- Tổng quan
- Nội dung
- VB gốc
- Tiếng Anh
- Hiệu lực
- VB liên quan
- Lược đồ
-
Nội dung hợp nhất
Tính năng này chỉ có tại LuatVietnam.vn. Nội dung hợp nhất tổng hợp lại tất cả các quy định còn hiệu lực của văn bản gốc và các văn bản sửa đổi, bổ sung, đính chính... trên một trang. Việc hợp nhất văn bản gốc và những văn bản, Thông tư, Nghị định hướng dẫn khác không làm thay đổi thứ tự điều khoản, nội dung.
Khách hàng chỉ cần xem Nội dung hợp nhất là có thể nắm bắt toàn bộ quy định hiện hành đang áp dụng, cho dù văn bản gốc đã qua nhiều lần chỉnh sửa, bổ sung.
- Tải về
Thông tư 32/2018/TT-BYT về việc đăng ký lưu hành thuốc nguyên liệu làm thuốc
| Cơ quan ban hành: | Bộ Y tế |
Số công báo:
Số công báo là mã số ấn phẩm được đăng chính thức trên ấn phẩm thông tin của Nhà nước. Mã số này do Chính phủ thống nhất quản lý.
|
Đang cập nhật |
| Số hiệu: | 32/2018/TT-BYT | Ngày đăng công báo: | Đang cập nhật |
| Loại văn bản: | Thông tư | Người ký: | Trương Quốc Cường |
|
Ngày ban hành:
Ngày ban hành là ngày, tháng, năm văn bản được thông qua hoặc ký ban hành.
|
12/11/2018 |
Ngày hết hiệu lực:
Ngày hết hiệu lực là ngày, tháng, năm văn bản chính thức không còn hiệu lực (áp dụng).
|
Đã biết
|
|
Áp dụng:
Ngày áp dụng là ngày, tháng, năm văn bản chính thức có hiệu lực (áp dụng).
|
Đã biết
|
Tình trạng hiệu lực:
Cho biết trạng thái hiệu lực của văn bản đang tra cứu: Chưa áp dụng, Còn hiệu lực, Hết hiệu lực, Hết hiệu lực 1 phần; Đã sửa đổi, Đính chính hay Không còn phù hợp,...
|
Đã biết
|
| Lĩnh vực: | Y tế-Sức khỏe |
TÓM TẮT THÔNG TƯ 32/2018/TT-BYT
Ngày 12/11/2018, Bộ Y tế ban hành Thông tư 32/2018/TT-BYT quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
Theo đó, hiệu lực, ký hiệu của giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc được quy định như sau:
- Thời hạn hiệu lực của giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc là 05 năm kể từ ngày cấp hoặc gia hạn;
- Thời hạn hiệu lực của giấy đăng ký lưu hành là 03 năm kể từ ngày cấp đối với các thuốc sau: Thuốc mới, vắc xin lần đầu cấp giấy đăng ký lưu hành, sinh phẩm tham chiếu, sinh phẩm tương tự lần đầu cấp giấy đăng ký lưu hành tại Việt Nam; thuốc cùng dược chất, nồng độ, hàm lượng, dạng bào chế với thuốc mà mới đó chưa được cấp giấy đăng ký lưu hành với thời hạn 05 năm; thuốc mà tại thời điểm nộp hồ sơ gia hạn giấy đăng ký lưu hành có báo cáo an toàn, hiệu quả tại thời điểm nộp hồ sơ gia hạn giấy đăng ký lưu hành mà chưa có báo cáo an toàn, hiệu quả vì lý do chưa lưu hành…
Ngoài ra, Thông tư còn quy định về việc gia hạn giấy đăng ký lưu hành thuốc, yêu cầu đối với đăng ký thuốc sản xuất tại Việt Nam theo hình thức chuyển giao công nghệ, yêu cầu bảo mật dữ liệu đối với hồ sơ đăng ký thuốc…
Thông tư có hiệu lực từ ngày 01/9/2019.
Văn bản này được thay thế bởi 08/2022/TT-BYT
Xem chi tiết Thông tư 32/2018/TT-BYT có hiệu lực kể từ ngày 01/09/2019
Tải Thông tư 32/2018/TT-BYT
 Thông tư 32/2018/TT-BYT PDF (Bản có dấu đỏ)
Thông tư 32/2018/TT-BYT PDF (Bản có dấu đỏ) Thông tư 32/2018/TT-BYT DOC (Bản Word)
Thông tư 32/2018/TT-BYT DOC (Bản Word)|
BỘ Y TẾ Số: 32/2018/TT-BYT |
CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM Hà Nội, ngày 12 tháng 11 năm 2018 |
THÔNG TƯ
QUY ĐỊNH VIỆC ĐĂNG KÝ LƯU HÀNH THUỐC, NGUYÊN LIỆU LÀM THUỐC
Căn cứ Luật dược ngày 06 tháng 4 năm 2016;
Căn cứ Nghị định số 54/2017/NĐ-CP ngày 08 tháng 5 năm 2017 của Chính phủ quy định chi tiết một số điều và biện pháp thi hành Luật dược;
Căn cứ Nghị định số 75/2017/NĐ-CP ngày 20 tháng 6 năm 2017 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Căn cứ Nghị định số 155/2018/NĐ-CP ngày 12 tháng 11 năm 2018 của Chính phủ sửa đổi, bổ sung một số quy định liên quan đến điều kiện đầu tư kinh doanh thuộc phạm vi quản lý nhà nước của Bộ Y tế;
Theo đề nghị của Cục trưởng Cục Quản lý dược,
Bộ trưởng Bộ Y tế ban hành Thông tư quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
Chương I
QUY ĐỊNH CHUNG
Điều 1. Phạm vi điều chỉnh
1. Thông tư này quy định chi tiết:
a) Hồ sơ, thủ tục cấp, gia hạn, thay đổi, bổ sung, thu hồi giấy đăng ký lưu hành thuốc hóa dược, vắc xin, sinh phẩm, thuốc dược liệu và nguyên liệu làm thuốc (dược chất, bán thành phẩm dược liệu, tá dược, vỏ nang) dùng cho người tại Việt Nam;
b) Yêu cầu về dữ liệu lâm sàng để bảo đảm an toàn, hiệu quả trong hồ sơ đăng ký thuốc;
c) Tiêu chí để xác định trường hợp miễn thử, miễn một số giai đoạn thử thuốc trên lâm sàng tại Việt Nam, thuốc phải yêu cầu thử lâm sàng giai đoạn 4;
d) Nguyên tắc tổ chức, hoạt động của chuyên gia thẩm định hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
đ) Nguyên tắc tổ chức, hoạt động của chuyên gia thẩm định hồ sơ đề nghị cấp phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành đối với các trường hợp quy định tại điểm a khoản 43 Điều 5 Nghị định số 155/2018/NĐ-CP;
e) Nguyên tắc tổ chức, hoạt động của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
g) Trình tự thẩm định hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc; trình tự thẩm định hồ sơ nhập khẩu thuốc chưa có giấy đăng ký lưu hành.
2. Thông tư này không áp dụng cho các trường hợp tại khoản 2 Điều 54 Luật dược quy định các nguyên liệu làm thuốc không phải đăng ký trước khi lưu hành tại Việt Nam, bán thành phẩm dược liệu do chính cơ sở sản xuất để sản xuất thuốc thành phẩm.
Điều 2. Giải thích từ ngữ
Trong Thông tư này, các từ ngữ dưới đây được hiểu như sau:
1. Hồ sơ kỹ thuật chung ASEAN (ACTD) là bộ tài liệu hướng dẫn hồ sơ đăng ký thuốc đáp ứng các yêu cầu kỹ thuật chung của Hiệp hội các nước Đông Nam Á (ASEAN) được quy định tại Phụ lục I ban hành kèm theo Thông tư này.
2. Hồ sơ kỹ thuật chung ICH-CTD là mẫu hồ sơ chung của Hội nghị quốc tế về hài hòa các thủ tục đăng ký dược phẩm sử dụng cho con người.
3. Thay đổi lớn là những thay đổi có ảnh hưởng rõ rệt, trực tiếp đến chất lượng, an toàn và hiệu quả của thuốc, được quy định tại Phụ lục II ban hành kèm theo Thông tư này.
4. Thay đổi nhỏ là những thay đổi không ảnh hưởng hoặc ảnh hưởng rất ít đến hiệu quả, chất lượng và an toàn của thuốc, được quy định tại Phụ lục II ban hành kèm theo Thông tư này.
5. Cơ sở đăng ký thuốc, nguyên liệu làm thuốc là cơ sở đứng tên nộp đơn đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
6. Cơ sở sản xuất thuốc là cơ sở thực hiện một, một số công đoạn hoặc toàn bộ quá trình sản xuất hoặc thực hiện việc xuất xưởng lô thuốc.
7. Cơ sở sản xuất nguyên liệu làm thuốc là cơ sở sản xuất ra nguyên liệu để sản xuất thuốc thành phẩm hoặc cơ sở thực hiện việc xuất xưởng lô nguyên liệu làm thuốc.
8. Chủ sở hữu giấy phép lưu hành thuốc (product license holder) hoặc chủ sở hữu sản phẩm (product owner) đối với thuốc nước ngoài là cơ sở hợp pháp chịu trách nhiệm về sản phẩm và được ghi trên Giấy chứng nhận sản phẩm dược phẩm (CPP).
9. Cơ quan quản lý tham chiếu quy định trong Thông tư này bao gồm: Cơ quan quản lý Dược phẩm Châu Âu (EMA), Mỹ, Nhật Bản, Pháp, Đức, Thụy Điển, Anh, Thụy Sỹ, Úc, Canada, Bỉ, Áo, Ai Len, Đan Mạch và Hà Lan.
10. Cơ quan quản lý dược chặt chẽ (SRA - Stringent Regulatory Authorities) là các cơ quan quản lý dược được Tổ chức Y tế thế giới (WHO) phân loại thuộc danh sách SRA, bao gồm:
a) Thành viên ICH trước 23 tháng 10 năm 2015, bao gồm: Cơ quan Quản lý Dược và thực phẩm Mỹ (US-FDA), Cơ quan quản lý dược của các nước thuộc Ủy ban Châu Âu (European Commission), Cơ quan quản lý thuốc và các sản phẩm y tế của Vương quốc Anh (MHRA), Cơ quan quản lý dược và trang thiết bị y tế Nhật Bản (PMDA);
b) Thành viên quan sát của ICH trước ngày 23 tháng 10 năm 2015 bao gồm: Cơ quan quản lý Dược thuộc Hiệp hội thương mại tự do Châu Âu (EFTA- European Free Trade Association) với đại diện Cơ quan quản lý Dược Thụy Sỹ (Swissmedic) và Bộ Y tế Canada (Health Canada);
c) Thành viên có hiệp định liên kết, công nhận lẫn nhau với Thành viên ICH trước ngày 23 tháng 10 năm 2015 bao gồm: Úc, Ai xơ len, Liechtenstein và Na Uy.
11. Giấy chứng nhận sản phẩm dược phẩm (CPP) là giấy chứng nhận được cấp theo Hệ thống chứng nhận chất lượng các sản phẩm dược phẩm lưu hành trong thương mại quốc tế của Tổ chức Y tế thế giới (WHO).
12. Bán thành phẩm dược liệu là nguyên liệu để sản xuất thuốc có nguồn gốc dược liệu dưới dạng cao, cốm, bột, dịch chiết, tinh dầu, nhựa, gôm, thạch.
Điều 3. Trách nhiệm đối với cơ sở đăng ký thuốc, nguyên liệu làm thuốc
1. Chịu trách nhiệm về việc thay đổi, bổ sung nội dung nhãn, hướng dẫn sử dụng thuốc theo đúng nội dung trong văn bản yêu cầu cập nhật của Bộ Y tế (Cục Quản lý Dược) trong thời gian giấy đăng ký lưu hành thuốc còn hiệu lực mà không phải nộp hồ sơ đăng ký thay đổi, bổ sung.
2. Thực hiện đăng ký thay đổi, bổ sung theo quy định tại khoản 4 Điều 28 và Điều 40 ban hành kèm Thông tư này trong thời gian giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc còn hiệu lực.
3. Bảo đảm chất lượng, an toàn, hiệu quả của thuốc, nguyên liệu làm thuốc đúng với hồ sơ đăng ký.
4. Chịu trách nhiệm về tính pháp lý và tính chính xác của tất cả các tài liệu trong hồ sơ đăng ký. Phối hợp với cơ sở sản xuất, cơ quan có thẩm quyền của nước ngoài trong việc trả lời các văn bản của Cục Quản lý Dược đề nghị kiểm tra tính xác thực của các giấy tờ pháp lý có liên quan trong hồ sơ đăng ký thuốc.
5. Thông báo bằng văn bản cho Cục Quản lý Dược trong thời hạn 30 ngày kể từ ngày có quyết định thu hồi giấy đăng ký lưu hành tại bất kỳ nước nào trên thế giới đối với thuốc, nguyên liệu làm thuốc đã được cấp giấy đăng ký lưu hành tại Việt Nam còn hiệu lực và nêu rõ lý do bị thu hồi.
6. Phối hợp chặt chẽ với cơ sở sản xuất thuốc để bảo đảm ít nhất một trong hai cơ sở này phải thực hiện nghiên cứu hoặc cung cấp thêm thông tin liên quan đến thuốc đăng ký khi có thông tin hoặc bằng chứng liên quan đến tính an toàn và hiệu quả của thuốc trong quá trình lưu hành theo yêu cầu của cơ quan quản lý nhà nước có thẩm quyền.
7. Phối hợp với cơ sở sản xuất, nhập khẩu, phân phối thuốc thực hiện việc theo dõi, giám sát, thu thập, tổng hợp, đánh giá và gửi báo cáo về Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc (Trung tâm DI &ADR Quốc gia) thông tin các trường hợp phản ứng sau tiêm chủng, phản ứng có hại của thuốc theo quy định tại khoản 5 Điều 77 Luật dược, hướng dẫn Thực hành tốt cảnh giác dược (Good Pharmacovigilance Practices), các hướng dẫn quốc gia về cảnh giác dược và các quy định có liên quan.
8. Bảo đảm duy trì điều kiện hoạt động của cơ sở đăng ký trong thời hạn hiệu lực của giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc. Trong trường hợp không còn đáp ứng đủ điều kiện hoạt động, cơ sở đăng ký phải có trách nhiệm thực hiện thay đổi cơ sở đăng ký theo quy định tại của khoản 4 Điều 28 và Điều 40 Thông tư này trong thời hạn 30 ngày kể từ ngày cơ sở đăng ký không còn đủ điều kiện hoạt động.
9. Chịu trách nhiệm về các vấn đề liên quan đến quyền sở hữu trí tuệ đối với thuốc, nguyên liệu làm thuốc do cơ sở đăng ký lưu hành tại Việt Nam.
10. Phối hợp cơ sở sản xuất cập nhật tiêu chuẩn chất lượng thuốc, nguyên liệu làm thuốc theo quy định tại Thông tư số 11/2018/TT-BYT ngày 04 tháng 5 năm 2018 của Bộ trưởng Bộ Y tế quy định về chất lượng thuốc, nguyên liệu làm thuốc (sau đây viết tắt là Thông tư số 11/2018/TT-BYT) và Thông tư số 13/2018/TT-BYT ngày 15 tháng 5 năm 2018 của Bộ trưởng Bộ Y tế quy định về chất lượng dược liệu, thuốc cổ truyền.
11. Thực hiện kế hoạch quản lý nguy cơ đã được phê duyệt trong hồ sơ đề nghị cấp, gia hạn giấy đăng ký lưu hành đối với vắc xin.
12. Chịu trách nhiệm theo quy định tại khoản 2 Điều 57 Luật dược và các quy định tại Điều này đối với thuốc, nguyên liệu làm thuốc do cơ sở đăng ký kể từ ngày Cục Quản lý Dược ký công văn cho phép thay đổi cơ sở đăng ký, bao gồm cả các thuốc, nguyên liệu làm thuốc đã được đưa ra lưu hành trước thời điểm Cục Quản lý Dược có công văn cho phép chuyển đổi cơ sở đăng ký, theo các quy định của pháp luật hiện hành.
13. Chịu các trách nhiệm khác quy định tại Thông tư này và các quy định của pháp luật khác có liên quan.
Điều 4. Trách nhiệm đối với cơ sở sản xuất thuốc, nguyên liệu làm thuốc
1. Phải sản xuất thuốc, nguyên liệu làm thuốc tại đúng cơ sở có giấy phép sản xuất do cơ quan nhà nước có thẩm quyền cấp.
2. Đề nghị thu hồi giấy đăng ký lưu hành do cơ sở sản xuất trong trường hợp thuốc, nguyên liệu làm thuốc có vấn đề về chất lượng, an toàn và hiệu quả ảnh hưởng đến sức khoẻ của người sử dụng theo Mẫu 1/TT ban hành kèm theo Thông tư này.
3. Phối hợp với cơ sở đăng ký thuốc, nguyên liệu làm thuốc thực hiện quy định tại khoản 1, 2 và 3 Điều 3 Thông tư này.
4. Phối hợp với cơ sở đăng ký thuốc thực hiện các yêu cầu về kiểm tra, đánh giá cơ sở sản xuất khi có yêu cầu của cơ quan quản lý có thẩm quyền.
5. Bảo đảm các điều kiện hoạt động của cơ sở sản xuất trong thời hạn hiệu lực của giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
6. Thực hiện việc thay đổi cơ sở đăng ký của thuốc, nguyên liệu làm thuốc do cơ sở sản xuất đã được cấp giấy đăng ký lưu hành trong thời hạn 30 ngày kể từ ngày Cục Quản lý Dược ký công văn thông báo về việc cơ sở đăng ký không còn đủ điều kiện hoạt động.
7. Thực hiện cập nhật tiêu chuẩn chất lượng thuốc, nguyên liệu làm thuốc theo quy định tại Thông tư số 11/2018/TT-BYT.
Điều 5. Quy định về báo cáo theo dõi, đánh giá an toàn, hiệu quả
1. Cơ sở kinh doanh dược, cơ sở khám bệnh, chữa bệnh phải thực hiện việc theo dõi, giám sát, thu thập, tổng hợp, đánh giá và báo cáo cơ quan quản lý nhà nước có thẩm quyền thông tin các trường hợp phản ứng sau tiêm chủng, phản ứng có hại của thuốc theo quy định tại Điều 77, Điều 78 Luật dược, hướng dẫn Thực hành tốt cảnh giác dược (Good Pharmacovigilance Practices), các hướng dẫn quốc gia về cảnh giác dược và các quy định có liên quan.
2. Cơ sở đăng ký thuốc phải báo cáo đánh giá an toàn, hiệu quả đối với thuốc quy định tại khoản 2 Điều 8 Thông tư này theo Mẫu 2A/TT (đối với thuốc) hoặc Mẫu 2B/TT (đối với vắc xin):
a) Định kỳ 06 tháng một lần trong thời hạn giấy đăng ký lưu hành còn hiệu lực gửi về Trung tâm DI &ADR Quốc gia để tổng hợp, đánh giá và báo cáo Cục Quản lý Dược;
b) Khi nộp hồ sơ đăng ký gia hạn giấy đăng ký lưu hành tại Cục Quản lý Dược.
3. Cơ sở khám bệnh, chữa bệnh có sử dụng thuốc báo cáo tình hình sử dụng thuốc theo Mẫu 2C/TT (đối với thuốc) ban hành kèm theo Thông tư này định kỳ 06 tháng một lần trong thời hạn giấy đăng ký lưu hành còn hiệu lực đối với thuốc quy định tại khoản 2 Điều 8 Thông tư này và gửi về Trung tâm DI & ADR Quốc gia để tổng hợp, đánh giá và báo cáo Cục Quản lý Dược.
Điều 6. Ngôn ngữ, hình thức hồ sơ, số lượng hồ sơ, tài liệu
1. Ngôn ngữ sử dụng trong hồ sơ đăng ký
Hồ sơ đăng ký thuốc, nguyên liệu làm thuốc phải được viết bằng tiếng Việt hoặc tiếng Anh. Tờ hướng dẫn sử dụng thuốc và Tóm tắt đặc tính sản phẩm phải được viết bằng tiếng Việt.
2. Hồ sơ đăng ký thuốc, nguyên liệu làm thuốc phải được chuẩn bị trên khổ giấy A4, đóng chắc chắn (trừ hồ sơ nộp trực tuyến). Hồ sơ phải có trang bìa (Mẫu 3/TT), tờ thông tin sản phẩm (Mẫu 4/TT) được sắp xếp theo đúng trình tự của mục lục (Mẫu 5/TT), có phân cách giữa các phần. Các phần phân cách phải được đánh số thứ tự để dễ tham khảo và có dấu xác nhận của cơ sở đăng ký hoặc cơ sở sản xuất thuốc, nguyên liệu làm thuốc ở trang đầu tiên của mỗi phần trong toàn bộ hồ sơ (chấp nhận dấu của văn phòng đại diện đối với thuốc nước ngoài).
Các tài liệu sau phải đóng thành các phần riêng và kèm 01 tờ thông tin sản phẩm:
a) Tài liệu nghiên cứu tương đương sinh học;
b) Tài liệu tiền lâm sàng, lâm sàng;
c) Tài liệu đánh giá việc đáp ứng GMP theo quy định tại Điều 95, 98 Nghị định số 54/2017/NĐ-CP ngày 08 tháng 5 năm 2017 của Chính phủ đối với cơ sở sản xuất thuốc, nguyên liệu làm thuốc nước ngoài khi đăng ký lưu hành tại Việt Nam.
3. Các thuốc (trừ vắc xin) có thể đăng ký trong cùng một hồ sơ khi có chung các yếu tố sau: tên thuốc; dạng bào chế; đường dùng; tiêu chuẩn chất lượng thuốc; tên và địa chỉ nhà sản xuất; cùng công thức bào chế, trong đó: có cùng hàm lượng dược chất tính theo đơn vị chia liều đối với các thuốc dạng rắn có chia liều; có cùng nồng độ hoặc hàm lượng dược chất đối với các thuốc dạng rắn không chia liều, dạng lỏng hoặc bán rắn; có cùng nồng độ hoặc hàm lượng dược chất và chất liệu bao bì tiếp xúc trực tiếp với thuốc đối với các thuốc dạng tiêm, truyền.
4. Số lượng các tài liệu phải nộp trong hồ sơ đề nghị cấp, gia hạn giấy đăng ký lưu hành, cụ thể như sau:
a) 01 (một) bộ có đầy đủ hồ sơ theo quy định tại khoản 1, 2, 3, 5, 6, 7 Điều 28 Thông tư này đối với thuốc hóa dược, vắc xin, sinh phẩm và hồ sơ quy định tại khoản 1, 2 Điều 31, khoản 1, 2 Điều 33 Thông tư này đối với thuốc dược liệu, nguyên liệu làm thuốc;
b) 01 (một) bản sao đầy đủ hồ sơ đối với vắc xin; 02 (hai) bản sao các tài liệu gồm đơn đăng ký, tiêu chuẩn chất lượng và phương pháp kiểm nghiệm thuốc, nguyên liệu làm thuốc đối với các trường hợp còn lại;
c) 02 (hai) bộ mẫu nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc dự kiến lưu hành có dấu xác nhận của cơ sở đăng ký (chấp nhận dấu của văn phòng đại diện đối với thuốc nước ngoài) hoặc cơ sở sản xuất. Các nhãn thuốc, nguyên liệu làm thuốc được gắn, thiết kế trên khổ giấy A4.
5. Số lượng các tài liệu phải nộp trong hồ sơ đăng ký thay đổi, bổ sung giấy đăng ký lưu hành:
a) 01 (một) bộ có đầy đủ các tài liệu theo quy định tại khoản 4 Điều 28 Thông tư này đối với thuốc hóa dược, vắc xin, sinh phẩm và khoản 3 Điều 31, khoản 3 Điều 33 Thông tư này đối với thuốc dược liệu, nguyên liệu làm thuốc.
b) 02 (hai) bộ mẫu nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc đề nghị thay đổi đối với trường hợp thay đổi nhãn, hướng dẫn sử dụng, có dấu xác nhận của cơ sở đăng ký (chấp nhận dấu của văn phòng đại diện đối với thuốc nước ngoài) hoặc cơ sở sản xuất. Các nhãn thuốc, nguyên liệu làm thuốc được gắn, thiết kế trên khổ giấy A4.
6. Quy định về việc áp dụng hồ sơ trực tuyến:
a) Số lượng, thành phần hồ sơ: 01 (một) bộ hồ sơ đầy đủ theo quy định tại Thông tư này đăng ký trực tuyến và gửi thêm 01 bản giấy của hồ sơ hành chính (trừ nhãn, tờ hướng dẫn sử dụng thuốc) tới Cục Quản lý Dược;
b) Lộ trình áp dụng hồ sơ trực tuyến theo công bố của Bộ trưởng Bộ Y tế.
Điều 7. Phí đăng ký thuốc, nguyên liệu làm thuốc
Cơ sở đăng ký thuốc, nguyên liệu làm thuốc phải nộp phí liên quan đến đăng ký thuốc, nguyên liệu làm thuốc theo quy định của pháp luật hiện hành về phí và lệ phí.
Điều 8. Hiệu lực, ký hiệu của giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc và thời hạn nộp hồ sơ đăng ký gia hạn
1. Thời hạn hiệu lực của giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc là 05 năm kể từ ngày cấp hoặc gia hạn, trừ trường hợp quy định tại khoản 2 Điều này.
2. Thời hạn hiệu lực của giấy đăng ký lưu hành là 03 năm kể từ ngày cấp đối với các thuốc sau:
a) Thuốc mới, vắc xin lần đầu cấp giấy đăng ký lưu hành, sinh phẩm tham chiếu, sinh phẩm tương tự lần đầu cấp giấy đăng ký lưu hành tại Việt Nam;
b) Thuốc cùng dược chất, nồng độ, hàm lượng, dạng bào chế với thuốc mới mà thuốc mới đó chưa được cấp giấy đăng ký lưu hành với thời hạn 5 năm;
c) Thuốc không thuộc trường hợp quy định tại điểm a và b khoản này nhưng tại thời điểm nộp hồ sơ gia hạn giấy đăng ký lưu hành mà chưa có báo cáo an toàn, hiệu quả vì lý do chưa lưu hành hoặc có báo cáo an toàn, hiệu quả nhưng số lượng thuốc sử dụng, số lượng bệnh nhân, thời gian sử dụng còn hạn chế theo ý kiến của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc hoặc có khuyến nghị của cơ sở khám chữa bệnh về việc cần tiếp tục theo dõi an toàn, hiệu quả;
d) Các trường hợp tiếp tục theo dõi an toàn, hiệu quả theo ý kiến tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
3. Trong thời hạn 12 tháng trước khi giấy đăng ký lưu hành hết hiệu lực, cơ sở đăng ký có thể nộp hồ sơ gia hạn giấy đăng ký lưu hành. Sau ngày giấy đăng ký lưu hành hết hiệu lực, cơ sở nộp hồ sơ theo hình thức cấp giấy đăng ký lưu hành.
4. Mỗi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc có mã số riêng để phân biệt: thuốc, nguyên liệu làm thuốc sản xuất trong nước, nhập khẩu, vắc xin, sinh phẩm, thuốc chuyển giao công nghệ, thuốc thực hiện đóng gói thứ cấp tại Việt Nam.
5. Trong thời hạn hiệu lực của giấy đăng ký lưu hành cũ, mà cơ sở đăng ký được gia hạn giấy đăng ký lưu hành mới thì hiệu lực giấy đăng ký lưu hành cũ được tiếp tục có hiệu lực đồng thời với giấy đăng ký lưu hành mới trong 06 tháng kể từ ngày giấy đăng ký lưu hành mới có hiệu lực.
Điều 9. Tiêu chí đối với thuốc đề nghị phân loại biệt dược gốc
1. Thuốc đề nghị được phân loại biệt dược gốc (không áp dụng đối với sinh phẩm) phải được xác định trong đơn đề nghị đăng ký thuốc và phải đáp ứng đồng thời các tiêu chí sau:
a) Có đầy đủ dữ liệu lâm sàng về an toàn, hiệu quả theo quy định tại Điều 13 Thông tư này;
b) Được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9, 10 Điều 2 Thông tư này, trừ thuốc mới sản xuất tại Việt Nam.
Biệt dược gốc trước khi có thay đổi cơ sở sản xuất hoặc chuyển giao công nghệ tại Việt Nam theo quy định tại khoản 2, 3 Điều này vẫn được công nhận là biệt dược gốc.
2. Đối với thuốc đã được Bộ Y tế công bố biệt dược gốc theo quy định tại khoản 1 Điều này sau đó chuyển giao công nghệ sản xuất một, một số hoặc toàn bộ các công đoạn tại cơ sở sản xuất thuốc tại Việt Nam phải bảo đảm biệt dược gốc và thuốc sản xuất tại Việt Nam đáp ứng đồng thời các tiêu chí sau:
a) Cùng công thức bào chế;
b) Cùng quy trình sản xuất;
c) Cùng tiêu chuẩn chất lượng nguyên liệu;
d) Cùng tiêu chuẩn chất lượng thuốc thành phẩm;
đ) Nếu có bất kỳ thay đổi nào liên quan đến yêu cầu tại các điểm a, b, c, d khoản này, cơ sở đăng ký phải cung cấp dữ liệu chứng minh thuốc sản xuất tại Việt Nam tương đương về chất lượng so với biệt dược gốc trước khi chuyển giao.
3. Thuốc đã được công bố là biệt dược gốc có thay đổi cơ sở sản xuất thì thuốc được cấp giấy đăng ký lưu hành mới của cơ sở sản xuất thay đổi cũng được công bố là biệt dược gốc trên cơ sở có đề nghị bằng văn bản của cơ sở đăng ký nếu đáp ứng đồng thời các tiêu chí sau đây:
a) Thuốc được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9, 10 Điều 2 Thông tư này;
b) Thuốc đáp ứng đồng thời các quy định tại điểm a, b, c, d khoản 2 Điều này.
Điều 10. Yêu cầu đối với đăng ký thuốc sản xuất tại Việt Nam theo hình thức chuyển giao công nghệ, thuốc thực hiện đóng gói thứ cấp
1. Thuốc đăng ký theo hình thức chuyển giao công nghệ phải đáp ứng đồng thời các yêu cầu sau:
a) Việc chuyển giao công nghệ sản xuất thuốc được thực hiện theo hình thức chuyển giao một, một số hoặc toàn bộ các công đoạn của quy trình sản xuất thuốc thành phẩm, không áp dụng đối với trường hợp chỉ chuyển giao công đoạn đóng gói thứ cấp;
b) Thuốc đăng ký và thuốc trước khi chuyển giao công nghệ phải đáp ứng đồng thời các tiêu chí sau:
- Cùng công thức bào chế;
- Cùng quy trình sản xuất;
- Cùng tiêu chuẩn chất lượng nguyên liệu;
- Cùng tiêu chuẩn chất lượng thuốc thành phẩm;
Nếu có bất kỳ thay đổi nào liên quan đến yêu cầu nêu trên, cơ sở đăng ký phải cung cấp dữ liệu chứng minh thuốc sản xuất tại Việt Nam tương đương về chất lượng so với thuốc trước khi chuyển giao công nghệ.
c) Riêng đối với thuốc generic có tác dụng toàn thân, thuốc trước khi chuyển giao công nghệ phải đã được chứng minh tương đương sinh học theo quy định, trừ trường hợp thuốc có dạng bào chế được miễn thử đương sinh học theo quy định tại Thông tư số 08/2010/TT-BYT ngày 26 tháng 4 năm 2010 của Bộ trưởng Bộ Y tế Hướng dẫn báo cáo số liệu nghiên cứu sinh khả dụng/tương đương sinh học trong đăng ký thuốc;
d) Hồ sơ đáp ứng quy định tại khoản 5 Điều 28 Thông tư này.
2. Thuốc thực hiện đóng gói thứ cấp tại Việt Nam
a) Trong vòng 05 năm kể từ ngày được cấp giấy đăng ký lưu hành, cơ sở đăng ký, cơ sở sản xuất thực hiện xong chuyển giao công nghệ toàn bộ các công đoạn sản xuất theo các điều kiện quy định tại khoản 1 Điều này. Ba (03) năm kể từ ngày được cấp giấy đăng ký lưu hành, cơ sở đăng ký phải báo cáo tiến độ thực hiện việc chuyển giao công nghệ theo Mẫu 6/TT ban hành kèm theo Thông tư này;
b) Hồ sơ đáp ứng quy định tại khoản 6 Điều 28 Thông tư này.
3. Thuốc trước chuyển giao công nghệ, đóng gói thứ cấp được tiếp tục lưu hành theo hiệu lực của giấy đăng ký lưu hành đã được cấp.
Điều 11. Yêu cầu bảo mật dữ liệu đối với hồ sơ đăng ký thuốc
Cơ sở đăng ký thuốc có nhu cầu bảo mật dữ liệu đối với hồ sơ đăng ký thuốc thực hiện theo quy định tại Thông tư 05/2010/TT-BYT ngày 01 tháng 3 năm 2010 của Bộ trưởng Bộ Y tế hướng dẫn bảo mật dữ liệu thử nghiệm trong đăng ký thuốc và phải nêu rõ đề nghị trong đơn đăng ký theo Mẫu 6/TT ban hành kèm Thông tư này.
Điều 12. Quy định về kiểm tra tính xác thực của các thông tin trên giấy tờ pháp lý
1. Cục Quản lý Dược phối hợp với các cơ quan ngoại giao và cơ quan có liên quan trong nước, nước ngoài thực hiện việc kiểm tra tính xác thực của các giấy tờ pháp lý trong hồ sơ đăng ký thuốc, cụ thể như sau:
a) CPP của tất cả các hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc;
b) Giấy tờ pháp lý do cơ quan quản lý nhà nước có thẩm quyền nước ngoài cấp đối với cơ sở đăng ký nước ngoài lần đầu đăng ký thuốc tại Việt Nam.
2. Việc kiểm tra được thực hiện đồng thời với thủ tục thẩm định hồ sơ đăng ký thuốc và trong thời hạn quy định tại khoản 5 Điều 56 Luật dược.
Chương II
YÊU CẦU VỀ DỮ LIỆU LÂM SÀNG ĐỂ BẢO ĐẢM AN TOÀN, HIỆU QUẢ VÀ TIÊU CHÍ XÁC ĐỊNH TRƯỜNG HỢP MIỄN THỬ, MIỄN MỘT SỐ GIAI ĐOẠN THỬ THUỐC TRÊN LÂM SÀNG, THUỐC PHẢI THỬ LÂM SÀNG GIAI ĐOẠN 4 TẠI VIỆT NAM
Điều 13. Quy định về dữ liệu lâm sàng trong hồ sơ đăng ký lưu hành đối với thuốc hóa dược mới, vắc xin, sinh phẩm
1. Yêu cầu về dữ liệu lâm sàng để bảo đảm an toàn, hiệu quả trong hồ sơ đăng ký lưu hành đối với thuốc hóa dược mới, vắc xin, sinh phẩm
a) Các nghiên cứu lâm sàng của thuốc, các dữ liệu trong hồ sơ lâm sàng phải phù hợp với hướng dẫn của ICH, Bộ Y tế Việt Nam hoặc hướng dẫn của các tổ chức khác mà Việt Nam công nhận (bao gồm: hướng dẫn của tổ chức quốc tế mà Việt Nam là thành viên, hướng dẫn của cơ quan quản lý tham chiếu quy định tại khoản 9 Điều 2 Thông tư này), trừ trường hợp quy định tại khoản 2 Điều này;
b) Dữ liệu lâm sàng (trừ sinh phẩm tương tự với sinh phẩm tham chiếu và vắc xin tương tự với vắc xin đã được cấp giấy đăng ký lưu hành tại Việt Nam) phải có đủ thông tin để phân tích, biện giải được về ảnh hưởng của yếu tố chủng tộc người châu Á liên quan đến an toàn, hiệu quả của thuốc nhằm ngoại suy dữ liệu lâm sàng trên chủng tộc người châu Á theo các hướng dẫn quy định tại điểm a khoản 1 Điều này hoặc phải có dữ liệu nghiên cứu bắc cầu theo hướng dẫn của ICH-E5 nhằm ngoại suy dữ liệu lâm sàng trên chủng tộc người châu Á;
c) Vắc xin đã được cấp phép lưu hành đáp ứng quy định tại điểm g khoản 4 Điều 23 Thông tư này và có đầy đủ dữ liệu lâm sàng về an toàn, hiệu quả theo quy định tại điểm a, b khoản 1 Điều này nhưng chưa được sản xuất toàn bộ các công đoạn trên dây chuyền của các nước là thành viên quy định tại khoản 10 Điều 2 Thông tư này thì phải có dữ liệu lâm sàng liên quan đến đánh giá tính an toàn và tính sinh miễn dịch trên quần thể đích tại Việt Nam trước khi được cấp phép lưu hành.
d) Vắc xin có đầy đủ dữ liệu lâm sàng đánh giá tính an toàn, hiệu quả quy định tại điểm a, b khoản 1 Điều này nhưng chưa đáp ứng quy định tại điểm g khoản 4 Điều 23 Thông tư này thì phải có dữ liệu lâm sàng liên quan đến đánh giá tính an toàn và tính sinh miễn dịch trên quần thể đích tại Việt Nam trước khi được cấp phép lưu hành.
2. Trong trường hợp nghiên cứu được thực hiện trước thời điểm có quy định, hướng dẫn về nghiên cứu phát triển thuốc quy định tại điểm a khoản 1 Điều này thì được xem xét chấp nhận dữ liệu của nghiên cứu để thẩm định.
Điều 14. Yêu cầu về dữ liệu lâm sàng để bảo đảm an toàn, hiệu quả trong hồ sơ đăng ký lưu hành đối với thuốc có sự kết hợp mới của các dược chất, sinh phẩm tương tự
1. Thuốc có sự kết hợp mới của các dược chất phải có đầy đủ dữ liệu lâm sàng theo hướng dẫn của US FDA, EMA hoặc WHO về phát triển lâm sàng thuốc phối hợp cố định liều thực hiện theo quy định tại Phụ lục IV ban hành kèm theo Thông tư này.
2. Sinh phẩm tương tự phải có đầy đủ dữ liệu lâm sàng theo hướng dẫn về phát triển sinh phẩm tương tự do Bộ Y tế Việt Nam ban hành hoặc hướng dẫn của WHO. Chấp nhận hướng dẫn của US FDA, EMA và các hướng dẫn được xây dựng trên cơ sở các hướng dẫn này. Các hướng dẫn của WHO, US FDA, EMA theo Phụ lục IV ban hành kèm theo Thông tư này.
Điều 15. Yêu cầu về dữ liệu lâm sàng để bảo đảm an toàn, hiệu quả trong hồ sơ đăng ký lưu hành đối với thuốc hóa dược mới không phải là biệt dược gốc
1. Đối với thuốc được cấp phép lưu hành ở nước sở tại là thuốc kê đơn (trừ trường hợp thuốc sản xuất tại Việt Nam) và đã có ít nhất một thuốc tương tự (cùng hoạt chất, nồng độ, hàm lượng, dạng bào chế, đường dùng) được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 10 Điều 2 Thông tư này phải có dữ liệu lâm sàng đáp ứng một trong trường hợp sau:
a) Có dữ liệu lâm sàng của chính thuốc tương tự đó được chủ sở hữu cho phép sử dụng. Dữ liệu lâm sàng của thuốc tương tự phải đáp ứng quy định tại Điều 13 Thông tư này;
b) Có dữ liệu lâm sàng tập hợp từ các công trình nghiên cứu công bố trong y văn và dữ liệu về nghiên cứu tương đương sinh học (trừ trường hợp thuốc không có yêu cầu phải thử tương đương sinh học theo quy định của cơ quan quản lý nước sở tại).
2. Đối với thuốc không kê đơn theo quy định của nước sở tại (trừ trường hợp thuốc sản xuất tại Việt Nam và trường hợp quy định tại khoản 3 Điều này) và đã có ít nhất một thuốc tương tự (cùng hoạt chất, nồng độ, hàm lượng, dạng bào chế, đường dùng) được cấp phép lưu hành bởi ít nhất một nước trên thế giới phải có dữ liệu lâm sàng đáp ứng một trong trường hợp sau:
a) Có dữ liệu lâm sàng của chính thuốc tương tự đó được chủ sở hữu cho phép sử dụng. Dữ liệu lâm sàng của thuốc tương tự phải đáp ứng quy định tại Điều 13 của Thông tư này;
b) Có dữ liệu lâm sàng tập hợp từ các công trình nghiên cứu công bố trong y văn và dữ liệu về nghiên cứu tương đương sinh học (trừ trường hợp thuốc không có yêu cầu phải thử tương đương sinh học theo quy định của cơ quan quản lý nước sở tại).
3. Đối với thuốc được cấp phép lưu hành và phân loại là thuốc không kê đơn bởi ít nhất một trong các cơ quan quản lý tham chiếu quy định tại khoản 9 Điều 2 Thông tư này thì phải có tài liệu thuyết minh và bằng chứng chứng minh việc sử dụng các dược chất trong thành phần của thuốc (về chỉ định, liều dùng, đường dùng, đối tượng sử dụng) đã được ghi rõ trong Dược thư Quốc gia Việt Nam, Dược điển Việt Nam, Dược thư hoặc các tài liệu được chấp nhận bởi một trong các cơ quan quản lý tham chiếu quy định tại khoản 9 Điều 2 Thông tư này.
Điều 16. Yêu cầu về dữ liệu lâm sàng trong hồ sơ đăng ký lưu hành đối với thuốc hóa dược có hàm lượng, nồng độ, đường dùng, cách dùng, liều dùng, chỉ định, đối tượng bệnh nhân khác so với biệt dược gốc đã được cấp phép lưu hành tại Việt Nam
Đối với thuốc hóa dược có hàm lượng, nồng độ, đường dùng, cách dùng, liều dùng, chỉ định, đối tượng bệnh nhân khác so với biệt dược gốc đã được cấp phép lưu hành tại Việt Nam hoặc có dạng bào chế mới ảnh hưởng đến sinh dược học của thuốc phải có hồ sơ lâm sàng theo quy định tại Điều 13 Thông tư này.
Điều 17. Yêu cầu về dữ liệu lâm sàng đối với thuốc đã được cấp giấy đăng ký lưu hành tại Việt Nam nhưng có thay đổi, bổ sung liên quan đến dữ liệu lâm sàng so với hồ sơ đăng ký thuốc đã được phê duyệt
Thuốc hóa dược, vắc xin, sinh phẩm, thuốc dược liệu đã được cấp giấy đăng ký lưu hành tại Việt Nam có thay đổi, bổ sung liên quan đến dữ liệu lâm sàng so với hồ sơ đăng ký thuốc đã được phê duyệt, cơ sở đăng ký phải bổ sung dữ liệu lâm sàng theo quy định tại Phụ lục II ban hành kèm theo Thông tư này.
Điều 18. Tiêu chí xác định miễn một, một số giai đoạn thử thuốc hóa dược mới, vắc xin, sinh phẩm trên lâm sàng trước khi cấp phép lưu hành
Thuốc chưa đáp ứng quy định tại Điều 13 Thông tư này được Bộ trưởng Bộ Y tế xem xét quyết định miễn một, một số giai đoạn thử thuốc trên lâm sàng (bao gồm cả miễn giảm dữ liệu lâm sàng) trên cơ sở ý kiến tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc khi thuộc một trong các trường hợp sau:
1. Thuốc đáp ứng nhu cầu cấp bách cho quốc phòng, an ninh, phòng, chống dịch bệnh, khắc phục hậu quả thiên tai, thảm họa mà trên thị trường chưa có sẵn các thuốc khác có khả năng thay thế.
2. Thuốc đã được cấp phép lưu hành bởi ít nhất hai trong số các cơ quan quản lý tham chiếu quy định tại khoản 9 Điều 2 Thông tư này hoặc đã được cấp phép lưu hành bởi Mỹ (US FDA) hoặc bởi EMA dựa trên hồ sơ lâm sàng miễn giảm theo quy định của các cơ quan này.
3. Thuốc dùng để điều trị các bệnh hiếm gặp; bệnh hiểm nghèo.
4. Vắc xin, sinh phẩm được sản xuất tại Việt Nam theo hình thức chuyển giao công nghệ một, một số hoặc toàn bộ các công đoạn của quy trình sản xuất thành phẩm mà vắc xin, sinh phẩm trước chuyển giao công nghệ có dữ liệu lâm sàng đáp ứng quy định tại khoản 1 Điều 13, Điều 14 Thông tư này.
Điều 19. Yêu cầu về dữ liệu lâm sàng trong hồ sơ đăng ký lưu hành đối với thuốc dược liệu mới
1. Yêu cầu về dữ liệu lâm sàng để bảo đảm an toàn, hiệu quả trong hồ sơ đăng ký lưu hành đối với thuốc dược liệu mới
a) Các nghiên lâm sàng của thuốc, các dữ liệu trong hồ sơ lâm sàng phải phù hợp với Hướng dẫn nghiên cứu tiền lâm sàng và lâm sàng thuốc dược liệu của Bộ Y tế hoặc của tổ chức khác mà Việt Nam công nhận, bao gồm: Hướng dẫn nghiên cứu đánh giá an toàn và hiệu quả của thuốc dược liệu của Tổ chức Y tế Thế giới (Research guidelines for evaluating the safety and efficacy of herbal medicines) hoặc cơ quan quản lý quy định tại khoản 10 Điều 2 Thông tư này. Trong trường hợp nghiên cứu được thực hiện trước thời điểm có quy định, hướng dẫn nêu trên về nghiên cứu phát triển thuốc thì được xem xét chấp nhận dữ liệu của nghiên cứu để thẩm định;
b) Thuốc dược liệu có dữ liệu trích từ các tài liệu sau được chấp nhận là dữ liệu lâm sàng để xem xét tính an toàn, hiệu quả của thuốc:
- Các chuyên luận liên quan đến tính an toàn, hiệu quả của thuốc được đề cập trong các dược điển, dược thư của Việt Nam hoặc của các nước trên thế giới;
- Các bài báo đánh giá về tính an toàn, hiệu quả của thuốc được đăng tải trên các tạp chí thuộc danh mục SCI (Science Citation Index) - Chỉ số trích dẫn khoa học và các dữ liệu lâm sàng tập hợp từ các công trình nghiên cứu công bố trong y văn khác;
- Báo cáo đánh giá tính an toàn, hiệu quả của đề tài khoa học và công nghệ cấp quốc gia, cấp bộ hoặc cấp tỉnh đã được nghiệm thu.
2. Thuốc dược liệu không yêu cầu phải nộp dữ liệu lâm sàng theo quy định tại khoản 1 Điều này nếu đáp ứng một trong các điều kiện sau:
a) Thuốc dược liệu có cùng thành phần, khối lượng dược liệu, chỉ định, đường dùng với một thuốc dược liệu khác đã được cấp giấy đăng ký lưu hành (bao gồm cả trường hợp giấy đăng ký lưu hành đã hết hiệu lực) trừ các thuốc đã được xác định là thuốc cổ truyền và không có chỉ định đối với các bệnh thuộc Danh mục bệnh do Bộ trưởng Bộ Y tế ban hành theo quy định tại điểm b khoản 1 Điều 89 Luật dược;
b) Trường hợp thuốc dược liệu có cùng thành phần, khối lượng dược liệu, chỉ định, đường dùng với một thuốc dược liệu mới được cấp phép lưu hành tại Việt Nam trên cơ sở có đầy đủ dữ liệu lâm sàng theo quy định tại khoản 1 Điều này và không có thêm chỉ định đối với các bệnh thuộc Danh mục bệnh do Bộ trưởng Bộ Y tế ban hành theo quy định tại điểm b khoản 1 Điều 89 Luật dược thì chỉ được xem xét cấp phép lưu hành khi thuốc dược liệu khác đó đã được cấp phép lưu hành tối thiểu 05 năm.
Điều 20. Tiêu chí xác định trường hợp được miễn một, một số giai đoạn thử thuốc dược liệu trên lâm sàng trước khi cấp phép lưu hành
Thuốc dược liệu chưa đáp ứng quy định tại Điều 19 Thông tư này được Bộ trưởng Bộ Y tế xem xét quyết định việc miễn một, một số giai đoạn thử thuốc trên lâm sàng (bao gồm cả miễn giảm dữ liệu lâm sàng) trên cơ sở ý kiến tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc khi thuộc một trong các trường hợp sau:
1. Thuốc đáp ứng nhu cầu cấp bách cho quốc phòng, an ninh, phòng, chống dịch bệnh, khắc phục hậu quả thiên tai, thảm họa mà trên thị trường chưa có sẵn các thuốc khác có khả năng thay thế.
2. Thuốc đã được cấp phép lưu hành bởi ít nhất một trong số các cơ quan quản lý tham chiếu quy định tại khoản 9, 10 Điều 2 Thông tư này dựa trên hồ sơ lâm sàng miễn giảm theo quy định của các cơ quan này.
3. Thuốc có chỉ định đối với các bệnh thuộc Danh mục bệnh do Bộ trưởng Bộ Y tế ban hành theo quy định tại điểm b khoản 1 Điều 89 Luật dược nhưng không thuộc trường hợp được miễn thử lâm sàng quy định tại khoản 3 Điều 21 Thông tư này.
4. Thuốc có sự phối hợp mới của các dược liệu đã từng sử dụng làm thuốc tại Việt Nam và không có chỉ định đối với các bệnh thuộc Danh mục bệnh do Bộ trưởng Bộ Y tế ban hành quy định tại điểm b khoản 1 Điều 89 Luật dược.
Điều 21. Tiêu chí xác định trường hợp được miễn thử lâm sàng tại Việt Nam trước khi cấp phép lưu hành
1. Thuốc generic có cùng dược chất, hàm lượng, nồng độ, đường dùng, cách dùng, liều dùng, chỉ định, đối tượng bệnh nhân, dạng bào chế với một thuốc khác đã được cấp giấy đăng ký lưu hành.
2. Thuốc mới (trừ vắc xin) đã được cấp phép lưu hành tại ít nhất một nước trên thế giới và có đầy đủ dữ liệu lâm sàng về an toàn, hiệu quả theo quy định tại Điều 13, Điều 19 Thông tư này.
3. Thuốc dược liệu đã được cấp giấy đăng ký lưu hành trước ngày Luật dược có hiệu lực và không có chỉ định đối với các bệnh thuộc Danh mục bệnh do Bộ trưởng Bộ Y tế ban hành.
4. Vắc xin đã được cấp phép lưu hành đáp ứng quy định tại điểm g khoản 4 Điều 23 Thông tư này, được sản xuất toàn bộ các công đoạn trên dây chuyền của các nước là thành viên quy định tại khoản 10 Điều 2 Thông tư này và có đầy đủ dữ liệu lâm sàng về an toàn, hiệu quả theo quy định tại Điều 13 Thông tư này.
Điều 22. Tiêu chí để xác định trường hợp phải thử lâm sàng giai đoạn IV tại Việt Nam
Thuốc đã được cấp giấy đăng ký lưu hành nhưng cần đánh giá thêm về an toàn, hiệu quả trên cơ sở ý kiến tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
Chương III
HỒ SƠ ĐĂNG KÝ THUỐC, NGUYÊN LIỆU LÀM THUỐC
Mục 1. QUY ĐỊNH CHUNG VỀ HỒ SƠ ĐỀ NGHỊ CẤP, GIA HẠN, THAY ĐỔI, BỔ SUNG GIẤY ĐĂNG KÝ LƯU HÀNH THUỐC, NGUYÊN LIỆU LÀM THUỐC
Điều 23. Quy định đối với các tài liệu trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
1. Giấy tờ do cơ quan quản lý nước ngoài cấp phải được hợp pháp hoá lãnh sự theo quy định của pháp luật về hợp pháp hóa lãnh sự, trừ các trường hợp được miễn theo quy định của pháp luật.
2. Giấy phép, giấy chứng nhận, giấy xác nhận, giấy đăng ký (gọi chung là giấy tờ pháp lý) trong hồ sơ phải còn hiệu lực tại thời điểm tiếp nhận ghi trên Phiếu tiếp nhận hồ sơ và phải được thể hiện bằng tiếng Anh hoặc tiếng Việt. Trường hợp CPP không ghi thời hạn hiệu lực thì thời hạn hiệu lực được tính là 24 tháng kể từ ngày cấp.
3. Giấy tờ pháp lý là bản chính hoặc bản sao có chứng thực:
a) Bản chính phải có đầy đủ chữ ký, tên người ký và dấu xác nhận của cơ quan quản lý nhà nước có thẩm quyền của nước cấp;
b) Bản sao có chứng thực phải do cơ quan, tổ chức có thẩm quyền của Việt Nam chứng thực theo quy định của pháp luật Việt Nam về chứng thực bản sao từ bản chính. Trong trường hợp cần thiết phải xuất trình bản chính để đối chiếu;
c) Trường hợp giấy tờ pháp lý được cấp là bản điện tử không thể hiện đầy đủ chữ ký, tên người ký và dấu xác nhận của cơ quan quản lý nhà nước có thẩm quyền của nước cấp giấy tờ pháp lý thì cơ sở đăng ký phải có văn bản cung cấp thông tin về đường dẫn truy cập website (website tiếng Anh) của cơ quan cấp giấy tờ pháp lý nêu trên và cam kết chịu trách nhiệm về tính hợp pháp của các giấy tờ pháp lý này.
4. Quy định đối với CPP:
a) CPP phải có chữ ký, tên người ký, ngày cấp và dấu của cơ quan cấp CPP;
b) CPP phải được cấp bởi cơ quan quản lý về dược phẩm có thẩm quyền cấp quốc gia.
Trường hợp CPP được cấp bởi cơ quan quản lý dược phẩm nhưng không phải là cơ quan quản lý dược phẩm cấp quốc gia: Cơ sở đăng ký thuốc phải cung cấp tài liệu pháp lý chứng minh cơ quan này là cơ quan có thẩm quyền và cơ quan quản lý dược phẩm quốc gia tại nước đó không thực hiện việc cấp CPP theo quy định của pháp luật nước sở tại.
Trường hợp CPP được cấp bởi cơ quan không phải cơ quan quản lý dược phẩm: Cơ sở đăng ký thuốc phải cung cấp tài liệu chứng minh cơ quan này là cơ quan có thẩm quyền và cơ quan quản lý dược phẩm tại nước đó không thực hiện việc cấp CPP theo quy định của pháp luật nước sở tại.
c) Chữ ký, tên người ký và dấu của cơ quan cấp CPP phải được chứng thực bởi cơ quan có thẩm quyền; Trường hợp nội dung xác nhận này không được thể hiện bằng tiếng Anh thì phải dịch công chứng sang tiếng Việt hoặc tiếng Anh;
d) Nội dung của CPP phải có đầy đủ thông tin theo quy định tại Mẫu 7/TT ban hành kèm theo Thông tư này và các nội dung sau:
- Công thức bào chế của thuốc, trong đó nêu rõ tên, thành phần, nồng độ, hàm lượng của từng dược chất, dược liệu, tá dược; đối với dạng bào chế viên nang mềm, viên nang cứng phải có thêm thông tin về thành phần công thức của vỏ nang;
- Tiêu chuẩn thành phẩm, tiêu chuẩn dược chất, dược liệu, tên, địa chỉ cơ sở sản xuất dược chất, dược liệu;
- Trường hợp thuốc có sự tham gia sản xuất bởi nhiều cơ sở sản xuất khác nhau thì CPP phải ghi rõ tên, địa chỉ, vai trò của từng cơ sở;
- Trường hợp CPP không có thông tin cơ sở sản xuất thuốc đáp ứng thực hành tốt sản xuất thuốc (GMP), cơ sở đăng ký phải nộp kèm giấy chứng nhận GMP của tất cả các cơ sở sản xuất đáp ứng quy định tại khoản 1, 2, 3 Điều này;
- Các phụ lục kèm theo CPP (nếu có) phải có xác nhận của cơ quan cấp CPP.
đ) Đối với thuốc generic, thuốc dược liệu, sinh phẩm probiotics (men tiêu hóa), thuốc gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành: CPP xác nhận thuốc được cấp phép và lưu hành ở nước sản xuất. Trường hợp thuốc không được cấp phép lưu hành ở nước sản xuất hoặc được cấp phép nhưng không lưu hành thực tế ở nước sản xuất, cơ sở đăng ký phải cung cấp CPP có xác nhận thuốc được cấp phép và lưu hành ở một trong các nước quy định tại khoản 10 Điều 2 Thông tư này;
e) Đối với thuốc hóa dược mới và sinh phẩm nhập khẩu, trừ sinh phẩm probiotics (men tiêu hóa): CPP được cấp bởi nước sản xuất và CPP được cấp bởi một trong các cơ quan quản lý khác quy định tại khoản 10 Điều 2 Thông tư này có xác nhận thuốc được cấp phép và lưu hành thực tế;
g) Đối với vắc xin nhập khẩu: CPP được cấp bởi nước sản xuất và CPP được cấp bởi một trong các cơ quan quản lý khác quy định tại khoản 9 Điều 2 Thông tư này có xác nhận vắc xin được cấp phép và lưu hành thực tế;
h) Đối với thuốc generic có báo cáo nghiên cứu tương đương sinh học: CPP được cấp bởi một trong các cơ quan quản lý quy định tại khoản 10 Điều 2 Thông tư này có xác nhận thuốc được cấp phép và lưu hành thực tế;
Trường hợp không có CPP đáp ứng quy định này, phải có báo cáo nghiên cứu tương đương sinh học của thuốc được thực hiện tại cơ sở kinh doanh dịch vụ thử tương đương sinh học của thuốc tại Việt Nam hoặc tại các cơ sở thử tương tương sinh học mà Việt Nam công nhận theo quy định của Bộ trưởng Bộ Y tế hoặc theo các thỏa thuận quốc tế mà Việt Nam đã tham gia ký kết.
i) Đối với thuốc đề nghị được phân loại biệt dược gốc, CPP được cấp bởi một trong các cơ quan quản lý quy định tại khoản 9, 10 Điều 2 Thông tư này, trừ thuốc sản xuất tại Việt Nam;
k) Đối với thuốc, vắc xin, sinh phẩm nhập khẩu không cung cấp được CPP đáp ứng quy định tại điểm đ, e, g, h khoản này, Bộ trưởng Bộ Y tế xem xét quyết định trên cơ sở ý kiến tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc khi thuốc được cấp phép lưu hành bởi ít nhất một cơ quan quản lý trên thế giới và thuộc một trong các trường hợp sau:
- Thuốc, vắc xin, sinh phẩm để đáp ứng nhu cầu cho quốc phòng, an ninh; phòng, chống dịch, bệnh, khắc phục hậu quả thiên tai, thảm họa, thuốc phục vụ cho chương trình y tế của nhà nước;
- Vắc xin dùng cho chương trình tiêm chủng mở rộng quốc gia mà trên thị trường không sẵn có vắc xin khác có khả năng thay thế về mặt số lượng, chất lượng, an toàn, hiệu quả hoặc chi phí sử dụng vắc xin;
- Các trường hợp đặc biệt khác có văn bản thỏa thuận, công nhận lẫn nhau giữa các cơ quan quản lý Nhà nước về Dược về điều kiện sản xuất, lưu hành thuốc, vắc xin, sinh phẩm.
l) Các thông tin thể hiện trên CPP phải thống nhất với các thông tin có liên quan trong hồ sơ đăng ký thuốc.
5. Đơn đăng ký và các hồ sơ, tài liệu khác trong phần hồ sơ hành chính có liên quan phải do một trong các chức danh Chủ tịch hội đồng thành viên, hội đồng quản trị, tổng giám đốc, giám đốc cơ sở hoặc người được những người có chức danh nêu trên ủy quyền trực tiếp để ký và đóng dấu, không sử dụng chữ ký dấu.
6. Giấy ủy quyền thực hiện theo Mẫu 8/TT ban hành kèm theo Thông tư này và được yêu cầu nộp trong các trường hợp sau đây:
a) Ủy quyền được đứng tên cơ sở đăng ký theo Mẫu 8A/TT ban hành kèm theo Thông tư này. Giấy ủy quyền đứng tên cơ sở đăng ký đối với thuốc nước ngoài phải được chứng thực chữ ký và hợp pháp hóa lãnh sự theo quy định.
Mỗi hồ sơ phải nộp kèm một giấy ủy quyền bản chính hoặc bản sao có chứng thực.
b) Ủy quyền ký tên trên hồ sơ đăng ký theo Mẫu 8B/TT ban hành kèm theo Thông tư này; trường hợp người được ủy quyền ký tên trên hồ sơ không phải trưởng văn phòng đại diện, trên giấy ủy quyền phải có dấu và chữ ký xác nhận của trưởng văn phòng đại diện tại Việt Nam.
Mỗi hồ sơ phải nộp kèm một giấy ủy quyền bản chính hoặc bản sao có dấu xác nhận của văn phòng đại diện (trường hợp là cơ sở đăng ký nước ngoài) hoặc dấu xác nhận của cơ sở đăng ký trong nước.
7. Bản sao hợp đồng chuyển giao công nghệ có dấu xác nhận của cơ sở đăng ký hoặc cơ sở sản xuất hoặc văn phòng đại diện (trường hợp là cơ sở đăng ký của nước ngoài).
8. Giấy chứng nhận đủ điều kiện kinh doanh dược với một trong các hình thức kinh doanh: sản xuất, bán buôn, xuất khẩu, nhập khẩu thuốc, nguyên liệu làm thuốc (đối với cơ sở đăng ký của Việt Nam).
9. Giấy phép thành lập Văn phòng đại diện tại Việt Nam.
Trường hợp tên, địa chỉ của cơ sở đăng ký trên Giấy phép thành lập Văn phòng đại diện tại Việt Nam khác với tên, địa chỉ trên Giấy tờ pháp lý của cơ sở đăng ký do cơ quan quản lý nhà nước có thẩm quyền nước ngoài cấp thì phải cung cấp tài liệu chứng minh.
10. Giấy tờ pháp lý do cơ quan quản lý nhà nước có thẩm quyền nước ngoài cấp cho phép thực hiện ít nhất một trong các hình thức kinh doanh sau: sản xuất, bán buôn, xuất khẩu, nhập khẩu thuốc, nguyên liệu làm thuốc (đối với cơ sở đăng ký của nước ngoài).
Trường hợp cơ sở đăng ký thuốc đồng thời là cơ sở sản xuất thuốc ghi trên CPP thì không yêu cầu phải nộp Giấy tờ pháp lý theo quy định tại khoản này.
Trường hợp các nước không cấp giấy phép sản xuất, bán buôn, xuất khẩu, nhập khẩu thuốc, nguyên liệu làm thuốc thì phải có giấy phép thành lập hoặc đăng ký kinh doanh có phạm vi kinh doanh là ít nhất một trong các hình thức sau: sản xuất, bán buôn, xuất khẩu, nhập khẩu thuốc, nguyên liệu làm thuốc kèm theo giấy chứng nhận của cơ quan quản lý có thẩm quyền chứng nhận cơ sở đáp ứng điều kiện và đang hoạt động về dược hoặc một trong các giấy chứng nhận thực hành tốt sản xuất thuốc, thực hành tốt phân phối thuốc, thực hành tốt cung cấp thuốc, thực hành tốt bảo quản thuốc.
Đối với cơ sở đăng ký nguyên liệu làm thuốc, trường hợp nước sở tại không cấp giấy phép kinh doanh dược cho các cơ sở kinh doanh nguyên liệu làm thuốc, chấp nhận các giấy phép theo quy định của nước sở tại trong đó có nội dung xác định phạm vi kinh doanh của cơ sở là một trong các hình thức: sản xuất, bán buôn, xuất khẩu, nhập khẩu nguyên liệu làm thuốc.
11. Trường hợp cơ sở đăng ký đã có tên trong danh sách cơ sở đăng ký thuốc, nguyên liệu làm thuốc được công bố trên trang thông tin điện tử của Cục Quản lý Dược thì không phải nộp giấy tờ quy định tại khoản 8, 9, 10 Điều này.
12. Giấy tờ pháp lý của cơ sở sản xuất dược chất, tá dược, vỏ nang, bán thành phẩm dược liệu và dược liệu (để sản xuất thuốc dược liệu) chứng minh đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) có thể là một trong các loại giấy tờ sau:
a) Giấy chứng nhận GMP;
b) Giấy phép sản xuất có xác nhận nội dung cơ sở sản xuất đáp ứng GMP;
c) CPP đối với dược chất có nội dung đáp ứng GMP;
d) Chứng nhận phù hợp chuyên luận Dược điển Châu Âu (CEP).
13. Mẫu nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc lưu hành thực tế tại nước sản xuất hoặc nước cấp CPP có dấu xác nhận của văn phòng đại diện hoặc cơ sở đăng ký hoặc cơ sở sản xuất. Trường hợp tờ hướng dẫn sử dụng thuốc thực tế tại nước sở tại không phải bằng tiếng Anh yêu cầu nộp bản dịch sang tiếng Việt có dấu xác nhận của văn phòng đại diện hoặc cơ sở đăng ký hoặc cơ sở sản xuất.
14. Mẫu nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc dự kiến lưu hành tại Việt Nam thực hiện theo quy định của Bộ trưởng Bộ Y tế quy định ghi nhãn thuốc, nguyên liệu làm thuốc và các yêu cầu cụ thể sau:
a) Mẫu nhãn, tờ hướng dẫn sử dụng dự kiến lưu hành phải có dấu xác nhận của văn phòng đại diện hoặc cơ sở đăng ký hoặc cơ sở sản xuất;
b) Nhãn bao bì ngoài của thuốc, nguyên liệu làm thuốc phải được in mã vạch (Bar code) hoặc mã QR (Quick response) hoặc mã DataMatrix Code (DMC) theo lộ trình quy định tại điểm 1 khoản 1 Điều 50 Thông tư này.
15. Trường hợp cơ sở sản xuất thuốc, nguyên liệu làm thuốc có tên trong danh mục cơ sở sản xuất được công bố trên trang thông tin điện tử của Cục Quản lý Dược về việc đã được đánh giá đáp ứng GMP thì không yêu cầu phải nộp hồ sơ đánh giá việc đáp ứng Thực hành tốt sản xuất trong hồ sơ đăng ký thuốc, nguyên liệu làm thuốc.
16. Tiêu chuẩn chất lượng, phương pháp kiểm nghiệm, phiếu kiểm nghiệm và hồ sơ nghiên cứu độ ổn định (áp dụng đối với cả phần hồ sơ dược chất và thuốc thành phẩm) phải là bản chính có chữ ký và dấu xác nhận của cơ sở sản xuất; trường hợp nộp bản sao thì phải có dấu xác nhận của cơ sở đăng ký (chấp nhận dấu của văn phòng đại diện đối với thuốc nước ngoài).
Phiếu kiểm nghiệm phải bao gồm các thông tin sau: thông tin hành chính (tên, địa chỉ cơ sở sản xuất, số phiếu kiểm nghiệm, tên và chữ ký của người được giao trách nhiệm, ngày phát hành phiếu kiểm nghiệm) và thông tin về mẫu thuốc, nguyên liệu làm thuốc (tên sản phẩm, số lô, hạn dùng, tiêu chuẩn chất lượng áp dụng, chỉ tiêu chất lượng, yêu cầu chất lượng, kết quả kiểm nghiệm, kết luận về chất lượng lô sản phẩm).
17. Quy định đối với phiếu kiểm nghiệm, kết quả thẩm định tiêu chuẩn chất lượng, phương pháp kiểm nghiệm bằng thực nghiệm tại Việt Nam:
Phiếu kiểm nghiệm, kết quả thẩm định tiêu chuẩn chất lượng, phương pháp kiểm nghiệm bằng thực nghiệm (đối với cơ sở sản xuất chưa đáp ứng GMP theo lộ trình của Bộ Y tế hoặc những trường hợp được Cục Quản lý Dược thông báo theo quy định tại Phụ lục III ban hành kèm theo Thông tư này) có xác nhận của cơ sở kiểm nghiệm thuốc của nhà nước đáp ứng GLP hoặc cơ sở kinh doanh dịch vụ kiểm nghiệm thuốc, nguyên liệu làm thuốc đã được cấp giấy chứng nhận đủ điều kiện kinh doanh phù hợp với phạm vi hoạt động phải là bản chính hoặc bản sao có chứng thực.
18. Giấy chứng nhận nguyên liệu làm thuốc được phép sản xuất hoặc lưu hành ở nước sản xuất, bao gồm các thông tin bắt buộc sau: tên nguyên liệu; tên và địa chỉ cơ sở sản xuất; nước sản xuất; chữ ký, dấu và họ tên của người ký giấy xác nhận.
Điều 24. Quy định chung về tài liệu hành chính trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
Tài liệu hành chính bao gồm:
1. Đơn đăng ký theo Mẫu 6/TT ban hành kèm theo Thông tư này.
2. Giấy ủy quyền (nếu có) theo Mẫu 8/TT ban hành kèm theo Thông tư này.
3. Giấy chứng nhận đủ điều kiện kinh doanh dược đối với cơ sở đăng ký của Việt Nam.
4. Giấy tờ pháp lý đối với cơ sở đăng ký của nước ngoài.
5. Giấy phép thành lập văn phòng đại diện tại Việt Nam đối với cơ sở đăng ký của nước ngoài.
6. Giấy chứng nhận CPP theo Mẫu 7/TT ban hành kèm theo Thông tư này.
7. Mẫu nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc dự kiến lưu hành.
8. Mẫu nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc lưu hành thực tế tại nước sản xuất hoặc nước cấp CPP.
9. Tóm tắt đặc tính sản phẩm đối với thuốc hoá dược mới, vắc xin, sinh phẩm theo Mẫu 9/TT ban hành kèm theo Thông tư này.
10. Tài liệu đánh giá việc đáp ứng GMP đối với các trường hợp quy định tại Điều 95 Nghị định số 54/2017/NĐ-CP đối với cơ sở sản xuất thuốc, nguyên liệu làm thuốc nước ngoài khi đăng ký lưu hành tại Việt Nam.
11. Giấy tờ pháp lý của cơ sở sản xuất dược chất, tá dược, vỏ nang, bán thành phẩm dược liệu, dược liệu.
12. Giấy chứng nhận nguyên liệu làm thuốc được phép sản xuất hoặc lưu hành ở nước sản xuất.
13. Giấy chứng nhận GLP của cơ sở kiểm nghiệm đối với trường hợp quy định tại khoản 17 Điều 23 Thông tư này.
14. Kế hoạch quản lý nguy cơ (đối với vắc xin) theo Mẫu 10/TT ban hành kèm theo Thông tư này.
15. Hợp đồng chuyển giao công nghệ đối với thuốc chuyển giao công nghệ.
16. Báo cáo an toàn, hiệu quả, sử dụng thuốc theo Mẫu 2/TT ban hành kèm theo Thông tư này.
17. Báo cáo lưu hành thuốc, nguyên liệu làm thuốc theo Mẫu 11/TT ban hành kèm theo Thông tư này.
18. Giấy chứng nhận, văn bằng bảo hộ, hợp đồng chuyển giao quyền đối tượng sở hữu công nghiệp, giấy tờ chứng nhận nguồn gốc nguyên liệu (GACP, CEP, nguồn dược liệu trong nước, nguồn dược liệu nhập khẩu,...) và các tài liệu có liên quan (nếu có).
19. Bản sao giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc tại Việt Nam.
Mục 2. HỒ SƠ ĐỀ NGHỊ CẤP, GIA HẠN, THAY ĐỔI, BỔ SUNG GIẤY ĐĂNG KÝ LƯU HÀNH THUỐC HÓA DƯỢC, VẮC XIN, SINH PHẨM
Điều 25. Tài liệu chất lượng trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc hóa dược, vắc xin, sinh phẩm
Tài liệu chất lượng thực hiện theo hướng dẫn tại Phần II - ACTD hoặc Hợp phần 3-ICH-CTD và các quy định sau:
1. Đối với vắc xin, huyết thanh có chứa kháng thể, dẫn xuất của máu và huyết tương người:
a) Giấy chứng nhận xuất xưởng lô được cấp bởi cơ quan có thẩm quyền nước cấp CPP theo quy định;
b) Phiếu kiểm nghiệm, tiêu chuẩn chất lượng và phương pháp kiểm nghiệm có xác nhận bởi Viện Kiểm định quốc gia vắc xin và sinh phẩm y tế;
2. Đối với thuốc hiếm và thuốc cho nhu cầu điều trị đặc biệt:
a) Thuốc hiếm để điều trị bệnh hiếm gặp: dữ liệu nghiên cứu độ ổn định sẵn có theo hướng dẫn của ASEAN hoặc của ICH;
b) Thuốc cần thiết cho nhu cầu điều trị đặc biệt: dữ liệu nghiên cứu độ ổn định sẵn có theo hướng dẫn của ASEAN hoặc của ICH được Bộ trưởng Bộ Y tế quyết định trên cơ sở ý kiến của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc trong trường hợp cơ sở đăng ký chứng minh thuốc không thể bảo quản ở điều kiện khí hậu vùng IVb theo hướng dẫn của ASEAN.
3. Trường hợp cơ sở sản xuất sử dụng nguyên liệu làm thuốc đã được cấp giấy đăng ký lưu hành tại Việt Nam:
a) Không yêu cầu phải nộp tài liệu chất lượng liên quan đến nguyên liệu và tài liệu quy định tại khoản 11 Điều 24 Thông tư này trong hồ sơ đăng ký thuốc thành phẩm.
b) Cơ sở đăng ký phải nộp:
- 01 phiếu kiểm nghiệm nguyên liệu làm thuốc do cơ sở sản xuất thuốc thành phẩm kiểm nghiệm phải có đầy đủ các chỉ tiêu chất lượng với mức chất lượng tương đương hoặc chặt chẽ hơn mức chất lượng trong tiêu chuẩn của cơ sở sản xuất nguyên liệu;
- 01 phiếu kiểm nghiệm nguyên liệu làm thuốc do cơ sở sản xuất nguyên liệu kiểm nghiệm.
4. Đối với thuốc chuyển giao công nghệ tại Việt Nam
a) Toàn bộ phần hồ sơ chất lượng thực hiện theo hướng dẫn tại Phần II - ACTD quy định tại Phụ lục I ban hành kèm theo Thông tư này hoặc Hợp phần 3-ICH-CTD của thuốc trước khi chuyển giao công nghệ (trong trường hợp thuốc trước khi chuyển giao công nghệ chưa được cấp giấy đăng ký lưu hành tại Việt Nam);
b) Bảng so sánh chi tiết các thay đổi, bổ sung (nếu có) giữa thuốc trước khi chuyển giao công nghệ và thuốc đăng ký theo hướng dẫn tại Phụ lục II ban hành kèm Thông tư này;
c) Hồ sơ phần Dược chất của thuốc đăng ký do bên nhận chuyển giao công nghệ cung cấp khi có thay đổi nhà sản xuất nguyên liệu dược chất so với thuốc trước khi chuyển giao công nghệ;
d) Hồ sơ phần thành phẩm của thuốc đăng ký, do bên nhận chuyển giao công nghệ thực hiện, bao gồm:
- Quy trình sản xuất thuốc đăng ký.
- Báo cáo thẩm định quy trình sản xuất (đối với các công đoạn sản xuất thực hiện tại bên nhận chuyển giao công nghệ).
- Báo cáo đánh giá sự phù hợp của quy trình phân tích (có thể thay thế bằng hồ sơ chuyển giao quy trình phân tích do bên chuyển giao công nghệ và bên nhận công nghệ phối hợp thực hiện).
- Số liệu phân tích lô (Phiếu kiểm nghiệm thành phẩm).
- Báo cáo nghiên cứu độ ổn định của thuốc đăng ký. Trường hợp thuốc trước khi chuyển giao công nghệ đã được cấp giấy đăng ký lưu hành tại Việt Nam hoặc đã có số liệu nghiên cứu độ ổn định phù hợp với các quy định tại Hướng dẫn ASEAN về nghiên cứu độ ổn định, chấp nhận dữ liệu nghiên cứu độ ổn định áp dụng đối với đăng ký thay đổi lớn hoặc thay đổi nhỏ (tùy thuộc vào nội dung thay đổi giữa thuốc trước khi chuyển giao công nghệ và thuốc đăng ký) theo hướng dẫn của ASEAN về nghiên cứu độ ổn định.
- Báo cáo nghiên cứu tương đương sinh học của thuốc đăng ký (đối với thuốc đề nghị công bố là biệt dược gốc, thuốc có yêu cầu phải báo cáo nghiên cứu tương đương sinh học theo quy định tại Thông tư số 08/2010/TT-BYT ngày 26 tháng 4 năm 2010 của Bộ trưởng Bộ Y tế Hướng dẫn báo cáo nghiên cứu sinh khả dụng, tương đương sinh học khi đăng ký thuốc hoặc thuốc không có yêu cầu phải báo cáo nghiên cứu tương đương sinh học theo quy định của Bộ trưởng Bộ Y tế nhưng cơ sở đăng ký có đơn đề nghị phân loại là thuốc có chứng minh tương đương sinh học). Trường hợp đáp ứng đồng thời các điều kiện sau có thể thay thế bằng báo cáo nghiên cứu tương đương độ hòa tan giữa thuốc đăng ký và thuốc trước khi chuyển giao công nghệ:
+ Thuốc trước khi chuyển giao công nghệ đã được cấp giấy đăng ký lưu hành tại Việt Nam và đã được công bố là biệt dược gốc, thuốc có chứng minh tương đương sinh học.
+ Thuốc đăng ký phải tương tự thuốc trước khi chuyển giao công nghệ về công thức bào chế thuốc, nhà sản xuất nguyên liệu dùng trong sản xuất thuốc, tiêu chuẩn chất lượng và quy trình phân tích các nguyên liệu dùng trong sản xuất thuốc, quy trình sản xuất, loại trang thiết bị dùng trong sản xuất thuốc, điều kiện môi trường trong quá trình sản xuất thuốc. Các thay đổi liên quan đến các nội dung này nếu có phải thuộc các mức không yêu cầu phải nộp báo cáo nghiên cứu tương đương sinh học của thuốc sau khi thay đổi theo các quy định tại các hướng dẫn nâng cỡ lô và thay đổi sau khi được cấp giấy đăng ký lưu hành đối với các thuốc dạng rắn dùng đường uống của US-FDA (SUPACs) và trong hồ sơ phải cung cấp kèm theo các tài liệu phù hợp với mỗi thay đổi quy định tại các hướng dẫn này.
5. Đối với thuốc đóng gói thứ cấp tại Việt Nam
Toàn bộ phần hồ sơ chất lượng của thuốc trước khi chuyển sang đóng gói thứ cấp tại Việt Nam theo hướng dẫn tại Phần II - ACTD hoặc Hợp phần 3-ICH-CTD trong trường hợp thuốc trước khi được chuyển giao chưa được cấp giấy đăng ký lưu hành tại Việt Nam.
6. Đối với thuốc đề nghị thực hiện theo quy trình thẩm định rút gọn
a) Phần hồ sơ dược chất:
- Tên dược chất (ghi theo tên chung quốc tế);
- Tên và địa chỉ cơ sở sản xuất dược chất, bán thành phẩm chứa dược chất;
- Tiêu chuẩn chất lượng và phương pháp kiểm nghiệm dược chất, bán thành phẩm chứa dược chất. Trường hợp thuốc đăng ký theo tiêu chuẩn Dược điển Việt Nam hoặc tiêu chuẩn dược điển tham chiếu theo quy định của Bộ Y tế thì chỉ ghi tên dược điển, phiên bản dược điển áp dụng hoặc ghi dược điển hiện hành;
- 01 Phiếu kiểm nghiệm dược chất, bán thành phẩm của cơ sở sản xuất dược chất, bán thành phẩm và 01 Phiếu kiểm nghiệm dược chất, bán thành phẩm của cơ sở sản xuất thuốc thành phẩm;
- Đối với dược chất ở dạng bán thành phẩm thì phải có thêm công thức bào chế và quy trình sản xuất bán thành phẩm chứa dược chất của cơ sở sản xuất bán thành phẩm.
b) Phần hồ sơ thành phẩm:
- Mô tả và thành phần theo hướng dẫn tại Phần P.1-ACTD;
- Tiêu chuẩn chất lượng và phương pháp kiểm nghiệm thuốc thành phẩm. Trường hợp đăng ký theo tiêu chuẩn Dược điển Việt Nam hoặc tiêu chuẩn dược điển tham chiếu theo quy định của Bộ Y tế thì ghi tên dược điển, phiên bản dược điển hoặc ghi dược điển hiện hành;
- Sản xuất thành phẩm, bao gồm: công thức lô sản xuất; quy trình sản xuất và kiểm soát quy trình; kiểm soát các bước quan trọng và các sản phẩm trung gian.
- Phiếu kiểm nghiệm thành phẩm;
- Bao bì đóng gói sơ cấp: Mô tả hình thức, chất liệu và tiêu chuẩn chất lượng bao bì đóng gói sơ cấp.
- Độ ổn định của thuốc thành phẩm.
c) Các tài liệu còn lại của phần hồ sơ chất lượng thực hiện theo hướng dẫn tại Phần II - ACTD hoặc Hợp phần 3-ICH-CTD và lưu tại cơ sở đăng ký, cơ sở sản xuất.
7. Tài liệu quy định tại Điều này phải thực hiện theo các quy định sau:
a) Áp dụng theo các quy định tại Phụ lục I ban hành kèm theo Thông tư này, bao gồm:
- Hồ sơ kỹ thuật chung ASEAN (ACTD);
- Hướng dẫn nghiên cứu độ ổn định;
- Hướng dẫn thẩm định quy trình sản xuất;
- Hướng dẫn thẩm định phương pháp phân tích;
- Hướng dẫn nghiên cứu sinh khả dụng và tương đương sinh học;
b) Đối với hồ sơ thuốc đã chuẩn bị theo mẫu ICH-CTD và hướng dẫn kỹ thuật tương ứng của ICH thì không yêu cầu chuyển đổi hồ sơ theo quy định tại điểm a khoản này;
Điều 26. Tài liệu tiền lâm sàng trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc hóa dược, vắc xin, sinh phẩm
Tài liệu tiền lâm sàng thực hiện theo hướng dẫn tại Phần III-ACTD hoặc Hợp phần 4-ICH-CTD.
Đối với sinh phẩm probiotics (men tiêu hóa) có nguồn gốc, chủng vi khuẩn, nồng độ, hàm lượng, chỉ định, liều dùng tương tự sinh phẩm được cấp phép bởi một trong các cơ quan quản lý quy định tại khoản 9, 10 Điều 2 Thông tư này thì không phải nộp tài liệu tiền lâm sàng.
Điều 27. Tài liệu lâm sàng trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc hóa dược, vắc xin, sinh phẩm
Tài liệu lâm sàng thực hiện theo hướng dẫn tại Phần IV-ACTD hoặc Hợp phần 5-ICH-CTD.
Đối với sinh phẩm probiotics (men tiêu hóa) có nguồn gốc, chủng vi khuẩn, nồng độ, hàm lượng, chỉ định, liều dùng tương tự sinh phẩm được cấp phép bởi một trong các cơ quan quản lý quy định tại khoản 9, 10 Điều 2 Thông tư này thì không yêu cầu tài liệu lâm sàng.
Điều 28. Hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc hóa dược, vắc xin, sinh phẩm
1. Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc hóa dược mới, vắc xin, sinh phẩm, bao gồm:
a) Các tài liệu hành chính theo quy định tại khoản 1, 2, 7, 9, 11, 13, 14, 18 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 6, 8, 10 Điều 24 Thông tư này đối với hồ sơ đăng ký thuốc nước ngoài.
b) Tài liệu chất lượng quy định tại Điều 25 Thông tư này;
c) Tài liệu tiền lâm sàng quy định tại Điều 26 Thông tư này;
d) Tài liệu lâm sàng quy định tại Điều 27 Thông tư này;
đ) Trường hợp cơ sở đăng ký có đề nghị phân loại biệt dược gốc khi nộp hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc thì thực hiện theo quy định tại điểm a, b, c, d khoản 1 Điều này và điểm b khoản 1 Điều 9 Thông tư này.
2. Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc generic, bao gồm:
a) Các tài liệu hành chính theo quy định tại khoản 1, 2, 7, 11, 13, 18 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 6, 8, 10 Điều 24 Thông tư này đối với hồ sơ đăng ký thuốc nước ngoài.
b) Tài liệu chất lượng quy định tại Điều 25 Thông tư này.
3. Hồ sơ đề nghị gia hạn giấy đăng ký lưu hành thuốc:
a) Các tài liệu hành chính theo quy định tại khoản 1, 2, 14, 16, 17, 18, 19 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 6, 10 Điều 24 Thông tư này đối với hồ sơ đăng ký thuốc nước ngoài.
b) Các tài liệu liên quan theo quy định tại Phụ lục II ban hành kèm Thông tư này đối với các trường hợp thuốc có thay đổi về hồ sơ hành chính tại thời điểm gia hạn giấy đăng ký lưu hành.
Trường hợp cơ sở đăng ký đã nộp thay đổi về hồ sơ hành chính trước thời điểm nộp hồ sơ gian hạn nhưng chưa được phê duyệt thì không phải nộp lại phần hồ sơ này trong hồ sơ gia hạn giấy đăng ký lưu hành.
4. Hồ sơ đề nghị thay đổi, bổ sung giấy đăng ký lưu hành thuốc, bao gồm:
a) Đơn đề nghị thay đổi, bổ sung giấy đăng ký lưu hành thuốc theo Mẫu 6/TT ban hành kèm Thông tư này;
b) Các tài liệu tương ứng với các nội dung thay đổi lớn, thay đổi nhỏ quy định tại Phụ lục II ban hành kèm Thông tư này. Đối với vắc xin của cùng chủ sở hữu sản phẩm hoặc cùng một cơ sở sản xuất hoặc chủ sở hữu giấy phép lưu hành thuốc chấp nhận thay đổi địa điểm cơ sở sản xuất ở cùng quốc gia hoặc ngoài quốc gia đã được cấp giấy đăng ký lưu hành.
5. Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc chuyển giao công nghệ
a) Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc theo hình thức chuyển giao công nghệ đối với thuốc chuyển giao có giấy đăng ký lưu hành tại Việt Nam còn hiệu lực:
- Các tài liệu hành chính theo quy định tại khoản 1, 2, 7, 13, 14, 15, 16, 17, 18, 19 Điều 24 Thông tư này và các tài liệu sau:
+ Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
+ Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài.
- Tài liệu chất lượng thực hiện theo quy định tại khoản 4 Điều 25 Thông tư này;
- Các tài liệu liên quan theo quy định tại Phụ lục II ban hành kèm Thông tư này đối với các trường hợp thuốc có thay đổi so với thuốc chuyển giao đã được cấp giấy đăng ký lưu hành.
b) Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc theo hình thức chuyển giao công nghệ đối với thuốc chuyển giao chưa có giấy đăng ký lưu hành tại Việt Nam hoặc có giấy đăng ký lưu hành tại Việt Nam đã hết hiệu lực:
- Hồ sơ của thuốc trước khi chuyển giao công nghệ thực hiện theo quy định tại khoản 1 hoặc khoản 2 Điều này và khoản 15 Điều 24 Thông tư này;
- Tài liệu chất lượng thực hiện theo quy định tại khoản 4 Điều 25 Thông tư này.
6. Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc đóng gói thứ cấp tại Việt Nam
a) Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc thực hiện đóng gói thứ cấp tại Việt Nam đối với thuốc có giấy đăng ký lưu hành tại Việt Nam còn hiệu lực: Thực hiện theo quy định đối với thay đổi cơ sở đóng gói thứ cấp quy định tại Phụ lục II ban hành kèm theo Thông tư này;
b) Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc thực hiện đóng gói thứ cấp tại Việt Nam đối với thuốc chưa có giấy đăng ký lưu hành tại Việt Nam hoặc có giấy đăng ký lưu hành tại Việt Nam đã hết hiệu lực:
- Hồ sơ của thuốc trước khi đóng gói thứ cấp: Thực hiện theo quy định tại khoản 1 hoặc khoản 2 Điều này;
- Giấy chứng nhận GMP của cơ sở đóng gói thứ cấp tại Việt Nam;
- Tài liệu chất lượng thực hiện theo quy định tại khoản 5 Điều 25 Thông tư này.
7. Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc theo quy trình thẩm định rút gọn
a) Các tài liệu hành chính theo quy định tại khoản 1, 2, 7, 11, 13, 18 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 6, 8 Điều 24 Thông tư này đối với hồ sơ đăng ký thuốc nước ngoài.
b) Tài liệu chất lượng thực hiện theo quy định tại điểm a, b khoản 6 Điều 25 Thông tư này.
Mục 3. HỒ SƠ ĐỀ NGHỊ CẤP, GIA HẠN, THAY ĐỔI, BỔ SUNG GIẤY ĐĂNG KÝ LƯU HÀNH THUỐC DƯỢC LIỆU
Điều 29. Tài liệu chất lượng trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc dược liệu
1. Nguyên liệu
a) Quy trình sản xuất (chỉ áp dụng đối với nguyên liệu dược liệu): Mô tả chi tiết, đầy đủ quá trình sơ chế, chế biến nguyên liệu dược liệu. Nếu nguyên liệu là bán thành phẩm dược liệu, cao dược liệu phải mô tả chi tiết quy trình sản xuất bán thành phẩm dược liệu, cao dược liệu từ nguyên liệu dược liệu (trừ trường hợp bán thành phẩm dược liệu, cao dược liệu đã được cấp giấy đăng ký lưu hành);
b) Tiêu chuẩn chất lượng và phương pháp kiểm nghiệm
- Đối với dược liệu không phải dạng bán thành phẩm dược liệu: thực hiện theo quy định tại Thông tư số 13/2018/TT-BYT ngày 15 tháng 5 năm 2018 của Bộ Y tế quy định về chất lượng dược liệu, thuốc cổ truyền.
- Đối với bán thành phẩm dược liệu áp dụng tương tự quy định về tiêu chuẩn chất lượng và phương pháp kiểm nghiệm đối với dược liệu không phải dạng bán thành phẩm dược liệu quy định tại Thông tư số 13/2018/TT-BYT ngày 15 tháng 5 năm 2018 của Bộ Y tế quy định về chất lượng dược liệu, thuốc cổ truyền.
c) Phiếu kiểm nghiệm nguyên liệu
- 01 Phiếu kiểm nghiệm dược liệu của cơ sở sản xuất thuốc thành phẩm.
- 01 Phiếu kiểm nghiệm bán thành phẩm dược liệu, cao dược liệu của cơ sở sản xuất bán thành phẩm dược liệu, cao dược liệu và 01 Phiếu kiểm nghiệm bán thành phẩm dược liệu, cao dược liệu của cơ sở sản xuất thuốc thành phẩm.
2. Thành phẩm
a) Quy trình sản xuất
- Công thức cho đơn vị đóng gói nhỏ nhất: tên, hàm lượng, nồng độ, khối lượng, tiêu chuẩn áp dụng của từng thành phần có trong công thức cho một đơn vị đóng gói nhỏ nhất. Trường hợp sản xuất từ bán thành phẩm dược liệu, cao dược liệu phải ghi rõ khối lượng dược liệu tương ứng với bán thành phẩm dược liệu, cao dược liệu hoặc tỷ lệ cao dược liệu, bán thành phẩm dược liệu so với dược liệu ban đầu hoặc kèm theo hàm lượng (%) của dược chất, nhóm hợp chất đã định lượng được theo từng dược liệu.
- Công thức cho một lô sản xuất thuốc thành phẩm: ghi rõ tên, khối lượng, thể tích của từng thành phần trong công thức lô thuốc.
- Sơ đồ quy trình sản xuất thuốc: thể hiện đầy đủ các giai đoạn trong quá trình sản xuất thuốc bao gồm đường đi của nguyên liệu và phù hợp với mô tả quy trình sản xuất.
- Mô tả quy trình sản xuất thuốc: mô tả đầy đủ, chi tiết các bước thực hiện trong từng giai đoạn của quy trình sản xuất bao gồm đầy đủ các thông số kỹ thuật của từng giai đoạn.
- Danh mục trang thiết bị: tên thiết bị, thông số, mục đích sử dụng.
- Kiểm soát trong quá trình sản xuất: Mô tả đầy đủ, chi tiết các chỉ tiêu kiểm tra, kiểm soát đối với mỗi giai đoạn gồm tên chỉ tiêu, tiêu chuẩn chấp nhận, phương pháp kiểm soát, tần suất kiểm soát, số lượng mẫu lấy để kiểm soát.
b) Tiêu chuẩn chất lượng và phương pháp kiểm nghiệm
- Công thức cho đơn vị đóng gói nhỏ nhất: tên, hàm lượng, nồng độ, khối lượng, tiêu chuẩn áp dụng của từng thành phần có trong công thức cho một đơn vị đóng gói nhỏ nhất. Trường hợp sản xuất từ bán thành phẩm dược liệu, cao dược liệu phải ghi rõ khối lượng dược liệu tương ứng với bán thành phẩm dược liệu, cao dược liệu hoặc tỷ lệ cao dược liệu, bán thành phẩm dược liệu so với dược liệu ban đầu hoặc kèm theo hàm lượng (%) của dược chất, nhóm hợp chất đã định lượng được theo từng dược liệu.
- Tiêu chuẩn thành phẩm: thực hiện theo quy định tại Thông tư số 11/2018/TT-BYT ngày 04 tháng 5 năm 2018 của Bộ Y tế quy định về chất lượng thuốc, nguyên liệu làm thuốc.
c) Phiếu kiểm nghiệm thành phẩm.
d) Tiêu chuẩn của bao bì đóng gói: Mô tả đầy đủ, chi tiết chất liệu bao bì, chỉ tiêu chất lượng, mức chất lượng và phương pháp kiểm nghiệm.
đ) Báo cáo nghiên cứu độ ổn định theo hướng dẫn nghiên cứu độ ổn định quy định tại Phụ lục I ban hành kèm theo Thông tư này.
Điều 30. Tài liệu an toàn, hiệu quả trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc dược liệu
1. Tài liệu an toàn, hiệu quả đối với thuốc dược liệu thực hiện theo quy định của Phụ lục V ban hành kèm theo Thông tư này hoặc theo quy định của ASEAN (ACTD), ICH-CTD.
2. Các tài liệu quy định tại điểm b khoản 1 Điều 19 Thông tư này (nếu có).
Điều 31. Hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc dược liệu
1. Hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc dược liệu, bao gồm:
a) Các tài liệu hành chính theo quy định tại khoản 1, 2, 7, 11, 13, 18 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 6, 8, 10 Điều 24 Thông tư này đối với hồ sơ đăng ký thuốc nước ngoài.
b) Tài liệu chất lượng quy định tại Điều 29 Thông tư này;
c) Tài liệu an toàn, hiệu quả quy định tại Điều 30 Thông tư này;
2. Hồ sơ đề nghị gia hạn giấy đăng ký lưu hành thuốc dược liệu:
a) Các tài liệu hành chính theo quy định tại khoản 1, 2, 16, 17, 18, 19 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 6, 10 Điều 24 Thông tư này đối với hồ sơ đăng ký thuốc nước ngoài.
b) Các tài liệu liên quan theo quy định tại Mục D Phụ lục II ban hành kèm Thông tư này đối với các trường hợp thuốc có thay đổi về hồ sơ hành chính tại thời điểm gia hạn giấy đăng ký lưu hành.
Trường hợp cơ sở đăng ký đã nộp thay đổi về hồ sơ hành chính trước thời điểm nộp hồ sơ gian hạn nhưng chưa được phê duyệt thì không phải nộp lại phần hồ sơ này trong hồ sơ gia hạn giấy đăng ký lưu hành.
3. Hồ sơ đề nghị thay đổi, bổ sung giấy đăng ký lưu hành thuốc dược liệu, bao gồm:
a) Đơn đề nghị thay đổi, bổ sung giấy đăng ký lưu hành thuốc dược liệu theo Mẫu 6/TT ban hành kèm theo Thông tư này;
b) Tài liệu tương ứng với các nội dung thay đổi lớn, thay đổi nhỏ quy định tại Mục D Phụ lục II ban hành kèm theo Thông tư này.
Mục 4. HỒ SƠ ĐĂNG KÝ NGUYÊN LIỆU LÀM THUỐC
Điều 32. Tài liệu chất lượng trong hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành nguyên liệu làm thuốc
1. Đối với nguyên liệu dược chất: Thực hiện theo hồ sơ ACTD phần dược chất. Trường hợp dược chất đăng ký chất lượng theo tiêu chuẩn nhà sản xuất, phải nộp kèm theo hồ sơ tổng thể dược chất (Drug Master File).
2. Đối với nguyên liệu ở dạng bán thành phẩm chứa dược chất: Thực hiện theo hồ sơ ACTD như đăng ký thuốc thành phẩm, trong đó phần hồ sơ về thành phẩm được thay bằng hồ sơ bán thành phẩm đăng ký; các công thức cho một đơn vị liều, đơn vị đóng gói nhỏ nhất thay bằng công thức lô sản xuất.
3. Đối với nguyên liệu bán thành phẩm dược liệu, tá dược, vỏ nang:
a) Công thức bào chế đối với bán thành phẩm dược liệu, tá dược ở dạng trộn sẵn, vỏ nang: thành phần, khối lượng, thể tích, tiêu chuẩn chất lượng của từng thành phần trong công thức. Trường hợp sử dụng nguyên liệu có nguồn gốc từ động vật phải cung cấp thông tin về các chất ngẫu nhiên (các số liệu an toàn vi rút).
b) Quy trình sản xuất
- Sơ đồ quy trình sản xuất: thể hiện đầy đủ các giai đoạn trong quá trình sản xuất bao gồm đường đi của nguyên liệu và phù hợp với mô tả quy trình sản xuất.
- Mô tả quy trình sản xuất: mô tả đầy đủ, chi tiết các bước thực hiện trong từng giai đoạn của quy trình sản xuất bao gồm đầy đủ các thông số kỹ thuật của từng giai đoạn.
- Danh mục trang thiết bị: tên thiết bị, thông số, mục đích sử dụng.
- Kiểm soát trong quá trình sản xuất: Mô tả đầy đủ, chi tiết các chỉ tiêu kiểm tra, kiểm soát đối với mỗi giai đoạn gồm tên chỉ tiêu, tiêu chuẩn chấp nhận, phương pháp kiểm soát, tần suất kiểm soát, số lượng mẫu lấy để kiểm soát.
c) Tiêu chuẩn chất lượng và phương pháp kiểm nghiệm
- Đối với bán thành phẩm dược liệu áp dụng tương tự quy định về tiêu chuẩn chất lượng và phương pháp kiểm nghiệm đối với dược liệu không phải dạng bán thành phẩm dược liệu quy định tại Thông tư số 13/2018/TT-BYT ngày 15 tháng 5 năm 2018 của Bộ Y tế quy định về chất lượng dược liệu, thuốc cổ truyền.
- Đối với tá dược, vỏ nang: thực hiện theo quy định tại Thông tư số 11/2018/TT-BYT ngày 04 tháng 5 năm 2018 của Bộ Y tế quy định về chất lượng thuốc, nguyên liệu làm thuốc.
d) Phiếu kiểm nghiệm.
đ) Tiêu chuẩn của bao bì đóng gói: Mô tả đầy đủ, chi tiết chất liệu bao bì, chỉ tiêu chất lượng, mức chất lượng và phương pháp kiểm nghiệm.
e) Báo cáo nghiên cứu độ ổn định, bao gồm đề cương nghiên cứu độ ổn định; số liệu nghiên cứu độ ổn định; kết quả và bàn luận.
Điều 33. Hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành nguyên liệu làm thuốc
1. Hồ sơ đề nghị cấp giấy đăng ký lưu hành nguyên liệu làm thuốc, bao gồm:
a) Phần I. Hồ sơ hành chính
- Các tài liệu hành chính theo quy định tại khoản 1, 2, 7, 11, 13, 18 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 8, 10, 12 Điều 24 Thông tư này đối với hồ sơ đăng ký nguyên liệu sản xuất tại nước ngoài.
b) Tài liệu chất lượng quy định tại Điều 32 Thông tư này;
2. Hồ sơ đề nghị gia hạn giấy đăng ký lưu hành nguyên liệu làm thuốc:
a) Các tài liệu hành chính theo quy định tại khoản 1, 2, 11, 17, 18, 19 Điều 24 Thông tư này và các tài liệu sau:
- Tài liệu quy định tại khoản 3 Điều 24 Thông tư này đối với cơ sở đăng ký của Việt Nam;
- Tài liệu quy định tại khoản 4, 5 Điều 24 Thông tư này đối với cơ sở đăng ký của nước ngoài;
- Tài liệu quy định tại khoản 10, 12 Điều 24 Thông tư này đối với trường hợp nguyên liệu sản xuất tại nước ngoài.
b) Các tài liệu liên quan theo quy định tại Mục B Phụ lục II ban hành kèm Thông tư này đối với các trường hợp nguyên liệu làm thuốc có thay đổi về hồ sơ hành chính tại thời điểm gia hạn giấy đăng ký lưu hành.
Trường hợp cơ sở đăng ký đã nộp thay đổi về hồ sơ hành chính trước thời điểm nộp hồ sơ gian hạn nhưng chưa được phê duyệt thì không phải nộp lại phần hồ sơ này trong hồ sơ gia hạn giấy đăng ký lưu hành.
3. Hồ sơ đề nghị thay đổi, bổ sung giấy đăng ký lưu hành nguyên liệu làm thuốc, bao gồm:
a) Đơn đề nghị thay đổi, bổ sung giấy đăng ký lưu hành thuốc theo Mẫu 06/TT ban hành kèm theo Thông tư này;
b) Tài liệu tương ứng với các nội dung thay đổi lớn, thay đổi nhỏ theo quy định tại Phần B Phụ lục II ban hành kèm theo Thông tư này.
Chương IV
THỦ TỤC CẤP, GIA HẠN, THAY ĐỔI, BỔ SUNG GIẤY ĐĂNG KÝ LƯU HÀNH THUỐC, NGUYÊN LIỆU LÀM THUỐC; TRÌNH TỰ THẨM ĐỊNH HỒ SƠ NHẬP KHẨU THUỐC CHƯA CÓ GIẤY ĐĂNG KÝ LƯU HÀNH
Điều 34. Các trường hợp được thực hiện theo quy trình thẩm định nhanh
1. Thuốc thuộc Danh mục thuốc hiếm do Bộ trưởng Bộ Y tế ban hành.
2. Thuốc đáp ứng nhu cầu cấp bách cho quốc phòng, an ninh, quốc phòng, chống dịch bệnh, khắc phục hậu quả thiên tai, thảm họa.
