩnh mạch ngày 1-5;
+ Cytarabin 2.000 mg/m2 da, truyền tĩnh mạch ngày 1-5;
+ G-CSF 5 mcg//kg cân nặng/ngày tiêm dưới da từ ngày 0 đến khi phục hồi bạch cầu hạt trung tính (> 1,5G/L);
+ Daunorubicin 40-60 mg/m2 da/ngày truyền tĩnh mạch ngày 1-3 hoặc ngày 1,3,5.
- Phác đồ Mito- FLAG
+ Fludarabin 25-30 mg/m2 da, truyền tĩnh mạch ngày 1-5;
+ Cytarabin 2.000 mg/m2 da, truyền tĩnh mạch ngày 1-5;
+ G-CSF 5 mcg//kg cân nặng/ngày tiêm dưới da từ ngày 0 đến khi phục hồi bạch cầu hạt trung tính (> 1,5G/L);
+ Mitoxantrone 10 mg/m2 da/ngày truyền tĩnh mạch ngày 1-3.
4.5. Điều trị hỗ trợ
- Chống thiếu máu, xuất huyết bằng các chế phẩm máu (khối hồng cầu, khối tiểu cầu).
- Chống nhiễm trùng: Người bệnh được điều trị trong điều kiện vô trùng; dùng kháng sinh phổ rộng khi có nhiễm trùng (imipenem, meropenem, cephalosporin thế hệ 3-4, phối hợp vancomycin, aminosid, kháng sinh chống nấm); chuyển sang kháng sinh đặc hiệu sau khi có kết quả cấy vi sinh và kháng sinh đồ; dùng các yếu tố kích thích sinh máu như GM-CSF, G-CSF (xin xem chi tiết trong bài Điều trị bệnh nhân suy giảm miễn dịch do giảm bạch cầu đoạn trung tính).
- Điều trị DIC bằng chế phẩm máu và heparin hoặc heparin trọng lượng phân tử thấp.
- Phòng ngừa hội chứng tiêu khối u và tăng acid uric bằng thuốc allopurinol, truyền dịch, tăng cường bài niệu, kiềm hoá nước tiểu. Gạn tách bạch cầu bằng máy tách thành phần máu tự động khi số lượng bạch cầu quá cao (trên 100 G/L).
5. THEO DÕI ĐIỀU TRỊ
5.1. Bilan xét nghiệm trước điều trị
- Tổng phân tích tế bào máu ngoại vi.
- Sinh hóa máu: Chức năng gan, thận, Axit Uric, LDH, điện giải đồ.
- Đông máu huyết tương: Fibrinogen, PT, APTT, TT, D-Dimer. Bệnh nhân có kèm theo rối loạn đông máu cần làm thêm xét nghiệm (nếu cần): Nghiệm pháp rượu, Von-Kaulla, Đàn hồi cục máu đồ (ROTEM), Fibrin monomer.
- Xét nghiệm chẩn đoán hình ảnh: X-quang tim phổi, siêu âm ổ bụng, điện tâm đồ, siêu âm tim và các xét nghiệm khác như: CT-scanner, MRI nếu có nghi ngờ thâm nhiễm.
- Xét nghiệm virus: HIV, HBV, HCV nếu có truyền máu và chế phẩm máu.
- Huyết tủy đồ, phân loại miễn dịch, công thức nhiễm sắc thể và PCR xác định gen bệnh máu ác tính dòng tủy.
- Sinh thiết tủy xương: Có thể dược chỉ định trong trường hợp tủy nghèo tế bào, tỷ lệ blast < 20% tế bào có nhân trong tủy.
- Định nhóm máu hệ ABO, Rh(D).
- Khám răng hàm mặt, tai mũi họng trước khi điều trị.
5.2. Bilan xét nghiệm trong quá trình điều trị hóa chất
- Tổng phân tích tế bào máu ngoại vi.
- Sinh hóa máu: Chức năng gan, thận, điện giải…
- Đông máu huyết tương.
- Các xét nghiệm khác tùy theo diễn biến bệnh.
5.3. Bilan xét nghiệm sau mỗi đợt điều trị hóa chất
- Tủy đồ sau mỗi đợt điều trị.
- MRD bằng phân loại miễn dịch, PCR (có gen đặc hiệu).
- Các xét nghiệm theo dõi điều trị và đánh giá chức năng cơ quan: Tổng phân tích tế bào máu ngoại vi, sinh hóa máu, đông máu cơ bản, siêu âm ổ bụng, siêu âm tim, X- quang tim phổi thẳng, dịch não tủy các xét nghiệm khác như CT-scanner, MRI nếu có nghi ngờ thâm nhiễm.
- Khi bệnh ở giai đoạn ổn định, theo dõi xét nghiệm tổng phân tích tế bào máu ngoại vi, sinh hóa máu, đông máu huyết tương hàng tháng, làm Huyết tủy đồ nếu nghi ngờ tái phát.
TÀI LIỆU THAM KHẢO
1. Rubnitz JE et al. Current management of childhood acute myeloid leukemia. Paediatr Drugs. 2017;19:1-10.
2. Sharifah Aida Alhabshi. Treatment Outcome of Children with Acute Myeloid Leukaemia Using the MASPORE AML 2006 Protocol in UMMC. Department of Paediatrics, Faculty of Medicine, University of Malaya
3. U Creutzig, J Ritter and G Schellong. Identification of two risk groups in childhood acute myelogenous leukemia after therapy intensification in study AML-BFM-83 as compared with study AML-BFM-78. AML-BFM Study Group. Blood, Vol 75, No 10 (May 15). 1990: pp 1932-1940.
4. Wiernik PH et al. Cytarabine plus idarubicin or daunorubicin as induction and consolidation therapy for previously untreated adult patients with acute myeloid leukemia. Blood 1992; 79:313.
5. Pastore D et al. FLAG-IDA in the treatment of refractory/relapsed acute myeloid leukemia: single-center experience. Ann Hematol 2003; 82:231.
6. Suh JK, Lee SW, Koh KN, et al. Hematopoietic stem cell transplantation in pediatric patients with acute myeloid leukemia without favorable cytogenetics. Pediatr Transplant. 2017;21:e13004
7. Powell BL et al. Effect of consolidation with arsenic trioxide (As2O3) on event-free survival (EFS) and overall survival (OS) among patients with newly diagnosed acute promyelocytic leukemia (APL): North American Intergroup Protocol C9710. 2007 ASCO annual meeting. Abstract 2.
8. Sievers EL et al. Efficacy and safety of gemtuzumab ozogamicin in patients with cd33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001; 19:3244.
9. Raul C. Ribeiro, Eduardo Rego. Management of APL in Developing Countries: Epidemiology, Challenges and Opportunities for International Collaboration. ASH2006p65.
10. Soignet SL et al. United states multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol 2001; 19:3852.
11. Min Lih et al. 2018. Advances in the drug therapies of acute myeloid leukemia (except acute promyelocytic leukemia). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5933364/.
23. LƠ XÊ MI TỦY MẠN (Bệnh bạch cầu mạn dòng tủy)
1. ĐẠI CƯƠNG
Lơ xê mi tủy mạn (Chronic myeloid leukemia - CML) là bệnh thuộc hội chứng tăng sinh tuỷ mạn ác tính, đặc trưng bởi sự tăng sinh các tế bào dòng bạch cầu hạt có biệt hóa trưởng thành, hậu quả là số lượng bạch cầu tăng cao ở máu ngoại vi với đủ các tuổi của dòng bạch cầu hạt. Bệnh thường diễn biến qua 3 giai đoạn mà cuối cùng là chuyển lơ xê mi cấp.
2. NGUYÊN NHÂN
Lơ xê mi tủy mạn là một bệnh mắc phải. Phóng xạ có thể là một nguyên nhân gây bệnh. Nhiễm sắc thể Philadelphia (NST Ph) và gen kết hợp BCR-ABL1 là khâu quan trọng nhất trong cơ chế bệnh sinh của bệnh.
3. TRIỆU CHỨNG
3.1. Lâm sàng
a. Giai đoạn mạn tính
- Lách to, gặp ở 85-90% người bệnh. Gan to gặp trên 50% người bệnh.
- Mệt mỏi, kém ăn, sụt cân, ra mồ hôi đêm.
- Thiếu máu mức độ nhẹ hoặc vừa.
- Tắc mạch (tắc mạch lách, tắc mạch chi, tắc tĩnh mạch dương vật, phù gai thị, giảm hoặc mất thị giác một bên, giảm thính giác…).
- Biểu hiện của bệnh Goute do tăng axit uric máu gặp trên một số người bệnh.
b. Giai đoạn tăng tốc
- Biểu hiện lâm sàng nặng lên (thiếu máu, xuất huyết, nhiễm trùng).
- Lách to không đáp ứng với điều trị.
c. Giai đoạn chuyển lơ xê mi cấp
- Trong giai đoạn này thường gặp biểu hiện lâm sàng đặc trưng cho lơ xê mi cấp như triệu chứng thiếu máu, xuất huyết, nhiễm trùng, hội chứng thâm nhiễm.
3.2. Xét nghiệm
a. Giai đoạn mạn tính
- Máu ngoại vi: (1) Thiếu máu bình sắc, kích thước hồng cầu bình thường; (2) Số lượng bạch cầu tăng cao (trung bình 80 G/l); (3) Gặp đủ các tuổi dòng bạch cầu hạt trong máu ngoại vi; (4) Tỷ lệ tế bào blast thường < 2%; (5) Tăng số lượng bạch cầu đoạn ưa acid và bạch cầu đoạn ưa base; (6) Số lượng tiểu cầu tăng trên 450 G/l (gặp trong khoảng 50-70% trường hợp).
- Tuỷ xương: (1) Tuỷ giàu tế bào (số lượng tế bào tuỷ trên 100 G/l); (2) Tăng sinh dòng bạch cầu hạt đủ các lứa tuổi; tỷ lệ M:E trên 10:1 (bình thường là 3-4:1); (3) Tỷ lệ tế bào blast thường < 5% (nếu ≥ 10% gợi ý giai đoạn muộn của bệnh).
- NST Ph và/hoặc gen bcr-abl: Dương tính trong khoảng 95% trường hợp.
- Nồng độ axit uric máu: Tăng trên 40-60% trường hợp.
b. Giai đoạn tăng tốc
Có ít nhất 1 trong các tiêu chuẩn sau:
- Tỷ lệ tế bào blast từ 10-19% trong máu ngoại vi hoặc tủy xương.
- Tỷ lệ bạch cầu ưa bazo ≥ 20% trong máu ngoại vi.
- Tiểu cầu < 100 G/l không do điều trị.
- Tiểu cầu > 1.000 G/l không đáp ứng điều trị.
- Lách to và tăng bạch cầu không đáp ứng điều trị.
- Xuất hiện thêm bất thường nhiễm sắc thể đơn dòng ở những tế bào Ph (+): NST Ph thứ 2, trisomy 8, đẳng NST 17q, trisomy 19, bất thường NST phức tạp, bất thường 3q26.2.
c. Giai đoạn chuyển lơ xê mi cấp
- Tỷ lệ tế bào blast ≥ 20% trong máu ngoại vi hoặc tủy xương. Hoặc
- Xuất hiện blast ngoài tủy xương (myeloid sarcoma).
d. Xét nghiệm sử dụng trước và trong điều trị
- Tổng phân tích tế bào máu.
- Hóa sinh máu: Chức năng gan, thận, acid uric, LDH, điện giải đồ.
- Đông máu cơ bản, D-Dimer.
- Marker virus viêm gan B: do nguy cơ virus tái hoạt động khi điều trị thuốc ức chế tyrosin kinase.
- Điện tâm đồ.
- Siêu âm ổ bụng đánh giá kích thước lách.
4. CHẨN ĐOÁN
a. Chẩn đoán xác định
- Dựa vào các triệu chứng lâm sàng, xét nghiệm tế bào máu ngoại vi, tuỷ đồ, NST Ph và/hoặc gen BCR-ABL1.
b. Chẩn đoán phân biệt
- Phản ứng giả lơ xê mi gặp trong nhiễm trùng nặng: Phát hiện ổ nhiễm trùng, không có NST Ph và gen kết hợp BCR-ABL1.
- Các bệnh khác trong hội chứng tăng sinh tuỷ mạn ác tính, theo bảng xếp loại của WHO năm 2016.
5. ĐIỀU TRỊ
5.1. Giai đoạn mạn tính
a. Điều trị nhắm đích bằng thuốc ức chế hoạt tính tyrosin kinase
- Lựa chọn điều trị thứ nhất là các thuốc ức chế hoạt tính tyrosin kinase (tyrosin kinase inhibitor - TKI) thế hệ 1 và 2, cụ thể là:
+ Thuốc ức chế hoạt tính tyrosin kinase thế hệ 1: Imatinib, liều dùng ban đầu: 400 mg/ngày ở giai đoạn mạn tính;
+ Thuốc ức chế hoạt tính tyrosin kinase thế hệ 2: nilotinib liều khởi đầu được khuyến cáo là 300 mg x 2 lần/ngày.
- Nếu xuất hiện kháng imatinib, có thể sử dụng các thuốc ức chế hoạt tính tyrosin kinase thế hệ 2, hoặc ghép tế bào gốc tạo máu đồng loài.
- Theo dõi đáp ứng điều trị bằng xét nghiệm:
+ Tủy đồ và NST Ph (công thức NST và/hoặc FISH) sau mỗi 3 tháng kể từ lúc bắt đầu điều trị;
+ Định lượng gen BCR-ABL1 (kỹ thuật PCR định lượng) từ lúc chẩn đoán và mỗi 3 tháng trong quá trình điều trị để lượng hóa mức độ lui bệnh phân tử;
+ Phát hiện đột biến gen BCR-ABL1 kháng thuốc, khi người bệnh không đáp ứng sau 3 tháng (%IS > 10), sau 12 tháng (%IS > 1), hoặc mất đáp ứng điều trị, tái phát. Xét nghiệm chẩn đoán đột biến kháng thuốc bằng phương pháp AS-PCR sẽ được chỉ định trước đối với đột biến T315I. Nếu âm tính với T315I, chỉ định tiếp xét nghiệm giải trình tự gen NGS để chẩn đoán các đột biến kháng thuốc còn lại.
b. Các thuốc khác
- Hydroxyurea: Liều khởi đầu 30-60 mg/kg cân nặng cơ thể/ ngày. Giảm liều tuỳ theo số lượng bạch cầu rồi chuyển sang điều trị duy trì liều thấp (10-20 mg/ngày) khi số lượng bạch cầu trở về giá trị bình thường.
- Interferon-α: Liều khởi đầu 5 MU/m2/ngày. Điều trị trong vòng 3 năm sau khi đạt lui bệnh về tế bào di truyền. Sau đó có thể giảm liều Interferon-α rồi dừng thuốc và xét nghiệm NST Ph mỗi 6 tháng.
5.2. Giai đoạn tăng tốc
- Lựa chọn hàng 1 là thuốc ức chế hoạt tính tyrosin kinase thế hệ 2 (nilotinib). Nếu không có khả năng sử dụng thuốc ức chế hoạt tính tyrosin kinase thế hệ 2, có thể dùng imatinib với liều 600 mg/ngày.
- Nếu bệnh nhân kháng hoặc không dung nạp thuốc ức chế hoạt tính tyrosin kinase khởi đầu và không có đột biến T315I, chuyển sang thuốc ức chế hoạt tính tyrosin kinase khác mà bệnh nhân chưa dùng.
- Nếu bệnh nhân đủ điều kiện để ghép tế bào gốc đồng loài, nên tiến hành ghép ngay khi đạt lui bệnh ở mức độ di truyền tế bào.
- Bệnh nhân có đột biến T315I nên ghép tế bào gốc đồng loài.
5.3. Giai đoạn chuyển cấp
- Trong giai đoạn chuyển cấp của lơ xê mi tủy mạn, cần điều trị như đối với lơ xê mi cấp đa hoá trị liệu phối hợp với điều trị nhắm đích bằng các thuốc ức chế hoạt tính tyrosin kynase, sau khi đạt lui bệnh cần ghép tế bào gốc đồng loài.
5.4. Điều trị hỗ trợ
- Truyền máu nếu bệnh nhân có thiếu máu nhưng cần hạn chế chỉ định truyền máu khi số lượng bạch cầu còn cao trên 100 G/l để tránh làm tăng nguy cơ tắc mạch.
- Bổ sung dịch bằng đường uống (2-3 lít nước/m2 hàng ngày), kiềm hóa nước tiểu, lợi niệu cưỡng bức phòng ngừa hội chứng tiêu khối u.
- Allopurinol đường uống 300 mg/ ngày phòng ngừa và điều trị tăng axit uric máu.
- Có thể gạn tách bạch cầu khi SLBC cao trên 100 G/l hoặc có triệu chứng ứ trệ bạch cầu (leukostasis).
5.5. Các xét nghiệm dõi trong qui trình điều trị:
- Tổng phân tích tế bào máu ngoại vi, được làm ít nhất 2 ngày 1 lần giai đoạn điều trị nội trú và hàng tháng khi bệnh ổn định.
- Sinh hóa máu: Chức năng gan, thận, điện giải, acid uric, photpho, LDH..., ít nhất 1 tuần/lần giai đoạn điều trị nội trú và hàng tháng khi bệnh ổn định.
- Đông máu huyết tương cơ bản (Fibrinogen, PT, APTT, TT), D-Dimer, ít nhất 1 tuần/lần giai đoạn điều trị nội trú.
- Vi sinh: Virus viêm gan, HIV… làm sàng lọc trước khi điều trị, lặp lại theo quy định. Các xét nghiệm đánh giá nhiễm trùng: Cấy vi khuẩn, vi nấm, CRP, PCT, CMV, EBV, kháng sinh đồ…, nếu lâm sàng có dấu hiệu nhiễm trùng.
- Huyết thanh học nhóm máu, làm lúc chẩn đoán hoặc khi cần truyền máu.
- Xét nghiệm tế bào và sinh hóa nước tiểu, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Chẩn đoán hình ảnh, thăm dò chức năng, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Xét nghiệm Huyết tủy đồ, STTX làm lúc chẩn đoán và có thể làm lại khi nghi ngờ bệnh tiến triển hoặc chuyển cấp; phát hiện gen đột biến BCR-ABL; Công thức nhiễm sắc thể hoặc FISH thể được làm lúc chẩn đoán. Định lượng đột biến gen BCR-ABL 3 tháng/lần để theo dõi đáp ứng điều trị nhắm đích.
- Một số xét nghiệm khác được chỉ định tùy theo bệnh nền và bệnh mắc kèm của người bệnh.
6. ĐÁNH GIÁ ĐÁP ỨNG
- Tiêu chuẩn đáp ứng hoàn toàn về huyết học: Số lượng bạch cầu < 10 G/l, không còn bạch cầu hạt tuổi trung gian; số lượng tiểu cầu < 450 G/l; lách không to, lâm sàng ổn định.
- Tiêu chuẩn đáp ứng về tế bào di truyền: Đáp ứng hoàn toàn: Ph(+) 0%; Đáp ứng nhiều: Ph(+) 1%-35%; Đáp ứng một phần: Ph(+) 36%-65%; Đáp ứng tối thiểu: Ph(+) 66%-95%; Không đáp ứng: Ph(+) > 95%.
- Tiêu chuẩn đáp ứng mức độ phân tử: Đáp ứng sâu: xét nghiệm PCR định lượng có kết quả BCR-ABL1/ABL1 đạt ít nhất MR4.5 (tương đương %IS ≥ 0,0032 hoặc 4,5 log dưới mức đường chuẩn); Đáp ứng tốt: kết quả định lượng BCR-ABL1/ABL1 đạt ít nhất MR3 (tương đương %IS ≥ 0,01, hoặc giảm 3 log dưới mức đường chuẩn). Hiện nay, khi xác định mức đáp ứng ở mức độ phân tử, thang điểm quốc tế (International Scale - IS) được thống nhất sử dụng để tính mức độ giảm số copy mRNA của gen BCR-ABL1 so với gen tham chiếu ABL1, theo tỷ lệ %).
7. TIÊN LƯỢNG
Trước đây, giai đoạn mạn tính của lơ xê mi tủy mạn thường kéo dài 3-5 năm, rồi chuyển thành lơ xê mi cấp (tiên lượng xấu, thời gian sống thêm thường không quá 1 năm). Hiện nay, với việc ứng dụng ghép tế bào gốc tạo máu đồng loài và điều trị nhắm đích bằng các thuốc TKI, tiên lượng người bệnh lơ xê mi tủy mạn được cải thiện mạnh mẽ.
TÀI LIỆU THAM KHẢO
1. Baccarani M, Deininger MW, Rosti G et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013; Prepublished online June 26 doi: 10.1182/blood-2013-05-501569.
2. Hehlmann R, Hochhaus A, Baccarani M. European LeukemiaNet. Chronic myeloid leukaemia. Lancet 2007; 370: 342-350.
3. National Comprehensive Cancer Network. NCCN guidelines Version 3.2020 Panel members: Chronic myelogenous leukemia.
4. Vardiman JW, Melo JV, Baccarani M, Thiele J. Chronic myelogenous leukemia, BCR-ABL1 positive. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues 2008; 32-37.
24. LƠ XÊ MI TỦY MONO MẠN (Bệnh bạch cầu mạn dòng tủy-mono)
1. ĐẠI CƯƠNG
- Lơ xê mi tủy mono mạn (Chronic Myelomonocytic Leukemia - CMML) là một bệnh máu ác tính ít gặp; biểu hiện đặc trưng là sự tăng cao bạch cầu mono trong máu và tủy xương. CMML mang đặc trưng của 2 nhóm bệnh khác nhau (Hội chứng rối loạn sinh tủy - MDS và tăng sinh tủy - MPNs); theo tổ chức y tế Thế giới (WHO - 2008) CMML như là một bệnh pha trộn giữa MDS và MPNs.
- Tuổi mắc bệnh trung bình là 65-70, nam gặp nhiều hơn nữ.
- Hầu hết các trường hợp bệnh không rõ nguyên nhân; môi trường và một số yếu tố như tia xạ, tiếp xúc hóa chất (benzen, thuốc trừ sâu...), thuốc lá có thể tác động làm xuất hiện bệnh.
2. CHẨN ĐOÁN
2.1. Lâm sàng: Các triệu chứng lâm sàng có thể gặp:
- Thiếu máu: Là triệu chứng hay gặp.
- Xuất huyết do giảm tiểu cầu: Xuất huyết dưới da nhiều nơi, chảy máu chân răng hoặc đường tiêu hóa…
- Có thể gặp gan to, lách to, thâm nhiễm da (khoảng 50%).
- Ngoài ra, còn gặp các triệu chứng như: Mệt mỏi, gầy sút cân, đổ mồ hôi về đêm, sốt kéo dài dùng kháng sinh không có hiệu lực.
2.2. Xét nghiệm
a. Tế bào máu:
Bất thường một hoặc nhiều dòng, cụ thể:
- Bạch cầu có thể tăng (gặp khoảng 50% trường hợp), bình thường hoặc giảm; bạch cầu mono tăng cao dai dẳng ít nhất 3 tháng (> 1 G/l), tỷ lệ mono > 10%, thường là các tế bào mono trưởng thành, có bất thường về hình thái (bất thường về nhân, nguyên sinh chất…); tỉ lệ blasts trong máu hoặc tủy xương < 20%; bạch cầu ưa base và bạch cầu ưa acid tăng nhẹ. Bạch cầu hạt trung tính bất thường hình thái, hay gặp là dạng nhiều nhân, nhân chia đôi còn cầu nối (thể Pelger Huët), giảm hoặc mất hạt đặc hiệu.
- Huyết sắc tố giảm; hồng cầu giảm, kích thước to, nhiều hồng cầu có chấm ưa base.
- Tiểu cầu thường giảm; to, nhỏ không đều.
b. Tế bào và mô bệnh học tủy xương:
- Tăng sinh dòng bạch cầu hạt, có tăng số lượng bạch cầu mono.
- Giảm dòng hồng cầu (50% các trường hợp), có thể gặp hồng cầu khổng lồ, nguyên hồng cầu sắt vòng.
- Tăng sinh xơ trên 75% các trường hợp, có thể gặp ở các mức độ khác nhau.
- Mẫu tiểu cầu giảm sinh (có thể gặp tới 80%), hình thái rối loạn, kích thước lớn, có một nhân hoặc nhiều nhân, nhân nhỏ. Có gặp mẫu tiểu cầu còi cọc.
c. Các xét nghiệm khác bao gồm:
- X-quang, siêu âm ổ bụng và/hoặc CT scanner bụng, để phát hiện lách và gan to.
- Xét nghiệm NST: NST Ph âm tính; hay gặp monosomy 7 và trisomy 8; bất thường 12p, t(5;12)(q31;p12), thường kèm tăng bạch cầu ưa acid; bất thường 11q23 ít gặp (hay chuyển thành lơ xê mi cấp sớm).
- Xét nghiệm FISH: Gen BCR-ABL1 âm tính.
- Đột biến gen: Khoảng 1-4 % bệnh nhân có chuyển đoạn gen PDGFR-β và TEL. Những bệnh nhân có đột biến PDGFR-β và TEL có thể đáp ứng với imatinib. Khoảng 35% bệnh nhân CMML có đột biến gen RAS (như K-RAS, N-RAS).
- Xét nghiệm miễn dịch: Dương tính với CD33, CD13, CD14, CD64, CD68; nếu số lượng tế bào CD34 dương tính tăng thì bệnh có thể chuyển thành lơ xê mi cấp sớm.
- Xét nghiệm sinh hóa máu: LDH và β2 microglobulin tăng cao.
2.3. Chẩn đoán xác định:
a. Tiêu chuẩn chẩn đoán CMML theo FAB (1982):
- Tăng bạch cầu mono máu ngoại vi và tủy xương trên 1 G/l.
- Rối loạn sinh sản dòng hồng cầu và/ hoặc bạch cầu hạt và/ hoặc bạch cầu đơn nhân.
- Blast máu ngoại vi < 5%.
- Blast trong tủy < 20%.
- Sự vắng mặt các thể Auer trong tế bào tủy.
b. Tiêu chuẩn chẩn đoán CMML theo WHO (2008):
- Tăng bạch cầu mono máu ngoại vi dai dẳng (trên 1 G/l).
- Không có NST Philadelphia hoặc gen BCR-ABL1.
- Không có gen PDGFRA hoặc PDGFRB (đặc biệt, trong các trường hợp tăng bạch cầu ưa acid).
- Blast ở máu ngoại vi và trong tủy < 20%.
- Có ít nhất một trong các biểu hiện sau:
+ Rối loạn sinh sản của một hoặc nhiều dòng tế bào tủy;
+ Xuất hiện bất thường về di truyền tế bào hoặc bất thường di truyền phân tử mắc phải trong các tế bào tạo máu;
+ Tăng bạch cầu mono dai dẳng ít nhất 3 tháng mà các nguyên nhân tăng bạch cầu mono khác đã được loại trừ (như nhiễm trùng, viêm, hoặc bệnh ác tính).
c. Tiêu chuẩn chẩn đoán CMML theo WHO (2016):
- Tăng bạch cầu mono máu ngoại vi dai dẳng (≥ 1 G/l), với tỷ lệ bạch cầu mono ≥ 10% tỷ lệ bạch cầu.
- Loại trừ CML, xơ tủy nguyên phát, đa hồng cầu nguyên phát, tăng tiểu cầu nguyên phát theo WHO (như các đặc trưng trong trong tủy xương, sự có mặt của các đột biến JAK2, CALR, MPL có NST Philadelphia hoặc gen BCR-ABL1).
- Không có gen PDGFRA (4q12) hoặc PDGFRB (5q31-33) hoặc FGFR1, PCM1-JAK2 (đặc biệt, trong các trường hợp tăng bạch cầu ưa acid).
- Blast ở máu ngoại vi và trong tủy xương < 20%.
- Rối loạn sinh sản của một hoặc nhiều dòng tế bào tủy.
- Xuất hiện bất thường về di truyền tế bào hoặc bất thường di truyền phân tử mắc phải trong các tế bào tạo máu như trisomy 8, mất NST Y, monosomy, del 7q…
- Tăng bạch cầu mono dai dẳng ít nhất 3 tháng mà các nguyên nhân tăng bạch cầu mono khác đã được loại trừ (như nhiễm trùng, viêm, hoặc bệnh ác tính).
- Đếm tế bào mono máu ngoại vi bằng phương pháp flow cytometry có sự xuất hiện của CD14+ và CD16-.
2.4. Chẩn đoán thể bệnh:
- CMML-0: Blast < 2% ở máu ngoại vi và < 5% trong tủy xương.
- CMML-1: Blast < 2-4% ở máu ngoại vi và < 5-9% trong tủy xương.
- CMML-2: Blast từ 5-19% ở máu ngoại vi và từ 10-19% trong tủy xương.
- CMML có tăng bạch cầu ưa acid: Tiêu chuẩn chẩn đoán CMML + bạch cầu ưa acid máu ngoại vi > 1,5G/l; xét nghiệm NST thường thấy có t(5;12).
2.5. Chẩn đoán phân biệt:
- Tăng sinh mono nguyên nhân không phải bệnh lý ác tính (lao, bệnh nấm mạn tính, viêm nội tâm mạc…), thường có các triệu chứng biểu hiện bệnh kèm theo.
- Phân biệt bệnh tăng sinh tủy ác tính: Xơ tủy tiên phát, lơ xê mi tủy mạn (CML).
Bảng 21. Phân biệt CMML, CML, CML không điển hình
|
Đặc điểm |
CML |
CMML |
CML không điển hình |
|
Loạn sản |
< 10% |
+ |
++ |
|
Tăng sinh monocyt |
< 3% số lượng tuyệt đối |
+ |
+ |
|
Bạch cầu hạt non |
Chủ yếu dòng tủy |
< 15% |
>15% |
|
Bạch cầu ưa acid |
+ |
- |
± |
|
NST Ph |
++ |
- |
- |
|
Đột biến RAS |
- |
40% |
± |
RAS: guamine nucleotide - Releasing factor
3. ĐIỀU TRỊ
3.1. Đánh giá tình trạng bệnh trước khi lựa chọn liệu pháp điều trị dựa vào các yếu tố:
- Đặc điểm và mức độ nặng của các triệu chứng.
- Các biện pháp điều trị chính là: Hóa trị liệu, sử dụng các chất cảm ứng biệt hóa, các điều kiện để ghép tế bào gốc (cho từng trường hợp cụ thể); điều trị bổ trợ cho thiếu máu, xuất huyết, nhiễm trùng.
- Tiên lượng: Các yếu tố tiên lượng nặng bao gồm:
+ Thiếu máu, Hb < 10g/l;
+ Tỷ lệ blast ≥ 10%;
+ Số lượng bạch cầu ≥ 13G/l;
+ Số lượng tiểu cầu < 100G/l;
+ LDH cao;
+ Kích thước lách lớn.
Hệ thống điểm đánh giá tiên lượng CMML (theo Onida F, Kantarjian HM, Smith TL, et all, Blood 2002)
|
Nhóm nguy cơ |
Tổng số các yếu tố bất lợi |
Thời gian sống trung bình (tháng) |
|
Thấp |
0-1 |
24 |
|
Nguy cơ trung bình- 1 |
2 |
15 |
|
Nguy cơ trung bình-2 |
3 |
8 |
|
Nguy cơ cao |
4 |
5 |
Trong đó: Các yếu tố bất lợi, bao gồm:
- Hb < 120g/l.
- Có bạch cầu chưa trưởng thành trong máu ngoại vi.
- Số lượng lympho > 2,5G/l.
- Tế bào tủy chưa trưởng thành và blast trong tủy > 10%.
Mỗi yếu tố trên được tính là 1 điểm.
3.2. Điều trị cụ thể:
a. Các thuốc diệt tế bào:
- Mục đích kiểm soát số lượng bạch cầu trong giới hạn bình thường.
- Thuốc cụ thể:
+ Hydroxyurea uống 0,5g đến 1g/ngày;
+ Mercaptopurin uống 50-100mg/ ngày;
+ Etoposide uống 50mg/ ngày trong 21 ngày, chu kỳ 4 tuần;
+ Những bệnh nhân CMML (khoảng 1-4%) có gen đột biến PDGFR-β và TEL có thể điều trị với imatinib.
b. Ghép tế bào gốc đồng loài trong điều trị CMML:
Đây là phương pháp điều trị duy nhất có thể chữa khỏi bệnh. Áp dụng ở những bệnh nhân trẻ tuổi có bệnh đang tiến triển cấp, thất bại hoặc đáp ứng kém với các phương pháp điều trị khác và tìm được người hiến tế bào gốc phù hợp HLA.
c. Các thuốc nhóm giảm methyl hóa DNA (hypomethylating agents):
Dùng 1 trong 2 loại thuốc sau:
- Azacitidin 75mg/m2/ngày tiêm dưới da 7 ngày, chu kỳ 28 ngày, 6 chu kỳ.
- Decitabine 20mg/m2 truyền tĩnh mạch hoặc tiêm dưới da trong 5 ngày, chu kỳ 4 tuần, duy trì điều trị nếu bệnh nhân vẫn có đáp ứng tốt với thuốc.
3.3. Điều trị hỗ trợ:
- Thiếu máu:
+ Truyền khối hồng cầu khi HST < 70g/l, truyền đến khi HST đạt > 100g/l;
+ Sử dụng thuốc kích thích sinh hồng cầu Erythropoetin.
● Erythropoetin 30.000U/tuần (dùng cách ngày) x 8 tuần, có thể lên 60.000U/ tuần nếu đáp ứng kém;
- Truyền khối tiểu cầu nếu tiểu cầu giảm ≤ 20G/l, hoặc khi SLTC ≤ 50G/l nhưng có xuất huyết trên lâm sàng.
- Chú ý vấn đề quá tải sắt nếu truyền KHC nhiều lần và kéo dài (xét nghiệm ferritin huyết thanh ≥ 1000µg/l hoặc sau truyền ≥ 20 đơn vị máu), dùng các thuốc thải sắt, như Desferrioxamin hoặc Deferasirox:
+ Desferrioxamin liều 20-50mg/kg truyền tĩnh mạch hoặc dưới da 8 đến 12 giờ dùng 5 đến 7 ngày/tuần;
+ Deferasirox 10-40mg/kg/ngày, uống 1 lần/ngày.
- Dùng kháng sinh khi có nhiễm trùng (sốt, viêm họng, viêm phổi, viêm cơ…). Cần làm kháng sinh đồ để chỉ định kháng sinh cho phù hợp.
4. TIẾN TRIỂN
Thời gian sống trung bình là từ 12-24 tháng, khoảng 15-30% bệnh nhân CMML tiến triển thành Lơ xê mi cấp.
5. XÉT NGHIỆM TRƯỚC VÀ TRONG QUÁ TRÌNH ĐIỀU TRỊ
5.1. Đánh giá trước điều trị
Ngoài các xét nghiệm để chẩn đoán bệnh đã nêu ở trên, cần làm các xét nghiệm để đánh giá toàn trạng bệnh nhân trước điều trị, bao gồm:
- Sinh hóa máu: Chức năng gan, thận, đường máu, acid uric, LDH, điện giải đồ, protein, albumin...
- Đông máu cơ bản vòng 1: Fibrinogen, PT, APTT, TT, D-Dimer. Nếu có bất thường sẽ làm thêm các xét nghiệm chuyên sâu.
- Tế bào nước tiểu, sinh hóa nước tiểu.
- HBsAg, HCVAb, HIV Ab với bệnh nhân vào viện lần đầu và khi bệnh nhân có truyền máu nhiều lần.
- Định nhóm ABO, Rh khi bệnh nhân vào viện lần đầu và khi bệnh nhân có chỉ định truyền máu.
- Điện tâm đồ, siêu âm tim với bệnh nhân có chỉ định điều trị hóa chất.
5.2. Theo dõi trong điều trị
5.2.1 Xét nghiệm theo dõi thường quy
- Tổng phân tích tế bào máu ít nhất 3 lần/ tuần.
- Sinh hóa máu: Chức năng gan, thận, điện giải đồ… ít nhất 1 lần/ tuần.
- Đông máu cơ bản ít nhất 1 lần/ tuần.
- Làm huyết tủy đồ, sinh thiết tủy xương sau mỗi chu kì điều trị hóa chất. Đối với bệnh nhân không điều trị hóa chất kiểm tra huyết tủy đồ, sinh thiết tủy xương mỗi 6 tháng hoặc khi có biểu hiện bệnh tiến triển.
5.2.2 Xét nghiệm theo dõi khi có bất thường: Tùy thuộc diễn biến của bệnh nhân để chỉ định phù hợp.
TÀI LIỆU THAM KHẢO
1. Đỗ Trung Phấn, Tế bào gốc và Bệnh lý tế bào gốc tạo máu, 2008: tr. 227-245.
2. Nguyễn Anh Trí, Tiền Lơ xê mi và Lơ xê mi cấp, Nhà xuất bản Y học, 2010 : tr.59-140.
3. Am J Hematol. 2012 Jun;87(6):610-619.
4. Alan F. ListAvery A. Sandberg Donald C. Doll. Myelodysplastic Syndromes. Wintrobe’s Clinical Hematology 12th Edition. 2009: 1957-1977.
5. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
6. Bacher U, Haferlach T, Schnittgeer S, et al. Recent advances in diagnosis, molecular pathology and therapy or chronic myelomonocytic leukemia. Br J Hematol. 2011;153:149-167.
7. David G Oscier and Sally B Killick. The myelodysplastic syndromes. Postgraduate - Haematology. 2005: 662-678.
8. Eric Solary and Raphael Itzykson. How I treat chronic myelomonocytic leukemia (Blood. 2017;130(2):126-136)
9. Malcovati L, Cazzola M. Myelodysplastic/myeloproliferative disorders. Hematological. 2008;93(1);4-6.
10. Orazi A, Germing U, Brunning R, Brain B, Thiele J. Chronic Myelomonocytic Leukemia. Lyon: IARC press 2008.
11. Sameer A. Parikh and Ayalew Tefferi. Chronic myelomonocytic leukemia: 2012 update on diagnosis,risk stratification, and management. Am. J. Hematol. 87:611-619, 2012.
12. Symeonidi A, Vanblezen A, Mufti G, et all. Allogeneic stem cell transplantation in patients with chronic myelomonocytic leukemia: The impact of WHO classfication and of the conditioning regimen on the transplantation outcome. Annual Meeting of the EBMT 2010. Bone marrow transplantation 2010;45(S240, Abtract P803).
25. ĐA HỒNG CẦU NGUYÊN PHÁT
1. ĐẠI CƯƠNG
- Đa hồng cầu nguyên phát là bệnh tăng sinh quá mức dòng hồng cầu, thuộc nhóm các bệnh tăng sinh tủy mạn ác tính (myeloproliferative neoplasms - MPNs) một nhóm bệnh ác tính hệ tạo máu tương đối thường gặp. Đặc điểm chung của nhóm bệnh lý này là diễn biến mạn tính, lách to kèm theo tăng một hoặc nhiều dòng tế bào tủy.
- Cơ chế bệnh sinh: Đột biến gen của protein tyrosine kinase thuộc Janus kinase family (JAK) gây giảm chức năng tự ức chế sinh sản tế bào. Đột biến gen JAK2-V617F gây tăng khả năng phosphoryl hoá của JAK2, dẫn tới tăng sinh tế bào tạo máu vạn năng (tế bào CD34+). Đột biến này gặp trên 90% người bệnh đa hồng cầu nguyên phát.
2. NGUYÊN NHÂN GÂY BỆNH
Nguyên nhân gây bệnh chưa được biết rõ, một số yếu tố được cho là làm tăng nguy cơ mắc bệnh: Tiếp xúc với bức xạ ion hóa, hóa chất (benzen), yếu tố gia đình. Tuy nhiên, nhiều bệnh nhân bị bệnh mà không phát hiện được yếu tố nguy cơ nào.
3. TRIỆU CHỨNG
3.1. Lâm sàng
- Biểu hiện lâm sàng chủ yếu do tăng độ quánh máu như: Đau đầu, chóng mặt, rối loạn thị lực, đau thắt ngực; đau, tê bì đầu ngón chân, ngón tay; hay gặp biến chứng tắc mạch máu nhỏ, một số trường hợp gặp tắc mạch lớn như huyết khối tĩnh mạch sâu; có thể biến chứng chảy máu: Xuất huyết niêm mạc, chảy máu chân răng, xuất huyết tiêu hoá.
- Các triệu chứng chính theo thứ tự gặp là: Lách to (75% người bệnh), có thể có nhồi máu lách; gan to (30%); biểu hiện ngứa (trên 40% người bệnh), gặp sau tắm nước nóng; cao huyết áp; nóng bừng mặt; ngoài ra có thể đau bụng vì viêm loét dạ dày do tăng tiết histamine và tăng tiết acid hay do tắc mạch (tắc mạch mạc treo, tĩnh mạch cửa).
- Giai đoạn cuối có thể kiệt quệ, biểu hiện chủ yếu là thiếu máu, số lượng tiểu cầu có thể tăng, tình trạng xơ tủy tăng dần và kết thúc bằng chuyển thành lơ xê mi cấp.
3.2. Xét nghiệm
- Máu ngoại vi: Tăng tất cả các tế bào thuộc dòng tuỷ (hồng cầu, bạch cầu hạt, tiểu cầu); tăng hematocrit; tăng acid uric; erythropoietin huyết thanh không tăng.
- Tủy xương: Tăng sinh các dòng tế bào tủy (hồng cầu, bạch cầu hạt, mẫu tiểu cầu). Có hiện tượng xơ hóa tủy từng phần.
- Có đột biến JAK2 V617F, hoặc đột biến JAK2 exon 12.
4. CHẨN ĐOÁN
4.1. Chẩn đoán xác định theo tiêu chuẩn WHO 2016
a. Tiêu chuẩn chính
- Hb > 165 g/l (nam) và > 160 g/l (nữ) hoặc hematocrit > 0,49 l/l (nam) và > 0,48 l/l (nữ) hoặc tăng thể tích khối hồng cầu toàn thể > 25% trị số bình thường.
- Xét nghiệm mô bệnh học tủy xương cho thấy tình trạng tăng mật độ tế bào với sự tăng sinh cả 3 dòng tế bào tủy trong đó chủ yếu là dòng hồng cầu, tăng sinh bạch cầu hạt và mẫu tiểu cầu (biểu hiện bằng sự đa dạng về hình thái, kích thước và mức độ trưởng thành). Tuy nhiên, nếu bệnh nhân có tăng hồng cầu kéo dài với mức độ Hb > 185 g/l (nam) và 165 g/l (nữ) thì tiêu chuẩn này không bắt buộc phải áp dụng.
- Có đột biến JAK2 V617F hoặc JAK2 exon 12.
b. Tiêu chuẩn phụ
- Nồng độ erythropoietin huyết thanh giảm.
Chẩn đoán xác định đa hồng cầu nguyên phát khi có cả 3 tiêu chuẩn chính hoặc 2 tiêu chuẩn chính số 1, 2 và tiêu chuẩn phụ.
4.2. Chẩn đoán phân biệt
a. Các bệnh khác thuộc nhóm các bệnh tăng sinh tủy mạn ác tính
- Tăng tiểu cầu tiên phát: Tủy tăng sinh mẫu tiểu cầu trưởng thành, rối loạn hình thái, gen JAK2 V617F (+) trong 50% trường hợp, có thể có đột biến CALR hoặc MPL ở bệnh nhân JAK2 V617F (-).
- Xơ tủy nguyên phát: Thường thiếu máu, tủy tăng sinh mẫu tiểu cầu rối loạn hình thái tập trung thành đám, cùng với tăng sinh xơ reticulin hoặc collagen, gen JAK2 V617F (+) trong 50% trường hợp, có thể có đột biến CALR hoặc MPL ở bệnh nhân JAK2 V617F (-).
- Lơ xê mi tủy mạn: Tủy tăng sinh đủ các lứa tuổi dòng bạch cầu hạt, NST Ph+, gen BCR-ABL+.
b. Đa hồng cầu thứ phát
Trong trường hợp này phân biệt bằng nghiệm pháp erythropoietin huyết thanh: Nồng độ erythropoietin giảm trong đa hồng cầu nguyên phát; tuỷ xương: Giàu tế bào, tăng sinh các dòng tế bào tủy. Trường hợp tăng hồng cầu thứ phát thì số lượng hồng cầu và nồng độ hemoglobin sẽ trở về bình thường nếu bệnh lý gây tăng hồng cầu được điều trị hiệu quả.
Một số nguyên nhân dưới đây có thể gây tình trạng đa hồng cầu thứ phát:
- Mất nước: Có thể làm tăng hematocrit giả.
- Tăng sinh hồng cầu thứ phát do tăng EPO trong các trường hợp: Tăng EPO thứ phát do thiếu oxy; bệnh phổi; người sinh sống ở vùng cao; hút thuốc lá (8-carboxyhemoglobin); bệnh tim gây xanh tím; hemoglobin ái lực cao oxy; nhiễm Coban; tăng sản xuất EPO; khối u ở thận, não, gan, u xơ tử cung, u crom; hẹp động mạch thận; nang thận…
- Các nguyên nhân khác: Thụ thể EPO quá mẫn cảm; tăng sinh hồng cầu di truyền; điều trị androgen; u thượng thận; hoàn máu tự thân (doping máu); tiêm EPO.
c. Đột biến receptor erythropoietin
Bệnh có tính chất gia đình, thường được phát hiện khi còn trẻ, nuôi cấy cụm hồng cầu in vitro thấy tăng nhạy cảm với EPO (trái ngược với đa hồng cầu nguyên phát tạo cụm hồng cầu không phụ thuộc EPO).
4.3. Phân nhóm nguy cơ theo NCCN
a. Nguy cơ cao
- Tuổi > 60. Và/hoặc
- Có tiền sử huyết khối.
b. Nguy cơ thấp
- Tuổi < 60. Và
- Không có tiền sử huyết khối.
5. ĐIỀU TRỊ
5.1. Điều trị bệnh chính
a. Nhóm nguy cơ cao
- Rút máu tĩnh mạch duy trì hematocrit < 45%.
- Aspirin liều thấp 81-100 mg/ngày.
- Thuốc diệt tế bào: Lựa chọn hàng 1 thường là hydroxyurea; interferons alpha thường dùng cho bệnh nhân dưới 40 tuổi (do không làm tăng nguy cơ xuất hiện lơ xê mi cấp) và phụ nữ mang thai.
+ Hydroxyurea uống liều khởi đầu 15-20 mg/kg x 2 lần/ngày, giảm liều nếu bệnh nhân suy thận; hydroxyurea thường cần 3-5 ngày mới có tác dụng;
+ Interferons alpha liều khởi đầu 3 triệu đơn vị tiêm dưới da x 3 lần/tuần; hoặc
+ Pegylated interferons alpha-2a liều khởi đầu 45 mcg/tuần tiêm dưới da tuần đầu tiên, tăng dần liều tùy theo đáp ứng về tế bào máu của bệnh nhân, liều tối đa 180 mcg/tuần.
Trường hợp bệnh nhân không dung nạp hoặc đáp ứng kém với điều trị hàng 1 (hydroxyurea hoặc interferons alpha), có thể dùng:
- Ruxolitinib uống 10 mg x 2 lần/ngày, hoặc
b. Nhóm nguy cơ thấp
- Rút máu tĩnh mạch duy trì hematocrit < 45%.
- Aspirin liều thấp 81-100 mg/ngày.
Thông thường bệnh nhân nhóm nguy cơ thấp chưa cần sử dụng ngay các thuốc diệt tế bào (do nguy cơ chuyển dạng lơ xê mi cấp hay xơ tủy lớn hơn lợi ích mang lại), chỉ dùng trong những trường hợp sau:
- Không kiểm soát được những triệu chứng của bệnh.
- Bạch cầu và/hoặc tiểu cầu tăng cao.
- Lách to có triệu chứng hoặc lách to nhiều.
- Đáp ứng kém với rút máu đơn thuần.
5.2. Điều trị hỗ trợ
- Bệnh nhân đa hồng cầu nguyên phát cần được tư vấn và quản lý tốt các yếu tố nguy cơ bệnh tim mạch: ngừng hút thuốc, duy trì hoạt động thể chất giảm béo phì, điều trị rối loạn mỡ máu, đái tháo đường, kiểm soát huyết áp (nên phối hợp bác sỹ chuyên khoa tim mạch).
- Điều trị hỗ trợ chế phẩm sắt cho các người bệnh rút máu nhiều lần có tình trạng thiếu sắt (giảm nồng độ sắt và Ferritin huyết thanh). Chỉ nên bổ sung sắt khi bệnh nhân có triệu chứng thiếu sắt (ví dụ: pica, dị cảm ở miệng, khó nuốt do viêm thực quản, hội chứng restless leg).
5.3. Các xét nghiệm đánh giá và theo dõi điều trị
- Tổng phân tích tế bào máu ngoại vi, được làm ít nhất 2 ngày 1 lần, làm lại ngay sau rút máu điều trị.
- Sinh hóa máu: Chức năng gan, thận, điện giải, acid uric, photpho, Fe, Ferritin, EPO, LDH..., ít nhất 1 tuần/lần.
- Xét nghiệm đông máu: Fibrinogen, PT, APTT, TT, D-Dimer, rotem… ít nhất 1 tuần/lần. Xét nghiệm bệnh von-Willebrand mắc phải (nếu người bệnh có chảy máu hoặc số lượng tiểu cầu > 1.000 G/l hoặc APTT kéo dài).
- Xét nghiệm vi sinh: Virus viêm gan, HIV… làm sàng lọc trước khi điều trị, lặp lại theo quy định; Các xét nghiệm đánh giá nhiễm trùng: Cấy vi khuẩn, vi nấm, CRP, PCT, CMV, EBV, kháng sinh đồ… nếu lâm sàng có dấu hiệu nhiễm trùng.
- Huyết thanh học nhóm máu, làm lúc chẩn đoán hoặc khi cần truyền máu.
- Xét nghiệm tế bào và sinh hóa nước tiểu, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Chẩn đoán hình ảnh, thăm dò chức năng, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Xét nghiệm Huyết tủy đồ, STTX, nhuộm sợi xơ làm lúc chẩn đoán và có thể làm lại khi nghi ngờ bệnh tiến triển hoặc chuyển thể; phát hiện gen đột biến JAK2 (V617F, exon 12), CALR, MPL, Công thức nhiễm sắc thể được làm lúc chẩn đoán. Định lượng đột biến gen JAK2 để theo dõi điều trị.
- Một số xét nghiệm khác được chỉ định tùy theo bệnh nền và bệnh mắc kèm của người bệnh.
6. ĐÁNH GIÁ ĐÁP ỨNG
Tiêu chuẩn đáp ứng theo hệ thống lơ xê mi châu Âu (ELN - 2013)
|
Mức độ đáp ứng |
Tiêu chuẩn |
|
Đáp ứng hoàn toàn |
1. Không còn triệu chứng của bệnh và thuyên giảm triệu chứng theo thang điểm MPN-SAF TSS (giảm 10 điểm) trong ít nhất 12 tuần |
|
2. Tế bào máu ngoại vi trở về bình thường (BC < 10 G/l, TC < 400 G/l, Hct < 45% không cần rút máu) trong ít nhất 12 tuần |
|
|
3. Không có biến cố mạch máu và bệnh tiến triển |
|
|
4. Không còn các bất thường trong tủy xương liên quan đến bệnh (mật độ tế bào tủy bình thường, không còn tăng sinh các dòng tế bào tủy, không có xơ hóa reticulin > mức độ 1) |
|
|
Đáp ứng một phần |
1. Thỏa mãn 3 tiêu chí đầu tiên của đáp ứng hoàn toàn và |
|
2. Còn tình trạng tăng sinh cả 3 dòng tế bào tủy xương |
|
|
Bệnh tiến triển |
Chuyển dạng xơ tủy sau đa hồng cầu, lơ xê mi cấp, rối loạn sinh tủy |
Ghi chú: Tiêu chuẩn này thường được áp dụng trong các thử nghiệm lâm sàng.
7. TIÊN LƯỢNG
Đa hồng cầu nguyên phát có tiên lượng tương đối tốt. Người bệnh có thể có thời gian sống thêm kéo dài gần bằng người bình thường nếu được điều trị phù hợp. Nguyên nhân tử vong chủ yếu là do tắc mạch, tai biến mạch máu não do tăng huyết áp. Một số người bệnh có thể chuyển thành lơ xê mi cấp.
TÀI LIỆU THAM KHẢO
1. Nordic MPN study group (2013), “Nordic guidelines on the diagnosis and treatment of patients with Myeloproliferative Neoplasms”.
2. Francesco P (2012), “How I treat polycythemia vera”, Blood, 120, pp. 2275-284.
3. Tefferi A, Juergen Thiele J, and Vardiman JW, MD3. The 2008 World Health Organization Classification System for Myeloproliferative Neoplasms, Cancer 2009, September 1: 3842-3847.
4. Tefferi A & Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies, Journal of clinical oncology 2011, 29(5): 573-581.
5. Tefferi A. Esential thrombocythemia, polycythemia vera, and myelofibrosis: current management and prospect of targeted therapy, Am J Hematol 2008, 83: 491-497.
26. TĂNG TIỂU CẦU TIÊN PHÁT
1. ĐẠI CƯƠNG
Tăng tiểu cầu tiên phát là bệnh máu ác tính hiếm gặp (khoảng 0,1 - 1,5/100.000 người/năm) do tăng sinh không kiểm soát các mẫu tiểu cầu trong tủy xương; đây là bệnh thuộc nhóm các bệnh tăng sinh tủy mạn ác tính (myeloproliferative neoplasms - MPNs).
2. NGUYÊN NHÂN GÂY BỆNH
Chưa được biết chính xác trong hầu hết trường hợp. Tuy nhiên, các đột biến của gen JAK2 ở tế bào gốc dẫn tới tăng sinh tế bào tạo máu gặp trên 50 - 55% người bệnh tăng tiểu cầu tiên phát.
3. TRIỆU CHỨNG
3.1. Lâm sàng
- Triệu chứng lâm sàng bao gồm tắc mạch và đôi khi có xuất huyết.
- Tắc mạch vừa và lớn (mạch máu não, mạch vành, mạch ngoại biên, tĩnh mạch sâu); tắc mạch tái đi tái lại gặp trên 15-20% người bệnh.
- Biểu hiện chảy máu ít gặp; tuy nhiên, khi số lượng tiểu cầu tăng trên 1.000 G/L thì tỷ lệ biến chứng chảy máu tăng lên; biểu hiện chảy máu hơi giống với bệnh von Willebrand: Chảy máu chân răng, xuất huyết tiêu hoá, chảy máu sau phẫu thuật.
- Phối hợp với triệu chứng rối loạn vận mạch: Thiếu máu đầu ngón tay, chân; đau, tê bì đầu ngón; đau đầu, đau nửa đầu, thiếu máu não thoáng qua, xây xẩm, đột ngột giảm hoặc mất thị lực từng bên.
3.2. Xét nghiệm
a. Máu ngoại vi
- Số lượng tiểu cầu tăng trên 450 G/L.
- Số lượng bạch cầu thường tăng.
- Số lượng hồng cầu, nồng độ hemoglobin bình thường hoặc giảm.
b. Tuỷ xương
- Tuỷ giàu tế bào.
- Tăng sinh dòng mẫu tiểu cầu.
- Có thể gặp các mẫu tiểu cầu khổng lồ, nhân nhiều múi, không đồng bộ.
- Thường gặp kèm tăng sinh dòng bạch cầu hạt.
- Có thể thấy hiện tượng xơ tuỷ, tăng sinh reticulin trên xét nghiệm mô bệnh học tủy.
c. Xét nghiệm NST, gen
- Có thể có một trong số các đột biến gen JAK2 (V617F, exon 12) hoặc CALR (exon 9) hoặc MPL.
- Khảo sát gen kết hợp BCR-ABL1 (loại trừ CML, khi JAK2 V617F âm tính).
4. CHẨN ĐOÁN
4.1. Chẩn đoán xác định theo tiêu chuẩn WHO 2016
a. Tiêu chuẩn chính:
- Số lượng tiểu cầu ≥ 450 G/l.
- Sinh thiết tủy xương cho thấy tăng sinh chủ yếu dòng mẫu tiểu cầu với các đặc điểm hình thái như mẫu tiểu cầu kích thước lớn, nhân nhiều múi. Không có biểu hiện công thức dòng bạch cầu hạt hoặc dòng hồng cầu chuyển trái. Đôi khi kèm theo tăng xơ reticulin nhẹ (độ 1).
- Không đáp ứng tiêu chuẩn chẩn đoán của WHO đối với LXM tủy mạn, đa hồng cầu nguyên phát, xơ tủy nguyên phát, hội chứng rối loạn sinh tủy và các bệnh lý ác tính dòng tủy khác.
- Có đột biến JAK2 (V617F, exon 12), CARL hoặc MPL. Trình tự chẩn đoán đột biến gen: (01) xét nghiệm đột biến gen JAK2 V617F, (02) xét nghiệm gen kết hợp BCR- ABL1 (khi JAK2 V617F âm tính), (03) xét nghiệm đột biến gen CALR type 1 và type 2 (khi xét nghiệm 01 và 02 âm tính), (04) xét nghiệm giải trình tự gen NGS xác định đột biến gen CALR và MPL (khi xét nghiệm 01, 02, 03 âm tính).
b. Tiêu chuẩn phụ:
- Có dấu ấn đơn dòng hoặc không có bằng chứng tăng tiểu cầu phản ứng.
Chẩn đoán xác định tăng tiểu cầu tiên phát khi có cả 4 tiêu chuẩn chính hoặc 3 tiêu chuẩn chính đầu và tiêu chuẩn phụ.
4.2. Chẩn đoán phân biệt
- Tăng tiểu cầu tiên phát cần được chẩn đoán phân biệt với tình trạng tăng tiểu cầu thứ phát (nhiễm khuẩn hoặc viêm, sau cắt lách, ung thư, chấn thương, mất máu, thiếu máu thiếu sắt, vô căn). Dấu hiệu phân biệt chủ yếu là đột biến gen JAK2 V617F, tình trạng tăng tiểu cầu và xơ tủy kéo dài không tìm được nguyên nhân. Ngoài ra, nếu là tăng tiểu cầu thứ phát thì số lượng tiểu cầu sẽ trở về bình thường khi bệnh lý gây tăng tiểu cầu được điều trị khỏi.
- Tăng tiểu cầu tiên phát cần được chẩn đoán phân biệt với các bệnh tăng sinh tủy mạn ác tính khác:
+ Đa hồng cầu nguyên phát ở giai đoạn kiệt quệ khi biểu hiện chủ yếu không còn là tăng số lượng hồng cầu và nồng độ hemoglobin (thậm chí các chỉ số này còn giảm) mà chủ yếu là tăng bạch cầu dòng hạt, tiểu cầu có thể tăng nhưng không cao lắm và kèm theo là tình trạng xơ tủy. Việc chẩn đoán phân biệt chủ yếu dựa vào biểu hiện lâm sàng và xét nghiệm lúc ban đầu của bệnh. Đối với tăng tiểu cầu tiên phát, biểu hiện ban đầu này chính là tình trạng tăng tiểu cầu điển hình kèm theo xơ tủy mức độ vừa phải trong một khoảng thời gian dài trong tiến trình của bệnh;
+ Lơ xê mi tủy mạn, nhất là ở giai đoạn tăng tốc, khi số lượng bạch cầu không còn tăng cao nhưng số lượng tiểu cầu cao đi kèm với tình trạng xơ tủy rõ. Dấu hiệu phân biệt chủ yếu là NST Ph và/ hoặc gen BCR-ABL1 dương tính cùng với biểu hiện lâm sàng và xét nghiệm ban đầu của bệnh khi chẩn đoán xác định;
+ Xơ tủy nguyên phát giai đoạn sớm (tiền xơ): Chủ yếu dựa vào sự tăng sinh và đặc điểm rối loạn hình thái của mẫu tiểu cầu, mức độ xơ tủy xương và lách to, xuất hiện tuổi trung gian dòng bạch cầu hạt và hồng cầu non ở máu ngoại vi;
+ Hội chứng rối loạn sinh tủy có tăng tiểu cầu: thường có bất thường NST 5q-, 3q21q26, hội chứng rối loạn sinh tủy/tăng sinh tủy có sideroblast vòng và tăng tiểu cầu;
+ Tăng tiểu cầu có tính chất gia đình: bệnh rất hiếm gặp, di truyền trội trên NST thường; đột biến gen thrombopoietin.
4.3. Phân nhóm nguy cơ
Thang điểm IPSET - thrombosis sửa đổi theo WHO
(The revised international prognostic score of thrombosis for ET)
|
Nhóm nguy cơ |
Tiêu chuẩn |
|
Cao |
Có tiền sử huyết khối và/ hoặc tuổi > 60, có đột biến JAK2 V617F |
|
Trung bình |
Tuổi > 60 và không có tiền sử huyết khối, không có đột biến JAK2 V617F |
|
Thấp |
Tuổi ≤ 60 và không có tiền sử huyết khối, có đột biến JAK2 V617F |
|
Rất thấp |
Tuổi ≤ 60 và không có tiền sử huyết khối, không có đột biến JAK2 V617F |
5. ĐIỀU TRỊ
5.1. Điều trị bệnh chính
a. Thuốc điều trị
- Hydroxyurea
+ Liều khởi đầu 15 mg/kg/ngày;
+ Chỉnh liều để duy trì số lượng tiểu cầu trong giới hạn bình thường và không làm giảm số lượng bạch cầu.
- Hoặc dùng 1 trong các thuốc sau:
+ Interferon-α: Liều trung bình 3.000.000 IU/ngày x 3 lần/tuần.
+ Pegylated interferons alpha-2a: Liều khởi đầu 45 mcg/tuần tiêm dưới da trong 2 tuần đầu, điều chỉnh liều theo đáp ứng của bệnh nhân, liều tối đa 180 mcg/tuần.
+ Anagrelide:
● Là tác nhân imidazoquinazoline đường uống;
● Hoạt tính kháng cAMP phosphodiesterase qua đó làm giảm số lượng tiểu cầu và ức chế ngưng tập tiểu cầu;
● Liều khởi đầu: 0,5 mg/ngày x 2-4 lần/ngày, điều chỉnh liều theo số lượng tiểu cầu. Liều duy trì thường là 1-4 mg/ngày.
+ Aspirin:
● Dùng liều thấp 81-100 mg/ngày chừng nào còn có tăng số lượng tiểu cầu;
● Cần cân nhắc khi sử dụng cho người bệnh có tiền sử chảy máu hoặc số lượng tiểu cầu rất cao (> 1.000 G/L) vì với số lượng tiểu cầu này người bệnh có tăng nguy cơ biểu hiện tình trạng bệnh von Willebrand mắc phải.
- Hoặc các thuốc ức chế ngưng tập tiểu cầu khác: Dipyridamole, ticlopidine, clopidogrel.
b. Phác đồ điều trị
- Nhóm nguy cơ rất thấp và thấp:
+ Aspirin liều thấp (81-100 mg) hàng ngày;
+ Chỉ dùng thuốc làm giảm số lượng tiểu cầu cho nhóm này khi: Xuất hiện huyết khối mới, bệnh von Willebrand mắc phải, chảy máu nặng liên quan đến bệnh; lách to, bạch cầu và tiểu cầu tăng cao; triệu chứng toàn thân (mệt mỏi, ngứa, ra mồ hôi đêm); rối loạn vi tuần hoàn không đáp ứng với aspirin.
- Nhóm trung bình và cao:
+ Thuốc giảm số lượng tiểu cầu: Lựa chọn hàng 1 là hydroxyurea, hàng 2 là anagrelide, interferons alpha;
+ Thuốc chống đông và hoặc chống ngưng tập tiểu cầu tùy thuộc tiền sử huyết khối của bệnh nhân:
● Bệnh nhân có tiền sử huyết khối tĩnh mạch: Thuốc chống đông đường uống (DOAC, warfarin), heparin trọng lượng phân tử thấp; có thể kết hợp aspirin nếu cần;
● Bệnh nhân có tiền sử huyết khối động mạch: Aspirin liều thấp;
● Bệnh nhân > 60 tuổi và không có tiền sử huyết khối: Aspirin liều thấp.
5.2. Điều trị hỗ trợ
- Theo dõi sự xuất hiện của huyết khối mới, bệnh von Willebrand mắc phải, hoặc chảy máu nặng liên quan đến bệnh.
- Điều trị các yếu tố nguy cơ bệnh tim mạch kèm theo (tăng huyết áp, rối loạn lipid máu, hút thuốc, béo phì).
- Gạn tiểu cầu khi số lượng tiểu cầu > 1.000 G/l.
5.3. Các xét nghiệm đánh giá và theo dõi điều trị
- Tổng phân tích tế bào máu ngoại vi, được làm ít nhất 2 ngày 1 lần, làm lại ngay sau gạn tách tiểu cầu điều trị.
- Sinh hóa máu: Chức năng gan, thận, điện giải, acid uric, photpho, LDH... ít nhất 1 tuần/ lần.
- Xét nghiệm đông máu: Fibrinogen, PT, APTT, TT, D-Dimer, Rotem… ít nhất 1 tuần/lần, xét nghiệm bệnh von-Willebrand mắc phải (nếu người bệnh có chảy máu hoặc số lượng tiểu cầu > 1000 G/l hoặc APTT kéo dài).
- Các xét nghiệm vi sinh: Virus viêm gan, HIV… làm sàng lọc trước khi điều trị, lặp lại theo quy định. Các xét nghiệm đánh giá nhiễm trùng: cấy vi khuẩn, vi nấm, CRP, PCT, CMV, EBV, kháng sinh đồ… nếu lâm sàng có dấu hiệu nhiễm trùng.
- Huyết thanh học nhóm máu, làm lúc chẩn đoán hoặc khi cần truyền máu.
- Xét nghiệm tế bào và sinh hóa nước tiểu, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Chẩn đoán hình ảnh, thăm dò chức năng, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Xét nghiệm Huyết tủy đồ, STTX, nhuộm sợi xơ làm lúc chẩn đoán và có thể làm lại khi nghi ngờ bệnh tiến triển hoặc chuyển thể; phát hiện gen đột biến JAK2 (V617F, exon 12), CALR, MPL, Công thức nhiễm sắc thể được làm lúc chẩn đoán. Định lượng đột biến gen JAK2 để theo dõi điều trị.
- Một số xét nghiệm khác được chỉ định tùy theo bệnh nền và bệnh mắc kèm của người bệnh.
6. ĐÁNH GIÁ ĐÁP ỨNG
Tiêu chuẩn đáp ứng của hệ thống lơ xê mi châu Âu (ELN 2013)
|
Mức độ đáp ứng |
Tiêu chuẩn |
|
Đáp ứng hoàn toàn |
1. Không còn triệu chứng của bệnh bao gồm gan lách không to, thuyên giảm triệu chứng theo thang điểm MPN-SAF TSS (giảm 10 điểm) trong ít nhất 12 tuần |
|
2. Tế bào máu ngoại vi trở về bình thường (BC < 10 G/l, TC < 400 G/l, không có tế bào tuổi trung gian ở máu ngoại vi) trong ít nhất 12 tuần |
|
|
3. Không có huyết khối và xuất huyết mới, không có dấu hiệu bệnh tiến triển |
|
|
4. Đáp ứng về mô học tủy xương: mẫu tiểu cầu không tăng sinh, rối loạn hình thái, không có xơ hóa reticulin > mức độ 1 |
|
|
Đáp ứng một phần |
1. Thỏa mãn 3 tiêu chí đầu tiên của đáp ứng hoàn toàn và |
|
2. Không đáp ứng về mô học tủy xương: còn tăng sinh mẫu tiểu cầu rối loạn hình thái |
|
|
Bệnh tiến triển |
Chuyển dạng đa hồng cầu, xơ tủy sau tăng tiểu cầu, lơ xê mi cấp, rối loạn sinh tủy |
7. TIÊN LƯỢNG
Người bệnh tăng tiểu cầu tiên phát, nhất là nhóm nguy cơ thấp, có tiên lượng tương đối tốt. Thời gian sống thêm gần với người bình thường cùng lứa tuổi. Nguyên nhân tử vong chủ yếu là do tắc mạch. Một số trường hợp có thể chuyển thành Lơ xê mi cấp.
TÀI LIỆU THAM KHẢO
1. Tefferi A, Juergen Thiele J, and Vardiman JW, MD3, 2008 World Health Organization Classification System for Myeloproliferative Neoplasms, Cancer 2009, September 1: 3842-3847.
2. Tefferi A & Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies, Journal of clinical oncology 2011, 29(5): 573-581.
3. Tefferi A. Esential thrombocythemia, polycythemia vera, and myelofibrosis: current management and prospect of targeted therapy, Am J Hematol 2008, 83: 491-497.
4. Barosi G et al, 2013, “Revised response criteria for polycythemia and essential thrombocythemia: an ELN and IWG-MRT consensus project”. Blood 121 (23): 4778-4781.
5. Barbui T el al, 2018, “Philadelphia chromosome negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet”. Leukemia 32: 1057-1069.
27. XƠ TỦY NGUYÊN PHÁT
1. ĐẠI CƯƠNG
Xơ tủy nguyên phát là bệnh máu, đặc trưng là tăng sinh xơ trong tủy xương và sinh máu ngoài tủy, thuộc nhóm các bệnh tăng sinh tủy mạn ác tính.
2. NGUYÊN NHÂN GÂY BỆNH
Nguyên nhân chính xác gây bệnh chưa được biết rõ. Người ta cho rằng xơ tủy nguyên phát cùng với các bệnh khác thuộc nhóm bệnh tăng sinh tủy mạn ác tính từ đột biến soma của các tế bào gốc sinh máu đa tiềm năng. Đột biến gen JAK2 gây giảm tự ức chế sinh sản tế bào của JAK2 - một protein tyrosin kinase thuộc họ Janus kinase (JAK), dẫn tới tăng sinh tế bào tạo máu. Sự khu trú của tế bào gốc bị đột biến tại các ổ sinh máu trong tủy xương là điều kiện cần để hình thành bệnh xơ tủy. Một số trường hợp xơ tủy nguyên phát có liên quan đến yếu tố nguy cơ: tiếp xúc hóa chất benzen, toluen, thorium dioxide và tia xạ.
3. TRIỆU CHỨNG
3.1. Lâm sàng
- Triệu chứng thường gặp là lách to, thiếu máu; Xuất huyết do giảm tiểu cầu có thể thấy ở những người bệnh có giảm số lượng tiểu cầu dưới 50G/l; Có thể có biểu hiện nhiễm trùng, nhất là nếu người bệnh có giảm số lượng bạch cầu hạt.
3.2. Xét nghiệm
a. Máu ngoại vi
- Giảm số lượng hồng cầu; Có biểu hiện bất thường về hình thái hồng cầu, thường gặp nhất là hồng cầu hình giọt lệ, mảnh hồng cầu, gặp hồng cầu có nhân; Tăng bạch cầu chủ yếu là dòng bạch cầu hạt, công thức bạch cầu chuyển trái (gặp một số tuổi trung gian dòng bạch cầu hạt); Có thể gặp tiểu cầu khổng lồ. LDH huyết thanh tăng.
b. Tuỷ xương
- Tủy đồ: Khó hút dịch tuỷ; tủy nghèo tế bào; tăng mẫu tiểu cầu.
- Mô bệnh học tuỷ xương: Tăng sinh xơ (reticulin); các đảo tế bào chứa nguyên bào sợi (fibroblast) và nguyên mẫu tiểu cầu bất thường.
- Lách đồ: Sinh máu ngoài tuỷ (gặp mẫu tiểu cầu trên tiêu bản lách đồ).
c. NST và gen: Có thể đột biến gen JAK2 V617F hoặc CALR exon 9 hoặc MPL (khi JAK2 V617F âm tính); có thể gặp bất thường NST: +8, -7/7q-, i(17q), -5/5q-, 12p-, bất thường 11q23. Không có gen kết hợp BCR-ABL; không có tái sắp xếp gen PDGFRA và PDGFRB.
4. CHẨN ĐOÁN
4.1. Tiêu chuẩn chẩn đoán của WHO 2016 (bảng 22)
Bảng 22. Tiêu chuẩn chẩn đoán xơ tủy nguyên phát theo WHO - 2016
|
Giai đoạn tiền xơ (prefibrotic/ early) |
1. Tăng sinh mẫu tiểu cầu và mẫu tiểu cầu không điển hìnhb, không có xơ hóa reticulin > mức 1c, kèm theo tăng mật độ tế bào tủy xương (điều chỉnh) theo tuổi, tăng sinh bạch cầu hạt và thường giảm sinh hồng cầu. 2. Không thỏa mãn các tiêu chuẩn chẩn đoán của WHO về CML, PV, ET, MDS hay các bệnh ác tính dòng tủy khác 3. Có đột biến JAK2, CALR, hoặc MPL; trong trường hợp không có các đột biến này, phải có một dấu ấn dòng khácd hoặc không có bằng chứng xơ hóa tủy xương phản ứnge |
|
Tiêu chuẩn phụ: Ít nhất 1 trong các tiêu chuẩn sau, được xác định trong 2 lần xét nghiệm liên tiếp: - Thiếu máu không do bệnh kèm theo - Tăng bạch cầu ≥11 G/L - Lách to sờ thấy được - LDH tăng cao hơn giới hạn trên theo khoảng tham chiếu đang được áp dụng tại cơ sở |
|
|
Giai đoạn xơ toàn phát (overt) |
Tiêu chuẩn chính: 1. Tăng sinh mẫu tiểu cầu và mẫu tiểu cầu không điển hìnhb kèm theo xơ hóa collagen và/hoặc reticulin (mức 2 hoặc 3) 2. Không thỏa mãn các tiêu chuẩn chẩn đoán của WHO về CML, PV, ET, MDS hay các bệnh ác tính dòng tủy khác 3. Có đột biến JAK2, CALR, hoặc MPL hoặc trong trường hợp không có các đột biến này, sự có mặt của một dấu ấn dòng khácd hoặc không có bằng chứng của xơ hóa tủy xương phản ứngf |
|
Tiêu chuẩn phụ: Ít nhất 1 trong các tiêu chuẩn sau, được xác định trong 2 lần xét nghiệm liên tiếp: - Thiếu máu không do bệnh kèm theo - Tăng bạch cầu ≥11 G/L - Lách to sờ thấy được - LDH tăng cao hơn giới hạn trên theo khoảng tham chiếu đang được áp dụng tại cơ sở - Tăng bạch cầu và/ hoặc hồng cầu non |
Ghi chú:
aChẩn đoán tiền xơ tủy/xơ tủy giai đoạn sớm đòi hỏi thỏa mãn 3 tiêu chuẩn chính và ít nhất 1 tiêu chuẩn phụ. Chẩn đoán xơ tủy giai đoạn toàn phát đòi hỏi thỏa mãn 3 tiêu chuẩn chính và ít nhất 1 tiêu chuẩn phụ.
bMẫu tiểu cầu nhỏ đến lớn với tỷ lệ nhân/NSC bất thường và tăng chất nhiễm sắc và nhân cuộn bất thường và co cụm thành đám dày đặc.
cTrong trường hợp xơ reticulin mức độ 1, mẫu tiểu cầu thay đổi nhưng phải kèm theo tăng mật độ tế bào tủy xương, tăng sinh dòng bạch cầu hạt, và thường giảm sinh hồng cầu (tiền xơ tủy).
dTrong trường hợp thiếu bất kỳ một trong 3 đột biến chính (JAK2, CALR, MPL), việc tìm kiếm một trong các đột biến đi kèm (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1) là có ích trong việc xác định bản chất dòng tế bào của bệnh.
eXơ hóa reticulin tối thiểu (mức độ 1) thứ phát sau nhiễm khuẩn, bệnh lý tự miễn hay các tình trạng viêm mạn tính khác, LXM tế bào tóc hoặc các tăng sinh lympho khác, ung thư di căn, hoặc bệnh tủy xương (mạn tính) nhiễm độc.
fXơ hóa tủy xương thứ phát do nhiễm khuẩn, bệnh lý tự miễn hay các tình trạng viêm mạn tính khác, LXM tế bào tóc hoặc các tăng sinh lympho khác, ung thư di căn, hoặc bệnh tủy xương (mạn tính) nhiễm độc.
4.2. Chẩn đoán phân biệt
- Đa hồng cầu nguyên phát ở giai đoạn kiệt quệ khi biểu hiện chủ yếu không còn là tăng số lượng hồng cầu và nồng độ hemoglobin (thậm chí các chỉ số này còn giảm) mà chủ yếu là tăng tiểu cầu và xơ tủy.
- Tăng tiểu cầu tiên phát, nhất là sau một thời gian điều trị, khi số lượng tiểu cầu không còn tăng cao nhưng biểu hiện xơ tủy thì lại thể hiện rất rõ.
Việc chẩn đoán phân biệt chủ yếu dựa vào biểu hiện lâm sàng và xét nghiệm lúc ban đầu của bệnh. Đối với xơ tủy nguyên phát, biểu hiện ban đầu này chính là tình trạng xơ tủy điển hình và biểu hiện sinh máu ngoài tủy trong một khoảng thời gian dài trong tiến trình của bệnh.
4.3. Phân nhóm nguy cơ
a. Hệ thống IPSS và DIPSS
|
Dấu hiệu |
IPSS (International Prognostic Scoring System): sử dụng tại thời điểm chẩn đoán bệnh |
DIPSS (dynamic IPSS): sử dụng tại bất kỳ giai đoạn nào của bệnh |
|
Tuổi > 65 tuổi |
1 điểm |
1 điểm |
|
Có các triệu chứng toàn thân (sụt cân, sốt, ra mồ hôi đêm) |
1 điểm |
1 điểm |
|
Thiếu máu rõ rệt (Hb < 100 g/L) |
1 điểm |
2 điểm |
|
Số lượng bạch cầu > 25G/l |
1 điểm |
1 điểm |
|
Blast máu ngoại vi ≥ 1% |
1 điểm |
1 điểm |
Trị số hemoglobin < 100 g/L phải được xác định trên người bệnh tại thời điểm chẩn đoán và chưa phụ thuộc truyền máu.
b. Hệ thống DIPSS-plus
|
Dấu hiệu |
Điểm DIPSS-plus tính dựa trên DIPSS |
Các yếu tố nguy cơ độc lập xuất hiện thêm, được tính điểm bổ sung cho DIPSS-plus |
|
DIPSS nguy cơ thấp |
0 |
|
|
DIPSS nguy cơ trung bình-1 |
1 |
|
|
DIPSS nguy cơ trung bình-2 |
2 |
|
|
DIPSS nguy cơ cao |
3 |
|
|
Số lượng tiểu cầu < 100 G/L |
|
1 |
|
Phụ thuộc truyền máu |
|
1 |
|
Có bất thường NST tiên lượng xấu: +8, -7/7q-, i(17q), -5/5q-, 12p-, bất thường 11q23 |
|
1 |
c. Cách tính điểm và phân nhóm nguy cơ theo các hệ thống tiên lượng trên
|
Nhóm nguy cơ |
IPSS |
DIPSS |
DIPSS-plus |
|||
|
Điểm |
Thời gian sống thêm trung bình (năm) |
Điểm |
Thời gian sống thêm trung bình (năm) |
Điểm |
Thời gian sống thêm trung bình (năm) |
|
|
Thấp |
0 |
11,3 |
0 |
Chưa kết thúc theo dõi |
0 |
15,4 |
|
Trung bình-1 |
1 |
7,9 |
1-2 |
14,2 |
1 |
6,5 |
|
Trung bình-2 |
2 |
4 |
3-4 |
4 |
2-3 |
2,9 |
|
Cao |
≥ 3 |
2,3 |
5-6 |
1,5 |
≥ 4 |
1,3 |
5. ĐIỀU TRỊ
5.1. Nguyên tắc điều trị
- Bệnh nhân không có triệu chứng lâm sàng: Chưa cần điều trị ngay, theo dõi lâm sàng và xét nghiệm hàng tháng.
- Bệnh nhân có triệu chứng sau đây cần bắt đầu điều trị:
+ Sốt > 38ºC;
+ Ra mồ hôi đêm;
+ Gầy sút cân trong vòng 6 tháng;
+ Ngứa;
+ Đau xương;
+ Mệt mỏi nhiều;
+ Ăn kém, đầy bụng do lách to;
+ Lách to nhiều (> 10 cm dưới bờ sườn);
+ Hb < 100 g/l;
+ BC > 25 G/l;
+ TC > 1.000 G/l.
- Các thuốc sử dụng trong điều trị:
+ Hydroxyurea: Liều khởi đầu 500-1000 mg cách ngày, điều chỉnh theo đáp ứng tế bào máu của bệnh nhân;
+ Ruxolitinib: Liều khởi đầu phụ thuộc số lượng TC của bệnh nhân.
- 20mg x 2 lần/ngày nếu tiểu cầu > 200 G/l;
- 15mg x 2 lần/ngày nếu tiểu cầu 100-200 G/l;
- 5mg x 2 lần/ngày nếu tiểu cầu 50-100 G/l.
5.2. Điều trị theo nhóm nguy cơ
a. Nhóm nguy cơ thấp và nguy cơ trung bình-1
- Không có triệu chứng lâm sàng: Theo dõi định kỳ hàng tháng.
- Có triệu chứng lâm sàng và/hoặc lách to > 10 cm dưới bờ sườn, BC > 25 G/l, TC > 1000 G/l:
+ Hydroxyurea là lựa chọn hàng 1;
+ Ruxolitinib nếu bệnh nhân có lách to nhiều và không đáp ứng hoặc dung nạp với hydroxyurea.
b. Nhóm nguy cơ trung bình-2 và nguy cơ cao
- Ghép tế bào gốc tạo máu đồng loài: Nếu bệnh nhân đủ điều kiện, có người hiến tế bào gốc phù hợp HLA (tham khảo bài Ghép tế bào gốc tạo máu đồng loài).
- Bệnh nhân không đủ điều kiện ghép tế bào gốc:
+ Ruxolitinib là lựa chọn hàng 1;
+ Tham gia thử nghiệm lâm sàng nếu không có khả năng điều trị hoặc thất bại với ruxolitinib.
5.2. Điều trị hỗ trợ
- Thiếu máu:
+ Truyền khối hồng cầu khi Hb < 80 g/L;
+ Dùng thuốc kích thích sinh hồng cầu (erythropoietin) nếu EPO huyết thanh < 500 mU/ml, tuy nhiên hiệu quả kém ở các bệnh nhân phụ thuộc truyền máu;
+ Dùng danazol nếu EPO huyết thanh ≥ 500 mU/ml;
+ Dùng methylprednisone kết hợp thalidomide hoặc lenalidomide nếu không đáp ứng với danazol.
- Sinh máu ngoài tủy: Tia xạ.
- Cắt lách: Cân nhắc khi bệnh nhân có lách quá to không đáp ứng điều trị hoặc bệnh nhân thường xuyên phải truyền > 3 đơn vị máu trong vòng 2 tuần, nhồi máu lách tái diễn, giảm TC dai dẳng, tăng áp lực tĩnh mạch cửa, suy tim cung lượng cao.
5.3. Các xét nghiệm đánh giá và theo dõi điều trị
- Tổng phân tích tế bào máu ngoại vi, được làm ít nhất 2 ngày 1 lần.
- Sinh hóa máu: Chức năng gan, thận, điện giải, acid uric, photpho, Fe, Ferritin, LDH... ít nhất 1 tuần/lần.
- Xét nghiệm đông máu: Fibrinogen, PT, APTT, TT, D-Dimer, Rotem… ít nhất 1 tuần/lần.
- Xét nghiệm vi sinh: Virus viêm gan, HIV… làm sàng lọc trước khi điều trị, lặp lại theo quy định; Các xét nghiệm đánh giá nhiễm trùng: Cấy vi khuẩn, vi nấm, CRP, PCT, CMV, EBV, kháng sinh đồ… nếu lâm sàng có dấu hiệu nhiễm trùng.
- Huyết thanh học nhóm máu, làm lúc chẩn đoán hoặc khi cần truyền máu.
- Xét nghiệm tế bào và sinh hóa nước tiểu, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Chẩn đoán hình ảnh, thăm dò chức năng, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Xét nghiệm Huyết tủy đồ, STTX, nhuộm sợi xơ làm lúc chẩn đoán và có thể làm lại khi nghi ngờ bệnh tiến triển hoặc chuyển thể; phát hiện gen đột biến JAK2 V617F hoặc CALR exon 9, MPL, Công thức nhiễm sắc thể được làm lúc chẩn đoán. Định lượng đột biến gen JAK2 để theo dõi điều trị.
- Một số xét nghiệm khác được chỉ định tùy theo bệnh nền và bệnh mắc kèm của người bệnh.
6. ĐÁP ỨNG ĐIỀU TRỊ
Bảng 23. Tiêu chuẩn đáp ứng điều trị theo IWG
(Nhóm làm việc quốc tế - International Working Group)
|
STT |
Mức độ đáp ứng |
|
Dấu hiệu lâm sàng và cận lâm sàng |
|
1 |
Đáp ứng hoàn toàn (CR: Complete remission) |
i |
Hết triệu chứng của bệnh bao gồm cả biểu hiện gan lách to. |
|
ii |
Lui bệnh về huyết học gồm Hb ≥ 110 G/L; số lượng tiểu cầu ≥ 100 G/L; neutrophil ≥ 1,0 G/L; số lượng 3 dòng tế bào máu không tăng cao hơn giới hạn trên của giá trị bình thường. |
||
|
iii |
Công thức bạch cầu bình thường, bao gồm không xuất hiện hồn cầu có nhân, tế bào blast và tế bào dòng tủy không trưởng thành trên tiêu bản máu ngoại vi (trường hợp không cắt lách). |
||
|
iv |
Lui bệnh về mặt tế bào trong tủy xương, blast < 5% và xơ hóa tủy ≤ 1. |
||
|
2 |
Đáp ứng một phần (PR: Partial remission) |
|
Gồm tất cả các tiêu chuẩn của CR, ngoại trừ tiêu chuẩn lui bệnh về mặt tế bào trong tủy xương. Tuy nhiên, cần lặp lại sinh thiết tủy xương để đánh giá. |
|
3 |
Cải thiện về lâm sàng (CI: Clinical improvement) |
|
Cần có 1 trong những tiêu chuẩn sau khi không đánh giá được CR, PR và bệnh không tiến triển. |
|
i |
Hb tăng thêm ≥ 20 G/L hoặc người bệnh không còn phụ thuộc truyền máu (chỉ dùng cho người bệnh có Hb < 100 G/L). |
||
|
ii |
Giảm ≥ 50% kích thước lách to trong trường hợp lách to ≥ 10 cm dưới bờ sườn hoặc không còn sờ thấy lách to nếu trước đó lách to ≥ 5 cm dưới bờ sườn. |
||
|
iii |
Tăng gấp đôi số lượng tiểu cầu và số lượng tiểu cầu ≥ 50 G/L (chỉ dùng cho người bệnh có số lượng tiểu cầu < 50 G/L). |
||
|
iv |
Tăng gấp đôi số lượng bạch cầu đoạn trung tính và số lượng bạch cầu đoạn trung tính ≥ 0,5 G/L (chỉ dùng cho trường hợp bạch cầu đoạn trung tính < 1,0 G/L). |
||
|
4 |
Bệnh tiến triển (PD: Progressive disease) |
|
Khi có 1 trong các tiêu chuẩn sau: |
|
i |
Lách to tiến triển (mới xuất hiện lách to ≥ 5 cm nếu trước đó không sờ thấy; kích thước lách dưới bờ sườn tăng gấp đôi trong trường hợp lách to 5-10 cm hoặc tăng gấp 1,5 lần trong trường hợp lách to > 10 cm. |
||
|
ii |
Chuyển lơ xê mi cấp với tỷ lệ blast ≥ 20% trong tủy xương. |
||
|
iii |
Tăng tỷ lệ blast ≥ 20% trong máu ngoại vi, thời gian dài ≥ 8 tuần. |
||
|
5 |
Bệnh ổn định (SD: Stable disease) |
|
Không có các tiêu chuẩn của CR, PR, CI hoặc PD. |
|
6 |
Tái phát (Relapse) |
|
Mất CR, PR hoặc CI. Bệnh nhân đạt CR và PR được xem là tái phát khi bệnh nhân không đạt cả tiêu chuẩn của CI. |
7. TIÊN LƯỢNG
Xơ tủy nguyên phát là bệnh có tiên lượng kém nhất trong các bệnh thuộc hội chứng tăng sinh tủy mạn ác tính. Thời gian sống thêm trung bình của người bệnh xơ tủy nguyên phát là 3,5-5,5 năm. Biến chứng nghiêm trọng nhất là khả năng chuyển thành Lơ xê mi cấp. Một số yếu tố nguy cơ khác dẫn tới thời gian sống thêm bị rút ngắn bao gồm: Tuổi cao, tình trạng thiếu máu nhiều, giảm bạch cầu hạt, giảm tiểu cầu nặng,…
TÀI LIỆU THAM KHẢO
1. Harrison C, Kiladjian J-J, Al-Ali H et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis, The New England Journal of Medicine 2012, 366(9): 787-797.
2. McLornan DP, Mead AJ, Jackson G et al. State of the art review - allogeneic stem cell transplantation for myelofibrosis in 2012, British Journal of Haematology 2012: 1-11.
3. Tefferi A, Juergen Thiele J, and Vardiman JW, MD3. The 2008 World Health Organization Classification System for Myeloproliferative Neoplasms, Cancer 2009, September 1: 3842-3847.
4. Tefferi A & Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies, Journal of clinical oncology 2011, 29(5): 573-581.
5. Verstovsek S, Mesa RA, Gotlib J et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis, The New England Journal of Medicine 2012, 366(9): 799-807.
6. Tiziano Barbui, Ayalew Tefferi et al, (2018), “Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet”, Leukemia: 32(5): 1057-1069.
28. HỘI CHỨNG RỐI LOẠN SINH TỦY
1. ĐẠI CƯƠNG
- Hội chứng rối loạn sinh tủy (Myelodysplastic Syndrome - MDS) là một nhóm các bệnh ác tính tế bào sinh máu có đặc điểm: Tủy sinh máu không hiệu lực; bất thường hình thái và chức năng tế bào máu; bệnh tiến triển âm ỉ, dai dẳng và thường kết thúc bằng lơ xê mi cấp nên còn gọi là tiền lơ xê mi (pre-leukemia).
- Nguyên nhân chưa rõ, được cho là tương tự các nguyên nhân gây ra bệnh lơ xê mi cấp.
- Chẩn đoán và tiên lượng dựa vào các tiêu chuẩn của FAB 1982, WHO 2001 và WHO 2008, WHO 2016, tùy vào điều kiện chẩn đoán của cơ sở y tế hiện có.
2. CHẨN ĐOÁN
Căn cứ vào lâm sàng và xét nghiệm.
2.1. Lâm sàng
- Thường gặp trên 50 tuổi, nam nhiều hơn nữ.
- Thiếu máu (> 90% người bệnh) thường là biểu hiện đầu tiên, dai dẳng và đáp ứng kém với điều trị.
- Nhiễm trùng (gặp ở khoảng 15% người bệnh).
- Có thể gặp xuất huyết do giảm tiểu cầu.
- Gan lách có thể to (gặp 10 - 15% người bệnh).
2.2. Xét nghiệm
a. Máu ngoại vi
- Giảm hồng cầu (90%) và/ hoặc tiểu cầu (25%), đôi khi có giảm cả 3 dòng. MCV thường cao, thường thiếu máu bình sắc, có thể gặp hồng cầu non ra máu (10%).
- Hình thái bạch cầu hạt có thể bất thường: Giảm, mất hạt đặc hiệu, giảm đoạn, dị thường kiểu Pelger Huët, hoặc nhân dạng vòng (Ring- Slaped nucle), nhân hình gậy, hình ảnh nhân bị đứt đoạn; hoặc tăng hạt đặc hiệu, tăng đoạn (ít gặp hơn). Có thể có tế bào non ác tính (blast).
- Có 25% người bệnh lúc chẩn đoán giảm tiểu cầu từ nhẹ đến trung bình, nhưng cũng có gặp tăng tiểu cầu; hay gặp tiểu cầu kích thước to (tiểu cầu khổng lồ); có thể giảm độ tập trung của tiểu cầu.
b. Tủy đồ: Rất có giá trị trong chẩn đoán, thể hiện tủy sinh máu không hiệu lực:
- Số lượng tế bào tủy: Đa số bình thường, nhưng có thể tăng hay giảm; nếu tăng, thường không quá cao như lơ xê mi; nếu giảm, cũng không quá thấp như suy tủy xương.
- Dòng hồng cầu: Thường gặp tăng sinh, đủ các tuổi hồng cầu non, các hồng cầu non có nhân chấm, hoặc hồng cầu non ít tạo hemoglobin (so với lứa tuổi); có thể tăng tiền nguyên hồng cầu nhưng ít gặp các cụm, có thể gặp nguyên hồng cầu nhiều nhân, nhân có vệ tinh, thấy ở giai đoạn đa sắc và ưa acid, bào tương có hốc hoặc ít tạo huyết sắc tố.
Nhuộm hồng cầu sắt (nhuộm Perls) có thể phát hiện được sideroblast vòng (là một tiêu chuẩn quan trọng trong xếp thể bệnh của hội chứng rối loạn sinh tủy).
- Dòng bạch cầu: Thường tăng bạch cầu dòng hạt và dòng mono; các bất thường là: giảm hoặc mất hạt đặc hiệu, tế bào méo mó, nứt vỡ, có vệ tinh, xuất hiện tế bào một nhân của bạch cầu đoạn trung tính, bất thường kiểu Pelger Huët; có thể gặp tăng tế bào tiền tủy bào và tủy bào; bao giờ cũng gặp tế bào blast nhưng tỷ lệ gần bình thường (dưới 2%), có thể gặp tăng cao, đến xấp xỉ 20%, ở thể có tăng quá mức tế bào blast.
- Mẫu tiểu cầu: Gặp mẫu tiểu cầu còi cọc, không chia thùy hoặc hai thùy, nhiều múi hoặc ít múi.
c. Sinh thiết tủy xương
- Rất có giá trị trong việc chẩn đoán sớm khi bệnh chuyển thành lơ xê mi cấp.
- Phát hiện thấy khu trú bất thường của các tế bào đầu dòng chưa trưởng thành (ALIP: Abnormal localization of immature precursors). Khi gặp ALIP thì tiên lượng bệnh dễ chuyển thành lơ xê mi cấp.
- Có thể có tăng sinh xơ, gặp ở thể ít blast.
d. Đặc điểm tế bào di truyền-Sinh học phân tử
- Di truyền tế bào: Mất NST: 5, 7, 20; thêm NST: 8, 21; mất đoạn nhánh dài NST: 5, 7, 20; trong đó mất nhánh dài NST 5 được sử dụng để xếp loại bệnh; chuyển đoạn: t(1;3); (p36;q21); t(1;7); (p11;q11); t(2;11); (p21;q23). Không gặp bất thường đặc hiệu của lơ xê mi dòng tủy: t(8;21); t(15;17), inv(16).
- Sinh học phân tử:
+ Phát hiện các đột biến gen có giá trị tiên lượng bệnh:
● Tiên lượng xấu: ASXL1, EZH2, SRSF2, U2AF1, RUNX1, ZRSR2, TP53, STAG2, NRAS , ETV8, GATA2, BCOR, IDH2, NPM1, WT1, PRPF8, FLT3, CBL;
● Tiên lượng tốt: SF3B1.
+ Phát hiện các gen khác có ý nghĩa chẩn đoán bệnh: IDH1, NEDD, ICPI, TET2, DNMT3A, NF1, JAK2, CALR, MPL, DDX41, SETBP1, PHF6, STAT3, PPM1D, PDGFRβ.
đ. Xét nghiệm phân loại miễn dịch (FCM): Nên được cân nhắc trong trường hợp cần phân biệt với bệnh tế bào lympho dạng hạt lớn (LGL) và bệnh đái huyêt sắc tố kịch phát (PNH), hay phát hiện các kháng thể đơn dòng nhắm đích (CD47, CD3, CD123,…).
e. Sinh hóa máu
- Đo nồng độ Erythropoietin trước khi truyền khối hồng cầu.
- Folat hồng cầu, Vitamin B12 huyết thanh.
- Ferritin huyết thanh, sắt huyết thanh và TIBC.
- Hormon tuyến giáp: T3, T4, FT3, FT4, TSH.
- Xét nghiệm LDH, acid uric.
f. Vi sinh:
- Xét nghiệm HIV, HCV, HBsAg, Anti-HBc IgG.
- Xét nghiệm: CMV (IgG,IgM), đo tải lượng CMV.
2.3. Chẩn đoán
a. Tiêu chuẩn chẩn đoán hội chứng rối loạn sinh tủy: Dựa trên lâm sàng, xét nghiệm tế bào học (bảng xếp loại mục 2.4).
b. Chẩn đoán phân biệt Hội chứng rối loạn sinh tủy với
- Hội chứng tăng sinh tủy: Cùng là bệnh đơn dòng, nhưng tăng sinh tế bào mạnh hơn, định hướng dòng rõ, có thể thấy tổn thương đặc hiệu như gen Jak2 V617F.
- Suy tủy xương: Thiếu máu, xuất huyết, nhiễm trùng thường nặng hơn, diễn biến nhanh hơn, tủy giảm sinh nặng hơn thậm chí tuyệt sản, ít bất thường về hình thái hơn.
- Thiếu máu hồng cầu to do thiếu axit folic và vitamin B12: Thường có nguyên nhân gây thiếu axit folic và vitamin B12 như cắt dạ dày, nhiễm trùng ruột nặng, thể tích hồng cầu rất to (trên 130fl), giảm axit folic và vitamin B12 trong huyết thanh, điều trị thử bằng axit folic và vitamin B12 có cải thiện tốt.
- Thiếu máu có sideroblast bẩm sinh: Thường ở trẻ em, MCV nhỏ, không bất thường về hình thái đối với các dòng tế bào khác.
2.4. Xếp loại thể bệnh hội chứng rối loạn sinh tủy: Tùy khả năng thực hiện một số xét nghiệm của cơ sở y tế mà lựa chọn cách xếp loại cho phù hợp sau đây.
a. Xếp loại theo FAB (1982)
|
Nhóm |
Blast máu |
Blast tuỷ |
Nguyên hồng cầu sắt vòng |
Tế bào Mono/ máu |
Rối loạn hồng cầu |
Rối loạn dòng bạch cầu |
Rối loạn dòng tiểu cầu |
|
TMDD* (Retractory anemia = R.A) |
< 1% |
< 5% |
< 15% |
< 1G/L |
++ |
+ |
+ |
|
TMDD có Sideroblast vòng (R.A. With ring sederoblast = RARS) |
< 1% |
< 5% |
> 15% |
< 1G/L |
++ |
+ / - |
+ / - |
|
TMDD có tăng quá mức tế bào blast (R.A. With excess of blast = RAEB) |
< 5% |
5-20% |
Thay đổi |
< 1G/L |
++ |
++ |
++ |
|
TMDD có tăng quá mức tế bào blast đang chuyển cấp (RAEB in transformation = RAEB-t) |
> 5% |
21-< 30% |
Thay đổi |
Thay đổi |
++ |
++ |
++ |
|
Lơ xê mi tủy mono mạn (Choronic Myelo - Monoleukemia = CMML) |
< 5% |
5-20% |
Thay đổi |
≥ 1G/L |
+/- |
+/- |
+/- |
TMDD*: Thiếu máu dai dẳng.
b. Xếp loại của tổ chức y tế thế giới WHO (2001)
|
TT |
Thể bệnh |
Máu ngoại vi |
Tủy xương |
|
1 |
Thiếu máu dai dẳng (Refractory anemia: RA) |
Thiếu máu. Không có blasts. |
- Chỉ rối loạn dòng hồng cầu. - Blasts < 5% - Nguyên hồng cầu sắt vòng < 15% |
|
2 |
Thiếu máu dai dẳng tăng nguyên hồng cầu sắt vòng (Refractory anemia with ringed sideroblasts: RARS) |
Thiếu máu. Không có blasts. |
- Chỉ rối loạn dòng hồng cầu - Nguyên hồng cầu sắt vòng ≥ 15% - Blasts < 5% |
|
3 |
Giảm tế bào dai dẳng có rối loạn nhiều dòng tế bào (Refractory cytopenia with multilineage dysplastic: RCMD) |
Giảm tế bào (2 hoặc 3 dòng) Không có hoặc hiếm gặp blasts. Không thấy thể Auer Tế bào mono < 1G/L |
- Rối loạn ≥ 10% tế bào của ít nhất 2 dòng tế bào tuỷ - Blasts tuỷ < 5% - Không thấy thể Auer - Nguyên hồng cầu sắt vòng < 15% |
|
4 |
Giảm tế bào dai dẳng có rối loạn nhiều dòng tế bào và tăng nguyên HC sắt vòng(Refractory cytopenia with multilineage dysplastic and ringed sideroblasts: RCMD - RS) |
Giảm tế bào (2 hoặc 3 dòng) Không có hoặc hiếm gặp blasts Không thấy thể Auer Tế bào mono < 1G/L |
- Rối loạn ≥ 10% tế bào của ít nhất 2 dòng tế bào tủy - Blasts tuỷ < 5% - Nguyên hồng cầu sắt vòng ≥ 15% - Không thấy thể Auer |
|
5 |
Thiếu máu dai dẳng có tăng quá mức blast - 1(Refractory anemia with exccess blasts1: RAEB - 1) |
Giảm tế bào Blasts < 5% Tế bào mono < 1G/L |
- Rối loạn một dòng hay nhiều dòng tế bào tủy - Blasts 5% đến 9% - Không thấy thể Auer |
|
6 |
Thiếu máu dai dẳng có tăng quá mức blast - 2 (Refractory anemia with excces blasts2: RAEB - 2) |
Giảm tế bào Blasts 5% đến 19% Có khi thấy thể Auer Tế bào mono < 1G/L |
- Rối loạn một dòng hay nhiều dòng tế bào tủy - Blasts 10% đến 19% - Có khi thấy thể Auer |
|
7 |
Hội chứng rối loạn sinh tủy không xếp loại (Myelodysplastic syndrome unclassified: MDS - U) |
Giảm tế bào Không có hoặc hiếm gặp blast Không thấy thể Auer |
- Chỉ rối loạn dòng bạch cầu hạt hoặc mẫu tiểu cầu. - Blasts < 5% - Không thấy thể Auer |
|
8 |
Hội chứng rối loạn sinh tủy có kết hợp mất nhánh dài nhiếm sắc thể số 5(del 5q) (MDS associated with isolatsed del(5q)) |
Thiếu máu. Blasts < 5% Tiểu cầu bình thường hoặc tăng |
- Mẫu tiểu cầu bình thường hoặc tăng, giảm chia thùy - Blasts < 5% - Không thấy thể Auer - del (5q) đơn độc |
c. Xếp loại của tổ chức y tế thế giới WHO (2008)
|
Nhóm |
Thể bệnh |
Các yếu tố về tủy xương |
|
1 |
Giảm tế bào dai dẳng loạn sản đơn dòng bao gồm: thiếu máu dai dẳng, giảm bạch cầu trung tính, giảm tiểu cầu (Refractory cytopenias with unilineage dysplasia: RCUD) |
≥ 10% tế bào một dòng bị rối loạn < 5% blast < 15% nguyên hồng cầu sắt vòng (RS) |
|
2 |
Thiếu máu dai dẳng tăng nguyên hồng cầu sắt vòng (Refractory anemia with ringed sideroblasts: RARS) |
< 5% blast ≥ 15% RS |
|
3 |
Giảm tế bào dai dẳng có rối loạn nhiều dòng tế bào (Refractory cytopenia with multilineage dysplastic: RCMD) |
≥ 10% tế bào bất thường ở ≥ 2 dòng tế bào < 5% blast Không kèm hoặc kèm tăng RS |
|
4 |
Hội chứng rối loạn sinh tủy có kết hợp mất nhánh dài nhiếm sắc thể số 5(del 5q) (MDS associated with isolatsed del(5q)) |
< 5% blast del(5q) |
|
5 |
- Thiếu máu dai dẳng có tăng quá mức blast -1 (Refractory anemia with exccess blasts1: RAEB - 1) - Thiếu máu dai dẳng có tăng quá mức blast - 2 (Refractory anemia with excces blasts2: RAEB - 2) |
5-9% blast, thể Auer (-)
10-19% blast, thể Auer (±) |
|
6 |
MDS-U: rối loạn sinh tủy chưa xếp loại |
<10% tế bào bất thường + < 5% blast + gen bất thường |
Theo xếp loại WHO 2008 thể RAEB-t của xếp loại FAB (1982) được coi là lơ xê mi tủy cấp với tổn thương đa dòng, và CMML được xếp là MDS/MPS (rối loạn sinh tủy/ tăng sinh tủy); có bất thường NST như t(15;17), t(8;21), inv16, t(16;16), được xếp là lơ xê mi tủy cấp, không tính đến tỉ lệ blast tủy và nguyên hồng cầu sắt vòng.
d. Xếp loại của tổ chức y tế thế giới WHO sửa đổi (2016)
|
- Hội chứng rối loạn sinh tủy có loạn sản đơn dòng (MDS with single lineage dysplasia). - Hội chứng rối loạn sinh tủy có nguyên hồng cầu sắt vòng (MDS with ring sideroblasts - MDS - RS). - MDS-RS có loạn sản đơn dòng (MDS-RS and single lineage dysplasia). - MDS-RS có loạn sản đa dòng (MDS-RS and multilineage dysplasia). - Hội chứng rối loạn sinh tủy có loạn sản đa dòng (MDS with multilineage dysplasia). - Hội chứng rối loạn sinh tủy có tăng quá mức tế bào blasts (MDS with excess blasts). - Hội chứng rối loạn sinh tủy có del(5q) đơn độc (MDS with isolated del 5q). - Hội chứng rối loạn sinh tủy không phân loại được theo cách khác (MDS unclassifiable). - Thể bệnh đề xuất bổ sung: Giảm tất cả các dòng tế bào dai dẳng ở trẻ em (refractory cytopenia of childhood). |
Lựa chọn sử dụng xếp loại nào cho phù hợp cần căn cứ vào khả năng làm được các xét nghiệm:
- Nhuộm hồng cầu sắt là bắt buộc phải có trong khi sử dụng cả 3 cách xếp loại.