3. Thuốc trong nước sản xuất trên những dây chuyền mới đạt tiêu chuẩn GMP hoặc trên dây chuyền nâng cấp đạt tiêu chuẩn GMP-EU, GMP-PIC/S và tương đương trong thời hạn không quá 18 tháng kể từ ngày được cấp giấy chứng nhận GMP.
4. Vắc xin đã được Tổ chức Y tế thế giới tiền đánh giá đạt yêu cầu, vắc xin dùng trong chương trình tiêm chủng mở rộng quốc gia.
5. Thuốc chuyên khoa đặc trị, thuốc có dạng bào chế đặc biệt chỉ có không quá 02 (hai) thuốc tương tự (cùng hoạt chất, cùng dạng bào chế, cùng hàm lượng, nồng độ) có giấy đăng ký lưu hành thuốc tại Việt Nam còn hiệu lực tại thời điểm nộp hồ sơ, bao gồm:
a) Thuốc điều trị ung thư;
b) Thuốc điều trị kháng vi rút thế hệ mới;
c) Kháng sinh thế hệ mới;
d) Thuốc dùng trong điều trị sốt xuất huyết, lao, sốt rét.
6. Thuốc sản xuất trong nước, bao gồm:
a) Thuốc được sản xuất gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam đối với thuốc điều trị ung thư, vắc xin, sinh phẩm, thuốc điều trị kháng vi rút thế hệ mới, kháng sinh thế hệ mới;
b) Thuốc dược liệu có đề tài khoa học và công nghệ cấp quốc gia, cấp bộ hoặc cấp tỉnh đã được nghiệm thu đạt yêu cầu, thuốc được sản xuất toàn bộ từ nguồn dược liệu trong nước đạt thực hành tốt nuôi trồng thu hái dược liệu theo tiêu chuẩn WHO (GACP);
c) Thuốc mới sản xuất trong nước đã hoàn thành thực hiện thử nghiệm lâm sàng tại Việt Nam;
7. Thuốc mới (điều trị ung thư, kháng vi rút thế hệ mới, kháng sinh thế hệ mới), sinh phẩm.
8. Biệt dược gốc được sản xuất gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam.
Điều 35. Các trường hợp được thẩm định theo quy trình rút gọn
Hồ sơ đăng ký thuốc được thẩm định theo quy trình thẩm định rút gọn khi đáp ứng đồng thời các điều kiện sau:
1. Thuốc được sản xuất tại cơ sở được Cục Quản lý Dược định kỳ đánh giá việc đáp ứng thực hành tốt sản xuất thuốc.
2. Thuốc thuộc Danh mục thuốc không kê đơn.
3. Thuốc không phải dạng bào chế giải phóng biến đổi.
4. Thuốc không dùng trực tiếp trên mắt.
Điều 36. Thẩm quyền cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
1. Bộ trưởng Bộ Y tế giao Cục Quản lý Dược tổ chức thẩm định hồ sơ, cấp, gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, phê duyệt thay đổi lớn giấy đăng ký lưu hành thuốc bao gồm thay đổi chỉ định, liều dùng, đối tượng dùng thuốc và công bố biệt dược gốc, thuốc có báo cáo nghiên cứu tương đương sinh học trên cơ sở ý kiến thẩm định hồ sơ, tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
2. Bộ trưởng Bộ Y tế giao Cục Quản lý Dược thẩm định hồ sơ và phê duyệt thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, trừ các trường hợp thay đổi lớn quy định tại khoản 1 Điều này.
Điều 37. Quy định chung
1. Hồ sơ được nộp trực tuyến, nộp trực tiếp hoặc gửi qua đường bưu điện đến Cục Quản lý Dược.
2. Sau khi nhận được đầy đủ hồ sơ, Cục Quản lý Dược trả cho cơ sở đăng ký Phiếu tiếp nhận hồ sơ theo Mẫu số 12/TT ban hành kèm theo Thông tư này.
3. Đối với hồ sơ nhập khẩu thuốc chưa có giấy đăng ký lưu hành, việc tiếp nhận hồ sơ thực hiện theo quy định tại điểm b khoản 1 Điều 77 Nghị định số 54/2017/NĐ-CP.
4. Tổ chức thẩm định hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc và hồ sơ nhập khẩu thuốc chưa có giấy đăng ký lưu hành:
a) Cục Quản lý Dược chuyển hồ sơ cho các chuyên gia thẩm định hoặc các đơn vị do Bộ trưởng Bộ Y tế quyết định (sau đây viết tắt là đơn vị thẩm định) để tổ chức thẩm định trên cơ sở danh sách chuyên gia thẩm định do Cục Quản lý Dược hoặc các đơn vị thẩm định thành lập, phê duyệt.
b) Trên cơ sở tổng hợp ý kiến thẩm định của chuyên gia thẩm định, đơn vị thẩm định quy định tại điểm a khoản này và xem xét các thông tin liên quan, Cục Quản lý Dược chịu trách nhiệm đề xuất việc cấp, gia hạn, thay đổi, bổ sung hoặc chưa cấp, gia hạn, thay đổi, bổ sung hoặc không cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc; cấp, chưa cấp hoặc không cấp giấy phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành. Ý kiến đề xuất của Cục Quản lý Dược được thể hiện trên biên bản thẩm định.
c) Cục Quản lý Dược trình Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc thẩm định, tư vấn về các ý kiến đề xuất của Cục Quản lý Dược quy định tại điểm b khoản này đối với các trường hợp sau:
- Cấp, không cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
- Gia hạn, không gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
- Phê duyệt, không phê duyệt thay đổi lớn giấy đăng ký lưu hành thuốc đối với nội dung thay đổi về chỉ định, liều dùng, đối tượng dùng thuốc;
- Công bố, không công bố biệt dược gốc, thuốc có báo cáo nghiên cứu tương đương sinh học;
- Cấp, không cấp giấy phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành;
- Các trường hợp khác do Cục Quản lý Dược đề xuất để đáp ứng nhu cầu cấp thiết trong phòng bệnh, chữa bệnh.
Điều 38. Thủ tục cấp giấy đăng ký lưu hành thuốc, trình tự thẩm định hồ sơ nhập khẩu thuốc chưa có giấy đăng ký lưu hành
1. Trong thời hạn tối đa 12 tháng kể từ ngày nhận đủ hồ sơ đối với hồ sơ đề nghị cấp giấy đăng ký lưu hành thuốc (trừ trường hợp quy định tại Điều 41 Thông tư này), Cục Quản lý Dược cấp giấy đăng ký lưu hành thuốc. Trường hợp không cấp hoặc chưa cấp, Cục Quản lý Dược có văn bản trả lời và nêu rõ lý do. Thời gian giải quyết các bước được quy định, cụ thể như sau:
a) Trong thời hạn 03 tháng kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược tiến hành rà soát, phân loại và gửi hồ sơ cho chuyên gia hoặc các đơn vị thẩm định. Trong thời hạn 06 tháng kể từ ngày nhận được hồ sơ từ Cục Quản lý Dược, chuyên gia và các đơn vị thẩm định phải hoàn thành biên bản thẩm định và gửi Cục Quản lý Dược tổng hợp, đề xuất ý kiến trên biên bản thẩm định theo quy định tại khoản 4 Điều 37 Thông tư này;
b) Trong thời hạn 02 tháng kể từ ngày nhận được biên bản thẩm định, Cục Quản lý Dược có văn bản trả lời đối với hồ sơ thẩm định chưa đạt hoặc không đạt và nêu rõ lý do. Đối với hồ sơ được Cục Quản lý Dược đề xuất cấp hoặc đề xuất cần xin ý kiến thẩm định, tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược trình Hội đồng trong phiên họp gần nhất;
c) Trong thời hạn 30 ngày kể từ ngày họp Hội đồng, Cục Quản lý Dược ban hành quyết định cấp giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; Cục Quản lý Dược có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
2. Trong thời hạn 36 tháng đối với trường hợp có yêu cầu bổ sung tài liệu tiền lâm sàng và lâm sàng, tài liệu tương đương sinh học, tài liệu nghiên cứu độ ổn định hoặc trong thời hạn 12 tháng đối với các trường hợp bổ sung tài liệu khác, kể từ ngày Cục Quản lý Dược có văn bản thông báo, cơ sở đăng ký phải nộp tài liệu bổ sung theo yêu cầu. Sau thời hạn này, cơ sở đăng ký không nộp tài liệu bổ sung thì hồ sơ đã nộp không còn giá trị.
Cơ sở đăng ký thông báo bằng văn bản về Cục Quản lý Dược trường hợp có các thông tin cập nhật liên quan đến an toàn, hiệu quả của thuốc so với hồ sơ đăng ký đã nộp và đang trong thời gian thẩm định.
Thời gian kể từ khi có văn bản thông báo của Cục Quản lý Dược đến khi cơ sở đăng ký nộp tài liệu bổ sung không được tính vào thời hạn quy định tại khoản 5 Điều 56 Luật dược.
3. Trong thời hạn 06 tháng kể từ ngày nhận đủ tài liệu bổ sung, Cục Quản lý Dược ban hành quyết định cấp giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; Cục Quản lý Dược có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
Trình tự xem xét tài liệu bổ sung được thực hiện theo quy định tại khoản 1 Điều này.
4. Trình tự thẩm định hồ sơ nhập khẩu thuốc chưa có giấy đăng ký lưu hành:
a) Trong thời hạn 05 ngày làm việc kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược tiến hành chuyển hồ sơ cho các chuyên gia thẩm định hoặc đơn vị thẩm định.
Thời gian thẩm định là không quá 30 ngày đối với các hồ sơ không yêu cầu dữ liệu lâm sàng, tài liệu chứng minh tương tự so với sinh phẩm tham chiếu, hoặc không quá 60 ngày đối với đối với các hồ sơ yêu cầu dữ liệu lâm sàng hoặc tài liệu chứng minh tương tự so với sinh phẩm tham chiếu kể từ ngày Cục Quản lý Dược chuyển hồ sơ cho chuyên gia thẩm định hoặc đơn vị thẩm định;
b) Trong vòng 20 ngày kể từ ngày nhận được biên bản đã được thẩm định theo đúng quy chế hoạt động của chuyên gia:
- Cục Quản lý Dược tổng hợp ý kiến thẩm định của chuyên gia thẩm định hoặc đơn vị thẩm định và xem xét các thông tin liên quan để đề xuất việc cấp, chưa cấp hoặc không cấp giấy phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành.
- Đối với hồ sơ cần phải trình Hội đồng theo quy định tại điểm c khoản 4 Điều 37 Thông tư này, Cục Quản lý Dược trình Hội đồng trong phiên họp gần nhất;
- Đối với hồ sơ Cục Quản lý Dược đề xuất chưa cấp, Cục Quản lý Dược có văn bản trả lời và nêu rõ lý do.
c) Trong vòng 5 ngày làm việc kể từ ngày họp Hội đồng hoặc có ý kiến kết luận của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược cấp Giấy phép nhập khẩu đối với hồ sơ đạt yêu cầu; hoặc có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ chưa đạt hoặc không đạt yêu cầu và nêu rõ lý do.
d) Sau khi nhận được hồ sơ sửa đổi, bổ sung của cơ sở nhập khẩu, Cục Quản lý Dược thực hiện theo quy định tại điểm a, b và c khoản này.
Đối với hồ sơ Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc có yêu cầu sửa đổi, bổ sung và không yêu cầu phải trình lại Hội đồng lần sau, Cục Quản lý Dược thông báo cho cơ sở sửa đổi, bổ sung; trường hợp hồ sơ sửa đổi, bổ sung thẩm định đạt yêu cầu, Cục Quản lý Dược thực hiện việc cấp phép mà không phải trình lại Hội đồng.
Điều 39. Thủ tục gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
1. Trong thời hạn 03 tháng kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược gia hạn giấy đăng ký lưu hành lưu hành thuốc, nguyên liệu làm thuốc. Trường hợp không gia hạn hoặc chưa gia hạn, Cục Quản lý Dược có văn bản trả lời và nêu rõ lý do. Thời gian các bước được quy định, cụ thể như sau:
a) Trong thời hạn 10 ngày làm việc kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược tiến hành rà soát, phân loại và gửi hồ sơ cho các tiểu ban thẩm định. Trong thời hạn 01 tháng kể từ ngày nhận được hồ sơ từ Cục Quản lý Dược, các tiểu ban thẩm định phải hoàn thành biên bản thẩm định và gửi Cục Quản lý Dược tổng hợp, kết luận biên bản thẩm định theo quy định tại khoản 4 Điều 37 Thông tư này;
b) Trong thời hạn 15 ngày làm việc kể từ ngày nhận được biên bản thẩm định của các tiểu ban, Cục Quản lý Dược có văn bản trả lời đối với hồ sơ thẩm định chưa đạt hoặc không đạt và nêu rõ lý do. Đối với hồ sơ thẩm định đạt yêu cầu hoặc những trường hợp khác cần xin ý kiến thẩm định, tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược trình Hội đồng trong phiên họp gần nhất;
c) Trong thời hạn 15 ngày làm việc kể từ ngày họp Hội đồng, Cục Quản lý Dược ban hành quyết định gia hạn giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; Cục Quản lý Dược có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
2. Trong thời hạn 12 tháng đối với các trường hợp bổ sung tài liệu, kể từ ngày Cục Quản lý Dược có văn bản thông báo, cơ sở đăng ký phải nộp tài liệu bổ sung theo yêu cầu. Sau thời hạn này, cơ sở đăng ký không nộp tài liệu bổ sung thì hồ sơ đã nộp không còn giá trị.
Cơ sở đăng ký thông báo bằng văn bản về Cục Quản lý Dược trường hợp có các thông tin cập nhật liên quan đến an toàn, hiệu quả của thuốc so với hồ sơ đăng ký đã nộp và đang trong thời gian thẩm định.
Thời gian kể từ khi có văn bản thông báo của Cục Quản lý Dược đến khi cơ sở đăng ký nộp tài liệu bổ sung không được tính vào thời hạn quy định tại khoản 5 Điều 56 Luật dược.
3. Trong thời hạn 03 tháng kể từ ngày nhận đủ tài liệu bổ sung, Cục Quản lý Dược ban hành quyết định gia hạn giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; Cục Quản lý Dược có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
Trình tự xem xét tài liệu bổ sung được thực hiện theo quy định tại khoản 1 Điều này.
Điều 40. Thủ tục thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc trong thời gian giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc còn hiệu lực
1. Thay đổi, bổ sung giấy đăng ký lưu hành thuốc đối với nội dung thay đổi lớn về chỉ định, liều dùng, đối tượng dùng thuốc; phân loại biệt dược gốc, thuốc có báo cáo nghiên cứu tương đương sinh học
Trong thời hạn 03 tháng kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược công bố biệt dược gốc, thuốc có báo cáo nghiên cứu tương đương sinh học, Cục Quản lý Dược phê duyệt nội dung thay đổi lớn về chỉ định, liều dùng, đối tượng dùng thuốc. Trường hợp không phê duyệt hoặc chưa phê duyệt, Cục Quản lý Dược có văn bản trả lời và nêu rõ lý do. Thời gian các bước được quy định, cụ thể như sau:
a) Trong thời hạn 10 ngày làm việc kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược tiến hành rà soát, phân loại và gửi hồ sơ cho các tiểu ban thẩm định. Trong thời hạn 01 tháng kể từ ngày nhận được hồ sơ từ Cục Quản lý Dược, các tiểu ban thẩm định phải hoàn thành biên bản thẩm định và gửi Cục Quản lý Dược tổng hợp, kết luận biên bản thẩm định theo quy định tại khoản 4 Điều 37 Thông tư này;
b) Trong thời hạn 15 ngày làm việc kể từ ngày nhận được biên bản thẩm định của các tiểu ban, Cục Quản lý Dược có văn bản trả lời đối với hồ sơ thẩm định chưa đạt hoặc không đạt và nêu rõ lý do. Đối với hồ sơ thẩm định đạt yêu cầu hoặc những trường hợp khác cần xin ý kiến thẩm định, tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược trình Hội đồng trong phiên họp gần nhất;
c) Trong thời hạn 15 ngày làm việc kể từ ngày họp Hội đồng, Cục Quản lý Dược công bố biệt dược gốc, thuốc có báo cáo nghiên cứu tương đương sinh học, Cục Quản lý Dược phê duyệt nội dung thay đổi lớn về chỉ định, liều dùng, đối tượng dùng thuốc đối với hồ sơ đạt yêu cầu. Cục Quản lý Dược có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
2. Thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, trừ trường hợp quy định tại khoản 1, 3 Điều này
Trong thời hạn 03 tháng kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược phê duyệt nội dung thay đổi, bổ sung. Trường hợp không phê duyệt hoặc chưa phê duyệt, Cục Quản lý Dược có văn bản trả lời và nêu rõ lý do. Thời gian các bước được quy định, cụ thể như sau:
a) Trong thời hạn 10 ngày kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược tiến hành rà soát, phân loại và gửi hồ sơ cho các tiểu ban thẩm định. Trong thời hạn 60 ngày kể từ ngày nhận được hồ sơ từ Cục Quản lý Dược, các tiểu ban thẩm định phải hoàn thành biên bản thẩm định và gửi Cục Quản lý Dược tổng hợp, kết luận biên bản thẩm định theo quy định tại khoản 4 Điều 37 Thông tư này;
b) Trong thời hạn 20 ngày kể từ ngày nhận được biên bản thẩm định của các tiểu ban, Cục Quản lý Dược phê duyệt nội dung thay đổi, bổ sung giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; có văn bản trả lời đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
3. Thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc đối với nội dung thay đổi nhỏ chỉ yêu cầu thông báo (Notification)
Trong thời hạn 15 ngày làm việc kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược phê duyệt thay đổi, bổ sung giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; có văn bản trả lời đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
4. Trong thời hạn 36 tháng đối với trường hợp có yêu cầu bổ sung tài liệu tiền lâm sàng và lâm sàng, tài liệu tương đương sinh học, tài liệu nghiên cứu độ ổn định hoặc trong thời hạn 12 tháng đối với các trường hợp bổ sung tài liệu khác, kể từ ngày Cục Quản lý Dược có văn bản thông báo, cơ sở đăng ký phải nộp tài liệu bổ sung theo yêu cầu. Sau thời hạn này, cơ sở đăng ký không nộp tài liệu bổ sung thì hồ sơ đã nộp không còn giá trị.
Cơ sở đăng ký thông báo bằng văn bản về Cục Quản lý Dược trường hợp có các thông tin cập nhật liên quan đến an toàn, hiệu quả của thuốc so với hồ sơ đăng ký đã nộp và đang trong thời gian thẩm định.
Thời gian kể từ khi có văn bản thông báo của Cục Quản lý Dược đến khi cơ sở đăng ký nộp tài liệu bổ sung không được tính vào thời hạn quy định tại khoản 5 Điều 56 Luật dược.
5. Trong thời hạn 02 tháng kể từ ngày nhận đủ tài liệu bổ sung đối với hồ sơ quy định tại khoản 1 Điều này, 01 tháng kể từ ngày nhận đủ tài liệu bổ sung đối với hồ sơ quy định tại khoản 2 Điều này, 10 ngày làm việc kể từ ngày nhận đủ tài liệu bổ sung đối với hồ sơ quy định tại khoản 3 Điều này, Cục Quản lý Dược phê duyệt thay đổi, bổ sung giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; có văn bản trả lời đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
Trình tự xem xét tài liệu bổ sung được thực hiện theo quy định tại khoản 1, 2, 3 Điều này.
6. Thời hạn phải thực hiện đối với các nội dung thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc: không quá 12 tháng đối với vắc xin, sinh phẩm hoặc không quá 06 tháng đối với các thuốc, nguyên liệu làm thuốc khác kể từ ngày Cục Quản lý Dược ký ban hành công văn phê duyệt đối với các trường hợp thay đổi, bổ sung, trừ trường có yêu cầu khác của Cục Quản lý Dược.
7. Một số trường hợp thay đổi, bổ sung, cơ sở đăng ký thuốc, cơ sở sản xuất thuốc tự cập nhật trên nhãn, hướng dẫn sử dụng thuốc và không yêu cầu phải nộp hồ sơ hoặc thông báo cho Cục Quản lý Dược, bao gồm các trường hợp sau đây:
a) Thực hiện việc ghi nhãn thuốc, nguyên liệu làm thuốc, hướng dẫn sử dụng thuốc theo quy định tại khoản 2 Điều 35 Thông tư 01/2018/TT-BYT ngày 18 tháng 01 năm 2018 của Bộ Y tế Quy định ghi nhận thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc;
b) Thực hiện việc thay đổi, bổ sung nội dung nhãn, hướng dẫn sử dụng thuốc theo đúng nội dung trong văn bản yêu cầu của Cục Quản lý Dược;
c) Ngoài những trường hợp phải nộp lại mẫu nhãn, hướng dẫn sử dụng thuốc khi có thay đổi, bổ sung theo quy định tại Phụ lục II ban hành kèm theo Thông tư này, các thay đổi khác liên quan đến các thông tin trên nhãn, hướng dẫn sử dụng thuốc cơ sở đăng ký, cơ sở sản xuất phải tự cập nhật khi đã được Cục Quản lý Dược phê duyệt đối với các thay đổi, bổ sung này;
d) Các nội dung khác:
- Thay đổi thông tin cơ sở nhập khẩu thuốc, nguyên liệu làm thuốc ghi trên nhãn hoặc tờ hướng dẫn sử dụng thuốc;
- Sửa lỗi chính tả trên nhãn, tờ hướng dẫn sử dụng thuốc;
- Thay đổi bố cục trình bày các mục trong tờ hướng dẫn sử dụng thuốc nhưng không thay đổi nội dung tờ hướng dẫn sử dụng thuốc đã được phê duyệt;
- Bổ sung thông tin về tiêu chuẩn chất lượng trên nhãn, hướng dẫn sử dụng thuốc theo hồ sơ đã được Cục Quản lý Dược phê duyệt;
- Các nội dung thay đổi, bổ sung theo đúng văn bản của Cục Quản lý Dược về việc thông báo kết quả thẩm định hồ sơ đăng ký thuốc, nguyên liệu làm thuốc.
Điều 41. Trình tự cấp giấy đăng ký lưu hành thuốc thực hiện theo quy trình thẩm định nhanh, thẩm định rút gọn và cấp giấy đăng ký lưu hành nguyên liệu làm thuốc
1. Trong thời hạn 06 tháng kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc. Trường hợp không cấp hoặc chưa cấp, Cục Quản lý Dược có văn bản trả lời và nêu rõ lý do. Thời gian giải quyết các bước được quy định, cụ thể như sau:
a) Trong thời hạn 10 ngày làm việc kể từ ngày nhận đủ hồ sơ, Cục Quản lý Dược phân loại và gửi hồ sơ cho các tiểu ban thẩm định. Trong thời hạn 03 tháng kể từ ngày nhận được hồ sơ từ Cục Quản lý Dược, chuyên gia và các đơn vị thẩm định phải hoàn thành biên bản thẩm định và gửi Cục Quản lý Dược tổng hợp, kết luận biên bản thẩm định theo quy định tại khoản 4 Điều 37 Thông tư này;
b) Trong thời hạn 20 ngày làm việc kể từ ngày nhận được biên bản thẩm định của các tiểu ban, Cục Quản lý Dược có văn bản trả lời đối với hồ sơ thẩm định chưa đạt hoặc không đạt và nêu rõ lý do. Đối với hồ sơ thẩm định đạt yêu cầu hoặc những trường hợp khác cần xin ý kiến thẩm định, tư vấn của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược trình Hội đồng trong phiên họp gần nhất;
c) Trong thời hạn 30 ngày làm việc kể từ ngày họp Hội đồng, Cục Quản lý Dược ban hành quyết định cấp giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; Cục Quản lý Dược có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
2. Trong thời hạn 36 tháng đối với trường hợp có yêu cầu bổ sung tài liệu tiền lâm sàng và lâm sàng, tài liệu tương đương sinh học, tài liệu nghiên cứu độ ổn định hoặc trong thời hạn 12 tháng đối với các trường hợp bổ sung tài liệu khác, kể từ ngày Cục Quản lý Dược có văn bản thông báo, cơ sở đăng ký phải nộp tài liệu bổ sung theo yêu cầu. Sau thời hạn này, cơ sở đăng ký không nộp tài liệu bổ sung thì hồ sơ đã nộp không còn giá trị.
Cơ sở đăng ký thông báo bằng văn bản về Cục Quản lý Dược trường hợp có các thông tin cập nhật liên quan đến an toàn, hiệu quả của thuốc, nguyên liệu làm thuốc so với hồ sơ đăng ký đã nộp và đang trong thời gian thẩm định.
Thời gian kể từ khi có văn bản thông báo của Cục Quản lý Dược đến khi cơ sở đăng ký nộp tài liệu bổ sung không được tính vào thời hạn quy định tại khoản 5 Điều 56 Luật dược.
3. Trong thời hạn 03 tháng kể từ ngày nhận đủ tài liệu bổ sung, Cục Quản lý Dược ban hành quyết định cấp giấy đăng ký lưu hành đối với hồ sơ đạt yêu cầu; Cục Quản lý Dược có văn bản trả lời theo kết luận của Hội đồng đối với hồ sơ thẩm định chưa đạt, không đạt và nêu rõ lý do.
Trình tự xem xét tài liệu bổ sung được thực hiện theo quy định tại khoản 1 Điều này.
Chương V
THU HỒI GIẤY ĐĂNG KÝ LƯU HÀNH, NGỪNG NHẬN HỒ SƠ CẤP, GIA HẠN GIẤY ĐĂNG KÝ LƯU HÀNH
Điều 42. Hồ sơ, thủ tục, thẩm quyền thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
1. Thẩm quyền thu hồi và trách nhiệm thông báo thu hồi giấy đăng ký lưu hành:
a) Bộ trưởng Bộ Y tế giao Cục Quản lý Dược thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc thuộc các trường hợp quy định tại khoản 1 Điều 58 Luật dược;
b) Sở Y tế các tỉnh, thành phố trực thuộc Trung ương, Y tế các ngành thông báo quyết định của Cục Quản lý Dược về việc thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc thuộc địa bàn quản lý.
2. Hồ sơ thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc đối với trường hợp quy định tại điểm g khoản 1 Điều 58 Luật dược, bao gồm:
- Đơn đề nghị thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc tại Việt Nam của cơ sở sản xuất hoặc cơ sở đăng ký thuốc, nguyên liệu làm thuốc theo Mẫu 1/TT ban kèm theo Thông tư này;
- Bản chính giấy đăng ký lưu hành;
- Các tài liệu chứng minh (nếu có).
3. Thủ tục thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc quy định tại điểm a, b khoản 1 Điều 58 Luật dược
Trong thời hạn không quá 30 ngày kể từ ngày có quyết định thu hồi thuốc của cơ quan quản lý có thẩm quyền, Cục Quản lý Dược ra quyết định thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
4. Thủ tục thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc quy định tại điểm c và e khoản 1 Điều 58 Luật dược
Trong thời hạn không quá 10 ngày kể từ ngày cơ quan quản lý có thẩm quyền của Việt Nam hoặc kể từ ngày nhận được thông báo của Tổ chức Y tế Thế giới hoặc của nước xuất xứ khuyến cáo thuốc không an toàn, hiệu quả cho người sử dụng hoặc cơ quan có thẩm quyền của nước ngoài thu hồi giấy chứng nhận sản phẩm, Cục Quản lý Dược ra quyết định thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
5. Thủ tục thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc quy định tại điểm d, đ khoản 1 Điều 58 Luật dược
Trong thời hạn không quá 30 ngày kể từ ngày có kết luận bằng văn bản của cơ quan quản lý có thẩm quyền về việc hồ sơ của thuốc đã được cấp giấy đăng ký lưu hành là hồ sơ giả mạo hoặc thuốc, nguyên liệu làm thuốc được sản xuất không đúng địa chỉ theo hồ sơ đăng ký, Cục Quản lý Dược ra quyết định thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
6. Thủ tục thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc quy định tại điểm g khoản 1 Điều 58 Luật dược
Trong thời hạn không quá 20 ngày kể từ ngày nhận đủ hồ sơ theo quy định tại khoản 2 Điều này, Cục Quản lý Dược ra quyết định thu hồi giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc. Trường hợp chưa đồng ý với đề nghị thu hồi của cơ sở, Cục Quản lý Dược có văn bản trả lời và nêu rõ lý do.
Điều 43. Quy định về việc ngừng nhận hồ sơ cấp, gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
1. Việc ngừng nhận hồ sơ cấp, gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc thực hiện theo quy định tại các khoản 2, 3 và 4 Điều 100 của Nghị định số 54/2017/NĐ-CP ngày 08 tháng 5 năm 2017 của Chính phủ quy định chi tiết một số điều và biện pháp thi hành Luật dược.
2. Cục Quản lý Dược thông báo việc ngừng nhận hồ sơ cấp, gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
Chương VI
NGUYÊN TẮC TỔ CHỨC, HOẠT ĐỘNG CỦA CHUYÊN GIA THẨM ĐỊNH VÀ HỘI ĐỒNG TƯ VẤN CẤP GIẤY ĐĂNG KÝ LƯU HÀNH THUỐC, NGUYÊN LIỆU LÀM THUỐC
Điều 44. Tổ chức, hoạt động của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
1. Bộ trưởng Bộ Y tế thành lập Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc. Hội đồng bao gồm các thành viên là các chuyên gia có trình độ chuyên môn và kinh nghiệm phù hợp bảo đảm khả năng thẩm định hồ sơ, phản biện ý kiến của chuyên gia thẩm định và ý kiến đề xuất của Cục Quản lý Dược và tư vấn cho Bộ trưởng Bộ Y tế các vấn đề liên quan đến pháp chế dược, hồ sơ về chất lượng, an toàn, hiệu quả của thuốc, nguyên liệu làm thuốc.
2. Hội đồng có trách nhiệm thẩm định hồ sơ, tư vấn cho Bộ trưởng Bộ Y tế trong việc cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc; cấp phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành tại Việt Nam trên cơ sở ý kiến đề xuất của Cục Quản lý Dược và các vấn đề liên quan do Bộ trưởng Bộ Y tế yêu cầu. Hội đồng chịu trách nhiệm trước Bộ trưởng Bộ Y tế về các ý kiến thẩm định, tư vấn.
3. Nguyên tắc hoạt động của Hội đồng:
a) Hội đồng hoạt động theo nguyên tắc đồng thuận, tập trung dân chủ, khách quan, công khai, minh bạch. Ý kiến của Hội đồng phải bảo đảm cơ sở pháp lý, cơ sở khoa học, xem xét kết quả thẩm định hồ sơ của các chuyên gia thẩm định, căn cứ thực tiễn lâm sàng, đề xuất của Cục Quản lý Dược.
b) Hội đồng họp khi có từ 2/3 thành viên Hội đồng tham dự, trường hợp thành viên Hội đồng không tham dự buổi họp nhưng có gửi ý kiến bằng văn bản thì được xem là tham dự họp;
Chủ tịch Hội đồng hoặc người được Chủ tịch Hội đồng ủy quyền chủ trì họp Hội đồng kết luận trên cơ sở có ít nhất 2/3 ý kiến của các thành viên tham dự hợp đồng thuận. Các ý kiến chưa đồng thuận với kết luận của Hội đồng được bảo lưu.
Ý kiến của các thành viên Hội đồng và kết luận của Hội đồng phải được thể hiện trong biên bản họp Hội đồng, kể cả ý kiến chưa đồng thuận với kết luận của Hội đồng.
c) Trường hợp không tổ chức họp Hội đồng, Chủ tịch Hội đồng thực hiện việc lấy ý kiến bằng văn bản đến các thành viên Hội đồng;
Trường hợp đã quá thời hạn gửi xin ý kiến, Chủ tịch Hội đồng hoặc người được ủy quyền đưa ra kết luận của Hội đồng khi có ít nhất 2/3 số thành viên đã gửi ý kiến gửi về Thường trực Hội đồng để tổng hợp.
Ý kiến kết luận của Hội đồng dựa trên cơ sở ý kiến đồng thuận của ít nhất 2/3 thành viên đã có ý kiến gửi về Thường trực Hội đồng và trên cơ sở báo cáo tổng hợp và đề xuất của Cục Quản lý Dược;
Ý kiến kết luận của Hội đồng được thể hiện bằng Phiếu trình ghi ý kiến kết luận của Chủ tịch Hội đồng hoặc của người được Chủ tịch Hội đồng ủy quyền.
d) Trong trường hợp cần thiết, Chủ tịch Hội đồng có quyền tham vấn thêm ý kiến từ các chuyên gia độc lập ngoài các thành viên trong Hội đồng trước khi đưa ra kết luận cuối cùng. Các chuyên gia này có thể trực tiếp tham dự phiên họp Hội đồng hoặc cho ý kiến bằng văn bản, có trách nhiệm và quyền lợi như các thành viên chính thức của Hội đồng;
đ) Không vi phạm các nguyên tắc về xung đột lợi ích.
4. Cục Quản lý Dược tham mưu trình Bộ trưởng Bộ Y tế ban hành quy chế về tổ chức, hoạt động của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, cơ chế phối hợp giữa Hội đồng và chuyên gia thẩm định trong quá trình cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, giấy phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành tại Việt Nam.
5. Kinh phí hoạt động của Hội đồng được thực hiện theo quy định của pháp luật.
6. Thường trực Hội đồng đặt tại Cục Quản lý Dược.
Điều 45. Tổ chức, hoạt động của chuyên gia thẩm định hồ sơ đăng ký thuốc, nguyên liệu làm thuốc, thẩm định hồ sơ đề nghị cấp giấy phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành
1. Cục Quản lý Dược căn cứ chức năng, nhiệm vụ được giao có trách nhiệm thành lập các tiểu ban chuyên gia thẩm định hồ sơ đăng ký thuốc, nguyên liệu làm thuốc, thẩm định hồ sơ đề nghị cấp giấy phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành (sau đây gọi tắt là tiểu ban chuyên gia thẩm định). Cơ cấu các tiểu ban chuyên gia thẩm định phải phù hợp với phân loại sản phẩm đăng ký và hình thức đăng ký hoặc sản phẩm đề nghị cấp phép nhập khẩu và hình thức đề nghị cấp phép nhập khẩu.
2. Chuyên gia thẩm định hoạt động theo nguyên tắc: các ý kiến thẩm định phải bảo đảm căn cứ pháp lý, cơ sở khoa học và phải được thể hiện trong biên bản thẩm định hồ sơ đăng ký thuốc, nguyên liệu làm thuốc hoặc biên bản thẩm định hồ sơ đề nghị cấp phép nhập khẩu thuốc chưa có giấy đăng ký lưu hành. Chuyên gia thẩm định chịu trách nhiệm trước Cục trưởng Cục Quản lý Dược về các nội dung thẩm định, ý kiến đề xuất liên quan đến công tác thẩm định hồ sơ đăng ký thuốc, nguyên liệu làm thuốc, thẩm định hồ sơ nhập khẩu thuốc chưa có giấy đăng ký lưu hành.
3. Cục Quản lý Dược căn cứ chức năng, nhiệm vụ được giao xây dựng và ban hành quy chế tổ chức và hoạt động của các nhóm chuyên gia thẩm định hồ sơ đăng ký thuốc, nguyên liệu làm thuốc, hồ sơ nhập khẩu thuốc chưa có giấy đăng ký lưu hành; ký hợp đồng với chuyên gia thẩm định hoặc đơn vị tham gia tổ chức thẩm định hồ sơ; tổ chức các khoá tập huấn, đào tạo cho chuyên gia thẩm định; tiến hành đánh giá năng lực chuyên môn và sự tuân thủ các quy định để có điều chỉnh, bổ sung chuyên gia thẩm định phù hợp.
4. Kinh phí tổ chức thẩm định hồ sơ được thực hiện theo quy định của pháp luật.
Chương VII
ĐIỀU KHOẢN THI HÀNH
Điều 46. Hiệu lực thi hành
1. Thông tư này có hiệu lực thi hành kể từ ngày 01 tháng 9 năm 2019.
2. Thông tư số 44/2014/TT-BYT ngày 25 tháng 11 năm 2014 của Bộ trưởng Bộ Y tế quy định việc đăng ký thuốc hết hiệu lực kể từ ngày Thông tư này có hiệu lực thi hành, trừ các nội dung quy định việc đăng ký sinh phẩm chẩn đoán in vitro.
Điều 47. Điều khoản chuyển tiếp
1. Các hồ sơ đăng ký nộp trước ngày Thông tư này có hiệu lực thi hành được tiếp tục thực hiện theo quy định tại Thông tư số 44/2014/TT-BYT ngày 25 tháng 11 năm 2014 của Bộ trưởng Bộ Y tế quy định việc đăng ký thuốc, trừ trường hợp cơ sở đăng ký tự nguyện thực hiện theo quy định kể từ ngày ký ban hành Thông tư này.
2. Thuốc, nguyên liệu làm thuốc hết hiệu lực giấy đăng ký lưu hành từ ngày 01 tháng 1 năm 2018 đến ngày 30 tháng 6 năm 2020 được tiếp tục duy trì hiệu lực giấy đăng ký lưu hành 12 tháng khi đáp ứng tất cả các quy định sau:
a) Thuốc không thuộc trường hợp tạm ngừng nhận hồ sơ cấp, gia hạn giấy đăng ký thuốc, nguyên liệu làm thuốc theo quy định tại khoản 2 Điều 100 Nghị định số 54/2017/NĐ-CP ngày 08 tháng 5 năm 2017 của Chính phủ quy định chi tiết một số điều và biện pháp thi hành Luật dược; khoản 54 Điều 4 và điểm a khoản 53 Điều 5 Nghị định số 155/2018/NĐ-CP ngày 12 tháng 11 năm 2018 của Chính phủ sửa đổi, bổ sung một số quy định liên quan đến điều kiện đầu tư kinh doanh thuộc phạm vi quản lý nhà nước của Bộ Y tế;
b) Thuốc, nguyên liệu làm thuốc không có khuyến cáo của Tổ chức Y tế thế giới, cơ quan quản lý dược Việt Nam về an toàn, hiệu quả của thuốc, nguyên liệu làm thuốc;
c) Cơ sở đăng ký có đơn đề nghị được tiếp tục duy trì hiệu lực giấy đăng ký lưu hành theo Mẫu 6/TT ban hành kèm theo Thông tư này;
Trong thời hạn 20 ngày, kể từ ngày nhận đơn đề nghị, Cục Quản lý Dược có văn bản trả lời.
3. Trong thời hạn hiệu lực duy trì của giấy đăng ký lưu hành cũ, mà cơ sở đăng ký được gia hạn giấy đăng ký lưu hành mới thì hiệu lực giấy đăng ký lưu hành cũ được tiếp tục có hiệu lực đồng thời với giấy đăng ký lưu hành mới trong 06 tháng kể từ ngày giấy đăng ký lưu hành mới có hiệu lực.
4. Biệt dược gốc đã được Cục Quản lý Dược công bố trước thời điểm Thông tư này có hiệu lực tiếp tục được công nhận. Cơ sở đăng ký thực hiện việc cập nhật phân loại biệt dược gốc theo quy định tại Phụ lục II ban hành kèm theo Thông tư này trong thời hạn giấy đăng ký lưu hành thuốc còn hiệu lực.
Điều 48. Lộ trình thực hiện
1. Thời hạn đăng ký lưu hành tá dược, vỏ nang thực hiện theo quy định tại khoản 8 Điều 143 Nghị định số 54/2017/NĐ-CP ngày 08 tháng 5 năm 2017 của Chính phủ quy định chi tiết một số điều và biện pháp thi hành Luật dược.
2. Thời hạn đăng ký lưu hành bán thành phẩm dược liệu thực hiện theo quy định tại điểm c khoản 78 Điều 5 Chương II Nghị định số 155/2018/NĐ-CP ngày 12 tháng 11 năm 2018 của Chính phủ sửa đổi, bổ sung một số quy định liên quan đến điều kiện đầu tư kinh doanh thuộc phạm vi quản lý nhà nước của Bộ Y tế.
3. Đối với các hồ sơ gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc nộp trước ngày 01 tháng 1 năm 2020: Không yêu cầu CPP phải có đầy đủ thông tin về tiêu chuẩn thuốc thành phẩm; tiêu chuẩn dược chất, dược liệu; tên, địa chỉ cơ sở sản xuất dược chất, dược liệu.
4. Đối với các hồ sơ đăng ký nguyên liệu làm thuốc dạng bán thành phẩm dược liệu, tá dược, vỏ nang và các nguyên liệu tá dược, vỏ nang, dược liệu, bán thành phẩm dược liệu có trong hồ sơ đăng ký thuốc nộp trước ngày 01 tháng 1 năm 2021: Không yêu cầu phải nộp giấy tờ pháp lý quy định tại khoản 11 Điều 24 Thông tư này.
Điều 49. Điều khoản tham chiếu
Trong trường hợp các văn bản quy phạm pháp luật và các quy định được viện dẫn trong Thông tư này có sự thay đổi, bổ sung hoặc được thay thế thì áp dụng theo văn bản quy phạm pháp luật mới.
Điều 50. Trách nhiệm thi hành
1. Cục Quản lý Dược căn cứ chức năng, nhiệm vụ được giao và lộ trình hòa hợp ASEAN trong đăng ký thuốc, có trách nhiệm:
a) Tổ chức hướng dẫn và thực hiện các quy định của Thông tư này;
b) Cập nhật danh mục các thuốc, nguyên liệu làm thuốc được cấp, gia hạn giấy đăng ký lưu hành trong thời hạn 05 ngày kể từ ngày cấp, gia hạn giấy đăng ký lưu hành và các thông tin đăng ký thuốc, nguyên liệu làm thuốc khác trên trang thông tin điện tử của Cục Quản lý Dược;
c) Cập nhật danh mục các thuốc có chứng minh tương đương sinh học trong thời hạn 05 ngày kể từ ngày cấp giấy đăng ký lưu hành và các thông tin thay đổi, bổ sung của thuốc có chứng minh tương đương sinh học trong thời hạn 07 ngày kể từ ngày phê duyệt thay đổi, bổ sung giấy đăng ký lưu hành thuốc trên trang thông tin điện tử của Cục Quản lý Dược;
d) Cập nhật danh mục các biệt dược gốc trong thời hạn 05 ngày kể từ ngày cấp giấy đăng ký lưu hành và các thông tin thay đổi, bổ sung biệt dược gốc trong thời hạn 07 ngày kể từ ngày phê duyệt thay đổi, bổ sung giấy đăng ký lưu hành thuốc trên trang thông tin điện tử của Cục Quản lý Dược;
đ) Xem xét, rà soát thuốc đã được công bố biệt dược gốc khi có cơ sở xác định thuốc được công bố biệt dược gốc không còn đáp ứng các tiêu chí theo quy định;
e) Xây dựng, ban hành tổ chức triển khai thực hiện các quy trình chuẩn (SOPs) trong đăng ký thuốc và sổ tay hướng dẫn đăng ký thuốc (QM);
g) Phối hợp với Cục Quản lý Y, Dược cổ truyền trong việc gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc cổ truyền, dược liệu đã được cấp giấy đăng ký lưu hành theo quy định tại Thông tư số 44/2014/TT-BYT ngày 25 tháng 11 năm 2014 của Bộ trưởng Bộ Y tế quy định việc đăng ký thuốc;
h) Trường hợp cơ sở đăng ký thuốc có hành vi giả mạo hoặc tự ý sửa chữa hồ sơ, tài liệu, giấy tờ pháp lý của các cơ quan chức năng của Việt Nam hoặc của nước ngoài; sử dụng con dấu giả hoặc giả mạo chữ ký hoặc dấu của cơ sở đăng ký, cơ sở sản xuất và các cơ sở liên quan trong hồ sơ đăng ký thuốc thì Cục Quản lý Dược sẽ có công văn cảnh báo cơ sở và ngừng nhận hồ sơ cấp, gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc theo quy định tại khoản 2, 3 và 4 Điều 100 Nghị định 54/2007/NĐ-CP ngày 08 tháng 5 năm 2017 của Chính phủ quy định chi tiết một số điều và biện pháp thi hành Luật dược.
Ngoài các hình thức trên, Cục Quản lý Dược công khai nội dung vi phạm của cơ sở trên trang thông tin điện tử của Cục Quản lý Dược, đồng thời thông báo tới cơ quan Thanh tra và các cơ quan chức năng có thẩm quyền để xem xét, xử lý theo quy định của pháp luật;
i) Trong trường hợp cần thiết, Cục Quản lý Dược tổ chức cuộc họp với cơ sở đăng ký, cơ sở sản xuất, chuyên gia thẩm định nhằm làm rõ các vướng mắc liên quan đến việc thẩm định hồ sơ đăng ký thuốc, nguyên liệu làm thuốc;
k) Công bố trên trang thông tin điện tử của Cục Quản lý Dược danh mục các cơ sở đăng ký, cơ sở sản xuất thuốc, nguyên liệu làm thuốc theo quy định tại khoản 11, 15 Điều 23 Thông tư này.
l) Xây dựng quy định và lộ trình triển khai việc in mã vạch (Bar code), mã QR, mã DataMatrix Code (DMC) trên bao bì ngoài của thuốc, nguyên liệu làm thuốc nhằm quản lý, nhận diện và truy xuất nguồn gốc thuốc, nguyên liệu làm thuốc lưu hành trên thị trường trình Bộ trưởng Bộ Y tế ban hành;
m) Trong thời hạn 30 ngày kể từ ngày cấp, gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược trả nhãn, hướng dẫn sử dụng thuốc cho cơ sở đăng ký;
n) Trong thời hạn 15 ngày kể từ ngày cấp, gia hạn giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, 07 ngày kể từ ngày phê duyệt thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược công bố nguồn nguyên liệu làm thuốc đối với các thuốc sản xuất tại Việt Nam trên trang thông tin điện tử của Cục Quản lý Dược.
2. Sở Y tế các tỉnh, thành phố trực thuộc Trung ương chịu trách nhiệm kiểm tra, thanh tra việc thực hiện Thông tư này đối với các đơn vị sản xuất, kinh doanh dược phẩm trong phạm vi quản lý.
3. Các đơn vị trực thuộc Bộ Y tế, Tổng Công ty Dược Việt Nam, các cơ sở kinh doanh thuốc có trách nhiệm thực hiện Thông tư này.
Trong quá trình thực hiện nếu có khó khăn, vướng mắc, các cơ quan, tổ chức, cá nhân phản ánh về Bộ Y tế (Cục Quản lý Dược) để xem xét giải quyết./.
|
Nơi nhận: |
KT. BỘ TRƯỞNG |
PHỤ LỤC I
BỘ HỒ SƠ KỸ THUẬT CHUNG ASEAN (ACTD) VÀ CÁC HƯỚNG DẪN KỸ THUẬT
(Ban hành kèm theo Thông tư số /2018/TT-BYT ngày tháng năm 2018)
MỤC LỤC TÀI LIỆU
PHẦN I. BỘ HỒ SƠ KỸ THUẬT CHUNG ASEAN (ACTD)
I. Lời mở đầu
II. Bố cục bộ hồ sơ kỹ thuật chung ASEAN (ACTD)
III. Hồ sơ hành chính
IV. Hồ sơ chất lượng
V. Hồ sơ tiền lâm sàng
VI. Hồ sơ lâm sàng
PHẦN II. CÁC HƯỚNG DẪN KỸ THUẬT CỦA ASEAN VÀ CÁC THUẬT NGỮ TRONG HỒ SƠ KỸ THUẬT
I. Hướng dẫn nghiên cứu độ ổn định của thuốc.
II. Hướng dẫn thẩm định quy trình sản xuất.
III. Hướng dẫn thẩm định phương pháp phân tích.
IV. Hướng dẫn nghiên cứu sinh khả dụng và tương đương sinh học.
V. Các thuật ngữ dùng trong hồ sơ kỹ thuật.
LỜI MỞ ĐẦU
Hồ sơ kỹ thuật chung ASEAN (ACTD) là một hướng dẫn về một mẫu thống nhất trong chuẩn bị các hồ sơ kỹ thuật chung (CTD) có bố cục tốt để nộp cho các cơ quan quản lý của các nước ASEAN để đăng ký dược phẩm dùng cho người. Hướng dẫn này mô tả một mẫu CTD giúp làm giảm đáng kể thời gian và nguồn lực cần thiết cho việc chuẩn bị những hồ sơ đăng ký thuốc, và trong tương lai sẽ giúp giảm gánh nặng trong việc chuẩn bị hồ sơ điện tử. Việc xét duyệt và liên lạc của các cơ quan quản lý với các cơ sở đăng ký sẽ được hỗ trợ bằng một bộ tài liệu chuẩn với các nội dung thống nhất.
Hướng dẫn này chỉ minh họa một mẫu phù hợp các dữ liệu sẽ nộp. Tuy nhiên, cơ sở đăng ký có thể điều chỉnh nếu cần thiết để có được một dạng trình bày tối ưu các thông tin kỹ thuật nhằm tạo điều kiện cho việc đọc hiểu và thẩm định kết quả trong đăng ký thuốc.
Trong suốt toàn bộ ACTD, việc trình bày thông tin không được phép đa nghĩa mập mờ mà phải rõ ràng, sao cho có thể thẩm định những dữ liệu cơ bản và giúp các chuyên gia thẩm định nhanh chóng tiếp cận và nắm bắt nội dung của hồ sơ. Các bảng biểu và văn bản phải được trình bày canh lề sao cho có thể in ra được trên giấy khổ A4 hoặc cỡ 8,5x11cm. Lề bên trái phải đủ rộng để thông tin không bị che lấp khi đóng gáy. Kiểu chữ và cỡ chữ (Times New Roman, cỡ 12) áp dụng cho phần văn bản và phần bảng biểu phải đủ lớn để dễ đọc, kể cả sau khi photo. Mỗi trang phải được đánh số, trang đầu ở mỗi phần được đánh số 1. Với mỗi phần tài liệu, cần có phần chú giải từ viết tắt và thuật ngữ kỹ thuật nếu chúng được sử dụng lần đầu ở mỗi phần. Mục lục tài liệu tham khảo phải được trích dẫn theo đúng Tuyên ngôn Vancouver 1979 về Quy định thống nhất đối với các trích dẫn trên Tạp chí Y – Sinh học.
BỐ CỤC ACTD
ACTD được bố cục thành bốn phần như sau:
Phần I: Hồ sơ hành chính
Mục A: Lời giới thiệu
Mục B: Mục lục tài liệu tổng quan của Hồ sơ Kỹ thuật chung ASEAN
Mục C: Đơn xin đăng ký, mẫu nhãn, thông tin kê đơn
Phần II: Hồ sơ chất lượng
Mục A: Mục lục tài liệu
Mục B: Tóm tắt tổng quan về chất lượng
Mục C: Nội dung số liệu
Phần III: Hồ sơ tiền lâm sàng
Mục A: Mục lục tài liệu
Mục B: Tổng quan về đánh giá tiền lâm sàng
Mục C: Tóm tắt bằng văn bản và bảng biểu về tiền lâm sàng
Mục D: Các báo cáo nghiên cứu tiền lâm sàng
Phần IV: Hồ sơ lâm sàng
Mục A: Mục lục tài liệu
Mục B: Tổng quan về lâm sàng
Mục C: Tóm tắt về lâm sàng
Mục D: Bảng danh mục tất cả các nghiên cứu lâm sàng
Mục E: Các báo cáo nghiên cứu lâm sàng
Mục F: Danh mục các tài liệu tham khảo chủ yếu
Theo mô hình thì hồ sơ ACTD như sau:
Các báo cáo nghiên cứu lâm sàng và tiền lâm sàng có thể được miễn đối với những sản phẩm đã được cấp đăng ký lưu hành ở “các nước tham khảo” – là những nước có hệ thống thẩm định thuốc và được công nhận bởi cơ quan quản lý dược các nước ASEAN.
GIỚI THIỆU TỔNG QUAN
Phần I: Mục lục tài liệu, tài liệu hành chính và thông tin sản phẩm
Phần I đầu tiên sẽ có phần Mục lục tài liệu tổng quan của toàn bộ bộ hồ sơ kỹ thuận chung ASEAN (ACTD) để cung cấp về cơ bản những nội dung thông tin có trong hồ sơ. Tiếp đến phần thứ hai là Tài liệu hành chính trong đó phải có các tài liệu cụ thể chi tiết đi cùng nhau, ví dụ như đơn xin đăng ký lưu hành, mẫu nhãn, tờ hướng dẫn sử dụng… Phần cuối là Thông tin sản phẩm trong đó có các thông tin cần thiết, kể cả thông tin cho kê đơn, cơ chế tác động, tác dụng phụ của sản phẩm …
Phần này cũng nên có phần giới thiệu chung về dược phẩm, bao gồm nhóm dược lý và cơ chế tác động của thuốc.
Phần II: Hồ sơ chất lượng
Phần II cần đưa ra một phần Tóm tắt chung sau đó đến các Báo cáo nghiên cứu. Tài liệu về kiểm tra chất lượng phải được trình bày càng chi tiết càng tốt.
Phần III: Hồ sơ tiền lâm sàng
Phần III cần cung cấp một Tổng quan về tiền lâm sàng, sau đó là các Tóm tắt về tiền lâm sàng bằng văn bản và bảng biểu. Tài liệu của phần này không yêu cầu đối với sản phẩm generic, sản phẩm có thay đổi nhỏ và một số sản phẩm có thay đổi lớn. Đối với các nước thành viên ASEAN, có thể không cần quy định các báo cáo nghiên cứu trong phần này đối với các sản phẩm có chứa dược chất mới (NCE), sản phẩm công nghệ sinh học và các sản phẩm có thay đổi lớn khác nếu sản phẩm gốc đã được đăng ký và cấp phép lưu hành ở các nước tham khảo. Vì thế, nếu cơ quan quản lý có nhu cầu về báo cáo nghiên cứu cụ thể nào thì có thể yêu cầu nộp tài liệu đó.
Phần IV: Hồ sơ lâm sàng
Phần IV cần đưa ra được Tổng quan về lâm sàng và Tóm tắt lâm sàng. Tài liệu trong phần này không cần quy định đối với sản phẩm generic, sản phẩm có những thay đổi nhỏ và một số sản phẩm có thay đổi lớn. Đối với các nước thành viên ASEAN, có thể không cần quy định các báo cáo nghiên cứu trong phần này đối với các sản phẩm có chứa dược chất mới (NCE), sản phẩm công nghệ sinh học và các sản phẩm có thay đổi lớn khác nếu sản phẩm gốc đã được đăng ký và cấp phép lưu hành ở các nước tham khảo. Vì thế, nếu cơ quan quản lý nào có nhu cầu báo cáo nghiên cứu cụ thể nào thì có thể yêu cầu nộp tài liệu đó.
HỒ SƠ KỸ THUẬT ASEAN (ACTD) TRONG ĐĂNG KÝ THUỐC DÙNG CHO NGƯỜI
PHẦN I: HỒ SƠ HÀNH CHÍNH VÀ THÔNG TIN SẢN PHẨM
(Phần này không thuộc phạm vi hòa hợp của ASEAN. Các nước có hướng dẫn riêng. Đề nghị xem hướng dẫn cụ thể tại Điều 28 Thông tư quy định việc đăng ký thuốc)
HỒ SƠ KỸ THUẬT CHUNG CỦA ASEAN (ACTD) SỬ DỤNG CHO ĐĂNG KÝ DƯỢC PHẨM DÙNG CHO NGƯỜI
PHẦN II: CHẤT LƯỢNG
MỤC LỤC HỒ SƠ
Phạm vi áp dụng của hướng dẫn
Chương A: Mục Lục
Chương B: Tóm tắt tổng thể về chất lượng
Chương C: Phần nội dung chính
1. Dược chất
2. Thành phẩm
Chương D: tài liệu tham khảo chủ yếu
MỘT SỐ TỪ VIẾT TẮT TRONG TÀI LIỆU
NCE : Chất hóa học mới (dược chất mới)
BIOTECH : Sản phẩm công nghệ sinh học
MaV : Thay đổi lớn
MiV : Thay đổi nhỏ
G : Thuốc Generic
PHẠM VI ÁP DỤNG CỦA HƯỚNG DẪN
Tài liệu này nhằm đưa ra một hướng dẫn về một mẫu hồ sơ đăng ký dược phẩm theo các yêu cầu kỹ thuật chung của ASEAN (ACTR). Mẫu này dùng cho các dược chất mới (NCE), sản phẩm công nghệ sinh học (Biotech), thay đổi lớn (MaV), thay đổi nhỏ (MiV) và sản phẩm generic (G). Để xác định tính khả thi của mẫu này đối với một loại sản phẩm cụ thể, cơ sở đăng ký cần tham khảo ý kiến của các cơ quan quản lý thuốc quốc gia có liên quan. Phần “Nội dung chính” của hướng dẫn này chỉ đơn thuần cho biết các thông tin về sản phẩm phải đặt ở đâu. Hướng dẫn này không đề cập đến loại hình cũng như phạm vi của số liệu hỗ trợ, mà chúng tuỳ thuộc vào hướng dẫn quốc gia cũng như các tài liệu tham khảo quốc tế chủ yếu (dược điển)
Đối với NCE và Biotech, xin tham khảo thêm các hướng dẫn của ICH có liên quan.
CHƯƠNG A: MỤC LỤC
Cần có một mục lục nội dung của hồ sơ xin đăng ký.
CHƯƠNG B: TÓM TẮT TỔNG THỂ VỀ CHẤT LƯỢNG
|
Số TT |
CÁC THÔNG SỐ |
NỘI DUNG CỦA CÁC THÔNG SỐ |
YÊU CẦU |
||||
|
NCE |
Biotech |
MaV |
MiV |
G |
|||
|
|
|
|
|
|
|
|
|
|
S |
Dược chất |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
S 1 |
Thông tin chung |
|
|
|
|
|
|
|
|
1.1 Danh pháp |
- Thông tin từ S 1 |
v |
v |
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
|
1.2 Cấu trúc |
- Công thức cấu trúc, bao gồm cả hóa học lập thể tuyệt đối và tương đối, công thức phân tử và khối lượng phân tử tương đối. |
v |
|
|
|
v |
|
|
|
- Chuỗi axit amin chỉ rõ vị trí các nhóm glycosyl hóa hoặc các biến đổi hậu dịch mã khác và khối lượng phân tử tương đối. |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1.3 Đặc tính chung |
- Đặc tính lý hóa và các đặc tính có liên quan khác kể cả hoạt tính sinh học đối với sản phẩm công nghệ sinh học. |
v |
v |
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
S 2 |
Sản xuất |
|
|
|
|
|
|
|
|
2.1 Nhà sản xuất |
- Tên và địa chỉ của nhà sản xuất. |
v |
v |
|
|
v |
|
|
|
|
|
|
|
|
|
|
|
2.2 Mô tả quá trình sản xuất và kiểm soát quy trình |
- Mô tả quy trình sản xuất dược chất và kiểm soát quy trình thể hiện cam kết của cơ sở đăng ký trong việc sản xuất ra các dược chất đó. |
v |
v |
|
|
|
|
|
|
- Thông tin về quy trình sản xuất mà đặc trưng là xuất phát từ một (một số) lọ ngân hàng tế bào, bao gồm mẫu cấy tế bào, thu hoạch, tinh chế, phản ứng biến đổi tế bào, các điều kiện đóng gói, bảo quản và vận chuyển. |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.3 Kiểm soát nguyên liệu |
- Nguyên liệu ban đầu, dung môi, thuốc thử, chất xúc tác và các nguyên liệu khác dùng sản xuất dược chất, cần nêu rõ mỗi nguyên liệu đó được dùng vào thời điểm nào trong quá trình sản xuất. Các phép thử và tiêu chuẩn chấp nhận của các nguyên liệu này. |
v |
v |
|
|
|
|
|
|
- Kiểm soát nguồn gốc và nguyên liệu ban đầu có nguồn gốc sinh học. |
|
v |
|
|
|
|
|
|
- Nguồn gốc, lịch sử và sự hình thành dòng tế bào sản xuất. |
|
v |
|
|
|
|
|
|
- Hệ thống ngân hàng tế bào, mô tả đặc điểm và phương pháp kiểm nghiệm. |
|
v |
|
|
|
|
|
|
- Đánh giá an toàn về virút. |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.4 Kiểm soát các bước quan trọng và sản phẩm trung gian |
- Các bước quan trọng: các phép thử và chỉ tiêu chấp nhận, có thuyết minh các dữ liệu thực nghiệm thu được từ việc đánh giá các bước quan trọng của quá trình sản xuất để chắc chắn rằng quy trình này đã được kiểm soát. |
v |
v |
|
|
|
|
|
|
- Sản phẩm trung gian: tiêu chuẩn chất lượng và quy trình phân tích, nếu có, đối với các sản phẩm trung gian được phân lập trong quá trình sản xuất. |
v |
v |
|
|
|
|
|
|
- Số liệu về độ ổn định làm căn cứ đưa ra các điều kiện bảo quản. |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.5 Đánh giá và/ hoặc Thẩm định quy trình. |
Các nghiên cứu đánh giá và/ hoặc thẩm định đối với quy trình chế biến vô trùng và tiệt trùng. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.6 Phát triển quy trình sản xuất |
- Mô tả và bàn luận về những thay đổi quan trọng đối với quy trình sản xuất và/hoặc cơ sở sản xuất dược chất dùng trong sản xuất các lô sản phẩm để nghiên cứu tiền lâm sàng, lâm sàng, lô thí nghiệm và, cả lô sản xuất thực tế nếu có. |
v |
|
|
|
|
|
|
|
- Lịch sử phát triển của quy trình sản xuất như mô tả ở S 2.2. |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
S 3 |
Đặc tính |
|
|
|
|
|
|
|
|
3.1 Giải thích cấu trúc và/ hoặc các đặc tính khác |
- Xác nhận cấu trúc dựa trên cơ sở quá trình tổng hợp và các phân tính phổ. |
v |
|
|
|
|
|
|
|
Quy định trong dược điển hoặc thông tin tương từ nhà sản xuất. |
|
|
|
|
v |
|
|
|
- Chi tiết về cấu trúc sơ cấp, thứ cấp hoặc cao hơn và thông tin về hoạt tính sinh học, độ tinh khiết và đặc tính hóa học miễn dịch (nếu có liên quan). |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.2 Tạp chất |
- Tóm tắt về các tạp chất đã được theo dõi hoặc thử nghiệm trong và sau khi sản xuất dược chất. |
v |
v |
|
|
|
|
|
|
Quy định trong dược điển hoặc thông tin tương đương từ nhà sản xuất. |
|
|
|
|
v |
|
|
|
|
|
|
|
|
|
|
S 4 |
Kiểm tra dược chất |
|
|
|
|
|
|
|
|
4.1 Tiêu chuẩn chất lượng |
- Chi tiết về tiêu chuẩn chất lượng, các phép thử và chỉ tiêu chấp nhận. |
v |
v |
|
|
|
|
|
|
- Tiêu chuẩn dược điển hoặc thông tin tương đương từ nhà sản xuất. |
|
|
|
|
v |
|
|
|
- Chỉ rõ nguồn gốc, kể cả loài động vật thích hợp, chủng vi sinh vật, … |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2 Quy trình phân tích |
- Quy trình phân tích được dùng để thử dược chất. |
v |
v |
|
|
|
|
|
|
Quy định trong dược điển và thông tin tương đương từ nhà sản xuất. |
|
|
|
|
v |
|
|
|
|
|
|
|
|
|
|
|
4.3 Thẩm định quy trình phân tích |
- Thông tin về thẩm định phép phân tích, bao gồm các dữ liệu thực nghiệm về quy trình phân tích được dùng để thử dược chất. |
v |
v |
|
|
|
|
|
|
Các phương pháp không có trong dược điển. |
|
|
|
|
v |
|
|
|
|
|
|
|
|
|
|
|
4.4 Phân tích lô |
Mô tả lô và kết quả phân tích để thiết lập tiêu chuẩn chất lượng. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.5 Thuyết minh tiêu chuẩn chất lượng |
Thuyết minh tiêu chuẩn chất lượng của dược chất. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
S 5 |
Chất chuẩn hoặc nguyên liệu đối chiếu |
- Thông tin về chất chuẩn hoặc nguyên liệu đối chiếu được dùng để thử dược chất. |
v |
v |
|
|
|
|
|
|
- Chất chuẩn đối chiếu theo dược điển hoặc thông tin thích hợp từ nhà sản xuất |
|
|
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
S 6 |
Hệ thống bao bì đóng gói |
Mô tả hệ thống bao bì đóng gói. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
S 7 |
Độ ổn định |
- Báo cáo độ ổn định. |
v |
v |
|
|
|
|
|
|
- Tài liệu khoa học. |
|
|
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
P |
Thành phẩm thuốc |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
P 1 |
Mô tả và thành phần |
- Mô tả Dạng bào chế và đặc tính, Dung môi để pha chế đi kèm theo sản phẩm, Loại bao bì đóng gói của dạng bào chế và dung môi kèm theo, nếu có. |
v |
v |
v* |
v* |
v |
|
|
|
- Thành phần: Tên, lượng công bố bằng khối lượng hay thể tích, chức năng và tham khảo tiêu chuẩn chất lượng. |
v |
v |
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
P 2 |
Phát triển dược học |
|
|
|
|
|
|
|
|
2.1 Thông tin về những nghiên cứu phát triển |
- Dữ liệu về các nghiên cứu phát triển được tiến hành để xác định rằng dạng bào chế, công thức, quy trình sản xuất, hệ thống bao bì đóng gói, các thuộc tính về vi sinh vật và hướng dẫn sử dụng phù hợp với mục đích ghi trong hồ sơ đăng ký. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.2 Thành phần của thành phẩm thuốc |
- Hoạt chất ● Chứng minh tính tương hợp của hoạt chất với tá dược được ghi ở mục P 1. ● Trong trường hợp thuốc đa thành phần, cần chứng minh tính tương hợp giữa các hoạt chất với nhau. |
v |
v |
|
|
|
|
|
|
● Tài liệu khoa học. |
|
|
v* |
|
v |
|
|
|
- Tá dược Chứng minh việc lựa chọn tá dược ghi ở mục P 1 là những tá dược có ảnh hưởng đến tác dụng của thành phẩm thuốc. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.3 Thành phẩm |
- Phát triển công thức bào chế Mô tả tóm tắt, ngắn gọn về sự phát triển thành phẩm (có tính đến đường dùng và cách sử dụng dự kiến đối với NCE và Biotech). |
v |
v |
|
|
v |
|
|
|
- Lượng đóng dư Thuyết minh về bất kỳ lượng đóng dư trong công thức ghi ở mục P1. |
v |
v |
|
|
v |
|
|
|
- Đặc tính hóa lý và sinh học Các thông số có liên quan đến khả năng tác dụng của thành phẩm thuốc như pH, độ hòa tan. |
v |
v |
|
|
v |
|
|
|
|
|
|
|
|
|
|
|
2.4 Phát triển quy trình sản xuất |
- Lựa chọn và tối ưu hóa quy trình sản xuất. |
v |
v |
|
|
|
|
|
|
- Sự khác nhau giữa những quy trình dùng để sản xuất những lô thuốc lâm sàng thiết yếu với quy trình mô tả ở mục P 3.2 nếu có. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.5 Hệ thống bao bì đóng gói |
Sự thích hợp của hệ thống bao bì đóng gói dùng trong bảo quản, vận chuyển (đường biển) và sử dụng thành phẩm. |
v |
v |
|
|
v |
|
|
|
|
|
|
|
|
|
|
|
2.6 Thuộc tính vi sinh vật |
Nêu thuộc tính vi sinh vật của dạng bào chế. |
v |
v |
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
|
2.7 Tính tương hợp |
Tính tương hợp của thành phẩm thuốc với dung môi pha loãng hoặc dụng cụ để phân liều. |
v |
v |
v* |
|
|
|
|
|
Tài liệu khoa học. |
|
|
|
|
v |
|
|
|
|
|
|
|
|
|
|
P 3 |
Sản xuất |
|
|
|
|
|
|
|
|
3.1 Công thức lô |
Tên và hàm lượng của tất cả các thành phần. |
v |
v |
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
|
3.2 Quy trình sản xuất và kiểm soát quy trình |
Mô tả quy trình sản xuất và kiểm soát quy trình. |
v |
v |
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
|
3.3 Kiểm soát các bước quan trọng và các sản phẩm trung gian |
Các phép thử và chỉ tiêu chấp nhận. |
v |
v |
|
|
v |
|
|
|
|
|
|
|
|
|
|
|
3.4 Thẩm định và/hoặc đánh giá quy trình |
Mô tả, dẫn chứng bằng tư liệu và kết quả của các nghiên cứu thẩm định và/hoặc đánh giá đối với những bước quan trọng hoặc các phép định lượng quan trọng sử dụng trong quy trình sản xuất. |
v |
v |
|
|
v |
|
|
|
|
|
|
|
|
|
|
P 4 |
Kiểm tra tá dược |
|
|
|
|
|
|
|
|
4.1 Tiêu chuẩn chất lượng |
- Tiêu chuẩn chất lượng của tá dược. |
v |
v |
|
|
|
|
|
|
Quy định trong dược điển hoặc thông tin thích hợp từ nhà sản xuất. |
|
|
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
|
4.2 Quy trình phân tích |
- Quy trình phân tích dùng để thử các tá dược khi thích hợp. |
v |
v |
|
|
|
|
|
|
- Quy định trong dược điển hoặc thông tin thích hợp từ nhà sản xuất. |
|
|
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
|
4.3 Tá dược có nguồn gốc từ người và động vật |
- Thông tin về nguồn gốc và/hoặc các chất ngẫu nhiên. - Quy định trong dược điển hoặc thông tin thích hợp từ nhà sản xuất. |
v |
v |
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
|
4.4 Tá dược mới |
- Đối với những tá dược được sử dụng lần đầu trong một thành phẩm hoặc đường dùng mới, cung cấp đầy đủ chi tiết về sản xuất, đặc tính và biện pháp kiểm tra, có tham khảo chéo những dữ liệu an toàn hỗ trợ (tiền lâm sàng hoặc lâm sàng). |
v |
v |
|
|
|
|
P 5 |
Kiểm tra thành phẩm |
|
|
|
|
|
|
|
|
5.1 Tiêu chuẩn chất lượng |
- Tiêu chuẩn chất lượng của thành phẩm. |
v |
v |
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
|
5.2 Quy trình phân tích |
- Quy trình phân tích dùng để kiểm nghiệm thành phẩm. |
v |
v |
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
|
5.3 Thẩm định quy trình phân tích |
- Thông tin bao gồm dữ liệu thực nghiệm đối với quy trình phân tích dùng để kiểm nghiệm thành phẩm. |
v |
v |
|
|
|
|
|
|
Phương pháp không có trong dược điển. |
v |
v |
v* |
v* |
v |
|
|
|
Xác minh khả năng áp dụng được của phương pháp có trong dược điển. |
|
|
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
|
5.4 Phân tích lô |
- Mô tả việc thử nghiệm và kết quả thử của tất cả các lô liên quan. |
v |
v |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5.5 Đặc tính của tạp chất |
- Thông tin về đặc tính của tạp chất. |
v |
v |
|
|
|
|
|
|
Quy định trong dược điển hoặc thông tin thích hợp từ nhà sản xuất. |
|
|
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
|
5.6 Thuyết minh tiêu chuẩn chất lượng |
- Thuyết minh tiêu chuẩn chất lượng dự kiến của thành phẩm. |
v |
v |
|
|
|
|
|
|
Quy định trong dược điển hoặc thông tin thích hợp từ nhà sản xuất. |
|
|
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
P 6 |
Chất chuẩn hoặc chất đối chiếu |
- Thông tin về chất chuẩn hoặc chất đối chiếu được dùng để kiểm nghiệm thành phẩm. |
v |
v |
|
|
|
|
|
|
Quy định trong dược điển hoặc thông tin thích hợp từ nhà sản xuất. |
|
|
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
P 7 |
Hệ thống bao bì đóng gói |
- Tiêu chuẩn chất lượng và phương pháp kiểm tra bao bì sơ cấp và thứ cấp, loại bao bì và kích thước bao bì, chi tiết phụ liệu (ví dụ: chất làm khô, vv). |
v |
v |
v* |
v* |
v |
|
|
|
|
|
|
|
|
|
|
P 8 |
Độ ổn định |
Báo cáo độ ổn định: dữ liệu chứng minh rằng sản phẩm ổn định trong suốt tuổi thọ dự kiến. Cam kết về việc theo dõi độ ổn định sau khi được phép lưu hành. |
v |
v |
v* |
|
v |
|
|
|
|
|
|
|
|
|
|
P 9 |
Khả năng thay thế lẫn nhau của sản phẩm |
- In Vitro Nghiên cứu độ hòa tan so sánh như yêu cầu. |
|
|
v* |
|
v |
|
|
|
- In Vivo Nghiên cứu tương đương sinh học như yêu cầu. |
|
|
v* |
|
v |
|
|
|
|
|
|
|
|
|
* : Nếu yêu cầu
NCE : Chất hóa học mới (dược chất mới)
BIOTECH : Sản phẩm công nghệ sinh học.
MaV : Thay đổi lớn
MiV : Thay đổi nhỏ
G : Thuốc generic
CHƯƠNG C: NỘI DUNG CHÍNH
S DƯỢC CHẤT
S 1 Thông tin chung
S 1.1 Danh pháp
● Tên chung quốc tế (INN)
● Tên rút gọn, nếu có
● Số đăng ký tra cứu trích dẫn hóa học (CAS)
● Mã phòng thí nghiệm (nếu có quy định)
● Tên hóa học
S 1.2 Công thức cấu tạo
NCE
Phải có cấu trúc kể cả hóa lập thể tương đối và tuyệt đối, công thức phân tử và khối lượng phân tử tương đối
Biotech
Phải có sơ đồ chuỗi acid amin chỉ rõ vị trí các nhóm glycosyl hóa hoặc các biến đổi hậu dịch mã khác và khối lượng phân tử tương đối, nếu thích hợp.
Generic
Quy định trong dược điển hoặc những thông tin tương đương của nhà sản xuất.
S 1.3 Đặc tính chung
Cần phải có một danh mục liệt kê các đặc tính hóa lý và các đặc tính có liên quan khác của dược chất, kể cả hoạt tính sinh học đối với các sản phẩm công nghệ sinh học (Biotech).
Tham khảo hướng dẫn ICH: NCE: Q6A; Biotech: Q6B.
S 2 Sản xuất
S2.1 Nhà sản xuất
Tên và địa chỉ đầy đủ, kể cả tên thành phố và nước của cơ sở sản xuất hoạt chất.
S 2.2 Mô tả quy trình sản xuất và kiểm soát quy trình
Mô tả quy trình sản xuất dược chất để thể hiện cam kết của cơ sở đăng ký trong việc sản xuất ra các dược chất đó. Cần cung cấp những thông tin sau để mô tả một cách đầy đủ quy trình sản xuất và các biện pháp kiểm soát quy trình:
NCE:
● Cần cung cấp sơ đồ miêu tả theo trình tự quy trình tổng hợp, gồm có công thức phân tử, khối lượng và sản lượng, cấu trúc hóa học của nguyên liệu ban đầu, sản phẩm trung gian, thuốc thử và dược chất phản ánh hóa lập thể, xác định điều kiện thao tác và dung môi.
● Mô tả quy trình sản xuất, nêu rõ lượng nguyên liệu, dung môi, chất xúc tác, phản ánh quy mô của lô đại diện, và nêu các biện pháp kiểm soát quy trình, trang thiết bị và điều kiện thao tác, ví dụ như nhiệt độ, áp suất, độ pH, thời gian, vv…
● Quy trình dùng thay thế phải được giải thích và mô tả ở mức độ chi tiết như quy trình gốc. Phải xác định và thuyết minh các bước chế biến tái lặp.
Biotech
● Những thông tin về quy trình sản xuất, mà đặc trưng là xuất phát từ một (một số) lọ ngân hàng tế bào, bao gồm mẫu cấy tế bào, thu hoạch, tinh chế, phản ứng biến đổi tế bào, điều kiện đóng gói, bảo quản và vận chuyển.
Tham khảo Hướng dẫn ICH: Q5A, Q5B và Q6B.
S 2.3 Kiểm soát nguyên liệu
Những nguyên liệu dùng trong sản xuất dược chất (ví dụ nguyên liệu thô, nguyên liệu ban đầu, dung môi, thuốc thử, chất xúc tác) cần được liệt kê, trong đó cần nêu rõ mỗi nguyên liệu đó được dùng vào thời điểm nào trong quá trình sản xuất. Cần cung cấp các thông tin về chất lượng và việc kiểm soát chất lượng của các nguyên vật này. Nếu cần, phải có thông tin chứng minh là những nguyên liệu (bao gồm các nguyên liệu có nguồn gốc sinh học, ví dụ như các thành phần môi trường, các kháng thể đơn dòng, enzyme) đạt tiêu chuẩn phù hợp với mục đích sử dụng của chúng (kể cả việc loại trừ hoặc kiểm soát các yếu tố ngoại lai). Đối với các nguyên liệu có nguồn gốc sinh học, thì còn phải có cả những thông tin về nguồn gốc, việc sản xuất và định tính của chúng.
Tham khảo hướng dẫn ICH: NCE: Q6A; Biotech: Q6B
Biotech:
● Kiểm soát nguồn gốc và nguyên liệu ban đầu có nguồn gốc sinh học
Cần có các tóm tắt những thông tin an toàn về virut đối với các nguyên vật liệu có nguồn gốc sinh học.
● Nguồn gốc, lịch sử và sự hình thành dòng tế bào sản xuất
Cần cung cấp những thông tin về nguồn gốc của dòng tế bào sản xuất và phân tích cơ cấu biểu hiện được dùng để biến đổi tế bào về mặt di truyền, và được đưa vào dòng tế bào ban đầu dùng để phát triển thành Ngân hàng Tế bào Mẹ, như quy định trong các phần Q5B và Q5D của hướng dẫn ICH.
● Hệ thống ngân hàng tế bào, mô tả đặc điểm và phương pháp kiểm nghiệm
Cần cung cấp các thông tin về hệ ngân hàng tế bào; các hoạt động kiểm tra chất lượng và độ ổn định dòng tế bào trong quá trình sản xuất và bảo quản (bao gồm cả các quy trình tạo ra Ngân hàng Tế Bào Mẹ và Ngân hàng Tế Bào Sản Xuất) như quy định trong các phần Q5B và Q5D của hướng dẫn ICH.
Tham khảo hướng dẫn ICH: Q5A, Q5B, Q5C và Q5D
S 2.4 Kiểm soát các bước quan trọng và sản phẩm trung gian
Các bước quan trọng: Các phép thử và chỉ tiêu chấp nhận cùng với thuyết minh nêu rõ các dữ liệu thực nghiệm đã được thực hiện ở các bước quan trọng trong quá trình sản xuất, để chắc chắn rằng quy trình này đã được kiểm soát.
Sản phẩm trung gian: Nêu rõ tiêu chuẩn chất lượng và quy trình phân tích, nếu có, đối với sản phẩm trung gian được phân lập trong quá trình sản xuất.
Tham khảo hướng dẫn ICH: Q6A, Q6B
Ngoài ra đối với Biotech: cung cấp số liệu về độ ổn định làm căn cứ đưa ra các điều kiện bảo quản.
Tham khảo hướng dẫn ICH: Q5C
S 2.5 Đánh giá và/ hoặc thẩm định quy trình
Nêu các nghiên cứu đánh giá hoặc thẩm định quy trình đối với quy trình chế biến vô trùng hoặc tiệt trùng.
Biotech:
Cần có đủ thông tin về các nghiên cứu đánh giá và thẩm định nhằm chứng minh rằng quy trình sản xuất (kể cả các bước chế biến lặp lại) là phù hợp cho mục đích sử dụng dự kiến và chứng minh cho việc lựa chọn các biện pháp kiểm soát quy trình quan trọng (các thông số vận hành và kiểm nghiệm trong quá trình sản xuất) và các giới hạn của chúng cho các bước sản xuất quan trọng (ví dụ nuôi cấy tế bào, thu hoạch, tinh chế và biến đổi).
Những thông tin này phải bao gồm một bản mô tả kế hoạch tiến hành nghiên cứu và kết quả, phân tích và kết luận của các nghiên cứu đã thực hiện. Việc thẩm định các phương pháp định lượng và phân tích tương ứng phải có tham chiếu chéo hoặc cung cấp dưới dạng thuyết minh cho việc lựa chọn các biện pháp kiểm soát quy trình quan trọng và các giới hạn.
Đối với các bước sản xuất nhằm loại bỏ hoặc bất hoạt các tác nhân gây nhiễm là virut, cần cung cấp thông tin về các nghiên cứu đánh giá quy trình.
Tham khảo hướng dẫn ICH: Q5A, Q5D và Q6B
S 2.6 Phát triển quy trình sản xuất
NCE
Mô tả và bàn luận về những thay đổi quan trọng đối với quy trình sản xuất hoặc cơ sở sản xuất dược chất dùng trong việc sản xuất các óngản phẩm để nghiên cứu tiền lâm sàng, lâm sàng, lô thử nghiệm, và cả lô sản xuất thực tế nếu có.
Tham khảo hướng dẫn ICH: Q3A
Biotech
Cần cung cấp lịch sử phát triển của quy trình sản xuất như mô tả ở S 2.2. Mô tả những thay đổi trong việc sản xuất các lô dược chất dùng cho các nghiên cứu để hoàn thiện hồ sơ đăng ký lưu hành (ví dụ các nghiên cứu tiền lâm sàng và lâm sàng), bao gồm những thay đổi về quy trình và thiết bị quan trọng. Cần giải thích lý do thay đổi. Các thông tin liên quan đến lô dược chất được sản xuất trong quá trình nghiên cứu phát triển, ví dụ số lô, quy mô sản xuất và việc sử dụng (ví dụ nghiên cứu độ ổn định, nguyên liệu tham khảo trong nghiên cứu tiền lâm sàng) liên quan đến sự thay đổi đó.
Ý nghĩa của thay đổi cần phải được kiểm tra bằng cách đánh giá khả năng ảnh hưởng đối với chất lượng dược chất (và/hoặc sản phẩm trung gian, nếu có). Đối với những thay đổi về sản xuất được coi là quan trọng, thì cần phải có số liệu từ các thí nghiệm phân tích so sánh trên dược chất có liên quan. Cần phải có phần bàn luận về số liệu, trong đó có cả các thuyết minh về việc lựa chọn phép thử và đánh giá kết quả.
Phép thử dùng để đánh giá ảnh hưởng của những thay đổi trong sản xuất đối với dược chất và thành phẩm thuốc tương ứng cũng có thể bao gồm cả các nghiên cứu lâm sàng và tiền lâm sàng có trong các phần hồ sơ được nộp khác.
Tham khảo hướng dẫn ICH: Q6B.
S 3 Đặc tính
S 3.1 Giải thích cấu trúc và các đặc tính khác
NCE
Xác định cấu trúc dựa trên cơ sở quá trình tổng hợp và các phân tích phổ. Cũng cần đến thông tin về khả năng của hiện tượng đồng phân, việc xác định hóa lập thể hoặc khả năng hình thành hiện tượng đa hình.
Tham khảo hướng dẫn ICH: Q6A
Biotech
Các chi tiết về cấu trúc sơ cấp, thứ cấp hoặc cao hơn và thông tin về hoạt tính sinh học, độ tinh khiết và đặc tính hóa miễn dịch (nếu có liên quan).
Tham khảo hướng dẫn ICH: Q6B
MaV, MiV, G
Qui định trong dược điển hoặc thông tin tương đương của nhà sản xuất.
S 3.2 Tạp chất
Phải cung cấp thông tin về các tạp chất
Phải tham khảo hướng dẫn ICH: Q3A, Q3C, Q5C, Q6A, Q6B
Generic
Quy định trong dược điển hoặc thông tin tương đương của nhà sản xuất.
S 4 Kiểm tra dược chất:
Tiêu chuẩn chất lượng và việc thuyết minh các tiêu chuẩn.
Tóm tắt phương pháp phân tích và thẩm định phương pháp phân tích.
S 4.1 Tiêu chuẩn chất lượng
Phải cung cấp chi tiết tiêu chuẩn chất lượng, các phép thử và các chỉ tiêu chấp nhận của dược chất.
Tham khảo hướng dẫn ICH, NCE: Q6A
Biotech
Nêu nguồn gốc, kể cả loài động vật phù hợp, chủng vi sinh vật
Tham khảo hướng dẫn ICH: Q6B
MaV, MiV, G
Nêu tiêu chuẩn dược điển là được. Phải chỉ rõ dược chất có được mua dựa trên tiêu chuẩn chất lượng kèm theo phiếu kiểm nghiệm hoặc đã được kiểm nghiệm bởi cơ sở đăng ký hay không.
S 4.2 Quy trình phân tích
Quy trình phân tích được dùng để thử dược chất phải có đầy đủ chi tiết để có thể tiến hành thử lại tại các phòng thí nghiệm khác.
Tham khảo hướng dẫn ICH: NCE: Q2A; Biotech: Q6B.
MaV, MiV, G
Quy định trong dược điển hoặc thông tin tương đương của nhà sản xuất.
S 4.3 Thẩm định quy trình phân tích
Phải có thông tin về thẩm định phép phân tích, bao gồm các dữ liệu thực nghiệm về quy trình phân tích được dùng để thử dược chất. Những điểm đặc trưng cần đánh giá là tính chọn lọc, độ chính xác (độ lặp lại, độ chính xác trung gian, độ tái lặp), độ đúng, tính tuyến tính, khoảng xác định, giới hạn định lượng, giới hạn phát hiện, độ thô và tính tương thích của hệ thống.
Tham khảo hướng dẫn ICH: NCE, Q2A, Q2B; Biotech: Q6B.
MaV, MiV, G
Chỉ yêu cầu đối với phương pháp phân tích không có trong dược điển. Tham khảo hướng dẫn của ASEAN về thẩm định quy trình phân tích.
S 4.4 Phân tích lô
Phải có sự mô tả lô, và các kết quả phân tích lô.
Tham khảo hướng dẫn ICH: NCE: Q3A, Q3C, Q6A; Biotech: Q6B
S 4.5 Thuyết minh tiêu chuẩn chất lượng
Phải có sự thuyết minh tiêu chuẩn chất lượng của dược chất.
Tham khảo hướng dẫn ICH: NCE: Q6A; Biotech: Q6B.
S 5 Chất chuẩn hoặc nguyên liệu đối chiếu
Phải cung cấp thông tin về chất lượng của các chất chuẩn hoặc nguyên liệu đối chiếu được dùng cho việc thử dược chất.
Tham khảo Hướng dẫn ICH: NCE: Q6A; Biotech: Q6B
MaV, MiV, G
Quy định trong dược điển hoặc thông tin tương đương của nhà sản xuất.
S 6 Hệ thống bao bì đóng gói:
NCE và Biotech:
Phải có mô tả hệ thống bao bì đóng gói bao gồm cả đặc điểm vật liệu chế tạo từng loại bao bì sơ cấp và tiêu chuẩn kỹ thuật của mỗi loại. Các tiêu chuẩn kỹ thuật phải có mô tả và định dạng (và những kích thước cơ bản thì nên thể hiện bằng hình vẽ khi có thể). Phải nêu các phương pháp không có trong dược điển (cùng kết quả thẩm định) nếu có thể.
Đối với bao bì thứ cấp không có chức năng bảo vệ (ví dụ những bao bì không có chức năng bảo vệ bổ trợ hoặc không giữ vai trò gì trong vận chuyển phân phối sản phẩm) thì chỉ cần miêu tả tóm tắt. Nếu bao bì thứ cấp có chức năng bảo vệ thì cần bổ sung thêm thông tin.
Nên bàn luận về tính phù hợp, ví dụ việc lựa chọn chất liệu, đối với việc bảo vệ khỏi ảnh hưởng của ẩm và ánh sáng, đến tính tương hợp của các chất liệu chế tạo với dược chất, kể cả tính hấp phụ của bao bì, tính thấm và/hoặc độ an toàn của vật liệu chế tạo.
S 7 Độ ổn định
Tóm tắt độ ổn định và kết luận
Phải tóm tắt các loại nghiên cứu đã tiến hành, các đề cương đã sử dụng và các kết quả nghiên cứu. Phần tóm tắt phải bao gồm cả kết quả nghiên cứu, ví dụ nghiên cứu ở điều kiện thúc đẩy sự phân hủy và các điều kiện khắc nghiệt, cũng như các kết luận liên quan đến điều kiện bảo quản và ngày kiểm tra lại hoặc tuổi thọ, nếu thích hợp.
Tham khảo hướng dẫn ICH: Q1A (R2), Q1B, Q5C
Đề cương và cam kết nghiên cứu độ ổn định sau khi được phép lưu hành
Cần có đề cương nghiên cứu độ ổn định sau khi được phép lưu hành và bản cam kết về độ ổn định.
Tham khảo hướng dẫn ICH: Q1A (R2), Q5C
Dữ liệu độ ổn định
Các kết quả nghiên cứu độ ổn định (như nghiên cứu ở điều kiện thúc đẩy sự phân
hủy và các điều kiện khắc nghiệt) phải được trình bày dưới dạng thích hợp như bảng biểu, đồ thị hoặc bài tường thuật. Phải có cả thông tin về các quy trình phân tích được dùng để có được các số liệu đó và việc thẩm định các quy trình này.
Tham khảo Hướng dẫn ICH: Q1A (R2), Q1B, Q2A, Q2B, Q5C.
MaV, MiV, G
Số liệu độ ổn định của nhà sản xuất hoặc thông tin tương đương.
P THÀNH PHẨM THUỐC
P 1 Mô tả và thành phần:
Phải có sự mô tả về thành phẩm thuốc và thành phần của nó. Thông tin cần cung cấp gồm có:
- Mô tả dạng bào chế;
- Thành phần, nghĩa là nêu tên của tất cả các thành phần có trong dạng bào chế và hàm lượng có trong mỗi đơn vị (kể cả lượng đóng dư, nếu có), chức năng của các thành phần và tham khảo tiêu chuẩn chất lượng của chúng (ví dụ như các chuyên luận trong dược điển hoặc tiêu chuẩn chất lượng của nhà nhà sản xuất)
- Mô tả dung môi để pha chế đi kèm theo sản phẩm; và
- Loại bao bì đóng gói của dạng bào chế và dung môi pha chế đi kèm theo sản phẩm (nếu có).
Tham khảo Hướng dẫn ICH: NCE: Q6A; Biotech: Q6B.
P 2 Sự phát triển dược học
P 2.1 Thông tin về những nghiên cứu phát triển
NCE và Biotech:
Phần phát triển dược học thể hiện thông tin và dữ liệu về các nghiên cứu phát triển được tiến hành để xác định rằng dạng bào chế, công thức, quy trình sản xuất, hệ thống bao bì đóng gói, các thuộc tính về vi sinh vật và hướng dẫn sử dụng là phù hợp với mục đích nêu trong hồ sơ đăng ký. Các nghiên cứu được mô tả ở đây được phân biệt với những kiểm nghiệm thường quy được tiến hành theo tiêu chuẩn. Ngoài ra, phần này cũng xác định và miêu tả công thức bào chế và các thuộc tính của quy trình (các thông số lâm sàng) có thể ảnh hưởng đến khả năng tái lặp lô mẻ, khả năng tác dụng của sản phẩm và chất lượng của thành phẩm thuốc. Các số liệu hỗ trợ và kết quả thu được từ những nghiên cứu đặc biệt hoặc tài liệu đã xuất bản có thể nằm trong hoặc đi kèm với phần phát triển dược học. Các số liệu hỗ trợ bổ sung có thể tham khảo ở các phần thích hợp khác ngoài phần lâm sàng trong hồ sơ đăng ký.
P 2.2 Thành phần của thành phẩm thuốc
P 2.2.1 Hoạt chất:
NCE và Biotech:
Phải bàn luận về tính tương hợp của các dược chất với tá dược được liệt kê trong mục 2.1. Hơn nữa, các tính chất lý hóa chủ yếu (như hàm lượng nước, độ hòa tan, phân bố kích cỡ hạt, trạng thái rắn hoặc đa hình) của dược chất có thể ảnh hưởng đến tác dụng của thành phẩm thuốc cũng phải được thảo luận.
MaV, MiV, G:
Các số liệu đăng tải trong các tài liệu khoa học là đủ.
P 2.2.2 Tá dược
Sự lựa chọn các tá dược được ghi ở mục P 1, nồng độ và các tính chất của chúng có ảnh hưởng đến tác dụng của thành phẩm thuốc cần được bàn luận liên quan đến chức năng tương ứng của chúng.
P 2.3 Thành phẩm thuốc
P 2.3.1 Phát triển công thức bào chế
Mô tả tóm tắt ngắn gọn về sự phát triển thành phẩm thuốc, có tính đến đường dùng và cách sử dụng dự kiến. Phải có bàn luận về sự khác nhau giữa các công thức bào chế dùng trong lâm sàng và công thức bào chế (nghĩa là thành phần) được miêu tả trong mục P 1 và P 2. Kết quả của các nghiên cứu so sánh in-vitro (ví dụ thử độ hòa tan) và nghiên cứu so sánh in-vivo (ví dụ thử tương đương sinh học) phải được bàn luận khi có thể.
P 2.3.2 Lượng đóng dư
Phải thuyết minh về lượng đóng dư trong công thức nêu ở mục P 1.
P 2.3.3 Đặc tính lý hóa và sinh học
Các thông số có liên quan đến khả năng tác dụng của thành phẩm thuốc như pH, hàm lượng Ion, độ hòa tan, độ khuyếch tán, sự tái tạo, sự phân bố cỡ hạt, sự kết tập, tính đa hình, tính lưu biến, hoạt tính sinh học, hoạt lực và hoạt tính miễn dịch cần phải được nêu rõ.
P 2.4 Sự phát triển quy trình sản xuất
Phải giải thích sự chọn lựa và tối ưu hóa quy trình sản xuất được mô tả trong mục P 3.2, đặc biệt ở những khía cạnh thiết yếu. Phương pháp tiệt trùng phải được giải thích và thuyết minh nếu có liên quan.
Phải thảo luận về sự khác nhau giữa những quy trình dùng để sản xuất các lô thuốc thử lâm sàng chủ yếu với quy trình được mô tả ở mục P 3.2 mà có thể ảnh hưởng tới việc phát huy tác dụng của sản phẩm.
P 2.5 Hệ bao bì đóng gói
Nếu cần, phải bàn luận về sự thích hợp hệ bao bì đóng gói dùng trong bảo quản, vận chuyển (đường biển) và sử dụng thành phẩm. Việc bàn luận này nên lưu ý đến những vấn đề như sự lụa chọn các vật liệu, việc bảo vệ khỏi ảnh hưởng của ẩm và ánh sáng, tính tương hợp của vật liệu chế tạo với dạng bào chế, kể cả việc hấp thụ đối với bao bì, tính an toàn không bị rò rỉ của vật liệu đóng gói và việc phát huy tác dụng ví dụ như khả năng tái lặp trong phân phối liều lượng từ dụng cụ phân liều nếu như đó là một phần của thành phẩm thuốc.
P 2.6 Thuộc tính vi sinh vật
Khi phù hợp, cần bàn luận về các thuộc tính vi sinh vật của dạng bào chế, kể cả tính lập luận cho việc không tiến hành thử giới hạn vi khuẩn đối với thành phẩm không vô trùng và việc lựa chọn cũng như hiệu quả của hệ thống chất bảo quản trong các sản phẩm có chứa chất bảo quản chống vi khuẩn. Đối với sản phẩm vô trùng, cần bàn luận về tính toàn vẹn của hệ bao bì đóng gói nhằm ngăn ngừa nhiễm khuẩn.
P 2.7 Tính tương hợp
Cần phải bàn luận về tính tương hợp của thành phẩm thuốc với dung môi pha loãng hoặc dụng cụ để phân liều, ví dụ như để kết tủa dược chất trong dung dịch, sự hấp thu của ống dẫn tiêm truyền và độ ổn định, mục đích là để cung cấp các thông tin phù hợp và hỗ trợ cho việc ghi nhãn.
MaV, MiV, G:
Có thể chấp nhận các dữ liệu đăng tải trong các tài liệu khoa học
P 3 Sản xuất
P 3.1 Công thức lô
Công thức bào chế, có tên và hàm lượng của tất cả các thành phần (thành phần hoạt tính và thành phần khác), kể cả những chất sẽ bị loại bỏ trong quá trình sản xuất, bao gồm:
- Lượng thực dùng của mỗi thành phần (bằng gam, kilôgam, lit);
- Lượng đóng dư: Phải có số liệu hỗ trợ và giải thích lý do đóng dư.
- Tổng lượng đơn vị liều dùng của một lô
- Cần mô tả tất cả các công đoạn trong quá trình sản xuất dạng bào chế.
Tham khảo Hướng dẫn ICH: Biotech: Q6B.
P 3.2 Quy trình sản xuất và kiểm soát quy trình
Phải có sơ đồ mô tả các công đoạn trong quy trình và chỉ rõ ở công đoạn nào thì các nguyên liệu được đưa vào. Phải xác định ở các bước quan trọng nào và ở thời điểm nào thì tiến hành kiểm soát quy trình, kiểm tra sản phẩm trung gian và sản phẩm cuối cùng.
- Mô tả đầy đủ qui trình sản xuất với đủ các chi tiết bao quát các điểm thiết yếu ở mỗi giai đoạn sản xuất.
- Đối với các sản phẩm vô trùng thì việc mô tả phải bao quát cả việc pha chế và tiệt trùng các thành phần (như bao bì, nắp nút, …).
P 3.3 Kiểm soát các bước quan trọng và sản phẩm trung gian
● Các bước quan trọng: Cần nêu các phép thử và chỉ tiêu chấp nhận (có thuyết minh, kể cả các số liệu thực nghiệm) được thực hiện ở các bước quan trọng của quy trình sản xuất như xác định ở mục P 3.3 để đảm bảo rằng quy trình đã được kiểm tra.
● Sản phẩm trung gian: phải cung cấp thông tin về chất lượng và việc kiểm tra các sản phẩm trung gian phân lập được trong quy trình.
Tham khảo Hướng dẫn ICH:Q2A, Q2B, Q6A và Q6B.
P 3.4 Thẩm định và/hoặc đánh giá quy trình
Phải có mô tả, dẫn chứng bằng tài liệu và kết quả của các nghiên cứu thẩm định ở những bước sản xuất quan trọng hoặc các phép định lượng quan trọng sử dụng trong quy trình sản xuất (như thẩm định quy trình tiệt trùng, quy trình chế biến hoặc đóng chai vô trùng).
Tham khảo Hướng dẫn ICH: NCE: Q6B; Biotech: Q6A.
MaV, MiV, G:
Tham khảo Hướng dẫn của ASEAN về thẩm định quy trình sản xuất.
P 4 Kiểm tra tá dược
P 4.1 Tiêu chuẩn chất lượng
Phải cung cấp tiêu chuẩn chất lượng của các tá dược.
Tham khảo Hướng dẫn ICH: NCE: Q6A; Biotech: Q6B.
MaV, MiV, G:
Các quy định trong dược điển hoặc thông tin tương đương từ nhà sản xuất.
P 4.2 Quy trình phân tích
Phải cung cấp quy trình phân tích dùng để thử các tá dược, khi thích hợp.
Tham khảo Hướng dẫn ICH: NCE: Q2A; Biotech: Q6B.
MaV, MiV, G:
Các quy định trong dược điển hoặc thông tin tương đương từ nhà sản xuất.
P 4.3 Tá dược có nguồn gốc từ người và động vật:
Đối với tá dược có nguồn gốc từ người và động vật phải cung cấp thông tin về các chất ngẫu nhiên (ví dụ: nguồn gốc, các tiêu chuẩn chất lượng, mô tả các phép thử, các số liệu an toàn về virut).
Tham khảo hướng dẫn ICH: NCE: Q5A, Q5D; Biotech: Q6B.
MaV, G:
Áp dụng các yêu cầu của dược điển nếu có, nếu không thì áp dụng các yêu cầu tương tự khác.
P 4.4 Tá dược mới:
Đối với các tá dược mới được dùng lần đầu trong thành phẩm thuốc hoặc sử dụng đường dùng mới, phải cung cấp đầy đủ các chi tiết về sản xuất, đặc tính và biện pháp kiểm tra, có tham khảo chéo những số liệu an toàn hỗ trợ (tiền lâm sàng hoặc lâm sàng).
P 5 Kiểm tra thành phẩm:
Tiêu chuẩn kỹ thuật và thuyết minh tiêu chuẩn kỹ thuật, tóm tắt quy trình phân tích và thẩm định quy trình, xác định đặc điểm các tạp chất.
P 5.1 Tiêu chuẩn chất lượng
Phải cung cấp tiêu chuẩn chất lượng của thành phẩm
Tham khảo hướng dẫn ICH: NCE: Q6A; Biotech: Q6B.
P 5.2 Quy trình phân tích
Phải cung cấp các quy trình phân tích dùng để kiểm nghiệm thành phẩm. Tham khảo hướng dẫn ICH: NCE: Q2A; Biotech: Q6B.
P 5.3 Thẩm định quy trình phân tích
Phải có thông tin về thẩm định quy trình phân tích bao gồm các dữ liệu thực nghiệm đối với quy trình phân tích dùng để kiểm nghiệm thành phẩm.
Tham khảo hướng dẫn ICH: NCE: Q2A và Q2B; Biotech: Q6B.
MaV, MiV, G:
Chỉ yêu cầu đối với các phương pháp không có trong dược điển, tuy nhiên, đối với phương pháp có trong dược điển, cần phải xác minh khả năng áp dụng được.
P 5.4 Phân tích lô
Cung cấp thông tin mô tả (bao gồm cỡ lô, nguồn gốc và việc sử dụng) và kết quả thử của tất cả các lô liên quan (ví dụ: lô thí nghiệm dùng để nghiên cứu lâm sàng và tiền lâm sàng, điều chỉnh cỡ lô, và lô ở quy mô sản xuất nếu có) dược dùng để thiết lập tiêu chuẩn chất lượng và đánh giá tính ổn định trong sản xuất.
Tham khảo hướng dẫn ICH: NCE: Q3A, Q3C và Q6A; Biotech: Q6B.
Generic: tham khảo P 3.4
MaV, MiV, G:
Cần cung cấp tóm tắt phân tích lô (trình bày bằng dạng bảng biểu), cùng với đồ thị nếu có.
P 5.5 Đặc tính của tạp chất
Cần cung cấp thông tin về đặc tính của các tạp chất nếu chưa được nêu ra trong mục S 3.2 Tạp Chất.
Tham khảo hướng dẫn ICH: NCE: Q3B và Q6A; Biotech: Q6B
MaV, MiV, G:
Các quy định trong dược điển hoặc thông tin thích hợp từ nhà sản xuất.
P 5.6 Thuyết minh tiêu chuẩn chất lượng
Cung cấp thuyết minh tiêu chuẩn chất lượng dự kiến của thành phẩm.
Tham khảo hướng dẫn ICH: NCE: Q3B và Q6A; Biotech: Q6B.
MaV, MiV, G:
Các quy định trong dược điển hoặc thông tin tương đương từ nhà sản xuất.
P 6 Chất chuẩn hoặc chất đối chiếu
Yêu cầu: thông tin về chất lượng và bảng biểu trình bày về chất chuẩn và chất đối chiếu được dùng để thử thành phẩm.
Tham khảo hướng dẫn ICH: NCE: Q6A, Biotech: Q6B.
MaV, MiV, G:
Các quy định trong dược điển hoặc thông tin tương đương từ nhà sản xuất.
P 7 Hệ thống bao bì đóng gói
Phải có mô tả hệ thống bao bì đóng gói bao gồm cả đặc điểm của vật liệu chế tạo của từng loại bao bì sơ cấp và bao bì thứ cấp và tiêu chuẩn kỹ thuật của mỗi loại. Các tiêu chuẩn kỹ thuật phải có mô tả và định dạng (và những kích thước cơ bản thì nên thể hiện bằng hình vẽ, nếu có thể). Các phương pháp không có trong dược điển (với kết quả thẩm định) cũng phải nêu ra khi có thể.
Đối với bao bì thứ cấp không có chức năng bảo vệ (như bao bì không có tác dụng bảo vệ hỗ trợ hoặc không có vai trò trong phân phối vận chuyển sản phẩm) chỉ cần miêu tả tóm tắt. Đối với loại bao bì thứ cấp có chức năng bảo vệ thì phải có thêm thông tin.
Thông tin về tính phù hợp phải nêu ở mục P 2.
P 8 Độ ổn định của sản phẩm
Cần có bằng chứng chứng minh rằng sản phẩm ổn định, đáp ứng được các tiêu chuẩn chất lượng của thành phẩm trong suốt tuổi thọ dự kiến của nó, rằng không có sản phẩm phân hủy độc hại được tạo ra ở mức có ý nghĩa trong thời gian này, và hoạt lực cũng như hiệu quả của chất bảo quản, vv…vẫn được duy trì.
Tóm tắt và kết luận về độ ổn định
NCE và Biotech:
Tất cả các tiêu chuẩn phù hợp với hướng dẫn ICH đều có thể chấp nhận được, trừ điều kiện bảo quản thực phải ở nhiệt độ 30oC và độ ẩm tương đối 75%. Phải lưu ý đến biện pháp chống ẩm của bao bì đóng gói.
Tham khảo Hướng dẫn ICH: Q1A (R2), Q1B, Q2A, Q2B và Q5C.
MaV, G:
Theo hướng dẫn ASEAN về nghiên cứu độ ổn định của thuốc.
Đề cương theo dõi độ ổn định sau khi được phép lưu hành và cam kết về độ ổn định.
Cần có đề cương theo dõi độ ổn định sau khi được phép lưu hành và cam kết về độ ổn định sản phẩm.
Tham khảo Hướng dẫn ICH: NCE, Biotech: Q1A (R2) và Q5C.
Generic:
Hướng dẫn ASEAN về nghiên cứu độ ổn định của thuốc.
Dữ liệu độ ổn định
Các kết quả nghiên cứu độ ổn định phải được trình bày dưới dạng phù hợp (như bảng biểu, đồ thị, bài tường thuật). Phải có thông tin về quy trình phân tích dùng để thu được các số liệu và việc thẩm định các quy trình này.
Tham khảo: Hướng dẫn ASEAN về nghiên cứu độ ổn định của thuốc, Hướng dẫn ASEAN về thẩm định quy trình phân tích.
P 9 Khả năng thay thế lẫn nhau của sản phẩm
Quy định này áp dụng đối với MaV, G
Trình bày loại nghiên cứu đã được thực hiện, đề cương đã dùng và kết quả nghiên cứu trong báo cáo nghiên cứu.
Loại nghiên cứu đã thực hiện cần đề cập đến các quy định của ASEAN (dự kiến) về tương đương sinh học và sinh khả dụng, Hướng dẫn về nghiên cứu tương đương sinh học và sinh khả dụng hoặc sổ tay hướng dẫn của Tổ Chức Y Tế Thế Giới dành cho cơ quan quản lý thuốc.
Tham khảo:
- Báo cáo hỗ trợ quản lý của WHO, bộ số 5: “Nghiên cứu tương đương sinh học trên người”.
- Hướng dẫn ASEAN về Nghiên Cứu Tương Đương Sinh Học.
CHƯƠNG D: TÀI LIỆU THAM KHẢO CHỦ YẾU
Cần cung cấp danh mục tài liệu tham khảo nếu có
HỒ SƠ KỸ THUẬT CHUNG ASEAN VỀ ĐĂNG KÝ THUỐC SỬ DỤNG CHO NGƯỜI
PHẦN III: HỒ SƠ TIỀN LÂM SÀNG
Lời mở đầu:
Phần III sẽ cung cấp tổng quan về tiền lâm sàng, tiếp theo là các tóm tắt nghiên cứu tiền lâm sàng bằng văn bản và bảng biểu. Tài liệu của phần này không quy định đối với các sản phẩm generic, các sản phẩm thay đổi nhỏ và một vài sản phẩm có sự thay đổi lớn. Đối với các nước thành viên ASEAN, các báo cáo nghiên cứu trong phần này không quy định cho các hoạt chất mới, các sản phẩm công nghệ sinh học và các sản phẩm có sự thay đổi lớn khác khi các sản phẩm gốc đã được đăng ký và cấp phép lưu hành ở các nước tham khảo. Do vậy, cơ quan quản lý nào có nhu cầu về các báo cáo nghiên cứu thì sẽ yêu cầu các tài liệu cần thiết.
Mục A: Mục lục
Cần có mục lục tài liệu trong hồ sơ đăng ký
Mục B: Tổng quan về tiền lâm sàng
1. Các vấn đề chung
2. Nội dung và cấu trúc
Mục C: Tóm tắt tiền lâm sàng bằng văn bản và bảng biểu
1. Các tóm tắt tiền lâm sàng bằng văn bản
1.1 Mở đầu
1.2 Các vấn đề chung
2. Nội dung các tóm tắt tiền lâm sàng bằng văn bản và bảng biểu
2.1 Dược lý học
2.1.1 Tóm tắt bằng văn bản
2.1.1.1 Dược lực học tổng quan
2.1.1.2 Dược lực học trên hệ cơ quan
2.1.1.3 Dược lý học về tính an toàn
2.1.1.4 Các tương tác thuốc về dược lực học
2.1.2 Tóm tắt bằng bảng biểu
2.2 Dược động học
2.2.1 Tóm tắt bằng văn bản
2.1.1.1 Sự hấp thu
2.1.1.2 Sự phân phối
2.1.1.3 Sự chuyển hóa
2.1.1.4 Sự thải trừ
2.1.1.5 Tương tác thuốc về dược động học (tiền lâm sàng)
2.2.2 Tóm tắt bằng bảng biểu
2.3 Độc tính
2.3.1 Tóm tắt bằng văn bản
2.3.1.1 Độc tính liều duy nhất
2.3.1.2 Độc tính liều lặp lại
2.3.1.3 Độc tính trên gen
2.3.1.4 Khả năng gây ung thư
2.3.1.5 Độc tính trên sự sinh sản và phát triển
2.3.1.5.1 Trên khả năng sinh sản và sự phát triển phôi đoạn đầu
2.3.1.5.2 Trên sự phát triển phôi thai
2.3.1.5.3 Trên sự phát triển trước và sau sinh
2.3.1.6 Dung nạp tại chỗ
2.3.1.7 Các nghiên cứu độc tính khác (nếu có)
2.3.2. Tóm tắt bằng bảng biểu
3. Các tóm tắt tiền lâm sàng bằng bảng biểu
Mục D: Báo cáo nghiên cứu tiền lâm sàng
1. Mục lục
2. Dược lý học
2.1 Các báo cáo nghiên cứu bằng văn bản
2.1.1 Dược lực học tổng quan
2.1.2 Dược lực học trên các cơ quan
2.1.3 Dược lý học về tính an toàn
2.1.4 Tương tác thuốc về dược lực học
3. Dược động học
3.1 Các báo cáo nghiên cứu bằng văn bản
3.1.1 Các phương pháp phân tích và báo cáo thẩm định
3.1.2 Sự hấp thu
3.1.3 Sự phân phối
3.1.4 Sự chuyển hóa
3.1.5 Sự thải trừ
3.1.6 Tương tác thuốc về dược động học (tiền lâm sàng)
3.1.7 Các nghiên cứu dược động học khác
4. Độc tính
4.1 Các báo cáo nghiên cứu bằng văn bản
4.1.1 Độc tính liều duy nhất
4.1.2 Độc tính liều lặp lại
4.1.3 Độc tính trên gen
4.1.3.1 Các báo cáo kết quả in vitro
4.1.3.2 Các báo cáo kết quả in vivo
4.1.4 Khả năng gây ung thư
4.1.4.1 Các nghiên cứu dài hạn
4.1.4.2 Các nghiên cứu ngắn hoặc trung hạn
4.1.4.3 Các nghiên cứu khác
4.1.5 Độc tính trên khả năng sinh sản và phát triển
4.1.5.1 Khả năng sinh sản và sự phát triển phôi giai đoạn đầu
4.1.5.2 Sự phát triển phôi thai
4.1.5.3 Sự phát triển trước và sau sinh
4.1.5.4 Nghiên cứu độc tính qua 2 thế hệ (thế hệ con được tiếp tục cho dùng thuốc và/hoặc được tiếp tục theo dõi)
4.1.6 Sự dung nạp tại chỗ
4.1.7 Các nghiên cứu độc tính khác (nếu có)
4.1.7.1 Tính kháng nguyên
4.1.7.2 Độc tính trên hệ miễn dịch
4.1.7.3 Sự lệ thuộc thuốc
4.1.7.4 Các chất chuyển hóa
4.1.7.5 Tạp chất (độ tinh khiết)
4.1.7.6 Các nghiên cứu độc tính khác
Mục E: Danh mục các tài liệu tham khảo chính
Cần có danh mục các tài liệu tham khảo trình bày theo đúng “Tuyên ngôn Vancouver 1979” về “Quy định thống nhất đối với các bản thảo nộp cho các tạp chí y–sinh học”, hoặc theo hệ thống sử dụng trong “Các bài tóm tắt về hóa học”. Cần cung cấp bản chụp các tài liệu tham khảo quan trọng nêu trong phần Tổng quan tiền lâm sàng ở phần này. Phải có sẳn bản chụp tất cả các tài liệu tham khảo chưa đưa vào hồ sơ để có thể cung cấp khi có yêu cầu.
HỒ SƠ KỸ THUẬT CHUNG ASEAN CHO ĐĂNG KÝ THUỐC SỬ DỤNG CHO NGƯỜI
PHẦN III: TÀI LIỆU TIỀN LÂM SÀNG*2
Mục A: Mục lục
1. Hướng dẫn về tổng quan và tóm tắt tiền lâm sàng
Mục B: Tổng quan tiền lâm sàng
1. Vấn đề chung
2. Nội dung và cấu trúc
Mục C: Tóm tắt tiền lâm sàng bằng văn bản và bảng biểu
1. Các tóm tắt tiền lâm sàng bằng văn bản
1.1. Giới thiệu
1.2. Các vấn đề chung về cách trình bày
2. Nội dung của các tóm tắt tiền lâm sàng bằng văn bản và bảng biểu
2.1. Dược lý học
2.2. Dược động học
2.3. Độc tính
3. Hướng dẫn về tóm tắt tiền lâm sàng bằng bảng biểu
Mục D: Các báo cáo nghiên cứu tiền lâm sàng
Mục E: Danh mục các tài liệu tham khảo chính
Phụ lục A: Các biểu mẫu về tóm tắt tiền lâm sàng bằng bảng biểu
2* Cập nhật từ ICH- CTD về tổng quan tiền lâm sàng
HƯỚNG DẪN VỀ TỔNG QUAN VÀ TÓM TẮT TIỀN LÂM SÀNG
Hướng dẫn này đưa ra các khuyến cáo để hòa hợp phần Tổng quan tiền lâm sàng, phần Tóm tắt tiền lâm sàng bằng văn bản và bảng biểu.
Mục đích chính của các tóm tắt tiền lâm sàng bằng văn bản và bảng biểu là cung cấp các tóm tắt sát thực và toàn diện về các dữ liệu tiền lâm sàng. Cần trình bày các diễn giải về các dữ liệu thu thập được, mối tương quan về mặt lâm sàng, sự liên kết chéo với các khía cạnh về chất lượng dược phẩm và những ảnh hưởng lên việc sử dụng an toàn dược phẩm (nghĩa là những chi tiết cần được nêu trên nhãn) của các phát hiện tiền lâm sàng.
PHẦN B. TỔNG QUAN TIỀN LÂM SÀNG
Phần tổng quan tiền lâm sàng cần đưa ra một phân tích toàn diện, tổng hợp về những thông tin ở trong tài liệu Kỹ thuật chung.
1. NHỮNG VẤN ĐỀ CHUNG
Phần này phải trình bày được một đánh giá then chốt và thống nhất đối với các thẩm định về dược lý, dược động học và độc tính. Nếu có các hướng dẫn phù hợp về tiến hành các nghiên cứu, thì phải cân nhắc thực hiện các hướng dẫn này, và phải bàn luận và giải trình về bất kỳ thay đổi nào trong thực hiện các hướng dẫn này. Chiến lược thử nghiệm tiền lâm sàng phải được bàn luận và giải trình. Cần có các diễn giải về thực hành phòng thí nghiệm tốt đối với các nghiên cứu đã nộp. Nên nêu rõ mối quan hệ giữa các phát hiện từ nghiên cứu tiền lâm sàng và đặc tính về chất lượng của thuốc dùng cho người, kết quả thử nghiệm lâm sàng, hoặc các tác dụng đã biết ở các sản phẩm có liên quan, nếu thích hợp.
Ngoại trừ sản phẩm công nghệ sinh học, cần trình bày các đánh giá về các tạp chất và các chất phân hủy hiện diện trong dược chất và thuốc thành phẩm cùng với các tác động dược lý và độc tính đã được biết hoặc có thể xảy ra của các chất này. Đánh giá này là một phần trong nội dung giải trình về giới hạn tạp chất đề nghị cho dược chất và thuốc thành phẩm và rất phù hợp để tham chiếu trong phần hồ sơ về chất lượng. Phải bàn luận về mối liên quan của bất kỳ sự khác biệt nào về tính đồng phân đối hình (chirality), cấu trúc hóa học và tạp chất giữa hợp chất dùng trong các nghiên cứu tiền lâm sàng và sản phẩm sẽ được lưu hành trên thị trường. Đối với các sản phẩm công nghệ sinh học, cần đánh giá tính tương đương của các sản phẩm dùng trong thử nghiệm tiền lâm sàng và lâm sàng với sản phẩm dự kiến đưa ra thị trường. Nếu sản phẩm thuốc có chứa một tá dược mới thì phải đánh giá các thông tin liên quan đến độ an toàn của tá dược này.
Cần phải xem xét đến các tài liệu khoa học thích hợp và các đặc tính của các sản phẩm có liên quan. Nếu sử dụng chi tiết các tài liệu tham khảo đã được đăng tải để thay thế cho các nghiên cứu do cơ sở đăng ký tiến hành, thì cần phải có phần giải trình phù hợp đánh giá về thiết kế nghiên cứu và bất kỳ sai lệch nào so với các hướng dẫn hiện hành. Bên cạnh đó, phải bàn luận về chất lượng những lô dược chất dùng trong các nghiên cứu tham khảo này.
Phần tổng quan tiền lâm sàng cần có các tham chiếu phù hợp đến các bảng tóm tắt tiền lâm sàng: trình bày theo mẫu sau (Bảng X.X, Nghiên cứu/báo cáo số).
2. NỘI DUNG VÀ CẤU TRÚC
Tổng quan tiền lâm sàng nên được trình bày theo trình tự sau:
TỔNG QUAN TIỀN LÂM SÀNG
1. Tổng quan về chiến lược nghiên cứu tiền lâm sàng
2. Dược lý
3. Dược động học
4. Độc tính
5. Tổng hợp và kết luận
6. Danh mục tài liệu tham khảo đã sử dụng
Cần đánh giá các nghiên cứu nhằm xác lập các tác dụng dược lực, cơ chế tác dụng và các tác dụng phụ có thể xảy ra và xem xét tầm quan trọng của các vấn đề phát sinh.
Khi đánh giá về dược động học, động học của độc chất, và chuyển hóa của thuốc nghiên cứu phải đề cập đến tính phù hợp của phương pháp phân tích đã sử dụng, các mô hình dược động học và các thông số đã lựa chọn. Có thể cần tham khảo chéo đến một số vấn đề nhất định trong các nghiên cứu dược lý hoặc độc tính (ví dụ như ảnh hưởng của trạng thái bệnh lý, sự thay đổi sinh lý, các kháng thể kháng lại thuốc và động học của độc chất giữa các loài). Nếu số liệu không đồng nhất thì phải có bàn luận. Cần bàn luận về những nghiên cứu so sánh về chuyển hóa giữa các loài và so sánh về nồng độ thuốc trong cơ thể trên người và động vật (AUC, Cmax và các thông số thích hợp khác) và nêu rõ những lợi ích cũng như hạn chế của các nghiên cứu tiền lâm sàng trong dự đoán các tác động bất lợi trên người.
Thời gian khởi phát độc tính, mức độ trầm trọng, thời gian thể hiện độc tính, sự lệ thuộc vào liều lượng sử dụng, mức độ hồi phục (hoặc không hồi phục) và sự khác nhau liên quan đến loài, giới phải được đánh giá và bàn luận những điểm nổi bật, đặc biệt về:
- Dược lực học
- Các dấu hiệu ngộ độc
- Nguyên nhân tử vong
- Các phát hiện về bệnh học
- Độc tính trên gen - dựa trên cấu trúc hóa học của hợp chất, cơ chế tác dụng và mối liên quan với các hợp chất đã biết là độc đối với gen.
- Khả năng gây ung thư trên cơ sở cấu trúc hóa học của hợp chất, mối liên quan của hợp chất với những chất gây ung thư đã biết, nguy cơ độc hại đối với gen và các dữ liệu về phơi nhiễm.
- Nguy cơ gây ung thư cho người - cần phải xem xét đến các dữ liệu dịch tễ học (nếu có).
- Độc tính trên khả năng sinh sản, sự phát triển của phôi thai, độc tính trước và sau khi sinh.
- Nghiên cứu trên động vật trước giai đoạn trưởng thành
- Hậu quả của việc sử dụng thuốc trước và trong khi có thai, trong thời gian cho con bú và đối với sự phát triển của trẻ nhỏ.
- Sự dung nạp tại chỗ
- Các nghiên cứu độc tính khác và/hoặc các nghiên cứu làm sáng tỏ những vấn đề đặc biệt.
Các đánh giá về độc tính phải được sắp xếp theo trình tự hợp lý sao cho những số liệu chứng minh cho một tác dụng và/hoặc hiện tượng phải được trình bày cùng nhau. Ngoại suy các kết quả thử nghiệm từ động vật lên người phải cân nhắc đến:
- Loài động vật được thử
- Số lượng động vật đã thử
- Đường dùng thuốc
- Liều dùng
- Thời gian điều trị hoặc thời gian nghiên cứu
- Nồng độ thuốc trong cơ thể ở các loài nghiên cứu độc tính ở mức liều không ghi nhận có tác dụng bất lợi và mức liều gây độc, và sự tương quan với nồng độ thuốc trong cơ thể ở người khi sử dụng liều khuyến cáo tối đa cho người. Nên sử dụng bảng biểu và hình vẽ để tóm tắt các thông tin này.
- Các tác dụng của dược chất trong các nghiên cứu tiền lâm sàng và sự tương quan đến tác dụng dự kiến hoặc quan sát được trên người.
Nếu sử dụng các thử nghiệm thay thế cho các thử nghiệm trên toàn thân động vật thì phải bàn luận về kết quả thẩm định khoa học của các phương pháp thay thế.
Phần tổng quan và kết luận nên xác định rõ đặc tính của dược phẩm dùng cho người được thể hiện qua các nghiên cứu tiền lâm sàng, và đi đến các kết luận hợp lý, có lập luận chặt chẽ chứng minh cho tính an toàn của thuốc trong các chỉ định lâm sàng dự kiến. Cần chú ý đến các kết quả về dược lý, dược động học và độc tính và bàn luận về ảnh hưởng của các phát hiện tiền lâm sàng đối với việc sử dụng an toàn thuốc cho người (như áp dụng cho việc ghi nhãn thuốc).
PHẦN C. TÓM TẮT TIỀN LÂM SÀNG DƯỚI DẠNG VĂN BẢN VÀ BẢNG BIỂU
1. HƯỚNG DẪN VỀ TÓM TẮT TIỀN LÂM SÀNG BẰNG VĂN BẢN
1.1 Mở đầu
Phần này hướng dẫn cách trình bày các tóm tắt về dược lý, dược động học và độc tính tiền lâm sàng bằng văn bản một cách phù hợp. Hướng dẫn này không nhằm chỉ ra các nghiên cứu nào cần có. Nó chỉ đơn thuần chỉ ra các trình bày phù hợp các dữ liệu tiền lâm sàng đã thu thập được.
Trình tự và nội dung của tóm tắt tiền lâm sàng bằng văn bản được nêu dưới đây. Cần lưu ý là không một hướng dẫn nào có thể bao trùm mọi tình huống, người viết cần nhận định những xu hướng chung và tập trung rõ ràng vào những yêu cầu của ban thẩm định hồ sơ cấp số đăng ký, coi đó là các chỉ dẫn rõ ràng nhất trong việc chuẩn bị hồ sơ. Vì thế, cơ sở đăng ký có thể thay đổi cách trình bày nếu cần, để có được phần trình bày thông tin tốt nhất và tạo thuận tiện cho việc đọc hiểu và thẩm định kết quả nghiên cứu.
Khi cần thiết, nên bàn luận đến những tác dụng liên quan đến tuổi và giới. Cần nêu những phát hiện liên quan đến các đồng phân lập thể và/hoặc các chất chuyển hóa, nếu thích hợp. Việc thống nhất đơn vị sử dụng trong toàn bộ các bản tóm tắt tiền lâm sàng sẽ tạo điều kiện cho thuận lợi cho người thẩm định. Bảng chuyển đổi đơn vị cũng có thể hữu ích.
Trong phần bàn luận và kết luận, các thông tin phải được phân tích xuyên suốt qua các nghiên cứu và các loài động vật sử dụng trong thử nghiệm, và việc sử dụng thuốc trên động vật thử cũng phải được liên hệ với việc sử dụng thuốc ở người sử dụng liều dùng tối đa dự kiến.
1.2 Những vấn đề chung về cách trình bày
Trình tự trình bày các thông tin trong phần này
Các nghiên cứu in vitro nên trình này trước các nghiên cứu in vivo, khi có thể. Khi tóm tắt nhiều nghiên cứu cùng loại trong phần Dược động học và Độc tính, thì các nghiên cứu phải được trình bày theo loài, đường dùng rồi sau đó là theo thời gian (những thử nghiệm trong thời gian ngắn nhất trình bày trước).
- Các loài được trình bày theo thứ tự sau:
. Chuột nhắt
. Chuột cống
. Chuột đồng
. Các động vật gặm nhấm khác
. Thỏ
. Chó
. Các loài linh trưởng không phải là người
. Các động vật có vú không thuộc loài gặm nhấm
. Các động vật không có vú.
Đường dùng được trình bày theo trình tự sau:
. Đường dùng dự kiến trên người
. Đường uống
. Đường tĩnh mạch
. Tiêm bắp
. Tiêm màng bụng
. Tiêm dưới da
. Hít
. Tại chỗ
. Đường dùng khác
Sử dụng bảng và hình vẽ
Mặc dù phần tóm tắt tiền lâm sàng này chủ yếu bằng văn viết nhưng cũng có thể sử dụng bảng và hình vẽ để trình bày hiệu quả và/hoặc rõ ràng hơn về những thông tin cần thiết.
Để cho tác giả được linh hoạt trong việc xác định cấu trúc tối ưu của phần tóm tắt bằng văn bản, tốt nhất là nên có bảng và hình vẽ. Thay vì làm như vậy, có thể tập hợp các bảng và hình vẽ ở cuối mỗi phần tóm tắt tiền lâm sàng bằng văn bản.
Trong suốt báo cáo, những trích dẫn tham khảo đến các Tóm tắt bằng bảng biểu cũng phải trình bày theo mẫu: (Bảng X.X, số nghiên cứu/báo cáo).
Độ dài của tóm tắt tiền lâm sàng bằng văn bản
Mặc dù không có một giới hạn chính thức nào đối với độ dài của tóm tắt tiền lâm sàng bằng văn bản, khuyến cáo tổng cả ba Tóm tắt tiền lâm sàng bằng văn bản không quá 100-150 trang.
Trình tự của tóm tắt tiền lâm sàng bằng văn bản và bảng biểu
Khuyến cáo nên trình bày theo thứ tự sau:
- Mở đầu
- Tóm tắt về dược lý học bằng văn bản
- Tóm tắt về dược lý học bằng bảng biểu
- Tóm tắt về dược động học bằng văn bản
- Tóm tắt về dược động học bằng bảng biểu
- Tóm tắt về độc tính bằng văn bản
- Tóm tắt về độc tính bằng bảng biểu
2. NỘI DUNG CÁC TÓM TẮT TIỀN LÂM SÀNG BẰNG VĂN BẢNG VÀ BẢNG BIỂU
Mở đầu
Mục đích của phần này là để giới thiệu cho cán bộ thẩm định về dược phẩm và mục đích sử dụng lâm sàng của nó. Phần này cần có những điểm chính sau:
- Thông tin tóm tắt về cấu trúc hóa học của dược chất (tốt nhất nên có cấu trúc hóa học dưới dạng biểu đồ), và đặc tính dược lý.
- Các thông tin liên quan đến chỉ định lâm sàng, liều dùng và thời gian điều trị đề nghị.
2.1. DƯỢC LÝ
2.1.1. TÓM TẮT BẰNG VĂN BẢN
Trong tóm tắt về Dược lý bằng văn bản, các dữ liệu cần trình bày theo trình tự sau:
- Tóm tắt nội dung
- Dược lực học tổng quan (Primary Pharmacodynamics)
- Dược lực học trên các cơ quan (Secondary Pharmacodynamics)
- Dược lý về tính an toàn
- Các tương tác thuốc về dược lực học
- Bàn luận và kết luận
- Bảng và hình (ở cuối phần này hoặc lồng vào trong phần văn bản)
Tóm tắt nội dung
Những phát hiện chính từ các nghiên cứu dược lý nên được tóm tắt ngắn gọn trong khoảng 2-3 trang, bắt đầu bằng một mô tả ngắn gọn nội dung toàn bộ dữ liệu dược lý, chỉ ra những mặt đáng chú ý ví dụ như việc chấp nhận hoặc loại bỏ các dữ liệu cụ thể nào đó (thí dụ thiếu mô hình thử nghiệm trên động vật).
2.1.1.1. Dược lực học tổng quan
Cần tóm tắt và đánh giá các nghiên cứu về dược lực cơ bản. Nếu có thể, nên tìm một mối liên quan giữa dược lý của chế phẩm thử với những dữ liệu đã có (như tính chọn lọc, độ an toàn, hoạt lực) của những thuốc khác trong cùng nhóm.
2.1.1.2. Dược lực học trên các cơ quan
Trong phần này, cần tóm tắt các nghiên cứu dược lực học theo hệ cơ quan nếu thích hợp, và đánh giá kết quả.
2.1.1.3. Dược lý về tính an toàn
Phần này cần tóm tắt và đánh giá các nghiên cứu dược lý liên quan đến tính an toàn của thuốc. Trong một số trường hợp; nghiên cứu dược lý trên các cơ quan cũng có thể đóng góp vào việc đánh giá độ an toàn khi sử dụng để dự đoán hoặc đánh giá các tác dụng bất lợi có thể xảy ra ở người. Trong những trường hợp đó, cần xem xét các nghiên cứu dược lực học trên hệ cơ quan cùng với các nghiên cứu dược lý về tính an toàn.
2.1.1.4. Các tương tác thuốc về dược lực học
Nếu có tiến hành các nghiên cứu về tương tác dược lực học, thì cần tóm tắt ngắn gọn các nghiên cứu đó trong phần này.
Bàn luận và kết luận
Phần này bàn luận những đánh giá dược lý và xem xét ý nghĩa của những vấn đề nảy sinh.
Bảng và hình
Bảng và hình có thể đưa vào văn bản ở những vị trí phù hợp. Hoặc có thể trình bày các bảng và hình ở cuối bản tóm tắt.
2.1.2. TÓM TẮT VỀ DƯỢC LÝ BẰNG BẢNG BIỂU (XEM PHỤ LỤC A)
2.2. DƯỢC ĐỘNG HỌC
2.2.1 TÓM TẮT BẰNG VĂN BẢN
Trình tự của Tóm tắt dược động học bằng văn bản được trình bày như sau:
- Phần tóm lược
- Phương pháp phân tích
- Hấp thu
- Phân phối
- Chuyển hóa
- Thải trừ
- Các tương tác thuốc về dược động học
- Các nghiên cứu dược động học khác
- Bàn luận và kết luận
- Bảng và hình (ở đây hoặc ở trong phần văn bản)
Phần tóm lược
Những kết quả chính từ các nghiên cứu dược động học nên trình bày tóm tắt ngắn gọn trong khoảng 2-3 trang. Phần này nên bắt đầu bằng việc miêu tả mục tiêu đánh giá dược động học, có nhấn mạnh ở các nội dung như: những loài, giống nghiên cứu có phải là loài, giống dùng trong nghiên cứu dược lý và độc tính hay không và công thức bào chế dùng trong các nghiên cứu có tương tự hoặc giống nhau không.
Phương pháp phân tích
Phần này cần nêu tóm tắt ngắn gọn các phương pháp phân tích các mẫu sinh học, kể cả các giới hạn về phát hiện và định lượng của các phương pháp phân tích. Nếu có thể, phần này nên bàn luận về các dữ liệu thẩm định phương pháp phân tích và độ ổn định của mẫu sinh học. Khả năng ảnh hưởng của các phương pháp phân tích khác nhau đối với việc diễn giải kết quả nên được bàn luận ở những phần thích hợp tiếp theo.
2.2.1.1 Hấp thu
Cần tóm tắt những dữ liệu sau ở phần này:
- Sự hấp thu (mức độ và tốc độ hấp thu qua các nghiên cứu in vivo và in situ)
- Các thông số động học, tương đương sinh học và/hoặc độ khả dụng sinh học (các nghiên cứu về dược động học trong huyết thanh/ huyết tương/ máu)
2.2.1.2 Phân phối
Cần tóm tắt những dữ liệu sau ở phần này:
- Nghiên cứu sự phân phối vào mô
- Sự gắn kết với protein và phân phối vào trong tế bào máu.
- Sự chuyển thuốc qua nhau thai
2.2.1.3 Chuyển hóa (so sánh giữa các loài)
Cần tóm tắt những dữ liệu sau ở phần này :
- Cấu trúc hóa học và lượng chất chuyển hóa trong mẫu sinh học
- Các con đường chuyển hóa có thể xảy ra
- Chuyển hóa trước khi vào vòng tuần hoàn chung (tác dụng chuyển hóa lần đầu trong hệ tiêu hóa/ ở gan).
- Chuyển hóa in vitro, kể cả các nghiên cứu trên Cytochrom P450
- Cảm ứng và ức chế men
2.2.1.4 Thải trừ
Cần tóm tắt những dữ liệu sau ở phần này :
- Đường và mức độ thải trừ
- Bài tiết qua sữa
2.2.1.5. Tương tác thuốc về dược động học
Nếu đã thực hiện các nghiên cứu tiền lâm sàng về tương tác dược động học (in vitro và/hoặc in vivo), thì cần tóm tắt ngắn gọn trong phần này.
2.2.1.6. Các nghiên cứu dược động học khác
Các nghiên cứu đã được thực hiện trên mô hình bệnh tật ở động vật (ví dụ các động vật bị tổn thương thận) phải được tóm tắt trong phần này.
Bàn luận và kết luận
Phần này bàn luận về các đánh giá dược động học và xem xét ý nghĩa của các vấn đề nảy sinh.
Các bảng biểu và hình vẽ
Các bảng biểu và hình vẽ có thể được đưa vào trong văn bản xếp ở vị trí thích hợp trong toàn bộ phần tóm tắt hoặc có thể trình bày ở phần cuối của văn bản.
2.2.2. TÓM TẮT VỀ DƯỢC ĐỘNG HỌC BẰNG BẢNG BIỂU (XEM PHỤ LỤC A)
2.3. ĐỘC TÍNH
2.3.1. TÓM TẮT BẰNG VĂN BẢN
Tóm tắt về độc tính bằng văn bản nên được trình bày theo trình tự như sau:
- Tóm tắt ngắn gọn
- Độc tính liều đơn
- Độc tính liều lặp lại
- Độc tính gen
- Khả năng gây ung thư
- Độc tính trên sự sinh sản và phát triển
- Các nghiên cứu ở động vật chưa trưởng thành
- Sự dung nạp tại chỗ
- Các nghiên cứu độc tính khác
- Bàn luận và kết luận
- Các bảng biểu và hình vẽ (có thể xếp ở đây hoặc đưa vào bài tóm tắt).
Tóm tắt ngắn gọn
Các phát hiện chủ yếu qua các nghiên cứu độc tính cần được tóm tắt ngắn gọn trong vài trang (nói chung không quá 6 trang). Trong phần này, mức độ đánh giá độc tính có thể được trình bày dưới dạng bảng liệt kê các nghiên cứu độc tính chủ yếu (không trình bày các kết quả trong bảng này), ví dụ như:
Chương trình thử độc tính:
|
Dạng nghiên cứu và thời hạn |
Đường dùng thuốc |
Loài súc vật |
Hợp chất nghiên cứu* |
|
Độc tính liều duy nhất |
Uống và tiêm tĩnh mạch |
Chuột cống và chuột nhắt |
Thuốc gốc |
|
Độc tính liều duy nhất |
Uống và tiêm tĩnh mạch |
Chuột cống và chuột nhắt |
Chất chuyển hóa X |
|
Độc tính liều lặp lại 1 tháng 6 tháng 9 tháng |
Uống Uống Uống |
Chuột cống và chó Chuột cống Chó |
Thuốc gốc Thuốc gốc Thuốc gốc |
* Cột này chỉ đưa vào khi có nghiên cứu các chất chuyển hóa.
Mục tiêu đánh giá độc tính phải được mô tả kèm theo mối liên hệ với việc dự định sử dụng thuốc trong lâm sàng. Cần diễn giải về điều kiện thực hiện thực hành phòng thí nghiệm tốt (GLP) trong khi tiến hành nghiên cứu.
2.3.1.1. Độc tính liều duy nhất
Các số liệu về nghiên cứu độc tính liều duy nhất cần được tóm tắt hết sức ngắn gọn theo thứ tự loài động vật và đường dùng.
2.3.1.2. Độc tính liều lặp lại
Các nghiên cứu cần phải được tóm tắt theo loài động vật, đường dùng, thời gian dùng, nêu chi tiết ngắn gọn về phương pháp nghiên cứu và nêu bật các phát hiện quan trọng (ví dụ như: Bản chất và mức độ độc tính trên cơ quan đích; liều sử dụng và/hoặc tương quan giữa liều lượng và đáp ứng, mức liều không ghi nhận tác dụng có hại (NOAEL). Các nghiên cứu không then chốt có thể tóm tắt sơ lược hơn (các nghiên cứu then chốt là những nghiên cứu đã hoàn tất, tiến hành theo nguyên tắc thực hành phòng thí nghiệm tốt được chỉ rõ trong hướng dẫn ICH M3)
2.3.1.3. Độc tính gen
Các nghiên cứu cần được tóm tắt ngắn gọn theo trình tự sau:
- Nghiên cứu in vitro trên hệ thống tế bào động vật không có vú.
- Nghiên cứu in vitro trên hệ thống tế bào động vật có vú
- Nghiên cứu in vivo trên động vật có vú (kể cả nghiên cứu đánh giá động học của độc chất)
- Nghiên cứu trên các hệ thống khác.
2.3.1.4. Khả năng gây ung thư (bao gồm cả đánh giá động học của độc chất)
Cần có một lập luận ngắn gọn giải thích tại sao các nghiên cứu được lựa chọn và cơ sở cho sự lựa chọn liều cao. Các nghiên cứu riêng biệt cần được tóm tắt theo trình tự sau:
- Các nghiên cứu dài hạn (xếp thứ tự theo loài), bao gồm các nghiên cứu phát hiện phạm vi liều lượng không thích hợp để đưa vào mục độc tính liều lặp lại hoặc nghiên cứu dược động học.
- Các nghiên cứu ngắn hoặc trung hạn (bao gồm các nghiên cứu phát hiện phạm vi liều lượng không thích hợp để đưa vào mục độc tính liều lặp lại hoặc nghiên cứu dược động học).
- Các nghiên cứu khác.
2.3.1.5. Độc tính trên sự sinh sản và phát triển (bao gồm các nghiên cứu phát hiện phạm vi liều lượng và các đánh giá động học của độc chất)
Các nghiên cứu cần được tóm tắt theo trình tự sau, trình bày chi tiết ngắn gọn về phương pháp nghiên cứu và nêu bật các phát hiện quan trọng:
- Khả năng sinh sản và sự phát triển phôi giai đoạn sớm
- Sự phát triển phôi thai
- Sự phát triển trước và sau khi sinh, bao gồm cả chức năng làm mẹ
- Các nghiên cứu trên cả thế hệ con được cho dùng thuốc và/hoặc đánh giá thêm nếu như các nghiên cứu như vậy được thực hiện
Khi sử dụng các thiết kế nghiên cứu có sửa đổi thì phải thay đổi các đề mục nghiên cứu ở trên cho phù hợp.
2.3.1.6. Sự dung nạp tại chỗ
Khi đã có các nghiên cứu về sự dung nạp tại chỗ thì các nghiên cứu này phải được tóm tắt theo trình tự: loài nghiên cứu, đường dùng, thời gian nghiên cứu, tóm tắt các chi tiết của phương pháp nghiên cứu và nêu các phát hiện quan trọng.
2.3.1.7. Các nghiên cứu độc tính khác (nếu có)
Nếu đã tiến hành các nghiên cứu độc tính khác, thì cần tóm tắt trong phần này. Nếu cần thiết, cần có phần lập luận về lý do thực hiện các nghiên cứu đó.
- Tính kháng nguyên
- Độc tính trên miễn dịch
- Các nghiên cứu về cơ chế (nếu chưa được báo cáo ở chỗ khác)
- Sự lệ thuộc thuốc
- Các nghiên cứu về các chất chuyển hóa
- Các nghiên cứu về độ tinh khiết (tạp chất)
- Các nghiên cứu khác
Bàn luận và kết luận
Phần này cần bàn luận về việc đánh giá độc tính và ý nghĩa của các vấn đề nảy sinh. Nên có các bảng và hình vẽ để tóm tắt lại những thông tin đã có.
Bảng và hình vẽ
Các bảng và hình vẽ có thể đưa vào trong văn bản ở những vị trí phù hợp. Nếu không thì có thể trình bày các bảng và hình vẽ ở cuối phần tóm tắt này.
2.3.2 TÓM TẮT VỀ ĐỘC TÍNH BẰNG BẢNG BIỂU (XEM PHỤ LỤC A)
3. HƯỚNG DẪN TÓM TẮT TIỀN LÂM SÀNG BẰNG BẢNG BIỂU
Nên trình bày các bảng tóm tắt các thông tin tiền lâm sàng trong Hồ sơ tài liệu kỹ thuật chung theo mẫu đã nêu trong hướng dẫn này. Nếu cần, cơ sở đăng ký có thể thay đổi mẫu để có thể trình bày hiệu quả nhất những thông tin đã có và tạo điều kiện cho việc đọc hiểu và thẩm định kết quả.
Hướng dẫn này không nhằm chỉ ra là cần những nghiên cứu nào, mà chỉ tư vấn xem làm thế nào để lập bảng tóm tắt kết quả các nghiên cứu đã thực hiện. Cơ sở đăng ký có thể thêm vào hoặc bớt đi một vài mục trong mẫu hướng dẫn nếu thấy thích hợp. Trong một bảng có thể bao gồm các kết quả từ nhiều nghiên cứu khác nhau. Hoặc kết quả từ một nghiên cứu có thể được trích dẫn vào trong nhiều bảng.
Hình thức các bảng trình bày trong phần Tóm tắt tiền lâm sàng bằng bảng biểu có trong phụ lục A. Phụ lục A bao gồm các bảng mẫu dùng để chuẩn bị lập bảng biểu.
Những bảng mẫu này có phần chú thích (bằng chữ in nghiêng) để hướng dẫn cho người lập bảng. (Nhưng phải xoá phần hướng dẫn in nghiêng này đi khi bảng đã lập xong). Tuy nhiên, cơ sở đăng ký có trách nhiệm tự quyết định cách trình bày tốt nhất các dữ liệu của mỗi sản phẩm. Tác giả cũng phải luôn ghi nhớ là ở một nước, việc thẩm định phần Tóm tắt tiền lâm sàng bằng bảng biểu (cùng với Tóm tắt tiền lâm sàng bằng văn bản) là thẩm định chủ yếu đối với những thông tin tiền lâm sàng. Việc trình bày các dữ liệu theo mẫu và biểu mẫu sẽ đảm bảo thông tin có đủ chi tiết cần thiết cho cán bộ thẩm định và trình bày được một tổng quan chính xác những thông tin liên quan.
Nếu có tiến hành nghiên cứu trên động vật chưa trưởng thành, thì kết quả nghiên cứu phải được trình bày theo mẫu bảng biểu thích hợp cho loại nghiên cứu này.
Trình tự của phần Tóm tắt tiền lâm sàng bằng bảng biểu nên theo trình tự của phần
Tóm tắt tiền lâm sàng bằng văn bản.
PHẦN D: CÁC BÁO CÁO NGHIÊN CỨU TIỀN LÂM SÀNG
Đối với các nước thành viên ASEAN, các báo cáo nghiên cứu trong phần này có thể không quy định đối với sản phẩm có chứa dược chất mới (NCE), các sản phẩm công nghệ sinh học và các sản phẩm có thay đổi lớn, nếu như các sản phẩm gốc đã được đăng ký và được cấp phép lưu hành ở các nước tham khảo. Hướng dẫn này trình bày thể thức thống nhất về bố cục của báo cáo nghiên cứu tiền lâm sàng trong hồ sơ kỹ thuật chung của Hồ sơ đăng ký thuốc nộp cho cơ quan quản lý thuốc. Hướng dẫn này không chỉ định phải có những nghiên cứu gì, mà chỉ đơn thuần chỉ ra một mẫu thích hợp cho việc trình bày các dữ liệu tiền lâm sàng thu thập được.
Vị trí phù hợp để trình bày các dữ liệu riêng cho từng động vật là ở trong báo cáo nghiên cứu hoặc ở phần phụ lục của báo cáo nghiên cứu.
1. MỤC LỤC
Mục lục tài liệu cần liệt kê tất cả các báo cáo nghiên cứu tiền lâm sàng và vị trí của mỗi báo cáo nghiên cứu trong phần hồ sơ kỹ thuật chung.
2. DƯỢC LÝ HỌC
2.1 Báo cáo nghiên cứu bằng văn bản
Các báo cáo nghiên cứu sẽ trình bày theo thứ tự sau:
2.1.1 Dược lực học tổng quan (Primary pharmacodynamics)
2.1.2 Dược lực học trên hệ cơ quan (Secondary pharmacodynamics)
2.1.3 Dược lý về tính an toàn
2.1.4 Các tương tác thuốc về dược lực học
3. DƯỢC ĐỘNG HỌC
3.1 Các báo cáo nghiên cứu bằng văn bản
Các báo cáo nghiên cứu sẽ phải trình bày theo thứ tự sau:
3.1.1 Các phương pháp phân tích và các báo cáo thẩm định (nếu có báo cáo riêng)
3.1.2 Sự hấp thu
3.1.3 Sự phân phối
3.1.4 Sự chuyển hóa
3.1.5 Sự thải trừ
3.1.6 Các tương tác thuốc về dược động học (tiền lâm sàng)
3.1.7 Các nghiên cứu dược động học khác
4. ĐỘC TÍNH
4.1. Các báo cáo nghiên cứu
Các báo cáo nghiên cứu cần phải trình bày theo thứ tự sau:
4.1.1. Độc tính của liều đơn (xếp thứ tự theo loài và đường dùng).
4.1.2. Độc tính của liều lặp lại (thứ tự theo loài, đường dùng, thời gian nghiên cứu trong đó bao gồm cả các đánh giá về động học của độc chất).
4.1.3. Độc tính gen
4.1.3.1. In vitro
4.1.3.2. In vivo (bao gồm các đánh giá về động học của độc chất)
4.1.4. Khả năng gây ung thư (kể cả các đánh giá về động học của độc chất)
4.1.4.1. Các nghiên cứu dài hạn (xếp theo thứ tự loài động vật, kể cả các nghiên cứu xác định phạm vi liều lượng không thích hợp để đưa vào mục độc tính liều lặp lại hoặc dược động học).
4.1.4.2. Các nghiên cứu ngắn và trung hạn (kể cả các nghiên cứu xác định phạm vi liều lượng không thích hợp để đưa vào mụcđộc tính liều lặp lại hoặc dược động học).
4.1.4.3. Các nghiên cứu khác
4.1.5. Độc tính trên sự sinh sản và phát triển (kể cả các nghiên cứu xác định phạm vi liều lượng và các đánh giá bổ trợ vê động học của độc chất). (Khi sử dụng các thiết kế nghiên cứu có thay đổi thì các đề mục sau đây cũng phải thay đổi phừ hợp theo)
4.1.5.1. Khả năng sinh sản và sự phát triển phôi giai đoạn sớm
4.1.5.2. Sự phát triển của phôi thai
4.1.5.3. Sự phát triển trước và sau sinh, bao gồm cả chức năng làm mẹ
4.1.5.4. Nghiên cứu độc tính trên 2 thế hệ (thế hệ con- động vật chưa trưởng thành được tiếp tục cho sử dụng thuốc và/hoặc đánh giá sâu thêm)
4.1.6. Sự dung nạp tại chỗ
4.1.7. Các nghiên cứu độc tính khác (nếu có)
4.1.7.1. Tính kháng nguyên
4.1.7.2. Độc tính trên hệ miễn dịch
4.1.7.3. Các nghiên cứu về cơ chế (nếu chưa đưa vào mục khác)
4.1.7.4. Sự lệ thuộc thuốc
4.1.7.5. Các chất chuyển hóa
4.1.7.6. Độ tinh khiết (tạp chất)
4.1.7.7. Các vấn đề khác
PHẦN E: DANH MỤC CÁC TÀI LIỆU THAM KHẢO CHÍNH
PHỤ LỤC A: MẪU TÓM TẮT TIỀN LÂM SÀNG BẰNG BẢNG BIỂU
2.1.2 Dược lý học
2.1.2.1. Dược lý học: Tổng quan
2.1.2.2. Dược lực học tổng quan (Primary Pharmacodynamics)*
2.1.2.3. Dược lực học trên hệ cơ quan (Secondary Pharmacodynamics)*
2.1.2.4. Dược lực học về tính an toàn
2.1.2.5. Tương tác thuốc về dược lực học*
2.2.2. Dược động học
2.2.2.1. Dược động học: Tổng quan
2.2.2.2. Các phương pháp phân tích và các báo cáo thẩm định*
2.2.2.3. Dược động học: Sự hấp thu sau khi dùng liều duy nhất
2.2.2.4. Dược động học: Sự hấp thu sau khi dùng liều lặp lại
2.2.2.5. Dược động học: Sự phân phối thuốc đến các cơ quan
2.2.2.6. Dược động học: Sự gắn kết với protein huyết tương
2.2.2.7. Dược động học: Nghiên cứu trên động vật mang thai hoặc cho con bú
2.2.2.8. Dược động học: Các nghiên cứu khác về sự phân phối thuốc
2.2.2.9. Dược động học: Sự chuyển hóa in vivo
2.2.2.10. Dược động học: Sự chuyển hóa in vitro
2.2.2.11. Dược động học: Các đường chuyển hóa
2.2.2.12. Dược động học: Sự cảm ứng/ức chế men chuyển hóa thuốc
2.2.2.13. Dược động học: Sự thải trừ
2.2.2.14. Dược động học: Sự thải trừ vào mật
2.2.2.15. Dược động học: Các tương tác thuốc – thuốc
2.2.2.16. Dược động học: Các nghiên cứu khác
2.3.2. Độc tính
2.3.2.1. Độc tính: Tổng quan
2.3.2.2. Động học của độc chất: Tổng quan về các nghiên cứu động học của độc chất
2.3.2.3. Động học của độc chất: Tổng quan các dữ liệu về động học của độc chất
2.3.2.4. Độc tính: Dược chất
2.3.2.5. Độc tính liều duy nhất
2.3.2.6. Độc tính của liều lặp lại: các nghiên cứu bổ trợ
2.3.2.7. Độc tính của liều lặp lại: các nghiên cứu cơ bản
2.3.2.8. Độc tính gen: in vitro
2.3.2.9. Độc tính gen: in vivo
2.3.2.10. Khả năng gây ung thư
2.3.2.11. Độc tính trên sự sinh sản và phát triển: Các nghiên cứu bổ trợ
2.3.2.12. Độc tính trên sự sinh sản và phát triển: Khả năng sinh sản, sự phát triển phôi sớm và sự làm tổ (các nghiên cứu cơ bản)
2.3.2.13. Độc tính trên sự sinh sản và phát triển: Các tác động trên sự phát triển phôi thai (các nghiên cứu cơ bản)
2.3.2.14. Độc tính trên sự sinh sản và phát triển: Các tác dụng trên sự phát triển trước và sau khi sinh, kể cả chức năng làm mẹ (các nghiên cứu cơ bản)
2.3.2.15. Sự dung nạp
2.3.2.16. Các nghiên cứu độc tính khác
*: Tóm tắt bằng bảng biểu là không bắt buộc. Tốt nhất là lồng ghép thêm các bảng và hình vẽ vào trong phần tóm tắt tiền lâm sàng bằng văn bản.
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.1.2. Dược lý học |
Tổng quan |
Chất thử (1) |
|||
|
Loại nghiên cứu |
Hệ thống thử nghiệm |
Cách dùng |
Phương tiện thử nghiệm |
Số nghiên cứu (4) |
Vị trí trong hồ sơ Tập Trang (3) |
|
Dược lực học tổng quan (2) Dược lực học trên hệ cơ quan Dược lý học về tính an toàn Các tương tác thuốc về dược lực học |
|
|
|
|
|
Ghi chú: (1) Tên chung quốc tế (INN: International Nonproprietary Name).
(2) Mỗi báo cáo dược lý viết 1 dòng, theo thứ tự như trong hồ sơ kỹ thuật chung. Các báo cáo có tuân thủ GLP cần được ghi chú ở cuối trang.
(3) Nêu vị trí của báo cáo kỹ thuật trong hồ sơ kỹ thuật chung.
(4) Hoặc số của báo cáo (trên tất cả các bảng).
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||||
|
2.1.2.4. Dược lý học về tính an toàn (1) |
Chất thử (2) |
||||||
|
Hệ thống cơ quan được đánh giá |
Loài/chủng súc vật |
Cách dùng |
Liều lượnga (mg/kg) |
Giới tính và số lượng súc vật mỗi nhóm |
Các phát hiện đáng lưu ý |
Việc tuân thủ thực hành thí nghiệm tốt (GLP) |
Nghiên cứu số (3) |
Ghi chú: (1) Phải tóm tắt tất cả nghiên cứu dược lý về an toàn.
(2) Tên chung quốc tế (INN: International Nonproprietary Name).
(3) Hoặc số báo cáo (trên tất cả các bảng).
a- liều đơn trừ khi có chỉ định đặc biệt khác.
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.2.2. Dược động học |
Tổng quan |
Chất thử (1) |
|||
|
Loại nghiên cứu |
Hệ thống thử nghiệm |
Cách dùng |
Phương tiện thử nghiệm |
Nghiên cứu số |
Vị trí trong hồ sơ (3) Tập Trang |
|
Sự hấp thu (2) Sự phân phối Sự chuyển hóa Sự thải trừ Các tương tác dược động học Các nghiên cứu khác |
|
|
|
|
|
Ghi chú: (1) Tên chung quốc tế (INN: International Nonproprietary Name).
(2) Mỗi báo cáo dược động học trình bày 1 dòng theo thứ tự như trong hồ sơ kỹ thuật chung. Các báo cáo tuân thủ thực hành thí nghiệm tốt (GLP) phải được ghi chú ở cuối trang.
(3) Chỉ rõ vị trí của báo cáo kỹ thuật trong hồ sơ kỹ thuật chung.
|
Hồ sơ kỹ thuật chung ASEAN – Tính an toàn |
|||||
|
2.2.2.3. Dược động học: Sự hấp thu sau khi dùng liều duy nhất |
Chất thử (1) |
||||
|
|
|
|
|
Vị trí trong hồ sơ: Tập Trang Nghiên cứu số: |
|
|
|
________________ |
________________ |
________________ |
________________ |
|
|
Loài Giống (đực/cái)/ số lượng súc vật Tình trạng dinh dưỡng Tá dược dẫn/ công thức Cách dùng Liều lượng (mg/kg) Mẫu thử (như máu toàn phần, huyết tương, huyết thanh) Chất phân tích Kết quả định lượng (2) Các thông số dược động học |
|
(4) |
|
|
|
|
Các thông tin bổ sung (3) |
|
|
|
|
|
Ghi chú (1) Tên chung quốc tế (INN: International Nonproprietary Name).
(2) Ví dụ: HPLC, LSC với chất đánh dấu bằng C14
(3) Tóm tắt kết quả, sự khác nhau giữa các loài, giống/giới, sự phụ thuộc liều lượng hoặc các nhận xét đặc biệt khác.
(4) Nên trình bày mỗi nghiên cứu một cột. Để so sánh, cần chỉ rõ thông tin tiêu biểu trên người khi dùng liều tối đa khuyến cáo.
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|
|
2.2.2.4. Dược động học: Sự hấp thu sau khi dùng liều lặp lại |
Chất thử: |
(Các dữ liệu có thể tập hợp thành bảng như trong mục 2.3)
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||||||||
|
Mẫu A |
|
|
|
Chất thử: |
|
||||||
|
2.2.2.5. Dược động học: Sự phân phối trong các cơ quan |
|
Vị trí trong hồ sơ: Tập Trang Số nghiên cứu: |
|||||||||
|
Loài Giống (đực/cái)/số lượng súc vật Tình trạng dinh dưỡng Tá dược dẫn/ công thức Cách dùng Liều lượng (mg/kg) Nuclide phóng xạ Tác động đặc hiệu Thời gian thu thập mẫu |
|
|
|
|
|||||||
|
|
|
Nồng độ (đơn vị) |
|
|
|
||||||
|
Mô/Cơ quan |
|
|
T(1) |
T(2) |
T(3) |
T(4) |
T(5) |
T(1/2) |
|||
|
Các thông tin bổ sung: |
|
|
_______ |
_______ |
_________ |
________ |
_________ |
__________ |
|||
|
1[Mô] /[Huyết tương] |
|
|
|
|
|
|
|
|
|||
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||||||||||
|
Mẫu B |
|
|
|
Chất thử: Vị trí trong hồ sơ: Tập Trang Số nghiên cứu |
|||||||||
|
2.6.5.5. Dược động học: Sự phân phối trong các cơ quan |
|
||||||||||||
|
Loài Giống (đực/cái)/số lượng súc vật Tình trạng dinh dưỡng Tá dược dẫn/ công thức Cách dùng Liều lượng (mg/kg) Nuclide phóng xạ Tác động đặc hiệu Chất phân tích/định lượng (đơn vị) Thời gian thu thập mẫu |
|
|
|
|
|||||||||
|
Mô/Cơ quan |
|
C1 |
|
|
Thời gian cuối cùng |
||||||||
|
|
|
Nồng độ |
T/P1 |
|
Nồng độ |
T/P1 |
Thời gian |
AUC |
t1/2 |
||||
|
Các thông tin bổ sung: |
|
|
|
|
|
|
|
|
|||||
|
1[Mô] /[Huyết tương] |
|
|
|
|
|
|
|
|
|||||
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.2.2.6. Dược động học: Sự gắn kết với protein huyết tương |
|
|
|
||
|
|
|
|
|
Tên chất thử: |
|
|
Hệ thống thử nghiệm: |
|
|
|
|
|
|
Mô đích, hệ thống và phương pháp thử nghiệm |
|
|
|
|
|
|
Loài súc vật |
Nồng độ chất thử |
% gắn kết |
Số nghiên cứu |
Vị trí trong hồ sơ: Tập Trang |
|
|
|
|
|
|
|
|
|
Các thông tin bổ sung: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.2.2.7. Dược động học: Nghiên cứu ở súc vật có thai hoặc đang cho con bú (1) |
Tên chất thử: (2) |
||||
|
|
|
|
|
Vị trí trong hồ sơ: Tập Trang Số nghiên cứu: |
|
|
Sự vận chuyển qua nhau thai Loài Thai kỳ/số lượng súc vật Tá dược dẫn/ công thức Cách dùng Liều lượng (mg/kg) Chất phân tích Định lượng Thời gian (giờ) Nồng độ/lượng (% liều lượng) Mẹ (3) Bào thai (3) |
|
|
|
|
|
|
Các thông tin bổ sung: |
|
|
|
|
|
|
|
|
|
Vị trí hồ sơ: Tập Trang |
|
|
|
Sự bài tiết qua sữa Loài Ngày cho con bú/số lượng súc vật Tình trạng dinh dưỡng Tá dược dẫn/công thức Cách dùng Liều lượng (mg/kg) Chất phân tích Định lượng: Thời gian (giờ) Nồng độ: Sữa Huyết tương Sữa / Huyết tương Động vật mới sinh |
|
|
|
|
|
|
Các thông tin bổ sung: |
|
|
|
|
|
Ghi chú: cho bảng 2.6.5.7
(1) Ngay cả các dữ liệu thu thập được từ các nghiên cứu độc tính trên sự sinh sản cũng phải được trình bày trong bảng này.
(2) Tên chung quốc tế (INN).
(3) Cần mô tả các mẫu mô (huyết tương từ mẹ, bào thai, nồng độ trong bào thai).
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|
|
2.2.2.8. Dược động học: Nghiên cứu khác về sự phân phối |
Tên chất thử |
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
||||||||||
|
2.2.2.9. Dược động học: sự chuyển hóa in vivo |
Tên chất thử |
|||||||||
|
Giống (đực/cái)/ số lượng súc vật Tình trạng dinh dưỡng Tá dược dẫn/ công thức Cách dùng Liều lượng (mg/kg) Nuclide phóng xạ Tác động đặc hiệu |
|
|
|
|
||||||
|
|
% chất phân tích trong mẫu thử |
Vị trí trong hồ sơ Tâp Trang |
||||||||
|
Loài |
Mẫu thử |
Thời gian lấy mẫu |
% liều trong mẫu thử |
Chất ban đầu |
Chất chuyển hóa 1 |
Chất chuyển hóa 2 |
Số nghiên cứu |
|||
|
|
Huyết tương Nước tiểu Mật Phân Huyết tương Nước tiểu Mật Phân Huyết tương Nước tiểu Mật Phân |
|
|
|
|
|
||||
Các thông tin bổ sung:
Ghi chú: Các dữ liệu trên người cần phải đưa vào để so sánh, nếu có thể.
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.2.2.10. Dược động học: Sự chuyển hóa in vitro |
Tên chất thử |
||||
|
|
|
|
|
Vị trí trong hồ sơ: Tập Trang Số nghiên cứu: |
|
|
Hệ thống nghiên cứu |
_____________ |
________________ |
_______________ |
______________ |
_______________ |
|
Thời gian Nồng độ: Hợp chất: Chất ban đầu |
|
|
|
|
|
|
Chất chuyển hóa 1 Chất chuyển hóa 2 |
|
|
|
|
|
|
Các thông tin bổ sung: |
|
|
|
|
|
Ghi chú: Các dữ liệu trên người cần phải đưa vào để so sánh (nếu có thể)
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|
|
2.2.2.11. Dược động học: Các đường chuyển hóa có thể có |
Tên chất thử: |
(Cung cấp các sơ đồ chuyển hóa có thể xảy ra và chỉ rõ những loài có xuất hiện các phản ứng chuyển hóa)
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
|||||
|
2.2.2.12. Dược động học: sự cảm ứng/ức chế của các men chuyển hóa thuốc |
Tên chất thử: |
||||
|
|
|
|
|
Vị trí trong hồ sơ: Tập Trang Số nghiên cứu: |
|
|
Ghi chú: Chỉ các nghiên cứu tiền lâm sàng |
|
|
|
||
|
Loại nghiên cứu: Phương pháp: |
|
|
|
|
|
|
Bảng kết quả: |
|
|
|
|
|
|
Thông tin bổ sung: |
|
|
|
|
|
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.2.2.13 Dược động học: Sự thải trừ |
Tên chất thử: (1) |
||||
|
|
|
________________ |
_______________ |
_______________ |
_______________ |
|
Loài Giống (đực/cái)/số lượng súc vật Tình trạng dinh dưỡng Tá dược dẫn/ công thức Cách dùng Liều lượng (mg/kg) Chất phân tích Định lượng |
|
(3’) |
|
|
|
|
Đường thải trừ (4) |
Nước tiểu Phân Tổng số Nước tiểu Phân Tổng số Nước tiểu Phân Tổng số Nước tiểu Phân Tổng số |
||||
|
Thời gian 0-T giờ |
|
|
|
|
|
|
Nghiên cứu số Vị trí trong hồ sơ |
|
|
|
|
|
Các thông tin bổ sung: (2)
Ghi chú:
(1) Tên chung quốc tế (INN).
(2) Ví dụ như tóm tắt các kết quả bằng văn bản, sự khác nhau giữa các loài, giống, sự lệ thuộc thuốc hoặc các nhận xét đặc biệt khác.
(3) Dùng một cột riêng cho từng nghiên cứu . Để so sánh, có thể đưa vào các thông tin đại diện ở người cho liều tối đa khuyến cáo. Có thể kết hợp với bảng dữ liệu hấp thu nếu thích hợp.
(4) Có thể thêm vào các đường thải trừ khác (ví dụ như đường mật, đường hô hấp) nếu có tiến hành khảo sát.
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
||||
|
2.2.2.14. Dược động học: Sự thải trừ qua mật |
|
|
Tên chất thử: |
|
[Nếu có dữ liệu, có thể lập bảng trình bày như mục 2.6.5.13]
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.2.2.15. Dược động học: Tương tác giữa các thuốc |
Tên chất thử |
|
|
||
|
|
|
|
Vị trí trong hồ sơ: Tập Trang Số nghiên cứu: |
|
|
|
Loại nghiên cứu: Phương pháp: |
|
|
|
|
|
|
Bảng kết quả: |
|
|
|
|
|
|
Thông tin bổ sung: |
|
|
|
|
|
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.2.2.16. Dược động học: Các thông số khác |
|
|
Tên chất thử: |
|
|
|
|
|
|
|
Vị trí trong hồ sơ: Tập Trang Số nghiên cứu: |
|
|
Loại nghiên cứu: Phương pháp: |
|
|
|
|
|
|
Bảng kết quả: |
|
|
|
|
|
|
Thông tin bổ sung: |
|
|
|
|
|
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
||||||||||||
|
2.3.2. Độc tính |
|
Tổng quan |
|
Chất thử: (1) |
|
|||||||
|
Loại nghiên cứu |
Loài và chủng súc vật (2) |
Cách dùng |
Thời gian sử dụng |
Liều lượng (mg/kga) |
Tuân thủ GLP |
Phương tiện thử nghiệm |
Số nghiên cứu |
Vị trí trong hồ sơ Tập Trang (3) |
||||
|
|
|
|
|
|
|
|||||||
|
Độc tính của liều duy nhất Độc tính của liều lặp lại Độc tính trên gen Khả năng gây ung thư Độc tính trên sự sinh sản và phát triển Sự dung nạp tại chỗ Các nghiên cứu độc tính khác |
|
|
|
|
|
|||||||
Ghi chú:
(1) Tên chung quốc tế (INN).
(2) Sử dụng 1 dòng cho mỗi báo cáo về độc tính, theo thứ tự như trong hồ sơ kỹ thuật chung.
(3) Nêu vị trí của báo cáo kỹ thuật trong hồ sơ kỹ thuật chung.
a- Trừ khi được nêu rõ. Đối với độc tính liều lặp lại, phải gạch chân dưới liều cao nhất không ghi nhận tác dụng có hại (NOAEL).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.3. Động học của độc chất (toxicokinetics) |
Tổng quan các nghiên cứu về động học của độc chất |
Chất thử: (1) |
|
||||||||
|
Loại nghiên cứu |
Hệ thống thử nghiệm |
Cách dùng |
Liều lượng (mg/kg) |
Việc tuân thủ GLP |
Số thử nghiệm |
Vị trí trong hồ sơ Tập Trang |
|||||
|
(2) |
|
|
|
|
(3) |
||||||
Ghi chú:
(1) Tên chung quốc tế (INN).
(2) Nên sử dụng 1 dòng riêng cho mỗi báo cáo về động học của độc chất và trình bày theo trình tự như trong hồ sơ kỹ thuật chung (phần 3, Độc tính).
(3) Nêu vị trí của báo cáo kỹ thuật trong hồ sơ kỹ thuật chung.
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.3. Động học của độc chất: |
Tổng quan về các dữ liệu động học của độc chất (2) |
Tên chất thử: (1) |
Ghi chú: (1) Tên chung quốc tế (INN).
(2) Cần tóm tắt (1 –3 trang) các dữ liệu động học của độc chất ở trạng thái ổn định (dạng bảng hoặc hình vẽ) với hình thức trình bày sao cho có thể dễ dàng so sánh giữa các loài, kể cả với người.
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||
|
2.3.2.4. Độc tính |
Dược chất |
|
|
Tên chất thử (1) |
|
|
Số lô |
Độ tinh khiết (%) |
Các tạp chất cụ thể |
Số nghiên cứu |
Loại nghiên cứu (3) |
|
|
Tiêu chuẩn đề nghị (2) |
|
|
|
|
|
Ghi chú (1) Tên chung quốc tế (INN).
(2) Liệt kê tất cả các lô thuốc sử dụng trong các nghiên cứu độc tính theo thứ tự thời gian.
(3) Xác định các nghiên cứu độc tính sử dụng trên từng lô thuốc.
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||||||||
|
2.3.2.5. Độc tính của liều duy nhất (1) |
|
|
|
|
|||||||
|
Cách dùng |
|
|
|
|
|
||||||
|
Loài/ chủng |
Tá dược dẫn / công thức |
Liều lượng (mg/kg) |
Giống và số lượng súc vật mỗi nhóm |
Liều tối đa dung nạp quan sát được (mg/kg) |
Ước lượng Liều gây chết (mg/kg) |
Các phát hiện đáng lưu ý |
Số nghiên cứu |
||||
Ghi chú (1) Tất cả các nghiên cứu độc tính liều đơn cần phải được tóm tắt theo thứ tự như trong hồ sơ kỹ thuật chung. Cần sử dụng các chú thích để chỉ ra những đặc tính đặc biệt ví dụ như thời gian dùng thuốc bất thường, tốc độ truyền hoặc tuổi của đối tượng nghiên cứu.
(2) Tên chung quốc tế (INN).
|
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng |
|||||||||
|
2.3.2.6. Độc tính của liều lặp lại |
Các nghiên cứu không cơ bản (1) |
Tên chất thử (2) |
|||||||
|
Cách dùng |
|
|
|
|
|
|
|
||
|
Loại hoặc chủng súc vật |
Tá dược dẫn / công thức |
Thời gian dùng thuốc |
Liều lượng (mg/kg) |
Giống và số lượng súc vật mỗi nhóm |
NOAELa (mg/kg) |
Các phát hiện đáng lưu ý |
Số nghiên cứu |
||
Ghi chú
(1) Tất cả các nghiên cứu độc tính liều lặp lại (bao gồm tất cả các nghiên cứu xác định mức liều gây độc tính) trừ các nghiên cứu đạt GLP theo hướng dẫn ICH M3 Nghiên cứu an toàn tiền lâm sàng đối với việc thực hiện các thử nghiệm lâm sàng dược phẩm ở người (11/1997) nên được tóm tắt theo trình tự như trong hồ sơ kỹ thuật chung. Cần sử dụng các ghi chú để chỉ rõ các đặc tính đặc biệt như độ tuổi bất thường của đối tượng nghiên cứu.
(2) Tên chung quốc tế (INN).
a- Mức liều không ghi nhận tác dụng có hại.
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.7 (1) Độc tính liều lặp lại (2) |
Tên báo cáo: |
|
Tên chất thử: (3) |
|
|||||
|
Loài/ chủng súc vật: Tuổi bắt đầu dùng: Ngày dùng liều đầu tiên: |
|
Thời gian dùng thuốc: Khoảng cách giữa các liều dùng: Cách dùng: Tá dược dẫn/ công thức: |
Số nghiên cứu Vị trí trong hồ sơ: Tập Trang Việc tuân thủ GLP: |
||||||
|
Điểm đặc biệt: |
|
|
|
|
|
||||
|
NOAEL (mức liều không ghi nhận tác dụng có hại) |
O (chứng) |
|
|
|
|||||
|
Liều hàng ngày (mg/kg) Số lượng súc vật Động học của độc chất: AUC ()(4) Các phát hiện đáng lưu ý Chết hoặc hấp hối Trọng lượng cơ thể (%) Sự tiêu thụ thức ăn (%) Sự tiêu thụ thức nước (%) Các quan sát lâm sàng Soi đáy mắt Điện tâm đồ |
Đực (5)
(5) (5) |
Cái |
Đực |
Cái |
Đực |
Cái |
Đực |
Cái |
|
|
- Không có phát hiện đáng chú ý +Nhẹ ++Trung bình +++Đáng chú ý (6) (7) *-p<0,05 **-p<0.01 |
|||||||||
a- Khi ngưng thuốc. Đối với nhóm chứng, nêu số trung bình của nhóm. Đối với nhóm điều trị, nêu tỷ lệ % khác biệt so với nhóm chứng. ý nghĩa thống kê dựa trên số liệu thực (không dựa trên tỷ lệ % khác biệt).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.7 (1) Độc tính liều lặp lại |
|
Số nghiên cứu (tiếp) |
|
|
|||||
|
Liều hàng ngày (mg/kg) Số lượng súc vật |
|
O (chứng) |
|
|
|
||||
|
|
Đực |
Cái |
Đực |
Cái |
Đực |
Cái |
Đực |
Cái |
|
|
|
|
|
|
|
|
||||
|
Chỉ số huyết học Chỉ số sinh hóa huyết thanh Phân tích nước tiểu Trọng lượng các cơ quana (%) Bệnh học đại thể Mô bệnh học Các kiểm tra bổ sung Đánh giá sau khi dùng thuốc: Số lượng được đánh giá (8) (9) - Các phát hiện đáng chú ý |
|
|
|
|
|||||
(7) *-p<0,05 **-p<0,01
a- Cả trọng lượng tuyệt đối và tương đối khác biệt so với nhóm chứng theo hướng dẫn. Số lượng cho thấy sự khác biệt % đối với trọng lượng tuyệt đối của cơ quan.
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
Chú thích cho Bảng 2.6.7.7.
(1) Các bảng cần được đánh thứ tự liên tiếp nhau (ví dụ:2.6.7.7A, 2.6.7.7.B, 2.6.7.7C).
(2) Cần lập 1 bảng cho mỗi nghiên cứu độc tính liều lặp lại như hướng dẫn ICH: M3 Nghiên cứu An toàn tiền lâm sàng để thực hiện các nghiên cứu lâm sàng trên người đối với dược phẩm (11/1997), cũng như bất kỳ nghiên cứu độc tính nào khác có thể được xem là cơ bản.
(3) Tên chung quốc tế (INN).
(4) Các chỉ số AUC, Cmax, Css ở trạng thái ổn định hoặc các thông tin về động học của độc chất khác hỗ trợ cho nghiên cứu. Nếu là các thông số từ một nghiên cứu riêng biệt, thì cần nêu số nghiên cứu ở phần chú thích.
(5) Chỉ trình này những phát hiện đáng chú ý. Nếu những chỉ số bổ sung (ngoài các chỉ số nêu trong mẫu) cho thấy những thay đổi đáng chú ý, thì cần đưa thêm vào trong bảng. Nói chung, có thể nêu các số liệu thu được khi kết thúc liều dùng; tuy nhiên, nếu có những phát hiện đáng chú ý ở những thời điểm sớm hơn, thì cũng phải đưa vào. Khi cần nên sử dụng các chú thích để cung cấp những thông tin bổ sung về thử nghiệm hoặc kết quả.
(6) Hoặc cách đánh giá khác nếu thích hợp.
(7) Cần chỉ ra phương pháp phân tích thống kê.
(8) Cần liệt kê tất cả các thông số cho thấy những thay đổi liên quan đến thuốc. Phần này nên xoá đi nếu nghiên cứu không bao gồm việc đánh giá sau khi dùng thuốc.
(9) Nếu thích hợp, cần trình bày riêng biệt các thông tin về những súc vật được giải phẫu tử thi sớm.
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.8. (1) Độc tính trên gen: in vitro |
Tên báo cáo: |
Tên chất thử (2) |
|||
|
Thử nghiệm cảm ứng đối với: Chủng: Hệ thống chuyển hóa: Tá dược dẫn: Cho chất thử: Điều trị: Tác dụng độc trên tế bào: Tác dụng độc trên gen: |
Số các định lượng độc lập Số môi trường nuôi cấy tái tạo Số tế bào được phân tích/môi trường Cho chất đối chứng có hoạt tính: |
Số nghiên cứu: Vị trí trong hồ sơ: Trang Tập Việc tuân thủ GLP: Ngày điều trị: |
|||
|
Kích hoạt chuyển hóa |
Chất thử nghiệm |
Nồng độ hoặc mức độ liều lượng (3) _________ ___________ __________ __________ __________ |
|||
|
Không kích hoạt Kích hoạt |
|
(4) |
|
|
|
Ghi chú: (1) Các bảng cần phải đánh số thứ tự liên tục (ví dụ như 2.6.7.8A, 2.6.7.8B). Kết quả định lượng tái tạo phải được trình bày ở các trang tiếp theo.
(2) Tên chung quốc tế (INN).
(3) Phải có đơn vị đo lường.
(4) Nếu quan sát thấy sự kết tủa, cần nêu trong phần chú thích. (5) Nêu phương pháp phân tích thống kê.
(5)*-p<0,05 **-p<0,01
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.9. (1) Độc tính trên gen: in vivo |
Tên báo cáo: |
|
Tên chất thử (2) |
|
|
|
Thử nghiệm gây cảm ứng trên: Loài/Chủng: Tuổi: Các tế bào được đánh giá: Số lượng tế bào được phân tích/ súc vật: Các điểm đặc biệt: Độc tính hoặc tác dụng độc với tế bào: Các tác dụng độc trên gen: Các biểu hiện phơi nhiễm: |
Quy trình điều trị: Thời gian lấy mẫu: Cách dùng: Tá dược dẫn/công thức |
|
Số nghiên cứu Vị trí trong hồ sơ: Trang Tập Sự tuân thủ GLP: Ngày sử dụng thuốc: |
||
|
Chất thử nghiệm |
Liều dùng (mg/kg) |
Số các súc vật |
_______ _______ _______ _______ ________ |
||
|
|
|
|
|
|
|
Ghi chú: (1) Các bảng cần phải đánh số thứ tự liên tục (ví dụ như 2.6.7.9A, 2.6.7.9B).
(2) Tên chung quốc tế (INN).
(3) Nêu phương pháp phân tích thống kê.
(3)*-p<0,05 **-p<0,01
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.10. (1) Khả năng gây ung thư |
Tên báo cáo: |
|
Tên chất thử: (3) |
|
|||||
|
Loài/ chủng súc vật: Tuổi bắt đầu dùng: Ngày dùng liều đầu tiên: |
|
Thời gian dùng thuốc: Khoảng cách giữa các liều dùng: Cách dùng: Tá dược dẫn/ công thức: |
Số nghiên cứu Vị trí trong hồ sơ: Tập Trang Việc tuân thủ GLP: |
||||||
|
Cơ sở để lựa chọn liều cao: (3) Điểm đặc biệt: |
|
|
|
|
|||||
|
Liều hàng ngày (mg/kg) Giống súc vật Động học của độc chất: AUC()(4) Số lượng súc vật: Khi bắt đầu Chết/hấp hối Số lượng chết khi kết thúc: Tỷ lệ sống còn Trọng lượng cơ thể (%a) Sự tiêu thụ thức ăn (%a) Sự tiêu thụ nước (%a) |
0 (chứng) |
|
|
|
|||||
|
Đực
(5)
(5) (5) |
Cái |
Đực |
Cái |
Đực |
Cái |
Đực |
Cái |
||
(6): *-p<0,05 **-p<0,01
a- Tại thời điểm 6 tháng. Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm điều trị, nêu tỷ lệ % khác biệt so với nhóm chứng. ý nghĩa thống kê dựa trên số liệu thực (không dựa trên tỷ lệ % khác biệt).
(còn tiếp)
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.10. (1) Khả năng gây ung thư |
|
Số nghiên cứu (tiếp) |
|
|
||||
|
Liều hàng ngày (mg/kg) Số lượng súc vật được đánh giá: Số lượng súc vật có sang thương tân sinh: (7) Những phát hiện đáng chú ý: Bệnh lý học đại thể Mô bệnh học – không phải sang thương tân sinh |
(chứng) |
0 (chứng) |
|
|
||||
|
Đực |
Cái |
Đực |
Cái |
Đực |
Cái |
Đực |
Cái |
|
*-p<0,05 **-p<0,01
a- Tại thời điểm 6 tháng. Đối với nhóm chứng, nêu trị số trung bình. Đối với nhóm điều trị, nêu tỷ lệ % khác biệt so với nhóm chứng. ý nghĩa thống kê dựa trên số liệu thực (không dựa trên tỷ lệ % khác biệt).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
Chú thích Bảng 2.6.7.10
(1) Bảng phải được đánh số thứ tự liên tục (ví dụ 2.6.7.10A, 2.6.7.10B). Lập một bảng cho mỗi nghiên cứu về khả năng gây ung thư.
(2) Tên chung quốc tế (INN).
(3) Lấy từ Hướng dẫn ICH: Lựa chọn Liều SIC cho các nghiên cứu về khả năng gây ung thư của dược phẩm (3/1995).
(4) Các chỉ số AUC, Cmax, Css ở trạng thái ổn định, hoặc các thông tin động học của độc chất khác hỗ trợ cho nghiên cứu. Nếu thông tin được lấy từ một nghiên cứu riêng biệt, thì cần nêu số báo cáo ở phần chú thích.
(5) Nếu những chỉ số bổ sung cho thấy những thay đổi đáng chú ý liên quan đến thuốc, thì cần đưa thêm chúng vào trong bảng. Khi cần nên sử dụng các chú thích để cung cấp những thông tin bổ sung về thử nghiệm hoặc kết quả.
(6) Cần chỉ ra phương pháp phân tích thống kê.
(7) Cần liệt kê các tổn thương liên quan đến thuốc trước. Sau đó những thương tổn khác cần được liệt kê theo cơ quan/ mô xếp theo thứ tự bảng chữ cái.
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.11. Độc tính trên sự sinh sản và phát triển |
Các nghiên cứu không cơ bản (1) |
Chất thử nghiệm (2) |
|||||||
|
Cách dùng |
|
|
|
|
|
|
|
||
|
Loài/chủng |
Tá dược dẫn / công thức |
Thời gian dùng thuốc |
Liều lượng (mg/kg) |
Số lượng súc vật ở mỗi nhóm |
Các phát hiện đáng lưu ý |
Số nghiên cứu |
|||
Ghi chú:
(1) Tóm tắt tất cả các nghiên cứu về độc tính trên sự sinh sản (bao gồm tất cả các nghiên cứu phát hiện khoảng liều lượng thích hợp) ngoại trừ các nghiên cứu thực hiện theo GLP theo M3 Các nghiên cứu an toàn tiền lâm sàng để tiến hành Thử nghiệm lâm sàng trên người đối với dược phẩm, 11/1997. Tuy nhiên, các nghiên cứu điều tra cần được tóm tắt dưới hình thức chi tiết hơn.
(2) Tên chung quốc tế (INN).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.12. (1) Độc tính trên sự sinh sản và phát triển- |
Tên báo cáo: |
Tên chất thử (2) |
|
Khả năng sinh sản và phát triển phôi giai đoạn sớm cho tới sự làm tổ (3) Thiết kế nghiên cứu: Loài/chủng: Ngày giao phối:(8)(giống cái) Tuổi bắt đầu được nghiên cứu: Ngày dùng liều đầu tiên: Các đặc điểm đặc biệt: Mức liều không ghi nhận các tác dụng có hại: Số lượng con đực ở F0 Số lượng con cái ở F0 Lứa F1 Liều hàng ngày (mg/kg) Con đực Động học của độc chất: AUC()(4) Số lượng được đánh giá Số lượng chết hoặc hấp hối Các quan sát lâm sàng Các quan sát qua giải phẫu tử thi Trọng lượng cơ thể (%a) Sự tiêu thụ thức ăn (%a) Số ngày trung bình trước khi giao phối Số con đực được giao phối Số con đực có khả năng sinh sản |
Thời gian dùng thuốc: Con đực Vị trí trong hồ sơ: Tập Trang Ngày vào phần C: Cách dùng: Tá dược dẫn/công thức: |
Số nghiên cứu: Việc tuân thủ GLP: 0(nhóm chứng) |
|
|
|
|
- Không có phát hiện đáng chú ý + Nhẹ ++Trung bình +++ Đáng chú ý
(7)* -p<0,05 **-p<0,01 (6)
a- 4 tuần sau khi bắt đầu liều dùng. Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm điều trị, nêu % khác biệt so với nhóm chứng. ý nghĩa thống kê dựa trên số liệu thực (chứ không trên % khác biệt). (còn tiếp)
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.13. (1) Độc tính trên sự sinh sản và phát triển |
|
Nghiên cứu số: (tiếp theo) |
|
|
Liều hàng ngày (mg/kg) |
0 (nhóm chứng) |
|
|
|
Con cái |
Động học của độc chất: AUC() (4) Số lượng được đánh giá Số lượng chết hoặc hấp hối Các quan sát lâm sàng Các quan sát qua giải phẫu tử thi Trọng lượng cơ thể trước khi giao phối (%a) Trọng lượng cơ thể thời kỳ mang thai (%a) Sự tiêu thụ thức ăn thời kỳ mang thai (%a) Số chu kỳ động đực trung bình /14 ngày Số ngày trung bình trước khi giao phối Số lượng con cái có xét nghiệm tinh trùng dương tính Số lượng con cái mang thai Số lượng sẩy thai hoặc sự tiêu thai toàn bộ lứa Số lượng thể vàng (hoàng thể) trung bình Số lượng phôi làm tổ trung bình Tỷ lệ % trung bình mất trước khi làm tổ Số lượng thai sống trung bình Số lượng tiêu thai trung bình Số lượng thai chết Tỷ lệ trung bình mất sau khi làm tổ |
|
|
- Không có phát hiện đáng chú ý + Nhẹ ++Trung bình +++Đáng chú ý (6)
(7) *-p<0,05 **-p<0,01
a- Vào cuối thời kỳ trước khi giao phối hoặc mang thai. Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm được điều trị, nêu % khác biệt so với nhóm chứng. Đánh giá ý nghĩa thống kê dựa trên số liệu thực (không dựa trên % khác biệt).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
Chú thích cho các bảng 2.6.7.1 2, 2.6.7.1 3, và 2.6.7.14
(1) Nếu có nhiều nghiên cứu cùng loại này, các bảng phải được đánh số thứ tự liên tiếp (ví dụ: 2.6.7.12A, 2.6.7.12B, 2.6.7.13A, 2.6.7.13B).
(2) Tên chung quốc tế (INN).
(3) Nếu sử dụng thiết kế nghiên cứu có sửa đổi, thì bảng cũng phải sửa đổi theo.
(4) Các chỉ số AUC, Cmax ở trạng thái ổn định, hoặc các thông tin về động học của độc chất khác hỗ trợ cho nghiên cứu. Nếu thông tin đó được lấy từ một nghiên cứu riêng biệt, cần nêu số nghiên cứu ở phần chú thích
(5) MẪU NÀY NÊU NHỮNG CÁCH TRÌNH BÀY KẾT QUẢ CÓ THỂ CÓ. VIỆC TRÌNH BÀY CÁC DỮ LIỆU CẦN PHẢI LINH HOẠT VÀ PHÙ HỢP VƠI PHÉP PHÂN TÍCH THỐNG KÊ VÀ THIẾT KẾ NGHIÊN CỨU TỐI ƯU. Nếu còn có các chỉ số khác cho thấy những thay đổi liên quan đến thuốc, thì các chỉ số này phải được trình bày trong bảng. Nên sử dụng các chú thích khi cần để cung cấp thêm thông tin về các thử nghiệm hoặc kết quả.
(6) Hoặc có thể dùng các phép đo lường khác.
(7) Cần nêu phương pháp phân tích thống kê.
(8) Cần nêu ngày giao phối (ví dụ Ngày 0 hoặc Ngày 1).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.13. (1) Độc tính trên sự sinh sản và phát triển Tác động trên sự phát triển bào thai (3) |
Tên báo cáo: |
Tên chất thử: (2) |
|
|
Thiết kế nghiên cứu: Loài/chủng: Tuổi bắt đầu được nghiên cứu: Ngày dùng liều đầu tiên: Mức không ghi nhận tác dụng có hại (NOAEL): Con cái F0 Lứa F1 |
Thời gian dùng thuốc: Ngày giao phối: (8) Ngày vào phần C Cách dùng Tá dược dẫn/công thức |
Số nghiên cứu: Vị trí trong hồ sơ: Tập Trang Sự tuân thủ GLP |
|
|
Liều hàng ngày (mg/kg) |
0 (nhóm chứng) |
|
|
|
Động vật mẹ |
Động học của độc chất: AUC()(4) Số lượng có thai Số lượng chết hoặc hấp hối Số lượng sẩy thai hoặc tiêu thai cả lứa Các quan sát lâm sàng Các quan sát qua giải phẫu tử thi Trọng lượng cơ thể (%a) Sự tiêu thụ thức ăn (%a) Số lượng hoàng thể trung bình Số lượng làm tổ trung bình Tỷ lệ % mất trước làm tổ |
(5) |
|
- Không có phát hiện đáng chú ý + Nhẹ ++Trung bình +++Đáng chú ý (6) G (Gestation day)= Thai kỳ
(7) *-p<0,05 **-p<0,01
a- Vào cuối thời kỳ dùng thuốc. Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm được điều trị, nêu % khác biệt so với nhóm chứng. Đánh giá ý nghĩa thống kê dựa trên số liệu thực (không dựa trên % khác biệt). (còn tiếp)
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.13. (1) Độc tính trên sự sinh sản và phát triển |
|
Số nghiên cứu: (tiếp theo) |
|
|
Liều hàng ngày (mg/kg) |
0 (nhóm chứng) |
|
|
|
Các lứa đẻ |
Số lứa đẻ được đánh giá Số bào thai sống sót Trung bình số lứa bị tiêu thai Số lượng lứa đẻ có thai chết Tỷ lệ % mất sau khi làm tổ Trọng lượng bào thai trung bình (g) Tỷ lệ giới tính của bào thai Các bất thường của bào thai Bất thường bên ngoài đại thể Bất thường về nội tạng Bất thường về xương Tổng số bào thai (lứa đẻ) bị ảnh hưởng |
|
|
- Không có phát hiện đáng chú ý
*-p<0,05 **-p<0,01
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.14. (1) Độc tính trên sự sinh sản và phát triển |
Tên báo cáo: |
Tên chất thử (2) |
|
|
Các tác dụng trên sự phát triển trước và sau sinh, bao gồm cả chức năng làm mẹ (3) |
|||
|
Thiết kế nghiên cứu: Loài/ chủng: Tuổi lúc bắt đầu nghiên cứu: Ngày dùng thuốc đầu tiên: |
Thời gian dùng thuốc: Ngày giao phối: (8) Cách dùng: Tá dược dẫn/công thức: Số lứa đẻ bị loại/ không bị loại: |
Số nghiên cứu: Vị trí trong hồ sơ: Tập Trang Việc tuân thủ GLP: |
|
|
Mức liều không ghi nhận tác dụng có hại (NOAEL): |
|
|
|
|
Con cái thế hệ F0: Con đực thế hệ F1: Con cái thế hệ F1: Liều hàng ngày (mg/kg) |
|
0 (nhóm chứng) |
|
|
Con cái thế hệ F0: |
Động học của độc chất: AUC() (4) Số lượng có thai Số lượng chết hoặc hấp hối Số lượng bị sẩy thai hoặc tiêu thai toàn bộ Các quan sát lâm sàng Các quan sát qua giải phẫu tử thi Trọng lượng cơ thể kỳ mang thai (%a) Trọng lượng cơ thể kỳ cho con bú (%a) Sự tiêu thụ thức ăn kỳ mang thai (%a) Sự tiêu thụ thức ăn kỳ cho con bú (%a) Thời gian mang thai trung bình (ngày) Sự sinh đẻ bất thường |
|
|
- Không có phát hiện đáng chú ý + Nhẹ ++Trung bình +++Đáng kể (6)
(7) *-p<0,05 **-p<0,01
a- Vào cuối thời kỳ mang thai hoặc cho con bú. Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm được điều trị, nêu % khác biệt so với nhóm chứng. Đánh giá ý nghĩa thống kê dựa trên số liệu thực (không dựa trên % khác biệt). (còn tiếp)
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.14. (1) Độc tính trên sự sinh sản và phát triển |
|
Số nghiên cứu (tiếp theo) |
|
|
Liều hàng ngày (mg/kg) |
|
0 (nhóm chứng) |
|
|
Lứa đẻ thế hệ F1 (trước cai sữa) |
Số lứa đẻ được đánh giá Số làm tổ trung bình Số lượng con trung bình/lứa đẻ Số lượng trung bình con sống lúc sinh/lứa đẻ Số lượng lứa đẻ có con chết Số con sống sót sau sinh đến ngày thứ 4 Số con sống sót sau sinh đến khi cai sữa Số lứa đẻ mất hoàn toàn Sự thay đổi trọng lượng con a (g) Tỷ lệ giới tính trong số các con Các dấu hiệu lâm sàng của các con Các quan sát qua giải phẫu tử thi các con |
|
|
|
Con đực thế hệ F1 (sau cai sữa) |
Số súc vật được đánh giá sau cai sữa ở mỗi lứa đẻ Số súc vật chết hoặc hấp hối Các quan sát lâm sàng Các quan sát qua giải phẫu tử thi Sự thay đổi trọng lượng cơ thể b (g) Sự tiêu thụ thức ăn (%c) Sự tách bao quy đầu Chức năng cảm giác Hoạt động vận động Học hỏi và trí nhớ Số ngày trung bình trước giao phối Số con đực giao phối Số con đực có khả năng sinh sản |
|
|
- Không có phát hiện đáng chú ý + Nhẹ ++Trung bình +++Đáng chú ý (6)
(7) *-p<0,05 **-p<0,01
a- Từ lúc sinh đến lúc cai sữa
b- Từ cai sữa đến khi giao phối
c- ở cuối thời kỳ cai sữa: Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm được điều trị, nêu % khác biệt so với nhóm chứng. Đánh giá ý nghĩa thống kê dựa trên số liệu thực (không dựa trên % khác biệt). (còn tiếp)
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.14. (1) Độc tính trên sự sinh sản và phát triển |
|
Số nghiên cứu (tiếp theo) |
|
|
Liều hàng ngày (mg/kg) |
|
0 (nhóm chứng) |
|
|
Con cái thế hệ F1 (sau cai sữa) |
Số con cái được đánh giá sau cai sữa Số lượng chết hoặc hấp hối Các quan sát lâm sàng Các quan sát qua giải phẫu tử thi Sự thay đổi trọng lượng cơ thể trước khi giao phối a (g) Sự thay đổi trọng lượng cơ thể kỳ mang thai (g) Sự tiêu thụ thức ăn trước khi giao phối (%b) Sự tiêu thụ thức ăn kỳ mang thai (%b) Tuổi trung bình có dấu hiệu mở âm đạo (ngày) Chức năng cảm giác Hoạt động vận động Học hỏi và trí nhớ Số ngày trung bình trước khi giao phối Số con cái có xét nghiệm tinh trùng dương tính Số lượng con cái có thai Số lượng hoàng thể trung bình Số lượng làm tổ trung bình Tỷ lệ phần trăm trung bình mất trước khi làm tổ |
|
|
|
Lứa đẻ thế hệ F2 |
Số lượng thai sống trung bình trên mỗi lứa đẻ Số lứa đẻ bị tiêu thai trung bình Số lứa đẻ có thai chết Số thai chết Tỷ lệ phần trăm trung bình mất sau khi làm tổ Trọng lượng bào thai (g) Tỷ lệ giới tính thai (% giống đực) Bào thai bất thường |
|
|
- Không có phát hiện đáng chú ý + Nhẹ ++Trung bình +++Đáng chú ý (6)
(7) *-p<0,05 **-p<0,01
a- Từ cai sữa đến khi giao phối
b- ở cuối thời kỳ giao phối hoặc mang thai. Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm được điều trị, nêu % khác biệt so với nhóm chứng. Đánh giá ý nghĩa thống kê dựa trên số liệu thực (không dựa trên % khác biệt).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.14. (1) Độc tính trên sự sinh sản và phát triển |
|
Số nghiên cứu (tiếp theo) |
||
|
Liều hàng ngày (mg/kg) |
|
0 (nhóm chứng) |
||
|
Con cái thế hệ F1 (sau cai sữa) |
Số con cái được đánh giá sau cai sữa Số súc vật chết hoặc hấp hối Các quan sát lâm sàng Các quan sát qua giải phẫu tử thi Sự thay đổi trọng lượng cơ thể trước khi giao phối (g) Sự thay đổi trọng lượng cơ thể kỳ mang thai (g) Sự tiêu thụ thức ăn trước khi giao phối (%b) Sự tiêu thụ thức ăn kỳ mang thai (%b) Tuổi trung bình có dấu hiệu mở âm đạo (ngày) |
|
||
|
Chức năng cảm giác Hoạt động vận động Học hỏi và trí nhớ |
Lưu ý: có mẫu riêng cho trường hợp sinh đẻ tự nhiên |
|
||
|
Số ngày trung bình trước khi giao phối Số con cái có xét nghiệm tinh trùng dương tính Số con cái có thai Thời gian trung bình của thai kỳ Sự sinh đẻ bất thường |
|
|||
|
Lứa đẻ thế hệ F2 |
Số lứa đẻ được đánh giá Số lượng làm tổ trung bình Số con trung bình/mỗi lứa đẻ Số con sống khi sinh trung bình mỗi lứa đẻ Số con chết khi sinh trung bình mỗi lứa đẻ Số con sống sót sau sinh đến ngày thứ 4 Số con sống sót sau sinh đến khi cai sữa Sự thay đổi trọng lượng của con (g) (a) Tỷ lệ giới tính trong các con Các dấu hiệu lâm sàng của các con Các quan sát qua giải phẫu tử thi của các con |
|
||
- Không có phát hiện đáng chú ý + Nhẹ ++Trung bình +++Đáng chú ý (6)
(7) *-p<0,05 **-p<0,01
a- Từ cai sữa đến khi giao phối
b- ở cuối thời kỳ giao phối hoặc mang thai. Đối với nhóm chứng, nêu trị số trung bình của nhóm. Đối với nhóm được điều trị, nêu % khác biệt so với nhóm chứng. Đánh giá ý nghĩa thống kê dựa trên số liệu thực (không dựa trên % khác biệt).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.16. Sự dung nạp tại chỗ (1) |
|
Tên chất thử (2) |
|||
|
Loại/chủng |
Cách dùng |
Liều lượng (mg/kg) |
Giới tính và Số lượng súc vật của mỗi nhóm |
Các phát hiện đáng lưu ý |
Số nghiên cứu |
Ghi chú: (1) Tất cả các nghiên cứu về sự dung nạp tại chỗ cần phải được tóm tắt.
(2) Tên chung quốc tế (INN).
Hồ sơ kỹ thuật chung ASEAN – Các dữ liệu tiền lâm sàng
|
2.3.2.17. Các nghiên cứu độc tính tại chỗ (1) |
|
Tên chất thử (2) |
||||
|
Loại/chủng |
Cách dùng |
Thời gian dùng |
Liều lượng (mg/kg) |
Giới tính và Số lượng súc vật của mỗi nhóm |
Các phát hiện đáng lưu ý |
Số nghiên cứu |
Ghi chú: (1) Tất cả các nghiên cứu về độc tính tại chỗ cần phải được tóm tắt.
(2) Tên chung quốc tế (INN).
DANH MỤC KIỂM TRA THEO PHÂN LOẠI
(Hồ sơ kỹ thuật chung về các dữ liệu nghiên cứu tiền lâm sàng trong đăng ký thuốc)
|
PHẦN III: TÀI LIỆU |
NCE |
BIOTECH |
MaV |
MiV |
|
||
|
RT |
S/P |
IND |
|||||
|
Phần A: Mục lục |
✓ |
✓ |
✓ |
✓ |
✓ |
|
|
|
Phần B: Tổng quan tiền lâm sàng |
✓ |
✓ |
|
|
|
|
|
|
1. Vấn đề chung 2. Nội dung và cấu trúc |
✓ ✓ |
✓ ✓ |
|
|
|
|
|
|
Phần C: Tóm tắt tiền lâm sàng (bằng văn bản và bảng biểu) |
✓ |
✓ |
|
|
|
|
|
|
1. Các tóm tắt tiền lâm sàng bằng văn bản |
|
|
|
|
|
|
|
|
1.1 Dược lý học 1.1.1. Dược lực học tổng quan 1.1.2. Dược lực học trên hệ cơ quan 1.1.3. Dược lý học về tính an toàn 1.1.4. Các tương tác thuốc về dược lực học |
✓ ✓ ✓ ✓ |
✓ ✓ ✓ ✓ |
|
|
|
|
|
|
1.2 Dược động học 1.2.1. Sự hấp thu 1.2.2. Sự phân phối 1.2.3. Sự chuyển hóa 1.2.4. Sự thải trừ 1.2.5. Tương tác thuốc về dược động học 1.2.6. Các nghiên cứu dược động học khác |
✓ ✓ ✓ ✓ ✓ ✓ |
v v v v
v |
v v v v |
|
|
|
|
|
1.3 Độc tính 1.3.1. Độc tính liều duy nhất 1.3.2. Độc tính liều lặp lại 1.3.3. Độc tính gen 1.3.4. Khả năng gây ung thư 1.3.5. Độc tính trên sự sinh sản và phát triển 1.3.5.1. Khả năng sinh sản và sự phát triển phôi giai đoạn đầu 1.3.5.2. Sự phát triển của phôi thai 1.3.5.3. Sự phát triển trước và sau sinh 1.3.6. Sự dung nạp tại chỗ 1.3.7. Các nghiên cứu độc tính khác nếu có |
✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ v v |
✓ ✓
t ✓ ✓ ✓ ✓ v v |
v v |
v v |
v v |
|
|
|
2. Tóm tắt tiền lâm sàng bằng bảng biểu |
✓ |
✓ |
v |
v |
v |
|
|
|
Mục D: Báo cáo nghiên cứu tiền lâm sàng (khi có yêu cầu) |
|
|
|
|
|
|
|
|
1. Mục lục |
✓ |
✓ |
|
|
|
|
|
|
2. Dược lý học 2.1. Dược lực học tổng quan 2.2. Dược lực học trên hệ cơ quan 2.3. Dược lý học về an toàn 2.4. Các tương tác thuốc về dược lực học |
✓ ✓ ✓ ✓ |
✓ ✓ ✓ ✓ |
|
|
|
|
|
|
3. Dược động học 3.1. Các phương pháp phân tích và các báo cáo thẩm định 3.2. Sự hấp thu 3.3. Sự phân bố 3.4. Sự chuyển hóa 3.5. Sự thải trừ 3.6. Tương tác thuốc về dược động học 3.7. Các nghiên cứu dược động học khác |
✓ ✓ ✓ ✓ ✓ ✓ ✓ |
v v v v v v v |
v v v v
v |
v v v v |
|
|
|
|
4. Độc tính 4.1. Độc tính liều đơn 4.2. Độc tính liều lặp lại 4.3. Độc tính gen 4.3.1. In vitro 4.3.2. In vivo 4.4. Khả năng gây ung thư 4.4.1. Nghiên cứu dài hạn 4.4.2. Các nghiên cứu ngắn hoặc trung hạn 4.4.3. Các nghiên cứu khác 4.5. Độc tính trên sự sinh sản và phát triển 4.5.1. Khả năng sinh sản và sự phát triển phôi trong giai đoạn sớm 4.5.2. Phát triển phôi - thai 4.5.3. Sự phát triển trước và sau sinh 4.5.4. Các nghiên cứu độc tính qua 2 thế hệ (thế hệ con được tiếp tục dùng thuốc và/hoặc đánh giá) 4.6. Sự dung nạp tại chỗ 4.7. Các nghiên cứu độc tính khác nếu có 4.7.1. Tính kháng nguyên 4.7.2. Độc tính miễn dịch 4.7.3. Sự lệ thuộc thuốc 4.7.4. Các chất chuyển hóa 4.7.5. Tạp chất 4.7.6. Vấn đề khác |
✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ v v |
✓ ✓
t t t t ✓ ✓ ✓ ✓ ✓ v v |
v v |
v v |
|
|
|
|
Mục E: Danh mục các tài liệu tham khảo chính |
✓ |
✓ |
v |
v |
v |
|
|
NCE - Dược chất mới
Biotech - Sản phẩm công nghệ sinh học
MaV - Thay đổi lớn (Dược phẩm có thay đổi ảnh hưởng đến một hoặc nhiều yếu tố sau: đường dùng, hàm lượng và liều dùng, các chỉ định. Yêu cầu phải nộp dữ liệu bổ sung và cần thiết phải xác lập chất lượng, tính an toàn và hiệu quả của công thức mới sau khi thay đổi).