- Muốn sử dụng xếp loại theo WHO 2001 thì phải làm được xét nghiệm tìm bất thường nhiễm sắc thể (mất nhánh dài NST số 5).
- Muốn sử dụng được xếp loại WHO 2008 phải làm được thêm nhiều xét nghiệm tìm tổn thương nhiễm sắc thể trong hội chứng rối loạn sinh tủy (cả del5q và các xét nghiệm khác như đã nêu ở trên t(15;17), t(8;21), inv16, t(16;16)…
2.5. Chẩn đoán nhóm nguy cơ bệnh theo di truyền
2.5.1. Di truyền tế bào
Bảng 24. Bảng tiên lượng bệnh theo đột biến NST
|
Rất tốt |
Tốt |
Trung bình |
Xấu |
Rất xấu |
|
del (11q) -Y |
CT-NST: bình thường der (1;7) del (5q) del (12p) del (20q) |
-7/7q- +8 iso (17q) + 19 + 21 Tổn thương đơn độc khác |
der(3)(q21)/ der(3)(q26) |
> 3 tổn thương bất kỳ |
|
|
2 tổn thương bao gồm del (5q) |
2 tổn thương bất kỳ |
2 tổn thương bao gồm -7/7q- |
|
|
|
|
|
3 tổn thương bất kỳ |
|
2.5.2. Sinh học phân tử:
- Tiên lượng xấu: ASXL1, EZH2, SRSF2, U2AF1, RUNX1, ZRSR2, TP53, STAG2, NRAS , ETV6, GATA2, BCOR, IDH2, NPM1, WT1, PRPF8, FLT3, CBL.
- Tiên lượng tốt: SF3B1.
3. ĐIỀU TRỊ
3.1. Nguyên tắc chung
- Điều trị căn cứ theo thể bệnh hội chứng rối loạn sinh tủy (xếp loại FAB 1982 và WHO), yếu tố nguy cơ, tuổi, toàn trạng.
- Các biện pháp điều trị chính là: Hóa trị liệu, sử dụng các chất cảm ứng biệt hóa, ghép tủy xương, điều trị bổ trợ cho thiếu máu, xuất huyết, nhiễm trùng.
3.2. Tiên lượng
- Tiên lượng phụ thuộc vào nhiều yếu tố: Mức độ giảm tế bào, tỷ lệ blast ở máu và tủy, tăng tế bào mono trong CMML, có mặt của ALIP trong tủy, bất thường NST…
Bảng 25. Hệ thống điểm tiên lượng theo tiêu chuẩn quốc tế
|
Tiêu chuẩn |
Điểm |
|
A. Blast trong tủy xương ■ < 5% ■ 5-10% ■ 11-20% ■ 21-30% |
0 1 1,5 2 |
|
B. Bất thường di truyền học tế bào ■ Bình thường, Y-, 5q-, 20q- ■ Bất thường NST số 7, hoặc số 3, hoặc có nhiều bất thường NST phối hợp. ■ Có các bất thường di truyền khác. |
0 1 0,5 |
|
C. Suy giảm các dòng tế bào máu: được định nghĩa là khi Hb < 10g/dl, Số lượng tuyệt đối bạch cầu trung tính < 1,5 G/L, số lượng tiểu cầu < 100G/L. |
|
|
■ Không suy giảm hay chỉ bị suy giảm một dòng tế bào máu. |
0 |
|
■ Suy giảm 2 hoặc 3 dòng tế bào máu. |
0,5 |
Đánh giá tiên lượng như sau:
|
Điểm |
Nhóm nguy cơ |
Thời gian sống trung bình |
|
0 |
Thấp |
5,7 năm |
|
0,5-1,0 |
Trung bình 1 |
3,5 năm |
|
1,5-2,0 |
Trung bình 2 |
1,2 năm |
|
2,5-5,0 |
Cao |
0,4 năm |
Bảng 26. Hệ thống điểm tiên lượng theo tiêu chuẩn quốc tế sửa đổi (IPSS-R: revised):
|
Chỉ số tiên lượng |
Điểm |
||||||
|
0 |
0,5 |
1 |
1,5 |
2 |
3 |
4 |
|
|
Blasts tủy (%) |
< 2 |
- |
2-5 |
- |
5-10 |
> 10 |
- |
|
Công thức NST |
Rất tốt |
- |
Tốt |
- |
Trung bình |
Xấu |
Rất xấu |
|
Hemoglobin (g/l) |
≥ 10 |
- |
8-10 |
< 8 |
- |
- |
- |
|
Số lượng bạch cầu trung tính tuyệt đối (G/l) |
≥ 0,8 |
< 0,8 |
- |
- |
- |
- |
- |
|
Số lượng tiểu cầu (G/l) |
≥ 100 |
50-100 |
< 50 |
- |
- |
- |
- |
Ghi chú: Công thức NST được đánh giá theo bảng 19
Đánh giá tiên lượng theo tiêu chuẩn quốc tế sửa đổi (IPSS-R) như sau:
|
Điểm |
Nhóm nguy cơ |
Thời gian sống trung bình |
|
≤ 1,5 |
Rất thấp |
8,8 năm |
|
2,0-3,0 |
Thấp |
5,3 năm |
|
3,5-4,5 |
Trung bình |
3,0 năm |
|
5,0-6,0 |
Cao |
1,6 năm |
|
> 6,0 |
Rất cao |
0,8 năm |
- Đánh giá tiên lượng theo di truyền:
+ Tiên lượng xấu: ASXL1, EZH2, SRSF2, U2AF1, RUNX1, ZRSR2, TP53, STAG2, NRAS , ETV6, GATA2, BCOR, IDH2, NPM1, WT1, PRPF8, FLT3, CBL;
+ Tiên lượng tốt: SF3B1.
3.3. Điều trị cụ thể:
3.3.1. Rối loạn sinh tủy nhóm nguy cơ thấp (IPSS thấp/trung bình 1, IPSS-R rất thấp/thấp/trung bình ≤ 3,5 điểm).
Liệu pháp điều trị chính là cải thiện các tế bào máu, tránh biến chứng thiếu máu, xuất huyết, nhiễm trùng ảnh hưởng đến chất lượng cuộc sống của bệnh nhân.
3.3.1.1. Theo dõi:
- Các trường hợp bệnh không có triệu chứng.
- Có các chiến lược cụ thể với các bệnh nhân có đột biến di truyền cụ thể.
3.3.1.2. Điều trị thiếu máu:
a. Chất kích thích sinh hồng cầu (ESA)
- Erythropoietin (EPO):
+ EPO huyết thanh ≤ 500 mU/ml và/hoặc truyền dưới 2 đơn vị hồng cầu/tháng: 40.00 - 60.000 UI x 1-2 lần/tuần và / hoặc thêm G-CSF (theo liệu trình phần dưới);
+ EPO huyết thanh > 500: Không có chỉ định dùng tiêm EPO.
- G-CSF: Liều 1-2 mcg/kg/lần nên bắt đầu bằng liều 300mcg. Theo dõi công thức máu hàng tuần và điều chỉnh liều lượng để giữ số lượng bạch cầu từ 6 đến 10 G/l.
b. Rối loạn sinh tủy có đột biến del (5q)
- Chất kích thích sinh hồng cầu (ESA) vẫn là lựa chọn hàng đầu (liều như trên).
- Lenalidomide: 10mg, uống ngày 1 lần x 21 ngày/tháng.
- Chú ý: Xem xét các đột biến TP53, TET2 và RUNX1 để đánh giá và theo dõi tiến triển của bệnh.
c. Rối loạn sinh tủy không có del (5q)
- Ức chế miễn dịch:
+ Antithymocyte globulin (hATG hoặc rATG):
● hATG: 40mg/kg/ngày truyền tĩnh mạch 4 ngày;
● rATG: 3,75 mg/kg/ngày truyền tĩnh mạch 5 ngày.
+ Cyclosporine: 3 - 6 mg/kg /ngày, uống;
+ Kháng CD33 trong trường hợp có biểu hiện bất thường của CD33 trên FCM;
+ Kháng TLR2,…
- Chất hypomethyl hóa (HMA):
+ Azacitidine: 75 mg/m2 tiêm dưới da hoặc truyền tĩnh mạch x 7 ngày liên tục (tổng liều 525 mg/m2);
+ Decitabine:
● Phác đồ 3 ngày: 15 mg/m² truyền tĩnh mạch trong 3 giờ mỗi 8 giờ x 3 ngày; lặp lại chu kỳ mỗi 6 tuần;
● Phác đồ 5 ngày: 20 mg/m² truyền tĩnh mạch trong 1 giờ mỗi ngày x 5 ngày, lặp lại chu kỳ mỗi 4 tuần.
- Ghép tế bào gốc đồng loài: Cân nhắc ghép sớm cho các trường hợp có đột biến tiên lượng xấu.
3.3.1.3. Điều trị giảm tiểu cầu
a. Chất hypomethyl hóa (HMA): Như trên.
b. Thuốc chủ vận thụ thể TPO:
- Romiplostim: Tiêm dưới da 300-1500 μg mỗi tuần.
- Eltrombopag: 50mg/ngày đến 150mg/ngày, uống.
3.3.2. Rối loạn sinh tủy nhóm nguy cơ cao
- Liệu pháp điều trị chính là hạn chế tiến triển của bệnh, cải thiện tỷ lệ sống.
- Trước khi điều trị phải đánh giá khả năng ghép TBG đồng loài của bệnh nhân.
3.3.2.1. Chất hypomethyl hóa (HMA)
Là lựa chọn đầu tiên cho nhóm rối loạn sinh tủy nguy cơ cao
- Azacitidine: 75 mg/m2 tiêm dưới da hoặc truyền tĩnh mạch x 7 ngày liên tục 1 chu kì điều trị (tổng liều 525 mg/m2) x tối thiểu 6 chu kì, mỗi chu kì cách nhau 4 tuần; có thể dùng liên tục cho đến khi bệnh tiến triển.
- Decitabine:
+ Phác đồ 3 ngày: 15 mg/m² truyền tĩnh mạch trong 3 giờ mỗi 8 giờ x 3 ngày; lặp lại chu kỳ mỗi 6 tuần; tối thiểu 4 chu kì, có thể dùng liên tục cho đến khi bệnh tiến triển.
+ Phác đồ 5 ngày: 20 mg/m² truyền tĩnh mạch trong 1 giờ mỗi ngày x 5 ngày, lặp lại chu kỳ mỗi 4 tuần; tối thiểu 4 chu kì, có thể dùng liên tục cho đến khi bệnh tiến triển.
3.3.2.2. Ghép tế bào gốc đồng loài
- Tiêu chí IPSS nên được xem xét ngay tại thời điểm chẩn đoán cho ghép tế bào gốc đồng loài.
- Ghép tế bào gốc đồng loài nên được thực hiện cho nhóm: Rối loạn sinh tủy IPSS giai đoạn trung bình 2 / cao hoặc IPSS-R cao / rất cao.
- Tiêu chí lựa chọn: Có nguồn cho phù hợp HLA, tuổi < 70, không có bệnh lý nền nặng, thể trạng tốt có thể trải qua cuộc ghép.
- Cần đánh giá cụ thể các tổn thương di truyền cho bệnh nhân để lựa chọn thời điểm ghép phù hợp và có chiến lược duy trì sau ghép trên những nhóm có tổn thương di truyền tiên lượng xấu.
- Duy trì sau ghép: Rất quan trọng trong ghép rối loạn sinh tủy (có thể duy trì bằng HMA hoặc các nhóm thuốc mới).
3.3.2.3. Hướng mới trong điều trị rối loạn sinh tủy nguy cơ cao
a. Các tác nhân có thể kết hợp với HMA:
- Azacitidine kết hợp với lenalidomide... hoặc eltrombopag.
- Azacitidine kết hợp với chất ức chế HDAC.
- Azacitidine kết hợp với chất ức chế enzym kích hoạt NEDD8.
- Azacitidine kết hợp với đơn chất trong HRMDS và oligoblastic AML.
- Azacitidine kết hợp với chất ức chế IDH1 hoặc IDH2 .
- Azacitidine kết hợp với các thuốc nhắm đích khác (kháng CD47, kháng CD3/CD123, CTLA - 4,…).
b. Nhắm đích TP53:
- Điều chế hoạt động phiên mã của đột biến TP53 bằng các loại thuốc như APR-246.
c. Các lựa chọn ở bệnh nhân thất bại liệu pháp HMA
- CPX-351 là công thức liposom mới với tỷ lệ Cytarabine/Daunorubicin cố định 5/1.
- Chất ức chế IDH1, IDH2, FLT3.
- Liệu pháp CAR T-cell, DARTs.
3.4. Điều trị hỗ trợ khác
3.4.1. Điều trị thiếu máu
- Truyền khối hồng cầu: Khi huyết sắc tố < 70g/l, đến khi đạt > 100g/l. Nếu có kế hoạch ghép tế bào gốc tạo máu thì hạn chế truyền máu; chế phẩm máu được lọc bạch cầu, chiếu xạ và sàng lọc CMV trước truyền; lưu ý kháng thể bất thường để chọn máu phù hợp phenotype.
- Lưu ý thiếu các nguyên liệu tạo máu như sắt, vitamin B12, acid folic.
3.4.2. Phòng/điều trị xuất huyết
- Truyền khối tiểu cầu cùng nhóm khi số lượng tiểu cầu ≤ 20G/L hoặc khi số lượng tiểu cầu ≤ 50 G/L nhưng có xuất huyết.
- Tranexamic acid 0,25 g x 1-2 ống; tiêm bắp hoặc tĩnh mạch.
- Mazipredone 30mg x 1 ống, tiêm tĩnh mạch; dùng 5-7 ngày.
3.4.3. Chống nhiễm trùng
- Điều trị kháng sinh khi có nhiễm khuẩn, cần làm kháng sinh đồ để chỉ định kháng sinh cho phù hợp.
- Nếu số lượng bạch cầu hạt trung tính quá thấp (≤ 1G/L) thì có thể dùng G-CFS hoặc truyền khối bạch cầu trung tính cùng nhóm (cân nhắc kỹ).
- Giữ vệ sinh ăn uống, vệ sinh răng miệng, cơ thể…
3.4.4. Thải sắt:
- Chỉ định: Khi Ferritin huyết thanh ≥ 1000 ng/ml hoặc khi truyền > 20-30 đơn vị khối hồng cầu.
- Thuốc thải sắt:
+ Deferoxamin
Liều lượng:
● Trẻ em 20 - 40 mg/kg/ngày;
● Người lớn: 40 - 60 mg/kg/ngày.
Cách dùng: Tiêm dưới da hoặc truyền tĩnh mạch liên tục 8-12 giờ/ngày, 5-6 ngày/ tuần. Trường hợp quá tải sắt nặng thì dùng liên tục cả tuần.
+ Deferipron: Sử dụng thuốc này khi thuốc deferoxamin không hiệu quả.
Liều lượng: 75mg/kg/ngày;
Cách dùng: Uống, chia 3 lần/ngày.
+ Deferasirox: Nếu có thể thì nên lựa chọn điều trị ngay từ đầu.
Liều lượng: 20-30 mg/kg mỗi ngày;
Cách dùng: Uống trước ăn 30 phút.
- Thải sắt là quy trình bắt buộc cho những bệnh nhân có kế hoạch ghép tế bào gốc tạo máu đồng loài.
4. TIẾN TRIỂN VÀ TIÊN LƯỢNG
- Hội chứng rối loạn sinh tủy là bệnh khá nặng, dai dẳng và rất khó điều trị. Người bệnh có thể tử vong do các biến chứng như: Nhiễm trùng, chảy máu, ứ sắt gây suy chức năng các cơ quan hoặc do chuyển lơ xê mi cấp.
- Thời gian sống thêm trung bình của nhóm tiên lượng xấu là 8 tháng, trong khi đó nhóm tiên lượng tốt có thể trên 5 năm và thời gian chuyển lơ xê mi cấp cũng chậm hơn.
5. XÉT NGHIỆM ĐÁNH GIÁ TRƯỚC VÀ TRONG QUÁ TRÌNH ĐIỀU TRỊ
5.1. Xét nghiệm đánh giá trước điều trị
Ngoài các xét nghiệm để chẩn đoán bệnh đã nêu ở trên, cần làm các xét nghiệm để đánh giá toàn trạng bệnh nhân trước điều trị, bao gồm:
- Sinh hóa máu: Chức năng gan, thận, đường máu, điện giải đồ, protein, albumin, EPO, bộ xét nghiệm sắt (Fe huyết thanh, ferritin, transferin, độ bão hòa transferin).
- Đông máu cơ bản vòng 1: Fibrinogen, PT, APTT, TT, D-Dimer, nếu có bất thường làm các xét nghiệm chuyên sâu.
- Tổng phân tích nước tiểu, tế bào nước tiểu.
- Định nhóm hệ ABO, Rh với bệnh nhân vào viện lần đầu và khi bệnh nhân có chỉ định truyền máu.
- Sàng lọc kháng thể bất thường, coombs với bệnh nhân có truyền máu nhiều lần.
- Điện tâm đồ, siêu âm tim với bệnh nhân có chỉ định điều trị hóa chất.
- Siêu âm ổ bụng, X-quang tim phổi.
5.2. Xét nghiệm theo dõi trong điều trị
5.2.1 Xét nghiệm theo dõi thường quy
- Tổng phân tích tế bào máu 2-3 lần/ tuần.
- Sinh hóa máu: Chức năng gan, thận, điện giải đồ và bộ xét nghiệm bilan sắt 1 lần/ tuần.
- Đông máu cơ bản 1 lần/ tuần.
- Làm huyết tủy đồ, sinh thiết tủy xương sau mỗi chu kì điều trị hóa chất. Đối với bệnh nhân không điều trị hóa chất kiểm tra HTĐ, STTX mỗi 6 tháng hoặc khi có biểu hiện bệnh tiến triển.
- Tế bào tủy, nhuộm hồng cầu sắt, NST, tổn thương gen bệnh bằng các kỹ thuật sinh học phân tử, mô bệnh học tủy xương: 3 tháng/1 lần.
- Nghiệm pháp Coombs (trực tiếp, gián tiếp), sàng lọc và định danh kháng thể bất thường: Khi truyền máu nhiều lần.
- Định lượng erythropoietin, thrombopoietin.
2.2 Xét nghiệm theo dõi khi có bất thường: Tùy thuộc diễn biến của bệnh nhân để chỉ định phù hợp.
TÀI LIỆU THAM KHẢO
1. Nguyễn Anh Trí: Tiền Lơ xê mi và Lơ xê mi cấp, Nhà xuất bản Y học, 2010: p.59-140.
2. Nguyễn Thị Quỳnh Nga: Hội chứng rối loạn sinh tủy. Bài giảng sau đại học Huyết học - Truyền máu, Nhà xuất bản Y học, 2006: p.115-127.
3. Đỗ Trung Phấn: Hội chứng rối loạn sinh tủy. Tế bào gốc và bệnh lý tế bào gốc tạo máu chẩn đoán, phân loại, điều trị, Nhà xuất bản Y học, 2008: p.227-244.
4. Phạm Quang Vinh: Các hội chứng rối loạn sinh tủy và bất thường tế bào di truyền trong bệnh máu ác tính ; Nhà xuất bản Y học. 2013: p.119-136.
5. Vũ Đức Bình, Nguyễn Anh Trí, Bùi Thị Mai An, Nguyễn Bá Khanh: Cập nhật xếp loại và điều trị hội chứng rối loạn sinh tủy. Một số chuyên đề Huyết học Truyền máu tập IV, Nhà xuất bản Y học. 2012: p. 277-295.
6. Nguyễn Thị Quỳnh Nga: Tổng quan về các phương pháp điều trị hội chứng rối loạn sinh tủy. Một số chuyên đề Huyết học Truyền máu tập I, Nhà xuất bản Y học. 2004: p. 112-127.
7. Nguyễn Công Khanh: Hội chứng loạn sản tủy. Huyết học lâm sàng nhi khoa, Nhà xuất bản Y học. 2004: p.195-202.
8. Nguyễn Đạt Anh, Nguyễn Lân Việt, Phạm Quang Vinh, Nguyễn Quốc Anh: Các thang điểm thiết yếu sử dụng trong lâm sàng, Nhà xuất bản Y học 2011: p.83-88.
9. Jason Gotlib, Lenn Fechter: 100 questions and answers about Myelodysplastic Symdromes: p.77-99.
10. Pocket Medicine Fourth edition, Myelodysplastic symdromes: p.5-14.
11. Hoffbrand AV, Moss PAH(2016). Myelodysplasia. IN Hofbrand’ Essential Haematology. 7th Edition. John Wiley & Sons Ltd.,pp.177-185.
12. ArberD, Orazi A, Hasserjian R, et al. (2016) The 2016 revision to the World Heath Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20), pp.2391-2405.
13. NCCN guidelines (Myelodysplastic Syndrome) version 1.2021.
14. Uwe Platzbecker. Treatment of MDS. Blood (2019) 133 (10): 1096-1107.
15. Guillermo Garcia-Manero, Kelly S. Chien, Guillermo Montalban‐Bravo. Myelodysplastic syndromes: 2021 update on diagnosis, risk stratification and management. American journal of Hematology (2020), 95 (11), pp:1399-1420.
29. U LYMPHO HODGKIN
1. ĐẠI CƯƠNG
U lympho ác tính là nhóm bệnh ác tính của tổ chức lympho, bao gồm 2 nhóm: U lympho Hodgkin (chiếm khoảng 20-30%) và u lympho không Hodgkin (70-80%).
Nguyên nhân gây bệnh chưa rõ ràng. Tuy nhiên, nhiều nghiên cứu đưa ra một số yếu tố nguy cơ cao: Nhiễm EBV, suy giảm miễn dịch (sau ghép tạng, HIV…), bệnh tự miễn (viêm khớp dạng thấp, lupus ban đỏ hệ thống, sarcoidosis…) và yếu tố gia đình (nguy cơ mắc u lympho Hodgkin cao hơn 3 - 4 lần ở các cá thể có cùng quan hệ huyết thống với người bệnh này).
2. CHẨN ĐOÁN
2.1. Chẩn đoán xác định
a. Lâm sàng
- Biểu hiện lâm sàng không đặc hiệu và phù hợp với tình trạng nhiễm khuẩn hơn là bệnh lý ác tính.
- Hạch to chiếm khoảng 70% người bệnh; thường gặp tại vùng cổ, nách, bẹn, trung thất và hạch ổ bụng. Số lượng hạch nhiều, mật độ chắc, thường không đau.
- Khối u trung thất hay gặp thứ hai nhưng hầu hết người bệnh không có biểu hiện lâm sàng hoặc biểu hiện không đặc hiệu như: Đau sau xương ức, ho, thở nông, hiếm gặp hơn là tràn dịch màng phổi, màng tim, chèn ép tĩnh mạch chủ trên...
- Triệu chứng toàn thân khá thường gặp như: Ngứa, mệt mỏi, triệu chứng B (gồm có sốt, ra mồ hôi đêm, sút cân trên 10% trọng lượng cơ thể trong 6 tháng không giải thích được nguyên nhân).
- Gan hoặc lách có thể to nhưng ít khi to nhiều.
- Biểu hiện ban đầu ngoài hạch hiếm gặp. Nếu có thì vị trí hay gặp là: Phổi, gan, xương và tủy xương.
- Một vài hội chứng cận u hiếm gặp nhưng khá đặc hiệu như: Đau do rượu (đau xương hoặc ở vị trí hạch xảy ra sau vài phút sử dụng rượu dù với lượng nhỏ); tổn thương da (Mày đay, hồng ban, nốt ban đỏ, vết loét...); hội chứng thận hư do sự giải phóng các lymphokine như IL-3.
- Bệnh lan tràn đầu tiên là các hạch kế cận (theo đường bạch huyết đặc biệt là ống ngực), sau đó là các cơ quan khác như lách, tủy xương. Ở giai đoạn muộn của bệnh, thường xuất hiện các biểu hiện chèn ép, xâm lấn của tổ chức lympho, u trung thất như: Hội chứng trung thất, chèn ép tĩnh mạch chủ trên, tràn dịch màng phổi...; có thể xuất hiện thiếu máu, nhiễm khuẩn hoặc xuất huyết.
b. Cận lâm sàng
- Hạch đồ: Hạch tăng sinh, đa hình thái, ngoài dòng lympho còn gặp nhiều bạch cầu đoạn trung tính, bạch cầu đoạn ưa acid, tế bào plasmo, đại thực bào, tế bào xơ. Trong trường hợp điển hình có gặp tế bào Reed-Sternberg.
- Mô bệnh học, hóa mô miễn dịch (hoặc đếm tế bào dòng chảy) hạch hoặc khối u thấy đảo lộn cáu trúc và dấu ấn tế bào Sternberg là tiêu chuẩn vàng chẩn đoán bệnh.
- Xét nghiệm khác:
+ Tổng phân tích tế bào máu ngoại vi: Có thể có dấu hiệu thiếu máu. Một số người bệnh tăng bạch cầu trung tính, bạch cầu ưa acid; Số lượng tiểu cầu bình thường;
+ Tốc độ máu lắng và protein C phản ứng thường tăng;
+ LDH tăng trong khoảng 30% trường hợp, có thể tăng calci và giảm albumin. Beta 2 microglobulin thường tăng;
+ Các phương pháp chẩn đoán hình ảnh như siêu âm, X-quang, CT, MRI, PET, PET-CT giúp phát hiện hạch sâu, vị trí di căn khác và theo dõi sau điều trị;
+ Tủy đồ, sinh thiết tủy xương và nhuộm hóa mô miễn dịch giúp phát hiện u lympho xâm lấn tủy;
+ Xét nghiệm dấu ấn EBV trên mảnh sinh thiết hạch/tổ chức lympho bằng nhiều phương pháp: Hóa mô miễn dịch EBV màng (LMP1, LMP2), EBV kháng nguyên nhân (EBNA1; FISH RNA nhân của EBV (EBER);
+ Xét nghiệm di truyền có thể thấy đột biến gen như: TP53, PDL1, PDL2...
+ Một số đột biến khác có thế phát hiện bằng kỹ thuật di truyền sinh học phân tử như: TP53, PDL1, PDL2...;
+ Xét nghiệm virus EBV, CMV IgG, IgM và đo tải lượng EBV, CMV để xác định nguyên nhân gây bệnh (mẫu máu);
+ Xét nghiệm virus: HBV, HCV, HIV với bệnh nhân vào viện lần đầu, trước can thiệp thủ thuật, truyền máu nhiều lần và trước điều trị các trường hợp phải dùng thuốc ảnh hưởng đến hệ miễn dịch như kháng CD20,…;
+ Xét nghiệm: Anti-HBc và anti-HBs nếu trường hợp HBsAg âm tính để có kế hoạch dự phòng thuốc kháng virus trước khi điều trị thuốc ức chế miễn dịch và hóa chất tránh virus tái hoạt động.
2.2. Chẩn đoán thể bệnh: WHO (2016) bổ sung thêm thể bệnh
Bảng 27. Chẩn đoán thể bệnh theo Tổ chức y tế thế giới - WHO (năm 2016) dựa vào mô bệnh học
|
Thể bệnh |
Đặc điểm |
|
|
Cổ điển |
Giàu tế bào lympho |
Dạng nốt hoặc lan tỏa. Trên nền nhiều lympho bào (chủ yếu lympho B), rải rác tế bào Reed-Sternberg. |
|
Nghèo tế bào lympho |
Dạng sợi võng trông giống như sarcoma nhưng chứa nhiều tế bào R-S/ tế bào Hodgkin và nhiều dạng đa hình khác. Dạng xơ hóa lan tỏa chứa ít tế bào R-S/ tế bào Hodgkin nằm trên nền nghèo tế bào, bắt màu acid, dương tính với PAS. |
|
|
Hỗn hợp tế bào |
Nhiều tế bào Reed-Sternberg điển hình, tế bào dạng Hodgkin trên nền. Nền gồm nhiều lympho bào T, bạch cầu ưa acid, trung tính, tương bào và mô bào. Mô bào có thể tạo thành viêm hạt thưa. |
|
|
Xơ nốt |
Xơ phát triển chia cắt nhu mô hạch thành nhiều nốt, có nhiều tế bào Reed-Sternberg, tế bào dạng Hodgkin và biến thể dạng tế bào khuyết, có thể thấy hoại tử trung tâm. |
|
|
Mới |
Dạng nốt, ưu thế tế bào lympho |
Gặp chủ yếu biến thể dạng lympho-histocytic - có nhân chia thùy (tế bào L-H, tế bào bỏng ngô). Tế bào nền chủ yếu lymphocyte T nhỏ và mô bào. Không có bạch cầu trung tính hoặc ưa acid. Hiếm gặp tương bào. Có thể gặp xơ hóa. |
2.3. Chẩn đoán giai đoạn
Cho đến nay, chẩn đoán giai đoạn của u lympho Hodgkin vẫn dựa theo các tiêu chuẩn của Ann Arbor, năm 1971 (bảng 28).
Bảng 28. Chẩn đoán giai đoạn của u lympho Hodgkin
|
Giai đoạn |
Biểu hiện |
|
I |
Tổn thương một vùng hạch hoặc một vị trí ngoài hạch (IE). |
|
II |
Tổn thương hai vùng hạch trở lên trên cùng một phía cơ hoành. Có thể bao gồm cả lách (IIS), vị trí ngoài hạch (IIE) hoặc cả hai (IIES) nhưng vẫn nằm một phía cơ hoành. |
|
III |
Tổn thương nằm hai phía cơ hoành. Có thể tổn thương ở lách (IIIS), hoặc vị trí ngoài hạch (IIIE), hoặc cả hai (IIIES). |
|
IV |
Tổn thương lan tỏa rải rác nhiều tạng hoặc mô ngoài hạch (như: Tủy xương, gan, phổi…), có kèm hoặc không kèm tổn thương hạch. |
|
- B là khi có biểu hiện: Sốt, ra mồ hôi đêm, sút cân trên 10% trọng lượng cơ thể trong 6 tháng. - A là khi không có các triệu chứng trên. |
|
2.4. Chẩn đoán phân biệt
- U lympho không Hodgkin: Trên hạch đồ và sinh thiết hạch có hình ảnh đồng nhất về mặt hình thái, không tìm thấy tế bào Reed-Sternberg hoặc các biến thể.
- Hạch tăng sinh phản ứng: Hạch to, thường đau. Hạch diễn biến cấp nhưng lành tính và trở lại bình thường sau khi khỏi bệnh chính.
- Hạch lao: Thường gặp chuỗi hạch dọc cơ ức đòn chũm, không đau, diễn biến mềm dần, vỡ và chảy ra chất bã đậu. Xét nghiệm hạch thấy tổn thương đặc hiệu: tế bào bán liên, tế bào khổng lồ Langerhans, chất hoại tử bã đậu.
- Ung thư di căn hạch: Xét nghiệm tế bào và mô bệnh học hạch thấy tế bào ung thư: kích thước lớn, nhân to, mịn, thường có nhiều hạt nhân, nguyên sinh chất rộng, đôi khi có hốc chế tiết, thường đứng thành đám. Đa số trường hợp có thể phát hiện được cơ quan ung thư nguyên phát.
3. ĐIỀU TRỊ
3.1. Điều trị với trường hợp mới chẩn đoán
- Đa hóa trị liệu: Lựa chọn phác đồ hàng đầu dựa trên giai đoạn bệnh, có hoặc không có khối trung thất, số lượng vị trí tổn thường (hạch và ngoài hạch, máu lắng). Các phác đồ có thể là: ABVD, BEACOPP esc (escalated), Stanford V, MOPP, Brentuximab vedotin (theo NCCN - National Comprehensive Cancer Network, 1.2020).
- Trường hợp u lympho Hodgkin dạng nốt, ưu thế lymphocyte: Sử dụng phác đồ CHOP, ABVD, EPOCH, CVP kết hợp Rituximab.
- Xạ trị:
+ Kết hợp với hóa chất trong trường hợp giai đoạn I, II và đặc biệt khi có khối u;
+ Xạ trị đơn độc: Ít dùng. Không sử dụng với u lympho Hodgkin dạng nốt, ưu thế lymphocyte.
3.2. Với trường hợp không lui bệnh hoặc tái phát
- Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Deauville trên PET/ CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2020).
- Phải sinh thiết hạch làm lại chẩn đoán.
- Sử dụng phác đồ đa hóa trị hàng hai như:
+ Brentuximab vedotin;
+ Brentuximab vedotin + Bendamustin;
+ Bendamustin + carboplastin + etoposid;
+ C-MOPP (cyclophosphamid, vincristin, procarbazine, prednisone);
+ DHAP (dexamethasone, cisplatin, high-dose cytarabine);
+ ESHAP (etoposide, methylprednisonlone, high-dose cytarabine, cisplatin);
+ GCD (gemcitabine, carboplastin, dexamethasone);
+ GVD (gemcitabine, vinorelbine, liposomal doxorubicin);
+ GEMOX (gemcitabine, Oxaliplatin);
+ ICE (ifosfamid, carboplastin, etoposid);
+ IGEV (ifosfamid, gemcitabine, vinorelbine);
+ MINE (etoposide, ifosfamid, mesna, mitoxantrone);
+ Kết hợp với một số thuốc như: bendamustine, everolimus, lenalidomide, pembrolizumab.
Với u lympho Hodgkin dạng nốt, ưu thế lymphocyte tái phát, dùng Rituximab kết hợp với các phác đồ hàng 2.
- Cân nhắc điều trị bằng phương pháp ghép tế bào gốc tạo máu.
3.3. Phác đồ cụ thể của đa hóa trị liệu hàng đầu
- ABVD
|
Ngày |
Liều |
Đường dung |
Ngày |
|
Doxorubicin |
25mg/m2 |
Truyền TM |
1, 14 hoặc 15 |
|
Bleomycin |
10mg/m2 |
Truyền TM |
1, 14 hoặc 15 |
|
Vinblastin |
6mg/m2 |
Truyền TM |
1, 14 hoặc 15 |
|
Dacarbazine |
375mg/m2 |
Truyền TM |
1, 14 hoặc 15 |
- BEACOPP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cyclophosphamid |
650mg/ m2 |
Truyền TM |
1 |
|
Doxorubicin |
25mg/m2 |
Truyền TM |
1 |
|
Etoposide |
100mg/m2 |
Truyền TM |
1→3 |
|
Procarbazine |
100mg/m2 |
Uống |
1→7 |
|
Methylprednisolone |
40mg/m2 |
Uống/ TM |
1→14 |
|
Vincristine |
1,4mg/m2 |
Truyền TM |
8 |
|
Bleomycin |
10mg/m2 |
Truyền TM |
8 |
- Stanford V
|
Thuốc |
Liều |
Đường dùng |
Ngày 1 trong các tuần |
|
Doxorubicin |
25mg/m2 |
Truyền TM |
Tuần 1,3,5,7,9,11. |
|
Vinblastin |
6mg/m2 |
Truyền TM |
Tuần 1,3,5,7,9,11. |
|
Nitrogen mustard |
6mg/m2 |
Truyền TM |
Tuần 1,5,9 |
|
Vincristine |
1,4mg/m2 |
Truyền TM |
Tuần 2,4,6,8,10,12 |
|
Bleomycin |
5mg/m2 |
Truyền TM |
Tuần 2,4,6,8,10,12 |
|
Etoposide |
60mg/m2 |
Truyền TM |
Tuần 3, 7,11 |
|
Methylprednisolone |
40mg/m2 |
Uống/ TM |
Hàng ngày liên tục trong 12 tuần |
+ Vinblastin giảm xuống 4 mg/m2 trong mũi thứ 2, 3 và 1 mg/m2 trong tuần 10-12;
+ Methylprednisolone giảm liều dần trong 12 tuần.
- MOPP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Nitrogen mustard |
6mg/m2 |
Truyền TM |
1,8 |
|
Vincristine |
1.4mg/m2 |
Truyền TM |
1,8 |
|
Procarbazin |
100mg/m2 |
Uống |
1→14 |
|
Methylprednisolone |
40mg/m2 |
Uống/ TM |
1→14, chỉ ở chu kỳ 1 và 4. |
Chú ý:
- Với các phác đồ ABVD, BEACOPP, MOPP sử dụng 4-6 đợt. Trong đó: 3 đợt đầu có thể dùng cách nhau từ 14-21 ngày; 3 đợt tiếp theo cách nhau 21-28 ngày; Thời gian cách bao nhiêu phụ thuộc vào lâm sàng (hạch, toàn trạng…) và xét nghiệm về chức năng gan, thận, tim mạch và đặc biệt là tủy sinh máu.
- Phác đồ Stanford V sử dụng 3 đợt vào ngày đầu tiên của mỗi tuần.
- Trì hoãn điều trị khi máu ngoại vi có số lượng bạch cầu đoạn trung tính < 1 G/L hoặc số lượng tiểu cầu < 100G/L.
- Các phác đồ CHOP, EPOCH, CVP kết hợp Rituximab (tham khảo bài u lympho không Hodgkin).
3.4. Phác đồ cụ thể của đa hóa trị liệu hàng hai
- GCD
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Gemcitabine |
1000 mg/m2 |
Truyền TM |
1,8 |
|
Carboplatin |
AUC 5 |
Truyền TM |
1 |
|
Dexamethasone |
40 mg/ngày |
uống |
1→4 |
Chu kỳ 21 ngày, sử dụng 4 chu kỳ
- GVD
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Gemcitabine |
1000 mg/m2 |
Truyền TM |
1,8 |
|
Vinorelbine |
20 mg/m2 |
Truyền TM |
1,8 |
|
Doxorubicin liposomal |
15 mg/m2 |
Truyền TM |
1,8 |
Chu kỳ 21 ngày, 2-6 chu kỳ.
- IGEV
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Gemcitabine |
800 mg/m2 |
Truyền TM |
1,4 |
|
Ifosfamide |
2000 mg/m2 |
Truyền TM trong 2h |
1→4 |
|
Vinorelbine |
25 mg/m2 |
Truyền TM |
1 |
|
Prednisolone |
100 mg/ngày |
Uống |
1→4 |
Thuốc hỗ trợ:
+ 2 lít NaCl 0,9%/ ngày, ngày 1-4;
+ Mesna (Mesnex) 2.600 mg/m2, truyền TM, ngày 1-4;
+ Filgrastim (Neupogen): Có thể 5 mcg/kg, TDD 1 lần/ngày, ngày 7-12.
Chu kỳ 21 ngày, sử dụng 4 chu kỳ.
- Brentuximab Vedotin
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Brentuximab |
1,8 mg/kg |
Truyền TM trong 30 phút |
1 |
Chu kỳ 21 ngày, tiếp tục điều trị cho đến tối đa 16 chu kỳ, bệnh tiến triển hoặc không chịu được độc tố của thuốc.
- Pembrolizumab
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Pembrolizumab |
200mg/ngày |
Truyền TM |
1 |
Chu kỳ 21 ngày, cho đến 24 tháng hoặc bệnh tiến triển, không chịu được độc tố của thuốc.
- Bendamustine
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Bendamustin |
120mg/m2 |
Truyền TM |
1,2 |
Chu kỳ 28 ngày, sử dụng 6 chu kỳ
- Lenalidomide
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Lenalidomide |
25mg/ngày |
Uống |
1→ 21 |
Chu kỳ 28 ngày
- Everolimus
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Everolimus |
10mg/ngày |
Uống |
Hàng ngày |
3.5. Trường hợp người già (≥ 60 tuổi)
Phác đồ điều trị cụ thể phụ thuộc vào từng bệnh nhân, đặc biệt là các biểu hiện toàn thân và bệnh lý kèm theo.
- Các phác đồ đa hóa trị có thể sử dụng như: ABVD/ AVD, CHOP, VEPEMB (vinblastine, cyclophosphamide, prednisolone, procarbazine, etoposide, mitoxantrone, bleomycine); PVAG (prednisolone,vinblastine, doxorubincin, gemcitabine).
- Có thể kết hợp với xạ trị.
- Trường hợp không lui bệnh hoặc tái phát, cân nhắc phác đồ hàng hai tùy theo cá thể. Có thể cân nhắc điều trị giảm nhẹ với các thuốc như: bendamustine, brentuximab.
Phác đồ cụ thể:
- PVAG
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Prednisolone |
40 mg/ngày |
Uống/ TM |
1-5 |
|
Vinblastin |
6 mg/m2 |
Truyền TM |
1 |
|
Doxorubicin |
50 mg/m2 |
Truyền TM |
1 |
|
Gemcitabin |
1.000 mg/ngày |
Truyền TM |
1 |
4. XÉT NGHIỆM ĐÁNH GIÁ TRƯỚC VÀ TRONG QUÁ TRÌNH ĐIỀU TRỊ
4.1. Đánh giá trước điều trị
Ngoài các xét nghiệm để chẩn đoán bệnh đã nêu ở trên, cần làm các xét nghiệm để đánh giá toàn trạng bệnh nhân trước điều trị, bao gồm:
- Sinh hóa máu: Chức năng gan, thận, đường máu, điện giải đồ, protein, albumin, fe huyết thanh, ferritin.
- Đông máu cơ bản vòng 1: Fibrinogen, PT, APTT, TT, D-Dimer. Nếu có bất thường làm các xét nghiệm chuyên sâu.
- Tổng phân tích nước tiểu, tế bào nước tiểu.
- Định nhóm hệ ABO, Rh bệnh nhân vào viện lần đầu và khi bệnh nhân có chỉ định truyền máu.
- HBsAg, HCV Ab, HIV Ab, HBc Ab với bệnh nhân vào viện lần đầu và khi bệnh nhân có truyền máu nhiều lần.
- EBV, CMV (IgG, IgM). Đo tải lượng EBV, CMV
- Điện tâm đồ, siêu âm tim với bệnh nhân có chỉ định điều trị hóa chất.
4.2. Theo dõi trong điều trị
- Tổng phân tích tế bào máu 2 lần/ tuần.
- Sinh hóa máu: Chức năng gan, thận, điện giải đồ 1 lần/ tuần.
- Đông máu cơ bản 1 lần/ tuần.
- Làm huyết tủy đồ, sinh thiết tủy xương sau mỗi 3 chu kì điều trị hóa chất nếu có xâm lấn tuỷ xương hoặc u lympho thể tuỷ.
- Đánh giá đáp ứng sau 2-3 đợt điều trị bằng chẩn đoán hình ảnh: CT-scan, MRI, PET-CT.
4.3. Theo dõi sau điều trị
- Các trường hợp đặc biệt như: Hạch to, sốt, gầy sút cân… phải tái khám ngay.
- Tái khám: 3 tháng/ lần trong 2 năm đầu. Sau đó, 6 tháng/ lần trong 3 năm tiếp và theo dõi hàng năm.
- Với mỗi lần tái khám:
+ Khám lâm sàng: Chú ý các triệu chứng lâm sàng, hạch to, hội chứng B;
+ Xét nghiệm: Tổng phân tích tế bào máu ngoại vi, sinh hóa máu (bao gồm LDH, chức năng gan, thận), máu lắng; Chức năng tuyến giáp nếu có xạ trị vùng trước đó. CT bụng ngực, MRI hoặc PET, PET/CT mỗi 6 tháng trong 2 năm đầu hoặc khi nghi ngờ bệnh tái phát/tiến triển. Làm lại sinh thiết khi xuất hiện hạch to trở lại hoặc có tổn thương mới.
TÀI LIỆU THAM KHẢO
1. Đỗ Trung Phấn (2008), “U lympho ác tính”, Tế bào gốc và bệnh lý tế bào gốc tạo máu, Nhà xuất bản y học, Tr 358-374.
2. Nguyễn Anh Trí (2004), “Điều trị bệnh Hodgkin”, Điều trị các bệnh ác tính cơ quan tao máu, Nhà xuất bản y học. Tr 15-21.
3. Hodgkin lymphoma - NCCN guidelines version 1.2020.
4. WHO classification of tumours of haematopoietic and lymphoid tissues 2008.
5. “Neoplasic lymphoid diseases”, William hematology 8th - 2010, chapter 99.
6. Jonathan Sive and David Linch (2011), “Hodgkin lymphoma”, Postgraduate hematology, 6th ed, chapter 34.
7. Richard S.Stein, David S. Morgan (2009), “Hodgkin Lymphoma”, Wintrobes clinical hematology 12th editition, 2313-2343.
8. Volker Diehl, Beate Klimm (2008), “Hodgkin lymphoma: Clinical manifestations, staging and therapy”, Hoffman: Hematology: Basic Principles and Practice, 5th ed, chapter 77.
30. U LYMPHO KHÔNG HODGKIN
1. ĐẠI CƯƠNG
U lympho không Hodgkin chiếm khoảng 70-80% bệnh u lympho-là nhóm bệnh ác tính của mô lympho, có thể biểu hiện tại hạch hoặc ngoài hạch.
Nguyên nhân sinh bệnh chưa được chứng minh một cách rõ ràng. Cho đến nay, người ta chỉ đưa ra các giả thuyết: yếu tố nhiễm khuẩn: HIV, EBV, HTLV-1, HHV8…; yếu tố miễn dịch: Suy giảm miễn dịch bẩm sinh hay mắc phải HIV/AIDS, sau ghép tạng…; bệnh lý tự miễn; môi trường: Thuốc trừ sâu, dioxin, phóng xạ…
2. CHẨN ĐOÁN
2.1. Chẩn đoán xác định
a. Lâm sàng
- 60-100% người bệnh có hạch to, số lượng nhiều và không đau; thường gặp hạch ở vùng cổ, hố thượng đòn, nách, bẹn, có thể gặp hạch trung thất, hạch ổ bụng.
- Tổn thương ngoài hạch tiên phát nghĩa là u xuất hiện đầu tiên, thậm chí là duy nhất ở ngoài các hạch lympho, chiếm khoảng 40% như: Dạ dày, amydal, hốc mắt, da...
- Lách thường to độ I/II. Tuy nhiên, trong u lympho thể lách hoặc giai đoạn muộn của bệnh, lách có thể to độ III/IV.
- Gan to ít gặp hơn và thường kèm theo hạch to và/ hoặc lách to.
- Khoảng < 25% trường hợp có triệu chứng “B” gồm: Sốt, ra mồ hôi đêm, sút cân trên 10% trọng lượng cơ thể trong 6 tháng không giải thích được nguyên nhân.
- Có thể thiếu máu do xâm lấn tủy xương, tan máu tự miễn, cường lách hoặc hiếm hơn là do hiện tượng thực bào tế bào máu.
- Giai đoạn muộn, thường có biểu hiện chèn ép, xâm lấn của mô lympho như: Hội chứng trung thất; liệt do chèn ép tủy sống; lồi mắt; tắc ruột nếu u ống tiêu hóa…
b. Cận lâm sàng
- Tế bào hạch: tăng sinh, khá đồng nhất, chủ yếu là lymphoblast hoặc prolymphocyte: Kích thước to, nhân lớn (có thể có hạt nhân, nhân chẻ, nhân chia, nhân quái), nguyên sinh chất hẹp, không tạo đám; ít gặp bạch cầu đoạn trung tính, bạch cầu đoạn ưa acid, tế bào plasmo, đại thực bào, tế bào xơ.
- Mô bệnh học hạch hoặc khối u, kết hợp xét nghiệm miễn dịch (hóa mô miễn dịch hoặc đếm tế bào dòng chảy) và di truyền học là tiêu chuẩn vàng giúp chẩn đoán bệnh.
- Xét nghiệm và thăm dò khác:
+ Tế bào máu ngoại vi (huyết đồ) có thể gặp giảm lượng huyết sắc tố, giảm số lượng tiểu cầu, số lượng bạch cầu có thể tăng hoặc giảm;
+ LDH tăng trong khoảng 30% trường hợp; tăng calci máu. Chức năng gan thận có thể có biểu hiện rối loạn. β2-microglobulin thường tăng. Định lượng các globulin miễn dịch có thể thay đổi (tùy từng trường hợp);
+ Các phương pháp hình ảnh: Siêu âm, X-quang, CT, MRI..., đặc biệt là PET, PET- CT giúp phát hiện các hạch sâu ở trung thất, ổ bụng... các vị trí di căn khác và theo dõi sau điều trị;
+ Tủy đồ, sinh thiết tủy xương (có thể làm tại 2 vị trí) và nhuộm hóa mô miễn dịch có thể phát hiện u lympho xâm lấn tủy;
+ Xét nghiệm vi sinh có thể phát hiện Helicobacter Pylori trong ULKH tại dạ dày;
+ Xét nghiệm di truyền-sinh học phân tử: Tùy từng loại u lympho không Hodgkin;
+ Xét nghiệm điện di miễn dịch protein huyết thanh tùy loại u lympho Hodgkin;
+ Xét nghiệm vi khuẩn Helicobacter Pylori trong ULKH tại dạ dày;
+ Xét nghiệm virus EBV, CMV IgG, IgM và đo tải lượng EBV, CMV để xác định nguyên nhân gây bệnh;
+ Xét nghiệm virus: HBV, HCV, HIV với bệnh nhân vào viện lần đầu, trước can thiệp thủ thuật, truyền máu nhiều lần và trước điều trị các trường hợp phải dùng thuốc ảnh hưởng đến hệ miễn dịch như kháng CD20,…;
+ Xét nghiệm: Anti-HBc và anti-HBs nếu trường hợp HBsAg âm tính để có kế hoạch dự phòng thuốc kháng virus trước khi điều trị thuốc ức chế miễn dịch và hóa chất tránh virus tái hoạt động.
2.2. Chẩn đoán thể bệnh
a. Xếp loại u lympho không Hodgkin theo Tổ chức y tế thế giới (WHO 2016)
Dựa trên ứng dụng miễn dịch và di truyền học, Tổ chức y tế thế giới (WHO) đã đưa ra hệ thống xếp loại u lympho không Hodgkin mới năm 2016 (Xem bảng 29).
Bảng 29. Xếp loại u lympho không Hodgkin theo WHO (2016)
|
Tế bào B |
Tế bào T |
|
U lympho/Lơ xê mi tiền B, không phân loại. U lympho/Lơ xê mi tiền B với bất thường di truyền đặc thù |
U lympho/Lơ xê mi lymphoblast tiền T U lympho/Lơ xê mi lymphoblast NK |
|
Tế bào B trưởng thành Lơ xê mi lympho kinh / u lympho tế bào nhỏ U lympho của tổ chức lympho niêm mạc (MALT) U lympho vùng rìa tại hạch U lympho vùng rìa tại lách Lơ xê mi/u lympho tại lách, không đặc hiệu. U lympho tế bào áo nang U lympho thể nang U lympho thể nang type trẻ em U lympho dạng lymphoplasmatic U lympho trung tâm nang ở da tiên phát. U lympho tế bào B lớn lan tỏa (DLBCL), không đặc hiệu U lympho tế bào B lớn với tái sắp xếp IRF4 U lympho tế bào B lớn giàu tế bào T/mô bào DLBCL tiên phát ở thần kinh trung ương. DLBCL tiên phát ở da, thể chân. DLBCL EBV dương tính, không đặc hiệu DLBCL liên quan đến viêm mãn tính. U lympho tế bào B lớn ở trung thất tiên phát. U lympho tế bào B lớn nội mạch U lympho tế bào B lớn ALK+ U lympho lan tỏa tiên phát U lympho nguyên tương bào Rối loạn tăng sinh lympho liên quan HHV8 U lympho thể Burkitt U lympho thể Burkitt với đột biến 11q U lympho B tiến triển U lympho tế bào B không phân loại, với đặc trưng trung gian DLBCL và bệnh Hodgkin cổ điển |
Tế bào T/NK trưởng thành U lympho tế bào T, EBV+ hệ thống ở trẻ em Lơ xê mi/ U lympho tế bào T người lớn U lympho tế bào T/NK ngoài hạch, thể mũi U lympho tế bào T liên quan đến bệnh lý đường ruột U lympho tế bào T biểu mô đơn hình thái ở ruột U lympho tế bào T ở ruột không phân loại U lympho tế bào T thể gan lách. U lympho tế bào T dạng panniculitis dưới da Mycois fungoides Sérazy syndrome U lympho tế bào T gamma/delta ở da tiên phát U lympho tế bào T ngoại vi, không đặc hiệu U lympho lympho T nguyên bào miễn dịch mạch U lympho tế bào lớn kém biệt hóa, ALK- U lympho tế bào lớn kém biệt hóa, ALK+ U lympho tế bào T dạng nang U lympho tế bào T ngoại vi tại hạch với dạng tế bào T hỗ trợ nang U lympho tế bào lớn kém biệt hóa ALK- liên quan cấy ghép ngực |
2.3. Chẩn đoán giai đoạn
- Cho đến nay, chẩn đoán giai đoạn chủ yếu vẫn dựa theo các tiêu chuẩn của Ann Arbor (năm 1971).
|
Giai đoạn |
Biểu hiện |
|
I |
Tổn thương một vùng hạch hoặc một vị trí ngoài hạch (IE). |
|
II |
Tổn thương hai vùng hạch trở lên trên cùng một phía cơ hoành. Có thể bao gồm cả lách (IIS), vị trí ngoài hạch (IIE) hoặc cả hai (IIES) nhưng vẫn nằm một phía cơ hoành. |
|
III |
Tổn thương nằm hai phía cơ hoành. Có thể tổn thương ở lách (IIIS), hoặc vị trí ngoài hạch (IIIE), hoặc cả hai (IIIES). |
|
IV |
Tổn thương lan tỏa rải rác nhiều tạng hoặc mô ngoài hạch (như: Tủy xương, gan, phổi…), có kèm hoặc không kèm tổn thương hạch. |
|
- B là khi có biểu hiện triệu chứng “B”: Sốt, ra mồ hôi đêm, sút cân trên 10% trọng lượng cơ thể trong 6 tháng. - A là khi không có các triệu chứng trên. |
|
- Một số thể có chẩn đoán giai đoạn riêng như:
+ ULKH ở đường tiêu hóa theo xếp loại của Lugano;
+ ULKH tế bào lớn kém biệt hóa liên quan cấy ghép ngực theo xếp loại TNM (NCCN 1.2020);
+ Mycois fungoides, Sérazy syndrome theo xếp loại TNMB (NCCN 1.2020).
2.4. Chẩn đoán phân biệt
- U lympho Hodgkin: Trên hạch đồ và sinh thiết hạch tìm thấy tế bào Reed- Sternberg hoặc các biến thể.
- Hạch tăng sinh phản ứng: Hạch to, thường đau, ở gần nơi tổn thương. Hạch to diễn biến cấp nhưng lành tính và trở lại bình thường sau khi khỏi bệnh chính.
- Hạch lao: Thường gặp hạch dọc cơ ức đòn chũm, thành chuỗi, không đau, nếu kéo dài thường vỡ và chất bã đậu chảy ra ngoài. Hạch đồ và sinh thiết hạch thường thấy tổn thương gồm: Tế bào bán liên, tế bào khổng lồ Langerhans, chất hoại tử bã đậu.
- Hạch ung thư di căn: Trên hạch đồ và sinh thiết hạch thường thấy các tế bào ung thư: kích thước lớn, nhân to, mịn, thường có nhiều hạt nhân, nguyên sinh chất rộng, đôi khi có hốc chế tiết, thường đứng thành đám. Đa số trường hợp phát hiện được cơ quan ung thư nguyên phát.
3. ĐIỀU TRỊ.
3.1. Điều trị với từng thể bệnh
a. U lympho thể nang
Lựa chọn điều trị tùy thuộc vào tuổi, giai đoạn bệnh, tiên lượng bệnh và lựa chọn điều trị tiếp theo như có hoặc không có ghép tủy.
Trường hợp mới chẩn đoán:
+ Theo dõi sát bệnh nhân: khi không có chỉ định hóa trị;
+ Xạ trị: Giai đoạn I, II không có khối u hoặc kết hợp hóa trị ở giai đoạn I,II kèm có khối u;
+ Hóa trị: Chỉ định điều trị khi có 1 trong các tiêu chuẩn GELF-1998:
● Có hạch hoặc tổn thương ngoài hạch có kích thước > 7cm;
● Có ít nhất 3 hạch với mỗi hạch có kích thước > 3cm;
● Có hội chứng B;
● Lách to;
● Tràn dịch lan tỏa hoặc tràn dịch màng bụng;
● Giảm tế bào máu (BC < 1 G/L, và/hoặc TC <100 G/L).
Các phác đồ hóa trị:
- Lựa chọn hàng đầu:
+ Bendamustine + Rituximab;
+ Bendamustine + Obinutuzumab;
+ R-CHOP (Rituximab, cyclophosphamide, doxorubicine, vincristine, prednisone);
+ CHOP + Obinutuzumab;
+ R-CVP (Rituximab, cyclophosphamide, vincristine, prednisone);
+ CVP + Obinutuzumab;
+ Rituximab 375mg/m2 hàng tuần, trong 4 tuần;
+ Lenalidomide + Rituximab.
- Lựa chọn hàng đầu cho người già (> 60 tuổi) hoặc thể trạng yếu:
+ Rituximab 375mg/m2 hàng tuần, trong 4 tuần;
+ Tác nhân alkyl hóa như: Chlorambucil, cyclophosphamide + Rituximab;
+ Phương pháp miễn dịch phóng xạ.
Trường hợp không lui bệnh, tái phát:
- Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Lugano trên PET/CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2020).
- Phải sinh thiết hạch làm lại chẩn đoán.
- Sử dụng phác đồ đa hóa trị hàng hai như:
+ Lenalidomide + Rituximab;
+ Bendamustine + Obinutuzumab; hoặc Rituximab;
+ Phương pháp miễn dịch phóng xạ;
+ Copanlisib;
+ RFND (Rituximab, Fludarabine, Mitoxantrone, dexamethasone).
Ghi chú:
+ Cân nhắc ghép tế bào gốc tạo máu;
+ Trường hợp chuyển dạng u lympho khác thì sử dụng phác đồ theo thể bệnh;
+ Điều trị bổ trợ và chăm sóc.
Điều trị duy trì:
- Rituximab 375mg/m2, 8 tuần/lần, tổng 12 lần.
- Obinutuzumab 1.000mg, 8 tuần/lần, tổng 12 lần.
- Nếu điều trị khởi đầu bằng Rituximab đơn thuần thì duy trì Rituximab 375mg/m2, 8 tuần/lần, tổng 12 lần.
- Miễn dịch phóng xạ.
b. U lympho vùng rìa
Lựa chọn điều trị tùy thuộc vào tuổi, giai đoạn bệnh, tiên lượng bệnh và lựa chọn điều trị tiếp theo như có hoặc không có ghép tế bào gốc tạo máu.
Trường hợp mới chẩn đoán
- Theo dõi sát bệnh nhân: Khi không có chỉ định hóa trị.
- Hóa trị: Chỉ định điều trị khi có 1 trong các dấu hiệu, triệu chứng đe dọa chức năng các cơ quan, giảm các dòng tế bào máu ngoại vi, có khối u, bệnh tiến triển.
+ Lựa chọn hàng đầu:
● Bendamustine + Rituximab;
● R-CHOP (Rituximab, cyclophosphamide, doxorubicine, vincristine, prednisone);
● R-CVP (Rituximab, cyclophosphamide, vincristine, prednisone);
● Rituximab 375mg/m2 hàng tuần, trong 4 tuần.
+ Lựa chọn hàng đầu cho người già (> 60 tuổi) hoặc thể trạng yếu:
● Rituximab 375mg/m2 hàng tuần, trong 4 tuần;
● Tác nhân alkyl hóa như: chlorambucil, cyclophosphamide + Rituximab.
- Xạ trị: Đơn thuần hoặc kết hợp hóa trị.
- Một số phương pháp điều trị khác:
+ Cắt dạ dày khi có chảy máu cấp tính;
+ Phẫu thuật cắt khi ULKH thể MALT khu trú ở cơ quan xác định như: phổi, tuyến giáp, tuyến vú...;
+ Điều trị kháng sinh diệt Helicobacter Pylori nếu dương tính;
+ Cắt lách với ULKH vùng rìa tại lách;
+ Điều trị viêm gan C nếu có chỉ định trước khi điều trị hóa chất.
- Trường hợp không lui bệnh, tái phát:
+ Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Lugano trên PET/CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2020);
+ Phải sinh thiết hạch làm lại chẩn đoán;
+ Sử dụng phác đồ đa hóa trị hàng hai như:
● Lenalidomide + Rituximab;
● Bendamustine + Obinutuzumab;
● RFND (Rituximab, Fludarabine, Mitoxantrone, dexamethasone);
● Ibrutinib;
● Fludarabine + Rituximab.
+ Cân nhắc ghép tế bào gốc tạo máu;
+ Trường hợp chuyển dạng thì sử dụng phác đồ theo thể bệnh;
+ Điều trị bổ trợ và chăm sóc.
- Điều trị duy trì:
+ Rituximab 375mg/m2, 8 tuần/lần, tổng 12 lần;
+ Obinutuzumab 1000mg, 8 tuần/lần, tổng 12 lần;
+ Nếu điều trị khởi đầu bằng Rituximab đơn thuần thì duy trì liều 375mg/m2, 8 tuần/lần x 12 lần.
c. U lympho tế bào áo nang
Lựa chọn điều trị tùy thuộc vào giai đoạn bệnh, tuổi và lựa chọn điều trị tiếp theo có hoặc không có ghép tế bào gốc tạo máu.
Trường hợp mới chẩn đoán:
- Hóa trị: Điều trị cảm ứng.
+ Phác đồ tấn công:
● CALGB: Đợt 1, 2, 2.5: Rituximab + Methotrexate + CHOP (điều trị đợt 2.5 kết quả sinh thiết tủy xương trước đợt 3 có xâm lấn > 15% tế bào u lympho); Đợt 3: Etoposide, Cytarabine, Rituximab; Đợt 4: duy trì Rituximab;
● Hyper CVAD + Rituximab;
● NORDIC;
● R-DHAP: Rituximab, Cytarabine, cisplastin, dexamethasone;
● R-CHOP/R-DHAP luân phiên;
● R-CHOP/RICE (Rituximab, ifosfamide, carboplastin, etoposide) liên tiếp.
+ Phác đồ ít tấn công hơn:
● Bendamustin + Rituximab;
● VR-CAP (Bortezomib, Rtuximab, Cyclophosphmid, Doxorubicin, Prednisone);
● R-CHOP;
● Lenalidomide + Rituximab;
● Rituximab + Ibrutinib.
Trường hợp không lui bệnh hoặc tái phát:
- Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Lugano trên PET/CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2020).
- Phải sinh thiết hạch làm lại chẩn đoán.
- Hóa trị: Sử dụng phác đồ đa hóa trị hàng hai như:
+ Acalabrutinib;
+ Bendamustine + Rituximab;
+ Bendamustine + Rituximab + Bortezomib;
+ Rituximab + Bortezomib;
+ FC (Fludarabine, Cyclophosphamid) + Rituximab;
+ Ibrutinib;
+ Ibrutinib + Lenalidomide + Rituximab;
+ Lenalidomide + Rituximab;
+ PEPC (Prednisone, Etoposide, Procarbazine, Cyclophosphamide) + Rituximab;
- Ghép tế bào gốc tạo máu.
- Trường hợp chuyển dạng thì sử dụng phác đồ theo thể bệnh.
- Điều trị bổ trợ và chăm sóc.
Điều trị duy trì: Rituximab 375mg/m2, 8 tuần/lần x 12 lần.
d. U lympho tế bào B lớn
Lựa chọn phác đồ điều trị tùy thuộc vào thể bệnh, giai đoạn bệnh, tuổi, tiên lượng bệnh theo IPI và lựa chọn điều trị tiếp theo như ghép tế bào gốc tạo máu.
Trường hợp mới chẩn đoán
- Hóa trị:
+ Phác đồ đa hóa trị liệu hàng đầu như:
● R-CHOP (chu kỳ 14 hoặc 21);
● R-EPOCH (Rituximab, Etoposide, Prednisone, Vincristine, Cyclophosphamide, Doxorubicine).
+ Phác đồ đa hóa trị với trường hợp có suy tim, bệnh lý tim mạch:
● R-CEPP (Rituximab, Etoposide, Prednisone, Procarbazine, Cyclophosphamide);
● R-CDOP (Rituximab, Prednisone, Vincristine, Cyclophosphamide, Liposomal Doxorubicine);
● DA-EPOCH-R (Rituximab, Etoposide, Prednisone, Vincristine, Cyclophosphamide, Doxorubicine);
● R-CROP (Rituximab, Etoposide, Prednisone, Vincristine, Cyclophosphamide);
● R-GCVP (Rituximab, Gemcitabine, Prednisone, Vincristine,Cyclophosphamide).
+ Phác đồ đa hóa trị với trường hợp ULKH tế bào B tiến triển (Double hit, tripble hit):
● DA-EPOCH-R;
● R-Hyper-CVAD;
● R-CODOX-M/R-IVAC (Rituximab, Cyclophosphamide, Vincristine, Doxorubicine, methotrexat/ Ifosfamide, Etoposide, cytarabine).
+ Phác đồ đa hóa trị cho bệnh nhân > 80 tuổi.
● R-mini-CHOP;
● R-GCVP;
● Lenalidimide.
- Xạ trị có thể sử dụng kết hợp hóa trị với giai đoạn I, II hoặc xạ trị vùng khi tổn thương khu trú.
Trường hợp không lui bệnh hoặc tái phát:
- Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Lugano trên PET/CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2020).
- Phải sinh thiết hạch làm lại chẩn đoán.
- Hóa trị:
+ Sử dụng phác đồ đa hóa trị hàng hai với trường hợp có chỉ định ghép tế bào gốc tạo máu:
● DHAP (Dexamethasone, Cisplastin, Cytarabine) + Rituximab;
● ESHAP (Etoposide, Methylprednisone, Cytarabine, Cisplastin) + Rituximab;
● GDP (Gemcitabine, Dexamethasone, Cisplastin/Carboplastin) + Rituximab;
● GemOx (Gemcitabine, Oxaliplatin) + Rituximab;
● ICE (Ifosfamide, Carboplastin, Etoposide) + Rituximab;
● MINE (Mesna, Ifosfamide, Mitoxantrone, Etoposide) + Rituximab.
+ Sử dụng phác đồ đa hóa trị hàng hai với trường hợp không có chỉ định ghép tế bào gốc tạo máu:
● Bendamustine + Rituximab;
● Brentuximab với trường hợp có CD30+;
● CEPP (Cyclophosphamide, Etoposide, Vincristine, Procarbazine)+ Rituximab;
● CEOP (Cyclophosphamide, Etoposide, Vincristine, Prednisone)+ Rituximab;
● DA-EPOCH + Rituximab;
● GDP (Gemcitabine, Dexamethasone, Carboplatin) + Rituximab;
● GVD (Gemcitabine, vinorelbine + Dexamethasone) + Rituximab;
● Lenalidomide + Rituximab.
- Ghép tế bào gốc tạo máu nếu có chỉ định.
- Sử dụng tế bào lympho T chứa thụ thể kháng nguyên dạng dạng khảm (CAR - T cell therapy): Axicabtagene ciloleucel.
- Điều trị bổ trợ và chăm sóc.
đ. Burkitt lymphoma (u lympho Burkitt)
Lựa chọn điều trị tùy thuộc vào thể bệnh, giai đoạn bệnh, tuổi, tiên lượng bệnh theo IPI và lựa chọn điều trị tiếp theo như có hoặc không có ghép tủy.
Trường hợp mới chẩn đoán
- Hóa trị: Phác đồ đa hóa trị liệu hàng đầu.
+ CALGB 10002;
+ CODOX-M (Cyclophosphamide, Doxorubicin, Vincristine) tiếp sau đó Methotrexate liều cao) + Rituximab. Với nhóm nguy cơ cao, CODOX-M kế tiếp IVAC (Ifosfamide, Cytarabine, Doxorubicin);
+ EPOCH + Rituximab;
+ Hyper-CVAD, kế tiếp bằng Methotrexat liều cao và Cytarabine + Rituximab.
Trường hợp không lui bệnh hoặc tái phát
- Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Lugano trên PET/CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2020).
- Phải sinh thiết hạch làm lại chẩn đoán.
- Phác đồ đa hóa trị liệu hàng hai:
+ EPOCH + Rituximab;
+ R-ICE (Rituximab, Ifosfamide, Carboplastin, Etoposide);
+ R-IVAC (Rituximab, Ifosfamide, Cytarabin, Etoposide);
+ R-GDP (Rituximab, Gemcitabine, Dexamethasone, Cisplastin);
+ Cytarabine liều cao + Rituximab.
- Ghép tế bào gốc tạo máu.
- Điều trị bổ trợ và chăm sóc.
e. U lympho nguyên tương bào (lymphoplasmacytic lymphoma)
Điều trị ban đầu
- Nhóm thuốc không độc với TBG gồm:
+ Bortezomib ± Rituximab;
+ Bortezomib/Dexamethasone;
+ Bortezomib/Dexamethasone/Rituximab;
+ R - CHOP;
+ Ibrutinib;
+ Rituximab;
+ Rituximab/ cyclophosphamide/ prednisone (Dexamethasone);
+ Thalidomide ± Rituximab.
- Nhóm thuốc độc với TBG và có thể làm chuyển dạng:
+ Bendamustin ± Rituximab;
+ Chlorambucin;
+ Fludarabin ± Rituximab;
+ Fludarabin/ Cyclophosphamide/ Rituximab.
- Ghép tế bào gốc tạo máu: Được chỉ định trong những trường hợp thích hợp:
+ Hóa chất liều cao và ghép tế bào gốc tạo máu;
+ Ghép tế bào gốc đồng loài.
f. U lympho tế bào T
Lựa chọn điều trị tùy thuộc vào thể bệnh, giai đoạn bệnh, tuổi, tiên lượng bệnh theo IPI, PIT (với u lympho tế bào T ngoại vi) và lựa chọn điều trị tiếp theo như có hoặc không có ghép tủy.
Trường hợp mới chẩn đoán
- Hóa trị: Phác đồ đa hóa trị liệu hàng đầu.
+ CHOP;
+ CHOP-E;
+ Dose-adjusted EPOCH;
+ CHOP, sau đó là IVE (Ifosfamide, Etoposide, Epirubicin);
+ Hyper-CVAD.
- Xạ trị.
Trường hợp tái phát hoặc không lui bệnh
- Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Lugano trên PET/CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2020).
- Phải sinh thiết hạch làm lại chẩn đoán.
- Phác đồ đa hóa trị liệu hàng hai với trường hợp có chỉ định ghép tế bào gốc tạo máu.
+ Brentuximab với trường hợp có CD30+;
+ Pralatrexate;
+ DHAP;
+ ESHAP;
+ GDP;
+ GemOx;
+ ICE.
+ GVD
- Phác đồ đa hóa trị liệu hàng hai với trường hợp không có chỉ định ghép tế bào gốc:
+ Brentuximab với trường hợp có CD30+;
+ Pralatrexate;
+ Bendamustine;
+ Bortezomib;
+ Lenalidomide;
- Xạ trị.
- Ghép tế bào gốc tạo máu.
- Điều trị bổ trợ và chăm sóc.
g. Mycosis Fungoides, Hội chứng Sezary
- Điều trị trực tiếp trên da: Steroids, PUVA, nb-UVB hoặc mechlorethamine, xạ trị tại chỗ liều khuyến cáo 20-24 Gy.
- Điều trị hệ thống:
+ Retinoid;
+ Interferon (alpha, gama);
+ Methotrexate;
+ Brentuximab Vedotin;
+ Gemcitabin;
+ Liposomal doxorubicin;
+ Pralatrexate liều thấp hoặc liều chuẩn;
+ Chlorambucin;
+ Cyclophosphamide;
+ Pembrolizumab;
+ Bortezomib.
- Điều trị kết hợp ở da và điều trị hệ thống.
h. U lympho tế bào T/NK hốc mũi
- Các phác đồ hóa chất kết hợp:
+ Modified - SMILE trong 4 - 6 chu kỳ trong giai đoạn tiến triển;
+ VIPD (Etoposide, Ifosfamide, Cisplatin, Dexamethasone).
- Điều trị đồng thời xạ trị và hóa trị liệu:
+ Xạ trị 50 Gy và 3 chu kỳ DeVIC (Dexamethasone, Etoposide, Ifosfamide, Carboplatin);
+ Xạ trị 40 - 52.8 Gy và 3 chu kỳ VIPD (Etoposide, Ifosfamide, Cisplatin, Dexamethasone).
- Điều trị hóa trị và xạ trị nối tiếp: Trong giai đoạn I, II điều trị 2 - 4 đợt Modified-SMILE sau đó xạ trị 45-50,4 Gy.
- Điều trị hóa trị và xạ trị xen kẽ: P-GEMOX 2 chu kỳ sau đó xạ trị 56Gy sau đó lại điều trị P-GEMOX 2-4 chu kỳ.
- Chỉ điều trị bằng xạ trị.
- Ghép tế bào gốc tạo máu.
3.2. Một số trường hợp đặc biệt
a. Điều trị u lympho thần kinh trung ương nguyên phát
- Không dùng phác đồ CHOP.
- Sử dụng Methotrexate liều cao đơn trị liệu hoặc kết hợp cùng Cytarabin hoặc Temozolomide, hoặc với procarbazin và Vincristin. Liều Methotrexate 3,5 - 8g/m2. Nếu CD 20 (+), kết hợp cùng Rituximab 2 tuần/chu kỳ x 6- 8 chu kỳ.
- Có thể kết hợp xạ trị.
b. Điều trị hoặc dự phòng thâm nhiễm thần kinh trung ương
- Các thể bệnh cần dự phòng thâm nhiễm thần kinh trung ương: U lympho tế bào B lớn lan tỏa (tại các vị trí: Mắt, thận và thượng thận, vùng sọ và mặt, bệnh nhân có gây tê ngoài màng cứng, u lympho tại tinh hoàn, vú); double hit; tripple hit; u lympho tế bào B lớn lan tỏa có điểm CNS - IPI cao, u lympho có liên quan với nhiễm HIV; u lympho tế bào B tại trung thất nguyên phát; u lympho tế bào áo nang, u lympho Burkit, anaplastic large cell lymphoma, lymphoblastic lymphoma...
- Các thuốc sử dụng trong điều trị dự phòng thâm nhiễm thần kinh trung ương: Tiêm tủy sống dự phòng hoặc truyền tĩnh mạch Methotrexate, liều ≥ 3g/m2, truyền TM trong 4 - 6h.
c. U lympho không Hodgkin trên người bệnh HIV/AIDS
Sử dụng phác đồ hóa trị liệu tương tự kèm G-CSF, tiêm thuốc nội tủy. Nếu CD20(+), có thể dùng rituximab (trừ khi CD4 < 100/μl).
3.3. Các phác đồ cụ thể
- CHOP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cyclophosphamid |
750mg/m2 da |
Truyền tĩnh mạch |
1 |
|
Doxorubicin |
50mg/m2 da |
Truyền/tiêm tĩnh mạch |
1 |
|
Vincristin |
1,4mg/m2 da (max 2mg) |
Tiêm/truyền tĩnh mạch |
1 |
|
Methylprednisolone |
45mg/m2 da |
Uống/ TM |
1→5 |
Có thể kết hợp với tiêm tủy sống: Methotrexat 12,5 mg, ngày 1 (khi có thâm nhiễm thần kinh trung ương).
- CVP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cyclophosphamid |
750mg/m2 da |
Truyền tĩnh mạch |
1 |
|
Vincristin |
1,4mg/m2 da (Max 2mg) |
Tiêm/truyền tĩnh mạch |
1 |
|
Methylprednisolone |
45mg/m2 da |
Uống/ TM |
1-5 |
- EPOCH
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Etoposid |
50mg/m2 da |
Truyền tĩnh mạch |
1-4 |
|
Vincristin |
0,4mg/m2 da |
Tiêm/truyền tĩnh mạch |
1-4 |
|
Doxorubicin |
10mg/m2 da |
Truyền/tiêm tĩnh mạch |
1-4 |
|
Cyclophosphamid |
750mg/m2 da |
Truyền tĩnh mạch |
6 |
|
Methylprednisolone |
60mg/m2 da |
Uống/ TM |
1-6 |
- CHOP-E
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cyclophosphamid |
750mg/m2 da |
Truyền tĩnh mạch |
1 |
|
Doxorubicin |
50mg/m2 da |
Truyền/tiêm tĩnh mạch |
1 |
|
Vincristin |
1,4mg/m2 da (max 2mg) |
Tiêm/truyền tĩnh mạch |
1 |
|
Methylprednisolone |
45mg/m2 da |
Uống/ TM |
1-5 |
|
Etoposid |
100 mg/m2 da |
Truyền tĩnh mạch |
1-3 |
- CHOP-Bleo
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cyclophosphamid |
750mg/m2 da |
Truyền tĩnh mạch |
1 |
|
Doxorubicin |
50mg/m2 da |
Truyền/tiêm tĩnh mạch |
1 |
|
Vincristin |
1,4mg/m2 da (max 2mg) |
Tiêm/truyền tĩnh mạch |
1, 5 |
|
Methylprednisolone |
45mg/m2 da |
Uống/ TM |
1-5 |
|
Bleomycin |
10 đơn vị/m2 da |
Truyền tĩnh mạch |
1, 5 |
- FC
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Fludarabin |
25mg/m2 da |
Truyền tĩnh mạch |
1-3 |
|
Cyclophosphamid |
250 mg/m2 da |
Truyền tĩnh mạch |
1-3 |
- DHAP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cisplatin |
100mg/m2 da |
Truyền tĩnh mạch chậm trong 24 giờ |
1 |
|
Cytarabine |
2g/m2 da/12 giờ |
Truyền tĩnh mạch trong 3 giờ, 2 lần/ngày |
2 |
|
Dexamethasone |
40 mg |
Uống hoặc truyền tĩnh mạch |
1-4 |
+ Nhỏ mắt bằng dexamethasone trước và sau dùng cytarabine liều cao.
+ Điều trị 3 đợt, mỗi đợt cách nhau 28 ngày.
- ESHAP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cisplatin |
25mg/m2 da |
Truyền tĩnh mạch liên tục |
1-4 |
|
Etoposid |
40mg/m2 da |
Truyền tĩnh mạch trong 1 giờ |
1-4 |
|
Cytarabine |
2g/m2 da |
Truyền tĩnh mạch trong 3 giờ |
5 |
+ Nhỏ mắt bằng dexamethasone trước và sau khi dùng cytarabine liều cao.
+ Bổ sung kali và magie trước và sau truyền.
+ Điều trị 3-6 đợt, mỗi đợt cách nhau 28 ngày.
- ICE
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Ifosfamide |
5g/m2 da |
Truyền tĩnh mạch liên tục 24 giờ |
2 |
|
Etoposid |
100mg/m2 da |
Truyền tĩnh mạch trong 2 giờ |
1-3 |
|
Carboplatin |
AUC 5 |
Truyền tĩnh mạch trong 1 giờ |
2 |
+ Điều trị 3 đợt, mỗi đợt cách nhau 14 ngày.
+ Dùng mesna trước và sau truyền ifosfamid.
- Chlorambucil: 0,1-0,2mg/kg/ngày x 7-14 ngày, chu kỳ 28 ngày. Hoặc 0,4-0,6mg/kg mỗi 2 tuần có thể kết hợp với prednisolone.
- Bendamustin + Rituximab
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Bendamustin |
90/m2 |
Truyền TM |
1, 2 |
|
Rituximab |
375mg/m2 |
Truyền TM |
1 |
Chu kỳ 28 ngày, 6 - 8 chu kỳ.
- Lenalidomide + Rituximab
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Lenalidomide |
25mg/ngày |
Uống |
1 - 21 |
|
Rituximab |
375mg/m2 |
Truyền TM |
1 |
Chu kỳ 28 ngày, 12 chu kỳ.
- Bendamustin + Obinutuzumab
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Bendamustin |
90/m2 |
Truyền TM |
1,2 |
|
Obinutuzumab |
1000 mg/ngày |
Truyền TM |
Chu kỳ 1: ngày 1,8,15 Chu kỳ 2- 6: ngày 1 |
Chu kỳ 28 ngày, 6 chu kỳ.
- Bortezomib + Rituximab
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Bortezomib |
1,6mg/m2 |
TM |
1, 8, 15, 22 |
|
Rituximab |
375mg/m2 |
Truyền TM |
Chu kỳ 1: ngày 1, 8, 15, 22 Chu kỳ 2 và 3: ngày 1 |
Chu kỳ 35 ngày, sử dụng 3 chu kỳ.
- Bortezomib đơn độc
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Bortezomib |
1,5mg/m2 |
TM |
1, 4, 8, 11 |
Chu kỳ 28 ngày, 8 chu kỳ.
- Bendamustin + Rituximab + Bortezomib
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Bendamustin |
90/m2 |
Truyền TM |
1,4 |
|
Rituximab |
375mg/m2 |
Truyền TM |
1 |
|
Bortezomib |
1,3 mg/m2 |
Tiêm TM |
1,4,8,11 |
Chu kỳ 28 ngày.
- VR - CAP (Bortezomib, Rituximab, Cyclophosphamide, Doxorubicin, Prednisone)
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Bortezomib |
1,3 mg/m2 |
Tiêm TM |
1, 4, 8, 11 |
|
Rituximab |
375mg/m2 |
Truyền TM |
1 |
|
Cyclophosphamide |
750 mg/m2 |
Truyền TM |
1 |
|
Doxorubicin |
5mg/m2 |
Truyền TM |
1 |
|
Prednisone |
100mg/ngày |
Uống/ TM |
1-5 |
Chu kỳ 21 ngày cho đến 8 chu kỳ.
- RGCVP
|
Thuốc |
Liều dùng |
Đường dùng |
Ngày |
|
Rituximab |
375mg/m2 |
Truyền TM |
1 |
|
Gemcitabine |
Chu kỳ 1: 750mg/m2 Chu kỳ 2: 875mg/m2 Chu kỳ 3 - 6: 1000mg/m2 |
|
1, 8 |
|
Cyclophosphamide |
750 mg/m2 |
Truyền TM |
1 |
|
Vincristin |
1,4mg/m2 |
Truyền TM |
1 |
|
Prednisone |
100mg/ngày |
Uống/ TM |
1- 5 |
Chu kỳ 21 ngày, 6 chu kỳ.
- GDP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Gemcitabine |
1000 mg/m2 |
Truyền TM |
1,8 |
|
Dexamethasone |
40 mg/ngày |
uống |
1-4 |
|
Cisplatin |
75mg/m2 |
Truyền TM |
1 |
Chu kỳ 21 ngày, 3 chu kỳ.
- GVD
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Gemcitabine |
1000 mg/m2 |
Truyền TM |
1, 8 |
|
Vinorelbine |
20 mg/m2 |
Truyền TM |
1, 8 |
|
Doxorubicin liposomal |
15 mg/m2 |
Truyền TM |
1, 8 |
Chu kỳ 21 ngày, 2 -6 chu kỳ.
- P - GEMOX
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Gemcitabine |
1000 mg/m2 |
Truyền TM |
1, 8 |
|
Asparaginase |
6000 UI/m2 |
Truyền TM |
1-7 |
|
Oxaliplatin |
130 mg/m2 |
Truyền TM |
1 |
Chu kỳ mỗi 3 tuần, ít nhất 2 chu kỳ, sau đó xạ trị.
- DeVIC
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Dexamethasone |
40mg/ngày |
Truyền TM |
1-3 |
|
Etoposide |
67 - 100 mg/m2 |
Truyền TM |
1-7 |
|
Ifosfamide |
1000 mg/m2 1500mg/m2 |
Truyền TM |
1-3 |
|
Carboplatin |
200 mg/m2 300mg/m2 |
Truyền TM |
1 |
Chu kỳ 21 ngày trong 3 chu kỳ.
- AspMetDex
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Asparaginase |
6000 UI/m2 |
Truyền TM |
2, 4, 6, 8 |
|
Methotrexate |
3000 mg/m2 > 70t: 2000mg/m2 |
Truyền TM |
1 |
|
Dexamethasone |
40 mg/ngày > 70t: 20mg/ngày |
Truyền TM |
1 - 4 |
Chu kỳ 21 ngày trong 3 chu kỳ.
- VIPD (Etoposid, Ifosfamide, Cisplatin, Dexamethasone)
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Dexamethasone |
40mg/ngày |
Truyền TM hoặc uống |
1 - 4 |
|
Etoposide |
100 mg/m2 |
Truyền TM trong 90 phút |
1 - 3 |
|
Ifosfamide |
1200 mg/m2 |
Truyền TM trong 60 phút |
1 - 3 |
|
Cispaltin |
33 mg/m2 |
Truyền TM trong 60 phút |
1 - 3 |
Chu kỳ 21 ngày, sử dụng 3 chu kỳ.
Thuốc hỗ trợ:
- Mesna 240 mg/m2, truyền TM ngày 1 - 3.
- G - CSF khi có giảm bạch cầu trung tính độ 3 hoặc 4.
Chú ý:
- Các phác đồ gồm CHOP, CVP, EPOCH, CHOP-E, CHOP-Bleo, FC có thể dùng đến 9 đợt. Trong đó: 3 đợt đầu có thể dùng cách nhau từ 14 -21 ngày; 3 đợt tiếp theo cách nhau 21-30 ngày; thời gian cách bao nhiêu phụ thuộc vào lâm sàng (hạch, toàn trạng… và xét nghiệm về chức năng gan, thận và đặc biệt là tủy sinh máu).
- Trì hoãn điều trị khi máu ngoại vi có số lượng bạch cầu đoạn trung tính < 1 G/L hoặc số lượng tiểu cầu < 100G/L.
- Đánh giá đáp ứng điều trị (theo NCCN 7.2017) sau 3 đợt để tiếp tục duy trì hoặc chuyển phác đồ điều trị.
- Rituximab 1.400mg/m2 tiêm dưới da trong 5 phút, bắt đâu từ liệu trình thứ 2, được sử dụng cho các bệnh nhân u lympho không Hodgkin tế bào B/ CD 20 (+).
4. XÉT NGHIỆM ĐÁNH GIÁ TRƯỚC VÀ TRONG QUÁ TRÌNH ĐIỀU TRỊ
4.1. Đánh giá trước điều trị
Ngoài các xét nghiệm để chẩn đoán bệnh đã nêu ở trên, cần làm các xét nghiệm để đánh giá toàn trạng bệnh nhân trước điều trị, bao gồm:
- Sinh hóa máu: Chức năng gan, thận, đường máu, điện giải đồ, protein, albumin, Fe huyết thanh, ferritin, định lượng các Ig, Free Kappa, Free Lambda đối với u lympho tế bào B.
- Đông máu cơ bản vòng 1: Fibrinogen, PT, APTT, TT, D-Dimer, nếu có bất thường làm các xét nghiệm chuyên sâu.
- Tổng phân tích nước tiểu, tế bào nước tiểu.
- Định nhóm hệ ABO, Rh bệnh nhân vào viện lần đầu và khi bệnh nhân có chỉ định truyền máu.
- Xét nghiệm virus: HBV, HCV, HIV với bệnh nhân vào viện lần đầu, trước can thiệp thủ thuật, truyền máu nhiều lần và trước điều trị các trường hợp phải dùng thuốc ảnh hưởng đến hệ miễn dịch như kháng CD20,…
- Xét nghiệm: Anti-HBc và anti-HBs nếu trường hợp HBsAg âm tính để có kế hoạch dự phòng thuốc kháng virus trước khi điều trị thuốc ức chế miễn dịch và hóa chất tránh virus tái hoạt động.
- EBV, CMV (IgG, IgM). Đo tải lượng EBV, CMV
- Điện tâm đồ, siêu âm tim với bệnh nhân có chỉ định điều trị hóa chất.
- Điện di protein, điện di miễn dịch huyết thanh với các thể bệnh Lymphoplasmacytic lymphoma
4.2. Theo dõi trong điều trị
- Tổng phân tích tế bào máu 2 lần/ tuần.
- Sinh hóa máu: Chức năng gan, thận, điện giải đồ 1 lần/ tuần.
- Đông máu cơ bản 1 lần/ tuần.
- Làm huyết tủy đồ, sinh thiết tủy xương sau mỗi 3 chu kì điều trị hóa chất nếu có xâm lấn tuỷ xương hoặc u lympho thể tuỷ.
- Đánh giá đáp ứng sau 2-3 đợt điều trị bằng chẩn đoán hình ảnh: CT-scan, MRI, PET-CT.
4.3. Theo dõi sau điều trị
- Khi xuất hiện hạch to, sốt, gầy sút cân… phải tái khám ngay.
- Với nhóm tiến triển nhanh:
+ Tái khám: 1 tháng/lần trong năm đầu, 3 tháng/lần trong năm thứ 2;
+ Sau đó 6 tháng/lần trong 3 năm tiếp;
+ Sau đó 1 năm/lần.
- Với nhóm tiến triển chậm:
+ Tái khám: 3 tháng/lần trong năm đầu;
+ 4 tháng/lần trong năm thứ 2, 6 tháng/lần trong năm thứ 3;
+ Sau đó 1 năm/lần.
- Với mỗi lần tái khám:
+ Khám lâm sàng: Chú ý các triệu chứng lâm sàng, hạch to, gan to, lách to;
+ Xét nghiệm: Tổng phân tích tế bào máu, sinh hóa máu (LDH, chức năng gan, thận), chức năng tuyến giáp nếu có xạ trị vùng trước đó; CT bụng ngực hoặc PET, PET/CT mỗi 6 tháng trong 2 năm đầu hoặc khi nghi ngờ bệnh tái phát/tiến triển. Làm lại chẩn đoán trong trường hợp bệnh tái phát, tiến triển không đáp ứng điều trị.
TÀI LIỆU THAM KHẢO
1. Đỗ Trung Phấn (2008), “U lympho ác tính”, Tế bào gốc và bệnh lý tế bào gốc tạo máu, Nhà xuất bản y học, Tr 358-374.
2. Nguyễn Anh Trí (2004), “Điều trị u lymph ác tính không Hodgkin”, Điều trị các bệnh ác tính cơ quan tạo máu, Nhà xuất bản y học. Tr 22-38.
3. Non Hodgkin lymphoma - NCCN guidelines 2.2020.
4. WHO classification of tumours of haematopoietic and lymphoid tissues 2008.
5. “Neoplasic lymphoid diseases”, William hematology 8th- 2010, chapter 92, 93, 94, 99, 100, 101, 102, 103, 104, 105, 106.
6. John P. Greer, Michael E. Williams (2009),“Non-Hodgkin Lymphoma in Adults”, Wintrobes clinical hematology 12th editition, 2145-2194.
7. John G. Gribben (2009), “Clinical manifestation, staging, and treatment of indolent non Hodgkin lymphoma”, Hoffman: Hematology: Basic Principles and Practice, 5th ed, chapter 79.
8. Kieron Dunleavy, Wyndham H. Wilson (2009), “Diagnosis and treatment of aggressive non Hodgkin lymphoma”, Hoffman: Hematology: Basic Principles and Practice, 5th ed, chapter 80.
31. U LYMPHO HODGKIN (TRẺ EM)
1. ĐẠI CƯƠNG
U lympho ác tính là nhóm bệnh ác tính của tổ chức lympho, bao gồm: U lympho Hodgkin (chiếm khoảng 20%-30%) và U lympho không Hodgkin (70%-80%).
U lympho Hodgkin hiếm gặp ở trẻ em; nam có xu hướng mắc bệnh nhiều hơn nữ, nhạy cảm hơn với hóa trị liệu nên thường có phác đồ điều trị riêng nhằm giảm bớt độc tính.
Căn nguyên và dịch tễ
- Căn nguyên không rõ.
- Chiếm 8,8% trong tất cả các bệnh ung thư ở trẻ em và thanh thiếu niên.
- Tỷ lệ mới mắc hàng năm ở Hoa Kỳ là 12,1/ 1 triệu đối với trẻ em, 32/ 1 triệu đối với thanh thiếu niên 15 - 19 tuổi.
- Liên quan tới Epstein-Barr virus (EBV).
- Bệnh Hodgkin gia đình: Chiếm 4,5% trong tổng số ca Hodgkin.
2. CHẨN ĐOÁN
2.1. Chẩn đoán xác định
a. Lâm sàng
- Biểu hiện lâm sàng không đặc hiệu và phù hợp với tình trạng nhiễm khuẩn hơn là bệnh lý ác tính.
- Hạch to chiếm khoảng 70%. Thường gặp tại vùng cổ, nách, bẹn, trung thất và hạch ổ bụng. Số lượng hạch nhiều, mật độ chắc, thường không đau.
- Khối u trung thất hay gặp thứ hai nhưng hầu hết không có biểu hiện lâm sàng hoặc biểu hiện không đặc hiệu như: Đau sau xương ức, ho, thở nông, hiếm gặp hơn là tràn dịch màng phổi, màng tim, chèn ép tĩnh mạch chủ trên...
- Triệu chứng toàn thân khá thường gặp như: Ngứa, mệt mỏi, triệu chứng B (gồm có sốt, ra mồ hôi đêm, sút cân trên 10% trọng lượng cơ thể trong 6 tháng không giải thích được nguyên nhân).
- Gan hoặc lách có thể to nhưng ít khi to nhiều.
- Biểu hiện ban đầu ngoài hạch hiếm gặp.
- Một vài hội chứng cận u hiếm gặp nhưng khá đặc hiệu như: Tổn thương da (Mày đay, hồng ban, nốt ban đỏ, vết loét...); Hội chứng thận hư do sự giải phóng các lymphokine như IL-3.
- Bệnh lan tràn đầu tiên là các hạch kế cận (theo đường bạch huyết đặc biệt là ống ngực), sau đó là các cơ quan khác như lách, tủy xương. Ở giai đoạn muộn của bệnh, thường xuất hiện các biểu hiện chèn ép, xâm lấn của tổ chức lympho, u trung thất như:
Hội chứng trung thất, chèn ép tĩnh mạch chủ trên, tràn dịch màng phổi...; có thể xuất hiện thiếu máu, nhiễm khuẩn hoặc xuất huyết.
Bảng 30: Tỷ lệ các biểu hiện lâm sàng ở trẻ em bị U lympho Hodgkin
|
Biểu hiện lâm sàng |
Phần trăm |
|
Hạch to |
90% |
|
Hạch trung thất + Thanh thiếu niên và Thanh niên + Trẻ em dưới 10 tuổi |
75% 25% |
|
Gan- lách to |
25% |
|
Triệu chứng B (Sụt cân, đổ mồ hôi trộm, sốt) |
20% |
|
Ngứa |
15% |
|
Triệu chứng phổi (ho, khó thở) |
10% |
b. Cận lâm sàng
- Hạch đồ: Hạch tăng sinh, đa hình thái, ngoài dòng lympho còn gặp nhiều bạch cầu đoạn trung tính, bạch cầu đoạn ưa acid, tế bào plasmo, đại thực bào, tế bào xơ. Trong trường hợp điển hình có gặp tế bào Reed-Sternberg.
- Sinh thiết hạch hoặc tổ chức lympho nhằm làm xét nghiệm mô bệnh học kết hợp miễn dịch học (hóa mô miễn dịch hoặc đếm tế bào dòng chảy) và di truyền học là tiêu chuẩn vàng giúp chẩn đoán bệnh.
- Xét nghiệm khác:
+ Tế bào máu ngoại vi (huyết đồ) có thể gặp giảm lượng huyết sắc tố, giảm số lượng tiểu cầu, số lượng bạch cầu có thể tăng hoặc giảm;
+ Tốc độ máu lắng và protein C phản ứng thường tăng;
+ LDH tăng trong khoảng 30% trường hợp, có thể tăng calci và giảm albumin. Beta 2 microglobulin thường tăng;
+ Các phương pháp chẩn đoán hình ảnh như siêu âm, X-quang, CT, PET, PET- CT, MRI giúp phát hiện hạch sâu, vị trí di căn khác và theo dõi sau điều trị. Điện tâm đồ, siêu âm tim trước khi điều trị hóa chất;
+ Xét nghiệm phát hiện virus: EBV, CMV, HIV, HBV, HCV;
+ Tủy đồ, sinh thiết tủy xương và nhuộm hóa mô miễn dịch giúp phát hiện u lympho xâm lấn tủy;
+ Xét nghiệm phát hiện dấu ấn EBV trên mảnh sinh thiết hạch/tổ chức lympho bằng nhiều phương pháp: Hóa mô miễn dịch EBV màng (LMP1, LMP2), EBV kháng nguyên nhân (EBNA1; FISH RNA nhân của EBV (EBER);
+ Một số đột biến khác có thế phát hiện bằng kỹ thuật di truyền sinh học phân tử như: TP53, PDL1, PDL2...
2.2. Chẩn đoán thể bệnh
Bảng 31: Chẩn đoán thể bệnh U lympho Hodgkin theo WHO 2016
|
Thể bệnh |
Đặc điểm |
|
|
Cổ điển |
Giàu lymphocyte |
Dạng nốt hoặc lan tỏa. Trên nền nhiều lympho bào (chủ yếu lympho B), rải rác tế bào Reed-Sternberg. |
|
Nghèo lymphocyte |
Dạng sợi võng trông giống như sarcoma nhưng chứa nhiều tế bào R-S/ tế bào Hodgkin và nhiều dạng đa hình khác. Dạng xơ hóa lan tỏa chứa ít tế bào R-S/ tế bào Hodgkin nằm trên nền nghèo tế bào, bắt màu acid, dương tính với PAS. |
|
|
Hỗn hợp tế bào |
Nhiều tế bào Reed-Sternberg điển hình, tế bào dạng Hodgkin trên nền. Nền gồm nhiều lympho bào T, bạch cầu ưa acid, trung tính, tương bào và mô bào. Mô bào có thể tạo thành viêm hạt thưa |
|
|
Xơ nốt |
Xơ phát triển chia cắt nhu mô hạch thành nhiều nốt, có nhiều tế bào Reed-Sternberg, tế bào dạng Hodgkin và biến thể dạng tế bào khuyết, có thể thấy hoại tử trung tâm |
|
|
Mới |
Dạng nốt, ưu thế lymphocyte |
Gặp chủ yếu biến thể dạng lympho-histocytic - có nhân chia thùy (tế bào L-H, tế bào bỏng ngô) Tế bào nền chủ yếu lymphocyte T nhỏ và mô bào. Không có bạch cầu trung tính hoặc ưa acid. Hiếm gặp tương bào. Có thể gặp xơ hóa |
Bảng 32: Tỷ lệ các thể bệnh mô bệnh học của U lympho Hodgkin theo tuổi
|
Tỷ lệ thể bệnh (%) Nhóm tuổi |
Giàu lymphocyte |
Nghèo lymphocyte |
Hỗn hợp tế bào |
Xơ nốt |
Dạng nốt, ưu thế lymphocyte |
|
< 10 tuổi |
14 |
0 |
32 |
45 |
9 |
|
11 - 16 tuổi |
7 |
1 |
11 |
78 |
3 |
|
≥ 17 tuổi |
5 |
1 |
17 |
72 |
5 |
2.3. Chẩn đoán giai đoạn
Cho đến nay, chẩn đoán giai đoạn của U lympho Hodgkin vẫn dựa theo các tiêu chuẩn của Ann Arbor năm 2017.
Bảng 33: Chẩn đoán giai đoạn bệnh U lympho Hodgkin theo Ann Arbor, 1971.
|
Giai đoạn |
Biểu hiện |
|
I |
Tổn thương một vùng hạch hoặc một vị trí ngoài hạch (IE). |
|
II |
Tổn thương hai vùng hạch trở lên trên cùng một phía cơ hoành. Có thể bao gồm cả lách (IIS), vị trí ngoài hạch (IIE) hoặc cả hai (IIES) nhưng vẫn nằm một phía cơ hoành. |
|
III |
Tổn thương nằm hai phía cơ hoành. Có thể tổn thương ở lách (IIIS), hoặc vị trí ngoài hạch (IIIE), hoặc cả hai (IIIES). |
|
IV |
Tổn thương lan tỏa rải rác nhiều tạng hoặc mô ngoài hạch (như: Tủy xương, gan, phổi…), có kèm hoặc không kèm tổn thương hạch. |
|
- B là khi có biểu hiện: Sốt, ra mồ hôi đêm, sút cân > 10% trọng lượng cơ thể trong 6 tháng. - A là khi không có các triệu chứng trên. |
|
2.4. Chẩn đoán phân biệt
- U lympho không Hodgkin: Trên hạch đồ và sinh thiết hạch có hình ảnh đồng nhất về mặt hình thái, không tìm thấy tế bào Reed-Sternberg hoặc các biến thể.
- Hạch tăng sinh phản ứng: Hạch to, thường đau. Hạch diễn biến cấp nhưng lành tính và trở lại bình thường sau khi khỏi bệnh chính.
- Hạch lao: Thường gặp hạch dọc cơ ức đòn chũm, thành chuỗi, không đau và diễn biến theo trình tự: mềm dần, vỡ và chất bã đậu chảy ra ngoài. Hạch đồ và sinh thiết hạch thường thấy tổn thương đặc hiệu: tế bào bán liên, tế bào khổng lồ Langerhans, chất hoại tử bã đậu.
- Hạch ung thư di căn: Trên hạch đồ và sinh thiết hạch thường thấy các tế bào ung thư: kích thước lớn, nhân to, mịn, thường có nhiều hạt nhân, nguyên sinh chất rộng, đôi khi có hốc chế tiết, thường đứng thành đám. Đa số trường hợp có thể phát hiện được cơ quan ung thư nguyên phát.
3. ĐIỀU TRỊ
3.1. Điều trị với trường hợp mới chẩn đoán
Điều trị tiêu chuẩn bao gồm: Hóa trị liệu kết hợp xạ trị liều thấp vùng liên quan (low-dose involved field radiation therapy - LD-IFRT). Liều xạ trị và hóa trị được xác định dựa vào các yếu tố tiên lượng và biểu hiện bệnh: Triệu chứng B, giai đoạn bệnh, có hoặc không có khối trung thất, số lượng vị trí tổn thường (hạch và ngoài hạch).
3.1.1. Hóa trị
Các phác đồ đa hóa trị hạn chế số chu kỳ, giảm liều tích luỹ các chất alkylating và anthracyclines để tránh nguy cơ vô sinh, bệnh bạch cầu thứ phát và độc tính trên phổi.
Một số phác đồ đa hóa trị liệu thường dùng:
a. Phác đồ EuroNet-PHL C1
- Điều trị hàng 1: Với các bệnh nhân mới chẩn đoán. Bệnh nhân được xếp vào các phân nhóm điều trị:
+ Nhóm 1: Bệnh nhân giai đoạn I A/B và IIA;
+ Nhóm 2: Bệnh nhân giai đoạn IEA/B, IIEA, II B hoặc III A;
+ Nhóm 3: Bệnh nhân giai đoạn IIEB, IIIEA/B, III B hoặc IV A/B.
- Điều trị hàng 2: Khi tái phát hoặc bệnh dai dẳng/ không lui bệnh.
Tóm tắt các bước điều trị của phác đồ EuroNet- PHL C1 theo sơ đồ 6 và 7 như sau:
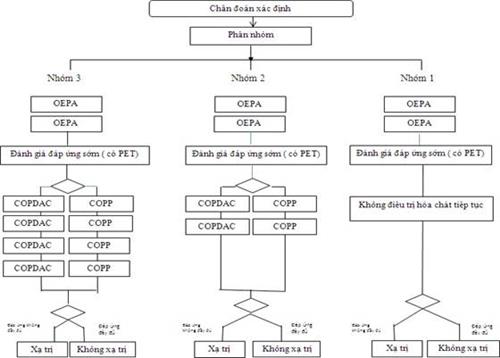
297
Sơ đồ 6: Phác đồ điều trị hàng 1 U lympho Hodgkin theo phân nhóm bệnh
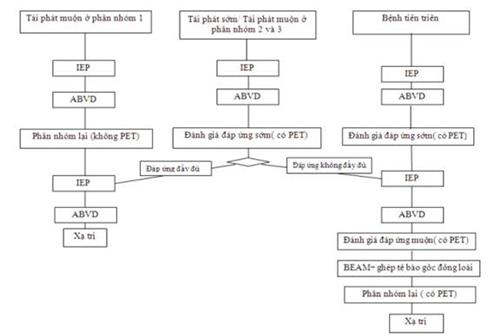
Sơ đồ 7: Phác đồ điều trị hàng 2 U lympho Hodgkin
b. Phác đồ của FriedmanD, Scharts, Hodgkin Lymphoma in The Lymphoid Neoplasms, 3rd edition, 2008
Chia 3 nhóm:
- Nhóm nguy cơ thấp (IA, IIA):
+ COPP/ABV x 4 chu kỳ (chu kỳ 28 ngày) + LD-IFRT;
+ DBVE x 4 chu kỳ (chu kỳ 28 ngày) + LD-IFRT;
+ OEPA (nam) hoặc OPPA (nữ) x 2 chu kỳ (chu kỳ 28 ngày) + LD-IFRT.
- Nguy cơ trung bình (Các trường hợp giai đoạn I, II không phân được vào nhóm giai đoạn sớm; IIIA, IVA):
+ COPP/ABV hybrid x 6 chu kỳ (chu kỳ 28 ngày) + LD-IFRT;
+ DBVE-PC x 3-5 chu kỳ (chu kỳ 21 ngày) + LD-IFRT;
+ OEPA (nam) hoặc OPPA (nữ) x 2 chu kỳ (chu kỳ 28 ngày) + COPP x 2 chu kỳ + LD-IFRT.
- Nhóm nguy cơ cao (Giai đoạn IIIB, IVB);
+ DBVE-PC x 3-5 chu kỳ (chu kỳ 21 ngày) + LD-IFRT;
+ BEACOPP x 8 chu kỳ (chu kỳ 21 ngày) + LD-IFRT;
+ BEACOPP x 8 chu kỳ (chu kỳ 21 ngày) + ABVD x 2 chu kỳ (chu kỳ 28 ngày)
+ LD-IFRT;
+ OEPA (nam) hoặc OPPA (nữ) x 2 chu kỳ (chu kỳ 28 ngày) + COPP x 4 chu kỳ + LD-IFRT.
c. Các phác đồ cụ thể
- OEPA
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Vincristine |
1.5 mg/m2/ngày |
Tĩnh mạch |
1,8,15 |
|
Etoposide |
125 mg/m2/ngày |
Tĩnh mạch |
3-6 |
|
Prednisone |
60 mg/m2/ngày |
Uống/ TM |
1-15 |
|
Doxorubicin |
40 mg/m2/ngày |
Tĩnh mạch |
1,15 |
- OPPA
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Vincristine |
1.5 mg/m2/ngày |
Tĩnh mạch |
1,8,15 |
|
Procarbazine |
100 mg/m2/ngày |
Uống |
1-15 |
|
Prednisone |
60 mg/m2/ngày |
Uống/ TM |
1-15 |
|
Doxorubicin |
40 mg/m2/ngày |
Tĩnh mạch |
1, 15 |
- COPP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Procarbazine |
100mg/m2 |
Uống |
1→15 |
|
Cyclophosphamide |
500mg/m2 |
Tĩnh mạch |
1,8 |
|
Vincristine |
1,5mg/m2 |
Tĩnh mạch |
1,8 |
|
Prednisone |
40 mg/m2 |
Uống/tĩnh mạch |
1→15 |
- COPDAC
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Dacarbazine |
250mg/m2 |
Tĩnh mạch |
1→3 |
|
Cyclophosphamide |
500mg/m2 |
Tĩnh mạch |
1,8 |
|
Vincristine |
1,5mg/m2 |
Tĩnh mạch |
1,8 |
|
Prednisone |
40 mg/m2 |
Uống/tĩnh mạch |
1-15 |
- ABVD
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Doxorubicin |
25mg/m2 |
Tĩnh mạch |
1, 14 hoặc 15 |
|
Bleomycin |
10mg/m2 |
Tĩnh mạch |
1, 14 hoặc 15 |
|
Vinblastin |
6mg/m2 |
Tĩnh mạch |
1, 14 hoặc 15 |
|
Dacarbazine |
375mg/m2 |
Tĩnh mạch |
1, 14 hoặc 15 |
- IEP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Ifosfamide |
2000 mg/m2 |
Tĩnh mạch/22h |
1-5 |
|
Mesna Mesna |
700 2000 |
Bolus Tĩnh mạch/24h |
1 1-7 |
|
Etoposide |
125 |
Tĩnh mạch/1- 2h |
1-5 |
|
prednisolone |
100 |
Uống, chia 3 liều |
1-5 |
- BEACOPP
|
Thuốc |
Liều |
Đường dùng |
Ngày |
|
Cyclophosphamid |
650mg/m2 |
Tĩnh mạch |
1 |
|
Doxorubicin |
25mg/m2 |
Tĩnh mạch |
1 |
|
Etoposide |
100mg/m2 |
Tĩnh mạch |
1→3 |
|
Procarbazine |
100mg/m2 |
Uống |
1→7 |
|
Prednisone |
40mg/m2 |
Uống/ Tĩnh mạch |
1→14 |
|
Vincristine |
1.4mg/m2 |
Tĩnh mạch |
8 |
|
Bleomycin |
10mg/m2 |
Tĩnh mạch |
8 |
- Các thuốc khác:
+ Brentuximab vedotin (chỉ với U lympho Hodgkin cổ điển) hoặc bendamustine;
+ ICE (ifosfamid, carboplastin, etoposid);
+ Kết hợp với một số thuốc như: Bendamustine, everolimus, lenalidomide, pembrolizumab…
3.1.2. Xạ trị
- U lympho Hodgkin nhạy cảm với xạ trị. Hầu hết các bệnh nhân trẻ em và thanh thiếu niên bị U lympho Hodgkin đều được điều trị bằng Hóa trị theo nhóm nguy cơ có hoặc không kết hợp với Xạ trị liều thấp. Liều xạ trị chung là 15-25 Gy, điều chỉnh dựa vào tuổi, kích thước khối u/ hạch, các cơ quan liên quan và nguy cơ tác dụng phụ cấp tính hoặc dài hạn.
- Xạ trị để điều trị (low-dose involved field radiation therapy - LD-IFRT, xạ trị liều thấp ở vùng liên quan) bao gồm các hạch bạch huyết ban đầu, xem xét bổ sung liên quan đến vị trí của bệnh (màng ngoài tim, thành ngực). Khi cần xạ trị xương chậu, cần chú ý đến buồng trứng và tinh hoàn. Nếu có thể, buồng trứng phải được di dời (di chuyển ngang theo xương chậu hoặc di chuyển về trung tâm phía sau tử cung), lê tưởng nhất là buồng trứng chỉ nên tiếp xúc dưới 3 Gy để bảo tồn khả năng sinh sản; tinh hoàn có thể tiếp xúc từ 5 đến 10% liều dùng cho vùng chậu, liều này cũng có thể gây ra chứng vô sinh nam tùy thuộc vào tổng liều xạ trị khung chậu. Cần tập trung xạ trị ở những khu vực biểu hiện bệnh mà vẫn bảo vệ được mô bình thường. Xạ trị liều cao có thể gây ảnh hưởng chức năng các cơ quan và làm giảm hiệu quả của các phác đồ cứu vãn khi điều trị tái phát.
- Xạ trị hàng ngày được thực hiện bằng cách sử dụng các trường trước và sau có trọng số bằng nhau với liều phân đoạn 1,5 - 1,8 Gy mỗi ngày, 5 lần một tuần.
- Xạ trị đơn độc: Ít dùng. Không sử dụng với u lympho Hodgkin dạng nốt, ưu thế lymphocyte.
3.2. Với trường hợp không lui bệnh hoặc tái phát
Đánh giá lui bệnh hoặc tái phát dựa trên: Lâm sàng (hạch to, hội chứng B) và thang điểm Deauville trên PET/CT, PET (theo NCCN - National Comprehensive Cancer Network, 1.2017).
Tái phát thường xảy ra trong vòng 4 năm đầu sau điều trị. Sự hiện diện của các triệu chứng B và biểu hiện ngoài hạch tại thời điểm tái phát là những đặc điểm tiên lượng kém. Một nghiên cứu được tiến hành bởi Hiệp hội Ung thư và Huyết học Nhi khoa (GPOH) cho thấy kết quả sau:
- Bệnh nhân tái phát sớm (trong khoảng 1- 3 tháng kể từ khi kết thúc điều trị) có tỷ lệ sống sót sau 10 năm (EFS) là 55% và tỷ lệ sống chung (OS) là 78%.
- Bệnh nhân tái phát muộn (xảy ra muộn hơn 12 tháng kể từ khi kết thúc điều trị) có EFS và OS 10 năm lần lượt là 86% và 90%.
- Bệnh nhân có yếu tố tiên lượng tốt khi chẩn đoán (giai đoạn IA hoặc giai đoạn IIA; không có khối u/hạch kích thước lớn; không có triệu chứng B), nếu tái phát giới hạn trong một khu vực có liên quan hạch ban đầu sau khi hóa trị và không xạ trị, nói chung có thể được cứu vãn bằng hóa trị theo phác đồ tái phát và LD-IFRT.
Hiện tại vẫn chưa có phác đồ tái tấn công tiêu chuẩn cho U lympho Hodgkin tái phát, tuy nhiên có 2 lựa chọn khả năng chữa bệnh bao gồm:
- Các liệu pháp hóa trị liệu thông thường và xạ trị LD-IFRT cho những bệnh nhân tái phát nguy cơ thấp.
- Hóa trị tái tấn công và sau đó là ghép tế bào gốc tạo máu tự thân đối với những bệnh nhân nguy cơ cao hơn.
Ghép tế bào gốc tự thân được ưu tiên sử dụng hơn ghép tế bào gốc đồng loại do tỷ lệ mắc bệnh và tử vong liên quan đến ghép đồng loài cao hơn. Sau khi ghép tế bào gốc tự thân, tỷ lệ sống dự kiến là 45 - 70% và tỷ lệ sống không tiến triển bệnh là 30 - 65%.
Cũng có thể lựa chọn ghép tế bào gốc đồng loài diệt tủy hoặc ghép giảm cường độ liều. Đối với những bệnh nhân ghép tế bào gốc tự thân thất bại hoặc không thể huy động đủ số lượng tế bào, có thể sử dụng ghép tế bào gốc đồng loài, tuy nhiên mức độ thành công khá thay đổi. Xạ trị thường được sử dụng sau khi hóa trị liệu liều cao và ghép tế bào gốc.
Bệnh nhân được điều trị bằng ghép tế bào gốc có thể bị tái phát muộn nhất là 5 năm; các bệnh nhân này nên được theo dõi chặt chẽ để phát hiện tái phát cùng với các tác dụng phụ muộn.
4. TIÊN LƯỢNG
- Các yếu tố tiên lượng xấu theo IPS-International Prognostic Score (NCCN 1.2017):
+ Albumin máu < 40 g/L;
+ Hemoglobin < 105 g/L;
+ Nam giới;
+ Giai đoạn bệnh (theo Ann Arbor): IV;
+ Số lượng bạch cầu máu ngoại vi > 15G/L;
+ Số lượng tế bào lympho máu ngoại vi < 0,6 G/L hoặc tỷ lệ tế bào lympho < 8%.
- Một số yếu tố khác:
+ Thể bệnh: Thể nốt ưu thế lymphocyte cơ tiên lượng tốt hơn các thể cổ điển;
+ Nồng độ các markers huyết thanh: Yếu tố hoại tử u, CD30 hòa tan, beta 2 macroglobulin, IL-10 (tiên lượng xấu); nồng độ caspase 3 trong các tế bào Reed-Sternberg cao có tiên lượng tốt;
+ Đáp ứng ban đầu với hóa trị liệu: Đáp ứng sớm và PET âm tính sau 2 chu kỳ hóa trị liệu có tiên lượng tốt.
5. ĐÁNH GIÁ, THEO DÕI
5.1. Khi chẩn đoán và trước điều trị
- Tiền sử, bệnh sử, lâm sàng.
- Xét nghiệm:
+ Tổng phân tích tế bào máu, hồng cầu lưới máu.
+ Sinh hóa máu: Chức năng gan, thận, Axit Uric, LDH, điện giải đồ...;
+ Đông máu huyết tương: Fibrinogen, PT, APTT, TT, D-Dimer. Bệnh nhân có kèm theo rối loạn đông máu cần làm thêm xét nghiệm: Nghiệm pháp rượu, Von-Kaulla, Đàn hồi cục máu đồ (ROTEM), Fibrin monomer;
+ Chẩn đoán hình ảnh: Xquang tim phổi, siêu âm ổ bụng, điện tâm đồ, siêu âm tim , CT scan, FDG - PET scan, MRI…;
+ Hạch đồ/ u đồ / lách đồ;
+ Sinh thiết hạch, sinh thiết khối u, sinh thiết lách… làm giải phẫu bệnh;
+ Hóa mô miễn dịch mảnh sinh thiết để chẩn đoán xác định, chẩn đoán thể bệnh (marker bệnh lý theo chỉ định của khoa Tế bào tổ chức học dựa trên định hướng từ lam nhuộm HE);
+ Xét nghiệm vi sinh: HIV, HBV, HCV...;
+ Huyết tủy đồ, sinh thiết tủy xương: đánh giá tình trạng tủy sinh máu, tình trạng xâm lấn tủy;
+ Công thức nhiễm sắc thể;
+ Gen bệnh;
+ Định nhóm máu hồng cầu: ABO, Rh(D);
+ Khám răng hàm mặt, tai mũi họng và các chuyên khoa khác(nếu bệnh nhân có biểu hiện bệnh lý ở các cơ quan khác) trước khi điều trị.
5.2. Trong qui trình điều trị
- Tổng phân tích tế bào máu, hồng cầu lưới máu.
- Sinh hóa máu: Chức năng gan, thận, Axit Uric, LDH, điện giải đồ...
- Đông máu huyết tương: Fibrinogen, PT, APTT, TT, D-Dimer, nghiệm pháp rượu, Von-Kaulla, Đàn hồi cục máu đồ (ROTEM), Fibrin monomer...
- Chẩn đoán hình ảnh: Xquang tim phổi, siêu âm ổ bụng, điện tâm đồ, siêu âm tim , CT scan, FDG - PET scan, MRI… trong quá trình điều trị (tùy theo phác đồ, ví dụ phác đồ EURONET- PHL- C1 yêu cầu đánh giá bằng PET-CT sau mỗi 2 đợt điều trị,...), kết thúc điều trị và trước khi xạ trị.
- Nếu có tổ chức hạch/ u mới xuất hiện hoặc ở vị trí cũ nhưng tăng kích thước hoặc có biểu hiện nghi ngờ tái phát/ không lui bệnh/ chuyển thể bệnh: Hạch đồ/ u đồ/ lách đồ, sinh thiết hạch, sinh thiết khối u làm giải phẫu bệnh lại và làm hóa mô miễn dịch (marker bệnh lý theo chỉ định của khoa Tế bào tổ chức học dựa trên định hướng từ lam nhuộm HE).
- Xét nghiệm vi sinh: HIV, HBV, HCV... (nếu có truyền máu và chế phẩm: HBV mỗi 1 tháng, HCV, HIV mỗi 3 tháng).
- Huyết tủy đồ, sinh thiết tủy xương, công thức nhiễm sắc thể, gen bệnh: Nghi ngờ tình trạng xâm lấn tủy, chuyển Lơ xê mi cấp...
- Định nhóm máu hồng cầu: ABO, Rh(D) (mỗi khi có chỉ định truyền máu
- Khám các chuyên khoa khác (răng hàm mặt, tai mũi họng...) nếu bệnh nhân có biểu hiện bệnh lý ở các cơ quan đó.
- Theo dõi độc tính trên các cơ quan: đánh giá chuyên sâu chức năng tim, chức năng phổi... (siêu âm tim, điện tim, đo chức năng hô hấp...).
5.3. Theo dõi sau điều trị
- Các trường hợp đặc biệt như: Hạch to, sốt, gầy sút cân… phải tái khám ngay.
- Tái khám: 1- 3 tháng/ lần trong 2 năm đầu. Sau đó, 6 tháng/ lần trong 3 năm tiếp và theo dõi hàng năm.
- Với mỗi lần tái khám:
+ Khám lâm sàng: Chú ý các triệu chứng lâm sàng, hạch to, hội chứng B;
+ Xét nghiệm: Tổng phân tích tế bào máu ngoại vi; Sinh hóa máu (bao gồm LDH, chức năng gan, thận, beta2-microglobulin, điện giải đồ...), máu lắng; Đông máu huyết tương; Chức năng tuyến giáp (đặc biệt nếu có xạ trị vùng trước đó); Chức năng tim (siêu âm tim màu, siêu âm doppler , điện tâm đồ...), chức năng phổi (đo chức năng hô hấp, XQ phổi, CT phổi...),...; CT bụng ngực; PET, PET/CT mỗi 6 tháng trong 2 năm đầu, sau đó chụp khi có biểu hiện lâm sàng; Xét nghiệm tủy đồ ít nhất 2 năm/ lần;
+ Làm lại sinh thiết khi xuất hiện hạch to trở lại hoặc có tổn thương mới.
5.4. Theo dõi di chứng lâu dài
Nói chung, tất cả bệnh nhân nên được theo dõi sức khỏe suốt đời. Các xét nghiệm bao gồm: Xét nghiệm chức năng tuyến giáp cho bệnh nhân xạ trị vùng cổ, hormon sinh dục cho những bệnh nhân đã điều trị bằng tác nhân kiềm hóa hoặc xạ trị vùng chậu, đánh giá chức năng tim cho những bệnh nhân điều trị bằng anthracycline hoặc xạ trị lồng ngực, đánh giá chức năng phổi cho những bệnh nhân điều trị bằng với bleomycin hoặc xạ trị lồng ngực. Tần suất theo dõi phụ thuộc vào nhiều yếu tố như phác đồ đã điều trị, độ tuổi và giới tính của bệnh nhân.
TÀI LIỆU THAM KHẢO
1. Đỗ Trung Phấn (2008), “U lympho ác tính”, Tế bào gốc và bệnh lý tế bào gốc tạo máu, Nhà xuất bản y học, Tr 358-374.
2. Nguyễn Anh Trí (2004), “Điều trị bệnh Hodgkin”, Điều trị các bệnh ác tính cơ quan tạo máu, Nhà xuất bản y học. Tr 15-21.
3. Hodgkin lymphoma - NCCN guidelines version 1.2017.
4. WHO classification of tumours of haematopoietic and lymphoid tissues 2008.
5. Debra L. Friedman, M.D., M.S. Manual of Pediatric Hematology and Oncology, Fifth Edition, Chapter19, p 599- 623.
6. Jonathan Sive and David Linch (2011), “Hodgkin lymphoma”, Postgraduate hematology, 6th ed, chapter 34.
7. Richard S.Stein, David S. Morgan (2009), “Hodgkin Lymphoma”, Wintrobes clinical hematology 12th editition, 2313-2343.
8. Volker Diehl, Beate Klimm (2008), “Hodgkin lymphoma: Clinical manifestations, staging and therapy”, Hoffman: Hematology: Basic Principles and Practice, 5th ed, chapter 77.
9. Christine Mauz-Körholz et al (2015). Pediatric Hodgkin lymphoma. Journal of clinical oncology, Volume 33, p 2975-2985.
10. Dieter Körholz et al (2010). First international Inter-Group Study for classical Hodgkin’s Lymphoma in Children and Adolescents. EuroNet-PHL-C1, Version 3
32. U LYMPHO KHÔNG HODGKIN (TRẺ EM)
1. ĐẠI CƯƠNG
U lympho ác tính là nhóm bệnh của tổ chức lympho, bao gồm U lympho Hodgkin (chiếm khoảng 20%-30%) và U lympho không Hodgkin (70%-80%).
U lympho không Hodgkin ở trẻ em thường khu trú ở các tổ chức lympho như hạch, mảng Peyer, lách, có thể xâm lấn tủy xương, nhưng ít gặp u lympho nguyên phát ở thần kinh trung ương và thể tủy.
Tỷ lệ mắc bệnh:
- U lympho không Hodgkin chiếm khoảng 8-10% các khối u ác tính ở trẻ em từ 5 - 19 tuổi.
- Chiếm khoảng 60% của tất cả các u lympho ở trẻ em và thanh thiếu niên.
- Đây là bệnh ác tính ở trẻ em phổ biến thứ ba với tỷ lệ mắc hàng năm là 750 - 800 mỗi năm ở trẻ em dưới 19 tuổi ở Hoa Kỳ.
- Tỷ lệ bệnh ở trẻ em dưới 15 tuổi không thay đổi nhiều trong hai thập kỷ qua, nhưng ở thanh thiếu niên từ 15 -19 tuổi tỷ lệ này tăng lên 50%.
Dịch tễ học:
- Giới tính: Tỷ lệ nam: Nữ là 2,5: 1, ở độ tuổi 5-14 tỷ lệ này là 3:1.
- Tuổi: Tương đối giống nhau ở mọi lứa tuổi, nhưng đỉnh điểm ở độ tuổi 15-19, phần lớn là do sự gia tăng đáng kể tỷ lệ mắc U lympho tế bào B lớn lan tỏa (DLBCL) ở độ tuổi này.
- Các yếu tố nguy cơ:
+ Di truyền: Khiếm khuyết miễn dịch (thiếu gammaglobulin miễn dịch typ Bruton liên quan đến giới tính, hội chứng Bloom, hội chứng Wiskott- Aldrich, hội chứng tăng sinh lympho tự miễn ALPS và 1 số hội chứng suy giảm miễn dịch bẩm sinh khác);
+ Suy giảm miễn dịch sau ghép tủy xương (đặc biệt là sử dụng tủy diệt tế bào T), sau ghép tạng đặc;
+ Bệnh u nhú lympho ở trẻ em có thể hoặc tiến triển hoặc cùng tồn tại với u lympho tế bào lớn bất thục sản (anaplastic large cell lymphoma- ALCL);
+ Thuốc: Diphenylhydantoin, Infliximab và các thuốc ức chế miễn dịch khác;
+ Phóng xạ: Trẻ em được điều trị bằng hóa trị và xạ trị U lympho Hodgkin trước đó;
+ Virus: EBV, HIV, HTLV.
2. CHẨN ĐOÁN
2.1. Chẩn đoán xác định
a. Lâm sàng
Biểu hiện lâm sàng của U lympho không Hodgkin ở trẻ em phụ thuộc chủ yếu vào thể bệnh và các vị trí tổn thương. Khoảng 70% trẻ em có bệnh ở giai đoạn tiến triển có biểu hiện ngoài hạch như đường tiêu hóa, tủy xương và hệ thần kinh trung ương.
- Bụng:
+ Bệnh nhân mắc U lympho Burkitt (Burkitt lymphoma-BL) và U lympho tế bào B lớn lan tỏa thường có khối u nguyên phát ở bụng;
+ Khoảng 35% U lympho không Hodgkin ở trẻ em có khối u nguyên phát ở bụng, liên quan đến vùng hồi tràng, ruột thừa, đại tràng hoặc các vị trí liên quan;
+ Triệu chứng: Đau bụng, chướng bụng, buồn nôn và nôn, sờ thấy khối, co thắt, gan lách to, vàng da tắc mật, cổ trướng hoặc viêm phúc mạc. Có thể xảy ra chảy máu và thủng ruột, mặc dù hiếm;
+ U lympho là nguyên nhân thường gặp nhất gây lồng ruột ở trẻ trên 6 tuổi.
- Đầu và cổ:
Biểu hiện bệnh ở vùng đầu và cổ chiếm 13% các trường hợp: Sưng hạch cổ, sưng tuyến mang tai, u xương hàm và phì đại amidan. Bệnh nhân có thể bị tắc nghẽn mũi, chảy nước mũi, khó nghe và liệt dây thần kinh sọ. U lympho Burkitt có tỷ lệ biểu hiện trên vùng mặt cổ cao hơn các thể khác, khoảng 50%-60%.
- Trung thất:
+ Tỷ lệ có biểu hiện ở trung thất là 26%. Bệnh nhân có khối u trung thất lớn có thể có hội chứng trung thất trước xuất hiện với cổ bị lệch, phù cổ và mặt, khó thở rõ rệt, khó thở khi nằm, chóng mặt, nhức đầu, khó thở, chảy máu cam, thay đổi trạng thái tâm thần;
+ Lymphoblastic lymphoma (LL): Thường biểu hiện với một khối trong lồng ngực hoặc trung thất cùng với sự lây lan đến các vị trí khác như tủy xương, màng não và tuyến sinh dục. U lympho Burkitt và Anaplastic large cell lymphoma ít có biểu hiện ở trung thất hơn. U lympho tế bào B trung thất nguyên phát (Primary mediastinal B-cell lymphoma - PMBL) hiện đang được WHO phân loại là một dưới nhóm của U lympho tế bào B lớn lan tỏa, mһc dù có nhiều dữ liệu cho thấy rằng đây là 1 thể riêng biệt mang cả đặc điểm lâm sàng, hình thái tế bào học và di truyền của cả U lympho Hodgkin và không Hodgkin. U lympho tế bào B trung thất nguyên phát thường biểu hiện ở thanh thiếu niên và thanh niên, tiên lượng xấu hơn các thể khác;
+ Các khối u trung thất có thể gây ra tràn dịch màng phổi, màng tim do xâm lấn hoặc chèn ép.
- Tủy xương: Cả Burkitt lymphoma và Lymphoblastic lymphoma đều có xu hướng thâm nhiễm đến tủy xương hơn so với anaplastic large cell lymphoma và U lympho không Hodgkin tế bào B lớn lan tỏa. Bệnh nhân Burkitt lymphoma có tiên lượng xấu hơn đáng kể nếu đã xâm lấn tủy xương, tuy nhiên trong Lymphoblastic lymphoma không có sự khác biệt về tiên lượng.
- Hệ thần kinh trung ương: Cũng như xâm lấn tủy, U lympho Burkitt và Lymphoblastic lymphoma cũng thường có biểu hiện ở thần kinh trung ương. Trong U lympho không Hodgkin tế bào B lớn lan tỏa hoặc anaplastic large cell lymphoma, điều này rất hiếm. Biểu hiện ở hệ thần kinh trung ương có tiên lượng rất xấu.
- Các vị trí khác: U lympho nguyên phát ở các vị trí khác (11%) bao gồm da và mô dưới da, hốc mắt, tuyến giáp, xương (có hoặc không tăng calci máu), thận, ngoài màng cứng, vú và tuyến sinh dục.
- Các triệu chứng toàn thân: Sốt và sụt cân tương đối hiếm gặp ngoại trừ trong anaplastic large cell lymphoma. Sụt cân cũng có thể xảy ra thứ phát sau tắc ruột.
b. Cận lâm sàng
- Hạch đồ: Hạch tăng sinh, khá đồng nhất, chủ yếu là lymphoblast hoặc prolymphocyte: Kích thước to, nhân lớn (có thể có hạt nhân, nhân chẻ, nhân chia, nhân quái), nguyên sinh chất hẹp, không tạo đám; ít gặp bạch cầu đoạn trung tính, bạch cầu đoạn ưa acid, plasmocyte, đại thực bào, tế bào xơ.
- Sinh thiết hạch hoặc tổ chức lympho nhằm làm xét nghiệm mô bệnh học kết hợp miễn dịch học (hóa mô miễn dịch hoặc đếm tế bào dòng chảy) và di truyền học là tiêu chuẩn vàng giúp chẩn đoán bệnh.
- Xét nghiệm khác:
+ Tổng phân tích tế bào máu ngoại vi (huyết đồ) có thể gặp giảm lượng huyết sắc tố, giảm số lượng tiểu cầu, số lượng bạch cầu có thể tăng hoặc giảm;
+ LDH tăng trong khoảng 30% trường hợp; tăng calci máu. Chức năng gan thận có thể có biểu hiện rối loạn. Beta 2 microglobulin thường tăng;
+ Các phương pháp chẩn đoán hình ảnh: Siêu âm, X-quang, CT, MRI..., đặc biệt là PET, PET-CT giúp phát hiện các hạch sâu như hạch trung thất, hạch ổ bụng... các vị trí di căn khác và theo dõi sau điều trị;
+ Tủy đồ, sinh thiết tủy xương (có thể làm tại 2 vị trí) và nhuộm hóa mô miễn dịch nhằm phát hiện u lympho xâm lấn tủy;
+ Xét nghiệm di truyền: Phát hiện bất thường tùy từng thể bệnh;
+ Xét nghiệm điện di miễn dịch phát hiện thay đổi protein huyết thanh tùy thể bệnh;
+ Xét nghiệm vi khuẩn Helicobacter Pylori trong U lympho không Hodgkin tại dạ dày;
+ Xét nghiệm viêm gan B, C.
2.2. Chẩn đoán thể bệnh
Xếp loại u lympho không Hodgkin theo Tổ chức y tế thế giới (WHO 2016)
- Phân loại của Tổ chức y tế Thế giới năm 2016 đã bổ sung thêm các điểm: Cập nhật các tiêu chuẩn chẩn đoán với thể u lympho đã có như: U lympho dạng lymphoplasmatic…; Cập nhật, thay đổi tên gọi một số thể như: U lympho tế bào B nguy cơ cao với MYC+ và BCL2+ và/hoặc BCL6+…; Bổ sung các thể mới như: U lympho tế bào B lớn với tái sắp xếp IRF4…
- Bảng xếp loại U lympho không Hodgkin theo WHO 2016 ở trẻ em cũng tương tự như ở người lớn và được trình bày ở bài U lympho không Hodkin người lớn.
2.3. Chẩn đoán giai đoạn
Bảng 34: Chẩn đoán giai đoạn theo Ann Arbor.
|
Giai đoạn |
Biểu hiện |
|
I |
Tổn thương một vùng hạch hoặc một vị trí ngoài hạch (IE). |
|
II |
Tổn thương hai vùng hạch trở lên trên cùng một phía cơ hoành. Có thể bao gồm cả lách (IIS), vị trí ngoài hạch (IIE) hoặc cả hai (IIES) nhưng vẫn nằm một phía cơ hoành. |
|
III |
Tổn thương nằm hai phía cơ hoành. Có thể tổn thương ở lách (IIIS), hoặc vị trí ngoài hạch (IIIE), hoặc cả hai (IIIES). |
|
IV |
Tổn thương lan tỏa rải rác nhiều tạng hoặc mô ngoài hạch (như: Tủy xương, gan, phổi…), có kèm hoặc không kèm tổn thương hạch. |
|
- B là khi có biểu hiện triệu chứng “B”: Sốt, ra mồ hôi đêm, sút cân trên 10% trọng lượng cơ thể trong 6 tháng. - A là khi không có các triệu chứng trên. |
|
Bảng 35: Chẩn đoán giai đoạn theo Murphy (nghiên cứu St’Jude)
|
Giai đoạn |
Tiêu chuẩn |
|
I |
- Một khối u duy nhất ngoài hạch hoặc một vị trí hạch duy nhất không bao gồm hạch trung thất và hạch ổ bụng |
|
II |
- Một khối u duy nhất ngoài hạch cộng thêm các hạch vùng ở cùng bên cơ hoành. - Hai hay nhiều hạch cùng bên cơ hoành. - Hai khối u ngoài hạch có hay không có hạch cùng bên cơ hoành. - U đường tiêu hóa, thường ở vị trí iléocaecale (gốc hồi manh tràng), có hoặc không có các hạch mạc treo có thể cắt bỏ được. |
|
III |
- Hai khối u ngoài hạch ở mỗi bên cơ hoành có sự tham gia của hai hay nhiều hạch ở hai bên cơ hoành. - Các khối u ở ngực (trung thất, phổi, tuyến ức) - Tất cả các khối u lan rộng ở bụng và không thể phẫu thuật được. - Tất cả các khối u gần xương sống hay ngoài màng cứng |
|
IV |
- Xâm lấn não, dịch não tủy hay tủy xương. |
Bảng 36: Chẩn đoán giai đoạn theo FAB ((French- American- Bristish) cho DLBCL, BL, BLL (Burkitt- like lymphoma) trẻ em .
|
Nhóm A: giai đoạn I và II theo Murphy (tổn thương đã cắt bỏ hoàn toàn). Nhóm B: Tất cả các bệnh nhân không đủ điều kiện tham gia Nhóm A hoặc Nhóm C Nhóm C: Bất kỳ sự tham gia nào của CNS ** và/hoặc liên quan đến tủy xương (≥ 25% blast) ** CNS: Khi có bất kì 1 hay nhiều dấu hiệu sau: - Hiện diện tế bào blast L3 trong dịch não tủy - Tổn thương một hoặc nhiều cặp thần kinh sọ (không do khối u ngoài sọ) - Có dấu hiệu lâm sàng của chèn ép tủy sống - U nội sọ khu trú - Giãn khoang dưới màng cứng (sọ não/tủy sống) |
2.4. Chẩn đoán phân biệt
- U lympho Hodgkin: Trên hạch đồ và sinh thiết hạch tìm thấy tế bào Reed- Sternberg hoặc các biến thể.
- Hạch tăng sinh phản ứng: Hạch to, thường đau, ở gần nơi tổn thương. Hạch to diễn biến cấp nhưng lành tính và trở lại bình thường sau khi khỏi bệnh chính.
- Hạch lao: Thường gặp chuỗi hạch dọc cơ ức đòn chũm, không đau, nếu kéo dài thường vỡ và chảy chất bã đậu. Hạch đồ và sinh thiết hạch thường thấy tổn thương gồm: tế bào bán liên, tế bào khổng lồ Langerhans, chất hoại tử bã đậu.
- Hạch ung thư di căn: Trên hạch đồ và sinh thiết hạch thường thấy các tế bào ung thư: kích thước lớn, nhân to, mịn, thường có nhiều hạt nhân, nguyên sinh chất rộng, đôi khi có hốc chế tiết, thường đứng thành đám. Đa số trường hợp có thể phát hiện được cơ quan ung thư nguyên phát.
3. ĐIỀU TRỊ
3.1. Điều trị cấp cứu
Ba biểu hiện lâm sàng cần được chú ý ngay lập tức bao gồm:
- Hội chứng tĩnh mạch chủ trên thứ phát sau một khối trung thất lớn cản trở lưu lượng máu.
- Chèn ép đường hô hấp.
- Hội chứng ly giải khối u.
3.2. Hóa trị
Phác đồ điều trị được chia thành các nhóm sau:
a. Lymphoblastic lymphoma (LL): Bệnh cục bộ và tiến triển được điều trị giống nhau.
b. U lympho Hodgkin dòng B (Burkitt lymphoma-BL), u lympho giống Burkitt (Burkitt - like lymphoma - BLL) và u lympho tế bào B lớn lan tỏa (DLCL).
- Nguy cơ thấp.
- Nguy cơ trung gian.
- Nguy cơ cao (CNS hoặc liên quan đến tủy xương).
c. Anaplastic large cell lymphoma
- Giai đoạn thấp.
- Giai đoạn tiến triển.
3.3. Phác đồ điều trị với từng nhóm bệnh
a. Lymphoblastic lymphoma
- Đối tượng áp dụng:
+ Lymphoblastic lymphoma dòng tế bào B hoặc T.
+ Bệnh khu trú hoặc tiến triển.
Phác đồ BFM-NHL
Sơ đồ chung:
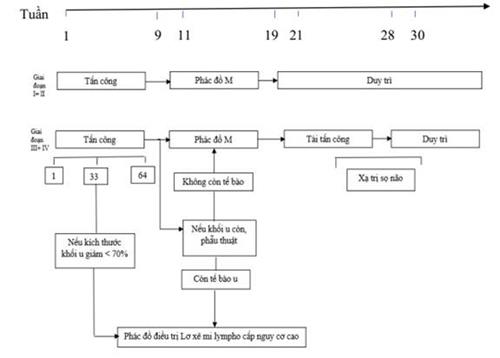
Thuốc cụ thể:
|
Thuốc |
Liều lượng |
Ngày dùng |
|
Tấn công |
|
|
|
Methylprednisone (uống) |
60 mg/m2 |
1-28, giảm liều trong 9 ngày |
|
Vincristine (tĩnh mạch) |
1.5 mg/m2 ( tối đa 2mg) |
8,15,22,29 |
|
Daunorubicin(tĩnh mạch/1h) |
30 mg/m2 |
8, 15, 22,29 |
|
Asparaginase (tĩnh mạch/ tiêm bắp) |
10,000 UI /m2 |
12,15,18,21,24,27,30,33 |
|
Cyclophosphamide(tĩnh mạch/1h) |
1,000 mg/m2 |
36,64 |
|
Cytarabine (tĩnh mạch) |
75 mg/m2 |
38-41, 45- 48, 52- 55, 59- 62 |
|
Mercaptopurine (uống) |
60 mg/m2 |
36- 63 |
|
MTX (tiêm tủy sống) |
12 mg |
1,15,29,45,59 |
|
Củng cố (phác đồ M) (sau pha Tấn công 2 tuần) |
|
|
|
Mercaptopurine (uống) |
25 mg/m2 |
1-56 |
|
MTX (tĩnh mạch/24h) |
5,000 mg/m2 |
8,22,36,50 |
|
MTX (tiêm tủy sống) |
12 mg |
8,22,36,50 |
|
Tái tấn công (sau pha Phác đồ M 2 tuần) |
|
|
|
Dexamethasone (uống) |
10 mg/m2 |
1-21, giảm liều trong 9 ngày |
|
Vincristine (tĩnh mạch) |
1.5 mg/m2 (tối đa 2mg) |
8,15,22,29 |
|
Doxorubicin (tĩnh mạch) |
30 mg/m2 |
8,15,22,29 |
|
Asparaginase (tĩnh mạch/ tiêm bắp) |
10,000UI/m2 |
8,11,15,18 |
|
Cyclophosphamide (tĩnh mạch) |
1,000mg/m2 |
36 |
|
Cytarabine (tĩnh mạch) |
75 mg/m2 |
38- 41, 45- 48 |
|
Mercaptopurine (uống) |
60mg/m2 |
36- 49 |
|
MTX (tiêm tủy sống) |
12 mg |
38, 45 |
|
Duy trì (sau Phác đồ M/ hoặc sau pha Tái tấn công 2 tuần) |
|
|
|
Mercaptopurine (uống) |
50mg/m2/ngày |
Hàng ngày |
|
Methotrexate (uống) |
20 mg/m2/ngày |
Hàng tuần |
312
Lưu ý:
- Bệnh nhân Lymphoblastic lymphoma khu trú: Điều trị Tấn công -> Củng cố -> Duy trì.
- Bệnh nhân Lymphoblastic lymphoma tiến triển có thêm xạ trị màng não dự phòng và tái tấn công.
- Ở ngày thứ 33, kích thước khối u giảm dưới 70% thì chuyển điều trị như Lơ xê mi lympho cấp nhóm nguy cơ cao.
- Nếu sau giai đoạn tấn công, khối u vẫn còn thì phẫu thuật cắt bỏ. Nếu sau phẫu thuật vẫn còn tế bào u, điều trị như Lơ xê mi lympho cấp nhóm nguy cơ cao.
- Truyền Methotrexat (MTX) và leucovorine (CF) rescue.
+ MTX: 5g/m2/24h, 10% tổng liều truyền trong 30 phút đầu, 90% tổng liều truyền trong 23,5 giờ còn lại;
+ CF rescue: 30mg/m2 IV giờ thứ 42, sau đó 6 liều 15mg/m2 mỗi 6h cho đến khi định lương MTX < 0,4µ□.
- Xạ trị sọ não (CRT):
+ Chỉ áp dụng cho bệnh nhân giai đoạn III và IV, không áp dụng trẻ dưới 1 tuổi;
+ Xạ trị dự phòng cho tất cả bệnh nhân không có thâm nhiễm TKTW với liều 12Gy;
+ Xạ trị điều trị (Bệnh nhân có thâm nhiễm TKTW) cho trẻ từ trên 1- 2 tuổi là 18Gy, trẻ lớn hơn là 24 Gy. Thời điểm: CRT ở pha 2 của Tái tấn công (tuần thứ 27- 29).
- Trường hợp có thâm nhiễm tinh hoàn: Xạ trị tinh hoàn liều 20 Gy sau khi điều trị xong Giai đoạn củng cố (phác đồ M).
- Liều Methotrexat tiêm tủy sống theo tuổi:
|
Tuổi |
Liều ( mg) |
|
> 3 tuổi |
12 |
|
2-3 tuổi |
10 |
|
1-2 tuổi |
8 |
|
Dưới 1 tuổi |
6 |
b. U lympho không Hodgkin tế bào B (Burkitt lymphoma [BL], u lympho giống Burkitt [BLL] và u lympho tế bào lớn lan tỏa [DLCL])
 Phác đồ LMB - BFM 2001: điều trị theo phân nhóm nguy cơ FAB như sau:
Phác đồ LMB - BFM 2001: điều trị theo phân nhóm nguy cơ FAB như sau:
|
Nhóm |
Giai đoạn |
Phác đồ |
|
A |
- Giai đoạn I đã cắt khối u hoàn toàn - Giai đoạn II khôi u ổ bụng đã cắt bỏ hoàn toàn |
COPAD - COPAD |
|
B |
- Giai đoạn I chưa cắt bỏ hoàn toàn khối u - Giai đoạn II (ngoại trừ khối u ổ bụng giai đoạn II đã cắt bỏ hoàn toàn) - Giai đoạn III - Giai đoạn IV (không thâm nhiễm TKTW, tủy xương < 25% blast) |
COP - COPADM1 - COPADM2 - CYM1 - CYM2 |
|
C |
- Thâm nhiễm tủy và/hoặc thâm nhiễm não-màng não |
COP-COPADM1-COPADM2-CYVE1-CYVE2-M1-M2 -M3-M4 * Bệnh nhân có CNS (+) điều trị thêm methotrexat 8g/m2 và 3 liều tiêm hóa chất khoang nội tủy |
 Các thuốc cụ thể:
Các thuốc cụ thể:
 Nhóm A: 2 đợt COPAD ( chu kỳ 21 ngày), đợt 2 bắt đầu khi số lượng tế bào máu hồi phục (BCTT > 1,000/cm3 và TC >100,000/cm3).
Nhóm A: 2 đợt COPAD ( chu kỳ 21 ngày), đợt 2 bắt đầu khi số lượng tế bào máu hồi phục (BCTT > 1,000/cm3 và TC >100,000/cm3).
- COPAD:
|
Thuốc |
Liều lượng, đường dùng, ngày dùng |
|
Cyclophosphamide |
250 mg/m2/ 12 giờ (500 mg/m2/ngày), tĩnh mạch, N1→N3. Hydrat hóa nên được duy trì với tốc độ 3000 ml /m2/ngày (125 ml/ m2/h) cho đến 12giờ sau liều cyclophosphamide cuối cùng. |
|
Vincristine |
2.0 mg/m2 (liều tối đa 2.0 mg), tĩnh mạch, ngày 1 và 6 |
|
Methylprednisolone |
60 mg/m2/ngày (chia làm 2 lần), đường uống/ tĩnh mạch, từ N1→ N5, sau đó giảm liều và ngưng trong 3 ngày |
|
Doxorubicin |
60 mg/m2, tĩnh mạch, truyền trong 6h giữa 2 liều cyclophosphamide |
|
G-CSF |
5 mcg/kg/ngày, dưới da, ngày thứ 7 cho đến khi BCTT > 3,000/mm3, |
Nhóm B
|
Thuốc |
Liều lượng, đường dùng |
Ngày dùng |
|
Pha trước tấn công: COP |
||
|
Cyclophosphamide |
300 mg/m2, tĩnh mạch |
Ngày 1 |
|
Vincristine |
1.0 mg/m2 (tối đa 2.0 mg), tĩnh mạch |
Ngày 1 |
|
Methylprednisolone |
60 mg/m2/day (chia 3 liều), đường uống/tĩnh mạch |
Ngày 1- 7 |
|
Methotrexate, Hydrocortisone |
15mg, tiêm tủy sống* |
Ngày 1 |
|
Tấn công: 2 chu kỳ COPADM, COPADM1 được bắt đầu từ ngày thứ 8 của COP nếu không có chống chỉ định; COPADM2 bắt đầu khi tế bào máu ngoại vi hồi phục ( BCTT > 1 G/L, SLTC > 100 G/L), không nên bắt đầu trước ngày thứ 16 tính từ ngày đầu của COPADM1 và nên trì hoãn sau 48 giờ kể từ liều G- CSF cuối cùng COPADM 1&2 |
||
|
Vincristine |
2.0 mg/m2 (tối đa 2.0 mg), tĩnh mạch |
Ngày 1 |
|
Methylprednisolone |
60 mg/m2/day (chia 3 liều), đường uống/ tĩnh mạch |
Ngày 1- 5 |
|
Methotrexate |
3 g/m2 trong 3h, tĩnh mạch (Axit folinic bắt đầu từ giờ 24 của MTX, 15 mg/m2 mỗi 6h, 12 liều) |
Ngày 1 |
|
Cyclophosphamide |
250 mg/m2/12 giờ ( tổng 6 liều), tĩnh mạch (trong chu kỳ COPADM2 liều 500 mg/m2/12 giờ) |
Ngày 2- 4 |
|
Doxorubicin |
60 mg/m2 (truyền trong 6 giờ), tĩnh mạch |
Ngày 2, sau liều cyclophosphamide đầu tiên |
|
Methotrexate & Hydrocortisone |
15mg, tiêm tủy sống * |
Ngày 2,6 |
|
G-CSF |
5 mcg/kg/ngày, tiêm dưới da |
ngày thứ 7 cho đến khi BCTT > 3,000 /mm3 |
|
CYM 1&2: CYM1 bắt đầu khi số lượng tế bào máu ngoại vi hồi phục sau COPADM2 (BCTT > 1,000/cm3 và TC >100,000/cm3); CYM2 bắt đầu khi số lượng tế bào máu ngoại vi hồi phục sau CYM1 (BCTT > 1,000/cm3 và TC >100,000/cm3). |
||
|
Methotrexate |
3 g/m2 trong 3h, tĩnh mạch (Axit folinic bắt đầu từ giờ 24 của MTX, 15 mg/m2 mỗi 6h, 12 liều) |
Ngày 1 |
|
Cytarabine |
100 mg/m2/ngày, tĩnh mạch/ 24h |
Ngày 2-6 |
|
Methotrexate |
15 mg, tiêm tủy sông * |
Ngày 2 |
|
Cytarabine |
15 mg, tiêm tủy sông * |
Ngày 7 |
|
Hydrocortisone |
30 mg, tiêm tủy sông * |
Ngày 2,7 |
|
G-CSF |
5 mcg/kg/ngày , tiêm dưới da |
ngày thứ 7 cho đến khi BCTT > 3,000 /mm3 |
Nhóm C**:
|
Thuốc |
Liều lượng, đường dùng |
Ngày dùng |
|
Pha trước tấn công: COP+ |
||
|
Cyclophosphamide |
300 mg/m2, tĩnh mạch |
Ngày 1 |
|
Vincristine |
1.0 mg/m2 (tối đa 2.0 mg), tĩnh mạch |
Ngày 1 |
|
Methylprednisolone |
60 mg/m2, đường uống/ tĩnh mạch |
Ngày 1- 7 (giảm liều trong 3 ngày) |
|
Methotrexate |
15mg, tiêm tủy sống* |
Ngày 1,3,5 |
|
Hydrocortisone |
15mg, tiêm tủy sống* |
Ngày 1,3,5 |
|
Cytarabine |
30mg, tiêm tủy sống* |
Ngày 1,3,5 |
|
Folinic acid |
15mg/m2, đường uống. mỗi 12h |
Ngày 2,4 |
|
Tấn công: |
||
|
COPADM 1&2:, COPADM1 được bắt đầu từ ngày thứ 8 của COP nếu không có chống chỉ định; COPADM2 bắt đầu khi tế bào máu ngoại vi hồi phục ( BCTT > 1,000/cm3 và TC >100,000/cm3), không nên bắt đầu trước ngày thứ 16 tính từ ngày đầu của COPADM1 và nên trì hoãn sau 48 giờ kể từ liều G- CSF cuối cùng |
||
|
Vincristine |
2.0 mg/m2 (tối đa 2.0mg), tĩnh mạch |
Ngày 1 |
|
Methylprednisolone |
60 mg/m2 ngày (chia 3 liều), đường uống/ tĩnh mạch |
Ngày 1- 5 (giảm dần liều trong 3 ngày) |
|
Methotrexate (HD) |
8 g/m2, tĩnh mạch, trong 4 h (Axit folinic bắt đầu từ giờ 24 của MTX, 15 mg/m2 mỗi 6h, 12 liều) |
Ngày 1 |
|
Cyclophosphamide |
250 mg/m2/12 giờ (500 mg/m2/ngày), tĩnh mạch |
Ngày 2- 4 |
|
Doxorubicin |
60 mg/m2, tĩnh mạch, trong 6h |
Ngày 2, sau liều cyclophosphamide đầu tiên |
|
Methotrexate Hydrocortisone Cytarabine G-CSF |
15mg, tiêm tủy sống* 15mg, tiêm tủy sống* 30mg, tiêm tủy sống* 5 mcg/kg/ngày, tiêm dưới da |
Ngày 2,4, 6 Ngày 2,4, 6 Ngày 2,4, 6 ngày thứ 7 cho đến khi BCTT > 3,000 /mm3 |
|
COPADM 2 bắt đầu khi BCTT > 1,000/cm3 và TC > 100,000/cm3 |
||
|
Củng cố: |
||
|
CYVE 1&2: bắt đầu khi BCTT > 1,000/cm3 và TC > 100,000/cm3 |
||
|
Cytarabine |
50 mg/m2/ngày, tĩnh mạch, liên tục trong 12 giờ (giờ 1- 12) |
Ngày 1-5 |
|
Cytarabine |
3000 mg/m2/ngày, tĩnh mạch, trong 3h (giờ 13- 15) |
Ngày 2-5 |
|
Etoposide |
200 mg/m2/ngày, tĩnh mạch, trong 2h (giờ 17- 19) |
Ngày 2-5 |
|
G-CSF |
5 mcg/kg/ngày, tiêm dưới da |
ngày thứ 7 cho đến khi BCTT> 3,000 /mm3 |
|
Duy trì: Khoảng cách giữa các đợt duy trì là 28 ngày |
||
|
Duy trì 1 (M1): bắt đầu khi BCTT > 1,000/cm3 và TC 100,000/cm3 |
||
|
Vincristine |
2.0 mg/m2 (tối đa 2.0 mg), tĩnh mạch |
Ngày 1 |
|
Methylprednisolone |
60 mg/m2/ngày (chia 3 liều), đường uống/ tĩnh mạch |
Ngày 1- 5 (giảm dần liều trong 3 ngày) |
|
Methotrexate (HD) |
8 g/m2, tĩnh mạch, trong 4h (Axit folinic bắt đầu từ giờ 24 của MTX, 15 mg/m2 mỗi 6h, 12 liều) |
Ngày 1 |
|
Cyclophosphamide |
500 mg/m2/ngày, tĩnh mạch |
Ngày 2,3 |
|
Doxorubicin |
60 mg/m2, tĩnh mạch, trong 6h |
Ngày 2, sau liều cyclophosphamide đầu tiên |
|
Methotrexate |
15mg, tiêm tủy sống* |
Ngày 2 |
|
Hydrocortisone |
15mg, tiêm tủy sống* |
Ngày 2 |
|
Cytarabine |
30mg, tiêm tủy sống * |
Ngày 2 |
|
Duy trì 2 (M2): bắt đầu khi BCTT > 1,000/cm3 và TC >100,000/ cm3 |
||
|
Cytarabine Etoposide |
50 mg/m2, tiêm dưới da, mỗi 12 giờ 150 mg/m2/ngày, tĩnh mạch, trong 90 phút |
Ngày 1-5 Ngày 1-3 |
|
Duy trì (M3): bắt đầu khi BCTT > 1,000/cm3 và TC >100,000/cm3 |
||
|
Methylprednisolone |
60 mg/m2/ngày, chia làm 2 lần/ngày, đường uống/tĩnh mạch |
Ngày 1-5 |
|
Vincristine |
2.0 mg/m2 (tối đa 2.0 mg), tĩnh mạch |
Ngày 1 |
|
Cyclophosphamide |
500 mg/m2/ngày, tĩnh mạch, trong 30 phút. |
Ngày 1,2 |
|
Doxorubicin |
60 mg/m2 (tĩnh mạch, trong 6 giờ). |
Ngày 1, sau cyclophosphamide |
|
Duy trì 4( M4): bắt đầu khi BCTT > 1,000/cm3 và TC > 100,000/cm3 |
||
|
Cytarabine Etoposide |
50 mg/m2/12 giờ, tiêm dưới da 150 mg/m2/ngày, tĩnh mạch, trong 90 phút |
Ngày 1-5 Ngày 1-3 |
* liều thay đổi theo tuổi
Liều lượng tiêm hóa chất khoang nội tủy theo tuổi
|
Tuổi |
MTX |
HC |
Cytarabine |
|
< 1 tuổi 1 tuổi 2 tuổi > 3 tuổi |
8 mg 10 mg 12 mg 15mg |
8 mg 10 mg 12 mg 15mg |
15 mg 20 mg 25 mg 30 mg |
** Đối với nhóm C: ngoài hóa trị như trên, có thể thêm Rituximab vào pha Tấn công và Củng cố
c. Anaplastic large cell lymphoma (ALCL)
 Phân nhóm điều trị trong ALCL
Phân nhóm điều trị trong ALCL
|
Nhóm |
Giai đoạn |
Phác đồ |
|
1 |
I, II, đã phẫu thuật cắt bỏ |
vA - B- A |
|
2 |
II- chưa phẫu thuật, III |
vA- B- A-B- A- B |
|
3 |
IV hoặc xâm lấn nhiều xương |
vAA - BB- CC- AA- BB- CC |
 Phác đồ BFM- NHL 90: gồm các đợt ( course) như sau
Phác đồ BFM- NHL 90: gồm các đợt ( course) như sau
|
Thuốc |
Liều lượng |
Ngày dùng |
|
Đợt điều trị giảm tế bào (Cytoreductive phase (course v)) |
|
|
|
Methylprednisone (uống/ tĩnh mạch) |
30 mg/m2 |
1-5 |
|
Cyclophosphamide, tĩnh mạch |
200 mg/m2 |
1-5 |
|
Course A Dexamethasone (uống) Ifosfamide (tĩnh mạch) Methotrexate (tĩnh mạch/ 24h) Methotrexate (tiêm tủy sống*) Cytarabine (tiêm tủy sống*) Methylprednisolone (tiêm tủy sống*) Cytarabine (tĩnh mạch/ 1h) Etoposide (tĩnh mạch/ 1h) |
10 mg/m2 800mg/m2 500 mg/m2 12 mg 30 mg 10 mg 150 mg/m2/12h 100 mg/m2 |
1-5 1-5 1 1 1 1 4,5 4,5 |
|
Course B Dexamethasone (uống) Cyclophosphamide (tĩnh mạch/1h) Methotrexate (tĩnh mạch/24h) Methotrexate (tiêm tủy sống*) Cytarabine (tiêm tủy sống*) Methylprednisolone (tiêm tủy sống*) Doxorubicin (tĩnh mạch/ 1h) |
10 mg/m2 200 mg/m2 500 mg/m2 12 mg 30 mg 10 mg 25 mg/m2 |
1-5 1-5 1 1 1 1 4,5 |
|
Course AA Dexamethasone (uống) Ifosfamide (tĩnh mạch) Vincristine (tĩnh mạch) Methotrexate (tĩnh mạch/24h) Methotrexate (tiêm tủy sống*) Cytarabine (tiêm tủy sống*) Methylprednisolone (tiêm tủy sống*) Etoposide (tĩnh mạch/1h) |
10 mg/m2 800 mg/m2 1,5mg/m2 (tối đa 2mg) 5000 mg/m2 6 mg 15 mg 5 mg 100 mg/m2 |
1-5 1-5 1 1 1,5 1,5 1,5 4,5 |
|
Course BB Dexamethasone (uống) Cyclophosphamide (tĩnh mạch/1h) Vincristine (tĩnh mạch) Methotrexate (tĩnh mạch/ 24h) Methotrexate (tiêm tủy sống*) Cytarabine (tiêm tủy sống*) Methylprednisolone (tiêm tủy sống*) |
10 mg/m2 200 mg/m2 1,5mg/m2 (tối đa 2mg) 5000 mg/m2 6 mg 15 mg 5 mg |
1-5 1-5 1 1 1,5 1,5 1,5 |
|
Doxorubicin (tĩnh mạch/1h) |
25 mg/m2 |
4,5 |
|
Course CC Dexamethasone (uống) Vinblastine (tĩnh mạch) Cytarabine (tĩnh mạch/3h) Methotrexate (tiêm tủy sống*) Cytarabine (tiêm tủy sống*) Methylprednisolone (tiêm tủy sống*) Etoposide (tĩnh mạch/1h) |
20 mg/m2 6 mg/m2 2000 mg/m2/12h 12 mg 30 mg 10 mg 150 mg/m2 |
1-5 1 1,2 5 5 5 3- 5 |
* Liều chỉnh theo tuổi
Liều thuốc tiêm khoang nội tủy
|
Tuổi |
Course AA, BB |
Course A,B, CC |
||||
|
MTX (mg) |
Ara-C (mg) |
Prednisolone (mg) |
MTX (mg) |
Ara-C (mg) |
Prednisolone (mg) |
|
|
< 1 tuổi |
3 |
8 |
2 |
6 |
16 |
4 |
|
1-2 tuổi |
4 |
10 |
3 |
8 |
20 |
6 |
|
2-3 tuổi |
5 |
13 |
4 |
10 |
26 |
8 |
|
≥ 3 tuổi |
5 |
15 |
5 |
12 |
30 |
10 |
d. U lympho không Hodgkin tế bào T: có thể dùng phác đồ Macao, phác đồ AALL 0434 ( của Children’s Oncology Group)...
Phác đồ MACAO: gồm các đợt hóa chất như sau, đợt kế tiếp bắt đầu khi tế bào máu ngoại vi hồi phục (BCTT > 1 G/L, SLTC > 100 G/L)
Đợt 1
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Methylprednisolone |
60mg/m2, tĩnh mạch |
1- 29 |
|
Vincristine |
1,5mg/m2, tĩnh mạch (tối đa 2mg) |
8,15,22,29 |
|
Cyclophosphamide |
1g/m2, tĩnh mạch |
8 |
|
Daunorubicin |
40mg/m2, tĩnh mạch |
15,22,29 |
|
L-asparaginase |
10 000UI/m2, tiêm bắp/ tĩnh mạch |
16, 18,20,22,24,26 |
|
Methotrexate |
3g/m2, tĩnh mạch, truyền trong 3h (giải độc bằng acid folic) |
8 |
|
Methotrexate Hydrocortisol |
8-15mg, tiêm tủy sông 8-15mg, tiêm tủy sống (liều lượng thay đổi theo tuổi) |
1, 8, 15 |
Đợt 2 và đợt 3: bắt đầu khi số lượng BC trung tính trên 1000 và Tiểu cầu trên 100 000. Ngày thứ 15 có thể hoãn tới ngày thứ 22 trong trường hợp có suy tủy (BC hạt dưới 500 và Tiểu cầu dưới 50 000) hoặc trong tình trạng lâm sàng không ổn định.
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Vincristine |
1,5mg/m2, tĩnh mạch (tối đa 2mg) |
15 |
|
Cyclophosphamide |
1g/m2, tĩnh mạch |
1 |
|
Cytarabin |
75mg/m2, tĩnh mạch |
2,3,4,5 và 8,9, 10,11 |
|
L-asparaginase |
25 000 UI/m2 , tiêm bắp/tĩnh mạch |
16, 23 |
|
Methotrexate |
3g/m2, tĩnh mạch, truyền trong 3h (giải độc bằng acid folic) |
1, 15 |
|
Methotrexate Hydrocortisol |
8-15mg, tiêm tủy sống 8-15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
2, 16 |
Đợt 4
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Mercaptopurin |
50 mg/m2, uống |
1-22 |
|
Methotrexate |
3g/m2, tĩnh mạch/3h (giải độc bằng acid folic) |
1, 15 |
|
Methotrexate Hydrocortisol |
8-15mg, tiêm tủy sống 8-15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
2, 16 |
Đợt 5
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Dexamethasone |
6 mg/m2 , uống/ tĩnh mạch |
1- 21 |
|
Vincristine |
1,5mg/m2, tĩnh mạch (tối đa 2mg) |
1, 8, 15 |
|
Adriamycine |
40 mg/m2, tĩnh mạch |
1, 8, 15 |
|
L-asparaginase |
10 000 UI/m2, tiêm bắp/ tĩnh mạch |
16, 18, 20 và 22, 24, 26 |
|
Cyclophosphamide |
1000 mg/m2, tĩnh mạch |
29 |
|
Cytarabin |
75mg/m2, tĩnh mạch/ tiêm dưới da |
29, 30, 31, 32 và 36, 37, 38, 39 |
|
Methotrexate Hydrocortisol |
8-15mg, tiêm tủy sống 8-15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
1, 29 |
Đợt 6:
6.1 : Phác đồ A vào tháng thứ 1, thứ 3 và thứ 5
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Methotrexate |
3g/m2, tĩnh mạch, truyền trong 3h (giải độc bằng acid folic) |
1 |
|
Vincristine |
1,5mg/m2, tĩnh mạch, (tối đa 2mg) |
1 |
|
L-asparaginase |
25 000 UI/m2, tiêm bắp/tĩnh mạch |
2 |
|
Mercaptopurin |
50 mg/m2, uống |
1-29 |
|
Methotrexate |
25 mg/m2, uống |
8, 15, 22 |
|
Methotrexate Hydrocortisol |
8-15mg, tiêm tủy sống 8-15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
2 |
6.2 : Phác đồ B vào tháng thứ 2, thứ 4 và thứ 6
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Methylprednisolone |
40 mg/m2, uống/ tĩnh mạch |
1-7 |
|
Cytarabine |
50 mg/m2, tiêm dưới da/ tĩnh mạch |
1-4 |
|
Mercaptopurin |
50 mg/m2, uống |
1-29 |
|
Methotrexate |
25 mg/m2, uống |
8, 15, 22 |
Đợt 7: ( điều trị duy trì)
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Mercaptopurin |
50 mg/m2/ ngày, uống |
Hàng ngày |
|
Methotrexate |
25 mg/m2/tuần 1 lần , uống |
Tuần 1 lần |
Thời gian điều trị duy trì: Giai đoạn điều trị duy trì bổ sung cho đợt điều trị để: Tổng thời gian điều trị của các giai đoạn I, II, III đủ 18 tháng hoặc tổng thời gian điều trị của các giai đoạn IV là 24 tháng
 Trong trường hợp bệnh giai đoạn IV có xân lấn não- màng não:
Trong trường hợp bệnh giai đoạn IV có xân lấn não- màng não:
- Tiêm tủy bằng 3 thuốc: MTX, ARAC, HSHC, liều theo lứa tuổi
- Điều trị tái tấn công (đợt 6):
Phác đồ A: tháng 1,2,3
Phác đồ B: tháng 4,5,6
- Xạ trị thần kinh: 18 Gy tới C2, giữa tháng thứ 5 và thứ 6
 Sử dụng Bactrim ( sulfamethoxazole - trimethoprim) để dự phòng viêm phổi do Pneumocystis carinii
Sử dụng Bactrim ( sulfamethoxazole - trimethoprim) để dự phòng viêm phổi do Pneumocystis carinii
Liều thuốc tiêm tủy sống theo lứa tuổi trong trưởng hợp xâm lấn não- màng não:
|
Tuổi |
Thuốc |
Liều lượng |
|
< 1 tuổi |
HSHC |
8mg |
|
Methotrexate |
8mg |
|
|
Aracytine ( Cytarabine) |
15mg |
|
|
1-2 tuổi |
HSHC |
10mg |
|
Methotrexate |
10mg |
|
|
Aracytine ( Cytarabine) |
20mg |
|
|
2- 3 tuổi |
HSHC |
12mg |
|
Methotrexate |
12mg |
|
|
Aracytine ( Cytarabine) |
25mg |
|
|
>3 tuổi |
HSHC |
15mg |
|
Methotrexate |
15mg |
|
|
Aracytine ( Cytarabine) |
30mg |
 Phác đồ AALL 0434: gồm các đợt hóa chất như sau, đợt kế tiếp bắt đầu khi tế bào máu ngoại vi hồi phục (BCTT > 0.75 G/L, SLTC > 75 G/L)
Phác đồ AALL 0434: gồm các đợt hóa chất như sau, đợt kế tiếp bắt đầu khi tế bào máu ngoại vi hồi phục (BCTT > 0.75 G/L, SLTC > 75 G/L)
 Nguy cơ thường
Nguy cơ thường
Đợt 1
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Vincristine |
1,5mg/m2, tĩnh mạch (tối đa 2mg) |
1, 8, 15, 22 |
|
Methylprednisone |
60mg/m2, tĩnh mạch |
1- d28 |
|
Daunorubicin |
25 mg/m2, tĩnh mạch |
1,8, 15, 22 |
|
L- asparaginase |
6000 UI/m2, tiêm bắp/ tĩnh mạch (6 mũi) |
4,6,8,10,12,14 |
|
Cytarabine |
30- 70mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
1 |
|
Methotrexate |
8- 15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
8 , 29 |
Đợt 2
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Intrathecal Methotrexate (IT MTX) |
8- 15mg, Tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
1, 8, 15, 22 |
|
Cyclophosphamide |
1000 mg/m2, tĩnh mạch |
1, 29 |
|
Cytarabine |
75mg/m2, tĩnh mạch/ dưới da |
1-4, 8-11, 29-32, 36-39 |
|
Mercaptopurine |
60 mg/m2, uống |
1-14, 29-42 |
|
Vincristine |
1,5mg/m2, tĩnh mạch (tối đa 2mg) |
15, 22, 43, 50 |
|
L- asparaginase |
2500 UI/m2, tiêm bắp/ tĩnh mạch |
15, 17, 19, 21, 23, 25, 27, 29, 31 |
Đợt 3
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Vincristine |
1,5mg/m2, tĩnh mạch, (tối đa 2mg) |
1, 11, 21, 31, 41 |
|
Methotrexate |
100 mg/m2/liều, sau đó tăng 50 mg/m2/ mỗi liều sau , tĩnh mạch |
1, 11, 21, 31, 41 |
|
L- asparaginase |
6000 UI/m2 tiêm bắp/ tĩnh mạch ( 9 mũi) |
2,4,6,8,10, 12,14,16,18 |
|
Methotrexate |
8- 15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
1, 31 |
Đợt 4
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Vincristine |
1,5mg/m2, tĩnh mạch (tối đa 2mg) |
1, 8, 15, 43, 50 |
|
Dexamethasone |
10 mg/m2, uống/ tĩnh mạch |
1-7,15-21 |
|
Doxorubicin |
25 mg/m2, tĩnh mạch |
1, 8, 15 |
|
L- asparaginase |
6000 UI/m2, tiêm bắp/ tĩnh mạch |
4,6,8,10,12,14 41,43,45,47,49, 51 |
|
Methotrexate |
8- 15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
1, 29, 36 |
|
Cyclophosphamide |
1000 mg/m2, tĩnh mạch |
29 |
|
Cytarabine (AraC) |
75mg/m2, tĩnh mạch/ dưới da |
29-32, 36-39 |
|
Mercaptopurine |
60 mg/m2, uống |
29- 42 |
Đợt 5
|
THUỐC |
LIỀU LƯỢNG, ĐƯỜNG DÙNG |
NGÀY DÙNG |
|
Vincristine |
1,5mg/m2, tĩnh mạch, (tối đa 2mg) |
1, 29, 57 |
|
Methylprednisone |
40mg/m2, uống, chia 3 lần/ngày |
1-5, 29-33, 57-61 |
|
Mercaptopurine |
75mg/m2, uống |
1-84 |
|
Methotrexate |
25mg/m2/liều, uống, hàng tuần |
8, 15, 22, 29, 36, 43, 50, 57, 64, 71,78 |
|
Methotrexate |
8- 15mg, tiêm tủy sống (Liều lượng thay đổi theo tuổi) |
1, 29 của 4 chu kỳ đầu |
 Nguy cơ cao: Theo phác đồ này các trường hợp nguy cơ cao sẽ được điều trị với 1 số thuốc mới
Nguy cơ cao: Theo phác đồ này các trường hợp nguy cơ cao sẽ được điều trị với 1 số thuốc mới
3.4. Các phác đồ khác
- Hiện nay có nhiều nhóm nghiên cứu đã đưa ra các phác đồ điều trị cho từng thể U lympho không Hodgkin có kết quả tốt, đưa lại tỷ lệ lui bệnh cao và kéo dài:
+ U lympho không Hodgkin dòng tế bào B: Điều trị như Lơ xê mi cấp dòng lympho tế bào B - phác đồ ANHL01P1/ COG...;
+ Anaplastic large cell lymphoma: International ALCL, ALCL 0311...
- Một số phác đồ khác và một số thuốc mới đang được nghiên cứu (đa số áp dụng ở người lớn và trẻ lớn): CHOP, CHOPE, CVP, EPOCH, DHAP, ICE (có hoặc không kết hợp với Rituximab)... (xem phần điều trị U lumpho không Hodgkin người lớn).
3.5. Xạ trị
Nói chung, xạ trị không được sử dụng rộng rãi. Xạ trị chỉ được chỉ định khi có các biến chứng cấp tính đe dọa tính mạng như hội chứng trung thất trước, hoặc trong phác đồ điều trị lymphoblastic lymphoma giai đoạn tiến triển có biểu hiện thâm nhiễm TKTW. Trong U lympho không Hodgkin dòng tế bào B có biểu hiện TKTW, xạ trị cũng không được sử dụng để điều trị hoặc dự phòng.
3.6. Điều trị bệnh tái phát
Bệnh nhân tái phát có tiên lượng xấu, bất kể vị trí tái phát, mô học khối u, các yếu tố tiên lượng ban đầu, phác đồ điều trị trước đó, hoặc thời gian từ lúc chẩn đoán đến khi tái phát. Vì lê do này, việc lựa chọn phác đồ điều trị ban đầu sao cho có hiệu quả nhất là rất quan trọng. Bệnh nhân tái phát được điều trị lại để tạo ra sự lui bệnh. Đối với lymphoblastic lymphoma và u lympho tế bào B, hóa trị liệu với ifosfamide, carboplatin và etoposide (VP-16) là một phác đồ điều trị cứu vãn để sau đó thu hoạch tế bào gốc tự thân. Việc bổ sung Rituximab ở những bệnh nhân U lympho tế bào B CD20 (+) có thể có hiệu quả để đạt được lui bệnh.
Thuốc:
- Rituximab 375 mg/m2 IV, ngày 1 và 3 của Course 1 và 2; và ngày 1 của course 3 (nếu có).
- Ifosfamide 3,000 mg/m2/ngày truyền tĩnh mạch trong 2 giờ trong 3 ngày (ngày 3,4,5), Mesna 600 mg/m2 IV truyền ngay cùng ifosfamide và giờ 3, 6, 9 and 12 sau ifosfamide.
- Carboplatin 635 mg/m2 truyền tĩnh mạch trong 1 giờ vào ngày 3.
- VP-16 100 mg/m2/ngày truyền tĩnh mạch trong 1 giờ vào ngày 3 (ngày 3,4,5).
- Lặp lại chu kỳ thứ hai 3 tuần kể từ chu kỳ đầu tiên, sau đó đánh giá lại bằng phương pháp chẩn đoán hình ảnh:
+ Nếu lui bệnh hoàn toàn hoặc lui bệnh một phần rất tốt thì tiến hành củng cố bằng ghép tế bào gốc tạo máu;
+ Nếu có đáp ứng, có thể điều trị thêm chu kỳ thứ 3 trước khi ghép, sau đó ghép tế bào gốc tạo máu;
+ Nếu kháng trị, chỉ định liệu pháp thay thế.
Sau khi lui bệnh hoàn toàn hoặc lui bệnh 1 phần rất tốt, phải củng cố bằng ghép tế bào gốc tạo máu. Bệnh nhân kháng hóa trị thường cũng không đáp ứng với ghép tế bào gốc tự thân. Bệnh nhân tái phát lần thứ hai có thể dùng các thuốc thay thế như Vinblastine (6 mg/m2/tuần trong 12 - 18 tháng) hoặc Cis-retinoic acid (1 mg/kg/ngày, uống chia thành 2 đến 3 lần trong 8 tuần), có hoặc không kèm interferon alpha (4.5 mega units (MU) tiêm dưới da cách ngày trong 8 tuần). Các thuốc này đã được báo cáo có thể duy trì lui bệnh ở một số bệnh nhân.
Ghép tế bào gốc đồng loài về mặt lê thuyết là thích hợp hơn vì nhiều nghiên cứu chứng minh tỷ lệ tái phát thấp hơn đáng kể nhờ hiệu ứng mảnh ghép chống lymphoma. Ghép đồng loài có xu hướng cải thiện tỷ lệ sống sót so với cấy ghép tự thân; tuy nhiên, nguy cơ tử vong liên quan đến ghép cao hơn. Tỷ lệ sống sót không mắc bệnh của ghép đồng loài dao động từ 24-68% và trong cấy ghép tự thân từ 22-46%. Phác đồ điều kiện hóa diệt tủy như CBV hoặc BEAM đã được sử dụng khi ghép tế bào gốc. Các phác đồ diệt tủy khác bao gồm chiếu xạ toàn thân, etoposide và cyclophosphamide. Sử dụng Interleukin-2 sau ghép có thể giảm tỷ lệ tái phát. Các phác đồ điều kiện hóa giảm liều với Busulfan và Fludarabine giúp giảm đáng kể tỷ lệ tử vong liên quan đến ghép và cải thiện kết quả lâu dài nói chung.
4. YẾU TỐ TIÊN LƯỢNG
Tiên lượng phụ thuộc vào kết quả từng phác đồ điều trị. Ngoài ra có các chỉ số sau có vài trò tiên lượng bệnh ban đầu (khi chưa tái phát).
4.1. Thuận lợi:
- Khu trú (giai đoạn I và II).
- LDH thấp (dưới 500 hoặc hai lần mức bình thường).
- Anaplastic large cell lymphoma không có biểu hiện ở tạng (không có khối trung thất, gan/ lách hoặc da).
4.2. Không thuận lợi:
- LDH cao (lớn hơn 500 hoặc gấp đôi mức bình thường).
- U lympho không Hodgkin dòng tế bào B thâm nhiễm cả tủy xương và sọ não.
- Đáp ứng kém với cyclophosphamide, vincristine, prednison (COP) trong điều trị U lympho không Hodgkin dòng tế bào B.
- U lympho tế bào B trung thất nguyên phát.
- Anaplastic large cell lymphoma có biểu hiện ở tạng (khối trung thất, gan/ lách hoặc da).
- Biểu hiện bệnh tối thiểu ở tủy xương lúc chẩn đoán.
5. ĐÁNH GIÁ, THEO DÕI
5.1. Khi chẩn đoán và trước điều trị
- Tiền sử, bệnh sử, lâm sàng.
- Xét nghiệm:
+ Tổng phân tích tế bào máu, hồng cầu lưới máu;
+ Sinh hóa máu: Chức năng gan, thận, Axit Uric, LDH, điện giải đồ...;
+ Đông máu huyết tương: Fibrinogen, PT, APTT, TT, D-Dimer. Bệnh nhân có kèm theo rối loạn đông máu cần làm thêm xét nghiệm : nghiệm pháp rượu, Von-Kaulla, Đàn hồi cục máu đồ (ROTEM), Fibrin monomer;
+ Chẩn đoán hình ảnh: X-quang tim phổi, siêu âm ổ bụng, điện tâm đồ, siêu âm tim, CT scan, FDG - PET scan, MRI…;
+ Hạch đồ/ u đồ / lách đồ;
+ Sinh thiết hạch, sinh thiết khối u, sinh thiết lách… làm giải phẫu bệnh;
+ Hóa mô miễn dịch mảnh sinh thiết để chẩn đoán xác định, chẩn đoán thể bệnh (marker bệnh lý theo chỉ định của khoa Tế bào tổ chức học dựa trên định hướng từ lam nhuộm HE);
+ Xét nghiệm vi sinh: HIV, HBV, HCV...;
+ Huyết tủy đồ, sinh thiết tủy xương: Đánh giá tình trạng tủy sinh máu, tình trạng xâm lấn tủy;
+ Công thức nhiễm sắc thể;
+ Gen bệnh;
+ Định nhóm máu hồng cầu: ABO, Rh(D);
+ Khám răng hàm mặt, tai mũi họng và các chuyên khoa khác (nếu bệnh nhân có biểu hiện bệnh lý ở các cơ quan khác) trước khi điều trị .
5.2. Trong qui trình điều trị
- Tổng phân tích tế bào máu, hồng cầu lưới máu.
- Sinh hóa máu: Chức năng gan, thận, Axit Uric, LDH, điện giải đồ...
- Đông máu huyết tương: Fibrinogen, PT, APTT, TT, D-Dimer, nghiệm pháp rượu, Von-Kaulla, Đàn hồi cục máu đồ (ROTEM), Fibrin monomer...
- Chẩn đoán hình ảnh: Xquang tim phổi, siêu âm ổ bụng, điện tâm đồ, siêu âm tim, CT scan, FDG - PET scan, MRI… trong quá trình điều trị, kết thúc điều trị và trước khi xạ trị.
- Nếu có tổ chức hạch/ u mới xuất hiện hoặc ở vị trí cũ nhưng tăng kích thước hoặc có biểu hiện nghi ngờ tái phát/ không lui bệnh/ chuyển thể bệnh: Hạch đồ/ u đồ/ lách đồ, sinh thiết hạch, sinh thiết khối u làm giải phẫu bệnh lại và làm hóa mô miễn dịch (marker bệnh lý theo chỉ định của khoa Tế bào- Tổ chức học dựa trên định hướng từ lam nhuộm HE).
- Xét nghiệm vi sinh: HIV, HBV, HCV... (nếu có truyền máu và chế phẩm: HBV mỗi 1 tháng, HCV, HIV mỗi 3 tháng).
- Huyết tủy đồ, sinh thiết tủy xương, công thức nhiễm sắc thể, gen bệnh: nghi ngờ tình trạng xâm lấn tủy, chuyển Lơ xê mi cấp...
- Định nhóm máu hồng cầu: ABO, Rh(D) (mỗi khi có chỉ định truyền máu).
- Khám các chuyên khoa khác (răng hàm mặt, tai mũi họng...) nếu bệnh nhân có biểu hiện bệnh lý ở các cơ quan đó.
- Theo dõi độc tính trên các cơ quan: Đánh giá chuyên sâu chức năng tim, chức năng phổi... (siêu âm tim, điện tim, đo chức năng hô hấp...).
5.3. Theo dõi sau điều trị
- Các trường hợp đặc biệt như: Hạch to, sốt, gầy sút cân… phải tái khám ngay.
- Tái khám: 1- 3 tháng/ lần trong 2 năm đầu. Sau đó, 6 tháng/ lần trong 3 năm tiếp và theo dõi hàng năm.
- Với mỗi lần tái khám:
+ Khám lâm sàng: Chú ý các triệu chứng lâm sàng, hạch to, hội chứng B;
+ Xét nghiệm: Tổng phân tích tế bào máu ngoại vi; Sinh hóa máu (bao gồm LDH, chức năng gan, thận, beta2-microglobulin, điện giải đồ...), máu lắng; Đông máu huyết tương; Chức năng tuyến giáp (đặc biệt nếu có xạ trị vùng trước đó); Chức năng tim (siêu âm tim màu, siêu âm doppler , điện tâm đồ...), chức năng phổi ( đo chức năng hô hấp, XQ phổi, CT phổi...)...; CT bụng ngực; PET, PET/CT mỗi 6 tháng trong 2 năm đầu, sau đó chụp khi có biểu hiện lâm sàng, Xét nghiệm tủy đồ ít nhất 2 năm/ lần;
+ Làm lại sinh thiết khi xuất hiện hạch to trở lại hoặc có tổn thương mới.
5.4. Theo dõi di chứng lâu dài
Nói chung, tất cả bệnh nhân nên được theo dõi sức khỏe suốt đời. Các xét nghiệm bao gồm: Xét nghiệm chức năng tuyến giáp cho bệnh nhân xạ trị vùng cổ, hormon sinh dục cho những bệnh nhân đã điều trị bằng tác nhân kiềm hóa hoặc xạ trị vùng chậu, đánh giá chức năng tim cho những bệnh nhân điều trị bằng anthracycline hoặc xạ trị lồng ngực, đánh giá chức năng phổi cho những bệnh nhân xạ trị lồng ngực. Tần suất theo dõi phụ thuộc vào nhiều yếu tố như phác đồ đã điều trị, độ tuổi và giới tính của bệnh nhân.
- Các trường hợp đặc biệt: Xuất hiện hạch to, sốt, gầy sút cân… phải tái khám ngay.
PHỤ LỤC
Hướng dẫn dùng Methotrexat liều cao
 Lưu ý:
Lưu ý:
- Tránh gây mê toàn thân trong 24 giờ trước đó.
- Tránh các loại thuốc có thể thay thế MTX-albumin như các loại thuốc kháng viêm như NSAIDS, salicylat, sulfonamid, ketoconazolevà ciprofloxacin.
- Tránh kết hợp với các thuốc gây độc cho thận.
- Trong trường hợp dự phòng Bactrim, tạm hoãn ít nhất 48 giờ trước và 5 ngày sau khi MTX liều cao.
Cách dùng HD-MTX 3g/m2/3h và HD-MTX 8 g/ m2/4h:
a. Kiềm hóa:
- Dịch truyền 3 lít/m²/ngày với tỷ lệ 1/3 bicar 1.4 % và 2/3 dịch (2/3 G 5% và 1/3 NaCl 0.9%).
- Thời gian kiềm hóa: 48h đối với HD-MTX 3g/m²/3h và 72h đối với HDMTX 8 g/m2/4h.
- Kiềm hóa: bắt đầu trước 2h dùng HD-MTX, truyền tĩnh mạch bicarbonate 1.4% khoảng 125 ml/m²/h, duy trì nước tiểu 100 ml/m2 + /h, giữ pH nước tiểu > 7 trước khi bắt đầu MTX liều cao và suốt quá trình truyền MTX. Theo dõi pH nước tiểu/6h hay sau mỗi lần đi tiểu, nếu pH < 7, truyền tiếp bicarbonate (1 mEq / kg ở dạng viên nang, hoặc 6 ml / kg bicar 1.4% = 1ml/kg bicar 8.4%).
- Xét nghiệm điện giải và creatinine hàng ngày trong trong suốt quá trình đa truyền, kiềm hóa.
b. Thuốc giải Acid folic:
- Liều dùng 15 mg/m2/lần cách mỗi 6h, có thể dùng đường uống hoặc tĩnh mạch.
- Thời gian bắt đầu 24h sau dùng MTX, tiêm kênh tủy thực hiện trước khi sử dụng thuốc giải.
- Tổng liều: 12 liều, tuy nhiên chỉnh liều thuốc giải theo nồng độ MTX ( xem phụ lục phác đồ FRALLE 2000).
Cách dùng HD-MTX TTM 24H:
a. Kiềm hóa:
- Dịch truyền 3 lít/m²/ ngày với tỷ lệ 1/3 bicar 1,4 % và 2/3 dịch (2/3 G 5% và 1/3 NaCl 0,9%).
- Thời gian kiềm hóa: liên tục 72h.
- Kiềm hóa: bắt đầu trước 2h dùng HD-MTX, truyền tĩnh mạch bicarbonate 1,4% khoảng 125 ml/m²/h, duy trì nước tiểu 100 ml/m2 + /h, giữ pH nước tiểu > 7 trước khi bắt đầu MTX liều cao và suốt quá trình đi MTX. Theo dõi pH nước tiểu/6h hay sau mỗi lần đi tiểu, nếu pH < 7 truyền tiếp bicarbonate (1 mEq/kg ở dạng viên nang, hoặc 6 ml/kg bicar 1,4% = 1ml / kg bicar 8,4%).
- Điện giải đồ và creatinine hàng ngày trong trong suốt quá trình đa truyền, kiềm hóa.
- MTX liều cao 8 g/m²/24h được sử dụng như sau: 1,6 g/m2 pha trong 100-250 mL G5% truyền tĩnh mạch trong 30 phút, sau đó 6.4 g/m² pha trong 500-1000 mL G5% truyền tĩnh mạch trong 23h30p (chỉnh liều theo cân nặng ở trẻ em, khoảng 125 ml/m2/h).
b. Thuốc giải Acid folinic:
- Liều dùng 15 mg/m²/lần cách mỗi 6h, có thể dùng đường uống (hay TMC hay TB).
- Tổng liều: 12 liều, tuy nhiên chỉnh liều thuốc giải theo nồng độ MTX (xem phụ lục phác đồ FRALLE 2000).
TÀI LIỆU THAM KHẢO
1. Đỗ Trung Phấn (2008), “U lympho ác tính”, Tế bào gốc và bệnh lý tế bào gốc tạo máu, Nhà xuất bản y học, Tr 358-374.
2. Nguyễn Anh Trí (2004), “Điều trị u lymph ác tính không Hodgkin”, Điều trị các bệnh ác tính cơ quan tạo máu, Nhà xuất bản y học. Tr 22-38.
3. Non Hodgkin lymphoma - NCCN guidelines 2.2013.
4. WHO classification of tumours of haematopoietic and lymphoid tissues 2008.
5. “Neoplasic lymphoid diseases”, William hematology 8th- 2010, chapter 92, 93, 94, 99, 100, 101, 102, 103, 104, 105, 106.
6. John P. Greer, Michael E. Williams (2009),“Non-Hodgkin Lymphoma in Adults”, Wintrobes clinical hematology 12th editition, 2145-2194.
7. John G. Gribben (2009), “Clinical manifestation, staging, and treatment of indolent non Hodgkin lymphoma”, Hoffman: Hematology: Basic Principles and Practice, 5th ed, chapter 79.
8. Kieron Dunleavy, Wyndham H. Wilson (2009), “Diagnosis and treatment of aggressive non Hodgkin lymphoma”, Hoffman: Hematology: Basic Principles and Practice, 5th ed, chapter 80.
9. United Kingdom Children's Cancer Study Group (UKCCSG) UKCCSG FAB LMB 96 9601 protocol.
10. French LMT 96 Protocol.
11. CHILDREN’S ONCOLOGY GROUP AALL0434 Intensified Methotrexate, Nelarabine (Compound 506U78; IND # 52611) and Augmented BFM Therapy for Children and Young Adults with Newly Diagnosed T-cell Acute Lymphoblastic Leukemia (ALL) or T-cell Lymphoblastic Lymphoma.
12. Katherine A. Janeway, M.D.Manual of Pediatric Oncology & Hematology 5ed 2010, chapter 19, 20.
33. LƠ XÊ MI LYMPHO MẠN (Bệnh bạch cầu mạn dòng lympho)
1. ĐẠI CƯƠNG
- Lơ xê mi lympho mạn (Chronic Lymphocytic Leukemia - CLL) là bệnh lý tăng sinh lympho ác tính đặc trưng bởi sự tích lũy các tế bào lympho B trưởng thành, kích thước nhỏ trong máu ngoại vi, tủy xương và hạch. Theo xếp loại của Tổ chức Y tế Thế giới (WHO-2016), lơ xê mi lympho mạn và u lympho tế bào nhỏ (small lymphocytic lymphoma) là 2 biểu hiện khác nhau của cùng một bệnh.
- Bệnh gặp nhiều hơn ở châu Âu và Mỹ, ít gặp ở châu Á; chiếm khoảng 0,8% trong tổng số các bệnh ung thư nói chung. Tại Mỹ tỷ lệ mắc Lơ xê mi lympho mạn trung bình là 2,7/100.000 người, tỷ lệ này dao động từ < 1 đến 5,5/100.000 người trên toàn Thế giới. Bệnh hay gặp ở người lớn tuổi, nam nhiều hơn nữ (gấp khoảng 2,8 lần).
2. NGUYÊN NHÂN GÂY BỆNH
Đến nay, chưa thấy yếu tố đơn độc nào là nguyên nhân gây bệnh. Các nghiên cứu cho rằng việc tiếp xúc lâu với các hóa chất nông nghiệp, chất độc màu da cam và làm việc lâu dài trong môi trường điện từ trường, nhiễm các virus viêm gan C, Epstein-Barr virus có thể làm tăng nguy cơ mắc bệnh. Đã có một vài báo cáo về các gia đình có nhiều người cùng mắc lơ xê mi lympho mạn.
3. TRIỆU CHỨNG
3.1. Lâm sàng
- Có đến 25% người bệnh không có triệu chứng lúc chẩn đoán, thường được phát hiện tình cờ là tăng tế bào lympho trong máu ngoại vi không giải thích được. Một số khác chỉ có dấu hiệu mệt mỏi hoặc hạn chế hoạt động thể lực. Ở giai đoạn muộn, người bệnh có thể sụt cân, nhiễm trùng tái diễn, xuất huyết do giảm tiểu cầu và thiếu máu.
- Trên 80% có hạch to, phần lớn ở vùng cổ, thượng đòn và nách; có thể từ một hạch rất nhỏ cho đến nhiều hạch lớn. Hạch không đau, không dính, mật độ chắc. Một tỷ lệ khá lớn có hạch ổ bụng có thể phát hiện được trên phim chụp cắt lớp vi tính.
- Lách to ở 50% người bệnh, thường to nhẹ đến vừa. Đôi khi lách to có thể gây cường lách, dẫn đến thiếu máu và giảm tiểu cầu.
- Ít gặp gan to và thâm nhiễm ngoài hạch, một số có biểu hiện viêm mũi mạn tính do thâm nhiễm các tế bào dòng lympho.
Các triệu chứng tiến triển từ từ, mờ nhạt trong thời gian dài, người bệnh ít để ý.
3.2. Xét nghiệm
a. Máu ngoại vi
- Tế bào lympho trên 5 G/L, hầu hết tăng trên 10 G/L, một số trên 100 G/L. Hình thái tế bào giống tế bào lympho trưởng thành, ngoài ra có thể có tế bào lympho kích thước lớn dạng tiền lympho nhưng không vượt quá 55% tổng số tế bào lympho.
- Hemoglobin giảm, càng về sau càng rõ; hồng cầu bình sắc, kích thước bình thường; tiểu cầu lúc đầu có thể bình thường, về sau giảm dần; bạch cầu hạt giảm nặng.
b. Tủy xương
Dòng lympho chiếm trên 30% các tế bào có nhân trong tủy xương. Hình ảnh các tế bào lympho xâm lấn một phần hoặc toàn bộ mô tủy sinh máu. Ở giai đoạn muộn thường thấy kiểu xâm lấn lan tỏa.
c. Hạch
Cấu trúc hạch bị phá hủy bởi sự xâm lấn của các tế bào lympho nhỏ hình dạng như trong máu ngoại vi. Mô bệnh học hạch tương tự như u lympho độ ác tính thấp.
d. Xét nghiệm miễn dịch
- Các tế bào lơ xê mi lympho mạn thường dương tính mạnh với CD5, CD19, CD23; dương tính yếu với CD20, CD79b, immunoglobulin bề mặt và một trong hai loại chuỗi nhẹ kappa hoặc lambda; âm tính với CD10, CD103.
- Nghiệm pháp Coombs trực tiếp dương tính ở những người bệnh có biểu hiện hoặc tiềm ẩn nguy cơ tan máu tự miễn.
- Có thể giảm nồng độ các globulin miễn dịch và suy giảm chức năng của tế bào T trong giai đoạn bệnh tiến triển.
- Điện di protein huyết thanh có thể gặp hình ảnh tăng gamma globulin miễn dịch đơn dòng (khoảng 5% người bệnh) hoặc đa dòng (15%).
đ. Một số bất thường di truyền tế bào trong Lơ xê mi lympho mạn
- Nhóm tiên lượng tốt: Đột biến gen IGHV, del 13q; đột biến gen Rb, Mir-15a, Mir-16-1.
- Nhóm tiên lượng xấu: Trisomy 12, del 11q, del 17p, del 6q; đột biến gen TP53, ATM, mdm2.
4. CHẨN ĐOÁN
4.1. Chẩn đoán xác định
Theo tiêu chuẩn của Viện Ung thư Quốc gia Hoa kỳ (NCCN phiên bản 3.2018), chẩn đoán CLL bao gồm 2 tiêu chuẩn sau:
- Số lượng tế bào lympho B trưởng thành, kích thước nhỏ trong máu ngoại vi tăng > 5G/l, tỷ lệ prolymphocyte ≤ 55%.
- Chứng minh được tính chất đơn dòng của lympho B trong máu ngoại vi bằng kỹ thuật flow cytometry (CD5+, CD19+, CD20+, CD23+, ZAP-70, CD10-, CD103-, chuỗi nhẹ Kappa hoặc Lambda).
4.2. Chẩn đoán giai đoạn
Bảng 37. Giai đoạn theo Rai (1975)
|
Nguy cơ |
Giai đoạn |
Triệu chứng |
|
Thấp |
0 |
Tăng tế bào lympho |
|
Trung bình |
1 |
Tăng tế bào lympho và hạch to |
|
2 |
Tăng tế bào lympho và gan/lách to kèm theo hạch to hoặc không |
|
|
Cao |
3 |
Tăng tế bào lympho và Hb < 11 g/dL kèm theo gan, lách, hạch to hoặc không |
|
4 |
Tăng tế bào lympho và tiểu cầu < 100 G/L kèm theo gan, lách, hạch to hoặc không |
Bảng 38. Giai đoạn theo Binet (1981)
|
Giai đoạn |
Triệu chứng |
|
A |
Hb ≥ 10 g/dL, Tiểu cầu ≥ 100 G/L, < 3 nhóm hạch to |
|
B |
Hb ≥ 10 g/dL, Tiểu cầu ≥ 100 G/L, ≥ 3 nhóm hạch to |
|
C |
Hb < 10 g/dL hoặc Tiểu cầu < 100 G/L bất kể gan lách hạch to hay không |
4.3. Chẩn đoán phân biệt
- U lympho tế bào nhỏ: Hạch to nhiều, lách to, tế bào lympho máu < 5 G/L, chẩn đoán xác định bằng sinh thiết và hóa mô miễn dịch mô hạch.
- Tăng sinh lympho B đơn dòng: Tăng sinh lympho B máu ngoại vi nhưng < 5 G/L, không thiếu máu, không giảm tiểu cầu, hạch và lách không to.
- Lơ xê mi tế bào tiền lympho B: Tỷ lệ tế bào tiền lympho B > 55% ở máu ngoại vi và tủy xương, hình thái tế bào lớn hơn, non hơn, có thể có hạt nhân.
- Lơ xê mi tế bào tóc: Có tế bào “tóc” ở máu ngoại vi, CD5 âm tính, bạch cầu mono thường giảm.
- U lympho tế bào lympho-plasmo: Tế bào lympho máu ngoại vi có thể bình thường hoặc tăng ít, sinh thiết hạch thấy tế bào lympho-plasmo, CD5 và CD23 âm tính.
5. ĐIỀU TRỊ
5.1. Điều trị bệnh chính
- Với những người bệnh giai đoạn sớm, bệnh ổn định (Binet A, B và Rai 0, I, II không có biểu hiện tiến triển của bệnh) thì không cần thiết phải điều trị ngay, theo dõi các dấu hiệu lâm sàng và xét nghiệm 3 tháng một lần.
- Chỉ định điều trị hóa chất với những người bệnh giai đoạn Binet A, B và Rai 0, I, II có dấu hiệu bệnh tiến triển; Binet C và Rai III, IV.
- Biểu hiện tiến triển của bệnh gồm: Triệu chứng B, thiếu máu và giảm tiểu cầu do xâm lấn tủy xương, gan lách hạch to nhiều, thời gian tăng gấp đôi số lượng tế bào lympho dưới 6 tháng (với người bệnh có số lượng tế bào lympho > 30 G/L), tan máu và giảm tiểu cầu miễn dịch đáp ứng kém với corticoid.
5.1.1. Bệnh nhân có đột biến TP53 và hoặc del(17p): dùng thuốc điều trị đích
- Ibrutinib hoặc thuốc ức chế Bruton’s tyrosin kinase (BTK) khác đến khi bệnh tiến triển hoặc phải dừng thuốc do tác dụng phụ.
- Ibrutinib hoặc thuốc ức chế BTK khác + rituximab đến khi bệnh tiến triển.
5.2.1. Bệnh nhân không có đột biến TP53 và del(17p)
a. Không có đột biến IGHV: dùng thuốc điều trị đích
- Ibrutinib hoặc thuốc ức chế BTK khác đến khi bệnh tiến triển hoặc phải dừng thuốc do tác dụng phụ.
- Ibrutinib hoặc thuốc ức chế BTK khác + rituximab đến khi bệnh tiến triển.
b. Có đột biến IGHV
b1. Phác đồ cho người bệnh dưới 65 tuổi, chức năng tim gan thận bình thường (dùng một trong các phác đồ)
- FCR (fludarabine + cyclophosphamide + rituximab).
- FCO (fludarabine + cyclophosphamide + obinutuzumab).
- BR (bendamustine + rituximab).
- BO (bendamustine + obinutuzumab).
- Ibrutinib hoặc thuốc ức chế BTK khác.
- Ibrutinib hoặc thuốc ức chế BTK khác + rituximab.
Lưu ý:
- Các phác đồ trên có thể dùng tối đa đến 6 đợt (tùy theo đáp ứng của từng người bệnh). Khoảng cách giữa 2 đợt điều trị thường là 28 ngày.