RT - Đường dùng
S/P - Hàm lượng và liều dùng
IND - Chỉ định
MiV - Thay đổi nhỏ (Dược phẩm có thay đổi ảnh hưởng đến một hoặc nhiều yếu tố sau: đường dùng, hàm lượng và liều dùng, các chỉ định hoặc hoạt chất. Yêu cầu phải nộp dữ liệu bổ sung và cần thiết phải xác lập chất lượng, tính an toàn và hiệu quả của công thức mới sau khi thay đổi).
G - Sản phẩm generic
v - Khi thích hợp, ví dụ như thay đổi đường dùng do thay đổi công thức
t - Thường không thích hợp đối với các sản phẩm công nghệ sinh học, tuy nhiên việc đánh giá một sản phẩm đặc biệt về khả năng gây ung thư có thể cần thiết tùy thuộc thời gian dùng thuốc trên lâm sàng, dân số bệnh nhân và/hoặc hoạt tính sinh lý của sản phẩm (ví dụ như yếu tố tăng trưởng, chất ức chế miễn dịch, ...)
HỒ SƠ KỸ THUẬT CHUNG ASEAN (ACTD) VỀ ĐĂNG KÝ THUỐC SỬ DỤNG CHO NGƯỜI
PHẦN IV. HỒ SƠ LÂM SÀNG
Phần A. Mục Lục Của Hồ Sơ
Cần có bảng mục liệt kê những nội dung trình bày trong hồ sơ.
Phần B. Tổng Quan Lâm Sàng
Mở Đầu
Tổng quan lâm sàng nhằm cung cấp một phân tích về các dữ liệu lâm sàng trong "Hồ sơ kỹ thuật chung ASEAN (ACTD)". Tổng quan lâm sàng chủ yếu được các cơ quan xét duyệt xem xét để cấp phép lưu hành thuốc sử dụng trong quá trình đánh giá phần lâm sàng của một hồ sơ đăng ký lưu hành thuốc. Đây cũng là tài liệu tham khảo hữu ích về những phát hiện lâm sàng nói chung cho các nhân viên của cơ quan xét duyệt cấp phép lưu hành thuốc tham gia trong quá trình đánh giá, xem xét các nội dung khác của hồ sơ đăng ký lưu hành thuốc. Nội dung của Tổng quan lâm sàng cần trình bày các điểm mạnh và những mặt hạn chế trong chương trình phát triển sản phẩm và các kết quả nghiên cứu, phân tích được các lợi ích và nguy cơ (rủi ro) trong chỉ định điều trị đề nghị của sản phẩm và mô tả các kết quả nghiên cứu đã hỗ trợ cho các phần chủ yếu của thông tin kê đơn như thế nào.
Để đạt được các mục đích này, tổng quan lâm sàng phải bao gồm các nội dung sau:
_ Miêu tả và giải thích tổng quan về phương pháp phát triển lâm sàng sản phẩm thuốc, bao gồm cả các quyết định thiết kế nghiên cứu chủ yếu.
_ Đánh giá chất lượng thiết kế nghiên cứu và thực hiện nghiên cứu, trong đó cần có một tuyên bố tuân thủ theo Thực hành lâm sàng tốt (GCP).
_ Nêu tóm lược các phát hiện về lâm sàng đã đạt được, bao gồm cả những mặt hạn chế quan trọng (thí dụ: thiếu so sánh với hoạt chất liên quan, thiếu thông tin về thử nghiệm trên một số dân số bệnh nhân, thiếu thông tin đánh giá kết quả thử nghiệm sau cùng hợp lý, và sử dụng chế phẩm trong điều trị phối hợp).
_ Cung cấp đánh giá về hiệu quả và nguy cơ dựa trên kết luận của các nghiên cứu lâm sàng liên quan, bao gồm giải thích các phát hiện về hiệu quả và tính an toàn hỗ trợ cho mức liều đề nghị và chỉ định điều trị như thế nào, cũng như đánh giá thông tin kê đơn và các khuyến cáo giúp tối ưu hóa hiệu quả và kiểm soát nguy cơ như thế nào.
_ Trình bày các vấn đề đặc biệt về hiệu quả và tính an toàn gặp phải trong quá trình phát triển sản phẩm và nêu rõ các vấn đề đó đã được đánh giá và giải quyết như thế nào.
_ Nêu ra các vấn đề còn chưa được giải quyết, giải thích tại sao những vấn đề đó không được coi là các trở ngại trong việc xem xét, cho phép đăng ký và miêu tả kế hoạch nhằm giải quyết các vấn đề đó.
_ Giải thích cơ sở của các nội dung quan trọng hoặc khác thường của thông tin kê đơn.
Phần tổng quan lâm sàng nên được viết ngắn gọn (khoảng 30 trang). Tuy nhiên, mức độ dài ngắn còn phụ thuộc vào tính phức tạp của thuốc đăng ký. Nên sử dụng các hình ảnh và các bảng cô đọng trong phần tổng quan để nêu bật được vấn đề và giúp người đọc dễ hiểu hơn. Trong phần tổng quan lâm sàng, không nên nhắc lại toàn bộ các nội dung đã trình bày trong các phần khác của hồ sơ; nên có các tham chiếu đến các nội dung chi tiết trong phần Tóm Tắt Lâm Sàng và Báo Cáo Nghiên Cứu Lâm Sàng.
Mục lục Phần Tổng Quan Lâm Sàng
1. Cơ sở phát triển sản phẩm
2. Tổng quan về Sinh dược học
3. Tổng quan về Dược lý lâm sàng
4. Tổng quan về Hiệu quả
5. Tổng quan về Tính an toàn
6. Kết luận về Lợi ích và Nguy cơ
Nội Dung Bàn Luận Chi Tiết Của Phần Tổng Quan Lâm Sàng.
1. Cơ sở phát triển sản phẩm
Phần bàn luận về cơ sở của việc phát triển sản phẩm bao gồm các nội dung chính sau đây:
+ Xác định nhóm dược lý của sản phẩm.
+ Mô tả những bệnh lý lâm sàng/sinh lý bệnh mà sản phẩm dự kiến dùng điều trị, dự phòng hoặc chẩn đoán (các chỉ định mục tiêu).
+ Nêu tóm tắt cơ sở khoa học cho việc đánh giá sản phẩm đối với các chỉ định đã dược nghiên cứu.
+ Miêu tả tóm tắt chương trình phát triển sản phẩm về mặt lâm sàng, bao gồm những nghiên cứu lâm sàng đang tiến hành và dự kiến cũng như cơ sở của quyết định đăng ký sản phẩm tại thời điểm này.
+ Liệt kê và giải thích những điểm phù hợp hoặc chưa phù hợp với quy định của nghiên cứu chuẩn hiện hành về các mặt thiết kế nghiên cứu, cách tiến hành và phân tích kết quả. Cần tham khảo các tài liệu thích hợp đã công bố.
2. Tổng quan về Sinh dược học
Mục đích của phần này là trình bày các phân tích chủ yếu về các thông tin quan trọng có liên quan đến sinh khả dụng có thể ảnh hưởng đến hiệu quả và/hoặc tính an toàn của các dạng bào chế đăng ký lưu hành (thí dụ: dạng bào chế/ tỷ lệ hàm lượng, sự khác biệt giữa dạng bào chế đăng ký lưu hành và dạng bào chế dùng trong các thử nghiệm lâm sàng, ảnh hưởng của thức ăn đến nồng độ thuốc trong cơ thể).
3. Tổng quan về Dược lý lâm sàng
Mục đích của phần này là trình bày các phân tích chủ yếu về dược động học (PK), dược lực học (PD) và các dữ liệu in vitro có liên quan trong hồ sơ kỹ thuật chung ASEAN (ACTD). Các phân tích phải xem xét đến tất cả dữ liệu có liên quan và giải thích được tại sao và làm thế nào để các dữ liệu này có thể làm cơ sở cho các kết luận đã nêu. Đặc biệt phải nhấn mạnh vào các kết quả bất thường và những vấn đề đã biết hoặc có thể xảy ra, hay chú thích lý do trong trường hợp không có. Phần này phải tập trung vào các kết quả sau:
. Dược động học (PK), thí dụ so sánh dược động học giữa những đối tượng khỏe mạnh, bệnh nhân và các dân số đặc biệt; dược động học liên quan đến các yếu tố nội tại (như tuổi, giới tính, chủng tộc, rối loạn chức năng gan, thận) và các yếu tố ngoại lai (như hút thuốc, thuốc dùng đồng thời, chế độ ăn); tỷ lệ và mức độ hấp thu, sự phân phối bao gồm khả năng gắn kết với protein huyết tương, các con đường chuyển hóa đặc hiệu, kể cả những tác động của hiện tượng đa hình thái di truyền có thể xảy ra và sự hình thành các chất chuyển hóa có và không có hoạt tính; sự thải trừ; những biến đổi về dược động học do yếu tố thời gian; các vấn đề về hóa học lập thể; các tương tác dược động học với các thuốc khác hoặc các chất khác có ý nghĩa lâm sàng.
. Dược lực học (PD), thí dụ: thông tin về cơ chế tác dụng như sự gắn kết với các thụ thể (receptor); thời gian khởi đầu tác dụng và/hoặc chấm dứt tác dụng; mối quan hệ giữa các tác dụng có lợi và bất lợi với liều dùng hoặc nồng độ thuốc trong huyết tương (nghĩa là mối quan hệ dược động học/dược lực học); Các kết quả về dược lực học phục vụ cho việc đề nghị liều điều trị và khoảng thời gian giữa các liều; các tương tác dược lực học có ý nghĩa lâm sàng với các thuốc và hoạt chất khác, những khác biệt về gen có thể ảnh hưởng đến đáp ứng.
. Giải thích các kết quả và kết luận rút ra từ các nghiên cứu về miễn dịch di truyền, vi sinh lâm sàng hoặc các nghiên cứu dược lực học đặc hiệu khác.
4. Tổng quan về hiệu quả
Mục đích của phần này là trình bày các phân tích chủ yếu về các dữ liệu lâm sàng phù hợp với hiệu quả của sản phẩm trên dân số dự định điều trị. Phân tích phải bao quát mọi dữ liệu thích hợp, dù cho kết quả tích cực hay tiêu cực và phải giải thích các kết quả này phục vụ thế nào cho chỉ định điều trị và thông tin kê đơn. Cần nêu được các nghiên cứu thích hợp cho việc đánh giá hiệu quả và những lý do tại sao các nghiên cứu được kiểm soát tốt và rõ ràng là thích hợp lại không được coi là có liên quan. Cần lưu ý những nghiên cứu bị ngưng lại khi chưa hoàn thành và xem xét tác động của chúng.
Các nội dung sau đây cần được xem xét
. Đặc điểm liên quan của dân số bệnh nhân nghiên cứu, bao gồm cả các đặc điểm về nhân khẩu học, giai đoạn bệnh, các đồng biến số quan trọng có thể có, bất kỳ dân số bệnh nhân quan trọng nào bị loại ra khỏi các nghiên cứu chính, sự tham gia của trẻ em và người cao tuổi (ICH E11 và E7). Phải bàn luận về sự khác nhau giữa quần thể tham gia nghiên cứu và dân số là đối tượng điều trị dự kiến khi thuốc được lưu hành.
. Hàm ý của thiết kế nghiên cứu, bao gồm việc lựa chọn bệnh nhân, thời gian nghiên cứu, lựa chọn nhóm chứng và những tiêu chí nghiên cứu. Cần đặc biệt lưu ý tới những tiêu chí nghiên cứu mới mà kinh nghiệm còn hạn chế. Cần giải thích nếu có sử dụng những tiêu chí nghiên cứu thay thế. Cần bàn luận về việc thẩm định các thang điểm đánh giá.
. Với những thử nghiệm để chứng minh hiệu quả tương đương hoặc tốt hơn phải đưa ra bằng chứng về độ nhạy của thử nghiệm và lý do lựa chọn khoảng giới hạn đánh giá tương đương hoặc tốt hơn (non-inferiority margin)(ICH E10)
. Các phương pháp thống kê và bất cứ những vấn đề nào có thể ảnh hưởng đến việc nhận định kết quả (thí dụ những thay đổi quan trọng trong việc thiết kế nghiên cứu, bao gồm cả việc đánh giá tiêu chí nghiên cứu và các phân tích theo kế hoạch, vốn đã được xác định trước trong đề cương nguyên bản; lý do ủng hộ các phân tích không có trong kế hoạch; các quy trình để xử lý các dữ liệu bị thất lạc và những điều chỉnh đối với nghiên cứu có nhiều tiêu chí).
. Sự giống nhau và khác nhau về kết quả trong các nghiên cứu hoặc sự khác nhau giữa các nhóm bệnh nhân trong dân số nghiên cứu, và tác động của chúng đến việc nhận định kết quả.
. Nhận xét về mối quan hệ giữa hiệu quả, liều dùng và chế độ liều cho từng chỉ định cụ thể, trong dân số nghiên cứu chung và các nhóm bệnh nhân khác nhau (ICH4).
. Đối với sản phẩm dự định dùng cho điều trị lâu dài, cần nêu các kết quả nghiên cứu về hiệu quả phù hợp cho việc duy trì hiệu quả kéo dài và việc thiết lập liều dùng kéo dài. Cần xem xét tiến triển của hiện tượng lờn thuốc.
. Các dữ liệu gợi ý rằng kết quả điều trị sẽ được cải thiện thông qua việc theo dõi nồng độ thuốc trong huyết tương (nếu có) và các tài liệu về khoảng nồng độ thuốc tối ưu trong huyết tương.
. ý nghĩa lâm sàng của mức tác dụng quan sát được.
. Nếu có sử dụng các tiêu chí nghiên cứu thay thế, cần nêu bản chất và mức độ của các lợi ích lâm sàng dự tính và cơ sở cho những dự tính này.
. Hiệu quả điều trị ở những đối tượng đặc biệt. Nếu chưa đủ dữ liệu lâm sàng chứng minh hiệu quả đối với dân số đặc biệt, cần cung cấp các yếu tố hỗ trợ các hiệu quả ngoại suy từ hiệu quả trong dân số chung.
5. Tổng quan về tính an toàn
Mục đích của phần này là đưa ra các phân tích quan trọng, cô đọng đối với các dữ liệu về tính an toàn, nêu rõ các kết quả nghiên cứu hỗ trợ và chứng minh như thế nào cho các thông tin kê đơn dự kiến. Một phân tích quan trọng về an toàn cần cân nhắc những vấn đề sau:
. Các đặc tính về các biến cố ngoại ý của nhóm thuốc. Cần mô tả phương pháp giám sát các biến cố tương tự.
. Các phương pháp theo dõi các biến cố đặc biệt (thí dụ: đối với mắt, kéo dài khoảng QT)
. Các thông tin về độc tính trên động vật và thông tin về chất lượng sản phẩm có liên quan. Cần xem xét các phát hiện ảnh hưởng hoặc có thể ảnh hưởng đến việc đánh giá tính an toàn trong sử dụng thuốc trên lâm sàng.
. Bản chất của dân số bệnh nhân nghiên cứu và mức độ tiếp xúc với thuốc, cả thuốc nghiên cứu và thuốc đối chứng. Cần xem xét những hạn chế của cơ sở dữ liệu về độ an toàn, ví dụ như các hạn chế liên quan tới các tiêu chuẩn chọn hoặc loại trừ đối tượng nghiên cứu và nhân khẩu học đối tượng nghiên cứu; cần bàn luận rõ về mối liên quan của các hạn chế đó trong việc dự đoán độ an toàn của thuốc trên thị trường.
. Những biến cố ngoại ý phổ biến và không nghiêm trọng, có tham chiếu đến bảng trình bày các biến cố ngoại ý của thuốc nghiên cứu và thuốc đối chứng trong phần tóm tắt lâm sàng. Phần bàn luận nên ngắn gọn, tập trung vào các biến cố ngoại ý có tần suất tương đối cao, các biến cố ngoại ý xảy ra nhiều hơn so với giả dược và các biến cố ngoại ý đã biết là có xảy ra trong các nhóm chứng dùng thuốc có hoạt tính hoặc các thuốc khác thuộc cùng nhóm điều trị. Những trường hợp biến cố ngoại ý ở mức độ phổ biến hoặc phức tạp hơn, hoặc ít hơn đáng kể ở thuốc thử nghiệm so với thuốc chứng (xét về khoảng thời gian và mức độ của biến cố ghi nhận được) là nội dung được đặc biệt quan tâm.
. Các biến cố ngoại ý nghiêm trọng (nên trình bày bằng bảng và có tham chiếu chéo đến phần tóm tắt lâm sàng). Phần này nên bàn luận về số lượng tuyệt đối và tần số xuất hiện các biến cố ngoại ý nghiêm trọng, như tử vong hay các biến cố ngoại ý quan trọng khác (thí dụ các biến cố đưa đến việc thay đổi liều hay ngưng thuốc), và nên bàn luận về các kết quả ghi nhận được ở nhóm thuốc nghiên cứu so với nhóm chứng. Phải đưa ra tất cả các kết luận về mối liên quan nhân quả (hoặc không có mối liên hệ nhân quả) đến sản phẩm. Cần xem xét các kết quả cận lâm sàng phản ảnh các tác dụng nghiêm trọng đã hoặc có thể xảy ra.
. Sự giống nhau và khác nhau giữa kết quả trong các nghiên cứu và ảnh hưởng của điều này đến việc giải thích các dữ liệu về tính an toàn.
. Bất cứ sự khác biệt về tỷ lệ biến cố ngoại ý giữa các nhóm nghiên cứu: ví dụ như những khác biệt bởi các yếu tố nhân học khẩu học, trọng lượng, bệnh lý đi kèm, các thuốc dùng đồng thời hoặc các đa hình chuyển hóa.
. Mối liên quan của biến cố ngoại ý với liều dùng, chế độ điều trị, và thời gian điều trị.
. Sự an toàn khi dùng kéo dài (E1a)
. Các biện pháp để ngăn ngừa, giảm nhẹ và xử trí các biến cố ngoại ý
. Phản ứng do quá liều, khả năng lệ thuộc thuốc, phản ứng dội và lạm dụng thuốc, hoặc thiếu những dữ liệu về vấn đề này.
. Kinh nghiệm lưu hành trên thế giới. Nên đề cập ngắn gọn đến:
. Phạm vi lưu hành thuốc trên thế giới.
. Các vấn đề mới hoặc khác biệt về tính an toàn đã được xác định.
. Các biện pháp quản lý liên quan đến an toàn của sản phẩm.
6. Kết luận về lợi ích và nguy cơ
Mục tiêu của phần này là tổng hợp tất cả các kết luận trong các phần trước về sinh dược học, dược lý lâm sàng, hiệu quả và tính an toàn của sản phẩm và đưa ra đánh giá chung về lợi ích và nguy cơ trong thực hành lâm sàng. Đồng thời, những gì không đúng với các hướng dẫn pháp lý và các hạn chế quan trọng cũng cần được bàn luận ở đây.
Đánh giá này cần làm rõ các khía cạnh chủ yếu của thông tin kê đơn đề nghị. Phần này cũng phải cho nhận định rõ ràng về lợi ích và nguy cơ khi sử dụng sản phẩm này so với các biện pháp điều trị thay thế khác hoặc so với không điều trị trong các trường hợp bệnh lý chưa có thuốc điều trị được chấp thuận; và cần nêu rõ vị trí mong đợi của thuốc này trong số các trị liệu cho chỉ định đề nghị. Nếu có những nguy cơ, rủi ro cho các cá thể khác không phải là người dùng thuốc điều trị, cần thảo luận các nguy cơ đó (ví dụ như nguy cơ xuất hiện các chủng vi khuẩn kháng thuốc do việc sử dụng tràn lan các kháng sinh cho các bệnh lý nhẹ). Các phân tích đưa ra trong các phần trước không nên lặp lại ở đây. Phần này thường được viết ngắn lại khi không có các vấn đề gì đặc biệt và khi thuốc nằm trong nhóm dược lý quen thuộc.
Những phân tích về ích lợi và nguy cơ thường rất ngắn gọn nhưng cần đưa ra các kết luận quan trọng nhất và các vấn đề liên quan tới các điểm sau:
_ Hiệu quả của thuốc đối với từng chỉ định đề nghị.
_ Các phát hiện quan trọng về tính an toàn và các biện pháp có thể làm tăng độ an toàn.
_ Mối tương quan giữa liều lượng-đáp ứng và liều lượng-độc tính; phạm vi liều dùng tối ưu và chế độ liều điều trị tối ưu.
_ Hiệu quả và tính an toàn trong các nhóm dân số, ví dụ như các nhóm xếp theo tuổi, giới tính, chủng tộc, chức năng các cơ quan, mức độ trầm trọng của bệnh, và tính đa hình thái di truyền.
_ Dữ liệu ở trẻ em trong các nhóm tuổi khác nhau (nếu có) và bất cứ các kế hoạch nghiên cứu phát triển nào trên trẻ em.
_ Các nguy cơ đối với bệnh nhân đã được biết và các tương tác có thể xảy ra, bao gồm tương tác thuốc-thức ăn và tương tác thuốc-thuốc, và các khuyến cáo đối với việc sử dụng thuốc.
_ Bất cứ tác dụng nào của thuốc có thể ảnh hưởng tới khả năng lái xe hoặc vận hành máy móc nặng.
Nêu ví dụ về các vấn đề và mối quan ngại đòi hỏi phải có bàn luận chi tiết hơn về lợi ích và nguy cơ như:
_ Thuốc dùng để điều trị bệnh không gây tử vong nhưng có độc tính nghiêm trọng đã được xác định hay tiềm ẩn, ví dụ như có dấu hiệu rõ rệt về khả năng gây ung thư, gây quái thai, thúc đẩy loạn nhịp tim (ảnh hưởng lên khoảng QT), hoặc có thể gây độc cho gan.
_ Sử dụng đề nghị được căn cứ trên một tiêu chí nghiên cứu thay thế và đã có ghi nhận có độc tính quan trọng.
_ Việc sử dụng thuốc an toàn và/hoặc hiệu quả đòi hỏi phải có sự chọn lọc khắt khe hoặc kiểm soát nghiêm ngặt, đòi hỏi tính chuyên môn cao hoặc cần huấn luyện bệnh nhân một cách đặc biệt.
Phần C. Tóm Tắt Lâm Sàng
Mở Đầu
Hồ sơ phần này không yêu cầu đối với các thuốc generic, các sản phẩm có thay đổi nhỏ và một vài sản phẩm có thay đổi lớn so với sản phẩm gốc. Với các nước thuộc thành viên ASEAN, các báo cáo nghiên cứu lâm sàng ở phần này có thể không cần thiết đối với sản phẩm chứa dược chất mới (NCE), các sản phẩm công nghệ sinh học và các sản phẩm có sự thay đổi lớn khác khi các sản phẩm gốc đã dược đăng ký và đã được cấp phép lưu hành tại các nước tham chiếu. Vì thế, nếu nhà chức trách muốn xem xét các báo cáo nghiên cứu lâm sàng này thì họ sẽ đề nghị bổ sung.
Phần tóm tắt lâm sàng nhằm cung cấp bản tóm tắt những chi tiết, có căn cứ xác thực về tất cả những thông tin lâm sàng trong hồ sơ kỹ thuật chung ASEAN (ACTD). Phần này bao gồm các thông tin có trong báo cáo nghiên cứu lâm sàng; thông tin thu được từ các phân tích gộp (meta-analysis) hay bất kỳ phân tích từ các nghiên cứu chéo (cross-study analyses) mà báo cáo toàn văn của các nghiên cứu này đã được đưa vào trong phần báo cáo nghiên cứu lâm sàng và dữ liệu sau khi đưa thuốc ra thị trường ở các nước khác. Những so sánh và phân tích kết quả xuyên suốt các nghiên cứu nêu trong tài liệu này cần tập trung vào các quan sát thực. Ngược lại, tài liệu tổng quan lâm sàng của ACTD lại nên trình bày những phân tích chủ yếu về chương trình nghiên cứu lâm sàng và các kết quả của nó, bao gồm bàn luận và giải thích các kết quả lâm sàng và bàn luận về vị trí của thuốc thử nghiệm trong danh mục thuốc đã có.
Độ dài của phần tóm tắt lâm sàng dao động tuỳ thuộc nội dung cần chuyển tải nhưng thường khoảng từ 50-400 trang (không tính đến các bảng biểu đính kèm)
Nội Dung Phần Tóm Tắt Lâm Sàng
1. Tóm tắt các nghiên cứu về sinh dược học và phương pháp phân tích
1.1 Cơ sở nghiên cứu và tổng quan
1.2 Tóm tắt kết quả các nghiên cứu riêng lẻ
1.3 So sánh và phân tích các kết quả xuyên suốt các nghiên cứu
Phụ lục 1
2. Tóm tắt các nghiên cứu về dược lý lâm sàng
2.1 Cơ sở nghiên cứu và tổng quan
2.2 Tóm tắt kết quả các nghiên cứu riêng lẻ
2.3 So sánh và phân tích các kết quả xuyên suốt các nghiên cứu.
2.4 Các nghiên cứu đặc biệt Ví dụ 1: Tính sinh miễn dịch Ví dụ 2: Vi sinh học lâm sàng Phụ lục 2
3. Tóm tắt về hiệu quả lâm sàng
3.1 Cơ sở nghiên cứu và tổng quan về hiệu quả lâm sàng
3.2 Tóm tắt kết quả các nghiên cứu riêng lẻ
3.3 So sánh và phân tích các kết quả xuyên suốt các nghiên cứu
3.4 Phân tích các thông tin lâm sàng liên quan đến các khuyến cáo về liều dùng
3.5 Sự duy trì hiệu quả và/hoặc sự lờn thuốc
Phụ lục 3
4. Tóm tắt về tính an toàn lâm sàng
4.1 Mức độ sử dụng thuốc
4.2 Biến cố ngoại ý
4.3 Đánh giá kết quả xét nghiệm
4.4 Dấu hiệu sinh tồn, triệu chứng thực thể và các ghi nhận khác liên quan đến sự an toàn
4.5 Sự an toàn đối với các nhóm dân số đặc biệt và tình huống đặc biệt
4.6 Các dữ liệu sau khi đưa thuốc ra thị trường
Phụ lục 4
5. Bảng tóm tắt các nghiên cứu riêng lẻ
Hướng Dẫn Chi Tiết Các Mục Trong Phần Tóm Tắt Lâm Sàng
1. Tóm tắt các nghiên cứu về sinh dược học và phương pháp phân tích
1.1. Cơ sở nghiên cứu và tổng quan
Phần này cung cấp cho người thẩm định một cái nhìn tổng quát về quá trình phát triển dạng bào chế, hoạt lực của dạng bào chế in vitro và in vivo, phương pháp tổng quát và cơ sở lý luận dùng trong phát triển cơ sở dữ liệu về sinh khả dụng (BA), nghiên cứu so sánh sinh khả dụng và tương đương sinh học (BE) và thử độ hòa tan in vitro. Cần tham khảo các hướng dẫn hoặc tài liệu trong việc lập kế hoạch và tiến hành các nghiên cứu. Phần này cũng cần cung cấp cho người thẩm định một tổng quan về các phương pháp phân tích đã sử dụng, trong đó nhấn mạnh vào các khả năng thẩm định các nghiên cứu (như phạm vi tuyến tính, độ nhạy, tính đặc hiệu) và quản lý chất lượng (như độ đúng và độ chính xác). Mục này không nên bao gồm các thông tin chi tiết về các nghiên cứu riêng lẻ.
1.2. Tóm tắt kết quả các nghiên cứu riêng lẻ
Nhìn chung cần đưa ra một bảng liệt kê tất cả các nghiên cứu về sinh dược học (xem phụ lục 1), cùng với mô tả tường thuật các đặc điểm và kết quả có liên quan của từng nghiên cứu in vivo và in vitro liên quan đến sinh khả dụng (BA) và tương đương sinh học (BE) quan trọng khác. Phần mô tả tường thuật phải ngắn gọn, tương tự như phần tóm tắt của một bài báo đăng trên tạp chí, và nên mô tả những đặc điểm thiết kế chính và các kết quả chủ yếu. Có thể mô tả các nghiên cứu tương tự cùng với nhau, có lưu ý kết quả của từng nghiên cứu riêng lẻ và những điểm khác nhau quan trọng giữa các nghiên cứu. Phần tường thuật này có thể rút ra từ bản tóm tắt ICH E3. Phải dẫn tài liệu tham khảo hoặc địa chỉ liên kết điện tử để có thể tra cứu được báo cáo đầy đủ của mỗi nghiên cứu.
1.3. So sánh và phân tích các kết quả qua các nghiên cứu
Phần này cần trình bày một bản tóm tắt các dữ liệu thực của tất cả các nghiên cứu hòa tan in vitro, sinh khả dụng và các nghiên cứu so sánh sinh khả dụng thực hiện trên dược chất hoặc trên sản phẩm thuốc đó, đặc biệt lưu ý về sự khác nhau giữa kết quả trong các nghiên cứu. Phần tổng quan này cần tập trung tóm tắt những thông tin dưới dạng văn bản và bảng biểu (xem phụ lục 1) và cần cân nhắc những điểm sau:
. Bằng chứng về ảnh hưởng của các thay đổi trong công thức bào chế và các quá trình sản xuất lên thử nghiệm độ hòa tan in vitro, sinh khả dụng, và các kết luận liên quan tới tương đương sinh học. Khi có thay đổi về công thức bào chế và quá trình sản xuất đối với các sản phẩm chứa thành phần phức tạp (thí dụ: một protein), cần phải tiến hành các nghiên cứu về dược động học (PK) để so sánh sản phẩm trước và sau khi có thay đổi để đảm bảo rằng sự thay đổi về sản phẩm không làm thay đổi các đặc tính dược động học. Mặc dù các nghiên cứu như vậy đôi khi được coi là các nghiên cứu về tương đương sinh học (BE), song chúng thường không tập trung vào đánh giá sự giải phóng hoạt chất từ thành phẩm thuốc. Tuy nhiên, các nghiên cứu như vậy cần được đưa ra trong phần này. Cũng cần lưu ý là chỉ riêng các nghiên cứu dược động học thì có khi không đủ để chứng minh sự tương đương giữa các sản phẩm thuốc. Trong nhiều trường hợp, có thể cần thêm các nghiên cứu về dược lực học hoặc các thử nghiệm lâm sàng. Ngoài ra, tuỳ trường hợp, có thể cần các dữ liệu về tính kháng nguyên. Kết quả các nghiên cứu thêm này, khi cần thiết, phải được báo cáo trong những phần thích hợp của hồ sơ .
. Bằng chứng về mức độ ảnh hưởng của thức ăn đến sinh khả dụng và các kết luận về tương đương sinh học liên quan đến dạng thức ăn và thời gian dùng bữa ăn (nếu phù hợp).
. Bằng chứng về mối tương quan giữa độ hòa tan in vitro và sinh khả dụng, bao gồm ảnh hưởng của pH đến độ hòa tan, và các kết luận về tiêu chuẩn chất lượng, về độ hòa tan của sản phẩm.
. So sánh sinh khả dụng, gồm cả kết luận về tương đương sinh học của các dạng bào chế có hàm lượng khác nhau.
. So sánh sinh khả dụng của các dạng bào chế sử dụng trong nghiên cứu lâm sàng (đối với các nghiên cứu lâm sàng cho bằng chứng đáng kể về hiệu quả) với dạng bào chế sẽ đưa ra lưu hành.
. Nguồn gốc và mức độ của các thay đổi giữa các đối tượng nghiên cứu, và trong bản thân từng đối tượng nghiên cứu của mỗi dạng bào chế trong nghiên cứu so sánh sinh khả dụng.
Phụ lục 1
Các bảng và hình minh họa nên được đưa vào tài liệu ở vị trí thích hợp để giúp người đọc dễ hiểu. Những bảng dài phức tạp có thể để ở phần phụ lục cuối mục tóm tắt các nghiên cứu về sinh dược học.
Các bảng 1.1 và 1.2 đưa ra thí dụ về cách trình bày bảng báo cáo về các thông tin và kết quả liên quan đến các nghiên cứu đánh giá sinh khả dụng và thử độ hòa tan trong ống nghiệm (in vitro). Các ví dụ này đưa ra các kết quả cũng như xác định dạng và thiết kế nghiên cứu. Các bảng được chuẩn bị để báo cáo kết quả các nghiên cứu tương đương sinh học cũng có thể bao gồm các tỷ số trung bình (nhóm thử/nhóm chứng) của Cmax, AUC với khoảng tin cậy 90%, hoặc các khuyến cáo hiện hành áp dụng cho việc thẩm định về tương đương sinh học.
Những bảng này không phải là các mẫu cố định, chúng chỉ dùng để minh họa những thông tin mà cơ sở đăng ký nên cân nhắc khi thiết kế các bảng báo cáo nghiên cứu sinh dược học. Các cơ sở đăng ký cũng cần quyết định xem các kết quả và thông tin về những nghiên cứu này nên được trình bày ở dạng nào rõ ràng nhất: bảng biểu, văn bản hay hình ảnh. Ví dụ nếu như kết quả tốt nhất nên được trình bày ở dạng văn bản và hình, thì bảng biểu chỉ nên dùng để liệt kê các nghiên cứu.
2. Tóm Tắt Các Nghiên Cứu Về Dược Lý Lâm Sàng
2.1. Cơ sở nghiên cứu và tổng quan
Phần này nên cung cấp cho người thẩm định một tổng quan chung về các nghiên cứu dược lý lâm sàng. Các nghiên cứu này bao gồm các nghiên cứu lâm sàng được tiến hành để đánh giá dược động học và dược lực học ở người, và các nghiên cứu in vitro thực hiện trên các tế bào, mô người, hoặc các nguyên liệu từ người (từ đây gọi là nguyên liệu sinh học từ người) thích hợp với các quá trình dược động học. Đối với vaccin, phần này phải trình bày cho người thẩm định các dữ liệu về đáp ứng miễn dịch là cơ sở cho việc chọn liều, phác đồ liều và dạng bào chế của thành phẩm. Nếu thích hợp, có thể tham khảo các dữ liệu được tóm tắt trong mục 1, 3,4 của phần C để cung cấp một cách nhìn toàn diện về phương pháp và cơ sở lý luận trong việc phát triển cơ sở dữ liệu về dược động học, dược lực học, quan hệ dược động học/ dược lực học và nguyên liệu sinh học từ người. Phần này không cần nêu chi tiết thông tin của các nghiên cứu riêng lẻ. Nên bắt đầu bằng việc nêu tổng quan ngắn gọn các nghiên cứu trên nguyên liệu sinh học từ người đã được hiện với mục đích phục vụ cho việc diễn giải các dữ liệu về dược động học hoặc dược lực học. Đặc biệt cần nêu lên các nghiên cứu về tính thấm qua màng (như hấp thu thuốc qua ruột, vận chuyển thuốc qua hàng rào máu-não), sự gắn kết với protein huyết tương, sự chuyển hóa thuốc ở gan và các tương tác giữa các thuốc dựa trên chuyển hóa. Sau đó trình bày một tổng quan ngắn gọn về các nghiên cứu lâm sàng đã được tiến hành để xác định các đặc điểm về dược động học và dược lực học của thuốc, kể cả các nghiên cứu về mối quan hệ dược động học/ dược lực học trên người khỏe mạnh và trên bệnh nhân. Cần lưu ý những khía cạnh then chốt của thiết kế nghiên cứu và phân tích dữ liệu, thí dụ: việc lựa chọn liều duy nhất hay liều lặp lại, dân số nghiên cứu, sự lựa chọn tiêu chí nghiên cứu về dược lực học, phương pháp tiếp cận truyền thống hay tiếp cận dựa trên dân số trong thu thập và phân tích dữ liệu nhằm đánh giá dược động học và dược lực học.
2.2. Tóm tắt kết quả các nghiên cứu riêng lẻ
Thông thường cần đưa ra một bảng liệt kê tất cả các nghiên cứu về dược lý lâm sàng (xem phụ lục 2) cùng với mô tả tường thuật các đặc điểm và kết quả có liên quan của từng nghiên cứu then chốt cung cấp các dữ liệu in vitro và in vivo liên quan đến dược động học, dược lực học và mối liên hệ dược động học/ dược lực học. Phần mô tả tường thuật phải ngắn gọn như phần tóm tắt của các bài báo, và cần mô tả đặc điểm thiết kế nghiên cứu và các kết quả chủ yếu. Các nghiên cứu tương tự có thể trình bày cùng nhau song cần nêu các kết quả của từng nghiên cứu riêng lẻ và các điểm khác nhau quan trọng giữa các nghiên cứu. Phải dẫn đầy đủ các nguồn tài liệu tham khảo hoặc trang liên kết mạng trong phần mô tả để có thể tra cứu được báo cáo đầy đủ của mỗi nghiên cứu.
Trong phần này thường nêu tóm tắt các nghiên cứu về đáp ứng với liều dùng hay đáp ứng với nồng độ (dược động học/dược lực học) cùng với các tiêu chí nghiên cứu về dược lực học. Tuy nhiên, những nghiên cứu chặt chẽ về sự tương quan giữa đáp ứng dược lực học với liều lượng hoặc sự tương quan dược động học/ dược lực học nhằm cung cấp những bằng chứng quan trọng về hiệu quả hoặc tính an toàn thì nên được trình bày trong mục 3 hoặc 4, chứ không trình bày ở phần tóm tắt này.
2.3 So sánh và phân tích kết quả qua các nghiên cứu
Phần này sử dụng kết quả của tất cả các nghiên cứu in vitro trên nguyên liệu sinh học từ người và các nghiên cứu về dược động học, dược lực học và sự tương quan dược động học/dược lực học để nêu lên được đặc tính dược động học, dược lực học và sự tương quan dược động học/dược lực học của chế phẩm. Cần có bàn luận về những kết quả nghiên cứu liên quan đến sự thay đổi giữa các đối tượng nghiên cứu và trong từng đối tượng nghiên cứu, ảnh hưởng tới sự tương quan dược động học này.
Phần này (thường được trình bày dưới dạng văn bản và bảng biểu), cần trình bày tất cả các dữ liệu qua các nghiên cứu liên quan tới các nội dung:
. Các nghiên cứu về chuyển hóa thuốc in vitro, tương tác thuốc-thuốc in vitro và các ảnh hưởng đến lâm sàng của chúng.
. Các nghiên cứu về dược động học trên người, bao gồm những ước lượng tốt nhất về các thông số chuẩn và nguồn gốc của sự biến thiên. Cần tập trung vào các bằng chứng hỗ trợ liều dùng và việc cá nhân hóa liều dùng trong dân số bệnh nhân mục tiêu cũng như trong dân số bệnh nhân đặc biệt như: người già, trẻ em, người suy giảm chức năng gan hoặc thận.
. So sánh dược động học khi dùng liều đơn và liều lặp lại
. Phân tích trị số dược động học của dân số, ví dụ như phân tích kết quả dựa trên việc lấy mẫu rải rác qua các nghiên cứu để làm rõ sự biến thiên thay đổi giữa các cá thể về dược động học hoặc dược lực học của các hoạt chất.
. Sự tương quan giữa đáp ứng-liều lượng hoặc đáp ứng-nồng độ. Phần này cần nhấn mạnh các bằng chứng làm cơ sở cho việc lựa chọn liều và khoảng cách liều trong các thử nghiệm lâm sàng quan trọng. Ngoài ra, những thông tin làm cơ sở cho các hướng dẫn về liều sử dụng đề nghị nêu trên nhãn nên được bàn luận ở mục 3.4.
. Những điểm không nhất quán chính trong cơ sở dữ liệu về dược động học, dược lực học và nghiên cứu trên nguyên liệu sinh học từ người.
2.4. Các nghiên cứu đặc biệt
Phần này nên bao gồm các nghiên cứu cung cấp các số liệu liên quan đặc biệt đến những loại chế phẩm thuốc đặc biệt. Đối với các nghiên cứu về sự gây đáp ứng miễn dịch và các nghiên cứu khác có thể liên quan đến các dữ liệu về dược động học, dược lực học, tính an toàn và/hoặc hiệu quả của thuốc, cần đưa ra những lý giải tóm tắt về mối tương quan đó ở đây. Tất cả các tác dụng quan sát được hoặc có thể ảnh hưởng đến dược động học, dược lực học, tính an toàn và/hoặc hiệu quả cũng cần được xem xét trình bày ở những mục thích hợp khác của phần tóm tắt lâm sàng cùng với tham khảo chéo vào mục này. Không nên trình bày các nghiên cứu chuyên biệt về độ an toàn trên người ở đây, mà nên trình bày ở mục 4-tóm tắt về an toàn lâm sàng .
Thí dụ 1. Nghiên cứu về sự gây đáp ứng miễn dịch
Với các sản phẩm có bản chất là protein hoặc các chế phẩm khác có thể gây phản ứng miễn dịch đặc hiệu, các dữ liệu về sự gây đáp ứng miễn dịch cần được tóm tắt trong phần này. Với vắccin hoặc các chế phẩm tạo ra đáp ứng miễn dịch đặc hiệu, các dữ liệu về khả năng gây đáp ứng miễn miễn dịch nên được mô tả trong phần hiệu quả. Cần mô tả tóm tắt các thử nghiệm định lượng và các thông tin khi tiến hành định lượng (ví dụ độ nhạy, tính đặc hiệu, độ tin cậy, hiệu lực). Cần có chỉ dẫn tham khảo chéo về vị trí của thông tin chi tiết ở trong hồ sơ đăng ký.
Nên tóm tắt các số liệu liên quan đến tần suất xuất hiện, độ chuẩn, thời điểm khởi phát tác dụng và thời gian duy trì kháng thể cho từng loại thử nghiệm kháng thể đã sử dụng (ví dụ: IgG trong thử nghiệm ELISA, phản ứng trung hòa). Cần khảo sát và tóm tắt mối liên quan giữa sự hình thành kháng thể với bệnh đang mắc, thuốc phối hợp, liều lượng, thời gian điều trị, phác đồ điều trị và dạng bào chế . Đối với thuốc dự định dùng cho điều trị dài hạn, liên tục, tất cả các dữ liệu về ảnh hưởng của sự gián đoạn điều trị lên sự sinh kháng thể cần được phân tích và tóm tắt lại.
Điều đặc biệt quan trọng là phải tóm tắt các phân tích về khả năng gây đáp ứng miễn dịch trên lâm sàng, thí dụ xác định mức độ hiện diện một loại kháng thể liên quan tới sự thay đổi các thông số dược động học, dược lực học, sự mất hiệu quả, sự mất hoặc phát triển các biến cố ngoại ý. Cần đặc biệt lưu ý đến các hiện tượng có thể xảy ra qua trung gian miễn dịch (ví dụ: bệnh huyết thanh ) và các hiện tượng có thể là hậu quả của sự gắn kết của các chất nội sinh phản ứng chéo (bởi kháng thể) với thuốc.
Thí dụ 2. Vi sinh lâm sàng
Với các sản phẩm là thuốc kháng vi khuẩn hoặc kháng virus, các nghiên cứu in vitro về phổ tác dụng là một phần quan trọng trong chương trình nghiên cứu về hiệu quả lâm sàng. Các nghiên cứu về hiệu quả lâm sàng về độ nhạy cảm của các chủng vi khuẩn phân lập được trên lâm sàng là một phần của việc xác định hiệu quả của thuốc nên được trình bày ở mục 3- Tóm Tắt Hiệu Quả Lâm Sàng. Tuy nhiên, các nghiên cứu để đánh giá về tính nhạy cảm của một số chủng vi khuẩn in vitro ở nhiều khu vực trên thế giới (không thuộc khuôn khổ của nghiên cứu hiệu quả lâm sàng) có thể đưa vào mục này.
Phụ lục 2.
Bảng và hình minh họa có thể đưa vào tài liệu ở vị trí thích hợp nếu giúp cho người đọc dễ hiểu. Những bảng số liệu dài có thể để ở phần phụ lục cuối mục tóm tắt các nghiên cứu về dược lý lâm sàng.
Bảng 2.1. cho ví dụ về cách trình bày bảng biểu dùng để báo cáo các thông tin và kết quả liên quan đến các nghiên cứu về tương tác thuốc-thuốc về dược động học. Có thể dùng các bảng tương tự để trình bày kết quả nghiên cứu về dược động học/dược lực học, nghiên cứu về đáp ứng-liều, nghiên cứu về tác dụng trên nguyên liệu sinh học từ người, nghiên cứu về dược động học trên dân số. Các bảng này chỉ dùng để minh họa những thông tin mà các nhà tài trợ nghiên cứu cần lưu ý khi thiết kế bảng biểu của mình. Cơ sở đăng ký thuốc cũng cần quyết định xem nên trình bày các thông tin và kết quả của những nghiên cứu dược lý học lâm sàng như thế nào là tốt nhất: bảng biểu, văn bản hay hình ảnh để làm rõ nội dung. Ví dụ, nếu trình bày các kết quả bằng văn bản và hình ảnh là tốt nhất, thì chỉ dùng các bảng đơn thuần vào việc liệt kê các nghiên cứu.
Khi thiết kế các bảng cho các loại nghiên cứu dược lý học lâm sàng khác nhau được liệt kê ở dưới đây (nếu có), cơ sở đăng ký cần lưu ý nêu các thông tin sau đây. Các ví dụ này chỉ có mục đích minh họa và cơ sở đăng ký cần quyết định nên trình bày thông tin nào.
. Các nghiên cứu về chuyển hóa sử dụng nguyên liệu sinh học từ người: Các nguyên liệu sinh học đã sử dụng (thí dụ: vi lập thể (microsom), tế bào gan), các thuốc thăm dò, các kiểu chuyển hóa enzyme, tỷ lệ % đóng góp và các thông số động học liên quan (thí dụ: Vmax, Km).
. Các nghiên cứu in vitro về tương tác thuốc-thuốc sử dụng nguyên liệu sinh học từ người: đối với các nghiên cứu sự ức chế của các thuốc khác đối với thuốc mới, phải nêu được các chất chuyển hóa bị ức chế, ảnh hưởng đối với chuyển hóa qua men, giới hạn nồng độ chất ức chế sử dụng, giá trị IC50 và Ki, giả thuyết về cơ chế ức chế. Với các nghiên cứu về sự ức chế của thuốc mới đối với các thuốc khác, phải nêu được các thuốc và các chuyển hóa bị ức chế, cùng với các thông tin như nêu ở trên.
. Các nghiên cứu về dược động học trên dân số: các đồng biến được nghiên cứu, số lượng và dạng đối tượng nghiên cứu hay bệnh nhân nghiên cứu, tóm tắt các thông số thống kê và ước lượng giá trị trung bình (± độ lệch chuẩn) của các thông số dược động học.
3. Tóm Tắt Hiệu Quả Lâm Sàng
Trong trường hợp một thuốc có hiệu quả với nhiều chỉ định thì hiệu quả lâm sàng cho từng chỉ định cần được trình bày thành một phần riêng biệt (phần 3), tuy nhiên các chỉ định liên quan mật thiết đến nhau có thể trình bày cùng nhau. Khi có nhiều phần 3 như vậy, phải viết thành các phần 3A, 3B, 3C...
3.1. Cơ sở nghiên cứu và tổng quan về hiệu quả lâm sàng.
Phần này nên mô tả chương trình của các nghiên cứu có đối chứng và các nghiên cứu phù hợp khác để đánh giá các tác dụng đặc hiệu cho các chỉ định đề nghị. Tất cả các kết quả của những nghiên cứu này mà thích hợp cho việc đánh giá tính an toàn của thuốc cần được bàn luận ở mục 4: Tóm tắt về an toàn lâm sàng.
Phần này nên mở đầu với tổng quan ngắn gọn về thiết kế của các nghiên cứu có đối chứng tiến hành để đánh giá hiệu quả của thuốc. Những nghiên cứu này bao gồm đáp ứng với liều dùng, so sánh hiệu quả điều trị, các nghiên cứu hiệu quả trong điều trị kéo dài và hiệu quả trên các phân nhóm dân số. Cần bàn luận về các đặc điểm chủ yếu của thiết kế nghiên cứu: ví dụ tính ngẫu nhiên, chế độ mù, chọn điều trị đối chứng, chọn dân số bệnh nhân, các đặc điểm bất thường trong thiết kế nghiên cứu: ví dụ thiết kế chéo (crossover) hoặc thiết kế rút khỏi nghiên cứu ngẫu nhiên, thiết kế có trải qua giai đoạn dẫn (run-in), các phương pháp tăng lượng mẫu (enrichment) khác, các tiêu chí nghiên cứu, thời gian nghiên cứu và các kế hoạch phân tích kết quả dự kiến. Mặc dù phần này nhằm tập trung vào các đánh giá lâm sàng nhưng các dữ liệu tiền lâm sàng và dược lý lâm sàng cũng có thể được đề cập đến để giúp phần tóm tắt về hiệu quả lâm sàng trên người dễ hiểu hơn. Phần này không nên bao gồm các thông tin chi tiết về các nghiên cứu riêng lẻ.
3.2. Tóm tắt kết quả của các nghiên cứu riêng lẻ.
Nhìn chung trong phần này nên lập bảng liệt kê tất cả các nghiên cứu nhằm cung cấp (hoặc được thiết kế để cung cấp) các thông tin liên quan đến hiệu quả của thuốc (xem phụ lục 3) cùng với mô tả tường thuật các nghiên cứu quan trọng. Phần mô tả tường thuật nên ngắn gọn như phần tóm tắt trong các bài báo khoa học, có mô tả các đặc điểm thiết kế chủ yếu và các kết quả chính. Các nghiên cứu tương tự nhau có thể đề cập cùng nhau song cần nêu lên các kết quả của từng nghiên cứu riêng lẻ cũng như tất cả các điểm khác nhau quan trọng giữa các nghiên cứu. Đối với các nghiên cứu cũng đóng góp có ý nghĩa cho đánh giá phân tích tính an toàn, tóm tắt nghiên cứu cần bao gồm các thông tin về mức độ phơi nhiễm (tiếp xúc) của đối tượng nghiên cứu với thuốc thử nghiệm hoặc chất đối chứng, và cách thu thập số liệu về tính an toàn. Phần này có thể trích từ phần tóm tắt của các báo cáo nghiên cứu lâm sàng (ICH E3). Phải nêu đầy đủ tài liệu tham khảo hoặc kết nối điện tử để có thể tra cứu được báo cáo đầy đủ của mỗi nghiên cứu.
3.3 So sánh và phân tích các kết quả qua các nghiên cứu.
Có thể sử dụng văn bản, hình ảnh và các bảng biểu phù hợp để trình bày (xem phụ lục 3); mục 3.3. nên tóm tắt tất cả dữ liệu sẵn có đặc trưng cho hiệu quả của thuốc nghiên cứu. Phần tóm tắt này nên có sự phân tích tất cả các dữ liệu, bất kể chúng có hỗ trợ cho kết luận tổng thể hay không và do đó nên bàn luận mức độ hỗ trợ lẫn nhau giữa các kết quả của các nghiên cứu có liên quan. Nên đề cập đến những điểm không nhất quán chính trong dữ liệu về hiệu quả và chỉ ra những phần cần được nghiên cứu thêm.
Phần này thường được sử dụng 2 loại phân tích: so sánh kết quả giữa các nghiên cứu riêng lẻ và phân tích các số liệu kết hợp lại từ nhiều nghiên cứu. Những chi tiết phân tích quá rộng nên được trình bày riêng thành một báo cáo tóm tắt trong phần "Báo cáo nghiên cứu lâm sàng".
Phần này cũng nên tham chiếu chéo những bằng chứng quan trọng từ mục 2, ví dụ như các dữ liệu hỗ trợ cho phần liều lượng và cách sử dụng ghi trên nhãn thuốc. Các dữ liệu này bao gồm: liều lượng và khoảng cách liều khuyến cáo, các bằng chứng thích hợp liên quan tới việc cá nhân hóa liều dùng, và nhu cầu điều chỉnh liều dùng cho các nhóm bệnh nhân đặc biệt (như trẻ em hoặc người cao tuổi, người suy giảm chức năng gan, thận), và các dữ liệu liên quan đến mối tương quan giữa đáp ứng–liều lượng hoặc đáp ứng-nồng độ (dược động học/ dược lực học).
3.3.1 Dân số nghiên cứu
Cần mô tả các đặc điểm về nhân khẩu học và đặc điểm của các bệnh nhân khi bắt đầu tham gia nghiên cứu trong tất cả các nghiên cứu về hiệu quả. Cần nêu các nội dung sau:
● Các đặc điểm về bệnh (như mức độ trầm trọng, thời gian mắc bệnh) và các điều trị trước đó của đối tượng thử nghiệm, và tiêu chuẩn chọn/loại trừ bệnh nhân.
● Những điểm khác nhau về các đặc tính khi bắt đầu tham gia thử nghiệm của dân số nghiên cứu trong các nghiên cứu hoặc trong các nhóm nghiên cứu.
● Phải nêu rõ những điểm khác nhau giữa nhóm dân số nghiên cứu trong các phân tích quan trọng về hiệu quả với toàn bộ dân số bệnh nhân dự kiến điều trị khi chế phẩm được lưu hành.
● Đánh giá số lượng bệnh nhân rút khỏi nghiên cứu, thời gian (ngày đã tham gia nghiên cứu hoặc ngày đến khám trong đợt điều trị hoặc giai đoạn theo dõi), nguyên nhân ngưng tham gia nghiên cứu.
Có thể có ích nếu trình bày bằng bảng tổng hợp và so sánh các dân số nghiên cứu qua các nghiên cứu.
3.3.2. So sánh kết quả về hiệu quả qua các nghiên cứu.
Các kết quả từ tất cả các nghiên cứu được thiết kế để đánh giá hiệu quả của thuốc, nên được tổng hợp và so sánh, kể cả các nghiên cứu không đi đến kết luận hoặc cho kết quả âm tính. Những điểm khác nhau quan trọng trong thiết kế nghiên cứu như tiêu chí nghiên cứu, nhóm đối chứng, thời gian nghiên cứu, phương pháp thống kê, dân số bệnh nhân nghiên cứu, liều dùng phải được nêu ra.
So sánh kết quả giữa các nghiên cứu nên tập trung vào các tiêu chí nghiên cứu chủ yếu đã được xác định từ trước. Tuy nhiên, khi các tiêu chí nghiên cứu chủ yếu bao gồm nhiều biến số khác nhau hoặc được ghi nhận ở các các thời điểm khác nhau trong các nghiên cứu khác nhau về hiệu quả, thì việc so sánh chéo giữa các nghiên cứu về các yếu tố dữ liệu quan trọng thu được trong tất cả các nghiên cứu sẽ có ích. Nếu kết quả thu được theo yếu tố thời gian có tính chất quan trọng thì cần phải trình bày một dạng biểu đồ minh họa sự thay đổi theo thời gian của mỗi nghiên cứu.
Phải nêu rõ khoảng tin cậy của hiệu quả điều trị để giúp cho việc diễn giải các kết quả ước lượng. Nếu giữa thuốc nghiên cứu và giả dược (placebo) có sự tác động khác nhau trên các trị số gốc ban đầu, thì cần trình bày các trị số này và mức độ tác dụng của tất cả các nhóm điều trị, kể cả nhóm dùng placebo và nhóm chứng sử dụng chất đối chứng có hoạt tính (nếu có dùng) ở dạng bảng biểu hoặc văn bản kèm theo hình minh họa. Nếu mục tiêu của thử nghiệm có so sánh với chất đối chứng có hoạt tính nhằm để chứng minh hiệu quả tương đương hoặc tốt hơn giữa thuốc nghiên cứu và thuốc đối chứng thì sự khác nhau hoặc tỉ số của kết quả thu được giữa các nhóm điều trị cần được đưa ra kèm theo khoảng tin cậy. Các kết quả cần được đánh giá bằng cách sử dụng các tiêu chuẩn định trước để xác định sự tương đương và cần phải nêu cơ sở hợp lý của các tiêu chuẩn đó cũng như cơ sở xác định (các) nghiên cứu có đủ độ nhạy cho việc đánh giá (xem ICH E10).
Cần nêu và bàn luận về những điểm khác nhau quan trọng trong kết quả giữa các nghiên cứu có thiết kế tương tự. Phải nêu rõ các so sánh chéo giữa các nghiên cứu về những yếu tố có thể gây ra sự khác nhau trong kết quả nghiên cứu.
Nếu thực hiện một phân tích gộp (meta-analysis) của các nghiên cứu lâm sàng, cần phải nêu rõ xem phân tích này được thực hiện theo thiết kế nghiên cứu đã được định trước hay một phân tích ngẫu nhiên. Bất kỳ một sự khác nhau nào về thiết kế thử nghiệm hay dân số thử nghiệm, hoặc khác nhau về việc đo lường hiệu quả trong các thử nghiệm đều phải được trình bày để có thể đánh giá được mối liên quan và tính thích hợp và giữa kết quả và kết luận (xem ICH E9). Mô tả chi tiết về phương pháp và kết quả của phân tích gộp (meta-analysis) nói chung nên được trình bày trong một báo cáo riêng (các báo cáo nghiên cứu lâm sàng).
3.3.3 So sánh kết quả của các phân nhóm dân số nghiên cứu
Kết quả của các nghiên cứu riêng rẽ hoặc phân tích tổng quan về hiệu quả trên các nhóm dân số đặc biệt nên được nêu tóm tắt trong phần này. Mục đích của các so sánh này nhằm cho thấy các tác dụng điều trị quan sát được là chắc chắn ở tất cả các phân nhóm dân số nghiên cứu liên quan, đặc biệt các nhóm có lý do cụ thể cần quan tâm. Các so sánh này cần nêu rõ những khác biệt rõ ràng về hiệu quả đòi hỏi phải nghiên cứu và bàn luận thêm. Tuy nhiên, cần nhận ra các hạn chế của các phân tích (ICH E9) và điều quan trọng cần lưu ý là mục đích của các so sánh không nhằm đưa ra cơ sở cho các kết luận cụ thể cũng như không nhằm củng cố các bằng chứng về hiệu quả khi mà các kết quả tổng quát không tốt.
Nếu từng nghiên cứu riêng lẻ tiến hành với cỡ mẫu nhỏ, nên có phân tích gộp nhiều nghiên cứu để đánh giá được ảnh hưởng của các yếu tố nhân khẩu học chủ yếu (như tuổi, giới, chủng tộc) lên hiệu quả của thuốc. Một số yếu tố cần đặc biệt quan tâm có thể xuất phát từ những mối quan ngại chung (như người cao tuổi) hoặc từ những vấn đề liên quan đến dược lý của thuốc, hay từ giai đoạn phát triển thuốc ban đầu. Hiệu quả của thuốc trên trẻ em trong các hồ sơ đăng ký thường được phân tích theo các chỉ định đề nghị cho đối tượng này. Tuỳ theo dữ liệu, nếu rộng, thì thực hiện các phân tích chi tiết về hiệu quả và trình bày trong phần "Báo cáo nghiên cứu lâm sàng" còn các kết quả phân tích thì báo cáo trong phần này.
3.4 Phân tích các thông tin lâm sàng liên quan đến các khuyến cáo về liều
Phần này cần cung cấp một tóm tắt và phân tích tổng hợp tất cả các dữ liệu liên quan đến mối tương quan giữa đáp ứng-liều lượng hay giữa đáp ứng với nồng độ thuốc trong máu (bao gồm cả mối liên quan liều dùng- nồng độ thuốc trong máu), và do vậy góp phần vào việc chọn liều và khoảng cách giữa các liều. Có thể tham khảo các số liệu của các nghiên cứu tiền lâm sàng, và cần tóm tắt các dữ liệu liên quan từ các nghiên cứu dược động học, các nghiên cứu dược lý lâm sàng khác, và các nghiên cứu lâm sàng có đối chứng hoặc không có đối chứng để minh họa cho các mối tương quan giữa đáp ứng- liều lượng hoặc đáp ứng- nồng độ thuốc trong máu. Trong phần tóm tắt này có thể sử dụng các số liệu về dược động học và dược lực học đã tổng hợp ở mục 2.2, đồng thời có tham chiếu chéo với các tóm tắt ở mục 2.2 nhưng không trình bày lặp lại.
Phần giải thích về việc các dữ liệu này phục vụ như thế nào cho các khuyến cáo về liều dùng nên được nêu trong tài liệu tổng quan lâm sàng, nhưng trong phần này nên tóm tắt kết quả của từng nghiên cứu và các phân tích tổng hợp qua các nghiên cứu dùng để giải thích cho các khuyến cáo liều dùng (trong đó có cả liều khởi đầu và liều tối đa khuyến cáo, phương pháp chuẩn liều, và các hướng dẫn liên quan đến việc xác định liều cho từng cá thể). Cần trình bày tất cả những thay đổi ghi nhận được về sự liên quan tương đối đơn giản giữa đáp ứng- liều hoặc giữa đáp ứng- nồng độ thuốc trong máu do sự không tuyến tính về mặt dược động, do tác dụng bị làm chậm lại, do lờn thuốc, hay do cảm ứng men gây ra.
Cần mô tả bất kỳ sự khác biệt nào về sự tương quan đáp ứng-liều lượng do tuổi tác, do giới, do chủng tộc, do bệnh lý của bệnh nhân hay những yếu tố khác gây ra. Cũng cần phải bàn luận về tất cả những khác biệt về các đáp ứng dược động học và dược lực học, hoặc tham khảo chéo đến phần bàn luận ở mục 2. Cần mô tả phương pháp để tìm những sự khác biệt như vậy, dù cho không có sự khác biệt nào được tìm thấy (ví dụ các nghiên cứu đặc trưng trong các phân nhóm dân số nghiên cứu, phân tích về hiệu quả của thuốc theo phân nhóm dân số nghiên cứu, hay xác định nồng độ thuốc thử nghiệm trong máu).
3.5 Sự duy trì tác dụng và/ hoặc sự lờn thuốc
Nên tóm tắt các thông tin hiện có về sự duy trì tác dụng của thuốc theo thời gian. Phải nêu rõ số lượng bệnh nhân có dữ liệu về hiệu quả trong điều trị kéo dài và thời gian dùng thuốc. Nên trình bày tất cả các trường hợp lờn thuốc (sự mất hiệu quả điều trị theo thời gian) ghi nhận được. Việc xem xét mối liên quan rõ ràng giữa việc thay đổi liều theo thời gian và hiệu quả trong điều trị kéo dài có thể có ích.
Phần này cần tập trung chủ yếu vào các nghiên cứu có đối chứng được thiết kế đặc biệt dành cho thu thập dữ liệu về hiệu quả trong điều trị kéo dài, các nghiên cứu như vậy cần được phân biệt rõ ràng với các nghiên cứu khác không nghiêm ngặt bằng, các nghiên cứu mở rộng. Cần áp dụng sự phân biệt này đối với các nghiên cứu đặc hiệu được thiết kế để đánh giá sự lờn thuốc và phản ứng cai thuốc. Các dữ liệu về phản ứng cai thuốc hoặc phản ứng dội liên quan đến tính an toàn của thuốc nên được trình bày ở mục an toàn lâm sàng (xem mục 4).
Trong các thử nghiệm về hiệu quả trong điều trị kéo dài, cần xem xét đến các tác động khi ngưng điều trị sớm hoặc chuyển sang các trị liệu khác khi đánh giá kết quả. Các tác động này cũng quan trọng đối với các thử nghiệm ngắn hạn và nên đề cập đến khi bàn luận kết quả của các thử nghiệm này, nếu phù hợp.
Phụ lục 3
Có thể sử dụng bảng và hình ảnh trong bản trình bày ở vị trí thích hợp nếu chúng giúp người đọc dễ hiểu hơn. Những bảng dài nên đưa vào phần phụ lục mục tóm tắt về hiệu quả lâm sàng.
Các bảng cần phải nêu ra tất cả các nghiên cứu phù hợp với việc đánh giá hiệu quả (bao gồm cả các nghiên cứu đã hoàn thành và chưa hoàn thành, nghiên cứu không chứng minh được hiệu quả vì bất kỳ lý do gì, các nghiên cứu đăng trên các ấn phẩm, các nghiên cứu được trình bày dưới dạng báo cáo kỹ thuật toàn văn (ICH E3), các nghiên cứu được mô tả dưới dạng báo cáo tóm tắt); và cần nêu những kết quả quan trọng nhất của các nghiên cứu này. Tuy nhiên cần chú ý là các phân tích tạm thời không có kế hoạch trước về các nghiên cứu đang diễn ra thường không cần thiết và không được khuyến khích. Khi có nhiều phần 3 được trình bày khi xin đăng ký một thuốc với nhiều chỉ định, thì mỗi phần nên trình bày riêng rẽ và đi kèm với phụ lục và bảng riêng.
Chúng tôi cung cấp kèm theo đây các bảng biểu minh họa cho một thuốc chống tăng huyết áp, nhưng những ví dụ này không phải thích hợp cho tất cả các thuốc. Nói chung, hồ sơ đăng ký cần các bảng và/hoặc hình ảnh riêng cho nhóm thuốc đó và cho các nghiên cứu thực hiện.
Bảng 3.1. Mô tả về các nghiên cứu về hiệu quả lâm sàng và an toàn lâm sàng
Bảng 3.2. Các kết quả về nghiên cứu hiệu quả điều trị
4. Tóm tắt về an toàn lâm sàng
Phần này tóm tắt các dữ liệu liên quan đến tính an toàn của thuốc đối với dân số bệnh nhân dự kiến, tổng hợp kết quả của các báo cáo nghiên cứu lâm sàng riêng lẻ cũng như các báo cáo phù hợp khác, thí dụ các phân tích tổng hợp về an toàn được nộp thường xuyên ở một số nước.
Các số liệu liên quan đến sự an toàn được xếp theo 3 mức độ (ICH E3):
- Cần xem xét mức độ sử dụng (liều lượng, thời gian điều trị, số lượng bệnh nhân, nhóm bệnh nhân) để xác định mức độ an toàn từ cơ sở dữ liệu.
- Cần xác định và phân loại các biến cố ngoại ý thường gặp và các thay đổi chỉ số xét nghiệm, và tóm tắt các biến cố xảy ra.
- Cần xác định các biến cố ngoại ý nghiêm trọng (theo định nghĩa trong ICH E2A) và các biến cố ngoại ý đáng chú ý khác (theo định nghĩa trong ICH E3), và tóm tắt các biến cố xảy ra. Nên kiểm tra tần suất xuất hiện, đặc biệt với các thuốc có thể phải dùng mạn tính.
Hồ sơ về tính an toàn của thuốc được mô tả trên cơ sở phân tích tất cả các dữ liệu về an toàn lâm sàng và cần được trình bày chi tiết, rõ ràng, khách quan, kết hợp với việc sử dụng các bảng và hình ảnh để minh họa.
4.1 Mức độ sử dụng thuốc
4.1.1. Kế hoạch đánh giá tổng thể về tính an toàn và mô tả các nghiên cứu về tính an toàn.
Nên mô tả ngắn gọn kế hoạch đánh giá tổng thể về an toàn, bao gồm những điểm lưu ý và ghi nhận đặc biệt về các dữ liệu tiền lâm sàng, mọi tác dụng theo nhóm dược lý liên quan, nguồn gốc của các dữ liệu về an toàn (các thử nghiệm có đối chứng, các nghiên cứu mở,...). Cần đưa ra một bảng liệt kê tất cả các nghiên cứu cung cấp dữ liệu về tính an toàn, có chia nhóm một cách thích hợp (xem phụ lục 4). Bên cạnh các nghiên cứu đánh giá tính an toàn và hiệu quả và các nghiên cứu không có đối chứng để cung cấp những thông tin về an toàn, phần này còn bao gồm các nghiên cứu xem xét những vấn đề đặc biệt về tính an toàn của thuốc. Ví dụ các nghiên cứu đánh giá tỷ lệ biến cố ngoại ý chuyên biệt giữa hai trị liệu, đánh giá an toàn trên các nhóm dân số đặc biệt, đánh giá hiện tượng cai thuốc hoặc phản ứng dội, hay để đánh giá các biến cố ngoại ý đặc biệt (thí dụ: an thần, chức năng tình dục, ảnh hưởng đối với lái tàu-xe, hoặc không có tác dụng phụ nào đó như thuốc cùng nhóm). Có thể nêu các nghiên cứu đối với các chỉ định không được đăng ký ở hồ sơ này và các nghiên cứu đang tiến hành nếu chúng có cung cấp thông tin cho việc phân tích độ an toàn.
Trong phần này cũng nên trình bày các tường thuật mô tả những nghiên cứu nêu ở trên, trừ những nghiên cứu cung cấp các thông tin về cả tính an toàn và hiệu quả của thuốc nên đưa vào mục 3.2 và có tham chiếu chéo ở đây. Nên mô tả đủ chi tiết giúp cho cán bộ thẩm định hồ sơ hiểu được mức độ sử dụng của các đối tượng nghiên cứu với thuốc thử hay chất đối chứng, cách thu nhập số liệu về an toàn (bao gồm cả các phương pháp đã sử dụng và mức độ theo dõi về tính an toàn đối với các đối tượng tham gia trong các nghiên cứu riêng lẻ). Nếu một số nghiên cứu không được phân tích riêng mà nhóm lại để phân tích về tính an toàn thì cần có ghi chú rõ và đưa ra một mô tả tường thật chung.
4.1.2. Mức độ sử dụng thuốc tổng thể
Nên tóm tắt mức độ sử dụng thuốc tổng thể ở tất cả các giai đoạn thử nghiệm lâm sàng trong một bảng (xem ví dụ ở phụ lục 4) và văn bản thích hợp. Bảng cần có mục nêu rõ số lượng bệnh nhân tham gia các nghiên cứu khác nhau với các liều dùng, đường dùng và thời gian dùng thuốc khác nhau. Nếu sử dụng nhiều mức liều và khoảng thời gian dùng thuốc khác nhau thì nên chia nhóm phù hợp với thuốc thử nghiệm. Theo cách đó, đối với mỗi liều dùng hoặc khoảng liều, thời gian dùng thuốc có thể được tóm tắt bằng số lượng các đối tượng nghiên cứu dùng thuốc trong những khoảng thời gian nhất định, ví dụ như 1 ngày trở xuống, từ 2 ngày đến 1 tuần, 1 tuần đến 1 tháng, 1 tháng đến 6 tháng, 6 tháng đến 1 năm, trên 1 năm (ICH3). Trong một số hồ sơ đăng ký, có thể rất cần phải xác định phân nhóm theo chẩn đoán và/hoặc các nhóm sử dụng các trị liệu đồng thời cụ thể được cho là có liên quan đặc biệt đến việc đánh giá tính an toàn đối với chỉ định dự kiến.
Các mức liều dùng ở đây có thể là liều tối đa dùng cho đối tượng nghiên cứu đó, liều dùng lâu nhất và/hoặc liều hàng ngày trung bình tuỳ trường hợp. Trong đó một số trường hợp có thể tính đến liều tích luỹ. Liều có thể được tính theo liều thực tế hàng ngày hoặc theo mg/kg hoặc mg/m2 cơ thể. Nên trình bày các dữ liệu về nồng độ thuốc (nồng độ lúc xảy ra biến cố ngoại ý, nồng độ tối đa trong huyết tương, diện tích dưới đường cong - AUC) nếu có, vì chúng có thể có ích đối với từng đối tượng về mối liên quan với các biến cố ngoại ý hoặc các biến đổi trong chỉ số xét nghiệm.
Khi phân tích, ta giả định rằng tất cả đối tượng được lựa chọn tham gia nghiên cứu và đã dùng ít nhất 1 liều thuốc điều trị đều được đưa vào phân tích về độ an toàn. Nếu không làm được như vậy thì phải giải thích.
4.1.3. Các đặc điểm về nhân khẩu học và các đặc điểm khác của dân số nghiên cứu
Cần có một bảng tóm tắt để người đọc có được một khái niệm tổng quan về đặc điểm nhân khẩu học (bảng 4.2) của dân số nghiên cứu đã được sử dụng thuốc điều trị trong quá trình phát triển thuốc. Khi lựa chọn giới hạn tuổi thử nghiệm cần xem xét đến những vấn đề được nêu trong ICH E7 [Các nghiên cứu trên các dân số đặc biệt: người cao tuổi] và ICH E11 [Đánh giá lâm sàng chế phẩm thuốc trên trẻ em]. Nếu mức độ sử dụng tương đối của các nhóm bệnh nhân nghiên cứu trong các thử nghiệm lâm sàng có đối chứng khác với mức độ sử dụng chung, thì cần trình bày trong các bảng riêng.
Ngoài ra, cần một hoặc một số bảng để trình bày các đặc điểm thích hợp của dân số nghiên cứu, số lượng các đối tượng nghiên cứu có các đặc tính đặc biệt. Các đặc tính này có thể gồm:
- Mức độ nghiêm trọng của bệnh
- Sự nhập viện
- Suy chức năng thận
- Bệnh mắc kèm
- Thuốc dùng đồng thời
- Vị trí địa lý cư trú
Nếu có những đặc điểm trên được phân bố khác nhau trong các thử nghiệm lâm sàng có đối chứng so với cơ sở dữ liệu chung thì nên trình bày bảng số liệu cho cả 2 nhóm.
Phần diễn giải đi kèm với bảng cần nêu tất cả những điểm mất cân đối về đặc điểm nhân khẩu học giữa nhóm dùng thuốc thử nghiệm và nhóm sử dụng placebo và/hoặc dùng thuốc so sánh, đặc biệt nếu chúng có thể dẫn đến sự khác nhau trong kết quả an toàn.
Nếu có các đối tượng bị loại khỏi nghiên cứu (do có bệnh mắc kèm, mức độ bệnh, thuốc dùng kèm) thì cần nêu rõ điều này.
Mỗi chỉ định điều trị được nghiên cứu cần có một bảng riêng về nhân khẩu học, dù vậy những chỉ định gần nhau có thể xem xét cùng với nhau nếu các đặc điểm của đối tượng nghiên cứu cho thấy họ có cùng nguy cơ.
4.2. Biến cố ngoại ý
4.2.1. Phân tích các biến cố ngoại ý
Các số liệu về tần số xuất hiện các biến cố ngoại ý nên được trình bày dưới dạng văn bản và bảng biểu. Phần văn bản cần được trình bày trong mục 4.2.1 và các bảng biểu không nên chèn vào phần văn bản, chỉ nên trình bày ở phần phụ lục 4.
Tất cả các biến cố ngoại ý đã xảy ra hoặc trở nên xấu hơn sau khi bắt đầu trị liệu ("các dấu hiệu và triệu chứng xuất hiện trong quá trình điều trị", các biến cố ngoại ý không thấy trước khi bắt đầu dùng thuốc và các biến cố ngoại ý diễn biến xấu hơn so với trước khi dùng thuốc) cần được tóm tắt thành các bảng liệt kê từng tác dụng, số lượng đối tượng có biến cố đó, và tần suất xuất hiện biến cố ở đối tượng được điều trị bằng thuốc nghiên cứu, ở nhóm dùng thuốc so sánh hoặc nhóm dùng placebo. Các bảng đó nên trình bày kết quả theo mỗi liều và có thể điều chỉnh để phản ánh được tỷ lệ biến cố ngoại ý theo mức độ bệnh, theo thời gian bắt đầu trị liệu hoặc theo đánh giá nguyên nhân.
Khi hầu hết các dữ liệu về tính an toàn thích hợp đều xuất phát từ một số ít các nghiên cứu (thí dụ trên 1 hoặc 2 nghiên cứu), hoặc khi nghiên cứu tiến hành trên những đối tượng rất khác nhau thì việc trình bày rõ dữ liệu theo từng nghiên cứu thường rất cần thiết. Tuy nhiên, nếu dữ liệu về mức độ sử dụng thuốc không tập trung trong một số ít nghiên cứu thì nên nhóm các nghiên cứu lại và phân tích gộp các kết quả để cải thiện tính chính xác của các ước lượng qua nghiên cứu và cần xem xét đến độ nhạy đối với những khác biệt.
Phân tích gộp các dữ liệu an toàn của các nghiên cứu thường là rất hữu ích nhưng nên được tiến hành thận trọng vì trong một số trường hợp việc diễn giải kết quả rất khó, và có thể che lấp những khác biệt thật sự. Trong các trường hợp mà sự khác biệt là rõ ràng, thì trình bày dữ liệu theo từng nghiên cứu sẽ thích hợp hơn. Các vấn đề sau cần được xem xét:
● Thích hợp nhất là kết hợp các dữ liệu của các nghiên cứu có thiết kế tương tự nhau, thí dụ tương tự về liều lượng, thời gian điều trị, phương pháp xác định biến cố ngoại ý và dân số nghiên cứu.
● Nếu tần suất của một biến cố ngoại ý khác nhau đáng kể giữa các nghiên cứu riêng lẻ trong một nhóm phân tích gộp, thì giá trị ước lượng từ phân tích gộp thường ít có giá trị.
● Bất kỳ nghiên cứu nào có một kiểu biến cố ngoại ý bất thường cần được trình bày riêng.
● Mức độ phân tích phụ thuộc vào mức độ nghiêm trọng của biến cố ngoại ý, độ mạnh của bằng chứng về quan hệ nhân quả với thuốc. Cần nghiên cứu kỹ hơn khi có sự khác biệt trong tỷ lệ các biến cố ngoại ý nghiêm trọng liên quan tới thuốc, hoặc các biến cố ngoại ý dẫn tới việc ngừng thuốc hay phải thay đổi liều lượng, trong khi những tác dụng phụ khác có thể không đòi hỏi phải phân tích tỉ mỉ.
● Việc kiểm tra các đối tượng có những bất thường quá mức về trị số xét nghiệm ("các giá trị nằm ngoài giới hạn thông thường") có thể hữu ích trong việc xác định các phân nhóm dân số có nguy cơ đặc biệt đối với các biến cố ngoại ý nào đó.
Nhóm các nghiên cứu để đánh giá gộp về tính an toàn bao gồm:
● Tất cả các nghiên cứu có đối chứng hay các phần của nghiên cứu có đối chứng, ví dụ như tất cả các nghiên cứu có đối chứng với placebo, nghiên cứu có đối chứng với bất kỳ chất có hoạt tính hoặc với chất có hoạt tính đặc biệt, các nghiên cứu về những chỉ định đặc biệt (và vì thế tiến hành trên các dân số nghiên cứu khác nhau). Việc nhóm các nghiên cứu như thế này được coi là nguồn thông tin tốt nhất về các biến cố ngoại ý thường gặp hơn và có thể phân biệt được các biến cố ngoại ý liên quan đến thuốc với các biến cố tự phát. Phải so sánh tỷ lệ giữa nhóm nghiên cứu và nhóm chứng.
● Tất cả các nghiên cứu trừ các nghiên cứu ngắn hạn trên người tình nguyện khỏe mạnh. Cách phân nhóm này hữu ích nhất cho việc đánh giá các biến cố ngoại ý hiếm gặp hơn.
● Tất cả các nghiên cứu sử dụng một đường dùng đặc biệt hay phác đồ liều đặc biệt, hay dùng một trị liệu đồng thời đặc biệt.
● Các nghiên cứu trong đó việc báo cáo các biến cố ngoại ý được tiến hành bằng việc đánh dấu vào bảng liệt kê hoặc hỏi trực tiếp, hoặc các nghiên cứu mà trong đó việc báo cáo các biến cố ngoại ý là tự nguyện.
● Nhóm các nghiên cứu theo vùng.
Thường sẽ là rất hữu ích khi thực hiện 2 cách phân nhóm đầu tiên, các cách phân nhóm khác có thể được lựa chọn tuỳ thuộc vào thuốc và vào việc kiểm tra kết quả của từng nghiên cứu. Dù cho dùng bất cứ phương pháp nào, cũng đều cần công nhận rằng, tỷ lệ qua kết quả phân tích thường chỉ là ước lượng thô của tỷ lệ thực tế, tương tự như các kết quả của các nghiên cứu đơn lẻ.
Khi quyết định gộp dữ liệu của nhiều nghiên cứu để phân tích, cần mô tả lý luận cho việc lựa chọn phương pháp sử dụng để gộp số liệu. Thông thường, người ta hay gộp các biến cố của phần tử số và các biến cố của phần mẫu số của các nghiên cứu được chọn. Các phương pháp khác để gộp kết quả các nghiên cứu hiện nay có thể sử dụng như: gia trọng cho các dữ liệu từ các nghiên cứu dựa trên cỡ mẫu hoặc nghịch đảo phương sai của chúng.
Nếu thấy có sự khác nhau đáng kể về tỷ lệ các biến cố ngoại ý giữa các thử nghiệm lâm sàng thì phải nêu rõ những khác biệt này và phải bàn luận về các nguyên nhân có thể có (thí dụ: sự khác nhau về dân số nghiên cứu, về liều dùng, về phương pháp thu thập các thông tin về biến cố ngoại ý).
Các biến cố ngoại ý cần được mô tả như đã nêu trong báo cáo nghiên cứu riêng lẻ (ICH E3). Khi kết hợp dữ liệu từ nhiều nghiên cứu, vấn đề quan trọng là phải sử dụng các thuật ngữ được chuẩn hóa để mô tả các biến cố và dùng một thuật ngữ được ưa thích cho tất cả các thuật ngữ đồng nghĩa. Điều này có thể thực hiện bằng một từ điển tiêu chuẩn quốc tế và cần xác định rõ thuật ngữ nào sẽ được sử dụng. Cần nêu tần số sử dụng thuật ngữ ưa dùng và các thuật ngữ thích hợp thuộc nhóm đó. Việc kiểm tra xem các biến cố ngoại ý nào dẫn đến thay đổi điều trị (như ngưng sử dụng thuốc, thay đổi liều dùng, cần phải bổ sung trị liệu) có thể giúp cho việc đánh giá tầm quan trọng lâm sàng của biến cố ngoại ý đó. Có thể bổ sung các tỷ lệ này vào bảng tỷ lệ các biến cố ngoại ý, hoặc trình bày trong một bảng riêng. Tỷ lệ ngưng trị liệu chung của từng nghiên cứu có thể hữu ích nhưng quan trọng là phải xác định các biến cố ngoại ý nào dẫn đến ngưng trị liệu trong một bảng riêng. Các thuật ngữ ưa dùng cần được nhóm lại theo hệ cơ quan trong cơ thể và sắp xếp theo tần suất giảm dần.
4.2.1.1. Các biến cố ngoại ý thường gặp
Cần sử dụng các bảng trình bày về tỷ lệ các biến cố ngoại ý (xem phụ lục 4) để so sánh các tỷ lệ giữa nhóm nghiên cứu và nhóm chứng. Đối với các phân tích này, có thể kết hợp các biến cố ngoại ý theo mức độ và theo nguyên nhân; nếu kết hợp nêu trên được sử dụng, sẽ có một so sánh đơn giản hơn từng cặp một giữa các nhóm điều trị. Cũng cần chú ý rằng, khi báo cáo các biến cố ngoại ý theo nguyên nhân, nếu có, thì dữ liệu trình bày cần nêu tất cả các biến cố ngoại ý (cho dù có liên quan hoặc không liên quan đến trị liệu); việc đánh giá nguyên nhân vốn mang tính chủ quan và có thể bỏ qua những biến cố ngoại ý không dự kiến mà trong thực tế liên quan đến trị liệu. Ngoài ra, nên tóm tắt phần so sánh tỷ lệ các biến cố ngoại ý giữa nhóm nghiên cứu và nhóm chứng trong từng nghiên cứu riêng lẻ. Việc sử dụng bảng tỷ lệ cho các nghiên cứu được lựa chọn thường rất có ích (xem thí dụ bảng 4.4, trong phụ lục 4).
Thường rất có ích nếu kiểm tra kỹ hơn các biến cố ngoại ý thường gặp mà dường như có liên quan đến thuốc (thí dụ: những biến cố ngoại ý có tỷ lệ đáp ứng với liều và/hoặc những tỷ lệ khác khác nhau rõ rệt giữa nhóm dùng thuốc và nhóm placebo) về sự liên quan với các yếu tố sau:
- Liều dùng
- Liều mg/kg hoặc mg/m2
- Chế độ liều
- Thời gian điều trị
- Tổng liều
- Đặc tính nhân khẩu học như tuổi, giới tính, chủng tộc
- Thuốc điều trị đồng thời
- Các đặc điểm trước khi dùng thuốc như chức năng thận
- Kết quả về hiệu quả
- Nồng độ thuốc (nếu có)
Sẽ rất có ích nếu tóm tắt các kết quả đánh giá thời gian xuất hiện và khoảng thời gian kéo dài của các biến cố liên quan đến thuốc.
Các đánh giá thống kê nghiêm ngặt về mối quan hệ có thể có giữa các biến cố ngoại ý đặc trưng với các yếu tố nêu trên thường không cần thiết. Có thể rõ ràng ngay từ biểu hiện ban đầu là không có bằng chứng về mối quan hệ có ý nghĩa đến các đặc điểm nhân khẩu học hay đặc điểm trước khi bắt đầu dùng thuốc khác. Trong trường hợp như vậy, không cần phải phân tích thêm về các yếu tố này. Hơn nữa, không cần thiết phải trình bày tất cả các phân tích như vậy sở đây. Khi các phân tích về tính an toàn quá sâu rộng để có thể trình bày chi tiết ở phần này, chúng có thể được trình bày ở một báo cáo riêng trong phần báo cáo nghiên cứu lâm sàng và chỉ tóm tắt ở đây.
Trong một số trường hợp nhất định, nên có bảng thống kê tỷ lệ sống còn (life table) hoặc những phân tích tương tự vì điều này sẽ có ý nghĩa hơn so với việc chỉ báo cáo tỷ lệ các biến cố ngoại ý đơn thuần.
4.2.1.2. Tử vong
Bảng ở phụ lục 4 nên liệt kê tất cả các trường hợp tử vong xảy ra trong nghiên cứu (bao gồm cả các trường hợp tử vong sau khi ngừng điều trị một thời gian ngắn, thí dụ khoảng 30 ngày hoặc trong khoảng thời gian đã xác định trong đề cương nghiên cứu, cũng như các trường hợp tử vong muộn hơn nhưng có thể do một quá trình bệnh lý khởi đầu trong quá trình nghiên cứu). Chỉ không đưa vào danh mục này những trường hợp tử vong rõ ràng là do bệnh theo định nghĩa trong đề cương nghiên cứu mà không liên quan đến sản phẩm thuốc đang nghiên cứu, có thể trong các nghiên cứu đối với những bệnh có tỷ lệ tử vong cao như ung thư tiến triển hay trong các nghiên cứu mà tử vong do bệnh là một tiêu chí nghiên cứu chính (tuy nhiên, những trường hợp tử vong này vẫn cần được báo cáo trong phần báo cáo nghiên cứu riêng lẻ ICH E3). Thậm chí những trường hợp tử vong này cần được nghiên cứu xem xét có sự khác biệt ngoài dự định giữa các nhánh nghiên cứu và cần phân tích sâu hơn nếu nhận thấy có các khác biệt mà không giải thích được. Các trường hợp tử vong cần được xem xét riêng và phân tích dựa trên tỷ lệ của các nghiên cứu riêng lẻ hoặc tỷ lệ phân tích gộp hợp lý của các nghiên cứu, về tổng số tử vong và theo nguyên nhân gây tử vong. Cũng nên xem xét mối liên quan tiềm tàng với các yếu tố đã liệt kê trong mục 4.2.1.1. Mặc dù tỷ lệ tử vong cụ thể theo nguyên nhân thường khó xác định, song một số trường hợp tử vong tương đối dễ giải thích. Theo đó, những trường hợp tử vong do những nguyên nhân tiên liệu trước trong dân số bệnh nhân (như do cơn đau tim, đột tử ở các bệnh nhân bị đau thắt ngực) thì về mặt cá thể không được xem là có giá trị, nhưng thậm chí chỉ một ca tử vong do loạn nhịp tim liên quan đến kéo dài quãng QT, do thiếu máu bất sản, hay do tổn thương gan lại có thể có giá trị. Cần đặc biệt lưu ý đến trường hợp tử vong được quy cho bệnh lý phối hợp.
4.2.1.3. Các biến cố ngoại ý nghiêm trọng khác.
Cần trình bày tóm tắt tất cả các biến cố ngoại ý nghiêm trọng (không kể tử vong nhưng có thể liên quan về thời gian hoặc xuất hiện trước khi tử vong). Các biến cố ngoại ý nghiêm trọng xảy ra sau khi ngừng thuốc cũng cần đưa vào phần này. Phần trình bày cần bao gồm những bất thường về chỉ số xét nghiệm, bất thường về các dấu hiệu sống còn và các bất thường về các dấu hiệu lâm sàng được xếp vào nhóm biến cố ngoại ý nghiêm trọng theo định nghĩa từ ICH E2A. Phải trình bày kết quả phân tích hoặc đánh giá về biến cố ngoại ý nghiêm trọng qua tất cả các nghiên cứu. Cần xem xét tần suất các biến cố ngoại ý nghiêm trọng theo thời gian, đặc biệt đối với những thuốc dùng kéo dài. Nên xem xét khả năng liên quan với các yếu tố đã liệt kê ở mục 4.2.1.1.
4.2.1.4. Các biến cố ngoại ý đáng chú ý khác:
Cần trình bày những bất thường về xét nghiệm huyết học và các xét nghiệm khác (ngoài những bất thường được định nghĩa là nghiêm trọng) và bất kỳ biến cố nào dẫn tới những can thiệp đáng kể (ngừng dùng thuốc nghiên cứu trước thời hạn, giảm liều, hoặc bổ sung thêm trị liệu đồng thời) ngoài những biến cố ngoại ý đã được báo cáo ở phần biến cố ngoại ý nghiêm trọng.
Những biến cố dẫn tới ngừng thuốc nghiên cứu trước thời hạn cho thấy có vấn đề quan trọng về an toàn và cần được chú ý đặc biệt trong phân tích về tính an toàn của thuốc vì 2 lý do. Một là, ngay cả với những biến cố đã được tiên lượng (dựa trên tác động dược lý), việc cần phải dừng điều trị (hoặc thay thế thuốc) phản ánh sự nghiêm trọng và tầm quan trọng của biến cố đối với bệnh nhân và thầy thuốc. Thứ hai, việc dừng thuốc có thể thể hiện một biến cố ngoại ý liên quan đến thuốc nhưng chưa được công nhận là có liên quan đến thuốc. Các biến cố ngoại ý dẫn đến việc ngừng điều trị cần được coi là có thể liên quan đến thuốc ngay cả khi điều này không được công nhận từ đầu và ngay cả khi biến cố này được coi là biểu hiện của bệnh gian phát. Cần bàn luận về nguyên nhân dừng điều trị trước thời hạn và cần so sánh tỷ lệ dừng thuốc giữa các nghiên cứu và so sánh với tỷ lệ ở nhóm dùng placebo và/ hoặc điều trị với chất đối chứng có hoạt tính. Ngoài ra, cần đánh giá các dữ liệu nghiên cứu về mối quan hệ có thể có với các yếu tố được liệt kê ở mục 4.2.1.1.
4.2.1.5. Phân tích các biến cố ngoại ý theo hệ cơ quan hoặc hội chứng
Việc đánh giá nguyên nhân và các yếu tố nguy cơ gây tử vong, gây các biến cố ngoại ý nghiêm trọng, hoặc các biến cố ngoại ý đáng chú ý khác thường phức tạp vì đây là các biến cố không thường gặp. Vì vậy, khi xem xét các biến cố liên quan đến nhau trong một nhóm, bao gồm cả những biến cố ít quan trọng hơn có thể liên quan đến sinh lý bệnh có thể có giá trị đáng kể giúp hiểu được về tính an toàn của thuốc. Thí dụ: mối quan hệ của một trường hợp đột tử riêng biệt với điều trị có thể rõ ràng hơn khi được xem xét trong bối cảnh của các trường hợp ngất, tăng nhịp tim hoặc loạn nhịp không triệu chứng.
Nhìn chung sẽ có ích nếu tóm tắt các biến cố ngoại ý theo các hệ cơ quan để cho chúng được xem xét trong phạm vi các biến cố có thể liên quan, kể cả các bất thường về chỉ số xét nghiệm. Nên trình bày các biến cố ngoại ý theo hệ cơ quan ở mục 4.2.1.5 và đánh số 4.2.1.5.1, 4.2.1.5.2 với tiêu đề là hệ cơ quan đang xem xét.
Danh sách các hệ cơ quan cần được đề cập và phương pháp nhóm các biến cố ngoại ý nên được lựa chọn thích hợp để trình bày tối ưu các dữ liệu về biến cố ngoại ý của thuốc. Nếu một số biến cố ngoại ý có khuynh hướng xảy ra theo hội chứng (ví dụ hội chứng giống cúm, hội chứng giải phóng cytokin), cơ sở tài trợ nghiên cứu có thể tạo thêm một số phần 4.2.1.5 để trình bày theo hội chứng thay vì theo hệ cơ quan.
Các dữ liệu và tóm tắt trùng nhau không nên trình bày lặp lại trong các phần khác nhau của mục 4.2.1. Thay vào đó, có thể đưa một trình bày tóm tắt vào một tiểu mục và nêu tham chiếu chéo ở phần khác nếu cần thiết.
4.2.2. Tường thuật
Để thuận lợi cho các chuyên gia xem xét hồ sơ, ở phần này, cần chỉ rõ vị trí tham khảo trong hồ sơ đăng ký thuốc cho những bản mô tả các trường hợp bệnh nhân tử vong, các biến cố ngoại ý nghiêm trọng khác hoặc các biến cố ngoại ý đáng chú ý khác thường được quan tâm đặc biệt do tầm quan trọng lâm sàng (như đã nêu trong các báo cáo nghiên cứu riêng lẻ ICH E3). Bản thân các phần tường thuật đã là một phần của các báo cáo nghiên cứu riêng lẻ, nếu có báo cáo như vậy. Trong trường hợp không có báo cáo nghiên cứu riêng (ví dụ nếu nhiều nghiên cứu mở được gộp lại trong phần phân tích tính an toàn và không có mô tả riêng từng nghiên cứu) thì phần tường thuật có thể được trình bày ở mục 5.3." Báo cáo nghiên cứu lâm sàng". Không nên đưa vào đây phần tường thuật trừ khi tường thuật tóm tắt các biến cố ngoại ý đặc biệt được coi là tối quan trọng đối với đánh giá tóm tắt về thuốc.
4.3. Đánh giá các kết quả xét nghiệm (KQXN)
Phần này cần trình bày những thay đổi về kết quả xét nghiệm khi có sử dụng thuốc.
Những bất thường đáng chú ý về KQXN và những bất thường dẫn đến can thiệp quan trọng cần được trình bày trong mục 4.2.1.3 hoặc 4.2.1.4. Nếu các số liệu đó cũng được trình bày trong phần này thì việc lặp lại đó phải được nêu rõ để cán bộ thẩm định hồ sơ được biết. Những đánh giá thích hợp về kết quả xét nghiệm sẽ được xác định một phần bởi các kết quả quan sát được, song nhìn chung cần cung cấp những phân tích mô tả dưới đây. Đối với từng phân tích, cần có so sánh giữa nhóm nghiên cứu với nhóm chứng, khi thích hợp và khi có tương thích về cỡ mẫu nghiên cứu. Ngoài ra, cần đưa ra khoảng giới hạn chỉ số xét nghiệm bình thường cho mỗi phân tích (ICH E3). Nếu có thể, cần cung cấp các giá trị xét nghiệm theo đơn vị quốc tế chuẩn.
Nên có tổng quan tóm tắt những thay đổi chính về KQXN qua các nghiên cứu lâm sàng. Các số liệu xét nghiệm bao gồm: huyết học, hóa lâm sàng (clinical chemistry), phân tích nước tiểu và các số liệu thích hợp khác. Mỗi thông số tại từng thời điểm qua quá trình nghiên cứu (thí dụ mỗi lần bệnh nhân đến khám) nên được trình bày theo ba mức độ sau đây:
● Khuynh hướng trung tâm, thí dụ: các giá trị trung bình và giá trị trung vị của nhóm.
● Giới hạn của trị số và số đối tượng có trị số bất thường hoặc có trị số bất thường theo mức độ nhất định (thí dụ: gấp 2 lần giới hạn trên của giá trị bình thường hoặc gấp 5 lần giới hạn trên; cần giải thích sự lựa chọn). Khi số liệu được gộp lại từ các trung tâm với các giới hạn giá trị xét nghiệm bình thường khác nhau, cần mô tả phương pháp gộp sử dụng. Việc phân tích các thay đổi của từng đối tượng nghiên cứu riêng lẻ theo nhóm điều trị có thể trình bày theo nhiều cách khác nhau (ví dụ: bảng biểu diễn sự thay đổi các giá trị xét nghiệm so với ban đầu (shift table), xem ICH E3 để minh họa).
● Những bất thường nghiêm trọng về lâm sàng của cá thể, kể cả những bất thường dẫn đến ngừng điều trị. Nên đánh giá ý nghĩa của những thay đổi về xét nghiệm và mối liên quan đến điều trị (thí dụ: qua phân tích các đặc điểm như mối quan hệ với liều dùng, liên hệ với nồng độ thuốc, sự biến mất khi điều trị tiếp tục, sự xuất hiện lại và bản chất của điều trị phối hợp). Cũng cần phải xem xét liên quan của các bất thường này với các yếu tố được liệt kê trong mục 4.2.1.1.
4.4. Dấu hiệu sinh tồn, các triệu chứng thực thể và các ghi nhận khác liên quan đến tính an toàn.
Cách trình bày các quan sát qua các nghiên cứu và cách so sánh các dấu hiệu sinh tồn (như nhịp tim, huyết áp, thân nhiệt, nhịp thở), thể trọng và các dữ liệu khác (như điện tâm đồ, X-quang) liên quan đến tính an toàn nên tương tự như cách trình bày so sánh đối với các KQXN. Nếu có bằng chứng về tác dụng của thuốc, cần chỉ ra tất cả sự tương quan giữa đáp ứng-liều lượng, sự tương quan giữa đáp ứng-nồng độ thuốc, hay các mối liên quan tới các đặc tính của cá thể (bệnh, nhân khẩu học, điều trị phối hợp) cũng như sự tương thích về lâm sàng của các quan sát đó. Cần lưu ý đặc biệt đến những thay đổi mà không được đánh giá là các thay đổi về hiệu quả của thuốc, và chú ý đến những thay đổi được xem là biến cố ngoại ý. Phải chú trọng đến những nghiên cứu được thiết kế để đánh giá những vấn đề về an toàn đặc biệt, ví dụ như nghiên cứu về kéo dài khoảng QT.
4.5. Tính an toàn đối với các nhóm dân số đặc biệt và trong tình huống đặc biệt.
4.5.1 Các nhóm bệnh nhân
Phần này tổng hợp các dữ liệu về tính an toàn thích hợp cho việc cá thể hóa điều trị hay quản lý bệnh nhân trên cơ sở nhân khẩu học, tuổi, giới, chiều cao, thể trọng, khối lượng gầy (lean body mass), đa hình thái di truyền, cấu trúc cơ thể, sự suy giảm chức năng cơ quan hay bệnh lý khác. Độ an toàn trên đối tượng trẻ em cần được đánh giá thường xuyên nếu trong hồ sơ có đăng ký các chỉ định đề nghị cho trẻ em. Phần phân tích về tác động lên độ an toàn được trình bày ở mục khác nhưng cần tóm tắt ở đây, cùng với những thông tin thích hợp về dược động học (PK) hoặc các thông tin khác, thí dụ ở bệnh nhân với bệnh lý gan hoặc bệnh lý thận, môi trường y tế, sử dụng thuốc khác (xem 4.5.2: Tương tác thuốc), hút thuốc lá, uống rượu, thói quen ăn uống. Ví dụ, nếu các số liệu về chuyển hóa, các kết quả nghiên cứu, các kinh nghiệm sau khi đưa thuốc ra thị trường, hoặc qua các thông tin về các thuốc tương tự gợi ý thấy có tương tác tiềm tàng với rượu thì cần cung cấp thông tin này ở đây. Nếu trong mẫu nghiên cứu có một lượng đủ lớn các đối tượng có một bệnh đi kèm nhất định như tăng huyết áp, bệnh tim hoặc đái tháo đường thì nên phân tích đánh giá ảnh hưởng của các bệnh đồng mắc đến tính an toàn của thuốc nghiên cứu. Cần đưa ra các tham chiếu chéo đến các bảng hoặc mô tả các biến cố ngoại ý khi phân tích các phân nhóm này.
4.5.2 Tương tác thuốc
Các nghiên cứu tương tác tiềm tàng thuốc-thuốc hoặc tương tác thuốc-thức ăn nên được tóm tắt trong phần "Tóm tắt nghiên cứu dược lý lâm sàng" của ACTD. Tác động tiềm tàng đến tính an toàn của những tương tác như vậy cần được tóm tắt ở đây, căn cứ trên những kết quả về dược động học, dược lực học, hoặc các quan sát lâm sàng. Bất kỳ thay đổi nào về biến cố ngoại ý, thay đổi về các nồng độ trong máu được cho là có liên quan đến nguy cơ, hoặc các thay đổi tác dụng của thuốc liên quan đến trị liệu khác cần được trình bày ở đây.
4.5.3. Sử dụng ở phụ nữ có thai và cho con bú
Bất cứ các thông tin về tính an toàn khi sử dụng thuốc trong quá trình mang thai hoặc cho con bú có được trong quá trình phát triển chế phẩm hoặc thu được từ các nguồn khác cần được trình bày ở đây.
4.5.4 Quá liều
Tất cả thông tin lâm sàng liên quan đến quá liều bao gồm các dấu hiệu/triệu chứng, KQXN, biện pháp điều trị, xử lý hoặc chất giải độc (nếu có) cần được trình bày và bàn luận ở đây. Thông tin về hiệu quả của chất giải độc và thẩm phân cũng trình bày ở đây nếu có.
4.5.5 Lạm dụng thuốc
Các nghiên cứu/thông tin về sự lạm dụng vào một thuốc mới trên động vật và trên người nên được tổng hợp lại và tham khảo chéo đến phần tóm tắt tiền lâm sàng. Chú ý xác định các dân số bệnh nhân nhạy cảm đặc biệt.
4.5.6. Cai thuốc và phản ứng dội (rebound)
Bất cứ các thông tin hoặc nghiên cứu liên quan tới phản ứng dội cần được trình bày ở đây. Các biến cố xảy ra, hoặc sự gia tăng độ nặng của bệnh sau khi ngừng thuốc trong các nghiên cứu mù đôi hay trong các nghiên cứu có sử dụng chất đối chứng có hoạt tính phải được xem xét để tìm hiểu có phải do ngừng thuốc không. Cần đặc biệt nhấn mạnh các nghiên cứu được thiết kế để đánh giá hiện tượng cai thuốc và/hoặc phản ứng dội.
Các dữ liệu liên quan đến sự lờn thuốc nên được tổng hợp trong mục 3.5 trong phần "Tóm tắt hiệu quả lâm sàng"
4.5.7 ảnh hưởng lên khả năng lái xe và vận hành máy móc hoặc suy giảm năng lực tâm thần
Cần tóm tắt các dữ liệu an toàn liên quan đến sự suy giảm cảm giác, khả năng phối hợp hoặc các yếu tố khác mà có thể làm giảm khả năng lái xe và vận hành máy móc hoặc làm giảm năng lực tinh thần. Các thông tin bao gồm các biến cố ngoại ý đã nêu trong phần theo dõi tính an toàn (thí dụ: buồn ngủ) và các nghiên cứu đặc hiệu về ảnh hưởng lên khả năng lái xe và vận hành máy móc hoặc suy giảm năng lực tâm thần.
4.6. Các dữ liệu sau khi đưa thuốc ra thị trường
Nếu thuốc đã được lưu hành trên thị trường, cần tóm tắt tất cả các dữ liệu sau khi đưa thuốc ra thị trường thích hợp sẵn có (đã công bố hoặc chưa công bố, bao gồm cả các báo cáo cập nhật định kỳ về độ an toàn nếu có). Các báo cáo cập nhật định kỳ về độ an toàn có thể đưa vào phần "Báo cáo nghiên cứu lâm sàng". Chi tiết về số lượng các đối tượng ước lượng đã dùng thuốc phải được nêu và phân loại theo chỉ định, liều dùng, đường dùng, thời gian điều trị và vị trí địa lý. Phương pháp đã dùng để ước lượng số lượng đối tượng đã dùng thuốc cần được mô tả. Nên cung cấp các ước lượng về nhân khẩu học có sẵn từ bất cứ nguồn nào.
Cần đưa ra một bảng tập hợp tất cả các biến cố nghiêm trọng đã được báo cáo sau khi đưa thuốc ra thị trường kể cả các tương tác thuốc nghiêm trọng có thể xảy ra.
Bất kỳ một phát hiện nào ở các phân nhóm bệnh nhân sau khi đưa thuốc ra thị trường đều cần được mô tả.
Phụ lục 4
Cần trình bày bảng tổng hợp các kết quả quan trọng từ tất cả các nghiên cứu đánh giá về tính an toàn, đặc biệt để hỗ trợ cho việc ghi nhãn. Có thể đưa bảng biểu và hình ảnh vào phần văn bản trình bày nếu giúp cho người đọc dễ hiểu hơn. Những bảng dài nên đưa vào phụ lục ở cuối phần tóm tắt về an toàn lâm sàng.
Nên có một số bảng minh họa nhưng một tóm tắt lâm sàng thường cần có bảng và hình ảnh đã được xây dựng đối với một thuốc, nhóm thuốc hoặc các chỉ định lâm sàng đặc biệt.
Xem mục 4.2.1., 4.2.2.3. và 4.3 của hướng dẫn này để thảo luận thêm về nội dung của các bảng trong phần 4
Bảng 4.1. Nghiên cứu mức độ sử dụng thuốc trên đối tượng nghiên cứu về liều trung bình hàng ngày và thời gian dùng thuốc.
Bảng 4.2. Các đặc điểm về nhân khẩu học của dân số bệnh nhân trong các thử nghiệm có đối chứng.
Bảng 4.3. Tỷ lệ biến cố ngoại ý qua phân tích gộp các thử nghiệm có đối chứng với placebo và chất đối chứng có hoạt tính.
Bảng 4.4. Tỷ lệ biến cố ngoại ý trong các thử nghiệm lớn nhất.
Bảng 4.5. Bệnh nhân rút khỏi nghiên cứu: các thử nghiệm có đối chứng
Bảng 4.6. Danh sách bệnh nhân tử vong
5. Tóm tắt các nghiên cứu riêng lẻ
Hướng dẫn ICH E3 (Cấu trúc và Nội dung của Báo cáo nghiên cứu lâm sàng") gợi ý về nội dung của tóm tắt nghiên cứu của mỗi báo cáo nghiên cứu lâm sàng và cho ví dụ về cách trình bày đối với các tóm tắt như vậy.
Phần này nên bao gồm bảng với tiêu đề "Danh sách các nghiên cứu lâm sàng" như đã nêu ở phần hướng dẫn "báo cáo nghiên cứu lâm sàng", tiếp theo sau là các tóm tắt nghiên cứu riêng lẻ được bố cục theo một trật tự giống như trong các báo cáo nghiên cứu lâm sàng.
Mỗi nghiên cứu nên có một bản tóm tắt sử dụng chung cho tất cả các nước và bản tóm tắt đó sẽ được nêu trong phần này như một phần của báo cáo nghiên cứu lâm sàng. Độ dài của tóm tắt thường tối đa là 3 trang, tuy nhiên nếu nội dung phức tạp và nghiên cứu quan trọng thì có thể dài hơn, thí dụ 10 trang. Với từng bản tóm tắt riêng lẻ, nên sử dụng bảng biểu và hình ảnh phù hợp giúp phần trình bày rõ ràng hơn.
Bảng 1.1 Tóm tắt các nghiên cứu về sinh khả dụng
|
Mã số nghiên cứu |
Mục tiêu nghiên cứu |
Thiết kế nghiên cứu |
Điều trị (liều, dạng bào chế, đường dùng) [Mã số sản phẩm] |
Đối tượng nghiên cứu (số lượng (nam/nữ), phân loại, tuổi trung bình (giới hạn tuổi) |
Các chỉ số trung bình (+/- độ lệch chuẩn) |
Vị trí của báo cáo nghiên cứu |
|||||
|
|
|
|
|
|
Cmax (mg/L) |
Tmax (giờ) |
AUC* (mg/L x giờ) |
Cmin** (mg/L) |
T1/2 (giờ) |
Các chỉ số khác |
|
|
192 (Nhật) |
Nghiên cứu pilot về sinh khả dụng tương đối so sánh sự hấp thu của lô viên nén 200mg với lô 200mg đối chứng |
Mở, ngẫu nhiên, bắt chéo, liều đơn 200mg |
200mg, viên nén, uống [17762]
200mg, viên nén, uống [19426] |
20 (10/10) Người tình nguyện khỏe mạnh 27 tuổi (20-35) |
83 ± 21
80 ± 32 |
1
0,5 |
217 ± 20
223 ± 19 |
|
3,1
2,9 |
|
|
|
195 (Nhật) |
Nghiên cứu về sinh khả dụng tương đối của xx uống khi đói và sau khi ăn |
Mở, ngẫu nhiên, chéo, liều đơn |
200mg, viên nén, uống [19426] |
30 (15/15) Người tình nguyện khỏe mạnh 32 tuổi (26-50) |
83 ± 21
120 ± 30 |
1
2 |
217 ± 20
350 ± 40 |
|
3,1
2,9 |
|
|
AUC*: AUCTAU hoặc AUCinf
Cmin**: sử dụng cho các nghiên cứu đa liều
Bảng 1.2. Tóm tắt các nghiên cứu về hòa tan in vitro
|
Mã số nghiên cứu |
Mã sản phẩm/ Số lô |
Dạng bào chế |
Phương pháp thử nghiệm |
Số đơn vị dùng |
Thời gian thu thập kết quả Trung bình % hòa tan (giới hạn) |
Vị trí của báo cáo nghiên cứu |
||
|
1821 |
979-03 |
Viên nang 25mg |
Sự hòa tan: bộ dụng cụ 2 (USP) Tốc độ quay: 50rpm Dung môi/ Nhiệt độ: Nước 37o |
12 |
10 42 (32-49) |
20 71 (58-85) |
30 (phút) 99 (96-100) (%) |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
Bảng 2.1 Tóm tắt nghiên cứu dược động học tương tác thuốc-thuốc về dược động học
|
NC/ Đề cương số (nước) |
Mã sản phẩm/ số lô (NME) |
Mục tiêu nghiên cứu |
Thiết kế nghiên cứu |
Số lượng đối tượng tham gia/ hoàn tất nghiên cứu (Nam/Nữ) |
Người tình nguyện khỏe mạnh/ Bệnh nhân1 (Tuổi: trung bình, khoảng giới hạn) |
Trị liệu |
Trung bình các thông số dược động học (%CV) của thuốc NC |
Trung bình tỷ lệ khoảng tin cậy |
Vị trí |
||||||
|
|
|
|
|
|
|
Thuốc NC |
Thuốc tương tác |
Cmax |
Tmax |
AUC |
T1/2 |
Độ thanh thải/kg |
Cmax |
AUC |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 Người tình nguyện khỏe mạnh= HV (Healthy Volunteers), Bệnh nhân= BN
2 Tỷ lệ giữa khoảng tin cậy của nhóm dùng thuốc nghiên cứu và nhóm sử dụng thuốc tương tác/ giả dược
Bảng 3.1 Mô tả các nghiên cứu lâm sàng về hiệu quả và an toàn
|
Mã số nghiên cứu |
Số lượng trung tâm NC, địa điểm NC |
Thời điểm bắt đầu NC Tình trạng tuyển đối tượng NC, ngày Số lượng đối tượng NC đã tuyển được/ số lượng đối tượng NC cần phải có |
Thiết kế NC Kiểu chứng |
Thuốc NC và thuốc chứng Liều, đường dùng Chế độ liều |
Mục tiêu nghiên cứu |
Số lượng đối tượng NC theo mục tiêu Số đối tượng tham gia NC/ số đối tượng hoàn tất NC |
Thời gian NC |
Giới (Nam/ Nữ) Tuổi trung vị (giới hạn) |
Chẩn đoán Tiêu chuẩn chấp nhận bệnh nhân |
Các tiêu chí nghiên cứu chính |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bảng 3.2 Kết quả của nghiên cứu về hiệu quả
|
Nghiên cứu |
Mục tiêu điều trị |
Số lượng đối tượng tham gia/ hoàn tất nghiên cứu |
Huyết áp tâm thu và huyết áp tâm trương trung bình |
Tiêu chí NC chính Sự khác nhau về HA tâm trương giữa nhóm chứng và nhóm nghiên cứu ở tuần 40 |
Phương pháp thống kê Giá trị p |
Các tiêu chí NC phụ % bình thường hóa ** (phân tích ITT) |
Các nhận xét khác |
||
|
|
|
|
Khi bắt đầu NC |
20 tuần |
40 tuần |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
** Định nghĩa về % bình thường hóa (normalised)
|
Bảng 4.1 Mức độ sử dụng thuốc của đối tượng nghiên cứu qua liều trung bình hàng ngày và thời gian sử dụng thuốc sử dụng đường tĩnh mạch |
|||||||||
|
N= |
Ngày kết thúc nghiên cứu: |
||||||||
|
Thời gian (tuần) |
Liều trung bình hàng ngày (mg) |
||||||||
|
|
0-5mg |
5-10mg |
10-20mg |
20-30mg |
30-50mg |
>50mg |
Tổng liều |
Phần trăm |
|
|
0-1 |
|
|
|
|
|
|
|
|
|
|
1-2 |
|
|
|
|
|
|
|
|
|
|
2-4 |
|
|
|
|
|
|
|
|
|
|
4-12 |
|
|
|
|
|
|
|
|
|
|
12-24 |
|
|
|
|
|
|
|
|
|
|
24-48 |
|
|
|
|
|
|
|
|
|
|
48-96 |
|
|
|
|
|
|
|
|
|
|
>96 |
|
|
|
|
|
|
|
|
|
|
Tổng thời gian |
|
|
|
|
|
|
|
|
|
|
Phần trăm |
|
|
|
|
|
|
|
|
|
Có thể sử dụng các bảng tương tự để trình bày về liều trung vị, phương thức liều và liều tối đa hoặc liều sử dụng lâu nhất. Có thể dùng cùng bảng như vậy để gộp các nghiên cứu và phân nhóm theo tuổi, giới, chủng tộc, bệnh đồng thời, thuốc dùng đồng thời hoặc sự kết hợp các yếu tố này.
Liều dùng có thể tính theo mg/kg, mg/m2, hoặc dưới dạng nồng độ trong huyết tương nếu có dữ liệu này.
|
Bảng 4.2 Các đặc điểm về nhân khẩu học của bệnh nhân trong các nghiên cứu có đối chứng Ngày kết thúc nghiên cứu |
|||
|
|
Nhóm điều trị |
||
|
|
Thuốc nghiên cứu N= |
Giả dược N= |
Thuốc đối chứng có hoạt tính N= |
|
Tuổi Trung bình ± độ lệch chuẩn Giới hạn Nhóm <18 18-40 40-64 65-75 >75 |
|
|
|
|
Giới Nữ Nam |
|
|
|
|
Chủng tộc Châu á Người da đen Người da trắng Các chủng tộc khác |
|
|
|
|
Các yếu tố khác |
|
|
|
|
Bảng 4.3 Tần suất các biến cố ngoại ý qua tổng hợp các dữ liệu từ các nghiên cứu có đối chứng với giả dược và đối chứng với thuốc có hoạt tính |
|||||||
|
Hệ cơ quan/ biến cố ngoại ý |
Thuốc nghiên cứu |
Giả dược |
Chất đối chứng có hoạt tính 1 |
Chất đối chứng có hoạt tính 2 |
|||
|
|
Tất cả các liều n= 1685 |
10mg n= 968 |
20mg n=717 |
n= 425 |
20mg n=653 |
50mg n=334 |
100mg n= 546 |
|
Tổng thể |
|
|
|
|
|
|
|
|
Chóng mặt |
|
|
|
|
|
|
|
|
... |
|
|
|
|
|
|
|
|
Tim mạch |
|
|
|
|
|
|
|
|
Hạ HA thế đứng |
|
|
|
|
|
|
|
|
... |
|
|
|
|
|
|
|
|
Tiêu hóa |
|
|
|
|
|
|
|
|
Táo bón |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bảng 4.4 Tần suất các biến cố ngoại ý của từng nghiên cứu |
||||||||
|
|
Tần suất biến cố ngoại ý của các nhóm điều trị được báo cáo |
|||||||
|
Hệ cơ quan/ biến cố ngoại ý |
Nghiên cứu 95-0403 |
Nghiên cứu 96-0011 |
Nghiên cứu97-0007 |
Nghiên cứu 98-0102s |
||||
|
|
Thuốc x 60mg bid N=104 |
Thuốc x 30mg bid N=102 |
Giả dược N=100 |
Thuốc x 60mg bid N=500 |
Giả dược n= 495 |
Thuốc x 60mg bid N=200 |
Thuốc y 100mg qd N=200 |
Thuốc x 60mg bid N=800 |
|
Tổng thể |
|
|
|
|
|
|
|
|
|
Chóng mặt |
|
|
|
|
|
|
|
|
|
... |
|
|
|
|
|
|
|
|
|
Tim mạch |
|
|
|
|
|
|
|
|
|
Hạ HA thế đứng |
|
|
|
|
|
|
|
|
|
... |
|
|
|
|
|
|
|
|
|
Tiêu hóa |
|
|
|
|
|
|
|
|
|
Táo bón |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bảng 4.5 Đối tượng rút khỏi nghiên cứu1: Nghiên cứu có đối chứng Ngày kết thúc nghiên cứu: |
|||||||||
|
Nghiên cứu |
Tổng số bệnh nhân rút khỏi nghiên cứu |
Lý do rút khỏi nghiên cứu |
Số lượng bệnh nhân được đưa vào dữ liệu về hiệu quả |
||||||
|
|
|
Tổng số |
Nữ/ nam |
Tuổi >65 |
Chủng tộc (xác định nhóm) /// |
Biến cố ngoại ý N (%) |
Không hiệu quả N (%) |
Các lý do khác N (%) |
N (%) |
|
Nghiên cứu |
Thuốc X |
N (%) |
N (%)/N (%) |
N (%) |
N (%)/N (%)/N (%) |
|
|
|
|
|
XXX |
Giả dược |
|
|
|
|
|
|
|
|
|
Nghiên cứu |
Thuốc X |
|
|
|
|
|
|
|
|
|
AAA |
Chất đối chứng A |
|
|
|
|
|
|
|
|
|
Nghiên cứu |
Thuốc X |
|
|
|
|
|
|
|
|
|
BBB |
Chất đối chứng B |
|
|
|
|
|
|
|
|
|
Nghiên cứu |
Thuốc X |
|
|
|
|
|
|
|
|
|
CCC |
Chất đối chứng C |
|
|
|
|
|
|
|
|
|
Tất cả NC |
|
|
|
|
|
|
|
|
|
Ghi chú: Dữ liệu về đối tượng rút khỏi nghiên cứu có thể chia nhóm theo liều dùng nếu như thấy hữu ích.
1 Đối tượng rút khỏi nghiên cứu là tất cả các đối tượng nghiên cứu đã được tuyển vào nghiên cứu nhưng không hoàn tất đợt điều trị theo kế hoạch (kể cả các đối tượng ngưng thuốc hoặc chuyển sang trị liệu khác sớm hơn dự kiến và/hoặc không được theo dõi.
|
Bảng 4.6 Danh sách các bệnh nhân tử vong Trị liệu: thuốc nghiên cứu Ngày kết thúc nghiên cứu |
||||||||||
|
NC/ nguồn khác1 |
Trung tâm |
Mã số bệnh nhân |
Tuổi |
Giới |
Thời gian sử dụng thuốc (ngày) |
Chẩn đoán |
Nguyên nhân tử vong |
Các thuốc sử dụng khác |
Các bệnh lý khác |
Vị trí của bản mô tả chi tiết |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 PM= tử vong ghi nhận được sau khi đưa thuốc ra thị trường
Danh sách này nên bao gồm tất cả các bệnh nhân tử vong trong các thử nghiệm lâm sàng hoặc ghi nhận được từ các nguồn khác như thông tin sau khi đưa thuốc ra thị trường. Đối với các hồ sơ điện tử cần cung cấp đường kết nối vào bản tường thuật tóm tắt về tử vong hoặc các tài liệu khác về biến cố này.
Cần có chú thích ở cuối trang mô tả nguyên tắc đưa các trường hợp tử vong vào trong bảng, ví dụ tất cả các trường hợp tử vong xảy ra trong quá trình sử dụng thuốc hoặc cho đến 30 ngày sau khi ngưng thuốc và cũng có thể bao gồm các trường hợp tử vong xảy ra trễ hơn nhưng do biến cố ngoại ý đã xuất hiện trong quá trình sử dụng thuốc hoặc trong vòng 30 ngày của quá trình theo dõi. Các nguyên tắc khác cũng có thể thích hợp.
Cũng cần cung cấp bảng danh sách các bệnh nhân sử dụng giả dược hoặc các thuốc đối chứng có hoạt tính.
Phần D. Bảng danh sách tất cả các nghiên cứu lâm sàng
Cần trình bày một bảng liệt kê tất cả các nghiên cứu lâm sàng và các thông tin liên quan. Đối với từng nghiên cứu, bảng liệt kê thường trình bày những thông tin đã được xác định trong bảng 1 của hướng dẫn này. Có thể thêm các thông tin khác vào bảng nếu cơ sở đăng ký thấy hữu ích. Trình tự liệt kê các nghiên cứu nên theo trình tự mô tả trong phần E: Báo Cáo Nghiên Cứu Lâm Sàng.
Bảng 1. Danh sách các nghiên cứu lâm sàng
|
Loại nghiên cứu |
Mã số nghiên cứu |
Vị trí của báo cáo nghiên cứu |
Mục tiêu nghiên cứu |
Thiết kế nghiên cứu và kiểu kiểm chứng |
Sản phẩm nghiên cứu; phác đồ liều; đường dùng |
Số lượng đối tượng nghiên cứu |
Người khỏe mạnh hoặc chẩn đoán của bệnh nhân |
Thời gian điều trị |
Tình trạng của nghiên cứu; loại báo cáo |
|
NC khả dụng sinh học |
001 |
Vol 3, phần 1.1, trang 183 |
Sinh khả dụng tuyệt đối: IV so với viên nén |
Thiết kế chéo (cross- over) |
Viên nén, 50mg, liều đơn, uống, 10mg IV |
20 |
Người khỏe mạnh |
Liều đơn |
NC đã hoàn tất; báo cáo tóm tắt |
|
NC tương đương sinh học |
|
|
|
|
|
|
|
|
|
|
NC dược động học |
|
|
|
|
|
|
|
|
|
|
NC dược lực học |
|
|
|
|
|
|
|
|
|
|
NC hiệu quả |
|
|
|
|
|
|
|
|
|
Phần E. Báo cáo nghiên cứu lâm sàng
Mở Đầu
Đối với các nước thành viên ASEAN, các báo cáo nghiên cứu trong phần này có thể không quy định đối với sản phẩm chứa hoạt chất mới (NCE), sản phẩm công nghệ sinh học và những sản phẩm có thay đổi lớn nếu các sản phẩm gốc đã được đăng ký và cấp phép lưu hành ở các nước tham khảo. Do đó, nếu như cơ quan quản lý nào yêu cầu báo cáo nghiên cứu cụ thể nào thì có thể yêu cầu nộp tài liệu cần thiết đó. Hướng dẫn ICH E3 có các chỉ dẫn về bố cục các báo cáo nghiên cứu lâm sàng, các dữ liệu lâm sàng khác và tài liệu tham khảo trong hồ sơ kỹ thuật chung ASEAN (ACTD) cho đăng ký dược phẩm dùng cho người. Trong trường hợp này, cơ sở đăng ký sẽ nộp hồ sơ ở phần A,B,C,D và F.
Bố cục của các báo cáo lâm sàng và các thông tin liên quan
A. Mục lục các báo cáo nghiên cứu lâm sàng B. Bảng liệt kê tất cả các nghiên cứu lâm sàng C. Báo cáo nghiên cứu lâm sàng
1. Báo cáo nghiên cứu sinh dược học
1.1. Báo cáo nghiên cứu sinh khả dụng (BA)
1.2. Báo cáo nghiên cứu so sánh sinh khả dụng (BA) và tương đương sinh học (BE)
1.3. Báo cáo nghiên cứu tương quan in vitro - in vivo
1.4. Báo cáo các phương pháp phân tích sinh học và phân tích sử dụng cho các nghiên cứu ở người
2. Báo cáo các nghiên cứu liên quan đến dược động học (PK) sử dụng nguyên liệu sinh học từ người
2.1. Các báo cáo nghiên cứu sự gắn kết với protein huyết tương
2.2. Các báo cáo nghiên cứu chuyển hóa ở gan và tương tác thuốc
2.3. Các báo cáo nghiên cứu sử dụng các nguyên liệu sinh học từ người khác
3. Báo cáo các nghiên cứu về dược động học (PK) trên người
3.1. Các báo cáo nghiên cứu về PK và sự dung nạp ban đầu trên người khỏe mạnh
3.2. Các báo cáo nghiên cứu về PK và sự dung nạp ban đầu trên bệnh nhân.
3.3. Các báo cáo nghiên cứu về PK trên dân số
4. Các báo cáo nghiên cứu về dược lực học (PD) trên người
4.1. Các báo cáo nghiên cứu về PD và PK/PD trên người khỏe mạnh
4.2. Các báo cáo nghiên cứu về PD và PK/PD trên bệnh nhân
5. Các báo cáo nghiên cứu về hiệu quả và tính an toàn
5.1. Báo cáo các nghiên cứu lâm sàng có đối chứng liên quan đến chỉ định đề nghị
5.2. Báo cáo các nghiên cứu lâm sàng không có đối chứng
5.3. Báo cáo phân tích các dữ liệu từ nhiều nghiên cứu, bao gồm tất cả các phân tích tích hợp chính thức (formal integrated analysis), phân tích gộp (meta-analysis) và phân tích bắc cầu (bridging analysis)
5.4. Các báo cáo nghiên cứu lâm sàng khác
6. Báo cáo các kinh nghiệm sau khi đưa thuốc ra thị trường
7. Mẫu báo cáo dữ liệu và danh sách các bệnh nhân
Hướng Dẫn Về Bố Cục Các Báo Cáo Nghiên Cứu Lâm Sàng Và Các Thông Tin Có Liên Quan
Hướng dẫn này khuyến nghị cách bố cục cụ thể để trình bày các báo cáo nghiên cứu lâm sàng và các thông tin có liên quan nhằm đơn giản hóa việc chuẩn bị và thẩm định các hồ sơ tài liệu và đảm bảo chúng đầy đủ. Vị trí của một báo cáo phải được xác định bởi mục tiêu chủ yếu của nghiên cứu. Mỗi báo cáo nghiên cứu chỉ nên xuất hiện ở một phần. Nếu nghiên cứu có nhiều mục tiêu, thì cần nêu tham chiếu chéo ở các phần khác nhau.
Cần có lời chú giải ví dụ "không áp dụng" hoặc "không có nghiên cứu" khi không có báo cáo hoặc thông tin nào cho 1 phần hay mục nào đó.
A. Mục lục các báo cáo nghiên cứu
Cần có mục lục cho tất cả các báo cáo nghiên cứu.
B. Bảng liệt kê tất cả các nghiên cứu lâm sàng
Cần có bảng liệt kê tất cả các nghiên cứu lâm sàng và thông tin có liên quan. Với mỗi nghiên cứu, bảng liệt kê thường chỉ nêu các thông tin đã được xác định ở bảng 1 của hướng dẫn này. Những thông tin khác có thể đưa vào trong bảng này nếu cơ sở đăng ký thấy hữu ích. Trình tự các báo cáo được liệt kê cần thực hiện theo trình tự nêu ở phần C dưới đây.
Nếu trình bày theo thứ tự khác thì cần có chú thích và giải thích ở phần giới thiệu của bảng.
C. Các báo cáo nghiên cứu lâm sàng
1. Các báo nghiên cứu sinh dược học
Các nghiên cứu sinh khả dụng (BA) đánh giá tỷ lệ và mức độ phóng thích hoạt chất từ sản phẩm thuốc. Các nghiên cứu so sánh sinh khả dụng hoặc tương đương sinh học có thể dùng các tiêu chí dược động học, dược lực học, lâm sàng hoặc tiêu chí hòa tan in vitro, và có thể là nghiên cứu đơn liều hay đa liều. Nếu như mục tiêu chủ yếu của nghiên cứu là đánh giá dược động học của thuốc, nhưng bao gồm cả những thông tin về sinh khả dụng thì báo cáo nghiên cứu này nên được trình bày trong mục 3.1 và nêu tham chiếu trong các phần 1.1 và/hoặc 1.2.
1.1 Các báo cáo nghiên cứu về sinh khả dụng
Các báo cáo nghiên cứu về sinh khả dụng trong phần này cần bao gồm: 1/ các nghiên cứu so sánh độ phóng thích dược chất và tỷ lệ dược chất trong tuần hoàn toàn thân của dạng bào chế rắn dùng đường uống, và sự có mặt trong tuần hoàn toàn thân của dược chất dùng đường tiêm tĩnh mạch hoặc là dạng bào chế lỏng dùng đường uống, 2/ các nghiên cứu về tỷ lệ thành phần của thuốc và 3/ các nghiên cứu về tác động của thức ăn.
1.2. Các báo cáo nghiên cứu so sánh sinh khả dụng và tương đương sinh học Các nghiên cứu trong phần này so sánh tỷ lệ và mức độ phóng thích dược chất từ các sản phẩm thuốc tương tự nhau (ví dụ viên nén so với viên nén, viên nén so với viên nang). Các nghiên cứu so sánh sinh khả dụng hoặc tương đương sinh học có thể bao gồm các so sánh giữa: 1) sản phẩm thuốc dùng trong các nghiên cứu lâm sàng đánh giá hiệu quả của thuốc và sản phẩm thuốc dự kiến đưa ra thị trường, 2) sản phẩm thuốc dùng trong các nghiên cứu lâm sàng đánh giá hiệu quả của thuốc và sản phẩm thuốc được sử dụng trong các lô nghiên cứu độ ổn định và 3) các sản phẩm thuốc tương tự của các nhà sản xuất khác nhau.
1.3. Các báo cáo nghiên cứu tương quan in vitro-in vivo
Cần trình bày các nghiên cứu độ hòa tan in vitro cung cấp thông tin về sinh khả dụng, kể cả các nghiên cứu nhằm đánh giá tương quan giữa các dữ liệu in vitro với các dữ liệu in vivo trong mục 1.3.
Các báo cáo về thử nghiệm hòa tan in vitro dùng để kiểm tra chất lượng lô sản phẩm và/hoặc để xuất lô thì trình bày trong phần hồ sơ chất lượng của ACTD.
1.4 Các báo cáo về phương pháp phân tích sinh học và phân tích dùng trong các nghiên cứu trên người
Thường thì nên cung cấp các phương pháp phân tích sinh học và/hoặc phân tích sử dụng trong các nghiên cứu sinh dược học hoặc các nghiên cứu độ hòa tan in vitro trong từng báo cáo nghiên cứu riêng rẻ. Khi một phương pháp được sử dụng trong nhiều nghiên cứu, thì nên trình bày một lần phương pháp này và kết quả thẩm định phương pháp ở trong mục 1.4. và đưa tham chiếu vào từng báo cáo nghiên cứu thích hợp.
2. Các báo cáo nghiên cứu liên quan đến sinh dược học sử dụng các nguyên liệu sinh học từ người
Nguyên liệu sinh học từ người là một thuật ngữ dùng để chỉ các protein, tế bào, mô và các mẫu sinh học khác có nguồn gốc từ con người dùng trong các nghiên cứu in vitro hoặc ex vivo nhằm đánh giá các đặc tính dược động học của dược chất. Ví dụ các tế bào kết tràng người được nuôi cấy dùng để đánh giá khả năng thấm qua màng sinh học và các quá trình vận chuyển, albumin người dùng để đánh giá sự gắn kết với protein huyết tương. Đặc biệt quan trọng là việc sử dụng các nguyên liệu sinh học từ người như các tế bào gan và/hoặc các vi lạp thể (microsome) của gan để nghiên cứu các đường chuyển hóa và đánh giá tương tác giữa các thuốc trong quá trình chuyển hóa này.
Các nghiên cứu có sử dụng nguyên liệu sinh học từ người để đánh giá những đặc tính khác (ví dụ như độ vô trùng hay dược lực học) không nên trình bày trong phần các báo cáo nghiên cứu lâm sàng, mà nên trình bày trong phần nghiên cứu tiền lâm sàng (Phần III).
2.1 Các báo cáo nghiên cứu về sự gắn kết với protein huyết tương
Cần cung cấp các báo cáo nghiên cứu về sự gắn kết với protein ex vivo ở phần này.
Cần cung cấp các dữ liệu về sự gắn kết với protein từ các nghiên cứu PK trong máu và/ hoặc huyết tương trong mục 3.
2.2 Các báo cáo nghiên cứu chuyển hóa ở gan và tương tác thuốc
Các báo cáo nghiên cứu về chuyển hóa ở gan và tương tác thuốc trong chuyển hóa sử dụng các mô gan cần được trình bày ở phần này.
2.3 Các nghiên cứu sử dụng các nguyên liệu sinh học từ người khác
Trong phần này cần trình bày các báo cáo nghiên cứu sử dụng các nguyên liệu sinh học từ người khác.
3. Các báo cáo nghiên cứu dược động học (PK) trên người
Việc đánh giá PK của một thuốc trên người khỏe mạnh và/hoặc bệnh nhân được coi là hết sức quan trọng trong việc thiết kế một chiến lược xác định liều lượng và các bước chuẩn liều, trong tiên đoán các tác động của các thuốc dùng đồng thời, trong việc diễn giải những khác biệt về dược lực học quan sát được. Những đánh giá này nên gồm mô tả vận mệnh của thuốc trong cơ thể theo thời gian, tập trung vào việc xác định nồng độ tối đa trong huyết tương (nồng độ đỉnh), diện tích dưới đường cong, sự thanh thải, và sự tích luỹ thuốc chưa chuyển hóa và chất chuyển hóa của nó, đặc biệt là những chất chuyển hóa có hoạt tính dược lý. Các nghiên cứu PK cần có báo cáo trình bày trong mục 3.1 và 3.2, nhìn chung được thiết kế để: (1) đo lường nồng độ thuốc và chất chuyển hóa trong huyết tương theo thời gian, (2) đo lường nồng độ thuốc và chất chuyển hóa trong nước tiểu hoặc trong phân khi phép đo lường này là cần thiết hoặc có ích, và /hoặc (3) đo lường sự gắn kết của thuốc và chất chuyển hóa với hồng cầu hoặc protein.
Đôi khi, các nghiên cứu PK có thể bao gồm phép đo lường sự phân bố của thuốc trong các mô khác, các cơ quan khác hoặc dịch khác của cơ thể (như hoạt dịch hoặc dịch não tủy), và kết quả của các nghiên cứu về sự phân bố trong mô này cần được đưa vào trong mục 3.1 đến 3.2, nếu thích hợp. Những nghiên cứu này cần xác định đặc tính PK của thuốc và cung cấp các thông tin về sự hấp thu, phân bố, chuyển hóa và thải trừ của một thuốc và bất kỳ chất chuyển hóa nào ở người khỏe mạnh và/hoặc bệnh nhân. Cần đặc biệt quan tâm đến các nghiên cứu về sự cân bằng khối lượng và những thay đổi trong PK liên quan đến liều lượng (ví dụ xác định tỷ lệ liều lượng) hoặc thời gian (ví dụ do sự cảm ứng men hoặc sự hình thành kháng thể) và nên đưa vào mục 3.1 và hoặc 3.2. Ngoài việc mô tả PK trung bình ở những bệnh nhân và người tình nguyện khỏe mạnh, các nghiên cứu PK còn cần mô tả khoảng biến thiên ở các cá thể.
3.1. Các báo cáo nghiên cứu PK và sự dung nạp ban đầu trên người khỏe mạnh
Các báo cáo nghiên cứu PK và sự dung nạp ban đầu trên người khỏe mạnh nên được đưa vào phần này.
3.2. Các báo cáo nghiên cứu PK và sự dung nạp ban đầu trên bệnh nhân
Các báo cáo nghiên cứu PK và sự dung nạp ban đầu trên bệnh nhân nên được đưa vào phần này.
3.3 Các báo cáo nghiên cứu PK trên dân số
Các báo cáo nghiên cứu PK trên dân số dựa trên các mẫu rãi rác thu được qua các thử nghiệm lâm sàng, kể cả các thử nghiệm đánh giá hiệu quả và tính an toàn cần được đưa vào phần này.
4. Các báo cáo nghiên cứu dược lực học (PD) trên người
Các báo cáo nghiên cứu có mục tiêu chủ yếu là xác định tác dụng dược lực học của một sản phẩm thuốc trên người nên được trình bày trong phần này. Tuy nhiên, các báo cáo về các nghiên cứu với mục tiêu chủ yếu là xác lập hiệu quả hoặc thu thập các dữ liệu về an toàn thì nên được trình bày trong mục 5.
Phần này nên trình bày các báo cáo về 1) các nghiên cứu về đặc tính dược lý đã biết hoặc được cho là có liên quan đến các tác dụng lâm sàng mong đợi (các dấu hiệu sinh học), 2) các nghiên cứu ngắn hạn về tác dụng lâm sàng chính, và 3) các nghiên cứu dược lực học về các đặc tính khác không liên quan đến tác dụng lâm sàng mong đợi. Vì mối quan hệ định lượng giữa các tác dụng dược lý này với liều dùng và/hoặc nồng độ thuốc và chất chuyển hóa trong huyết tương thường rất được quan tâm, cho nên các thông tin về dược lực học thường được thu thập trong các nghiên cứu về đáp ứng với liều dùng, hoặc cùng với các thông tin về nồng độ thuốc trong các nghiên cứu dược động học (các nghiên cứu đáp ứng- nồng độ hoặc nghiên cứu dược động học/dược lực học). Mối quan hệ giữa tác dụng dược động học và dược lực học không thu được từ các nghiên cứu kiểm soát chặt chẽ thường được đánh giá bằng phương pháp thích hợp và dùng làm cho cơ sở cho việc thiết kế các nghiên cứu tiếp theo về liều- đáp ứng, hoặc trong một số trường hợp dùng để diễn giải sự khác nhau về nồng độ trong các nhóm dân số.
Các nghiên cứu xác định liều lượng, dược lực học và/hoặc dược động–dược lực học có thể tiến hành trên những người khỏe mạnh và/hoặc bệnh nhân, và cũng có thể kết hợp trong các nghiên cứu đánh giá tính an toàn và hiệu quả trong một chỉ định lâm sàng. Các báo cáo về các nghiên cứu xác định liều lượng, dược lực học và/hoặc dược động- dược lực học tiến hành trên người khỏe mạnh nên trình bày trong mục 4.1, và các báo cáo của các nghiên cứu tiến hành trên bệnh nhân nên trình bày trong mục 4.2.
Trong một số trường hợp, những thông tin về dược lực học ngắn hạn, xác định liều và/hoặc dược động- dược lực học có được từ các nghiên cứu dược lực học tiến hành trên bệnh nhân sẽ cung cấp các số liệu đóng góp vào việc đánh giá hiệu quả, có thể vì chúng cho thấy một tác dụng trên một dấu hiệu thay thế chấp nhận được (ví dụ: huyết áp) hoặc trên một tiêu chí về lợi ích lâm sàng (ví dụ: giảm đau). Tương tự như vậy, một nghiên cứu dược lực học có thể bao gồm các thông tin về an toàn lâm sàng quan trọng. Khi những nghiên cứu này được dùng làm một phần minh họa cho hiệu quả và tính an toàn, thì chúng được coi là các nghiên cứu về hiệu quả và tính an toàn nên được trình bày trong mục 5, chứ không phải là mục 4.
4.1. Các báo cáo nghiên cứu dược lực học và dược động/dược lực học trên người khỏe mạnh
Các nghiên cứu dược lực học và/hoặc dược động/dược lực học có mục đích phi điều trị trên người khỏe mạnh cần được trình bày ở phần này.
4.2. Các báo cáo nghiên cứu dược lực học và dược động/dược lực học trên bệnh nhân
Các báo cáo nghiên cứu dược lực học và dược đông/dược lực học trên bệnh nhân cần được trình bày trong phần này.
5. Các báo cáo nghiên cứu về hiệu quả và tính an toàn
Phần này nên có các báo cáo của tất cả các nghiên cứu lâm sàng về hiệu quả và/hoặc tính an toàn đã tiến hành với thuốc, được thực hiện bởi cơ sở tài trợ hay có được bằng cách nào đó, bao gồm tất cả các nghiên cứu đã hoàn thành và các nghiên cứu đang tiến hành trên thuốc đó cho các chỉ định đề nghị hoặc không đề nghị phê duyệt. Báo cáo nghiên cứu cần cung cấp các chi tiết thích hợp với nghiên cứu và nêu rõ vai trò của chúng. Hướng dẫn ICH E3 mô tả nội dung một báo cáo nghiên cứu đầy đủ để đưa ra các bằng chứng về cả tính an toàn và hiệu quả. Có thể cung cấp báo cáo tóm tắt cho một số nghiên cứu (xem ICH E3 và các hướng dẫn riêng của từng nước).
Trong mục 5, các nghiên cứu nên được bố cục theo thiết kế (có đối chứng, không có đối chứng) và trong số các nghiên cứu có đối chứng thì trình bày theo dạng đối chứng. Trong mỗi phần, các nghiên cứu nên được phân nhóm nhỏ hơn theo loại nghiên cứu có báo cáo đầy đủ hoặc báo cáo tóm tắt (ICH E3), trong đó các nghiên cứu có báo cáo đầy đủ trình bày trước. Các báo cáo đã công bố mà không có hoặc chỉ có rất giới hạn dữ liệu bổ sung cho các báo cáo của cơ sở tài trợ thì nên được trình bày cuối cùng trong phần này.
Trong trường hợp hồ sơ nộp xin đăng ký nhiều chỉ định, thì các báo cáo phải được bố cục gồm nhiều mục 5 riêng cho mỗi chỉ định. Trong trường hợp đó, nếu một nghiên cứu về hiệu quả lâm sàng chỉ liên quan đến một trong những chỉ định đăng ký, thì nó nên được trình bày trong tiểu mục thích hợp ở mục 5; nếu một nghiên cứu về hiệu quả lâm sàng liên quan đến nhiều chỉ định thì báo cáo nghiên cứu nên được đưa vào trong mục 5 phù hợp nhất và nêu tham chiếu khi cần ở các mục khác, ví dụ mục 5A , mục 5B.
5.1. Các báo cáo nghiên cứu lâm sàng có đối chứng liên quan đến chỉ định đăng ký
Các báo cáo nghiên cứu lâm sàng có đối chứng cần được sắp xếp theo dạng đối chứng:
o Đối chứng với giả dược (placebo) (có thể bao gồm các nhóm chứng khác, ví dụ như nhóm so sánh dùng thuốc có hoạt lực hoặc các liều dùng khác)
o Đối chứng với nhóm không điều trị
o Liều-đáp ứng (không dùng placebo)
o Chất đối chứng có hoạt tính (không dùng placebo)
o Đối chứng với nghiên cứu khác (đã thực hiện), bất kể có điều trị hay không.
Trong mỗi dạng đối chứng, để thích hợp với việc đánh giá tác dụng của thuốc, các nghiên cứu cần được bố cục theo thời gian điều trị. Các nghiên cứu về các chỉ định khác ngoài các chỉ định đăng ký, nhưng có hỗ trợ cho hiệu quả và cách dùng dự kiến thì cũng nên trình bày ở mục 5.1.
Một nghiên cứu dược lực học đóng góp bằng chứng về hiệu quả thì nó nên được đưa vào mục 5.1. Thứ tự mà các nghiên cứu này được tiến hành không liên quan đến việc trình bày chúng. Vì thế, các thử nghiệm có đối chứng với placebo dù ở giai đoạn nào của nghiên cứu cũng nên được trình bày trong mục 5.1. Các nghiên cứu về tính an toàn có đối chứng kể cả các nghiên cứu trong các điều kiện không liên quan đến mục đích của việc xin đăng ký, cũng phải được báo cáo trong mục 5.1.
5.2. Các báo cáo nghiên cứu lâm sàng không có đối chứng
Các báo cáo nghiên cứu của các nghiên cứu lâm sàng không có đối chứng (ví dụ các báo cáo từ các nghiên cứu an toàn mở) cần được đưa vào phần này. Nó bao gồm cả các nghiên cứu trong những điều kiện không liên quan đến mục đích xin phép lưu hành.
5.3. Các báo cáo phân tích dữ liệu có được từ nhiều nghiên cứu
Nhiều vấn đề lâm sàng trong một hồ sơ đăng ký có thể được đề cập đến bằng việc phân tích các dữ liệu có được từ nhiều nghiên cứu. Kết quả của việc phân tích như vậy nhìn chung có thể tóm tắt trong các tài liệu tóm tắt lâm sàng, nhưng việc mô tả và trình bày chi tiết các kết quả phân tích đó lại được coi là cực kỳ quan trọng giúp cho việc diễn giải kết quả phân tích. Khi các phân tích đó gồm quá nhiều chi tiết không thể tóm lược vào trong một tài liệu tóm tắt, thì chúng cần phải được trình bày trong một báo cáo riêng. Những báo cáo như vậy cần được trình bày ở mục 5.3. Ví dụ về các báo cáo trong phần này gồm: một báo cáo về một phân tích gộp (meta-analysis) chính thức hoặc một phân tích thăm dò hiệu quả trên diện rộng nhằm xác định một ước đoán tổng thể về mức hiệu quả trên tất cả bệnh nhân và/hoặc trong một nhóm dân số đặc biệt, và một báo cáo phân tích tích hợp (integrated analysis) về tính an toàn trong đó đánh giá những yếu tố ví dụ như cơ sở dữ liệu về tính an toàn đã đầy đủ hay chưa, ước đoán tỷ lệ biến cố ngoại ý, và độ an toàn đối với những thay đổi, ví dụ thay đổi về liều dùng, về nhân khẩu học, và về các thuốc dùng đồng thời.
5.4. Các báo cáo nghiên cứu lâm sàng khác
Phần này có thể bao gồm:
- Các báo cáo phân tích tạm thời về các nghiên cứu liên quan đến chỉ định đăng ký.
- Các báo cáo từ các nghiên cứu có đối chứng về tính an toàn chưa trình bày ở những phần khác.
- Các báo cáo các nghiên cứu có hoặc không có đối chứng không liên quan đến chỉ định đăng ký.
- Các báo cáo đã công bố về các kinh nghiệm lâm sàng của sản phẩm thuốc này mà chưa được đưa vào mục 5.1. Tuy nhiên, khi các tài liệu khoa học có giá trị quan trọng trong việc minh họa hoặc là chứng minh về hiệu quả, thì chúng cần phải được trình bày trong phần 5.1.
- Các báo cáo về các nghiên cứu đang tiến hành.
6. Báo cáo về những kinh nghiệm sau khi đưa thuốc ra thị trường
Đối với những sản phẩm hiện đang được lưu hành, thì trong mục 6 cần trình bày các báo cáo tóm tắt những kinh nghiệm thu thập được trong quá trình lưu hành thuốc (kể cả những ghi nhận đáng chú ý về an toàn).
7. Mẫu báo cáo dữ liệu và danh sách dữ liệu của từng bệnh nhân
Các mẫu báo cáo dữ liệu và danh sách dữ liệu của từng bệnh nhân như mô tả trong phụ lục 16.3 và 16.4 trong hướng dẫn báo cáo nghiên cứu lâm sàng của ICH khi được nộp thì phải để ở phần này, theo thứ tự như các báo cáo nghiên cứu lâm sàng và chú dẫn theo nghiên cứu.
Bảng 1. Danh sách các nghiên cứu lâm sàng
|
Loại nghiên cứu |
Mả số nghiên cứu |
Vị trí của báo cáo nghiên cứu |
Mục tiêu nghiên cứu |
Thiết kế nghiên cứu và kiểu đối chứng |
Sản phẩm nghiên cứu; phác đồ liều; đường dùng |
Số lượng đối tượng nghiên cứu |
Người khỏe mạnh hoặc chẩn đoán của bệnh nhân |
Thời gian điều trị |
Tình trạng của nghiên cứu; loại báo cáo |
|
NC sinh khả dụng(BA) |
001 |
Vol 3,phần 1.1, trang 183 |
Sinh khả dụng tuyệt đối: IV so với viên nén |
Thiết kế chéo (cross- over) |
Viên nén,50mg, liều đơn, uống, 10mg IV |
20 |
Người khỏe mạnh |
Liều đơn |
NC đã hoàn tất; báo cáo tóm tắt |
|
NC tương đương sinh học (BE) |
|
|
|
|
|
|
|
|
|
|
NC dược động học(PK) |
|
|
|
|
|
|
|
|
|
|
NC dược lực học(PD) |
|
|
|
|
|
|
|
|
|
|
NC hiệu quả |
|
|
|
|
|
|
|
|
|
Phần F: Danh Mục Các Tài Liệu Tham Khảo Chủ Yếu
Phần này cần trình bày danh mục các tài liệu tham khảo, bao gồm các bài báo quan trọng đã công bố, biên bản các cuộc họp chính thức hoặc các hướng dẫn hay các thông báo về quản lý khác. Trong đó bao gồm tất cả các tài liệu tham khảo trích dẫn trong phần tổng quan lâm sàng và các tài liệu tham khảo trích dẫn trong phần tóm tắt lâm sàng hay trong từng báo cáo kỹ thuật có trong phần báo cáo nghiên cứu lâm sàng. Điểm cuối cùng cần lưu ý là phải có bản chụp các tài liệu tham khảo đó để trình nộp khi có yêu cầu.
DANH MỤC KIỂM TRA PHẦN TÀI LIỆU LÂM SÀNG TRONG ACTD THEO PHÂN LOẠI ĐĂNG KÝ THUỐC
(Hồ sơ kỹ thuật chung ASEAN-ACTD về đăng ký thuốc)
|
PHẦN IV: TÀI LIỆU LÂM SÀNG |
NCE |
BIOTECH |
MaV |
MiV |
GP |
||
|
RT |
ST/P |
IND |
|||||
|
Phần A: Mục lục |
ü |
ü |
ü |
ü |
ü |
- |
- |
|
Phần B: Tổng quan lâm sàng 1. Cơ sở phát triển sản phẩm 2. Tổng quan về Sinh dược học 3. Tổng quan về Dược lý lâm sàng 4. Tổng quan về Hiệu quả 5. Tổng quan về An toàn 6. Kết luận về Lợi ích và Nguy cơ |
ü |
ü |
ü |
ü |
ü |
- |
- |
|
Phần C: Tóm tắt lâm sàng 1. Tóm tắt các nghiên cứu về sinh dược học và phương pháp phân tích 1.1 Cơ sở nghiên cứu và tổng quan 1.2 Tóm tắt kết quả các nghiên cứu riêng lẻ 1.3 So sánh và phân tích kết quả xuyên suốt các nghiên cứu Phụ lục 1 2. Tóm tắt các nghiên cứu về dược lý lâm sàng 2.1 Cơ sở nghiên cứu và tổng quan 2.2 Tóm tắt kết quả các nghiên cứu riêng lẻ 2.3 So sánh và phân tích kết quả xuyên suốt các nghiên cứu 2.4 Các nghiên cứu đặc biệt Phụ lục 2 3. Tóm tắt về hiệu quả lâm sàng 3.1 Cơ sở nghiên cứu và tổng quan về hiệu quả lâm sàng 3.2 Tóm tắt kết quả các nghiên cứu riêng lẻ 3.3 So sánh và phân tích kết quả xuyên suốt các nghiên cứu 3.4 Phân tích các thông tin lâm sàng liên quan đến các khuyến cáo về liều dùng 3.5 Sự duy trì hiệu quả và/hoặc sự lờn thuốc Phụ lục 3 4. Tóm tắt về tính an toàn lâm sàng 4.1 Mức độ sử dụng thuốc 4.2 Biến cố ngoại ý 4.3 Đánh giá kết quả xét nghiệm 4.4 Dấu hiệu sinh tồn, triệu chứng thực thể và các ghi nhận khác liên quan đến tính an toàn 4.5 Sự an toàn đối với các nhóm bệnh nhân đặc biệt và các tình huống đặc biệt Phụ lục 4 5. Bản tóm tắt các nghiên cứu riêng lẻ |
ü |
ü |
ü |
ü |
ü |
- |
- |
|
Phần D: Bảng danh sách tất cả các nghiên cứu lâm sàng |
ü |
ü |
ü |
ü |
ü |
- |
- |
|
Phần E: Báo cáo nghiên cứu lâm sàng 1. Báo cáo các nghiên cứu sinh dược học Báo cáo nghiên cứu sinh khả dụng (BA) Báo cáo nghiên cứu so sánh sinh khả dụng hoặc tương đương sinh học (BE) Báo cáo nghiên cứu tương quan in vitro-in vivo Báo cáo các phương pháp phân tích sinh học và phương pháp phân tích sử dụng cho các nghiên cứu ở người 2. Báo cáo các nghiên cứu liên quan đến dược động học (PK) sử dụng nguyên liệu sinh học từ người Báo cáo nghiên cứu sự gắn kết với protein huyết tương Báo cáo nghiên cứu sự chuyển hóa ở gan và tương tác thuốc Báo cáo các nghiên cứu sử dụng nguyên liệu sinh học từ người khác 3. Báo cáo các nghiên cứu về dược động học (PK) trên người Báo cáo nghiên cứu về PK và sự dung nạp ban đầu trên người khỏe mạnh Báo cáo nghiên cứu về PK và sự dung nạp ban đầu trên bệnh nhân Báo cáo nghiên cứu PK trên dân số 4. Báo cáo các nghiên cứu về dược lực học (PD) trên người Báo cáo nghiên cứu về PK và PK/PD trên người khỏe mạnh Báo cáo nghiên cứu về PK và PK/PD trên bệnh nhân 5. Báo cáo các nghiên cứu về hiệu quả và tính an toàn Báo cáo các nghiên cứu lâm sàng có đối chứng liên quan đến chỉ định đề nghị Báo cáo các nghiên cứu lâm sàng không có đối chứng Báo cáo phân tích các dữ liệu từ nhiều nghiên cứu, bao gồm tất cả các phân tích tích hợp (integrated analysis), phân tích gộp (meta-analysis) và phân tích bắc cầu (bridging analysis) chính thức Các báo cáo nghiên cứu lâm sàng khác 6. Báo cáo các kinh nghiệm sau khi đưa thuốc ra thị trường 7. Mẫu báo cáo dữ liệu và danh sách các bệnh nhân |
ü |
ü |
ü |
ü |
ü |
- |
- |
|
Phần F: Danh mục các tài liệu tham khảo chủ yếu |
ü |
ü |
ü |
ü |
ü |
- |
- |
HƯỚNG DẪN CỦA ASEAN VỀ NGHIÊN CỨU ĐỘ ỔN ĐỊNH CỦA THUỐC
1. ĐẶT VẤN ĐỀ
1.1. Độ ổn định là nhân tố quan trọng có ảnh hưởng đến chất lượng, an toàn hiệu quả của thành phẩm thuốc. Thuốc kém ổn định có thể dẫn tới sự thay đổi các đặc tính vật lý (như độ cứng, tốc độ hòa tan, sự tách pha...) cũng như hóa học (sự tạo thành các chất phân hủy có hoạt tính mạnh). Sự kém ổn định về mặt vi sinh của thành phẩm thuốc vô khuẩn có thể dẫn tới các rủi ro.
1.2. Về nguyên tắc, thử nghiệm độ ổn định nên được thực hiện ở các điều kiện khắc nghiệt hơn là ở các điều kiện ít khắc nghiệt để đảm bảo sai số thừa ưu tiên cho hiệu quả điều trị thuốc trên bệnh nhân và để tăng xác suất phát hiện chất hay công thức bào chế có vấn đề về độ ổn định.
1.3. Mục tiêu của nghiên cứu độ ổn định là xác định tuổi thọ, đó là khoảng thời gian bảo quản ở một điều kiện xác định mà trong khoảng thời gian đó thành phẩm thuốc vẫn đạt tiêu chuẩn chất lượng đã được thiết lập.
1.4. Nghiên cứu độ ổn định bao gồm một chuỗi các thử nghiệm để đảm bảo độ ổn định của một thành phẩm thuốc, đó là khả năng duy trì các tiêu chuẩn chất lượng của thành phẩm thuốc được đóng gói trong bao bì phù hợp cho thành phẩm đó và bảo quản ở điều kiện đã thiết lập trong một khoảng thời gian xác định.
1.5. Điều kiện chung cho thử nghiệm độ ổn định dài hạn ở khu vực ASEAN là điều kiện Vùng IVB (nhiệt độ 30oC, độ ẩm tương đối 75%).
2. MỤC TIÊU
Hướng dẫn này đưa ra những gợi ý tổng quát cho nghiên cứu độ ổn định đối với các thành phẩm thuốc, tuy vậy vẫn có sự linh hoạt trong những hoàn cảnh thực tế khác nhau, có xem xét đến tính khoa học đặc thù và các đặc tính của các thành phẩm được đánh giá. Hướng dẫn này cũng có thể sử dụng để ước lượng tuổi thọ dựa trên các số liệu độ ổn định thu được từ nghiên cứu.
3. PHẠM VI
Hướng dẫn này đưa ra những thông tin phải có trong hồ sơ đăng ký lưu hành các thành phẩm thuốc ở các nước ASEAN, bao gồm cả các mẫu quy trình nghiên cứu độ ổn định, mẫu báo cáo, thiết kế rút gọn và việc ngoại suy số liệu, và ví dụ về các loại, độ dày, hệ số thấm của vật liệu bao gói đã được nêu trong các phụ lục.
Hướng dẫn này được áp dụng cho các thành phẩm thuốc chứa dược chất mới (New Chemical entity - NCE), các thuốc generics và các thuốc có sự thay đổi trong quá trình lưu hành (thay đổi lớn MaV và thay đổi nhỏ MiV) mà không áp dụng cho vắc xin, sinh phẩm và các thành phẩm thuốc chứa vitamin và muối khoáng.
4 THIẾT KẾ
4.1 Tổng quát
Việc thiết kế nghiên cứu độ ổn định cho các thành phẩm thuốc cần dựa trên kiến thức về bản chất và các tính chất của dược chất và dạng bào chế.
4.2. Thử nghiệm độ ổn định đối với ánh sáng.
Thử nghiệm độ ổn định đối với ánh sáng cần được thực hiện tối thiểu với một lô đầu tiên của thành phẩm thuốc nếu thích hợp. Những điều kiện chuẩn trong thử nghiệm độ ổn định đối với ánh sáng được mô tả trong ICH Q1B.
4.3. Lựa chọn lô thử
Vào thời điểm nộp hồ sơ đăng ký, phải cung cấp các dữ liệu thử độ ổn định trên các lô thuốc có cùng một công thức bào chế và cùng dạng bào chế trong hệ thống bao bì đóng gói như dự kiến lưu hành trên thị trường.
- Đối với NCE, phải cung cấp các dữ liệu độ ổn định của ít nhất ba lô đầu tiên.
- Đối với thuốc Generics và các thay đổi, có thể áp dụng những lựa chọn sau:
● Đối với các dạng bào chế qui ước (ví dụ: dạng thuốc rắn giải phóng ngay, dung dịch) và khi các dược chất là các chất bền vững, thì có thể chấp nhận số liệu độ ổn định thu được từ nghiên cứu thực hiện tối thiểu trên hai lô ở quy mô thử nghiệm (pilot).
● Đối với các dạng bào chế đặc biệt (ví dụ các dạng thuốc giải phóng kéo dài) hoặc đối với các dược chất không bền vững, thì dữ liệu về độ ổn định phải được thu thập trên ba lô đầu tiên. Hai trong số ba lô đó ít nhất cũng phải ở quy mô thử nghiệm, lô thứ ba có thể ở quy mô nhỏ hơn, nếu có giải trình.
- Quy trình sản xuất đã áp dụng cho những lô đầu tiên phải là quy trình sẽ áp dụng cho các lô sản xuất ở quy mô công nghiệp và phải cho ra sản phẩm có cùng chất lượng và đạt cùng tiêu chuẩn chất lượng như sản phẩm dự định lưu hành.
- Nếu có thể, các lô thành phẩm thuốc nên được sản xuất từ các lô nguyên liệu dược chất khác nhau.
- Các nghiên cứu độ ổn định phải được thực hiện trên mỗi hàm lượng và mỗi loại cỡ đóng gói của thành phẩm thuốc, trừ khi áp dụng thiết kế phân cực (ô trống) hoặc ma trận.
Có thể cung cấp các dữ liệu hỗ trợ khác.
4.4. Tiêu chuẩn chất lượng (Chỉ tiêu thử nghiệm)
i. Tiêu chuẩn chất lượng là danh sách các chỉ tiêu chất lượng, với quy trình phân tích kèm theo và các mức chất lượng bao gồm cả các mức chất lượng khác nhau giữa tiêu chuẩn chất lượng khi xuất xưởng và tiêu chuẩn chất lượng khi lưu hành.
ii. Mức chất lượng khi lưu hành nên xây dựng dựa trên việc xem xét tất cả các thông tin độ ổn định có sẵn. Sự khác nhau giữa mức chất lượng khi xuất xưởng và khi lưu hành có thể được điều chỉnh một cách hợp lý dựa trên đánh giá về độ ổn định và các biến đổi quan sát được trong quá trình bảo quản. Bất kỳ sự khác nhau nào giữa mức chất lượng khi xuất xưởng và khi lưu hành của hàm lượng chất kháng vi sinh vật phải được chứng minh bằng mối tương quan đã được thẩm định giữa hàm lượng hóa học và hiệu quả bảo quản trong công thức bào chế cuối cùng (trừ nồng độ chất kháng vi sinh vật) dự định thương mại hóa thu được từ quá trình phát triển sản phẩm. Hiệu quả của chất kháng vi sinh vật (bên cạnh hàm lượng chất kháng vi sinh vật) nên được đánh giá trên một lô đầu tiên của thành phẩm thuốc tại thời điểm hết hạn dùng đề xuất nhằm mục đích thẩm tra, không phụ thuộc vào việc có sự khác nhau giữa mức chất lượng khi xuất xưởng và khi lưu hành của hàm lượng chất kháng vi sinh vật hay không.
4.5. Các chỉ tiêu chất lượng cần đánh giá
i. Nghiên cứu độ ổn định phải bao gồm việc đánh giá các đặc tính của thành phẩm thuốc dễ biến đổi trong quá trình bảo quản và có thể ảnh hưởng đến chất lượng, độ an toàn và/hoặc hiệu quả. Các phép thử, nếu phù hợp, phải bao gồm các đặc tính vật lý, hóa học, sinh học, vi sinh học, hàm lượng chất bảo quản (ví dụ chất chống oxy hóa, chất kháng vi sinh vật) và các phép thử chức năng (ví dụ với hệ cung cấp thuốc). Quy trình phân tích phải được thẩm định đầy đủ và biểu thị được độ ổn định theo hướng dẫn của ASEAN về thẩm định phương pháp phân tích. Việc có phải lặp lại phép thử hay không và lặp lại ở mức độ nào sẽ phụ thuộc vào các kết quả từ các nghiên cứu thẩm định.
ii. Nhìn chung, đối với tất cả các dạng bào chế, cần phải đánh giá: tính chất, hàm lượng và các sản phẩm phân hủy. Với các thuốc Generic, mức chất lượng đối với sản phẩm phân hủy tối thiểu phải đáp ứng yêu cầu của dược điển. Danh sách các chỉ tiêu chất lượng cần đánh giá dưới đây cho mỗi dạng bào chế được xem như là gợi ý về các loại phép thử nên thực hiện trong nghiên cứu độ ổn định. Danh mục các phép thử đối với mỗi dạng bào chế không hẳn là đã đầy đủ và cũng không có nghĩa là mọi phép thử đã liệt kê đều phải đưa vào quy trình theo dõi độ ổn định đối với mỗi thành phẩm thuốc cụ thể (ví dụ: phép thử về mùi chỉ nên tiến hành khi cần thiết và phải xem xét đến tính an toàn cho người phân tích).
1. Viên nén
Viên nén cần được đánh giá về hình thức viên, mùi, màu sắc, định lượng, các sản phẩm phân hủy, độ hòa tan (hoặc độ rã, nếu có bàn luận), hàm lượng nước và độ cứng/độ bở.
2. Viên nang
Nang gelatin cứng cần được đánh giá về hình thức (kể cả sự rạn nứt), màu sắc, mùi của thành phần chứa trong nang, định lượng, các sản phẩm phân hủy, độ hòa tan, hàm lượng nước và giới hạn vi sinh vật.
Phép thử đối với nang mềm gelatin cần bao gồm các đánh giá về hình thức nang, màu sắc, mùi của thành phần chứa trong nang, định lượng, các sản phẩm phân hủy, độ hòa tan, giới hạn vi sinh vật, pH, độ rò rỉ, và sự hình thành màng. Thêm vào đó, cần kiểm tra sự kết tủa hay vẩn đục của thuốc đóng trong nang.
3. Nhũ tương
Nhũ tương cần được đánh giá về hình thức (kể cả sự tách pha), màu sắc, mùi, định lượng, các sản phẩm phân hủy, pH, độ nhớt, giới hạn vi sinh vật, hàm lượng chất bảo quản, kích thước trung bình và sự phân bố của giọt nhũ tương.
4. Dung dịch và hỗn dịch uống
Dung dịch và hỗn dịch uống cần được đánh giá về hình thức (kể cả sự hình thành kết tủa, độ trong của dung dịch), màu sắc, mùi, định lượng, các sản phẩm phân hủy, pH, độ nhớt, giới hạn vi sinh vật và hàm lượng chất bảo quản.
Thêm vào đó, đối với hỗn dịch, cần đánh giá khả năng tái phân tán, các tính chất lưu biến, kích thước trung bình và sự phân bố của các tiểu phân. Sau khi bảo quản, mẫu của các hỗn dịch cần được chuẩn bị cho việc định lượng theo chỉ dẫn ghi trên nhãn (chẳng hạn như lắc kỹ trước khi tiến hành định lượng).
5. Bột pha thành dịch lỏng khi uống
Bột pha thành dịch lỏng khi uống cần được đánh giá về hình thức, màu sắc, mùi, định lượng, các sản phẩm phân hủy, độ ẩm, và thời gian pha thành dịch lỏng.
Các sản phẩm đã pha thành dịch lỏng (dung dịch và hỗn dịch) cần được đánh giá các chỉ tiêu chất lượng như đã nêu ở mục Dung dịch và hỗn dịch uống sau khi pha lại như đã ghi trên nhãn trong thời gian sử dụng tối đa đã được ấn định.
6. Thuốc hít có van định liều và thuốc phun mù qua mũi
Thuốc hít có van định liều và thuốc phun mù qua mũi cần được đánh giá về hình thức (bao gồm chất chứa bên trong, bình/ống chứa thuốc, van và các thành phần của nó), màu sắc, vị, định lượng, các sản phẩm phân hủy, định lượng đồng dung môi (nếu có dùng), độ đồng đều hàm lượng phân liều, số lần ấn van một bình thuốc theo ghi trên nhãn đạt được độ đồng đều hàm lượng phân liều, phân bố kích thước tiểu phân khí lực học, đánh giá bằng kính hiển vi, hàm lượng nước, tốc độ rò rỉ, giới hạn vi sinh vật, sự phân phối thuốc của van (khối lượng thuốc được phun ra), các chất chiết/chất thôi ra từ các thành phần làm bằng chất dẻo và cao su. Các mẫu thành phẩm thử nghiệm cần được đặt cả theo hướng thẳng đứng và hướng lật ngược/nằm ngang.
Đối với thuốc phun mù dạng hỗn dịch, hình thức các bộ phận của van và chất chứa trong bình cần được đánh giá bằng kính hiển vi đối với các tiểu phân lớn và sự thay đổi hình thái bề mặt tiểu phân dược chất, mức độ kết tụ, sự hình thành tinh thể, cũng như tiểu phân lạ.
Những tiểu phân đó có thể gây tắc van hoặc làm cho sự phân liều không lặp lại. Sự ăn mòn mặt trong bình chứa hoặc sự thoái hóa của vòng đệm có thể ảnh hưởng không tốt đến thành phẩm thuốc.
7. Thuốc xịt mũi: Dung dịch và hỗn dịch
Dung dịch hay hỗn dịch thuốc xịt mũi có gắn bơm định liều cần được đánh giá về hình thức, màu sắc, độ trong đối với dung dịch, định lượng, các sản phẩm phân hủy, hàm lượng chất bảo quản và chất chống oxy hóa, giới hạn vi sinh vật, pH, tiểu phân lạ, độ đồng nhất về hàm lượng dược chất mỗi lần xịt, số lần xịt đạt sự đồng nhất về lượng xịt ra của một đơn vị đóng gói, sự phân bố kích thước giọt và/hoặc tiểu phân, sự giảm khối lượng, sự phân phối của bơm, soi kính hiển vi (đối với hỗn dịch), kích thước tiểu phân lạ, các chất chiết /chất thôi ra từ các thành phần của bao bì, nắp, bơm bằng chất dẻo và cao su.
8. Các thành phẩm thuốc dùng tại chỗ, tai và thuốc nhãn khoa
Nhóm này bao gồm các thuốc mỡ, kem, lotion, bột nhão, gel, dung dịch và thuốc phun mù không phân liều dùng trên da. Các thành phẩm thuốc dùng tại chỗ cần được đánh giá về hình thức, độ trong, màu sắc, độ đồng nhất, mùi, pH, khả năng phân tán lại (đối với lotion), độ đặc, độ nhớt, phân bố kích thước tiểu phân (đối với hỗn dịch, khi có thể), định lượng, các sản phẩm phân hủy, nồng độ chất bảo quản và chất chống oxy hóa (nếu có), giới hạn vi sinh vật/độ vô khuẩn và giảm khối lượng (khi thích hợp).
Việc đánh giá đối với các thành phẩm thuốc nhãn khoa hoặc thuốc dùng tại tai (như kem, thuốc mỡ, dung dịch và hỗn dịch) cần tiến hành thêm các chỉ tiêu sau: độ vô khuẩn, tiểu phân lạ và các chất thôi từ đồ bao gói.
Việc đánh giá các thuốc phun mù không phân liều dùng tại chỗ cần bao gồm các chỉ tiêu: hình thức, định lượng, các sản phẩm phân hủy, áp suất, sự giảm khối lượng, khối lượng thực được phun ra, tốc độ phun, giới hạn vi sinh vật, kiểu xịt, hàm lượng nước, và phân bố kích thước tiểu phân (đối với hỗn dịch)
9. Thuốc đặt
Thuốc đặt cần được đánh giá về hình thức, màu sắc, định lượng, các sản phẩm phân hủy, kích thước tiểu phân, khoảng nhiệt độ biến dạng, độ hòa tan (ở 37oC) và giới hạn vi sinh vật.
10. Thuốc tiêm thể tích nhỏ (Small Volume Parenterals, SVPs)
Thuốc tiêm thể tích nhỏ bao gồm một loạt các thành phẩm tiêm như thuốc tiêm, bột thuốc để pha tiêm, hỗn dịch tiêm và nhũ tương tiêm. Mẫu thuốc cần được lưu trữ ở các vị trí khác nhau như thẳng, lộn ngược, nằm ngang.
Thành phẩm thuốc tiêm cần được đánh giá về hình thức, độ trong, màu sắc, định lượng, hàm lượng chất bảo quản (nếu có), các sản phẩm phân hủy, tiểu phân lạ, pH, độ vô khuẩn và chất gây sốt/nội độc tố.
Thành phẩm bột pha tiêm cần được đánh giá về hình thức, màu sắc, thời gian pha lại, và hàm lượng nước. Độ ổn định của các thành phẩm thuốc sau khi pha theo như hướng dẫn trên nhãn cũng cần được đánh giá. Những chỉ tiêu chất lượng đặc trưng cần được kiểm tra vào những thời điểm thích hợp trong thời hạn sử dụng tối đa đã được ấn định của thành phẩm đã pha lại, được bảo quản đúng điều kiện đã ghi trên nhãn, nên bao gồm: hình thức, độ trong, mùi, màu sắc, pH, định lượng (hoạt lực), chất bảo quản (nếu có), các sản phẩm phân hủy/khối kết tủa, độ vô khuẩn, chất gây sốt/nội độc tố và tiểu phân lạ.
Ngoài các chỉ tiêu chất lượng như đã nêu ở mục thuốc tiêm và thuốc bột để pha tiêm, khi nghiên cứu độ ổn định đối với hỗn dịch thuốc tiêm và thuốc để pha hỗn dịch tiêm cần theo dõi thêm: phân bố kích thước tiểu phân, khả năng phân tán lại và tính chất lưu biến.
Khi nghiên cứu độ ổn định của các thành phẩm nhũ tương thuốc tiêm, ngoài các chỉ tiêu chất lượng như đã nêu đối với thuốc tiêm, cần tiến hành theo dõi thêm: sự tách pha, độ nhớt, kích thước giọt trung bình và sự phân bố của pha phân tán.
11. Thuốc tiêm thể tích lớn (Large Volume Parenterals, LVPs)
Thuốc tiêm thể tích lớn cần được đánh giá về hình thức, màu sắc, định lượng, hàm lượng chất bảo quản (nếu có), các sản phẩm phân hủy, kích thước tiểu phân, pH, độ vô khuẩn, chất gây sốt/nội độc tố, độ trong và thể tích.
12. Hợp dịch thuốc
Đối với bất kỳ thành phẩm thuốc nào hoặc chất pha loãng nào định dùng để thêm vào thành phẩm thuốc khác rất có thể xảy ra tương kỵ. Trong những trường hợp như vậy, thành phẩm thuốc đã ghi nhãn là được dùng bằng cách thêm vào thành phẩm thuốc khác (như thuốc tiêm, dung dịch xông hít) cần phải đánh giá về độ ổn định và mức độ tương hợp trong hợp dịch với các thành phẩm thuốc khác hoặc với chất pha loãng cả khi để theo chiều thẳng đứng và chiều lật ngược/nằm ngang, nếu được cảnh báo.
Quy trình thử độ ổn định với các thử nghiệm thích hợp cần được tiến hành vào các thời điểm 0, 6 đến 8 và 24 giờ hoặc phù hợp với khoảng thời gian sử dụng đã dự kiến ở nhiệt độ bảo quản/sử dụng đã nêu. Các phép thử cần thực hiện là hình thức, màu sắc, độ trong, định lượng, sản phẩm phân hủy, pH, kích thước tiểu phân, tương tác với bao bì/nắp đậy/dụng cụ và độ vô khuẩn. Cũng có thể đưa ra số liệu hỗ trợ thích hợp thay cho việc đánh giá về sự phân hủy bởi ánh sáng.
13. Miếng dán dùng qua da
Đối với các sản phẩm dán trực tiếp vào da với mục đích khuếch tán liên tục dược chất vào trong da qua lớp biểu bì, các chỉ tiêu chất lượng cần đánh giá gồm hình thức, định lượng, sản phẩm phân hủy, tốc độ giải phóng in vitro, độ rò rỉ, giới hạn vi sinh vật/độ vô khuẩn, lực tháo và dính, và tốc độ giải phóng thuốc.
14. Các sản phẩm đông khô
Các chỉ tiêu chất lượng cần đánh giá gồm hình thức của cả thành phẩm đông khô và sản phẩm thuốc pha lại, định lượng, sản phẩm phân hủy, pH, hàm lượng nước và tốc độ tạo thành dung dịch.
iii. Chất lượng về mặt vi sinh của các dạng bào chế vô khuẩn đa liều và các dạng bào chế không vô khuẩn nên được kiểm soát. Các thử nghiệm nên được thực hiện ít nhất tại thời điểm bắt đầu và kết thúc của tuổi thọ. Các thử nghiệm này thường được tiến hành như là một phần của kế hoạch phát triển sản phẩm, ví dụ, trong nghiên cứu độ ổn định của lô đầu tiên. Chúng không nhất thiết phải được lặp lại trong các nghiên cứu độ ổn định sau này trừ khi có sự thay đổi ảnh hưởng đến trạng thái vi sinh. Không nhất thiết là mọi thử nghiệm được liệt kê phải được thực hiện tại mỗi thời điểm. Điều này được áp dụng cụ thể cho phép thử độ vô khuẩn: hầu hết các sản phẩm vô khuẩn có thể thực hiện phép thử này tại thời điểm đầu và cuối của chu kỳ nghiên cứu độ ổn định. Phép thử chất gây sốt và nội độc tố vi khuẩn có thể chỉ cần thực hiện tại thời điểm xuất xưởng. Các dạng bào chế vô khuẩn có chứa các nguyên liệu khô (bột hay sản phẩm đông khô) và dung dịch đóng trong ống thủy tinh hàn kín có thể không cần thêm phép thử vi sinh ngoài thời điểm khởi đầu. Mức độ nhiễm vi sinh vật ở dạng thuốc lỏng đóng trong bao bì thủy tinh có nắp bằng chất dẻo hay trong bao bì bằng chất dẻo nên được đánh giá ít nhất tại thời điểm đầu và cuối của chu kỳ nghiên cứu độ ổn định; nếu dữ liệu nghiên cứu dài hạn cung cấp cho cơ quan có thẩm quyền về việc đăng ký thuốc không bao phủ đến hết hạn dùng của sản phẩm thì mức độ nhiễm vi sinh vật tại thời điểm cuối cùng phải được cung cấp. (WHO 2009, Annex 2, p. 124)
iv. Hướng của sản phẩm khi bảo quản (đúng chiều hay ngược chiều) có thể phải xem xét trong đề cương khi có sự thay đổi trong hệ thống bao bì đóng gói.
4.6. Tần số thử nghiệm
Khi nghiên cứu dài hạn, tần số thử nghiệm phải đủ để thiết lập tính ổn định của thành phẩm thuốc. Tần số thử nghiệm ở điều kiện bảo quản dài hạn thông thường là 3 tháng một lần trong năm đầu tiên và 6 tháng một lần trong năm thứ 2, và một năm một lần cho các năm sau đó cho đến hết tuổi thọ đề xuất.
Ở điều kiện cấp tốc, tối thiểu là 3 thời điểm, kể cả thời điểm đầu và thời điểm kết thúc (chẳng hạn: 0, 3, và 6 tháng) đối với thời gian thử nghiệm là 6 tháng. Trong trường hợp (dựa trên kinh nghiệm phát triển sản phẩm) các kết quả nghiên cứu cấp tốc cho thấy có sự biến đổi đáng kể của các chỉ tiêu theo dõi, cần thực hiện thêm thử nghiệm bằng cách thêm một số mẫu ở thời điểm kết thúc hoặc bằng cách thêm thời điểm thứ tư vào thiết kế nghiên cứu.
Các thiết kế rút gọn, như thiết kế ma trận hoặc phân cực, trong đó tần số thử nghiệm được giảm đi hoặc không nhất thiết phải kết hợp tất cả các yếu tố trong thử nghiệm, có thể được áp dụng, nếu phù hợp; xem Phụ lục 5.3.
|
Điều kiện bảo quản |
Các thành phẩm |
Tần số thử nghiệm |
|
Điều kiện dài hạn (Real time) |
NCE, Generics và các thay đổi (MaV và MiV) |
0, 3, 6, 9, 12, 18, 24 tháng, hàng năm cho đến hết hạn dùng đề xuất |
|
Cấp tốc (Accelerated) |
NCE, Generics, và các thay đổi (MaV và MiV) |
0, 3, và 6 tháng |
Ghi chú: NCE: New chemical entity (thuốc hóa dược mới); MaV: Major Variation (thay đổi lớn); MiV: Minor Variation (thay đổi nhỏ).
4.7. Điều kiện bảo quản
4.7.1. Trường hợp chung
i. Nói chung, một thành phẩm thuốc phải được đánh giá ở những điều kiện bảo quản (với sự dao động thích hợp) cho phép đánh giá tính ổn định với nhiệt và nếu có thể, độ nhạy cảm với ẩm hoặc khả năng mất dung môi của thành phẩm. Các điều kiện bảo quản và thời gian nghiên cứu đã chọn phải phù hợp việc bảo quản, chuyên chở và sử dụng sau này (ví dụ sau khi pha lại hoặc sau khi pha loãng như hướng dẫn ghi trên nhãn).
ii. Nghiên cứu độ ổn định được thực hiện ở điều kiện bảo quản như sau:
|
Loại bao bì/nghiên cứu |
Điều kiện bảo quản |
|
Dài hạn (cho các thành phẩm chứa trong bao bì sơ cấp bán thấm hơi nước) |
Nhiệt độ 30oC ± 2oC, Độ ẩm tương đối 75% ± 5% |
|
Dài hạn (cho các thành phẩm chứa trongbao bì không thấm hơi nước) |
Nhiệt độ 30oC ± 2oC Không cần chỉ rõ độ ẩm tương đối |
|
Nghiên cứu cấp tốc |
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 75% ± 5% |
|
Nghiên cứu khắc nghiệt* |
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 75% ± 5% Hoặc tại các điều kiện khắc nghiệt hơn |
* Nghiên cứu khắc nghiệt là cần thiết để thẩm định phương pháp phân tích, xây dựng công thức bào chế, xác định và kiểm soát các chất phân hủy có thể có trong nghiên cứu độ ổn định.
iii. Nghiên cứu ở điều kiện dài hạn phải được tiếp tục theo dõi với tần số thử nghiệm thích hợp trong khoảng thời gian tối thiểu bằng với hạn dùng.
iv. Có thể sử dụng số liệu thu được từ điều kiện bảo quản cấp tốc để đánh giá ảnh hưởng của việc tiếp xúc của thuốc trong thời gian ngắn với điều kiện vượt ra ngoài điều kiện bảo quản đã ghi trên nhãn (chẳng hạn điều kiện có thể xảy ra khi chuyên chở thuốc).
v. Nếu các số liệu trong hồ sơ đăng ký thuốc thu được từ các nghiên cứu ở các điều kiện ít khắc nghiệt hơn điều kiện yêu cầu (ví dụ nhiệt độ 30oC/độ ẩm tương đối 65%) thì cần phải bổ sung thêm các số liệu thích hợp để tiến hành các đánh giá khoa học phù hợp. Các yếu tố cần cân nhắc sẽ bao gồm:
1. Có quan sát thấy sự không ổn định nào không;
2. Có cung cấp các dữ liệu ở điều kiện cấp tốc không;
3. Có cần thiết dùng bao bì có khả năng bảo vệ tốt hơn không.
Có thể thêm một hướng dẫn trên nhãn như "Bảo quản dưới 30oC và tránh ẩm" nếu thích hợp.
vi. Các số liệu thu thập thêm trong thời gian xem xét cấp đăng ký phải được trình lên cơ quan có thẩm quyền nếu được yêu cầu.
vii. Các điều kiện bảo quản khác có thể được phép nếu có lý do chính đáng, ví dụ như các trường hợp dưới đây:
- Các thành phẩm thuốc nhạy cảm với nhiệt phải được bảo quản ở điều kiện nhiệt độ thấp hơn và nhiệt độ đó chính là nhiệt độ bảo quản dài hạn được chọn lựa.
* Đối với thành phẩm thuốc có các thành phần dược chất kém bền và các công thức không thích hợp cho việc nghiên cứu thực nghiệm khi bảo quản ở nhiệt độ nâng cao (ví dụ các thuốc đặt) thì cần nghiên cứu độ ổn định ở điều kiện dài hạn trong thời gian dài hơn.
- Cần xem xét đặc biệt đối với các thành phẩm có biến đổi về vật lý hoặc thậm chí cả về hóa học ở điều kiện nhiệt độ bảo quản thấp hơn, ví dụ như hỗn dịch hoặc nhũ tương có thể lắng cặn hoặc tách kem, dầu và các thành phẩm bán rắn có thể có độ nhớt tăng cao.
* Khi áp dụng điều kiện nhiệt độ thấp hơn, thì thử nghiệm cấp tốc trong 6 tháng phải được tiến hành ở nhiệt độ cao hơn nhiệt độ bảo quản thực đã chọn tối thiểu là 15oC (và điều kiện về độ ẩm tương đối phù hợp với nhiệt độ đó). Ví dụ, Một thành phẩm được bảo quản dài hạn trong tủ lạnh, thì thử nghiệm cấp tốc phải được thực hiện ở nhiệt độ 25oC ± 2oC, độ ẩm tương đối là 60% ± 5%. Các điều kiện thực của thử nghiệm đã lựa chọn phải được phản ánh trên nhãn và tuổi thọ (ngày hết hạn).
4.7.2. Đối với các thành phẩm thuốc đựng trong bao bì không thấm hơi nước
i. Các loại bao bì thường được xem là không thấm hơi nước gồm ống thủy tinh, vỉ nhôm/nhôm, polyethylen có tỷ trọng lớn (High Density Polyethylene, HDPE) hoặc chai thủy tinh có nắp bằng kim loại hay HDPE.
ii. Mức độ nhạy cảm với ẩm hay khả năng mất dung môi không phải là vấn đề lớn với các thành phẩm đựng trong bao bì không thấm đóng vai trò như một hàng rào ngăn cản sự thấm hơi ẩm hay sự mất dung môi. Vì vậy nghiên cứu độ ổn định cho các thành phẩm đựng trong bao bì không thấm có thể được thực hiện ở điều kiện độ ẩm bất kỳ (trong phòng hay có kiểm soát). (WHO 2009, p. 100)
4.7.3. Đối với các thành phẩm thuốc đựng trong bao bì bán thấm (dung môi là nước)
i. Đối với các sản phẩm có dung môi là nước đựng trong bao bì bán thấm, nên đánh giá khả năng mất nước bên cạnh các chỉ tiêu vật lý, hóa học, sinh học và vi sinh trong nghiên cứu độ ổn định. Đánh giá này có thể được thực hiện dưới điều kiện độ ẩm tương đối thấp, như bàn luận dưới đây. Phải chứng minh được rằng các sản phẩm có dung môi là nước đựng trong bao bì bán thấm có thể chịu được môi trường có độ ẩm tương đối thấp.
|
Loại nghiên cứu |
Điều kiện bảo quản |
Thời gian nghiên cứu tối thiểu phải thực hiện trong hồ sơ |
|
Dài hạn |
30°C ± 2°C/35% RH ± 5% RH |
12 tháng |
|
Cấp tốc |
40°C ± 2°C/không quá 25% RH |
6 tháng |
ii. Các sản phẩm đáp ứng các điều kiện nghiên cứu cả dài hạn và cấp tốc như đã chỉ ra trong bảng trên thì bao bì bán thấm chứa sản phẩm đó coi như đã được chứng minh là đạt yêu cầu.
iii. Việc mất đi 5% hoặc hơn lượng nước so với lượng ban đầu được xem là có ý nghĩa đối với một sản phẩm đựng trong bào bì bán thấm sau khi bảo quản trong khoảng thời gian tương đương 3 tháng ở điều kiện nhiệt độ 40°C/độ ẩm tương đối không quá 25%. Tuy nhiên, với bao bì có thể tích nhỏ (không quá 1 ml) hay các sản phẩm đơn liều, việc mất đi 5% lượng nước hoặc hơn sau khi bảo quản trong khoảng thời gian tương đương 3 tháng ở điều kiện nhiệt độ 40 °C/độ ẩm tương đối không quá 25% có thể được bỏ qua, nếu phù hợp.
iv. Một cách tiếp cận thay thế cho nghiên cứu ở độ ẩm tương đối thấp như đã đề xuất trong bảng trên (cả nghiên cứu dài hạn và cấp tốc) là thực hiện nghiên cứu độ ổn định ở điều kiện độ ẩm tương đối cao hơn rồi suy ra lượng nước mất đi ở điều kiện độ ẩm tương đối thấp bằng tính toán. Điều này có thể thực hiện được bằng cách xác định bằng thực nghiệm hệ số thấm cho hệ thống bao bì hoặc, như ví dụ dưới, sử dụng tỷ lệ tính toán của tốc độ mất nước giữa hai điều kiện độ ẩm ở cùng một nhiệt độ. Hệ số thấm của một hệ thống bao bì có thể xác định bằng thực nghiệm ở tình huống xấu nhất (chẳng hạn, nồng độ loãng nhất trong một dãy các nồng độ) của thành phẩm xin đăng ký.
● Ví dụ về cách tiếp cận xác định lượng nước mất đi:
- Với sản phẩm được đựng trong hệ thống bao bì, với kích cỡ và lượng xác định, một cách tiếp cận phù hợp để tính toán tốc độ mất nước ở độ tương đối ẩm thấp là nhân tốc độ mất nước đo được ở độ ẩm tương đối thay thế ở cùng một nhiệt độ cho tỷ lệ tốc độ mất nước được biểu diễn ở bảng dưới. Tính tuyến tính của tốc độ mất nước tại độ ẩm tương đối thay thế trong thời gian bảo quản cần được chứng minh.
- Ví dụ, tại nhiệt độ cho trước, chẳng hạn 40oC, tốc độ tính toán của lượng nước mất đi trong quá trình bảo quản ở độ ẩm tương đối không quá 25% là tốc độ mất nước đo được ở độ ẩm tương đối 75% nhân với 3 là tỷ lệ tốc độ mất nước tương ứng.
|
Điều kiện độ ẩm thấp |
Điều kiện thay thế |
Tỷ lệ tốc độ mất nước |
Tính toán |
|
30°C/35% RH |
30°C/75% RH |
2,6 |
(100-35)/(100-75) |
|
40°C/≤ 25% RH |
40°C/75% RH |
3,0 |
(100-25)/(100-75) |
Tỷ lệ tốc độ mất nước chính xác tại các điều kiện có độ ẩm tương đối khác với bảng trên có thể được sử dụng. (WHO 2009 p.100-102)
v. Các cách tiếp cận tương đương khác có thể được phát triển và báo cáo cho các sản phẩm có dung môi khác nước.
4.7.4 Các thành phẩm thuốc dự kiến bảo quản trong tủ lạnh
|
Nghiên cứu |
Điều kiện bảo quản |
Khoảng thời gian tối thiểu của dữ liệu khi nộp hồ sơ đăng ký |
Số lô thử |
|
Dài hạn |
Nhiệt độ 5oC ± 3oC |
12 tháng |
Tối thiểu 3 lô |
|
Cấp tốc |
Nhiệt độ 25oC ± 2oC Độ ẩm tương đối 60% ± 5% |
6 tháng |
Tối thiểu 3 lô |
Nếu thành phẩm thuốc được đóng gói trong bao bì bán thấm, phải cung cấp thông tin phù hợp để đánh giá mức độ mất nước. Các số liệu theo dõi khi bảo quản lạnh cần được đánh giá theo mục đánh giá của hướng dẫn này, trừ các trường hợp được ghi rõ dưới đây.
4.7.5. Các thành phẩm thuốc dự kiến bảo quản đông lạnh
|
Nghiên cứu |
Điều kiện bảo quản |
Khoảng thời gian tối thiểu của dữ liệu khi nộp hồ sơ đăng ký |
|
Dài hạn |
-20oC ± 5oC |
12 tháng |
Đối với các thành phẩm thuốc dự kiến bảo quản đông lạnh, nên dựa trên các dữ liệu ở điều kiện thực và bảo quản dài hạn để xác định tuổi thọ. Do không có số liệu ở điều kiện bảo quản cấp tốc đối với các thành phẩm thuốc dự định bảo quản đông lạnh, thử nghiệm trên một lô ở một nhiệt độ nâng cao (ví dụ 5oC ± 3oC hoặc 25oC ± 2oC) trong một khoảng thời gian thích hợp cần được thực hiện để chỉ rõ ảnh hưởng của việc chuyên chở ngắn hạn trong những điều kiện vượt ra ngoài điều kiện bảo quản đã ghi trên nhãn.
4.7.6 Các thành phẩm thuốc dự kiến bảo quản dưới -20oC
Các thành phẩm thuốc dự kiến bảo quản dưới -20oC cần được xem xét theo từng trường hợp cụ thể.
4.7.7. Thuốc hóa dược mới (NCE)
|
Nghiên cứu |
Điều kiện bảo quản |
Khoảng thời gian tối thiểu của dữ liệu khi nộp hồ sơ đăng ký |
Số lô thử |
|
Dài hạn |
Nhiệt độ 30oC ± 2oC Độ ẩm tương đối 75% ± 5% |
12 tháng |
Tối thiểu 3 |
|
Cấp tốc |
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 75% ± 5% |
6 tháng |
Tối thiểu 3 |
4.7.8. Thuốc Generics
|
Nghiên cứu |
Điều kiện bảo quản |
Khoảng thời gian tối thiểu của dữ liệu khi nộp hồ sơ đăng ký |
Số lô thử |
|
Dài hạn |
Nhiệt độ 30oC ± 2oC Độ ẩm tương đối 75% ± 5% |
6 tháng |
Tối thiểu 2 lô đối với dạng bào chế qui ước và dược chất bền vững |
|
12 tháng |
Tối thiểu 3 lô đối với dạng bào chế đặc biệt hoặc dược chất kém bền vững |
||
|
Cấp tốc |
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 75% ± 5% |
6 tháng |
Tối thiểu 2 lô đối với dạng bào chế qui ước và dược chất bền vững |
|
Tối thiểu 3 lô đối với dạng bào chế đặc biệt hoặc dược chất kém bền vững |
4.7.9. Thay đổi áp dụng đối với thuốc đã có số đăng ký (thay đổi lớn (MaV) và thay đổi nhỏ (MiV)
Khi thuốc đã được cấp số đăng ký, cần tiến hành thêm các nghiên cứu độ ổn định khi có sự thay đổi có thể ảnh hưởng đến độ ổn định của thuốc (xem Phụ lục II- Các thay đổi lớn, thay đổi nhỏ, thay đổi khác áp dụng đối với thuốc đã được cấp số đăng ký lưu hành).
Thay đổi lớn (MaV)
|
Nghiên cứu |
Điều kiện bảo quản |
Khoảng thời gian tối thiểu của dữ liệu khi nộp hồ sơ đăng ký |
Số lô thử |
|
Dài hạn |
Nhiệt độ 30oC ± 2oC Độ ẩm tương đối 75% ± 5% |
6 tháng |
Tối thiểu 2 lô đối với dạng bào chế qui ước và dược chất bền vững |
|
Tối thiểu 3 lô đối với dạng bào chế đặc biệt hoặc dược chất kém bền vững |
|||
|
Cấp tốc |
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 75% ± 5% |
6 tháng |
Tối thiểu 2 lô đối với dạng bào chế qui ước và dược chất bền vững |
|
Tối thiểu 3 lô đối với dạng bào chế đặc biệt hoặc dược chất kém bền vững |
Thay đổi nhỏ (MiV)
|
Nghiên cứu |
Điều kiện bảo quản |
Khoảng thời gian tối thiểu của dữ liệu khi nộp hồ sơ đăng ký |
Số lô thử |
|
Dài hạn |
Nhiệt độ 30oC ± 2oC Độ ẩm tương đối 75% ± 5% |
3 tháng* |
Tối thiểu 2 lô đối với dạng bào chế qui ước và dược chất bền vững |
|
6 tháng |
Tối thiểu 3 lô đối với dạng bào chế đặc biệt hoặc dược chất kém bền vững |
||
|
Cấp tốc |
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 75% ± 5% |
3 tháng* |
Tối thiểu 2 lô đối với dạng bào chế qui ước và dược chất bền vững |
|
6 tháng |
Tối thiểu 3 lô đối với dạng bào chế đặc biệt hoặc dược chất kém bền vững |
* Ví dụ: thay một tá dược bằng một tá dược tương đương, thay đổi thành phần (về lượng hay loại) vật liệu bao bì sơ cấp, thay đổi cỡ lô thành phẩm, thay đổi nhỏ trong quy trình sản xuất thành phẩm, thay đổi tá dược màu hoặc mùi trong thành phẩm, thay đổi khối lượng màng bao viên nén hoặc thay đổi khối lượng vỏ nang và bất kỳ thay đổi nhỏ khác được đề cập trong Phụ lục II- Các thay đổi lớn, thay đổi nhỏ, thay đổi khác áp dụng đối với thuốc đã được cấp số đăng ký lưu hành.
(WHO Expert Committee on Specifications forPharmaceutical Preparations, Annex 6: Guidance on Variations to a Prequalified Product Dossier, WHO Technical Report Series No. 943, 2007)
4.8. Độ ổn định trong khi sử dụng thuốc
i. Mục đích của việc nghiên cứu độ ổn định trong khi sử dụng thuốc là nhằm cung cấp thông tin cho việc ghi nhãn, điều kiện bảo quản và thời gian sử dụng của các thành phẩm đa liều sau khi mở, pha lại hoặc pha loãng dung dịch, ví dụ như thuốc tiêm kháng sinh được cung cấp dưới dạng bột để pha lại.
ii. Nghiên cứu nên được thiết kế sao cho mô phỏng giống nhất cách sử dụng của thành phẩm thuốc trong thực tế, chú ý đến thể tích đóng của bao bì và bất kỳ sự pha loãng hoặc pha lại nào trước khi sử dụng. Tại những thời điểm tương đương với thời điểm sử dụng thuốc trong thực tế, một lượng thuốc thích hợp nên được lấy ra khỏi thành phẩm bằng phương pháp thường được sử dụng và mô tả trong tờ hướng dẫn sử dụng thuốc.
iii. Đặc tính vật lý, hóa học và vi sinh của thành phẩm thuốc nhạy cảm với sự thay đổi trong quá trình bảo quản nên được kiểm soát cho đến hết hạn dùng trong khi sử dụng thuốc. Nếu có thể, các phép thử nên được thực hiện tại thời điểm giữa và cuối của hạn dùng trong khi sử dụng thuốc (khi lượng thuốc cuối cùng còn trong đồ bao gói). Các chỉ tiêu đặc biệt, ví dụ như chất bảo quản (cả hàm lượng và hiệu lực) đối với dạng thuốc lỏng và bán rắn, cũng cần được nghiên cứu.
iv. Nghiên cứu nên được thực hiện trên tối thiểu 2 lô, với cỡ lô tối thiểu là pilot. Ít nhất một trong các lô này nên được lựa chọn tại thời điểm cuối hạn dùng của nó. Nếu kết quả chưa có sẵn, một lô nên thử tại thời điểm cuối trước khi nộp dữ liệu nghiên cứu độ ổn định.
v. Nghiên cứu này nên được thực hiện trên thành phẩm pha loãng hoặc pha lại của lô đầu tiên trong suốt thời hạn sử dụng đề xuất như là một phần của các nghiên cứu độ ổn định tại thời điểm đầu và thời điểm cuối của hạn dùng hoặc thời điểm cuối trước khi nộp dữ liệu nghiên cứu độ ổn định (khi dữ liệu nghiên cứu dài hạn chưa có sẵn).
vi. Nói chung, nghiên cứu này không cần lặp lại trên các lô cam kết. (WHO 2009, p. 105 – 106).
4.9. Hệ thống bao bì đóng gói
i. Thử nghiệm độ ổn định phải tiến hành đối với dạng bào chế đã đóng gói trong bao bì dự kiến sẽ lưu hành trên thị trường (bao gồm cả bao bì thứ cấp và nhãn bao bì). Bất kỳ các nghiên cứu nào đã tiến hành trên sản phẩm không đóng trong bao bì trực tiếp hoặc trong các vật liệu bao bì khác lần lượt được xem là một phần của thử nghiệm khắc nghiệt của dạng bào chế hoặc như là các thông tin hỗ trợ.
ii. Các thông số yêu cầu để phân loại vật liệu bao bì là bán thấm hay không thấm phụ thuộc vào tính chất vật liệu làm bao bì như độ dày, hệ số thấm và các thông số liên quan khác. Sự thích hợp của vật liệu làm bao bì cho một sản phẩm đặc biệt được xác định bởi tính chất của sản phẩm. Một ví dụ về loại, độ dày và hệ số thấm của vật liệu làm bao bì được trình bày trong Phụ lục 5.4.
iii. Khi sử dụng bao bì thấm ẩm để đóng gói, cần phải cân nhắc độ ổn định của chất đựng bên trong dưới điều kiện độ ẩm cao.
iv. Độ ẩm có thể có các ảnh hưởng không mong muốn đến độ ổn định hóa học (ví dụ một số kháng sinh có thể bị thủy phân) và độ ổn định vật lý (ví dụ tốc độ hòa tan có thể thay đổi).
v. Cần chú ý đến khả năng thấm khác nhau của các loại vật liệu bao bì khác nhau, từ đó cần phải cụ thể hóa các thông số như độ dày của vật liệu và hệ số thấm. Nên có bàn luận thích hợp ở mục P2 Phát triển dược phẩm và P7 Hệ thống bao bì trong Hồ sơ ACTD.
vi. Ảnh hưởng của độ ẩm cao lên dạng bào chế rắn đóng gói trong bao bì có khả năng thấm ẩm phải được chứng minh bằng số liệu.
4.10. Đánh giá
Cần có một phương pháp hệ thống trong việc trình bày và đánh giá thông tin về độ ổn định, các thông tin cần có là kết quả từ các thử nghiệm vật lý, hóa học và vi sinh, bao gồm cả các chỉ tiêu đặc thù của từng dạng bào chế (ví dụ tốc độ hòa tan đối với các dạng thuốc rắn dùng đường uống).
Mục đích của nghiên cứu độ ổn định là dựa trên thử nghiệm tối thiểu với 2 hoặc 3 lô thành phẩm thuốc (xem mục 4.7. Điều kiện bảo quản) để xác lập tuổi thọ và ghi hướng dẫn bảo quản trên nhãn áp dụng cho tất cả các lô thành phẩm thuốc sau này được sản xuất và đóng gói dưới những điều kiện tương tự như các lô thử. Mức độ sai khác giữa các lô có ảnh hưởng đến mức độ tin cậy rằng một lô sản phẩm tương lai sẽ vẫn đạt các tiêu chuẩn chất lượng trong suốt hạn dùng của sản phẩm.
Các nghiên cứu đơn yếu tố so với đa yếu tố và nghiên cứu thiết kế đầy đủ so với thiết kế rút gọn có cùng khái niệm cơ bản trong việc đánh giá số liệu độ ổn định. Đánh giá số liệu từ các nghiên cứu độ ổn định và sử dụng các số liệu hỗ trợ nếu cần để xác định các chỉ tiêu chất lượng trọng yếu có thể ảnh hưởng tới chất lượng và hiệu quả của thành phẩm thuốc. Mỗi một chỉ tiêu cần đánh giá một cách vừa riêng biệt vừa tổng thể để ước lượng tuổi thọ. Tuổi thọ đề xuất của thành phẩm không được vượt quá tuổi thọ ước lượng tính theo từng chỉ tiêu đơn lẻ.
Cây quyết định ở Phụ lục 5.5 phác họa các bước đánh giá dữ liệu độ ổn định, khi nào thì thực hiện ngoại suy và mức độ ngoại suy là bao nhiêu có thể được xem xét cho tuổi thọ đề xuất. Phụ lục 5.6 cung cấp (1) thông tin làm thế nào để phân tích dữ liệu nghiên cứu dài hạn cho các chỉ tiêu định lượng thích hợp từ một thiết kế nghiên cứu đa yếu tố, đầy đủ hay rút gọn, (2) thông tin làm thế nào để sử dụng phân tích hồi quy để ước lượng tuổi thọ và (3) ví dụ các phân tích thống kê để kiểm định tính hợp nhất của dữ liệu từ các lô khác nhau hoặc các yếu tố khác nhau. Hướng dẫn bổ sung có thể được tìm thấy trong các tài liệu tham khảo được liệt kê. (ICH Q1E 6 Feb 03, p.2)
Nói chung, các chỉ tiêu hóa học định lượng được (ví dụ như hàm lượng, các sản phẩm phân hủy, hàm lượng chất bảo quản) đối với một thành phẩm thuốc có thể được giả định là biến đổi theo phương trình động học bậc 0 trong suốt thời gian bảo quản dài hạn. Vì vậy, các dữ liệu này tuân theo hồi quy tuyến tính. Mặc dù động học của một số chỉ tiêu định lượng khác (ví dụ: pH, độ hòa tan) nói chung không được rõ nhưng vẫn có thể áp dụng cùng loại phân tích thống kê, nếu phù hợp. Dữ liệu của các chỉ tiêu định tính và vi sinh không phù hợp với phép phân tích thống kê này.