- Khuyến cáo: Khi quyết định điều trị thì nên xác định rõ mục tiêu điều trị là gì (ví dụ để làm giảm số lượng bạch cầu lympho, hoặc để làm cho lách nhỏ lại…); sau khi giải quyết được mục tiêu đề ra thì có thể tạm ngừng và chuyển sang uống duy trì chlorambucil 2 mg/ngày.
b2. Phác đồ cho người bệnh trên 65 tuổi (dùng một trong các phác đồ)
- BR (bendamustine + rituximab).
- Chlorambucil + obinutuzumab trong 12 tháng.
- Ibrutinib hoặc thuốc ức chế BTK khác.
- Chlorambucil 2-4mg uống hàng ngày.
Lưu ý: Có thể điều trị 2-3 đợt tùy thuộc vào đáp ứng lâm sàng và xét nghiệm của từng người bệnh (ví dụ lách và hạch nhỏ lại, số lượng bạch cầu lympho giảm xuống…); sau khi giải quyết được mục tiêu điều trị thì nên tạm ngừng và chuyển sang uống duy trì Chlorambucil.
5.2. Điều trị bệnh tái phát/kháng thuốc
a. Tái phát trong vòng 24-36 tháng từ khi điều trị hàng 1:
- Nếu chưa điều trị thuốc ức chế BTK trước đó: Dùng thuốc ức chế BTK đơn trị hoặc phối hợp kháng thể đơn dòng kháng CD20.
- Nếu không có bất thường del(17p) hoặc đột biến TP53: có thể dùng các hóa chất chưa được điều trị trước đó kết hợp kháng thể đơn dòng kháng CD20 (ví dụ nếu ban đầu điều trị FCR thì có thể chuyển BR hoặc ngược lại).
- Cân nhắc ghép tế bào gốc đồng loài khi đạt lui bệnh.
b. Tái phát sau 24-36 tháng từ khi điều trị hàng 1:
- Nếu có bất thường del(17p) hoặc đột biến TP53: Dùng thuốc điều trị đích (ibrutinib hoặc thuốc ức chế BTK khác).
- Nếu không có bất thường del(17p) hoặc đột biến TP53: Có thể dùng lại phác đồ hóa trị ban đầu kết hợp kháng thể đơn dòng kháng CD20.
5.3. Điều trị biến chứng
a. Tan máu tự miễn và giảm tiểu cầu miễn dịch do xuất hiện tự kháng thể
- Methylprednisolone: 2-4mg/ kg cân nặng/ ngày, giảm dần liều và dừng khi người bệnh không còn biểu hiện tan máu và giảm tiểu cầu miễn dịch.
- Rituximab truyền tĩnh mạch 375mg/m2 da /tuần x 4 tuần.
- Cắt lách nếu điều trị nội khoa không đáp ứng.
b. Điều trị nhiễm trùng do suy giảm miễn dịch
- Kháng sinh, chống nấm, kháng virus.
- Có thể dự phòng bằng truyền globulin miễn dịch.
6. Các xét nghiệm chẩn đoán, đánh giá và theo dõi điều trị
- Tổng phân tích tế bào máu ngoại vi, huyết tủy đồ, sinh thiết tủy xương.
- Sinh hóa máu: Glucose, chức năng gan, thận, điện giải đồ...
- Đông máu toàn bộ, rotem…
- Các xét nghiệm miễn dịch, di truyền: flowcytometry, nhiễm sắc thể, đột biến gen…
- Vi sinh: Xét nghiệm virus, vi khuẩn, vi nấm, kháng sinh đồ…
- Huyết thanh học nhóm máu.
- Xét nghiệm tế bào và tổng phân tích nước tiểu.
- Chẩn đoán hình ảnh, thăm dò chức năng.
- Và các xét nghiệm khác tùy theo bệnh nền và bệnh mắc kèm của người bệnh.
6. ĐÁNH GIÁ ĐÁP ỨNG ĐIỀU TRỊ (theo bảng 39)
Bảng 39. Các tiêu chuẩn đánh giá đáp ứng điều trị
|
Thông số |
CR (lui bệnh hoàn toàn) |
PR (lui bệnh 1 phần) |
PD (bệnh tiến triển) |
SD (ổn định) |
|
Nhóm A: Hạch |
Không có hạch nào >1,5cm |
Giảm 50% trở lên |
Tăng 50% trở lên |
Không thay đổi quá 50% |
|
Gan, lách |
Gan không to Lách < 13 cm |
Giảm 50% trở lên |
Tăng 50% trở lên |
Không thay đổi quá 50% |
|
Triệu chứng toàn thân |
Không có |
Có một triệu chứng bất kỳ |
Có một triệu chứng bất kỳ |
Có một triệu chứng bất kỳ |
|
Lympho máu |
Số lượng bình thường |
Giảm trên 50% so với ban đầu |
Tăng 50% trở lên so với ban đầu |
Không thay đổi quá 50% |
|
Nhóm B Tiểu cầu |
> 100 G/L |
> 100 G/L hoặc tăng 50% trở lên so với ban đầu |
Giảm 50% trở lên so với ban đầu do CLL |
|
|
Hemoglobin |
> 110 g/L (không do truyền máu hoặc dùng EPO) |
> 110 g/L hoặc tăng 50% trở lên so với ban đầu |
Giảm trên 20 g/L so với ban đầu do CLL |
< 110 g/L hoặc không tăng quá 50% so với ban đầu hoặc giảm không quá 20 g/L |
|
Tủy xương (++) |
Số lượng tế bào tủy bình thường, không còn tế bào CLL, không xuất hiện các nang lympho B |
Xuất hiện tế bào CLL, hoặc các nang lympho B, hoặc không được làm xét nghiệm tủy xương |
Tăng số lượng tế bào CLL trên 50% (đánh giá trên sinh thiết tủy xương) |
Tình trạng thâm nhiễm tủy xương không thay đổi |
|
Bạch cầu hạt trung tính (không dùng G-CSF) |
> 1,5 G/L |
> 1,5 G/L hoặc cải thiện trên 50% so với ban đầu |
|
|
- Thông số nhóm A xác định gánh nặng của khối u.
- Thông số nhóm B xác định chức năng của hệ tạo máu (hoặc tủy xương).
- CR (Complete remission - Lui bệnh hoàn toàn): Đáp ứng tất cả các tiêu chuẩn.
- PR (partial remission - Lui bệnh một phần): Ít nhất 2 tiêu chuẩn nhóm A và cải thiện 1 tiêu chuẩn nhóm B (nếu trước đó có bất thường); nếu cả nhóm A và B chỉ có 1 tiêu chuẩn bất thường trước điều trị, chỉ cần cải thiện 1 trong 2 tiêu chuẩn đó.
- PD (bệnh tiến triển): Ít nhất 1 tiêu chuẩn nhóm A hoặc nhóm B.
- SD (bệnh ổn định): Đáp ứng tất cả các tiêu chuẩn.
7. TIÊN LƯỢNG (theo bảng 40)
Bảng 40. Các yếu tố có giá trị tiên lượng trong Lơ xê mi lympho mạn
|
Yếu tố tiên lượng tốt |
Yếu tố tiên lượng xấu |
|
Giai đoạn sớm theo Rai hoặc Binet |
Giai đoạn muộn theo Rai hoặc Binet |
|
Xâm lấn kiểu khe kẽ hoặc thành nốt của lympho B trong tủy xương |
Xâm lấn kiểu lan tỏa của lympho B trong tủy xương |
|
Thời gian tế bào lympho tăng gấp đôi > 6 tháng |
Thời gian tế bào lympho tăng gấp đôi < 6 tháng |
|
CD38 âm tính |
CD38 dương tính |
|
Đột biến các gen chuỗi nặng immunoglobulin |
Không có đột biến các gen chuỗi nặng immunoglobulin |
|
ZAP-70 âm tính |
ZAP-70 dương tính |
|
Del 13q14 |
Del 11q23 |
|
Đột biến IGHV |
Del 17p hoặc đột biến TP53 |
|
|
Tăng TNF-α, β2 microglobulin, LDH |
Phác đồ điều trị chi tiết
|
Phác đồ |
Cách dùng |
|
FCR (fludarabine + cyclophosphamide + rituximab) |
Fludarabine 25mg/m2 da/ ngày x 3 ngày (ngày 1,2,3); Cyclophosphamide 250mg/m2 da/ngày x 3 ngày (ngày 1,2,3); Rituximab 375mg/m2 da (ngày 0). |
|
FCO (fludarabine + cyclophosphamide + obinutuzumab) |
Fludarabine 25mg/m2 da/ngày x 3 ngày (ngày 1,2,3); Cyclophosphamide 250mg/m2 da/ngày x 3 ngày (ngày 1,2,3); Obinutuzumab truyền tĩnh mạch 100mg vào ngày 1, 900mg vào ngày 2 và 1000mg vào ngày 8, 15 của đợt điều trị đầu tiên; từ đợt 2 trở đi, Obinutuzumab dùng 1000mg duy nhất ngày 1 |
|
BR (bendamustine + rituximab) |
Bendamustine 90 mg/m2 da x 2 ngày (ngày 1, 2); Rituximab 375 mg/m2 da (ngày 0) |
|
BO (bendamustine + obinutuzumab) |
Bendamustine 90 mg/m2 da x 2 ngày (ngày 1, 2); Obinutuzumab truyền tĩnh mạch 100mg vào ngày 1, 900mg vào ngày 2 và 1000mg vào ngày 8, 15 của đợt điều trị đầu tiên; từ đợt 2 trở đi, Obinutuzumab dùng 1000mg duy nhất ngày 1 |
|
Ibrutinib đơn trị |
Ibrutinib 420mg, uống hàng ngày cho đến khi bệnh tiến triển hoặc phải dừng thuốc do tác dụng phụ |
|
Chlorambucil + obinutuzumab |
Obinutuzumab truyền tĩnh mạch 100mg vào ngày 1, 900mg vào ngày 2 và 1000mg vào ngày 8, 15 của đợt điều trị đầu tiên; từ đợt 2 trở đi, Obinutuzumab dùng 1000mg duy nhất ngày 1; Chlorambucil uống 0.5 mg/ kg cân nặng vào ngày 1, 15 |
|
Chlorambucil đơn trị |
Chlorambucil 2-4mg uống hàng ngày |
TÀI LIỆU THAM KHẢO
1. Hallek M, Cheson BD, Catovsky D et al. (2008), “Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute Working Group 1996 guidelines”. Blood; 111: 5446-5456.
2. Paolo Strati, Nitin Jain et al. (2018), ‘Chronic lymphocytic leukemia : diagnosis and treatment’. Mayo Clini Proc; 93(5): 651-664.
3. Hallek M, Fischer K, Fingerle-Rowson G et al. (2010), “Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukemia: a randomised, open-label, phase III trial”. Lancet; 376: 1164-1174.
4. Robak T, Dmoszynska A, Solal-Celigny P et al. (2010), “Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia”. J Clin Oncol; 28: 1756-1765.
5. Knauf WU, Lissichkov T, Aldaoud A et al. (2009), “Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia”. J Clin Oncol; 27: 4378-4384.
34. LƠ XÊ MI TẾ BÀO TÓC
1. ĐẠI CƯƠNG
- Lơ xê mi tế bào tóc (Hairy Cell Leukemia - HCL) là một bệnh ác tính của tế bào dòng lympho B. Bệnh có hai đặc điểm nổi bật là lách to và giảm các dòng tế bào máu.
- Tế bào bệnh có nguồn gốc từ lympho B với các kháng nguyên biệt hoá: CD19, CD20, CD22 và các Ig bề mặt, đặc biệt IgG3, IgA.
2. NGUYÊN NHÂN
Nguyên nhân chưa được biết rõ, nhiễm tia xạ, chất hóa học sử dụng trong nông nghiệp hoặc bụi gỗ, hay nhiễm trùng tăng bạch cầu đơn nhân cũng có thể là yếu tố nguy cơ gây bệnh.
3. TRIỆU CHỨNG
3.1. Lâm sàng
Một số bệnh nhân có thể không có triệu chứng tại thời điểm chẩn đoán. Các triệu chứng điển hình gồm:
- Thiếu máu (khoảng 25% người bệnh).
- Nhiễm trùng cơ hội.
- Lách to thường độ II, độ III, mật độ chắc.
- Gan to (khoảng 20% người bệnh).
- Hạch to chỉ có ở 10% người bệnh Lơ xê mi tế bào tóc, chủ yếu là các hạch ở sâu (trung thất và ổ bụng).
3.2. Cận lâm sàng
- Giảm các dòng tế bào máu: Trong đó có khoảng 50% người bệnh giảm cả 3 dòng, các người bệnh còn lại có biểu hiện giảm 1 hoặc 2 dòng tế bào máu. Chủ yếu giảm bạch cầu, thường gặp giảm bạch cầu mono. Chỉ 10% người bệnh có tăng bạch cầu.
- Tế bào tóc ở máu ngoại vi xuất hiện ở 90% các trường hợp với đặc điểm: Tế bào kích thước bằng hoặc gấp 2 lần lympho trưởng thành, nguyên sinh chất bắt màu kiềm nhạt, có các sợi nguyên sinh chất tỏa ra xung quanh gợi hình ảnh “sợi tóc”, nhân có các hạt nhân rõ.
- Tăng nồng độ các cytokin đặc biệt là TNF- α.
- Xét nghiệm tuỷ xương:
+ Tuỷ đồ: Thường rất khó hút được dịch tủy. Trên tiêu bản tủy có thể thấy tế bào “tóc” tăng sinh lấn át các dòng tế bào khác và tế bào mỡ, tế bào càng trưởng thành thì “tóc” càng rõ. Đôi khi gặp thể giảm sinh tuỷ;
+ Mô bệnh học tủy xương: Tủy thường giàu tế bào, tăng sinh tế bào tóc lan tỏa hoặc tập trung thành từng cụm lẫn giữa các khoang sinh máu. Các tế bào sinh máu khác như hồng cầu, bạch cầu hạt và mẫu tiểu cầu giảm sinh. Khoảng 10-20% người bệnh giảm mật độ tế bào tủy. Đôi khi gặp tủy mỡ hóa gần như hoàn toàn, xen kẽ là các tế bào tóc.
- Hoá học tế bào và hóa mô miễn dịch: Hầu hết các tế bào dương tính với acid phosphate kháng tartrate (Tartrate-resistant acid photphate - TRAP), CD20, DBA.44; dương tính đồng thời 2 marker TRAP và DBA.44 có độ đặc hiệu cao trong chẩn đoán.
- Sử dụng kỹ thuật đếm tế bào dòng chảy (Flow Cytometry): Cho thấy tế bào tóc dương tính mạnh với các marker: CD45, CD19, CD20, CD22, CD11c, CD25, CD103, CD123; âm tính với CD5 và CD23. Đây là xét nghiệm quan trọng giúp chẩn đoán cũng như theo dõi tồn dư tối thiểu của bệnh sau điều trị.
- Bất thường nhiễm sắc thể (NST): Gặp khoảng 2/3 người bệnh, thường là các NST 1, 2, 5, 6, 11, 14, 19 và 20. Trong đó, bất thường nhiễm sắc thể số 5 gặp ở 40% trường hợp, thường là trisomy 5 hoặc mất đoạn gen trong vùng 5q13.
- Đột biến gen BRAF V600E: Rất đặc hiệu cho chẩn đoán, gặp ở hầu hết người bệnh (khoảng 95%) và có thể được phát hiện bằng kỹ thuật giải trình tự hoặc gián tiếp thông qua protein BRAF bằng phương pháp nhuộm hóa mô miễn dịch.
- Các xét nghiệm khác: Tăng men gan (19%), tăng ure máu (27%), tăng gamma globulin đa dòng (18%). Giảm gamma globulin máu hiếm gặp.
- Các xét nghiệm sử dụng trước và trong điều trị:
+ Tổng phân tích tế bào máu;
+ Hóa sinh máu: Glucose, chức năng gan, thận, điện giải đồ;
+ Đông máu cơ bản;
+ Định lượng globulin miễn dịch;
+ Marker virus viêm gan B: Khi điều trị với Rituximab có nguy cơ làm virus tái hoạt động;
+ Siêu âm ổ bụng: Đánh giá kích thước lách.
4. CHẨN ĐOÁN BỆNH
4.1. Chẩn đoán xác định
- Lâm sàng: Lách to, thiếu máu, nhiễm trùng cơ hội.
- Xét nghiệm:
+ Giảm 3 dòng tế bào máu;
+ Tuỷ: Giàu tế bào, chủ yếu tế bào tóc; các tế bào dương tính với CD19, CD20, CD22, CD11c, CD25, CD103, CD123;
+ Hóa mô miễn dịch tủy xương: dương tính với CD20, TRAP, DBA.44.
4.2. Biến thể của Lơ xê mi tế bào tóc
Tỷ lệ gặp khoảng 10% tổng số ca bệnh; đặc điểm có tăng số lượng bạch cầu, không giảm bạch cầu mono, dịch tủy dễ hút hơn; đáp ứng với điều trị kém hơn so với lơ xê mi tế bào tóc thể thông thường.
4.3. Chẩn đoán phân biệt
Bảng 41. Phân biệt với những bệnh tăng sinh lympho B có lách to.
|
Bệnh |
Đặc điểm miễn dịch |
Triệu chứng khác |
|
Lơ xê mi tế bào tóc |
CD11c (+), CD25 (+), CD103 (+), CD123 (+), CD20 (+) |
Thường gặp giảm bạch cầu mono. |
|
Lơ xê mi tế bào tóc biến thể |
CD11c (+), CD103 (+), CD25 (-) |
Tăng bạch cầu, không gặp giảm bạch cầu mono. |
|
U lympho vùng vỏ lách |
CD11c (+), CD25 (+), CD24 (+), CD79b (+) |
Bạch cầu không tăng, không có “tóc”. |
|
Lơ xê mi lympho mạn |
CD5 (+), CD19 (+), CD23 (+) |
Tăng bạch cầu lympho, nhưng không có “tóc”. |
|
Lơ xê mi tế bào tiền lympho B |
CD19 (+), FMC7 (+), CD79b (+), CD20 (+), CD22 (+) |
Tăng bạch cầu lympho, nhưng không có “tóc”. |
5. ĐIỀU TRỊ
5.1. Điều trị bệnh chính
Tiến triển tự nhiên của bệnh thường kéo dài một vài năm. Những bệnh nhân không có triệu chứng lâm sàng cần được theo dõi định kỳ 3 tháng một lần: Khám lâm sàng, xét nghiệm tế bào máu ngoại vi. Chỉ định điều trị khi có biểu hiện:
- Triệu chứng toàn thân: Sốt, ra mồ hôi đêm, mệt nhiều, sụt cân không giải thích được (> 10% trọng lượng cơ thể trong 6 tháng).
- Lách to nhiều và đau, hạch to.
- Nhiễm trùng tái diễn.
- Thiếu máu: Hb < 110 g/L.
- Giảm tiểu cầu (< 100 G/L).
- Giảm bạch cầu hạt (< 1 G/L).
- Hạch to nhanh hoặc tăng nhanh số lượng lymphocyte.
5.2. Điều trị bệnh nhân mới chẩn đoán
- Fludarabine 40 mg/m2 truyền tĩnh mạch trong 30 phút từ ngày 1 đến 5; có thể kết hợp với rituximab 375 mg/m2 truyền tĩnh mạch ngày 1. Có thể điều trị 4 đến 6 đợt, mỗi đợt cách nhau 28 ngày. Lưu ý giảm liều fludarabine nếu bệnh nhân suy thận.
Trong trường hợp người bệnh cần truyền chế phẩm máu, nên chiếu xạ túi máu trước khi truyền nhằm tránh bệnh ghép chống chủ do truyền máu.
Khi số lượng bạch cầu lympho dưới 1 G/L, cần điều trị dự phòng:
- Acyclovir 600 mg/ngày để tránh tái hoạt virus Herpes.
- Co-trimoxazole 960 mg x 3 lần/tuần để phòng pneumocystis carini.
b. Interferon alpha (INF-α)
- Thường dùng cho những bệnh nhân đang bị giảm bạch cầu hạt trung tính quá thấp (< 0,2 G/l) hoặc phụ nữ có thai. Liều dùng 2 triệu đơn vị/m2 da, tiêm dưới da 3 lần một tuần trong 12-18 tháng.
c. Cắt lách trong trường hợp:
- Lách quá to và đau, nhồi máu lách, vỡ lách.
- Giảm tế bào máu nhiều do cường lách.
5.3. Điều trị bệnh nhân tái phát/ kháng thuốc
Một số phác đồ điều trị bệnh nhân tái phát/ kháng thuốc:
- Rituximab (kháng thể kháng CD20): Liều 375 mg/m2 da truyền tĩnh mạch mỗi tuần 1 lần, 4 - 8 tuần liên tục, thường dùng phối hợp với hóa chất nhóm purin.
- Rituximab 375 mg/m2 da, truyền tĩnh mạch ngày 1 + Fludarabine 40 mg/m2 da, uống từ ngày 1-5. Có thể dùng 4 đợt, mỗi đợt cách nhau 28 ngày.
- Rituximab 375 mg/m2 da, truyền tĩnh mạch ngày 1 + Bendamustine 70-90 mg/m2 da, truyền tĩnh mạch ngày 2 và 3.
- Ibrutinib (thuốc ức chế BTK): Liều 420 mg, uống hàng ngày.
5.4. Các xét nghiệm đánh giá và theo dõi điều trị
- Tổng phân tích tế bào máu ngoại vi, được làm ít nhất 2 ngày một lần.
- Sinh hóa máu: Chức năng gan, thận, điện giải, acid uric, photpho, LDH..., ít nhất 1 tuần/lần.
- Bộ xét nghiệm đông máu, D-Dimer, rotem…, ít nhất 1 tuần/lần.
- Các xét nghiệm vi sinh: virus viêm gan, HIV… làm sàng lọc trước khi điều trị, lặp lại theo quy định. Các xét nghiệm đánh giá nhiễm trùng: cấy vi khuẩn, vi nấm, CRP, PCT, CMV, EBV, kháng sinh đồ…, nếu lâm sàng có dấu hiệu nhiễm trùng.
- Huyết thanh học nhóm máu, làm lúc chẩn đoán hoặc khi cần truyền máu.
- Xét nghiệm tế bào và sinh hóa nước tiểu, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Chẩn đoán hình ảnh, thăm dò chức năng, làm để sàng lọc trước điều trị và làm lại khi có dấu hiệu lâm sàng bất thường liên quan.
- Xét nghiệm Huyết tủy đồ, Sinh thiết tủy xương làm lúc chẩn đoán và làm lại sau các đợt điều trị; Gen đột biến lơ xê mi cấp, Nhiễm sắc thể được làm lúc chẩn đoán và làm lại để đánh giá sau mỗi 2 đợt điều trị hóa chất.
- Xét nghiệm Flowcytometry được làm lúc chẩn đoán và làm lại sau mỗi đợt điều trị hóa chất để đánh giá bệnh tồn lưu tối thiểu.
- Một số xét nghiệm khác được chỉ định tùy theo bệnh nền và bệnh mắc kèm của người bệnh.
6. ĐÁNH GIÁ ĐÁP ỨNG ĐIỀU TRỊ
|
Lui bệnh hoàn toàn |
Hb > 110 g/l (không do truyền máu), TC > 100 G/l, ANC > 1,5 G/l; không tìm thấy tế bào tóc ở máu ngoại vi và tủy xương; Lách nhỏ lại trên lâm sàng. |
|
Lui bệnh một phần |
Hb > 110 g/l (không do truyền máu), TC > 100 G/l, ANC > 1,5 G/l; vẫn còn tế bào tóc xâm lấn tủy xương; lách nhỏ lại > 50% trên lâm sàng. |
|
Bệnh ổn định |
Không đáp ứng các tiêu chuẩn lui bệnh như trên sau khi kết thúc điều trị. |
|
Bệnh tiến triển |
Gia tăng các triệu chứng liên quan đến bệnh; tăng kích thước gan, lách, hạch > 25%; giảm > 25% các chỉ số tế bào máu ngoại vi so với tiêu chuẩn lui bệnh 1 phần (lưu ý phân biệt với tác dụng phụ ức chế tủy do thuốc điều trị). |
|
Bệnh tái phát |
Tái phát về hình thái: tái xuất hiện tế bào tóc ở máu ngoại vi, sinh thiết tủy xương mà chưa có thay đổi về chỉ số tế bào máu; Tái phát về huyết học: tái xuất hiện tình trạng giảm các dòng tế bào máu dưới mức lui bệnh một phần. |
7. TIÊN LƯỢNG
Trước giai đoạn có interferon, tỷ lệ sống đến năm thứ 4 chỉ 68%. Với sự xuất hiện của hóa chất nhóm purine, việc điều trị đã đạt được tình trạng lui bệnh bền vững, thậm chí điều trị sau khi tái phát vẫn cho kết quả khá tốt. Tỷ lệ sống sau 5 năm cao hơn 85%.
TÀI LIỆU THAM KHẢO
1. Else M, Dearden CE, Matutes E, et al. (2009), "Long-term follow-up of 233 patients with hairy cell leukemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol; 145 (6): 733-740
2. Falini B, Tiacci E, Liso A, et al. (2004), "Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1)". Lancet; 363 (9424): 1869-1870
3. Habermann TM. (2006), "Splenectomy, interferon, and treatments of historical interest in hairy cell leukemia". Hematol Oncol Clin North Am; 20 (5): 1075-1086
4. Matutes E. (2006), "Immunophenotyping and differential diagnosis of hairy cell leukemia". Hematol Oncol Clin North Am; 20 (5): 1051-1063.
5. Golomb HM. (2008), "Hairy cell leukemia: treatment successes in the past 25 years". J Clin Oncol; 26 (16): 2607-2609.
6. Grever MR et al (2017), “Consensus guidelines for the diagnosis and management of patients with classical hairy cell leukemia”. Blood, 129: 553-560.
35. LƠ XÊ MI TẾ BÀO PLASMO
1. ĐẠI CƯƠNG
- Lơ xê mi tế bào plasmo (plasma cell leukemia - PCL) là bệnh lý tế bào dòng plasmo tủy xương (plasma cell myeloma), xuất hiện tế bào dòng plasmo trong máu ngoại vi chiếm ≥ 20% hoặc ≥ 2 G/L.
- Lơ xê mi tế bào plasmo được chia thành 2 nhóm:
+ Lơ xê mi tế bào plasmo nguyên phát (primary plasma cell leukemia-pPCL): Chiếm khoảng 60%);
+ Lơ xê mi tế bào plasmo thứ phát (second plasma cell leukemia-sPCL): Là giai đoạn cuối của bệnh đa u tủy xương.
Trong bài này đề cập chi tiết đến lơ xê mi plasmo nguyên phát (pPCL).
2. CHẨN ĐOÁN
2.1. Lâm sàng
a. Thiếu máu: Khoảng 50% người bệnh mới chẩn đoán có thiếu máu.
b. Xuất huyết: Xuất huyết dưới da hoặc niêm mạc, nội tạng do giảm tiểu cầu.
c. Thâm nhiễm tế bào plasmo vào các cơ quan: Gan to (gặp khoảng 50%), lách to (gặp khoảng 50%), hạch to, tràn dịch màng phổi, triệu chứng thần kinh.
d. Tăng canxi máu: Táo bón, buồn nôn; đa niệu, suy thận; loạn thần, hôn mê; rối loạn nhịp tim. Nếu canxi máu > 11 mg/dl (2,75 mmol/l) cần phải xử trí cấp cứu.
đ. Triệu chứng tổn thương xương: Ít gặp.
2.2. Cận lâm sàng
a. Tổng phân tích tế bào máu: Thiếu máu, giảm tiểu cầu, bạch cầu có thể giảm, bình thường hoặc tăng nhưng có tế bào plasmo với tỷ lệ ≥ 20% hoặc ≥ 2G/L.
b. Tủy đồ và sinh thiết tủy xương: Tăng sinh dòng plasmo (> 10%); phân tích karyotype có thể thấy bất thường NST (hay gặp monosomy 13); xét nghiệm miễn dịch thấy có dấu ấn đặc trưng tế bào plasmo.
c. Điện di protein huyết thanh và nước tiểu: Phát hiện protein đơn dòng; điện di miễn dịch phát hiện thành phần đơn dòng của các chuỗi nặng và nhẹ.
d. Xét nghiệm sinh hoá: Protid máu toàn phần, albumin, globulin, β2- microglobulin, creatinine và canxi huyết thanh. Định lượng IgG, IgA, IgM, IgD, IgE huyết thanh, định lượng M-Protein/ nước tiểu 24h (Protein Bence Jones), đo chuỗi nhẹ tự do (free light-chain: FLC) trong huyết thanh và nước tiểu.
đ. Chẩn đoán hình ảnh
- Chụp X-quang xương (cột sống, xương chậu, xương sọ, xương sườn...): Đánh giá các tổn thương tiêu xương.
- Chụp cộng hưởng từ, chụp cắt lớp vi tính, chụp PET, CT-scan, chụp xạ hình xương: Cần thiết để đánh giá sự thâm nhiễm của tế bào plasmo tại các cơ quan, vì triệu chứng này khá thường gặp.
e. Các xét nghiệm sử dụng trước và trong điều trị
- Tổng phân tích tế bào máu.
- Sinh hóa máu: Chức năng gan, thận, điện giải đồ, acid uric, LDH.
- Đông máu cơ bản, D-Dimer.
- Siêu âm tim đánh giá chức năng tim trước khi điều trị hóa chất.
2.3. Chẩn đoán xác định: Tiêu chuẩn chẩn đoán PCL theo International Myeloma Working Group 2003.
- Có ≥ 20% tế bào dòng plasmo hoặc ≥ 2G/L tế bào dòng plasmo trong máu ngoại vi.
Nhiều tác giả nước ngoài (Mayo Clinic-Mĩ) đang đưa ra đề xuất tiêu chuẩn số lượng tế bào plasmo trong máu ngoại vi ≥ 5% để chẩn đoán PCL thay vì ≥ 20% như hiện tại.
2.4. Chẩn đoán phân biệt: Một số trường hợp có thể xuất hiện tế bào dòng plasmo trong máu ngoại vi như: Đa u tủy xương, nhiễm khuẩn huyết, nhiễm trùng tăng bạch cầu mono. Tuy nhiên, tỷ lệ plasmo < 20% hoặc < 2G/L.
3. ĐIỀU TRỊ
3.1. Điều trị ban đầu
- Điều trị ban đầu được chỉ định cho tất cả các người bệnh, có thể chọn một trong các phác đồ phối hợp thuốc, trong đó thuốc chính là bortezomib, như: VTD, VCD, PAD, RVD, VTD-PACE, HyperCVAD-VTD.
- Các thuốc điều trị nhắm đích như kháng thể đơn dòng chống CD38 như Daratumumab, các thuốc ức chế chọn lọc proteasome.. cũng được đưa vào phác đồ điều trị ban đầu.
- Số đợt điều trị ban đầu chưa có khuyến cáo cụ thể, nhưng để điều trị tiếp theo có hiệu quả, kết quả điều trị ban đầu phải đạt được tối thiểu là đáp ứng một phần.
- Điều trị thuốc hỗ trợ khi có các tác dụng phụ của dexamethasone.
3.2. Ghép tế bào gốc
- Sau điều trị ban đầu, tiến hành ghép tế bào gốc đồng loại đối với người bệnh < 45 tuổi hoặc ghép tế bào gốc tự thân đối với người bệnh < 70 tuổi.
- Để ghép tế bào gốc có hiệu quả, kết quả đạt được sau điều trị ban đầu tối thiểu phải là đáp ứng một phần (PR).
3.3. Điều trị củng cố
- Được chỉ định sau điều trị ban đầu với kết quả tối thiểu là đáp ứng một phần hoặc sau ghép tế bào gốc; phác đồ kết hợp 3 thuốc, thuốc chính là bortezomib: VRD hoặc VTD (Điều trị 2 đợt).
- Các thuốc điều trị nhắm đích như kháng thể đơn dòng chống CD38 như Daratumumab, các thuốc ức chế chọn lọc proteasome..., các dẫn xuất của thalidomide như cũng được đưa vào phác đồ điều trị củng cố.
3.4. Điều trị duy trì
- Được chỉ định sau điều trị củng cố đạt kết quả tối thiểu là đáp ứng một phần.
- Phác đồ 1 thuốc (bortezomib liều 1,3mg/m2 da mỗi 2 tuần) hoặc kết hợp với lenalidomide liều 10mg/ngày, ngày 1-21 mỗi 28 ngày hoặc uống liên tục.
- Các thuốc điều trị nhắm đích như kháng thể đơn dòng chống CD38 như Daratumumab, các thuốc ức chế chọn lọc proteasome. .. ,các dẫn xuất của thalidomide… cũng được đưa vào phác đồ điều trị duy trì.
Nếu sau mỗi đợt điều trị mà không đáp ứng thì xem xét thay đổi phác đồ, kết hợp thêm thuốc.
3.5. Điều trị hỗ trợ
a. Nhiễm trùng
- Phòng nhiễm trùng có thể dùng gammaglobulin, GCSF.
- Nếu có nhiễm trùng thì phải điều trị kháng sinh ngay.
b. Hội chứng tiêu khối u
Khá thường gặp, cần theo dõi creatinine, ure, LDH; điều trị dự phòng bằng truyền dịch, thuốc lợi tiểu, allopurinol.
c. Tổn thương xương
- Điều trị tăng canxi máu:
+ Truyền dịch, lợi tiểu;
+ Ức chế huỷ xương: Biphosphonate, calcitonine (4-8 UI/kg pha NaCl 0,9% truyền tĩnh mạch trong 6-8 giờ), methylprednisolon (50-100mg/ngày);
+ Lọc máu: Chỉ định cho trường hợp tăng canxi máu nặng đe doạ tính mạng, người bệnh có suy thận, phù phổi.
- Bisphosphonate: Làm tăng chuyển hoá của xương và tránh gây huỷ xương.
+ Zoledronic acid: Liều 4 mg/lần/tháng, ở những người bệnh có suy thận phải giảm liều acid zoledronic;
+ Pamidronate: Liều hàng tháng 90 mg truyền tĩnh mạch 2 giờ;
+ Hoại tử xương hàm là biến chứng muộn nghiêm trọng liên quan đến thời gian điều trị bisphosphonate (tỷ lệ cao hơn ở zoledronic acid so với với pamidronate).
- Khuyến khích người bệnh vận động nhẹ nhàng nếu có thể, nhưng tránh gây ra chấn thương.
- Trường hợp đau nhiều và có tính khu trú có thể xạ trị.
d. Thiếu máu
- Erythropoietin tái tổ hợp: Liều 4.000 UI/ngày.
- Truyền khối hồng cầu.
Lưu ý:
- Điều trị dự phòng Herpes zoster khi điều trị bằng các thuốc ức chế proteasome và kháng thể đơn dòng chống CD38.
- Xét nghiệm sàng lọc viêm gan B trước khi điều trị bằng Daratuximab.
- Điều trị dự phòng bằng kháng sinh đối với bệnh nhân có nguy cơ nhiễm trùng.
3.6. Đánh giá đáp ứng điều trị
Bảng 42. Tiêu chuẩn đánh giá đáp ứng điều trị của lơ xê mi tế bào dòng plasmo (được kết hợp từ tiêu chuẩn đáp ứng của lơ xê mi cấp và Đa u tuỷ xương).
|
Mức độ đáp ứng |
Tiêu chuẩn tuỷ xương |
Tiêu chuẩn máu ngoại vi |
Tiêu chuẩn huyết thanh |
Tiêu chuẩn khác |
|
Đáp ứng hoàn toàn triệt để |
Tế bào plasmo trong tuỷ xương: < 5%. Không phát hiện tế bào plasmo ác tính bằng kỹ thuật đếm tế bào dòng chảy (flowcytometry). |
Không có tế bào plasmo trong máu ngoại vi. |
Điện di miễn dịch cố định huyết thanh và nước tiểu: âm tính. Tỷ lệ kappa/lambda tự do trong giới hạn bình thường. |
Không có u plasmo phần mềm. |
|
Đáp ứng hoàn toàn |
Tế bào plasmo trong tuỷ xương: < 5%. |
Không có tế bào plasmo trong máu ngoại vi. |
Điện di miễn dịch cố định huyết thanh và nước tiểu: âm tính. |
Không có u plasmo phần mềm. |
|
Đáp ứng một phần rất tốt |
Tế bào dòng plasmo trong tuỷ xương: < 5%. |
Không có tế bào plasmo trong máu ngoại vi. |
Ig đơn dòng giảm ≥ 90%, protein niệu < 100mg/24h. |
Không có u plasmo phần mềm. |
|
Đáp ứng một phần |
Tế bào dòng plasmo trong tuỷ xương: 5-25%. |
Tế bào dòng plasmo: 1-5% |
Ig đơn dòng giảm ≥ 50%, protein niệu < 200mg/24h hoặc giảm ≥ 90%. |
Kích thước khối u plasmo phần mềm giảm ≥ 50%. |
|
Không đáp ứng |
Không gặp bất cứ tiêu chuẩn nào của đáp ứng một phần và tiêu chuẩn của bệnh tiến triển. |
|||
|
Bệnh tiến triển |
Tăng > 25% tế bào dòng plasmo trong tuỷ hoặc tăng số lượng tuyệt đối ≥ 10%. |
Tăng > 5% số lượng tuyệt đối tế bào dòng plasmo. |
Tăng > 25% Ig đơn dòng với số lượng tuyệt đối ≥ 5G/L, tăng > 25% protein niệu 24h với số lượng tuyệt đối ≥ 200mg/24h |
Tăng canxi huyết thanh, tăng tiêu huỷ xương, tăng kích thước khối u phần mềm. |
|
Bệnh tái phát |
Tăng > 10% tế bào dòng plasmo. |
Xuất hiện tế bào plasmo dù ở mức độ nào. |
Xuất hiện Ig đơn dòng trong huyết thanh hoặc nước tiểu. |
Có thâm nhiễm u plasmo ở phần mềm. |
4. THEO DÕI SAU ĐIỀU TRỊ
- Xét nghiệm hàng tháng: Tổng phân tích tế bào máu, định lượng các loại Ig, chuỗi nhẹ tự do trong huyết thanh, creatinine, canxi.
- Chụp cộng hưởng tự hoặc chụp cắt lớp vi tính nên được thực hiện để phát hiện các thâm nhiễm mới.
- Đánh giá đáp ứng sau mỗi 1 đợt điều trị để điều chỉnh phác đồ.
PHỤ LỤC
MỘT SỐ PHÁC ĐỒ CỤ THỂ ĐIỀU TRỊ LƠ XÊ MI TẾ BÀO DÒNG PLASMO
a. VTD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu, ngày 1, 8, 15, 22 trong 4 đợt tiếp.
- Thalidomide 100-200 mg uống ngày 1-21.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch ngày 1- 4.
b. VCD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu, ngày 1, 8, 15, 22 trong 4 đợt tiếp.
- Cyclophosphamide 300 mg/m2/ngày truyền tĩnh mạch 1, 8, 15 và 22.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch 1, 8, 15 và 22.
c. PAD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu và ngày 1, 8, 15 và 22 trong 4 đợt tiếp.
- Doxorubicin 10 mg/m2/ngày truyền tĩnh mạch 1-4.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch 1-4.
d. VRD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu và ngày 1, 8, 15 và 22 trong 4 đợt tiếp.
- Lenalidomide liều 25mg/ngày uống ngày 1 - 21.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch ngày 1, 8, 15 và 22.
Lưu ý: Khoảng cách tối thiểu giữa 2 liều bortezomib là 72 giờ. Điều chỉnh liều các thuốc đối với bệnh lớn tuổi, có suy thận.
TÀI LIỆU THAM KHẢO
1. C Fernández de Larea. “Plasma cell leukemia: consesus statement on diagnostic requirement, response criteria and treatment recommendations by the International Myeloma Working Group”. 2013. Leukemia, 1-12.
2. Kennet C. Anderson et al. “How I treat plasma cell leukemia”. Blood September 20.2012 vol.120 no. 12 2376-2389.
3. International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003; 121(5): 749-757.
4. NCCN guidelines version 3.2020 - Multiple Myeloma.
5. Ludwig H, Miguel JS, Dimopoulos MA, Palumbo A, et al, 2013. International Myeloma Working Group recommendations for global myeloma care. Leukemia, 1-12.
6. Bergsagel PL, Mateos MV, Gutierrez NC, Rajkumar SV and San Miguel JF, 2013. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood, 121(6): 884-892.
7.WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th Edition-2017); 250.
8. Praful Ravi et al. Blood Cancer Journal (2018)8:116. “ Revised diagnostic criteria for plasma cell leukemia: results of a Mayo Clinic study with comparison of outcome to multiple myeloma.
36. BỆNH WALDENSTRÖM
1. ĐẠI CƯƠNG
Bệnh tăng sinh globulin đại phân tử (Waldenström’s macroglobulinemia) là bệnh lý dòng tế bào lympho B đặc trưng bởi sự tăng sinh, tích lũy các tế bào lymphoplasmo đơn dòng trong tủy xương và tổ chức lympho kèm theo xuất hiện IgM đơn dòng trong huyết thanh. Theo Tổ chức Y tế thế giới, bệnh Waldenström được xếp vào nhóm u lympho tế bào lymphoplasmo, có độ ác tính thấp.
2. NGUYÊN NHÂN GÂY BỆNH
- Đến nay còn chưa rõ, một số yếu tố có thể làm tăng nguy cơ mắc bệnh: Viêm gan virus C, tiếp xúc với hóa chất nông nghiệp, thuốc nhuộm tóc, bụi gỗ…
3. TRIỆU CHỨNG
3.1. Lâm sàng
a. Triệu chứng do xâm lấn của tế bào u
- Triệu chứng B: Sốt, ra mồ hôi đêm, sút cân, mệt mỏi.
- Thiếu máu do xâm lấn tủy xương của tế bào u và do tan máu tự miễn.
- Có thể có gan lách hạch to, thâm nhiễm da, đường tiêu hóa và phổi.
b. Triệu chứng do tăng tiết IgM đơn dòng trong huyết thanh
- Hội chứng tăng độ nhớt huyết tương gặp: Đau đầu, nhìn mờ, chóng mặt, điếc đột ngột, chảy máu mũi, chảy máu chân răng. Soi đáy mắt có thể gặp hiện tượng ứ máu trong các tĩnh mạch võng mạc, xuất huyết võng mạc, phù gai thị.
- Xuất hiện globulin “lạnh” trong máu (cryoglobulinemia) gây ra hiện tượng ngưng kết lạnh: Xanh tím các đầu chi khi gặp lạnh.
- Amyloidosis: Lắng đọng dạng tinh bột ở các cơ quan.
- Bệnh lý thần kinh ngoại vi.
3.2. Xét nghiệm
a. Tế bào máu và tủy
- Hb giảm, số lượng hồng cầu giảm, có thể thấy hồng cầu ngưng kết, chuỗi tiền; Số lượng bạch cầu trung tính giảm, có thể < 1,0 G/L; Số lượng tiểu cầu giảm, có thể < 50 G/L; Tăng số lượng bạch cầu lympho và/ hoặc mono. Tăng tỷ lệ tế bào lymphoplasmo trong tủy (thường > 10%).
b. Mô bệnh học và hóa mô miễn dịch tủy xương
Mật độ tế bào tủy bình thường hoặc tăng. Hình ảnh xâm lấn của các tế bào lymphoplasmo và lymphoxit kích thước nhỏ trong tủy xương, có thể gặp các kiểu xâm lấn lan tỏa, thành nốt, hoặc xen kẽ giữa các khoang sinh máu. Các tế bào lymphoplasmo dương tính với CD19, CD20, IgM bề mặt, CD25, CD27, FMC7; âm tính với CD5, CD10, CD23, CD103, CD138.
c. Hóa sinh máu
- Tăng các thành phần: Protein máu toàn phần, gammaglobulin, IgM đơn dòng huyết thanh, β2 microglobulin, LDH, bilirubill gián tiếp. Điện di protein và điện di miễn dịch huyết thanh cho hình ảnh đỉnh đơn dòng IgM.
d. Các xét nghiệm thăm dò khác
Độ nhớt máu tăng cao (tăng > 2 mPas), tăng tốc độ máu lắng; nghiệm pháp Coombs trực tiếp và/ hoặc gián tiếp dương tính khi có tan máu tự miễn; giảm độ ngưng tập tiểu cầu với các chất kích tập (ADP, Collagen, Ristocetin); thời gian thrombin (TT) kéo dài; bất thường NST del 6q, gặp ở 50% người bệnh; đột biến gen MYD88 L265P: gặp ở > 90% người bệnh; chụp cắt lớp ngực, bụng, khung chậu: có thể phát hiện gan, lách, hạch to; sinh thiết tổ chức đệm mỡ và/ hoặc nhuộm đỏ Congo mô tủy xương để phát hiện lắng đọng dạng tinh bột (amyloid).
e. Các xét nghiệm sử dụng trước và trong điều trị
- Tổng phân tích tế bào máu.
- Hóa sinh máu: Chức năng gan, thận, điện giải đồ, acid uric, LDH, bilirubin.
- Đông máu cơ bản.
- Marker virus viêm gan B: Người bệnh điều trị với Rituximab có nguy cơ làm virus tái hoạt động.
- Siêu âm bụng đánh giá tình trạng gan lách hạch to.
4. CHẨN ĐOÁN
4.1. Chẩn đoán xác định
Tiêu chuẩn chẩn đoán của Hội thảo Quốc tế về bệnh Waldenström năm 2003
(Waldenström’s macroglobulinemia International Workshop - 2003)
- Tăng IgM đơn dòng trong huyết thanh (bất kỳ nồng độ nào).
- Xâm lấn tủy xương bởi các tế bào lymphoplasmo (tối thiểu 10%) và tế bào lympho kích thước nhỏ, tạo thành đám giữa các khoang sinh máu.
- Xét nghiệm hóa mô miễn dịch các tế bào này thấy: IgM bề mặt (+), CD5 (±), CD19 (+), CD20 (+), CD25 (+), CD27 (+), FMC7 (+).
Chẩn đoán xác định yêu cầu thỏa mãn tất cả các tiêu chuẩn trên.
4.2. Chẩn đoán phân biệt
- Lơ xê mi lympho mạn: Hạch to nhiều nơi, lách to, tăng sinh dòng lympho trong máu và tủy xương, không thấy tế bào lymphoplasmo, không tăng IgM đơn dòng trong huyết thanh.
- U lympho thể nang: Hạch to nhiều, tế bào u xâm lấn tủy xương tạo thành nhiều nang, không có tế bào lymphoplasmo, không tăng IgM đơn dòng trong huyết thanh.
- U lympho tế bào Mantle: Hạch và lách to nhiều, tế bào u xâm lấn tủy xương dương tính với CD5, CD20; âm tính với Ig bề mặt và CD25; không tăng IgM đơn dòng trong huyết thanh.
- Đa u tủy xương IgM: Tăng sinh tế bào plasmo trong tủy xương > 10%; nồng độ IgG, IgA giảm thấp; có tổn thương xương; bất thường nhiễm sắc thể t(11;14), del13q.
- Tăng IgM đơn dòng chưa có ý nghĩa về mặt lâm sàng (IgM MGUS): Xuất hiện IgM đơn dòng trong huyết thanh, nhưng thường < 3g/dL; không có biểu hiện xâm lấn tủy xương của tế bào lymphoplasmo và không có triệu chứng liên quan đến tăng IgM.
5. ĐIỀU TRỊ
5.1. Điều trị bệnh chính
- Đối với những người bệnh không có triệu chứng lâm sàng: Theo dõi 3 tháng một lần: lâm sàng, xét nghiệm tổng phân tích tế bào máu, protein máu, định lượng IgM, độ nhớt huyết tương.
- Chỉ định điều trị hóa chất khi người bệnh có: Hb < 100 g/L, Tiểu cầu < 100 G/L, IgM > 6.000 mg/dL, gan lách hạch to nhiều, tăng độ nhớt huyết tương có triệu chứng, tổn thương thần kinh ngoại vi mức độ vừa đến nặng, amyloidosis, có ngưng kết lạnh.
- Lựa chọn phác đồ điều trị cần dựa vào những yếu tố: Tuổi của người bệnh, bệnh lý khác kèm theo, mức độ nặng của triệu chứng, khả năng ghép tế bào gốc tự thân. Nếu có dự định ghép tủy, nên hạn chế dùng Chlorambucil, Fludarabine do ảnh hưởng đến khả năng huy động tế bào gốc.
Phác đồ điều trị cụ thể:
a. Người bệnh < 70 tuổi, chức năng tim gan thận bình thường có thể chọn 1 trong các phác đồ:
- Bortezomib + dexamethasone + rituximab (BDR).
- Cyclophosphamide + methylprednisone + rituximab (CPR).
- Dexamethasone + cyclophosphamide + rituximab (DRC).
- Rituximab + cyclophosphamide + doxorubicin + vincristine + methylprednisone (R-CHOP).
- Fludarabine + cyclophosphamide + rituximab (FCR).
- Ibrutinib đơn trị.
- Bendamustine + rituximab (BR).
b. Người bệnh > 70 tuổi có thể chọn 1 trong các phác đồ
- Chlorambucil uống mỗi ngày 2-4mg.
- Rituximab đơn trị liệu 375mg/m2 da/tuần x 4 tuần.
- Fludarabine đơn trị liệu 25mg/m2 da/ngày x 4 ngày.
- Bortezomib + Rituximab (VR).
- Ibrutinib đơn trị.
- Bendamustine + rituximab (BR).
Lưu ý:
- Các phác đồ trên, trừ rituximab đơn trị liệu, có thể điều trị 4 - 6 đợt (tùy từng người bệnh cụ thể). Khoảng thời gian giữa 2 đợt điều trị không sớm hơn 21 ngày và không muộn hơn 28 ngày.
- Khi điều trị các phác đồ có Rituximab, cần dự phòng thuốc kháng virus (Entecavir hoặc Tenofovir) nếu người bệnh có nhiễm virus HBV để tránh virus tái hoạt động.
5.2. Điều trị bệnh tái phát, điều trị cứu vãn
- Bệnh nhân tái phát sớm trong vòng 1 năm: Uống Ibrutinib 420 mg/ ngày cho đến khi bệnh tiến triển hoặc phải ngừng thuốc do tác dụng phụ.
- Đối với những trường hợp tái phát muộn sau 3 năm, có thể điều trị lại cho người bệnh bằng các phác đồ khởi đầu.
- Đối với người bệnh tái phát trong khoảng 2-3 năm, có thể sử dụng những thuốc mà trước đó người bệnh chưa điều trị trước đó, có thể tham khảo một số phác đồ sau:
+ Everolimus;
+ Thalidomide + Rituximab;
5.3. Điều trị duy trì
Người bệnh đáp ứng với các phác đồ trước đó có Rituximab nên được điều trị duy trì Rituximab 375mg/ m2 da cứ 3 tháng một lần trong 2 năm tiếp theo.
5.4. Điều trị hỗ trợ (hội chứng tăng độ nhớt huyết tương)
- Hội chứng tăng độ nhớt huyết tương đặc biệt hay gặp khi IgM tăng cao > 4.000mg/dL và cần được can thiệp mang tính cấp cứu bằng phương pháp gạn huyết tương (plasmapheresis) hoặc trao đổi huyết tương (plasma exchange).
- Cần tiến hành trao đổi huyết tương trước khi điều trị hóa chất cho những người bệnh có IgM cao > 4.000mg/dL; hạn chế truyền khối hồng cầu cho người bệnh trước khi trao đổi huyết tương bởi nguy cơ làm nặng thêm hội chứng tăng độ nhớt huyết tương.
6. TIÊU CHUẨN ĐÁNH GIÁ ĐÁP ỨNG (theo bảng 43)
Bảng 43. Đánh giá đáp ứng với điều trị của bệnh Waldenström)
|
Mức độ đáp ứng |
Tiêu chuẩn |
|
Đáp ứng hoàn toàn |
IgM huyết thanh trở về giới hạn bình thường, không thấy đỉnh protein đơn dòng khi điện di miễn dịch huyết thanh; tủy xương không còn biểu hiện xâm lấn, gan lách hạch trở về kích thước bình thường, không có triệu chứng toàn thân liên quan đến bệnh |
|
Đáp ứng một phần rất tốt |
IgM huyết thanh giảm ≥ 90% so với ban đầu, kích thước gan lách hạch nhỏ đi ít nhất 90% so với trước điều trị; không có biểu hiện tiến triển của bệnh. |
|
Đáp ứng một phần |
IgM huyết thanh giảm ≥ 50% nhưng < 90% so với ban đầu, kích thước gan lách hạch nhỏ đi ít nhất 90% so với trước điều trị; không có biểu hiện tiến triển của bệnh |
|
Đáp ứng tối thiểu |
IgM huyết thanh giảm ≥ 25% nhưng < 50% so với ban đầu; không có biểu hiện tiến triển của bệnh |
|
Bệnh ổn định |
IgM huyết thanh giảm < 25% hoặc tăng không quá 25% so với ban đầu; không có sự phát triển của gan lách hạch, không giảm tế bào máu hoặc xuất hiện thêm triệu chứng lâm sàng liên quan đến bệnh |
|
Bệnh tiến triển |
IgM huyết thanh tăng thêm ≥ 25% so với mức đáp ứng thấp nhất trước đó, gan lách hạch to lên, xuất hiện triệu chứng B và các biểu hiện lâm sàng khác liên quan đến bệnh |
7. TIÊN LƯỢNG (theo bảng 44)
Bảng 44. Chỉ số tiên lượng Quốc tế (International Prognostic Scoring System for Waldenström’s macroglobulinemia - 2009)
|
Yếu tố nguy cơ |
Điểm |
||
|
Tuổi ≥ 65 |
1 |
||
|
Hb ≤ 115 g/L |
1 |
||
|
Tiểu cầu ≤ 100 G/L |
1 |
||
|
β2 microglobulin > 3 mg/L |
1 |
||
|
IgM > 70 g/L |
1 |
||
|
Nhóm nguy cơ |
Thấp |
Trung bình |
Cao |
|
Điểm |
0-1 (trừ yếu tố tuổi ≥ 65) |
2 hoặc tuổi ≥ 65 |
≥ 3 |
|
Tỷ lệ sống toàn bộ 5 năm |
87% |
68% |
36% |
PHỤ LỤC
|
Phác đồ |
Thuốc, cách dùng |
|
Bortezomib + dexamethasone + rituximab (BDR) |
Bortezomib 1,3mg/m2 da (ngày 1,4,8,11); Dexamethasone 40mg/ngày (ngày 1-4 và ngày 8-11); Rituximab 375mg/m2 da (ngày 0) |
|
Cyclophosphamide + methylprednisone + rituximab (CPR) |
Cyclophosphamide 750mg/m2 da (ngày 1); Methylprednisone 60mg/m2 (ngày 1, 2, 3, 4, 5); Rituximab 375mg/m2 da (ngày 0) |
|
Dexamethasone + cyclophosphamide + rituximab (DRC) |
Rituximab 375mg/m2 da (ngày 0); Cyclophosphamide 750mg/m2 da (ngày 1); Dexamethasone 20mg/ ngày (ngày 1, 2, 3, 4, 5) |
|
Rituximab + cyclophosphamide + doxorubicin + vincristine + methylprednisone (R-CHOP) |
Rituximab 375mg/m2 da (ngày 0); Cyclophosphamide 750mg/ m2 da (ngày 1); Doxorubicin 50mg/m2 da (ngày 1); Vincristine 1,4mg/m2 da (tối đa 2mg) (ngày 1); Methylprednisone 60mg/m2 (ngày 1, 2, 3, 4, 5) |
|
Bendamustine + rituximab (BR) |
Bendamustine truyền tĩnh mạch trong 60 phút, liều 90mg/m2 da (ngày 1,2); Rituximab 375mg/m2 da truyền tĩnh mạch ngày 0 |
|
Fludarabine + cyclophosphamide + rituximab (FCR) |
Fludarabine 25 mg/m2 da/ ngày (ngày 1,2,3) Cyclophosphamide 250mg/m2 da/ ngày (ngày 1,2,3) Rituximab 375 mg/m2 da, ngày 0 |
|
Ibrutinib đơn trị |
420mg/ ngày, uống liên tục đến khi bệnh tiến triển hoặc không chấp nhận được tác dụng phụ của thuốc |
|
Bortezomib + Rituximab (VR) |
Bortezomib 1,3mg/m2 da (ngày 1, 4, 8, 11) Rituximab 375mg/m2 da (ngày 0) |
TÀI LIỆU THAM KHẢO
1. Morel P, Duhamel A, Gobbi P, et al. (2009), “International prognostic scoring system for Waldenstrom macroglobulinemia”. Blood; 113 (18): 4163-4170.
2. Treon SP, Ioakimidis L, Soumerai JD, et al. (2009), “Primary therapy of Waldenstrom macroglobulinemia with bortezomib, dexamethasone and rituximab: WMCTG clinical trial 05-180”. J Clin Oncol; 27(23): 3830-3835.
3. Owen RG, Kyle RA, Stone MJ, Rawstron AC, Leblond V, Merlini Get al. (2013), “Response assessment in Waldenstrom macroglobulinaemia: update from the 6th International Workshop”. Br J Haematol; 160: 171-176.
4. Ansell SM, Kyle RA, Reeder CB, Fonseca R, Mikhael JR, Morice WGet al. (2009), “Diagnosis and management of Waldenstrom macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines”. Mayo Clin Proc; 85: 824-833.
5. Treon SP, Hanzis C, Manning RJ, Ioakimidis L, Patterson CJ, Hunter ZRet al. (2011), “Maintenance rituximab is associated with improved clinical outcome in rituximab naive patients with Waldenstrom macroglobulinaemia who respond to a rituximab- containing regimen”. Br J Haematol; 154: 357-362.
6. Treon SP. (2009), “How I treat Waldenstrom macroglobulinemia”. Blood; 114: 2375-2385
37. ĐAU TUỶ XƯƠNG
1. ĐẠI CƯƠNG
Đa u tủy xương (Multiple Myeloma: MM), còn gọi là bệnh Kahler, là một bệnh máu ác tính, đặc trưng bởi sự tăng sinh ác tính tế bào dòng plasmo trong tuỷ xương, xuất hiện protein đơn dòng trong huyết thanh và nước tiểu, tổn thương các cơ quan đích.
2. CHẨN ĐOÁN
2.1. Lâm sàng
- Khoảng 70% có thiếu máu lúc chẩn đoán; khoảng 60% biểu hiện đau xương, gãy xương và u xương; có 20% suy thận, trong đó 10% người bệnh mới chẩn đoán suy thận nặng cần thận nhân tạo; ngoài ra gặp khoảng 15 - 20 % có táo bón, buồn nôn; đa niệu, loạn thần, hôn mê, rối loạn nhịp tim do tăng calci máu. Biểu hiện thần kinh: Chèn ép rễ - tuỷ sống; bệnh lý thần kinh ngoại biên; thâm nhiễm thần kinh trung ương. Biến chứng nặng là khối u chèn ép tủy sống (chiếm khoảng 10%), chẩn đoán dựa vào chụp cộng hưởng từ.
- Nhiễm trùng tái diễn, là nguyên nhân chính gây tử vong.
- Khó thở, có những cơn thiếu máu cơ tim thoáng qua, huyết khối tĩnh mạch sâu, chảy máu võng mạc, mũi do tăng độ quánh máu.
2.2. Cận lâm sàng
- Xét nghiệm tủy xương:
+ Tăng tỷ lệ tế bào dòng plasmo; có thể thấy tăng hủy cốt bào, giảm tạo cốt bào; hình ảnh rối loạn sinh tủy thứ phát…;
+ Xét nghiệm NST và FISH: Phát hiện bất thường thiểu bội, đa bội, bất thường nhiễm sắc thể số 1, t(14;16), t(11;14), t(6;14), t(4;14), t(14;20) và del 13; del 17…;
+ Hoá mô miễn dịch (IHC) và/ hoặc phân tích dấu ấn miễn dịch (FC): xác định dấu ấn tế bào dòng plasmo (để đánh giá chính xác tỷ lệ tế bào ác tính và chẩn đoán phân biệt). Các CD tiêu chuẩn xác định plasmo ác tính: CD38, CD138, CD56, CD19, CD45. Các CD giúp chẩn đoán phân biệt và làm kiểu hình miễn dịch cơ sở của tế bào myeloma (LAIP- MAIP) để theo dõi MRD: CD27, CD81, light chain K và L, CD20, CD117;
+ Sinh thiết mô, nhuộm hóa mô miễn dịch, chẩn đoán u tương bào (khi có u);
+ Nhuộm đỏ Congo/ mô mỡ, tủy xương nếu cần loại trừ Amyloid.
- Điện di protein huyết thanh và nước tiểu/24h: Phát hiện protein đơn dòng; điện di miễn dịch phát hiện thành phần đơn dòng của các chuỗi nặng và nhẹ.
- Xét nghiệm sinh hoá: Có thể tăng protid máu toàn phần; giảm albumin; tăng globulin, β2-microglobulin, LDH, acid uric, men gan, ure/creatinine và canxi huyết thanh. Định lượng globulin miễn dịch IgG, IgA, IgM và đo chuỗi nhẹ tự do (free light-chain: FLC) trong huyết thanh và nước tiểu/24h thấy thay đổi; protein niệu 24h.
- Chẩn đoán hình ảnh:
+ Chụp cắt lớp vi tính liều thấp toàn thân: Là kỹ thuật chuẩn mới để phát hiện tổn thương tiêu xương;
+ Chụp X-quang xương (cột sống, xương chậu, xương sọ, xương sườn...): Chỉ định khi không thể chụp cắt lớp vi tính toàn thân liều thấp, phát hiện tổn thương tiêu xương;
+ Chụp cộng hưởng từ toàn thân: cần thiết trong những trường hợp có biểu hiện đau xương nhưng chụp X-quang không thấy tổn thương hoặc phát hiện tổn thương chèn ép tuỷ sống;
+ Chụp PET/CT: Phát hiện những tổn thương xương, tổn thương ngoài tuỷ.
2.3. Tiêu chuẩn chẩn đoán
Hiệp hội Đa u tủy xương quốc tế năm 2014 đưa tiêu chuẩn chẩn đoán bệnh Đa u tủy xương và các rối loạn dòng plasmo ở bệnh nhân có một hay nhiều triệu chứng lâm sàng và cận lâm sàng nêu trên (Bảng 45), gồm:
- Bệnh lý gamma đơn dòng có ý nghĩa không xác định (monoclonal gammopathy of undetermined significance: MGUS).
- Đa u tủy xương tiềm tàng (smouldering multiple myeloma: SMM).
- Đa u tủy xương có triệu chứng (multiple myeloma: MM).
Bảng 45. Tiêu chuẩn chẩn đoán cho các thể bệnh Đa u tủy xương
|
Thể bệnh |
Tiêu chuẩn |
|
MGUS |
Tất cả 3 tiêu chuẩn sau: 1) Protein đơn dòng trong huyết thanh < 3 g/dl, 2) Tế bào dòng plasmo trong tuỷ xương < 10%, và 3) Không thấy tổn thương cơ quan (tăng calci máu, suy thận, thiếu máu và tổn thương xương) mà có thể là do rối loạn tăng sinh tế bào dòng plasmo. |
|
SMM |
Cả hai tiêu chuẩn sau: 1) Protein đơn dòng trong huyết thanh (IgG hoặc IgA) ≥ 3 g/dl, hoặc protein đơn dòng trong nước tiểu ≥ 500mg/24h và/hoặc các tế bào dòng plasmo trong tuỷ xương ≥ 10%-60%. 2) Không có đủ tiêu chuẩn chẩn đoán đa u tuỷ xương hoặc amyloid. |
|
MM |
Cả hai tiêu chuẩn sau: 1) Tế bào dòng plasmo trong tuỷ xương ≥ 10% hoặc sinh thiết chứng minh có u tế bào dòng plasmo ngoài tuỷ hoặc xương. 2) Có 1 hay nhiều biểu hiện của đa u tuỷ xương: - Có bằng chứng tổn thương cơ quan là hậu quả do rối loạn tăng sinh tế bào dòng plasmo: + Tăng calci máu: calci huyết thanh cao hơn giới hạn cao của bình thường > 0,25 mmol/l (> 1 mg/dl) hoặc 2,75 mmol/l (> 11 mg/dl). + Suy thận: Creatinine huyết thanh > 177 µmol/l (> 2 mg/dl) hoặc độ thanh thải creatinin < 40 ml/phút. + Thiếu máu: hemoglobin giảm > 20 g/l dưới mức giới hạn thấp bình thường hoặc hemoglobin < 100 g/l. + Tổn thương xương: có 1 hay nhiều tổn thương tiêu xương trên Xq xương sọ, CT scanner hay PET-CT. - Tế bào dòng plasmo trong tuỷ xương ≥ 60%. - Tỷ lệ chuỗi nhẹ tự do bệnh lý/không bệnh lý trong huyết thanh ≥ 100 (nồng độ chuỗi nhẹ tự do bệnh lý ≥ 100mg/L). - Có >1 ổ tổn thương trên MRI với kích thước ≥ 5mm. |
|
MGUS chuỗi nhẹ |
Tất cả các tiêu chuẩn sau: 1) Bất thường tỷ lệ chuỗi nhẹ tự do (< 0,26 hay > 1,65). 2) Tăng nồng độ chuỗi nhẹ tự do bệnh lý (tăng chuỗi nhẹ tự do kappa với tỷ lệ > 1,65 và tăng tăng chuỗi nhẹ tự do lambda với tỷ lệ < 0,26). 3) Không có biểu hiện chuỗi nặng trên điện di miễn dịch cố định. 4) Không thấy tổn thương cơ quan là hậu quả của rối loạn tăng sinh tế bào dòng plasmo. 5) Tế bào dòng plasmo trong tuỷ xương < 10%. 6) Protein đơn dòng trong nước tiểu < 500mg/24h. |
|
U tương bào đơn độc |
Tất cả 4 tiêu chuẩn sau: 1) Tổn thương xương đơn độc hoặc tổn thương mô mềm được chứng minh trên sinh thiết có tế bào dòng plasmo. 2) Tuỷ xương bình thường không tăng sinh tế bào dòng plamo. 3) Kết quả kiểm tra xương sọ và MRI hay CT cột sống, khung chậu bình thường (trừ tổn thương đơn độc nguyên phát). 4) Không có tổn thương cơ quan như tăng calci máu, suy thận, thiếu máu hay tổn thương xương (CRAB) là hậu quả do rối loạn tăng sinh tế bào dòng plasmo - lympho. |
|
U tương bào đơn độc với tổn thương tối thiểu tuỷ xương |
Tất cả 4 tiêu chuẩn sau: 1) Tổn thương xương đơn độc hoặc tổn thương mô mềm được chứng minh trên sinh thiết có tế bào dòng plasmo. 2) Tế bào dòng plasmo trong tuỷ xương < 10%. 3) Kết quả kiểm tra xương sọ và MRI hay CT cột sống, khung chậu bình thường (trừ tổn thương đơn độc nguyên phát). 4) Không có tổn thương cơ quan như tăng canxi máu, suy thận, thiếu máu hay tổn thương xương (CRAB) là hậu quả do rối loạn tăng sinh tế bào dòng plasmo - lympho. |
2.4. Phân chia giai đoạn
Theo hệ thống phân chia giai đoạn quốc tế (The International Staging System: ISS) (Bảng 46), ISS giai đoạn III được coi là bệnh có tiên lượng xấu.
Bảng 46. Hệ thống phân chia giai đoạn quốc tế ISS
|
Giai đoạn |
Tiêu chuẩn |
|
I |
β2M * < 3,5 mG/L. Albumin ≥ 3,5 g/dl. |
|
II |
β2M < 3,5 mG/L và albumin < 3,5 g/dl, hoặc: β2M 3,5 - 5,5 mG/L và nồng độ albumin bất kỳ. |
|
III |
β2M ≥ 5,5 mG/L. |
* β2M: β2 microglobulin huyết thanh.
2.5. Phân nhym nguy cơ theo di truyền tế bào
Phân nhóm theo tổn thương di truyền của Mayo Clinic giúp định hướng điều trị (bảng 47).
Bảng 47. Các nhóm nguy cơ theo di truyền tế bào
|
Nguy cơ thấp |
Nguy cơ trung bình |
Nguy cơ cao |
|
- Thêm bội (Hyperdiploidy) - t(11;14) (q13;q32) - t(6;14) (p21;q32) - Không có tổn thương |
- t(4;14) (q32;q23) - Del 13,14 hay thiểu bội (Hypodiploidy) - Gain(1q21) |
- Del 17p - t(14;16) (q32;q23) - t(14;20) (q32;q11) |
2.6. Cic xét nghiệm đánh giá tồn dư tối thiểu bệnh (MRD)
- Định lượng hoá sinh: FLC và chuỗi nặng/chuỗi nhẹ.
- Phát hiện MRD ở tuỷ xương bằng phương pháp đếm tế bào dòng chảy hay phân tử; ghi nhận tổn thương bằng PET/CT, MRI.
2.7. Chẩn đoán phân biệt
- U tế bào dòng plasmo ngoài tuỷ: Khối u xương hay u phần mềm khu trú. Tăng sinh tế bào dòng plasmo thể hiện trên sinh thiết tổn thương ở xương hay phần mềm, không có bằng chứng của tăng sinh tế bào dòng plasmo trong tuỷ xương trên tuỷ đồ và sinh thiết. Không có tổn thương xương, ngoại trừ khối u xương đơn độc.
- Lơ xê mi tế bào dòng plasmo: Có thể nguyên phát hay thứ phát sau Đa u tủy xương, được chẩn đoán khi máu ngoại vi có tỷ lệ tế bào dòng plasmo trên 20% hay số lượng tuyệt đối > 2 G/L.
- Bệnh Waldenstrom: Tăng IgM > 3 g/dl, tăng sinh lympho và tế bào lympho dạng tế bào dòng plasmo trong tuỷ xương. Không có tổn thương xương. Thường biểu hiện triệu chứng của tăng độ quánh huyết tương; người bệnh hay có gan, lách và hạch to.
3. ĐIỀU TRỊ
3.1. Điều trị ban đầu cho bệnh nhân mới chẩn đoán
- U tương bào đơn độc hoặc u tương bào đơn độc có tổn thương tuỷ xương tối thiểu điều trị: Xạ trị và/hoặc phẫu thuật, theo dõi mỗi 3-6 tháng trong vòng 5 năm.
- Người bệnh thuộc nhóm MGUS và SMM: Không có chỉ định điều trị ngay. Nguy cơ thấp theo dõi 3-6 tháng, nguy cơ cao theo dõi mỗi 3 tháng hoặc điều trị Lenalidomide tuỳ theo tình trạng bệnh nhân.
- Chỉ định điều trị cho người bệnh Đa u tủy xương có triệu chứng: Calci máu > 2,75 mmol/L, creatinine > 2,0 mg/ml, Hb < 100 g/l, tổn thương xương hoạt động.
- Cần dựa vào khả năng thực hiện ghép tế bào gốc tự thân và nhóm nguy cơ theo phân loại di truyền cho từng ca bệnh để lựa chọn các phác đồ điều trị cho thích hợp và hiệu quả (xem bảng 48).
Bảng 48. Phác đồ điều trị ban đầu Đa u tủy xương cy triệu chứng
|
Khả năng ghép tế bào gốc tự thân |
|
|
Có khả năng ghép tế bào gốc |
Không có khả năng ghép tế bào gốc |
|
Phác đồ thuốc điều trị tấn công: - VTD: Bortezomib, thalidomide, dexamethasone. - VCD: Bortezomib, cyclophosphamide, dexamethasone. - PAD: Bortezomib, doxorubicin, dexamethasone. - VRd: Bortezomib, lenalidomide, dexamethasone liều thấp. - DRd: Daratumumab, lenalidomide dexamethasone liều thấp. - D kết hợp với các phác đồ:VRd, KRd, VTD.... |
Lựa chọn đầu tiên: - MPT: Melphalan, methylprednisone, thalidomide hoặc: - VMP: Bortezomib, melphalan, methylprednisone. Lựa chọn thứ hai: Bendamustine, methylprednisone. Các lựa chọn khác: - CTD: Cyclophosphamide, thalidomide, dexamethasone. - MP: Melphalan, methylprednisone. - VRd: Bortezomib, lenalidomide, dexamethasone liều thấp. - DRd: Daratumumab, lenalidomide dexamethasone liều thấp. - Rd: Lenalidomide dexamethasone liều thấp. |
3.1.1. Người bệnh không có chỉ định ghép tế bào gốc (thường > 65 tuổi và thể trạng bệnh kém). Thể trạng bệnh có ý nghĩa lựa chọn ghép hơn tuổi của người bệnh.
a. Điều trị tấn công
- MP: Melphalan và Methylprednisone:
|
Melphalan |
0,25mg/kg/ngày |
Uống |
Ngày 1-4 |
|
Methylprednisone |
2mg/kg/ngày |
Uống |
Ngày 1-4 |
Cách 4-6 tuần/đợt x 12 đợt. Điều chỉnh liều melphalan theo số lượng bạch cầu (BC) và tiểu cầu (TC).
Kết hợp uống melphalan và methylprednisone (MP) với các thuốc mới:
- MPT
|
Melphalan |
0,25mg/kg hoặc 4mg/m2/ngày |
Uống |
Ngày 1-4 |
|
Methylprednisone |
2mg/kg/ngày |
Uống |
Ngày 1-4 |
|
Thalidomide |
100-200mg/ngày |
Uống |
Liên tục |
MP: Cách 4-6 tuần/đợt x 12 đợt. Điều chỉnh liều melphalan theo số lượng bạch cầu và tiểu cầu. Thalidomide: Kéo dài 72 tuần.
- VMP
|
Melphalan |
9mg/m2/ngày |
Uống |
Ngày 1-4 |
|
Methylprednisone |
60mg/m2/ngày |
Uống |
Ngày 1-4 |
|
Bortezomib |
1,3mg/m2 |
Tiêm tĩnh mạch hoặc tiêm dưới da |
Ngày 1, 4, 8, 11 (4 đợt đầu); ngày 1, 8, 15, 22 (4 đợt tiếp) |
Cách 5 tuần/đợt x 12 đợt.
- Rd
|
Lenalidomide |
25mg/ngày |
Uống |
Ngày 1-21 |
|
Dexamethasone |
40mg/ngày |
Uống hoặc truyền tĩnh mạch |
Ngày 1-4 |
Cách 4 tuần/ đợt (đến khi bệnh tiến triển).
- Bendamustine kết hợp methylprednisone: Chỉ định cho những người bệnh không thể ghép tuỷ và có biểu hiện bệnh lý thần kinh ngoại biên nên chống chỉ định điều trị bortezomib và thalidomide.
* Một số phác đồ khác:
- Bortezomib/lenalidomide/dexamethasone (VRD).
- Bortezomib/cyclophosphamide/dexamethasone (VCD).
- Bortezomib/dexamethasone (Vd).
b. Điều trị duy trì: Tuỳ theo nhóm tiên lượng.
- Thalidomide: Uống 100mg/ngày.
- Lenalidomide: Uống 10mg/ngày x 28 ngày x 3 chu kỳ; sau đó liều 15mg ở các chu kỳ sau.
Hoặc: 10mg/ngày x 21 ngày.
- Mỗi chu kỳ cách nhau 4 tuần cho đến khi bệnh tiến triển hay có độc tính không uống được lenalidomide. Mỗi chu kỳ có thể kết hợp dexamethasone.
- Với nhóm nguy cơ trung bình và cao cần điều trị bổ sung: Phác đồ có Bortezomib: 1,3mg/m2, ngày 1, 4, 8, 11. Lặp lại mỗi chu kỳ sau 2 tuần trong 2 năm hoặc cho đến khi bệnh tiến triển hay có độc tính phải dừng Bortezomib.
Hoặc:
Bortezomib: 1,6mg/m2, ngày 1, 8, 15 và 22. Lặp lại mỗi chu kỳ sau 5 tuần trong 6 tháng hoặc cho đến khi bệnh tiến triển hay có độc tính phải dừng Bortezomib.
3.1.2. Người bệnh có khả năng ghép tế bào gốc (< 65 tuổi, có thể tới 70 tuổi nếu thể trạng tốt)
a. Điều trị tấn công: Tuỳ theo nhóm tiên lượng và khả năng có đủ các nhóm thuốc để có chiến lược lựa chọn phác đồ điều trị, thường kết hợp các loại thuốc của các nhóm: nhóm thuốc ức chế proteasome, điều hoà miễn dịch, kháng thể đơn dòng và corticoid .
* Một số phác đồ cụ thể:
- VRD:
|
Bortezomib |
1,3 mg/m2 |
Tiêm dưới da hay tĩnh mạch |
Ngày 1,4,8,11 (4 đợt đầu), ngày 1,8,15,22 (4 đợt tiếp) |
|
Lenalidomide |
25m/ngày |
Uống |
Ngày 1-14 |
|
Dexamethasone |
40mg/ngày |
Truyền tĩnhmạch/uống |
Ngày 1,8,15 |
- VTD:
|
Bortezomib |
1,3 mg/m2 |
Tiêm dưới da hay tĩnh mạch |
Ngày 1,4,8,11 (4 đợt đầu), ngày 1,8,15,22 (4 đợt tiếp). |
|
Thalidomide |
100-200mg |
Uống |
Liên tục |
|
Dexamethasone |
40mg/ngày |
Truyền tĩnh mạch/uống |
Ngày 1-4 |
- VCD:
|
Bortezomib |
1,3 mg/m2 |
Tiêm dưới da hay tĩnh mạch |
Ngày 1,4,8,11 (4 đợt đầu), ngày 1,8,15,22 (4 đợt tiếp) |
|
Cyclophosphamide (Một trong 3 kiểu) |
300mg/m2/ngày |
Truyền tĩnh mạch/uống |
Ngày 1,8,15,22 |
|
500mg/m2/ngày |
Truyền tĩnh mạch/uống |
Ngày 1,8,15 |
|
|
900mg/m2/ngày |
Truyền tĩnh mạch |
Ngày 1 |
|
|
Dexamethasone |
40mg/ngày |
Truyền tĩnh mạch/uống |
Ngày 1-4, 9-12, 17-20 |
|
40mg/ngày |
Truyền tĩnh mạch/uống |
Ngày 1,8,15 |
|
|
40mg/ngày |
Truyền tĩnh mạch |
Ngày 1-4 |
- PAD:
|
Bortezomib |
1,3 mg/m2 |
Tiêm dưới da hay tĩnh mạch |
Ngày 1,4,8,11 (4 đợt đầu), ngày 1,8,15,22 (4 đợt tiếp) |
|
Doxorubicin |
9mg/m2/ngày |
Truyền tĩnh mạch |
Ngày 1-4 |
|
Dexamethasone |
40mg/ngày |
Truyền tĩnh mạch |
Ngày 1-4, 9-12, 17-20 (chu kỳ 1) và Ngày 1-4 (chu kỳ 2-4). Hoặc cả 4 chu kỳ: Ngày 1-4, 9-12, 17-20 |
* Một số phác đồ khác:
- Daratuzumab kết hợp các nhóm thuốc.
- Bortezomib/ dexamethasone.
- Lenalidomide/ dexamethasone.
- VTD-PACE.
* Thường điều trị từ 4 đến 6 đợt cách nhau 10-14 ngày, trước khi thực hiện huy động và thu gom tế bào gốc. Sau mỗi đợt điều trị phải đánh giá đáp ứng để cân nhắc phác đồ tiếp theo.
* Lưu ý tác dụng phụ của các thuốc mới:
- Bortezomib: Tổn thương thần kinh ngoại biên (đau buốt chi), giảm tiểu cầu (5%), biến chứng muộn nhiễm vi rút Zoster. Biến chứng độc với thần kinh ngoại biên của bortezomib giảm đáng kể khi thay đổi tiêm tuần 2 lần sang mỗi tuần một lần hoặc chuyển từ tiêm tĩnh mạch sang tiêm dưới da tuần 1 lần.
- Thalidomide: Suy thận, tổn thương thần kinh ngoại biên, tắc mạch.
- Lenalidomide: Gây ức chế tuỷ (giảm bạch cầu, tiểu cầu) nên có kế hoạch lấy tế bào gốc ngay sau 4 đợt điều trị và thường phải huy động tế bào gốc bằng Cyclophosphamid; tắc mạch, táo bón.
- Với tác dụng phụ của thalidomide và lenalidomide gây tắc mạch, dự phòng bằng aspirin, heparin trọng lượng phân tử thấp hay courmarin nếu người bệnh ở nhóm nguy cơ cao tắc tĩnh mạch sâu.
b. Điều trị ghép tế bào gốc tự thân
- Phác đồ điều kiện hoá chuẩn: Melphalan (200 mg/ m2 tĩnh mạch). Liều melphalan cần được điều chỉnh theo mức độ suy thận.
- Sử dụng nguồn tế bào gốc máu ngoại vi là chính. Có thể truyền tươi hoặc được bảo quản âm sâu -196ºC.
- Sau truyền tế bào gốc theo dõi mọc mảnh ghép và các biến chứng như nhiễm trùng, xuất huyết... Chăm sóc người bệnh theo chế độ chăm sóc cấp 1.
- Ghép tự thân hai lần: Được chỉ định ở người bệnh không đạt được lui bệnh một phần rất tốt sau ghép đầu tiên hoặc sau tái phát.
c. Điều trị duy trì sau ghép
- Mục đích: Giúp đạt lui bệnh thêm, kéo dài thời gian bệnh không tiến triển và tái phát.
- Nhóm nguy cơ tốt: Duy trì thalidomide 100 mg/ngày cho đến khi bệnh tiến triển hay có độc tính không uống được thalidomide.
- Nhóm nguy cơ tốt không đạt CR hoặc VGPR: Lenalidomide cho đến khi bệnh tiến triển hay có độc tính không uống được lenalidomide.
- Nhóm nguy cơ cao và trung bình: Điều trị duy trì bằng phác đồ có bortezomib tối đa trong 2 năm; hoặc cho đến khi bệnh tiến triển hoặc có các tác dụng phụ do bortezomib.
3.2. Điều trị bệnh tái phát và khing thuốc
a. Nguyên tắc:
- Khi tái phát và kháng thuốc cần xét nghiệm lại đánh giá yếu tố nguy cơ và tổng thể bilan bệnh như ban đầu chẩn đoán vì có sự chuyển đổi clone để có kế hoạch điều trị.
- Lựa chọn điều trị trong bệnh cảnh tái phát phụ thuộc vào nhiều yếu tố như: tuổi, thể trạng người bệnh, các bệnh đi kèm, thể bệnh, hiệu quả và dung nạp của điều trị trước, số lần điều trị trước, các lựa chọn điều trị có sẵn còn lại và khoảng thời gian của lần điều trị gần nhất; yếu tố kinh tế các phác đồ, sự thuận tiện trong điều trị...
- Ở những người bệnh trẻ tuổi tái phát, chỉ định ghép tự thân lần hai (tham khảo phác đồ ghép tự thân cho đa u tuỷ xương).
b. Các phác đồ điều trị khi tái phát/ kháng thuốc:
- Có thể quay trở lại các phác đồ tấn công (VTD, VCD...) đã điều trị trước đây nếu tái phát sau 6 tháng và đánh giá đáp ứng sau mỗi đợt điều trị để điều chỉnh phác đồ.
- Các phác đồ kết hợp thuốc mới được ưu tiên chỉ định:
+ Bortezomib/lenalidomide/dexamethasone;
+ Daratumumab/bortezomib/dexamethasone;
+ Daratumumab/lenalidomide/dexamethasone;
- Một số phác đồ khác có thể chỉ định:
+ Bendamustine/bortezomib/dexamethasone;
+ Bendamustine/lenalidomide/dexamethasone;
+ Bendamustine/pegylated liposomal doxorubicin /dexamethasone;
+ Bortezomib/cyclophosphamide/dexamethasone;
+ Cyclophosphamide/lenalidomide/dexamethasone;
+ Bortezomib/dexamethasone;
+ Daratumumab;
+ Lenalidomide/dexamethasone;
+ Lenalidomide/PLD/VD;
+ PLD-VTD;
+ Selinexor: Khi kháng với tất cả các thuốc.
- Một số phác đồ khác có thể hiệu quả trong một số trường hợp:
+ Bendamustine;
+ Dexamethasone/ cyclophosphamide/ etoposide/ cisplatin (DCEP);
+Dexamethasone/thalidomide/cisplatin/doxorubicin/cyclophosphamide/etoposide (DT-PACE) +/- bortezomib (VTD-PACE);
+ Liều cao cyclophosphamide.
- Một số phác đồ cụ thể:
+ Phác đồ bortezomib kết hợp với pegylated liposomal doxorubicin (PLD) như sau:
|
Bortezomib |
1,3 mg/m2 |
Tiêm dưới da hay tĩnh mạch |
Ngày 1,4,8,11 (4 đợt đầu), ngày 1,8,15,22 (4 đợt tiếp) |
|
PLD |
30mg/m2/ngày |
Truyền tĩnh mạch |
ngày 4 |
Cách 21 ngày/đợt x 8 đợt.
+ L-PLD-VD:
● Lenalidomide: 10mg/ngày, uống 21ngày, chu kỳ 28 ngày;
● Pegylated liposomal Doxorubicin: 40mg/m2/ngày da, truyền tĩnh mạch ngày 1 (truyền trong 2 giờ);
● Bortezomib: 2mg/ngày, tiêm dưới da hoặc tĩnh mạch ngày 1;
● Dexamethasone: 40mg/ngày, truyền tĩnh mạch ngày 1.
+ PLD-VTD:
● Pegylated liposomal Doxorubicin: 40mg/m2/ngày da, truyền tĩnh mạch ngày 1 (truyền trong 2 giờ);
● Bortezomib: 1,3mg/m2 da, tiêm dưới da hoặc tĩnh mạch ngày 1, 4, 8 và 11;
● Thalidomide 50-200mg/ngày, uống hàng ngày trước khi ngủ;
● Dexamethasone: 40mg/ngày, truyền tĩnh mạch hay uống ngày 1-4.
- VTD-PACE
Tấn công:
+ Bortezomib: 1mg/m2 da, tiêm dưới da hoặc tĩnh mạch ngày 1, 4, 8 và 11;
+ Ngày 4-7: Thalidomide 50-200mg, uống hàng ngày trước khi ngủ + dexamethasone 40mg/ ngày, truyền tĩnh mạch hay uống;
+ Ngày 4-7: Cyclophosphamide 400mg/m2 + etoposide 40mg/m2 + cisplatin 10mg/m2 + doxorubicin 10mg/m2 (tất cả truyền tĩnh mạch hàng ngày trong 24h).
Củng cố:
Chu kỳ 1: Bắt đầu 6 tuần - 4 tháng sau ghép:
Ngày 1, 4, 8, and 11: Bortezomib 1mg/m2 tiêm tĩnh mạch hay dưới da.
1-4: Thalidomide 50-200mg uống, tối + dexamethasone 40mg/ ngày.
Ngày 1-4: Cyclophosphamide 300mg/m2 truyền tĩnh mạch mỗi ngày trong 24h + etoposide 30mg/m2 truyền tĩnh mạch mỗi ngày trong 24h + cisplatin 7,5mg/m2 truyền tĩnh mạch mỗi ngày trong 24h + doxorubicin 7,5mg/m2 truyền tĩnh mạch mỗi ngày trong 24h.
Chu kỳ 2: Sau chu kỳ 1: 2-4 tháng.
Ngày 1, 4, 8, and 11: Bortezomib 1mg/m2 IV tiêm tĩnh mạch hay dưới da.
Ngày 1-4: Thalidomide 50-200mg uống, tối + dexamethasone 40mg/ ngày.
Ngày 4-7: Cyclophosphamide 300mg/m2 truyền tĩnh mạch mỗi ngày trong 24h + etoposide 30mg/m2 truyền tĩnh mạch mỗi ngày trong 24 giờ + cisplatin 7,5mg/m2 truyền tĩnh mạch mỗi ngày trong 24 giờ + doxorubicin 7,5mg/m2 truyền tĩnh mạch mỗi ngày trong 24 giờ.
Lưu ý cho tất cả các phác đồ: đánh giá lại kết quả sau mỗi đợt điều trị, nếu không đáp ứng thì chuyển sang phác đồ khác.
3.3. Điều trị hỗ trợ
a. Suy thận
- Bệnh nhân Đa u tủy xương mới chẩn đoán có suy thận: Nên trao đổi huyết tương, chọn phác đồ điều trị có bortezomib và không cần điều chỉnh liều bortezomib. Có thể kết hợp lenalidomide và dexamethasone, tuy nhiên liều lenalidomide phải được điều chỉnh theo mức độ suy thận.
- Chăm sóc chức năng thận: Giảm canxi, giảm acid uric.
- Phòng suy thận:
+ Một số thuốc có nguy cơ gây độc đối với thận nên hạn chế dùng như: Kháng sinh nhóm aminoglycoside, thuốc chống viêm non-steroid, tránh sử dụng thuốc cản quang...;
+ Theo dõi chức năng thận khi điều trị kéo dài thuốc nhóm Bisphosphonate.
b. Thiếu máu
- Erythropoietin tái tổ hợp:
+ Chỉ định cho các trường hợp thiếu máu Hb < 100g/L, đặc biệt bệnh nhân có suy thận;
+ Liều 4.000 UI/ ngày hoặc liều 10.000UI/ 1 lần x 3 lần/ tuần. Mục đích huyết sắc tố đạt trên 120g/L, nhưng < 140g/L.
- Truyền khối hồng cầu.
c. Tổn thương xương
- Tất cả các bệnh nhân cần điều trị tổn thương xương với thuốc nhóm Bisphosphonate hoặc denosumab, nhưng cần lưu ý:
+ Khám răng;
+ Theo dõi chức năng thận khi điều trị kéo dài Bisphosphonate;
+ Theo dõi biến chứng hoại tử xương hàm.
- Điều trị tăng canxi máu:
+ Truyền dịch, lợi tiểu;
+ Nếu canxi máu > 11 mg/dl (2,75 mmol/l) cần phải xử trí cấp cứu;
+ Ức chế huỷ xương: Biphosphonate (ưu tiên zoledronic acid), calcitonine (4-8 UI/kg pha NaCl 0,9% truyền tĩnh mạch trong 6-8 giờ hoặc denosumab;
+ Lọc máu: Chỉ định cho trường hợp tăng canxi máu nặng đe doạ tính mạng, người bệnh có suy thận, phù phổi.
- Bisphosphonate: Làm tăng chuyển hoá của xương và tránh gây huỷ xương, tác dụng giảm đau và cải thiện chất lượng cuộc sống. Bệnh nhân mới: Điều trị không quá 2 năm; bệnh nhân tái phát: Điều trị lại bisphosphonate kết hợp điều trị tích cực bệnh.
+ Zoledronate: Liều 4 mg/lần/tháng, truyền tĩnh mạch trên 30 phút;
+ Pamidronate: Liều 90 mg/lần/tháng, truyền tĩnh mạch trong 2 giờ.
- Bệnh nhân có suy thận mức độ trung bình (độ thanh thải creatinin 30-60mL/p):
+ Zoledronate: giảm đến liều tối đa 3mg/lần, vẫn truyền trên 30 phút;
+ Pamidronate: 90 mg/lần, kéo dài truyền TM trong 4 giờ.
- Lưu ý khi điều trị Bisphosphonate và denosumab:
+ Khám răng;
+ Theo dõi chức năng thận khi điều trị kéo dài Bisphosphonate;
+ Theo dõi biến chứng hoại tử xương hàm: BC muộn, nghiêm trọng liên quan thời gian ĐT bisphosphonate (gặp ở zoledronate > pamidronate).
- Khuyến khích người bệnh vận động nhiều nhất nếu có thể, nhưng tránh gây ra chấn thương.
- Trường hợp đau nhiều và có tính khu trú có thể: Tia xạ liều thấp để giảm đau cho trường hợp không kiểm soát được đau, nguy cơ gãy xương bệnh lý hay nguy cơ chèn ép tuỷ.
- Xem xét đeo khung hỗ trợ cột sống.
- Bệnh nhân đau lưng mức độ nặng do gẫy xẹp thân sống nên tạo hình thân sống bằng đổ xi măng (vertebroplasty) hoặc nong bóng đổ xi măng (kyphoplasty).
d. Dự phòng nhiễm trùng
- Gammaglobulin: Chỉ định khi nhiễm trùng tái diễn, nhiễm trùng nặng đe doạ tính mạng bệnh nhân.
- Tiêm phòng vắc xin phòng viêm phổi do cầu khuẩn, H.influenza.
- Phòng viêm phổi do Pneumocystis carinii (PCP), herpes và chống nấm khi điều trị hoá chất liều cao.
- Khi điều trị nhóm ức chế proteasome (bortezomib và daratumumab): Dự phòng mắc herpes zoster bằng aciclovir.
e. Tổn thương hệ thống thần kinh
- Ép tủy: Cần cấp cứu kịp thời để ngăn ngừa tổn thương không hồi phục như liệt.
+ Chống phù tủy: Dexamethasone liều cao, ban đầu 100mg sau đó 25mg mỗi 6 giờ, rồi giảm dần liều;
+ Tia xạ tại chỗ càng sớm càng tốt, đồng thời kết hợp dexamethasone liều cao;
+ Nếu các biện pháp trên không hiệu quả: Phẫu thuật để giảm áp lực lên tủy sống.
- Thâm nhiễm thần kinh trung ương: Điều trị tiêm tủy sống, tia xạ và điều trị toàn thân, tiên lượng nhóm này rất xấu với thời gian sống trung bình 3 tháng.
g. Tăng độ quánh máu
- Xử trí: Chỉ định trao đổi huyết tương cho những người bệnh có biểu hiện triệu chứng của tăng độ quánh máu như: Chảy máu niêm mạc, triệu chứng thần kinh (đau đầu, chóng mặt hoặc co giật, hôn mê)... hoặc độ quánh huyết tương tăng trên 4 CP (centipoise).
h. Phòng và điều trị huyết khối
- Dự phòng huyết khối:
+ Nguy cơ gặp biểu hiện huyết khối tĩnh mạch (VTE) hay gặp nhất trong giai đoạn 6 đến 12 tháng đầu điều trị, sau đó giảm hơn và thấp hơn ở những bệnh nhân tái phát so với những bệnh nhân chưa điều trị trước đó;
+ Khi điều trị phác đồ có thuốc điều hoà miễn dịch (thalidomide, lenalidomide...), đặc biệt khi kết hợp với glucocorticoid, doxorubicin, erythropoietin cần điều trị dự phòng huyết khối thường quy;
+ Dựa trên nhóm nguy cơ để lựa chọn điều trị dự phòng: Aspirin khi điều trị thuốc điều hoà miễn dịch kết hợp dexamethazone. Khi điều trị liều cao dexamethazone hoặc doxorubicin hoặc bệnh nhân có thêm yếu tố nguy cơ như: Béo phì, bị chấn thương nên dự phòng heparin trọng lượng phân tử thấp (LMWH), ví dụ enoxaparin 40mg tiêm dưới da hàng ngày hoặc đủ liều warfarin.
- Đánh giá các yếu tố nguy cơ sau:
+ Chỉ số khối cơ thể (BMI) ≥ 30 kg/m2;
+ Tiền sử VTE trước đó;
+ Khi điều trị phác đồ có thuốc điều hoà miễn dịch, dexamethazone, doxorubicin...
+ Có catheter tĩnh mạch trung tâm hoặc máy tạo nhịp tim;
+ Bệnh tim (bệnh động mạch vành có triệu chứng, suy tim sung huyết hoặc tiền sử đһt stent/CABG);
+ Bệnh thận mạn tính (mức lọc cầu thận ước tính dưới 30 ml/phút);
+ Đái tháo đường;
+ Nhiễm trùng cấp tính;
+ Bất động;
+ Điều trị erythropoietin;
+ Huyết khối di truyền;
+ D-dimer tăng cao.
- Phác đồ dự phòng:
+ Không có yếu tố nguy cơ hay 1 yếu tố nguy cơ: Aspirin 81 đến 325mg, uống 1 lần hàng ngày tuỳ quyết định của bác sỹ;
+ Có trên 2 yếu tố nguy cơ: LMWH (ví dụ: enoxaparin 40mg/ngày) hoặc warfarin đủ liều (đích INR 2-3).
- Điều trị thuốc chống đông khi có nguy cơ cao tắc mạch:
+ Enoxaparin 40 mg tiêm dưới da (SC) một lần/ngày;
+ Dalteparin 5,000 tiêm dưới da (SC) một lần/ngày;
+ Warfarin (khuyến cáo khi INR 2-3);
+ Aspirin 81-325 mg hàng ngày.
Ghi chú:
+ Nhóm nguy cơ cao (≥ 2 điểm);
+ Nhóm nguy cơ thấp (< 2 điểm).
3.4. Đánh giá đáp ứng điều trị
- Tiêu chuẩn đáp ứng điều trị chuẩn: do nhóm điều trị đa u tuỷ xương Quốc tế (the International Myeloma Working Group) đề xuất năm 2006 và được sửa đổi năm 2016 (bảng 49).
Bảng 49. Tiêu chuẩn đáp ứng điều trị chuẩn (IMWG-2016)
|
Mức độ |
Tiêu chuẩn đáp ứng |
|
Đáp ứng hoàn toàn nghiêm ngặt (stringent CR) |
Tiêu chuẩn như của CR kết hợp với: Tỷ lệ kappa/lamda bình thường và Không phát hiện thấy tế bào dòng plasmo bất thường bằng xét nghiệm hóa mô miễn dịch (immunohistochemistry) hoặc bằng đếm tế bào dòng chảy 6-10 màu. |
|
Đáp ứng hoàn toàn (Complete response-CR) |
Miễn dịch cố định huyết thanh và nước tiểu âm tính và Biến mất các khối u tế bào dòng plasmo mô mềm và Tỷ lệ tế bào dòng plasmo trong tuỷ xương <5%. |
|
Đáp ứng một phần rất tốt (Very good partial response) |
Phát hiện được protein đơn dòng trong huyết thanh và nước tiểu bằng điện di miễn dịch cố định (immunofixation) nhưng không phát hiện được bằng điện di thường hoặc Giảm ≥ 90% protein đơn dòng trong huyết thanh và protein đơn dòng trong nước tiểu <100 mg/ 24 giờ. |
|
Đáp ứng một phần (Partial response) |
Giảm ≥ 50% protein đơn dòng trong huyết thanh và giảm protein đơn dòng trong nước tiểu ≥ 90% hoặc < 200 mg / 24 giờ. Nếu protein đơn dòng trong huyết thanh và nước tiểu không đo được, thì tiêu chuẩn Ig đơn dòng được thay thế bằng giảm ≥ 50% nồng độ chuỗi nhẹ tự do liên quan và không liên quan. Nếu protein đơn dòng trong huyết thanh và nước tiểu không đo được, và không định lượng được chuỗi nhẹ tự do huyết thanh, thì giảm ≥ 50% tế bào dòng plasmo thay thế cho protein đơn dòng, với tỷ lệ tế bào dòng plasmo tuỷ xương trước đó ≥ 30%. Ngoài các tiêu chuẩn trên, nếu có ngưỡng ban đầu, cần có tiêu chuẩn giảm ≥ 50% kích thước của u tế bào dòng plasmo mô mềm. |
|
Bệnh ổn định (Stable disease) |
Không đủ tiêu chuẩn của CR, VGPG, PR hoặc PD. |
|
Bệnh tiến triển (Progress disease) |
Tăng 25% từ giá trị đáp ứng thấp nhất của bất kỳ các tiêu chuẩn sau: Protein đơn dòng trong huyết thanh (trị số tuyệt đối phải ≥ 0,5 g/dl), và/hoặc Protein đơn dòng trong nước tiểu (trị số tuyệt đối phải ≥ 200 mg/24h), và/ hoặc Trên người bệnh không đo được nồng độ protein đơn dòng trong huyết thanh và nước tiểu: Có sự khác biệt giữa nồng độ chuỗi nhẹ tự do liên quan và không liên quan (trị số tuyệt đối phải ≥ 10 mg/dl) Trên người bệnh không đo được nồng độ protein đơn dòng trong huyết thanh và nước tiểu và người bệnh không đo được nồng độ chuỗi nhẹ tự do, tỷ lệ tế bào dòng plasmo trong tuỷ xương phải ≥ 10% Xuất hiện những tổn thương mới hoặc tăng rõ về kích thước của những tổn thương cũ ở ở xương hoặc u plasmo ở mô mềm Tăng can xi máu (can xi huyết thanh được hiệu chỉnh > 11,5 mg/dl) chỉ do rối loạn tăng sinh dòng plasmo. |
CR: Complete response; sCR: strigent complete response; VGPR: very good partial response; PR: partial response; SD: stable disease; PD: progress disease.
- Đánh giá mức độ đáp ứng của IMWG dựa trên tồn dư bệnh tối thiểu (MRD) năm 2018 (NCCN 2018) (theo bảng 50)
Bảng 50. Đánh giá đáp ứng điều trị Đa u tủy xương dựa trên MRD
|
Mức độ |
Tiêu chuẩn đáp ứng |
|
MRD âm tính bền vững |
MRD tủy xương âm tính bền vững (bằng đếm tế bào dòng chảy thế hệ mới (độ phân giải cao, nhiều màu - NGF) hoặc giải trình tự gen thế hệ mới và bằng chẩn đoán hình ảnh, như mô tả dưới đây, được tính khi đạt tối thiểu 1 năm. Những xét nghiệm tiếp theo đó có thể được sử dụng để đánh giá mức độ bền vững của MRD âm tính (v/d MRD âm tính 5 năm) |
|
MRD âm tính với đếm tế bào dòng chảy (FC) |
Không phát hiện thấy các tế bào bất thường dòng plasmo với xét nghiệm đếm tế bào dòng chảy thế hệ mới (NGF) trên mẫu dịch tủy xương, sử dụng quy chình chuẩn EuroFlow cho xét nghiệm đa u tủy xương (hoặc phương pháp đã được kiểm chuẩn) với độ nhạy tối thiểu cần đạt là 1/10^5 tế bào có nhân, hoặc cao hơn. |
|
MRD âm tính với xét nghiệm giải trình tự gen |
Âm tính với kết quả giải trình tự thế hệ mới (NGS) với dòng plasmo tủy xương. Trong đó kết quả giải trình tự ADN tách chiết từ tế bào plasma tủy xương có ít hơn 2 lần đọc trùng nhau, sử dụng hệ thống LymphoSIGHT (hoặc phương pháp khác đã được kiểm chuẩn) với độ nhạy tối thiểu cần đạt là 1/10^5 tế bào có nhân, hoặc cao hơn. |
|
Kết hợp đáp ứng trên chẩn đoán hình ảnh và MRD âm tính |
MRD âm tính được xác định bởi NGF hoặc NGS kết hợp với sự biến mất của tất cả các vùng tăng hoạt tính phóng xạ so với nền cơ sở hoặc tổn thương đã có trên PET/CT trước đó hoặc giảm thấp hơn SUV vùng máu trung thất hoặc giảm thấp hơn mô bình thường xung quanh. |
3.5. Theo dõi sau điều trị: Sau mỗi đợt điều trị
- Tổng phân tích tế bào máu.