Các gợi ý về phân tích thống kê trong hướng dẫn này không ngụ ý rằng sử dụng đánh giá thống kê là bắt buộc trong khi nó có thể được biện luận là không cần thiết. Tuy nhiên, phân tích thống kê có thể hữu ích trong hỗ trợ tính toán ngoại suy tuổi thọ trong một vài trường hợp và có thể được yêu cầu để thẩm tra tuổi thọ đề xuất trong các trường hợp khác. (ICH Q1E 6 Feb 03, p.2)
4.10.1. Trình bày dữ liệu
Dữ liệu cho tất cả các chỉ tiêu nên được trình bày dưới định dạng phù hợp (ví dụ như bảng, biểu đồ, mô tả) và việc đánh giá dữ liệu này nên được gắn kèm trong hồ sơ đăng ký. Giá trị của các chỉ tiêu định lượng tại tất cả các thời điểm nên được báo cáo dưới dạng số liệu đo đếm được (ví dụ như hàm lượng tính theo phần trăm so với hàm lượng trên nhãn). Nếu một phân tích thống kê được thực hiện, phương pháp sử dụng và các giả thiết của mô hình nên được chỉ rõ và bàn luận. Bảng tóm tắt các kết quả phân tích thống kê và/hoặc biểu đồ biểu diễn dữ liệu nghiên cứu dài hạn nên được gắn kèm. (ICH Q1E 6 Feb 03, p.3)
4.10.2. Ngoại suy dữ liệu
Thực tế của việc ngoại suy là sử dụng một tập dữ liệu đã biết để suy ra thông tin về tập dữ liệu tương lai. Phương pháp ngoại suy giới hạn được sử dụng để suy rộng chu kỳ tái kiểm hay tuổi thọ có thể được đề xuất trong hồ sơ đăng ký vượt quá phạm vi quan sát của dữ liệu nghiên cứu dài hạn có sẵn, đặc biệt khi không có sự biến đổi có ý nghĩa quan sát được ở điều kiện cấp tốc. Khi ngoại suy phải luôn xem xét đến tình huống xấu nhất có thể xảy ra tại thời điểm xuất xưởng.
Việc ngoại suy dữ liệu độ ổn định sử dụng giả thiết rằng mô hình biến đổi tương tự sẽ tiếp tục được áp dụng tại các thời điểm sau khoảng thời gian của dữ liệu dài hạn có sẵn. Sau đó, việc sử dụng ngoại suy sẽ được bàn luận ở các khía cạnh, ví dụ, cơ chế của quá trình phân hủy là gì, mức độ phù hợp của các mô hình toán học và sự tồn tại của các dữ liệu hỗ trợ có liên quan.
Tính đúng của giả thiết mô hình biến đổi sẽ quyết định rằng việc ngoại suy dựa trên dữ liệu dài hạn có sẵn có đáng tin cậy hay không. Ví dụ, khi ước lượng một đường thẳng hay đường cong hồi quy với dữ liệu có sẵn, dữ liệu tự nó cung cấp một kiểm định tính đúng của giả thiết mô hình biến đổi, và phương pháp thống kê có thể được sử dụng để kiểm định mức độ phù hợp của dữ liệu với đường thẳng hay đường cong giả thiết. Không có một kiểm định nội tại có sẵn nằm ngoài phạm vi dữ liệu quan sát được. Vì vậy, tuổi thọ được tính trên cơ sở ngoại suy luôn luôn cần được thẩm tra bằng dữ liệu độ ổn định dài hạn bổ sung ngay khi dữ liệu có sẵn. Cần chú ý đưa vào trong đề cương nghiên cứu thời điểm tương ứng với tuổi thọ ngoại suy của lô cam kết.
Nếu dữ liệu dài hạn được hỗ trợ bởi những kết quả từ những nghiên cứu cấp tốc, tuổi thọ có thể được suy rộng vượt quá thời điểm cuối cùng của nghiên cứu dài hạn. Tuổi thọ ngoại suy có thể tới gấp đôi, nhưng không vượt quá 12 tháng so với khoảng thời gian nghiên cứu dài hạn, phụ thuộc vào sự biến đổi theo thời gian, mức độ dao động của dữ liệu quan sát được, điều kiện bảo quản đề xuất và mức độ phân tích thống kê đã thực hiện.
4.10.3. Đánh giá dữ liệu cho việc ước lượng tuổi thọ của thành phẩm thuốc dự định bảo quản ở nhiệt độ phòng
Với thành phẩm thuốc dự định bảo quản ở nhiệt độ phòng, việc đánh giá nên bắt đầu khi có bất kỳ sự biến đổi có ý nghĩa ở điều kiện cấp tốc và tiếp tục theo xu hướng và sự dao động của dữ liệu dài hạn. Nên phác họa các trường hợp phù hợp cho việc ngoại suy tuổi thọ vượt quá khoảng thời gian của dữ liệu nghiên cứu dài hạn. Cây quyết định ở Phụ lục 5.5 có thể được sử dụng như công cụ hỗ trợ.
4.10.3.1. Không có sự biến đổi có ý nghĩa ở điều kiện cấp tốc
Khi không có sự biến đổi có ý nghĩa ở điều kiện cấp tốc, tuổi thọ phụ thuộc vào bản chất của dữ liệu dài hạn và cấp tốc. (ICH Q1E 6 Feb 03, p.3)
a. Dữ liệu dài hạn và cấp tốc ít biến đổi hoặc không biến đổi theo thời gian và ít hoặc không dao động
Khi dữ liệu dài hạn và cấp tốc của một chỉ tiêu chất lượng ít biến đổi hoặc không biến đổi theo thời gian và ít hoặc không dao động, thì có thể rõ ràng rằng thành phẩm thuốc giữ được các chỉ tiêu chất lượng nằm trong mức chất lượng trong suốt tuổi thọ đề xuất. Trong trường hợp này, thường không cần thiết phải thực hiện phân tích thống kê mà nên có những bàn luận cho việc bỏ qua. Bàn luận có thể bao gồm cơ chế của sự phân hủy hoặc không có sự phân hủy, sự liên quan của dữ liệu cấp tốc, cân bằng khối, và/hoặc dữ liệu hỗ trợ khác.
b. Dữ liệu dài hạn hoặc cấp tốc biến đổi theo thời gian và/hoặc có dao động
Nếu dữ liệu dài hạn hoặc cấp tốc của một chỉ tiêu chất lượng biến đổi theo thời gian và/hoặc dao động theo một yếu tố hoặc giữa các yếu tố, phân tích thống kê dữ liệu dài hạn có thể hữu ích để thiết lập tuổi thọ. Khi có sự khác nhau ở độ ổn định quan sát được giữa các lô hoặc giữa các yếu tố khác (ví dụ như hàm lượng, kích cỡ bao bì và/hoặc lượng thuốc đóng gói) hoặc sự kết của các yếu tố (ví dụ như hàm lượng theo kích cỡ bao bì và/hoặc lượng thuốc đóng gói) mà ngăn cản sự hợp nhất dữ liệu, tuổi thọ đề xuất không nên vượt quá thời hạn ngắn nhất được rút ra từ bất kỳ lô, yếu tố khác hoặc sự kết hợp các yếu tố nào. Nói một cách khác, khi sự khác nhau chắc chắn là do một yếu tố cụ thể (như hàm lượng), tuổi thọ khác nhau có thể xuất hiện ở các mức khác nhau của yếu tố đó (như hàm lượng khác nhau). Nên có các bàn luận để chỉ ra nguyên nhân của sự khác nhau và ý nghĩa chung của sự khác nhau đó đối với thành phẩm thuốc. Sự ngoại suy vượt quá khoảng thời gian nghiên cứu dài hạn có thể được đề xuất; tuy nhiên, mức độ ngoại suy sẽ phụ thuộc vào dữ liệu nghiên cứu dài hạn của chỉ tiêu chất lượng có thể đem phân tích thống kê được hay không.
● Dữ liệu không thể đem phân tích thống kê
Khi dữ liệu nghiên cứu dài hạn không thể đem phân tích thống kê, nhưng các dữ liệu hỗ trợ liên quan được cung cấp, tuổi thọ đề xuất có thể gấp tới 1,5 lần, nhưng không vượt quá 6 tháng, so với khoảng thời gian nghiên cứu dài hạn. Dữ liệu có liên quan hỗ trợ dữ liệu nghiên cứu dài hạn thu được từ các lô dùng để phát triển thành phẩm gồm (1) dữ liệu từ công thức bào chế có liên quan chặt chẽ, (2) dữ liệu từ lô có quy mô sản xuất nhỏ hơn, hoặc (3) dữ liệu từ lô được đóng gói trong hệ thống bao bì tương tự với lô đầu tiên dùng để nghiên cứu độ ổn định.
● Dữ liệu có thể đem phân tích thống kê
Khi dữ liệu nghiên cứu dài hạn có thể đem phân tích thống kê nhưng phân tích thống kê không được thực hiện, mức độ ngoại suy sẽ tương tự như khi dữ liệu không thể đem phân tích thống kê. Tuy nhiên, nếu phân tích thống kê được thực hiện, có thể hợp lý nếu đề xuất tuổi thọ tới gấp đôi, nhưng không vượt quá 12 tháng, so với khoảng thời gian nghiên cứu dài hạn, khi đề xuất được củng cố bằng kết quả phân tích và dữ liệu hỗ trợ có liên quan.
4.10.3.2. Biến đổi có ý nghĩa ở điều kiện cấp tốc
Nếu có "biến đổi có ý nghĩa" xảy ra trong trong vòng 3 đến 6 tháng khi thử nghiệm ở điều kiện cấp tốc, thì tuổi thọ đề xuất nên dựa trên các số liệu thu được khi bảo quản ở điều kiện dài hạn.
Biến đổi có ý nghĩa
Nói chung, “biến đổi có ý nghĩa” đối với một thành phẩm thuốc được định nghĩa như sau:
1. Hàm lượng giảm 5% so với giá trị ban đầu hoặc vượt quá mức chất lượng;
2. Có bất kỳ sản phẩm phân hủy nào đó vượt quá mức chất lượng;
3. Không đạt các chỉ tiêu về hình thức, tính chất vật lý và các thử nghiệm chức năng (ví dụ như màu sắc, tách pha, khả năng tái phân tán, đóng bánh, độ cứng, phân phối liều mỗi lần xịt), tuy nhiên, một vài biến đổi về tính chất vật lý (ví dụ như thuốc đặt bị mềm, kem bị chảy) có thể gặp ở điều kiện cấp tốc thì được xem như là bình thường đối với các dạng bào chế này;
4. Không đạt mức chất lượng pH;
5. Không đạt giới hạn về độ hòa tan đối với 12 đơn vị phân liều (nang cứng hoặc viên nén).
Nếu “biến đổi đáng kể” xảy ra trong vòng 3 tháng đầu của thử nghiệm ở điều kiện bảo quản cấp tốc, thì cần có sự bàn luận để chỉ rõ ảnh hưởng của việc tiếp xúc ngắn hạn với những điều kiện vượt ra ngoài điều kiện bảo quản đã ghi trên nhãn, ví dụ như trong khi chuyên chở bằng hoặc bốc dỡ. Nếu phù hợp, thì việc bàn luận này có thể được làm rõ hơn bằng cách thử nghiệm thêm trên một lô thành phẩm trong khoảng thời gian dưới 3 tháng nhưng với tần số thử nghiệm lớn hơn bình thường. Không cần tiếp tục thử nghiệm thành phẩm thuốc đến hết 6 tháng khi mà “biến đổi có ý nghĩa” đã xuất hiện trong vòng 3 tháng đầu tiên.
Cách làm này có thể áp dụng cho các thành phẩm thuốc như thuốc mỡ, kem hoặc thuốc đặt là những thành phẩm không thể thử nghiệm ở điều kiện cấp tốc và chỉ yêu cầu thử nghiệm ở điều kiện dài hạn.
*Ghi chú: Những biến đổi vật lý sau có thể xảy ra ở điều kiện cấp tốc và không được xem là biến đổi có ý nghĩa với thuốc bảo quản ở điều kiện dài hạn nếu không có các biến đổi khác kèm theo:
a. Sự mềm ra của thuốc đặt được thiết kế để nóng chảy ở 37oC, nếu điểm chảy được chứng minh rõ ràng.
b. Không đạt giới hạn độ hòa tan với 12 viên nang cứng gelatin hoặc viên nén được bao bằng tá dược tạo gel nếu chắc chắn rằng sự tạo liên kết chéo gây ra sự không đạt này.
Tuy nhiên, sự tách pha xuất hiện ở một dạng bào chế bán rắn tại điều kiện cấp tốc, thử nghiệm ở điều kiện dài hạn nên được thực hiện. Ảnh hưởng của sự tương tác nên được xem xét để chắc chắn rằng không có sự biến đổi có ý nghĩa khác.
4.10.4. Đánh giá dữ liệu để ước lượng tuổi thọ của thành phẩm thuốc dự kiến bảo quản ở nhiệt độ dưới nhiệt độ phòng
4.10.4.1. Thành phẩm thuốc dự kiến bảo quản trong tủ lạnh
Dữ liệu từ thành phẩm thuốc dự kiến bảo quản trong tủ lạnh nên được đánh giá theo các nguyên tắc tương tự như đã mô tả ở mục 4.10.3 cho thành phẩm thuốc dự kiến bảo quản ở nhiệt độ phòng, ngoại trừ các lưu ý dưới đây. Cây quyết định ở Phụ lục 5.5 có thể được sử dụng như công cụ hỗ trợ.
a. Không có sự biến đổi có ý nghĩa ở điều kiện cấp tốc
Khi không có sự biến đổi có ý nghĩa ở điều kiện cấp tốc, việc ngoại suy tuổi thọ vượt quá thời gian nghiên cứu dài hạn có thể được đề xuất dựa trên các nguyên tắc phác họa ở mục 4.10.3, ngoại trừ mức độ ngoại suy sẽ bị giới hạn hơn.
Nếu dữ liệu dài hạn và cấp tốc cho thấy ít có sự biến đổi theo thời gian và ít dao động, tuổi thọ đề xuất có thể gấp tới 1,5 lần, nhưng không vượt quá 6 tháng, so với khoảng thời gian nghiên cứu dài hạn mà thông thường không cần phân tích thống kê hỗ trợ.
Nếu dữ liệu dài hạn và cấp tốc cho thấy sự biến đổi theo thời gian và/hoặc dao động, tuổi thọ đề xuất có thể vượt tới 3 tháng so với khoảng thời gian nghiên cứu dài hạn nếu (1) dữ liệu dài hạn có thể đem phân tích thống kê nhưng phân tích thống kê không được thực hiện, hoặc (2) dữ liệu dài hạn không thể đem phân tích thống kê nhưng dữ liệu hỗ trợ có liên quan được cung cấp.
Nếu dữ liệu dài hạn và cấp tốc cho thấy sự biến đổi theo thời gian và/hoặc dao động, tuổi thọ đề xuất có thể gấp tới 1,5 lần, nhưng không vượt quá 6 tháng, so với khoảng thời gian nghiên cứu dài hạn nếu (1) dữ liệu dài hạn có thể đem phân tích thống kê nhưng phân tích thống kê được thực hiện, hoặc (2) dữ liệu dài hạn không thể đem phân tích thống kê nhưng dữ liệu hỗ trợ có liên quan được cung cấp, và (3) đề xuất được củng cố bằng kết quả phân tích và dữ liệu hỗ trợ có liên quan.
b. Biến đổi có ý nghĩa ở điều kiện cấp tốc
Nếu biến đổi có ý nghĩa xuất hiện giữa 3 và 6 tháng của thử nghiệm ở điều kiện cấp tốc, nên dựa trên dữ liệu dài hạn để đề xuất tuổi thọ. Việc ngoại suy được xem xét là không phù hợp. Hơn nữa, tuổi thọ ngắn hơn khoảng thời gian nghiên cứu dài hạn có thể được xem xét. Nếu dữ liệu dài hạn có sự dao động, nên thẩm tra tuổi thọ đề xuất bằng phân tích thống kê.
Nếu biến đổi có ý nghĩa xuất hiện trong 3 tháng đầu của thử nghiệm ở điều kiện cấp tốc, nên dựa trên dữ liệu dài hạn để đề xuất tuổi thọ. Việc ngoại suy được xem xét là không phù hợp. Tuổi thọ ngắn hơn khoảng thời gian nghiên cứu dài hạn có thể được xem xét. Nếu dữ liệu dài hạn có sự dao động, nên thẩm tra tuổi thọ đề xuất bằng phân tích thống kê. Hơn nữa, nên có sự bàn luận để chỉ rõ ảnh hưởng của việc tiếp xúc ngắn hạn với những điều kiện vượt ra ngoài điều kiện bảo quản đã ghi trên nhãn (ví dụ như trong khi chuyên chở bằng hoặc bốc dỡ). Bàn luận này nên được hỗ trợ, nếu phù hợp, bằng thử nghiệm tiếp theo trên một lô thành phẩm ở điều kiện cấp tốc trong thời gian ngắn hơn 3 tháng.
4.10.4.2. Thành phẩm thuốc dự kiến bảo quản đông lạnh
Với thuốc dự kiến bảo quản đông lạnh, nên dựa trên dữ liệu dài hạn để xác định tuổi thọ. Khi không tiến hành thử nghiệm cấp tốc cho thành phẩm thuốc dự kiến bảo quản đông lạnh, thử nghiệm một lô ở điều kiện nhiệt độ nâng cao (ví dụ như 5°C ± 3°C or 25°C ± 2°C) trong một khoảng thời gian phù hợp nên được thực hiện để chỉ rõ ảnh hưởng của việc tiếp xúc ngắn hạn với những điều kiện vượt ra ngoài điều kiện bảo quản đã ghi trên nhãn (ví dụ như trong khi chuyên chở bằng hoặc bốc dỡ).
4.10.4.3. Thành phẩm thuốc dự kiến bảo quản dưới -20oC
Với thuốc dự kiến bảo quản dưới -20oC, nên dựa trên dữ liệu dài hạn để xác định tuổi thọ và nên đánh giá theo từng trường hợp cụ thể. (ICH Q1E 6 Feb 03, p.4-7)
4.10.5. Các phương pháp thống kê tổng quát
Nếu có thể, nên sử dụng một phương pháp thống kê thích hợp để phân tích dữ liệu độ ổn định dài hạn của lô đầu tiên trong hồ sơ đăng ký gốc. Mục đích của phân tích này là để thiết lập, với mức độ tin cậy cao, tuổi thọ mà ở đó các chỉ tiêu định lượng sẽ duy trì trong mức chất lượng cho các lô được sản xuất, đóng gói và bảo quản ở các điều kiện tương tự trong tương lai. Phương pháp tương tự có thể được áp dụng đối với các lô cam kết để thẩm tra hoặc suy rộng tuổi thọ đã được chấp nhận bạn đầu.
Trong trường hợp phân tích thống kê được thực hiện để đánh giá dữ liệu dài hạn do có sự biến đổi theo thời gian và/hoặc dao động, phương pháp thống kê tương tự nên được sử dụng cho các lô cam kết để thẩm tra hoặc suy rộng tuổi thọ đã được chấp nhận bạn đầu. (ICH Q1E 6 Feb 03, p.7)
Phân tích hồi quy được xem xét là cách tiếp cận phù hợp để đánh giá dữ liệu độ ổn định của các chỉ tiêu định lượng và để thiết lập tuổi thọ. Bản chất mối quan hệ giữa một chỉ tiêu chất lượng và thời gian sẽ quyết định có nên chuyển đổi dữ liệu trước khi phân tích hồi quy hay không. Thông thường, mối quan hệ có thể được biểu diễn bởi một hàm số tuyến tính hoặc phi tuyến trên thang số học hoặc logarit. Đôi khi hồi quy phi tuyến có thể được mong đợi là phản ánh chính xác hơn mối quan hệ thực sự.
Một cách tiếp cận hợp lý để ước lượng tuổi thọ là phân tích các chỉ tiêu định lượng để xác định thời điểm sớm nhất mà ở đó giới hạn khoảng tin cậy 95% của giá trị trung bình quanh đường hồi quy giao với đường biểu diễn mức chất lượng đề xuất.
Với một chỉ tiêu có xu hướng giảm theo thời gian, cận dưới của giới hạn khoảng tin cậy 95% nên được so sánh với mức chất lượng. Với một chỉ tiêu có xu hướng tăng theo thời gian, cận trên của giới hạn khoảng tin cậy 95% nên được so sánh với mức chất lượng. Với một chỉ tiêu có thể tăng lẫn giảm theo thời gian, hoặc xu hướng biến đổi của nó là chưa rõ, cả hai cận của giới hạn khoảng tin cậy 95% nên được tính và so sánh với cận trên và cận dưới của mức chất lượng.
Nếu kết quả phân tích cho thấy sự dao động giữa các lô là nhỏ, thì nên hợp nhất các dữ liệu để ước lượng chung. Điều này có thể được thực hiện trước tiên bằng cách áp dụng các kiểm định thống kê thích hợp (ví dụ giá trị p đối với mức ý nghĩa để bác bỏ giả thiết lớn hơn 0,25) đối với độ dốc của đường thẳng hồi quy và giá trị cắt trục tung tại thời điểm 0 cho các lô riêng rẽ. Nếu việc hợp nhất các dữ liệu của một vài lô là không thích hợp, thì tuổi thọ chung cần dựa trên thời gian tối thiểu mà một lô có thể được mong đợi là vẫn đạt mức chất lượng. Bất kỳ sự đánh giá nào cũng nên xem xét không chỉ chỉ tiêu hàm lượng mà còn cả chỉ tiêu các sản phẩm phân hủy và các chỉ tiêu thích hợp khác.
Phương pháp tính thống kê dùng để phân tích dữ liệu phải phù hợp với thiết kế nghiên cứu độ ổn định để đưa ra một kết luận thống kê có giá trị cho việc ước lượng tuổi thọ. Phương pháp đã mô tả trên có thể được sử dụng để ước lượng tuổi thọ cho một lô đơn lẻ hoặc cho nhiều lô hợp nhất hợp sau khi đánh giá bằng một kiểm định thống kê thích hợp. Các ví dụ về các phương pháp thống kê để phân tích dữ liệu độ ổn định từ thiết kế nghiên cứu được trình bày ở Phụ lục 5.6. (ICH Q1E 6 Feb 03, p.7)
4.11. Cam kết về độ ổn định
4.11.1. Khi dữ liệu về độ ổn định ở điều kiện dài hạn của các lô đầu tiên chưa đạt đủ thời gian của tuổi thọ đề xuất được phê duyệt tại thời điểm cấp phép lưu hành, cần có một cam kết tiếp tục nghiên cứu độ ổn định sau khi được cấp phép lưu hành để xác định tuổi thọ một cách chắc chắn.
4.11.2. Nếu trong hồ sơ đăng ký đã có các số liệu về độ ổn định ở điều kiện dài hạn của số lô sản xuất ít nhất bằng số lô tối thiểu theo quy định mà bao quát được tuổi thọ dự định, thì cam kết sau khi được cấp phép lưu hành được xem là không cần thiết. Ngược lại, thì một trong số những cam kết sau cần phải làm:
a. Nếu trong hồ sơ đăng ký đã có các số liệu nghiên cứu độ ổn định của số lô sản xuất ít nhất bằng số lô tối thiểu theo quy định, cần cam kết tiếp tục các nghiên cứu dài hạn đến hết tuổi thọ đề xuất và các nghiên cứu cấp tốc trong 6 tháng.
b. Nếu trong hồ sơ đăng ký đã có các số liệu nghiên cứu độ ổn định của ít hơn 3 lô sản xuất, cần cam kết tiếp tục nghiên cứu ở điều kiện thực trong tuổi thọ đề xuất và nghiên cứu cấp tốc trong 6 tháng, cam kết có thêm các lô sản xuất để có đủ ít nhất là số lô tối thiểu theo quy định, thực hiện nghiên cứu độ ổn định ở điều kiện dài hạn đến hết tuổi thọ đề xuất và nghiên cứu cấp tốc trong 6 tháng trên các lô này.
c. Nếu trong hồ sơ đăng ký không có các số liệu về độ ổn định của các lô sản xuất, cần cam kết thực hiện nghiên cứu độ ổn định ở điều kiện dài hạn đến hết tuổi thọ đề xuất và nghiên cứu cấp tốc trong 6 tháng trên 3 lô sản xuất đầu tiên.
Đề cương nghiên cứu độ ổn định áp dụng cho các lô cam kết phải giống như đã thực hiện đối với các lô đầu tiên, trừ khi có những biện luận có tính khoa học.
4.11.3. Nếu báo cáo nghiên cứu độ ổn định đã nộp được thực hiện ở những điều kiện khác và không chứng minh được rằng thành phẩm thuốc vẫn đạt các chỉ tiêu chấp nhận được nêu ra trong hướng dẫn này, cơ sở đăng ký phải nộp cam kết và đề cương nghiên cứu về độ ổn định sau khi được cấp phép lưu hành. Trong những trường hợp như vậy, phải cân nhắc các lựa chọn sau đây: (1) rút ngắn tuổi thọ, (2) dùng hệ thống bao bì đóng gói có khả năng bảo vệ tốt hơn hoặc (3) đưa thêm các chú ý trên nhãn
4.11.4. Nghiên cứu độ ổn định sau khi cấp phép lưu hành có thể được thực hiện ở bất kỳ nước thành viên nào thuộc ASEAN, nước xuất xứ hoặc bất kỳ nước nào có điều kiện bảo quản như đã yêu cầu.
4.12. Cách ghi điều kiện bảo quản
Cách ghi điều kiện bảo quản trên nhãn phải theo đúng yêu cầu của quốc gia/khu vực có liên quan. Điều kiện bảo quản phải dựa trên đánh giá về độ ổn định của thành phẩm thuốc. Nếu có thể, nên đưa ra chỉ dẫn cụ thể, đặc biệt là đối với các thành phẩm thuốc không bảo quản được ở điều kiện đông lạnh. Nên tránh dùng các thuật ngữ như “điều kiện phòng” hoặc “nhiệt độ phòng”.
Phải có sự liên quan trực tiếp giữa điều kiện bảo quản ghi nhãn với các đặc tính về độ ổn định đã được chứng minh của thành phẩm thuốc.
Điều kiện bảo quản (nhiệt độ, ánh sáng, độ ẩm) được nêu ra phải dựa trên các yêu cầu liên quan của quốc gia/khu vực hoặc tuân theo đề xuất dưới đây. Khoảng biến đổi phải dựa trên đánh giá về độ ổn định của thành phẩm thuốc.
Bảng 1
Đề xuất ghi nhãn cho thành phẩm thuốc
|
Điều kiện nghiên cứu mà ở đó độ ổn định của thành phẩm thuốc đã được chứng minh |
Đề xuất ghi nhãna |
|
30°C/75% RH (dài hạn) 40°C/75% RH (cấp tốc) |
“Không bảo quản ở nhiệt độ trên 30°C” |
|
5°C ± 3°C |
“Bảo quản trong tủ lạnh (2°C đến 8°C)” |
|
-20°C ± 5 °C |
“Bảo quản đông lạnh” |
a Trong quá trình bảo quản, vận chuyển và phân phối thành phẩm thuốc, phải giám sát việc thực hiện Thực hành tốt phân phối thuốc hiện hành (GDP).
Nếu điều kiện nghiên cứu khác với bảng trên, đề xuất điều kiện bảo quản trên nhãn cần được chứng minh bằng các nghiên cứu độ ổn định hỗ trợ.
Về nguyên tắc, thành phẩm thuốc nên được đóng trong bao bì đảm bảo độ ổn định và bảo vệ thuốc khỏi sự biến chất. Không nên sử dụng việc trình bày điều kiện bảo quản để bù đắp cho sự chưa hợp lý hay chưa hoàn chỉnh của việc đóng gói. Các điều kiện bảo quản bổ sung trên nhãn có thể được áp dụng trong các trường hợp kết quả nghiên cứu độ ổn định chứng minh được ảnh hưởng của các yếu tố được liệt kê ở bảng 2 dưới đây.
Bảng 2
Các điều kiện bảo quản bổ sung trên nhãn khi kết quả nghiên cứu độ ổn định chứng minh được các yếu tố ảnh hưởng.
|
Các yếu tố ảnh hưởng |
Điều kiện bảo quản bổ sung trên nhãn tương ứng |
|
Thành phẩm thuốc không thể bảo quản trong tủ lạnh |
“Không bảo quản trong tủ lạnh hoặc đông lạnh”a |
|
Thành phẩm thuốc không thể bảo quản đông lạnh |
“Không bảo quản đông lạnh”a |
|
Thành phẩm thuốc nhạy cảm với ánh sáng |
“Tránh ánh sáng” |
|
Thành phẩm thuốc không thể bảo quản ở nhiệt độ cao, ví dụ như thuốc đặt |
“Không bảo quản và vận chuyển ở nhiệt độ trên 30°C” |
|
Thành phẩm thuốc dễ hút ẩm |
“Bảo quản ở điều kiện khô ráo” |
a Phụ thuộc vào dạng bào chế và đặc tính của thành phẩm thuốc mà có thể có nguy cơ thuốc bị biến chất do các biến đổi vật lý khi bảo quản ở nhiệt độ thấp, ví dụ như dạng thuốc lỏng hoặc bán rắn. Nhiệt độ thấp còn có thể ảnh hưởng đến đồ bao gói trong một vài trường hợp. Các điều kiện bảo quản bổ sung có thể cần thiết khi xem xét đến các khả năng này. (WHO, 2009, Appendix 3, p. 128-129)
1. Các cụm từ như “điều kiện phòng” hoặc “nhiệt độ phòng” không được chấp nhận.
2. Phải ghi các yêu cầu như hạn dùng và điều kiện bảo quản sau khi mở nắp, sau khi pha loãng hoặc sau khi pha lại thành dung dịch, nếu có. Ví dụ, thuốc tiêm kháng sinh hoặc hỗn dịch được bào chế ở dạng bột pha lại.
5. PHỤ LỤC
5.1. Đề cương nghiên cứu độ ổn định (ví dụ)
5.1.1. VIÊN NÉN PARACETAMOL 500 mg ÉP TRONG VỈ PVC (10 viên)
1. Mục đích
Để đánh giá độ ổn định của sản phẩm do việc nâng quy mô từ nghiên cứu và phát triển sang quy mô sản xuất.
2. Thiết kế nghiên cứu
Sản phẩm được ép trong vỉ PVC và được bảo quản theo điều kiện bảo quản được nêu trong hướng dẫn của nhà sản xuất.
2.1. Nguyên liệu thử
- Màng dính (push - through foil):
Màng nhôm dày 20 µm, mặt trong phủ keo dính nhiệt, mặt ngoài phủ PVC (8g/m2), cứng, mặt ngoài sáng ánh thiếc bạc.
- Màng tạo hình:
Màng PVC dày 250 µm.
|
Lô số |
Kiểu đóng gói |
Điều kiện/thời hạn bảo quản |
|
001 002 003 |
Vỉ PVC Vỉ PVC Vỉ PVC |
Điều kiện dài hạn (60 tháng); Cấp tốc (6 tháng) Điều kiện dài hạn (60 tháng); Cấp tốc (6 tháng) Điều kiện dài hạn (60 tháng); Cấp tốc (6 tháng) |
2.2. Kế hoạch thử nghiệm
2.2.1. Điều kiện bảo quản và khoảng thời gian lấy mẫu
Viên nén paracetamol được đưa vào ép vỉ PVC, 10 vỉ được đựng trong một hộp giấy carton và bảo quản ở các điều kiện sau:
|
Điều kiện bảo quản |
Khoảng thời gian lấy mẫu |
|
Bảo quản ở điều kiện dài hạn Nhiệt độ 30oC, độ ẩm tương đối 75% |
0, 3, 6, 9, 12, 18, 24, 36, 48, 60 tháng |
|
Cấp tốc Nhiệt độ 40oC, độ ẩm tương đối 75% |
0, 1, 3, 6 tháng |
Thời gian biểu chi tiết được đính kèm.
2.2.2. Thử nghiệm và tiêu chuẩn thử
Phòng đảm bảo chất lượng/kiểm tra chất lượng chịu trách nhiệm bảo quản và thử nghiệm mẫu tuân theo điều kiện bảo quản và phương pháp thử đã được thẩm định.
Các mẫu được lấy ra khỏi nơi bảo quản trước ngày thử như đã ghi trong thời gian biểu và để ở 5oC cho đến khi phân tích.
Công việc phân tích phải được tiến hành không muộn hơn 4 tuần kể từ khi lấy mẫu ra khỏi nơi bảo quản.
Quy trình thử: Số XXXX. Các thông số được thử nghiệm là:
a. Thử nghiệm vật lý
- Tính chất
- Khối lượng trung bình
- Độ hòa tan
- Thời gian rã
- Độ cứng
- Độ bở
- Hàm lượng nước
b. Hàm lượng paracetamol
c. Sản phẩm phân hủy: p-aminophenol
3. Số lượng mẫu thử (của một lô/một điều kiện bảo quản)
Nghiên cứu cấp tốc:
- Hình thức: 0 viên*
- Hàm lượng nước : 10 viên
- Thời gian rã: 6 viên
- Độ hòa tan: 6 viên
- Hàm lượng và tạp chất: 10 viên
- Độ cứng: 10 viên
- Độ bở: 50 viên
* Việc quan sát được thực hiện trên các viên dùng thử các tiêu chuẩn khác
Tổng số viên cho 1 lần thử = 92 viên (làm tròn thành 100 viên)
Số thử nghiệm: 4 lần
Số lượng cần dùng:
= 4 x 100 viên
= 400 viên
= 40 vỉ x 10 viên
= 4 hộp
Nghiên cứu dài hạn:
- Hình thức: 0 viên*
- Hàm lượng nước : 10 viên
- Thời gian rã: 6 viên
- Độ hòa tan: 6 viên
- Hàm lượng và tạp chất: 10 viên
- Độ cứng: 10 viên
- Độ bở: 50 viên
* Việc quan sát được thực hiện trên các viên dùng thử các tiêu chuẩn khác
Tổng số viên cho 1 lần thử = 92 viên (làm tròn thành 100 viên)
Số thử nghiệm: 9 lần
Số lượng cần dùng:
= 9 x 100 viên
= 900 viên
= 90 vỉ x 10 viên
= 9 hộp
Tổng số thuốc cần cho cả 2 thử nghiệm cần = 4 hộp + 9 hộp =
= 13 hộp x 10 vỉ
4. Nội dung báo cáo
1. Người chịu trách nhiệm
2. Tóm tắt
3. Mục đích
4. Nguyên liệu thử
5. Thành phần
6. Đóng gói
7. Điều kiện bảo quản và thời gian biểu của quá trình thử
8. Quy trình phân tích
9. Chuẩn đối chiếu
10. Kết quả:
10.1. Độ ổn định vật lý
10.2. Độ ổn định hóa học
10.2.1. Độ ổn định trong điều kiện bảo quản dài hạn
10.2.2. Độ ổn định trong điều kiện bảo quản cấp tốc
11. Bàn luận/kết luận:
12. Kết quả thử ở dạng bảng
Người xét duyệt Người kiểm tra Người soạn thảo
5.1.2. THỜI GIAN BIỂU NGHIÊN CỨU ĐỘ ỔN ĐỊNH
Viên nén paracetamol 500 mg
Ngày 02.7.1997
|
Bảo quản |
Thời gian biểu |
|||
|
Thời điểm |
Điều kiện |
Lô số 001 |
Lô số 002 |
Lô số 003 |
|
Bắt đầu |
Cấp tốc |
02.07.1997 |
09.07.1997 |
16.07.1997 |
|
Dài hạn |
04.07.1997 |
12.07.1997 |
18.07.1997 |
|
|
1 tháng |
Cấp tốc |
02.08.1997 |
09.08.1997 |
16.08.1997 |
|
3 tháng |
Cấp tốc |
02.10.1997 |
09.10.1997 |
16.10.1997 |
|
Dài hạn |
04.10.1998 |
12.10.1997 |
18.10.1997 |
|
|
6 tháng |
Cấp tốc |
02.01.1998 |
09.01.1998 |
16.01.1998 |
|
Dài hạn |
04.01.1998 |
12.01.1998 |
18.01.1998 |
|
|
9 tháng |
Dài hạn |
04.04.1998 |
12.04.1998 |
18.04.1998 |
|
12 tháng |
Dài hạn |
04.07.1998 |
12.07.1998 |
18.07.1998 |
|
18 tháng |
Dài hạn |
04.01.1999 |
12.01.1999 |
18.01.1999 |
|
24 tháng |
Dài hạn |
04.07.1999 |
12.07.1999 |
18.07.1999 |
|
36 tháng |
Dài hạn |
04.07.2000 |
12.07.2000 |
18.07.2000 |
|
48 tháng |
Dài hạn |
04.07.2001 |
12.07.2001 |
18.07.2001 |
|
60 tháng |
Dài hạn |
04.07.2002 |
12.07.2002 |
18.07.2002 |
|
Ghi chú: Cấp tốc: Nhiệt độ 40oC ± 2oC/độ ẩm tương đối 75% ± 5% Điều kiện dài hạn: Nhiệt độ 30oC ± 2oC/độ ẩm tương đối 75% ± 5% |
||||
|
Người xét duyệt |
Người kiểm tra |
Người soạn thảo |
5.2. Mẫu báo cáo (ví dụ)
THÀNH PHẨM THUỐC: VIÊN NÉN PARACETAMOL
|
HÀM LƯỢNG: |
500 mg |
Ngày 23/07/02 |
|
Hồ sơ số: |
XXXX |
Trang: 1/20 |
|
Loại nghiên cứu: |
Độ ổn định trước và sau khi lưu hành |
|
|
Mục tiêu: |
Độ ổn định của thành phẩm thuốc được bảo quản ở điều kiện dài hạn và điều kiện cấp tốc |
|
|
Thời gian nghiên cứu: |
60 tháng |
|
|
Đóng gói: |
Ép vỉ PVC |
|
|
Xuất xứ: |
MMM Ltd, Jakarta, Indonesia |
|
|
Đơn vị nghiên cứu độ ổn định: |
Phòng nghiên cứu và phát triển John Doe |
|
|
Đảm bảo chất lượng: |
Tom Smith |
|
1. Chịu trách nhiệm
|
Người chịu trách nhiệm |
Phòng/Địa điểm |
Chịu trách nhiệm |
|
John Doe |
Nghiên cứu và phát triển |
Phép thử vật lý và hóa học |
|
John Doe |
Nghiên cứu và phát triển |
Phép thử vi sinh |
2. Tóm tắt
Báo cáo này trình bày số liệu về độ ổn định của viên nén paracetamol 500 mg được bảo quản tới 60 tháng trong bao bì đóng gói sơ cấp như đã lưu hành trên thị trường.
Mọi biến đổi có liên quan đến bảo quản xảy ra trong sản phẩm cuối cùng đã được theo dõi bằng các phép thử kiểm tra độ ổn định chuyên biệt. Thiết kế thử nghiệm được dựa trên đặc tính ổn định của dược chất paracetamol và những yêu cầu cụ thể của dạng bào chế.
Tuổi thọ:
Thành phẩm có tuổi thọ 5 năm.
Hướng dẫn bảo quản:
Thành phẩm không có nhãn ghi hướng dẫn bảo quản.
3. Mục tiêu
Mục tiêu của nghiên cứu này là đánh giá độ ổn định của viên nén paracetamol 500 mg được bảo quản ở điều kiện dài hạn và điều kiện cấp tốc. Các mẫu thuốc được lật ngược để thuốc chắc chắn tiếp xúc với hệ thống bao bì đóng gói.
4. Vật liệu thử
Thông tin chi tiết về các lô đem thử nghiệm độ ổn định được liệt kê trong bảng sau:
4.1. Nguyên liệu
|
Nguyên liệu |
Số lô |
Nguồn gốc |
||
|
001 |
002 |
003 |
||
|
Paracetamol Lactose monohydrat Tinh bột ngô Tinh bột ngô thủy phân Talc Silic dioxyd keo khan (Aerosil 200) Magnesi stearat |
Ghi chú: DC .................. .................. .................. ................. ................. ................. |
................ ................. ................. ................. ................. ................. ................. |
................ ................. ................. ................ ................. ................. ................. |
................ ................ ................ ................ ................ ................ ................ |
4.2. Thành phẩm
|
Hàm lượng |
Lô số |
Sản xuất |
Quy mô |
Cỡ lô (viên) |
|
|
Ngày sản xuất |
Nơi sản xuất |
||||
|
500 mg/viên 500 mg/viên 500 mg/viên |
001 002 003 |
02.07.1997 09.07.1997 16.07.1997 |
Jakarta Jakarta Jakarta |
Sản xuất Sản xuất Sản xuất |
280000 280000 280000 |
5. Thành phần
1 viên nén paracetamol có chứa:
|
Thành phần |
Khối lượng (mg) |
Nguồn gốc |
|
Paracetamol Lactose monohydrat Tinh bột ngô Tinh bột ngô thủy phân Talc Silic dioxyd keo khan (Aerosil 200) Magnesi stearat |
500,00 79,00 65,50 5,00 3,00 2,00 0,50 |
.................... .................... .................... .................... .................... .................... .................... |
|
Tổng cộng |
655,00 |
|
6. Đóng gói
Các thử nghiệm về độ ổn định đối với các lô thuốc đã nêu trên được đóng gói trong bao bì sơ cấp như sau:
Thuốc được ép trong vỉ PVC gồm các lớp sau:
● Màng dính: Màng nhôm dày 20 µm, mặt trong được phủ keo dính nhiệt, mặt ngoài phủ PVC (8 g/m2), cứng, mặt ngoài sáng ánh thiếc bạc.
● Màng tạo hình: Màng PVC dày 250 µm.
7. Điều kiện bảo quản và các thời điểm thử
Các mẫu thuốc khác nhau của các thành phẩm thuốc đã được đóng gói đã được thử theo lịch trình sau đây:
|
Điều kiện bảo quản |
Khoảng thời gian thử (tháng) |
||||||||||
|
0 |
1 |
3 |
6 |
9 |
12 |
18 |
24 |
36 |
48 |
60 |
|
|
Nhiệt độ 30oC ± 2oC Độ ẩm tương đối 75% ± 5% |
x |
- |
x |
x |
x |
x |
x |
x |
x |
x |
X |
|
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 75% ± 5% |
x |
x |
x |
x |
- |
- |
- |
- |
- |
- |
- |
8. Quy trình phân tích
Các phép thử độ ổn định của viên nén paracetamol đã được tiến hành theo phương pháp thử của USP.
Trong quá trình thử nghiệm về độ ổn định, các chỉ tiêu chính để đánh giá độ ổn định được ghi ở bảng dưới đây:
|
Chỉ tiêu thử |
Phương pháp thử |
Giới hạn |
|
Độ cứng |
USP |
≥ 70 N |
|
Độ bở |
USP |
≤ 2% |
|
Sản phẩn phân hủy ● p-aminophenol |
USP |
≤ 0,005% |
|
Độ nhiễm khuẩn |
USP |
Tổng cộng ≤ 102 khuẩn lạc E. coli: không có |
|
Hàm lượng (Sắc ký lỏng) |
USP |
95,0 - 105,0% |
Chú ý: Như đã đề cập ở mục 2.1.2, 3.1 và 3.2, cần thêm thử nghiệm thời gian rã và độ hòa tan.
9. Chuẩn đối chiếu
Đã dùng paracetamol chuẩn theo USP hàm lượng 99,5%.
10. Kết quả
Các kết quả thử được trình bày trong các bảng kèm theo.
10.1. Độ ổn định vật lý
Nghiên cứu độ ổn định vật lý của viên nén paracetamol 500 mg đã chứng tỏ viên nén không bị biến đổi sau 60 tháng bảo quản ở nhiệt độ 30oC/độ ẩm tương đối 75% và sau 6 tháng trong điều kiện cấp tốc ở nhiệt độ 40oC/độ ẩm tương đối 75%.
Hình thức viên không có biến đổi gì đáng kể.
10.2. Độ ổn định hóa học
Độ ổn định ở điều kiện dài hạn
Bảo quản sau 60 tháng ở nhiệt độ 30oC/độ ẩm tương đối 75% không có ảnh hưởng gì đáng kể lên tính ổn định hóa học của thành phẩm thuốc. Riêng chỉ tiêu “tạp chất hữu cơ” chỉ có sự biến đổi rất nhỏ. Nồng độ p-aminophenol dưới 0,005%.
Hàm lượng paracetamol đã không thay đổi đáng kể sau khi bảo quản ở điều kiện dài hạn trong 60 tháng so với hàm lượng ban đầu của các lô.
Độ ổn định ở điều kiện cấp tốc
Bảo quản ở điều kiện cấp tốc trong 6 tháng không ảnh hưởng đến độ ổn định hóa học.
Hàm lượng paracetamol đã không thay đổi đáng kể so với giá trị ban đầu của các lô.
11. Bàn luận/kết luận
Bảo quản ở điều kiện thử nghiệm dài hạn không làm thay đổi kết quả định lượng paracetamol. Không nhận thấy có biến đổi đáng kể nào về độ ổn định vật lý và hóa học. Vì các dữ liệu thử nghiệm dài hạn và cấp tốc cho thấy sự thay đổi rất nhỏ hoặc không thay đổi theo thời gian và sự dao động rất thấp nên việc phân tích thống kê được xem là không cần thiết.
Tuổi thọ:
Căn cứ vào các số liệu về kết quả nghiên cứu đã xác định được tuổi thọ của thành phẩm là 5 năm.
Hướng dẫn bảo quản:
Thành phẩm có thể được ghi nhãn "Bảo quản dưới 30oC".
Tóm tắt kết quả nghiên cứu độ ổn định
Bảng 1
|
Thành phẩm: |
Paracetamol |
Lô số: 001 |
|
|
Hàm lượng: |
500 mg/viên nén |
|
|
|
Đóng gói: |
Vỉ bấm PVC |
|
|
|
Bảo quản |
Hình thức |
Độ cứng (N) |
Độ bở (%) |
Hàm lượng: paracetamol 500 mg |
Sản phẩm phân hủy |
Giới hạn vi sinh vật |
|
|
Thời gian (tháng) |
Điều kiện |
p-aminophenol (%) |
|||||
|
Chỉ tiêu |
Viên nén màu trắng, hình trụ dẹt |
≥ 70 N |
≤ 2 % |
95,0 - 105,0% |
≤ 0,005% |
Tổng số ≤ 102 CFU E. coli: không có |
|
|
Ban đầu |
|
Đạt |
80 |
1 |
98,8 |
0,001 |
Đạt |
|
3 6 9 12 18 24 36 48 60 |
Nhiệt độ 30oC ± 2oC Độ ẩm tương đối 70% ± 5% |
Đạt Đạt Đạt Đạt Đạt Đạt Đạt Đạt Đạt |
80 85 90 85 97 94 87 98 93 |
1 0,5 0,5 1 1 0,5 1 1 0,5 |
101,4 98,3 99,6 98,9 99,0 98,9 99,1 99,5 99,3 |
0,002 0,004 0,001 0,003 0,003 0,004 0,002 0,001 0,001 |
Đạt Đạt Đạt Đạt Đạt Đạt Đạt Đạt |
|
3 6 |
Nhiệt độ 40oC ± 2oC Độ ẩm tương đối 70% ± 5% |
Đạt Đạt |
96 80 |
0,5 0,5 |
100,05 99,6 |
0,004 0,004 |
Đạt Đạt |
Chú ý: - Cần thêm dữ liệu về thời gian rã hoặc độ hòa tan cho mỗi lô
- Với lô 002 và 003, các kết quả nghiên cứu được báo cáo theo cùng mẫu như lô 001.
5.3. Thiết kế rút gọn (phân cực và ma trận)
Một thiết kế nghiên cứu đầy đủ là một thiết kế mà trong đó ở các mẫu thử có sự kết hợp của tất cả các yếu tố được thử nghiệm ở tất cả các thời điểm. Một thiết kế rút gọn là một thiết kế mà trong đó ở các mẫu thử không có sự kết hợp của tất cả các yếu tố tại tất cả các thời điểm thử nghiệm. Một thiết kế rút gọn có thể là sự thay thế thích hợp cho một thiết kế đầy đủ khi có nhiều yếu tố cần xem xét. Bất kỳ một thiết kế rút gọn nào cũng cần có đủ khả năng để ước lượng tuổi thọ. Trước khi xem xét một thiết kế rút gọn, các giả thiết cần được đánh giá và lý giải. Cần xem xét nguy cơ phải thiết lập một tuổi thọ ngắn hơn tuổi thọ được xác định từ thiết kế đầy đủ vì lượng dữ liệu thu được bị giảm đi.
Trong khi thực hiện một nghiên cứu theo thiết kế rút gọn, việc chuyển đổi thành thử nghiệm đầy đủ hoặc thành một thiết kế rút gọn ít hơn có thể được xem xét nếu đưa ra được các lý giải và các nguyên tắc của thiết kế đầy đủ hoặc thiết kế rút gọn được tuân theo. Tuy nhiên, việc điều chỉnh đúng phải được thực hiện bằng phân tích thống kê, để giải thích cho việc tăng cỡ mẫu thử do sự thay đổi. Khi thay đổi thiết kế, thử nghiệm đầy đủ hoặc thử nghiệm rút gọn ít hơn phải cần được tiến hành ở tất cả các thời điểm còn lại của nghiên cứu độ ổn định.
Khả năng áp dụng thiết kế rút gọn
Thiết kế rút gọn có thể được áp dụng để nghiên cứu độ ổn định của hầu hết các loại thành phẩm thuốc, mặc dù vậy việc lý giải thêm phải được đưa ra đối với một vài hệ cung cấp thuốc phức tạp mà ở đó có thể có nhiều tương tác giữa thuốc - dụng cụ cung cấp thuốc.
Thiết kế phân cực
Thiết kế phân cực là thiết kế về một lịch trình độ ổn định trong đó chỉ những mẫu ở về các cực của các yếu tố thiết kế nào đó (ví dụ như hàm lượng, cỡ bao bì và/hoặc lượng đóng) được thử nghiệm tại tất cả các thời điểm như trong thiết kế đầy đủ. Thiết kế giả thiết rằng độ ổn định của bất kỳ hàm lượng trung gian nào được đại diện bởi độ ổn định của các cực thử.
Ví dụ về thiết kế:
Bảng 1 là một ví dụ về thiết kế phân cực. Thí dụ này được dựa trên một thành phẩm có 3 loại hàm lượng và 3 cỡ bao bì (P1, P2 và P3). Trong ví dụ này, 2 cỡ bao bì bằng polyethylen tỷ trọng cao có dung tích 15 ml (P1) và 500 ml (P3) đại diện cho 2 cực. Các lô đối với mỗi kết hợp đã chọn cần được thử nghiệm tại mỗi một thời điểm giống như thiết kế đầy đủ.
Bảng 1: Thí dụ về một thiết kế phân cực
|
Hàm lượng |
50 mg |
75 mg |
100 mg |
|||||||
|
Lô |
1 |
2 |
3 |
1 |
2 |
3 |
1 |
2 |
3 |
|
|
Kích cỡ bao bì |
15 ml |
T |
T |
T |
|
|
|
T |
T |
T |
|
100 ml |
|
|
|
|
|
|
|
|
|
|
|
500 ml |
T |
T |
T |
|
|
|
T |
T |
T |
|
Chú thích: T là mẫu được thử
Thiết kế phân cực giả thiết rằng độ ổn định với hàm lượng hoặc kích cỡ trung gian được biểu diễn thông qua độ ổn định ở các cực thiết kế. Nếu phân tích thống kê chỉ ra rằng độ ổn định với hàm lượng hoặc kích cỡ ở các cực thiết kế là khác nhau thì độ ổn định với hàm lượng hoặc kích cỡ trung gian không được xem là ổn định hơn cực kém ổn định nhất. Ví dụ, nếu P1 trong thiết kế phân cực ở trên kém ổn định hơn P3 thì tuổi thọ với P2 không được vượt quá P1. Sự nội suy giữa P1 và P3 là không được xem xét. (ICH Q1E 6 Feb 03, p.13)
Thiết kế ma trận
Là thiết kế cho một chương trình nghiên cứu độ ổn định trong đó chỉ có một nhóm mẫu được chọn trong tổng số mẫu có sự kết hợp tất cả các yếu tố sẽ được thử nghiệm tại một thời điểm xác định. Ở thời điểm kế tiếp, một nhóm mẫu khác có sự kết hợp tất cả các yếu tố sẽ được thử nghiệm. Thiết kế giả định rằng tại từng thời điểm thử nghiệm, độ ổn định của mỗi nhóm mẫu đã được thử nghiệm sẽ đại diện cho độ ổn định của toàn bộ mẫu. Các yếu tố khác nhau giữa các mẫu của cùng một thành phẩm thuốc phải được xác định, ví dụ khác nhau về lô sản xuất, hàm lượng, cỡ đóng gói của cùng hệ thống bao bì đóng gói, và trong một số trường hợp, có thể khác hệ thống bao bì đóng gói.
Khi hệ bao bì thứ cấp có tác động đến độ ổn định của thành phẩm thuốc thì thiết kế ma trận có thể được thực hiện chéo giữa các hệ bao bì. Mỗi điều kiện bảo quản phải được xử lý riêng bằng thiết kế ma trận riêng của nó. Thiết kế ma trận không được thực hiện chéo giữa các thuộc tính thử. Tuy nhiên, những thiết kế ma trận thay thế khác cho các thuộc tính thử khác nhau có thể áp dụng nếu được chứng minh.
Các ví dụ về thiết kế:
Các ví dụ về thiết kế ma trận dựa trên thời điểm đối với một sản phẩm có hai hàm lượng (S1 và S2) như trình bày ở bảng 2. Thuật ngữ “rút gọn một nửa” và “rút gọn một phần ba” nói đến chiến lược rút gọn áp dụng đầu tiên với thiết kế nghiên cứu đầy đủ. Ví dụ, rút gọn một nửa là loại đi 1 trong 2 thời điểm từ thiết kế nghiên cứu đầy đủ và rút gọn 1/3 bớt đi 1 trong 3 thời điểm. Trong các ví dụ trình bày ở bảng 2, sự rút gọn ở đây ít hơn một nửa và 1/3 vì bao gồm thử nghiệm đủ ở một vài thời điểm như thời điểm bắt đầu, thời điểm 12 tháng và thời điểm kết thúc. Chính vì thế sự rút gọn ở đây ít hơn 1/2 (24/48) và 1/3 (16/48) thực tế rút gọn tương ứng là 15/48 và 10/48.
Bảng 2: Các ví dụ về thiết kế ma trận dựa trên thời điểm cho một sản phẩm có hai hàm lượng
“Rút gọn một nửa”
|
Thời điểm (tháng) |
0 |
3 |
6 |
9 |
12 |
18 |
24 |
36 |
||
|
Hàm lượng |
S1 |
Lô 1 |
T |
T |
|
T |
T |
|
T |
T |
|
Lô 2 |
T |
T |
|
T |
T |
T |
|
T |
||
|
Lô 3 |
T |
|
T |
|
T |
T |
|
T |
||
|
S2 |
Lô 1 |
T |
|
T |
|
T |
|
T |
T |
|
|
Lô 2 |
T |
T |
|
T |
T |
T |
|
T |
||
|
Lô 3 |
T |
|
T |
|
T |
|
T |
T |
||
T là mẫu được thử
“Rút gọn một phần ba”
|
Thời điểm (tháng) |
0 |
3 |
6 |
9 |
12 |
18 |
24 |
36 |
||
|
Hàm lượng |
S1 |
Lô 1 |
T |
T |
|
T |
T |
|
T |
T |
|
Lô 2 |
T |
T |
T |
|
T |
T |
|
T |
||
|
Lô 3 |
T |
|
T |
T |
T |
T |
T |
T |
||
|
S2 |
Lô 1 |
T |
|
T |
T |
T |
T |
T |
T |
|
|
Lô 2 |
T |
T |
|
T |
T |
|
T |
T |
||
|
Lô 3 |
T |
T |
T |
|
T |
T |
|
T |
||
T là mẫu được thử
Chi tiết hơn được mô tả trong ICH Q1D.
5.4. Ví dụ về kiểu, độ dày và hệ số thấm của vật liệu làm bao bì trình bày trong bảng 1 và khả năng thấm hơi nước của các loại vật liệu bao bì khác nhau được trình bày trong hình 1
Bảng 1. Ví dụ về kiểu, độ dày và hệ số thấm của vật liệu làm bao bì
|
TT |
Vật liệu |
Độ dày |
Độ dày thường dùng (µm) |
Tiêu chuẩn về khả năng thấm |
Khả năng chịu nhiệt |
|
|
Ở 23oC/ 85% RH (g/m2.ngày) |
Ở 38oC/ 90% RH (g/m2.ngày) |
|||||
|
1 |
PVC (Polivinyl clorid) |
250 µm |
200-250 µm |
1,6-1,8 |
3,0-3,2 |
Tốt |
|
2 |
Duplex (PVC+PVDC) PVC (Polivinyl clorid) PVDC (Polivinyliden clorid) |
|
270 µm |
|
Tốt/ Xuất sắc |
|
|
200 - 250 µm |
|
|||||
|
5 µm cho bề rộng 10g/m2 (40-60-80 g/m2) |
40 g/m2 |
0,15 |
0,6 |
|||
|
60 g/m2 |
0,1 |
0,4 |
||||
|
80 g/m2 |
0,05 |
0,3 |
||||
|
3 |
Triplex (PVC + PE + PVDC) PVC (Polivinyl clorid) PE (Polyethylen) PVDC (Polivinyliden clorid) |
|
300 µm |
|
Tốt/ Xuất sắc (tuỳ theo độ dày) |
|
|
200 -250 µm |
|
|
||||
|
25 µm |
||||||
|
5 µm cho bề rộng 10 g/m2 (40-60-90 g/m2) |
40 g/m2 |
0,12 |
0,55 |
|||
|
60 g/m2 |
0,06 |
0,35 |
||||
|
90 g/m2 |
0,02 |
0,2 |
||||
|
4 |
Starflex (PVC+TE +PVDC) PVC (Polivinyl clorid) TE (Thermolast) PVDC (Polivinyliden clorid) |
|
Tối đa 300 µm |
|
Tốt/ Xuất sắc (tuỳ theo độ dày) |
|
|
200 -250 µm |
|
|||||
|
Trải rộng TE (bao phủ) 5 g/m2 |
||||||
|
5 µm cho bề rộng 10 g/m2 (60-90-120 g/m2) |
60 g/m2 |
0,06 |
0,35 |
|||
|
90 g/m2 |
0,03 |
0,2 |
||||
|
120 g/m2 |
0,01 |
0,15 |
||||
|
5 |
PVC +ACLAR PVC (Polivinyl clorid) ACLAR (Polyfluor carbonat) |
|
270 µm |
|
|
Xuất sắc |
|
200 -250 µm |
|
|
|
|||
|
15 -23-51 µm |
15 g/m2 |
- |
0,39 |
|||
|
|
23 g/m2 |
- |
0,22 |
|||
|
|
51 g/m2 |
- |
0,11 |
|||
|
6 |
PVC/PE/ACLAR PVC (Polivinyl clorid) PE (Polyethylen) ACLAR (Polyfluor carbonat) |
|
280 µm |
|
|
Xuất sắc |
|
200 -250 µm |
|
|
|
|||
|
25 µm |
|
|
|
|||
|
15 -51 µm |
15 µm |
- |
<0.32 |
|||
|
51 µm |
- |
<0.11 |
||||
|
7 |
Aluminum Cold Forming Nhôm PVC cứng OPA |
|
130 µm |
- |
0 |
Xuất sắc |
|
40 -45 µm |
|
- |
- |
|||
|
60 µm |
|
- |
- |
|||
|
25 µm |
|
- |
- |
|||
|
8 |
Màng nhôm luyện cứng (màng che phủ Vỉ nhôm cho màng PVC - Nhôm - PVC Vỉ nhôm cho màng PVC-PVDC - Nhôm - PVDC |
|
20 µm |
|
|
|
|
|
|
- |
- |
|||
|
20 µm |
|
- |
- |
|||
|
tối thiểu 7g/m2 |
|
- |
- |
|||
|
|
30 µm |
- |
- |
|||
|
20 µm |
|
- |
- |
|||
|
15 g/m2 |
|
- |
- |
|||
|
9 |
Màng nhôm luyện mềm - Nhôm - PVDC |
|
40 µm |
- |
- |
|
|
30 µm |
|
|
|
|||
|
15 g/m2 |
|
|
|
|||
Hình 1 Độ thấm hơi nước của một số loại vật liệu làm bao bì
(Phương pháp ASTM F1249, 38oC/độ ẩm tương đối 90%)
5.5. Cây quyết định để đánh giá dữ liệu nhằm ước lượng tuổi thọ của thành phẩm thuốc (trừ thành phẩm bảo quản đông lạnh)
5.6. Ví dụ về các phương pháp thống kê để phân tích dữ liệu độ ổn định
Hồi quy tuyến tính, kiểm định tính hợp nhất và mô hình hóa thống kê được mô tả dưới đây là các ví dụ về các phương pháp và quy trình thống kê có thể được sử dụng trong phân tích dữ liệu độ ổn định với loại dữ liệu có thể đem phân tích như các chỉ tiêu định lượng với các mức chất lượng đề xuất.
Phân tích dữ liệu cho một lô
Nói chung, mối liên hệ giữa một vài chỉ tiêu định lượng và thời gian được giả thiết là tuyến tính1. Hình 1 biểu diễn đường hồi quy của hàm lượng của một thành phẩm thuốc có mức chất lượng trên và dưới lần lượt là 105% và 95% so với hàm lượng trên nhãn, với dữ liệu dài hạn 12 tháng và tuổi thọ đề xuất là 24 tháng. Trong ví dụ này, giới hạn tin cậy 95% về hai phía của giá trị trung bình được sử dụng vì chưa rõ hàm lượng sẽ giảm hay tăng theo thời gian (ví dụ như trong trường hợp thành phẩm có dung môi nước đóng trong bao bì bán thấm). Đường biểu diễn giới hạn tin cậy dưới cắt đường biểu diễn cận dưới của mức chất lượng ở thời điểm 30 tháng, trong khi đó đường biểu diễn giới hạn tin cậy trên không cắt đường biểu diễn cận trên của mức chất lượng tại các thời điểm trở về sau. Vì vậy, tuổi thọ đề xuất 24 tháng có thể được hỗ trợ bởi phân tích thống kê dữ liệu hàm lượng theo các hướng dẫn được ghi ở mục 4.10.1 và 4.10.2.
Hình 1
Khi dữ liệu cho một chỉ tiêu chỉ có một mức chất lượng trên hoặc dưới được phân tích, giới hạn tin cậy 95% về một phía tương ứng được sử dụng. Hình 2 biểu diễn đường hồi quy của một sản phẩm phân hủy trong một thành phẩm thuốc có dữ liệu dài hạn 12 tháng và hạn dùng đề xuất 24 tháng, trong khi mức chất lượng là không quá 1,4%. Đường biểu diễn giới hạn tin cậy 95% về phía trên của giá trị trung bình cắt đường biểu diễn mức chất lượng ở thời điểm 31 tháng. Vì vậy, tuổi thọ đề xuất 24 tháng có thể được hỗ trợ bởi phân tích thống kê dữ liệu sản phẩm phân hủy theo các hướng dẫn được ghi ở mục 4.10.1 và 4.10.2.
Nếu phương pháp trên được sử dụng, giá trị trung bình của chỉ tiêu định lượng (ví dụ như hàm lượng, các sản phẩm phân hủy) có thể được mong đợi là sẽ nằm trong mức chất lượng cho đến hết tuổi thọ với độ tin cậy 95%.
Hình 2
6. THUẬT NGỮ
Thử nghiệm cấp tốc (Accelerated testing)
Thử nghiệm được thiết kế để tăng tốc độ phân hủy hóa học hoặc biến đổi vật lý của một dược chất hoặc một thành phẩm thuốc bằng cách sử dụng điều kiện bảo quản khắc nghiệt như là một phần của các nghiên cứu độ ổn định chính thức. (Dữ liệu thu được từ các thử nghiệm này cùng với các thử nghiệm độ ổn định dài hạn có thể được sử dụng để đánh giá các ảnh hưởng hóa học ở điều kiện không cấp tốc trong thời gian dài hơn và để đánh giá tác động của việc tiếp xúc ngắn hạn với những điều kiện vượt ra ngoài điều kiện bảo quản ghi trên nhãn, chẳng hạn điều kiện có thể xảy ra khi chuyên chở. Các kết quả thu được từ nghiên cứu thử nghiệm cấp tốc không phải lúc nào cũng dự đoán được những biến đổi vật lý; xem thêm độ ổn định và các thuật ngữ có liên quan.)
Lô (Batch)
Một lượng xác định nguyên liệu ban đầu, vật liệu bao gói hay thành phẩm được xử lý trong một quy trình đơn lẻ hay một loạt các quy trình sao cho nó được mong đợi là đồng nhất. Đôi khi cần thiết để chia một lô thành một số mẻ nhỏ, mà những mẻ này cùng nhau tạo thành một lô đồng nhất cuối cùng. Trong trường hợp thành phẩm có tiệt khuẩn ở giai đoạn cuối, cỡ lô được xác định bởi dung lượng của nồi hấp. Trong sản xuất liên tục, lô phải tương ứng với một tỷ lệ xác định trước của quá trình sản xuất, được đặc trưng bởi độ đồng nhất dự kiến của nó. Cỡ lô có thể được định nghĩa cả theo một lượng xác định lẫn một lượng được sản xuất trong một khoảng thời gian xác định. (WHO, 2009, p.109)
Thiết kế phân cực (Bracketing)
Thiết kế cho một chương trình nghiên cứu độ ổn định trong đó chỉ những mẫu thử ở về các cực của các yếu tố thiết kế nào đó (ví dụ như hàm lượng, cỡ đóng gói) sẽ được thử nghiệm tại tất cả các thời điểm như trong thiết kế đầy đủ. (Thiết kế giả định rằng độ ổn định của các mức trung gian sẽ được đại diện bởi độ ổn định của các cực thử. Khi một dãy các hàm lượng được thử nghiệm, thiết kế phân cực được áp dụng nếu hàm lượng các chất giống nhau hoặc thành phần công thức gần như nhau [ví dụ như đối với một dãy viên nén được dập với những khối lượng khác nhau từ một loại cốm cơ bản tương tự nhau hoặc một dãy viên nang được đóng với các khối lượng khác nhau từ cùng thành phần cơ bản vào các cỡ vỏ nang khác nhau]. Thiết kế ô trống cũng có thể được dùng cho các cỡ bao bì khác nhau hoặc các lượng đóng gói khác nhau trong cùng hệ thống bao bì đóng gói).
Vùng khí hậu (Climatic zones)
|
Vùng khí hậu |
Định nghĩa |
Điều kiện nghiên cứu dài hạn |
|
I |
Khí hậu ôn đới |
21°C/45% RH |
|
II |
Khí hậu Địa trung hải và cận nhiệt đới |
25°C/60% RH |
|
III |
Khí hậu nóng và khô |
30°C/35% RH |
|
IVA |
Khí hậu nóng và ẩm |
30°C/65% RH |
|
IVB |
Khí hậu nóng và rất ẩm |
30°C/75% RH |
Lô cam kết (Commitment batches)
Lô sản xuất của một dược chất hay một thành phẩm thuốc mà trên những lô này, các nghiên cứu độ ổn định được bắt đầu thực hiện hoặc hoàn thiện sau khi được cấp phép theo một cam kết trong hồ sơ đăng ký.
Hệ thống bao bì đóng gói (Container closure system)
Tất cả các thành phần đóng gói dùng để chứa đựng và bảo vệ dạng bào chế. Hệ thống bao gồm bao bì sơ cấp và bao bì thứ cấp (nếu bao bì thứ cấp với mục đích bảo vệ thêm cho thành phẩm thuốc). Thuật ngữ hệ thống bao gói (packaging system) tương đương với hệ thống bao bì đóng gói (container closure system).
Dạng bào chế (Dosage form)
Một dạng thành phẩm thuốc (ví dụ viên nén, viên nang, dung dịch, kem) có chứa dược chất, thường được phối hợp, nhưng không nhất thiết, với các tá dược.
Thành phẩm thuốc/Dược phẩm (Drug product/Pharmaceutical product)
Bất kỳ thành phẩm nào được dùng cho người với mục đích làm thay đổi hoặc thăm dò các hệ sinh lý hoặc các tình trạng bệnh lý vì lợi ích của người dùng.
Dược chất (Drug substance)
Dược chất chưa được pha chế mà sau đó có thể kết hợp với các tá dược để tạo ra dạng bào chế (Xem thêm Dược chất trong phần thuật ngữ của ACTD Quality).
Tá dược (Excipient)
Một thành phần được chủ định thêm vào dược chất mà không có các tác dụng dược lý ở lượng sử dụng.
Ngày hết hạn (Expire date)
Ngày được ghi trên bao bì của thành phẩm thuốc mà trước ngày này thành phẩm vẫn đạt tiêu chuẩn chất lượng trong suốt hạn dùng đã được phê duyệt nếu được bảo quản trong các điều kiện đã định. (Sau ngày hết hạn, sẽ không có gì đảm bảo là thành phẩm vẫn còn đạt các chỉ tiêu chất lượng đã được phê duyệt và do đó thành phẩm có thể không thích hợp và không nên sử dụng).
Nghiên cứu độ ổn định chính thức (Formal stability studies)
Nghiên cứu dài hạn hoặc cấp tốc được thực hiện trên các lô ban đầu và/hoặc các lô cam kết theo đề cương nghiên cứu độ ổn định đã viết để thiết lập hoặc xác nhận tuổi thọ của thành phẩm thuốc.
Bao bì không thấm (Impermeable containers)
Bao bì đóng vai trò như một hàng rào vĩnh viễn ngăn cản các chất khí hoặc dung môi, ví dụ như tuýp nhôm hàn kín đựng thuốc bán rắn, ống thủy tinh hàn kín đựng dung dịch hoặc vỉ nhôm/nhôm cho dạng bào chế rắn.
Thử nghiệm dài hạn (Long term testing)
Thử nghiệm độ ổn định được thực hiện dưới điều kiện bảo quản gợi ý trong chu kỳ tái kiểm hoặc hạn dùng đề xuất (hay phê duyệt) để ghi nhãn.
Thay đổi lớn (Major variations)
Thay đổi đối với một thành phẩm thuốc đã được cấp phép lưu hành ảnh hưởng đến một hoặc một số điểm sau:
- Đường dùng,
- Hàm lượng, liều dùng,
- Chỉ định,
- Hoặc những điểm không nằm trong định nghĩa thay đổi nhỏ.
(Hồ sơ xin phép cho các thay đổi lớn thường phải có các dữ liệu cần thiết về chất lượng, độ an toàn và hiệu quả của công thức mới do các thay đổi mang lại).
Cân bằng khối (Mass balance)
Quá trình cộng gộp kết quả xác định hàm lượng và lượng các sản phẩm phân hủy để thấy được độ xấp xỉ của giá trị này với 100% giá trị ban đầu, có xem xét đến sai số của quy trình phân tích.
Thiết kế ma trận (Matrixing)
Thiết kế cho một chương trình nghiên cứu độ ổn định trong đó chỉ có một nhóm mẫu được chọn trong tổng số mẫu có sự kết hợp tất cả các yếu tố sẽ được thử nghiệm ở một thời điểm xác định. (Ở một thời điểm kế tiếp, một nhóm mẫu khác có sự tổ hợp tất cả yếu tố sẽ được thử nghiệm. Thiết kế giả định rằng tại từng thời điểm thử nghiệm, độ ổn định của mỗi nhóm mẫu đã được thử sẽ đại diện cho độ ổn định của toàn bộ mẫu. Sự khác nhau giữa các mẫu của cùng một thành phẩm thuốc phải đại diện cho, ví dụ: sự khác nhau về lô sản xuất, hàm lượng, cỡ đóng gói của cùng hệ thống bao bì đóng gói, và trong một số trường hợp, có thể là sự khác hệ thống bao bì đóng gói).
Thay đổi nhỏ (Minor variations)
Thay đổi đối với một thành phẩm thuốc đã được cấp phép lưu hành không ảnh hưởng tới một hoặc một số điểm sau:
- Đường dùng,
- Hàm lượng, liều dùng,
- Chỉ định, và
- Dược chất
(Hồ sơ xin phép cho các thay đổi nhỏ thường phải có các dữ liệu cần thiết để chứng minh chất lượng của công thức mới do các thay đổi mang lại)
Lô ở quy mô thử nghiệm (Pilot scale batch)
Một lô dược chất hoặc thành phẩm thuốc được sản xuất bởi một quy trình đại diện và mô phỏng cho quy trình áp dụng ở quy mô sản xuất. (Với các dạng thuốc rắn dùng đường uống, quy mô thử nghiệm thông thường tối thiểu phải băng 1/10 quy mô sản xuất hoặc 100.000 viên nén hoặc viên nang, tuỳ theo số lượng nào lớn hơn trừ khi có giải trình khác).
Lô đầu tiên (Primary batch)
Lô dược chất hoặc thành phẩm thuốc được dùng trong nghiên cứu độ ổn định mà các số liệu về độ ổn định của nghiên cứu này được cung cấp trong hồ sơ đăng ký lần lượt với mục đích thiết lập chu kỳ tái kiểm hoặc tuổi thọ.
Với thành phẩm thuốc, hai trong số ba lô ít nhất phải là lô ở quy mô thử nghiệm và lô thứ ba có thể nhỏ hơn nếu lô này đại diện cho các bước sản xuất trọng yếu. Tuy nhiên, một lô đầu tiên có thể là lô sản xuất.
Lô sản xuất (Production batch)
Lô thành phẩm thuốc được sản xuất ở quy mô sản xuất bằng cách sử dụng các thiết bị sản xuất trong cơ sở sản xuất như mô tả trong hồ sơ đăng ký.
Bao bì bán thấm (Semi-impermeable containers)
Bao bì cho phép dung môi, thường là nước đi qua, trong khi ngăn cản sự mất chất hòa tan. Cơ chế của việc vận chuyển dung môi là hấp thụ lên một bề mặt bao bì, khuếch tán vào chất liệu làm bao bì và thoát ra bề mặt kia. Sự vận chuyển là do gradient áp suất riêng. Các ví dụ về bao bì bán thấm bao gồm các túi nhựa và túi bán cứng bằng polyethylen tỷ trọng thấp (LDPE) dùng cho thuốc tiêm truyền thể tích lớn (LVPs) và ống tiêm, chai và lọ thuốc tiêm bằng LDPE.
Tuổi thọ (Shelf-life, expiration dating period)
Khoảng thời gian một thành phẩm thuốc vẫn đạt các tiêu chuẩn chất lượng đã được phê duyệt khi được bảo quản ở điều kiện ghi trên nhãn bao bì.
Tiêu chuẩn chất lượng (Specifications)
Danh mục các phép thử, còn gọi là các quy trình phân tích và các mức chất lượng được biểu thị dưới dạng các giới hạn, các khoảng bằng số hoặc các hình thức khác cho các phép thử được mô tả.
(Tiêu chuẩn chất lượng thiết lập ra tập hợp các tiêu chuẩn mà một dược chất, thành phẩm thuốc hoặc nguyên liệu ở các giai đoạn của quá trình sản xuất phải đáp ứng để được xem là chấp nhận được cho mục đích sử dụng. "Đạt tiêu chuẩn chất lượng" có nghĩa là dược chất hoặc thành phẩm thuốc, khi được thử nghiệm theo các quy trình phân tích, đạt các mức chất lượng cho phép. Các chỉ tiêu chất lượng là các tiêu chuẩn chất lượng trọng yếu do nhà sản xuất đề nghị và chứng minh, và được các cơ quan quản lý phê duyệt).
Tiêu chuẩn chất lượng - Xuất xưởng (Specification - Release)
Tiêu chuẩn chất lượng quyết định sự phù hợp của một thành phẩm thuốc tại thời điểm xuất xưởng của thành phẩm đó (xem thêm Tiêu chuẩn chất lượng).
Tiêu chuẩn chất lượng - Tuổi thọ/lưu hành (Specification - Shelf life)
Tiêu chuẩn chất lượng quyết định sự thích hợp của một dược chất trong suốt chu kỳ tái kiểm hoặc tiêu chuẩn chất lượng của một thành phẩm thuốc trong suốt tuổi thọ của nó.
Độ ổn định (Stability)
Khả năng một dược chất hoặc một thành phẩm thuốc duy trì được các đặc tính của nó ở những giới hạn đã định trong suốt tuổi thọ. (Các tính chất hóa học, vật lý, vi sinh và sinh dược phải được xem xét).
Nghiên cứu độ ổn định (Stability studies)
Nghiên cứu ở điều kiện dài hạn và cấp tốc (và trung gian) trên các lô đầu tiên và/hoặc lô cam kết theo một chương trình thử nghiệm độ ổn định để thiết lập hoặc khẳng định chu ký tái kiểm của một dược chất hoặc tuổi thọ của một thành phẩm thuốc.
Dung sai điều kiện bảo quản (Storage condition tolerances)
Các thay đổi chấp nhận được về nhiệt độ và độ ẩm tương đối của các thiết bị bảo quản trong các nghiên cứu độ ổn định chính thức (Thiết bị phải có khả năng điều chỉnh được điều kiện bảo quản trong giới hạn được nêu ra trong các hướng dẫn hiện hành có liên quan. Nhiệt độ và độ ẩm thực - khi được điều chỉnh - phải được giám sát trong suốt quá trình bảo quản của thử nghiệm độ ổn định. Những biến động trong thời gian ngắn do mở cửa thiết bị bảo quản được chấp nhận vì không thể tránh được. Ảnh hưởng của việc sai lệch do hỏng thiết bị phải được ghi nhận và báo cáo nếu có ảnh hưởng đến các kết quả độ ổn định. Các sai lệch vượt quá các dung sai đã đưa ra trong thời gian hơn 24 giờ phải được mô tả trong báo cáo nghiên cứu và đánh giá ảnh hưởng của chúng).
Thử nghiệm khắc nghiệt - Thành phẩm (Stress testing - Drug product)
Các nghiên cứu được tiến hành để đánh giá ảnh hưởng của điều kiện khắc nghiệt lên thành phẩm thuốc. (Các nghiên cứu này bao gồm thử nghiệm độ ổn định với ánh sáng - xem ICH Q1B - và thử nghiệm đặc thù cho các thành phẩm nhất định, ví dụ như khí dung định liều, kem, nhũ dịch, dung dịch nước đông lạnh).
Thử nghiệm khắc nghiệt - Dược chất (Stress testing - Drug substance)
Các nghiên cứu được tiến hành để làm rõ độ ổn định thực chất của một dược chất. Thử nghiệm này là một phần của chiến lược phát triển và thông thường được tiến hành ở các điều kiện khắc nghiệt hơn điều kiện dùng trong thử nghiệm cấp tốc.
Số liệu hỗ trợ (Supporting data)
Các số liệu, không phải là số liệu thu được từ nghiên cứu độ ổn định chính thức, mà là các số liệu hỗ trợ cho các quy trình phân tích, chu kỳ tái kiểm dự kiến hoặc tuổi thọ, và các điều kiện bảo quản ghi trên nhãn (Các số liệu này bao gồm (1) Số liệu độ ổn định của các lô dược chất ở giai đoạn tổng hợp đầu, lô nguyên liệu ở quy mô nhỏ, các công thức nghiên cứu không có ý định lưu hành trên thị trường và các công thức có liên quan, thành phẩm được trình bày trong dạng bao bì đóng gói khác với loại lưu hành trên thị trường; (2) Các thông tin liên quan đến các kết quả thử nghiệm trên các loại bao bì; và (3) Các cơ sở khoa học khác).
TÀI LIỆU THAM KHẢO
1. Note for Guidance on Stability Testing of Existing Active Substance and Related Finished Product (Draft), February 2002, The European Agency for The Evaluation of Medicinal Product (EMEA)
2. ICH Q1A (R2) Guideline on Stability Testing of New Drug Substances and Product, February 2003 and its annexes (Q1B Photostability Testing of New Drug Substances and Producsts, Q1C Stability Testing : Requirements for New Dosage Forms, Q1D Bracetting and Matrixing Designs for Stability Testing of New Drug Substances and Products, Q1E Evaluation for Stability Data, Q1F Stability Data Package for Registration Applications in Climatic Zones III and IV).
3. Guidelines for Stability Testing of Pharmaceutical Products Containing Well Established Drug Substances in Conventional Dosage Form, WHO Technical Report Series No. 863, 1996.
5. Carstensen, J.T., “Stability and Dating of Solid Dosage Forms,” Pharmaceutics of Solids and Solid Dosage Forms, Wiley-Interscience, 182-185, 1977.
6. Ruberg, S.J. and Stegeman, J.W., “Pooling Data for Stability Studies: Testing the Equality of Batch Degradation Slopes,” Biometrics, 47:1059-1069, 1991.
7. Ruberg, S.J. and Hsu, J.C, “Multiple Comparison Procedures for Pooling Batches in Stability Studies,” Technometrics, 34:465-472, 1992.
8. Shao, J. and Chow, S.C., “Statistical Inference in Stability Analysis,” Biometrics, 50:753-763, 1994.
9. Murphy, J.R. and Weisman, D., “Using Random Slopes for Estimating Shelf-life,” Proceedings of American Statistical Association of the Biopharmaceutical Section, 196-200, 1990.
10. Yoshioka, S., Aso, Y, and Kojima, S., “Assessment of Shelf-life Equivalence of Pharmaceutical Products,” Chem. Pharm. Bull., 49:1482-1484, 1997.
11. Chen, J.J., Ahn, H., and Tsong, Y., “Shelf-life Estimation for Multifactor Stability Studies,” Drug Inf. Journal, 31:573-587, 1997.
12. Fairweather, W., Lin, T.D., and Kelly, R., “Regulatory, Design, and Analysis Aspects of Complex Stability Studies,” J. Pharm. Sci., 84 (11): 1322 – 1326, 1995.
13. WHO Expert Committe on Specifications for Pharmaceutical Preparations, Annex 2 : Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products, WHO Technical Report Series No. 953, 2009.
14. WHO Expert Committee on Specifications for Pharmaceutical Preparations, Annex 6: Guidance on Variations to a Prequalified Product Dossier, WHO Technical Report Series No. 943, 2007.
HƯỚNG DẪN CỦA ASEAN VỀ VIỆC CUNG CẤP DỮ LIỆU THẨM ĐỊNH QUY TRÌNH SẢN XUẤT TRONG ĐĂNG KÝ THUỐC
MỤC LỤC
1. LỜI MỞ ĐẦU
2. PHẠM VI ÁP DỤNG
3. YÊU CẦU VỀ DỮ LIỆU THẨM ĐỊNH CẦN NỘP
4. NỘI DUNG PHÁT TRIỂN DƯỢC HỌC
5. NỘI DUNG CỦA ĐỀ CƯƠNG THẨM ĐỊNH
6. NỘI DUNG CỦA BÁO CÁO THẨM ĐỊNH
7. MỘT SỐ LƯU Ý VỀ THẨM ĐỊNH HỒI CỨU VÀ THẨM ĐỊNH ĐỒNG THỜI
8. KIỂM SOÁT THAY ĐỔI
9. MỤC LỤC CỦA TÀI LIỆU THẨM ĐỊNH QUY TRÌNH
10. CÁCH TIẾP CẬN MỚI TRONG THẨM ĐỊNH QUY TRÌNH SẢN XUẤT: QUALITY BY DESIGN HAY CHẤT LƯỢNG THEO THIẾT KẾ
11. THUẬT NGỮ
12. DANH MỤC CÁC PHIÊN BẢN CŨ CỦA HƯỚNG DẪN NÀY
PHỤ LỤC A1
HƯỚNG DẪN XÂY DỰNG ĐỀ CƯƠNG THẨM ĐỊNH QUY TRÌNH SẢN XUẤT CHO CÁC DẠNG THUỐC RẮN DÙNG ĐƯỜNG UỐNG
PHỤ LỤC A2
HƯỚNG DẪN XÂY DỰNG ĐỀ CƯƠNG THẨM ĐỊNH QUY TRÌNH SẢN XUẤT CỦA CÁC THUỐC ĐƯỢC SẢN XUẤT TRONG ĐIỀU KIỆN VÔ KHUẨN
PHỤ LỤC A3
HƯỚNG DẪN THẨM ĐỊNH QUY TRÌNH SẢN XUẤT CỦA CÁC THUỐC ĐƯỢC TIỆT KHUẨN CUỐI
PHỤ LỤC B
MỤC LỤC CỦA HỒ SƠ THẨM ĐỊNH QUY TRÌNH SẢN XUẤT
PHỤ LỤC C
HƯỚNG DẪN THẨM ĐỊNH QUY TRÌNH SẢN XUẤT DỰA TRÊN CHẤT LƯỢNG THEO THIẾT KẾ HAY QUALITY BY DESIGN
PHỤ LỤC D
THUẬT NGỮ
HƯỚNG DẪN CỦA ASEAN VỀ VIỆC CUNG CẤP DỮ LIỆU THẨM ĐỊNH QUY TRÌNH SẢN XUẤT TRONG ĐĂNG KÝ THUỐC
1. LỜI MỞ ĐẦU
Thẩm định quy trình sản xuất là biện pháp để đảm bảo rằng các quy trình sản xuất có đủ khả năng tạo ra các sản phẩm có chất lượng đạt yêu cầu một cách ổn định. Thẩm định quy trình sản xuất bao gồm việc cung cấp bằng chứng dưới dạng văn bản về tính ổn định và lặp lại của các bước trọng yếu của quy trình sản xuất. Một quy trình sản xuất được thẩm định là quy trình đã được chứng minh là có khả năng thực hiện được mục tiêu đề ra.
Thuật ngữ "thẩm định" được xem như là lần thẩm tra cuối cùng ở quy mô sản xuất. Thông thường, tối thiểu 03 lô sản xuất liên tiếp cần được thẩm định trước khi sản phẩm được lưu hành.
2. PHẠM VI ÁP DỤNG
Hướng dẫn này tóm tắt các yêu cầu pháp lý liên quan đến các nghiên cứu thẩm định quy trình sản xuất khi tiến hành đăng ký thuốc, các hướng dẫn xây dựng hồ sơ đăng ký để xin cấp giấy phép lưu hành thuốc cũng như xây dựng hồ sơ đăng ký thay đổi sau khi đã được cấp giấy phép lưu hành. Hướng dẫn này không hướng đến việc quản lý các quy trình sản xuất hoạt chất hay các nguyên liệu ban đầu khác mà chỉ áp dụng cho quy trình sản xuất thuốc thành phẩm. Đối với các thuốc có nguồn gốc sinh học hoặc sản xuất bằng công nghệ sinh học, cơ quan quản lý có thể sẽ yêu cầu các dữ liệu bổ sung nằm ngoài phạm vi của hướng dẫn này.
3. YÊU CẦU VỀ DỮ LIỆU THẨM ĐỊNH CẦN NỘP
Phương án 1 - Các dữ liệu thẩm định phải nộp bao gồm báo cáo thẩm định của 03 lô liên tiếp đã được thẩm định đạt yêu cầu (xem mục Nội dung của Báo cáo thẩm định).
Phương án 2 - Trong trường hợp không đủ dữ liệu của 03 lô sản xuất liên tiếp tại thời điểm nộp hồ sơ, các tài liệu sau có thể được nộp cho cơ quan quản lý dược phẩm để xin cấp giấy phép lưu hành:
a) Báo cáo phát triển dược học; và
b) Dữ liệu thẩm định của 01 lô ở quy mô pilot và đề cương thẩm định ở quy mô sản xuất thực tế.
Ngoài ra, cơ sở đăng ký cần phải thực hiện các cam kết sau:
● Đảm bảo rằng 3 lô sản xuất thực tế đã được thẩm định đạt yêu cầu trước khi sản phẩm được lưu hành trên thị trường với sự cho phép của cơ quan quản lý dược phẩm.
● Cần phải nộp báo cáo cho cơ quan quản lý dược phẩm sau một khoảng thời gian cụ thể được quy định hoặc chuẩn bị sẵn sàng các dữ liệu của các nghiên cứu thẩm định để cơ quan quản lý dược phẩm tiến hành kiểm tra sau khi cấp phép lưu hành, tuỳ theo quy định của từng quốc gia.
Ghi chú:
Không nên áp dụng phương án 2 đối với các sản phẩm có nguồn gốc sinh học hoặc được sản xuất bằng công nghệ sinh học, các sản phẩm được sản xuất theo các phương pháp không được chuẩn hóa (chẳng hạn: các phương pháp tiệt khuẩn hoặc các quy trình bào chế vô khuẩn không được chuẩn hóa) và các sản phẩm đặc biệt khác như các dạng bào chế điều chỉnh giải phóng.
Phương án 3: Với sản phẩm đã được cấp phép bởi một cơ quan quản lý tham chiếu, cơ sở đăng ký phải cam kết bằng văn bản rằng hồ sơ được nộp cho cơ quan quản lý dược phẩm (trong đó chứa phần thẩm định quy trình sản xuất) để thẩm định có nội dung giống với hồ sơ đã được nộp trước đó nhằm mục đích xin giấy phép lưu hành của cơ quan quản lý tham chiếu. Trong một số trường hợp mà các tài liệu thẩm định không nằm trong hồ sơ xin cấp phép trước đó, cơ quan quản lý dược phẩm có thể yêu cầu cơ sở đăng ký nộp báo cáo thẩm định hoặc đề cương thẩm định. Thêm vào đó, cơ sở đăng ký cần phải đảm bảo rằng 3 lô sản xuất thực tế liên tiếp sẽ được thẩm định đạt yêu cầu trước khi sản phẩm được đưa ra lưu hành trên thị trường và cơ sở đăng ký phải nộp báo cáo thẩm định cho cơ quan quản lý dược phẩm khi được yêu cầu.
4. NỘI DUNG PHÁT TRIỂN DƯỢC HỌC
Báo cáo phát triển dược học cần bao gồm các phần sau:
a) Lý do lựa chọn dạng bào chế
b) Lý do lựa chọn các thành phần trong công thức
● Tính tương hợp - tương kỵ giữa dược chất - tá dược,
● Đặc tính hóa lý.
c) Xây dựng công thức
● Lượng dôi ra so với công thức,
● Tác động của pH và các thông số khác,
● Tác động của chất chống oxy hóa, dung môi, tác nhân tạo phức, loại và nồng độ của tác nhân diệt khuẩn,
● Độ ổn định, độ đồng nhất và tính lặp lại của các lô sản xuất.
d) Lựa chọn quy trình sản xuất, quy trình tiệt khuẩn
e) Lựa chọn bao bì và vật liệu đóng gói
● Tính toàn vẹn của bao bì đóng gói
● Các vấn đề hấp phụ/rò rỉ nếu có khi sử dụng bao bì đóng gói được lựa chọn
f) Tiêu chí về mức độ nhiễm vi sinh vật của dạng bào chế
g) Tính tương thích của thuốc với các tá dược dùng để pha loãng hoặc với dụng cụ chia liều (ví dụ: sự kết tủa của dược chất trong dung dịch hoặc sự hấp phụ dược chất lên thành của dụng cụ...) trong suốt hạn dùng của thuốc.
Báo cáo phát triển dược học cần phải chứng minh được rằng dạng bào chế và công thức bào chế được lựa chọn là phù hợp với mục đích sử dụng đã được nêu rõ khi nộp hồ sơ đăng ký thuốc. Báo cáo phát triển dược học cũng cần phải nêu rõ được những khía cạnh của công thức và quy trình sản xuất cần được theo dõi chặt chẽ và có ảnh hưởng lớn đến tính đồng nhất / độ lặp lại giữa các lô trong quá trình sản xuất. Nội dung của báo cáo phát triển dược học và báo cáo thẩm định quy trình sản xuất ở quy mô pilot cần có sự tương quan và liên kết chặt chẽ với đề cương thẩm định dự kiến của quy trình sản xuất ở quy mô sản xuất thực tế.
5. NỘI DUNG CỦA ĐỀ CƯƠNG THẨM ĐỊNH
Nội dung của đề cương thẩm định bao gồm các nghiên cứu thẩm định được tiến hành trên các lô sản phẩm được sản xuất ở quy mô sản xuất thực tế theo đúng quy định. Đề cương thẩm định nên bao gồm, nhưng không bị giới hạn bởi các phần sau:
a) Mô tả quy trình sản xuất và sơ đồ quy trình
b) Bản tổng hợp các bước trọng yếu trong quy trình, các thông số được kiểm soát và lý do lựa chọn các thông số đó
c) Tiêu chuẩn xuất xưởng của thành phẩm
d) Các thông tin về các phương pháp phân tích (kèm chỉ dẫn đến các phần chi tiết tương ứng trong hồ sơ)
e) Kiểm soát trong quá trình và các giới hạn chấp nhận đề xuất cho các thông số đó
f) Các phép thử khác cần được tiến hành (ví dụ: giới hạn chấp nhận của các tiêu chí đánh giá và phương pháp thẩm định quy trình phân tích phù hợp)
g) Kế hoạch lấy mẫu (khi nào, lúc nào và mẫu được lấy như thế nào)
h) Thông tin chi tiết về cách lưu lại và đánh giá kết quả
i) Thời gian dự kiến để tiến hành nghiên cứu thẩm định
j) Các máy móc thiết bị và cơ sở vật chất trọng yếu được sử dụng (chẳng hạn: thông tin về các máy đo/ghi kết quả cần phải đi kèm với trạng thái của máy móc khi tiến hành kiểm tra/hiệu chuẩn).
6. NỘI DUNG CỦA BÁO CÁO THẨM ĐỊNH
Nội dung của báo cáo thẩm định cần bao gồm, mà không bị giới hạn bởi các nội dung sau:
a) Tóm tắt
b) Giới thiệu
c) Thông tin về các lô sản xuất được dùng trong thẩm định (ví dụ, ngày sản xuất, cỡ lô)
d) Các thiết bị dùng trong sản xuất
e) Các bước và thông số trọng yếu trong quá trình sản xuất
f) Giới hạn chấp nhận
g) Kế hoạch lấy mẫu
h) Kết quả thẩm định dưới dạng bảng
i) Phân tích kết quả của từng lô thẩm định
j) Đánh giá kết quả thu được, bao gồm cả việc phân tích thống kê số liệu của kiểm soát trong quá trình k) Đánh giá kết quả và so sánh với giới hạn chấp nhận
l) Đánh giá mức độ biến thiên của kết quả cũng như các kết quả nằm ngoài giới hạn cho phép
m) Kết luận và đề xuất
Cơ quan quản lý dược phẩm có thể yêu cầu cung cấp mô tả quy trình sản xuất và sơ đồ quy trình trong trường hợp cần thiết.
Vui lòng xem chi tiết ở các phụ lục dưới đây:
a) Phụ lục A1 hướng dẫn xây dựng đề cương thẩm định quy trình sản xuất cho các dạng thuốc rắn dùng đường uống,
b) Phụ lục A2 hướng dẫn xây dựng đề cương thẩm định quy trình sản xuất các thuốc được sản xuất trong điều kiện vô khuẩn,
c) Phụ lục A3 hướng dẫn xây dựng đề cương thẩm định quy trình sản xuất các thuốc có tiệt khuẩn cuối.
7. MỘT SỐ LƯU Ý VỀ THẨM ĐỊNH HỒI CỨU VÀ THẨM ĐỊNH ĐỒNG THỜI
7.1 Thẩm định hồi cứu
Thẩm định hồi cứu nên được thực hiện đối với những sản phẩm đã được lưu hành trên thị trường trong một khoảng thời gian nhất định. Thẩm định hồi cứu bao gồm việc phân tích các xu hướng (sử dụng biểu đồ kiểm soát...) của các dữ liệu trong quá trình sản xuất trước đây cùng với các dữ liệu của quá trình kiểm tra chất lượng (Quality control - chẳng hạn như kết quả định lượng, kết quả thử độ hòa tan, pH...) của thành phẩm. Dữ liệu của 10 đến 20 lô được sản xuất bằng cùng một quy trình ổn định nên được phân tích để chứng minh rằng quy trình sản xuất này được kiểm soát tốt và "đảm bảo yêu cầu". Chỉ số Cpk (Process Capability – Hiệu quả quy trình) và/hoặc Ppk (Process Performance – Hiệu năng quy trình) có giá trị là 1,0; 1,33 và 2,0 lần lượt tương ứng với 3, 4 và 6 sigma. Giá trị tính toán được của Cp, Cpk, Pp hay Ppk được chấp nhận như một công cụ thống kê để phân tích việc kiểm soát trong quy trình.
7.2 Thẩm định đồng thời
Đối với các thuốc đặc trị cho các bệnh hiếm gặp, việc thẩm định đồng thời được chấp nhận do số lượng lô sản xuất trong một năm thường ít. Các loại thuốc vốn có tuổi thọ ngắn (chẳng hạn thuốc phóng xạ) nhưng việc sử dụng là cần thiết vì các lý do y tế (ví dụ: thuốc được sử dụng để phòng ngừa hoặc điều trị các bệnh hoặc tình trạng nghiêm trọng có thể gây nguy hiểm đến tính mạng hoặc các thuốc mà nguồn cung không đủ và không có nguồn thay thế) sẽ được xem xét theo từng trường hợp cụ thể. Cơ sở đăng ký cần nhận được sự đồng thuận của cơ quan quản lý dược phẩm trước khi tiến hành nộp hồ sơ đăng ký bất kỳ thuốc nào sử dụng phương pháp thẩm định đồng thời.
8. KIỂM SOÁT THAY ĐỔI
Việc quản lý, lập kế hoạch và hồ sơ tài liệu về các thay đổi được đề xuất trong quá trình sản xuất cần được thực hiện theo quy trình. Cần có đầy đủ dữ liệu để chứng minh rằng quy trình thay đổi vẫn duy trì được chất lượng sản phẩm theo yêu cầu và thỏa mãn được các tiêu chuẩn đã đề ra.
Các thay đổi nhỏ trong quy trình thao tác chuẩn, thiết bị, môi trường thường không cần phải được chấp thuận bởi cơ quan quản lý nếu những thay đổi này không ảnh hưởng nhiều đến chất lượng thành phẩm.
Các loại thay đổi khác có ảnh hưởng đáng kể đến chất lượng của thành phẩm sẽ dẫn đến yêu cầu tái thẩm định, chẳng hạn như các thay đổi trong quy trình (thời gian trộn, nhiệt độ sấy, quy trình tiệt khuẩn), thay thế thiết bị bằng các thiết bị khác có các thông số / nguyên tắc thiết kế và cách vận hành khác nhau. Cơ sở đăng ký phải nộp các dữ liệu thích hợp để chứng minh rằng những thay đổi này là cần thiết.
9. MỤC LỤC CỦA TÀI LIỆU THẨM ĐỊNH QUY TRÌNH
Biểu mẫu ở phụ lục B nên được điền bởi cơ sở đăng ký nhằm mục đích kiểm tra và tránh sai sót.
10. CÁCH TIẾP CẬN MỚI TRONG THẨM ĐỊNH QUY TRÌNH SẢN XUẤT: QUALITY BY DESIGN HAY CHẤT LƯỢNG THEO THIẾT KẾ
Cách tiếp cận truyền thống của thẩm định quy trình sản xuất dựa trên việc thẩm định quy trình sản xuất của 3 lô liên tiếp ở quy mô sản xuất thực tế. Việc thẩm định quy trình được coi là đã đạt yêu cầu khi mà kết quả thẩm định của 3 lô nằm trong giới hạn chấp nhận được nêu trong đề cương thẩm định.
Một biện pháp thay thế cho cách tiếp cận truyền thống là cách tiếp cận dựa trên việc thẩm tra liên tục quy trình dựa trên nền tảng của QbD (Quality by Design - Chất lượng theo thiết kế). Cách tiếp cận này được thực hiện trong suốt vòng đời của sản phẩm và quy trình liên tục được thẩm tra ngay cả khi việc thẩm định của các lô thẩm định đầu tiên đã được hoàn thành. Chi tiết tham khảo ở phụ lục C.
11. THUẬT NGỮ
Các thuật ngữ được dùng trong hướng dẫn này được định nghĩa ở phụ lục D
12. DANH MỤC CÁC PHIÊN BẢN CŨ CỦA HƯỚNG DẪN NÀY
Phiên bản 1.0: Bắt đầu có hiệu lực từ tháng 1 năm 2005
Phiên bản 2.0: Bản thảo sử dụng cho hội nghị ACCSQ-PPWG lần thứ 18 (tháng 6 năm 2011)
Phiên bản 3.0: Bản thảo sử dụng cho hội nghị ACCSQ-PPWG lần thứ 19 (tháng 7 năm 2012)
PHỤ LỤC A1 HƯỚNG DẪN THẨM ĐỊNH QUY TRÌNH SẢN XUẤT CHO CÁC DẠNG THUỐC RẮN DÙNG ĐƯỜNG UỐNG
MỤC LỤC
1. MỤC TIÊU
2. PHẠM VI
3. THÔNG TIN CHUNG
4. ĐỀ CƯƠNG THẨM ĐỊNH QUY TRÌNH SẢN XUẤT CÁC DẠNG THUỐC RẮN DÙNG ĐƯỜNG UỐNG
4.1. CÔNG THỨC LÔ
4.2. CÁC THIẾT BỊ QUAN TRỌNG VÀ LOẠI THIẾT BỊ TRONG QUY TRÌNH SẢN XUẤT
4.3 MÔ TẢ QUY TRÌNH SẢN XUẤT VÀ CÁC THÔNG SỐ TRONG QUY TRÌNH SẢN XUẤT
4.4. KẾ HOẠCH LẤY MẪU VÀ GIỚI HẠN CHẤP NHẬN
4.5. THỜI GIAN LƯU TRỮ
5. THUẬT NGỮ
1. MỤC TIÊU
Tài liệu này nhằm hướng dẫn xây dựng đề cương thẩm định quy trình sản xuất các dạng thuốc rắn dùng đường uống.
Hướng dẫn này nên được nghiên cứu đồng thời với các hướng dẫn như dưới đây:
● ASEAN Guidelines for Validation of Analytical Procedures
● Current United States Pharmacopeia, European Pharmacopoeia and Japanese Pharmacopoeia
● Guidance for Industry, Process Validation: General Principles and Practices (US-FDA, January 2011)
● CPG Sec. 490.100 Process Validation Requirements for Drug Products and Active Pharmaceutical Ingredients Subject to Pre-Market Approval
● SUPAC-IR: Immediate-Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation (US-FDA, 1995)
● SUPAC-IR/MR: Immediate Release and Modified Release Solid Oral Dosage Forms Manufacturing Equipment Addendum (US-FDA, 1999)
● SUPAC-MR: Modified Release Solid Oral Dosage Forms Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation (US-FDA, 1997)
● Dissolution Testing of Immediate Release Solid Oral Dosage Forms (US-FDA, 1997)
2. PHẠM VI
Nội dung của hướng dẫn này được áp dụng cho các dạng thuốc rắn dùng đường uống: viên nang, viên nén và bột / cốm pha dung dịch hoặc hỗn dịch.
3. THÔNG TIN CHUNG
Các dạng thuốc rắn dùng đường uống thường là viên nang, viên nén và bột / cốm pha dung dịch / hỗn dịch. Các dạng thuốc rắn dùng đường uống có thể được đóng gói dưới dạng đơn liều (dạng vỉ, túi) hoặc dạng đa liều (trong lọ thuốc đa liều).
Viên nang là dạng thuốc rắn, trong đó thuốc được đóng vào vỏ nang cứng hoặc mềm hòa tan được. Các nang này thường được làm từ gelatin hoặc tinh bột hoặc các chất thích hợp khác. Viên nang có thể được bào chế với mục đích giải phóng dược chất ngay hoặc khả năng giải phóng dược chất điều chỉnh (thuốc được chứa trong nang có thể ở dưới dạng bột, chất lỏng hoặc chất bán rắn). Viên nang cũng có thể chứa các viên nén nhỏ, bột hoặc cốm, pellet bao hoặc không bao giúp cho thuốc được vận chuyển qua dạ dày và đến được ruột non trước khi thuốc được giải phóng để giảm thiểu sự mất hoạt tính của dược chất hoặc sự kích thích niêm mạc dạ dày nếu có.
Viên nén là dạng thuốc rắn chứa dược chất cùng với các tá dược thích hợp, được bào chế bằng phương pháp dập thẳng hỗn hợp bột hoặc cốm dưới áp suất lớn được tạo bởi chày và cối thép. Viên nén có thể có nhiều kích thước, khối lượng, hình dạng, màu sắc và có thể có các ký hiệu / chi tiết trên bề mặt viên. Viên nén cũng có thể được bao màng mỏng hoặc mang ký hiệu đặc biệt.
Quy cách đóng gói của bột hoặc cốm pha dung dịch / hỗn dịch có thể là dạng đơn liều hoặc đa liều và cần phải được hoàn nguyên với nước trước khi uống. Quy cách đóng gói đa liều được sử dụng khi liều của mỗi lần sử dụng không cần quá chính xác.
Việc thẩm định quy trình sản xuất các dạng thuốc rắn dùng đường uống cần phải dựa trên tính đặc trưng của công thức lô và các nguyên tắc hoạt động của các thiết bị dùng trong sản xuất. Các thông số quy trình cần được kiểm tra, kiểm soát và / hoặc theo dõi cũng như các phép thử cần được tiến hành khi thẩm định quy trình sản xuất của bán thành phẩm khi sản xuất các dạng thuốc rắn phụ thuộc vào phương pháp sản xuất và dạng thành phẩm (viên nén, viên bao, viên nang hay bột / cốm). Tiêu chuẩn chấp nhận cần phải được xây dựng dựa trên đặc điểm của dạng thuốc đó, chẳng hạn như đặc điểm giải phóng dược chất (thuốc giải phóng ngay hay thay đổi giải phóng). Đề cương thẩm định được trình bày dưới đây có thể được tham khảo khi thẩm định quy trình sản xuất. Tuy vậy, đề cương thẩm định cần phải được đánh giá và kiểm tra tuỳ theo từng trường hợp cụ thể.
4. ĐỀ CƯƠNG THẨM ĐỊNH QUY TRÌNH SẢN XUẤT CÁC DẠNG THUỐC RẮN DÙNG ĐƯỜNG UỐNG
Các yếu tố sau nên được xem xét khi tiến hành thẩm định quy trình sản xuất các dạng thuốc rắn dùng đường uống
4.1. Công thức lô
Cần xác định rõ công thức lô khi tiến hành thẩm định quy trình sản xuất. Với mỗi dạng bào chế, tất cả các thành phần cũng như lượng dùng của từng thành phần trong mỗi lô được sản xuất cần phải được nêu đầy đủ (bao gồm cả lượng dôi dư để bù lại phần hư hao trong quá trình sản xuất, nếu có).
4.2. Các thiết bị quan trọng và loại thiết bị trong quy trình sản xuất
Các thiết bị quan trọng được sử dụng trong quy trình sản xuất cần phải được nêu rõ và được phân loại vào từng nhóm thiết bị cụ thể. Thông thường, các thiết bị sẽ được phân loại theo các công đoạn trong quy trình sản xuất (ví dụ: nhào trộn, sấy, giảm kích thước tiểu phân, tạo hạt, phân liều, bao, đóng thuốc, đóng nang, in, đóng gói). Các nhóm thiết bị này sẽ được phân thành các loại nhỏ hơn dựa trên nguyên tắc hoạt động.
Dưới đây là một số ví dụ của các nhóm thiết bị tương ứng với các bước trong quy trình sản xuất:
|
Thiết bị |
Loại thiết bị |
|
Nồi trộn |
Máy khuấy trộn đối lưu |
|
Máy nhào |
Máy nhào khuếch tán Máy nhào kiểu đối lưu Máy nhào kiểu khí nén |
|
Máy nghiền |
Máy nghiền bột siêu mịn dùng khí nén Máy nghiền theo cơ chế va đập Máy nghiền theo cơ chế cắt chẻ Máy nghiền theo cơ chế nén ép Máy nghiền kết hợp với rây Máy nghiền bi |
|
Máy tạo hạt |
Máy tạo hạt khô Máy tạo hạt ướt tốc độ cao Máy tạo hạt ướt tốc độ thấp Máy tạo hạt hình trống tốc độ thấp Máy tạo hạt theo cơ chế đùn Máy tạo hạt quay tròn Máy tạo hạt tầng sôi Máy tạo hạt phun sấy |
|
Máy sấy |
Gia nhiệt trực tiếp, nguyên liệu sấy tĩnh Gia nhiệt trực tiếp, nguyên liệu sấy chuyển động Gia nhiệt trực tiếp, sấy tầng sôi Gia nhiệt trực tiếp, nguyên liệu được sấy bị pha loãng, phun sấy Gia nhiệt trực tiếp, nguyên liệu được sấy bị pha loãng, sấy tức thời Dẫn nhiệt gián tiếp, nguyên liệu sấy chuyển động Dẫn nhiệt gián tiếp, nguyên liệu sấy đứng yên Dẫn nhiệt gián tiếp, đông khô Sấy bằng phương pháp thổi khí Sấy bằng bức xạ gián tiếp, nguyên liệu được sấy chuyển động |
|
Máy phân tách |
Máy rung, lắc Máy ly tâm |
|
Máy dập viên |
Máy dập viên - bột, cốm được phân liều nhờ trọng lực Máy dập viên - bột, cốm được phân liều nhờ trợ lực Máy dập viên quay tròn - bột, cốm được phân liều nhờ lực ly tâm Máy bao dập |
|
Máy bao |
Máy bao sử dụng nồi bao Máy bao hỗn dịch khí Máy bao màng mỏng chân không Máy bao nhúng Máy bao tĩnh điện |
|
Máy đóng nang |
Máy đóng nang dùng phễu và trục xoắn Máy đóng nang chân không Máy đóng nang có đĩa nhựa đục lỗ rung Máy đóng nang sử dụng đĩa phân liều Máy đóng nang sử dụng ống phân liều |
|
Máy đóng nang mềm |
Máy bơm dịch nhờ trọng lực hoặc đóng nhờ lực Máy khuấy trộn, bể trộn Thiết bị chống kết tụ Thiết bị loại khí Thùng lưu trữ |
|
Máy đóng bột |
Kiểu chân không Kiểu dùng phễu và trục xoắn |
|
Máy ép vỉ |
Máy ép vỉ kiểu tấm mỏng |
|
Máy đóng lọ |
Các loại |
Cơ sở đăng ký sẽ tự quyết định mức độ chi tiết của các thông tin về quy trình sản xuất được cung cấp cho cơ quan quản lý. Các thông tin này nên bao gồm cả công suất tối đa của thiết bị. Nếu cơ quan quản lý thấy các thông tin này cần thiết nhưng chưa được trình bày trong tài liệu đã nộp thì cơ quan quản lý có quyền yêu cầu cơ sở đăng ký bổ sung các thông tin nói trên.
4.3 Mô tả quy trình sản xuất và các thông số trong quy trình sản xuất
Quy trình sản xuất nên được mô tả hoặc trình bày dưới dạng sơ đồ.
Các thông số kỹ thuật ở bảng sau được nên được kiểm soát và theo dõi khi tiến hành thẩm định, tuỳ vào dạng bào chế và phương pháp sản xuất. Một số ví dụ được liệt kê trong bảng sau. Cần lưu ý rằng các ví dụ này không phản ánh được đầy đủ thực tế sản xuất, chỉ nên được sử dụng để tham khảo và có thể thay đổi tuỳ theo loại thiết bị được sử dụng.
|
Các bước trong quá trình |
Viên nén |
Viên nang |
Bột cốm |
Các thông số trong quy trình |
|
Rây nguyên liệu nếu cần thiết |
ü |
ü |
ü |
● Cỡ rây / đường kính mắt rây |
|
Trộn bột kép |
ü |
ü |
ü |
● Thời gian trộn, tốc độ trộn, khối lượng mẻ trộn |
|
Khuấy trộn - hòa tan dịch đóng nang |
Không áp dụng |
ü |
Không áp dụng |
● Thời gian khuấy trộn, tốc độ khuấy trộn, thể tích khuấy trộn |
|
Nghiền khô, phân loại kích thước tiểu phân (nếu có) |
TK |
TK |
TK |
● Đường kính rây ● Tốc độ nghiền ● Tốc độ cấp bột |
|
Trộn cuối |
ü |
ü |
ü |
● Thời gian trộn, khối lượng mẻ trộn, tốc độ trộn ● Cỡ rây |
|
Chuẩn bị tá dược dính |
TU |
TU |
TU |
● Lượng tá dược dính, nồng độ tá dược dính, ● Nhiệt độ |
|
Tạo hạt |
TU |
TU |
TU |
● Khối lượng mẻ nhào ● Thời gian nhào, tốc độ nhào ● Nhiệt độ nhào ● Tốc độ cấp dịch ● Phương pháp phun |
|
Nghiền ướt (nếu có) |
TU |
TU |
TU |
● Số vòng trên phút ● Áp suất ● Nhiệt độ |
|
Xát hạt ướt (nếu có) |
TU |
TU |
TU |
● Cỡ rây, cỡ lô |
|
Sấy |
TU |
TU |
TU |
● Thời gian sấy ● Phân bố nhiệt độ |
|
Làm lạnh |
TU |
TU |
TU |
● Tốc độ làm lạnh ● Nhiệt độ làm lạnh |
|
Dập viên (bao gồm cả việc phát hiện vết kim loại và loại bỏ bụi) |
ü |
Không áp dụng |
Không áp dụng |
● Các thông số cài đặt của máy dập viên ● Tốc độ dập viên (số viên/giờ) |
|
Chuẩn bị dịch bao (dung dịch / hỗn dịch) |
ü |
ü |
Không áp dụng |
● Nhiệt độ ● Thời gian, tốc độ khuấy trộn |
|
Bao |
ü |
ü |
Không áp dụng |
● Khối lượng mẻ bao ● Đặc điểm nồi bao ● Nhiệt độ ● Tốc độ phun dịch ● Tốc độ quay của nồi bao (số vòng trên phút) ● Tốc độ thổi khí ● Khoảng cách và vị trí súng phun |
|
In trên nang (nếu cần thiết) |
ü |
ü |
Không áp dụng |
● Tốc độ in (đơn vị / giờ) ● Nhiệt độ |
|
Đóng nang (bao gồm cả loại bụi) |
Không áp dụng |
ü |
Không áp dụng |
● Các thông số cài đặt của thiết bị ● Công suất đóng (số nang/giờ) ● Hệ thống cấp nang |
|
Đóng bao bì sơ cấp (có thể tiến hành như là một phần của thẩm định thiết bị) |
ü |
ü |
ü |
● Các thông số cài đặt của thiết bị ● Tốc độ đóng bao bì ● Tốc độ cấp bao bì |
|
Kiểm soát môi trường trong suốt quá trình sản xuất (áp dụng đối với các sản phẩm nhạy cảm với nhiệt và ẩm) |
ü |
ü |
ü |
● Nhiệt độ ● Độ ẩm tương đối |
Ký hiệu:
TK: Chỉ áp dụng với quá trình trộn khô
TU Chỉ áp dụng với quá trình trộn ướt
ü : Áp dụng được
Cơ sở đăng ký sẽ tự quyết định mức độ chi tiết của các thông tin về quy trình sản xuất được cung cấp cho cơ quan quản lý. Nếu cơ quan quản lý thấy có các thông số quan trọng nhưng chưa được trình bày trong tài liệu đã nộp thì cơ quan quản lý có quyền yêu cầu cơ sở đăng ký bổ sung.
4.4. Kế hoạch lấy mẫu và giới hạn chấp nhận
Cơ sở sản xuất phải có trách nhiệm đảm bảo rằng kế hoạch lấy mẫu và giới hạn chấp nhận được đăng ký là đủ để đảm bảo rằng quá trình sản xuất được kiểm soát tốt và đủ khả năng sản xuất được sản phẩm đạt yêu cầu về chất lượng. Các kế hoạch lấy mẫu và các giới hạn chấp nhận được trình bày dưới đây có thể được sử dụng để tham khảo khi tiến hành thẩm định quy trình sản xuất một dạng thuốc rắn dùng đường uống thông thường với mức độ rủi ro trung bình.
|
Giai đoạn |
Kế hoạch lấy mẫu |
Phép thử |
Giới hạn chấp nhận |
|
Sấy, nếu cần thiết |
Ít nhất 3 mẫu tại ít nhất 3 vị trí khác nhau trong tủ sấy hoặc 3 thời điểm khác nhau trong suốt quá trình sấy (1). |
Mất khối lượng do làm khô (mỗi vị trí thử một mẫu) |
Phụ thuộc vào tiêu chuẩn độ mất khối lượng do làm khô được yêu cầu đối với từng sản phẩm cụ thể |
|
Trộn cuối |
Ít nhất 3 mẫu ở ít nhất 10 vị trí được phân bố đều nhau trong thiết bị trộn (1) (20 vị trí trong trường hợp máy trộn theo kiểu đối lưu) |
Độ đồng đều hàm lượng (Tiến hành định lượng 1 mẫu cho mỗi vị trí) |
Mức yêu cầu 1 - Kết quả riêng lẻ: giá trị trung bình ± 10% (giá trị tuyệt đối) Tất cả các kết quả phải có RSD ≤ 5,0% |
|
Nếu cần thiết, ● Độ trơn chảy ● Tỷ trọng ● Cảm quan / hình thức |
Cơ sở sản xuất tự xây dựng |
||
|
Mẫu hỗn hợp Có thể được tiến hành như là một phần của kiểm nghiệm xuất xưởng |
● *Hình thức ● *Độ đồng nhất ● *Định lượng (Hoạt lực) ● *Chỉ tiêu tạp chất ● *Độ nhiễm vi sinh ● Các tiêu chuẩn nội bộ khác * Có thể bỏ qua nếu như các bước sau đó là đóng nang hoặc dập viên |
Độ đồng nhất (theo yêu cầu của dược điển) Giới hạn nhiễm vi sinh vật: theo yêu cầu của dược điển Các chỉ tiêu khác: theo yêu cầu của dược điển hoặc cơ sở sản xuất xây dựng |
|
|
Dập viên |
Lấy mẫu phân tầng |
● Độ đồng nhất ● Các yêu cầu nội bộ khác nếu có |
Độ đồng nhất: theo yêu cầu của dược điển Các chỉ tiêu khác: theo yêu cầu của dược điển hoặc cơ sở sản xuất |
|
Mẫu hỗn hợp (có thể tiến hành như là một phần của kiểm nghiệm xuất xưởng) |
● Hình thức ● Độ đồng nhất ● Định lượng (Hoạt lực) ● Độ bở ● **Độ cứng ● **Độ rã ● **Kích thước viên ● **Độ hòa tan ● **Tạp chất ● **Độ nhiễm vi sinh vật ● Các tiêu chuẩn nội bộ khác ** Tiến hành đánh giá sau khi đóng nang hoặc bao (nếu có). |
Độ đồng nhất (theo yêu cầu của dược điển) Giới hạn nhiễm vi sinh vật: theo yêu cầu của dược điển Các chỉ tiêu khác: theo yêu cầu của dược điển hoặc cơ sở sản xuất |
|
|
Đóng nang |
Lấy mẫu phân tầng |
● Độ đồng nhất ● Hình thức ● Độ dài của viên nang |
Độ đồng nhất: theo yêu cầu của dược điển Các chỉ tiêu khác: theo yêu cầu của dược điển hoặc cơ sở sản xuất |
|
Mẫu hỗn hợp (có thể tiến hành như là một phần của kiểm nghiệm xuất xưởng) |
● Hình thức ● Độ đồng nhất ● Định lượng (hoạt lực) ● Kích thước ● Độ hòa tan/Độ rã ● Tạp chất ● Độ nhiễm vi sinh vật ● Các tiêu chuẩn nội bộ khác |
Độ đồng nhất (theo yêu cầu của dược điển) Giới hạn nhiễm vi sinh vật: theo yêu cầu của dược điển Các chỉ tiêu khác: theo yêu cầu của dược điển hoặc cơ sở sản xuất |
|
|
Bao |
Một mẫu với mỗi nồi bao |
● Định lượng (chỉ áp dụng đối với trường hợp bao dược chất) ● Hàm ẩm / tồn dư dung môi |
Định lượng: Cơ sở sản xuất tự xây dựng Độ ẩm / dung môi tồn dư (theo hướng dẫn của ICH) |
|
Với mỗi một nồi bao, tiến hành lấy mẫu một lần tại ít nhất 10 vị trí phân bố đồng đều trong tất cả các phần nhỏ của lô sản xuất (1). |
Độ đồng nhất |
Theo quy định của dược điển |
|
|
Mẫu hỗn hợp (có thể được tiến hành như là một phần của kiểm nghiệm xuất xưởng) |
● Kiểm tra cảm quan bằng mắt thường ● Độ đồng nhất (chỉ trong trường hợp bao dược chất) ● Định lượng (hoạt lực) ● ***Độ cứng ● ***Độ rã ● ***Độ hòa tan ● ***Tạp chất ● ***Độ nhiễm vi sinh vật ● Các chỉ tiêu nội bộ khác *** Có thể bỏ qua nếu đóng nang |
Độ đồng nhất: theo tiêu chuẩn dược điển Các chỉ tiêu khác: theo dược điển hoặc cơ sở sản xuất |
|
|
In |
Lấy mẫu phân tầng |
Hình thức |
Cơ sở sản xuất tự xây dựng |
|
Phân liều bột / cốm vào lọ |
Lấy mẫu phân tầng |
Độ đồng đều khối lượng |
Chênh lệch (± 5%) so với lượng ghi trên nhãn (giá trị tuyệt đối) |
|
Đóng gói sơ cấp (có thể được thực hiện khi kiểm tra thiết bị) |
Lấy mẫu phân tầng |
● Hình thức ● Độ kín của bao bì, nếu cần |
Cơ sở sản xuất tự xây dựng |
|
Kiểm soát môi trường (Áp dụng đối với các sản phẩm nhạy cảm với nhiệt và ẩm) |
Trong suốt quá trình sản xuất |
● Nhiệt độ ● Độ ẩm tương đối |
Cơ sở sản xuất tự xây dựng |
Chú thích: RSD - Relative Standard Deviation - Độ lệch chuẩn tương đối
ICH - International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use - Hội nghị quốc tế về sự hòa hợp của những yêu cầu kỹ thuật đối với các đăng ký thuốc sử dụng trên người
(1) Chú ý: có thể thiết lập các kế hoạch lấy mẫu khác, miễn là tính hợp lý của các kế hoạch lấy mẫu đó có thể chứng minh được dựa trên các phép phân tích thống kê.
Quy mô lấy mẫu, thử và giới hạn chấp nhận cần phải được xây dựng dựa trên mức độ rủi ro mà bệnh nhân phải chịu khi sử dụng thuốc (ví dụ: khi thuốc được sản xuất bằng các loại thiết bị khác nhau, với công suất khác nhau) và cần được đánh giá đối với từng sản phẩm cụ thể.
Lý do lựa chọn chỉ tiêu của thuốc thành phẩm cần phải được chứng minh và quy trình phân tích cần phải được thẩm định theo Hướng dẫn thẩm định quy trình phân tích của ASEAN.
4.5. Thời gian lưu trữ
Nếu trong quy trình sản xuất, bán thành phẩm được lưu trữ tạm thời (ví dụ: bán thành phẩm sau giai đoạn trộn sơ bộ hoặc sau một số bước trung gian), cơ sở sản xuất cần chứng minh rằng khoảng thời gian lưu trữ là hợp lý. Dữ liệu về độ ổn định thu được (độ ổn định về hóa học và vi sinh vật) ứng với mỗi khoảng thời gian lưu trữ nêu trên nên được cung cấp để chứng minh. Các khảo sát về thời gian lưu trữ có thể được thực hiện một cách riêng rẽ hoặc được thực hiện như một phần của đề cương thẩm định quy trình sản xuất. Thời gian lưu trữ nên được lựa chọn dựa trên các kết quả phân tích các mẫu hoặc các lô được giữ trong các khoảng thời gian lưu trữ xác định khác nhau. Thời gian lưu trữ cũng có thể được xây dựng dựa vào dữ liệu của sản xuất thực tế (thời gian lưu trữ phát sinh theo từng mẻ sản xuất).
Trong trường hợp thông tin về thời gian lưu trữ không được nêu trong hồ sơ đăng ký, nếu cơ quan quản lý dược phẩm yêu cầu, cơ sở sản xuất cần bổ sung các dữ liệu có sẵn hoặc phải các phải đưa ra lời giải thích thỏa đáng cho việc bỏ qua các thông tin này.
5. THUẬT NGỮ
Trì hoãn giải phóng - Delayed release (DR):
Quá trình giải phóng thuốc không diễn ra tức thì sau khi uống
Giải phóng kéo dài - Extended Release (ER):
Các dạng thuốc giải phóng kéo dài được thiết kế sao cho sự có mặt dược chất được duy trì trong một khoảng thời gian kéo dài sau khi uống. Điều này cho phép giảm tần suất sử dụng thuốc so với khi sử dụng thuốc ở dạng bào chế quy ước (chẳng hạn thuốc dưới dạng dung dịch hoặc thuốc giải phóng ngay)
Giải phóng ngay - Immediate release (IR):
Cho phép dược chất được hòa tan trong đường tiêu hóa, không nhằm mục đích trì hoãn hoặc kéo dài quá trình hòa tan hoặc hấp thu của thuốc.
Dạng bào chế điều chỉnh giải phóng - Modified release dosage forms:
Là dạng bào có mà đặc điểm giải phóng thuốc sao cho thời gian giải phóng và / hoặc vị trí giải phóng thuốc được thiết kế để đạt được mục đích điều trị hoặc các mục tiêu có lợi khác mà các dạng bào chế quy ước như dung dịch hoặc các thuốc giải phóng ngay không có được. Các dạng thuốc rắn điều chỉnh giải phóng bao gồm cả giải phóng trì hoãn và giải phóng kéo dài.
Lấy mẫu phân tầng - Stratified sampling:
Quá trình lấy mẫu có cân nhắc từ các vị trí khác nhau trong một mẻ hoặc lô hoặc trong các giai đoạn hay thời điểm khác nhau của quy trình sản xuất để thu được mẫu.
Với hỗn hợp trộn hoặc đơn vị phân liều, lấy mẫu phân tầng tập trung chủ yếu vào các vị trí khác nhau ở trong máy nhào trộn hoặc các bước có nguy cơ cao gây mất đồng đều hàm lượng trong suốt quá trình dập / đóng bột.
PHỤ LỤC A2 HƯỚNG DẪN THẨM ĐỊNH QUY TRÌNH SẢN XUẤT CỦA CÁC THUỐC ĐƯỢC SẢN XUẤT TRONG ĐIỀU KIỆN VÔ KHUẨN
MỤC LỤC
1. MỤC ĐÍCH
2. PHẠM VI
3. THÔNG TIN CHUNG
4. CÁC THÔNG TIN ĐƯỢC YÊU CẦU ĐỐI VỚI VIỆC THẨM ĐỊNH QUY TRÌNH PHA CHẾ VÔ KHUẨN
4.1. NHÀ XƯỞNG
4.2. TIỆT KHUẨN VÀ LOẠI CHẤT GÂY SỐT CHO BAO BÌ, NẮP, THIẾT BỊ VÀ CÁC THÀNH PHẦN THUỐC
4.3. LỌC VÀ THỜI GIAN LƯU TRỮ
4.4. NGHIÊN CỨU MÔ PHỎNG ĐỂ KIỂM TRA KHẢ NĂNG ĐẢM BẢO ĐỘ VÔ KHUẨN
4.5. TÍNH TOÀN VẸN CỦA BAO BÌ
5. THUẬT NGỮ
1. MỤC ĐÍCH
Tài liệu này được biên soạn nhằm hướng dẫn nộp các thông tin và dữ liệu để chứng minh hiệu quả của quá trình tiệt khuẩn được yêu cầu trong hồ sơ đăng ký khi tiến hành xin giấy phép kinh doanh thuốc.
Tài liệu này nên được đọc cùng với các tài liệu dưới đây:
● Note for Guidance on Process Validation (EMA, 2001)
● Guidance for Industry for the Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products (FDA, 1994)
● Annex 4 WHO Good Manufacturing Practices for Sterile Pharmaceutical Products (Technical Report Series No. 957, 2010)
● Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice (US-FDA, September 2004)
● Recommendation on the Validation of Aseptic Process (PIC/S, January 2011)
● Guide To Good Manufacturing Practice For Medicinal Products Annexes (PIC/S, September 2009)
● EC Guide to Good Manufacturing Practice (Annex 1) March 2009
2. PHẠM VI
Hướng dẫn này được áp dụng cho các thuốc được pha chế trong điều kiện vô khuẩn.
3. THÔNG TIN CHUNG
Quá trình tiệt khuẩn có thể được thực hiện bằng phương pháp nhiệt khô, nhiệt ẩm, chiếu xạ ion hóa, bằng khí ethylen oxyd hoặc bằng phương pháp lọc và đóng thuốc vào bao bì vô khuẩn trong điều kiện vô khuẩn.
Phương pháp tiệt khuẩn bằng nhiệt khô nên được ưu tiên lựa chọn nếu có thể.
Việc lựa chọn phương pháp pha chế thuốc trong điều kiện vô khuẩn cần được giải thích hoặc chứng minh là cần thiết (Ví dụ: chế phẩm dễ mất ổn định hoặc bao bì không tương thích với các phương pháp tiệt khuẩn).
4. CÁC THÔNG TIN ĐƯỢC YÊU CẦU ĐỐI VỚI VIỆC THẨM ĐỊNH QUY TRÌNH PHA CHẾ VÔ KHUẨN
Các thông tin dưới đây nên được nộp đối với việc thẩm định quy trình pha chế trong điều kiện vô khuẩn:
4.1. Nhà xưởng
Theo khuyến cáo, thông tin về sơ đồ mặt bằng bao gồm các nội dung như dưới đây:
● Các khu vực sản xuất quan trọng, chẳng hạn như nơi tiến hành pha chế và lưu trữ, lọc và đóng thuốc, phòng thay đồ và nêu rõ cấp sạch của từng khu vực.
● Các hệ thống cách ly hoặc ngăn cách nếu có.
● Vị trí của các thiết bị trọng yếu, bao gồm nhưng không giới hạn bởi: tủ hood thổi khí theo dòng, nồi hấp tiệt khuẩn, máy đông khô và nơi đóng thuốc.
● Đường đi của nguyên liệu và nhân viên.
Tham khảo phụ lục 4 của WHO về thực hành tốt sản xuất thuốc đối với các loại thuốc vô khuẩn (báo cáo kỹ thuật số 957 2010) để xem chi tiết về yêu cầu về tiêu chuẩn của các cấp độ sạch khi tiến hành sản xuất thuốc vô khuẩn.
4.2. Tiệt khuẩn và loại chất gây sốt cho bao bì, nắp, thiết bị và các thành phần thuốc
4.2.1. Mô tả quy trình
Quy trình tiệt khuẩn và loại chất gây sốt trong bình chứa, bao bì, nắp, thiết bị và các thành phần thuốc cần được mô tả tóm tắt.
4.2.2. Thẩm định quy trình
a. Đối với tiệt khuẩn bằng nhiệt khô hoặc loại chất gây sốt, báo cáo thẩm định cần phải được cung cấp và bao gồm các thông tin như dưới đây:
● Nghiên cứu về phân bố nhiệt và truyền nhiệt bao gồm, nhưng không giới hạn bởi, sơ đồ sắp xếp vật cần tiệt khuẩn trong buồng và xác định rõ các vị trí có nhiệt độ thấp.
● Báo cáo thử khả năng kháng nhiệt của vi sinh vật
Nếu một dung dịch thuốc bán thành phẩm được bào chế vô khuẩn từ các nguyên liệu đã được tiệt khuẩn riêng biệt từ trước thì cần phải nộp báo cáo thẩm định của tất cả các quy trình tiệt khuẩn đã nêu.
Đối với các quá trình loại chất gây sốt, cần cung cấp các thông tin về phương pháp thử chất gây sốt và kết quả chuẩn độ chất gây sốt phải giảm không dưới 3 log.
b. Với quá trình tiệt khuẩn bằng chiếu xạ, cần cung cấp các thông tin như dưới đây trong báo cáo thẩm định:
● Cơ sở vật chất của khu vực chiếu xạ.
● Nguồn chiếu xạ, phương pháp cho tiếp xúc với bức xạ (ví dụ: sự di chuyển của đối tượng được chiếu xạ so với nguồn phát xạ).
● Loại và vị trí của các thiết bị theo dõi lượng bức xạ.
● Dữ liệu về đặc tính bao bì.
● Nghiên cứu về phân bố đa liều tại các vị trí khác nhau .
● Các phương pháp vi sinh và phương pháp kiểm soát trong xây dựng, thẩm định và kiểm tra tính hiệu quả của quy trình.
c. Nếu một biện pháp tiệt khuẩn nào khác tiệt khuẩn bằng nhiệt hoặc tiệt khuẩn bằng chiếu xạ được sử dụng thì các dữ liệu thẩm định liên quan đến quá trình tiệt khuẩn cần phải được cung cấp. Tham khảo phụ lục A3 (mục 4.2) để biết thêm chi tiết.
4.3. Lọc và thời gian lưu trữ
a. Quá trình lọc dung dịch bán thành phẩm cần được mô tả và bao gồm các nội dung sau:
● Các quá trình lọc và chỉ tiêu tương ứng
● Hệ thống lọc nối tiếp nếu có, màng tiền lọc và màng lọc loại khuẩn nếu có.
Việc sử dụng màng với kích thước lỗ lọc nhỏ hơn hoặc bằng 0,2 µm được chấp nhận để tiệt khuẩn mà không cần chứng minh gì thêm. Nếu quá trình tiệt khuẩn được thực hiện bằng cách lọc với kích thước màng lớn hơn 0,2 µm kết hợp với một biện pháp tiệt khuẩn bổ trợ thì cả quá trình tiệt khuẩn cần phải được thẩm định và chứng minh.
Thông tin về các phép thử tính toàn vẹn của màng tiền lọc và lọc cản khuẩn cần được cung cấp. Trong trường hợp không sử dụng màng tiền lọc, cần đưa ra lý giải cho việc không dùng.
Thông tin về tính tương hợp và khả năng loại vi sinh vật của màng lọc đối với dịch lọc cần phải được cung cấp. Tác động của màng lọc lên sản phẩm cần phải được mô tả nếu có.
b. Các tiêu chí về thời gian lưu trữ sau khi tiến hành pha chế dung dịch bán thành phẩm và trước khi đóng vào bao bì cuối cần được cung cấp và bao gồm:
● Thùng lưu trữ
● Thời gian lưu trữ
● Nhiệt độ
● Các điều kiện bảo quản khác nếu có
4.4. Nghiên cứu mô phỏng để kiểm tra khả năng đảm bảo độ vô khuẩn
Phương pháp và tiêu chuẩn sử dụng cho nghiên cứu mô phỏng cũng như tóm tắt các kết quả đánh giá gần nhất tính từ thời điểm nộp hồ sơ (bao gồm 3 lần đánh giá liên tiếp độc lập đạt yêu cầu), kể cả các kết quả đánh giá không đạt cần phải được cung cấp.
Các kết quả thu được này cần phải là kết quả tiến hành trên cùng dây chuyền đóng thuốc được sử dụng thường xuyên để đóng thuốc thành phẩm.
Số lượng bao bì được đóng trong nghiên cứu mô phỏng nên nằm trong khoảng 5000 đến 10000 đơn vị. Với các quá trình đóng với ít hơn 5000 đơn vị sản phẩm, số lượng đơn vị được đóng nên bằng số lượng của cỡ lô lớn nhất được sản xuất trên dây chuyền đó.
Nhìn chung, các thông tin sau nên được cung cấp với mỗi nghiên cứu mô phỏng:
a. Ngày tiến hành
b. Danh mục thiết bị, phòng tiến hành đóng thuốc
c. Loại và kích thước bao bì
d. Thể tích và loại môi trường được sử dụng trong mỗi loại bao bì đựng thuốc
e. Số lượng đơn vị được đóng, loại bỏ, ủ và cho kết quả dương tính quan sát được
f. Thông tin về quá trình ủ, ví dụ như thời gian, nhiệt độ và hướng sắp xếp bao bì
g. Mô phỏng lại quy trình 1
h. Các thông số quy trình 2
i. Kết quả dưới dạng bảng và kết luận về việc theo dõi mức độ kiểm soát vi sinh vật trong môi trường.
Ghi chú 1: Cần mô tả bất kì quy trình nào được thực hiện để mô phỏng lại các bước trong một quy trình đóng thuốc thông thường, chẳng hạn như về việc giảm tốc độ đóng thuốc, thay ca của nhân viên, thiết bị gặp trục trặc trong quá trình đóng và được khắc phục, sửa chữa, mô phỏng quá trình đông khô và thổi khí vào lọ.
Ghi chú 2: Các thông số được sử dụng trong quá trình kiểm tra khả năng đảm bảo độ vô khuẩn cần phải được so sánh với sản xuất thực tế (ví dụ như tốc độ đóng, thể tích đóng, số lượng đơn vị được đóng hoặc thời gian đóng thuốc).
4.5. Tính toàn vẹn của bao bì
Cần cung cấp các dữ liệu, bao gồm mô tả ngắn gọn phương pháp thử và tổng hợp các kết quả thử nghiệm chứng minh tính toàn vẹn và khả năng ngăn chặn vi sinh vật của bao bì.
5. THUẬT NGỮ
Sản xuất trong điều kiện vô khuẩn - Aseptic Processing:
Việc sản xuất thuốc ở cấp sạch A hoặc một môi trường tương đương mà thường bao gồm công đoạn lọc vô khuẩn và đóng thuốc.
Vi sinh vật tạp nhiễm - Bioburden:
Tổng số lượng các vi khuẩn hiếu khí, nấm mốc và nấm men còn sống được biểu thị bằng số đơn vị tạo khuẩn lạc (colony forming units - CFU) trên một đơn vị sản phẩm hoặc một gam sản phẩm.
Loại chất gây sốt - Depyrogenation:
Quá trình loại hoặc phá hủy chất gây sốt (ví dụ: nội độc tố).
Nghiên cứu mô phỏng - Media fills:
Là phương pháp để đánh giá quy trình sản xuất thuốc vô khuẩn sử dụng môi trường nuôi cấy vi khuẩn. Nghiên cứu mô phỏng được hiểu đồng nghĩa với mô phỏng quá trình đóng thuốc, thử môi trường và đóng môi trường...
PHỤ LỤC A3 HƯỚNG DẪN XÂY DỰNG ĐỀ CƯƠNG THẨM ĐỊNH QUY TRÌNH CHO SẢN PHẨM TIỆT KHUẨN CUỐI.
MỤC LỤC
1. MỤC ĐÍCH
2. PHẠM VI
3. THÔNG TIN CHUNG
4. THÔNG TIN VỀ CÁC QUÁ TRÌNH TIỆT KHUẨN CUỐI
4.1. TIỆT KHUẨN CUỐI BẰNG NHIỆT ẨM
4.2. CÁC QUY TRÌNH TIỆT KHUẨN CUỐI KHÁC
4.3. TÍNH TOÀN VẸN CỦA HỆ THỐNG BAO BÌ - NẮP
5. THUẬT NGỮ
1. MỤC ĐÍCH
Tài liệu này được xây dựng nhằm hướng dẫn nộp các dữ liệu về tính hiệu quả của quy trình tiệt khuẩn cuối khi xin giấy phép lưu hành sản phẩm.
Hướng dẫn này nên được đọc cùng với các hướng dẫn được nêu dưới đây:
● Note for Guidance on Process Validation (EMA, 2001)
● Guidance for Industry for the Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products (FDA, 1994)
● Annex 4 WHO Good Manufacturing Practices for Sterile Pharmaceutical Products (Technical Report Series No. 957, 2010)
● EC Guide to Good Manufacturing Practice (Annex 1) March 2009
● Guide To Good Manufacturing Practice For Medicinal Products Annexes (PIC/S, September 2009)
2. PHẠM VI
Hướng dẫn này được áp dụng cho thuốc vô khuẩn được tiệt khuẩn cuối.
3. THÔNG TIN CHUNG
Quá trình tiệt khuẩn có thể được thực hiện bằng nhiệt khô, nhiệt ẩm, chiếu xạ ion hóa, tiệt khuẩn bằng khí hoặc lọc và sau đó đóng vô khuẩn vào bao bì vô khuẩn
Tiệt khuẩn bằng nhiệt nên là phương pháp được ưu tiên lựa chọn nếu điều kiện cho phép.
4. THÔNG TIN VỀ CÁC QUÁ TRÌNH TIỆT KHUẨN CUỐI
Nhìn chung, việc mô tả quá trình tiệt khuẩn và kết quả thẩm định quy trình cần được cung cấp đối với:
● Thuốc thành phẩm được chứa trong hệ thống bao bì
● Bao bì, nắp, thiết bị và các thành phần thuốc
● Sản phẩm trung gian
Trong trường hợp sản phẩm có thể được xử lý thêm (chẳng hạn xử lý thêm bằng nhiệt độ), cần cung cấp các dữ liệu minh chứng cho việc xử lý thêm đó.
4.1. Tiệt khuẩn cuối bằng nhiệt ẩm
4.1.1. Mô tả quá trình tiệt khuẩn bằng nhiệt ẩm
Phần mô tả quá trình tiệt khuẩn bằng nhiệt ẩm nên bao gồm các phần sau:
● Thông tin nhận dạng của nồi hấp (ví dụ: mã số, nhà sản xuất, model của thiết bị sử dụng).
● Loại chu trình được sử dụng (ví dụ: sử dụng hơi bão hòa, ngâm nước và phun nước).
● Các thông số trong chu trình và các chỉ tiêu trong quá trình chạy, bao gồm nhiệt độ, thời gian, giá trị lớn nhất và nhỏ nhất của F0.
● Các phương pháp và biện pháp kiểm soát được sử dụng để theo dõi các chu trình trong sản xuất (ví dụ: nhiệt độ đầu dò, chỉ thị vi sinh và hóa học, phép thử độ rò) bao gồm số lượng, vị trí cũng như chỉ tiêu chấp nhận và chỉ tiêu loại trừ Tuỳ chọn.
4.1.2. Thẩm định quy trình và / hoặc đánh giá quy trình
a. Nghiên cứu phân bố nhiệt và truyền nhiệt
Phương pháp và tiêu chuẩn để đánh giá phân bố nhiệt và truyền nhiệt cần được cung cấp và đi kèm với bản tóm tắt các kết quả gần thời điểm nộp:
● Phương pháp và tiêu chuẩn
● Sơ đồ thể hiện số lượng đầu đo nhiệt, chất chỉ thị hóa học và/hoặc sinh học nếu có - nếu được sử dụng cũng như vị trí của chúng trong buồng hấp tiệt khuẩn.
● Sơ đồ thể hiện sự sắp xếp sản phẩm ở khối lượng tối thiểu và tối đa cũng như các điểm có nhiệt độ thấp.
● Kết quả của tối thiểu 3 chu trình tiệt khuẩn đạt liên tiếp.
b. Phép kiểm tra khả năng kháng nhiệt của vi sinh vật
Các thành phẩm được coi là vô khuẩn khi mức độ đảm bảo vô khuẩn (Sterility assurance level - SAL) đạt được là 10-6 hoặc thấp hơn.
Báo cáo tổng hợp về kết quả nghiên cứu khả năng kháng nhiệt của vi sinh vật cần được cung cấp và bao gồm các nội dung sau:
● Mức độ tạp nhiễm vi sinh vật (bioburden), đặc biệt là khi các điều kiện tiệt khuẩn có khả năng tận diệt vi khuẩn (overkill) không được sử dụng.
● Phiếu kiểm nghiệm của chỉ thị vi sinh vật được sử dụng, bao gồm có phân loại chủng chỉ thị, mức độ kháng và độ ổn định.
● Thông tin về khả năng kháng của chỉ thị vi sinh vật
Thông tin về khả năng kháng của chỉ thị vi sinh vật trong hoặc trên sản phẩm (ví dụ: trong dung dịch thuốc, trên các bề mặt của bao bì, nắp và các mặt ngăn cách) hoặc các đối tượng được dùng để thay thế sản phẩm cần phải được chỉ rõ. Cần phải có sự so sánh giữa khả năng kháng của bào tử khi sử dụng giá mang bào tử (ví dụ: băng mang bào tử) để nuôi cấy thay vì nuôi cấy trực tiếp.
● Kết quả và kết luận của nghiên cứu thẩm định vi sinh nhằm đưa ra quy trình tiệt khuẩn với điều kiện tối thiểu mà duy trì được mức độ đảm bảo vô khuẩn (SAL) tối thiểu là 10-6 cho những sản phẩm được tiệt khuẩn ở điều kiện khó khăn nhất.
4.2. Các quy trình tiệt khuẩn cuối khác
Các loại thông tin được yêu cầu đối với tiệt khuẩn bằng nhiệt ẩm, nhìn chung, có thể được áp dụng cho tiệt khuẩn bằng nhiệt khô, khí (chẳng hạn ethylen oxyd) hay tiệt khuẩn bằng chiếu xạ (chẳng hạn tia gamma và electron).
Dưới đây là các thông tin tối thiểu cần phải cung cấp:
● Mô tả lượng và cách sắp xếp, phân bố của vật cần tiệt khuẩn.
● Các số liệu thẩm định nhằm chứng minh mức độ hiệu quả của chu trình tiệt khuẩn ở điều kiện tối thiểu.
● Tính toàn vẹn của bao bì, nắp.
● Các thông tin về việc tái xử lý nếu có.
● Các ảnh hưởng của quy trình tiệt khuẩn tới đặc tính hóa học và vật lý của dược chất hoặc thuốc thành phẩm nếu có.
Các yêu cầu đặc biệt đối với tiệt khuẩn bằng ethylen oxyd và chiếu xạ được trình bày dưới đây.
4.2.1. Etylen oxyd (EO)
a. Giải thích rõ lý do sử dụng ethylen oxyd để tiệt khuẩn.
b. Thiết bị tiệt khuẩn và môi trường được kiểm soát để làm ẩm ban đầu và thông khí cho khối sản phẩm.
c. Các thông số và giới hạn của từng bước trong chu trình tiệt khuẩn, ví dụ như làm ẩm ban đầu, nồng độ khí sử dụng, chu kì chân không và áp suất khí, thời gian tiếp xúc, nhiệt độ, độ ẩm, đuổi khí, thông khí và xác định khí tồn dư.
d. Các phương pháp vi sinh (môi trường nuôi cấy, nhiệt độ và thời gian ủ) và nuôi cấy bào tử từ mẫu được ủ khi tiến hành thẩm định quy trình.
4.2.2. Chiếu xạ
a. Cơ sở vật chất của nơi chiếu xạ.
b. Nguồn chiếu xạ và phương pháp tiếp xúc (sự di chuyển của sản phẩm so với nguồn phát xạ).
c. Loại và vị trí thiết bị để theo dõi thường xuyên khối lượng sản phẩm được tiệt khuẩn.
d. Đặc tính bao bì.
e. Nghiên cứu về phân bố đa liều tại các vị trí khác nhau.
f. Phương pháp đánh giá và kiểm soát vi sinh được sử dụng để thiết lập, thẩm định, kiểm tra hiệu quả của chu trình.
4.3. Tính toàn vẹn của hệ thống bao bì - nắp
Nhìn chung, các loại dữ liệu và thông tin sau về tính toàn vẹn đối với vi sinh vật của các thành phần trong bao bì cần phải được cung cấp:
a. Mô phỏng lại các điều kiện khắc nghiệt trong sản xuất
Việc thiết kế thí nghiệm cần mô phỏng được các điều kiện khắc nghiệt trong quá trình tiệt khuẩn, vận chuyển và bảo quản thuốc cũng như ảnh hưởng của các điều kiện này lên độ kín và tính toàn vẹn của hệ thống bao bì - nắp. Việc nghiên cứu độ ổn định về hóa học, vật lý và vi sinh vật trong các điều kiện này có thể là cần thiết.
b. Chứng minh tính toàn vẹn của bao bì sau tiếp xúc tối đa
Độ kín và tính toàn vẹn của hệ thống bao bì – nắp nên được kiểm chứng bằng đặc tính của sản phẩm đã được tiệt khuẩn ở một hoặc nhiều chu kỳ ở điều kiện khắc nghiệt nhất. Nếu sản phẩm được xử lý bằng nhiều phương pháp thì thiết kế thí nghiệm cần phải được tiến hành trên các chu kỳ ở điều kiện khắc nghiệt nhất của tất cả các phương pháp xử lý.
c. Độ nhạy của phương pháptuỳ chọn
Độ nhạy của phương pháp thử tính toàn vẹn của hệ thống bao bì – nắp cần được xác định rõ và được cung cấp.
5. THUẬT NGỮ
Chỉ thị vi sinh vật - Biological Indicator:
Một quần thể vi sinh vật được ủ trong một môi trường phù hợp và được đặt trong một vị trí nhất định ở khoang / nơi tiệt khuẩn của thiết bị tiệt khuẩn để xác định hiệu quả của chu trình tiệt khuẩn bằng phương pháp vật lý hoặc hóa học.
Thành phần thuốc - Component:
Bất kể một thành phần nào được sử dụng với mục đích sản xuất thuốc, kể cả các thành phần không xuất hiện trong thành phẩm cuối cùng.
Giá trị F0:
Thể hiện hiệu quả tiệt khuẩn của một chu trình cụ thể (giá trị F0 chính là số phút cần thiết khi tiệt khuẩn ở 121°C để đạt được hiệu quả tiệt khuẩn tương đương với chu trình đó).
Ví dụ: chu trình tiệt khuẩn ở điều kiện ở 111°C trong 15 phút có hiệu quả tiệt khuẩn tương đương với chu trình tiệt khuẩn ở 121 °C trong 1,5 phút.
Tiệt khuẩn cuối - Terminal Sterilization:
Tiệt khuẩn một sản phẩm ở công đoạn cuối cùng bằng hơi nước và/hoặc nhiệt khô hoặc chiếu xạ.
PHỤ LỤC B MỤC LỤC CỦA HỒ SƠ THẨM ĐỊNH QUY TRÌNH SẢN XUẤT
I. TÀI LIỆU ĐƯỢC NỘP (Đánh dấu nếu nộp):
|
Loại tài liệu |
Đánh dấu |
Đính kèm |
Trang |
|
a) Phát triển dược học b) Đề cương thẩm định |
□ □ |
___________ ___________ |
_____________ _____________ |
|
c) Báo cáo thẩm định |
|
|
|
|
o Lô pilot o 3 lô sản xuất thực tế |
□ □ |
___________ ___________ |
___________ ___________ |
II. Thông tin thẩm định chi tiết:
a) Cơ sở sản xuất nơi tiến hành thẩm định:
|
STT |
Tên nhà sản xuất |
Quốc gia |
|
|
|
|
|
|
|
|
|
|
|
|
b) Loại thẩm định:
□ Thẩm định hồi cứu
□ Thẩm định trước
□ Thẩm định đồng thời
□ Khác; đề nghị nêu rõ: _______________________________
c) Số lượng lô được thẩm định: __________
d) Thông tin chi tiết của từng lô:
|
Số lô |
Ngày sản xuất |
Cỡ lô |
Loại lô (pilot / lô thực tế) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PHỤ LỤC C HƯỚNG DẪN THẨM ĐỊNH QUY TRÌNH SẢN XUẤT DỰA TRÊN CÁC TIẾP CẬN CHẤT LƯỢNG THEO THIẾT KẾ (QUALITY BY DESIGN)
MỤC LỤC
1. MỤC ĐÍCH
2. PHẠM VI
3. THÔNG TIN CHUNG
4. KHUYẾN CÁO
4.1 BƯỚC 1 - THIẾT KẾ QUY TRÌNH (PROCESS DESIGN)
4.2 BƯỚC 2 - THẨM ĐỊNH QUY TRÌNH (PROCESS QUALIFICATION)
4.3 BƯỚC 3 – THẨM TRA LIÊN TỤC QUY TRÌNH (CONTINUED PROCESS VERIFICATION)
5. QUY ĐỊNH NỘP TÀI LIỆU THEO YÊU CẦU CỦA HỒ SƠ KỸ THUẬT CHUNG CỦA ASEAN (ACTD)
6. THUẬT NGỮ
1. MỤC ĐÍCH
Nhằm hướng dẫn nộp các thông tin và dữ liệu thẩm định quy trình dựa trên cách tiếp cận Chất lượng theo Thiết kế (Quality by Design hay QbD).
Hướng dẫn này nên được đọc cùng với các tài liệu sau:
● Process Validation: General Principles and Practices (US-FDA, Jan 2011)
● Pharmaceutical Development Q8(R2) (ICH, August 2009)
● ICH Quality Risk Management Q9 (ICH, Nov 2005)
● ICH Pharmaceutical Quality System Q10 (ICH, June 2008)
● ICH Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological /Biological Entities) Q11 (ICH, May 2012)
● ICH Quality Implementation Working Group on Q8, Q9 and Q10 Questions & Answers (R4) (ICH, Nov 2010)
● ICH Quality Implementation Working Group Points To Consider (R2) (ICH, Dec 2011)
2. PHẠM VI
Hướng dẫn này được áp dụng cho cả thuốc có nguồn gốc hóa học hoặc sinh học cũng như nguyên liệu làm thuốc.
3. THÔNG TIN CHUNG
FDA đã ban hành tài liệu “Guideline on General Principles of Process Validation - Hướng dẫn về các nguyên tắc chung trong thẩm định quy trình vào năm 1987”, trong đó nhấn mạnh rằng việc thẩm định quy trình được hoàn thành với 3 lô sản xuất thực tế. Một phương pháp khác có thể được áp dụng đó là thẩm tra quy trình liên tục, hay còn gọi là phương pháp dựa trên vòng đời sản phẩm vốn là một nội dung trọng tâm của khái niệm Chất lượng theo Thiết kế (Quality by Design - QbD).
Tháng 8 năm 2009, ICH đã đưa ra hướng dẫn Q8R (2) (Bước 4) để hướng dẫn ngành công nghiệp dược phẩm thực hiện Chất lượng theo Thiết kế (QbD) trong mục 3.2.P.2 (Phát triển Dược học) cho các sản phẩm thuốc như đã nêu trong phạm vi của Module 3 trong Tài liệu Kỹ thuật Chung (Hướng dẫn ICH M4). Chất lượng theo thiết kế, hay QbD (theo ICH Q8 (R2)) được định nghĩa là "một cách tiếp cận có hệ thống trong phát triển dược phẩm được bắt đầu với các mục tiêu xác định trước và nhấn mạnh vào những hiểu biết về sản phẩm, quy trình và kiểm soát quy trình, dựa trên cơ sở khoa học và quản lý rủi ro về chất lượng”. Đây là một cách tiếp cận có hệ thống đối với quá trình phát triển dược phẩm và bao gồm việc sử dụng các kiến thức đã thu được, các kết quả của các nghiên cứu sử dụng phương pháp Thiết kế thí nghiệm, quản lý rủi ro (ICH Q9), quản lý kiến thức (ICH Q10) trong suốt vòng đời của sản phẩm.
Sau đó, tài liệu "Questions and Answers" (Hỏi và đáp) lần thứ tư nhằm tạo thuận lợi cho việc thực hiện Hướng dẫn Q8 (R2), Hướng dẫn Q9 và Q10 đã được ban hành vào tháng 11 năm 2010 (Q8 / Q9 / Q10 Q&As (R4)). Nhóm thực thi về chất lượng ICH (ICH Quality IWG) cũng đã đưa ra tài liệu "Points to Consider" (Những điểm cần xem xét) bao gồm các chủ đề liên quan đến việc thực hiện Q8 (R2), Q9 và Q10 để bổ sung cho tài liệu "Questions and Answers" đã lưu hành tại thời điểm đó vào tháng 12 năm 2011. Cùng với sự phát triển của QbD, việc thẩm định quy trình và các lĩnh vực có liên quan cũng phát triển theo. Cuối cùng, FDA đã công bố tài liệu "Process Validation: General Principles and Practices" (Thẩm định quy trình: Nguyên tắc chung và thực hành) vào tháng 1 năm 2011. Tài liệu này đã đề cập đến QbD, Công nghệ phân tích Quy trình (PAT), quản lý rủi ro và khái niệm thẩm định quy trình với cách tiếp cận theo vòng đời sản phẩm. Khái niệm này nhấn mạnh về một cách tiếp cận toàn diện hơn đối với thẩm định quy trình.
Trong hướng dẫn mới của FDA, thẩm định quy trình được định nghĩa là "sự thu thập và đánh giá dữ liệu từ giai đoạn nghiên cứu tới khi sản xuất sản phẩm thương mại, từ đó thiết lập bằng chứng khoa học rằng quy trình đủ khả năng để liên tục đảm bảo chất lượng của sản phẩm sản xuất ra". Thẩm định quy trình bao gồm một loạt các hoạt động xảy ra liên tục trong quy trình cũng như xuyên suốt vòng đời của sản phẩm. Hướng dẫn này mô tả hoạt động thẩm định quy trình qua 3 bước:
● Bước 1 - Thiết kế quy trình - Process Design (PD): Quy trình ở quy mô sản xuất thực tế sẽ được sẽ được thiết lập ở bước này dựa trên các kiến thức thu được từ quá trình phát triển sản phẩm và nâng cấp quy mô.
● Bước 2: Thẩm định quy trình - Process Qualification (PQ): Ở giai đoạn này, quy trình theo thiết kế sẽ được kiểm tra về khả năng sản xuất được chế phẩm thương mại với độ lặp lại cao.
● Bước 3: Thẩm tra liên tục quy trình - Continued Process Verification (CPV): Thẩm tra liên tục quy trình để bảo đảm rằng quy trình sản xuất nằm trong tầm kiểm soát.
4. KHUYẾN CÁO
Các mục dưới đây sẽ mô tả các hoạt động cụ thể của mỗi bước trong vòng đời sản phẩm:
4.1 Bước 1 - Thiết kế quy trình (Process Design)
Mục đích của bước này là cung cấp các hiểu biết cơ bản về sản phẩm và quy trình sản xuất. Các hoạt động phát triển dược phẩm đóng vai trò rất quan trọng trong bước thiết kế quy trình. Các hoạt động này bao gồm việc xác định các yếu tố ảnh hưởng đến việc thiết kế quy trình, chẳng hạn như dạng bào chế dự kiến, đặc tính chất lượng và quy trình sản xuất tổng quát. Ở bước đầu tiên này, cần xem xét tới chức năng và giới hạn / hạn chế của thiết bị cũng như sự thay đổi / khác biệt ở quy mô sản xuất thực tế, chẳng hạn như việc sử dụng các lô khác nhau của các thành phần công thức, sự khác biệt về người vận hành, các thông số về môi trường và hệ thống đo lường. Việc sử dụng các thiết kế thí nghiệm dựa trên các phép phân tích thống kê chẳng hạn như Thiết kế Thí nghiệm (Design of Experiment - DoE) rất hữu dụng cho việc xác định các mối tương quan, bao gồm cả các tương tác đa biến, giữa các biến đầu ra và biến đầu vào. Các công cụ phân tích rủi ro có thể được sử dụng để sàng lọc sơ bộ các biến tiềm năng được sử dụng cho thiết kế thí nghiệm (DoE), nhằm làm giảm tối đa số lượng thí nghiệm trong khi thu được tối đa thông tin. Kết quả thu được có thể được sử dụng làm minh chứng cho việc thiết lập các khoảng cho phép của nguyên liệu đầu vào, thông số kỹ thuật của thiết bị, các đặc tính của bán thành phẩm cũng như thiết lập không gian thiết kế.
4.1.1 Thiết kế và phát triển quy trình
Các điểm dưới đây là những điểm quan trọng cần được xem xét khi tiến hành thiết kế và phát triển một quy trình:
● Mục tiêu chất lượng của sản phẩm - Quality Target Product Profile (QTPP) - Các mục tiêu này nên được xác định sớm ở giai đoạn đầu của việc phát triển sản phẩm và quy trình. Các yếu tố của mục tiêu chất lượng sản phẩm bao gồm mục tiêu sử dụng thuốc trên lâm sàng, dạng bào chế, đường dùng, mức liều, hệ thống bao bì đóng gói, dược động học...
● Chỉ tiêu chất lượng trọng yếu - Critical Quality Attribute (CQA) - là các đặc tính vật lý, hóa học, sinh học hoặc vi sinh cần phải nằm dưới một giới hạn, trong một khoảng hoặc một phạm vi phân bố nhất định nhằm đạt được chất lượng sản phẩm như mong muốn. CQA được xây dựng từ QTPP và các CQA được lựa chọn dựa trên với những lý giải mang tính khoa học. Những lý giải này thường liên quan đến dược chất, tá dược, các sản phẩm trung gian và thành phẩm (theo ICH). CQA phụ thuộc vào dạng thuốc và chính dạng thuốc sẽ quyết định các yêu cầu về chất lượng tương ứng, chẳng hạn như đặc tính khí động học đối với thuốc xông hít hoặc khả năng bám dính đối với các miếng dán ngoài da... Một ví dụ khác: CQA của viên nén giải phóng ngay gồm có hình thức, các đặc tính vật lý, độ hòa tan, định lượng, độ đồng đều hàm lượng, tạp chất, giới hạn vi sinh vật...
● Xây dựng công thức và phát triển quy trình – Phần lớn các công việc được thực hiện để hiểu về quy trình được tiến hành trong giai đoạn xây dựng công thức và phát triển sản phẩm. Các công việc này bao gồm có việc nghiên cứu ở quy mô phòng thí nghiệm, quy mô pilot và quy mô sản xuất thực tế. Các nguyên liệu phù hợp cũng như hàm lượng của chúng được xác định. Mỗi một công đoạn của quá trình sản xuất được xác định và cần phải thống nhất với khả năng sản xuất thực tế của cơ sở tiến hành sản xuất thực tế trong tương lai. Các công cụ đánh giá nguy cơ được mô tả trong ICH Q9 có thể được dùng để xác định các tác động có thể ảnh hưởng tới đặc tính của nguyên liệu hoặc thông số quy trình lên các CQA. Có thể xếp hạng các thông số này theo mức độ nguy cơ dựa trên các kiến thức đã biết hoặc kết các quả thí nghiệm sơ bộ.
Dưới đây là một trong số những yếu tố chính cần lưu ý:
a. Dược chất (API):
Nhiều đặc tính của dược chất có thể ảnh hưởng đến quá trình sản xuất và CQA của sản phẩm thuốc. Ví dụ, kích thước hạt, hình dạng, tính đa hình, độ tan, độ trơn chảy, khả năng chịu nén, khả năng tương hợp với các loại tá dược... Các rủi ro tiềm ẩn xuất phát từ các đặc tính của dược chất ảnh hưởng lên CQA của sản phẩm thuốc cần được đánh giá dựa trên các kiến thức sẵn có hoặc dựa trên các cơ sở khoa học. Kết quả phân tích rủi ro của tất cả các đặc tính của tá dược nên được bàn luận một cách cụ thể, trong đó, các yếu tố rủi ro cao nên được tiếp tục nghiên cứu sâu hơn. Khi các ảnh hưởng đã được xác định, cần phải có chiến lược phù hợp để kiểm soát các đặc tính của dược chất nhằm đảm bảo đạt được CQA. Ví dụ về cách các đặc tính của API ảnh hưởng đến CQAs cho cả thuốc hóa học và sinh học có thể được tìm thấy trong ICH Q11.
b. Xây dựng công thức:
Các tá dược được lựa chọn dựa trên mức chất lượng / mức độ ảnh hưởng đến CQA hoặc khả năng sản xuất. Các chức năng của tá dược cũng như tính tương hợp của dược chất và tá dược cần phải được xác định và chỉ rõ. Khi sử dụng phương thức QbD, ảnh hưởng của các thành phần trong công thức lên CQA cần phải được bàn luận và trình bày một cách chi tiết. Các ảnh hưởng này có thể được nghiên cứu dựa trên cơ sở khoa học hoặc dựa trên kinh nghiệm. Việc hiểu rõ các ảnh hưởng này có thể giúp giải thích được việc lựa chọn loại và tiêu chuẩn của các tá dược. Chẳng hạn, các tá dược mà cấu trúc và tính chất hóa học có thể làm phân hủy dược chất cần phải tránh được sử dụng. Trong trường hợp việc sử dụng tá dược đó là không thể tránh khỏi, cần phải tìm ra các biện pháp để làm giảm sự phân hủy của dược chất (chẳng hạn như giảm lượng tá dược sử dụng hoặc giảm khả năng tiếp xúc giữa dược chất tá dược).
Ở giai đoạn đầu của quá trình phát triển dược học, quy trình sản xuất chi tiết chưa được thiết lập. Nhà sản xuất có thể đề xuất một quy trình phù hợp dựa trên các kiến thức sẵn có về các sản phẩm tương tự, các công thức tương tự và/hoặc các nghiên cứu tiền công thức. Đánh giá phân loại rủi ro ban đầu được thực hiện nhằm phân loại rủi ro dựa trên khả năng thay đổi để thích ứng của các công đoạn sản xuất với những sự thay đổi nhỏ (nếu có) của công thức bào chế. Việc đánh giá rủi ro được thực hiện dựa trên các giả định và hoàn cảnh (assumptions and context). Nhà sản xuất cần cung cấp lý giải cho kết quả đánh giá rủi ro ban đầu và chỉ rõ những nguyên nhân nào sẽ được nghiên cứu thực sự trong quá trình phát triển sản phẩm. Sau khi hoàn thành các nghiên cứu thực nghiệm để xây dựng công thức, thường là ở quy mô phòng thí nghiệm, có thể từ đó xem xét lại quá trình đánh giá rủi ro ban đầu. Từ các kết quả đó, tiến hành đề xuất công thức sản phẩm và phát triển công thức đó cũng như phát triển quy trình.
c. Phát triển quy trình:
Các thông số quy trình trọng yếu ảnh hưởng đến CQA và các thông số hoặc biến này cần phải được nghiên cứu dựa trên đánh giá rủi ro hoặc các thí nghiệm được xây dựng dựa trên các phương pháp thống kê. Các phương pháp đánh giá rủi ro được mô tả trong ICH Q9. Đánh giá rủi ro ban đầu có thể được tiến hành để nghiên cứu ảnh hưởng của các công đoạn sản xuất lên CQA. Số lượng các thông số nguy cơ có ảnh hưởng lớn có thể rất lớn, nhưng có thể được sàng lọc lại bằng thí nghiệm. Phương pháp quy ước sử dụng trong nghiên cứu ảnh hưởng của từng yếu tố riêng lẻ không nên sử dụng trong QbD. Thay vào đó, phương pháp Thiết kế thí nghiệm (DoE) nên được tiến hành để sàng lọc các yếu tố trọng yếu của quy trình trong khi có thể làm giảm được số thí nghiệm. Một khi các thông số trọng yếu đã được xác định, phương pháp thiết kế thí nghiệm ở mức độ chi tiết và cụ thể hơn có thể được tiến hành để xác định rõ hơn đặc điểm của quy trình, từ đó xây dựng chiến lược để kiểm soát quy trình.
Các quy mô sản xuất khác nhau được nâng cấp và hướng dẫn đến quy mô sản xuất thực tế có thể được đề xuất dựa trên các kiến thức đã biết hoặc các kết quả thí nghiệm thực tế. Sau đó, ảnh hưởng của việc nâng cấp quy mô của từng công đoạn sản xuất nên được nghiên cứu và bàn luận. Hệ số nâng quy mô có thể được sử dụng cho một số loại thiết bị nếu khả năng áp dụng hệ số này được lý giải một cách hợp lý. Sau khi đã thu được đủ các hiểu biết về sản phẩm và quy trình từ kết quả của việc nghiên cứu ở quy mô phòng thí nghiệm và quy mô pilot, cần phải áp dụng được các hiểu biết này vào điều kiện thực tế ở cơ sở sản xuất. Việc sản xuất ở quy mô phòng thí nghiệm có thể khác biệt đáng kể so với việc sản xuất ở quy mô sản xuất thực tế. Trên thực tế, một số yếu tố ảnh hưởng đến quy trình chỉ có thể được nghiên cứu khi đưa ra quy mô sản xuất thực tế. Khả năng nâng cấp thành công lên quy mô sản xuất là một phần rất quan trọng của việc thẩm định và theo dõi quy trình sản xuất trong tương lai. Các nhà sản xuất có thể tiến hành nâng cấp quy mô từng phần để dự đoán khả năng nâng cấp quy mô của toàn bộ quy trình. Sau đó, kết quả thẩm định của các lô đạt yêu cầu sẽ là cơ sở để khẳng định sự thành công của việc sử dụng QbD trong phát triển sản phẩm và nâng cấp quy mô.
d. Không gian thiết kế:
Trong ICH Q8(R2), không gian thiết kế được định nghĩa là "sự kết hợp và tương tác đa chiều của các biến đầu vào (ví dụ: đặc tính của nguyên liệu) và thông số quy trình đã được chứng minh là giúp đảm bảo chất lượng". Nếu quy trình sản xuất được thực hiện trong phạm vi của không gian này thì những sự khác biệt xảy ra trong quy trình không được coi là thay đổi cần được chấp thuận bởi cơ quan quản lý. Không gian thiết kế được mô tả bằng các khoảng biến thiên của các đặc tính của nguyên liệu, khoảng biến thiên của thông số quy trình hoặc được mô tả bằng các mối liên hệ toán học phức tạp hơn. Không gian thiết kế thường được xác định bằng các thiết kế dựa trên toán thống kê chẳng hạn như Thiết kế thí nghiệm (DoE). Việc xác định này cho phép thu được lượng tối đa thông tin với số thí nghiệm tối thiểu. Không gian thiết kế chỉ bao gồm các Thông số quy trình trọng yếu (Critical process parameters - CPP) hoặc các đặc tính quan trọng của nguyên liệu có ảnh hưởng trực tiếp tới CQA của sản phẩm. Không gian thiết kế có thể được thiết lập cho từng công đoạn sản xuất riêng lẻ hoặc nhóm của một vài công đoạn của toàn bộ quy trình.
Khi Thiết kế thí nghiệm (DoE) được tiến hành để thiết lập CPP và/hoặc không gian thiết kế, nhà sản xuất cần phải giải thích một cách hợp lý lý do lựa chọn các biến cũng như khoảng của chúng, giải thích cách lựa chọn loại thiết kế thí nghiệm dựa trên độ mạnh của thiết kế đó, liệu các yếu tố trong thiết kế có phụ thuộc vào quy mô thí nghiệm hay không, mức độ phù hợp của phương pháp phân tích được sử dụng, kết quả phân tích dựa trên phân tích thống kê của Thiết kế thí nghiệm (DoE) cho thấy ảnh hưởng có ý nghĩa thống kê của từng yếu tố riêng lẻ cũng như tương tác giữa các yếu tố và khả năng dự đoán khi đã xét tới quy mô sản xuất và sự khác biệt về thiết bị. Dưới đây là ví dụ về không gian thiết kế (tuyến tính và không tuyến tính) của 2 CPP trong giai đoạn tạo hạt ảnh hưởng tới độ hòa tan (trích từ ICH Q8(R2))
Ví dụ 1: Sơ đồ thể hiện mặt đáp (Hình 1a) và đường đồng mức (Hình 1b) của tốc độ hòa tan. Thông số 1 và 2 là các yếu tố của quá trình tạo hạt có ảnh hưởng tới tốc độ hòa tan của viên nén (chẳng hạn như đặc tính tá dược, lượng nước, kích thước hạt cốm).
|
|
|
|
Hình 1a: Mặt đáp của tốc độ hòa tan là một hàm số của hai thông số trong quá trình tạo hạt. Tốc độ hòa tan mong muốn là trên 80%. |
Hình 1b: Đường đồng mức của độ hòa tan từ ví dụ 1a. |
|
|
|
|
Hình 1c: Không gian thiết kế của các thông số trong quá trình tạo hạt (được xác định bởi tổ hợp không tuyến tính giữa các khoảng biến thiên của các thông số) cho kết quả tốc độ hòa tan mong muốn (> 80%). |
Hình 1d: Không gian thiết kế của các thông số trong quá trình tạo hạt (được xác định bởi tổ hợp tuyến tính của các khoảng biến thiên của các thông số) cho kết quả độ hòa tan mong muốn (> 80%). |

Việc xác định mức độ phù hợp của không gian thiết kế được xây dựng ở quy mô nhỏ hoặc pilot đối với việc sản xuất ở quy mô thực tế là rất quan trọng. Các rủi ro tiềm năng khi tiến hành nâng cấp quy mô cần phải được bàn luận. Không gian thiết kế cần phải được thẩm tra lại và vận hành ở quy mô sản xuất thực tế, mặc dù việc xây dựng không gian thiết kế ở quy mô sản xuất thực tế là không bắt buộc. Không nên nhầm lẫn và đánh đồng giữa việc thẩm tra lại một không gian thiết kế và thẩm định quy trình. Tuy vậy, việc thẩm tra không gian thiết kế có thể bao gồm việc theo dõi và kiểm tra các CQA bị ảnh hưởng khi nâng cấp quy mô sản xuất. Các yếu tố có thể dẫn đến sự cần thiết phải thẩm tra lại không gian thiết kế có thể là việc thay đổi thiết bị, thay đổi cơ sở sản xuất...
Việc tiến hành sản xuất các lô thẩm định (qualification batches) với các điều kiện nằm ngoài giới hạn của không gian thiết kế trong khi tiến hành thẩm định ở quy mô sản xuất là không cần thiết. Không gian thiết kế cần phải được nghiên cứu một cách kỹ càng ngay từ trước đó trong giai đoạn đầu của việc nghiên cứu phát triển. Việc nghiên cứu các giới hạn có thể làm chất lượng sản phẩm không đạt yêu cầu được khuyến khích mặc dù đó không phải là nội dung trọng tâm khi thiết lập không gian thiết kế.
Tổ hợp của các Khoảng chấp nhận đã được chứng minh (Proven acceptable ranges - PARs) từ cách thiết kế thí nghiệm đơn biến không thể được sử dụng để xây dựng không gian thiết kế vì các thí nghiệm kiểu này không xét tới tương tác giữa các thông số trong quy trình và/hoặc các đặc tính của nguyên liệu.
Việc mô hình hóa bằng các mối liên hệ toán học là không cần thiết khi xây dựng Không gian thiết kế. Tuy vậy, nếu sử dụng mô hình hóa, mô hình được lựa chọn cần phải được thẩm định, cập nhật và duy trì. Một trong số các biện pháp để thẩm định mô hình chính là phương pháp thẩm định chéo nội sử dụng cùng một bộ số liệu. Tính chính xác và mức độ dao động / thay đổi do bản chất các công đoạn trong quy trình và các phương pháp phân tích cần phải được giải thích. Mô hình không được phụ thuộc vào quy mô cũng như thiết bị. Không gian thiết kế cần được cập nhật trong suốt vòng đời của sản phẩm với các kiến thức mới thu được. Việc vận hành trong phạm vi của không gian thiết kế được coi là một phần của chiến lược kiểm soát và không bị coi là thay đổi cần có sự chấp thuận của cơ quan quản lý.
4.1.2 Xây dựng chiến lược Kiểm soát quy trình
Mục đích của việc Kiểm soát quy trình là kiểm soát sự biến thiên của quy trình, có thể được thực hiện bằng các giảm thiểu sự thay đổi của các yếu tố đầu vào và/hoặc điều chỉnh mức độ biến thiên của các yếu tố đầu vào trong quá trình sản xuất. Việc xác định các biến thông số quy trình và các biến công thức chính là bước chìa khóa đối với phương pháp thẩm định trong suốt vòng đời sản phẩm và bao gồm việc xác định những sự thay đổi trong từng công đoạn sản xuất. Ví dụ của các biến đầu vào là nguyên liệu, thiết bị, quy trình, hệ thống đo lường, nhân viên, môi trường... Chiến lược kiểm soát các biến này cần phải được giải thích dựa trên hiểu biết về sản phẩm và quy trình sản xuất.
Một quy trình có độ vững chắc là một quy trình có khả năng sản xuất ra được sản phẩm đạt yêu cầu chất lượng mặc dù có thể có sự thay đổi đáng kể của các yếu tố đầu vào quy trình. Nhà sản xuất cần phải nghiên cứu các biến này và thẩm tra lại chiến lược kiểm soát quy trình khi sản xuất ở quy mô thực tế. Việc kiểm tra lại bước thiết kế quy trình và chiến lược kiểm soát quy trình là cần thiết nếu quy trình không có độ vững chắc. Chiến lược kiểm soát cần phải được xây dựng trong hồ sơ tổng thể về sản xuất và kiểm soát quy trình.
Các chiến lược kiểm soát tiên tiến hơn có thể được sử dụng, chẳng hạn như việc sử dụng Công nghệ phân tích quy trình (Process analytical technology - PAT), cho phép tiến hành phân tích theo thời gian thực và kiểm soát chất lượng đầu ra. Việc sử dụng PAT được khuyến khích nhưng cách thẩm định quy trình (process qualification) sẽ khác với các cách thiết kế quy trình khác. PAT được coi như một công cụ đắc lực của QbD vì nó giúp tăng cường hiểu biết về quy trình. Việc sử dụng PAT cho phép nhà sản xuất thực hiện việc xuất xưởng theo thời gian thực (Real Time Release) mà không cần phải kiểm nghiệm thành phẩm sau quá trình sản xuất (End Product Testing). Tuy vậy, việc kiểm nghiệm sản phẩm theo thời gian thực (Real time release testing - RTRT) không thay thế được việc kiểm tra và kiểm soát chất lượng khi xuất xưởng theo quy định của GMP. Nếu việc kiểm nghiệm theo thời gian thực (RTRT) trong quá trình sản xuất được đưa vào tiêu chuẩn thành phẩm thì các kết quả kiểm nghiệm của RTRT phải được sử dụng làm căn cứ để ra quyết định xuất xưởng và khi sản phẩm không đạt theo các tiêu chuẩn được đề ra bởi kiểm nghiệm theo thời gian thực thì không được dựa trên kết quả kiểm nghiệm thành phẩm sau quá trình sản xuất để làm cơ sở xuất xưởng. Khả năng xuất xưởng của các lô đó phụ thuộc vào kết quả thanh tra tiến hành trên các lô đó. Bên cạnh đó, nghiên cứu độ ổn định vẫn cần phải được tiến hành khi áp dụng phương pháp kiểm nghiệm theo thời gian thực.
4.2 Bước 2 - Thẩm định quy trình (Process Qualification)
Mục tiêu của Thẩm định quy trình (process qualification) là để xác định xem liệu việc thiết kế quy trình có đảm bảo được khả năng sản xuất sản phẩm đạt chất lượng ở quy mô sản xuất thương mại một cách lặp lại hay không. Kiểm tra quá trình bao gồm 2 phần: (1) thiết kế cơ sở vật chất cùng với năng lực của trang thiết bị và hệ thống phụ trợ và (2) Thẩm định hiệu năng của quy trình (Process Performance Qualification - PPQ).
Việc thẩm định hệ thống phụ trợ và thiết bị nhằm đảm bảo rằng hệ thống và thiết bị hoạt động được một cách bình thường và phù hợp với mục đích sử dụng. Việc kiểm tra được thực hiện bằng cách thay đổi khối lượng sản xuất theo thiết kế, can thiệp trong quá trình, dừng và tái khởi động quá trình sản xuất. Đây là một yêu cầu trước khi bắt đầu Thẩm định hiệu năng quy trình (PPQ).
Thẩm định hiệu năng quy trình (PPQ) được dùng để kiểm định lại thiết kế quy trình được xây dựng cho việc sản xuất ở quy mô sản xuất thực tế và cần phải được thực hiện thành công trước khi sản phẩm được đưa ra lưu hành trên thị trường. Thông thường, việc nghiên cứu toàn bộ các công đoạn trong quy trình là không cần thiết nếu như các dữ liệu của bước thiết kế quy trình đã đủ để chứng minh được hiệu quả. Tuy vậy, tốt nhất là quá trình kiểm tra vẫn nên được tiến hành một cách kỹ lưỡng bằng thí nghiệm bổ sung và với số lượng mẫu kiểm tra lớn. Cần tiếp tục kiểm tra cho tới giai đoạn thẩm tra lại quy trình nếu được. Việc xem xét thời gian lấy mẫu tăng cường và thời gian theo dõi nên được dựa trên các lý giải khoa học chẳng hạn như các hiểu biết trước đó về sản phẩm, sản lượng, mức độ phức tạp của quy trình... Việc áp dụng PAT khi tiến hành Thẩm định hiệu năng quy trình (PPQ) sẽ tập trung vào hệ thống đo lường và vòng lặp kiểm soát của các đặc tính được đo lường. Tuy vậy, các phương pháp lấy mẫu mới hoặc các phép thử mới không nên được thử nghiệm ở giai đoạn này mà nên được nghiên cứu từ trước đó ở giai đoạn xây dựng quy trình. Trong quá trình này không nên để bất kỳ một chỉ tiêu nào không được tiến hành kiểm tra, đồng thời không tiến hành kiểm tra thêm bất kỳ chỉ tiêu mới nào.
Trước khi tiến hành thẩm định, cần xây dựng Đề cương thẩm định gốc (Validation Master Plan -VMP), trong đó đề cập tới triết lý và nguyên tắc, cách tiếp cận của cơ sở sản xuất khi tiến hành thẩm định/kiểm tra. Trong đề cương thẩm định tổng thể (VMP), đề cương thẩm định của giai đoạn Thẩm định quy trình sản xuất cần phải bao gồm các lưu ý về kỹ thuật để minh chứng cho mức độ hiểu biết về quy trình, cách tiếp cận và chiến lược thẩm định cũng như hiểu biết về yêu cầu đối với hồ sơ tài liệu và các tài liệu tham khảo. Đề cương Thẩm định hiệu năng quy trình (PPQ) dưới dạng văn bản cần phải được xây dựng chi tiết và được duyệt bởi bộ phận phù hợp, có đủ thẩm quyền trước khi được thực thi. Đề cương bao gồm chi tiết về các điều kiện sản xuất, kiểm soát trong quá trình, kế hoạch lấy mẫu, thử nghiệm và các kết quả dự kiến đạt được. Số lượng lô được lựa chọn và kế hoạch lấy mẫu khi tiến hành Thẩm định hiệu năng quy trình (PPQ) cần phải dựa trên các bằng chứng khoa học và chứng minh bằng các phương pháp thống kê.
Các lô được sử dụng trong Thẩm định hiệu năng quy trình (PPQ), còn được gọi là lô thích hợp, là lô được sản xuất trong điều kiện sản xuất thông thường với nhân lực đúng theo dự kiến. Báo cáo Kiểm tra hiệu năng quy trình (PPQ) cần phải tổng hợp lại các dữ liệu và các phân tích, bàn luận về bất kỳ sự thay đổi hay sai lệch quan sát được cùng với bất kỳ hành động sửa chữa, thay đổi nào cũng như kết luận cuối cùng rằng liệu quy trình có nằm trong tầm kiểm soát hay không. Nếu sau quá trình kiểm tra mà kết quả kiểm tra không đạt, cơ sở sản xuất có thể tiến hành kiểm tra lại bước thiết kế quy trình để có thể hiểu rõ hơn về quy trình và độ tin cậy trước khi tiến hành lại Kiểm tra hiệu năng quy trình (PPQ). Phần bàn luận ở bước 3 của việc Thẩm tra liên tục quy trình (Continued Process Verification) cũng phải được đưa vào báo cáo Kiểm định hiệu năng quy trình (PPQ).
4.3 Bước 3 – Thẩm tra liên tục quy trình (Continued Process Verification)
Mục đích của bước này nhằm đảm bảo liên tục rằng quy trình nằm trong tầm kiểm soát khi tiến hành sản xuất ở quy mô thực tế. Cần thiết lập Hệ thống chất lượng (Quality System) để theo dõi dữ liệu quy trình, để phát hiện bất kỳ sự thay đổi không mong muốn nào trong quy trình cũng như các hành động cần thiết phải được thực hiện. Các dữ liệu thu được bao gồm các xu hướng thay đổi của quy trình và chất lượng của nguyên liệu đầu vào, nguyên liệu trong quá trình sản xuất và thành phẩm. Việc sử dụng các phần mềm thống kê hiện đại cho phép đánh giá dữ liệu ngay lập tức (chẳng hạn như biểu đồ kiểm soát và các chỉ số hiệu năng quy trình) được khuyến khích. Các dữ liệu này nên được phân tích thống kê để thấy được xu hướng và nên được kiểm tra định kì để xác nhận rằng hệ thống vẫn hợp lệ. Việc lấy mẫu tăng cường cũng như tăng cường kiểm tra thông số quy trình và đặc tính của nguyên liệu được khuyến khích cho tới khi thu được đủ dữ liệu để đưa ra ước tính cho mức độ biến thiên. Các hoạt động nêu trên là cơ sở để thiết lập mức độ và tần suất lấy mẫu cũng như mức độ / tần suất theo dõi thường xuyên của quy trình. Sự biến thiên của quy trình cũng cần được kiểm tra lại định kì. Các dữ liệu của quá trình sản xuất cần được đánh giá lại ít nhất một năm một lần. Mức độ và tần suất đánh giá nên được quyết định dựa trên việc đánh giá các rủi ro về sản phẩm / quy trình, trong đó các thông số trọng yếu có thể ảnh hưởng tới các chỉ tiêu quan trọng của chế phẩm nên được theo dõi thường xuyên hơn. Khi các dữ liệu về sản phẩm và quy trình đã thu được một cách đầy đủ và đáng tin cậy, việc kiểm tra định kì có thể được điều chỉnh tương ứng.
(ICH-PtC) Các CQA và CPP có thể thay đổi và phát triển theo vòng đời của sản phẩm, khi mà quy trình và đặc tính sản phẩm được hiểu biết một cách cụ thể và đầy đủ hơn (ví dụ: sự thay đổi trong quá trình sản xuất, sự thay đổi về nguyên liệu ban đầu...). Khi đó, chiến lược kiểm soát để đảm bảo rằng các CQA đều được đáp ứng sẽ thay đổi trong suốt vòng đời sản phẩm. Các công ty cần phải đăng ký thay đổi nếu chiến lược kiểm soát nằm ngoài không gian thiết kế đã được chấp thuận bởi cơ quan quản lý.
5. QUY ĐỊNH NỘP TÀI LIỆU THEO YÊU CẦU CỦA HỒ SƠ KỸ THUẬT CHUNG CỦA ASEAN (ACTD)
Thông tin thu được từ các nghiên cứu phát triển dược học có thể được đưa vào hồ sơ ACTD theo nhiều cách khác nhau. Một số khuyến nghị về cách sắp xếp các thông tin này khi nộp các tài liệu đăng ký được trình bày ở dưới đây. Cơ sở đăng ký cần chỉ rõ vị trí của các thông tin đó trong tài liệu để việc tra cứu được thuận tiện.
Đối với thành phẩm thuốc, đa phần các thông tin về phát triển thành phẩm và quy trình có thể được đưa vào phần tương ứng của mục P2 của Phần II. Chẳng hạn, các thông tin về tác động của các đặc tính của API lên CQA có thể được đưa vào P2.2.1 của Phần II. Phát triển công thức và quy trình lần lượt được đưa vào P2.3 và P2.4 Phần II, trong đó bao gồm cả các thông tin về quản lý rủi ro chất lượng, thiết kế thí nghiệm và các cơ sở để thiết lập không gian thiết kế trong quá trình phát triển. Tuy vậy, không gian thiết kế được đề xuất cho quy mô sản xuất thực tế có thể được đưa vào mục P3.2 và P3.3 như là một yếu tố của quá trình sản xuất và kiểm soát quá trình. Các thông tin về thẩm định quy trình (process qualification - bước 2) ở quy mô sản xuất nên được đưa vào phần II mục P3.4. Các chiến lược kiểm soát tổng thể chất lượng thuốc thành phẩm bao gồm cả việc kiểm soát liên tục có thể được đưa vào phần II mục P5.6 nhưng thông tin chi tiết về kiểm soát nguyên liệu đầu vào (tức Phần II mục P4) và kiểm soát trong quá trình (tức Phần 2 mục P3.3) nên được đưa vào các mục tương ứng của hồ sơ ACTD.
Mặc dù các phần bàn luận nêu trên tập trung chủ yếu vào việc phát triển dược học của thuốc thành phẩm (phần II mục P2), việc thẩm định quy trình sử dụng phương pháp chất lượng theo thiết kế (QbD) cũng có thể được áp dụng cho dược chất. Đối với dược chất, quy trình tổng hợp ở quy mô nhỏ bao gồm cả việc lựa chọn nguyên liệu ban đầu, tác nhân phản ứng, thiết bị, cách thí nghiệm theo DoE cũng như cơ sở để xây dựng không gian thiết kế có thể được đưa vào phần II mục S2.6. Việc thẩm tra lại bước thẩm định quy trình ở quy mô sản xuất thực tế có thể được đưa vào phần II mục S2.5. Không gian thiết kế được đề xuất cho quy mô sản xuất thực tế nên được mô tả ở Phần II mục S2.2 và S2.4. Chiến lược kiểm soát tổng thể đối với nguyên liệu, bao gồm cả phần Thẩm tra quy trình liên tục, có thể được đưa vào phần II S4.5 nhưng thông tin chi tiết về việc kiểm soát nguyên liệu đầu vào (tức phần II mục S2.3) và kiểm soát quy trình (tức phần II mục S2.4) nên được đưa vào các mục tương ứng trong hồ sơ ACTD.
6. THUẬT NGỮ
Thông số quy trình trọng yếu - Critical Process Parameter (CPP):
Thông số quy trình mà sự biến động của thông số đó có ảnh hưởng đến Đặc tính chất lượng trọng yếu và do đó cần được theo dõi và kiểm soát để đảm bảo quy trình cho phép sản xuất ra được chất lượng mong muốn.
Đặc tính chất lượng trọng yếu - Critical Quality Attribute (CQA):
Đặc tính, có thể là vật lý, hóa học, sinh học, vi sinh cần phải nằm trong một giới hạn, một khoảng hay một phân bố nhất định để đảm bảo chất lượng mong muốn của chế phẩm.
Không gian thiết kế - Design Space (DS):
Sự kết hợp đa chiều và tương tác của các biến đầu vào (chẳng hạn như đặc tính nguyên liệu) và các thông số quy trình đã được chứng minh là sẽ đảm bảo chất lượng của sản phẩm. Khi quy trình sản xuất vẫn được tiến hành trong phạm vi của không gian thiết kế thì khi đó quy trình không bị coi là có thay đổi. Nếu quy trình được tiến hành ngoài phạm vi của không gian thiết kế thì khi đó quy trình bị coi là đã có sự thay đổi, cần tiến hành đăng ký thay đổi. Không gian thiết kế được đề xuất bởi cơ sở đăng ký và cần được sự đánh giá và chấp thuận bởi cơ quan quản lý.
Khoảng chấp nhận được chứng minh - Proven Acceptable Range (PAR):
Một khoảng cụ thể của một số thông số quy trình mà khi tiến hành hoạt động sản xuất trong khoảng này trong khi giữ các thông số còn lại không đổi thì sản phẩm được sản xuất ra đạt chỉ tiêu chất lượng.
Chất lượng theo thiết kế - Quality by Design (QbD):
Một cách tiếp cận có hệ thống của quá trình phát triển sản phẩm, bắt đầu bằng việc xác định rõ các mục tiêu và chú trọng vào việc hiểu rõ và kiểm soát quá trình, dựa trên các cơ sở khoa học và quản lý rủi ro về chất lượng.
Mục tiêu chất lượng của sản phẩm - Quality Target Product Profile (QTPP):
Một bản tóm tắt đặc tính chất lượng của một thuốc thành phẩm cần đạt được để đảm bảo chất lượng sản phẩm mong muốn, bao gồm cả tính an toàn và hiệu quả của thuốc thành phẩm.
Kiểm tra chất lượng theo thời gian thực - Real Time Release Testing (RTRT):
Khả năng đánh giá và đảm bảo chất lượng của bán thành phẩm trong quá trình sản xuất và/hoặc thành phẩm dựa trên các dữ liệu về quy trình, thường là tổ hợp của hợp lý của các đặc tính được đo lường của nguyên vật liệu và kiểm soát trong quy trình.
PHỤ LỤC D THUẬT NGỮ
Thẩm định đồng thời - Concurrent Validation:
Việc thẩm định được tiến hành đồng thời với quá trình sản xuất các sản phẩm để đưa ra lưu hành trên thị trường.
Thuốc thành phẩm - Finished Product:
Sản phẩm đã trải qua tất cả các giai đoạn của quá trình sản xuất và kiểm tra chất lượng, bao gồm cả đóng gói và dán nhãn.
Lô Pilot - Pilot Batches:
Đây là các lô được sử dụng trong quá trình phát triển và tối ưu hóa quy trình sản xuất. Quy mô của lô pilot nên tối thiểu là 10% quy mô của lô sản xuất thực tế. Với các dạng thuốc rắn dùng đường uống, cỡ lô pilot cần phải tối thiểu là 10% của lô sản xuất thực tế hoặc 100.000 đơn vị phân liều (giá trị nào lớn hơn sẽ được lựa chọn nếu không có cách lý giải nào khác).
Lô sản xuất - Production Batches:
Lô của dược chất hoặc thuốc thành phẩm được sản xuất ở quy mô sản xuất thực tế với các thiết bị sản xuất được chỉ rõ trong hồ sơ đăng ký.
Thẩm định trước - Prospective Validation:
Bằng chứng được xây dựng dưới dạng hồ sơ các cho thấy rằng quy trình, hệ thống, thiết bị hoặc nguyên tắc được sử dụng trong quá trình sản xuất đáp ứng được các mục tiêu có trong đề cương thẩm định đã được xây dựng từ trước.
Thẩm định hồi cứu - Restropective Validation:
Thẩm định quy trình sản xuất của một thuốc đã được đưa ra lưu hành trên thị trường dựa vào các dữ liệu tích lũy được từ quá trình sản xuất, từ kiểm soát và kiểm tra chất lượng các lô.
HƯỚNG DẪN CỦA ASEAN VỀ THẨM ĐỊNH QUY TRÌNH PHÂN TÍCH
Được trích dẫn theo hướng dẫn của ICH (International Conference on Harmonisation):
-ICH Q2A: Thẩm định phương pháp phân tích: Định nghĩa và thuật ngữ, ngày 27 tháng 10 năm 1994
-ICH Q2B: Thẩm định quy trình phân tích: Phương pháp luận, ngày 6 tháng 11 năm 1996.
MỤC LỤC
1- Giải thích các thuật ngữ
2- Giới thiệu
3- Các loại quy trình phân tích cần thẩm định
4- Các chỉ tiêu trong thẩm định quy trình phân tích
4.1- Tính đặc hiệu (Specificity)
4.2- Tính tuyến tính (Linearity)
4.3- Khoảng xác định (Range)
4.4- Độ đúng (Accuracy)
4.5- Độ chính xác (Precision)
4.6- Giới hạn phát hiện (Detection Limit = DL)
4.7- Giới hạn định lượng (Quantitation Limit = QL)
4.8- Độ thô (Robustness)
4.9- Kiểm tra tính thích hợp của hệ thống (System Suitability Testing)
1- GIẢI THÍCH CÁC THUẬT NGỮ
1.1 Quy trình phân tích (Analytical Procedure)
Quy trình phân tích chỉ ra cách tiến hành phân tích, trong đó mô tả chi tiết các bước cần thiết để thực hiện từng phép thử phân tích. Quy trình có thể bao gồm cách pha mẫu thử, mẫu chuẩn và thuốc thử, cách sử dụng thiết bị, cách thiết lập đường chuẩn, sử dụng công thức để tính kết quả.....nhưng không chỉ giới hạn ở những phần này.
1.2 Tính đặc hiệu (Specificity)
Tính đặc hiệu là khả năng đánh giá chắc chắn một chất phân tích khi có mặt các thành phần khác có thể có trong mẫu thử. Thông thường các thành phần này gồm các tạp chất, sản phẩm phân hủy, chất nền... Một quy trình phân tích kém đặc hiệu có thể được bổ trợ bằng một hoặc nhiều quy trình phân tích khác. Định nghĩa này có liên quan đến các phép thử sau:
+ Định tính là để khẳng định sự có mặt của chất phân tích.
+ Thử tinh khiết là để khẳng định tất cả các quy trình phân tích cho phép xác định chính xác hàm lượng tạp chất trong chất phân tích ví dụ như phép thử tạp chất liên quan, kim loại nặng, hàm lượng của dung môi tồn dư ...
+ Định lượng (hàm lượng hoặc hoạt lực) là đưa ra kết quả chính xác về hàm lượng hoặc hoạt lực của chất phân tích trong mẫu thử.
1.3 Độ đúng (Accuracy)
Độ đúng của một quy trình phân tích biểu diễn sự đồng nhất giữa giá trị tìm thấy với giá trị thực hoặc giá trị đối chiếu được chấp nhận. Đôi khi khái niệm này còn gọi là độ xác thực (trueness).
1.4 Độ chính xác (Precision)
Độ chính xác của một quy trình phân tích diễn tả sự thống nhất (mức độ phân tán) kết quả giữa một loạt phép đo từ nhiều lần lấy mẫu trên cùng một mẫu thử đồng nhất dưới những điều kiện mô tả. Độ chính xác có thể chia thành 3 cấp: độ lặp lại, độ chính xác trung gian và độ tái lặp. Độ chính xác nên được thử trên một mẫu thử thực, đồng nhất. Tuy nhiên, nếu không có mẫu đồng nhất thì có thể dùng mẫu tự tạo hoặc một dung dịch mẫu thử. Độ chính xác thường được biểu thị dưới dạng độ dao động, độ lệch chuẩn hoặc hệ số độ dao động của một loạt phép đo.
1.4.1-Độ lặp lại (Repeatability)
Độ lặp lại diễn tả độ chính xác của một quy trình phân tích trong cùng điều kiện thí nghiệm trong khoảng thời gian ngắn. Độ lặp lại còn được gọi là độ chính xác trong cùng điều kiện định lượng.
1.4.2- Độ chính xác trung gian (Intermediate precision)
Độ chính xác trung gian diễn tả mức dao động của kết quả trong cùng một phòng thí nghiệm được thực hiện ở các ngày khác nhau, kiểm nghiệm viên khác nhau và thiết bị khác nhau.
1.4.3- Độ tái lặp (Reproducibility)
Độ tái lặp diễn tả độ chính xác giữa các phòng thí nghiệm (Các nghiên cứu phối hợp giữa các phòng thí nghiệm thường được áp dụng để tiêu chuẩn hóa phương pháp).
1.5 Giới hạn phát hiện (Detection Limit )
Giới hạn phát hiện của một quy trình phân tích là lượng nhỏ nhất của chất phân tích trong mẫu thử có thể phát hiện được nhưng không nhất thiết để có thể định lượng được.
1.6 Giới hạn định lượng (Quantitation Limit)
Giới hạn định lượng của một quy trình phân tích là lượng nhỏ nhất của chất phân tích trong mẫu thử để có thể định lượng được với độ đúng và độ chính xác thích hợp. Giới hạn định lượng là một thông số của phép định lượng các chất có nồng độ thấp trong mẫu thử, đặc biệt thường được dùng để xác định tạp chất và/hoặc sản phẩm phân hủy.
1.7 Tính tuyến tính (Linearity)
Tính tuyến tính của một quy trình phân tích diễn tả kết quả phân tích thu được tỷ lệ với nồng độ (trong khoảng nhất định) của chất phân tích trong mẫu thử.
1.8 Khoảng xác định (range)
Khoảng xác định của một quy trình phân tích là khoảng cách giữa nồng độ trên và dưới của chất phân tích trong mẫu thử (bao gồm cả các nồng độ này), trong khoảng nồng độ này, quy trình phân tích đã được chứng minh đáp ứng độ chính xác, độ đúng và tính tuyến tính.
1.9 Độ thô (Robustness)
Độ thô của quy trình phân tích nhằm đánh giá khả năng duy trì của quy trình phân tích không bị ảnh hưởng bởi những biến đổi nhỏ nhưng có tính chủ định trong các thông số của phương pháp và chỉ ra mức tin cậy của quy trình trong điều kiện sử dụng bình thường.
2- GIỚI THIỆU
Việc thẩm định quy trình phân tích là nhằm chứng minh quy trình đó có phù hợp với mục đích ứng dụng không.
Bản hướng dẫn này đưa ra các các hướng dẫn và gợi ý cho việc đánh giá các quy trình phân tích dùng trong hồ sơ đăng ký thuốc ở khu vực ASEAN. Tài liệu này chủ yếu được trích dẫn từ hai bản hướng dẫn của ICH “Q2A: Thẩm định phương pháp phân tích: Định nghĩa và thuật ngữ, ngày 27 tháng 10 năm 1994” và “Q2B: Thẩm định quy trình phân tích: Phương pháp luận, ngày 6 tháng 11 năm 1996”. Hệ phương pháp áp dụng cho các chế phẩm sinh học và công nghệ sinh học có thể khác so với các chế phẩm hóa học.
Tất cả các số liệu liên quan thu được trong quá trình thẩm định và các công thức được sử dụng để tính toán các đại lượng đặc trưng của việc thẩm định cần được đưa ra và thảo luận. Các chất đối chiếu được sử dụng trong quá trình thẩm định cần phải được đánh giá rõ ràng và kèm theo tài liệu về độ tinh khiết. Mức độ tinh khiết phụ thuộc vào mục đích sử dụng.
Trong thực tế, thường có thể phác thảo công việc thực nghiệm nhằm xem xét tiến hành đánh giá một cách thích hợp đồng thời nhiều thuộc tính để đưa ra những hiểu biết về khả năng của một quy trình phân tích, ví dụ: tính đặc hiệu, tuyến tính, khoảng xác định, độ đúng và độ chính xác. Những phương pháp phân tích theo dược điển không yêu cầu thẩm định nhưng tính phù hợp của chúng phải được kiểm chứng lại ở điều kiện sử dụng thực tế.
Theo yêu cầu của ASEAN : tất cả các dữ liệu liên quan đến các chỉ tiêu thẩm định cùng với các chỉ tiêu chấp nhận tương ứng phải nộp cho cơ quan quan lý dược phẩm.
3- CÁC LOẠI QUY TRÌNH PHÂN TÍCH CẦN THẨM ĐỊNH
Thẩm định quy trình phân tích liên quan đến 4 loại quy trình chung sau đây:
+ Định tính
+ Định lượng hàm lượng các tạp chất
+ Phép thử giới hạn tạp chất
+ Định lượng các hoạt chất trong mẫu nguyên liệu hoặc thành phẩm thuốc hoặc một hay nhiều thành phần được chọn khác trong thành phẩm thuốc.
Sau đây là mô tả ngắn gọn các loại phép thử được đề cập trong tài liệu này:
- Định tính: nhằm để khẳng định sự có mặt của các chất phân tích trong mẫu thử. Thông thường được thực hiện bằng cách so sánh các kết quả phân tích (ví dụ như : phổ đồ, đáp ứng sắc ký, phản ứng hóa học, vv....) của mẫu thử với chất chuẩn.
- Phép thử tạp chất: có thể là định lượng hoặc thử giới hạn tạp chất trong mẫu thử, nhưng đều nhằm mục đích phản ánh chính xác mức độ tinh khiết của mẫu thử. So với phép thử giới hạn tạp chất thì phép thử định lượng tạp chất còn yêu cầu thêm một số chỉ tiêu thẩm định khác.
- Định lượng: nhằm mục đích đo lượng chất phân tích có mặt trong mẫu thử. Trong tài liệu này, định lượng được hiểu là phép đo hàm lượng một hoặc nhiều thành phần chính của dược chất. Đối với thành phẩm thuốc, những chỉ tiêu thẩm định tương tự cũng được áp dụng khi định lượng các hoạt chất hoặc một hay nhiều thành phần được lựa chọn khác. Các chỉ tiêu thẩm định này cũng có thể áp dụng cho các phép định lượng liên quan đến các quy trình phân tích khác (ví dụ thử độ hòa tan).
Mục đích của quy trình phân tích phải được hiểu rõ ràng vì điều này sẽ quyết định những chỉ tiêu cần được đánh giá. Danh mục sau đây chỉ ra các chỉ tiêu điển hình trong thẩm định cần được xem xét:
Độ đúng
Độ chính xác
Độ lặp lại
Độ chính xác trung gian
Độ tái lặp
Tính đặc hiệu
Giới hạn phát hiện (DL
Giới hạn định lượng (QL)
Tính tuyến tính
Khoảng xác định
Độ thô
Các chỉ tiêu thẩm định này đã được định nghĩa trong phần giải thích các thuật ngữ. Bảng dưới đây liệt kê các chỉ tiêu được xem là quan trọng nhất cho việc thẩm định các loại quy trình phân tích khác nhau. Danh mục này được xem là điển hình đối với các quy trình phân tích đã nêu, tuy nhiên các trường hợp ngoại lệ phải được giải quyết theo từng trường hợp cụ thể. Cần chú ý rằng độ thô không được liệt kê trong bảng dưới đây nhưng cần được xem xét đến ở các giai đoạn thích hợp trong quá trình phát triển quy trình phân tích.