- Sinh hoá: Protein (toàn phần, albumin và globulin), các Ig, creatinin, canxi, LDH, beta2 microglobulin, acid uric, chức năng gan...
- Điện di protein và miễn dịch cố định huyết thanh.
- Protein nước tiểu 24h, điện di protein và miễn dịch cố định nước tiểu.
- Định lượng chuỗi nhẹ tự do tuỳ theo diễn biến lâm sàng.
- Kiểm tra tuỷ đồ hay sinh thiết tuỷ xương tuỳ theo diễn biến lâm sàng.
- Xét nghiệm FISH, công thức nhiễm sắc thể đánh giá sau 2-3 đợt điều trị để đánh giá hiệu quả và điều chỉnh phác đồ.
- Chụp kiểm tra CT scan liều thấp toàn thân hay hệ thống xương tuỳ theo diễn biến lâm sàng.
- Kiểm tra PET/CT tuỳ theo diễn biến lâm sàng.
- Đánh giá bệnh tồn dư tối thiểu tuỳ theo diễn biến lâm sàng.
4. TIÊN LƯỢNG
- Các yếu tố tiên lượng bệnh: β2-microglobulin, albumin và LDH huyết thanh.
- Di truyền tế bào, được đánh giá bằng kỹ thuật FISH. Các bất thường di truyền liên quan đến tiên lượng xấu là: t(4;14), del(17p) và t(14;16).
- Nếu kết hợp tổn thương di truyền cùng với phân chia giai đoạn ISS, có thể giúp đánh giá tiên lượng sống không tiến triển bệnh (progression-free survival: PFS) và thời gian sống toàn bộ (overall survival: OS).
TÀI LIỆU THAM KHẢO
1. Ludwig H, Miguel JS, Dimopoulos MA, Palumbo A, et al, 2013. International Myeloma Working Group recommendations for global myeloma care. Leukemia, 1-12.
2. Kyle RA, Rajkumar SV, 2009. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia, 23:3-9.
3. Avet-Loiseau H, Durie BGM, Cavo M et al. 2013. Combining fluorescent in situ hybridization data with ISS staging improves risk assessment in myeloma: an International Myeloma Working Group collaborative project. Leukemia, 27: 711-717.
4. Lemieux E, Hulin C, Caillot D, et al, 2013. Autologous stem cell transplantation: an effective salvage therapy in multiple myeloma. Biol Blood Marrow Transplant, 19:445-449.
5. Barlogie B, Angtuaco E, Bartel T, 2010. Chapter 109: Myeloma. Williams Hematology eighth edition (Marshall, Thomas J.Kipps, Uri Seligsohn, Kenneth Kaushansky, Josef T. Prachal).
6. Lyman GH, Khorana AA, Kuderer NM, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol 2013; 31:2189.
7. Rajkumar SV. Multiple Myeloma: 2016 update on Diagnosis, Risk-stratification and Management. Am J Hematol. 2016 July ; 91(7): 719-734.
8. Moreau P, Miguel J.S, Ludwig H, Schouten H, Mohty M, Dimopoulos M, Dreyling M. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology 28 (Supplement 4): iv52-iv61, 2017
9. David Dingli, S. Vincent Rajkumar, MD; and Asher A. Chanan Khan. Therapy for Relapsed Multiple Myeloma: Guidelines From the Mayo Stratification for Myeloma and Risk-Adapted Therapy. Mayo Clin Proc. 2017;92(4):578-598.
10. Tulio E. Rodriguez et all. Busulfan, Melphalan, and Bortezomib versus High-Dose Melphalan as a Conditioning Regimen for Autologous Hematopoietic Stem Cell Transplantation in Multiple Myeloma. Biol Blood Marrow Transplant. 2016 August ; 22(8): 1391-1396.
11. Gunjan L. Shah. Feasibility and Toxicity of Pharmacokinetic (PK)- Directed Dosing of Evomela® (propylene glycol free melphalan, PGF-MEL) for Multiple Myeloma (MM) and AL Amyloidosis (AL) Patients Undergoing Autologous Hematopoietic Stem Cell Transplant (AHCT). Abstracts / Biol Blood Marrow Transplant 24 (2018) S119-S290.
12. Shaji K. Kumar et all, NCCN Guidelines Version 2.2019 Multiple Myeloma.
13. S. Vincent Rajkumar. Multiple Myeloma: 2020 update on Diagnosis, Risk- stratification and Management.
38. U PLASMO ĐƠN ĐỘC
1. ĐẠI CƯƠNG
U plasmo đơn độc (solitary plasmacytoma - SP là bệnh lý tăng sinh đơn dòng tế bào plasmo tại chỗ, khu trú tại xương hoặc tại mô mềm, và không có triệu chứng lâm sàng, không có tiêu chuẩn chẩn đoán của đa u tủy xương.
U plasmo đơn độc được chia thành 2 nhóm, dựa theo vị trí: U plasmo tại xương (solitary plasmacytoma of bone: SBP) và U plasmo tại mô mềm (extramedullary plasmacytoma: EMP).
2. CHẨN ĐOÁN
2.1. Lâm sàng
a. Vị trí khối u:
- U plasmo tại xương thường gặp tại: Các đốt sống, xương sườn, xương sọ, xương khung chậu, xương đùi, xương ống chân, xương đòn, xương bả vai.
- U plasmo tại mô mềm thường gặp tại: Đường hô hấp trên, hệ tiêu hóa, hạch lympho, bàng quang, vú, tuyến giáp, tinh hoàn, tuyến mang tai, da, thần kinh trung ương.
b. Triệu chứng lâm sàng:
- U plasmo tại xương: Đau xương, gãy xương bệnh lý tại chỗ, liệt do chèn ép vào rễ thần kinh.
- U plasmo tại mô mềm: Tăng tiết đờm dãi, chèn ép đường thở, chảy máu mũi, đau bụng, đau đầu, buồn nôn… do khối u chèn ép tại chỗ.
2.2. Cận lâm sàng
a. Xét nghiệm mô bệnh học tổ chức khối u: Là xét nghiệm cơ bản để chẩn đoán. Hình thái của tế bào plasmo khá đặc trưng và được khẳng định bằng nhuộm hóa mô miễn dịch. Trường hợp hiếm gặp như u nguyên bào plasmo (plasmablastic) hoặc kém biệt hóa (anaplastic) có thể gây khó khăn cho chẩn đoán.
b. Xét nghiệm tủy xương
- Xét nghiệm huyết tủy đồ.
- Sinh thiết tủy xương.
Tỷ lệ tế bào plasmo trong tủy < 10%.
c. Điện di protein, điện di miễn dịch huyết thanh và nước tiểu: Phát hiện thành phần đơn dòng chuỗi nặng và nhẹ. Khoảng 20% bệnh nhân u tương bào có đỉnh đơn dòng protein (mức độ nhẹ), chủ yếu là IgA.
d. Chẩn đoán hình ảnh
- Chụp X-quang xương (cột sống, xương chậu, xương sọ, xương sườn...): Đánh giá tổn thương tiêu xương, kích thước khối u…
- Chụp cộng hưởng từ, chụp cắt lớp vi tính, chụp xạ hình xương, chụp PET CT-scan: Cần thiết để phát hiện, đánh giá tổn thương và kích thước khối u.
đ. Xét nghiệm sinh hoá: Protid máu toàn phần, albumin, globulin, β2-microglobulin, creatinine và canxi huyết thanh. Định lượng globulin miễn dịch IgG, IgA, IgD, IgM và đo chuỗi nhẹ tự do (free light-chain: FLC) trong huyết thanh, nước tiểu: Cần thiết để tiên lượng và theo dõi quá trình tiến triển của bệnh.
e. Tổn thương di truyền: Có thể gặp t(14;16), t(6;14), t(4;14), t(14;20), del13, del17…
2.3. Tiêu chuẩn chẩn đoán (bảng 51)
Bảng 51: Tiêu chuẩn chẩn đoán u tương bào đơn độc theo IMWG (International Myeloma Working Group (Rajkuma SV và cộng sự) (Theo WHO 2017).
|
U tương bào đơn độc Sinh thiết khối u tại xương hoặc tại mô mềm có tăng sinh dòng plasmo. Sinh thiết tủy xương ngẫu nhiên không có bằng chứng tăng sinh dòng plasmo. Ngoại trừ tổn thương khối u tại chỗ, thăm dò xương, chụp MRI hoặc CT cho hình ảnh bình thường. Không có tổn thương cơ quan đích, ví dụ như: tăng canxi máu, suy thận, thiếu máu, tổn thương xương do khối u tế bào plasmo. |
|
U tương bào đơn độc có liên quan tối thiểu với tủy xương Có tăng sinh tế bào dòng plasmo trong tủy xương nhưng <10% ( thường được xác định bằng đếm tế bào dòng chảy (flow cytometry). |
3. ĐIỀU TRỊ
3.1. Xạ trị
- Hiện nay, xạ trị là phương pháp điều trị chuẩn đối với u plasmo tại xương.
- Xạ trị vẫn là lựa chọn hàng đầu cho điều trị u plasmo mô mềm ở vùng đầu và cổ. Riêng đối với u plasmo tại dạ dày, ruột, đường hô hấp trên: nếu cần xạ trị phải lưu ý chảy máu niêm mạc do biến chứng của tia xạ.
- Xạ trị kết hợp với hóa trị.
|
Kích thước khối u |
Liều tia xạ |
Số đợt tia xạ |
|
< 5cm |
40Gy |
20 |
|
> 5cm |
50Gy |
25 |
3.2. Phẫu thuật
- Phẫu thuật bóc khối u để làm chẩn đoán mô bệnh học hoặc để giải phóng chèn ép do khối u gây ra.
- Phẫu thuật kết hợp với xạ trị, phẫu thuật kết hợp với hóa trị.
3.3. Hóa trị
- Chỉ định:
+ Người bệnh không đáp ứng với xạ trị;
+ Kết hợp cùng xạ trị khi kích thước khối u > 5cm, khối u có độ ác tính cao;
+ Người bệnh thuộc nhóm nguy cơ cao (xem mục 4).
- Có thể điều trị u plasm tại dạ dày ruột với phác đồ kết hợp bortezomib với dexamethasone.
4. TIẾN TRIỂN VÀ TIÊN LƯỢNG
- Có 3 dạng tiến triển xấu: Tiến triển thành bệnh đa u tủy xương, tái phát tại chỗ, có tổn thương xương mới dù chưa tiến triển thành đa u tủy xương.
- U plasmo tại xương có tiên lượng xấu hơn u tại mô mềm về mọi mặt: Nguy cơ tiến triển thành đa u tủy xương cao hơn, thời gian sống toàn thể ngắn hơn.
- Một số yếu tố có ảnh hưởng xấu đến kết quả điều trị cũng như có nguy cơ cao tiến triển thành đa u tủy xương:
+ Kích thước khối u > 5 cm;
+ Tổn thương tại gai cột sống;
+ Liều tia xạ (liều cao trên 45 Gy có thể xuất hiện tổn thương tại chỗ hoặc tổn thương lan toả);
+ Protein huyết thanh tăng cao, tăng chuỗi nhẹ, tăng Ig đơn dòng dai dẳng (1 năm) sau điều trị.
Dựa trên tỷ lệ chuỗi nhẹ và protein đơn dòng, Dengli phân các nhóm nguy cơ (bảng 52).
Bảng 52. Phân nhóm nguy cơ
|
Chỉ số |
Nhym nguy cơ |
Tỷ lệ tiến triển thành Đa u tủy xương sau 5 năm |
|
Tỷ lệ chuỗi nhẹ tự do kappa/lambda huyết thanh trong giới hạn bình thường. Protein đơn dòng < 5 g/L. |
Thấp |
13% |
|
Có một trong hai giá trị trên bất thường. |
Trung bình |
26% |
|
Tỷ lệ chuỗi nhẹ tự do kappa/lambda huyết thanh bất thường. Protein đơn dòng > 5 g/L. |
Cao |
62% |
Tỷ lệ chuỗi nhẹ tự do kappa/lambda bình thường: 0,26-1,65.
5. THEO DÕI SAU ĐIỀU TRỊ
- Xét nghiệm mỗi 2-3 tháng: Tổng phân tích tế bào máu, định lượng chuỗi nhẹ tự do huyết thanh, nước tiểu, creatinin và canxi…
- Chẩn đoán hình ảnh: X-quang xương, chụp cộng hưởng từ, chụp cắt lớp vi tính, đặc biệt là chụp xạ hình xương hoặc chụp PET CTscan để theo dõi, đánh giá và phát hiện các tổn thương mới.
PHỤ LỤC
Một số phác đồ điều trị tấn công cụ thể u plasmo đơn độc
a. VD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu và 1, 8, 15 và 22 trong 4 đợt tiếp.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch ngày 1- 4 và ngày 9-12 trong tất cả các đợt.
(Điều trị trong 8 đợt).
b. VTD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu và 1, 8, 15 và 22 trong 4 đợt tiếp.
- Thalidomide 100-200 mg uống ngày 1-21.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch ngày 1- 4.
(Điều trị trong 8 đợt).
c. VCD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu và 1, 8, 15 và 22 trong 4 đợt tiếp.
- Cyclophosphamide 300 mg/m2/ngày truyền tĩnh mạch 1, 8, 15 và 22.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch 1, 8, 15 và 22.
(Điều trị trong 8 đợt).
d. PAD
- Bortezomib 1,3 mg/m2 tiêm dưới da hay tiêm tĩnh mạch ngày 1, 4, 8, 11 trong 4 đợt đầu và 1, 8, 15 và 22 trong 4 đợt tiếp.
- Doxorubicin 10 mg/m2/ngày truyền tĩnh mạch 1- 4.
- Dexamethasone 40 mg/ngày truyền tĩnh mạch 1-4.
(Điều trị trong 8 đợt).
Lưu ý tác dụng phụ của các thuốc:
- Bortezomib: Đau buốt chi, giảm tiểu cầu, nhiễm virus Zoster (biến chứng muộn). Tùy thuộc vào biến chứng có thể thay đổi thành tiêm mỗi tuần một lần hoặc chuyển sang tiêm dưới da.
- Thalidomide: Suy thận, tổn thương thần kinh ngoại biên, tắc mạch.
- Lenalidomide: Gây ức chế tuỷ (giảm bạch cầu, tiểu cầu), tắc mạch, táo bón.
Với tác dụng phụ của thalidomide và lenalidomide gây tắc mạch, nên dự phòng bằng aspirin, heparin trọng lượng phân tử thấp hay courmarin nếu người bệnh ở nhóm nguy cơ cao tắc tĩnh mạch sâu.
- Dexamethasone: Nóng rát sau xương ức, tăng huyết áp… Dự phòng bằng thuốc bảo vệ niêm mạc dạ dày, theo dõi huyết áp để chỉ định thuốc điều chỉnh huyết áp.
TÀI LIỆU THAM KHẢO
1. Sevil Kilciksiz. “A Review for Plasmacytoma of Bone and Extramedularry Plasmacytoma”. 2012. The Scientific World Journal ID 895765;
2. M. Hughes. “Guidelines on the diagnosis and management of solitary of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: update 2009”. Group of UKMF Guidelines working group.
3. International Myeloma Working Group. Criteria for the classification of monoclonal gammapathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. B Br J Haematol. 2003; 121(5): 749-757.
4. NCCN guidelines version 3.2020 - Multiple Myeloma.
5. Ludwig H, Miguel JS, Dimopoulos MA, Palumbo A, et al, 2013. International Myeloma Working Group recommendations for global myeloma care. Leukemia, 1-12.
6. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th Edition-2017); 250-253.
CHƯƠNG III. TRUYỀN MÁU, CHỈ ĐỊNH XÉT NGHIỆM, THỦ THUẬT
39. CHỈ ĐỊNH VÀ SỬ DỤNG MÁU, CHẾ PHẨM MÁU TRONG LÂM SÀNG
1. LỜI NÓI ĐẦU
Chế phẩm máu bao gồm nhiều sản phẩm đa dạng dùng để điều trị thay thế cho một hoặc nhiều thành phần quan trọng bị thiếu hụt ở người bệnh. Chế phẩm máu có thể được điều chế giản đơn từ máu toàn phần bằng cách ly tâm phân lớp theo tỷ trọng hoặc gạn tách từ người hiến máu bằng máy tách thành phần máu tự động. Huyết tương có thể được sử dụng để truyền trong điều trị lâm sàng hoặc được tiếp tục sản xuất theo các công nghệ chiết tách công nghiệp tạo ra các dẫn xuất huyết tương. Ngoài ra chế phẩm máu còn được điều chế bằng công nghệ tái tổ hợp gen.
2. NHỮNG VẤN ĐỀ THIẾT YẾU TRONG TRUYỀN MÁU LÂM SÀNG
2.1. Truyền máu là một phần quan trọng trong hoạt động điều trị; chỉ định truyền máu hợp lý có thể cứu sống người bệnh; ngược lại, các tai biến trong truyền máu làm trầm trọng thêm tình trạng bệnh lý và có thể dẫn đến tử vong.
2.2. Tất cả các cơ sở y tế có thực hiện truyền máu phải xây dựng và phê duyệt quy trình thực hành chuẩn cho từng giai đoạn trong toàn bộ quá trình truyền máu và các nhân viên có liên quan đều phải được đào tạo, tuân thủ quy trình.
2.3. Chỉ định truyền máu là trách nhiệm của bác sĩ điều trị, dựa trên các quy định, hướng dẫn, phác đồ điều trị được phê duyệt, căn cứ tình trạng cụ thể ở từng người bệnh, cũng như tiên lượng diễn biến bệnh lý với khả năng điều trị can thiệp hiện có và những nguy cơ, bất lợi có thể gặp do truyền máu.
2.4. Cần lựa chọn đúng chế phẩm máu, phù hợp với tình trạng bệnh lý của mỗi người bệnh, vào đúng thời điểm cần thiết:
a. Đánh giá đúng tình trạng và tiên lượng.
b. Lựa chọn loại chế phẩm máu, thể tích truyền và tốc độ truyền phù hợp.
c. Thông báo cho người bệnh và/hoặc người nhà về chỉ định điều trị và có ghi bệnh án.
d. Điền đầy đủ mẫu phiếu dự trù; Nếu cần truyền cấp cứu, cần liên lạc ngay với đơn vị cung cấp bằng điện thoại để thông báo rõ ràng tình huống khẩn cấp.
2.5. Máu và chế phẩm máu phải được bảo quản đúng điều kiện trong lưu trữ, vận chuyển, tại bệnh phòng trước truyền nhằm ngăn ngừa bội nhiễm và giảm hoạt tính chế phẩm.
2.6. Đảm bảo hòa hợp miễn dịch truyền máu thông qua:
a. Xác định cẩn thận và chính xác người bệnh được chỉ định cần truyền máu.
b. Lấy mẫu, bàn giao mẫu máu xét nghiệm chính xác.
c. Xác định nhóm máu người bệnh, đơn vị máu và thực hiện các xét nghiệm xác nhận khả năng hòa hợp miễn dịch.
d. Cần xác nhận chính xác định danh người bệnh, nhóm máu của người bệnh và đơn vị máu ngay trước khi thực sự thực hiện truyền máu lâm sàng.
2.7. Kiểm soát, phát hiện các dấu hiệu hư hỏng và không được sử dụng túi máu, nếu:
a. Túi máu bị mở van từ trước, bị thủng, rách ở bất cứ vị trí nào làm cho dịch trong túi có thể rò rỉ ra ngoài.
b. Đổi màu, vẩn, tủa: Màu hồng, hoặc đỏ ở lớp huyết tương; lớp hồng cầu màu sẫm màu, tím, đen,…
c. Kiểm soát thấy quá hạn sử dụng của đơn vị máu:
Thông báo ngay cho đơn vị cung cấp máu khi phát hiện hoặc nghi ngờ có các dấu hiệu trên.
2.8. Truyền máu cần thực hiện với bộ dây kim truyền máu vô trùng, có kèm bầu lọc loại 170-200 μm, chỉ sử dụng một lần; thay dây truyền máu đã sử dụng trong vòng tối đa không quá 12 giờ; cần luôn sử dụng bộ dây truyền máu mới khi truyền khối tiểu cầu.
2.9. Lưu ý khi cần làm ấm máu truyền:
a. Cần đảm bảo giữ ấm cơ thể người bệnh khi: truyền máu khối lượng lớn; truyền thay máu, truyền thay huyết tương; nghi ngờ người bệnh có tự kháng thể lạnh.
b. Chỉ được làm ấm máu với thiết bị làm ấm chuyên dụng.
c. Không được làm ấm máu bằng cách đặt vào bình cách thủy hoặc bình đựng nước nóng: Đặc biệt nguy hiểm đối với khối hồng cầu, khối tiểu cầu.
2.10. Trước khi truyền máu cần phải hướng dẫn, khuyến khích người bệnh, người nhà chủ động thông báo cho nhân viên y tế nếu người bệnh cảm thấy các biểu hiện bất thường, như mới xuất hiện cảm giác khó chịu, sốt cao, rét run, nóng bừng, đau ngực, khó thở, đau cơ, đau thắt lưng,…
2.11. Nhân viên thực hiện truyền máu cần được đào tạo, đảm bảo theo dõi tình trạng lâm sàng người bệnh chặt chẽ trước, trong và sau truyền máu; phát hiện và xử trí kịp thời các tai biến có thể xảy ra.
2.12. Tiêm, truyền thuốc, dịch trong khi truyền máu:
a. Chỉ cho phép truyền dung dịch muối sinh lý NaCl 0,9% chung đường truyền tĩnh mạch với máu, chế phẩm máu.
b. Không được tiêm, truyền bất cứ thuốc, dịch truyền nào cùng đường tĩnh mạch khi đang truyền máu, đặc biệt là các dung dịch có canxi (Ringer lactat, canxi các loại…), dung dịch đường glucose các loại,…
2.13. Việc theo dõi người bệnh đang truyền máu:
a. Đối với mỗi đơn vị máu truyền, cần theo dõi người bệnh liên tục và xử trí, ghi hồ sơ khi có bất thường; trường hợp bình thường cần khám và ghi theo dõi vào các thời điểm sau:
- Ngay trước khi truyền máu.
- Ngay sau khi bắt đầu truyền máu.
- 15 phút sau khi bắt đầu truyền máu.
- Mỗi giờ trong khi truyền máu.
- Khi kết thúc truyền máu.
b. Tại mỗi thời điểm theo dõi, cần xác định và ghi hồ sơ các thông tin:
- Toàn trạng chung.
- Mạch, nhiệt độ, huyết áp, nhịp thở, tốc độ truyền.
- Cân bằng dịch: Dịch đưa vào gồm lượng dịch, chế phẩm máu truyền tĩnh mạch và nước uống với dịch mất từ lượng nước tiểu, mồ hôi, lượng máu mất,...
2.14. Tốc độ và giới hạn thời gian truyền máu:
a. Cần bắt đầu truyền máu trong vòng 30 phút từ lúc lấy đơn vị máu ra ngoài thiết bị hoặc hộp bảo quản quy định.
b. Nên truyền xong mỗi đơn vị máu trong vòng 4 giờ.
c. Tốc độ truyền máu:
Cần truyền không quá 20 giọt (1ml)/phút trong 15 phút đầu tiên; tốc độ truyền sau đó tùy thuộc đánh giá tiên lượng về khả năng dung nạp tuần hoàn, hô hấp của người bệnh:
- Trong trường hợp bệnh lý mạn tính, không có thiếu hụt khối lượng tuần hoàn: Cần truyền không quá 30 giọt (1,5ml)/phút; trong trường hợp truyền các chế phẩm huyết tương chứa yếu tố đông máu không bền vững (yếu tố V, VIII) có thể truyền nhanh, nhưng không quá 40 giọt (2ml)/phút.
- Khi có thiếu hụt thể tích lượng tuần hoàn: Có thể truyền nhanh, thành dòng, thậm chí truyền dưới áp lực và phải căn cứ vào tính toán cân bằng dịch ra vào cơ thể và tình trạng tuần hoàn, hô hấp.
- Cần thận trọng điều chỉnh tốc độ truyền chậm: Ở người bệnh có nguy cơ suy tim, suy hô hấp, suy thận, các bệnh lý tim-phổi khác, sốt cao, người suy kiệt, người già, trẻ nhỏ,...
2.15. Hồ sơ liên quan đến truyền máu: Hồ sơ truyền máu là văn bản chuyên môn có tính pháp lê rất quan trọng nhằm kiểm soát hoạt động truyền máu, tối thiểu bao gồm bệnh án, ý kiến đồng ý truyền máu của người bệnh/ người nhà người bệnh bằng văn bản, giấy dự trù, hồ sơ tiếp nhận mẫu máu, hồ sơ kỹ thuật xét nghiệm và cấp phát máu, phiếu truyền máu lâm sàng ghi nhận các thông tin có liên quan đến mỗi đơn vị máu, chế phẩm máu truyền. Các thông tin cần ghi hồ sơ gồm:
a. Việc thông báo cho người bệnh, người thân được về khả năng, lợi ích khi được truyền máu, cũng như nguy cơ có thể xảy ra liên quan đến truyền máu; người thực hiện và ghi hồ sơ.
b. Tình trạng lâm sàng, thông số xét nghiệm dẫn đến chỉ định truyền máu; người ghi hồ sơ.
c. Chỉ định truyền máu: Thời gian chỉ định, loại chế phẩm máu, thể tích, tốc độ truyền; người chỉ định truyền.
d. Các thông tin về dự trù: Thời gian dự trù, thời gian dự kiến cần được cung cấp, loại chế phẩm máu, nhóm máu, thể tích, tính chất cấp bách, các lưu ý đặc biệt về tiền sử truyền máu (nếu có), các yêu cầu bổ sung với đơn vị máu (cô đặc, rửa, lọc bạch cầu, chiếu xạ, bất hoạt vi rút, nếu có); người chỉ định truyền.
e. Các thông tin về xét nghiệm định nhóm máu ABO, Rh(D), xét nghiệm hòa hợp miễn dịch tối thiểu ở các điều kiện 22ºC, 37ºC, anti-globulin; nhân viên thực hiện kỹ thuật xét nghiệm.
f. Thông tin về đơn vị máu truyền: Loại chế phẩm, mã số định danh duy nhất của đơn vị máu, nhóm máu ABO, Rh(D), thể tích, ngày điều chế, hạn sử dụng, điều kiện bảo quản; người cấp phát và ghi hồ sơ.
g. Nhãn hòa hợp miễn dịch của đơn vị máu: Định danh người bệnh đã được xét nghiệm hòa hợp gồm tên, mã người bệnh, nhóm máu ABO, Rh(D), số giường bệnh, khoa phòng điều trị.
h. Thông tin tại giường bệnh: Định nhóm máu ABO của bệnh nhân và đơn vị máu truyền, mạch, huyết áp, tình trạng lâm sàng trước truyền, thời gian bắt đầu truyền; các thông số về tốc độ truyền, mạch, huyết áp và tình trạng lâm sàng theo dõi định kỳ trong khi truyền; các thuốc, dịch truyền được sử dụng trước, trong và sau truyền máu, cũng như việc sử dụng đường truyền chung hoặc riêng rẽ; người thực hiện truyền máu.
i. Tình trạng khi hoàn thành hoặc ngừng truyền bao gồm: Thời gian, lượng máu hoặc chế phẩm máu đã truyền và thể tích còn lại (nếu có); các thông số về mạch, huyết áp và tình trạng lâm sàng; người thực hiện và ghi hồ sơ.
k. Các tình trạng lâm sàng mới xuất hiện trong, sau truyền máu và các xử trí, các theo dõi lâm sàng và xét nghiệm; người phát hiện, chỉ định, thực hiện và ghi hồ sơ.
l. Trong trường hợp nghi ngờ có tai biến liên quan đến truyền máu, thì cần ngừng truyền, báo ngay cho bác sĩ, ghi nhận các dấu hiệu sinh tồn, lấy mẫu máu mới làm xét nghiệm bổ sung tìm nguyên nhân và đánh giá tình trạng bệnh lý. Trong trường hợp quyết định ngừng truyền máu, thì không được hủy đơn vị máu và bộ dây truyền mà phải bàn giao ngay cho đơn vị cấp phát máu để thực hiện điều tra nguyên nhân tai biến. Duy trì đường truyền tĩnh mạch bằng dung dịch muối NaCl 0,9%.
3. TRUYỀN CHẾ PHẨM HỒNG CẦU
Hồng cầu chứa hemoglobin có vai trò chính trong vận chuyển oxy cho các mô và tế bào. Thời gian bảo quản đơn vị chế phẩm tùy thuộc dạng chế phẩm và dung dịch chống đông - bảo quản. Quá trình điều chế, bảo quản các chế phẩm hồng cầu làm cho các các tế bào máu bị hủy hoại, mất chức năng và hơn nữa có nguy cơ gây nhiều phản ứng bất lợi cho người bệnh khi truyền chế phẩm còn tồn dư nhiều bạch cầu. Mỗi đơn vị chế phẩm hồng cầu còn tồn dư khoảng 20 - 100 ml huyết tương và do vậy, thường không đủ để bổ sung thể tích tuần hoàn trong trường hợp giảm khối lượng tuần hoàn, có thiếu hụt hoặc rối loạn đông cầm máu. Trong những trường hợp đó, cần bổ sung truyền huyết tương cho người bệnh.
3.1. Máu toàn phần (Whole Blood)
Máu toàn phần được lấy từ người hiến máu và chứa trong túi chất dẻo vô trùng có dung dịch chống đông - bảo quản bao gồm các chất citrate, phosphate, dextrose và có thể có adenine (CPD-A). Mỗi đơn vị máu toàn phần có thể tích 250-450 ml (không tính lượng dung dịch chống đông - bảo quản), nồng độ hemoglobin khoảng 12g/100 ml. Thời hạn bảo quản tối đa của máu toàn phần có dung dịch chống đông phù hợp ở điều kiện nhiệt độ khoảng 2-6ºC trong 35 ngày ở tủ lạnh chuyên dùng bảo quản máu.
Chỉ định truyền máu toàn phần:
- Mất máu cấp kèm giảm thể tích tuần hoàn (mất khối lượng lớn hơn 30% thể tích máu, tương ứng với hơn 1.500 ml máu ở người nặng khoảng 50kg).
- Truyền thay máu.
Lưu ý:
Máu toàn phần ngày càng ít được cung cấp, sử dụng trong lâm sàng và thường được thay thế bằng các loại khối hồng cầu.
- Trong trường hợp thực sự cần chỉ định máu toàn phần có thể thay thế bằng chỉ định khối hồng cầu kèm các chế phẩm huyết tương.
- Trong trường hợp cần truyền thay máu, có thể dự trù máu toàn phần hoàn nguyên từ khối hồng cầu trộn với huyết tương hòa hợp miễn dịch (thí dụ: có thể thay máu trẻ sơ sinh hoặc truyền máu trong tử cung bằng chế phẩm khối hồng cầu nhóm O trộn huyết tương nhóm AB). Truyền thay máu, đặc biệt là thay máu ở trẻ sơ sinh nên sử dụng máu toàn phần hoặc khối hồng cầu có thời gian bảo quản không quá 7 ngày kể từ lúc lấy máu.
3.2. Khối hồng cầu đậm đặc (Packed Red Cells)
Mỗi đơn vị khối hồng cầu đậm đặc điều chế từ một đơn vị máu toàn phần lấy từ một người hiến máu, chứa 150-250ml hồng cầu (tương ứng với 250-450 ml/đv máu toàn phần) đã loại bỏ hầu hết huyết tương với hematocrit khoảng 65-75%, nồng độ hemoglobin khoảng 15-20 g/100ml thể tích thực.
Chỉ định truyền Khối hồng cầu đậm đặc:
- Người bệnh bị thiếu hồng cầu có kèm nguy cơ quá tải tuần hoàn.
Lưu ý: Để tăng tốc độ truyền khối hồng cầu đậm đặc, có thể truyền dung dịch NaCl 0,9% qua chạc nối chữ Y khi truyền máu.
3.3. Khối hồng cầu có dung dịch bảo quản
Khối hồng cầu có dung dịch bảo quản được điều chế bằng cách loại bỏ huyết tương từ máu toàn phần bằng ly tâm, bổ sung dung dịch bảo quản giúp duy trì khả năng sống và chức năng hồng cầu (thành phần thường gồm muối NaCl, adenine, glucose và manitol,…).
Mỗi đơn vị khối hồng cầu có bổ sung khoảng 50-100 ml dung dịch hòa loãng (tương ứng với 250-450 ml/đv máu toàn phần), có thể tích 180-300 ml, hematocrit khoảng 50-70%, nồng độ hemoglobin khoảng 10-15 g/100 ml thực. Có thể bảo quản ở 2-6ºC trong thời gian tối đa 42 ngày.
Chỉ định điều trị khối hồng cầu có dung dịch bảo quản:
- Người bệnh thiếu hồng cầu các loại.
Lưu ý:
- Truyền khối hồng cầu cần phù hợp tối thiểu nhóm hệ ABO và Rh(D) đảm bảo bằng định nhóm máu, xét nghiệm hòa hợp và sàng lọc kháng thể bất thường. Trong trường hợp cấp cứu không thể trì hoãn, cho phép truyền khối hồng cầu nhóm O trước khi hoàn thành các xét nghiệm an toàn. Ngoài ra, cần truyền khối hồng cầu Rh(D) âm nếu đã biết trước, hoặc nghi ngờ người bệnh mang nhóm Rh(D) âm.
- Khối hồng cầu có thể được điều chế theo cách thức làm giảm số lượng bạch cầu tồn dư (có thể loại bỏ tới 70% bạch cầu) nhằm giảm bớt các phản ứng liên quan đến bạch cầu.
- Không truyền mỗi đơn vị khối hồng cầu quá 4 giờ. Nếu tiên lượng cuộc truyền kéo dài, cần dự trù để chia tách thành các đơn vị thể tích nhỏ và truyền lần lượt từng đơn vị nhỏ, trong khi bảo quản các đơn vị còn lại trong điều kiện quy định.
3.4. Khối hồng cầu xét nghiệm CMV âm tính/ lӑc bạch cầu/ chiếu xạ
CMV là vi rút thuộc nhóm Herpes có thể lây truyền từ người sang người. Người khỏe bị nhiễm có thể mang vi rút suốt đời. Truyền đơn vị máu từ người nhiễm CMV có thể gây nguy cơ cho người bệnh suy giảm hệ thống miễn dịch bẩm sinh hoặc mắc phải hoặc đang điều trị thuốc ức chế miễn dịch. Việc xét nghiệm bổ sung sàng lọc CMV đơn vị máu giúp giảm thiểu nguy cơ này.
Mỗi đơn vị máu toàn phần, khối hồng cầu có thể chứa trên 1 x 109 bạch cầu. Số bạch cầu này bị hủy hoại phần lớn trong 24 giờ sau khi hiến máu, có thể gây tắc nghẽn, tổn thương tuần hoàn vi mạch, gây ra một số phản ứng truyền máu và nguy cơ đồng miễn dịch ở người bệnh. Bạch cầu có thể mang các vi rút CMV lây truyền cho người bệnh. Việc lọc bạch cầu làm giảm bạch cầu dưới 1 x 106 trong mỗi đơn vị máu giúp ngăn ngừa nguy cơ đồng miễn dịch do bạch cầu và giảm thiểu nguy cơ nhiễm CMV cũng như giảm các nguy cơ khác do bạch cầu.
Những bạch cầu lympho có khả năng tồn tại khá lâu trong đơn vị máu và khi truyền vào cơ thể người bệnh đang trong tình trạng ức chế miễn dịch có thể tăng sinh, phát triển gây phản ứng “ghép chống chủ” nguy hiểm cho người bệnh. Việc chiếu xạ giúp ngăn ngừa khả năng tăng sinh, phát triển của các bạch cầu lympho có tiềm năng miễn dịch này. Chiếu xạ các đơn vị chế phẩm máu trong các thiết bị chuyên dụng với liều chiếu xạ tối thiểu 2.500 centigray (cGy hoặc rad).
Khối hồng cầu có thể được chỉ định thực hiện đồng thời xét nghiệm CMV âm tính, lọc bạch cầu và chiếu xạ hoặc có thể chỉ lọc bạch cầu đơn thuần (không chiếu xạ và xét nghiệm CMV).
Các đặc điểm về hemoglobin, hematocrit, điều kiện và thời gian bảo quản như mục III.3, ngoài ra,khối hồng cầu chiếu xạ phải sử dụng trong vòng 28 ngày sau chiếu xạ
Chỉ định điều trị khối hồng cầu xét nghiệm CMV âm tính, lọc bạch cầu và chiếu xạ đối với người bệnh thiếu hồng cầu có kèm theo:
- Cấy ghép cơ quan, tổ chức.
- Tình trạng giảm miễn dịch bẩm sinh hoặc thứ phát.
- Người bệnh đang điều trị các thuốc ức chế miễn dịch.
- Truyền máu trẻ sơ sinh, truyền máu thai nhi trong tử cung.
- Truyền máu, chế phẩm máu lấy từ cha, mẹ, anh chị em ruột cho người bệnh già yếu, trẻ nhỏ, suy kiệt.
Chỉ định điều trị khối hồng cầu lọc bạch cầu đối với người bệnh thiếu hồng cầu có kèm theo:
- Tình trạng đồng miễn dịch bạch cầu ở người bệnh có giảm bạch cầu, giảm tiểu cầu miễn dịch, xuất huyết giảm tiểu cầu.
- Phản ứng truyền máu (sốt cao, rét run, dị ứng nặng,…) tái diễn nhiều lần do bạch cầu khi truyền máu do bất cứ bệnh lý nào.
- Truyền máu khối lượng lớn.
3.5. Khối hồng cầu rửa (Washed Red Cells)
Khối hồng cầu rửa là khối hồng cầu được loại bỏ huyết tương tối đa bằng cách rửa nhiều lần (tối thiểu 3 lần) với dung dịch muối đẳng trương và được pha loãng trong dung dịch muối, dung dịch bảo quản hồng cầu.
Do có nguy cơ nhiễm khuẩn khi thực hiện quy trình rửa trong hệ thống hở, chế phẩm này có thời hạn bảo quản ngắn ở nhiệt độ 2 - 6ºC không quá 24 giờ kể từ khi điều chế.
Chỉ định điều trị khối hồng cầu rửa đối với người bệnh thiếu hồng cầu:
- Có nguy cơ miễn dịch với các thành phần trong huyết tương (đặc biệt với người bệnh có thiếu hụt IgA, nguy cơ sốc phản vệ, tổn thương phổi do truyền máu TRALI,…).
- Thiếu máu tan máu tự miễn có hoạt hóa bổ thể.
3.6. Khối hồng cầu hòa hợp nhóm máu ngoài hệ ABO
Kháng nguyên hồng cầu có thể gây miễn dịch tạo ra kháng thể khi truyền cho người bệnh không có kháng nguyên tương ứng. Ngoài hệ ABO, hồng cầu còn có các kháng nguyên khác quan trọng trong miễn dịch truyền máu. Một số kháng nguyên thuộc các hệ nhóm máu này có thể kích thích sinh kháng thể gây ra các phản ứng gây tan máu miễn dịch cấp hoặc tan máu miễn dịch chậm.
a. Chỉ định điều trị khối hồng cầu hòa hợp nhóm máu ngoài hệ ABO:
Người bệnh thiếu hồng cầu có kháng thể bất thường được định danh.
b. Lưu ý:
- Phát hiện kháng thể bất thường ngoài hệ ABO bằng xét nghiệm sàng lọc kháng thể bất thường. Cần thực hiện xét nghiệm này ở người bệnh có chỉ định truyền máu, đặc biệt lưu ý đối với phụ nữ đã có tiền sử thai nghén, sinh đẻ, người có tiền sử truyền máu nhiều lần. Ở người được truyền máu nhiều lần, nên thực hiện lặp lại xét nghiệm sàng lọc kháng thể bất thường sau mỗi đợt truyền máu trong khoảng 7 ngày.
- Khi phát hiện thấy có kháng thể bất thường, cần xét nghiệm định danh loại kháng thể; Có thể phát hiện được 1 đến nhiều loại kháng thể bất thường ở một người bệnh; Các kháng thể bất thường ở người bệnh có nguồn gốc chủng tộc Việt Nam hay gặp bao gồm các kháng thể tương ứng với một số kháng nguyên hệ Rh (E, c, C, e, D), hệ MNSs (Mia, S, M), Kidd (Jka, Jkb), Duffy (Fya), P (P1), Lewis (Lea, Leb),…
- Khi định danh được loại kháng thể bất thường, cần dự trù khối hồng cầu hòa hợp âm tính đối với kháng nguyên tương ứng với một hoặc nhiều kháng thể bất thường gặp ở người bệnh.
- Việc tìm kiếm và cung cấp các đơn vị khối hồng cầu hòa hợp hiện mới chỉ đáp ứng ở một vài trung tâm cung cấp máu lớn ở Hà Nội, thành phố Hồ Chí Minh thông qua việc xây dựng được cộng đồng người hiến máu được định nhóm máu hệ ABO và các hệ nhóm máu khác. Đối với những nhóm máu quá hiếm cần được phát hiện và lưu trữ dài ngày bằng kỹ thuật đông lạnh hồng cầu, được trình bày ở phần sau.
3.7. Khối hồng cầu đông lạnh (Frozen Red Cells)
Máu toàn phần và khối hồng cầu bảo quản ở 2 - 6ºC và có hạn sử dụng không quá 35 - 42 ngày. Khối hồng cầu có thể bảo quản đông lạnh nhằm kéo dài thời gian bảo quản. Khối hồng cầu đông lạnh được xử lý và bảo quản đông lạnh ở nhiệt độ không cao hơn - 60ºC trong dung dịch bảo vệ hồng cầu có glycerol với thời hạn bảo quản lên tới 10 năm.
Trước khi sử dụng để truyền cho người bệnh, khối hồng cầu đông lạnh cần được làm tan đông, rửa loại bỏ hết glycerol và hòa loãng trong dung dịch muối đẳng trương, dung dịch bảo quản hồng cầu hoặc trong huyết tương hòa hợp miễn dịch. Khối hồng cầu đông lạnh đã tan đông, loại bỏ glycerol cần bảo quản ở 2- 6ºC không quá 24 giờ trước khi sử dụng cho người bệnh.
Chỉ định điều trị Khối hồng cầu đông lạnh đối với người bệnh thiếu hồng cầu có nhóm máu hiếm gặp trong cộng đồng.
4. TRUYỀN CHẾ PHẨM TIỂU CẦU
4.1. Thông tin chung
- Khối tiểu cầu có thể điều chế từ máu toàn phần có chứa tối thiểu 35-45 x 109 tiểu cầu từ mỗi đơn vị máu toàn phần 250ml; để sử dụng có thể pool (tập hợp) 3-4 đơn vị cùng nhóm máu.
- Khối tiểu cầu gạn tách từ một người hiến bằng máy tách thành phần máu tự động thu nhận được thể tích khoảng 250-300ml và chứa khoảng 3 x 1011 tiểu cầu.
- Khối tiểu cầu được điều chế từ máu toàn phần và pool trong hệ thống kín vô trùng, bảo quản ở 20-24ºC, lắc liên tục trong thiết bị bảo quản tiểu cầu chuyên dụng có thời gian lưu trữ tới 5 ngày. Khối tiểu cầu được điều chế hoặc pool trong hệ thống hở cần sử dụng trong vòng 24 giờ. Khối tiểu cầu sau khi lĩnh từ đơn vị cấp phát máu, phải được đặt trong nhiệt độ phòng ở 20-24ºC và cần được truyền sớm sau khi cấp phát.
- Một người bệnh nặng khoảng 50 kg được truyền đơn vị tiểu cầu pool (chứa ít nhất 1,4 x 1011 tiểu cầu), có thể làm tăng số lượng tiểu cầu sau khi truyền lên thêm 20-40 G/l; nếu được truyền 1 đơn vị tiểu cầu gạn tách (chứa ít nhất 3,0 x 1011 tiểu cầu), có thể làm tăng số lượng tiểu cầu sau khi truyền lên thêm 40-80 G/l. Số lượng tiểu cầu tăng sau truyền còn tùy thuộc nhiều vào tình trạng chảy máu, đồng miễn dịch với bạch cầu, tiểu cầu, hiệu quả điều trị bệnh chính và điều trị rối loạn đông cầm máu,…
Chỉ định chung cho các loại chế phẩm khối tiểu cầu:
- Các tình trạng có xuất huyết do giảm số lượng tiểu cầu dưới 50G/L. Trong tình trạng xuất huyết có giảm tiểu cầu kèm giảm chất lượng tiểu cầu, thì có thể chỉ định truyền khi số lượng tiểu cầu còn cao trên 50G/L.
- Trường hợp người bệnh chưa có xuất huyết, cần xem xét chỉ định truyền khối tiểu cầu khi giảm số lượng tiểu cầu dưới 10G/L. Trong trường hợp số lượng tiểu cầu chưa giảm dưới 50G/L, cần cân nhắc chỉ định truyền nếu đồng thời có giảm chất lượng tiểu cầu của người bệnh và các yếu tố nguy cơ kèm theo như sốt, nhiễm trùng, rối loạn đông máu; nhu cầu phẫu thuật, thủ thuật có nguy cơ chảy máu; các tổn thương hiện có ở các cơ quan trọng yếu trong hộp sọ, lồng ngực, ổ bụng,...
Thận trọng khi chỉ định các loại chế phẩm khối tiểu cầu:
- Số lượng tiểu cầu lớn hơn 100G/L không có kèm các bất thường chức năng tiểu cầu (loại trừ giảm chức năng tiểu cầu do dùng thuốc, do các yếu tố ngoại sinh như ure máu cao, tăng globulin máu,…).
- Các bệnh lý có phá hủy tiểu cầu do nguyên nhân ngoại sinh như giảm tiểu cầu do heparin, ban xuất huyết giảm tiểu cầu tắc mạch, xuất huyết giảm tiểu cầu miễn dịch hoặc vô căn,… trừ khi bệnh nhân có xuất huyết đe dọa tính mạng.
- Có thể truyền khối tiểu cầu thay huyết tương hòa hợp, nếu có nguy cơ không hòa hợp do kháng thể có mặt trong huyết tương của đơn vị khối tiểu cầu.
Liều lượng và đánh giá hiệu quả truyền tiểu cầu:
- Chỉ số CCI (corected count increment) được sử dụng để đánh giá đáp ứng truyền tiểu cầu dựa trên lượng tiểu cầu được truyền (x1011), nồng độ tiểu cầu trước truyền (/μL), sau truyền (/μL) và diện tích da cơ thể - BSA (m2).
CCI = (nồng độ tiểu cầu sau truyền - trước truyền) x BSA / lượng tiểu cầu đã truyền.
Tính CCI vào thời điểm trong vòng 1giờ và 24 giờ sau truyền tiểu cầu.
Bình thường CCI trong vòng 1 giờ là > 7.500 và 24 giờ sau truyền là > 4.500.
Nếu CCI lúc 1 giờ thấp dưới 5.000 có thể nghi ngờ tăng phá hủy tiểu cầu do miễn dịch.
Nếu CCI lúc 1 giờ bình thường, nhưng CCI 24 giờ giảm có thể nghi ngờ tăng phá hủy tiểu cầu do nguyên nhân không miễn dịch.
4.2. Khối tiểu cầu pool từ máu toàn phần
Khối tiểu cầu pool điều chế từ máu toàn phần có 4 loại thể tích 40, 80, 120, 150 ml có số lượng tiểu cầu tương ứng là 0,35 x 1011 tiểu cầu/đơn vị 40ml, 0,7x1011 tiểu cầu/đơn vị 80ml, 1,0x1011 tiểu cầu/đơn vị 120 ml và 1,4x1011 tiểu cầu/đơn vị 150 ml.
Tất cả các đơn vị Khối tiểu cầu này được bảo quản tới 05 ngày ở 20-24ºC kèm lắc liên tục. Các đơn vị khối tiểu cầu pool có thể tích nhỏ được sử dụng cho các bệnh nhân nhi, trọng lượng cơ thể nhỏ.
Chỉ định khối tiểu cầu pool:
- Xem phần trên.
4.3. Khối tiểu cầu pool lӑc bạch cầu
Thể tích đơn vị Khối tiểu cầu pool lọc bạch cầu khoảng 250 ml điều chế từ 2.000 ml máu toàn phần trong hệ thống kín; có tối thiểu 3,0 x 1011 tiểu cầu/đơn vị, số lượng bạch cầu tồn dư thấp hơn 1 x 106/đơn vị, bảo quản tới 05 ngày ở 20-24ºC kèm lắc liên tục.
Chỉ định khối tiểu cầu pool lọc bạch cầu:
- Các tình trạng xuất huyết hoặc có nguy cơ xuất huyết nặng do giảm số lượng hoặc chất lượng tiểu cầu có kèm nguy cơ đồng miễn dịch bạch cầu, hoặc có tình trạng tái diễn phản ứng sốt, dị ứng sau truyền khối tiểu cầu.
4.4. Khối tiểu cầu gạn tách từ một người hiến
- Khối tiểu cầu gạn tách từ một người hiến máu có 3 loại thể tích 120 ml, 250-300 ml và 500 ml có số lượng tiểu cầu tương ứng là 1,5 x 1011 tiểu cầu/đơn vị 120ml, 3,0x1011 tiểu cầu/đơn vị 250-300ml, 6,0 x 1011 tiểu cầu/đơn vị 500ml. Loại đơn vị Khối tiểu cầu thể tích 500 ml sử dụng trong các trường hợp giảm tiểu cầu nặng và kéo dài, có nguy cơ đe dọa tính mạng.
- Khối tiểu cầu gạn tách từ một người hiến máu giảm thiểu tình trạng người bệnh phơi nhiễm với nhiều loại kháng nguyên bạch cầu của nhiều người hiến máu hạn chế tình trạng đồng miễn dịch với bạch cầu.
Chỉ định khối tiểu cầu gạn tách:
Các tình trạng xuất huyết hoặc có nguy cơ xuất huyết nặng do giảm số lượng hoặc chất lượng tiểu cầu
4.5. Khối tiểu cầu gạn tách lӑc bạch cầu, xét nghiệm CMV âm tính và chiếu xạ
Việc điều chế khối tiểu cầu lọc bạch cầu và/hoặc xét nghiệm CMV âm tính và/hoặc chiếu xạ nhằm mục đích ngăn ngừa đồng thời các tai biến đồng miễn dịch do bạch cầu, nguy cơ do nhiễm CMV và nguy cơ “ghép chống chủ” ở người bệnh suy giảm miễn dịch tự nhiên hoặc thứ phát; nguy cơ thải ghép nếu sau này người bệnh được ghép tạng hay tế bào gốc.
Chỉ định khối tiểu cầu gạn tách lọc bạch cầu, xét nghiệm CMV âm tính và chiếu xạ:
- Các tình trạng xuất huyết hoặc có nguy cơ xuất huyết nặng do giảm số lượng hoặc chất lượng tiểu cầu có kèm nguy cơ đồng miễn dịch do bạch cầu, nguy cơ do nhiễm CMV và nguy cơ “ghép chống chủ”, hoặc nguy cơ thải ghép sau này.
- Khối tiểu cầu gạn tách lọc bạch cầu, xét nghiệm CMV âm tính và chiếu xạ có 3 loại thể tích là 120 ml, 250-300 ml và 500 ml. Số lượng tiểu cầu trong các đơn vị này cũng tương tự như Khối tiểu cầu gạn tách cùng thể tích. Loại thể tích 500 ml sử dụng trong các trường hợp điều trị hóa chất, ức chế miễn dịch, cấy ghép cơ quan, tổ chức gây giảm tiểu cầu nặng và kéo dài có nguy cơ đe dọa tính mạng.
5. TRUYỀN CHẾ PHẨM BẠCH CẦU HẠT
Khối bạch cầu hạt pool thể tích 250 ml điều chế từ máu toàn phần có chứa tối thiểu 10 x 109 bạch cầu hạt từ 4.000 ml máu toàn phần.
Khối bạch cầu hạt gạn tách từ một người hiến bằng máy tách thành phần máu tự động thu nhận được thể tích khoảng 250-300 ml và chứa khoảng 10 x 109 bạch cầu hạt.
Các khối bạch cầu hạt được điều chế trong hệ thống kín vô trùng, bảo quản ở 20-24ºC, không lắc có thời gian lưu trữ tới 24 giờ. Khối bạch cầu hạt được điều chế trong hệ thống hở cần sử dụng trong vòng 6 giờ. Khối bạch cầu hạt sau khi lĩnh từ đơn vị cấp phát máu, phải được đặt trong nhiệt độ phòng ở 20-24ºC và cần được truyền sớm sau khi lĩnh.
Một người bệnh, cân nặng khoảng 50 kg được truyền 1 đơn vị khối bạch cầu (chứa ít nhất 10 x 109 bạch cầu hạt), có thể làm tăng số lượng bạch cầu sau khi truyền lên thêm 2-3 G/l. Tuy nhiên, số lượng bạch cầu tăng sau truyền còn tùy thuộc nhiều vào tình trạng nhiễm trùng, đồng miễn dịch với bạch cầu, hiệu quả điều trị bệnh chính,…
Chỉ định chung cho các loại chế phẩm khối bạch cầu hạt:
Các tình trạng nhiễm trùng không kiểm soát được bằng liệu pháp kháng sinh và có kèm giảm số lượng bạch cầu hạt dưới 0,5 G/l;
Chỉ định khối bạch cầu hạt gạn tách:
Các tình trạng nhiễm trùng không kiểm soát được bằng liệu pháp kháng sinh và có kèm giảm số lượng bạch cầu hạt dưới 0,5 G/l trên bệnh nhân có suy giảm miễn dịch bẩm sinh hoặc thứ phát.
Chỉ định khối bạch cầu hạt gạn tách xét nghiệm CMV âm tính và chiếu xạ:
Các tình trạng nhiễm trùng không kiểm soát được bằng liệu pháp kháng sinh và có kèm giảm số lượng bạch cầu hạt dưới 0,5G/l trên bệnh nhân suy giảm miễn dịch bẩm sinh hoặc thứ phát và cấy ghép cơ quan, tổ chức.
6. TRUYỀN CHẾ PHẨM HUYẾT TƯƠNG
6.1. Huyết tương đông lạnh (Frozen Plasma)
Huyết tương đông lạnh là chế phẩm huyết tương điều chế từ máu toàn phần có thời gian bảo quản quá 18 giờ kể từ khi lấy máu, hoặc từ các đơn vị huyết tươi đông lạnh đã được điều chế tủa lạnh. Huyết tương đông lạnh chứa phần lớn các thành phần như huyết tương tươi đông lạnh, ngoại trừ giảm thấp nồng độ các yếu tố đông máu không bền vững như yếu tố V, VIII.
Huyết tương đông lạnh thường được bảo quản ở -18ºC hoặc lạnh hơn trong thời gian 1 năm. Huyết tương tươi đông lạnh sau khi phá đông, được vận chuyển trong điều kiện giữ nhiệt độ trong khoảng 2-6ºC và cần được truyền trong vòng 30 phút. Trong trường hợp chưa sử dụng ngay, phải được bảo quản trong tủ lạnh ở nhiệt độ 2-6ºC không quá 14 ngày.
Chỉ định truyền huyết tương đông lạnh:
- Truyền máu khối lượng lớn: bổ sung với truyền khối hồng cầu.
- Giảm áp lực keo.
- Điều trị quá liều các thuốc kháng vitamin K.
- Truyền hoặc thay huyết tương trong ban xuất huyết giảm tiểu cầu tắc mạch (TTP).
- Điều trị người bệnh thiếu một trong các thành phần protein huyết tương (albumin, C1 inhibitor,…) trong khi không có các chế phẩm đặc hiệu thay thế.
6.2. Huyết tương tươi đông lạnh (Fresh Frozen Plasma)
Huyết tương tươi đông lạnh được điều chế bằng cách chiết tách từ máu toàn phần hoặc tách bằng máy tách thành phần máu tự động, làm đông nhanh đến -25ºC hoặc lạnh hơn trong thời gian 18 giờ kể từ lúc lấy máu hay tách huyết tương. Một đơn vị huyết tương tươi đông lạnh có thể tích khoảng 200-300ml có chứa các protein huyết tương bao gồm các yếu tố đông máu, albumin và immunoglobulin. Huyết tương tươi đông lạnh duy trì nồng độ các yếu tố đông máu không bền vững như yếu tố V, VIII.
Huyết tương tươi đông lạnh thường được bảo quản ở -18ºC hoặc lạnh hơn trong thời gian 1 năm. Huyết tương tươi đông lạnh sau khi phá đông, được vận chuyển trong điều kiện giữ nhiệt độ trong khoảng 2-6ºC và cần được truyền trong vòng 30 phút. Trong trường hợp chưa sử dụng ngay, phải được bảo quản trong tủ lạnh ở nhiệt độ 2-6ºC và truyền trong vòng 24 giờ. Nếu bảo quản ở 2-6ºC quá 24 giờ thì phải hủy hoặc được chuyển sang loại huyết tương đông lạnh.
Chỉ định truyền huyết tương tươi đông lạnh:
Các chỉ định tương tự như với huyết tương đông lạnh (mục VI.1). Ngoài ra, Huyết tương tươi đông lạnh còn thêm chỉ định, đó là:
- Tình trạng chảy máu do thiếu yếu tố đông máu.
- Tình trạng bệnh lý thiếu bẩm sinh hoặc thứ phát các yếu tố đông máu không bền vững (yếu tố V, VIII) mà không có các chế phẩm cô đặc thích hợp (như Khối cô đặc yếu tố VIII, tủa lạnh giàu yếu tố VIII,…).
6.3. Tủa lạnh giàu yếu tố VIII (Cryoprecipitate)
Bằng cách kiểm soát quá trình làm tan đông chậm huyết tương tươi đông lạnh và giữ lại khoảng 10-20 ml tủa hình thành trong huyết tương tan đông tạo ra chế phẩm tủa lạnh. Tủa lạnh giàu yếu tố V, VIII, XIII, fibrinogen, vWF và fibronectin trong một lượng nhỏ thể tích. Tủa lạnh yếu tố VIII thường được cấp phát dưới dạng túi đơn hoặc pool (thường trộn từ các đơn vị huyết tương tách từ 6-8 đơn vị máu toàn phần). Lượng yếu tố đông máu chủ yếu trong một túi tủa lạnh thể tích 80-120 ml gồm yếu tố 240-280 iu VIII/túi, 600-650 mg fibrinogen/túi. Tủa lạnh giàu yếu tố VIII bảo quản ở nhiệt độ thấp hơn -18ºC trong 12 tháng. Sau khi phá đông, tủa lạnh cần được truyền trong vòng 6 giờ.
Chỉ định điều trị tủa lạnh yếu tố VIII:
- Các bệnh thiếu hụt di truyền hoặc mắc phải yếu tố VIII (bệnh Hemophilia A), yếu tố vWF (bệnh von Willebrand), yếu tố XIII, yếu tố XIII, fibrinogen.
- Các rối loạn đông máu có giảm nặng fibrinogen và các yếu tố đông máu khác gặp trong đông máu rải rác nội mạch, tiêu sợi huyết tiên phát hoặc thứ phát.
Lưu ý:
- Có thể truyền tủa lạnh giàu yếu tố VIII không hòa hợp nhóm máu hệ ABO với liều lượng không vượt quá 10 ml/kg cân nặng cơ thể trong mỗi khoảng thời gian 12 giờ (với người bệnh 50 kg có thể truyền 500 ml - tương ứng với 4 đơn vị tủa lạnh loại 80-120 ml/đơn vị không hòa hợp ABO).
7. CÁC SẢN PHẨM CHIẾT TÁCH TỪ PROTEIN HUYẾT TƯƠNG VÀ CHẾ PHẨM TÁI TỔ HỢP
Các sản phẩm protein tinh chế từ nguồn gốc chiết tách từ huyết tương người hoặc công nghệ tái tổ hợp, bao gồm albumin, immunoglobulin và các yếu tố đông máu như yếu tố VIIa, VIII, IX, phức hợp prothrombin,...
7.1. Truyền dung dịch albumin
Chế phẩm albumin được điều chế bằng cách chiết tách công nghiệp từ pool huyết tương của nhiều người hiến máu. Chế phẩm albumin có thể đóng gói dưới dạng dung dịch albumin 5%, 20% hoặc các dung dịch protein giàu albumin (thành phần trên 95% là albumin) có nồng độ albumin khoảng 5%.
Chỉ định điều trị dung dịch albumin (người):
- Điều trị các tình trạng giảm albumin huyết thanh do nhiều nguyên nhân: Hội chứng thận hư, xơ gan cổ trướng, bỏng,…
Lưu ý:
- Không cần làm xét nghiệm hòa hợp miễn dịch khi truyền; truyền không cần bộ lọc.
- Thận trọng khi sử dụng dung dịch albumin với mục đích nuôi dưỡng.
- Truyền nhanh và nhiều dung dịch albumin 20% có thể có nguy cơ tăng thể tích tuần hoàn gây phù phổi.
7.2. Truyền các chế phẩm yếu tố đông máu
a. Khối cô đặc yếu tố VIII (Factor VIII Concentrates, AHF Concentrates)
- Khối cô đặc yếu tố VIII được điều chế từ huyết tương người hoặc công nghệ sinh học tái tổ hợp; được đông khô và bảo quản ở 2-6ºC trong thời hạn quy định của nhà sản xuất; đóng gói loại 250 hoặc 500 hoặc 1.000 iu/chai.
- Truyền 1 đơn vị yếu tố VIII cho mỗi kg cân nặng có thể tăng 2% nồng độ yếu tố VIII trong huyết tương.
- Tái huyền dịch trước khi tiêm truyền. Thời gian bán hủy sau khi tiêm truyền của yếu tố VIII là 8 - 15 giờ.
Chỉ định điều trị khối cô đặc yếu tố VIII:
- Điều trị bệnh Hemophilia A.
- Điều trị bệnh von Willebrand (tùy từng loại chế phẩm và theo hướng dẫn của nhà sản xuất).
Lưu ý:
- Chế phẩm có thể được chỉ định để sử dụng tại nhà với bệnh nhân đã được hướng dẫn.
- Chế phẩm đã tái huyền dịch cần được tiêm truyền qua bộ lọc đóng gói cùng chế phẩm trong vòng 2 giờ.
b. Khối cô đặc yếu tố IX (Factor IX Concentrates)
- Khối cô đặc yếu tố IX được điều chế từ huyết tương người hoặc công nghệ sinh học tái tổ hợp; được đông khô và bảo quản ở 2-6ºC trong thời hạn quy định của nhà sản xuất; đóng gói loại 350 - 600 iu/chai.
- Truyền 1 đơn vị yếu tố IX cho mỗi kg cân nặng có thể tăng 1% nồng độ yếu tố IX trong huyết tương.
- Tái huyền dịch trước khi tiêm truyền. Thời gian bán hủy sau khi tiêm truyền của yếu tố VIII là 11 - 27 giờ.
Chỉ định điều trị Khối cô đặc yếu tố IX:
- Điều trị bệnh Hemophilia B
Lưu ý: Tương tự như lưu ý với khối cô đặc yếu tố VIII (Mục VII.2.a Phần này)
c. Tổ hợp prothrombin cô đặc (prothrombin complex concentrates - PCC)
Tổ hợp cô đặc prothrombin điều chế từ huyết tương người có chứa yếu tố đông máu II, IX, X.
Chỉ định điều trị Tổ hợp prothrombin cô đặc:
- Điều trị bệnh Hemophilia B.
- Các bệnh lý gây kéo dài thời gian prothrombin.
Lưu ý:
- Không nên dùng cho bệnh nhân bị bệnh lý gan mật, các tình trạng bệnh lý có nguy cơ huyết khối.
d. Khối cô đặc yếu tố VIIa tái tổ hợp (Recombinant Factor VIIa- rFVIIa)
Khối cô đặc yếu tố VIIa là glycoprotein điều chế bằng công nghệ tái tổ hợp, có đặc tính tương tự yếu tố VII người. Yếu tố VIIa là dạng hoạt hóa của yếu tố VII có thể hỗ trợ con đường đông máu ngoại sinh, mà không cần sự tham gia của yếu tố VIII và IX.
Chỉ định điều trị Khối cô đặc yếu tố VIIa:
- Hemophilia A và B có xuất hiện chất ức chế (kháng thể kháng yếu tố VIII hoặc IX).
- Hemophilia mắc phải.
- Bệnh suy nhược tiểu cầu Glanzmann.
- Thiếu hụt bẩm sinh yếu tố VII.
- Có thể dùng để điều trị quá liều thuốc chống đông đường uống chống thrombin như dabigatran, rivaroxaban hoặc apixaban.
7.3. Truyền các chế phẩm globulin miễn dịch (Immunoglobulin)
a. Globulin miễn dịch dùng tiêm bắp
Là dung dịch đậm đặc chứa kháng thể IgG điều chế từ huyết tương có khả năng chống lại các tác nhân vi sinh vật mà quần thể người hiến máu đã phơi nhiễm và sinh kháng thể đặc hiệu. Có thể được điều chế từ huyết tương được tăng miễn dịch từ người được miễn dịch chủ động bằng vắc xin (viêm gan B) hoặc người trong giai đoạn hồi phục sau khi nhiễm bệnh (uốn ván, dại). Đóng gói dưới dạng dung dịch 1-2 ml/ống. Không được tiêm tĩnh mạch loại globulin miễn dịch dùng để tiêm bắp.
Chỉ định điều trị các loại Globulin miễn dịch dùng tiêm bắp:
- Giảm immunoglobulin máu bẩm sinh hoặc thứ phát.
- Phòng ngừa các bệnh lý miễn dịch thường gặp ở bệnh nhân có nguy cơ giảm miễn dịch.
- Phòng ngừa hoặc điều trị viêm gan B, uốn ván, dại,…
Chỉ định điều trị Globulin miễn dịch anti-D (anti-D RhIG):
Chế phẩm được điều chế từ huyết tương người hiến có nồng độ cao kháng thể anti-D do được mẫn cảm từ trước và được chỉ định sử dụng:
- Phòng ngừa tình trạng miễn dịch cho người mẹ nhóm máu Rh(D) âm sau khi sinh con, nạo phá thai có nhóm Rh(D) dương hoặc sau truyền các thành phần máu (như khối tiểu cầu) nhóm Rh(D) dương.
Liều thường dụng: Tiêm bắp 500 iu đơn vị ngay khi sinh. Tiêm liều bổ sung chỉ định tùy thuộc vào số lượng chảy máu qua nhau thai khi đánh giá bằng xét nghiệm Kleihauer; nếu chảy máu > 4 ml, thêm 125 iu cho mỗi ml hồng cầu.
Chỉ định điều trị Globulin miễn dịch anti-HBs
- Phòng ngừa tái nhiễm virus viêm gan B sau phẫu thuật cấy ghép gan đối với suy gan do viêm gan B: tiêm 2.000 IU ở người lớn mỗi 15 ngày khi điều trị dài hạn đáp ứng mục tiêu là duy trì nồng độ kháng thể chống HBsAg trong huyết thanh cao hơn 100 IU/l ở người bệnh có HBV-DNA âm tính và cao hơn 500 iu/L ở bệnh nhân HBV-DNA dương tính.
- Phòng ngừa viêm gan B trong trường hợp phơi nhiễm ở người chưa có miễn dịch: Tùy mức độ phơi nhiễm, tối thiểu là 500 iu tiêm càng sớm càng tốt sau phơi nhiễm, tốt nhất là trong vòng 24-72 giờ.
- Phòng ngừa viêm gan B ở người bệnh đang thẩm phân máu: 8-12 iu/kg, tối đa là 500 iu mỗi 2 tháng, cho đến khi có chuyển đổi huyết thanh sau khi tiêm vắc xin.
- Phòng ngừa viêm gan B trên trẻ sơ sinh có mẹ nhiễm virus viêm gan B: Tiêm ngay hoặc càng sớm càng tốt sau khi sinh: 30-100 iu/kg và tiêm nhắc lại cho đến khi chuyển đổi huyết thanh sau khi tiêm vắc xin.
Chỉ định điều trị Globulin miễn dịch chống uốn ván và Huyết thanh chống uốn ván:
Globulin miễn dịch chống uốn ván được điều chế từ huyết tương người trưởng thành đã được tăng miễn dịch bằng giải độc tố uốn ván, có thời gian bán hủy khoảng 28 ngày. Huyết thanh chống uốn ván điều chế từ huyết tương ngựa được tăng miễn dịch bằng giải độc tố và sau đó là độc tố uốn ván, thời gian bán hủy khoảng 2-3 ngày. Các chế phẩm này được chỉ định:
- Dự phòng cho người bệnh có nguy cơ uốn ván (vết thương bị nhiễm bụi bẩn, phân, đất hoặc nước bọt; vết thương do châm chích; nhổ răng; vết thương do vũ khí, dập nát, gãy xương hở, bỏng mà trước đó chưa tiêm phòng đủ 3 liều giải độc tố hoặc tiền sử tiêm phòng uốn ván không rõ.
- Phối hợp (với các liệu pháp kháng sinh, an thần, giãn cơ,…) trong điều trị bệnh uốn ván đang tiến triển.
Liều lượng:
- Dự phòng: Liều Globulin miễn dịch chống uốn ván 250 iu (hoặc 4 iu/kg). Đối với trường hợp vết thương nặng (nhiều nguy cơ bị uốn ván) hoặc có sự chậm trễ trong dự phòng thì liều có thể tăng lên đến 500 iu. Trong tình huống tiếp tục đe dọa bị uốn ván có thể tiêm các liều bổ sung với khoảng cách là 4 tuần.
Huyết thanh chống uốn ván được dùng theo phương pháp giải mẫn cảm Besredka: Tiêm 0,1 ml, chờ nửa giờ, tiêm 0,25 ml, chờ nửa giờ nữa, nếu không phản ứng, tiêm hết liều còn lại. Liều thông thường huyết thanh chống uốn ván dự phòng sau khi bị thương là 1.500 iu. Tăng liều gấp đôi đối với vết thương dễ gây uốn ván hoặc chậm trễ trong bắt đầu tiêm phòng hoặc người có thể trọng quá cao.
- Điều trị: Liều Globulin miễn dịch chống uốn ván là 3.000 - 6.000 iu.
Liều Huyết thanh chống uốn ván: Với uốn ván sơ sinh 5.000 - 10.000 iu; Trẻ em và người lớn 50.000 - 100.000 iu, tiêm dưới da 1/2 liều và nửa còn lại tiêm bắp.
Lưu ý:
- Thận trọng với người có tiền sử phản ứng dị ứng với Globulin miễn dịch từ người hoặc Huyết thanh chống uốn ván từ ngựa, phụ nữ mang thai, người thiếu hụt IgA, người giảm tiểu cầu hoặc rối loạn cầm máu.
- Cần chuẩn bị sẵn sàng xử trí nếu xảy ra phản ứng phản vệ.
b. Globulin miễn dịch dùng truyền tĩnh mạch
Là các chế phẩm immunoglobulin được tinh chế có thể an toàn khi tiêm truyền tĩnh mạch.
Chỉ định điều trị Globulin miễn dịch dùng tiêm tĩnh mạch:
- Điều trị các bệnh lý miễn dịch như xuất huyết giảm tiểu cầu miễn dịch, thiếu máu tan máu tự miễn, các trường hợp thiếu hụt yếu tố đông máu có chất ức chế và một số bệnh miễn dịch khác.
- Điều trị các tình trạng thiếu hụt miễn dịch gây nhiễm trùng nặng.
- Điều trị giảm immunoglobulin máu bẩm sinh hoặc thứ phát
TÀI LIỆU THAM KHẢO
1. Thông tư 26/2013/TT-BYT hướng dẫn hoạt động truyền máu.
2. Sổ tay sử dụng máu lâm sàng (tài liệu dịch của Tổ chức Y tế thế giới), 2008.
3. Technical manual, AABB, 19th edition, 2017.
4. Guide to the preparation, use and quality asurance of blood components, EDQM, 19th edition, 2017.
5. Platelet transfusion therapy, J.Sweeney, M.Lozano, AABB Press, 2013
6. Circular of information for the use of human blood and blood components, AABB, 2017.
40. CHỈ ĐỊNH GẠN TÁCH THÀNH PHẦN MÁU TRONG ĐIỀU TRỊ
1. ĐẠI CƯƠNG
Gạn tách thành phần máu là một phương pháp được sử dụng trong điều trị nhiều bệnh lý và hội chứng khác nhau. Phương pháp gạn tách thành phần máu điều trị (Therapeutic apheresis) được thực hiện qua sử dụng hệ thống máy tự động lấy máu của bệnh nhân ra ngoài cơ thể rồi phân tách máu thành các thành phần khác nhau và loại bỏ một hoặc nhiều thành phần bất thường trong máu để điều trị, đồng thời truyền trả các thành phần còn lại cho bệnh nhân; thông qua đó góp phần vào điều trị, cải thiện tình trạng nặng của bệnh.
Hiện nay, trên lâm sàng người ta thường sử dụng các hệ thống máy tự động để điều trị cho bệnh nhân như: Cobe Spectra, Optia Spectra, Comtec Fresenius...
2. CHỈ ĐỊNH GẠN TÁCH CÁC THÀNH PHẦN MÁU ĐIỀU TRỊ
Căn cứ vào thành phần loại bỏ, thay thế có thể phân chia chỉ định gạn tách thành phần máu điều trị theo các nhóm sau:
2.1. Trao đổi huyết tương điều trị (Therapeutic plasma exchange - TPE)
Thực hiện loại bỏ khối lượng lớn huyết tương của bệnh nhân và thay thế bằng nước muối sinh lý hoặc loại dịch thích hợp để giúp giảm kháng thể và phức hợp miễn dịch cũng như giảm protein máu trong một số bệnh lý.
Chỉ định:
a. Bệnh lý huyết học:
- Hội chứng tăng cao độ nhớt huyết tương: bệnh Waldenström, bệnh đa u tủy xương.
- Ban xuất huyết giảm tiểu cầu huyết khối (TTP).
- Kháng thể miễn chống hồng cầu trong quá trình mang thai (trong bất đồng nhóm máu mẹ con).
- Hội chứng tăng urê huyết tán.
- Ghép tủy không tương thích nhóm máu hệ ABO.
- Biến chứng của bệnh hồng cầu hình liềm.
- Tăng globulin ngưng kết lạnh (cryoglobulinemia).
- Tự kháng thể chống tế bào chất bạch cầu hạt trung tính.
- Giảm kháng thể chống HLA trong ghép tế bào gốc đồng loài.
b. Bệnh lý khác:
- Bệnh nhược cơ (Myasthenia gravis).
- Hội chứng Guillain-Barré cấp tính.
- Viêm hủy myelin cấp tính.
- Viêm đa dây thần kinh tiêu myelin mạn tính.
- Bệnh đa xơ cứng.
- Viêm não Rasmussen.
- Múa giật Sydenham: rối loạn tâm thần kinh tự miễn dịch ở trẻ em liên quan đến nhiễm liên cầu khuẩn.
- Bệnh lý viêm đa dây thần kinh liên quan đến gamma-globulin đơn dòng.
- Viêm cầu thận tiến triển nhanh liên quan đến kháng thể và phức hợp kháng nguyên- kháng thể.
- Tăng chuỗi nhẹ trong đa u tủy xương gây suy thận.
- Hội chứng Goodpasture.
- U bạch cầu hạt Wegener.
- Viêm mạch.
- Viêm khớp dạng thấp.
- Bệnh lupus ban đỏ hệ thống.
- Tăng cholesterol máu gia đình typ II đồng hợp tử.
- Bệnh tăng acid phytanic…
2.2. Gạn tách tế bào máu điều trị (Therapeutic cell depletion):
Mục đích để loại bỏ một lượng lớn các tế bào cao bất thường trong máu tuần hoàn, ngăn ngừa biến chứng huyết khối, tắc mạch, hội chứng tiêu khối u…
Chỉ định:
Khi kết quả xét nghiệm máu ngoại vi thấy tăng cao một hoặc nhiều thành phần sau:
- Bạch cầu cao hơn 100 G/l.
- Tiểu cầu cao hơn 1.000 G/l.
- Thể tích khối hồng cầu (hematocrit) cao hơn 50%.
Gặp trong các trường hợp bệnh lơ-xê-mi cấp và mạn; bệnh tăng sinh tủy ác tính; bệnh tăng tiểu cầu tiên phát; bệnh đa hồng cầu...
2.3. Trao đổi hồng cầu điều trị (Red blood cell exchange):
Mục đích là loại bỏ khối lượng lớn các hồng cầu bệnh lý trong máu bệnh nhân và truyền thay thế bằng hồng cầu bình thường từ người khỏe mạnh.
Chỉ định:
- Bệnh hồng cầu hình liềm: Đột quỵ, hội chứng ngực cấp mức độ nặng, phòng quá tải sắt do truyền máu.
- Bệnh thalassemia.
- Bệnh sốt rét nặng.
- Bệnh babesiosis thể nặng.
- Ghép tế bào gốc tạo máu đồng loài (tế bào gốc từ máu ngoại vi) có bất đồng nhóm máu ABO kiểu minor.
2.4. Gạn tách kết hợp hấp phụ miễn dịch (Immunoadsorption apheresis)
Mục đích loại bỏ chất trung gian gây bệnh trong huyết tương để điều trị cho bệnh nhân bằng cách sử dụng một cột hấp phụ miễn dịch.
Chỉ định:
- Giảm tiểu cầu miễn dịch.
- Kháng thể kháng yếu tố VIII.
2.5. Gạn tách lipoprotein tỷ trọng thấp (LDL apheresis)
Mục đích loại bỏ lipoprotein tỷ trọng thấp trong huyết tương của bệnh nhân thông qua xử lý thứ cấp.
Chỉ định:
- Mức LDL cholesterol lớn hơn 300 g/dl.
- Mức LDL cholesterol lớn hơn 200 g/dl và kèm bệnh lý tim mạch.
2.6. Gạn tách kết hợp kích hoạt bổ sung (Extracorporeal photopheresis)
Sử dụng kỹ thuật apheresis phối hợp với thuốc kích hoạt ánh sáng và tia cực tím để điều chỉnh hoạt động tế bào lympho sau đó truyền lại cho bệnh nhân.
Chỉ định:
- Pemphigus vulgaris thể nặng.
- U lympho tế bào T thể da.
- Viêm da cơ.
- Ghép tim: thải ghép tế bào, dự phòng thải ghép.
- Bệnh ghép chống chủ (GVHD) cấp hoặc mạn (da hoặc không ở da).
- Bệnh Crohn.
- Ghép phổi: thải ghép đồng loài (hội chứng viêm phế quản tắc nghẽn).
- Bệnh xơ cứng toàn thể.
3. KẾT LUẬN
Trong lâm sàng, gạn tách thành phần máu là một phương pháp điều trị quan trọng nhằm nhanh chóng làm giảm tình trạng nặng của bệnh cũng như các nguy cơ xảy ra biến chứng ở nhiều loại bệnh lý khác nhau. Hiện nay, phương pháp này đang được áp dụng rộng rãi tại nhiều cơ sở y tế lớn, đặc biệt trong lĩnh vực điều trị bệnh lý huyết học
TÀI LIỆU THAM KHẢO
1. Gambro BCT Inc. (2005); Essentials guide. Cobe Spectra apheresis system manual.
2. McLeod BC, Prince TH, Drew MJ (2007); Therapeutic Apheresis.Apheresis: Principles and Practice, Bethesda, MD: AABB Press, p.223-418.
3. Oxford Health Plans (2014); Apheresis: Clinical Policy (Effective 01/01/2014) ©1996-2014, LLC 19.
4. Szczepiorkowski ZM, Winters JL, et al (2010); Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher.; 25(3):83-177.
5. Joseph Schwartz, Anand Padmanabhan et al (2019), Guidelines on the use of therapeutic apheresis in clincal practice-evidence-based approach from the writing committee of the American Society for Apheresis: the eighth special issue. Journal of clinical apheresis, 171-354.
41. XỬ TRÍ TAI BIẾN TRUYỀN MÁU
1. ĐẠI CƯƠNG
- Tai biến hoặc phản ứng truyền máu là các phản ứng, biểu hiện xảy ra ở người bệnh có liên quan đến truyền máu hoặc các chế phẩm máu, là hậu quả của tương tác bất thường không mong muốn giữa cơ thể người bệnh với máu, chế phẩm được truyền.
- Các tai biến truyền máu xảy ra có thể là các phản ứng tự giới hạn, tự hồi phục; nhưng cũng có thể gây ra hậu quả nặng nề làm trầm trọng thêm tình trạng lâm sàng, dẫn đến tử vong; các tai biến có thể xảy ra cấp tính hoặc hậu quả có thể kéo dài nhiều năm.
- Các loại tai biến truyền máu khác nhau có thể xảy ra với các biểu hiện ban đầu gần giống nhau, khó phân biệt.
- Tính đa dạng của các tai biến truyền máu: Do nhiều nguyên nhân có thể là các sai sót, nhầm lẫn hành chính; tổ chức và phân công làm việc; năng lực, trình độ, kinh nghiệm chuyên môn; có nhiều cơ chế bệnh lý khác nhau; xảy ra cấp tính hoặc xuất hiện nhiều giờ hoặc nhiều ngày, thậm chí nhiều tháng sau khi kết thúc truyền máu; khác nhau về vị trí cơ quan bị tổn thương; khác nhau về mức độ tổn thương; có thể không ảnh hưởng đáng kể đến sức khỏe người bệnh hoặc có thể gây ra hậu quả nghiêm trọng, thậm chí tử vong; có thể chỉ xảy ra ở một người bệnh được truyền máu nhưng cũng có thể xảy ra ở nhiều người bệnh trong một ngày hoặc trong nhiều ngày,…
- Mục tiêu của xử trí tai biến truyền máu là: Phát hiện sớm các biểu hiện ban đầu của tai biến truyền máu, xác định loại tai biến; tập trung vào điều trị hậu quả, cũng như dự phòng sớm, giảm thiểu các nguy cơ, tiến triển có thể xảy ra tiếp theo.
- Cần coi trọng tính pháp lê khi xảy ra tai biến truyền máu, bảo vệ các hồ sơ tài liệu, vật phẩm có liên quan.
- Xây dựng và duy trì hoạt động của hệ thống bảo đảm chất lượng, giám sát và cải thiện chất lượng hoạt động chung nhằm dự phòng, ngăn ngừa tai biến.
2. XẾP LOẠI TAI BIẾN TRUYỀN MÁU
2.1. Xếp loại theo tính chất xuất hiện và diễn biến
- Tai biến truyền máu cấp: Có thể xảy ra ngay từ khi bắt đầu truyền máu và trong vòng 24 giờ sau truyền máu.
- Tai biến truyền máu chậm: Xảy ra sau 24 giờ đến nhiều ngày sau truyền máu.
2.2. Xếp loại theo mức độ triệu chứng lâm sàng của tai biến
- Phản ứng nhẹ: Thường biểu hiện ở phát ban, mẩn ngứa, mề đay ở da, thường do dị ứng quá mẫn nhẹ.
- Phản ứng trung bình: Lo lắng, mệt, đỏ da, rét run, mề đay, sốt, mạch nhanh, đau đầu, thường do dị ứng, quá mẫn mức độ trung bình, phản ứng sốt sau truyền máu không có tan máu, nhiễm chất gây sốt, nhiễm khuẩn nhẹ,…
- Phản ứng nặng, nguy hiểm tính mạng người bệnh: Sốt cao, đau ngực, đau đầu, đau lưng, khó thở, buồn nôn, nôn, huyết áp hạ, mạch nhanh, đái huyết sắc tố, chảy máu không cầm nơi vết thương hở, rối loạn tri giác,…
2.3. Xếp loại theo các nhóm dấu hiệu hội chứng lâm sàng: Tan máu; tai biến dị ứng, sốc phản vệ; rối loạn hoặc suy hô hấp, suy tuần hoàn, suy thận; rối loạn đông cầm máu; nhiễm trùng huyết; lây nhiễm các tác nhân vi sinh vật đặc hiệu (HIV, HCV, HIV, giang mai, sốt rét, sốt xuất huyết, ZIKA,…).
2.4. Xếp loại theo cơ chế bệnh sinh
- Cơ chế miễn dịch: Có thể gặp trong tai biến tan máu cấp do bất đồng miễn dịch, phản ứng sốt không có tan máu, phản ứng dị ứng, quá mẫn, phản vệ, phản ứng tan máu muộn, ban xuất huyết sau truyền máu, tổn thương phổi cấp do truyền máu (Transfusion-related acute lung injury-TRALI), phản ứng ghép chống chủ…
- Tan máu không do miễn dịch;
- Nhiễm khuẩn, virus: Nhiễm khuẩn máu, lây truyền giang mai, sốt rét, các virus viêm gan, HIV,….
- Rối loạn huyết động: Quá tải tuần hoàn, suy tim, phù phổi cấp,…
- Ứ sắt,…
2.5. Xếp loại hỗn hợp
- Phản ứng miễn dịch cấp tính: Tan máu cấp, sốc phản vệ, dị ứng-mề đay cấp, phản ứng do đồng miễn dịch bạch cầu,…
- Phản ứng cấp tính không do miễn dịch: Quá tải tuần hoàn, tan máu không do miễn dịch, tắc mạch khí, tan máu do các yếu tố hóa, lê,…
- Phản ứng miễn dịch muộn: Phản ứng tan máu muộn, phản ứng ghép chống chủ, ban xuất huyết do miễn dịch tiểu cầu,…
- Phản ứng muộn không do miễn dịch: Quá tải sắt,…
3. CHẨN ĐOÁN
Các dấu hiệu, triệu chứng sớm có vai trò quan trọng trong định hướng chẩn đoán.
3.1. Các nhóm dấu hiệu và biểu hiện sớm về lâm sàng của tai biến truyền máu
Các triệu chứng sớm không đặc hiệu, đa dạng, trùng lặp ở nhiều loại tai biến và có thể được xếp thành các nhóm biểu hiện sau:
- Thay đổi thân nhiệt: Sốt hoặc rét run.
- Dấu hiệu trên da: Sẩn, mẩn ngứa, mề đay.
- Bất thường về tuần hoàn, hô hấp: Mạch nhanh, huyết áp hạ, trụy mạch, khó thở, suy hô hấp.
- Bất thường về đông cầm máu: Đột ngột chảy máu ở các vết thương đã cầm từ trước, không cầm máu được các vết thương mới, máu chảy ra không đông.
- Bất thường về ý thức: Bồn chồn, lo âu, lơ mơ, mất tri giác.
- Thay đổi về cảm giác cơ thể: Khó chịu, bất an, đánh trống ngực, đau lưng, đau bụng, đau đầu, đau vùng tiêm,…
3.2. Định hướng chẩn đoán
- Việc điều trị kịp thời tai biến truyền máu cấp là rất cần thiết, không thể chờ đợi tới khi triệu chứng tai biến đã thể hiện quá rõ ràng, có đầy đủ tiêu chuẩn chẩn đoán.
- Các dấu hiệu, biểu hiện sớm về lâm sàng của tai biến truyền máu thường không đặc hiệu, chưa cho phép xác định chẩn đoán, nhưng rất giá trị giúp định hướng thực hiện các thăm khám, kiểm tra, chỉ định xét nghiệm nhằm xác định chẩn đoán, xử trí phù hợp với từng loại tai biến.
- Một số lưu đồ định hướng chẩn đoán, giúp định hướng loại tai biến truyền máu cấp được trình bày trong Phụ lục 1.
- Các chẩn đoán bệnh lý cụ thể cần được khẳng định khi tiếp tục tìm kiếm các bằng chứng lâm sàng, xét nghiệm phù hợp tiêu chuẩn chẩn đoán cho từng loại bệnh lý và được trình bày trong Phụ lục 2.
4. XÉT NGHIỆM
4.1. Các xét nghiệm hỗ trợ chẩn đoán nguyên nhân tai biến
- Đánh giá nguy cơ không hòa hợp miễn dịch: Định nhóm máu ABO, RhD, xét nghiệm antiglobulin trực tiếp và gián tiếp, sàng lọc và định danh kháng thể bất thường dòng hồng cầu, phát hiện tình trạng thiếu hụt bẩm sinh IgA, phát hiện kháng thể hệ HLA và kháng thể đặc hiệu bạch cầu, tiểu cầu….
- Đánh giá tình trạng tan máu: Định lượng huyết sắc tố, haptoglobin, bilirubin, tìm huyết sắc tố niệu,…
- Đánh giá nguy cơ nhiễm khuẩn máu, các nguy cơ lây nhiễm các tác nhân truyền qua đường máu: CRP, procalcitonin, cấy máu vi khuẩn hiếu khí và kỵ khí, HIV, HBV, HCV, sốt rét, giang mai,…
4.2. Các xét nghiệm đánh giá tiến triển, tiên lượng tình trạng bệnh lý
- Đánh giá tình trạng đông cầm máu, phát hiện nguy cơ đông máu rải rác nội mạch, tiêu sợi huyết,…: Số lượng tiểu cầu, PT, APTT, fibrinogen, D-Dimer, FDP monomer, nghiệm pháp rượu, đàn hồi cục máu đồ,…
- Đánh giá chức năng tuần hoàn, hô hấp,…
- Đánh giá chức năng gan, thận,…
- Tình trạng bất thường về điện giải (K+, canxi ion hóa,..), kiềm toan, quá tải sắt.
5. XỬ TRÍ, ĐIỀU TRỊ
5.1. Xử trí chung
Xử trí chung được áp dụng trong mọi trường hợp xảy ra tai biến và được điều chỉnh tùy theo mức độ tình trạng lâm sàng.
a) Xử trí ban đầu: Trước bất kỳ dấu hiệu bất thường mới xuất hiện ở người bệnh đang hoặc ngay sau truyền máu, cần phải:
- Khóa tạm dừng đường truyền máu.
- Khám và đánh giá các dấu hiệu sinh tồn của người bệnh.
- Xác định hoặc loại trừ nguy cơ truyền máu không hòa hợp nhóm hồng cầu thông qua: Kiểm tra, đối chiếu hồ sơ, định danh người bệnh, nhãn túi máu, chế phẩm máu, kết quả định nhóm máu tại giường bệnh,…; thực hiện lặp lại định nhóm máu ABO với mẫu máu mới lấy từ người bệnh và từ đơn vị máu truyền.
- Ngừng ngay các cuộc truyền máu ở bệnh nhân khác đang hoặc sắp thực hiện để kiểm soát đảm bảo chắc chắn không có tiếp tục nhầm lẫn.
- Đồng thời thông báo ngay cho khoa, phòng, bộ phận xét nghiệm cấp phát để phối hợp cùng kiểm soát các hồ sơ, mẫu phẩm, thuốc thử có liên quan với đơn vị máu có xảy ra tai biến, cũng như kiểm tra các đơn vị máu cấp phát trong cùng khoảng thời gian đó.
- Xác định tai biến mức độ theo mức độ nhẹ, trung bình hay nặng để tiếp tục xử lý bước sau.
b) Khi phản ứng nhẹ, tình trạng lâm sàng ổn định, khẳng định không có nguy cơ tai biến bất đồng nhóm máu, không có biểu hiện tan máu
- Chống dị ứng: Ví dụ diphenynhydramin 10-20 mg tiêm bắp hoặc các thuốc tương đương và theo dõi diễn biến.
- Khi tình trạng người bệnh có cải thiện, có thể bắt đầu cho truyền chậm (< 10 giọt/phút) và theo dõi sát tình trạng lâm sàng: Nếu tình trạng lâm sàng không cải thiện, hoặc tiến triển xấu dần cần xử lý như với phản ứng trung bình.
c) Khi phản ứng trung bình
- Ngừng truyền máu, nhưng không được rút bỏ đường truyền.
- Duy trì đường truyền bằng dung dịch muối sinh lý NaCl 0,9%.
- Điều trị triệu chứng phù hợp với triệu chứng được phát hiện:
+ Ủ ấm cơ thể khi rét run, hạ thân nhiệt;
+ Chống dị ứng: diphenynhydramin 10-20 mg tiêm bắp, methylprednisolon 20-80 mg tiêm, truyền tĩnh mạch hoặc các thuốc tương đương;
+ Chống sốt cao: Paracetamol 10 mg/kg cân nặng, khi thân nhiệt tăng trên 1,5ºC so với trước truyền máu.
- Theo dõi nước tiểu về màu sắc và lưu lượng.
- Nếu xác định không có nguy cơ truyền máu không hòa hợp và có cải thiện lâm sàng, xem xét, nếu cần thì truyền chậm đơn vị máu khác và tiếp tục theo dõi; Nếu tình trạng lâm sàng không cải thiện sau 30 phút, hoặc tiến triển xấu dần cần xử lý như với phản ứng nặng.
d) Khi phản ứng nặng, tình trạng lâm sàng không ổn định, có nguy cơ tai biến bất đồng nhóm máu, có thể nguy hiểm đến tính mạng người bệnh
- Ngừng truyền máu, nhưng không được rút bỏ đường truyền.
- Duy trì đường truyền tĩnh mạch bằng dung dịch muối sinh lý NaCl 0,9%. Lượng dịch truyền căn cứ vào thăng bằng dịch vào ra.
- Hỗ trợ tuần hoàn tăng sức bóp cơ tim với tiêm, truyền tĩnh mạch dopamin, dobutamin, adrenalin, noradrenalin.
- Duy trì hô hấp với hỗ trợ oxy, thở máy.
- Sử dụng corticoid tiêm, truyền tĩnh mạch và thuốc giãn phế quản nếu người bệnh có tiền sử hoặc xuất hiện dấu hiệu co thắt khí phế quản (ví dụ salbutamol khí dung).
- Lợi tiểu đường tĩnh mạch với furosemid.
- Điều trị đông máu rải rác nội mạch: Tùy theo giai đoạn tăng đông (dùng thuốc chống đông) hoặc có tình trạng giảm đông (sử dụng khối tiểu cầu, huyết tương, tủa lạnh).
- Chống nhiễm khuẩn bằng kháng sinh tĩnh mạch phổ rộng nếu nghi ngờ có nhiễm trùng máu.
- Xử trí phù hợp theo tình trạng lâm sàng và loại tai biến.
e) Đồng thời với thực hiện xử trí chung, cần nhanh chóng xác định loại tai biến để bổ sung bằng các điều trị đặc hiệu: Xem mục 5.2.
5.2. Xử trí, điều trị một số các trường hợp cụ thể
Xử lý, điều trị các trường hợp cụ thể, tham khảo theo phụ lục 2.
6. TỔ CHỨC THỰC HIỆN, GHI HỒ SƠ, BÁO CÁO VÀ CẢI THIỆN CHẤT LƯỢNG DỰ PHÒNG TAI BIẾN TRUYỀN MÁU
6.1. Phân công trách nhiệm
- Bác sĩ điều trị, điều dưỡng viên thực hiện truyền máu phải kiểm tra, đối chiếu hồ sơ, xác định danh tính người bệnh, định nhóm máu tại giường; theo dõi truyền máu, phát hiện, xử trí kịp thời các tai biến xảy ra trong và sau truyền máu; khi phát hiện thấy tai biến phải thực hiện ngay các xử trí ban đầu, báo cáo ngay với người phụ trách và phối hợp với các đơn vị liên quan để điều trị người bệnh; theo dõi tiến triển về lâm sàng, xét nghiệm, xử lý kịp thời các nguy cơ hiện có và điều trị dự phòng các biến chứng có thể xảy ra tiếp theo.
- Người phụ trách khoa lâm sàng cần xem xét, cập nhật, trình phê duyệt các quy trình, thực hiện đào tạo và giám sát việc tuân thủ của nhân viên trong khoa.
- Khoa, phòng xét nghiệm cấp phát máu có trách nhiệm xem xét, phân tích, thông báo cập nhật cho khoa lâm sàng về các nguy cơ và loại tai biến có liên quan đến bất đồng miễn dịch.
- Phòng kế hoạch tổng hợp có trách nhiệm giám sát việc tuân thủ của các khoa phòng liên quan và tổng hợp báo cáo lãnh đạo bệnh viện, hội đồng truyền máu bệnh viện, cũng như tổ chức phối hợp với các đơn vị bên ngoài có liên quan.
- Phòng quản lý chất lượng xem xét các quy trình, hồ sơ biểu mẫu kỹ thuật và nhân sự để phát hiện các vấn đề nguy cơ có tính hệ thống.
6.2. Một số công việc cần làm để xác định nguyên nhân
- Thực hiện kiểm tra, đối chiếu các loại hồ sơ có liên quan: Bệnh án, phiếu dự trù, phiếu truyền máu, danh tính người bệnh.
- Thực hiện tại giường: Định nhóm máu ABO các mẫu máu từ người bệnh và đơn vị máu sau khi có biểu hiện tai biến truyền máu, cũng như đối chiếu kết quả đã làm trước khi truyền máu tại giường.
- Thu thập các mẫu bệnh phẩm gồm: Mẫu máu người bệnh trước truyền và sau truyền, mẫu nước tiểu; túi máu, chế phẩm máu và các bộ dây truyền máu đính kèm (Tuyệt đối không được làm các việc sau: ghi thêm thông tin lên nhãn, bóc nhãn, chọc thủng, cắt các đầu van, tháo bộ dây truyền khỏi túi máu, đổ máu và chế phẩm máu ra khỏi túi đựng,…).
- Gửi mẫu bệnh phẩm làm xét nghiệm huyết học (tổng phân tích tế bào máu, xét nghiệm đông máu,…), xét nghiệm sinh hóa máu và nước tiểu (thăm dò chức năng gan, thận, điện giải,…), xét nghiệm vi sinh (nhuộm gram, cấy máu,…).
- Gửi túi máu, chế phẩm máu kèm bộ dây truyền máu và các bệnh phẩm có liên quan cho khoa, phòng xét nghiệm cấp phát máu làm các xét nghiệm về miễn dịch nhóm máu nhằm điều tra về nguy cơ bất đồng miễn dịch truyền máu cũng như nguy cơ về chất lượng của đơn vị máu truyền.
6.3. Báo cáo
- Các phát hiện tai biến truyền máu ở khoa, phòng điều trị lâm sàng cần được thông báo ngay cho phòng xét nghiệm cấp phát máu; thông báo cho phòng kế hoạch tổng hợp, lãnh đạo bệnh viện trong vòng 2 giờ đầu sau khi xác định có tai biến nghiêm trọng.
- Bệnh viện cần thông báo với trung tâm đã cung cấp đơn vị máu có liên quan để phối hợp tìm hiểu nguyên nhân tai biến trong vòng 2 giờ đầu sau khi xác định có tai biến nghiêm trọng.
- Bệnh viện cần báo cáo cơ quan quản lý cấp trên về các tai biến theo định kỳ; trong trường hợp có các tai biến gây tử vong cần báo cáo ngay cơ quan quản lý cấp trên trong vòng 24 giờ đầu.
- Các đơn vị cung cấp máu (do khoa xét nghiệm trong bệnh viện tự cung cấp hoặc do các trung tâm máu bên ngoài thực hiện cung cấp) cần báo cáo với bệnh viện và cơ quan quản lý cấp trên về những thông tin có liên quan đến chất lượng đơn vị máu và những phát hiện có liên quan đến tai biến truyền máu.
6.4. Xem xét, phân tích, xác định nguy cơ, thực hiện các biện pháp cải thiện chất lượng dự phòng nguy cơ
- Tùy theo phân công, tổ chức của từng bệnh viện, phòng Quản lý chất lượng hoặc phòng Kế hoạch tổng hợp cần có báo cáo tổng hợp, phân tích, xác định các nguyên nhân dẫn đến tai biến, cũng như hiệu quả của việc phát hiện, xử trí tai biến, đề xuất kế hoạch nhằm khắc phục các sai sót, đào tạo cập nhật, đánh giá, giám sát và dự phòng các nguy cơ hiện có.
Phụ lục 1
Một số lưu đồ định hướng chẩn đoán loại tai biến truyền máu cấp
1. Biểu hiện sớm với sốt và/hoặc rét run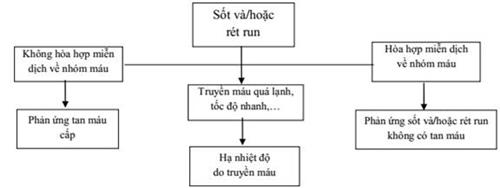
2. Biểu hiện sớm với ban, mề đay, nổi mẩn trên da
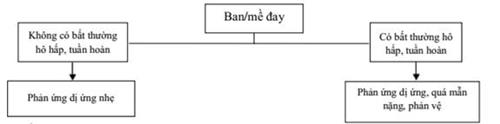
3. Biểu hiện sớm với khó thở
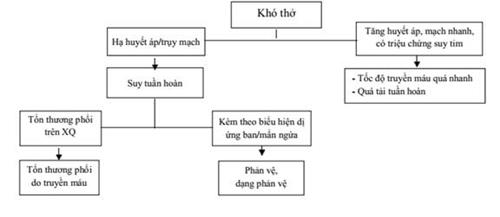
4. Biểu hiện sớm với hạ huyết áp/trụy mạch
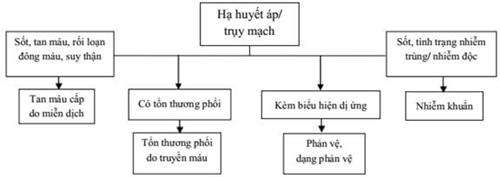
5. Biểu hiện sớm với máu chảy ra không đông, khó cầm máu, đột ngột chảy máu ồ ạt,…

6. Biểu hiện sớm với rối loạn ý thức
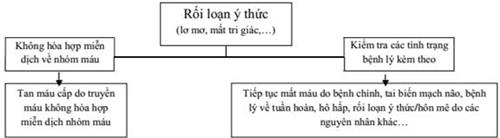
7. Biểu hiện sớm với bất thường về cảm giác cơ thể (khó chịu, cảm giác bất an, đau lưng, đau bụng, đau đầu,…)
Phụ lục 2
Xử trí, điều trị các tai biến truyền máu theo nguyên nhân
|
Loại tai biến/ nguyên nhân |
Dấu hiệu/ triệu chứng/chẩn đoán |
Xử trí/điều trị |
Dự phòng và các lưu ý khác |
|
Tan máu cấp do truyền máu không hòa hợp miễn dịch. Nguyên nhân thường do sai sót, nhầm lẫn gây truyền nhầm đơn vị máu không đúng người bệnh |
- Kích thích, lo lắng, đỏ da, buồn nôn, nôn, đau vị trí truyền máu, đau bụng, đau lưng, đau ngực, sốt/rét run, mạch nhanh, hạ huyết áp, chảy máu không cầm ở các vị trí tổn thương, nước tiểu đỏ hoặc sẫm màu,… - Khẳng định bằng phát hiện sự không hòa hợp nhóm máu của người bệnh và đơn vị máu truyền. |
- Duy trì đường truyền tĩnh mạch bằng dung dịch muối đẳng trương 0,9%; duy trì huyết áp; duy trì lưu lượng nước tiểu tối thiểu 100ml/h bằng truyền dịch và thuốc lợi tiểu đường tĩnh mạch; - Điều trị rối loạn đông máu rải rác trong lòng mạch theo xét nghiệm đông cầm máu; - Truyền máu hòa hợp nếu cần thiết. |
- Theo dõi tình trạng, tiến triển bằng xét nghiệm đông cầm máu, sinh hóa máu, nước tiểu. - Dự phòng bằng định nhóm máu các đơn vị máu và người bệnh tại giường, đối chiếu hồ sơ. |
|
Tan máu không do nguyên nhân miễn dịch, nhiễm khuẩn. Nguyên nhân tan máu có thể do quá hạn sử dụng, pha trộn dung dịch nhược trương, thuốc gây tan máu, bảo quản quá nóng hoặc đông lạnh. |
- Đái huyết sắc tố, có thể biểu hiện vô niệu; - Có thể kết hợp với các triệu chứng khác tùy trường hợp cụ thể; - Chẩn đoán khi kiểm tra đơn vị máu phát hiện tình trạng tan máu. |
- Điều trị triệu chứng; - Chống vô niệu, suy thận. |
- Cần kiểm tra hình thái, màu sắc đơn vị máu trước truyền (màu huyết tương, nước trong trên; màu phần khối hồng cầu,…) |
|
Sốt không có tan máu. Nguyên nhân do các thành phần từ bạch cầu thoái hóa trong quá trình điều chế, bảo quản hoặc do các kháng thể chống bạch cầu. |
- Sốt đơn thuần không quá 1,5ºC so với trước truyền; - Có thể kèm rét run, đau đầu, buồn nôn. - Khẳng định chẩn đoán bằng loại trừ các nguyên nhân khác. |
- Paracetamol 10 mg/kg uống, hoặc tiêm truyền. |
- Thường xảy ra sau khi bắt đầu truyền máu 30-60 phút, lúc cuối hoặc đã kết thúc truyền máu. - Dự phòng bằng chế phẩm máu lọc bạch cầu. |
|
Dị ứng nặng. Nguyên nhân do kháng thể trong huyết thanh người bệnh chống các thành phần protein trong huyết tương truyền |
- Mề đay, ngứa, đỏ mặt; - Tiêu chảy, nôn; - Ran rít, ran ngáy; - Khẳng định chẩn đoán bằng loại trừ. |
- Kháng histamin; - Corticoid. |
- Sử dụng các chế phẩm máu loại bỏ bạch cầu hoặc rửa khối hồng cầu; - Dự phòng bằng kháng histamin, corticoid. |
|
Sốc phản vệ. Nguyên nhân có thể gặp ở người bệnh có thiếu hụt bẩm sinh IgA và tạo ra kháng thể IgE chống kháng nguyên của IgA có mặt trong huyết tương được truyền. |
- Thường xuất hiện ngay sau khi bắt đầu truyền máu. - Khẳng định chẩn đoán khi xét nghiệm phát hiện thiếu hụt bẩm sinh IgA ở người bệnh và có kháng thể chống IgA. |
Nhanh chóng kiểm soát 3 nguy cơ chính (giãn mạch, tăng tính thấm thành mạch và co thắt tiểu phế quản): - Hỗ trợ hô hấp, thở oxy, chống co thắt khí- phế quản; - Truyền dịch; - Adrenalin, dopamin, corticoid, diphehydramin, ... |
- Sử dụng hồng cầu rửa hoặc tiểu cầu rửa để dự phòng; - Sử dụng chế phẩm điều chế từ người hiến máu có thiếu hụt bẩm sinh IgA. |
|
Nhiễm khuẩn máu. Nguyên nhân do máu nhiễm khuẩn khi lấy máu, người cho nhiễm khuẩn tiêu hóa, hiến máu sau khi ăn no, nhiễm khuẩn trong tiếp nhận hiến máu, điều chế, bảo quản máu |
- Các triệu chứng nhiễm trùng, nhiễm độc; - Các triệu chứng tan máu; - Cần nhuộm Gram, cấy máu để khẳng định. |
- Chống sốc, duy trì tuần hoàn, hô hấp, chống suy thận; - Kháng sinh phổ rộng và điều chỉnh theo kết quả kháng sinh đồ. |
- Cần kiểm tra màu sắc túi máu bằng mắt thường, hạn sử dụng, điều kiện nhiệt độ bảo quản; - Cần truyền đơn vị máu trong vòng 4 giờ. |
|
Qui tải tuần hoàn. Nguyên nhân do tốc độ truyền quá nhanh, thể tích truyền quá nhiều và tùy thuộc chức năng tim mạch |
- Ho, khó thở, xanh tím, nổi phồng tĩnh mạch cảnh, đau đầu, nhịp nhanh; - Biểu hiện suy tim, phù phổi cấp tùy mức độ. |
- Tư thế nửa ngồi; - Thở oxy; - Lợi tiểu; - Trợ tim. |
- Dễ gặp ở trẻ sơ sinh, trẻ nhỏ, người già, suy kiệt, suy tim |
|
Tổn thương phổi do truyền máu. Nguyên nhân do kháng thể chống bạch cầu người bệnh có mặt trong huyết tương truyền, gây tổn thương vi tuần hoàn ở phổi (lượng huyết tương còn lại trong khối hồng cầu cũng có thể đủ gây tai biến). |
- Xuất hiện khó thở, suy hô hấp trong vòng 6 giờ sau truyền máu. Có thể trụy mạch, sốt. - Bão hòa oxy máu giảm; - Biểu hiện thâm nhiễm phổi trên XQ. - Xét nghiệm phát hiện và định loại kháng thể chống bạch cầu (HLA class I, II, kháng nguyên đặc hiệu bạch cầu) ở huyết tương người hiến và người bệnh. |
- Hỗ trợ hô hấp, tim mạch. - Có thể sử dụng thêm corticoid. |
Có thể gặp nhiều trên thực tế, nhưng khó xác định chẩn đoán. |
|
Hạ thân nhiệt. Nguyên nhân do truyền máu khối lượng lớn. |
- Rét run; - Hạ nhiệt độ; - Có thể kèm theo các rối loạn hô hấp, tuần hoàn. |
- Giảm tốc độ truyền; - Ủ ấm cơ thể. |
- Cần làm ấm đơn vị máu truyền, điều chỉnh tốc độ truyền phù hợp |
|
Quá tải xitrat. Nguyên nhân do truyền máu khối lượng lớn, truyền quá nhanh. |
- Có biểu hiện lâm sàng giảm canxi; - Xét nghiệm có giảm canxi ion hóa. |
- Tiêm truyền tĩnh mạch canxi. |
- Dễ gặp ở trẻ sơ sinh, người bệnh có rối loạn chức năng gan, rối loạn chuyển hóa toan hóa máu, hạ thân nhiệt, tình trạng sốc. |
|
Tan máu muộn do miễn dịch. Nguyên nhân do đồng miễn dịch sau truyền máu, phụ nữ chửa đẻ nhiều lần; được kích thích miễn dịch sau truyền máu, tăng nhanh hiệu giá kháng thể đồng miễn dịch và có thể gây tan máu sau truyền máu nhiều ngày. |
- Sau truyền máu 7-10 ngày hoặc muộn hơn, xuất hiện sốt, thiếu máu nhanh, vàng da, đái huyết sắc tố. Tăng nồng độ LDH, bilirubin. Trong trường hợp nặng có thể suy thận, đông máu rải rác nội mạch; - Phát hiện được bằng xét nghiệm kháng globulin gián tiếp và xác định tính đặc hiện kháng thể miễn dịch. |
- Điều trị triệu chứng tùy thuộc tình trạng lâm sàng. |
- Dự phòng bằng cách thực hiện sàng lọc kháng thể bất thường; - Chọn lựa đơn vị máu hòa hợp, không có kháng nguyên tương ứng với kháng thể đồng loại đã xác định ở người bệnh. |
|
Ban xuất huyết sau truyền máu. Nguyên nhân do miễn dịch đồng loại với kháng nguyên tiểu cầu sau truyền máu, người chửa đẻ nhiều lần, ở người có kháng nguyên HPA-1a âm tính. |
- Giảm tiểu cầu đơn thuần và đột ngột sau truyền máu 5-12 ngày; - Biểu hiện xuất huyết tùy thuộc mức độ giảm tiểu cầu. - Khẳng định khi xác định kháng thể anti HPA-1a trong huyết thanh người bệnh. |
- Điều trị bằng immunoglobulin tiêm truyền tĩnh mạch 1g/kg cân nặng/ngày x 2 ngày. - Có thể chỉ định thay huyết tương khi cần. |
|
|
Tai biến ghép chống chủ. Nguyên nhân do tăng sinh tế bào lympho T có thẩm quyền miễn dịch từ đơn vị máu truyền; gặp ở người bệnh nhận máu có tình trạng giảm miễn dịch, hoặc khi truyền máu người thân có hòa hợp HLA một phần với người bệnh nhận máu. |
- Xảy ra sau truyền máu 4 - 30 ngày; - Sốt, nổi ban đỏ dát sần, đỏ da toàn thân, phỏng nước; - Nôn, tiêu chảy, viêm gan ứ mật, nổi hạch, giảm nhiều dòng tế bào máu. - Chẩn đoán xác định bằng xét nghiệm chimerism, xét nghiệm sinh học phân tử mảnh sinh thiết da tổn thương. |
- Điều trị triệu chứng |
- Dự phòng bằng chiếu xạ máu, chế phẩm máu. |
|
Quá tải sắt. Nguyên nhân ứ đọng sắt trong cơ thể người bệnh do truyền hồng cầu nhiều lần. |
- Xạm da; - Rối loạn chức năng tụy nội tiết, gan, tim; - Xét nghiệm sắt, ferritin tăng. |
- Thải sắt |
|
|
Lây truyền các virus, ký sinh trùng, giang mai, prion. |
Tham khảo các tài liệu chuyên sâu về các bệnh lý này. |
||
TÀI LIỆU THAM KHẢO
1. Sử dụng máu lâm sàng trong nội khoa chung, sản khoa, nhi khoa, phẫu thuật & gây mê, chấn thương & bỏng, WHO, 2011.
2. Bộ Y tế, Thông tư 26 “hướng dẫn hoạt động truyền máu”, 2013.
3. Circular of information for the use of human blood and blood components, AABB, 2017
42. GHÉP TẾ BÀO GỐC TỰ THÂN ĐIỀU TRỊ BỆNH MÁU
- Ghép tế bào gốc tạo máu tự thân là phương pháp lấy tế bào gốc của chính người bệnh ghép lại cho người bệnh, được chỉ định điều trị cho cho các bệnh máu ác tính không đáp ứng với các phác đồ đa hóa trị hoặc khi bệnh tái phát, hoặc các bệnh lành tính liên quan đến vấn đề tự miễn. Các tế bào gốc hỗ trợ và giúp phục hồi nhanh chóng hệ thống sinh máu của người bệnh sau hóa trị liệu liều cao, nhằm phòng tránh những biến chứng đe dọa tính mạng người bệnh. Hiệu quả của ghép tế bào gốc tự thân chủ yếu có được là nhờ hóa trị liệu liều cao nhằm tiêu diệt tối đa các tế bào ác tính.
- Khi so sánh với ghép tế bào gốc đồng loại, tỷ lệ tử vong liên quan đến ghép trong ghép tế bào gốc tự thân thấp hơn chỉ khoảng 5%.
- Vấn đề đáng lo ngại nhất của ghép tự thân (đặc biệt khi tiến hành đối với lơ xê mi cấp và u lympho ác tính) là nguy cơ nhiễm các tế bào ác tính trong khối tế bào gốc truyền lại cho người bệnh và nguy cơ tái phát cao.
2. CHỈ ĐỊNH GHÉP TẾ BÀO GỐC TỰ THÂN
- Các bệnh máu ác tính: Đa u tủy xương, u lympho không Hodgkin, u lympho Hodgkin, lơ xê mi tủy cấp...
- Các bệnh u bướu: Ung thư vú, buồng trứng, phổi, u tế bào mầm…
- Các bệnh tự miễn: Lupus ban đỏ rải rác, xơ hoá hệ thống, viêm khớp dạng thấp, xuất huyết giảm tiểu cầu miễn dịch…
2.1. Đau tủy xương
Chỉ định hàng đầu ghép tế bào gốc tự thân hiện nay là trong điều trị Đa u tuỷ xương. Tất cả các người bệnh Đa u tủy xương mới được chẩn đoán phải được đánh giá có đủ tiêu chuẩn để ghép tế bào gốc tự thân hay không. Quyết định dựa trên thể trạng người bệnh, chức năng các cơ quan và tình trạng bệnh hơn là tuổi của người bệnh.
- Tuổi: Hầu hết chỉ định ghép tế bào gốc tự thân đối với người bệnh dưới 65 tuổi; tuy nhiên nếu người bệnh trên 65 tuổi nhưng có thể trạng tốt thì vẫn có thể chỉ định ghép, còn nếu dưới 65 tuổi nhưng thể trạng kém thì không có chỉ định.
- Thời điểm ghép: Sau điều trị tấn công đạt lui bệnh hoàn toàn hoặc lui bệnh một phần, cần lựa chọn những người bệnh chưa điều trị các thuốc ảnh hưởng đến tế bào gốc như melphalan…:
+ Vấn đề ghép sớm ngay hay trì hoãn: Ghép tự thân nên cân nhắc cho tất cả các bệnh nhân có chỉ định và đủ điều kiện ghép; nhưng có thể trì hoãn ghép tự thân cho đến khi tái phát lần 1 ở nhóm nguy cơ chuẩn/tốt có đáp ứng với điều trị tấn công, nhưng đã được lấy tế bào gốc sau 4 đợt điều trị;
+ Ghép 2 lần liên tiếp (tandem): Được xem xét chỉ định ở bệnh nhân không đạt được VGPR sau ghép lần 1 hoặc một số trường hợp lựa chọn nhóm nguy cơ cao có del (17p)... ngay sau điều trị tấn công;
+ Ghép lần 2: Cân nhắc chỉ định ở bệnh nhân tái phát sau ghép lần 1 với các tiêu chuẩn sau:
● Bệnh nhân phải đạt thêm lui bệnh sau ghép lần 1 hay có đáp ứng sau ghép;
● Bệnh nhân dung nạp tốt với ghép lần 1;
● Thời gian sống không bệnh (PFS) sau ghép lần 1 ít nhất từ 18 tháng.
2.2. U lympho không Hogdkin
- Chẩn đoán xác định u lympho không Hogdkin;
- Tuổi: Dưới 65 tuổi;
- Phải có tiêu chuẩn đáp ứng với hoá chất trong đợt điều trị tấn công từ đầu hay sau khi tái phát điều trị cứu vãn trước khi ghép;
- Thời điểm chỉ định ghép là tùy từng thể như sau:
+ U lympho không Hogdkin thể tiến triển:
● U lympho không Hogdkin tế bào B lớn lan tỏa (DLBCL): Chỉ định ghép tự thân khi kháng với điều trị từ đầu hoặc sau khi tái phát điều trị hàng 2 có đáp ứng hoá chất cứu vãn hoặc ghép ngay sau khi lui bệnh đợt đầu nếu thuộc nhóm nguy cơ cao như: Double hit lymphoma (DHL) hoặc double express lymphoma (DEL);...
● U lympho không Hogdkin tế bào vùng rìa (MCL): Chỉ định ghép tự thân ngay sau khi điều trị tấn công đạt lui bệnh;
● U lympho tế bào T ngoại vi (PTCL): tất cả các trường hợp trừ U lympho tế bào lớn bất thục sản ALK (+) (ALCL): sau điều trị đáp ứng với hoá chất hàng 1 hoặc sau tái phát/kháng thuốc điều trị cứu vãn hàng 2 có đáp ứng với hoá chất sẽ chỉ định ghép tự thân ngay;
● U lympho tế bào T/NK: Chỉ định ghép tự thân cho những bệnh nhân U lympho tế bào T/NK ngoài hạch, thể mũi (ENKTL-NT) có chỉ số tiên lượng NKPI cao, nhưng có đáp ứng ngay từ đầu với điều trị tấn công bằng phác đồ hoá chất không dựa trên nhóm anthracycline.
+ U lympho không Hogdkin thể tiến triển chậm: U lympho thể nang, U lympho tế bào dạng lympho và plasmo, U lympho tổ chức lympho liên quan màng nhày, U lympho tế bào lympho nhỏ và U lympho thể vùng rìa lách: Chỉ định cho các trường hợp tái phát điều trị cứu vãn còn đáp ứng.
2.3. Bệnh Hodgkin
- Tuổi: Dưới 60 tuổi.
- Chẩn đoán xác định bệnh Hodgkin.
- Thời điểm chỉ định ghép: Bệnh nhân HL tái phát/kháng thuốc nhưng còn nhạy cảm với hoá chất cứu vãn.
2.4. Lơ xê mi tủy cấp
- Tuổi: Dưới 50 tuổi.
- Lơ xê mi cấp thể M3 tái phát sau điều trị đạt lui bệnh về di truyền (PML/RARα âm tính).
Ngoài các tiêu chuẩn trên, các người bệnh cần có thêm các điều kiện sau:
- Thể trạng lâm sàng tốt: Chỉ số toàn trạng bệnh nhân Karnofsky (KPS) 70-100% hoặc chỉ số toàn trạng bệnh nhân ECOG 0-2.
- Chức năng các cơ quan chính còn tốt:
+ Chức năng phổi: LVEF > 50%, PFTs [FVC, FEV1, DLCO] > 60%;
+ Thận: Creatinine < 150 µmol/L;
+ Gan: ALT < 2 lần giới hạn trên của mức bình thường (< 2 x ULN), Bilirubin < 2 lần giới hạn trên của mức bình thường (< 2 x ULN), không có bằng chứng xơ gan.
- Không bị nhiễm trùng dạng đang hoạt động: HIV, nhiễm lao, viêm gan HbeAg dương tính, nhiễm vi khuẩn/ nấm.
- Người bệnh và gia đình người bệnh sau khi được giải thích, cam kết đồng ý ghép tế bào gốc tự thân.
3. CÁC BƯỚC TRONG GHÉP TẾ BÀO GỐC TỰ THÂN
3.1. Lựa chӑn nguồn tế bào gốc
Có 2 nguồn tế bào gốc để ghép tự thân là từ tuỷ xương và máu ngoại vi. Tuy nhiên, trong ghép tự thân chủ yếu sử dụng nguồn tế bào gốc từ máu ngoại vi được thu gom sau khi huy động bằng G-CSF đơn thuần hay có thể phối hợp hoá chất để "mồi" nhằm huy động và thu gom được số lượng cao tế bào gốc.
3.2. Huy động, thu gom và lưu trữ bảo quản tế bào gốc máu ngoại vi
a. Huy động tế bào gốc ở người bệnh: Có một số phương pháp sau:
- Thuốc kích thích bạch cầu như:
+ G-CSF đơn thuần: Liều 10-16 µg/kg/ngày chia 2-3 lần cách đều nhau, đếm số lượng bạch cầu hàng ngày và số lượng tế bào CD34+ từ ngày thứ 4. Khi số lượng tế bào CD34+ ở máu ngoại vi > 10-20 tế bào/µl thì tiến hành gạn tế bào gốc máu ngoại vi. Thường đỉnh huy động đạt cao nhất sau huy động G-CSF đơn thuần là ngày thứ 4-5;
- Phối hợp hoá chất và G-CSF: Chỉ định khi tiên lượng huy động tế bào gốc kém nếu chỉ sử dụng G-CSF đơn thuần (tỷ lệ xảy ra từ 5-30%) và do một số nguyên nhân:
Đã điều trị trước đó bằng những thuốc ảnh hưởng đến tế bào gốc, người bệnh lớn tuổi, đã điều trị tia xạ và có tế bào ác tính xâm lấn tuỷ xương.
+ Với đa u tuỷ xương:
● Cyclophosphamide (liều 2-4g/m2, 1 lần) phối hợp với G-CSF (thường bắt đầu từ ngày thứ 5 sau điều trị hoá chất). Thời điểm đạt huy động tế bào gốc cao nhất phụ thuộc tính cá thể từng người bệnh, thông thường sau 10-20 ngày kể từ khi bắt đầu điều trị hoá chất;
● Hoặc sau đợt điều trị hoá chất mạnh phối hợp với G-CSF thường sau khi bạch cầu giảm và theo dõi xét nghiệm CD34+ máu ngoại vi nếu > 10 -20 tế bào/µl thì tiến hành gạn tế bào gốc.
+ Với U lympho Hodgkin và không Hodgkin: Sau đợt điều trị cứu vãn người bệnh tái phát hoặc sau đợt cuối điều trị tấn công bằng các phác đồ: ICE, DHAP, ESHAP, CE, EA, CBV... kết hợp G-CSF để huy động tế bào gốc;
+ Với Lơ xê mi cấp: Cytarabine 2g/m2 x 2 lần/ngày (cách 12 giờ) trong 4 ngày liên tiếp. Đến ngày 14 sau truyền hoá chất: G-CSF với liều 10 µg/kg/ngày chia 2 lần cách nhau mỗi 12 giờ cho đến khi bạch cầu máu ngoại vi hồi phục, tiến hành theo dõi số lượng tế bào CD34+ máu ngoại vi hàng ngày, khi đạt tối thiểu 10-20 tế bào/ ul thì có thể tiến hành gạn tế bào gốc; hoặc phác đồ etoposide và cytarabine (EA).
b. Gạn tách và thu gom tế bào gốc máu ngoại vi
- Tiến hành gạn tách bằng hệ thống máy tách tế bào như: COBE-Spectra...
- Kết thúc thu gom khi số lượng CD34+ ≥ 2 x 106/kg cân nặng người bệnh nếu kế hoạch ghép 1 lần; còn khi có kế hoạch ghép 2 lần thì liều thấp nhất cần thu gom là ≥ 4 x 106/kg cân nặng người bệnh.
c. Lưu trữ và bảo quản tế bào gốc
- Truyền tươi tế bào gốc: Khối tế bào gốc được bảo quản và lưu trữ ở nhiệt độ 2°C đến 8°C (trong thời hạn 72 giờ); khi đó phải tính toán thời gian sử dụng phác đồ điều kiện hóa trên người bệnh kết thúc trước 72 giờ.
- Truyền tế bào gốc được lưu trữ và bảo quản ở điều kiện âm sâu (-196°C): Tiến hành gạn tế bào gốc cho người bệnh trước, sau khi đã lấy đủ theo yêu cầu số lượng CD34+ ≥ 2 x 106/kg cân nặng người bệnh sẽ tiến hành điều kiện hoá cho người bệnh. Bảo quản âm sâu khối tế bào gốc đông lạnh bằng chất DMSO (dimethyl sulfoxide).
d. Đánh giá tổng thể bệnh nhân trước ghép
- Cộng hưởng từ sọ não để loại trừ vấn đề di căn, thâm nhiễm.
- Chức năng tim mạch: Siêu âm tim, men tim, điện tâm đồ...
- Chức năng hô hấp: CT lồng ngực, đo chức năng hô hấp...
- Tiêu hoá: Nội soi dạ dày, thực quản...
- Khám Răng hàm mặt, Tai mũi họng, Phụ khoa...
3.3. Điều kiện hóa cho người bệnh
- Mục đích của phác đồ điều kiện hoá: Nhằm tiêu diệt các tế bào ung thư kháng với hoá chất thông thường, nhưng không gây ra độc đối với các cơ quan không phải bệnh máu ở mức độ nguy hiểm tính mạng người bệnh.
- Đa u tủy xương:
+ Nếu tuổi dưới 65, thể trạng lâm sàng tốt và chức năng gan, thận bình thường: melphalan liều 200mg/m2 da;
+ Nếu tuổi trên 65, thể trạng lâm sàng tốt hoặc người bệnh có suy thận độ I-II: melphalan liều 140mg/m2 da.
Một số trường hợp đặc biệt có thể sử dụng một trong các phác đồ điều kiện hóa sau:
+ Mel/Bor/dex:
● Melphalan: Liều 200mg/m2 da, ngày -2;
● Bortezomib: Liều 1,3mg/m2 -4 và liều 1,6mg/m2 -1, tiêm tĩnh mạch nhanh;
● Dexamethazone: 20mg truyền tĩnh mạch cùng với mỗi lần tiêm bortezomib (ngày -4 và -1); 10mg truyền tĩnh mạch để chống nôn ngày truyền melphalan (-2).
+ Bu/Mel/Bor:
● Busulfan: Ngày-6 và -5: Liều 130mg/m2 da, truyền tĩnh mạch trên 3 giờ; ngày -4 và -3 truyền điều chỉnh liều để đạt nồng độ AUC đích là 20.000 µM/phút bằng phân tích PK sau liều Bu đầu tiên;
● Melphalan: Liều 140mg/m2 da, ngày -2;
● Bortezomib: Liều 1,6mg/m2, tiêm tĩnh mạch nhanh ngày -1.
- U lympho ác tính (Không Hodgkin và Hodgkin): Chọn 1 trong các phác đồ:
+ BeEAM:
|
Bendamustine |
200 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -7,-6 |
|
Etoposide |
200 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -5 đến -2 |
|
Cytarabine |
200 -400 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -5 đến -2 |
|
Melphalan |
140mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -1 |
+ LEED
|
Melphalan |
130mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -1 |
|
Etoposide |
250 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -4, -3,-2 |
|
Cyclophosphamide |
60 mg/kg/ngày |
Truyền tĩnh mạch |
Ngày -4, -3 |
|
Dexamethazone |
40mg/ngày |
Truyền tĩnh mạch |
Ngày -4 đến -1 |
+ Bu/Cy/Eto
|
Busulfan |
3,2 mg/kg x 3 ngày |
Truyền tĩnh mạch |
Ngày -7,-6,-5 |
|
Cyclophosphamide |
50 mg/kg/ngày |
Truyền tĩnh mạch |
Ngày -3, -2 |
|
Etoposide |
400 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -5, -4 |
+ Bu/Cy/Mel
|
Busulfan |
3,2 mg/kg x 4 ngày |
Truyền tĩnh mạch |
Ngày -7 đến -4 |
|
Cyclophosphamide |
60 mg/kg/ngày |
Truyền tĩnh mạch |
Ngày -3, -2 |
|
Melphalan |
140 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -1 |
+ Bu/Mel/Eto
|
Busulfan |
3,2 mg/kg x 3 ngày |
Truyền tĩnh mạch |
Ngày -8,-7,-6 |
|
Melphalan |
50 mg/kg/ngày |
Truyền tĩnh mạch |
Ngày -3, -2 |
|
Etoposide |
400 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -5, -4 |
+ Gem/Bu/Mel
|
Gemcitabine (liều tuỳ mức độ) |
Mức độ 8: 2.475mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -8 và -3 |
|
Busulfan |
105mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -8 đến -5 |
|
Melphalan (sau kết thúc gemcitabin) |
60 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -3, -2 |
* Lưu ý: Liều và cách dùng sẽ khác nhau tuỳ ghép nhóm U lympho không Hodgkin hay Hodgkin. Trường hợp có CNS: Chọn thuốc ngấm tốt qua hàng rào máu não (Busulphan...); thuốc ngấm kém: Melphalan và Etoposide.
- Lơ xê mi cấp dòng tuỷ:
|
Busulfan |
1mg/kg/lần x 4lần/ngày Hoặc 0,8mg/kg/lần x 4lần/ngày |
Uống Truyền tĩnh mạch |
Ngày -7,-6,-5,-4 Ngày -7,-6,-5,-4 |
|
Etoposide |
60 mg/m2/ngày |
Truyền tĩnh mạch |
Ngày -3 |
|
G-CSF |
10 µg/kg/ngày, chia 2 lần |
Tiêm dưới da |
Từ ngày 0 đến khi bạch cầu hồi phục |
3.4. Truyền tế bào gốc
3.4.1. Quy trình truyền tế bào gốc
- Truyền sau khi kết thúc điều kiện hóa 24 giờ. Nếu truyền tươi tế bào gốc nguồn gốc từ tuỷ xương hoặc máu ngoại vi: Tốc độ truyền 40-50 giọt/ phút; nếu truyền tế bào gốc bảo quản âm sâu: Truyền càng nhanh càng tốt sau khi rã đông.
- Dự phòng biến chứng và các phản ứng bằng methylprednisolon và thuốc kháng histamin.
3.4.2. Một số những biến chứng và hướng dẫn xử trí
a. Biến chứng chảy máu
- Do khối tế bào gốc tuỷ xương không xử lý chứa khoảng 20.000 đơn vị heparin và thường được truyền trong vòng 1-4 giờ. Những người bệnh có nguy cơ chảy máu là: Giảm tiểu cầu nặng, tiền sử mới phẫu thuật, viêm bàng quang chảy máu.
- Với người bệnh có nguy cơ cao, khối tế bào gốc tuỷ xương nên được cô đặc và rửa để loại heparin hoặc sử dụng ACD (acid citrate dextrose) thay thế heparin khi bảo quản.
- Xử trí khi có biến chứng: Trung hoà heparin bằng protamine: 1mg protamine có thể trung hoà được khoảng 100 đơn vị heparin, liều tối đa là 50mg và tốc độ truyền không được quá 5mg/ phút.
b. Sốt
- Do nhiễm trùng khối tế bào gốc: Thường sốt cao, có thể có sốc nhiễm khuẩn; cần xử trí khẩn trương: Cấy bệnh phẩm lấy từ túi tế bào gốc để tìm nguyên nhân và điều trị kháng sinh phổ rộng ngay từ đầu cho đến khi cấy máu âm tính hay cho đến khi xác định nguyên nhân gây bệnh.
- Do các cytokin được tiết ra trong quá trình thu gom, xử lý và bảo quản: Sốt mức độ nhẹ không kèm rét run, huyết áp tụt hay những triệu chứng gợi ý nhiễm trùng. Nên truyền hết khối tế bào gốc, xử trí hạ sốt.
c. Quá tải dịch: Tránh biến chứng quá tải dịch bằng cách truyền chậm và cho thêm lợi tiểu.
d. Độc chất bảo quản tế bào gốc (DMSO)
- Biểu hiện: Nôn, buồn nôn, ban đỏ ở da, mẩn ngứa, đau đầu, chóng mặt, thay đổi huyết áp và nhịp tim; xử lý bằng cách truyền chậm tế bào gốc. Nếu huyết áp tụt cần phải tăng truyền dịch muối với tốc độ nhanh. Nếu sau 20 phút triệu chứng còn nặng và không cải thiện cân nhắc cho dopamine. Nếu triệu chứng còn nặng phải loại chất bảo quản DMSO khỏi khối tế bào gốc.
- Có thể có loạn nhịp tim như rung nhĩ, nhịp tim chậm; có thể biểu hiện huyết áp tụt hoặc huyết áp tăng và suy thận. Tất cả các người bệnh phải được theo dõi chức năng tim trong quá trình truyền tế bào gốc.
3.5. Theo dõi sau ghép tế bào gốc tự thân
- Chăm sóc người nhận ghép: Chăm sóc toàn diện, điều dưỡng chăm sóc và theo dõi bệnh nhân 24/24, không có người nhà chăm sóc; Theo dõi xét nghiệm tổng phân tích tế bào máu hàng ngày, các xét nghiệm chức năng gan, thận 3 ngày/lần.
- Chống nhiễm khuẩn, nhiễm nấm: Người bệnh nằm trong phòng cách ly tuyệt đối. Nhân viên y tế và người chăm sóc người bệnh phải mang trang phục vô trùng tương tự trang phục phòng mổ. Sử dụng kháng sinh dự sinh phòng khi bạch cầu trung tính dưới 0,5G/L. Khi người bệnh có sốt: Đo nhiệt độ ở miệng trên 38ºC hoặc 2 lần liên tiếp trên 38ºC cần cấy máu ít nhất 3 lần liên tục ở ít nhất 2 vị trí, sau đó sử dụng kháng sinh phổ rộng và mạnh phối hợp; điều chỉnh kháng sinh theo kháng sinh đồ.
- Sử dụng thuốc kích bạch cầu (G-GSF): Khi số lượng tuyệt đối bạch cầu trung tính dưới 0,5G/L.
- Dinh dưỡng: Đảm bảo vô trùng và vệ sinh dinh dưỡng tuyệt đối. Tất cả thức ăn được hâm nóng bằng lò vi sóng trước khi cho người bệnh ăn, xây dựng chế độ ăn đủ chất dinh dưỡng và hợp lý, bổ sung dung dịch nuôi dưỡng đường tĩnh mạch khi người bệnh không ăn được hoặc có biểu hiện rối loạn tiêu hoá.
- Truyền chế phẩm máu: Truyền máu từng phần, truyền khối hồng cầu khi Hb < 80g/L, truyền khối tiểu cầu máy khi tiểu cầu < 20G/L hoặc khi có xuất huyết trên lâm sàng.
- Chăm sóc tinh thần.
3.6. Đánh giá người bệnh để ra viện
- Người bệnh không sốt, không cần dinh dưỡng qua đường tĩnh mạch.
- Không cần truyền tiểu cầu hay ít hơn 2 tuần/lần.
- Số lượng tuyệt đối bạch cầu trung tính (ANC) > 1G/L.
4. BIẾN CHỨNG CỦA GHÉP TẾ BÀO GỐC TỰ THÂN
- Tỷ lệ biến chứng và tử vong liên quan đến ghép tự thân thấp hơn rất nhiều so với ghép đồng loại, thường dưới 5%.
- Biến chứng sớm:
+ Tác dụng phụ của G-CSF: Đau cơ, đau xương và đau đầu…;
+ Hóa chất của phác đồ điều kiện hóa có thể gây tổn thương các cơ quan:
● Phổi: Chảy máu phế nang lan tỏa, viêm phổi kẽ đặc biệt ở người bệnh điều kiện hóa bằng bleomycin/carmustine và tia xạ toàn thân;
● Đường tiêu hóa: Viêm loét miệng, nôn, tiêu chảy;
● Gan: Tăng bilirubin và men gan không có triệu chứng, viêm tắc tĩnh mạch trên gan (veno-occlusive disease - VOD) đặc biệt ở người bệnh điều trị busulfan;
● Thận: Suy thận cấp trước thận, độc thận (kháng sinh độc ống thận), viêm thận kẽ;
● Tim: Suy tim, loạn nhịp tim (thường do cyclophosphamide).
+ Biến chứng do giảm bạch cầu trung tính và tiểu cầu;
+ Những biến chứng nhiễm trùng do vi khuẩn, nấm và thường xảy ra trong 28 ngày đầu sau ghép;
+ Biến chứng CMV tái hoạt động gây sốt, tiêu chảy, giảm các tế bào máu. Thuốc điều trị: Ganciclovir hoặc Foscarnet.
- Biến chứng muộn: Hiếm xảy ra, bao gồm:
+ Xơ phổi ở những người bệnh điều trị busulfan và carmustine;
+ Suy tuyến sinh dục ở những người bệnh điều kiện hóa bằng tia xạ toàn thân hoặc busulfan. Người bệnh nam điều kiện hóa bằng BEAM có thể hồi phục một số chức năng tuyến sinh dục;
+ Có thể có rối loạn sinh tủy thứ phát ở giai đoạn muộn sau ghép, đặc biệt ở người bệnh điều kiện hóa phác đồ dựa trên tia xạ toàn thân. Thông thường, MDS hoặc AML liên quan đến ghép xảy ra trong vòng 12 tháng sau ghép và thường liên quan đến bất thường NST số 5 hay 7 hoặc nhiều bất thường phối hợp.
5. ĐÁNH GIÁ KẾT QUẢ ĐIỀU TRỊ
- Đa u tuỷ xương: Đánh giá kết quả sau ghép 3 tháng/lần trong năm đầu, sau đó 6 tháng/ lần trong năm thứ 2, sau 2 năm kiểm tra định kỳ 1 năm/lần; hoặc khi có bất kỳ dấu hiệu lâm sàng hoặc cận lâm sàng nghi tái phát: dựa trên so sánh với trước điều trị; các xét nghiệm cần làm: Điện di miễn dịch huyết thanh, điện di protein và nước tiểu, định lượng các Ig và chuỗi nhẹ tự do trong huyết thanh, nước tiểu; tuỷ đồ và sinh thiết tuỷ xương, xét nghiệm FISH, công thức NST đánh giá chuyển clone của dòng và chụp PET/CT hoặc MRI toàn thân dựa trên tình trạng lâm sàng.
- U lympho ác tính: Sau ghép 3 tháng/ lần trong năm đầu, sau đó 6 tháng/ lần trong năm thứ 2, sau 2 năm kiểm tra định kỳ 1 năm/lần; hoặc khi có bất kỳ dấu hiệu lâm sàng hoặc cận lâm sàng nghi tái phát: So sánh với trước điều trị về khám lâm sàng và các xét nghiệm: Chụp cắt lớp vi tính, cộng hưởng từ hoặc PET-CT, xét nghiệm sinh thiết tủy xương.
- Lơ xê mi cấp: Dựa trên xét nghiệm tủy đồ, sinh thiết tủy xương, tồn dư tối thiểu và phân tích di truyền.
6. BẢNG XÉT NGHIỆM THEO DÕI SAU GHÉP
XÉT NGHIỆM THEO DÕI SAU GHÉP TẾ BÀO GỐC TỰ THÂN ĐIỀU TRỊ U LYMPHO ÁC TÍNH
|
Xét nghiệm |
Tháng sau ghép (từ ngày truyền TBG) |
||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
22 |
23 |
24 |
Xa hơn |
|
|
Tổng phân tích tế bào máu |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
Xét nghiệm tùy diễn biến (Nghi ngờ tái phát ) |
|
Sinh hoá máu cơ bản |
x |
x |
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
Đông máu cơ bản-D- Dimer |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
|
|
|
X |
|
|
|
|
|
X |
|
|
Huyết tủy đồ (tuỷ diễn biến bệnh) - nghi có xâm lấn |
|
|
X |
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
|
|
|
|
|
|
X |
|
|
Sinh thiết tuỷ xương (tuỳ diễn biến bệnh) - nghi có xâm lấn |
|
|
X |
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
|
|
|
|
|
|
X |
|
|
Xét nghiệm hormon |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
|
|
|
X |
|
|
|
|
|
X |
|
|
Xét nghiệm marker ung thư |
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
X |
|
|
|
|
|
X |
|
|
Beta 2 micro globulin/LDH |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
CMV/EBV (tuỳ BN) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Khám tai mũi họng (tuỳ BN) |
|
|
X |
|
|
X |
|
|
X |
|
|
X |
|
|
|
|
|
X |
|
|
|
|
|
X |
|
|
Siêu âm/ X- quang |
X |
|
X |
|
|
X |
|
|
X |
|
|
X |
|
|
|
|
|
X |
|
|
|
|
|
X |
|
|
Chụp PET/CT |
|
|
x |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
X |
|
|
Chụp CT, MRI (khi không chụp PET) |
Tuỳ bệnh nhân cân nhắc giữa PET và CT/MRI |
||||||||||||||||||||||||
HƯỚNG DẪN QUY TRÌNH KHÁM VÀ XÉT NGHIỆM THEO DÕI SAU GHÉP TẾ BÀO GỐC TỰ THÂN ĐA U TỦY XƯƠNG
|
|
Thời điểm các tháng sau ghép |
||||||||||||||||||||||||
|
Xét nghiệm |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
22 |
23 |
24 |
Chú thích |
|
Tổng phân tích tế bào máu |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
Xét nghiệm tùy diễn biến bệnh: tái phát hay ổn định trong giai đoạn sau 2 năm |
|
Sinh hoá cơ bản |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
Định lượng IgA,M,E,G; K, L |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
Đông máu cơ bản và D-Dimer |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
Tổng phân tích nước tiểu/ sinh hoá nước tiểu/ Free K, L nước tiểu |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
Beta 2 micro globulin/LDH |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
Điện di protein, miễn dịch |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
x |
|
|
Huyết tủy đồ |
|
|
x |
|
|
|
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
Sinh thiết tuỷ xương |
|
|
x |
|
|
|
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
Công thức NST |
|
|
x |
|
|
|
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
Bất thường di truyền đầy đủ (FISH) |
|
|
x |
|
|
|
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
Phân loại miễn dịch đánh giá tồn dư bệnh |
|
|
x |
|
|
|
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
PET/ CT, MRI toàn thân/X quang |
|
|
x |
|
|
|
|
|
|
|
|
x |
|
|
|
|
|
x |
|
|
|
|
|
x |
|
TÀI LIỆU THAM KHẢO
1. Omar S Aljitawi and Parameswaran Hari. Propylene glycol-free melphalan as conditioning regimen for autologous transplantation in myeloma. Int J Hematol Oncol. 2016 May; 5(1): 5-10.
2. Antin J.H. and Raley D.Y, 2013. Rationale for transplantation. In: Manual of Stem Cell and Bone Marow Transplantation. Cambridge University Press, Second edition, Ch.7: Stem cell infusion, pp.31-33.
3. Craddock C. and Chakraverty R., 2011. Stem cell transplantation. Postgraduate Hematology, Sixth Edition (Hoffbrand AV, Catovsky D, Edward G.D. Tuddenham, Green AR), Wiley-Blackwell, Ch.38, pp.722-745.
4. Wilson I. Gonsalves, Francis K. Buadi et all. Utilization of hematopoietic stem cell transplantation for the treatment of multiple myeloma: a Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus statement. Bone Marrow Transplantation, 11 May 2018.
5. Moreau P, MiguelJ.S, Ludwig H, Schouten H, Mohty M, Dimopoulos M, Dreyling (2013). Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 00: 1-5.
6. Yago Nieto, Uday Popat, Paolo Anderlini et all. Autologous Stem Cell Transplantation for Refractory or Poor-Risk Relapsed Hodgkin's Lymphoma: Effect of the Specific High-Dose Chemotherapy Regimen on Outcome. Biol Blood Marrow Transplant. 2013 Mar; 19(3): 410-417.
7. Yi-Bin Chen, Andrew A. Lane, Brent R. Logan et all. Impact of Conditioning Regimen on Outcomes for Patients with Lymphoma Undergoing High-Dose Therapy with Autologous Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant (2015) 1046-1053.
8. Noa Biran, MD et all. A Phase I/II Study of Escalating Doses of Bortezomib in Conjunction with High Dose Melphalan, as a Conditioning Regimen for Salvage Autologous Peripheral Blood Stem Cell Transplant in Patients with Multiple Myeloma. Biology of Blood and Marrow Transplantation (2016).
9. Kenneth C. Anderson, MD and Joseph R. Mikhael. Management of Multiple Myeloma: Using Emerging Therapies with ASCT. Biology of Blood and Marrow Transplantation. VOLUME 25, NO 1 2016.
43. GHÉP TẾ BÀO GỐC ĐỒNG LOÀI ĐIỀU TRỊ BỆNH MÁU
1. ĐẠI CƯƠNG
Ghép tế bào gốc đồng loài là một phương pháp điều trị được sử dụng rộng rãi trên thế giới, có thể chữa khỏi cho một số bệnh máu ác tính kháng với hoá chất cũng như một số bệnh máu khác. Đây là phương pháp truyền tế bào gốc tạo máu từ người hiến phù hợp HLA hoàn toàn hoặc không hoàn toàn, cùng hoặc không cùng huyết thống hoặc từ máu dây rốn cùng hoặc không cùng huyết thống, sau khi đã điều kiện hoá người bệnh bằng phác đồ diệt tuỷ hoặc không diệt tuỷ.
2. CHỈ ĐỊNH GHÉP TẾ BÀO GỐC ĐỒNG LOÀI
2.1. Các bệnh máu ác tính: Chiếm chủ yếu (75%).
- Lơ xê mi cấp (Dòng tuỷ, dòng lympho).
- Lơ xê mi mạn (Dòng bạch cầu hạt, dòng lympho).
- Rối loạn tăng sinh lympho (U lympho ác tính không Hodgkin, U lympho ác tính Hodgkin).
- Hội chứng rối loạn sinh tuỷ (Thể RA; RAEB, CMML...).
- Hội chứng thực bào tế bào máu.
- Hội chứng tăng sinh tuỷ (Xơ tuỷ, đa hồng cầu, tăng tiểu cầu tiên phát...).
2.2. Một số bệnh máu khác
- Những rối loạn sinh máu: Rối loạn tế bào gốc (suy tuỷ xương, thiếu máu Fanconi, đái huyết sắc tố kịch phát ban đêm, tuỷ giảm sinh dòng hồng cầu); hội chứng thiếu hụt miễn dịch (bệnh Chediak-Higashi, hội chứng thiếu hụt miễn dịch kết hợp mức độ nặng), bệnh tự miễn...
- Những rối loạn bẩm sinh của dòng hồng cầu: Thalassemia...
- Những khiếm khuyết về chuyển hoá ở trẻ sơ sinh: Bệnh rối loạn chuyển hoá đường (mucopolysaccharidose)...
Người bệnh từ các nhóm bệnh nói trên phải có đủ các điều kiện về: Tình trạng sức khoẻ, tình trạng bệnh và nguồn tế bào gốc phù hợp... thì mới có thể tiến hành ghép. Bởi vậy cần thực hiện các thăm khám lâm sàng và xét nghiệm cho người bệnh trước khi quyết định ghép tế bào gốc, cụ thể:
- Xét nghiệm HLA cho cả người nhận và người hiến/ hoặc đơn vị Tế bào gốc máu dây rốn để tìm được sự phù hợp giữa người hiến và người nhận.
- Khám lâm sàng và lập hồ sơ ghép tế bào gốc cho người bệnh.
- Xét nghiệm tổng thể cho người bệnh trước ghép, bao gồm:
+ Xét nghiệm tổng phân tích tế bào máu;
+ Các xét nghiệm sinh hóa cơ bản (chức năng gan, thận...);
+ Xét nghiệm HIV/ HCV/ HBV phương pháp PCR hoặc NAT; anti-HCV; HBsAg, anti-HBs, anti-HBc; anti-CMV (IgG, IgM) và anti-EBV; CMV, EBV đo tải lượng
+ Xét nghiệm kháng thể kháng HLA.
- Xét nghiệm tủy đồ và sinh thiết tủy xương.
- Các xét nghiệm về di truyền và miễn dịch (nếu cần).
- Các xét nghiệm về chẩn đoán hình ảnh:
+ CT-scaner: Bụng, ngực, xoang;
+ MRI sọ não.
- Khám kiểm tra chức năng tim: Điện tâm đồ, siêu âm tim, một số men tim...
- Khám kiểm tra chức năng hô hấp: Đo chức năng hô hấp...
- Khám kiểm tra các chuyên khoa: Tai mũi họng, răng hàm mặt, mắt...
- Khám đánh giá hệ tiêu hoá: Nội soi dạ dày, đại trực tràng...
3. CÁC BƯỚC KỸ THUẬT TRONG GHÉP TẾ BÀO GỐC ĐỒNG LOÀI
3.1. Lựa chӑn nguồn tế bào gốc
Nguồn tế bào gốc gồm: Từ tuỷ xương, từ máu ngoại vi hoặc từ máu dây rốn hoặc kết hợp máu dây rốn với các nguồn khác.
a. Nguồn tế bào gốc từ máu ngoại vi
- Huy động tế bào gốc từ máu ngoại vi của người hiến phù hợp HLA (ít nhất 5/6 hoặc 9/10) bằng các thuốc kích thích sinh bạch cầu hạt như:
+ G-CSF: Tiêm dưới da G-CSF (Neupogen) 10μg/kg cân nặng/ngày, chia hai lần, cách nhau 12 giờ;
+ Pegfilgrastim: 12mg/ lần, tiêm dưới da, thường gạn tách tế bào gốc vào ngày thứ 4.
- Kiểm tra kết quả huy động: Kiểm tra số lượng bạch cầu hàng ngày và số lượng tế bào CD34+ vào ngày thứ tư sau khi tiêm thuốc kích thích sinh bạch cầu hạt.
- Gạn tách tế bào gốc:
+ Thời điểm gạn: Khi số lượng tế bào CD34+ > 10-20 tế bào/µl ở máu ngoại vi;
+ Thiết bị gạn tách: Tuỳ theo kiểu ghép sẽ sản xuất khối tế bào gốc bằng:
● Hệ thống máy tách tế bào (COBE-Spectra...): Khối tế bào gốc không xử lý loại lympho T người hiến;
● Hệ thống máy CliniMACS: Gạn tách CD34+ chọn lọc, loại bỏ bớt tế bào lympho T người hiến.
+ Số buổi gạn: Khoảng 2-3 buổi, mỗi buổi gạn ≥ 3 lần thể tích máu người hiến;
+ Số lượng tế bào gốc cần gạn:
● Khối tế bào gốc không xử lý: CD34+ ≥ 3 x 106 tế bào/kg cân nặng người bệnh;
● Khối tế bào gốc CD34+ chọn lọc: CD34+ ≥ 2 x 106 tế bào/kg cân nặng người bệnh; CD3+ lympho T < 1 x 104 CD3+ tế bào/kg.
- Xử lý tế bào gốc: Khối tế bào gốc có thể cần được xử lý trước khi truyền cho người bệnh hoặc trước khi bảo quản âm sâu, nhằm giảm lượng hồng cầu hoặc huyết tương khi có bất đồng hệ nhóm máu ABO giữa người bệnh và người hiến; hoặc chiết tách tế bào CD34+ và loại lympho T của người hiến.
- Bảo quản khối tế bào gốc sau gạn tách bằng 1 trong 2 phương pháp sau:
+ Nhiệt độ 2-8°C: Chỉ trong thời hạn 72 giờ, nên phải tính toán sao cho khi kết thúc phác đồ điều kiện hóa cho người bệnh đồng thời cũng là thời gian kết thúc quá trình gạn tách tế bào gốc ở người hiến;
+ Điều kiện âm sâu (-196°C): Tiến hành gạn tách tế bào gốc cho người hiến trước để lấy đủ số lượng CD34+ ≥ 3x106/kg cân nặng người bệnh, sau đó sẽ tiến hành điều kiện hoá cho người bệnh.
b. Nguồn tế bào gốc từ tuỷ xương
- Vị trí lấy tế bào gốc: Ở gai chậu sau trên; hoặc trước trên hoặc ở xương ức trong trường hợp đặc biệt.
- Quy trình: Người hiến phải được gây mê toàn thân. Dịch tuỷ xương hút được cho vào hộp chứa vô trùng có chứa một lượng thuốc chống đông thích hợp (tỉ lệ 1/10, thường dùng là Heparin) hoặc trong bộ kít chuyên dụng. Dung dịch tuỷ sau thu thập được lọc qua phin lọc vô trùng để loại bỏ mỡ, mảnh xương nhỏ và các mảnh tế bào vỡ.
- Thể tích dịch tuỷ cần lấy dựa vào cân nặng người bệnh vì liều tối thiểu tế bào có nhân cần lấy là 2 x 108/kg cân nặng người bệnh. Thường lấy trung bình 10-15 ml/kg cân nặng người hiến, tuy nhiên phải lưu ý đảm bảo an toàn cho người hiến.
- Dịch tuỷ xương cần được xử lý trước khi truyền cho người bệnh, nhằm giảm lượng hồng cầu hoặc huyết tương khi có bất đồng hệ nhóm máu ABO giữa người bệnh và người hiến; hoặc chiết tách tế bào CD34+ và loại lympho T của người hiến.
- Tế bào gốc từ tuỷ xương được truyền tươi trong vòng 24 giờ sau khi thu thập.
c. Nguồn tế bào gốc từ máu dây rốn
- Chỉ định ghép tế bào gốc máu dây rốn: Ghép không cùng huyết thống; ghép cho thành viên trong gia đình, đặc biệt cho trẻ em.
- Tế bào gốc máu dây rốn được thu thập ngay sau khi trẻ tách khỏi dây rốn; sau thu thập và xử lý tế bào gốc máu dây rốn được bảo quản âm sâu.
- Liều tế bào gốc cần thiết truyền:
+ Liều tế bào có nhân (TNC): Ít nhất phải đạt 1,5-2 x 107 tế bào có nhân/kg cân nặng người bệnh;
+ Liều CD34+: Ít nhất phải đạt 1 x 105 tế bào CD+34/kg cân nặng người bệnh.
Có thể kết hợp 2 đơn vị máu dây rốn để đủ số lượng tế bào gốc.
- Lựa chọn đơn vị máu dây rốn phù hợp để ghép: Tốt nhất dựa trên xét nghiệm HLA độ phân giải cao giữa người bệnh và đơn vị máu dây rốn để chọn đơn vị phù hợp ít nhất 4/6 (HLA-A, B và DR). Có thể sử dụng xét nghiệm HLA-A, B bằng độ phân giải thấp, riêng đối với HLA-DR bắt buộc phải thực hiện bằng độ phân giải cao.
3.2. Điều kiện hóa cho người bệnh
- Điều kiện hoá nhằm mục đích tiêu diệt các tế bào ung thư, đồng thời ức chế hệ miễn dịch của người bệnh để đảm bảo mọc mảnh ghép và phòng hiện tượng thải ghép.
- Có rất nhiều loại phác đồ điều kiện hoá được sử dụng trong ghép tế bào gốc đồng loài. Lựa chọn phác đồ điều kiện hoá cho người bệnh được dựa trên chẩn đoán bệnh, tuổi của người bệnh, tình trạng bệnh và đặc điểm người hiến (đặc biệt mức độ phù hợp HLA) (Một số phác đồ được trình bày ở bảng 53).
- Phác đồ điều kiện hoá diệt tuỷ: Chỉ định cho các bệnh máu ác tính hoặc các rối loạn huyết học bẩm sinh như thalassemia...
- Phác đồ điều kiện hóa giảm liều: Chỉ định cho các bệnh máu ác tính không đủ điều kiện sử dụng phác đồ diệt tuỷ, ung thư tạng đặc và các bệnh rối loạn huyết học không phải ác tính như suy tuỷ xương, đái huyết sắc tố niệu...
Bảng 53. Một số phác đồ điều kiện hóa thường được sử dụng
|
(1) Phác đồ diệt tuỷ |
||
|
Cy/ TBI |
Cyclophosphamide |
120mg/kg truyền tĩnh mạch |
|
TBI |
1.000-1.575 centigray (cGy) |
|
|
Bu/Cy |
Busulfan |
16mg/kg uống hay 12,8 mg/kg truyền tĩnh mạch |
|
Cyclophosphamide |
120-200 mg/kg truyền tĩnh mạch |
|
|
Bu/Cy/Eto |
Busulfan |
3,2 mg/kg x 3 ngày, truyền tĩnh mạch |
|
Cyclophosphamide |
50mg/kg/ngày x 2 ngày, truyền tĩnh mạch |
|
|
Etoposid |
400mg/m2/ngày x 2 ngày, truyền tĩnh mạch |
|
|
Bu/Cy/Mel |
Busulfan |
3,2 mg/kg x 4 ngày, truyền tĩnh mạch |
|
Cyclophosphamide |
60mg/kg/ngày x 2 ngày, truyền tĩnh mạch |
|
|
Melphalan |
140 mg/m2 truyền tĩnh mạch 1 ngày |
|
|
Bu/Flu/Mel |
Busulfan |
3,2-6 mg/kg x 4 ngày, truyền tĩnh mạch |
|
Fludarabin |
30 mg/m2x 4 ngày, truyền tĩnh mạch |
|
|
Melphalan |
90 mg/m2 x 2 ngày, truyền tĩnh mạch |
|
|
Bu/Cy/ATG hoặc Flu |
Busulfan |
16mg/kg uống hay 12,8 mg/kg truyền tĩnh mạch |
|
Cyclophosphamide |
50 mg/kg/ngày x 4 ngày, truyền tĩnh mạch |
|
|
ATG/Flu |
hATG: 40 mg/kg x 4 ngày, truyền tĩnh mạch hoặc rATG: 1,5mg/kg/ngày x 4ngày Hoặc: Flu 30 mg/m2x 4 ngày, truyền tĩnh mạch |
|
|
rATG/Cy/Flu+Cy ngày 3,4 sau truyền TBG |
rATG |
rATG: 0,5mg/kg/ngày D-9 rATG: 2mg/kg/ngày D-8,-7 |
|
Cyclophosphamide |
60mg/kg/ngày D-7,-6, truyền tĩnh mạch Và 50mg/kg/ngày ngày 3,4 sau ghép |
|
|
Fludarabin |
30 mg/m2 D-5 đến D-1, truyền tĩnh mạch |
|
|
Bu/Flu/Cy + Cy ngày 3,4 sau truyền TBG |
Busulfan |
110-130mg/ m2/ngày x 4 ngày, truyền tĩnh mạch |
|
Fludarabin |
25 mg/m2x 4 ngày, truyền tĩnh mạch |
|
|
Cyclophosphamide |
14,5mg/kg/ngày x 2 ngày, truyền tĩnh mạch Và 50mg/kg/ngày ngày 3,4 sau ghép |
|
|
Bu/Mel |
Busulfan |
16 mg/kg uống hay 12,8 mg/kg truyền tĩnh mạch |
|
Melphalan |
120-200 mg/kg truyền tĩnh mạch |
|
|
(2) Phác đồ giảm cường độ liều (RIC) hay không diệt tuỷ |
||
|
Flu/TBI liều thấp |
Fludarabine |
90 mg/m2 truyền tĩnh mạch |
|
TBI |
200 cGy |
|
|
Flu/Mel |
Fludarabine |
125 mg/m2 truyền tĩnh mạch |
|
Melphalan |
180 mg/m2 truyền tĩnh mạch |
|
|
Flu/Bu/ATG |
Fludarabine |
180 mg/m2 truyền tĩnh mạch |
|
Busulfan |
8 mg/kg uống hoặc 6,4 mg/kg truyền tĩnh mạch |
|
|
ATG |
40 mg/kg truyền tĩnh mạch |
|
|
Cy/Flu |
Cyclophosphamide |
120 mg/kg truyền tĩnh mạch |
|
Fludarabine |
125 mg/m2 truyền tĩnh mạch |
|
|
Gem/Flu/Mel Allo R/R HL (G-FM140) |
Gemcitabine |
800 mg/m2 truyền tĩnh mạch trong 1 ngày |
|
Fludarabine |
33 mg/m2 truyền tĩnh mạch x 4 ngày |
|
|
Melphalan |
70 mg/m2 truyền tĩnh mạch x 2 ngày |
|
|
Cy/Flu/ATG |
Cyclophosphamide |
120 mg/m2 truyền tĩnh mạch |
|
Fludarabine |
125 mg/m2 truyền tĩnh mạch |
|
|
ATG |
40 mg/kg truyền tĩnh mạch |
|
TBI: Total body irradition (chiếu xạ toàn thân), RIC: Reduced intensity conditioning (giảm cường độ liều), ATG: Anti thymocyte glubulin.
Lưu ý trường hợp đặc biệt: Nhằm mục đích giảm việc tăng sinh hồng cầu để giảm biến chứng thải ghép cần điều trị ức chế miễn dịch, tăng cường thải sắt và tăng cường truyền máu trước ghép cho bệnh nhân thalassemia class 3 hoặc nhóm mức độ nặng (phụ thuộc truyền máu ngay trong giai đoạn 3 tuổi, Hb khi bắt đầu truyền máu < 7g/dl; gan lách to và bộ mặt thalassemia).
Phác đồ 1: Từ ngày -45 đến ngày -11 trước ghép:
- Deferoxamine: 40mg/kg/ngày truyền tĩnh mạch;
- Truyền máu khoảng mỗi 3 ngày để duy trì Hb từ 140-150g/L;
- Hydroxyurea: 30mg/kg/ngày và Azathioprine: 3mg/kg/ngày;
- G-CSF và Erythropoietin: 2 lần/tuần.
Hoặc:
Phác đồ 2: 2 đợt như sau vào ngày -68 đến ngày -64 và ngày -40 đến -36
- Fludarabine 40mg/m2/ngày x 5 ngày.
- Dexamethazone: 25mg/m2/ngày x 5 ngày.
3.3. Truyền tế bào gốc
3.3.1. Quy trình truyền tế bào gốc
- Truyền tĩnh mạch khối tế bào gốc sau khi kết thúc điều kiện hoá 24-48 giờ.
- Khối tế bào gốc tạo máu được kiểm tra lại về số lượng tế bào gốc trước khi truyền để đánh giá tỷ lệ tế bào sống chết. Quy trình đạt chất lượng nếu tỷ lệ tế bào gốc sống/chết đạt tối thiểu 80% và lượng tế bào gốc được truyền vào cho người bệnh phải đạt tiêu chuẩn tuỳ từ máu ngoại vi hay máu dây rốn.
- Dự phòng biến chứng và các phản ứng phụ bằng methylprednisolon và thuốc kháng histamin.
3.3.2. Một số những biến chứng và hướng dẫn xử trí
a. Biến chứng chảy máu
- Do khối tế bào gốc tuỷ xương không xử lý có chứa khoảng 20.000 đơn vị heparin và thường được truyền trong vòng 1-4 giờ. Những người bệnh có nguy cơ chảy máu là: Giảm tiểu cầu nặng, tiền sử mới phẫu thuật, viêm bàng quang chảy máu.
- Với người bệnh có nguy cơ cao, khối tế bào gốc tuỷ xương nên được cô đặc và rửa để loại heparin hoặc sử dụng ACD (acid citrate dextrose) thay thế heparin khi thu gom tế bào gốc.
- Xử trí khi có biến chứng: Trung hoà heparin bằng protamine: 1mg protamine có thể trung hoà được khoảng 100 đơn vị heparin, liều tối đa là 50mg và tốc độ truyền không được quá 5mg/phút.
b. Sốt
- Do nhiễm trùng khối tế bào gốc: Thường sốt cao, có thể có sốc nhiễm khuẩn; cần xử trí khẩn trương: Cấy bệnh phẩm lấy từ túi tế bào gốc để tìm nguyên nhân và điều trị kháng sinh phổ rộng ngay từ đầu cho đến khi cấy máu âm tính hay cho đến khi xác định nguyên nhân gây bệnh.
- Do các cytokin được tiết ra trong quá trình thu gom, xử lý và bảo quản: Sốt mức độ nhẹ không kèm rét run, tụt huyết áp hay những triệu chứng gợi ý nhiễm trùng. Nên truyền hết khối tế bào gốc, xử trí hạ sốt.
c. Quá tải dịch
Tránh biến chứng quá tải dịch bằng cách truyền chậm và cho lợi tiểu.
d. Độc chất bảo quản tế bào gốc (DMSO)
- Biểu hiện: Nôn, buồn nôn, ban đỏ ở da, mẩn ngứa, đau đầu, chóng mặt, thay đổi huyết áp và nhịp tim; xử lý bằng cách truyền chậm tế bào gốc. Nếu tụt huyết áp cần phải tăng truyền dịch muối với tốc độ nhanh. Nếu sau 20 phút triệu chứng còn nặng và không cải thiện cân nhắc cho dopamine. Nếu triệu chứng còn nặng phải loại chất bảo quản DMSO khỏi khối tế bào gốc.
- Có thể có loạn nhịp tim như rung nhĩ, nhịp tim chậm; có thể biểu hiện tụt huyết áp hoặc tăng huyết áp và suy thận. Tất cả các người bệnh phải được theo dõi chức năng tim trong quá trình truyền tế bào gốc.
4. XỬ TRÍ MỘT SỐ BIẾN CHỨNG CHÍNH
4.1. Hội chứng mọc mảnh ghép
- Biểu hiện: Sốt, ban đỏ ở da và tổn thương phổi; xảy ra vào khoảng ngày thứ 10-14 sau ghép.
- Điều trị bằng methylprednisolon 1mg/kg/ngày, thường đáp ứng tốt.
4.2. Biến chứng nhiễm trùng
4.2.1. Nhiễm vi khuẩn
Cần tham khảo tình hình vi khuẩn kháng thuốc ở cơ sở điều trị để xác định xử trí phù hợp. Nhiễm vi khuẩn ở giai đoạn muộn sau ghép thường liên quan đến thuốc ức chế miễn dịch và bệnh ghép chống chủ.
a. Dự phòng nhiễm vi khuẩn
- Phòng nhiễm pneumonia Carrini: Trimethoprim/sulfamethoxazole (TMP-SMX) 480mg/ lần x 2 lần/ ngày x 3 ngày/ tuần, uống từ ngày thứ 14 sau ghép cho đến khi ngừng thuốc ức chế miễn dịch.
- Người bệnh có ghép chống chủ mạn tiến triển: Trimethoprim/ sulfamethoxazole 480mg/lần x 2 lần/ngày, uống hàng ngày.
- Người bệnh có ghép chống chủ cấp đường ruột mức độ III-IV: Ampicillin/sulbactam 1g/lần x 2 lần/ngày, cách 12h; dùng đến khi hết ghép chống chủ đường ruột.
b. Sốt do giảm bạch cầu, khi nhiệt độ trên 38ºC
- Cần cấy tìm vi khuẩn và nấm:
+ Cấy máu: 3 lần liên tục cách nhau 1 giờ;
+ Cấy nước tiểu;
+ Cấy các ổ nhiễm trùng nếu có thể lấy được bệnh phẩm (dịch màng phổi, loét miệng...).
- Chỉ định kháng sinh: Tuỳ theo tính chất dịch tễ vi khuẩn và mức độ kháng kháng sinh của từng bệnh viện, tuỳ có nghi ổ nhiễm khuẩn như đặt đường truyền tĩnh mạch trung tâm, tổn thương loét niêm mạc, tổn thương phổi, tổn thương đường tiêu hoá...
+ Có thể khởi đầu bằng: Ceftazidime hoặc cefepime, piperacillin/ tazobactam hoặc carbapenem;
+ Điều chỉnh kết hợp kháng sinh dựa trên vị trí tổn thương, kết quả vi sinh và chẩn đoán hình ảnh. Bổ sung Vancomycin hay linezolid nếu có viêm mô tế bào hay viêm phổi; aminoglycoside hay chuyển carbapenem khi có viêm phổi hay nhiễm vi khuẩn gram âm; thêm metronidazole/hoặc vancomycin đường uống khi có biểu hiện triệu chứng trong ổ bụng hay nghi ngờ nhiễm C. difficile...;
+ Không đáp ứng sau 2-4 ngày điều trị kháng sinh theo kinh nghiệm: Cần đánh giá tuỳ nhóm nguy cơ, có bằng chứng nhiễm trùng hay không dựa trên kết quả cấy máu, tìm vị trí nhiễm trùng... để xem lại liều và phổ kháng sinh đã điều trị, điều chỉnh kháng sinh phổ rộng hơn, thêm thuốc chống nấm theo kinh nghiệm;
+ Trường hợp nặng nguy kịch: Kết hợp carbapenem và vancomycin hoặc teicoplanin hoặc linezolid, aminoglycoside hoặc quinolone; Nếu kháng với những kháng trên có thể cân nhắc dùng các kháng sinh thế hệ tiếp theo như Colistin, Tigecycline, Daptomycin, Ceftazidime/avibactam...
- Ngừng kháng sinh: Nếu điều trị như trên hết sốt > 24 giờ hoặc bạch cầu trung tính tăng > 1G/L.
- Trường hợp khác cần lưu ý:
+ Có ổ nhiễm trùng chỉ điểm: Cân nhắc kháng sinh phù hợp vị trí, ví dụ nhiễm trùng chân ống thông (catheter, hickman, piccline, port...): Phải rút catheter và cấy tìm vi khuẩn, bắt buộc điều trị vancomycin hoặc teicoplanin; nhiễm trùng đường mật, nhiễm trùng tiết niệu...;
+ Nếu có kết quả cấy và kháng sinh đồ, điều trị kháng sinh dựa trên kháng sinh đồ.
4.2.2. Nhiễm virus
4.2.2.1. CMV tái hoạt động sau ghép:
Bảng 54. Xếp loại nguy cơ CMV tái hoạt động
|
Yếu tố nguy cơ |
Phân tầng nguy cơ |
||
|
Nguy cơ cao |
Yếu tố nguy cơ (Được xác định bới các nhân tố khác nhau) |
Nguy cơ thấp |
|
|
Các yếu tố trước khi ghép TBG |
|||
|
CMV (IgG) người hiến và người nhận |
Người hiến (-)/ Người nhận (+) Người hiến (+)/ Người nhận (+) |
Người hiến (-)/ Người nhận (+) Người hiến (+)/ Người nhận (+) |
Người hiến (-)/ Người nhận (-) Người hiến (+)/ Người nhận (-) |
|
Kiểu ghép |
- Không cùng HT và không hòa hợp HLA hoàn toàn - Cùng HT và không hòa hợp HLA hoàn toàn - Ghép nửa hòa hợp - Ghép máu dây rốn |
- Không cùng huyết thống - Cùng huyết thống và không hòa hợp HLA hoàn toàn - Ghép nửa hòa hợp - Máu dây rốn |
- Cùng huyết thống và hòa hợp HLA hoàn toàn |
|
Giảm tế bào T trên ex-vivo hoặc in-vivo |
Suy giảm tế bào T trên ex-vivo |
- Suy giảm tế bào T trên ex-vivo - Liệu pháp huyết thanh (ATG) |
Không có suy giảm tế bào T |
|
Phác đồ điều kiện hóa |
|
Diệt tủy |
- Không diệt tủy - Giảm cường độ liều - Không có sử dụng liệu pháp huyết thanh |
|
Tuổi người nhận |
|
> 40-50 |
< 40 |
|
Khác |
|
- Trước ghép có CMV tái hoạt động - Trước ghép có thiếu hụt miễn dịch |
|
|
Các yếu tố sau ghép TBG |
|||
|
Ức chế miễn dịch |
|
UCMD mạnh (đặc biệt corticoid, với liều tương đương prenisolon >= 1mg/kg/ngày |
Tối thiểu/ không có dùng ƯCMD |
|
GVHD |
Từ độ 2 trở lên yêu cầu cần sử dụng corticoid với liều tương đương prednisolon >= 1mg/kg/ngày |
GVHD cấp >= độ 2 GVHD mạn |
Không có GVHD |
|
Khác |
|
- Giảm số lượng BC lympho (ví dụ: < ALC < 900 tế bào / L ở ngày 100) - Phục hồi miễn dịch chậm - Cyclophosphamide sau ghép - Liệu pháp GVHD với ibrutinib hoặc ruxolitinib |
|
a. Chỉ định đo tải lượng CMV trong máu: Tất cả các trường hợp ghép tế bào gốc tạo máu.
b. Thời điểm thực hiện đo tải lượng CMV trong máu
- Dựa vào tình trạng lâm sàng và nguy cơ nhiễm bệnh.
- Thời điểm bắt đầu theo dõi CMV:
+ Phụ thuộc vào phác đồ điều kiện hóa và nguy cơ tái hoạt động CMV (Bảng 54);
+ Thời điểm làm đầu tiên là trước khi bắt đầu điều kiện hóa;
+ Nếu CMV (IgG, IgM) dương tính trước ghép, cần theo dõi liên tục trong quá trình ghép và trước ghi mọc mảnh ghép;
+ Những bệnh nhân có nguy cơ tái hoạt động CMV cao hơn (Bảng 54) có thể cần theo dõi sớm hơn.
c. Tần suất theo dõi xét nghiệm đo tải lượng CMV trong máu
- Theo dõi ít nhất 1 lần/ tuần trong thời gian nghi ngờ, theo dõi thường xuyên (ít nhất 2 lần/tuần) khi CMV đã tái hoạt động để xác định việc trì hoãn hay điều trị thuốc kháng virus, cũng như đưa ra quyết định về việc giảm cường độ liều (chuyển từ liều tấn công sang liều duy trì).
- Mẫu xét nghiệm nên được thực hiện 2 lần/ tuần và cách nhau ít nhất 3 ngày để kịp điều chỉnh điều trị.
- Trước khi ghép TBG (nếu có chỉ định): 1 lần/tuần.
- Trong thời gian điều trị dự phòng (nếu có chỉ định dự phòng): 1 lần/tuần.
- Nếu có CMV tái hoạt động: 2 lần/tuần.
- Sau khi CMV trong máu âm tính: 1 lần/tuần.
- Bệnh nhân có nguy cơ cao (Bảng 54): 2 lần/tuần.
d. Thời gian theo dõi bằng đo tải lượng CMV trong máu
- Giai đoạn có nguy cơ cao tái hoạt động sau ghép: 100 ngày sau ghép.
- Theo dõi liên tục các trường hợp: Dùng thuốc ức chế miễn dịch liên tục hoặc đang có tình trạng ghép chống chủ hoặc thuộc nhóm nguy cơ cao (Bảng 54).
- Bệnh nhân ghép máu dây rốn cần được theo dõi ít nhất 6 tháng sau ghép.
- Bệnh nhân có thiếu hụt miễn dịch nên được theo dõi CMV kéo dài.
e. Phòng CMV tái hoạt động
- Dự phòng CMV tái hoạt động ở bệnh nhân nhi ghép máu dây rốn, nửa hoà hợp hoặc kết hợp cả 2:
+ Dự phòng đầu tiên: Acyclovir 500 mg/m2 truyền tĩnh mạch mỗi 8h (3 lần/ngày), từ ngày D -5 cho đến khi bạch cầu trung tính hồi phục; hoặc
+ Dự phòng thay thế:
● Bệnh nhân < 20kg: Acyclovir 600 mg/m2 uống mỗi 6h;
g. Điều trị
Đối với tất cả các kiểu ghép TBG đồng loài (trừ ghép máu dây rốn):
- Chỉ định:
+ Khi CMV > 1.000 bản copy/ml;
+ Hai lần kết quả PCR liên tiếp trong khoảng từ 250-1.000 bản copy/ml;
+ Kết quả PCR một lần < 250 bản sao/ ml, sau đó 1 lần trong trong khoảng từ 250-1.00 bản copy/ml;
+ Kết quả PCR một lần < 1.000 bản sao/ ml, nhưng có triệu chứng gợi ý CMV hoạt động: Tiêu chảy, tổn thương phổi; sốt, bất thường các xét nghiệm về chức năng gan và giảm các tế bào máu không rõ nguyên nhân.
- Thuốc điều trị:
Điều trị hàng đầu
+ Ganciclovir:
● Điều trị tấn công: Ganciclovir 5mg/kg truyền tĩnh mạch, mỗi 12h x 7 ngày;
● Điều trị duy trì: Ganciclovir 5mg/kg truyền tĩnh mạch 1 lần mỗi ngày cho đến khi xét nghiệm CMV (bằng phương pháp PCR) âm tính 2 lần liên tiếp.
+ Thay thế bằng foscarnet:
● Nếu bệnh nhân có giảm các tế bào máu vì Ganciclovir hay Valganciclovir gây ức chế tuỷ thì cân nhắc sử dụng Foscarnet ngay từ đầu;
● Khi điều trị Ganciclovir hay Valganciclovir gây giảm bạch cầu không đáp ứng với điều trị G-CSF, cân nhắc chuyển sang Foscarnet;
● Liều tấn công: 90mg/kg mỗi 12 giờ x 7 ngày hoặc 60mg/kg mỗi 8 giờ;
● Liều duy trì: 90mg/kg/ngày, cho đến khi đủ điều kiện dừng.
Điều trị hàng hai
+ Valganciclovir:
● Tấn công: 900mg (uống), mỗi 12 giờ trong 7 ngày;
● Duy trì: 900mg/ngày (uống) cho đến khi đủ điều kiện dừng.
Lưu ý:
- Mỗi bệnh nhân phải được điều trị tấn công tối thiểu 1 tuần và 1 tuần điều trị duy trì.
- Trong quá trình điều trị xét nghiệm CMV PCR 2 lần/tuần. Nếu CMV PCR tăng gấp rưỡi log (0,5 log10, tương đương 125 bản sao) trong 2 lần liên tiếp thì tiếp tục duy trì liều tấn công hoặc chuyển sang thuốc thay thế khác. Tiếp tục điều trị cho đến khi: PCR âm tính 1 lần hoặc 2 lần kết quả PCR dương tính < 3,08 log10 (IU / mL) tương đương 250 bản sao/ml.
Ghép máu dây rốn:
- Chỉ định:
+ Khi CMV > 250 bản copy/ml;
+ Hai lần kết quả PCR liên tiếp dương nhưng < 250 bản copy/ml (kiểm tra lần 2 sau 3 ngày);
+ Kết quả PCR một lần < 250 bản sao/ml, sau đó 1 lần > 250 bản copy/ml;
+ Kết quả PCR một lần < 250 bản sao/ml, nhưng có triệu chứng gợi ý CMV hoạt động: Tiêu chảy, tổn thương phổi, sốt, bất thường các xét nghiệm về chức năng gan và giảm các tế bào máu không rõ nguyên nhân.
- Thuốc điều trị:
Điều trị thuốc hàng đầu
+ Ganciclovir:
● Điều trị tấn công: Ganciclovir 5mg/kg truyền tĩnh mạch, mỗi 12h trong ít nhất 2 tuần;
● Điều trị duy trì: Ganciclovir 5mg/kg truyền tĩnh mạch 1 lần mỗi ngày.
+ Thay bằng foscarnet:
● Nếu bệnh nhân có giảm các tế bào máu vì Ganciclovir/Valganciclovir gây ức chế tuỷ thì cân nhắc sử dụng foscarnet ngay từ đầu;
● Khi điều trị Ganciclovir hay Valganciclovir gây giảm bạch cầu không đáp ứng với điều trị G-CSF, cân nhắc chuyển sang Foscarnet;
● Liều tấn công: 90mg/kg mỗi 12 giờ x 14ngày hoặc 60mg/kg mỗi 8 giờ;
● Liều duy trì: 90mg/kg/ngày cho đến khi xét nghiệm cho phép dừng điều trị.
Điều trị thuốc hàng hai
+ Valganciclovir:
● Tấn công: 900mg, uống, mỗi 12 giờ x 14 ngày;
● Duy trì: 900mg/ngày, uống, cho đến khi đủ điều kiện dừng.
Lưu ý:
- Chỉnh liều ganciclovir theo chức năng thận.
- Trong quá trình điều trị xét nghiệm CMV PCR 2 lần/tuần. Nếu CMV PCR tăng gấp đôi ở 2 lần kiểm tra liên tiếp thì tiếp tục duy trì liều tấn công hoặc chuyển sang thuốc thay thế khác.
- Nếu CMV không giảm được 0,5 log10 (125 bản sao) sau khi điều trị tấn công được 2 tuần, cân nhắc chuyển sang điều trị thuốc khác và xét nghiệm xác định CMV kháng thuốc.
- Chuyển điều trị duy trì chỉ khi PCR CMV < 250 bản sao. Tiếp tục điều trị duy trì đến khi: PCR âm tính trong 2 tuần liên tiếp.
h. Nhiễm các virus khác sau ghép (HHV6, HZV, HSV, BK, Adenovirus...)
Virus Herpes simplex (HSV):
- Dự phòng: Bằng thuốc kháng virus với hiệu quả gần 100%. Bắt đầu từ khi điều kiện hoá và kéo dài 1 năm hoặc hơn nếu còn tiếp tục điều trị ức chế miễn dịch.
+ Acyclovir: 400mg/ lần x 3 lần/ngày, uống, điều chỉnh liều theo chức năng thận;
- Điều trị loét miệng do Herpes:
+ Acyclovir: 400mg/lần x 5 lần/ngày, uống, điều chỉnh liều theo chức năng thận; hoặc
+ Famciclovir: 250mg/lần x 3 lần/ngày; hoặc
+ Foscarnet (nếu kháng acyclovir): 40mg/kg/lần x 2-3 lần/ngày, truyền tĩnh mạch, điều chỉnh theo chức năng thận.
- Điều trị loét niêm mạc nặng, tổn thương phỏng nước ngoài da:
+ Acyclovir: 250mg/m2 hay 5mg/kg/lần x 3 lần/ngày, truyền tĩnh mạch.
Virus Varicella zoster (VZV):
- Tái hoạt động ở giai đoạn muộn sau ghép thường ở người bệnh có GVHD mạn, tỷ lệ mắc khoảng 50-60%.
- Dự phòng: Hạn chế tiếp xúc với người bị nhiễm VZV. Nếu tiếp xúc với người bị nhiễm VZV, cần điều trị dự phòng bằng:
+ Acylovir: 400mg/lần x 3 lần/ngày, uống; hoặc
- Điều trị:
+ Famciclovir: 500mg/lần x 3 lần/ngày; hoặc
Virus Epstein - Barr (EBV):
- Rối loạn tăng sinh lympho liên quan EBV là một biến chứng nghiêm trọng, thường xảy ra vị trí ngoài hạch như não và hệ thống dạ dày ruột.
- Phòng bằng cách theo dõi số lượng EBV qua PCR.
- Điều trị:
+ Xạ trị tại chỗ; hoặc
+ Hoá chất toàn thân; hoặc
+ Rituximab: 375mg/m2 mỗi tuần trong 4 tuần, theo dõi chức năng tế bào B.
Virus Human herpes 6 (HHV 6):
- Virus tái hoạt động thường gây ra giảm các tế bào máu, viêm phổi và viêm não.
- Điều trị:
+ Ganciclovir: 5mg/kg mỗi 12 giờ;
+ Foscarnet: 60mg/kg mỗi 8 giờ hoặc 90mg/kg mỗi 12 giờ.
4.2.3. Nhiễm nấm
Nhiễm nấm xâm lấn là một trong những nguyên nhân quan trọng gây tử vong sau ghép. Nguyên nhân hay gặp là nấm candida; nhiễm aspergillus gặp khoảng 5-15%, thường do yếu tố môi trường trong giai đoạn giảm bạch cầu trung tính hay sử dụng corticoid.
a. Chẩn đoán
- Dựa vào kết quả vi sinh; có thể điều trị theo kinh nghiệm khi có các triệu chứng sau:
+ Sốt dai dẳng trên 4 ngày sau khi đã dùng kháng sinh phổ rộng;
+ Sốt tái phát sau khi đã ngừng sốt;
+ Tổn thương thâm nhiễm phổi mới trong khi đang điều trị kháng sinh phổ rộng;
+ Có ban đỏ trên da, giả mạc trắng ở miệng...
- Các xét nghiệm:
+ Xét nghiệm huyết thanh: Định lượng β-glucan và galactomannan;
+ Cấy các bệnh phẩm tìm nấm như: Máu, đờm, các chất dịch...;
+ Chẩn đoán hình ảnh: Hình ảnh điển hình cho nhiếm nấm.
b. Dự phòng: khi số lượng tuyệt đối của bạch cầu trung tính (ANC) dưới 0,5G/L cho đến khi ANC hồi phục và dửng corticoid....
- Itraconazol: 200mg/ngày, hoặc
- Fluconazole: 6-12mg/kg/ngày (tối đa 400mg/ngày), uống hàng ngày từ khi bắt đầu điều kiện hoá cho đến sau ghép 100 ngày, hoặc đến khi ngừng ức chế miễn dịch; hoặc;
- Voriconazole: 6mg/kg/lần x 2 lần/ngày, sau đó giảm 4mg/kg/lần x 2 lần/ngày truyền tĩnh mạch), sau đó chuyển 200mg/lần x 2 lần/ngày (uống); hoặc
- Posaconazole: 4mg/kg/lần (tối đa 200/lần) x 3 lần/ngày, uống sau ăn.
c. Điều trị
Nấm Candida: Chỉ định một trong các thuốc sau:
- Nhóm echinocandi:
+ Caspofungin: 70mg ngày đầu tiên, sau đó 50mg/ngày, truyền tĩnh mạch;
+ Micafungin: 100mg/ngày, truyền tĩnh mạch;
+ Anidulafungin: 200mg ngày đầu tiên, sau đó 100mg/ngày.
- Nhóm Azole:
+ Fluconazole: Nếu còn nhạy, liều ban đầu 12mg/kg/ngày (tối đa 600mg/ngày), sau đó 200-400mg/ngày, uống hàng ngày;
+ Voriconazole: Khởi đầu 6mg/kg/lần x 2 lần/ngày x 2 ngày, sau đó 3mg/kg/lần x 2 lần/ ngày;
+ Posaconazole: 400/lần x 2 lần/ngày;
+ Itraconazole: 200mg/lần x 2 lần/ngày (dạng viên uống hay siro) hoặc 200mg/lần x 2 lần/ngày x 2 ngày (truyền tĩnh mạch) sau đó chuyển uống 200mg/ngày.
- Nhóm amphotericin B: 1-1,5mg/kg/ngày (truyền tĩnh mạch) trong 8 giờ hoặc amphotericin B lipid: 3-5mg/kg/ngày, truyền tĩnh mạch ít nhất trong 2 giờ, điều chỉnh theo chức năng thận.
Nấm Aspergillus: Chỉ định một trong các thuốc sau:
- Nhóm Azole:
+ Voriconazole: Khởi đầu 6mg/kg/lần x 2 lần/ngày x 2 ngày, sau đó 4mg/kg/lần x 2 lần/ngày (truyền tĩnh mạch); sau đó chuyển uống: 200-300mg/lần x 2 lần ngày nếu người bệnh nặng trên 40kg, 100-150mg/lần x 2 lần/ngày nếu người bệnh nặng dưới 40kg;
+ Posaconazole: 400/lần x 2 lần/ngày; chỉ định khi người bệnh kháng với amphotericin hay voriconazole;
+ Itraconazole: 400mg/lần x 2 lần/ngày (dạng viên uống hay siro) hoặc 200mg/ lần x 2 lần/ ngày x 2 ngày (truyền tĩnh mạch) sau đó chuyển uống 200mg/ngày.
- Nhóm echinocandi: Chỉ định khi người bệnh kháng hay không dung nạp với amphotericin hay voriconazole.
+ Caspofungin: 70mg ngày đầu tiên, sau đó 50mg/ngày, truyền tĩnh mạch;
+ Micafungin: 150/ngày, truyền tĩnh mạch.
- Nhóm amphotericin B: 1-1,5mg/kg/ngày) truyền tĩnh mạch trong 8 giờ hoặc amphotericin B lipid: 3-5mg/kg/ngày, truyền tĩnh mạch ít nhất trong 2 giờ, điều chỉnh theo chức năng thận.
Lưu ý: Thuốc chống nấm khi phối hợp với các thuốc ức chế miễn dịch như cyclosporin A, tacrolimus... sẽ làm tăng nồng độ các thuốc này, do đó cần hiệu chỉnh liều thuốc khi có tương tác.
4.2.4. Truyền gammaglobulin (IgG)
- Theo dõi nồng độ IgG mỗi 4-12 tuần/lần và tuỳ vào tình trạng nhiễm trùng của người bệnh.
- Chỉ định truyền gammaglobulin:
+ IgG < 400mg/dl;
+ Tiền sử nhiễm trùng xoang và phổi tái diễn nhiều lần (2 đợt nhiễm trùng trong vòng 6 tháng phải điều trị kháng sinh), nhiễm trùng nặng khi không kiểm soát được bằng kháng sinh kinh nghiệm hoặc kháng sinh đồ hoặc nhiễm trùng nặng đe doạ tính mạng người bệnh;
+ Trường hợp viêm phổi do CMV;
+ Truyền dự phòng trong ghép máu dây rốn: IVIG 500mg/kg/ngày bắt đầu vào ngày D-6, sau đó mỗi 2 tuần cho đến ngày D+100 sau ghép.
- Liều: IgG 400-500mg/kg truyền tĩnh mạch, mỗi 2-4 tuần. Ngừng truyền khi IgG > 400 g/dl trong 3 tháng liên tiếp (Theo dõi IgG: mỗi 2-4 tuần sau khi bắt đầu điều trị IVIG).
4.3. Bệnh ghép chống chủ cấp (acute GVHD)
4.3.1. Phác đồ dự phòng bệnh ghép chống chủ cấp (aGVHD)
- Ghép cùng huyết thống:
+ CSA/MTX: Truyền CSA tĩnh mạch từ ngày -4 (D-4) với liều 3mg/kg/ngày, chia 2 lần cách 12 giờ. Khi chuyển sang uống với liều 5mg/kg/ngày. Duy trì nồng độ thuốc trong máu tuỳ từng bệnh. MTX: Liều dùng 5mg/m2 da, truyền tĩnh mạch vào ngày thứ 1, 3, 6 sau ghép;
+ Tacrolimus/MTX;
+ CSA/MMF.
- Ghép không cùng huyết thống: Tacrolimus/MMF...
- Ghép haplotype: Tac/MMF/Cy liều cao sau ghép...
4.3.2. Triệu chứng của ghép chống chủ cấp
a. Ghép chống chủ cấp biểu hiện ở da: Hay gặp nhất.
- Tổn thương ban đỏ, thường bắt đầu ở lòng bàn tay và gan bàn chân, có thể đau hay ngứa. Sau đó xuất hiện ở má, tai, cổ và trên người; thường ở dạng sần. Trường hợp nặng: Phỏng nước và hoại tử da:
- Giai đoạn:
1: < 25% diện tích da cơ thể bị tổn thương;
2: 25-50% diện tích da cơ thể bị tổn thương;
3: 50-100%, tổn thương đỏ da;
4: Tổn thương dạng mụn nước hay phỏng nước.
b. Ghép chống chủ cấp biểu hiện ở gan: Là cơ quan thường gặp tiếp sau tổn thương da.
- Biểu hiện vàng da tắc mật; thường không tiến triển thành suy gan.
- Giai đoạn:
1: Bilirubin: 34-50µmol/l;
2: Bilirubin: 51-102 µmol/l;
3: Bilirubin: 103-255 µmol/l;
4: Bilirubin: > 255 µmol/l.
c. Ghép chống chủ cấp biểu hiện ở ống tiêu hóa:
- Tiêu chảy (màu xanh, nước, nhầy; cũng có thể có tế bào và tổ chức phân khuôn), chảy máu ruột, đau co cứng bụng, tắc ruột.
- Tổn thương hệ tiêu hóa cao ít gặp hơn và thuờng gặp ở người bệnh nhiều tuổi. Cần sinh thiết để chẩn đoán xác định.
- Giai đoạn:
1: Thể tích 500-1.000 ml/ngày hay buồn nôn liên tục;
2: 1.000-1.500ml/ngày;
3: > 1.500ml/ngày;
4: Đau bụng +/- tắc ruột.
Phân độ chung của ghép chống chủ cấp (theo Glucksberg và cs)
|
Mức độ |
Biểu hiện tổn thương ở da |
Biểu hiện tổn thương ở gan |
Biểu hiện tổn thương ở ruột |
Chức năng bị tổn thương |
|
0 (không) |
0 |
0 |
0 |
0 |
|
I (nhẹ) |
1-2 |
0 |
0 |
0 |
|
II (trung bình) |
1-3 |
1 |
1 |
+ |
|
III (nặng) |
2-3 |
2-3 |
2-3 |
++ |
|
IV(nguy hiểm tính mạng) |
2-4 |
2-4 |
2-4 |
+++ |
4.3.3. Phác đồ điều trị ghép chống chủ cấp
a. Điều trị hàng 1: Corticoid đơn thuần hay phối hợp nhóm ức chế calcineurin (cyclosporin A, tacrolimus...) và tùy theo mức độ của ghép chống chủ cấp.
- Điều trị độ I: Chủ yếu ghép chống chủ cấp ở da, corticoid tại chỗ và duy trì nồng độ ức chế calcineurin không cần điều trị ức chế miễn dịch hệ thống.
|
Nồng độ Corticoid |
Đậm đặc (Dermovate) |
Mạnh (Betnovate) |
Trung bình (Eumovate) |
Nhẹ (1% Hydrocortisone) |
|
Mặt |
Không sử dụng |
2 lần/ngày; 4-12 tuần |
2 lần/ ngày ; 6-12 tháng |
2 lần/ngày; điều trị lâu dài. |
|
Thân mình |
2 lần/ngày; 4-12 tuần |
2 lần/ngày; điều trị lâu dài. |
|
|
|
Lòng bàn tay và chân |
2 lần/ ngày; có thể sử dụng dưới xoa bóp mạnh, điều trị lâu dài. |
2 lần/ ngày; điều trị lâu dài. |
|
|
- Điều trị độ II-IV: Methylprednisolone.
+ 1mg/kg/ngày chia 2- 3 lần (tĩnh mạch), thời gian 7 ngày cho người bệnh ghép chống chủ cấp độ II;
+ 2-10mg/kg/ngày chia 2- 3 lần (tĩnh mạch), thời gian 7 ngày cho người bệnh ghép chống chủ cấp độ III-IV.
Vẫn tiếp tục điều trị cyclosporin A hoặc tacrolimus, chuyển dạng truyền tĩnh mạch nếu đang uống.
- Giảm liều methylprednisolone với nhóm có đáp ứng: theo nguyên tắc giảm dần và tuỳ vào tình trạng lâm sàng.
b. Điều trị hàng 2: Khi đã kháng corticoid
- Đánh giá kháng corticoid: Ghép chống chủ cấp mức độ II-IV không đáp ứng với methylprednisolone với liều > 2mg/kg/ngày, đã điều trị tối thiểu 6 ngày liên tiếp; và thất bại sau điều trị thêm 3 ngày liều cao methylprednisolone: 3-10mg/kg/ngày (tổng liều có thể 500mg/ngày).
- Chuyển điều trị hàng 2: Giảm nhanh liều methylprednisolon ≤ 1mg/kg/ngày. Cyclosporin A hay tacrolimus kết hợp với các thuốc sau tuỳ từng trường hợp người bệnh:
+ ATG ngựa 15-20mg/kg/ngày x 5 ngày; hoặc ATG thỏ 3mg/kg/ngày x 5 ngày; hoặc:
+ Nhóm kháng thể đơn dòng kháng TNF: Đơn thuần hoặc phối hợp nhóm kháng thể kháng thụ thể Interleukin 2.
● Infliximab: 5-10mg/kg mỗi tuần x 4 tuần, truyền trong 2h, dùng fil lọc protein. Dự phòng trước truyền 30-60 phút: Acetaminophen 650 mg uống và diphenhydramin 50 mg uống hoặc truyền. Hoặc:
● Etanercept: 25mg tiêm dưới da 2 lần/tuần.
+ Nhóm kháng thể kháng thụ thể Interleukin 2:
● Basiliximab: 20mg/ngày, ngày 1, 4; có thể nhắc lại sau 2 tuần nếu triệu chứng chưa cải thiện.
+ MMF 2-3g/ngày (uống 3 lần/ngày).
c. Điều trị hàng 3:
+ Methotrexate: 5mg/m2/ngày mỗi tuần x 4 tuần;
+ Ghép tế bào trung mô.
Lưu ý:
+ ATG ưu tiên hơn nếu tổn thương gan chiếm ưu thế, nhóm kháng thể đơn dòng kháng TNF nếu biểu hiện đường tiêu hóa chiếm ưu thế;
+ Ghép chống chủ biểu hiện đường ruột: Không được ăn đường miệng;
+ Tất cả các người bệnh ghép chống chủ đường ruột mức độ III-IV: Điều trị dự phòng ampicillin/sulbactam: Liều 2g/ngày truyền tĩnh mạch cách 12 giờ.
4.4. Bệnh ghép chống chủ mạn (chronic GVHD)
4.4.1. Chẩn đoán ghép chống chủ mạn
- Chẩn đoán ghép chống chủ mạn cần phải có các yếu tố như sau:
+ Có ít nhất 1 triệu chứng xác định ghép chống chủ mạn hoặc 1 triệu chứng gợi ý ghép chống chủ mạn (được xác định lại bằng sinh thiết hoặc các xét nghiệm khác);
+ Chẩn đoán phân biệt với ghép chống chủ mạn;
+ Chẩn đoán phân biệt với bệnh khác (nhiễm trùng, dị ứng thuốc, ung thư thứ phát…).
- Một số cơ quan tổn thương:
+ Da: Dạng sừng hoá, xơ cứng; mất hoặc tăng sắc tố; ban sẩn;
+ Móng: Loạn dưỡng, khía và dễ gãy;
+ Miệng: Dạng sừng hoá, hạn chế há miệng; khô miệng, teo niêm mạc miệng, giả mạc và loét; ban đỏ, đau;
+ Mắt: Khô, viêm kết mạc, sợ ánh sáng, viêm bờ mi, tăng sắc tố quanh mắt;
+ Ống tiêu hoá: Ăn không ngon, buồn nôn, nôn; tiêu chảy; xơ hẹp thực quản;
+ Gan: Tăng bilirubin, tăng men gan;
+ Phổi: Viêm phế quản tắc nghẽn;
+ Cơ, khớp và dây chằng: Xơ cứng khớp, viêm cơ, viêm hoặc đau khớp;
+ Hệ máu và miễn dịch: Giảm tiểu cầu, tăng bạch cầu ưa acid; tăng hoặc giảm globulin.
4.4.2. Điều trị ghép chống chủ mạn
a. Điều trị ban đầu
- Corticoid đơn độc hoặc phối hợp với chất ức chế calcineurin (ciclosporin A hoặc tacrolimus). Sử dụng kết hợp ciclosporin A hoặc tacrolimus giúp giảm nhanh liều corticoid, hạn chế các biến chứng do coticoid.
+ Liều khởi đầu methylprednisolone 1mg/kg/ngày và CSA 10mg/kg/ngày, chia làm 2 lần trong ngày. Sau 2 tuần, nếu bệnh không tiến triển, giảm liều methylprednisolone: Giảm 25% liều mỗi tuần. Khi methylprednisolone được giảm liều hoàn toàn mà không có biểu hiện tái phát, ghép chống chủ, có thể giảm liều CSA: 25% liều mỗi tuần cho đến khi đáp ứng hoàn toàn. Liều duy trì có thể xen kẽ giữa methylprednisolone và CSA trong 9 tháng;
+ Người bệnh được đánh giá sau 3 tháng điều trị: Nếu đáp ứng hoàn toàn có thể ngừng thuốc, đáp ứng không hoàn toàn thì tiếp tục điều trị trong 3 tháng nữa, sau đó đánh giá lại. Nếu trong quá trình giảm liều methylprednisolone bệnh có xu hướng tăng lên thì tăng liều methylprednisolone trở lại và tiếp tục duy trì ít nhất 3 tháng trước khi giảm liều;
+ cGVHD kháng với methylprednisolone: Khi mà bệnh tiến triển với liều methylprednisolone 1mg/kg/ngày trong 2 tuần hoặc bệnh ổn định với methylprednisolone liều > 0,5mg/kg/ngày trong 4-8 tuần hoặc không thể giảm liều methylprednisolone dưới 0,5mg/kg/ngày. Trường hợp này cần chuyển sang lựa chọn hàng 2.
- Thalidomide: Liều 50-200mg x 2 lần/ngày.
b. Lựa chọn hàng 2: Corticoid, kết hợp với:
- Ciclosporin A.
- Tacrolimus: 0,05mg/kg mỗi 12 giờ, uống, duy trì nồng độ 5-12 ng/ml, hoặc:
- MMF: 15 mg/kg mỗi 12 giờ, uống đơn thuần hoặc kết hợp thuốc khác, nguy cơ nhiễm trùng và tái phát bệnh thấp.
- Rituximab: Chỉ định khi có ghép chống chủ mạn gây xơ cứng cơ khớp hoặc giảm tiểu cầu. Liều 375mg/m2/tuần x 4 tuần, có thể điều trị đợt 2 nếu đáp ứng chưa hoàn toàn.
- Thalidomide: Thường chỉ định cho bệnh nhân viêm niêm mạc và tổn thương dạng sừng hoá. Liều khởi đầu 50-200mg x 2 lần/ngày.
- PUVA: Chỉ định ở bệnh nhân cGVHD tổn thương da kháng thuốc.
- Extracorporeal photopheresis: Có hiệu quả cao trong điều trị cGVHD da, gan, mắt và miệng.
- Liều thấp tia xạ (100-cGy) toàn bộ lympho vùng bụng, ngực.
- Imatinib: Chỉ định cho bệnh nhân cGVHD kháng thuốc, liều 100-400mg/ngày.
- Ngoài ra có thể điều trị một số thuốc: Ibrutinib, Ruxolitinib.
c. Điều trị hỗ trợ có ý nghĩa quan trọng, giúp kéo dài thời gian sống và nâng cao chất lượng cuộc sống của người bệnh ghép chống chủ mạn bao gồm:
- Phòng ngừa nhiễm trùng:
+ Pneumocystis crinii: TMP-SMX (tham khảo phần dự phòng nhiễm trùng);
+ Vi khuẩn có vỏ bao như là pneumococcus với PNCV;
+ Chống nấm, chống virus khi có triệu chứng xảy ra;
+ IgIV sử dụng khi nồng độ IgG thấp và nhiễm trùng tái phát nhiều lần, duy trì nồng độ IgG > 400mg/dL;
+ Tiêm phòng sau 1 năm, sau khi điều trị ghép chống chủ hoàn tất.
- Điều trị triệu chứng: điều trị tại chỗ như ghép chống chủ ở da, miệng, mắt... bằng thuốc có corticoid, tacrolimus...
4.5. Biến chứng thải ghép, mảnh ghép mọc kém và tái phát bệnh
4.5.1. Thải ghép
- Là biến chứng hay gặp trong ghép các bệnh máu không phải ác tính.
- Thải ghép sớm (thải ghép nguyên phát) là không có hiện tượng mọc mảnh ghép thể hiện giảm tế bào máu dai dẳng.
- Thải ghép muộn (thải ghép thứ phát) xảy ra sau khi đã có hiện tượng mọc mảnh ghép, có nghĩa là đã có biểu hiện mọc các tế bào tạo máu của người hiến ở người bệnh, nhưng sau đó lại có hiện tượng giảm các tế bào máu của người hiến đã mọc.
- Theo dõi:
+ Các xét nghiệm: Chỉ số tế bào máu ngoại vi, chimerism tế bào dòng tuỷ và lympho T; các xét nghiệm virus như CMV, HHV6...;
+ Tình trạng nhiễm trùng, bệnh ghép chống chủ, biểu hiện CMV tái hoạt động...
- Xử trí thải ghép: Nên tiến hành ghép lần hai cho người bệnh, ghép tế bào gốc lần hai là một sự lựa chọn cho những người bệnh đã không mọc mảnh ghép hay thải ghép sau ghép lần một.
4.5.2. Mảnh ghép mọc kém
- Tiêu chuẩn:
+ Huyết sắc tố < 100 g/L, số lượng tuyệt đối bạch cầu trung tính < 1 G/L, tiểu cầu < 30 G/L ít nhất hai tuần liên tiếp tính từ ngày thứ 14 sau ghép;
+ Người bệnh tiếp tục phải truyền máu và có những biểu hiện giảm sinh tủy, trong khi xét nghiệm chimerism vẫn đạt gần hoàn toàn và không có biểu hiện của ghép chống chủ mức độ nặng hay tái phát bệnh.
- Xử trí:
+ G-CSF khi bạch cầu trung tính giảm nhưng chỉ có hiệu quả trong thời gian ngắn;
+ Erythropoietin tái tổ hợp cũng có thể có hiệu quả trong việc cải thiện nồng độ huyết sắc tố;
+ Bổ sung thêm tế bào gốc của người hiến mà không cần xử lý hoặc có thể truyền chọn lọc tế bào CD34+ cho người bệnh.
4.5.3. Tái phát bệnh
- Là biến chứng hay gặp khi ghép cho nhóm bệnh máu ác tính và là nguyên nhân chính gây tử vong sau ghép.
- Nhận biết người bệnh có nguy cơ tái phát bệnh cao sau ghép: Dựa trên bệnh được chẩn đoán, tình trạng bệnh lúc ghép, tổn thương di truyền và tồn dư tối thiểu bệnh máu ác tính trong giai đoạn điều trị hoá chất và thời điểm khi ghép.
- Theo dõi nguy cơ tái phát dựa trên các xét nghiệm sau:
+ Tồn dư tối thiểu bệnh máu ác tính: Theo dõi động học sau ghép cho các bệnh AML, CML và ALL để nhận biết người bệnh còn tồn tại bệnh sau ghép hay không để can thiệp sớm bằng cách tăng hiệu ứng ghép chống lơxêmi nhằm loại bỏ khả năng tiến triển dẫn đến tái phát bệnh;
+ Xét nghiệm đánh giá mọc mảnh ghép (chimerism): Qua biểu hiện giảm chimerism của người hiến có thể tiên lượng bệnh tái phát.
- Xử trí:
+ Dừng thuốc ức chế miễn dịch: là phương pháp đầu tiên khi bệnh tái phát sau ghép; có thể đạt trở lại sự lui bệnh với tỷ lệ khoảng 80% với CML giai đoạn mạn tính, nhưng chỉ đạt 10% với AML; với ALL và CML giai đoạn tăng tốc thì không có hiệu quả;
+ Truyền lympho người hiến (donor lymphocyte infusion - DLI): có thể tạo lại sự lui bệnh ở người bệnh tái phát sau ghép với hiệu quả đạt khoảng 80% với CML, 40% với đa u tuỷ xương, 20% với AML và NHL, 15% với ALL. Có thể DLI đơn thuần hay điều trị hoá chất rồi DLI;
+ Thuốc nhắm đích: Nhóm TKIs (imatinib...) trong CML, nhóm demethylating (decitabine, azacitidine...) trong AML/MDS, rituximab trong U lympho không Hodgkin tế bào B;
+ Ghép lần 2: Hiệu quả ghép lần 2 phụ thuộc vào bệnh, tuổi người bệnh, tình trạng người bệnh, phác đồ điều kiện hoá sử dụng trong ghép lần 1 và khoảng thời gian giữa 2 lần ghép. Tỷ lệ tử vong liên quan đến ghép ở lần hai khoảng 30%, tỷ lệ tái phát sau ghép lần 2 khoảng 42% và tỷ lệ sống sau 5 năm chỉ khoảng 28%;
+ Điều trị liệu pháp tế bào: Tế bào lympho T gây độc, tế bào diệt tự nhiên NK, tế bào liên võng...;
+ Theo dõi và điều trị hỗ trợ.
4.6. Theo dõi và xử trí bất đồng nhóm máu
4.6.1. Bất đồng nhóm máu thứ yếu (minor ABO incompatibility)
- Định nghĩa: Khi có mặt kháng thể chống A hoặc kháng thể chống B hay cả kháng thể chống A và B trong huyết tương của người hiến mà các kháng thể này có thể gây ngưng kết hồng cầu người bệnh.
- Từ ngày 4-14 sau ghép:
+ Kiểm tra hàng ngày: Tổng phân tích máu, LDH, bilirubin;
+ Truyền khối hồng cầu khi Hb dưới 9,5g/dl;
+ Làm xét nghiệm Coombs trực tiếp 4 ngày/lần;
+ Xác định lại nhóm máu hệ ABO, Rh, Mia và Coombs gián tiếp 4 ngày/lần.
- Lựa chọn truyền khối hồng cầu và chế phẩm máu cho người bệnh trong trường hợp có bất đồng nhóm ABO thứ yếu (bảng 55).
Bảng. 55 Lựa chọn nhóm máu trong truyền máu khi bất đồng nhóm ABO thứ yếu
|
Người hiến |
Bệnh nhân |
Pha I |
Pha II |
Pha III |
||
|
Tất cả chế phẩm |
KHC |
Tiểu cầu |
Huyết tương |
Tất cả chế phẩm |
||
|
Bất đồng thứ yếu |
||||||
|
O |
A |
Nhóm bệnh nhân |
O |
A;AB;B;O |
A;AB |
Nhóm người cho |
|
O |
B |
Nhóm bệnh nhân |
O |
B;AB;A;O |
B;AB |
Nhóm người cho |
|
O |
AB |
Nhóm bệnh nhân |
O |
AB;A;B;O |
AB |
Nhóm người cho |
|
A |
AB |
Nhóm bệnh nhân |
A |
AB;A;B;O |
AB |
Nhóm người cho |
|
B |
AB |
Nhóm bệnh nhân |
B |
AB;B;A;O |
AB |
Nhóm người cho |
Pha I: Từ khi bệnh nhân chuẩn bị đến khi ghép.
Pha II: Từ khi bắt đầu bắt đầu điều kiện hóa, đến khi xét nghiệm coombs trực tiếp âm tính, không còn kháng thể kháng hồng cầu người cho, hoặc hồng cầu bệnh nhân không còn.
Pha III: Khi chuyển đổi nhóm máu hoàn toàn sang nhóm máu bệnh nhân.
4.6.2. Bất đồng nhóm máu “chính” (major ABO incompatibility)
- Định nghĩa: Khi có mặt kháng thể chống A hoặc kháng thể chống B hay cả kháng thể chống A và B trong huyết tương của người bệnh mà các kháng thể này có thể gây ngưng kết hồng cầu người hiến.
- Biến chứng thường xảy ra muộn sau vài tháng với biểu hiện: dòng hồng cầu hồi phục chậm hay suy tủy một dòng hồng cầu.
- Lựa chọn truyền khối hồng cầu và chế phẩm máu cho người bệnh trong trường hợp có bất đồng nhóm ABO chính (bảng 56).
Bảng 56. Lựa chọn truyền khối hồng cầu và chế phẩm máu cho người bệnh trong trường hợp có bất đồng nhóm ABO chính (chủ yếu)
|
Người hiến |
Bệnh nhân |
Pha I |
Pha II |
Pha III |
||
|
Tất cả chế phẩm |
KHC |
Tiểu cầu |
Huyết tương |
Tất cả chế phẩm |
||
|
Bất đồng chủ yếu |
||||||
|
A |
O |
Nhóm bệnh nhân |
O |
A;AB;B;O |
A;AB |
Nhóm người cho |
|
B |
O |
Nhóm bệnh nhân |
O |
B;AB;A;O |
B;AB |
Nhóm người cho |
|
AB |
O |
Nhóm bệnh nhân |
O |
AB;A;B;O |
AB |
Nhóm người cho |
|
AB |
A |
Nhóm bệnh nhân |
A |
AB;A;B;O |
AB |
Nhóm người cho |
|
AB |
B |
Nhóm bệnh nhân |
B |
AB;B;A;O |
AB |
Nhóm người cho |
4.6.3. Bất đồng nhóm máu “hai chiều” (bidirectional ABO incompatibility).
- Định nghĩa: Cả người hiến và người bệnh đều sản xuất ra kháng thể chống lại hồng cầu của nhau.
- Biến chứng: Gây tan máu sớm và kéo dài.
- Lựa chọn truyền khối hồng cầu và chế phẩm máu trong trường hợp bất đồng 2 chiều (bảng 57).
Bảng 57. Lựa chọn truyền khối hồng cầu và chế phẩm máu trong trường hợp bất đồng 2 chiều
|
Người hiến |
Bệnh nhân |
Pha I |
Pha II |
Pha III |
||
|
Tất cả chế phẩm |
KHC |
Tiểu cầu |
Huyết tương |
Tất cả chế phẩm |
||
|
Bất đồng 2 chiều |
||||||
|
B |
A |
Nhóm bệnh nhân |
O |
AB;A;B;O |
AB |
Nhóm người cho |
|
A |
B |
Nhóm bệnh nhân |
O |
AB;B;A;O |
AB |
Nhóm người cho |
5. TRUYỀN MÁU HỖ TRỢ SAU GHÉP
5.1. Chỉ định
- Khi số lượng huyết sắc tố của người bệnh giảm < 90g/L hoặc tuỳ tình trạng bệnh phối hợp (tim, phổi...): Chỉ định truyền khối hồng cầu.
- Khi số lượng tiểu cầu của người bệnh giảm < 20 G/L hoặc khi có xuất huyết: Chỉ định truyền khối tiểu cầu.
5.2. Loại chế phẩm
- Truyền máu và chế phẩm máu đã được lọc bạch cầu và chiếu xạ.
- Chế phẩm máu có kết quả xét nghiệm CMV âm tính nếu người bệnh CMV âm tính.
6. DINH DƯỠNG
Chế độ dinh dưỡng cho người bệnh được thực hiện theo nguyên tắc:
- An toàn dinh dưỡng: Dinh dưỡng được đảm bảo vô trùng tuyệt đối. Tất cả thức ăn đều được đun nóng lại bằng lò vi sóng trước khi người bệnh ăn.
- Chế độ ăn: Có tư vấn và xây dựng thực đơn dinh dưỡng theo tư vấn của chuyên gia dinh dưỡng.
- Dinh dưỡng bằng truyền tĩnh mạch được chỉ định khi có biểu hiện:
+ Rối loạn chức năng hệ tiêu hóa;
+ Cho ruột nghỉ trong ghép chống chủ cấp đường tiêu hóa;
+ Suy dinh dưỡng nặng.
- Dung dịch nuôi dưỡng tĩnh mạch bao gồm: Albumin human; dung dịch đường, mỡ và đạm (túi 2 ngăn và 3 ngăn).
6. BẢNG THEO DÕI XÉT NGHIỆM SAU GHÉP
XÉT NGHIỆM THEO DÕI SAU GHÉP TẾ BÀO GỐC ĐỒNG LOÀI ĐIỀU TRỊ LƠ XÊ MI CẤP, LƠ XÊ MI KINH
|
Xét nghiệm |
Tháng sau ghép (từ ngày truyền TBG) |
|||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
15 |
18 |
21 |
24 |
27 |
30 |
33 |
3 năm |
3.5 Năm |
4 năm |
4.5 năm |
≥5 năm |
|
|
Tổng phân tích tế bào |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
Sinh hoá máu cơ bản/ IgG,A,M,E |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
Đông máu cơ bản |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
x |
x |
x |
x |
x |
|
|
|
|
|
|
|
|
|
Xét nghiệm hormon |
|
|
|
|
|
x |
|
|
x |
|
|
x |
x |
x |
x |
x |
|
|
|
x |
|
x |
|
x |
|
Xét nghiệm marker ung thư |
|
|
|
|
|
x |
|
|
x |
|
|
x |
x |
x |
x |
x |
|
|
|
x |
|
x |
|
x |
|
Định lượng Cyclo/Tacrolimus |
x |
x |
x |
x |
x |
x |
|
|
Tuỳ bệnh nhân có bị cGVHD đang ĐT UCMD. |
|||||||||||||||
|
Huyết tủy đồ |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
x |
|
|
|
x |
|
Sinh thiết tuỷ xương/CD3,4,8 (rốn, haplo) |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
Phân loại miễn dịch tồn dư tối thiểu |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
Gen bệnh (cho Lơ xê mi cấp có gen, CML chuyển cấp) |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
Công thức NST |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
Chimerism hoặc FISH (dòng tuỷ và lympho) |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
x |
|
x |
|
|
|
x |
Tuỳ tình trạng BN |
|||
|
CMV đo tải lượng |
x |
x |
x |
x |
x |
x |
|
|
x |
|
|
x |
|
|
|
|
*Tuỳ bệnh nhân có bị cGVHD đang ĐT UCMD nên có nguy cơ CMV tái hoạt động |
|||||||
|
Bộ nhóm máu (ABO-Rh, hiệu giá KT tự nhiên, miễn dịch, coombs trực tiếp) (nếu bất đồng nhóm máu) |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
EBV IgG/IgM |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
x |
x |
x |
|
x |
|
|
|
|
|
|
|
|
|
Định lượng IgA,M,G,E, CD3-4-8 |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
Đo chức năng hô hấp |
Thực hiện khi bệnh nhân có biến chứng đặc biệt (nghi cGVHD ở phổi, mắt, cơ quan sinh dục...) |
|||||||||||||||||||||||
|
Siêu âm, X-Quang |
||||||||||||||||||||||||
|
Khám mắt |
||||||||||||||||||||||||
|
Khám sinh dục |
||||||||||||||||||||||||
Lưu ý: Trong qui trình ghép tế bào gốc bệnh nhân có làm xét nghiệm tủy đồ, sinh thiết tủy xương, chimerism, fish XY, kết quả CTScaner, MIR (nếu có). Đề nghị bệnh nhân khi ra viện photo và giữ lại, khi đến khám lại bệnh nhân nhớ mang theo để bác sĩ khám theo dõi tiếp.
Cho thuốc sau ghép (đơn ra viện thông thường):
- Cyclosporin (neoral), tacrolimus (prograft): duy trì nồng độ thấp và ngừng thuốc sớm trong vòng 6 tháng (trừ khi có GvHD nhiều);
- Magie B6: luôn cho song song cùng với cyclosporin hoặc tacrolimus.
- Acyclovir duy trì đến 6 tháng-1 năm (liều 400-800 mg/ngày, uống);
- Chống nấm dự phòng (fluconazole): duy trì tối đa 75-100 ngày sau ghép (liều 150-300 mg/ngày); hoặc phải ĐT corticoid liều >1mg/kg/ngày.
- Uruso, bổ gan: duy trì tối đa 3 tháng sau ghép, trừ khi có vấn đề về gan thì lâu hơn (đặc biệt lưu ý khi dừng UCMD nguy cơ cao cGVHD ở gan)
- Biseptol (sulfaprim, cotrimoxazole 480 mg): duy trì 6 tháng-1 năm, uống 2 ngày mỗi tuần (thứ 7 và chủ nhật), mỗi ngày 2 viên 480 mg(sáng, chiều).
XÉT NGHIỆM THEO DÕI SAU GHÉP TẾ BÀO GỐC ĐỒNG LOÀI SUY TỦY XƯƠNG - ĐÁI HUYẾT SẮC TỐ KỊCH PHÁT BAN ĐÊM
|
Xét nghiệm |
Tháng sau ghép (từ ngày truyền TBG) |
|||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
15 |
18 |
21 |
24 |
27 |
30 |
33 |
3 năm |
3.5 Năm |
4 năm |
4.5 năm |
≥5 năm |
|
|
Tổng phân tích tế bào máu |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
Sinh hoá máu cơ bản (bộ đủ như vào viện) |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
Đông máu cơ bản |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
x |
x |
x |
x |
x |
|
|
|
|
|
|
|
|
|
Định lượng IgA,M,G,E, CD3-4-8 |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
Xét nghiệm hormon |
|
|
|
|
|
x |
|
|
x |
|
|
x |
x |
x |
x |
x |
|
|
|
|
|
|
|
|
|
Xét nghiệm marker ung thư |
|
|
|
|
|
x |
|
|
x |
|
|
x |
x |
x |
x |
x |
|
|
|
|
|
|
|
|
|
Định lượng Cyclo/Tacrolimus |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
Tuỳ bệnh nhân có bị cGVHD đang ĐT UCMD |
|||||||||||
|
Huyết tủy đồ |
x |
|
Khi có diễn biến đặc biệt thì chỉ định |
x |
|
|
|
x |
|
|
|
|
|
x |
|
|
||||||||
|
Sinh thết tuỷ xương |
x |
|
x |
|
|
|
x |
|
|
|
|
|
x |
|
|
|||||||||
|
Công thức NST |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
|
|
x |
|
|
|
Chimerism hoặc FISH (dòng tuỷ và lympho) |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
x |
|
x |
|
|
|
x |
|
x |
|
x |
|
CMV đo tải lượng |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
x |
x |
x |
|
x |
|
Tuỳ bệnh nhân có bị cGVHD đang ĐT UCMD nên có nguy cơ CMV tái hoạt động |
||||||
|
Bộ nhóm máu (ABO-Rh, hiệu giá KT tự nhiên, miễn dịch, coombs trực tiếp) (nếu bất đồng nhóm máu) |
x |
|
x |
|
|
x |
|
|
x |
|
|
x |
|
x |
|
x |
|
|
|
|
|
|
|
|
|
EBV IgG/IgM |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
|
x |
x |
x |
|
x |
|
|
|
|
|
|
|
|
|
CD55/59 Hồng cầu, Bạch cầu |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
x |
|
x |
|
x |
|
|
|
x |
|
x |
|
x |
|
Đo chức năng hô hấp |
Thực hiện khi bệnh nhân có biến chứng đặc biệt (nghi cGVHD ở phổi, mắt, cơ quan sinh dục...) |
|||||||||||||||||||||||
|
Siêu âm, X- Quang |
||||||||||||||||||||||||
|
Khám mắt |
||||||||||||||||||||||||
|
Khám sinh dục |
||||||||||||||||||||||||
Lưu ý: Trong qui trình ghép tế bào gốc bệnh nhân có làm xét nghiệm tủy đồ, sinh thiết tủy xương, chimerism, fish XY, kết quả CTScaner, MIR (nếu có). Đề nghị bệnh nhân khi ra viện photo và giữ lại, khi đến khám lại bệnh nhân nhớ mang theo để bác sĩ khám theo dõi tiếp.
Cho thuốc sau ghép (đơn ra viện thông thường):
- Cyclosporin (neoral), tacrolimus (prograft): duy trì nồng độ 200-400 và ngừng thuốc sớm trong vòng 12 tháng (trừ khi có GvHD nhiều), chú ý chỉnh liều khi suy thận;
- Magie B6: luôn cho song song cùng với cyclosporin hoặc tacrolimus;
- Acyclovir duy trì đến 6 tháng-1 năm (liều 400-800 mg/ngày, uống);
- Chống nấm dự phòng (fluconazole, vori, posa, itra…): duy trì tối đa 75-100 ngày sau ghép (liều 150-300 mg/ngày), điều chỉnh tùy theo có sử dụng; hoặc phải ĐT corticoid liều >1mg/kg/ngày.
- Uruso, bổ gan: duy trì tối đa 3 tháng sau ghép, trừ khi có vấn đề về gan thì lâu hơn; (đặc biệt lưu ý khi dừng UCMD nguy cơ cao cGVHD ở gan)
- Biseptol (sulfaprim, cotrimoxazole 480 mg): duy trì 6 tháng-1 năm, uống 2 ngày mỗi tuần (thứ 7 và chủ nhật), mỗi ngày 2 viên 480 mg chia sáng, chiều.
TÀI LIỆU THAM KHẢO
1. Joseph H. Antin, 2009. Common Uses of Hematopoietic Stem Cell Transplantation. In: Hematopoietic Stem Cell Transplantation A Hand book for Clinicians. Bethesda, MD: AABB, Ch.2, pp9-26.
2. Antin J.H. and Raley D.Y, 2013. Rationale for transplantation. In: Manual of Stem Cell and Bone Marow Transplantation. Cambridge University Press, Second edition, Ch.1, pp.1-2.
3. Childs R.W. and Srinivasan R., 2013. Hematopoietic Stem Cell Transplantation. In: The Bethesda Hand book of Clinical Hematology. Lippincott Williams & Wilkins, A Wolters Kluwer business, Third edition, Ch.18, pp 282-295.
4. U Anurathapan, S Hongeng et all. Hematopoietic stem cell transplantation for homozygous β- thalassemia and β-thalassemia/hemoglobin E patientsfrom haploidentical donors. Bone Marrow Transplantation (2016) 51, 813-818.
5. Childs R.W. Recommendations for the pre-emptive management of CMV after allogeneic stem cell or bone marrow transplant using PCR (2014).
6. Johan A. Maertens. Invasive Fungal Infections. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies [Internet]. 7th edition.
7. Stephen J. Forman, Robert S. Negrin, Joseph H. Antin, Frederick R. Appelbaum. Thomas’ Hematopoietic Cell Transplantation. Fifth Edition, 2016.
8. Helene M. Schoemans, Stephanie J. Lee et all. EBMT−NIH−CIBMTR Task Force position statement on standardized terminology & guidance for graft-versus-host disease assessment. Bone Marrow Transplantation (2018) 53:1401-1415.
9. Shannon R. McCurdyand and Leo Luznik. How we perform haploidentical stem cell transplantation with posttransplant cyclophosphamide. Blood, 21 November 2019, Vol 134, Num 21.
10. A.John Barrett and Minoo Battiwalla (2010), Relapse after Allogeneic Stem Cell Transplantation. Expert Rev Hematol. 2010 August ; 3(4): 429-441.
44. HƯỚNG DẪN CHỈ ĐỊNH VÀ ĐÁNH GIÁ MỘT SỐ XÉT NGHIỆM TẾ BÀO - MÔ BỆNH HỌC CƠ QUAN TẠO MÁU
Xét nghiệm tế bào - mô bệnh học cơ quan tạo máu: Là tất cả các xét nghiệm đánh giá về số lượng, hình thái của các tế bào trong các tổ chức tạo máu, sự phát triển và biệt hóa của các tế bào và cấu trúc mô bệnh học của các cơ quan tạo máu.
1. HUYẾT ĐỒ
1.1. Khái niệm: Là xét nghiệm quan sát về số lượng, hình thái cũng như thành phần các tế bào trong máu ngoại vi, từ đó giúp định hướng tình trạng sinh lý hoặc bệnh lý về máu của cơ thể.
1.2. Chỉ định:
- Khi có biểu hiện bất thường trên xét nghiệm tổng phân tích tế bào máu:
+ Tăng hoặc giảm bạch cầu, bất thường về thành phần bạch cầu;
+ Thiếu máu, giảm số lượng hồng cầu, hoặc tăng hồng cầu, tăng huyết sắc tố và hematocrit;
+ Tăng hoặc giảm tiểu cầu, tiểu cầu to, không đều;
+ Có các cảnh báo bất thường sau khi xét nghiệm tế bào máu bằng máy đếm tế bào: cảnh báo có tế bào blast, có bạch cầu chưa trưởng thành ra máu, có mảnh vỡ hồng cầu, tiểu cầu tạo đám, có hồng cầu non…
1.3. Cách lấy và xử lý mẫu:
- 1 ống máu được chống đông bằng EDTA để đếm số lượng các loại tế bào, đếm số lượng hồng cầu lưới (máy đếm tế bào hoặc phương pháp thủ công).
- Kéo lam và nhuộm Giemsa (bằng phương pháp thủ công hoặc máy nhuộm tự động). Tốt nhất nên lấy máu ngoại vi rồi kéo tiêu bản trực tiếp.
1.4. Nhận định kết quả:
Đánh giá các chỉ số và hình thái các tế bào trong huyết đồ có thể định hướng chẩn đoán một số bệnh:
- Thiếu máu, nhược sắc, hồng cầu nhỏ cùng với hình thái hồng cầu có thể định hướng thiếu máu do thiếu sắt, bệnh Thalassemia; từ đó đề xuất làm thêm xét nghiệm khác, như: Định lượng sắt huyết thanh, ferritin, điện di huyết sắc tố, sức bền hồng cầu...
- Thiếu máu, hồng cầu to, hồng cầu lưới tăng thường gặp trong bệnh tan máu tự miễn; hồng cầu lưới giảm thường gặp trong hội chứng rối loạn sinh tủy, thiếu vitamin B12, acid folic, suy tủy xương...
- Giảm số lượng và độ tập trung tiểu cầu: Trong xuất huyết giảm tiểu cầu, bệnh hệ thống, hội chứng Evans, xơ gan, suy tủy xương, rối loạn sinh tủy, bệnh lơ-xê-mi cấp…
- Tăng số lượng và độ tập trung tiểu cầu: Các bệnh tăng sinh tủy, bệnh nhân sau cắt lách, hoặc một số ung thư khác (K phổi, K di căn phổi)…
- Giảm bạch cầu trong các trường hợp nhiễm virus, suy tủy xương, lơ-xê-mi cấp, rối loạn sinh tủy, ung thư di căn tủy xương…
- Tăng bạch cầu: Trong nhiễm khuẩn, lơ-xê-mi cấp hoặc lơ-xê-mi mạn dòng hạt, dòng lympho…
- Hồng cầu lưới đánh giá sự phát triển dòng hồng cầu trong tủy xương. Hồng cầu lưới tăng trong các trường hợp thiếu máu lành tính; hồng cầu lưới giảm trong trường hợp tủy giảm sinh, suy tủy xương, rối loạn sinh tủy…
2. TỦY ĐỒ (Xét nghiệm tế bào học tủy xương)
2.1. Khái niệm:
Là xét nghiệm phân tích số lượng, hình thái các tế bào tủy xương để thăm dò chức năng tạo máu cũng như gợi ý các nguyên nhân gây rối loạn chức năng tạo máu tại tủy xương.
2.2. Chỉ định:
- Thiếu máu chưa tìm thấy nguyên nhân khi xét nghiệm máu ngoại vi.
- Tăng số lượng hồng cầu, lượng huyết sắc tố và hematocrit.
- Tăng hoặc giảm tiểu cầu.
- Tăng hoặc giảm bạch cầu máu ngoại vi.
- Bất thường trong thành phần bạch cầu: Có tế bào bất thường trong máu ngoại vi (có tế bào non ác tính, tế bào chưa trưởng thành, tăng tỷ lệ lympho, mono…).
- Khi lâm sàng và xét nghiệm khác nghi ngờ bệnh cơ quan tạo máu như: Đa u tủy xương, hội chứng tăng sinh lympho…
- Bệnh nhân U lympho (để đánh giá xem có hay không xâm lấn tủy xương).
- Các bệnh ung thư nghi di căn tủy xương.
- Các trường hợp sốt kéo dài chưa tìm được nguyên nhân.
- Đánh giá đáp ứng sau điều trị các bệnh máu.
- Đánh giá mọc mảnh ghép sau ghép tế bào gốc.
2.3. Cách lấy và xử lý mẫu:
- Dùng kim chọc hút tủy xương chuyên dụng (kim 1 lần, nhiều lần hoặc máy khoan cầm tay).
- Hút 0,5-1 ml dịch tủy xương (thường làm thủ thuật ở gai chậu sau trên), làm tiêu bản trực tiếp, nhuộm Giemsa để quan sát tế bào tủy xương bằng kính hiển vi quang học.
2.4. Nhận định kết quả
- Giá trị bình thường của tủy xương:
+ Số lượng tế bào tủy xương: 30 - 100 G/L, số lượng này tỷ lệ nghịch với số tuổi, tuổi càng già số lượng tế bào tủy càng giảm;
+ Tỷ lệ dòng hồng cầu/dòng bạch cầu trong tủy = 1/6, các dòng tế bào có sự cân đối giữa các lứa tuổi;
+ Tỷ lệ dòng lympho khoảng 8 - 25%;
+ Tỷ lệ dòng mono: 0 - 2%, dòng plasmo: 0 - 1%;
+ Mật độ mẫu tiểu cầu trong tủy: 30 - 100 mẫu tiểu cầu.
Ngoài ra có thể gặp đại thực bào vận chuyển sắt hoặc đang thực bào các tế bào thoái hóa, có thể gặp hủy cốt bào, tạo cốt bào.
- Đánh giá xét nghiệm tủy đồ: Dựa vào tiêu chuẩn của các bệnh lý và hình ảnh trên tủy đồ có thể chẩn đoán được:
+ Lơ xê mi tủy cấp, lơ xê mi lympho cấp: Khi tế bào blast ≥ 20 % tế bào có nhân trong tủy kết hợp với nhuộm hóa học tế bào và/hoặc phân loại miễn dịch;
+ Đa u tủy xương (có tăng sinh và rối loạn hình thái dòng plasmo), bệnh Waldenstrom (tăng sinh lympho-plasmo);
+ Tăng sinh lành tính dòng hồng cầu, tủy tăng sinh phản ứng trong nhiễm trùng, xuất huyết giảm tiểu cầu, hội chứng Evans…;
+ Hội chứng rối loạn sinh tủy: Dựa vào sự rối loạn của các dòng tế bào sinh máu: rối loạn về nhân, nguyên sinh chất, phát triển không đồng bộ giữa nhân và nguyên sinh chất…;
+ Góp phần chẩn đoán các bệnh trong hội chứng tăng sinh tủy, hội chứng tăng sinh lympho;
+ Hội chứng thực bào máu: Thấy hình ảnh tăng đại thực bào và đại thực bào đang thực bào tế bào máu;
+ Ung thư di căn tủy xương: Thấy hình ảnh tế bào K di căn vào tủy xương, thường gặp di căn tủy loại tế bào biểu mô, nhiều khi tình cờ phát hiện K di căn tủy khi bệnh nhân mới chỉ có dấu hiệu thiếu máu và/hoặc sốt kéo dài (trường hợp này nên sinh thiết tủy xương để nhuộm hóa mô miễn dịch. Từ đó, tìm ra ổ K nguyên phát);
+ U lympho xâm lấn tủy xương: Gặp hình ảnh tế bào ác tính dòng Lympho, khi này cần sinh thiết tủy xương để chẩn đoán xác định.
3. SINH THIẾT TỦY XƯƠNG (Xét nghiệm mô bệnh học tủy xương)
3.1. Khái niệm
Là xét nghiệm nhằm khảo sát một cách đầy đủ về cấu trúc mô bệnh học của tủy sinh máu cũng như số lượng, hình thái, thành phần của các dòng tế bào trong môi trường sinh máu.
3.2. Chỉ định: Trong các trường hợp sau:
- Tủy nghèo tế bào, hoặc có thể giảm sinh một hoặc nhiều dòng tế bào sinh máu (hồng cầu, bạch cầu và mẫu tiểu cầu) trên xét nghiệm tủy đồ.
- Hội chứng rối loạn sinh tủy.
- Hội chứng tăng sinh tủy (xơ tủy, tăng tiểu cầu tiên phát, đa hồng cầu..).
- Hội chứng rối loạn sinh tủy/ tăng sinh tủy.
- Hội chứng tăng sinh lympho.
- Đa u tủy xương.
- U lympho Hodgkin và không Hodgkin.
- Hội chứng thực bào máu, tăng sinh mô bào.
- Ung thư nghi ngờ di căn tủy xương.
- Các trường hợp cần làm xét nghiệm hóa mô miễn dịch tủy xương.
- Các trường sốt kéo dài chưa tìm được nguyên nhân.
- Đánh giá mọc mảnh ghép sau ghép tế bào gốc, đánh giá lui bệnh sau điều trị hóa chất bệnh nhân bệnh máu ác tính.
- Đánh giá đáp ứng sau điều trị các bệnh máu.
- Chọc hút tủy thất bại: do dịch tủy khô, không hút được dịch tủy;hay gặp trong các bệnh có tăng sinh xơ (xơ tủy, lơ-xê-mi cấp thể M7); K di căn…
3.3. Cách lấy và xử lý mẫu:
- Dùng kim sinh thiết tủy xương chuyên dụng (kim 1 lần, nhiều lần hoặc máy khoan cầm tay).
- Lấy 1 mảnh tủy xương ở gai chậu sau trên, dài khoảng 2 cm, sau đó dùng các kỹ thuật xử lý mảnh sinh thiết, cắt lát, cố định và nhuộm mô học để quan sát, đánh giá tủy sinh máu.
3.4. Nhận định kết quả
- Giá trị bình thường:
+ Mật độ và phân bố tế bào: Đánh giá sơ bộ tỷ lệ tế bào tạo máu và tế bào đệm (xơ, mỡ) trên toàn bộ tế bào tủy. Bình thường khu vực tạo máu chiếm 60 - 70 % diện tích sinh máu, càng nhiều tuổi tổ chức mỡ càng phát triển;
+ Thành phần tế bào: Bình thường tỷ lệ dòng hạt/dòng hồng cầu: 2/1-4/1; Dòng mẫu tiểu cầu: 1-4 MTC/vi trường; Lymphocyte chiếm khoảng 5% tế bào tủy; Plasmocyte < 5% tế bào tủy, thường nằm cạnh mạch máu và bè xương. Thường rất ít thấy đại thực bào.
- Xét nghiệm sinh thiết tủy xương giúp chẩn đoán các bệnh sau:
+ Hội chứng tăng sinh tủy: Tăng tiểu cầu tiên phát, đa hồng cầu nguyên phát: hình ảnh tủy giàu tế bào, tăng sinh mạnh cả 3 dòng tế bào, có thể tăng sinh xơ; Xơ tủy nguyên phát: Hình ảnh xơ hóa mạnh trong các khoang sinh máu (xơ nhiều hay ít phụ thuộc vào mỗi giai đoạn bệnh)…;
+ Đa u tủy xương: Hình ảnh tăng sinh dòng plasmo với hình thái bất thường, tạo thành từng ổ (u tương bào);
+ U lympho xâm lấn tủy: Thấy các nang hoặc các đám lớn tế bào lympho trong tủy xương;
+ Hội chứng tăng sinh lympho: Tăng sinh mạnh và rối loạn hình thái dòng lympho;
+ K di căn vào tủy xương: Thấy hình ảnh tế bào ung thư xâm lấn trong các khoang sinh máu (thường gặp loại K biểu mô như: K đường tiêu hóa, K phổi, K tiền liệt tuyến, K vú…);
+ Hội chứng thực bào tế bào máu: Tăng đại thực bào và thấy rõ hình ảnh đại thực bào đang ăn tế bào máu
+ Bệnh, mức độ bệnh suy tủy xương.
4. HẠCH ĐỒ (Xét nghiệm tế bào hạch)
4.1. Khái niệm:
Là một xét nghiệm thăm dò trực tiếp vào hạch để đánh giá thành phần và tỷ lệ tế bào trong hạch.
4.2. Chỉ định:
Tất cả các trường hợp hạch to chưa rõ nguyên nhân.
4.3. Cách lấy và xử lý mẫu:
Dùng kim nhỏ chọc hút tổ chức hạch, kéo tiêu bản, nhuộm Giemsa và quan sát trên kính hiển vi.
4.4. Nhận định kết quả:
- Quan sát toàn bộ tiêu bản để đánh giá thành phần tế bào hạch. Bình thường thành phần của hạch như sau: Tế bào lympho chiếm 90 - 99%, trong đó chủ yếu lymphocyte. Có thể gặp các tế bào sau với tỷ lệ rất thấp: Tế bào liên võng, monocyte, đại thực bào, plasmocyte.
- Kết quả hạch đồ trong một số bệnh lý:
+ Hạch viêm cấp do nhiễm khuẩn: Xuất hiện nhiều bạch cầu đoạn trung tính xen lẫn với lymphocyte và đại thực bào. Bạch cầu trung tính thường là dạng thoái hoá;
+ Bệnh Lơ-xê-mi lympho cấp và lơ xê mi lympho mạn: Hạch đồ giàu tế bào, tăng sinh dòng lympho, đơn dạng tế bào, hầu hết các tế bào giống nhau:
● Lơ-xê-mi lympho mạn: Gặp chủ yếu lymphocyte.
● Lơ-xê-mi lympho cấp: Gặp chủ yếu lymphoblast.
+ U lympho không Hodgkin: Tổn thương dạng nang hoặc lan toả. Có thể chủ yếu tế bào to hoặc tế bào nhỏ hoặc hỗn hợp to, nhỏ. Căn cứ đặc điểm tế bào, mô bệnh học và miễn dịch (miễn dịch tế bào, hóa mô miễn dịch) có thể xếp loại theo WHO 2008, WHO 2016...;
+ U lympho Hodgkin: Hình ảnh tổn thương đa dạng tế bào, có gặp tế bào Reed- Sternberg hoặc các biến thể (tế bào xác ướp, tế bào bỏng ngô…). Căn cứ đặc điểm mô bệnh học có thể xếp loại U lympho Hodgkin theo Tổ chức Y tế thế giới 2008, 2016 (typ mới: Ưu thế lympho bào dạng nốt; typ cổ điển: Giàu lympho bào, Hỗn hợp tế bào, Xơ nốt, nghèo lympho bào);
+ Hạch viêm lao: Hình ảnh nang lao điển hình có chất hoại tử bã đậu, nhiều tế bào bán liên, tế bào Langerhangs;
+ Ung thư di căn: Trên tiêu bản sinh thiết hạch thấy các đám lớn tế bào ung thư, tế bào có kích thước lớn, nhiều tế bào nhân quái, nhân chia...;
+ Các bệnh lý khác: Bệnh mô bào, các hạch viêm...
5. SINH THIẾT HẠCH (Xét nghiệm mô bệnh hӑc hạch)
5.1. Khái niệm:
Là xét nghiệm để quan sát cấu trúc hạch, hình thái và phân bố tế bào trong hạch.
5.2. Chỉ định:
- Khi kết quả hạch đồ không loại trừ được bệnh lý ác tính.
- Nghi ngờ hạch lao.
- Nghi ngờ ung thư di căn hạch.
- Hạch ở sâu không chọc hút được để làm hạch đồ.
5.3. Cách lấy và xử lý mẫu:
- Tốt nhất sinh thiết hạch ngoại biên (nếu có), chọn hạch kích thước vừa, hạch mới xuất hiện, nên chọn hạch vùng hiếm gặp như hạch thượng đòn.
- Nên lấy cả hạch, chỉ cắt một phần khi chỉ có một hạch quá to.
- Cố định, chuyển đúc, cắt nhuộm theo quy trình kỹ thuật làm tiêu bản mô mềm.
5.4. Nhận định kết quả
- Bình thường thành phần của hạch như sau: Cấu trúc vỏ hạch rõ, các nang lympho có tâm mầm rõ cực tính.
- Kết quả sinh thiết hạch trong một số bệnh lý:
+ U lympho không Hodgkin: Tổn thương dạng nang hoặc lan toả. Có thể chủ yếu tế bào to hoặc tế bào nhỏ hoặc hỗn hợp to, nhỏ. Căn cứ đặc điểm tế bào, mô bệnh học và miễn dịch (miễn dịch tế bào,hóa mô miễn dịch) có thể xếp loại theo WHO 2008, WHO 2016...;
+ U lympho Hodgkin: Hình ảnh tổn thương đa dạng tế bào, có gặp tế bào Reed-Sternberg hoặc các biến thể (tế bào xác ướp, tế bào bỏng ngô…). Căn cứ đặc điểm mô bệnh học có thể xếp loại U lympho Hodgkin theo Tổ chức Y tế thế giới 2008, 2016 (typ mới: Ưu thế lympho bào dạng nốt và typ cổ điển: Giàu lympho bào, Hỗn hợp tế bào, Xơ nốt, nghèo lympho bào);
+ Hạch viêm lao: Hình ảnh nang lao điển hình: Có chất hoại tử bã đậu, nhiều tế bào bán liên, tế bào Langerhangs;
+ Ung thư di căn: Trên tiêu bản sinh thiết hạch thấy các đám lớn tế bào ung thư, tế bào có kích thước lớn, nhiều tế bào nhân quái, nhân chia...;
+ Các bệnh lý khác: Bệnh mô bào, các hạch viêm...
6. LÁCH ĐỒ
6.1. Khái niệm
Là xét nghiệm chọc hút lách và làm tiêu bản nhằm quan sát thành phần tế bào trong lách giúp chẩn đoán một số bệnh.
6.2. Chỉ định
Cần cân nhắc rất kỹ do nguy cơ chảy máu ổ bụng khi chọc lách cao, chỉ định khi lách to trong bệnh sinh máu tại lách hoặc theo dõi u lympho thể lách.
6.3. Lấy mẫu xét nghiệm.
- Chọc hút bằng kim nhỏ tổ chức lách, làm tiêu bản như kỹ thuật máu đàn, nhuộm Gieemssa.
- Làm thêm tiêu bản máu ngoại vi để so sánh.
6.4. Nhận định kết quả.
- Bình thường thành phần của lách như sau:
+ Tế bào lympho: 50 - 60%;
+ Bạch cầu đoạn: 20 - 30%;
+ Tế bào liên võng: 10 - 15%.
- Trong thực tế, giá trị của lách đồ không phải dựa vào công thức đã nêu trên mà chủ yếu đánh giá có hay không có tình trạng sau:
+ Dị sản tế bào: Các tế bào chưa trưởng thành dòng hồng cầu, dòng bạch cầu hạt, có thể thấy mẫu tiểu cầu trong các bệnh tăng sinh tủy (đặc biệt trong bệnh xơ tủy).
+ Xuất hiện các tế bào bất thường:
● Tế bào Reed Sternberg (bệnh Hodgkin);
● Tế bào Gaucher (trong bệnh Gaucher);
● Tế bào ung thư (trong bệnh sarcom lách);
● Ký sinh trùng sốt rét (trong bệnh sốt rét);
● U lympho không Hodgkin...
TÀI LIỆU THAM KHẢO
1. Nguyễn Thị Minh An (2004), “Bệnh máu và cơ quan tạo máu”, Bài giảng Bệnh học Nội khoa tập 1, Nhà xuất bản Y học, tr. 111-200.
2. Trương Công Duẩn, Trần Hồng Thủy (2005),"Tế bào-Tổ chức học cơ quan tạo máu", Kỹ thuật xét nghiệm huyết học và truyền máu ứng dụng trong lâm sàng, Nhà xuất bản Y học, tr. 9-51.
3. Nguyễn Anh Trí (2004), “U lympho ác tính”, Bài giảng Huyết học-Truyền máu, Nhà xuất bản Y học, tr. 140-149.
4. Camille N. Abboud, Marshall A. Lichtman (2006), “Structure of the Marrow and the Hematopoietic Microenvironment, the Lymphoid tissues, Williams Hematology, Seventh Edition, McGraw-Hill Medical, pp. 35-80.
5. Daniel H. Ryan, Raymond E. Felgar (2006), “Examination of the Blood, Bone Marrow”, Williams Hematology, Seventh Edition, McGraw-Hill Medical, pp. 11-34.
6. Sheila L. Perkins (2004), “Examination of the Blood and Bone marrow”, Wintrobe’s Clinical Hematology, Volume 1, Lippincott Williams & Wilkins, pp. 3-26.
7. World Health Organization (2008), Classification of Tumours of Heamatopoietic and Lymphoid Tissues, Lyon 2008.
45. CHỈ ĐỊNH VÀ ĐÁNH GIÁ KẾT QUẢ MỘT SỐ XÉT NGHIỆM ĐÔNG CẦM MÁU
Để chẩn đoán chính xác các bất thường đông cầm máu, bên cạnh các thông tin về triệu chứng lâm sàng, tiền sử bản thân và gia đình người bệnh, kết quả các xét nghiệm đông cầm máu đóng vai trò rất quan trọng. Xét nghiệm đông cầm máu giúp chẩn đoán sớm, chính xác loại rối loạn, mức độ rối loạn cũng như tiến triển của các rối loạn đó. Tương ứng với các giai đoạn của quá trình đông cầm máu: Cầm máu kỳ đầu, đông máu huyết tương và tiêu sợi huyết, là các xét nghiệm để đánh giá các giai đoạn này.
Hình 1: Các giai đoạn chính của quá trình đông máu
1. ĐÁNH GIÁ GIAI ĐOẠN CẦM MÁU KỲ ĐẦU
Giai đoạn cầm máu kỳ đầu với sự tham gia của thành mạch, tiểu cầu và yếu tố von Willebrand để tạo nút tiểu cầu cầm máu. Các xét nghiệm đánh giá giai đoạn cầm máu kỳ đầu bao gồm:
- Xét nghiệm đếm số lượng tiểu cầu.
- Xét nghiệm đánh giá chức năng (chất lượng) tiểu cầu.
- Xét nghiệm đánh giá sức bền thành mao mạch.
- Xét nghiệm yếu tố von Willebrand (kháng nguyên, hoạt tính).
1.1. Các xét nghiệm tổng quát, hiện đang sử dụng rộng rãi ở các bệnh viện
1.1.1. Xét nghiệm: Thời gian máu chảy, nghiệm pháp dây thắt, co cục máu đông.
1.1.2. Chỉ định: Tất cả những trường hợp nghi ngờ có bất thường cầm máu kỳ đầu: Thiếu vitamin C, giảm số lượng và/hoặc chất lượng tiểu cầu, bệnh von Willebrand.
a. Thời gian máu chảy
- Khái niệm: Là thời gian từ khi tạo một vết thương chuẩn đến khi máu ngừng chảy từ vết thương đó.
- Đánh giá kết quả:
+ Giá trị bình thường: Tùy phương pháp, tùy từng phòng xét nghiệm, thường:
● Thời gian máu chảy theo phương pháp Duke: ≤ 5 phút;
● Thời gian máu chảy theo phương pháp Ivy: ≤ 7 phút.
+ Thời gian máu chảy đánh giá giai đoạn cầm máu kì đầu liên quan đến tiểu cầu và thành mao mạch. Thời gian máu chảy kéo dài trong các trường hợp sau:
● Giảm tiểu cầu;
● Bất thường chức năng tiểu cầu bẩm sinh hoặc mắc phải bao gồm cả các trường hợp dùng thuốc ức chế tiểu cầu như aspirin, clopidogrel...;
● Bệnh von Willebrand;
● Thiếu máu nặng: Bệnh nhân thiếu máu làm thay đổi sự phân bố của tiểu cầu và giảm sự tương tác của tiểu cầu và nội mạc, làm kéo dài thời gian máu chảy;
● Thiếu fibrinogen (hypofibrinogenemia): Fibrinogen cần thiết cho sự tương tác giữa các tiểu cầu, do vậy thời gian máu chảy kéo dài trong trường hợp hypofibrinogenemia;
● Giảm sức bền thành mạch;
● Rối loạn collagen: Hội chứng Ehlers Danlos;
b. Co cục máu đông
- Khái niệm: Là xét nghiệm được sử dụng để đánh giá khả năng co của cục máu sau khi đông.
- Đánh giá kết quả:
+ Bình thường cục máu co hoàn toàn;
+ Cục máu co không hoàn toàn hoặc bị tan trong những trường hợp giảm số lượng và/hoặc chất lượng tiểu cầu, fibrinogen, tiêu sợi huyết cấp.
c. Nghiệm pháp dây thắt
- Khái niệm: Là xét nghiệm được sử dụng để đánh giá sức bền của thành mao mạch.
- Bình thường nghiệm pháp dây thắt âm tính.
- Đặc điểm chung của các xét nghiệm tổng quát đánh giá cầm máu kỳ đầu nêu ở trên là khả năng áp dụng rộng rãi ở mọi cơ sở Y tế. Tuy nhiên, độ nhạy và độ đặc hiệu của các xét nghiệm này đều thấp, kết quả của xét nghiệm bị ảnh hưởng nhiều bởi người tiến hành kỹ thuật.
1.2. Các xét nghiệm đánh giá chức năng tiểu cầu
a. Đánh giá tổng quát chức năng tiểu cầu bằng máy tự động (Platelet Funtion Analyzer: PFA)
- Khái niệm: PFA là xét nghiệm đánh giá cả 2 khả năng dính và ngưng tập của tiểu cầu; Kết quả PFA trong giới hạn bình thường cho phép loại trừ hầu hết các bệnh lý gây bất thường chức năng tiểu cầu.
- Chỉ định: Những trường hợp nghi ngờ bệnh lý chức năng tiểu cầu bẩm sinh hoặc mắc phải, bệnh von Willebrand, nghi ngờ kháng aspirin khi điều trị thuốc này.
- Đánh giá kết quả:
+ Giá trị bình thường: Đánh giá khả năng ngưng tập và dính của tiểu cầu bằng thời gian tạo nút cầm máu tiểu cầu (closure time: CT);
● CT Collagen/ ADP: 68 - 121 giây;
v CT Collagen/ Epinephrin: 84 - 160 giây.
+ Bệnh von Willebrand, bất thường chức năng tiểu cẩu bẩm sinh như Glanzmann, Bernard soulier: Cả CT Collagen/ADP và CT Collagen/ EPI đều kéo dài;
+ Những trường hợp điều trị aspirin hoặc các chất chống viêm non-steroid: CT Collagen/ ADP bình thường, CT Collagen/ EPI kéo dài.
b. Đánh giá chức năng tiểu cầu bằng máy Verify Now
- Khái niệm: Dựa vào sự thay đổi mật độ quang của mẫu máu toàn phần khi tiểu cầu hoạt hóa bằng các chất như ADP (với bệnh nhân dùng thuốc kháng P2Y12) hoặc arachidonic (khi dùng aspirin), tiểu cầu sẽ gắn với các hạt được phủ fibrinogen, từ đó đánh giá được mức độ đáp ứng khi điều trị thuốc kháng tiểu cầu.
- Chỉ định: Đánh giá hiệu quả điều trị thuốc kháng tiểu cầu (aspirin, kháng P2Y12); Xét nghiệm trước khi can thiệp ở bệnh nhân đang dùng thuốc kháng tiểu cầu cần dừng thuốc.
- Đánh giá kết quả:
+ Verifynow Aspirin test: Đơn vị: ARU (aspirin reaction units);
< 550 ARU: Aspirin ức chế tiểu cầu.
+ Verifynow PRU test: Đơn vị PRU (P2Y12 reaction units);
< 180 PRU: P2Y12 ức chế tiểu cầu.
+ Ngoài ra còn có xét nghiệm đánh giá đáp ứng thuốc ức chế thụ thể IIb/IIIa của tiểu cầu: Verifynow IIb/IIIa test.
c. Đánh giá đáp ứng với thuốc chống ngưng tập tiểu cầu trên hệ thống Rotem sử dụng module tiểu cầu.
d. Đo độ ngưng tập tiểu cầu
- Khái niệm: Hiện tượng ngưng tập của tiểu cầu xảy ra khi cho chất kích tập như ADP, Collagen, Ristocetin, Epinephrine, Arachidonic, thrombin vào máu toàn phần hoặc huyết tương giàu tiểu cầu. Đo độ ngưng tập của tiểu cầu với những chất gây ngưng tập khác nhau giúp đánh giá chức năng của tiểu cầu. Có 2 phương pháp đo chính: quang học (sử dụng huyết tương giàu tiểu cầu) và trở kháng (sử dụng máu toàn phần).
- Chỉ định: Tất cả những trường hợp nghi ngờ giảm hoặc tăng ngưng tập tiểu cầu: Bệnh lý chức năng tiểu cầu, bệnh von Willebrand, huyết khối động mạch, nghi ngờ kháng aspirin.
- Đánh giá kết quả:
+ Giá trị bình thường: Giá trị này khác nhau giữa các phòng xét nghiệm, phụ thuộc loại máy tiến hành kỹ thuật, loại mẫu (máu toàn phần hay huyết tương giàu tiểu cầu), nồng độ chất kích tập...;
+ Độ ngưng tập tiểu cầu tăng: Thể hiện tình trạng tiểu cầu tăng hoạt hóa, nguy cơ huyết khối;
+ Độ ngưng tập tiểu cầu giảm: Tùy theo loại chất gây ngưng tập để chẩn đoán bệnh lý suy giảm chức năng tiểu. Ví dụ: Hội chứng Bernard Soulier ngưng tập với ristocetin giảm, bệnh Glanzmann giảm ngưng tập với ADP và collagen, axit arachidonic.
e. Các xét nghiệm khác đánh giá chức năng tiểu cầu
- Định lượng nucleotide tiểu cầu.
- Glycoprotein trên màng tiểu cầu.
- Đo độ dính tiểu cầu.
- Rotem tiểu cầu.
1.3. Xét nghiệm yếu tố von Willebrand huyết tương
Yếu tố von Willebrand (vWF) là cầu nối giúp tiểu cầu có thể gắn với collagen thành mạch, vì vậy thiếu hoặc bất thường chức năng của v WF có thể dẫn đến tiểu cầu không dính vào collagen thành mạch tổn thương. Xét nghiệm yếu tố von Willebrand cần được chỉ định ở những bệnh nhân nghi ngờ có bất thường giai đoạn cầm máu kỳ đầu. Chú ý ở những người có nhóm máu O, nồng độ v WF thường thấp hơn so với những nhóm máu khác, do đó cần cân nhắc trong chẩn đoán.
Xét nghiệm v WF bao gồm:
- Định lượng kháng nguyên yếu tố von Willebrand.
- Định lượng hoạt tính von Willebrand đồng yếu tố ristocetin.
- Định lượng hoạt tính von Willebrand gắn collagen (collagen binding).
- Định lượng von Willebrand multimers (ý nghĩa trong phân loại thể bệnh).
2. ĐÁNH GIÁ GIAI ĐOẠN ĐÔNG MÁU HUYẾT TƯƠNG
2.1. Các xét nghiệm vòng đầu (first - line tests)
2.1.1. Xét nghiệm: APTT, PT, TT, fibrinogen, số lượng tiểu cầu.
2.1.2. Chỉ định:
- Xét nghiệm trước khi can thiệp thủ thuật, phẫu thuật.
- Sàng lọc các bất thường đông cầm máu và theo dõi điều trị (do thuốc, hóa chất, truyền chế phẩm, trao đổi huyết tương…).
a. PT (Prothrombin Time: Thời gian prothrombin); Tên khác: Thời gian Quick, tỷ lệ prothrombin
- Khái niệm: PT là xét nghiệm đánh giá đường đông máu ngoại sinh; Thời gian prothrombin kéo dài khi giảm hoặc có chất ức chế các yếu tố II, V, VII, X và giảm nặng fibrinogen.
- Giá trị bình thường:
+ Thời gian: Thông thường trong khoảng 11-13 giây (tùy theo chứng thực tế khi làm xét nghiệm của từng labo), kéo dài khi PT bệnh dài hơn PT chứng 3 giây;
+ Tỷ lệ %: 70 - 140%, giảm khi < 70%;
+ INR: 0,8 - 1,2; INR là chỉ số được sử dụng để theo dõi điều trị chống đông loại kháng vitamin K.
- PT kéo dài (PT % giảm):
+ Thiếu yếu tố đông máu liên quan đến đường đông máu ngoại sinh do bệnh bẩm sinh hoặc mắc phải (DIC, suy gan, truyền máu khối lượng lớn), thiếu vitamin K, dùng thuốc chống đông nhóm kháng vitamin K...;
+ Có chất ức chế đông máu con đường ngoại sinh trong các bệnh tự miễn, nhiễm trùng.
- PT ngắn trong trường hợp điều trị rFVIIa, tăng đông.
b. APTT (Activated Partial Thromboplastin Time: Thời gian thromboplastin một phần hoạt hóa); Tên khác: TCK (Temps de Céphaline Kaolin: Thời gian Céphaline Kaolin)
- Khái niệm: APTT là xét nghiệm đánh giá đường đông máu nội sinh; APTT kéo dài khi giảm hoặc có chất ức chế các yếu tố II, V, VIII, IX, X, XI, XII và giảm nặng fibrinogen.
- Giá trị bình thường:
+ Thời gian: 25 - 35 giây;
+ Chỉ số rAPTT: 0,85 - 1,25 (rAPTT = APTT bệnh/APTTchứng).
- APTT kéo dài khi rAPTT > 1,25. Thường gặp trong các bệnh lý rối loạn đông máu do thiếu hụt yếu tố tham gia đường đông máu nội sinh (bệnh Hemophilia, Von Willebrand…) hoặc do có chất kháng đông lưu hành (ức chế yếu tố VIII, yếu tố IX, Kháng đông Lupus), người bệnh được điều trị Heparin hoặc NOACs...
c. Thời gian thrombin (TT)
- Khái niệm: Là thời gian đông của huyết tương khi cho thêm thombin; Xét nghiệm này đánh giá giai đoạn chuyển fibrinogen thành fibrin dưới tác dụng của thrombin.
- Giá trị bình thường:
+ Thời gian: 12 - 15 giây;
+ Chỉ số rTT: 0,85 - 1,25 (rTT = TT bệnh/TTchứng);
+ TT kéo dài khi rTT > 1,25. Gặp trong các trường hợp giảm nặng fibrinogen (<1g/L), như:
● Thiếu fibrinogen bẩm sinh: Thiếu hoặc không có fibrinogen; Rối loạn chức năng fibrinogen;
● Thiếu fibrinogen mắc phải: DIC, bệnh gan, điều trị thuốc tiêu sợi huyết;
● Tăng sản phẩm thoái giáng của fibrin (fibrinogen): D-dimer, FDPs;
● Điều trị thuốc chống đông: Thuốc ức chế trực tiếp thrombin (DTIs), heparin;
● Một số trường hợp tăng globulin, tăng fibrinogen cũng làm TT kéo dài.
d. Định lượng fibrinogen bằng phương pháp Clauss
- Khái niệm: Có nhiều phương pháp định lượng fibrinogen. Phương pháp Clauss đánh giá nồng độ fibrinogen có hoạt tính đông máu qua thời gian đông của huyết tương khi cho thêm thrombin nồng độ cao. So với phương pháp nội suy qua PT, phương pháp này có độ chính xác cao hơn và không bị ảnh hưởng của thuốc chống đông kháng vitamin K, thuốc tiêu sợi huyết và một số tình trạng bệnh lý như đông máu rải rác trong lòng mạch (DIC), bệnh gan…
- Giá trị bình thường: 2 - 4g/L.
- Khi fibrinogen < 1g/L, nguy cơ chảy máu cao. Fibrinogen giảm thường gặp trong các trường hợp:
+ Suy giảm chức năng gan;
+ DIC dẫn đến tiêu thụ các yếu tố đông máu;
+ Bệnh lý bất thường fibrinogen bẩm sinh: Thiếu hụt (Hypofibrinogenaemia) hoặc không có (afibrinogenaemia) và rối loạn chức năng fibrinogen (dysfibrinogenaemia);
+ Tiêu sợi huyết;
+ Truyền máu khối lượng lớn dẫn tới hòa loãng các yếu tố đông máu;
+ Điều trị thuốc tiêu sợi huyết, một số trường hợp điều trị L-asparaginase.
- Fibrinogen tăng trong tình trạng nhiễm trùng, bệnh lý tim mạch, phụ nữ có thai, dùng thuốc tránh thai…
2.1.3. Đánh giá kết quả xét nghiệm vòng đầu
- Người bệnh không có triệu chứng lâm sàng, không có tiền sử về rối loạn đông cầm máu, kết quả các xét nghiệm vòng đầu đều trong giới hạn bình thường: Ở thời điểm đó không nghĩ đến bất thường của hệ thống đông cầm máu.
- Người bệnh có triệu chứng lâm sàng và/hoặc có tiền sử chảy máu nhưng kết quả các xét nghiệm trong giới hạn bình thường. Trong thực tế lâm sàng, nhóm các bất thường này có thể gặp, dù với tỷ lệ thấp và thường do các bất thường hiếm gặp như: Thiếu yếu tố XIII, giảm nhẹ yếu tố đông máu, ban xuất huyết (Henoch-Schönlein purpura: HSP), giảm chức năng tiểu cầu, xuất huyết do thành mạch, xuất huyết không rõ nguyên nhân.
- Người bệnh có/ hoặc không có triệu chứng lâm sàng, có/ hoặc không có tiền sử chảy máu, kết quả xét nghiệm bất thường:
- Tùy theo xét nghiệm bất thường để hướng đến các loại rối loạn thường gặp, cụ thể:
+ PT kéo dài (PT% giảm) các xét nghiệm còn lại đều bình thường:
● Thiếu yếu tố VII;
● Suy gan;
● Giai đoạn đầu điều trị kháng vitamin K...;
● Có chất ức chế đông máu ngoại sinh.
+ APTT kéo dài, các xét nghiệm còn lại đều bình thường:
● Thiếu yếu tố VIII, IX, XI, XII, von Willebrand;
● Có chất ức chế đông máu đường nội sinh (thường gặp: Lupus anticoagulant)...
+ PT và APTT kéo dài, các xét nghiệm còn lại bình thường:
● Suy gan nặng;
● Thiếu vitamin K;
● Điều trị kháng vitamin K;
● Thiếu yếu tố V kết hợp thiếu yếu tố VIII;
● Thiếu yếu tố II kết hợp thiếu yếu tố V và yếu tố X;
● Dùng NOACs, heparin.
+ PT, APTT và TT đều kéo dài, fibrinogen bình thường hoặc giảm, số lượng tiểu cầu bình thường:
● Suy gan nặng;
● Điều trị heparin hoặc mẫu máu bị nhiễm heparin;
● Ức chế trùng hợp fibrin;
● Tiêu sợi huyết;
● Thiếu fibrinogen bẩm sinh.
+ Số lượng tiểu cầu giảm, các xét nghiệm còn lại bình thường: Giảm tiểu cầu;
+ TT bình thường, fibrinogen bình thường hoặc giảm, PT và APTT đều kéo dài, số lượng tiểu cầu giảm:
● Truyền máu khối lượng lớn;
● Suy gan.
+ Tất cả các xét nghiệm đều bất thường: PT, APTT, TT đều kéo dài, fibrinogen và số lượng tiểu cầu giảm:
● DIC;
● Suy gan cấp...
2.2. Các xét nghiệm chuyên sâu
2.2.1. Xét nghiệm phát hiện sự có mặt của chất ức chế (mix test)
a. Khái niệm
Đây là xét nghiệm nhằm mục đích xác định thời gian đông kéo dài bất thường do thiếu hụt yếu tố đông máu hay do có mặt chất ức chế; thông thường, các xét nghiệm đông máu vẫn trong giới hạn bình thường ngay cả khi nồng độ các yếu tố chỉ đạt 50%. Vì vậy, khi trộn huyết tương người bệnh với huyết tương bình thường theo tỷ lệ 1:1, thời gian đông sẽ trở về bình thường nếu thiếu hụt yếu tố đông máu và thời gian đông không về bình thường trong trường hợp có chất ức chế quá trình đông máu. Tùy theo xét nghiệm nào kéo dài (PT, APTT hay TT) mà chỉ định tiến hành mix test bằng xét nghiệm đó.
b. Đánh giá kết quả
- Thời gian đông của mẫu trộn huyết tương bệnh với huyết tương chứng điều chỉnh về bình thường: Thiếu hụt yếu tố đông máu; Trong trường hợp này, cần tiến hành tiếp xét nghiệm định lượng riêng rẽ các yếu tố đông máu.
- Thời gian đông của mẫu trộn huyết tương bệnh với huyết tương chứng không điều chỉnh về bình thường: Có chất ức chế đông máu. Nếu APTT kéo dài, cần tiến hành mix test ở 2 điều kiện: Tiến hành ngay sau khi trộn và tiến hành sau 2 giờ ủ ở bình cách thủy 37°C để xác định chất ức chế thuộc nhóm tác dụng ngay (thường là lupus anticoagulant) hay tác dụng phụ thuộc thời gian, nhiệt độ (thường là chất ức chế yếu tố VIII). Căn cứ vào các tình huống có thể đánh giá do thiếu hụt yếu tố đông máu hay do chất ức chế (Bảng 58).
Bảng 58. Tóm tắt đánh giá kết quả xét nghiệm mix test
|
Tình huống |
|
|
|
|
Huyết tương bình thường |
Bình thường |
Bình thường |
Bình thường |
|
Huyết tương bệnh |
Kéo dài |
Kéo dài |
Kéo dài |
|
Huyết tương bình thường + bệnh, ủ 2 giờ ở 37ºC |
Bình thường |
Kéo dài |
Kéo dài |
|
Huyết tương bình thường + bệnh, không ủ |
Bình thường |
Bình thường |
Kéo dài |
|
Đánh giá |
Thiếu hụt yếu tố |
Có chất ức chế phụ thuộc thời gian, nhiệt độ |
Có chất ức chế tác dụng ngay. |
2.2.2. Định lượng hoạt tính yếu tố đông máu
a. Khái niệm
Xét nghiệm này được sử dụng để đánh giá chính xác nồng độ hoạt tính của từng yếu tố đông máu riêng biệt, nhằm mục đích chẩn đoán chính xác loại bệnh lý, mức độ bệnh lý rối loạn đông máu: Xét nghiệm hoạt tính yếu tố đông máu được chỉ định khi kết quả của xét nghiệm mix test cho thấy thời gian đông của mẫu huyết tương trộn 1 thể tích huyết tương bệnh với 1 thể tích huyết tương chứng điều chỉnh về bình thường. Tùy vào thời gian đông của xét nghiệm nào kéo dài để chỉ định yếu tố cần định lượng phù hợp:
- APTT kéo dài trong khi PT và TT bình thường: Định lượng yếu tố VIII, IX, XI, XII;
- PT kéo dài trong khi APTT và TT bình thường: Định lượng yếu tố VII;
- Cả APTT và PT kéo dài: Định lượng các yếu tố II, V, VII, IX, X.
b. Đánh giá kết quả
So sánh kết quả thu được với giá trị bình thường (phụ thuộc kỹ thuật tiến hành, loại sinh phẩm,... của phòng xét nghiệm) để nhận định hoạt tính yếu tố bình thường hay tăng, giảm.
2.2.3. Phát hiện sự có mặt của chất ức chế đông máu dạng lupus (Lupus Anticoagulant: LA) và định lượng các kháng thể kháng phospholipid.
a. Khái niệm
LA là chất ức chế đông máu có đặc điểm làm kéo dài thời gian đông của những xét nghiệm có sử dụng phospholipid nhưng lại gây huyết khối tắc mạch trên lâm sàng.
b. Chỉ định: Thông thường, xét nghiệm này được chỉ định khi kết quả của xét nghiệm mix test cho thấy có chất ức chế tác dụng ngay.
- Chỉ định trong trường hợp nghi ngờ hội chứng anti phospholipid, bệnh tự miễn.
c. Đánh giá kết quả
LA dương tính gặp trong các bệnh lý tự miễn như thiếu máu tan máu tự miễn, lupus ban đỏ, hội chứng anti-phospholipid, nhiễm trùng, ung thư…
2.2.4. Phát hiện sự có mặt của chất ức chế yếu tố VIII
a. Khái niệm
Ức chế yếu tố VIII thường xuất hiện ở những người bệnh Hemophilia A di truyền thể nặng đã được điều trị thay thế hoặc những người bệnh Hemophilia mắc phải, ức chế yếu tố VIII tác động phụ thuộc thời gian và nhiệt độ.
b. Chỉ định
Tất cả những trường hợp nghi ngờ có ức chế VIII, người bệnh Hemophilia A di truyền điều trị không hiệu quả, người bệnh nghi ngờ Hemophilia A mắc phải, xét nghiệm mix test có chất ức chế phụ thuộc thời gian nhiệt độ.
c. Đánh giá kết quả
Nồng độ ức chế VIII được tính bằng đơn vị Bethesda: 1 đơn vị Bethesda là lượng chất ức chế yếu tố VIII trung hoà 50% yếu tố VIII trong thời gian 2 giờ ở điều kiện 37ºC.
475
3. ĐÁNH GIÁ TÌNH TRẠNG TIÊU SỢI HUYẾT
Xét nghiệm được sử dụng rộng rãi hiện nay ở Việt Nam là nghiệm pháp Von-Kaulla, định lượng D-Dimer và Rotem/TEG.
3.1. Nghiệm phip Von-kaulla
3.1.1. Khái niệm
Sử dụng kỹ thuật pha loãng và axit hóa huyết tương sau đó tiến hành ly tâm mạnh để loại bỏ các chất ức chế, thu nhận chất hoạt hóa quá trình tiêu sợi huyết và nhờ vậy sẽ bộc lộ tình trạng tăng tiêu sợi huyết ở người bệnh. Nghiệm pháp Von-Kaulla là xét nghiệm đơn giản, không cần máy móc thiết bị nên có khả năng áp dụng rộng rãi ở các bệnh viện, nhưng độ nhạy không cao.
3.1.2. Chỉ định
Đánh giá tình trạng tiêu sợi huyết (tiên phát, thứ phát).
3.1.3. Đánh giá kết quả
- Bình thường: Nghiệm pháp Von-Kaulla âm tính (thời gian tan cục đông > 60 phút);
- Khi thời gian tan cục đông < 60 phút: Kết luận dương tính; Tùy theo thời gian tan cục đông, đánh giá mức độ tiêu sợi huyết:
+ Tan trước 15 phút: Tiêu sợi huyết tối cấp;
+ Tan trong vòng 15 đến 30 phút: Tiêu sợi huyết cấp;
+ Tan trong vòng 30 đến 45 phút: Tiêu sợi huyết bán cấp;
+ Tan trong vòng 45 đến 60 phút: Tiêu sợi huyết tiềm tàng.
3.2. Đӏnh lượng D-Dimer
3.2.1. Khái niệm
D-Dimer là sản phẩm thoái giáng của fibrin dưới tác dụng của plasmin, nồng độ D-Dimer tăng phản ánh tình trạng tăng tiêu sợi huyết. Hiện nay, có rất nhiều phương pháp để tiến hành xét nghiệm này.
3.2.2. Chỉ định
Những trường hợp nghi ngờ tiêu sợi huyết, DIC, tăng đông huyết khối (ung thư, nhiễm trùng, virus, có thai…), theo dõi điều trị rối loạn đông máu (do bệnh nền hoặc dùng thuốc).
3.2.3. Đánh giá kết quả
- Giá trị bình thường: < 500ng/ml (hoặc < 0,5 µg/ml) FEU hoặc < 250ng/ml DDU (giá trị này có thể thay đổi tùy theo phương pháp, hóa chất sinh phẩm… của từng phòng xét nghiệm).
- D-Dimer tăng: Nhiễm khuẩn, DIC, tiêu sợi huyết tiên phát, huyết khối, điều trị thuốc tiêu cục đông, có thai, người cao tuổi...
- D-Dimer không tăng: Loại trừ huyết khối.
3.3. Một số xét nghiệm khác đánh giá hoạt động của hệ thống tiêu sợi huyết
- PIC: Plasmin - α plasmin inhibitor complex: Là xét nghiệm đánh giá gián tiếp sự hình thành plasmin. Trong một số nghiên cứu các tác giả nhận thấy rằng ở bệnh nhân DIC thường có PIC tăng, điều đó phản ánh có sự tăng hoạt động của hệ thống tiêu sợi huyết, đặc biệt ở nhóm DIC thể ưu thế tiêu sợi huyết.
- PAI - 1 (plasminogen activator inhibitor): Ức chế hoạt hóa plasminogen.
- Alpha 2 antiplasmin.
- Plasminogen.
4. ĐÁNH GIÁ TÌNH TRẠNG TĂNG ĐÔNG, HUYẾT KHỐI
4.1. Đánh giá nồng độ các chất ức chế đông máu sinh lý
4.1.1. Khái niệm
Những chất ức chế đông máu sinh lý bao gồm antithrombin/antithrombin III (AT/ATIII), protein S (PS), protein C (PC); thiếu hụt chất ức chế đông máu sinh lý là nguyên nhân gây tăng đông huyết khối.
4.1.2. Chỉ định
Những trường hợp huyết khối nghi ngờ do thiếu hụt chất ức chế đông máu sinh lý bẩm sinh (huyết khối khi còn trẻ, tái phát, có tính chất gia đình) hoặc mắc phải (xơ gan, DIC…).
4.1.3. Đánh giá kết quả
So sánh kết quả thu được với giá trị bình thường (phụ thuộc kỹ thuật tiến hành, loại sinh phẩm của phòng xét nghiệm) để nhận định.
- Nồng độ antithrombin giảm trong các trường hợp: Thiếu hụt antithrombin bẩm sinh, mắc phải (DIC, xơ gan...).
- Nồng độ PS, PC giảm trong những trường hợp: Thiếu hụt PS, PC bẩm sinh, thiếu hụt PS, PS mắc phải (tình trạng nhiễm trùng, xơ gan, điều trị kháng vitamin K…).
4.2. Xét nghiệm phit hiện fibrin monomer
Khi một lượng lớn thrombin tác động lên fibrinogen, rất nhiều fibrin monomer được tạo thành, và như vậy sự tồn tại các fibrin monomer trong máu gián tiếp cho thấy tình trạng tăng tạo thrombin - tăng hoạt hóa đông máu.
4.2.1. Nghiệm pháp rượu
a. Khái niệm
Các fibrin monomer trong máu sẽ tạo phức hệ với fibrinogen hoặc sản phẩm thoái giáng của fibrinogen: Nghiệm pháp rượu là xét nghiệm được sử dụng để phát hiện các phức hệ này, qua đó phát hiện tình trạng tăng hoạt hóa đông máu.
b. Chỉ định: Những trường hợp nghi ngờ DIC, tăng đông huyết khối (ung thư, nhiễm trùng, virus, có thai…), theo dõi điều trị rối loạn đông máu (do bệnh nền hoặc dùng thuốc).
c. Đánh giá kết quả
- Bình thường: Âm tính.
- Mức độ dương tính: +++, ++ hay +, phản ánh mức độ hoạt hóa đông máu mạnh hay yếu.
Nghiệm pháp rượu dương tính chứng tỏ hệ thống đông máu đang hoạt hóa mạnh, thường gặp trong DIC giai đoạn còn bù (non-overt DIC).
4.2.2. Định lượng fibrin monomer hòa tan (soluble fibrin monomer: SFM/FM)
- Khái niệm: Sử dụng phản ứng miễn dịch ngưng kết latex để phát hiện/ hoặc đánh giá nồng độ monomer fibrin, qua đó phát hiện tình trạng tăng hoạt hóa đông máu.
- Xét nghiệm định lượng monomer fibrin có độ nhạy và độ đặc hiệu cao hơn nghiệm pháp rượu, là 1 dấu ấn trong chẩn đoán, theo dõi DIC và tình trạng tăng đông.
- Chỉ định: Những trường hợp nghi ngờ DIC, tăng đông huyết khối (ung thư, nhiễm trùng, virus, có thai…), theo dõi điều trị rối loạn đông máu (do bệnh nền hoặc dùng thuốc).
4.3. Một số xét nghiệm khác trong đánh giá tình trạng tăng đông, huyết khối
- Xét nghiệm phát hiện kháng protein C hoạt hóa, hay gặp trong bệnh nhân có đột biến V- Leiden.
- Đột biến gene Prothrombin G20210A.
- Nồng độ homocystein.
- Nồng độ yếu tố đông máu.
- Nồng độ chất ức chế hoạt hóa plasminogen: PAI (Plasminogen activator inhibitor).
5. XÉT NGHIỆM ĐÁNH GIÁ TỔNG QUÁT VÀ ĐỘNG HỌC QUÁ TRÌNH ĐÔNG MÁU
5.1. Đàn hồi đồ cục máu (ThromboElastography: TEG)
a. Khái niệm
TEG là một trong những xét nghiệm được sử dụng để đánh giá nhanh, tổng thể cũng như động học của toàn bộ quá trình hình thành cục đông. Kết quả TEG giúp chỉ định đúng loại chế phẩm máu cần truyền trong những trường hợp chảy máu sau phẫu thuật (nhất là phẫu tim mạch) và sau chấn thương.
b. Đánh giá kết quả
6 chỉ số thông dụng được sử dụng khi nhận định kết quả TEG: CI, R, K, góc alpha, MA, Lys 30.
Bảng 59. Các chỉ số của xét nghiệm TEG
|
Chỉ số |
Giá trị bình thường |
Đánh giá kết quả |
|
CI (Coagulation Index): Chỉ số đông. |
Từ -3 đến + 3 |
< -3: Giảm đông; > +3: Tăng đông. |
|
R (Reaction time): thời gian phản ứng. |
Từ 2 đến 8 phút |
< 2: Tăng nồng độ yếu tố đông máu; > 8: Thiếu hụt hoặc có chất ức chế yếu tố đông máu. |
|
K (Clot kinetic): động học hình thành cục đông. |
Từ 1 đến 3 phút |
< 1: Tăng nồng độ yếu tố đông máu, tăng số lượng tiểu cầu; > 3: Giảm nồng độ yếu tố đông máu, giảm nặng số lượng tiểu cầu. |
|
Góc α |
Từ 55º đến 78º |
> 78º: Tăng nồng độ yếu tố đông máu, tăng số lượng tiểu cầu; < 55º: Giảm nồng độ yếu tố đông máu, giảm số lượng tiểu cầu. |
|
MA (Maximum Amplitude): biên độ tối đa. |
Từ 51 đến 69 mm |
< 51mm: Giảm số lượng và/hoặc chất lượng tiểu cầu, fibrinogen; > 69 mm: Tăng số lượng và/hoặc chất lượng tiểu cầu, fibrinogen. |
|
Lys 30 (Lysis at 30 minutes): tiêu cục đông ở thời điểm 30 phút sau khi có MA. |
Từ 0 đến 8% |
> 8%: Tiêu sợi huyết nặng. |
5.2. Động học đàn hồi cục máu ROTEM (Rotational Thromboelastometry)
a. Khái niệm
Là xét nghiệm ghi lại sự thay đổi động học quá trình đông máu của máu tĩnh mạch đã được chống đông bằng citrat thông qua hệ thống cup (chứa mẫu máu với sự có mặt của các chất hoạt hóa hoặc ức chế) và pin (ghi nhận hiện tượng đông máu). Các biến đổi động học được ghi lại thông qua hệ thống quang học và kết quả được biểu diễn bằng đồ thị và các chỉ số.
Các loại xét nghiệm: INTEM, EXTEM, FIBTEM, APTEM, HEPTEM:
- INTEM: Phân tích con đường đông máu nội sinh.
- EXTEM: Đánh giá con đường đông máu ngoại sinh.
- FIBTEM: Loại bỏ tiểu cầu, đánh giá vai trò của sợi huyết.
- APTEM: Ức chế tiêu sợi huyết, phát hiện tình trạng tiêu sợi huyết.
- HEPTEM: Sử dụng heparinase để trung hòa heparin.
b. Phân tích kết quả dựa vào các chỉ số: CT, CFT, A, MCF, ML, LY, TPI
|
Chỉ số |
Định nghĩa và ý nghĩa |
|
CTs |
Thời gian tính từ khi bắt đầu đến khi biên độ cục máu đạt 2mm CT kéo dài khi có thiếu yếu tố đông máu hoặc có chất ức chế đông máu |
|
CFTs |
Thời gian từ khi cục máu đạt từ 2mm đến 20mm CFT đánh giá vai trò của tiểu cầu, fibrinogen |
|
Góc Alpha |
Là góc được xác định giữa trục trục X và đường tiếp tuyến với đường cong đông máu qua điểm có biên độ 2mm. Mô tả động học của quá trình đông máu |
|
MCFs (mm) |
Biên độ tối đa của cục máu đạt được Phản ánh độ vững và chất lượng của cục máu đông Sự tham gia của tiểu cầu, sợi huyết và yếu tố XIII |
|
A(x) (mm) |
Biên độ của cục máu đông đạt được x (phút) sau CT |
|
LI30 (%) |
Chỉ số thể hiện mức độ tiêu fibrin tại thời điểm 30 phút sau CT (% là biên độ cục máu còn lại so với MCF) |
|
ML (%) |
ML là chỉ số ly giải tối đa, thể hiện tỷ lệ tiêu fibrin tối đa (% biên độ cục máu bị suy giảm so với MCF. |
c. Chỉ định
- Là công cụ chẩn đoán nhanh tình trạng rối loạn đông máu đặc biệt trong cấp cứu, sản khoa, ngoại khoa... và đưa ra hướng dẫn truyền chế phẩm trong vòng 15 phút.
- Đánh giá tình trạng đông máu tổng thể trong một số trường hợp như: tăng đông huyết khối, DIC, tiêu sợi huyết, xuất huyết, bệnh lý gan, ung thư, chấn thương...
- Phối hợp cùng các xét nghiệm khác trong chẩn đoán và điều trị các rối loạn đông máu.
6. XÉT NGHIỆM ĐÔNG MÁU KHI ĐIỀU TRỊ THUỐC CHỐNG ĐÔNG
6.1. Điều trị khing vitamin K
- Xét nghiệm cần tiến hành: PT, sử dụng chỉ số INR (International Normalized Ratio: chỉ số bình thường hóa quốc tế) của PT để điều chỉnh liều của thuốc;
- Giá trị cần đạt: Tùy loại bệnh lý, thường INR trong khoảng 2,0 - 3,0. Người bệnh có nguy cơ chảy máu cao khi INR > 5.
- Thời điểm lấy máu làm xét nghiệm:
+ Tuần đầu: 2 ngày/lần;
+ Tiếp theo: 1 tuần/lần trong tháng đầu;
+ Tháng thứ 2: 2 tuần/lần;
+ Từ tháng thứ 3: 1 tháng/lần;
+ Bất kỳ thời điểm nào khi bệnh nhân có dấu hiệu chảy máu.
6.2. Điều trị heparin
6.2.1. Heparin tiêu chuẩn: Các xét nghiệm cần tiến hành:
- Thời điểm lấy máu làm xét nghiệm:
+ Tiêm tĩnh mạch ngắt quãng: 30 phút trước mũi tiêm tiếp theo;
+ Truyền tĩnh mạch liên tục: Bất kỳ thời điểm nào trong quá trình truyền heparin.
a. APTT: Sử dụng chỉ số rAPTT (rAPTT= APTT bệnh /APTT chứng);
- Giá trị cần đạt: rAPTT trong khoảng 2,5 - 3,5.
b. Xét nghiệm anti Xa:
Cần theo dõi trong 1 số trường hợp sau:
- Bệnh nhân có bệnh lý nền APTT kéo dài ví dụ APS, Hemophilia.
- Điều trị heparin.
- Trẻ em dưới 12 tháng tuổi.
- Giá trị cần đạt nồng độ anti Xa trong khoảng 0,35 - 0,7 UI/ml (liều dùng trong điều trị); Anti Xa 0,1 - 0,4UI/ml (liều dự phòng huyết khối).
c. Kiểm tra số lượng tiểu cầu: 2 lần/ tuần.
d. Xét nghiệm phát hiện giảm tiểu cầu do heparin (Heparin Induced Thrombocytopenia: HIT): Khi số lượng tiểu cầu giảm 50% so với mức nền (trước điều trị).
e. Thời gian máu đông hoạt hóa (Activated Clotting Time: ACT): Đây là xét nghiệm tiến hành ngay tại giường bệnh hay phòng mổ, thực hiện tại các thời điểm trong và ngay sau quá trình phẫu thuật tim mạch, chạy thận nhân tạo…
- Kết quả ACT trong giới hạn bình thường (khoảng 80 - 160 giây) thể hiện trong máu người bệnh không có heparin tiêu chuẩn, thường là kết quả của việc trung hòa bằng protamine.
- Cần lưu ý kết quả xét nghiệm này bị ảnh hưởng bởi rất nhiều yếu tố: Độ pha loãng của máu, nhiệt độ, giảm số lượng tiểu cầu của người bệnh.
6.2.2. Heparin trọng lượng phân tử thấp: Các xét nghiệm cần tiến hành:
a. Định lượng anti Xa:
- Chỉ định anti Xa trong những trường hợp sau: Suy thận, bệnh nhân chảy máu hoặc có nguy cơ chảy máu cao, có thai, người già, béo, chỉnh liều điều trị.
- Giá trị cần đạt: Nồng độ anti Xa trong khoảng 0,5 - 1UI/ml với liều điều trị huyết khối và 0,2 - 0,4 UI/ml với liều điều trị dự phòng.
- Thời điểm lấy máu làm xét nghiệm: 4 - 6 giờ sau tiêm dưới da để đánh giá thời điểm heparin đạt nồng độ cao nhất.
b. Kiểm tra số lượng tiểu cầu: 2 lần/ tuần.
c. Xét nghiệm phát hiện giảm tiểu cầu do heparin (HIT) khi số lượng tiểu cầu giảm 50% so với mức nền (trước điều trị).
6.3. Điều trị các thuốc kháng tiểu cầu (antiplatelet therapy)
Mục đích chính của sử dụng xét nghiệm khi điều trị các thuốc kháng tiểu cầu là phát hiện tình trạng kháng thuốc, điều trị không hiệu quả;
Các xét nghiệm được sử dụng để đánh giá đáp ứng điều trị thuốc kháng tiểu cầu hiện nay:
- Ngưng tập tiểu cầu;
- PFA;
- Rotem tiểu cầu: ARATEM: với bệnh nhân điều trị aspirin, ADP TEM (điều trị kháng P2Y12), TRAPTEM (điều trị thuốc ức chế thụ thể IIb /IIIa);
- Verifynow với các kit cho bệnh nhân điều trị aspirin, kháng P2Y12 hoặc ức chế GPIIb/IIIa.
6.4. Các thuốc chống đông đường uống thế hệ mới
Hiện nay, NOACs ngày càng được sử dụng rộng rãi như anti IIa (dabigatran...), anti Xa (rivaroxaban..). Mục đích xét nghiệm khi điều trị thuốc là phát hiện tình trạng điều trị không hiệu quả, kháng thuốc hoặc quá liều để có điều chỉnh phù hợp.
- Với thuốc kháng Xa:
+ Xét nghiệm PT, APTT;
+ Xét nghiệm anti Xa với thuốc thử đặc hiệu.
- Với thuốc kháng IIa:
+ Xét nghiệm TT: Có sự có mặt của thuốc hay không;
+ Xét nghiệm PT, APTT;
+ Xét nghiệm diluted thrombin time;
+ Eucarin clotting assay (ECA).
TÀI LIỆU THAM KHẢO
1. Nguyễn Anh Trí (2008), Đông máu ứng dụng trong lâm sàng, NXB Y học.
2. Dacie and Lewis (2011), Investigation of haemostasis, Practical Haematology, Churchill Livingstone, 11th Edi, p. 392-446.
3. Dacie and Lewis (2011), Laboratory control of anticoagulant, thrombolytic and antiplatelet therapy, Practical Haematology, Churchill Livingstone, 11th Edi, p. 466-482.
4. Kitchen and Makris (2009), Laboratory tests of hemostasis, Practical Hemostasis and Thrombosis, Wiley-Blackwell scientific publication, 2th Edi,p.7-17.
5. Uri S., Kenneth K. (2010), Classification, Clinical Manifestations, and Evaluation of disorders of Hemostasis, Williams Hematology, 8th Edi, p. 1825-1846.
6. Neha Singh, Hara Prasad Pati et all (2107), Evaluation of the diagnostic performance of fibrin monomer in comparison to D- Dimer in patient with overt and novonert disseminated intravascular coagulation. Clinical and applied Thrombosis/Hemostasis 2017, Vol. 23 (5) 460-465.
7. Vries MJ, van der Meijden PE, Kuiper GJ (2018), Preoperative screening for bleeding disorders: a comprehensive laboratory assessment of clinical pratice. Res Pract Thromb Haemost. 2018, Jul 27;2(4):767-777.
46. CHỈ ĐỊNH MỘT SỐ XÉT NGHIỆM MIỄN DỊCH HUYẾT HỌC
1. XÉT NGHIỆM ĐỊNH NHÓM HLA (HLA typing)
1.1. Khái niệm
Là xác định kiểu hình kháng nguyên bạch cầu (HLA) của một người. HLA có vai trò quan trọng trong ghép tạng và ghép tế bào gốc. Trong ghép, sự khác biệt về HLA giữa người cho và người nhận là nguồn gốc của phản ứng thải ghép hoặc ghép chống chủ (trong trường hợp ghép tế bào gốc). Các locus HLA quan trọng hay được xét nghiệm là lớp I: HLA-A, HLA-B, HLA-C; lớp II: HLA-DR, HLA-DQ, HLA-DP.
1.2. Chỉ định xét nghiệm định nhóm HLA
a. Ghép tạng (ghép gan, ghép thận, ghép tim...)
- Chỉ định xét nghiệm cho cả người cho và cả người nhận.
- Chỉ cần xác định 3 locus HLA là HLA-A, HLA-B, HLA-DR.
- Chỉ cần làm với kỹ thuật xác định ở độ phân giải thấp hoặc trung bình là đủ.
- Việc chỉ định xét nghiệm có thể tiến hành dưới 2 hình thức: Chỉ định cho từng cһp người cho và người nhận cụ thể; hoặc chỉ định xét nghiệm cho người bệnh có nhu cầu ghép (mục đích quản lý danh sách người bệnh chờ ghép) và người tình nguyện cho tạng (mục đích quản lý danh sách người cho).
b. Ghép tế bào gốc tạo máu
- Chỉ chỉ định xét nghiệm HLA cho ghép tế bào gốc tạo máu đồng loài, không chỉ định xét nghiệm HLA với ghép tế bào gốc tạo máu tự thân.
- Chỉ định xét nghiệm cho cả người cho và người nhận.
- Cần xác định ít nhất 5 locus HLA là HLA-A, HLA-B, HLA-C, HLA-DR, và HLA-DQ. Một số trung tâm ghép có thể yêu cầu xác định cả HLA-DP.
- Yêu cầu phải xác định HLA bằng kỹ thuật có độ phân giải cao (với người cho không cùng huyết thống).
- Việc chỉ định xét nghiệm cũng có thể dưới 2 hình thức: Xét nghiệm cho người nhận (người bệnh) và người cho tiềm năng; hoặc xét nghiệm cho người bệnh (để làm cơ sở tìm kiếm người cho phù hợp), hoặc người đăng kê cho tế bào gốc tạo máu (để lập ngân hàng tế bào gốc tạo máu), hoặc xét nghiệm cho các mẫu máu cuống rốn thu thập (để lập ngân hàng tế bào gốc máu cuống rốn).
1.3. Nhận định kết quả xét nghiệm HLA
Kết quả phân tích HLA dùng để so sánh độ phù hợp kháng nguyên HLA giữa người cho và người nhận. Về nguyên tắc giữa người cho và người nhận càng có nhiều điểm giống nhau thì kết quả ghép càng tốt, hạn chế được tình trạng thải ghép và ghép chống chủ. Trong ghép tạng, do có nhiều thuốc chống thải ghép tốt, thậm chí không phù hợp về HLA vẫn có thể ghép được. Tuy nhiên, trong ghép tế bào gốc đồng loài từ máu dây rốn, yêu cầu phù hợp HLA tối thiểu phải 4/6 allen (HLA-A, HLA-B, HLA-DR), trong ghép tế bào gốc đồng loài từ người cho không cùng huyết thống, yêu cầu phù hợp phải tối thiểu là 5/6 (là HLA-A, HLA-B, HLA-DR) hoặc 9/10 (là HLA-A, HLA-B, HLA-C, HLA-DR và -DQ).
2. XÉT NGHIỆM ĐỌ CHÉO LYMPHO (Lympho cross match)
2.1. Khái niệm
Là xét nghiệm nhằm phát hiện xem liệu trong huyết thanh người nhận có sẵn kháng thể đặc hiệu chống lại HLA của người cho hay không. Xét nghiệm này có ý nghĩa phát hiện và tiên lượng khả năng thải ghép tối cấp sau ghép, giúp tránh được phản ứng thải ghép tối cấp. Xét nghiệm đọ chéo lympho được thực hiện bằng cách ủ huyết thanh người nhận với các tế bào lympho T và lympho B của người cho sau đó phát hiện và đánh giá mức độ phản ứng giữa kháng thể đặc hiệu có trong huyết thanh người nhận với HLA có trên bề mặt tế bào lympho người cho.
2.2. Chỉ định xét nghiệm đọ chéo lympho
- Khi đã tìm được cһp người cho và người nhận cho ghép tạng (gan, thận, tim, tụy…). Xét nghiệm này được làm càng gần với ngày ghép bao nhiêu càng tốt bấy nhiêu, đặc biệt khi người nhận được truyền máu nhiều lần hoặc người nhận là nữ có tiền sử chửa, đẻ nhiều lần.
- Chỉ cần chỉ định làm xét nghiệm với người nhận, tuy nhiên mẫu phải lấy ở cả người cho và người nhận.
2.3. Lấy mẫu để xét nghiệm đọ chéo lympho
- Người cho: Lấy 4 ml máu ngoại vi có chống đông bằng EDTA.
- Người nhận: Lấy 4 ml máu không chống đông.
2.4. Nhận định kết quả xét nghiệm đọ chéo lympho
- Nếu phản ứng dương tính: Trong huyết thanh người nhận có kháng thể chống HLA người cho, trường hợp này không ghép được, vì nếu ghép sẽ xảy ra thải ghép tối cấp.
- Nếu phản ứng âm tính: Chứng tỏ trong huyết thanh người nhận không có kháng thể chống HLA người cho, có thể tiến hành ghép, không xảy ra thải ghép tối cấp.
3. XÉT NGHIỆM ĐIỆN DI PROTEIN (Serum Protein Electrophoresis) VÀ ĐIỆN DI MIỄN DỊCH HUYẾT THANH (Serum immunofixation Electrophoresis)
3.1. Khái niệm
Là một xét nghiệm phân tích thành phần protein và thành phần miễn dịch của protein trong huyết thanh. Trong huyết học đây là một trong những xét nghiệm có giá trị trong chẩn đoán và theo dõi hiệu quả điều trị bệnh Đa u tủy xương. Tuy cùng là phân tích thành phần protein và các Ig miễn dịch nhưng khác với các xét nghiệm hóa sinh (định lượng protein huyết thanh, định lượng albumin, globulin, định lượng các Ig miễn dịch huyết thanh) ở chỗ nếu các xét nghiệm hóa sinh tập trung vào việc xác định các con số định lượng thì xét nghiệm điện di lại tập trung vào các đặc điểm hình ảnh.
3.2. Chỉ định xét nghiệm điện di protein và điện di miễn dịch huyết thanh
- Chỉ định cho các trường hợp nghi ngờ bệnh Đa u tủy xương.
- Đánh giá kết quả điều trị (sau các đợt hóa trị liệu hay ghép).
- Để bổ sung kết quả cần chỉ định đồng thời cả điện di protein và điện di miễn dịch.
3.3. Lấy mẫu để xét nghiệm điện di protein và điện di miễn dịch huyết thanh
2 ml máu ngoại vi không chống đông.
3.4. Nhận định kết quả xét nghiệm điện di protein và điện di miễn dӏch huyết thanh
a. Với điện di protein (Hình 1)
Hình 1. Kết quả điện di protein huyết thanh
- Nếu kết quả có hình ảnh đỉnh globulin đơn dòng (đỉnh nhọn và đáy hẹp) thì được coi là một tiêu chuẩn vàng để chẩn đoán đa u tủy xương, cho dù đỉnh đơn dòng có thể cao hay thấp, có thể nằm ở vị trí alpha-globulin, beta-globulin hay gama-globulin (hay gặp nhất là vị trí gama-globulin, ít gặp vị trí alpha-globulin và beta-globulin).
- Nếu kết quả có hình ảnh tăng gama-globulin đa dòng (đỉnh không nhọn và đáy rất rộng) thì thường không phải đa u tủy xương mà là một bệnh lý tăng gamaglobulin nào đó, tuy nhiên cũng không loại trừ một số trường hợp khó, bản chất là đa u tủy xương nhưng có kết hợp bệnh lý hoặc các viêm nhiễm khác làm cho hình ảnh đơn dòng bị che lấp bởi biểu hiện đa dòng. Khi đó cần phối hợp các chỉ tiêu lâm sàng và xét nghiệm khác để chẩn đoán hoặc có thể xét nghiệm lại sau khi đã hết viêm nhiễm.
b. Với điện di miễn dịch (Hình 2)
Hình 2. Kết quả điện di miễn dịch huyết thanh bình thường
Kết quả điện di miễn dịch cho biết typ miễn dịch của bệnh đa u tủy xương: Chuỗi nặng (IgA, IgG, IgM) kết hợp chuỗi nhẹ (Kappa, Lambda) hay chỉ có chuỗi nhẹ đơn thuần hoặc kiểu hỗn hợp giữa 2 kiểu này. Kết quả điện di miễn dịch có thể là:
- Kiểu hình chuỗi nặng kết hợp chuỗi nhẹ: Trên hình ảnh điện di chỉ có 1 đỉnh đơn dòng. Về bản chất miễn dịch, đỉnh này có thể là IgA-Kappa, IgA-Lambda, IgG-Kappa, IgG-Lambda, IgM-Kappa, hoặc IgM-Lambda.
- Kiểu hình chuỗi nhẹ đơn thuần: Trên hình ảnh điện di cũng chỉ có 1 đỉnh đơn dòng. Về bản chất miễn dịch, đỉnh này có thể là chuỗi nhẹ Kappa hoặc chuỗi nhẹ Lambda.
- Kiểu hỗn hợp: Trên hình ảnh điện di chỉ có từ 2 đỉnh đơn dòng trở lên. Về bản chất miễn dịch, các đỉnh này có thể là bất cứ dạng chuỗi nặng hoặc chuỗi nhẹ nào, có thể là sự kết hợp giữa các đỉnh chuỗi nặng, giữa các đỉnh chuỗi nhẹ hoặc kết hợp hỗn hợp vừa chuỗi nặng vừa chuỗi nhẹ.
4. KỸ THUẬT TẾ BÀO DÒNG CHẢY PHÂN TÍCH KHÁNG NGUYÊN BIỆT HÓA TẾ BÀO (CD)
4.1. Khái niệm
- Là một xét nghiệm dựa trên kỹ thuật phân tích tế bào dòng chảy căn cứ vào sự có mặt của các kháng nguyên (dấu ấn) đặc trưng dòng và giai đoạn tế bào để xác định chính xác bản chất và giai đoạn phát triển của tế bào ung thư (trong bệnh lơ xê mi cấp, u lympho, đa u tủy xương), để đánh giá quần thể lympho trong các bệnh lý miễn dịch (các tình trạng rối loạn miễn dịch, suy giảm miễn dịch, bệnh tự miễn, bệnh khớp, đái huyết sắc tố kịch phát ban đêm…) hoặc để đếm số lượng một loại tế bào máu cụ thể nào đó (lympho T, lympho B, tế bào NK, tế bào gốc tạo máu…).
- Tùy theo các tình trạng bệnh lý khác nhau mà lựa chọn phân tích các CD khác nhau. Hiện nay, có nhiều CD được dùng trong huyết học như CD2, CD3, CD4, CD5, CD7…
4.2. Chỉ định
- Các CD dùng cho phân loại miễn dịch (chỉ định trong lơ xê mi cấp, lơ xê mi mạn, u lympho, đa u tủy xương, suy tủy xương…).
- Đánh giá CD14, CD24, CD55, CD59, FLARE (chỉ định trong bệnh đái huyết sắc tố kịch phát ban đêm, suy tủy xương, thiếu máu tan máu có test Coomb âm tính…).
- Đánh giá quần thể lympho T (T-CD3, T-CD4, T-CD8), quần thể lympho B (CD19), quần thể tế bào giết tự nhiên - NK (CD56) trong các bệnh lý miễn dịch (chỉ định trong suy giảm miễn dịch, bệnh tự miễn, lupus ban đỏ…).
- Xác định HLA-B 27 (chỉ định trong viêm đa khớp dạng thấp, viêm màng bồ đào…).
- Đếm tế bào gốc tạo máu CD34 (chỉ định sau tiêm thuốc huy động tế bào gốc, đánh giá số lượng tế bào gốc trong túi tế bào gốc trước và sau bảo quản…).
- Đánh giá CD64 trên quần thể bạch cầu hạt trung tính (chỉ định khi cần chẩn đoán sớm tình trạng nhiễm trùng).
4.3. Lấy mẫu xét nghiệm
- Tủy xương:
+ Dùng cho phân tích CD để phân loại miễn dịch trong lơ xê cấp, lơ xê mi mạn, đa u tủy xương;
+ 0,5-2 ml dịch hút tủy xương chống đông bằng EDTA. Tốt nhất mẫu phải được xử lý ngay trong vòng 24 giờ sau chọc hút tủy. Trường hợp cần thiết có thể lưu mẫu ở 2-6ºC nhưng không quá 5 ngày sau chọc hút tủy.
- Máu ngoại vi:
+ Trong lơ xê cấp, lơ xê mi mạn, đa u tủy xương nếu không lấy được dịch hút tủy xương, có thể lấy mẫu máu ngoại vi để phân tích. Với đa phần các xét nghiệm còn lại chỉ cần dùng máu ngoại vi;
+ Lấy 2 ml máu ngoại vi chống đông bằng EDTA. Tốt nhất mẫu phải được xử lý ngay trong vòng 24 giờ sau chọc hút tủy. Trường hợp cần thiết có thể lưu mẫu ở 2 -6ºC nhưng không quá 5 ngày sau chọc hút tủy.
- Trong một số trường hợp đặc biệt u lympho, có dịch màng phổi, dịch màng bụng…, xâm lấn da, niêm mạc… có thể dùng bệnh phẩm là mẫu mô (niêm mạc, gan, lách, hạch…), hoặc các loại dịch (dịch màng phổi, dịch màng bụng,…).
4.4. Nhận định kết quả xét nghiệm bằng kỹ thuật tế bào dòng chảy
a. Nhận định quần thể tế bào blast bất thường: Căn cứ vào vị trí các quần thể tế bào bình thường trên đồ thị phân tích tế bào dòng chảy để xác định quần thể tế bào bất thường. Quần thể tế bào blast bất thường thường có các đặc điểm sau:
- Nằm trong vùng cửa sổ blast, có kích thước nhỏ, độ phức tạp nhân và bào tương thấp hoặc trung bình, CD45(-), CD45dim, SSC(-), SSC thấp, hoặc lympho có SSC trung bình,…
- Có xuất hiện các kháng nguyên khác dòng. Ví dụ: vừa có các kháng nguyên dòng tủy vừa có các kháng nguyên dòng lympho.
- Xuất hiện các kháng nguyên bất thường (bình thường không có hoặc có với mật độ thấp).
- Không đồng bộ về kháng nguyên. Ví dụ: Vừa có kháng nguyên non vừa có kháng nguyên trưởng thành trên cùng 1 quần thể tế bào.
- Giảm xuất hiện hoặc xuất hiện quá mức một kháng nguyên nào đó so với bình thường.
b. Nhận định dòng tế bào ung thư
- Các dấu ấn non: TdT (dòng lympho), CD34, HLA-DR.
- Dòng tủy: Có các dấu ấn dòng tủy như CD13, CD33, CD117, CD15, MPO.
- Dòng hồng cầu: Có các dấu ấn dòng hồng cầu như CD71, CD125a.
- Dòng tiểu cầu: Có các dấu ấn dòng tiểu cầu như CD41, CD42, CD61.
- Dòng tủy mono: Ngoài các dấu ấn dòng tủy còn có các dấu ấn dòng mono như CD11b, CD14, CD64.
- Dòng lympho T: Có các dấu ấn dòng lympho T như CD3, cyCD3, CD4, CD8, CD7, CD5.
- Dòng lympho B: Có các dấu ấn dòng lympho B như CD5, CD10, CD19, CD20, CD22, cyCD22.
- Dòng tương bào: Có các dấu ấn CD38, CD138, Kappa, Lambda.
- Ngoài ra tùy thuộc vào kiểu hình xuất hiện phối hợp các dấu ấn có thể xếp loại Lơ xê mi cấp thành các thể: Lai tủy- lympho, lai lympho T-B, dòng tủy có dấu ấn bất thường dòng lympho, dòng lympho có dấu ấn bất thường dòng tủy...
5. MỘT SỐ ỨNG DỤNG CỦA KỸ THUẬT TẾ BÀO DÒNG CHẢY TRONG BỆNH MÁU
5.1. Xét nghiệm tồn dư tối thiểu trong lơ xê mi cấp bằng kỹ thuật phân tích tế bào dòng chảy
5.1.1. Khái niệm
Là một xét nghiệm sử dụng kỹ thuật phân tích tế bào dòng chảy (flow cytometry), nhằm phát hiện tỷ lệ tế bào ung thư còn tồn dư sau điều trị hóa chất ở các người bệnh Lơ xê mi cấp. Xét nghiệm này có thể đánh giá mức độ lui bệnh về tế bào tốt hơn so với xét nghiệm đánh giá lui bệnh về huyết học sử dụng kết quả xét ngiệm tế bào máu và tủy.
5.1.2. Chỉ định
- Các người bệnh Lơ xê mi cấp trước điều trị đã được làm xét nghiệm phân loại miễn dịch xác định các tổ hợp dấu ấn miễn dịch đặc trưng riêng cho tế bào ác tính và có thể dùng các dấu ấn đó để theo dõi tồn dư tối thiểu của bệnh máu ác tính.
- Chỉ định sau mỗi đợt điều trị hóa chất ở các người bệnh đã đạt lui bệnh về huyết học.
- Cần phối hợp chặt chẽ với phòng xét nghiệm để xác định các dấu ấn có ý nghĩa ở từng người bệnh cụ thể, có thể dùng để theo dõi tồn dư tối thiểu bệnh máu ác tính.
- Lưu ý khi chỉ định phải cung cấp cho cơ sở xét nghiệm hồ sơ đầy đủ của xét nghiệm phân loại miễn dịch trước đây.
5.1.3. Lấy mẫu để xét nghiệm
- Tủy xương: 0,5-2 ml dịch hút tủy xương chống đông bằng EDTA. Tốt nhất mẫu phải được xử lý ngay trong vòng 24 giờ sau chọc hút tủy. Trường hợp cần thiết có thể lưu mẫu ở 2-6ºC nhưng không quá 5 ngày sau chọc hút tủy.
- Máu ngoại vi: Trong trường hợp không lấy được dịch hút tủy xương, có thể lấy mẫu máu ngoại vi để phân tích. Cần 2 ml máu ngoại vi chống đông bằng EDTA, mẫu cũng phải được xử lý trong vòng 24 giờ sau lấy máu. Trường hợp cần thiết có thể lưu mẫu ở 2-6ºC nhưng không quá 5 ngày.
5.1.4. Nhận định kết quả theo dõi tồn dư tối thiểu
- Tồn dư tối thiếu < 0,01%: Lui bệnh hoàn toàn ở mức độ phân tích tế bào dòng chảy.
- Tồn dư tối thiểu > 0,01% - <0,1%: Nguy cơ tái phát trung bình.
- Tồn dư tối thiểu 0,1% - 1,0%: Nguy cơ tái phát cao.
- Tồn dư tối thiếu > 1,0%: Chưa lui bệnh ở mức độ phân tích tế bào dòng chảy.
5.2. Chẩn đoán bệnh Đái huyết sắc tố kích phát ban đêm bằng kỹ thuật tế bào dòng chảy
5.2.1. Khái niệm
Là một xét nghiệm miễn dịch dựa trên sự phân tích các CD (có bản chất là các protein gắn màng hoặc các protein gắn với các protein gắn màng) như FLAER, CD55, CD59, CD24, CD14 trên bề mặt hồng cầu và/ hoặc bạch cầu để phát hiện trực tiếp sự thiếu hụt của các protein gắn màng này. Bình thường các kháng nguyên protein gắn màng này có vai trò ngăn cách không để bổ thể có trong huyết tương bám vào tế bào và làm tan vỡ các tế bào này. Bệnh đái huyết sắc tố kịch phát ban đêm có cơ chế bệnh sinh là do các đột biến gen PIG-A làm giảm hoặc mất các protein gắn màng, làm cho các tế bào máu (đặc biệt là tế bào hồng cầu) của người bệnh dễ dàng bị phá hủy do tác động của bổ thể có sẵn trong huyết tương người bệnh.
5.2.2. Chỉ định xét nghiệm bằng kỹ thuật tế bào dòng chảy
- Chẩn đoán xác định hoặc chẩn đoán phân biệt bệnh đái huyết sắc tố kịch phát ban đêm.
- Thiếu máu tan máu.
- Người bệnh được chẩn đoán rối loạn sinh tủy.
- Thiếu máu tan máu có test Coombs âm tính.
5.2.3. Lấy mẫu để xét nghiệm bằng kỹ thuật tế bào dòng chảy
Cần 2 ml máu ngoại vi chống đông bằng EDTA, mẫu cũng phải được xử lý trong vòng 24 giờ sau lấy máu. Trường hợp cần thiết có thể lưu mẫu ở 2-6ºC nhưng không quá 5 ngày sau chọc hút tủy.
5.2.4. Nhận định kết quả
- Bình thường: Trên bề mặt tế bào hồng cầu không có biểu hiện thiếu hụt CD55 và CD59 (trên 95% quần thể hồng cầu dương tính CD55 và CD59), trên bề mặt bạch cầu không thiếu hụt CD24, CD14.
- Bệnh đái huyết sắc tố kịch phát ban đêm: Khi phát hiện thấy có trên 0,5% quần thể hồng cầu hoặc bạch cầu có thiếu hụt FLAER hoặc trên 10% quần thể hồng cầu/bạch cầu có thiếu hụt một hoặc nhiều dấu ấn CD14, CD24, CD55, CD59.
5.3. Xác định HLA-B27 bằng kỹ thuật tế bào dòng chảy
5.3.1. Khái niệm
HLA-B27 liên quan đến các bệnh viêm cột sống dính khớp, bệnh viêm khớp phản ứng, viêm khớp vảy nến, hội chứng REITER (95% người bệnh dương tính với HLA-B27). Dùng kháng thể đơn dòng kháng HLA-B27 có thể phát hiện người mang kháng nguyên HLA-B27 bằng kỹ thuật tế bào dòng chảy.
5.3.2. Chỉ định xét nghiệm
Khi có dấu hiệu nghĩ tới bệnh viêm cột sống dính khớp, bệnh viêm khớp phản ứng, viêm khớp vảy nến, hoặc hội chứng REITER.
5.3.3. Lấy mẫu để xét nghiệm
2 ml máu ngoại vi chống đông bằng EDTA, mẫu cũng phải được xử lý trong vòng 24 giờ sau lấy máu.
5.3.4. Nhận định kết quả
Kết quả HLA-B27 có thể dương tính hoặc âm tính. Có tới 8% người bình thường có HLA-B27 dương tính. HLA-B27 dương tính không xác định bệnh nhưng có giá trị đánh giá nguy cơ. Ở những người có biểu hiện viêm khớp, viêm bồ đào mắt, kết quả HLA-B27 dương tính giúp bác sĩ nghĩ nhiều đến khả năng bị bệnh hơn so với người có HLA-B27 âm tính.
5.4. Đếm tế bào gốc tạo máu CD34
5.4.1. Khái niệm
Tế bào gốc tạo máu có trong máu cuống rốn, trong dịch hút tủy xương và trong máu ngoại vi sau huy động. Tế bào gốc tạo máu có kích thước nhỏ, trên bề mặt có dấu ấn nhận diện là CD34. Dựa trên đặc tính này có thể dùng kỹ thuật phân tích tế bào dòng chảy đếm số lượng tuyệt đối tế bào gốc tạo máu CD34+ trong các mẫu bệnh phẩm.
5.4.2. Chỉ định
- Theo dõi tỉ lệ tế bào gốc CD34 trong máu sau liệu trình huy động tế bào gốc tạo máu ra máu ngoại vi.
- Đếm tế bào gốc CD34+ thu được trong túi tế bào gốc (lấy từ tủy xương, máu cuống rốn, hoặc máu ngoại vi).
- Đếm tế bào gốc CD34+ trong các túi tế bào gốc rã đông sau thời gian bảo quản đông lạnh.
5.4.3. Lấy mẫu xét nghiệm
- Máu ngoại vi: Lấy 1-2 ml máu ngoại vi chống đông bằng EDTA. Xử lý và đếm tế bào ngay sau khi chuyển tới labo.
- Mẫu lấy từ túi máu cuống rốn, túi tế bào gốc tủy xương, túi tế bào gốc máu ngoại vi sau huy động: Lấy 0,5-1 ml mẫu. Xử lý và đếm tế bào ngay sau khi chuyển tới labo.
5.4.4. Nhận định kết quả
Kết quả đếm sẽ được tính toán và xuất ra dưới dạng số lượng tế bào gốc CD34+ trong 1 microlit mẫu và/ hoặc số lượng tế bào gốc CD34+ có trong túi tế bào gốc.
5.5. Đếm số lượng tế bào CD3, CD4,CD8, CD19, CD56.
5.5.1. Khái niệm
Bạch cầu lympho và tế bào giết tự nhiên (NK) là thành phần tế bào chính của hệ miễn dịch, có vai trò tham gia phản ứng miễn dịch bảo vệ cơ thể. Các dưới nhóm lympho có lympho B (có dấu ấn CD19 dương tính) có vai trò tham gia đáp ứng miễn dịch thể dịch, lympho T (có dấu ấn CD3+) có vai trò tham gia đáp ứng miễn dịch tế bào. Lympho T có 2 loại là lympho T hỗ trợ (có dấu ấn CD3+, CD4+) và lympho T gây độc/ ức chế (có dấu ấn CD3+, CD8+). Tế bào NK (có dấu ấn CD16+, CD56+) là tế bào tham gia vào cơ chế đáp ứng miễn dịch không đặc hiệu. Trong một số trường hợp bệnh lý như nhiễm trùng, tiêm vắc- xin, bệnh tự miễn… các thành phần tế bào miễn dịch khác nhau có thể tăng lên. Trái lại, trong các trường hợp suy giảm miễn dịch tiên phát hoặc mắc phải, các thành phần tế bào miễn dịch có thể giảm đi. Do vậy, kỹ thuật phân tích tế bào dòng chảy đếm tỷ lệ % và số lượng tuyệt đối các thành phần tế bào miễn dịch là một xét nghiệm cho phép bác sĩ đánh giá tình trạng miễn dịch của một người bệnh cụ thể.
5.5.2. Chỉ định
- Suy giảm miễn dịch tiên phát ở trẻ em.
- Suy giảm miễn dịch thứ phát do HIV/AIDS, do điều trị hóa chất, tia xạ hoặc thuốc ức chế miễn dịch khác.
- Các trường hợp cần khảo sát đánh giá tình trạng miễn dịch khác (nhiễm khuẩn, sau tiêm vắc xin…).
5.5.3. Lấy mẫu xét nghiệm
2 ml máu ngoại vi chống đông bằng EDTA, mẫu cũng phải được xử lý trong vòng 24 giờ sau lấy máu.
5.5.4. Nhận định kết quả
Giá trị bình thường:
|
Thành phần |
Phần trăm (%) |
Tuyệt đối (tế bào/μl) |
|
Lympho T-CD3 |
50 - 85 |
600 - 2.800 |
|
Lympho T-CD4 |
30 - 60 |
500 - 1.600 |
|
Lympho T-CD8 |
15 - 40 |
200 - 800 |
|
Lympho B (CD19+) |
7 - 25 |
80 - 900 |
|
Tế bào NK (CD56+) |
6 - 30 |
70 - 1.200 |
5.6. Đánh giá CD64 trên bạch cầu hạt trung tính
5.6.1. Khái niệm
Bạch cầu hạt trung tính bình thường không biểu hiện hoặc biểu hiện rất yếu CD64. CD64 chỉ biểu hiện mạnh trên tế bào bạch cầu hạt trung tính khi các tế bào này hoạt hóa, phản ứng lại với các tác nhân lây nhiễm. Do vậy, CD64 được dùng làm dấu ấn sinh học để chẩn đoán tình trạng nhiễm trùng và vãng khuẩn huyết. Kỹ thuật phân tích tế bào dòng chảy cho phép phát hiện được sự có mặt CD64 trên bạch cầu hạt trung tính.
5.6.2. Chỉ định
Người bệnh nghi ngờ có tình trạng vãng khuẩn huyết hoặc nhiễm trùng.
5.6.3. Lấy mẫu xét nghiệm
2 ml máu ngoại vi chống đông bằng EDTA, mẫu cũng phải được xử lý trong vòng 24 giờ sau lấy máu.
5.6.4. Nhận định kết quả
- CD64 âm tính: Bình thường.
- CD64 dương tính >10%: Có tình trạng nhiễm trùng.
TÀI LIỆU THAM KHẢO
1. Linssen A, Feltkamp T. E. W. 1988. B27 positive diseases verusus B27 negative disease. Annals of the rheumatic disease, 47, 431-439.
2. Barnett D., Janossy G, Lubenko A, Matutes E, Newland A., Reilly J. T. 1999. Guidline for the flow cytometric enumeration of CD34+ haemopoietic stem cell. Clin. Lab. Haem. 21:301-308.
3. Allen E, Bakke A. C, Purtzer M. Z, Deodhar A. 2002. Neutrophil CD64 expression: distinguishing acute inflammatory autoimmune disease from systemic infections. Ann Rheum Dis; 61:522-525.
4. Sutherland D. R, Kuek N, Davidson J, Barth D, Chang H, Yeo E, Bamford S, Chin-Yee I, Keeney M. 2007. Diagnosing PNH with FLAER and multiparameter flow cytometry. Cytometry part B (clinical cytometry) 72B:167-177.
5. Brown M, Wittwer C. 2000. Flow cytometry: Principles and clinical applications in hematology. Clin Chem 46 (8 Pt2):1221-9.
6. David F Keren. 2003. Protein electrophoresis in clinical diagnosis. Oxford University Press Inc.
7. Sheldon S., Poulton K. HLA typing and its influence on organ transplantation. 2006. Methods Mol Biol, 333:157-174.
8. Mulley W. R, Kanellis J. 2011. Understanding crossmatch testing in organ transplantation: a case-based guide for the general nephrplogist. Neuphrology. 16(2):125-133.
9. Baig M. M. The normal range reference for peripheral blood lymphocyte subsets in regional arab population. 1991. Barhrain Medical Bullentin. 13(2): 55-57.
47. CHỈ ĐỊNH XÉT NGHIỆM PHÁT HIỆN ĐỘT BIẾN GEN TRONG CÁC BỆNH MÁU DI TRUYỀN
Bệnh máu di truyền là bệnh lý về huyết học gây ra bởi đột biến gen và có tính chất di truyền, từ cha mẹ sang con cái và các thế hệ tiếp theo. Trong các bệnh máu di truyền, phổ biến nhất và có tác động lớn đến xã hội là bệnh thiếu máu tán máu (α/β thalassemia) và bệnh rối loạn chảy máu do thiếu hụt yếu tố đông máu VIII, IX (hemophilia A/B).
1. ĐẠI CƯƠNG
Xét nghiệm phân tích đột biến di truyền trong bệnh thalassemia và hemophilia cần phải sử dụng các kỹ thuật sinh học phân tử để xác định chính xác vị trí và loại đột biến gây bệnh. Kết quả xét nghiệm đối với người bệnh hoặc một người mang gen có thể chỉ điểm sàng lọc cho các thành viên khác trong gia đình nhằm phát hiện người mang gen tiềm ẩn, hay chẩn đoán trước sinh. Mục đích quan trọng của xác định người mang gen là cảnh báo, tư vấn tiền hôn nhân, tư vấn sinh sản, giúp cá nhân người mang gen có cơ hội sinh ra những đứa con khỏe mạnh và ngăn chһn sự lan rộng của gen bệnh trong cộng đồng. Đây là vấn đề rất quan trọng vì nếu không được kiểm soát và phòng ngừa nguồn gen bệnh thì cộng đồng hemophilia và thalassemia sẽ ngày càng lan rộng và trở thành gánh nặng của xã hội và ảnh hưởng đến chất lượng dân số.
2. ĐẶC ĐIỂM ĐỘT BIẾN GEN BỆNH HEMOPHILIA VÀ THALASSEMIA
2.1. Bệnh hemophilia
Bệnh hemophilia gây ra bởi đột biến gen mã hóa yếu tố VIII (FVIII - Hemophilia A) và yếu tố IX (FIX - Hemophialia B) gây giảm hoặc mất tổng hợp yếu tố VIII, IX trong máu. Các gen này là gen lặn, nằm trên nhiễm sắc thể X nên có tính chất di truyền liên kết giới tính. Phụ nữ nếu có đột biến gen này trên một nhiễm sắc thể X thì không biểu hiện bệnh và được xác định là người mang gen (carrier). Nam giới chỉ cần mang nhiễm sắc thể X đột biến sẽ biểu hiện bệnh.
Hiện nay, cơ sở dữ liệu thế giới đã thống kê được trên 2.000 đột biến gen FVIII và trên 1.000 đột biến gen FIX. Đồng thời, các nghiên cứu cũng ghi nhận nhiều kiểu và tần suất của các đột biến khác nhau ở các thể bệnh:
- Hemophilia A thể nặng: Chủ yếu gây ra bởi các đột biến đảo đoạn intron 22 và intron 1 (45-50%) ở gen FVIII, còn lại là do các đột biến điểm, đột biến chèn xóa đoạn lớn.
- Hemophilia A thể nhẹ và thể vừa: Chủ yếu gây ra bởi đột biến điểm và các mất đoạn nhỏ.
- Hemophilia B: Chủ yếu gây ra bởi các đột biến điểm.
2.2. Bệnh thalassemia
- Thalassemia là bệnh lý thiếu máu tán máu gây ra bởi bất thường tổng hợp chuỗi globin, gồm α-globin, β-globin và các beta-like globin khác. Các gen này nằm trên nhiễm sắc thể thường, di truyền theo định luật Mendel. Gen α-globin (HBA) nằm trên nhiễm sắc thể số 16 gồm 4 alen (2x HBA1 và 2x HBA2). Gen beta-globin (HBB) nằm trên nhiễm sắc thể 11, gồm 2 alen β.
- Tùy theo tình trạng thiếu hụt chuỗi globin và kiểu tổ hợp hemoglobin và thalassemia được phân loại thành alpha thalassemia, beta thalassemia (phổ biến) và delta thalassemia (hiếm).
- Hiện nay cơ sở dữ liệu thế giới đã ghi nhận được trên 400 loại đột biến gặp trong bệnh α-thalassemia (chủ yếu là các đột biến mất đoạn) và hơn 900 loại đột biếntrong β-thalassemia (chủ yếu là các đột biến điểm, thay thế, chèn, xóa). Các đột biến trong thalassemia có tính chất đặc trưng theo vùng miền và dân tộc.
- Tại Việt Nam, theo thống kê của Viện Huyết học - Truyền máu Trung ương, các loại đột biến gen gây bệnh thalassemia phổ biến gồm có: SEA, THAI, α4.2, α3.7, HbCs, HbQs, c.2delT,... (ở alpha thalassemia) và Codon 26, codon 17, codon 95, codon 71/72, IVS I-5, IVSI-1, IVSII-654, -28,... (ở beta thalassemia)
3. CÁC KỸ THUẬT ĐƯỢC SỬ DỤNG ĐỂ PHÁT HIỆN ĐỘT BIẾN GEN HEMOPHILIA VÀ THALASSEMIA
3.1. Kỹ thuật tổng hợp chuỗi (Polymerase Chain Reaction - PCR)
Dựa vào cơ sở dữ liệu về hệ gen để thiết kế cһp mồi đặc hiệu và sử dụng enzyme DNA-polymerase để khuếch đại số copy của vùng gen đích cần phân tích. Sản phẩm của phản ứng này có thể được đọc trên bản gel agarose hoặc giải trình tự gen để tìm kiếm các bất thường nucleotit. Có nhiều loại xét nghiệm PCR đang được sử dụng thường quy trong chẩn đoán hemophilia và thalassemia, bao gồm:
a. Kỹ thuật PCR-RFLP (PCR-Restriction Fragment Length Polymorphism)
Là kỹ thuật PCR phối hợp với enzyme cắt giới hạn (restriction enzyme), trong đó kỹ thuật PCR được sử dụng để nhân bản đoạn gen quan tâm, sau đó sản phẩm PCR được cắt bằng enzym giới hạn có trình tự nhận biết điểm cắt nằm trong đoạn gen. Phân tích chiều dài các đoạn gen thu được sau khi cắt sẽ nhận dạng được đột biến. Kỹ thuật này thường được dùng để phân tích các đa hình trong bệnh Hemophilia.
b. Kỹ thuật Multiplex-PCR
Dựa trên nguyên lê của phản ứng PCR, nhưng sử dụng cùng lúc nhiều cһp mồi đặc hiệu cho nhiều đoạn gen khác nhau. Như vậy, với một phản ứng Multiplex PCR, nhiều đoạn gen (hoặc đột biến gen) cần phân tích có thể đồng thời được khuếch đại và xác định. Kỹ thuật này giúp phân tích cùng lúc nhiều đột biến gen, rút ngắn thời gian trả kết quả. Tuy nhiên, để thiết lập được quy trình kỹ thuật này, người thực hiện cần có chuyên môn cao trong việc thiết kế, tối ưu phản ứng và phân tích kết quả.
c. Kỹ thuật Longrange-PCR
Dựa trên nguyên lê của phản ứng PCR, tuy nhiên trong phản ứng sử dụng enzym có khả năng tổng hợp ADN chuỗi dài, đoạn gen cần tổng hợp có thể từ 10-20kb. Kỹ thuật này chủ yếu được sử dụng để phát hiện các đảo đoạn gen.
3.2. Kỹ thuật lai ADN
Kỹ thuật lai ADN dựa trên nguyên lê bắt cһp của các đoạn ADN có trình tự tương đồng. Trong đó, các đoạn ADN có trình tự bổ sung với gen cần phân tích được phân lập, và gắn lên màng lai ở các vị trí đã được thiết kế. ADN từ mẫu bệnh phẩm sẽ được khuếch đại đoạn gen cần phân tích, biến tính và lai với ADN trên màng. Ở những vị trí có phản ứng lai ADN xảy ra sẽ được phát hiện bằng chỉ thị mầu và kết quả được đọc theo vị trí đã thiết kế trong thang chuẩn.
3.3. Kỹ thuật MLPA
Kỹ thuật MLPA (Multiplex Ligation-dependent Probe Amplification) là kỹ thuật phát hiện đột biến chèn/xóa đoạn gen có nguyên lê dựa trên phản ứng multiplex-PCR để khuếch đại nhiều đoạn gen đích dựa trên các đầu dò (probe) đã được thiết kế sẵn và chỉ sử dụng một cһp mồi duy nhất. Mỗi đầu dò được thiết kế gồm hai đoạn oligo tương đồng và đặc hiệu cho vùng gen cần phân tích. Các oligo này đồng thời mang trình tự tương đồng cho cặp mồi PCR. Như vậy chỉ khi hai oligo cùng lai với trình tự gen đích, phản ứng PCR mới được thực hiện và sản phẩm mới được hình thành. Xét nghiệm MLPA có độ chính xác cao trong việc phát hiện các chèn xóa đoạn ở vùng gen quan tâm cũng như phát hiện số copy của các vùng gen này.
3.4. Kỹ thuật giải trình tự gen (sequencing)
Là kỹ thuật đọc chính xác trình tự sắp xếp nucleotit trên chuỗi ADN hoặc ARN. Các vùng gen đích được phân tích có thể bao gồm một hoặc nhiều đoạn gen đã biết (resequencing), hệ phiên mã (RNA sequencing), hệ exome (whole exome sequencing) hoặc cả hệ gen (whole genome sequencing). Các kỹ thuật giải trình tự gen phổ biến bao gồm Sanger sequencing (theo nguyên lý Sanger), Pyrosequencing (nguyên lý phát hiệm nguyên tử Hydro tự do) Next-generation sequencing (NGS, nguyên lê đọc trình tự nucleotit cùng phản ứng kéo dài chuỗi liên tục). Các kỹ thuật này đều có ưu nhược điểm về độ chính xác, mức độ phức tạp, khả năng ứng dụng và giá thành xét nghiệm. Vì vậy, phòng xét nghiệm cần xác định đúng đối tượng và thời điểm để lựa chọn kỹ thuật phù hợp.
4. CHỈ ĐỊNH XÉT NGHIỆM PHÁT HIỆN ĐỘT BIẾN GEN HEMOPHILIA
Hiện nay có hai phương pháp phân tích di truyền để phát hiện người mang gen trong các gia đình có tiền sử hemophilia, đó là:
- Phân tích trực tiếp: Là phương pháp xác định trực tiếp loại và vị trí đột biến ở người bệnh hoặc người mang gen bằng các phương pháp sinh học phân tử. Phương pháp này gồm các kỹ thuật như PCR, giải trình tự gen, MLPA,… đang được sử dụng ở các nước phát triển và ở nhiều nước trên thế giới. Phương pháp này có lợi ích lớn khi xác định được đột biến ở người nam giới bị bệnh và sau đó sử dụng đột biến này như chỉ điểm để phát hiện tình trạng mang gen của các thành viên trong gia đình và chẩn đoán trước sinh, trước chuyển phôi.
- Phân tích gián tiếp: Sử dụng các đa hình (polymorphism) trong gen mã hóa yếu tố VIII và yếu tố IX để lần theo dấu vết của gen bệnh. Từ đó, sẽ xác định được người mang gen hoặc áp dụng cho chẩn đoán trước sinh. Phương pháp này được sử dụng nhiều ở các nước đang phát triển do kỹ thuật thực hiện đơn giản, không cần phải đầu tư nhiều trang thiết bị đắt tiền và chi phí để thực hiện xét nghiệm thấp. Tuy nhiên, phương pháp này có nhược điểm là không xác định chính xác loại đột biến và không thể thực hiện được khi không có đủ mẫu của những người liên quan hoặc không tìm được đa hình phù hợp.
4.1. Phương pháp phân tích trực tiếp các đột biến gây bệnh hemophilia
4.1.1. Hemophilia A
Dựa trên kết quả phân loại mức độ bệnh (nặng, trung bình, nhẹ) bằng định lượng các yếu tố đông máu (FVIII và FIX).
- Hemophilia A thể nặng: Cần sàng lọc đột biến đảo đoạn intron 22.
+ Nếu dương tính với đảo đoạn intron 22, sử dụng đột biến này để sàng lọc các thành viên nữ giới trong gia đình để phát hiện người mang gen và chẩn đoán trước sinh;
+ Nếu âm tính với đảo đoạn intron 22, cần tiếp tục sàng lọc với đảo đoạn intron 1.
(2 loại đột biến trên chiếm 45-50% ở bệnh nhân hemophilia A thể nặng).
+ Nếu âm tính với cả đảo đoạn intron 22 và intron 1, người bệnh cần được xét nghiệm giải trình tự gen FVIII để tìm các đột biến điểm và chèn xóa đoạn hiếm gặp khác.
- Hemophilia A thể nhẹ và thể vừa: Do không có đột biến đặc trưng nên cần phải giải trình tự gen FVIII để tìm các đột biến.
4.1.2. Hemophilia B
Gen mã hóa yếu tố F9 mang nhiều đột biến và không tập trung nên cần chỉ định xét nghiệm giải trình tự gen để tìm đột biến.
Lưu ý:
Phương pháp giải trình tự gen Sanger là phương pháp được sử dụng phổ biến để tìm đột biến trong vùng exon và vùng liên kết intron-exon của gen FVIII, FIX. Tuy nhiên, có khoảng 6-10% trường hợp sau giải trình tự gen vẫn không tìm được đột biến do đột biến có thể nằm sâu vùng không mã hóa (intron) của gen. Trong trường hợp này, cần chỉ định xét nghiệm NGS để phân tích toàn bộ gen FVIII (khoảng 40kb) hoặc gen FIX (khoảng 170kb). Trong một số ít trường hợp khác, khi xét nghiệm âm tính với giải trình tự gen (Sanger và NGS), cần thực hiện xét nghiệm MLPA để tìm các mất đoạn ADN hiếm gặp ở gen FVIII/FIX.
Sơ đồ 8. Tóm tắt hướng dẫn chỉ định xét nghiệm đột biến gen hemophilia
(tham khảo: UK Haemophilia Centre Doctors’ Organisation, Mayo Clinics)
4.2. Phương pháp phân tích liên kết (linkage analysis)
4.2.1. Yêu cầu mẫu: Phải có đủ mẫu của những người liên quan đến người cần xét nghiệm, bao gồm:
- Bố, mẹ của người cần xác định tình trạng mang gen.
- Bệnh nhân hemophilia có liên quan huyết thống (anh trai hoặc em trai hoặc một người nam giới liên quan khác như chú bác ruột về phía mẹ).
4.2.2. Hemophilia A:
Chỉ định xét nghiệm PCR-RFLP phân tích các đa hình phổ biến trên gen mã hóa yếu tố VIII bao gồm: G/A (exon 7), (CA)n (int22), (CA)n (int13), T/A (BclI), G/A (XbaI), C/T (BglI), A/G (HindIII), G/A (MspI).
4.2.3 Hemophilia B:
Chỉ định xét nghiệm PCR-RFLP phân tích các đa hình phổ biến trên gen mã hóa yếu tố IX bao gồm: DdeI, MseI, XmnI, MspI, TaqI, MnlI, HhaI.
Lưu ý:
- Xét nghiệm này chỉ thực hiện được khi mẹ của người làm xét nghiệm phải là người mang gen và phải dị hợp tử với một trong số các đa hình nêu trên.
- Trường hợp không thu thập được đủ mẫu hoặc mẫu không đáp ứng được theo yêu cầu tóm tắt hướng dẫn chỉ định xét nghiệm ở sơ đồ 7 của kỹ thuật thì cần phải sử dụng phương pháp phân tích trực tiếp.
5. CHỈ ĐỊNH XÉT NGHIỆM PHÁT HIỆN ĐỘT BIẾN GEN THALASSEMIA
Có nhiều dạng đột biến gen alpha và beta globin gây bệnh thalassemia được phát hiện và thống kê trên thế giơi. Theo nghiên cứu và thống kê của Viện Huyết học - Truyền máu TW ở gần 25.000 người bệnh và người mang gen, ở Việt Nam có khoảng 18 đột biến phổ biến trên tổng số hơn 40 đột biến ở 2 gen này được ghi nhận, bao gồm các đột biến điểm, chèn xóa và mất đoạn ADN. Ngoài ra, một số đột biến hiếm gặp cũng được phát hiện bằng phương pháp giải trình tự gen và MLPA.
5.1. Xác định đột biến ở người bệnh và người mang gen
Bước 1: Sàng lọc các đột biến phổ biến
Sử dụng xét nghiệm PCR-RFLP và lai ADN
- Đối với α-Thalassemia: SEA, THAI, α4.2, α3.7, HbCs, c.2delT, HbQs, HbPakse.
- Đối với β-Thalassemia: Codon 17, codon 26, codon 41/42, codon 71/72, codon 95, IVS I-5, IVSI-1, IVSII-654, -28, codon 8/9.
Bước 2: Sàng lọc các đột biến hiếm gặp
Sử dụng phương pháp giải trình tự gen Sanger, NGS hoặc phương pháp MLPA để xác định các đột biến hiếm gặp. Phương pháp giải trình tự gen Sanger cho phép xác định các đột biến trong vùng exon của gen alpha 1, alpha 2 và beta-globin, phương pháp NGS cho phép giải trình tự toàn bộ các gen (bao gồm intron và exon). Trong khi đó, phương pháp MLPA có thể phát hiện các mất đoạn mới ở các gen nói trên (tóm tắt ở sơ đồ 9).
5.2. Chẩn đoán trước sinh, trước chuyển phôi.
Kết quả phân tích đột biến gen của cһp vợ chồng có thể được dùng như chỉ thị trong xét nghiệm chẩn đoán trước sinh hoặc trước chuyển phôi.
- Đối với chẩn đoán trước sinh: Ở tuần thai 16-18 người phụ nữ mang thai có thể được thực hiện thủ thuật hút dịch ối để làm xét nghiệm chẩn đoán trước sinh. Tế bào dịch ối cần được nuôi cấy trong môi trường đặc hiệu (ví dụ: Amniomax) để loại bỏ tạp nhiễm máu mẹ và tăng sinh số lượng, sau đó ADN được tách chiết và thực hiện xét nghiệm sinh học phân tử theo chỉ định.
- Đối với xét nghiệm chẩn đoán trước chuyển phôi: Sinh thiết phôi có thể được thực hiện ở ngày 3 hoặc 5 và khuếch đại hệ gen (WGA) của tế bào sinh thiết để làm xét nghiệm sinh học phân tử. Tuy nhiên, đối với alpha thalassemia, việc xác định các mất đoạn lớn có thể là trở ngại lớn về kỹ thuật vì hệ gen WGA có kích thước rất nhỏ.
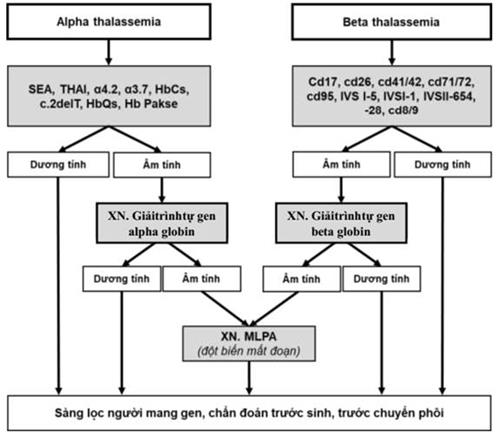
Sơ đồ 9. Hướng dẫn chẩn đoán đột biến gen gây bệnh thalassemia
TÀI LIỆU THAM KHẢO
1. Phạm Quang Vinh, Nguyễn Hà Thanh (2019). “Bệnh hemoglobin di truyền”, Bài giảng sau đại học huyết học truyền máu, Nhà xuất bản Y học, 204-223.
2. Fucharoen G (2004). A simplified screening strategy for Thalassemia and hemoglobin E in rural communities in South - East Asia, Bull World Health Organ, 82(5):364-72.
3. Kanokwan Sanchaisuriya. A Reliable Screening Protocol for Thalassemia and Hemoglobinopathies in Pregnancy, Am J Clin Pathol 2005;123:113-118.
4. D J Bowen. Haemophilia A and haemophilia B: molecular insights. J Clin Pathol: Mol Pathol 2002;55:1-18.
Doctors’ Organisation (UKHCDO), the Haemophilia Genetics Laboratory Network and the Clinical Molecular Genetics Society.
5. J. Oldenburg et at (2004). Molecular basis of haemophilia A. Haemophilia,10, (suppl. 4), 133-139.
6. Steve Keeney, Mike Mitchell and Anne Goodeve (2010). Practice Guidelines for the Molecular Diagnosis of Haemophilia A. UK Haemophilia Centre.
7. John Old, Prevention and diagnosis of haemoglobinopathies: A short guide for health professionals and laboratory scientists, TIF
Prevention of thalassaemias and other haemoglobin disorders. Volume 2: laboratory protocols, TIF 2012
48. CHỈ ĐỊNH XÉT NGHIỆM DI TRUYỀN TẾ BÀO VÀ SINH HỌC PHÂN TỬ TRONG CHẨN ĐOÁN VÀ THEO DÕI ĐIỀU TRỊ BỆNH MÁU ÁC TÍNH
1. ĐẠI CƯƠNG
Cùng với các bệnh lý ác tính khác như ung thư mô đặc, bệnh máu ác tính cũng gây nên bởi các bất thường di truyền phức tạp, gồm các bất thường nhiễm sắc thể, bất thường gen (genetics) và ngoài gen (epigenetics). Các bất thường di truyền này là các chỉ thị để chẩn đoán, xếp loại tiên lượng và theo dõi điều trị bệnh máu ác tính.
Tùy theo mức độ bất thường di truyền, có 2 nhóm xét nghiệm thường được dùng trong phát hiện gồm kỹ thuật di truyền tế bào (Công thức nhiễm sắc thể, FISH) và sinh học phân tử (ADN hybridization, PCR, Sequencing,…).
1.1. Kỹ thuật di truyền tế bào (DTTB)
a. Lập công thức nhiễm sắc thể (karyotyping)
- Lập công thức nhiễm sắc thể là kỹ thuật “chẩn đoán hình ảnh” bộ nhiễm sắc thể, trong đó bộ nhiễm sắc thể của tế bào được bộc lộ, nhuộm màu và phân tích số lượng cũng như cấu trúc. Đây là kỹ thuật cơ bản giúp đánh giá ban đầu bộ nhiễm sắc thể trong tế bào của đối tượng cần xét nghiệm. Phương pháp này có kỹ thuật đơn giản và lợi ích lớn, tuy nhiên có nhược điểm là độ nhạy thấp và không phát hiện được bất thường nhỏ trong hệ gen.
b. Kỹ thuật lai huỳnh quang tại chỗ (FISH)
- Kỹ thuật lai huỳnh quang tại chỗ là kỹ thuật phân tích di truyền phân tử, sử dụng đầu dò (probe) đặc hiệu đã được đánh dấu huỳnh quang, tương đồng với vùng ADN trên nhiễm sắc thể để phát hiện sự có mặt hay vắng mặt của vùng ADN đó thông qua phản ứng lai theo nguyên tắc bổ sung. Tín hiệu lai được đọc và phân tích trên hệ thống kính hiển vi huỳnh quang.
- Khác với kỹ thuật công thức nhiễm sắc thể, kỹ thuật FISH có độ nhạy cao hơn (1-10% so với 5-10%) và có thể phân tích tế bào ở kỳ trung gian và kỳ giữa.
- Trong chẩn đoán di truyền các bệnh máu ác tính, kỹ thuật FISH được ứng dụng để phát hiện các bất thường cụ thể, gắn với các bệnh lý cần chẩn đoán (ví dụ: t(9;22) ở bệnh lơ xê mi tủy mạn). Với độ nhạy 1-10%, kỹ thuật FISH cũng có thể được ứng dụng để phân tích tồn dư thiểu ở một số bệnh lý có bất thường đặc trưng.
- Tuy nhiên, kỹ thuật FISH không phát hiện được các bất thường ngoài thiết kế đặc hiệu của đầu dò. Do đó, kỹ thuật FISH cần được phối hợp với công thức nhiễm sắc thể để phát hiện các bất thường mới trong một số trường hợp cụ thể, như bệnh tiến triển hoặc mất đáp ứng điều trị.
1.2. Sinh hӑc phân tử (SHPT)
a. Kỹ thuật PCR (Polymerase Chain Reaction)
PCR là kỹ thuật cơ bản của sinh học phân tử sử dụng cһp mồi (primer) đặc hiệu để khuếch đại vùng ADN cần phân tích để phát hiện các thay đổi ở vị trí nucleotit xác định. Kỹ thuật PCR chỉ phân tích được vùng ADN đã biết trước trình tự sắp xếp nucleotit và chỉ dùng để xác định đột biến gen tại vị trí đã xác định. Ngoài cặp mồi đặc hiệu, kỹ thuật PCR cần sử dụng một số thành phần như nucleotit dự do (dNTPs), enzyme ADN polymerase và dung dịch đệm cho phản ứng. Phản ứng PCR được thực hiện trên hệ thống máy luân nhiệt (thermo-cycler) và kết quả được phân tích thông qua điện di sản phẩm trên bản gel agarose. Nếu đầu vào của phản ứng PCR là 1 sợi kép ADN đích, sau n chu kỳ luân nhiệt, số đoạn ADN đích thu được sẽ là 2n sản phẩm. Một phản ứng PCR thông thường được thiết kế từ 23-35 chu kỳ. Như vậy số sản phẩm thu được sẽ là 223 - 235 đoạn ADN đích.
b. Kỹ thuật Real-time PCR
- Real-time PCR là kỹ thuật dựa trên nền tảng kỹ thuật PCR, trong đó ngoài cһp mồi đặc hiệu còn bổ sung đầu dò (probe) đặc hiệu với sản phẩm PCR và được đánh dấu huỳnh quang để đọc kết quả ở thời gian thực (real-time). Ngoài ra, khác với PCR truyền thống, hệ thống máy luân nhiệt trong kỹ thuật real-time PCR được thiết kế bổ sung đầu đọc huỳnh quang để phát hiện tín hiệu của sản phẩm PCR được tổng hợp. Kỹ thuật này vì vậy vượt trội so với PCR truyền thống ở chỗ thời gian phân tích được rút ngắn và độ nhạy của phản ứng cũng được nâng cao đáng kể bởi việc sử dụng đầu đọc tín hiệu huỳnh quang.
- Hiện nay, có rất nhiều biến thể của kỹ thuật PCR và real-time PCR được thiết kế cho các mục đích khác nhau như; phát hiện đột biến ở vị trí cắt enzyme giới hạn (PCR- RFLP), tăng độ chính xác phát hiện đột biến (AS-PCR), phát hiện mất đoạn lớn (GAP- PCR), tăng độ nhạy của phản ứng (Nested-PCR), phát hiện đồng thời nhiều đột biến trong cùng một phản ứng (multiplex-PCR),… Ngoài ra, phản ứng PCR cũng có thể được thực hiện với gen đích là ARN tổng số (RT-PCR) thay vì ADN tổng số.
- Ngoài việc phát hiện đột biến theo kiểu định tính, kỹ thuật PCR định lượng (RQ-PCR, Real-time Quantitative PCR) được ứng dụng để đánh giá tồn dư tối thiểu các bệnh máu ác tính. Mấu chốt của kỹ thuật này là bổ sung mẫu chuẩn đã biết số bản copy ADN vào cùng một phản ứng với mẫu cần xét nghiệm để thông qua đó tính toán số copy ADN của mẫu cần phân tích.
- Để tăng độ nhạy và đặc hiệu của phản ứng PCR, real-time PCR, chúng ta có thể sử dụng kỹ thuật droplet-PCR (dd-PCR) thông qua việc chia nhỏ 1 phản ứng PCR (gồm hàng triệu đoạn ADN đích) thành hàng triệu phản ứng PCR (với chỉ 1 đến một vài đoạn ADN đích trong 1 phản ứng). Ngoài ra, kỹ thuật phân tích gen của từng tế bào riêng lẻ (single-cell analysis) cũng giúp tăng độ chính xác, giảm giá trị cut-off và giảm âm tính giả xuống mức đặc biệt thấp. Các kỹ thuật này có giá trị cao khi sàng lọc các dòng tế bào đột biến ở giai đoạn xuất hiện sớm hoặc tái phát sớm khi chưa có biểu hiện lâm sàng.
- Kỹ thuật sinh học phân tử dựa trên nền tảng PCR có độ chính xác và độ nhạy đặc biệt cao, từ 10-4 đến dưới 10-6 nên có thể phát hiện gen đột biến ở tần suất đặc biệt thấp (MR4.5 - MR5 hoặc thấp hơn).
c. Kỹ thuật MLPA (Multiplex Ligation-dependent Probe Amplification)
Kỹ thuật MLPA dựa trên nguyên lê thiết kế nhiều đầu dò (probe) cho vùng ADN (gen) cần xác định đột biến để tìm kiếm các đột biến về cấu trúc gen (chủ yếu là chèn xóa đoạn ADN). Nguyên lý của xét nghiệm này là lai ghép các đầu dò ADN vào trình tự ADN của gen đích cần phân tích và khuếch đại bằng phản ứng PCR đa mồi. Sau phản ứng PCR số copy của các sản phẩm sẽ được định lượng để xác định sự thêm vào hoặc mất đi của các nucleotit hoặc đoạn ADN trên gen. Xét nghiệm này có thể phân biệt được trạng thái đột biến kiểu đồng hợp tử hoặc dị hợp tử và chủ yếu sử dụng để phát hiện các đột biến mới hoặc tổ hợp các đột biến trên nhiều vùng gen khác nhau.
d. Kỹ thuật phân tích đoạn (Fragment Analysis)
Kỹ thuật phân tích đoạn được sử dụng để xác định các đột biến chèn xóa đoạn ngắn trên chuỗi ADN (gen) hoặc phân tích các chỉ thị đa hình di truyền. Nguyên lý của kỹ thuật này là sử dụng các cһp mồi đặc hiệu và phản ứng PCR để khuếch đại vùng ADN đích cần xác định đột biến hoặc chứa các đa hình di truyền. Sau PCR, sản phẩm sẽ được phân tích trên hệ thống điện di mao quản kết hợp với phần mềm để phân tích kết quả. Xét nghiệm này có thể phân biệt được chèn xóa 1 nucleotit trên chuỗi ADN hoặc định lượng đa hình di truyền ở tỷ lệ rất thấp, đến 0,1%.
đ. Kỹ thuật giải trình tự gen Sanger
- Kỹ thuật giải trình tự gen Sanger dựa trên nguyên lý sinh tổng hợp chuỗi ADN bổ sung với gen đích, trong đó các nucleotit tự do đã được sửa đổi (ddNTPs) để mỗi chu kỳ phản ứng chỉ duy nhất 1 nucleotit được gắn vào sợi bổ sung. Như vậy, sau bước PCR, thư viện ADN sẽ bao gồm hàng trăm sản phẩm PCR với độ dài cách nhau 1 nucleotit và việc điện di các sản phẩm này trên hệ thống điện di mao quản có độ chính xác cao sẽ giúp phần mềm phân tích lắp ráp được trình tự nucleotit của đoạn gen cần phân tích.
- Kỹ thuật này cho phép đọc chính xác trình tự sắp xếp của đoạn gen cần phân tích và khi so sánh với gen tham chiếu (ref. gen.), mọi sai khác sẽ được phát hiện. Kỹ thuật này có độ chính xác đặc biệt cao, tuy nhiên có một số nhược điểm gồm: công suất phân tích mẫu thấp, đoạn gen phân tích ngắn, không phù hợp với các đột biến gen ung thư (somatic) có tỷ lệ tế bào ác tính thấp. Ngược lại, kỹ thuật này rất hiệu quả khi phân tích các bệnh lý di truyền bẩm sinh (germline mutation) như thalassemia, hemophilia,…
e. Kỹ thuật giải trình tự gen thế hệ mới (NGS, Next-generation sequencing)
- Kỹ thuật NGS dựa trên nguyên lê đọc trình tự gen liên tục, song song với phản ứng kéo dài chuỗi ADN. Ở kỹ thuật này, 4 loại nucleotit (ATGC) được đánh dấu bằng các màu huỳnh quang khác nhau, khi phản ứng PCR được thực hiện liên tục, mỗi nucleotit gắn vào mạch ADN sẽ được đọc tức thì bằng đầu đọc huỳnh quang và ghi nhận bằng phần mềm phân tích. Chíp giải trình tự gen NGS thiết kế để hàng triệu phản ứng giải trình tự gen diễn ra cùng lúc, tín hiệu được đọc bằng camera có độ phân giải cao và phân tích bởi phần mềm chuyên dụng trên hệ thống máy tính hiệu xuất cao. Như vậy, với NGS, hàng trăm mẫu xét nghiệm có thể được phân tích cùng lúc để xác định các sai khác trên chuỗi ADN, ARN liên quan đến các bệnh lý.
- Tùy theo mục đích xét nghiệm, có nhiều hệ thống NGS và sinh phẩm để phân tích trình tự gen như: giải trình tự gen đích (targeted-sequencing), giải trình tự nhóm gen (targeted panel quenching), giải trình tự RNA (RNA-sequencing), giải trình tự hệ exome (Whole exome sequencing - WES) hoặc giải trình tự hệ gen (Whole genome sequencing - WGS).
f. Xét nghiệm theo dõi mọc mảnh ghép (chimerism)
Xét nghiệm theo dõi mọc mảnh ghép là xét nghiệm dùng bộ các chỉ thị phân tử (indels, STR, SNP,…) để đánh giá tỷ lệ % của tế bào người hiến và người nhận trong điều trị ghép tế bào gốc tạo máu đồng loài. Nguyên lê xét nghiệm là sử dụng các cặp mồi đặc hiệu và PCR (realtime) và điện di mao quản để tính toán tỷ lệ chỉ thị phân tử khác biệt (giữa người hiến và người nhận tế bào gốc) trong máu ngoại vi của người nhận. Thông qua tỷ lệ các chỉ thị phân tử này, tỷ lệ tế bào có xuất xứ từ người hiến trong mẫu máu ngoại vi của người nhận được xác định là tỷ lệ mọc mảnh ghép (% chimerism). Xét nghiệm này có độ nhạy rất cao và có thể phân biệt được ở mức 0,1%.
2. CÁC XÉT NGHIỆM DI TRUYỀN TẾ BÀO VÀ SINH HỌC PHÂN TỬ ĐANG ĐƯỢC SỬ DỤNG HIỆN NAY
- Xét nghiệm công thức nhiễm sắc thể.
- Xét nghiệm lai huỳnh quang tại chỗ (FISH).
- Xét nghiệm PCR-RFLP.
- Xét nghiệm multiplex-PCR.
- Xét nghiệm nested-PCR.
- Xét nghiệm RT-PCR.
- Xét nghiệm phân tích đoạn ADN (fragment analysis).
- Xét nghiệm định lượng gen RQ-PCR.
- Xét nghiệm MLPA.
- Xét nghiệm giải trình tự gen Sanger.
- Xét nghiệm giải trình tự gen NGS.
- Xét nghiệm theo dõi mọc mảnh ghép (chimerism).
3. DẤU ẤN DI TRUYỀN TẾ BÀO VÀ SINH HỌC PHÂN TỬ ĐƯỢC SỬ DỤNG ĐỂ CHẨN ĐOÁN VÀ THEO DÕI ĐIỀU TRỊ BỆNH MÁU ÁC TÍNH
- Bất thường di truyền trong các bệnh máu ác tính rất đa dạng, bao gồm bất thường nhiễm sắc thể (chuyển đoạn, mất/ thêm đoạn, đảo đoạn, mất hoặc thêm nhiễm sắc thể) và các đột biến gen (thay thế, chèn xóa,…). Một số bất thường di truyền điển hình có thể được sử dụng để chẩn đoán bệnh (BCR-ABL1 trong CML) hoặc để xếp loại tiên lượng (FLT3 trong AML). Ngoài ra, các bệnh lý huyết học có xu hướng phát sinh dòng tế bào mang đột biến mới trong quá trình điều trị, đặc biệt là điều trị không hiệu lực hoặc tái phát, ví dụ như đột biến kháng thuốc TKI ở bệnh CML. Do vậy, phối hợp các phương pháp di truyền tế bào và sinh học phân tử để chẩn đoán và theo dõi điều trị bệnh máu ác tính là rất quan trọng.
- Ngoài các bất thường di truyền phổ biến (bảng 60) được dùng là dấu ấn theo các hướng dẫn của Mạng lưới ung thư quốc gia Hoa kỳ (NCCN) hay Mạng lưới lơ xê mi châu Âu (ELN), các nghiên cứu trên thế giới tiếp tục làm rõ vai trò của sinh học phân tử trong cơ chế bệnh sinh và bổ sung nhiều đột biến mới vào phác đồ chẩn đoán và theo dõi điều trị các bệnh lý huyết học ác tính.
Bảng 60. Trình bày một số dấu ấn được sử dụng bất thường trong các bệnh máu ác tính:
|
Bệnh |
Nhiễm sắc thể |
ADN/gene |
Tần suất |
Tiên lượng |
Kỹ thuật xét nghiệm |
|
Lơ xê mi tủy mạn (CML) |
t(9;22) |
BCR-ABL |
90-95% |
|
Công thức nhiễm sắc thể, xét nghiệm FISH, xét nghiệm RT-PCR, xét nghiệm RQ-PCR, giải trình tự gen. |
|
Lơ xê mi cấp thể M2 |
t(8;21) |
AML1-ETO, cKIT |
29-40% |
Tốt |
Công thức nhiễm sắc thể, Xét nghiệm FISH, Xét nghiệm RT-PCR, Giải trình tự gen. |
|
Lơ xê mi cấp thể M3 |
t(15;17) |
PML-RARα |
65-70% |
Tốt |
|
|
Lơ xê mi cấp thể M4, M4eo |
t(16;16) hoặc inv(16) |
CBFB-MYH11, cKIT |
6% |
Tốt |
|
|
Lơ xê mi tủy cấp có kiểu hình NST bình thường (CN-AML) |
- |
NPM1 |
30-40% |
Tốt |
Xét nghiệm RT-PCR, Giải trình tự gen. |
|
- |
FLT3 |
~ 23% |
Xấu |
||
|
- |
CEBPA |
~10% |
Xấu |
||
|
Lơ xê mi lympho cấp (ALL) |
t(9;22) |
BCR-ABL |
25-30% |
Xấu |
Công thức nhiễm sắc thể, Xét nghiệm FISH, Xét nghiệm RT-PCR. |
|
t(12;21) |
TEL-AML1 |
25% |
Tốt |
||
|
t(4;11) |
MLL-AF4 |
50-75% |
Xấu |
||
|
t(1;19) |
E2A-PBX1 |
8-11% |
Xấu |
||
|
Đa u tủy xương (MM) |
t(11;14) |
CCND1 |
15-20% |
Tốt |
Xét nghiệm FISH |
|
t(4;14) |
FGFR3 và MMSET |
15% |
Xấu |
||
|
t(6;14) |
CCND3 |
3% |
Tốt |
||
|
t(14;16) |
C-MAF |
5-7% |
Xấu |
||
|
t(14;20) |
MAFB |
5% |
Xấu |
||
|
del(13q) |
- |
50% |
Tốt |
||
|
del(17p13) |
TP53 |
10% |
Xấu |
||
|
Rối loạn sinh tủy (MDS) |
-5/del(5q) |
- |
10-20% |
Tốt |
Công thức nhiễm sắc thể, Xét nghiệm FISH |
|
-7/del(7q) |
- |
10-20% |
Xấu |
||
|
Trisomy 8 |
- |
10% |
Trung bình |
||
|
17p-syndrome |
- |
7% |
Xấu |
||
|
del(20q) |
- |
5% |
Tốt |
||
|
≥ 3 tổn thương |
- |
10-20% |
Xấu |
||
|
|
SF3B1 |
20-30% |
Tốt |
Giải trình tự gen |
|
|
|
DMNT3A |
12-18% |
- |
||
|
|
TET2 |
20-25% |
- |
||
|
|
AXL1 |
15-25% |
Xấu |
||
|
|
TP53 |
8-12% |
Xấu |
||
|
Đa hồng cầu nguyên phát (PV) |
- |
JAK2 |
>90% |
|
Xét nghiệm PCR, Giải trình tự gen |
|
Tăng tiểu cầu tiên phát (ET) |
- |
JAK2/CALR/ MPN |
55%,15-24% 4% |
|
|
|
Xơ tủy nguyên phát (PMF) |
- |
JAK2/ CALR/ MPN |
65%, 25-35% 8% |
|
|
|
Lơ xê mi lympho mạn (CLL) |
+ 12 |
- |
20% |
Trung bình |
Công thức nhiễm sắc thể, Xét nghiệm FISH |
|
del(13q) |
- |
45-60% |
Tốt |
||
|
del(17p) |
- |
13-235 |
Rất xấu |
||
|
Lơ xê mi tế bào tóc |
- |
BRAF V600E IGHV4-34 |
- |
|
Công thức nhiễm sắc thể, Xét nghiệm PCR, Giải trình tự gen |
|
U lympho không Hodgkin |
t(11;14) |
CCND1/IGH |
|
|
Công thức nhiễm sắc thể, Xét nghiệm FISH, Xét nghiệm RT-PCR |
|
t(14;18) |
IgH-BclII |
|
|
||
|
t(8;14) |
c-MYC |
|
|
||
|
3q27 |
BCL6 |
|
|
||
|
- |
BCL2 |
|
|
||
|
- |
FN1 |
|
|
4. HƯỚNG DẪN CHỈ ĐỊNH VÀ PHỐI HỢP CÁC XÉT NGHIỆM DTTB VÀ SHPT TRONG CHẨN ĐOÁN, THEO DÕI ĐIỀU TRỊ MỘT SỐ BỆNH MÁU ÁC TÍNH
Để đảm bảo tính chính xác, phát huy thế mạnh của từng loại xét nghiệm; đồng thời để đảm bảo sự hợp lê, hiệu quả và chi phí hợp lê, các xét nghiệm cần được chỉ định theo các phác đồ chẩn đoán cụ thể theo từng giai đoạn và đặc điểm đáp ứng điều trị.
4.1. Bệnh lơ xê mi tủy mạn (chronic myeloid leukemia - CML)
Hiện nay, trong các phác đồ điều trị nhắm đích đối với lơ xê mi tủy mạn, số lượng các tế bào ác tính được coi như các tiêu chí để đánh giá mức độ lui bệnh. Ở mức độ phân tử, nhiễm sắc thể Ph và tỷ lệ bản sao mRNA của gen lai BCR-ABL1 so với gen tham chiếu ABL1 (%IS, hoặc MR) được sử dụng làm tiêu chuẩn quốc tế để đánh giá mức đáp ứng điều trị. Các tiêu chí đánh giá cụ thể như sau:
a. Đánh giá đáp ứng ở mức độ di truyền tế bào
- Đáp ứng hoàn toàn ở mức độ di truyền tế bào (CCyR): NST Ph âm tính.
- Đáp ứng nhiều ở mức độ di truyền tế bào: NST Ph: 1-35%.
- Đáp ứng một phần ở mức độ di truyền tế bào (PCyR): NST Ph: 36-65%.
- Đáp ứng tối thiểu ở mức độ di truyền tế bào: NST Ph: 66-95%
b. Đánh giá lui bệnh ở mức độ phân tử
- Đáp ứng sớm ở mức độ phân tử (EMR): BCR-ABL1 ≤ 10%IS trong khoảng 3-6 tháng.
- Đáp ứng phần lớn ở mức độ phân tử (MMR): BCR-ABL1 ≤ 0.1%IS hoặc giảm ≥ 3-log so với đường chuẩn ban đầu (nếu không thực hiện được RQ-PCR).
- Đáp ứng hoàn toàn ở mức độ phân tử (CMR): BCR-ABL1 ≤ 0.0032%IS (MR4.5)
c. Tái phát
- Tăng 1-log BCR-ABL1 kèm với mất đáp ứng MMR.
d. Chỉ định xét nghiệm di truyền trong bệnh lơ xê mi tủy mạn
Chẩn đoán ban đầu: - Xét nghiệm công thức nhiễm sắc thể hoặc FISH (NST Ph).
- Xét nghiệm RT-PCR (BCR-ABL1, kiểu gen).
Theo dõi đáp ứng TKI: - Xét nghiệm RQ-PCR mỗi 3 tháng sau điều trị (%IS).
Nguy cơ kháng TKI: - Xét nghiệm công thức nhiễm sắc thể hoặc FISH (đánh giá lui bệnh ở mức di truyền tế bào).
- Xét nghiệm RQ-PCR (đánh giá lui bệnh ở mức phân tử).
- Phân tích đột biến kháng TKI (AS-PCR hoặc giải trình tự gen).
Kháng TKI: - Đánh giá lui bệnh ở mức phân tử (RQ-PCR).
- Phân tích đột biến kháng TKI (AS-PCR hoặc giải trình tự gen).
Tóm tắt ở bảng 61 và bảng 62
Bảng 61. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh CML
|
BCR-ABL1 (%IS) |
3 Tháng |
6 Tháng |
12 Tháng |
≥ 15 Tháng |
|
> 10% |
Nguy cơ kháng TKI |
Kháng TKI |
||
|
> 1-10% |
Đáp ứng TKI |
Nguy cơ kháng TKI |
Kháng TKI |
|
|
≤ 1% |
Đáp ứng TKI |
|||
Bảng 62. Chỉ định phát hiện đột biến trên gen BCR-ABL1/KD và tình trạng kháng TKI tương ứng
|
TKI bị kháng |
phát hiện đột biến kháng thuốc |
|
Imatinib |
Các đột biến gen BCR-ABL1/KD |
|
Nilotinib |
T315I, Y253H, E255K/V, F359V/V/I, G250E |
|
... |
|
4.2. Bệnh Lơ xê mi tủy cấp (AML) (tóm tắt ở bảng 63)
Bảng 63. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh AML
|
Thể bệnh |
Giai đoạn |
Xét nghiệm |
Mục đích |
|
Lơ xê mi tủy cấp thể M3 |
Chẩn đoán ban đầu |
1. Công thức NST/ FISH t(15;17) |
Phát hiện bất thường NST đặc hiệu và các bất thường khác. |
|
2. RT-PCR phát hiện gen PML-RARα. |
Chẩn đoán phân tử bệnh và xác định khả năng điều trị nhắm đích. |
||
|
3. RT-PCR hoặc giải trình tự ADN phát hiện đột biến gen NPM1. |
Tiên lượng bệnh. |
||
|
4. RT-PCR hoặc giải trình tự ADN phát hiện gen đột biến gen FLT3. |
|||
|
Theo dõi điều trị |
1. Công thức NST và/hoặc FISH phát hiện t(15;17) 2. RT-PCR hoặc RQ-PCR hoặc giải trình tự gen phát hiện gen dương tính lúc chẩn đoán. 3. Làm lại toàn bộ xét nghiệm gen ban đầu nếu tái phát |
Theo dõi hiệu quả điều trị. |
|
|
Lơ xê mi cấp khác ngoài M3 |
Chẩn đoán ban đầu |
1. Công thức NST và/hoặc FISH. |
Phát hiện bất thường NST đặc hiệu và các bất thường khác. |
|
2. RT-PCR phát hiện gen AML1-ETO. |
Chẩn đoán phân tử bệnh. |
||
|
3. RT-PCR phát hiện gen CBFB-MYH11. |
Chẩn đoán phân tử bệnh. |
||
|
4. RT-PCR hoặc giải trình tự ADN phát hiện đột biến gen NPM1. |
Xếp loại tiên lượng bệnh. |
||
|
5. RT-PCR hoặc giải trình tự ADN phát hiện đột biến gen FLT3. |
|||
|
6. Giải trình tự ADN phát hiện đột biến gen CEBPA |
|||
|
7. Giải trình tự gen IDH1/ IDH2 với trường hợp gen âm hoặc kháng trị |
|
||
|
Theo dõi điều trị |
1. Công thức NST và/hoặc FISH 2. RT-PCR hoặc RQ-PCR hoặc giải trình tự gen phát hiện gen dương tính lúc chẩn đoán. 3. Làm lại toàn bộ xét nghiệm gen ban đầu nếu tái phát |
Theo dõi hiệu quả điều trị. |
4.3. Bệnh Lơ xê mi Lympho cấp (ALL) (bảng 64)
Bảng 64. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh ALL
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán ban đầu |
1. Công thức NST và/ hoặc FISH |
Phát hiện bất thường NST đặc hiệu và các bất thường khác |
|
2. RT-PCR phát hiện gen BCR-ABL |
Chẩn đoán phân tử bệnh. Tiên lượng bệnh. |
|
|
3. RT-PCR phát hiện gen TEL-AML1 |
||
|
4. RT-PCR phát hiện gen MLL-AF4 |
||
|
5. RT-PCR phát hiện gen E2A-PBX1 |
||
|
Theo dõi điều trị |
1. Công thức NST và/ hoặc FISH 2. RT-PCR hoặc RQ-PCR phát hiện gen dương tính lúc chẩn đoán. 3. Làm lại toàn bộ xét nghiệm gen ban đầu nếu tái phát 4. Giải trình tự gen kháng thuốc với trường hợp điều trị TKI không đáp ứng hoặc tiến triển. |
Theo dõi hiệu quả điều trị. |
4.4. Bệnh Đa u tủy xương (MM) (bảng 65)
Bảng 65. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh MM
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán ban đầu |
1. Công thức NST. |
Phát hiện bất thường NST |
|
2. FISH phát hiện các bất thường t(11;14), t(4;14), t(6;14), t(14;16), t(14;20), del(13q), del(17p) |
Xếp loại tiên lượng và theo dõi điều trị |
|
|
Theo dõi điều trị |
Xét nghiệm FISH Công thức NST |
Theo dõi hiệu quả điều trị |
4.5. Đa hồng nguyên phát (PV) hoặc Tăng tiểu cầu tiên phát (ET) hoặc Xơ tủy nguyên phát (PMF) (bảng 66)
Bảng 66. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh PV hoặc ET hoặc PMF
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán xác định |
1. Công thức NST hoặc FISH |
Phát hiện bất thường NST. |
|
2. PCR phát hiện đột biến JAK2 V617F (PV, ET, PMF) 3. Xét nghiệm tổ hợp gen BCR-ABL1 (loại trừ CML) |
Chẩn đoán |
|
|
4. Giải trình tự ADN xác định đột biến CALR, MPL (ET, PMF) hoặc JAK2 exon12 (PV) nếu JAK2 V617F âm tính |
||
|
5. Giải trình tự ADN xác định đột biến các gen ASXL1, EZH2, SRSF2, U2AF1, IDH1/2 (nếu âm tính cả 3 đột biến gen JAK2, CALR và MPL) |
4.6. Hội chứng rối loạn sinh tủy (MDS) (bảng 67)
Bảng 67. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh MDS
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán ban đầu |
1. Công thức NST. |
Phát hiện bất thường NST đặc hiệu và các bất thường khác. Xếp loại bệnh và đánh giá tiên lượng. |
|
2. FISH phát hiện các bất thường: del(5q), del(7q), del 20q, TET2 |
||
|
3. Xét nghiệm RT-PCR hoặc giải trình tự gen: ASXL1, EZH2, SRSF2, U2AF1, RUNX1, ZRSR2, TP53, STAG2, NRAS , ETV8, GATA2, BCOR, IDH2, NPM1, WT1, PRPF8, FLT3, CBL, SF3B1. IDH1, NEDD, ICPI, TET2, DNMT3A, NF1, JAK2, CALR, MPL, DDX41, SETBP1, PHF6, STAT3, PPM1D, PDGFRβ. |
||
|
Theo dõi điều trị |
Công thức NST Xét nghiệm FISH (đối với chỉ thị dương tính chẩn đoán ban đầu) Xét nghiệm giải trình tự gen (SF3B1, DMNT3A, TET2, AXL1, TP53) Xét nghiệm tổn thương di truyền giống LXM cấp và và làm lại toàn bộ xét nghiệm giống ban đầu nếu tái phát/kháng trị. |
Theo dõi hiệu quả điều trị |
4.7. Lơ xê mi lympho mạn (CLL) (bảng 68)
Bảng 68. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh CLL
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán ban đầu |
1. Công thức NST. |
Phát hiện bất thường NST đặc hiệu và các bất thường khác. |
|
2. FISH phát hiện các bất thường: +12, del (11q), del (13q), del (17p). |
Tiên lượng bệnh. |
|
|
3.Giải trình tự gen IGHV, TP53 |
||
|
Theo dõi điều trị |
Xét nghiệm công thức NST Xét nghiệm FISH (đối với chỉ thị dương tính chẩn đoán ban đầu) Làm lại toàn bộ xét nghiệm tổn thương di truyền ban đầu nếu tái phát/kháng trị |
Theo dõi hiệu quả điều trị. |
4.8. Lơ xê mi tế bào tyc (HCL) (bảng 69)
Bảng 69. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh HCL
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán ban đầu |
1. Công thức NST. |
- Phát hiện bất thường NST đặc hiệu và các bất thường khác. - Tiên lượng bệnh. |
|
2. FISH phát hiện các bất thường +1p, 5q13-q31, del (17p). |
||
|
Khi điều trị |
Công thức NST/FISH nếu có bất thường lúc chẩn đoán |
Theo dõi hiệu quả điều trị. |
4.9. U lympho không Hodgkin (bảng 70)
Bảng 70. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh u lympho không Hodgkin (sử dụng bệnh phẩm là mô hạch)
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán ban đầu |
1. Công thức NST. |
Phát hiện bất thường NST đặc hiệu và các bất thường khác. |
|
2. FISH phát hiện các gen: ALK, BCL6, MYC, CCND1/IgH t(11;14), IgH, IgH/BCL2 t(14;18), MALT1. |
Xếp loại bệnh. Tiên lượng bệnh. |
|
|
Theo dõi điều trị |
Xét nghiệm FISH (đối với chỉ thị dương tính chẩn đoán ban đầu) Làm lại toàn bộ xét nghiệm tổn thương di truyền ban đầu nếu tái phát/kháng trị |
Theo dõi hiệu quả điều trị |
4.10. Hội chứng tăng bạch cầu ưa axit (HES)
Bảng 70. Chỉ định xét nghiệm di truyền và sinh học phân tử trong bệnh u lympho không Hodgkin (sử dụng bệnh phẩm là mô hạch)
|
Giai đoạn |
Xét nghiệm cần làm |
Mục đích |
|
Chẩn đoán ban đầu |
1. Công thức NST. |
Phát hiện bất thường NST đặc hiệu và các bất thường khác. |
|
2. FISH phát hiện bất thường các gen: FGFR1, PDGFRa và PDGFRb. |
Xếp loại bệnh. Tiên lượng bệnh. |
|
|
Theo dõi điều trị |
Xét nghiệm FISH (đối với chỉ thị dương tính chẩn đoán ban đầu) |
Theo dõi hiệu quả điều trị |
Các hướng dẫn chỉ định các xét nghiệm di truyền và sinh học phân tử đề cập ở trên đây chủ yếu tập trung vào các bất thường di truyền thường gặp và đã biết. Tuy nhiên, các phác đồ chẩn đoán, tiên lượng và theo dõi điều trị liên tục được cập nhật trên thế giới và chuyên khoa Huyết học - Truyền máu tại Việt Nam. Do đó, trong một số trường hợp cần thiết, bác sĩ lâm sàng có thể chỉ định thêm để phát hiện các chỉ thị di truyền khác để đánh giá diễn biến lâm sàng và nâng cao chất lượng điều trị cho người bệnh.
TÀI LIỆU THAM KHẢO
1. Nguyễn Anh Trí, 2004. Điều trị các bệnh ác tính cơ quan tạo máu. Nhà xuất bản Y học.
2. Phạm Quang Vinh, 2013. Bất thường di truyền tế bào và bệnh máu ác tính. Nhà xuất bản y học.
3. Hướng dẫn chẩn đoán điều trị bệnh lý huyết học, Bộ Y tế, NXB Y học Việt Nam 2015.
4. R. Fonseca et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia (2009) 23, 2210-2221.
5. Kumar et al. Mayo Clin Proc 2009 84:1095-1110, Revised and updated: June 2010
6. NCCN guidelines (Hướng dẫn chẩn đoán điều trị của Mạng lưới ung thư quốc gia Hoa Kỳ) https://www.nccn.org/professionals/physician_gls/default.aspx
7. ENL recommendation ((Hướng dẫn chẩn đoán điều trị của Mạng lưới ung thư quốc gia Châu Âu) https://www.leukemia-net.org/content/physicians/recommendations/index_eng.html
8. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May 19;127(20):2391-405.
9. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 May 19; 127(20): 2375-2390.
49. HƯỚNG DẪN CHỈ ĐỊNH XÉT NGHIỆM HUYẾT THANH HỌC NHÓM MÁU
1. XÁC ĐỊNH NHÓM MÁU HỆ ABO
1.1. Khái niệm /định nghĩa
Nhóm máu hệ ABO được xác định dựa vào sự có mặt của kháng nguyên A và kháng nguyên B trên bề mặt hồng cầu và sự có mặt hoặc không có mặt của kháng thể chống A và kháng thể chống B trong huyết thanh. Người bình thường trong huyết thanh có kháng thể tự nhiên chống lại kháng nguyên mà kháng nguyên đó lại không có trên bề mặt hồng cầu của chính mình.
Nhóm máu hệ ABO được xác định bằng hai phương pháp là huyết thanh mẫu và hồng cầu mẫu:
- Phương pháp huyết thanh mẫu: sử dụng huyết thanh mẫu chống A, chống B, chống AB để xác định sự có mặt của kháng nguyên A và B trên bề mặt hồng cầu.
- Phương pháp hồng cầu mẫu: sử dụng hồng cầu A, hồng cầu B, hồng cầu O để xác định sự có mặt của kháng thể chống A và kháng thể chống B trong huyết thanh.
1.2. Chỉ định làm xét nghiệm
- Bệnh nhân vào điều trị nội trú hoặc ngoại trú lần đầu;
- Trước khi chỉ định truyền đơn vị máu, chế phẩm máu đầu tiên trong mỗi đợt điều trị.
- Định nhóm máu hệ ABO cho người bệnh và đơn vị máu khi thực hiện phát máu, chế phẩm máu;
- Định nhóm máu hệ ABO cho người hiến máu.
1.3. Lấy máu để làm xét nghiệm (lấy 2 ống)
(1) 2 ml máu tĩnh mạch được chống đông bằng EDTA;
(2) 4 - 5 ml máu tĩnh mạch không chống đông.
1.4. Nhận định kết quả định nhóm máu hệ ABO
- Nhóm A: Trên bề mặt hồng cầu người bệnh có kháng nguyên A và trong huyết thanh người bệnh có kháng thể chống B;
- Nhóm B: Trên bề mặt hồng cầu người bệnh có kháng nguyên B và trong huyết thanh người bệnh có kháng thể chống A;
- Nhóm AB: Trên bề mặt hồng cầu người bệnh có cả kháng nguyên A, B nhưng trong huyết thanh người bệnh lại không có cả kháng thể chống A, chống B;
- Nhóm O: Không có cả kháng nguyên A và kháng nguyên B trên bề mặt hồng cầu người bệnh nhưng trong huyết thanh người bệnh lại có cả kháng thể chống A và chống B.
2. XÁC ĐỊNH NHÓM MÁU HỆ ABO TRONG TRƯỜNG HỢP KHÔNG XÁC ĐỊNH ĐƯỢC BẰNG PHƯƠNG PHÁP THÔNG THƯỜNG (xác định nhóm máu khó hệ ABO)
2.1. Khái niệm/định nghĩa
Các trường hợp định nhóm máu hệ ABO mà có sự bất đồng giữa phương pháp huyết thanh mẫu và phương pháp hồng cầu mẫu gọi là trường hợp nhóm máu hệ ABO khó xác định hay không xác định được bằng phương pháp thông thường.
Tình trạng nhóm máu hệ ABO khó xác định thường chia làm 4 nhóm sau:
- Kháng nguyên yếu;
- Kháng nguyên bất thường trên bề mặt hồng cầu;
- Kháng thể yếu;
- Kháng thể bất thường trong huyết thanh.
Căn cứ vào kết quả định nhóm máu, tiền sử của bệnh nhân mà có các biện pháp xử lê thích hợp để khẳng định nhóm máu hệ ABO cho bệnh nhân/ người hiến máu.
2.2. Chỉ định làm xét nghiệm
Các trường hợp không xác định được nhóm máu hệ ABO bằng phương pháp thông thường.
3. XÁC ĐỊNH NHÓM MÁU HỆ Rh(D)
3.1. Khái niệm/định nghĩa: Sử dụng anti-D để xác định kháng nguyên D trên bề mặt hồng cầu.
3.2. Chỉ định làm xét nghiệm:
- Bệnh nhân vào điều trị nội trú hoặc ngoại trú lần đầu, có thể phải truyền máu;
- Trước khi chỉ định truyền đơn vị máu, chế phẩm máu đầu tiên trong mỗi đợt điều trị.
- Định nhóm máu hệ Rh(D) cho người bệnh khi thực hiện phát khối hồng cầu, máu toàn phần, khối bạch cầu, khối tiểu cầu;
- Định nhóm máu hệ Rh(D) cho đơn vị máu dán nhãn Rh(D) âm khi phát máu;
- Định nhóm máu hệ Rh(D) cho người hiến máu;
3.3. Lấy máu để làm xét nghiệm
- 2 ml máu tĩnh mạch được chống đông bằng EDTA;
3.4. Nhận định kết quả
- Nhóm máu Rh(D) dương: Có kháng nguyên D trên bề mặt hồng cầu;
- Nhóm máu Rh(D) âm: Không có kháng nguyên D trên bề mặt hồng cầu.
4. XÉT NGHIỆM HÒA HỢP MIỄN DỊCH Ở MÔI TRƯỜNG NƯỚC MUỐI SINH LÝ, NHIỆT ĐỘ PHÒNG
4.1. Khái niệm/định nghĩa: Là xét nghiệm dùng để phát hiện các kháng thể có trong huyết thanh của người nhận hoặc người cho mà các kháng thể này có khả năng gây ngưng kết trực tiếp hồng cầu của người cho hoặc người nhận trong môi trường nước muối sinh lý, nhiệt độ phòng xét nghiệm.
4.2. Chỉ định làm xét nghiệm:
Làm xét nghiệm hòa hợp miễn dịch ở môi trường nước muối sinh lý, nhiệt độ phòng giữa huyết tương/huyết thanh của bệnh nhân và hồng cầu của người cho và/hoặc giữa hồng cầu của bệnh nhân và huyết tương của người cho khi thực hiện phát máu toàn phần, các chế phẩm hồng cầu, các chế phẩm bạch cầu, các chế phẩm tiểu cầu, các chế phẩm huyết tương và tủa lạnh.
4.3. Lấy máu để làm xét nghiệm (lấy 2 ống máu)
(1) 2 ml máu tĩnh mạch được chống đông bằng EDTA;
(2) 4 - 5 ml máu tĩnh mạch không chống đông.
4.4. Nhận định kết quả
- Xét nghiệm hòa hợp miễn dịch ở môi trường nước muối sinh lý, nhiệt độ phòng dương tính: Máu người cho và người nhận không hòa hợp;
- Xét nghiệm hòa hợp miễn dịch ở môi trường nước muối sinh lý, nhiệt độ phòng âm tính: Máu người cho và người nhận hòa hợp một số hệ nhóm máu hồng cầu mà kháng thể có bản chất là IgM;
4.5. Chỉ định thêm xét nghiệm khi xét nghiệm hòa hợp miễn dịch ở môi trường nước muối sinh lý, nhiệt độ phòng dương tính
Xét nghiệm lựa chọn đơn vị máu phù hợp (sau khi kiểm tra lại nhóm máu hệ ABO, Rh(D) của người cho và người nhận).
5. XÉT NGHIỆM HÒA HỢP MIỄN DỊCH Ở 37ºC VÀ CÓ SỬ DỤNG KHÁNG GLOBULIN NGƯỜI
5.1. Khái niệm/định nghĩa: Là xét nghiệm ở 37ºC và sử dụng thuốc thử kháng globulin người để phát hiện sự có mặt của kháng thể có bản chất là IgG có trong huyết thanh của bệnh nhân mà đã được gắn lên bề mặt hồng cầu của người cho.
5.2. Chỉ định làm xét nghiệm:
Làm xét nghiệm hòa hợp miễn dịch ở 37°C và có sử dụng kháng globulin người giữa huyết tương/huyết thanh của bệnh nhân và hồng cầu của người cho khi thực hiện phát máu toàn phần, các chế phẩm hồng cầu, các chế phẩm bạch cầu.
5.3. Lấy máu để làm xét nghiệm
- 4 - 5 ml máu tĩnh mạch không chống đông.
5.4. Nhận định kết quả
- Xét nghiệm hòa hợp miễn dịch ở 37°C và có sử dụng kháng globulin người dương tính: Máu người cho và người nhận không hòa hợp;
- Xét nghiệm hòa hợp miễn dịch ở môi trường nước muối sinh lý, nhiệt độ phòng âm tính: Máu người cho và người nhận hòa hợp.
5.5. Chỉ định thêm xét nghiệm khi xét nghiệm hòa hợp miễn dịch ở 37°C và có sử dụng kháng globulin người dương tính
Xét nghiệm lựa chọn đơn vị máu phù hợp.
6. XÉT NGHIỆM LỰA CHỌN ĐƠN VỊ MÁU PHÙ HỢP
6.1. Khái niệm/định nghĩa: Là xét nghiệm hòa hợp miễn dịch giữa huyết thanh của bệnh nhân với 10 đơn vị máu phù hợp nhóm máu hệ ABO, Rh(D) với bệnh nhân ở các điều kiện: môi trường nước muối sinh lý, nhiệt độ phòng, 37ºC và có sử dụng kháng globulin người để lựa chọn đơn vị máu hòa hợp nhất với bệnh nhân.
6.2. Chỉ định làm xét nghiệm:
- Phản ứng hòa hợp dương tính.
- Không lựa chọn được đơn vị máu có kháng nguyên phù hợp với kết quả xác định kháng nguyên nhóm máu ngoài hệ ABO hoặc không lựa chọn được đơn vị máu có kháng nguyên phù hợp với kết quả định danh kháng thể bất thường của bệnh nhân.
6.3. Lấy máu để làm xét nghiệm
- 4 - 5 ml máu tĩnh mạch không chống đông.
6.4. Nhận định kết quả
- Chọn được đơn vị máu hòa hợp: Khi kết quả xét nghiệm hòa hợp miễn dịch giữa huyết thanh bệnh nhân với hồng cầu của đơn vị máu âm tính ở tất cả các điều kiện: môi trường nước muối sinh lý, nhiệt độ phòng, 37ºC và có sử dụng kháng globulin người;
- Không chọn được đơn vị máu hòa hợp: Khi kết quả xét nghiệm hòa hợp miễn dịch giữa huyết thanh bệnh nhân với hồng cầu của 10 đơn vị máu đều dương tính.
6.5. Chỉ định thêm xét nghiệm khi không chọn được đơn vị máu hòa hợp
- Sàng lọc kháng thể bất thường.
- Định danh kháng thể bất thường.
7. XÉT NGHIỆM COOMBS TRỰC TIẾP
7.1. Khái niệm/định nghĩa
Xét nghiệm Coombs trực tiếp là một xét nghiệm sử dụng thuốc thử kháng globulin người để phát hiện các kháng thể miễn dịch (đồng miễn dịch và tự miễn dịch) đã cảm nhiễm trên bề mặt hồng cầu.
7.2. Chỉ định làm xét nghiệm Coombs trực tiếp
- Bệnh nhân có tình trạng thiếu máu nghi tan máu do nguyên nhân miễn dịch (đồng miễn dịch và tự miễn dịch).
- Các trường hợp nghi ngờ bệnh lý tự miễn: lupus ban đỏ hệ thống, thiếu máu tan máu tự miễn, hội chứng Evans ...
7.3. Mẫu máu để làm xét nghiệm
2ml máu tĩnh mạch được chống đông bằng EDTA.
7.4. Nhận định kết quả
Xét nghiệm Coombs trực tiếp dương tính khi có kháng thể miễn dịch (đồng miễn dịch hoặc tự miễn dịch) cảm nhiễm trên bề mặt hồng cầu.
7.5. Chỉ định thêm xét nghiệm khi kết quả xét nghiệm Coombs trực tiếp dương tính
- Xác định bản chất kháng thể;
- Sàng lọc kháng thể bất thường;
- Định danh kháng thể bất thường.
8. XÉT NGHIỆM COOMBS GIÁN TIẾP
8.1. Khái niệm/định nghĩa
Xét nghiệm Coombs gián tiếp là xét nghiệm sử dụng thuốc thử kháng globulin người để phát hiện các các kháng thể miễn dịch (đồng miễn dịch và tự miễn dịch) có mặt tự do trong huyết thanh của người bệnh.
8.2. Chỉ định làm xét nghiệm Coombs gián tiếp
- Bệnh nhân có tình trạng thiếu máu nghi tan máu do nguyên nhân miễn dịch (đồng miễn dịch và tự miễn dịch)
- Các trường hợp nghi ngờ bệnh lý tự miễn: lupus ban đỏ hệ thống, thiếu máu tan máu tự miễn, hội chứng Evans ...
8.3. Lấy máu để xét nghiệm
4 - 5 ml máu tĩnh mạch không chống đông.
8.4. Nhận định kết quả
Xét nghiệm Coombs gián tiếp dương tính khi có kháng thể miễn dịch (đồng miễn dịch hoặc tự miễn dịch) lưu hành trong huyết thanh của người bệnh.
8.5. Chỉ định thêm xét nghiệm khi có kết quả xét nghiệm Coombs gián tiếp dương tính
- Sàng lọc kháng thể bất thường;
- Định danh kháng thể bất thường;
- Xác định hiệu giá kháng thể miễn dịch chống lại kháng nguyên hồng cầu.
9. SÀNG LỌC KHÁNG THỂ BẤT THƯỜNG
9.1. Khái niệm/định nghĩa: Sàng lọc kháng thể bất thường là một xét nghiệm được chỉ định để phát hiện các kháng thể bất thường có trong huyết thanh của người bệnh hoặc sản phụ hoặc người hiến máu.
9.2. Chỉ định
- Xét nghiệm cho đơn vị máu của người hiến;
- Xét nghiệm cho phụ nữ có thai; có tiền sử sinh con bị tan máu trẻ sơ sinh
- Bệnh nhân dự kiến cần truyền máu mà có tiền sử truyền máu hoặc phụ nữ có tiền sử chửa đẻ, sảy thai.
- Trong mỗi đợt điều trị có truyền máu nhiều lần, lặp lại xét nghiệm định kỳ 10 ngày/lần tính từ lần xét nghiệm gần nhất và có truyền máu sau lần xét nghiệm đó (loại sau 7 ngày gửi máu).
- Bệnh nhân truyền máu không hiệu quả, thể hiện bằng một trong các dấu hiệu sau:
+ Huyết sắc tố tăng không tương ứng với thể tích máu đã truyền;
+ Có tình trạng thiếu máu tiến triển nhanh;
+ Tăng bilirubin máu mà không phát hiện được nguyên nhân rõ ràng nào khác.
9.3. Lấy máu để làm xét nghiệm (lấy 2 ống máu)
(1) 2 ml máu tĩnh mạch được chống đông bằng EDTA;
(2) 4 - 5 ml máu tĩnh mạch không chống đông.
9.4. Nhận định kết quả
Xét nghiệm sàng lọc kháng thể bất thường dương tính: Có kháng thể bất thường trong huyết thanh.
9.5. Chỉ định thêm xét nghiệm khi có kết quả xét nghiệm sàng lọc kháng thể bất thường dương tính
- Định danh kháng thể bất thường;
- Xác định hiệu giá kháng thể miễn dịch chống lại kháng nguyên hồng cầu.
10. ĐỊNH DANH KHÁNG THỂ BẤT THƯỜNG
10.1. Khái niệm/định nghĩa
Định danh kháng thể bất thường là một xét nghiệm được chỉ định để xác định tên các loại kháng thể bất thường có trong huyết thanh của người bệnh, đơn vị máu hiến và sản phụ mà đã có kết quả xét nghiệm sàng lọc kháng thể bất thường dương tính. Mục đích của xét nghiệm định danh kháng thể bất thường là để lựa chọn đơn vị máu phù hợp nhất để truyền cho người bệnh.
10.2. Chỉ định làm xét nghiệm
- Khi xét nghiệm sàng lọc kháng thể bất thường cho kết quả dương tính.
- Bệnh nhân đã được truyền máu hòa hợp theo kết quả định danh kháng thể bất thường nhưng không hiệu quả (phần b, mục 3).
10.3. Lấy máu để làm xét nghiệm (lấy 2 ống máu)
(1) 2 ml máu tĩnh mạch được chống đông bằng EDTA;
(2) 4 - 5 ml máu tĩnh mạch không chống đông.
10.4. Nhận định kết quả
Xác định được tên của các kháng thể bất thường.
10.5. Chỉ định thêm xét nghiệm khi đã xác định được tên kháng thể bất thường
Xác định hiệu giá kháng thể miễn dịch chống lại kháng nguyên hồng cầu.
11. XÁC ĐỊNH KHÁNG NGUYÊN NHÓM HỒNG CẦU NGOÀI HỆ ABO
11.1. Khái niệm/định nghĩa
Sử dụng các kháng huyết thanh chuẩn để xác định kháng nguyên của các hệ nhóm máu hồng cầu (Ví dụ như xác định kháng nguyên Fya của hệ Duffy, xác định kháng nguyên M của hệ MNS…).
11.2. Chỉ định làm xét nghiệm
- Các trường hợp bệnh nhân phải điều trị chính bằng phương pháp truyền máu và/ hoặc bệnh có nguy cơ xuất hiện kháng thể bất thường cao như: thalassemia, suy tủy xương, hội chứng rối loạn sinh tủy...
- Để xác định kháng thể bất thường đã định danh (xác định sự vắng mặt kháng nguyên tương ứng trên hồng cầu người bệnh)
- Loại kháng nguyên nhóm máu ngoài hệ ABO cần xét nghiệm bao gồm các kháng nguyên: Rh (D, C, c, E, e), MNS (Mia, M, N, S, s), Kidd (Jka, Jkb), Duffy (Fya, Fyb), Lewis (Lea, Leb), P1PK (P1), Kell (K, k), Lutheran (Lua, Lub).
11.3. Lấy máu để làm xét nghiệm
2 ml máu tĩnh mạch được chống đông bằng EDTA.
11.4. Nhận định kết quả xác định kháng nguyên nhóm máu
- Kết quả xét nghiệm dương tính: Có kháng nguyên nhóm máu cần xác định trên bề mặt hồng cầu.
- Kết quả xét nghiệm âm tính: Không có kháng nguyên nhóm máu cần xác định trên bề mặt hồng cầu.
12. XÁC ĐỊNH BẢN CHẤT KHÁNG THỂ HỒNG CẦU
12.1. Khái niệm/định nghĩa: Là xét nghiệm sử dụng thuốc thử kháng globulin người đơn giá để xác định bản chất của kháng thể đã cảm nhiễm trên bề mặt hồng cầu.
12.2. Chỉ định làm xét nghiệm:
Bệnh nhân có kết quả xét nghiệm Coombs trực tiếp dương tính.
12.3. Lấy máu để làm xét nghiệm
2 ml máu tĩnh mạch chống đông bằng EDTA.
12.4. Nhận định kết quả
- Hồng cầu bệnh nhân cho phản ứng ngưng kết với anti-IgG: Kháng thể có bản chất là IgG.
- Hồng cầu bệnh nhân cho phản ứng ngưng kết với anti-IgM: Kháng thể có bản chất là IgM.
- Hồng cầu bệnh nhân cho phản ứng ngưng kết với anti-IgA: Kháng thể có bản chất là IgA.
- Hồng cầu bệnh nhân cho phản ứng ngưng kết với anti-C3d: Kháng thể có bản chất là C3d.
- Hồng cầu bệnh nhân cho phản ứng ngưng kết với anti-C3c: Kháng thể có bản chất là C3c.
13. XÉT NGHIỆM HÒA HỢP TIỂU CẦU
13.1. Khái niệm/định nghĩa:
Tiểu cầu của người cho được gắn vào bề mặt của giếng polystyrene microplate. Huyết thanh/huyết tương của bệnh nhân được thêm vào giếng và ủ với tiểu cầu của người cho, nếu trong huyết thanh của bệnh nhân có kháng thể kháng tiểu cầu thì các kháng thể này sẽ gắn với tiểu cầu của người cho. Các globulin miễn dịch không gắn lên tiểu cầu có trong giếng sẽ được rửa sạch, sau đó thêm vào giếng dung dịch hồng cầu chỉ thị đã cảm nhiễm với anti-IgG. Ly tâm để các hồng cầu chỉ thị tiếp xúc được với các kháng thể kháng tiểu cầu có trong huyết thanh bệnh nhân đã được gắn với các tiểu cầu của người cho. Trong trường hợp xét nghiệm dương tính, sự di chuyển của các hồng cầu chỉ thị xuống đáy giếng bị cản trở bởi cầu nối anti-IgG được hình thành giữa các hồng cầu chỉ thị và kháng thể kháng tiểu cầu đã gắn lên tiểu cầu từ trước. Với nguyên lê bắc cầu như vậy, các hồng cầu chỉ thị sẽ bao phủ lên toàn bộ bề mặt giếng. Ngược lại trong trường hợp xét nghiệm âm tính, các hồng cầu chỉ thị sẽ không bị cản trở trong quá trình di chuyển, theo lực ly tâm sẽ lắng chặt xuống đáy giếng.
13.2. Chỉ định làm xét nghiệm:
Bệnh nhân có tình trạng truyền khối tiểu cầu không hiệu quả hoặc có phản ứng với lần truyền trước đó:
- Số lượng tiểu cầu trong máu ngoại vi sau truyền tăng không quá 50% so với dự tính;
- Có phản ứng lâm sàng kiểu miễn dịch quá mẫn (mẩn ngứa, gai rét, tụt huyết áp...) hoặc vẫn tiếp tục xuất huyết.
13.3. Lấy máu để làm xét nghiệm
2 ml máu tĩnh mạch chống đông bằng EDTA hoặc 4-5 ml máu tĩnh mạch không chống đông.
13.4. Nhận định kết quả
- Phản ứng ngưng kết: Các hồng cầu chỉ thị sẽ bao phủ lên toàn bộ bề mặt giếng, kết luận phản ứng hòa hợp tiểu cầu dương tính, đơn vị tiểu cầu không hòa hợp với bệnh nhân;
- Phản ứng không ngưng kết: Các hồng cầu chỉ thị lắng chһt xuống đáy giếng, kết luận phản ứng hòa hợp tiểu cầu âm tính, đơn vị tiểu cầu hòa hợp với bệnh nhân.
13.5. Chỉ định thêm xét nghiệm khi xét nghiệm hòa hợp tiểu cầu dương tính
- Sàng lọc kháng thể kháng tiểu cầu.
- Định danh kháng thể kháng tiểu cầu.
14. XÁC ĐỊNH KHÁNG NGUYÊN NHÓM MÁU HỒNG CẦU BẰNG PHƯƠNG PHÁP SINH HỌC PHÂN TỬ
14.1. Khái niệm/định nghĩa:
Kháng nguyên nhóm máu là các glyco - protein trên màng hồng cầu; có thể xác định gián tiếp bằng cách phân tích sự có mặt gen mã hóa hay mARN các protein này bằng phương pháp sinh học phân tử. Dựa trên kỹ thuật PCR-SSP (PCR với những đoạn mồi có trình tự đặc hiệu):
Phản ứng chuỗi polymerase (PCR) cho phép khuếch đại trình tự ADN đã được xác định. Sau khi khuếch đại thành công, chuỗi ADN đích được nhân lên với lượng vừa đủ. PCR-SSP sử dụng các đoạn mồi có trình tự đặc hiệu với alen. Để có thể phân tích kết quả phản ứng PCR-SSP, nhiều phản ứng khuếch đại đã được thực hiện đồng thời. Những mẫu PCR gắn với đoạn mồi đặc hiệu sẽ được khuếch đại sau phản ứng PCR, trong khi những mẫu không gắn với đoạn mồi đặc hiệu thì sẽ không được khuếch đại sau phản ứng PCR. Việc phát hiện của các sản phẩm PCR được thực hiện bằng cách đo các tín hiệu huỳnh quang bằng máy đọc huỳnh quang.
14.2. Chỉ định làm xét nghiệm:
Xác định kháng nguyên nhóm máu bằng phương pháp sinh học phân tử trong trường hợp:
- Bệnh nhân có nhóm máu hệ ABO khó xác định bằng phương pháp huyết thanh học: Bệnh nhân có xét nghiệm Coombs trực tiếp dương tính, bệnh nhân mới truyền máu, truyền máu nhiều lần, bệnh nhân có nhóm A yếu, B yếu …
- Bệnh nhân có nhóm máu D yếu, D từng phần, D âm;
- Theo dõi sự chuyển đổi nhóm máu ở bệnh nhân ghép tế bào gốc đồng loài có bất đồng kháng nguyên nhóm máu với người cho;
14.3. Lấy máu để làm xét nghiệm
2 ml máu tĩnh mạch chống đông bằng EDTA.
14.4. Nhận định kết quả
- Phản ứng dương tính: Có sản phẩm PCR, phát tín hiệu huỳnh quang và được phát hiện bằng máy đọc huỳnh quang: Có kháng nguyên cần xác định trên bề mặt hồng cầu.
- Phản ứng âm tính: Không có sản phẩm PCR, không phát tín hiệu huỳnh quang: Không có kháng nguyên cần xác định trên bề mặt hồng cầu.
15. HIỆU GIÁ KHÁNG THỂ TỰ NHIÊN CHỐNG A/ CHỐNG B
15.1. Khái niệm/định nghĩa:
Hiệu kháng thể là một phương pháp bán định lượng để xác định nồng độ kháng thể. Huyết thanh bệnh nhân được pha loãng theo tỷ lệ 1:2, 1:4, 1:8 … sau đó cho phản ứng với hồng cầu mang kháng nguyên tương ứng với kháng thể cần hiệu giá. Nghịch đảo của độ pha loãng cao nhất của huyết thanh tạo ra phản ứng 1+ với hồng cầu được gọi là hiệu giá.
Hiệu giá kháng thể tự nhiên chống A/chống B là sử dụng hồng cầu mang kháng nguyên A/ kháng nguyên B cho phản ứng với huyết thanh đã pha loãng ở điều kiện nhiệt độ phòng xét nghiệm để xác định nồng độ kháng thể chống A/chống B tự nhiên trong huyết thanh của bệnh nhân.
15.2. Chỉ định làm xét nghiệm:
- Bệnh nhân ghép có bất đồng nhóm máu hệ ABO với người cho cần theo dõi sự chuyển đổi nhóm máu.
- Bệnh nhân sinh con vàng da tan máu nghi do bất đồng nhóm máu mẹ con hệ ABO.
15.3. Lấy máu để làm xét nghiệm
4-5 ml máu tĩnh mạch không chống đông.
15.4. Nhận định kết quả
- Âm tính: Huyết thanh bệnh nhân không cho phản ứng ngưng kết với hồng cầu ở tất cả các độ pha loãng.
- Dương tính: Huyết thanh bệnh nhân cho phản ứng ngưng kết với hồng cầu. Hiệu giá kháng thể là nghịch đảo của độ pha loãng cao nhất của huyết thanh tạo ra phản ứng 1+ với hồng cầu. Ví dụ: huyết thanh pha loãng 1:16 cho phản ứng ngưng kết 1+ với hồng cầu thì hiệu giá kháng thể là 16.
16. HIỆU GIÁ KHÁNG THỂ MIỄN DỊCH
16.1. Khái niệm/định nghĩa:
Hiệu kháng thể là một phương pháp bán định lượng để xác định nồng độ kháng thể. Huyết thanh bệnh nhân được pha loãng theo tỷ lệ 1:2, 1:4, 1:8… sau đó cho phản ứng với hồng cầu mang kháng nguyên tương ứng với kháng thể cần hiệu giá. Nghịch đảo của độ pha loãng cao nhất của huyết thanh tạo ra phản ứng 1+ với hồng cầu được gọi là hiệu giá.
Hiệu giá kháng thể miễn dịch là sử dụng hồng cầu mang kháng nguyên tương ứng với kháng thể cần hiệu giá (ví dụ: cần hiệu giá kháng thể miễn dịch chống D thì sử dụng hồng cầu Rh(D) dương) cho phản ứng với huyết thanh đã pha loãng ở điều kiện kháng globulin người để xác định nồng độ kháng thể miễn dịch trong huyết thanh của bệnh nhân.
16.2. Chỉ định làm xét nghiệm:
- Bệnh nhân ghép có bất đồng nhóm máu hệ ABO hoặc bất đồng kháng nguyên nhóm máu ngoài hệ ABO với người cho cần theo dõi sự chuyển đổi nhóm máu.
- Bệnh nhân (trẻ sơ sinh) và mẹ bệnh nhân vàng da tan máu nghi do bất đồng nhóm máu mẹ con.
16.3. Lấy máu để làm xét nghiệm
4-5 ml máu tĩnh mạch không chống đông.
16.4. Nhận định kết quả
- Âm tính: Huyết thanh bệnh nhân không cho phản ứng ngưng kết với hồng cầu ở tất cả các độ pha loãng.
- Dương tính: Huyết thanh bệnh nhân cho phản ứng ngưng kết với hồng cầu. Hiệu giá kháng thể là nghịch đảo của độ pha loãng cao nhất của huyết thanh tạo ra phản ứng 1+ với hồng cầu. Ví dụ: huyết thanh pha loãng 1:16 cho phản ứng ngưng kết 1+ với hồng cầu thì hiệu giá kháng thể là 16.
TÀI LIỆU THAM KHẢO
1. Đỗ Trung Phấn (2013), Kỹ thuật xét nghiệm huyết học và truyền máu ứng dụng trong lâm sàng (Tái bản lần 2), Nhà xuất bản y học năm 2013.
2. Denise M Harmening (1999) Modern blood banking and transfusion practices, fourth edition, Book Promotion & Service Co, LTD.
3. Geoff Daniels and Imelda Bromilow (2009), Essential Guide to Blood Groups, Blackwell.
4. Mark E. Brecher (2005), Technical Manual 15th Edition, AABB.
5. Capture-P - Solid Phase System for the Detection of IgG Antibodies to Platelets, Immuco Gamma.
6. Hướng dẫn sử dụng bộ kít E.Z.N.A Blood DNA kit của hãng Omega.
7. Hướng dẫn sử dụng bộ kít RBC-FluoGene ABO basic của hãng Inno-Train, Đức.
8. Hướng dẫn sử dụng bộ kít RBC-FluoGene VERYfy của hãng Inno-Train, Đức.
9. Hướng dẫn sử dụng bộ kít RBC-FluoGene D screen của hãng Inno-Train, Đức.
Bạn chưa Đăng nhập thành viên.
Đây là tiện ích dành cho tài khoản thành viên. Vui lòng Đăng nhập để xem chi tiết. Nếu chưa có tài khoản, vui lòng Đăng ký tại đây!
Bạn chưa Đăng nhập thành viên.
Đây là tiện ích dành cho tài khoản thành viên. Vui lòng Đăng nhập để xem chi tiết. Nếu chưa có tài khoản, vui lòng Đăng ký tại đây!