Ngoài ra việc thẩm định lại quy trình phân tích có thể cần thiết trong các trường hợp dưới đây:
- Thay đổi trong khâu tổng hợp dược chất
- Thay đổi thành phần của thành phẩm.
- Thay đổi quy trình phân tích.
Mức độ thẩm định lại được yêu cầu tuỳ thuộc vào bản chất của sự thay đổi. Một số thay đổi khác cũng có thể yêu cầu phải thẩm định lại.
|
Loại quy trình phân tích Các chỉ tiêu |
Định tính |
Xác định tạp chất |
Định lượng: - Độ hòa tan - Hàm lượng/ hoạt lực |
|
|
Định lượng |
Thử giới hạn |
|||
|
- Độ đúng - Độ chính xác + Độ lặp lại + Độ chính xác trung gian - Tính đặc hiệu (2) - Giới hạn phát hiện(LOD) - Giới hạn định lượng (LOQ) - Tính tuyến tính - Khoảng xác định |
-
- - + - - - - |
+
+ +(1) + -(3) + + + |
-
- - + + - - - |
+
+ +(1) + - - + + |
Dấu - nhằm chỉ các chỉ tiêu này thông thường không cần phải đánh giá.
Dấu + nhằm chỉ các chỉ tiêu này cần phải đánh giá
(1) trong trường hợp đã tiến hành kiểm tra độ tái lặp thì độ chính xác trung gian không cần phải xem xét.
(2) một quy trình phân tích kém đặc hiệu có thể được bổ trợ bằng một hay nhiều quy trình phân tích hỗ trợ khác.
(3) có thể cần trong một số trường hợp .
4- CÁC CHỈ TIÊU TRONG THẨM ĐỊNH QUY TRÌNH PHÂN TÍCH
4.1 Tính đặc hiệu.
Việc xác định tính đặc hiệu cần thiết được tiến hành trong khi thẩm định các phép thử định tính, xác định tạp chất và định lượng. Quy trình dùng để xác định tính đặc hiệu phụ thuộc vào mục tiêu đã định của quy trình phân tích. Không phải lúc nào cũng xác định được một quy trình phân tích đặc hiệu cho một chất phân tích nhất định (phân biệt hoàn toàn). Trong trường hợp này, cần thiết phải kết hợp hai hay nhiều quy trình phân tích để đạt được mức độ đặc hiệu cần thiết.
4.1.1- Định tính.
Những phép thử định tính phù hợp là phép thử có thể phân biệt được các hợp chất có cấu trúc tương tự cùng có mặt trong mẫu thử. Khả năng phân biệt của một quy trình định tính có thể được khẳng định bằng kết quả dương tính của mẫu có chứa chất phân tích (có thể bằng cách so sánh với chất đối chiếu đã biết) kết hợp với kết quả âm tính của mẫu thử không chứa chất phân tích. Thêm vào đó, phép thử định tính này có thể được áp dụng cho các chất có cấu trúc tương tự hoặc gần với với cấu trúc của chất phân tích để chứng tỏ phép thử định tính không cho kết quả dương tính với các chất này. Việc lựa chọn xem chất nào có khả năng lẫn vào mẫu phân tích cần dựa trên những đánh giá khoa học kết hợp cân nhắc xem chúng có khả năng có mặt không.
4.1.2- Định lượng và thử tạp chất.
Đối với quy trình sắc ký, các sắc ký đồ đại diện nên được sử dụng để chứng minh tính đặc hiệu và từng thành phần riêng biệt phải được ghi lại rõ ràng. Với những kỹ thuật phân tách khác cũng cần phải có những ghi chép tương tự. Giới hạn của phân tách trong sắc ký cần phải được xem xét ở mức độ phù hợp. Đối với giới hạn của phân tách trong sắc ký, chia tách quan trọng, tính đặc hiệu có thể được chứng minh bằng độ phân giải của hai thành phần được rửa giải gần nhau nhất. Trong trường hợp sử dụng phép định lượng không đặc hiệu, thì cần dùng các quy trình phân tích hỗ trợ khác để chứng minh tính đặc hiệu của chúng, ví dụ nếu dùng phương pháp chuẩn độ thể tích để định lượng các nguyên liệu khi xuất xưởng, thì có thể kết hợp phép định lượng này với phép thử tạp chất thích hợp. Cách đánh giá đều giống nhau đối với cả phép định lượng và thử tạp chất bao gồm:
4.1.2.1- Những tạp chất sẵn có.
Đối với phép định lượng, cần phải chứng minh phương pháp đã dùng phân biệt được chất cần phân tích khi có mặt của tạp chất và/hoặc các tá dược; trong thực tế, có thể thực hiện bằng cách thêm một lượng thích hợp tạp chất và/hoặc tá dược vào mẫu ban đầu cần định lượng (nguyên liệu hoặc thành phẩm) và chứng minh rằng kết quả định lượng không bị ảnh hưởng bởi sự có mặt của tạp chất và/hoặc tá dược (bằng cách so sánh với kết quả định lượng trên mẫu không thêm tạp chất và/hoặc tá dược).
Đối với phép thử tạp chất, sự phân biệt này có thể được thiết lập bằng cách thêm vào nguyên liệu hoặc thành phẩm một lượng thích hợp các tạp chất và chứng minh rằng từng tạp chất riêng biệt này được tách riêng rẽ ra khỏi nhau và/hoặc ra khỏi các thành phần khác có trong mẫu.
4.1.2.2- Những tạp chất không có sẵn.
Nếu không có tạp chất hoặc sản phẩm phân hủy chuẩn, tính đặc hiệu có thể được chứng minh bằng cách so sánh kết quả phân tích trên mẫu thử có chứa tạp chất hoặc các sản phẩm phân hủy bằng quy trình phân tích đã xây dựng với kết quả phân tích trên mẫu thử có chứa tạp chất hoặc chất phân hủy bằng quy trình chính thống khác ví dụ như phương pháp dược điển hoặc quy trình phân tích khác đã được thẩm định (quy trình độc lập). Nếu cần, thì bao gồm cả so sánh trên mẫu được lưu trữ ở các điều kiện khắc nghiệt có liên quan như: ánh sáng, nhiệt độ, độ ẩm, thủy phân bằng acid/kiềm và oxi hóa.
- Để định lượng cần so sánh hai kết quả
- Để thử tạp chất cần so sánh các hồ sơ tạp chất đã thu được.
Các phép thử độ tinh khiết của đỉnh cũng rất hữu ích để chỉ ra rằng đỉnh sắc ký của chất phân tích không chứa nhiều hơn một thành phần (ví dụ phép thử độ tinh khiết bằng detector dãy di-ốt, detector khối phổ).
4.2 Tính tuyến tính.
Cần đánh giá mối tương quan tuyến tính trong khoảng xác định (xem mục 4.3) của quy trình phân tích. Tuyến tính có thể thực hiện trực tiếp trên mẫu chuẩn (bằng cách pha loãng dung dịch chuẩn gốc) và/hoặc cân riêng biệt các hỗn hợp tự tạo chứa các thành phần dược chất dựa trên quy trình đã đặt ra. Cách sau có thể được dùng để nghiên cứu khoảng phân tích. Tính tuyến tính được đánh giá bằng cách quan sát đồ thị của tín hiệu ứng với nồng độ hoặc hàm lượng của chất phân tích. Nếu có tương quan tuyến tính thì kết quả thử phải được đánh giá băng phương pháp thống kê thích hợp, ví dụ bằng cách tính đường hồi quy dựa vào phương pháp bình phương tối thiểu. Trong một số trường hợp, để có được mối tương quan tuyến tính giữa định lượng và nồng độ của mẫu thử, các số liệu phân tích thu được cần phải qua một bước biến đổi toán học trước khi phân tích hồi quy. Các số liệu từ đường hồi quy có thể giúp đưa ra ước lượng toán học về mức độ tuyến tính. Cần phải đưa ra hệ số tương quan, giao điểm với trục tung, độ dốc của đường hồi quy và tổng hiệu các bình phương . Đồ thị của các số liệu cũng cần được đưa ra. Thêm vào đó, việc phân tích độ lệch khỏi đường hồi qui của các điểm dữ liệu thực tế cũng hữu ích cho việc đánh giá độ tuyến tính .
Một số quy trình phân tích như định lượng miễn dịch không thể hiện tính tuyến tính sau bất kỳ phép biến đổi nào. Trong trường hợp này, cần có một hàm thích hợp để biểu thị mối liên quan giữa đáp ứng thu được với nồng độ (lượng) chất phân tích trong mẫu.
Để xác định tính tuyến tính cần tiến hành ít nhất 5 nồng độ. Trong những trường hợp khác, cần nêu rõ lí do.
4.3 Khoảng xác định
Khoảng xác định thường được lấy từ những nghiên cứu tính tuyến tính và phụ thuộc vào việc ứng dụng dự định của quy trình. Khoảng xác định được thiết lập bởi việc khẳng định quy trình phân tích đã xây dựng có tính tuyến tính, độ đúng và độ chính xác chấp nhận được khi áp dụng để định lượng mẫu thử chứa chất phân tích với hàm lượng nằm trong khoảng hoặc ở 2 cực (cực đại và cực tiểu) của khoảng xác định của quy trình phân tích.
Sau đây là các khoảng xác định tối thiểu cần được cân nhắc:
- Để định lượng nguyên liệu hoặc thành phẩm thuốc: Thường từ 80 -120% của nồng độ thử.
- Đối với độ đồng đều hàm lượng: Trong khoảng từ 70 -130% nồng độ thử trừ trường hợp do bản chất của dạng bào chế (ví dụ ống hít định liều) thì cần khoảng xác định thích hợp rộng hơn.
- Để thử độ hòa tan: ± 20% khoảng quy định trong tiêu chuẩn, ví dụ nếu tiêu chuẩn yêu cầu cho chế phẩm giải phóng hoạt chất có kiểm soát là phải giải phóng hoạt chất trên một khoảng từ 20% sau 1 giờ đến 90% sau 24 giờ thì khoảng được đánh giá là từ 0% đến 110% hàm lượng ghi trên nhãn.
- Để xác định tạp chất: Từ giới hạn cho phép của một tạp chất1 đến 120% của tiêu chuẩn; đối với các tạp chất đã biết có độc tính bất thường hoặc sinh ra độc tính hoặc có tác dụng dược lý không mong muốn thì giới hạn phát hiện (LOD) và giới hạn định lượng (LOQ) của tạp chất phải tương ứng với giới hạn mà tạp chất đó cần được kiểm soát.
Ghi chú: Để thẩm định quy trình thử tạp chất được tiến hành trong phát triển sản phẩm có thể cần thiết phải cân nhắc khoảng xác định xung quanh một giới hạn đã được gợi ý.
Nếu định lượng và phép thử tinh khiết được thực hiện đồng thời trên cùng một phép thử và chỉ sử dụng một chuẩn 100% thì tính tuyến tính cần phải phủ toàn bộ khoảng xác định từ giới hạn cho phép tạp chất[1] đến 120% của tiêu chuẩn định lượng.
4.4 Độ đúng
Độ đúng cần được thiết lập trong khoảng phân tích xác định của quy trình phân tích.
4.4.1 Định lượng
4.4.1.1- Nguyên liệu
Một số phương pháp xác định độ đúng:
a- Áp dụng quy trình phân tích đối với chất phân tích đã biết rõ độ tinh khiết (ví dụ chất đối chiếu)
b- So sánh các kết quả của quy trình phân tích được đề xuất với kết quả của quy trình phân tích chính thống có độ đúng đã được công bố và/hoặc đã được xác định (quy trình độc lập xem mục 4.1.2)
c- Độ đúng có thể được suy ra một khi độ chính xác, tính tuyến tính và tính đặc hiệu đã được thiết lập.
4.4.1.2- Thành phẩm thuốc
Một số phương pháp xác định độ đúng:
a- Áp dụng quy trình phân tích đối với hỗn hợp mẫu tự tạo chứa các thành phần của thành phẩm thuốc mà trong đó có một lượng đã biết trước các dược chất cần phân tích được thêm vào.
b- Trong trường hợp không có đầy đủ các thành phần để làm mẫu tự tạo thì có thể chấp nhận cho thêm một lượng đã biết của chất cần phân tích vào chế phẩm hoặc so sánh kết quả thu được với một quy trình chính thống thứ hai có độ đúng đã được công bố và/ hoặc đã được xác định (quy trình độc lập xem mục 4.1.2)
c- Độ đúng có thể được suy ra một khi độ chính xác, tính tuyến tính và tính đặc hiệu đã được thiết lập.
4.4.2 Tạp chất (định lượng)
Độ đúng phải được tiến hành trên các mẫu thử (nguyên liệu hoặc thành phẩm thuốc) đã được thêm một lượng tạp chuẩn đã biết. Trong trường hợp không có tạp và/hoặc sản phẩm phân hủy chuẩn thì có thể chấp nhận so sánh kết quả thu được với một quy trình độc lập (xem mục 4.1.2). Hệ số đáp ứng của hoạt chất cũng có thể được sử dụng.
Trong mọi trường hợp, cần phải xác định rõ từng tạp chất hoặc tổng các tạp chất được tính như thế nào so với chất phân tích chính (ví dụ khối lượng/ khối lượng hoặc phần trăm diện tích).
4.4.3 Các dữ liệu cần có
Độ đúng phải được tính dựa trên tối thiểu 9 lần định lượng trên ít nhất 3 mức nồng độ khác nhau trong khoảng nồng độ đã được xác định của quy trình phân tích (ví dụ 3 nồng độ, mỗi nồng độ được tiến hành 3 lần).
Độ đúng được biểu thị dưới dạng phần trăm tìm thấy của chất phân tích trước đã biết được thêm vào mẫu thử hoặc độ lệch giữa giá trị trung bình đo được và giá trị thực cùng với khoảng tin cậy.
4.5 Độ chính xác
Thẩm định các phép thử định lượng và các phép thử xác định hàm lượng tạp chất cần xác định độ chính xác.
4.5.1 Độ lặp lại. (Repeatability)
Độ lặp lại có thể được đánh giá trên kết quả của:
a- Tối thiểu 9 lần định lượng trong khoảng nồng độ đã được xác định của quy trình (ví dụ 3 nồng độ, mỗi nồng độ được tiến hành 3 lần) hoặc
b- Tối thiểu 6 lần định lượng ở nồng độ thử 100%.
4.5.2 Độ chính xác trung gian( Intermediate Precision)
Việc xác định độ chính xác trung gian phụ thuộc vào tình hình cụ thể đối với từng quy trình phân tích được áp dụng. Cơ sở đăng ký cần chỉ ra ảnh hưởng của các biến cố ngẫu nhiên đến độ chính xác của quy trình phân tích. Những thay đổi điển hình cần xem xét bao gồm: ngày phân tích, kiểm nghiệm viên, thiết bị, v.v....Thực tế không cần phải nghiên cứu những ảnh hưởng này một cách riêng rẽ. Khuyến khích sử dụng thiết kế thực nghiệm (ma trận).
4.5.3 Độ tái lặp (Reproducibility).
Độ tái lặp được xác định bằng cách so sánh kết quả giữa các phòng thí nghiệm.
Độ tái lặp được tiến hành đánh giá trong trường hợp tiêu chuẩn hóa quy trình phân tích ví dụ như đối với các quy trình trong dược điển. Những số liệu này không nằm trong hồ sơ đăng ký thuốc.
4.5.4 Dữ liệu cần có.
Độ chính xác của mỗi một quy trình cần phải đưa ra các dữ liệu sau: Độ lệch chuẩn (standart deviation), độ lệch chuẩn tương đối (Relative standart deviation hay hệ số biến thiên = coefficient of variation) và khoảng tin cậy.
4.6 Giới hạn phát hiện (Detection Limit= DL)
Phương pháp xác định giới hạn phát hiện tuỳ thuộc vào quy trình phân tích là phương pháp phân tích dụng cụ hay không dụng cụ. Ngoài các phương pháp nêu ra ở dưới đây, các phương pháp khác cũng có thể được chấp nhận để xác định giới hạn phát hiện.
4.6.1 Dựa vào quan sát.
Phương pháp này thường dùng cho phương pháp phân tích không dụng cụ, nhưng cũng có thể dùng cho các phương pháp phân tích dụng cụ.
Giới hạn phát hiện được xác định bằng phân tích mẫu thử có chất phân tích đã biết nồng độ và xác định nồng độ tối thiểu mà tại đó có thể đọc được đáp ứng của chất phân tích.
4.6.2 Dựa vào tỉ lệ đáp ứng so với nhiễu.
Phương pháp này chỉ có thể áp dụng cho những phương pháp phân tích có nhiễu đường nền.
Việc xác định tỉ lệ đáp ứng trên nhiễu được tiến hành bằng cách so sánh đáp ứng đo được trên mẫu thử có nồng độ chất phân tích thấp đã biết với đáp ứng của mẫu trắng và từ đó tính được nồng độ tối thiểu của chất phân tích có thể phát hiện được. Tỷ lệ đáp ứng trên nhiễu nằm giữa 3:1 hoặc 2:1 thường được chấp nhận để thiết lập giới hạn phát hiện.
4.6.3 Dựa vào độ lệch chuẩn của đáp ứng và độ dốc.
Giới hạn phát hiện (DL) có thể được tính như sau:
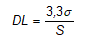
Trong đó: σ là độ lệch chuẩn của đáp ứng
S là độ dốc của đường chuẩn
độ dốc S có thể được tính dựa vào đường chuẩn của chất phân tích. Có thể xác định S theo nhiều cách khác , ví dụ như:
4.6.3.1-Dựa vào độ lệch chuẩn của mẫu trắng
Tiến hành một số lượng thích hợp các phân tích trên mẫu trắng, đo đáp ứng nền và tính độ lệch chuẩn của các đáp ứng này.
4.6.3.2- Dựa vào đường chuẩn
Dựa vào đường chuẩn đặc trưng của mẫu thử có chứa chất phân tích có nồng độ nằm trong khoảng DL. Số dư độ lệch chuẩn của đường hồi quy hoặc độ lệch chuẩn của giá trị giao điểm với trục tung của đường hồi quy có thể được sử dụng như là độ lệch chuẩn.
4.6.4 Các dữ liệu cần có
Cần đưa ra giới hạn phát hiện (DL) và cách xác định giới hạn phát hiện. Nếu DL được xác định dựa vào quan sát hoặc dựa vào tỷ lệ đáp ứng trên nhiễu thì cần đưa ra các sắc ký đồ có liên quan. Trong trường hợp ước tính giá trị DL bằng tính toán hoặc bằng ngoại suy thì sau đó những ước tính này cần được đánh giá bằng cách phân tích độc lập một số lượng mẫu thử thích hợp có nồng độ đã biết gần với giới hạn phát hiện hoặc bằng giới hạn phát hiện.
4.7 Giới hạn định lượng (Quantitation Limit = QL)
Phương pháp xác định giới hạn định lượng (QL) tuỳ thuộc vào quy trình phân tích là phương pháp phân tích dụng cụ hay không dụng cụ. Ngoài các phương pháp nêu ra ở dưới đây, các phương pháp khác cũng có thể được chấp nhận để xác định giới hạn định lượng.
4.7.1 Dựa vào quan sát
Phương pháp này thường dùng cho phương pháp phân tích không dụng cụ, nhưng cũng có thể dùng cho các phương pháp phân tích dụng cụ.
Giới hạn định lượng (QL) thường được xác định bằng phân tích mẫu thử có chất phân tích đã biết nồng độ và xác định nồng độ tối thiểu mà tại đó có thể định lượng được chất cần phân tích với độ đúng và độ chính xác có thể chấp nhận được.
4.7.2 Dựa vào tỉ lệ đáp ứng so với nhiễu
Phương pháp này chỉ dùng cho những phương pháp phân tích có thấy được sự nhiễu đường nền.
Việc xác định tỉ lệ đáp ứng trên nhiễu được tiến hành bằng cách so sánh đáp ứng đo được trên mẫu thử có nồng độ chất phân tích thấp đã biết so với đáp ứng của mẫu trắng và xác định nồng độ tối thiểu của chất phân tích có thể định lượng được. Tỷ lệ đáp ứng trên nhiễu thông thường là 10:1
4.7.3 Dựa vào độ lệch chuẩn của đáp ứng và độ dốc
Giới hạn định lượng (QL) có thể được tính như sau:
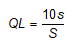
Trong đó: s là độ lệch chuẩn của đáp ứng
S là độ dốc của đường chuẩn
Độ dốc S có thể được tính dựa vào đường chuẩn của chất phân tích. Cũng có thể được tính theo nhiều cách khác ví dụ như:
4.7.3.1-Dựa vào độ lệch chuẩn của mẫu trắng
Tiến hành một số lượng thích hợp các phân tích trên mẫu trắng, đo dao động của đáp ứng nền và tính độ lệch chuẩn của các đáp ứng này.
4.7.3.2- Dựa vào đường chuẩn
Xây dựng đường chuẩn đặc trưng của mẫu thử có chứa chất phân tích có nồng độ nằm gần trong khoảng QL. Số dư độ lệch chuẩn của đường hồi quy hoặc độ lệch chuẩn của các giá trị giao điểm với trục tung của đường hồi quy có thể được sử dụng như là độ lệch chuẩn.
4.7.4 Các dữ liệu cần có
Cần đưa ra giới hạn định lượng (QL) và cách xác định giới hạn định lượng.
Giới hạn này sau đó cần được đánh giá bằng cách phân tích một số lượng mẫu thử thích hợp có nồng độ đã biết gần với giới hạn định lượng hoặc bằng giới hạn định lượng.
4.8 Độ thô (Robustness)
Việc đánh giá độ thô cần được xem xét trong giai đoạn phát triển phương pháp và tuỳ thuộc vào loại quy trình phân tích đang nghiên cứu. Độ thô chỉ ra được mức độ tin cậy của phương pháp khi có những thay đổi nhỏ có chủ định của các thông số của phương pháp. Nếu những phép đo nhạy cảm với những thay đổi điều kiện phân tích, thì điều kiện phân tích cần được kiểm soát thích hợp hoặc chỉ dẫn những điểm cần lưu ý trong quá trình phân tích. Kết quả đánh giá độ thô là kết quả đánh giá dãy các thông số phản ánh tính thích hợp của hệ thống (ví dụ phép thử độ phân giải) phải được thiết lập để đảm bảo duy trì được tính hiệu lực của quy trình phân tích bất kỳ khi nào sử dụng. Những ví dụ của các biến đổi thường gặp trong phân tích là:
- tính ổn định của các dung dịch phân tích
- thời gian chiết
Trong trường hợp sắc ký lỏng, các biến đổi thường gặp là:
- Ảnh hưởng của sự thay đổi pH trong pha động
- Ảnh hưởng của sự thay đổi thành phần trong pha động
- Các cột khác nhau (do nhà cung cấp và /hoặc lô khác nhau)
- Nhiệt độ
- Tốc độ dòng
Trong trường hợp sắc ký khí, các biến đổi thường gặp là:
- Các cột khác nhau (do nhà cung cấp và /hoặc lô khác nhau)
- Nhiệt độ
- Tốc độ dòng
4.9 Phép thử tính thích hợp của hệ thống
Kiểm tra tính tương thích hệ thống là một phần không thể tách rời trong nhiều quy trình phân tích. Đánh giá tính thích hợp của hệ thống là những phép thử nhằm đánh giá tính thích hợp của toàn hệ thống phân tích được cấu thành bởi các yếu tố như máy móc thiết bị, hệ thống điện, cách tiến hành phân tích và mẫu thử. Các thông số của phép thử tính tương thích của hệ thống được thiết lập cho từng quy trình riêng biệt phụ thuộc vào loại quy trình được thẩm định. Các thông số này đặc biệt quan trọng trong các phương pháp sắc ký, xem các dược điển để có thêm thông tin.
HƯỚNG DẪN THỰC HIỆN NGHIÊN CỨU TƯƠNG ĐƯƠNG SINH HỌC ASEAN
Được xây dựng trên cơ sở:
“HƯỚNG DẪN NGHIÊN CỨU TƯƠNG ĐƯƠNG SINH HỌC” (Cơ quan quản lý Dược phẩm châu Âu, London, ngày 20 tháng 1 năm 2010, CPMP/EWP/QWP/1401/98 Rev 1)
với một số thay đổi thích hợp để áp dụng tại các nước Asean.
Kiểm soát tài liệu
|
Phiên bản |
Ngày |
|
Chính thức |
Tháng 7 năm 2004 (Hội nghị PPWG lần thứ 8, Băng Cốc) |
|
Hiệu đính 1, Dự thảo 1 |
Tháng 6 năm 2011, Singapore |
|
Hiệu đính 1, Dự thảo 2 |
Tháng 7 năm 2012, Băng cốc, Thái Lan |
|
Hiệu đính 1, Dự thảo 3 |
Tháng 5 năm 2013, Bali, Indonesia |
|
Hiệu đính 1, Dự thảo 4 |
Tháng 6 năm 2014, Kuala Lumpur, Malaysia |
|
Hiệu đính 1, Dự thảo 4 Cuối cùng |
Tháng 3 năm 2015 , Viêng chăn, Lào |
MỤC LỤC
TÓM TẮT
1. GIỚI THIỆU
1.1 TỔNG QUAN
1.2 CÁC THUỐC GENERIC
1.3 CÁC LOẠI ĐĂNG KÝ KHÁC
2. PHẠM VI
3. NỘI DUNG CHÍNH CỦA HƯỚNG DẪN
3.1 THIẾT KẾ, THỰC HIỆN VÀ ĐÁNH GIÁ CÁC NGHIÊN CỨU TƯƠNG ĐƯƠNG SINH HỌC
3.1.1 Thiết kế nghiên cứu
3.1.2 Chế phẩm đối chiếu và chế phẩm thử
3.1.3 Người tình nguyện
3.1.4 Tiến hành nghiên cứu
3.1.5 Các thông số cần nghiên cứu
3.1.6 Hàm lượng nghiên cứu
3.1.7 Phương pháp phân tích sinh học
3.1.9 Thuốc có khoảng điều trị hẹp
3.1.10 Các thuốc hoặc chế phẩm thuốc có tính biến thiên cao
3.2 ĐÁNH GIÁ ĐỘ HÒA TAN INVITRO
3.2.1 Đánh giá độ hòa tan in vitro bổ sung cho các nghiên cứu tương đương sinh học
3.2.2 Đánh giá độ hòa tan in vitro để cung cấp dữ liệu xem xét miễn thử hàm lượng khác
3.3 BÁO CÁO NGHIÊN CỨU
3.3.1 Báo cáo nghiên cứu tương đương sinh học
3.3.2 Các dữ liệu khác cần đưa vào hồ sơ đăng ký
3.4 ĐĂNG KÝ THAY ĐỔI
CÁC KHÁI NIỆM
PHỤ LỤC I: THỬ ĐỘ HÒA TAN VÀ TÍNH TƯƠNG ĐỒNG CỦA BIỂU ĐỒ HÒA TAN
PHỤ LỤC II: YÊU CẦU VỀ NGHIÊN CỨU TƯƠNG ĐƯƠNG SINH HỌC CHO CÁC DẠNG BÀO CHẾ KHÁC
PHỤ LỤC III: MIỄN THỬ SINH HỌC DỰA TRÊN BCS
PHỤ LỤC IV: Mẫu báo cáo nghiên cứu tương đương sinh học ASEAN
TÓM TẮT
Hướng dẫn này đưa ra các yêu cầu về thiết kế, thực hiện và đánh giá, nghiên cứu tương đương sinh học đối với thuốc ở dạng bào chế giải phóng ngay có tác dụng toàn thân.
1. GIỚI THIỆU
1.1 Tổng quan
Hai chế phẩm thuốc chứa cùng một dược chất được coi là tương đương sinh học nếu hai chế phẩm này tương đương về mặt bào chế hoặc là những thế phẩm bào chế mà sinh khả dụng (mức độ và tốc độ hấp thu) của hai thuốc sau khi sử dụng với cùng một mức liều (tính theo lượng dược chất) nằm trong giới hạn chấp nhận đã định trước. Giới hạn này được thiết lập để đảm bảo các thuốc thể hiện đặc tính in vivo có thể so sánh được, tức là tương đương về hiệu quả và độ an toàn.
Trong các nghiên cứu tương đương sinh học, đường cong nồng độ thuốc trong huyết tương theo thời gian thường được sử dụng để đánh giá tốc độ và mức độ hấp thu. Các thông số dược động học đặc trưng và các giới hạn chấp nhận định trước là những cơ sở để đưa ra quyết định cuối cùng về sự tương đương sinh học của các thuốc đã thử. AUC (diện tích dưới đường cong biểu diễn nồng độ thuốc theo thời gian) phản ảnh mức độ phơi nhiễm thuốc. Cmax (nồng độ thuốc tối đa trong huyết tương hoặc đỉnh phơi nhiễm) và tmax (thời gian để thuốc đạt nồng độ tối đa trong huyết tương) là các thông số phản ánh tốc độ hấp thu.
Mục đích của hướng dẫn này là đưa ra những yêu cầu về thiết kế, thực hiện và đánh giá các nghiên cứu tương đương sinh học. Khả năng sử dụng các nghiên cứu in vitro để thay thế cho các nghiên cứu in vivo cũng được đề cập.
1.2 Các thuốc generic
Trong các hồ sơ đăng ký thuốc generic, dữ liệu tương đương sinh học là một dữ liệu quan trọng. Mục đích của việc thiết lập tương đương sinh học là chứng minh sự tương đương về đặc tính sinh dược học giữa chế phẩm generic và chế phẩm đối chứng nhằm cho phép bắc cầu dữ liệu, các nghiên cứu tiền lâm sàng và thử nghiệm lâm sàng của chế phẩm đối chứng. Thuốc generic là một chế phẩm có cùng thành phần định tính, định lượng các dược chất và dạng bào chế giống với thuốc tham chiếu, đồng thời chế phẩm này đã được chứng minh tương đương sinh học bằng các nghiên cứu sinh khả dụng thích hợp. Các dạng muối, ester, ether, đồng phân, hỗn hợp đồng phân, phức chất hoặc dẫn chất khác của một dược chất được coi là có cùng dược chất, trừ khi các dạng này thể hiện sự khác biệt đáng kể về đặc tính liên quan đến hiệu lực và độ an toàn.
1.3 Các loại đăng ký khác
Các loại hồ sơ đăng ký khác cũng có thể yêu cầu chứng minh tương đương sinh học bao gồm đăng ký thay đổi, phối hợp cố định liều và các đăng ký gia hạn.
Những khuyến cáo về thiết kế và thực hiện nghiên cứu tương đương sinh học đề cập trong hướng dẫn này cũng có thể áp dụng cho các nghiên cứu so sánh sinh khả dụng của những công thức khác nhau đã dùng trong quá trình phát triển một thuốc mới chứa dược chất mới và cho các nghiên cứu so sánh sinh khả dụng trong các trường hợp mở rộng khác khi hồ sơ đăng ký không chỉ dựa vào các dữ liệu, tương đương sinh học.
2. PHẠM VI
Hướng dẫn này tập trung vào việc đưa ra khuyến cáo cho các nghiên cứu tương đương sinh học của các thuốc có dạng bào chế giải phóng ngay có tác dụng toàn thân. Hướng dẫn này cũng đưa ra các tiêu chí thích hợp để không cần yêu cầu thực hiện các nghiên cứu sinh khả dụng (bao gồm miễn thử cho các hàm lượng khác- xem mục 3.1.6, Miễn thử với một dạng bào chế cụ thể- Phụ lục II hoặc Miễn thử sinh học dựa trên BCS- Phụ lục III).
Những khuyến cáo riêng cho các nghiên cứu tương đương sinh học các dạng thuốc khác như dạng thuốc giải phóng biến đổi, thuốc dùng qua da và dạng hít qua miệng v.v... được đề cập trong các hướng dẫn tương ứng và được trích dẫn trong phần dưới đây.
Phạm vi của hướng dẫn này chỉ giới hạn với các thuốc có nguồn gốc hóa dược. Khuyến cáo so sánh các sinh phẩm với chế phẩm tham chiếu tương ứng được đề cập trong những hướng dẫn riêng cho sinh phẩm tương tự.
Nếu không thể chứng minh tương đương sinh học bằng phương pháp định lượng nồng độ thuốc, có thể sử dụng các phương pháp đánh giá dược lực học hoặc lâm sàng cho trường hợp đặc biệt. Hướng dẫn này không đề cập tới các trường hợp kể trên, khi cần thì tham khảo các hướng dẫn riêng cho lĩnh vực điều trị.
Mặc dù khái niệm tương đương sinh học có thể xem xét áp dụng cho các thuốc có nguồn gốc dược liệu, những nguyên tắc chung được trình bày trong hướng dẫn này không thể áp dụng cho thuốc từ dược liệu do các thành phần có hoạt tính của loại thuốc này không được xác định đầy đủ như các thuốc hóa dược.
Hướng dẫn này cần được tham khảo cùng với các thông tin cần thiết khác được đề cập trong những hướng dẫn và quy định có liên quan hiện hành, bao gồm:
- Những lưu ý chung trong thử nghiệm lâm sàng (ICH topic E8,CPMP/ICH/291/95)
- Hướng dẫn thực hành tốt lâm sàng (ICH E6 (R1), CPMP/ICH/135/95)
- Nguyên lý thống kê trong thực hành lâm sàng (ICH E9, CPMP/ICH/363/96)
- Bố cục và nội dung của báo cáo nghiên cứu lâm sàng (ICH E3, CPMP/ICH/137/95)
- Nghiên cứu dược động học trên người (Eudralex, Volume 3, 3CC3a)
- Dạng bào chế giải phóng biến đổi dùng qua da hoặc đường uống: Mục I và II (CPMP/QWP/604/96, CPMP/EWP/280/96)
- Thuốc phối hợp cố định (CPMP/EWP/240/95 Rev 1)
- Những yêu cầu về tài liệu lâm sàng đối với các thuốc hít qua miệng (OIP), bao gồm yêu cầu chứng minh tương đương điều trị giữa hai thuốc hít dùng trong điều trị hen và bệnh phổi tắc nghẽn mạn tính (COPD)(CPMP/EWP/4151/00 Rev 1)
- Những yêu cầu lâm sàng đối với các thuốc dùng tại chỗ và tác dụng tại chỗ chứa các thành phần đã biết.(CPMP/EWP/239/95)
- Hồ sơ kỹ thuật chung ASEAN
- Hướng dẫn thẩm định phương pháp phân tích ASEAN
- Những thuốc đa nguồn (generic): Hướng dẫn đăng ký để thiết lập khả năng sử dụng thay thế lẫn nhau (WHO)
- Hướng dẫn thẩm định phương pháp phân tích thuốc trong mẫu sinh học (EMEA/CHMP/EWP/192217/2009)
Hướng dẫn này cũng cần được tham khảo cùng với các hướng dẫn liên quan về chất lượng thuốc. Chế phẩm thử dùng trong nghiên cứu tương đương sinh học phải được sản xuất theo quy chuẩn GMP.
3. NỘI DUNG CHÍNH CỦA HƯỚNG DẪN
3.1 Thiết kế, thực hiện và đánh giá các nghiên cứu tương đương sinh học
Số lượng nghiên cứu và thiết kế nghiên cứu phụ thuộc vào tính chất lý hóa, đặc tính dược động học của dược chất, tỷ lệ dược chất trong công thức và cần được biện giải thích hợp. Đặc biệt, có thể cần chỉ ra tính tuyến tính của các thông số dược động học, sự cần thiết thực hiện nghiên cứu ở cả trạng thái no và đói, phân tích chọn lọc đồng phân đối quang và khả năng miễn thử đối với các hàm lượng khác (xem mục 3.1.4, 3.1.5 và 3.1.6).
3.1.1 Thiết kế nghiên cứu
Nghiên cứu cần được thiết kế sao cho có thể phân biệt được ảnh hưởng về mặt bào chế với các yếu tố ảnh hưởng khác.
Thiết kế chuẩn
Trọng trường hợp so sánh hai công thức bào chế, khuyến cáo sử dụng thiết kế ngẫu nhiên, chéo đôi, đơn liều, hai giai đoạn, hai trình tự (nghiên cứu chéo đôi 2 x 2). Giữa các giai đoạn thử nghiệm cần có một thời gian nghỉ đủ dài để đảm bảo nồng độ thuốc giảm xuống thấp hơn giới hạn định lượng dưới của phương pháp phân tích ở tất cả các đối tượng trước khi bắt đầu giai đoạn 2. Thông thường, thời gian cần thiết để đạt được điều nay bằng tối thiểu 5 lần thời gian bán thải của thuốc.
Các kiểu thiết kế thay thế
Trong một số trường hợp, với điều kiện thiết kế nghiên cứu và phân tích thống kê phải dựa trên các căn cứ khoa học, có thể cân nhắc sử dụng các thiết kế thay thế được công nhận như thiết kế song song áp dụng cho các chất có thời gian bán thải rất dài và thiết kế lặp lại dành cho các thuốc có đặc tính dược động học biến thiên cao (xem mục 3.1.10).
Có thể sử dụng nghiên cứu đa liều trên bệnh nhân nếu không thể thực hiện được nghiên cứu đơn liều trên người tình nguyện khỏe mạnh do khả năng dung nạp và nghiên cứu đơn liều trên bệnh nhân không được chấp nhận.
Trường hợp hiếm gặp khi độ nhạy của phương pháp phân tích không cho phép định lượng chính xác nồng độ thuốc trong huyết tương sau khi dùng liều đơn, đồng thời nồng độ thuốc ở trạng thái ổn định đủ cao để có thể định lượng một cách chính xác, nghiên cứu đa liều có thể được sử dụng thay thế cho nghiên cứu đơn liều. Tuy nhiên, do nghiên cứu đa liều kém nhạy hơn trong việc phát hiện sự khác biệt về Cmax, điều này chỉ có thể được chấp nhận nếu cơ sở đăng ký đưa ra được các bằng chứng thuyết minh rằng không thể cải thiện được độ nhạy của phương pháp phân tích và không có phương pháp phân tích đáng tin cậy để định lượng chất có tác dụng sau khi sử dụng liều đơn, có tính đến cả phương án lựa chọn liều nghiên cứu cao hơn mức liều điều trị trong nghiên cứu tương đương sinh học (xem mục 3.1.6). Do những tiến bộ gần đây về phương pháp phân tích thuốc trong mẫu sinh học, hiếm gặp trường hợp không thể định lượng được chất cần phân tích một cách chính xác và tin cậy. Vì vậy, sử dụng nghiên cứu đa liều thay cho nghiên cứu đơn liều vì lí do độ nhạy của phương pháp phân tích hạn chế chỉ có thể được chấp nhận trong những trường hợp đặc biệt.
Trong các nghiên cứu ở trạng thái ổn định, thời gian tích lũy của giai đoạn thử thứ hai có thể được phép gối lên thời gian rửa giải của giai đoạn thử nghiệm trước đó với điều kiện thời gian tích lũy này phải đủ dài (tối thiểu bằng 5 lần thời gian bán thải).
3.1.2 Thuốc đối chứng và thuốc thử
Thuốc đối chứng
Thuốc thử là thuốc generic hoặc thuốc phát triển mở rộng tiếp theo của một thuốc generic sẵn có đang nộp hồ sơ đăng ký lưu hành thường được so sánh với dạng bào chế tương ứng của thuốc đối chứng. Cần lựa chọn thuốc đối chứng dựa trên các tiêu chí lựa chọn thuốc đối chứng của ASEAN (theo thứ tự ưu tiên) như sau:
i. Thuốc phát minh và thuốc từ các cơ sở sản xuất khác của cùng nhà phát minh đã được đăng ký lưu hành tại nước nơi nộp hồ sơ đăng ký thuốc generic.
ii. Trường hợp thuốc phát minh đã sử dụng để làm thuốc đối chứng không phải là thuốc phát minh đã đăng ký lưu hành tại nước nơi nộp hồ sơ đăng ký thuốc generic, nhà sản xuất thuốc generic cần chứng minh khả năng thay thế lẫn nhau giữa thuốc đối chứng dùng trong nghiên cứu với thuốc phát minh đã được đăng ký lưu hành tại nước nơi nộp hồ sơ đăng ký thuốc generic(in vitro hoặc in vivo).
iii. Nếu không thể xác định được thuốc phát minh, việc lựa chọn một thuốc làm thuốc đối chứng phải được thực hiện cẩn thận và cơ sở đăng ký phải giải thích đầy đủ về sự lựa chọn này. Tiêu chí lựa chọn thuốc đối chứng trong trường hợp này được áp dụng theo thứ tự ưu tiên như sau:
- Được phê duyệt bởi các nước thuộc ICH và các nước liên kết
- Đã được tiền thẩm định bởi WHO
Thuốc đối chứng được lựa chọn tốt phải có chất lượng đạt tiêu chuẩn của dược điển, nếu có các tiêu chuẩn này.
Cần làm rõ với cơ quan quản lý về việc lựa chọn thuốc đối chứng trước khi thực hiện nghiên cứu tương đương sinh học.
Lựa chọn lô thuốc đối chứng để sử dụng cho nghiên cứu tương đương sinh học cần dựa trên hàm lượng định lượng, dữ liệu độ hòa tan và điều này thuộc trách nhiệm của cơ sở đăng ký. Trừ trường hợp có lý do hợp lý, hàm lượng định lượng được của lô thuốc thử dùng trong nghiên cứu không được chênh lệch quá 5% so với lô thuốc đối chứng khi áp dụng quy trình định lượng đã đề xuất để đánh giá chất lượng của thuốc thử. Phiếu kiểm nghiệm (CoA) của thuốc đối chứng có thể kèm theo trong hồ sơ đăng ký để chứng minh hàm lượng định lượng được của lô thuốc thử đã dùng không chênh lệch quá 5% so với hàm lượng của lô đối chứng. Cơ sở đăng ký cần lưu trữ các tài liệu thể hiện tính đại diện của lô thuốc đối chứng thông qua dữ liệu độ hòa tan và kết quả định lượng. Nên kiểm tra một vài lô thuốc đối chứng để lựa chọn một lô thuốc phù hợp dùng trong nghiên cứu tương đương sinh học.
Thuốc thử
Thuốc thử đã dùng trong nghiên cứu cần đại diện cho sản phẩm dự kiến lưu hành trên thị trường và cơ sở đăng ký cần giải thích và bàn luận về vấn đề này.
Ví dụ, đối với dạng thuốc rắn dùng đường uống có tác dụng toàn thân:
a) Thông thường, cần chọn thuốc thử từ một lô có cỡ lô tối thiểu bằng 1/10 cỡ lô sản xuất hoặc 100 000 đơn vị (tùy thuộc số lượng nào lớn hơn), trừ trường hợp có lý do hợp lý.
b) Khi sản xuất các lô thuốc sử dụng trong nghiên cứu, cần đảm bảo chắc chắn rằng sản phẩm và quy trình sẽ có khả năng áp dụng trên quy mô công nghiệp.
Trường hợp cỡ lô sản xuất theo qui trình nhỏ hơn 100 000 đơn vị, mẫu thuốc dùng cho nghiên cứu cần được sản xuất theo đúng cỡ lô đăng ký.
c) Những đặc tính và tiêu chuẩn quan trọng của thuốc như độ hòa tan cần được thiết lập từ lô thử, là lô lâm sàng đã được chứng minh tương đương sinh học.
d) Mẫu thuốc sản xuất từ các lô thử nghiệm thêm và/ hoặc lô sản xuất chính thức (đã nộp kèm theo hồ sơ đăng ký) sẽ được so sánh với mẫu thuốc của lô dùng trong nghiên cứu tương đương sinh học và phải cho biểu đồ hòa tan in vitro tương đồng khi sử dụng phương pháp thử độ hòa tan thích hợp (xem Phụ lục I).
Thử nghiệm so sánh độ hòa tan với lô dùng trong nghiên cứu tương đương sinh học sẽ được thực hiện trên ba lô sản xuất đầu tiên. Các kết quả thu được sẽ được cung cấp cho cơ quan quản lý khi có yêu cầu hoặc khi không thu được sự tương đồng về biểu đồ hòa tan giữa các lô sản xuất với lô dùng trong nghiên cứu tương đương sinh học có kèm theo các hành động đề xuất cần thực hiện tiếp theo.
Đối với những dạng bào chế giải phóng ngay có tác dụng toàn thân khác, cần giải thích tính đại diện của lô thử tương tự như trên.
Đóng gói mẫu thuốc dùng trong nghiên cứu
Thuốc thử và thuốc đối chứng cần được đóng gói riêng cho từng đối tượng và từng giai đoạn của nghiên cứu trước khi vận chuyển đến nơi thử hoặc tại nơi thử nghiệm. Quy trình đóng gói (bao gồm ghi nhãn) cần được thực hiện tuân thủ thực hành tốt sản xuất thuốc.
Cần đảm bảo rằng có thể nhận diện một cách rõ ràng mã hóa của các thuốc đã dùng cho mỗi người tình nguyện ở mỗi giai đoạn thử. Do đó, cần ghi hồ sơ chi tiết việc đóng gói, ghi nhãn và cho đối tượng dùng thuốc. Việc ghi hồ sơ này cần hết sức thận trọng để tránh nhầm lẫn cũng như phát hiện được lỗi có thể xảy ra liên quan đến sử dụng thuốc trong nghiên cứu. Khuyến cáo sử dụng loại nhãn với một phần có thể xé rời.
3.1.3 Người tình nguyện
Số lượng người tình nguyện
Cần xác định số lượng người tình nguyện tham gia nghiên cứu dựa trên tính toán cỡ mẫu thích hợp.
Đối với một nghiên cứu chéo đôi chuẩn (hai trình tự, hai giai đoạn), số lượng người tình nguyện được xác định dựa trên:
a) Hệ số biến thiên giữa các cá thể của thuốc nghiên cứu được ước lượng từ một nghiên cứu thăm dò hoặc từ kết quả của các nghiên cứu lâm sàng trước đó hoặc từ các tài liệu đã được công bố.
b) Mức ý nghĩa mong muốn (α=0,05)
c) Sai lệch dự kiến của tỷ số T/R (delta trong khoảng 5% đến 10%)
d) Giới hạn cho phép (cần phù hợp với các mục 3.1.8, 3.1.9 & 3.1.10 tương ứng trong hướng dẫn này.)
e) Hiệu lực thống kê (power) của nghiên cứu nên tối thiểu là 80%
Các tiêu chuẩn phân tích và lâm sàng được áp dụng cũng có thể ảnh hưởng đến thống kê xác định số lượng người tham gia nghiên cứu. Tuy nhiên, số lượng người tham gia tối thiểu thường không được dưới 12 người.
Lựa chọn người tình nguyện
Cần lựa chọn người tình nguyện tham gia nghiên cứu tương đương sinh học sao cho có thể phát hiện được sự khác biệt giữa các chế phẩm thuốc. Để giảm thiểu những ảnh hưởng không liên quan đến sự khác biệt giữa các thuốc, nghiên cứu thường được thực hiện trên người tình nguyện khỏe mạnh, trừ khi thuốc này có vấn đề về độ an toàn và khía cạnh đạo đức không cho phép thực hiện nghiên cứu trên người tình nguyện khỏe mạnh. Thử nghiệm in vivo trên người tình nguyện khỏe mạnh được coi là phù hợp trong hầu hết các trường hợp để phát hiện sự khác biệt giữa các công thức bào chế và cho phép ngoại suy kết quả cho toàn bộ quần thể mà thuốc đối chứng được phê duyệt sử dụng (người già, trẻ em, bệnh nhân suy gan hoặc suy thận...).
Cần làm rõ tiêu chí chấp nhận/ loại trừ trong đề cương nghiên cứu. Đối tượng tham gia nghiên cứu cần trong độ tuổi từ 18-55 tuổi và ưu tiên những người có chỉ số khối lượng cơ thể (BMI) từ 18 đến 30 kg/m2.
Cần sàng lọc đối tượng tham gia bằng đánh giá các xét nghiệm cận lâm sàng, tiền sử bệnh và kiểm tra thể chất. Tùy thuộc vào nhóm điều trị và dữ liệu an toàn của thuốc, cần kiểm tra sức khỏe riêng và thực hiện các lưu ý phòng ngừa trước, trong và sau khi hoàn thành nghiên cứu. Người tham gia nghiên cứu có thể thuộc một trong hai giới; tuy nhiên, cần cân nhắc nguy cơ đối với phụ nữ có khả năng mang thai. Ưu tiên người không hút thuốc lá và không có tiền sử nghiện rượu hoặc ma túy. Kiểu hình và/ hoặc kiểu gen của các đối tượng cũng có thể được cân nhắc vì các lý do liên quan đến độ an toàn hoặc dược động học.
Trong các nghiên cứu được thiết kế song song, các nhóm thử nghiệm cần tương đồng về các yếu tố có thể ảnh hưởng đến dược động học của dược chất (như tuổi, cân nặng, giới tính, chủng tộc, thói quen hút thuốc, khả năng chuyển hóa mạnh/ kém). Đây là điều kiện tiên quyết để các kết quả của nghiên cứu này có giá trị.
Nếu dược chất cần nghiên cứu đã được xác định là có tác dụng không mong muốn và tác dụng dược lý hoặc nguy cơ không thể chấp nhận với người tình nguyện khỏe mạnh, có thể lựa chọn đối tượng tham gia nghiên cứu là bệnh nhân với các biện pháp phòng ngừa và giám sát thích hợp.
3.1.4 Tiến hành nghiên cứu
Tiêu chuẩn hóa
Cần chuẩn hóa điều kiện thử để hạn chế tối đa sự khác biệt về tất cả các yếu tố liên quan, ngoại trừ sự khác biệt giữa các thuốc được nghiên cứu. Do đó, khuyến cáo chuẩn hóa chế độ ăn, lượng nước uống và hoạt động thể lực.
Cần xác định rõ thời điểm ăn uống trong ngày. Người tình nguyện cần nhịn đói ít nhất 8 giờ trước khi dùng thuốc, trừ khi có lý do hợp lý. Do lượng chất lỏng sử dụng có thể ảnh hưởng đến tốc độ di chuyển từ dạ dày xuống ruột của các thuốc dạng uống, cần uống thuốc thử và thuốc đối chứng với cùng một thể tích nước xác định (tối thiểu 150 ml). Người tình nguyện được uống nước theo nhu cầu, trừ khoảng thời gian một giờ trước và một giờ sau khi dùng thuốc, và không được phép ăn trong vòng 4 giờ sau khi dùng thuốc. Chế độ ăn sau khi dùng thuốc cần được chuẩn hóa về thành phần và thời điểm dùng bữa trong một thời gian thích hợp (ví dụ, trong 12 giờ).
Nếu nghiên cứu được thực hiện trong tình trạng no, thời điểm dùng thuốc và bữa ăn nên dựa theo bản tóm tắt đặc tính sản phẩm của thuốc gốc (thuốc phát minh). Nếu trong tóm tắt đặc tính sản phẩm của thuốc gốc không có khuyến cáo cụ thể, người tình nguyện cần bắt đầu bữa ăn 30 phút trước khi dùng thuốc và nên ăn hết suất ăn này trong vòng 30 phút.
Do sinh khả dụng của chất có hoạt tính trong dạng bào chế có thể phụ thuộc vào thời gian di chuyển của thuốc trong đường tiêu hóa và lưu lượng máu tại chỗ, có thể cần chuẩn hóa tư thế và hoạt động thể lực của người tình nguyện.
Người tình nguyện tham gia thử nghiệm cần tránh sử dụng đồ ăn hoặc đồ uống có thể ảnh hưởng tới tuần hoàn, tiêu hóa, chức năng gan hoặc thận (như đồ uống có chứa cồn hoặc một số loại nước hoa quả như nước ép bưởi) trong một khoảng thời gian thích hợp trước và trong khi nghiên cứu. Các đối tượng tham gia cũng không nên dùng đồng thời bất kỳ thuốc nào khác (kể cả thuốc có nguồn gốc dược liệu) trong một khoảng thời gian thích hợp trước và trong thời gian nghiên cứu. Tuy nhiên, các thuốc tránh thai có thể được phép sử dụng. Trường hợp không thể tránh được và một người tình nguyện đã được phép sử dụng các thuốc khác (ví dụ dùng thuốc để điều trị các biến cố bất lợi như đau đầu), thì phải báo cáo việc sử dụng thuốc này (liều và thời gian dùng thuốc) và xem xét để chỉ ra khả năng ảnh hưởng đến kết quả nghiên cứu và cách xử lý. Trong một số trường hợp hiếm gặp, có thể cần dùng đồng thời một thuốc khác cho tất cả các đối tượng tham gia thử nghiệm vì lý do liên quan đến độ an toàn hoặc khả năng dung nạp (như các thuốc đối kháng opioid, thuốc chống nôn). Khi đó, nguy cơ xảy ra tương tác hoặc sự ảnh hưởng tới việc phân tích mẫu sinh học có thể gây ảnh hưởng đến kết quả nghiên cứu cần được chỉ rõ.
Các thuốc cần sử dụng phối hợp với một thuốc khác theo tóm tắt đặc tính sản phẩm của thuốc phát minh (ví dụ, một số thuốc ức chế protease thường dùng phối hợp với ritonavir) có thể được nghiên cứu dưới dạng phối hợp đã được phê duyệt hoặc không dùng đồng thời thuốc như khuyến cáo.
Trong các nghiên cứu tương đương sinh học của những chất nội sinh, cần kiểm soát các yếu tố có thể ảnh hưởng đến nồng độ ban đầu của chất nội sinh đang nghiên cứu, nếu có thể (ví dụ, kiểm soát chặt chẽ chế độ ăn).
Thời điểm lấy mẫu nghiên cứu
Số lượng mẫu lấy phải đủ để có thể mô tả đầy đủ đường biểu diễn nồng độ thuốc trong huyết tương theo thời gian. Kế hoạch lấy mẫu cần bao gồm lấy mẫu thường xuyên hơn quanh thời điểm đạt tmax dự kiến để ước lượng chính xác hơn nồng độ đỉnh. Đặc biệt, cần xây dựng kế hoạch lấy mẫu sao cho tránh điểm đạt Cmax là điểm đầu tiên của đường cong biểu diễn nồng độ thuốc theo thời gian. Kế hoạch lấy mẫu cũng cần đảm bảo đường cong nồng độ thuốc trong huyết tương theo thời gian đủ dài để có thể ước lượng một cách tin cậy mức độ phơi nhiễm, giới hạn cho phép là AUC(0-t) phải đạt tối thiểu 80% AUC(0-∞). Cần lấy tối thiểu ba đến bốn mẫu trong pha thải trừ tuyến tính (theo logarit nồng độ) để đảm bảo có thể ước lượng chính xác hằng số tốc độ thải trừ (thông số này được sử dụng để ước lượng AUC(0-∞) một cách tin cậy). Giá trị AUC tới 72 giờ (AUC(0-72h)) có thể được sử dụng thay thế cho AUC(0-t) để so sánh mức độ phơi nhiễm do thông số này đã phản ánh toàn bộ pha hấp thu của các dạng bào chế giải phóng ngay. Do đó, thời gian lấy mẫu dài hơn 72 giờ là không cần thiết đối với các dạng bào chế giải phóng ngay, bất kể thời gian bán thải của thuốc dài hay ngắn.
Trong các nghiên cứu đa liều, trong một khoảng liều, mẫu trước liều được lấy ngay trước khi dùng thuốc (trong vòng 5 phút trước khi dùng thuốc) và mẫu cuối cùng được lấy trong vòng 10 phút so với thời gian dự kiến của khoảng cách liều để đảm bảo có thể xác định chính xác AUC(0-t).
Trường hợp mẫu sinh học được chọn là nước tiểu, mẫu nước tiểu thường được lấy trong khoảng thời gian tối thiểu bằng ba lần thời gian bán thải. Tuy nhiên, tương tự các khuyến cáo về lấy mẫu huyết tương, thời gian lấy mẫu nước tiểu không cần vượt quá 72 giờ. Nếu cần xác định tốc độ thải trừ, các khoảng cách lấy mẫu trong pha thải trừ cũng phải ngắn đến mức có thể như trong pha hấp thu (xem mục 3.15).
Với các chất nội sinh, kế hoạch lấy mẫu cần đảm bảo khả năng xác định được dữ liệu nồng độ chất nội sinh ban đầu (mức cơ bản) của mỗi đối tượng tham gia trong mỗi giai đoạn. Thông thường, mức cơ bản được xác định từ 2-3 mẫu lấy trước khi dùng thuốc. Một số trường hợp cần lấy mẫu đều đặn trong thời gian 1-2 ngày trước khi sử dụng thuốc để xác định sự dao động của mức cơ bản liên quan đến nhịp sinh học.
Trạng thái đói hoặc no
Thông thường, nghiên cứu tương đương sinh học cần được thực hiện trong tình trạng đói vì đây được xem là điều kiện tốt nhất để phát hiện sự khác biệt giữa các công thức bào chế. Do đó, đối với các thuốc mà tóm tắt đặc tính sản phẩm của thuốc đối chứng khuyến cáo sử dụng thuốc vào lúc đói hoặc không phụ thuộc bữa ăn, cần nghiên cứu tương đương sinh học trong tình trạng đói. Đối với các thuốc mà tóm tắt đặc tính sản phẩm của thuốc đối chứng khuyến cáo chỉ sử dụng thuốc trong tình trạng no, nghiên cứu tương đương sinh học thường được tiến hành trong tình trạng no.
Tuy nhiên, đối với các thuốc có các đặc tính bào chế đặc biệt (ví dụ: vi nhũ tương, hệ phân tán rắn), cần thực hiện nghiên cứu tương đương sinh học trong cả trạng thái no và trạng thái đói, trừ khi có yêu cầu chỉ được dùng thuốc khi đói hoặc khi no.
Trường hợp cần có dữ liệu cho cả trạng thái no và trạng thái đói, có thể thực hiện hai nghiên cứu chéo đôi riêng rẽ hoặc một nghiên cứu chéo bốn.
Trong các nghiên cứu được thực hiện ở trạng thái no, thành phần bữa ăn sử dụng cần căn cứ vào tóm tắt đặc tính sản phẩm của thuốc phát minh. Nếu tóm tắt đặc tính sản phẩm của thuốc phát minh không đưa ra khuyến cáo cụ thể, nên sử dụng bữa ăn giàu chất béo (khoảng 50% so với tổng lượng calo của bữa ăn) và giàu năng lượng (khoảng 800 đến 1000 calo). Bữa ăn này cần có lượng protein, carbohydrat và chất béo theo thứ tự tương ứng khoảng 150, 250 và 500-600 calo. Cần mô tả thành phần của bữa ăn sử dụng trong nghiên cứu, chú ý tới lượng protein, carbohydrat và chất béo (số gam, lượng calo tổng cộng và tỷ lệ calo tương ứng của từng thành phần (%)).
3.1.5 Các thông số cần nghiên cứu
Thông số dược động học
Sử dụng thời gian lấy mẫu thực tế để tính toán các thông số dược động học. Trong các nghiên cứu đánh giá tương đương sinh học sau khi dùng liều đơn, cần xác định AUC(0-t), AUC(0-∞), Cmax và tmax. Các nghiên cứu với khoảng thời gian lấy mẫu là 72 giờ và có thể định lượng được nồng độ thuốc tại thời điểm 72 giờ, không cần báo cáo AUC(0-∞) và phần diện tích dưới đường cong còn lại; Trường hợp này, chỉ cần báo cáo diện tích dưới đường cong tính đến thời điểm 72 giờ (AUC(0-72h)). Có thể ghi nhận thêm các thông số khác bao gồm hằng số tốc độ thải trừ pha cuối λz, và t1/2.
Trong các nghiên cứu đánh giá tương đương sinh học các thuốc giải phóng ngay ở trạng thái ổn định, cần xác định AUC(0-t), Cmax,ss, và tmax,ss.
Khi sử dụng dữ liệu nồng độ thuốc trong nước tiểu, cần xác định các giá trị Ae(0-t) và  .
.
Cần sử dụng phương pháp không ngăn để xác định các thông số dược động học trong các nghiên cứu tương đương sinh học. Sử dụng phương pháp ngăn để xác định các thông số này không được chấp nhận.
Chất mẹ hoặc các chất chuyển hóa
Khuyến cáo chung
Về nguyên tắc, việc đánh giá tương đương sinh học cần dựa trên nồng độ chất mẹ (thuốc mẹ) đo được. Lý do là Cmax của chất mẹ thường là dữ liệu nhạy hơn để phát hiện sự khác biệt về tốc độ hấp thu giữa các thuốc nghiên cứu so với Cmax của chất chuyển hóa.
Tiền thuốc không có hoạt tính
Đối với các tiền thuốc không có hoạt tính, cũng khuyến cáo chứng minh tương đương sinh học dựa trên chất mẹ. Khi đó không cần thiết định lượng chất chuyển hóa có hoạt tính. Tuy nhiên, một số tiền thuốc có thể có nồng độ thấp trong huyết tương và bị thải trừ nhanh dẫn đến khó chứng minh tương đương sinh học dựa trên chất mẹ. Trong trường hợp này, chấp nhận chứng minh tương đương sinh học dựa trên chất chuyển hóa chính mà không cần định lượng chất mẹ. Trong phạm vi hướng dẫn này, một chất mẹ được coi là một tiền thuốc không có hoạt tính nếu chất này không có hoặc có đóng góp rất nhỏ vào hiệu quả lâm sàng của thuốc.
Sử dụng dữ liệu chất chuyển hóa để thay thế cho chất mẹ có hoạt tính
Không khuyến cáo sử dụng một chất chuyển hóa để thay thế cho chất mẹ có hoạt tính. Điều này chỉ có thể được xem xét nếu cơ sở đăng ký có thể chứng minh một cách thỏa đáng rằng không thể cải thiện được độ nhạy của phương pháp phân tích trên chất mẹ, vì vậy phương pháp phân tích là không đủ tin cậy để định lượng chất mẹ sau khi dùng liều đơn, ngay cả khi có thể lựa chọn nghiên cứu ở mức liều cao hơn (xem mục 3.1.6). Do những tiến bộ gần đây về phương pháp phân tích mẫu sinh học, thường ít xảy ra trường hợp không thể định lượng được chất mẹ một cách chính xác. Do đó, sử dụng một chất chuyển hóa thay thế cho chất mẹ có hoạt tính chỉ được chấp nhận trong những trường hợp cá biệt. Khi sử dụng dữ liệu nồng độ chất chuyển hóa thay thế cho nồng độ chất mẹ có hoạt tính, cơ sở đăng ký cần trình bày tất cả dữ liệu đã có để chứng minh rằng mức độ phơi nhiễm với chất chuyển hóa sẽ phản ánh mức độ phơi nhiễm với chất mẹ và sự hình thành chất chuyển hóa chưa đạt trạng thái bão hòa ở các mức liều điều trị.
Đồng phân đối quang
Nhìn chung, có thể sử dung các phương pháp phân tích sinh học đối xứng (không phân biệt đồng phân đối quang). Tuy nhiên, cần định lượng riêng từng loại đồng phân trong trường hợp có đồng thời tất cả các điều kiện sau:
(1) Các đồng phân đối quang có đặc điểm dược động học khác nhau
(2) Các đồng phân đối quang khác nhau rõ rệt về mặt dược lực học
(3) Tỷ lệ phơi nhiễm (AUC) của các đồng phân thay đổi khi tốc độ hấp thu thay đổi.
Cần định lượng các loại đồng phân đối quang riêng rẽ trong trường hợp các điều kiện trên thỏa mãn hoặc chưa được xác định. Nếu một đồng phân đối quang có tác dụng dược lý trong khi đồng phân còn lại không có hoạt tính hoặc chỉ có rất ít hoạt tính, chỉ cần chứng minh tương đương sinh học dựa trên đồng phân đối quang có hoạt tính.
Sử dụng dữ liệu trong nước tiểu
Có thể sử dụng dữ liệu thuốc bài tiết trong nước tiểu thay thế cho nồng độ thuốc trong huyết tương để xác định mức độ phơi nhiễm trong trường hợp không thể định lượng chính xác nồng độ thuốc trong huyết tương theo thời gian của chất mẹ. Tuy nhiên, cần biện giải một cách chặt chẽ khi sử dụng dữ liệu thuốc trong nước tiểu để tính toán mức độ phơi nhiễm. Nếu có thể xác định được Cmax trong huyết tương một cách đáng tin cậy, nên kết hợp cả dữ liệu này với dữ liệu trong nước tiểu (phản ánh mức độ phơi nhiễm) để đánh giá tương đương sinh học. Khi sử dụng dữ liệu thuốc trong nước tiểu, cơ sở đăng ký cần trình bày tất cả các dữ liệu đã có để chứng minh rằng dữ liệu thuốc bài tiết trong nước tiểu sẽ phản ánh sự phơi nhiễm thuốc trong huyết tương.
Các chất nội sinh
Nếu thuốc cần nghiên cứu là chất nội sinh, khi tính toán các thông số dược động học, cần hiệu chỉnh so với mức cơ bản để giá trị tính được có thể phản ánh đúng nồng độ thuốc tăng thêm sau thử nghiệm. Trong nghiên cứu tương đương sinh học các thuốc là chất nội sinh, có thể cân nhắc sử dụng liều thử cao hơn mức liều điều trị để xác định chính xác nồng độ tăng thêm sau thử nghiệm so với mức cơ bản, với điều kiện đã biết mức liều thử được dung nạp tốt. Nếu sự khác biệt về phơi nhiễm sau khi sử dụng các mức liều khác nhau của một chất nội sinh cụ thể chưa được thiết lập trước đó thì cần xác định trong một nghiên cứu thăm dò hoặc lồng ghép trong nghiên cứu tương đương sinh học chính thức sử dụng các mức liều khác nhau của thuốc đối chứng để đảm bảo mức liều sử dụng để đánh giá tương đương sinh học đủ nhạy để phát hiện được sự khác biệt tiềm tàng giữa các công thức bào chế.
Phương pháp chính xác để hiệu chỉnh so với mức cơ bản cần được xác định trước và phải được làm rõ trong đề cương nghiên cứu. Nhìn chung, phương pháp hiệu chỉnh được ưu tiên áp dụng là trừ đi giá trị cơ bản, tức là trừ đi giá trị trung bình nồng độ chất nội sinh trước khi dùng thuốc hoặc trừ đi AUC của chất nội sinh trước dùng thuốc của mỗi người tình nguyện. Một số ít trường hợp nồng độ chất nội sinh sau thử nghiệm đó được tăng hơn hẳn so với mức cơ bản (giá trị ban đầu), có thể không cần hiệu chỉnh theo giá trị ban đầu.
Trong các nghiên cứu tương đương sinh học với chất nội sinh, cần chú ý đảm bảo thời gian nghỉ giữa hai giai đoạn phải đủ dài do không thể trực tiếp xác định có xảy ra hiện tượng nhiễm chéo hay không.
3.1.6 Hàm lượng cần nghiên cứu
Khi đăng ký đồng thời một số hàm lượng khác nhau của thuốc thử, có thể chỉ cần thiết lập tương đương sinh học với một hoặc hai hàm lượng, phụ thuộc vào tỷ lệ thành phần giữa các hàm lượng khác nhau và các vấn đề khác liên quan đến sản phẩm được mô tả dưới đây. Hàm lượng sử dụng trong nghiên cứu phụ thuộc vào sự tuyến tính dược động học của dược chất.
Trường hợp dược động học không tuyến tính (tức là AUC tăng không tỷ lệ thuận với liều), có thể có sự khác nhau giữa các hàm lượng về độ nhạy trong việc phát hiện sự khác biệt tiềm tàng giữa các công thức bào chế. Trong phạm vi hướng dẫn này, dược động học được coi là tuyến tính nếu sự khác biệt về AUC trung bình đã hiệu chỉnh theo liều không vượt quá 25% khi so sánh hàm lượng đã nghiên cứu (hoặc hàm lượng đã có kế hoạch nghiên cứu tương đương sinh học) với (những) hàm lượng muốn được xem xét miễn thử tương đương sinh học. Để đánh giá tính tuyến tính, cơ sở đăng ký cần xem xét tất cả các dữ liệu sẵn có đã được công bố liên quan đến tỷ lệ theo liều và rà soát các dữ liệu một cách chặt chẽ.
Nếu đã chứng minh tương đương sinh học với (các) hàm lượng được coi là nhạy cảm nhất để phát hiện được sự khác biệt tiềm tàng giữa các thuốc, nghiên cứu tương đương sinh học in vivo cho (các) hàm lượng khác có thể được miễn.
Các tiêu chí chung để được xem xét miễn thử
Để đề nghị miễn thực hiện nghiên cứu cho (các) hàm lượng khác, phải đáp ứng các yêu cầu chung sau đây:
a) Các chế phẩm thuốc được sản xuất bởi cùng một quy trình sản xuất,
b) Thành phần định tính của các hàm lượng khác nhau là tương tự nhau,
c) Công thức của các hàm lượng phải tỷ lệ với nhau, tức là tỷ lệ giữa khối lượng mỗi tá dược so với khối lượng (các) dược chất phải giống nhau đối với tất cả các hàm lượng (quy định này không áp dụng đối với tá dược bao, vỏ nang, tá dược màu và tá dược điều hương của thuốc ở dạng bào chế giải phóng ngay),
Nếu có sai khác về tỷ lệ định lượng các thành phần, điều kiện c vẫn được coi là thỏa mãn nếu đáp ứng được điều kiện i) và ii) hoặc i) và iii) dưới đây đối với hàm lượng đã dùng trong nghiên cứu tương đương sinh học và (các) hàm lượng đề nghị xem xét miễn thử:
i. Lượng (các) dược chất nhỏ hơn 5% khối lượng viên nhân của viên nén hoặc khối lượng thuốc đóng trong nang
ii. Lượng của các tá dược trong viên nhân hoặc phần bột đóng trong nang giống nhau giữa các hàm lượng và chỉ khác nhau về lượng dược chất
iii. Lượng tá dược độn thay đổi theo sự thay đổi của lượng dược chất.
Lượng các tá dược khác trong viên nhân hoặc phần bột thuốc đóng trong nang giống nhau giữa các hàm lượng
d) Dữ liệu độ hòa tan in vitro phù hợp và đầy đủ cho việc miễn thử thêm tương đương sinh học in vivo (xem mục 3.2).
Dược động học tuyến tính
Đối với các chế phẩm đáp ứng tất cả các điều kiện từ a) đến d), chỉ cần thiết lập tương đương sinh học đối với một hàm lượng.
Nghiên cứu tương đương sinh học thường được thực hiện với hàm lượng cao nhất. Đối với các thuốc có dược động học tuyến tính và dược chất có độ tan cao (xem Phụ lục III), lựa chọn nghiên cứu với hàm lượng thấp hơn hàm lượng cao nhất cũng được chấp nhận. Cũng có thể xem xét nghiên cứu với hàm lượng thấp hơn nếu hàm lượng cao nhất không thể sử dụng được cho người tình nguyện khỏe mạnh vì lý do an toàn/ khả năng dung nạp. Ngoài ra, nếu phương pháp phân tích không đủ nhạy để định lượng chính xác nồng độ thuốc trong huyết tương sau khi dùng liều đơn của hàm lượng cao nhất, có thể lựa chọn một mức liều cao hơn (ưu tiên dùng nhiều viên nén có hàm lượng cao nhất). Liều được lựa chọn để nghiên cứu có thể cao hơn mức liều tối đa sử dụng trong điều trị, với điều kiện người tình nguyện khỏe mạnh phải dung nạp tốt mức liều đơn này và khả năng hấp thu hoặc độ tan không bị hạn chế ở mức liều này.
Dược động học không tuyến tính
Với các thuốc có dược động học không tuyến tính, đặc trưng bởi tỷ lệ tăng AUC cao hơn so với tỷ lệ tăng liều trong khoảng liều điều trị, nghiên cứu tương đương sinh học thường được thực hiện với hàm lượng cao nhất. Tương tự trường hợp các thuốc có dược động học tuyến tính, có thể nghiên cứu trên hàm lượng thấp hơn nếu không thể sử dụng hàm lượng cao nhất trên người tình nguyện khỏe mạnh vì lý do an toàn/ khả năng dung nạp. Cũng có thể nghiên cứu ở một liều cao hơn trong trường hợp độ nhạy của phương pháp phân tích không đáp ứng với các khuyến cáo tương tự cho thuốc có dược động học tuyến tính ở trên.
Đối với các thuốc có tỷ lệ tăng AUC thấp hơn so với tỷ lệ tăng liều trong khoảng liều điều trị, trong hầu hết các trường hợp, cần thiết lập tương đương sinh học cho cả hàm lượng cao nhất và hàm lượng thấp nhất (hoặc một hàm lượng nằm trong khoảng tuyến tính), tức là cần thực hiện hai nghiên cứu tương đương sinh học trong trường hợp này. Nếu sự không tuyến tính không phải do giới hạn về độ tan mà do các nguyên nhân khác như sự bão hòa các chất vận chuyển thuốc vào trong tế bào và các điều kiện từ a) đến d) trên đều thỏa mãn, đồng thời thuốc thử và thuốc đối chứng không chứa bất kì tá dược nào có thể ảnh hưởng đến nhu động đường tiêu hóa hoặc protein vận chuyển, chỉ cần chứng minh tương đương sinh học với hàm lượng thấp nhất (hoặc một hàm lượng nằm trong khoảng tuyến tính). Có thể cân nhắc lựa chọn các hàm lượng khác nếu độ nhạy của phương pháp phân tích không cho phép tiến hành nghiên cứu với hàm lượng thấp nhất hoặc không thể sử dụng hàm lượng cao nhất trên người tình nguyện khỏe mạnh vì lý do an toàn/ khả năng dung nạp.
Tiếp cận theo hướng phân cực
Khi cần đánh giá tương đương sinh học với nhiều hơn hai hàm lượng, chẳng hạn do sự chênh lệch về tỷ lệ các thành phần, có thể sử dụng cách tiếp cận theo hướng phân cực. Trong trường hợp này, có thể thực hiện hai nghiên cứu tương đương sinh học với hai hàm lượng được chọn đại diện cho hai điểm giới hạn, ví dụ, hàm lượng cao nhất và hàm lượng thấp nhất hoặc hai hàm lượng khác nhau nhất về công thức bào chế, sao cho hai nghiên cứu tương đương sinh học này có thể là đại diện để phát hiện bất kì sự khác biệt nào về công thức của các hàm lượng còn lại.
Trường hợp cần đánh giá tương đương sinh học ở cả trạng thái no và trạng thái đói với hai hàm lượng khác nhau do sự hấp thu không tuyến tính hoặc công thức không theo tỷ lệ, có thể chỉ cần thực hiện nghiên cứu tương đương sinh học ở cả trạng thái no và trạng thái đói với một trong hai hàm lượng. Việc miễn thử nghiệm ở trạng thái no hoặc trạng thái đói với (các) hàm lượng còn lại có thể được biện giải dựa trên dữ liệu đã có trước đó và/hoặc dữ liệu dược động học từ nghiên cứu thực hiện với một hàm lượng được thử ở cả hai trạng thái no và đói. Trạng thái được lựa chọn (đói hoặc no) để đánh giá (các) hàm lượng còn lại là trạng thái nhạy cảm nhất để phát hiện sự khác biệt giữa các chế phẩm.
Phối hợp cố định liều
Tất cả các dược chất trong công thức của dạng phối hợp cố định liều cần đáp ứng những điều kiện liên quan đến công thức tỷ lệ. Khi xem xét khối lượng của mỗi dược chất trong một công thức ở dạng phối hợp cố định liều, có thể coi (các) dược chất còn lại trong công thức như thành phần tá dược. Trường hợp viên nén có hai lớp, mỗi lớp có thể được coi là độc lập với nhau.
3.1.7 Phương pháp phân tích sinh học
Cần thực hiện phần phân tích thuốc trong mẫu sinh học của các thử nghiệm tương đương sinh học theo nguyên tắc Thực hành tốt phòng thí nghiệm (GLP). (EMA/OECD GLP/WHO GLP STANDARD/ISO/IEC 17025/2005). Nếu các yêu cầu về GLP của quốc gia phù hợp với của Tổ chức hợp tác và phát triển kinh tế (OECD), cơ quan quản lý dược phẩm có thể tiến hành thanh tra tại chỗ theo nguyên tắc của OECD.
Các phương pháp phân tích đã sử dụng phải được mô tả rõ ràng, được thẩm định, ghi chép đầy đủ để thu được kết quả đáng tin cậy và có thể được giải thích thỏa đáng. Thẩm định trong khi nghiên cứu cần được thực hiện bằng việc sử dụng các mẫu đối chiếu (mẫu QC) trong mỗi lô phân tích.
Các đặc điểm chính của một phương pháp phân tích cần có để đảm bảo khả năng chấp nhận và độ tin cậy của kết quả bao gồm: độ chọn lọc, giới hạn định lượng dưới, hàm đáp ứng (diễn tiến của đường chuẩn), độ chính xác, độ đúng và độ ổn định.
Giới hạn định lượng dưới phải nhỏ hơn hoặc bằng 1/20 của Cmax, do cần phát hiện được nồng độ thuốc trước khi dùng thuốc với giá trị ít nhất bằng 5% Cmax (xem mục 3.1.8. Sự nhiễm chéo).
Việc phân tích lại các mẫu nghiên cứu cần được xác định rõ trong đề cương nghiên cứu (và/hoặc SOP) trước khi bắt đầu tiến hành phân tích mẫu. Thông thường, việc phân tích lại mẫu thử từ người tình nguyện vì lý do dược động học không được chấp nhận. Đây là điều đặc biệt quan trọng trong các nghiên cứu tương sinh học do điều này có thể làm thiên lệch kết quả cuối cùng của nghiên cứu.
Cần thực hiện phân tích mẫu trong điều kiện mù thông tin về thuốc nghiên cứu (mẫu được mã hóa).
3.1.8 Đánh giá
Trong các nghiên cứu tương đương sinh học, nói chung là không cần hiệu chính các thông số dược động học theo sự khác nhau về hàm lượng thực tế (tức kết quả định lượng) giữa lô thuốc thử và lô thuốc đối chứng. Tuy nhiên, trong một số trường hợp ngoại lệ khi không thể tìm được một lô thuốc đối chứng có hàm lượng thực tế khác biệt dưới 5% so với lô thuốc thử (xem mục 3.1.2), hiệu chỉnh theo hàm lượng thực có thể được chấp nhận. Nếu có thực hiện hiệu chỉnh theo hàm lượng thực, việc hiệu chỉnh cần được chỉ rõ trong đề cương nghiên cứu kèm theo giải thích lý do dựa trên các kết quả định lượng lô thuốc thử và thuốc đối chứng.
Đối tượng dùng để tính thống kê
Trường hợp lý tưởng là dữ liệu của tất cả các đối tượng tham gia nghiên cứu được dùng để tính thống kê. Tuy nhiên, những người tình nguyện không có đủ dữ liệu của cả thuốc thử và thuốc đối chứng trong các nghiên cứu thiết kế chéo cũng như những người tình nguyện thiếu dữ liệu của một giai đoạn trong các nghiên cứu thiết kế song song không được sử dụng để tính thống kê.
Dữ liệu của tất cả các đối tượng tham gia nghiên cứu cần được xử lý như nhau. Không chấp nhận một đề cương nghiên cứu trong đó chỉ rõ các đối tượng ‘dự phòng’ sẽ chỉ được đưa vào phân tích khi cần để thay thế cho các đối tượng tham gia nghiên cứu khác đã bị loại ra. Trong kế hoạch nghiên cứu, dữ liệu của tất cả các đối tượng đã tham gia nghiên cứu cần được đưa vào phân tích, kể cả khi không có đối tượng nào bị loại.
Trong các nghiên cứu có nhiều hơn hai mục tiêu nghiên cứu (ví dụ, một nghiên cứu ba giai đoạn bao gồm hai thuốc đối chứng, một thuốc từ châu Âu và một thuốc khác từ Mỹ hoặc một nghiên cứu bốn giai đoạn bao gồm thuốc thử và thuốc đối chứng được đánh giá trong cả trạng thái no và trạng thái đói), phân tích cho mỗi cặp so sánh cần được thực hiện sau khi đã loại các dữ liệu không liên quan đến phép so sánh đang được đề cập.
Lý do loại dữ liệu
Để đảm bảo tính khách quan của kết quả trong các nghiên cứu ngẫu nhiên, tất cả các đối tượng tham gia phải được quan sát và áp dụng các bước nghiên cứu theo những quy tắc giống nhau. Những quy tắc này không phụ thuộc vào thuốc thử nghiệm hoặc kết quả. Do đó, quyết định loại một đối tượng ra khỏi phân tích thống kê phải được thực hiện trước khi tiến hành phân tích mẫu sinh học.
Về nguyên tắc, bất kì lý do loại bỏ dữ liệu nào có thể được chấp nhận nếu điều này được làm rõ trong đề cương nghiên cứu và quyết định loại bỏ dữ liệu được đưa ra trước khi tiến hành phân tích mẫu sinh học. Tuy nhiên, nên tránh loại bỏ dữ liệu để tránh làm giảm hiệu lực (power) của nghiên cứu và đảm bảo có tối thiểu 12 đối tượng tham gia nghiên cứu.
Ví dụ về lý do loại bỏ dữ liệu của một người tình nguyện trong một giai đoạn cụ thể bao gồm các biến cố như nôn và tiêu chảy dẫn đến làm giảm độ tin cậy của dữ liệu nồng độ thuốc trong huyết tương theo thời gian. Trong một số trường hợp ngoại lệ, sử dụng đồng thời với một thuốc khác cũng có thể là lý do để loại bỏ dữ liệu của người tình nguyện đó.
Các lý do cho phép được loại bỏ dữ liệu phải được cụ thể hóa trước trong đề cương nghiên cứu. Nếu một trong số các biến cố này xảy ra, cần ghi chú rõ trong Hồ sơ người tình nguyện (CRF) ngay khi đang thực hiện nghiên cứu. Cần liệt kê và mô tả rõ việc loại bỏ các đối tượng tham gia dựa trên những tiêu chí định trước trong báo cáo nghiên cứu.
Không chấp nhận việc loại bỏ dữ liệu dựa trên phân tích thống kê hoặc chỉ do các nguyên nhân liên quan đến dược động học, bởi vì không thể phân biệt giữa yếu tố về mặt bào chế với những yếu tố khác có ảnh hưởng đến dược động học.
Các trường hợp ngoại lệ bao gồm:
1) Người tình nguyện thiếu bất kì dữ liệu định lượng nồng độ thuốc nào hoặc nồng độ thuốc rất thấp khi dùng thuốc đối chứng. Người tình nguyện được coi là có nồng độ thuốc trong huyết tương rất thấp khi AUC nhỏ hơn 5% giá trị AUC trung bình nhân của thuốc đối chứng (giá trị trung bình nhân này được tính không bao gồm các dữ liệu của người tình nguyện bất thường). Việc loại dữ liệu trong trường hợp này sẽ chỉ được chấp nhận trong những trường hợp cá biệt và có thể bị nghi ngờ về giá trị của thử nghiệm.
2) Người tình nguyện có nồng độ trước khi dùng thuốc > 5% Cmax. Không sử dụng dữ liệu này để tính thống kê tương đương sinh học (xem ảnh hưởng của sự nhiễm chéo dưới đây).
Đối với các thuốc giải phóng ngay, hiện tượng trên có thể xảy ra do sự không tuân thủ của người tình nguyện hoặc khoảng cách giữa hai giai đoạn dùng thuốc không đủ dài và cần được tránh tối đa, có thể bằng cách kiểm tra miệng của người tình nguyện sau khi dùng thuốc nghiên cứu để đảm bảo các đối tượng này đã nuốt hoàn toàn thuốc nghiên cứu và thiết kế với khoảng cách nghỉ giữa hai giai đoạn dùng thuốc đủ dài. Các mẫu thu được từ người tình nguyện bị loại không dùng tính thống kê vẫn cần được định lượng và trích dẫn kết quả (xem Trình bày kết quả dưới đây).
Như đã đề cập tại mục 3.1.4, AUC(0-t) cần bằng tối thiểu 80% AUC(0-∞). Khi tính thống kê, không nên loại bỏ dữ liệu của người tình nguyện có giá trị AUC(0-t) thấp hơn 80% AUC(0-∞), tuy nhiên, nếu tỷ lệ dưới 80% xuất hiện với trên 20% số người tình nguyện tham gia nghiên cứu, cần có bàn luận phù hợp. Không áp dụng điều này nếu thời gian lấy mẫu là 72 giờ hoặc dài hơn và AUC(0-72h) được sử dụng thay thế cho AUC(0-t).
Các thông số cần phân tích và giới hạn cho phép
Trong các nghiên cứu xác định tương đương sinh học khi dùng liều đơn, các thông số được phân tích bao gồm AUC(0-t) hoặc AUC(0-72h) (nếu phù hợp) và Cmax. Đối với các thông số này, khoảng tin cậy 90% của tỷ số giữa thuốc thử và thuốc đối chứng cần nằm trong giới hạn chấp nhận là 80,00 - 125,00%.
Đối với các nghiên cứu xác định tương đương sinh học của các thuốc giải phóng ngay ở trạng thái ổn định, cần phân tích AUC(0-t) và Cmax,ss với cùng mức chấp nhận đã nêu ở trên.
Trong một số trường hợp hiếm gặp khi sử dụng dữ liệu nồng độ thuốc trong nước tiểu, Ae(0-t) được phân tích với cùng mức chấp nhận tương tự AUC(0-t) trên. Rmax cũng được phân tích với cùng mức chấp nhận tương tự Cmax.
Không yêu cầu phân tích thông kê đối với tmax. Tuy nhiên, nếu tốc độ giải phóng nhanh được xác định là có ý nghĩa lâm sàng và quan trọng là liên quan đến khởi phát tác dụng hoặc các tác dụng bất lợi, không nên có sự khác biệt rõ ràng về giá trị tmax trung bình và sự biến thiên của nó giữa thuốc thử và thuốc đối chứng.
Trong những trường hợp đặc biệt khi thuốc có khoảng điều trị hẹp, giới hạn chấp nhận đối với AUC có thể cần quy định chặt hơn (xem mục 3.1.9). Ngoại ra, đối với các thuốc có tính biến thiên lớn, giới hạn chấp nhận đối với Cmax có thể được mở rộng trong một số trường hợp (xem mục 3.1.10).
Phân tích thống kê
Đánh giá tương đương sinh học dựa trên khoảng tin cậy 90% của tỷ số giữa các giá trị trung bình nhân của quần thể (thuốc thử/ thuốc đối chứng) đối với các thông số quan tâm. Phương pháp này tương đương với hai kiểm định một phía sử dụng giả thuyết ‘không’ (null hypothesis) cho trường hợp không tương đương sinh học ở mức ý nghĩa 5%.
Cần phân tích các thông số dược động học quan tâm bằng kiểm định ANOVA. Cần chuyển dữ liệu sang dạng logarit trước khi tiến hành phân tích. Khoảng tin cậy của sự khác biệt giữa các thuốc theo dữ liệu log hóa được xác định từ mô hình ANOVA. Khoảng tin cậy này sau đó được chuyển lại dạng ban đầu (khử log) để thu được khoảng tin cậy mong muốn cho tỷ số tính theo thang đo ban đầu. Không chấp nhận phân tích phi tham số.
Mô hình chính xác được sử dụng trong phân tích cần được xác định trước trong đề cương nghiên cứu. Phân tích thống kê cần tính toán đến các nguồn biến thiên được giả định là có ảnh hưởng đến biến đáp ứng. Các yếu tố được sử dụng trong mô hình ANOVA thường bao gồm trình tự, đối tượng tham gia trong mỗi trình tự, giai đoạn và thuốc nghiên cứu. Nên sử dụng mô hình ảnh hưởng cố định thay vì mô hình ảnh hưởng ngẫu nhiên cho tất cả các yếu tố.
Sự nhiễm chéo (carry-over)
Thử nghiệm đánh giá sự nhiễm chéo không được xem là có ý nghĩa và không nên đưa ra các quyết định liên quan đến phân tích (ví dụ, chỉ phân tích giai đoạn đầu tiên) dựa trên thử nghiệm này. Có thể xác định trực tiếp khả năng nhiễm chéo bằng cách kiểm tra nồng độ thuốc trong huyết tương trước khi dùng thuốc trong giai đoạn thứ hai (và sau đó, nếu áp dụng).
Nếu có bất kì đối tượng tham gia nghiên cứu nào có nồng độ trước dùng thuốc lớn hơn 5% giá trị Cmax của cùng đối tượng trong cùng giai đoạn, cần loại bỏ dữ liệu ở giai đoạn này của đối tượng khỏi phân tích thống kê. Trong một thử nghiệm hai giai đoạn, điều này sẽ dẫn đến việc người tình nguyện bị loại khỏi phân tích. Thử nghiệm sẽ không được chấp nhận nữa nếu sự loại dữ liệu này làm cho số người tình nguyện còn lại thấp hơn 12 người. Cách xử lý này không áp dụng với các thuốc nội sinh.
Thiết kế hai đợt
Có thể cho phép áp dụng mô hình thiết kế hai đợt khi cố gắng để chứng minh tương đương sinh học. Một nhóm người tình nguyện ban đầu có thể đã dùng thuốc và dữ liệu đã được phân tích. Nếu chưa chứng minh được tương đương sinh học, có thể phải sử dụng thêm một nhóm người tình nguyện khác và gộp dữ liệu của cả hai nhóm trong phân tích cuối cùng. Nếu áp dụng cách này, các bước thích hợp phải được thực hiện để tránh sai lầm loại I nói chung trong thực nghiệm và tiêu chí dừng cần được xác định rõ trước khi tiến hành nghiên cứu. Phân tích dữ liệu của đợt đầu tiên cần được tiến hành như một phân tích tạm thời và cả hai phân tích được thực hiện với mức ý nghĩa hiệu chỉnh (với khoảng tin cậy thích hợp sử dụng xác xuất bao phủ hiệu chỉnh lớn hơn 90%). Ví dụ, có thể chấp nhận sử dụng khoảng tin cậy 94,12% cho cả phân tích của đợt 1 và phân tích dữ liệu gộp từ các đợt 1 và 2, tuy nhiên, có nhiều lựa chọn thay thế cũng được chấp nhận và lựa chọn giá trị alpha là bao nhiêu để dùng trong phân tích tạm thời tùy thuộc vào quyết định của cơ sở nghiên cứu. Kế hoạch sử dụng mô hình nghiên cứu hai đợt phải được làm rõ trong đề cương nghiên cứu cùng với mức ý nghĩa hiệu chỉnh được sử dụng cho mỗi phân tích.
Khi phân tích dữ liệu kết hợp từ hai đợt, yếu tố đợt nghiên cứu cần đưa vào mô hình ANOVA.
Trình bày dữ liệu
Tham khảo PHỤ LỤC IV (mẫu báo cáo nghiên cứu tương đương sinh học ASEAN)
3.1.9 Thuốc có khoảng điều trị hẹp
Trong những trường hợp đặc biệt khi thuốc có khoảng điều trị hẹp, giới hạn chấp nhận đối với AUC cần được quy định chặt chẽ hơn, trong khoảng 90,00-111,11%. Trong trường hợp Cmax có vai trò đặc biệt quan trọng đối với tính an toàn, hiệu quả hoặc việc theo dõi nồng độ thuốc, cũng cần áp dụng giới hạn chấp nhận 90,00-111,11% cho thông số này. Không thể đưa ra một bộ tiêu chí để xác định các thuốc có khoảng điều trị hẹp và điều này phải được xem xét trong từng trường hợp cụ thể dựa trên những lưu ý về lâm sàng.
3.1.10 Các thuốc hoặc chế phẩm thuốc có tính biến thiên cao
Các chế phẩm thuốc có tính biến thiên cao (HVDP) là các chế phẩm có sự biến thiên của một thông số dược động học trong cá thể lớn hơn 30%. Nếu cơ sở đăng ký cho rằng chế phẩm thuốc có thể được coi là có tính biến thiên cao về tốc độ/ mức độ hấp thu, có thể thực hiện một nghiên cứu thiết kế chéo lặp lại (replicate cross-over design).
Đối với những chế phẩm thuốc có tính biến thiên cao, mà sự khác nhau lớn hơn của trị số Cmax được coi là không có ý nghĩa lâm sàng dựa trên các bằng chứng lâm sàng thuyết phục, có thể đánh giá tương đương sinh học với giới hạn chấp nhận rộng hơn. Trong trường hợp này, giới hạn chấp nhận với Cmax có thể mở rộng tới khoảng tối đa 69,84 – 143,19%. Để áp dụng giới hạn chấp nhận mở rộng, nghiên cứu tương đương sinh học phải được thiết kế lặp lại và kết quả phải cho thấy sự biến thiên trong cá thể đối với Cmax của thuốc đối chứng trong nghiên cứu là lớn hơn 30%. Cơ sở đăng ký cần chứng minh sự biến thiên trong cá thể tính được là tin cậy và không phải do các giá trị sai khác cá biệt gây ra. Yêu cầu về khoảng giới hạn chấp nhận mở rộng phải được xác định trước trong đề cương nghiên cứu.
Mức độ mở rộng giới hạn chấp nhận đối với Cmax được xác định như sau:
|
Hệ số biến thiên trong cá thể (CV%)* |
Giới hạn dưới |
Giới hạn trên |
|
30 |
80,00 |
125,00 |
|
35 |
77,23 |
129,48 |
|
40 |
74,62 |
134,02 |
|
45 |
72,15 |
138,59 |
|
≥50 |
69,84 |
143,19 |
* CV (%) = 100 √ ℮s2WR – 1
Tỷ số trung bình nhân (GMR) cần nằm trong giới hạn chấp nhận thông thường 80,00-125,00%.
Không áp dụng việc mở rộng giới hạn chấp nhận với AUC trong trường hợp có biến thiên cao trong cá thể đối với AUC. Do đó, giới hạn chấp nhận của thông số này luôn duy trì ở mức 80,00-125,00%, không tính đến sự biến thiên.
Trong nghiên cứu được thiết kế lặp lại, có thể áp dụng thiết kế chéo 3 giai đoạn hoặc 4 giai đoạn.
3.2 Đánh giá độ hòa tan invitro
Các khía cạnh chung của đánh giá độ hòa tan in vitro được trình bày ngắn gọn tại phụ lục I, bao gồm những yêu cầu cơ bản về cách sử dụng hệ số tương đồng (f2-test).
3.2.1 Đánh giá độ hòa tan in vitro bổ sung cho các nghiên cứu tương đương sinh học
Cần báo cáo kết quả đánh giá độ hòa tan in vitro của lô thuốc thử và thuốc đối chứng đã dùng trong nghiên cứu tương đương sinh học trong ba dung dịch đệm khác nhau (thường là đệm pH 1,2; 4,5 và 6,8) và môi trường mong muốn thuốc giải phóng (môi trường QC, nếu cần và sẵn có). Đối với các dạng bào chế đặc biệt như ODT (viên nén phân tán trong miệng), có thể phải áp dụng những điều kiện thí nghiệm khác. Cần báo cáo kết quả dưới dạng tỷ lệ phần trăm lượng dược chất được hòa tan so với nhãn theo thời gian, bao gồm giá trị trung bình và thống kê tóm tắt thông số này.
Trừ khi có lý do hợp lý, các mức tiêu chuẩn về độ hòa tan in vitro được sử dụng để kiểm tra chất lượng thuốc cần được xác định từ dữ liệu độ hòa tan của lô thuốc thử đã được chứng minh tương đương sinh học với thuốc đối chứng (xem Phụ lục I).
Trường hợp kết quả so sánh độ hòa tan in vitro của các lô không phản ánh tương đương sinh học như được chứng minh bằng thử nghiệm in vivo, ưu tiên sử dụng kết quả của thử nghiệm in vivo. Tuy nhiên, cần chỉ rõ và giải thích nguyên nhân dẫn đến sự khác nhau giữa hai thử nghiệm này.
3.2.2 Đánh giá độ hòa tan in vitro để cung cấp dữ liệu cho xem xét miễn thử các hàm lượng khác
Dữ liệu độ hòa tan in vitro thích hợp được dùng để khẳng định cho sự phù hợp của việc miễn thử tương đương sinh học in vivo thêm cho các hàm lượng khác ngoài hàm lượng đã được nghiên cứu. Do đó, cần đánh giá độ hòa tan ở các môi trường pH khác nhau như đã đề cập ở phần trên đây (thường là pH 1,2; 4,5 và 6,8) trừ khi có lý do hợp lý. Cần chứng minh tính tương đồng về độ hòa tan in vitro (xem phụ lục I) ở tất cả các điều kiện đối với các hàm lượng khác nhau của thuốc đã thử, tức là giữa (các) hàm lượng khác còn lại so với hàm lượng (lô) đã dùng trong nghiên cứu tương đương sinh học in vivo.
Trong các môi trường pH mà điều kiện hòa tan thích hợp không thể đạt được với tất cả các hàm lượng, độ hòa tan in vitro có thể khác nhau giữa các hàm lượng. Tuy nhiên, cần so sánh với hàm lượng tương ứng của thuốc đối chứng để xác nhận rằng sự khác biệt này chỉ liên quan đến dược chất mà không liên quan đến công thức bào chế. Ngoài ra, cơ sở đăng ký có thể cung cấp biểu đồ hòa tan tương đồng ở cùng mức liều (ví dụ, hai viên nén 5 mg có thể được so sánh với một viên nén 10 mg).
3.3 Báo cáo nghiên cứu
3.3.1 Báo cáo nghiên cứu tương đương sinh học
Báo cáo nghiên cứu tương đương sinh học là bộ tài liệu đầy đủ gồm đề cương nghiên cứu, tiến hành nghiên cứu và đánh giá kết quả nghiên cứu. Báo cáo này cần được viết theo mẫu nêu tại PHỤ LỤC IV (mẫu báo cáo nghiên cứu tương đương sinh học ASEAN) và có chữ ký của nghiên cứu viên. (Các) nghiên cứu viên chịu trách nhiệm từng phần của nghiên cứu (nếu có) cần ký tên vào mục tương ứng của báo cáo.
Cần ghi rõ tên và vị trí của (các) nghiên cứu viên phụ trách nghiên cứu, nơi thực hiện và thời gian thực hiện nghiên cứu. Các bản xác nhận kiểm tra, thanh tra (nếu có) cần bao gồm trong báo cáo.
Báo cáo nghiên cứu cần ghi đầy đủ tên, hàm lượng, dạng bào chế, số lô sản xuất, nhà sản xuất, ngày hết hạn sử dụng và xuất xứ (tên quốc gia đã cung cấp mẫu thuốc) của thuốc đối chứng.
Đồng thời, cũng cần cung cấp đầy đủ tên và công thức của (các) thuốc thử đã sử dụng trong nghiên cứu. Thông tin về cỡ lô, số lô, ngày sản xuất và nếu có thể- ngày hết hạn của thuốc thử cũng cần được nêu trong báo cáo.
Phiếu phân tích của các lô thuốc thử và thuốc đối chứng dùng trong nghiên cứu cần đưa vào phần phụ lục của báo cáo nghiên cứu.
Cần trình bày chi tiết các dữ liệu về nồng độ thuốc, dữ liệu dược động học và phân tích thống kê.
3.3.2 Các dữ liệu khác cần đưa vào hồ sơ đăng ký
Cơ sở đăng ký cần nộp một bản tuyên bố đã ký tên xác nhận thuốc thử có cùng lượng các thành phần trong công thức và được sản xuất theo cùng một quy trình như thuốc nộp hồ sơ đăng ký lưu hành cho cơ quan quản lý. Cần xác nhận rõ chế phẩm thử đã được nâng cỡ lô sản xuất hay chưa. Cần cung cấp biểu đồ độ hòa tan so sánh (xem mục 3.2).
Các dữ liệu đầy đủ và chi tiết để có thể tính toán lại các thông số dược động học và phân tích thống kê, ví dụ dữ liệu về thời điểm lấy mẫu thực tế, nồng độ thuốc, giá trị các thông số dược động học của mỗi người tình nguyện trong từng giai đoạn và bảng phân bố ngẫu nhiên khi dùng thuốc cần được chuẩn bị sẵn dưới dạng bản điện tử thích hợp (như định dạng excel hoặc text được phân cách bởi các khoảng trống và dấu phẩy) để cung cấp cho cơ quan quản lý khi có yêu cầu.
3.4 Đăng ký thay đổi
Nếu một thuốc được thay đổi công thức so với thuốc ban đầu đã được phê duyệt hoặc thay đổi phương pháp sản xuất theo hướng có thể ảnh hưởng đến sinh khả dụng của thuốc, cần thực hiện nghiên cứu tương đương sinh học in vivo, trừ trường hợp có biện giải xác đáng. Bất kỳ biện giải nào được đưa ra cần dựa trên những sự cân nhắc chung, ví dụ như các căn cứ nêu tại phụ lục III, hoặc một tương quan in vitro/ in vivo có thể chấp nhận đã được thiết lập hay chưa.
Trong các trường hợp chế phẩm trước khi tiến hành thay đổi đã được nghiên cứu sinh khả dụng và mối tương quan thích hợp giữa độ hòa tan in vitro và dược động học in vivo đã được thiết lập, có thể miễn thực hiện tương đương sinh học in vivo đối với chế phẩm sau khi thay đổi nếu biểu đồ hòa tan in vitro của thuốc mới tương đồng với thuốc đã được phê duyệt trong các điều kiện thử giống như điều kiện sử dụng để thiết lập mối tương quan nói trên (xem Phụ lục I).
Khi thực hiện những thay đổi đối với một thuốc generic, thuốc được dùng để so sánh trong nghiên cứu tương đương sinh học thường là một lô thuốc đối chứng đang lưu hành. Nếu thuốc đối chứng theo qui định hiện không có lưu hành trên thị trường, có thể tiến hành so sánh với thuốc đã được phê duyệt trước đó (tức là thuốc generic trước khi thay đổi đã được phê duyệt), tuy nhiên, điều này cần được biện giải thích hợp. Đối với những thay đổi không yêu cầu nghiên cứu tương đương sinh học, cần thực hiện theo những khuyến cáo và những quy định đã được đưa ra trong hướng dẫn hiện hành khác của cơ quan quản lý.
CÁC KHÁI NIỆM
Tương đương bào chế
Các chế phẩm thuốc được coi là tương đương bào chế nếu chúng cùng chứa một (hoặc một số) dược chất với cùng hàm lượng, cùng dạng bào chế và đáp ứng các tiêu chuẩn chất lượng tương tự hoặc tương đương nhau.
Tương đương bào chế không nhất thiết phải bao hàm tương đương sinh học do những khác biệt về tá dược và/ hoặc quy trình sản xuất có thể thúc đẩy hoặc làm chậm quá trình hòa tan và/ hoặc hấp thu.
Thế phẩm bào chế
Thế phẩm bào chế là các chế phẩm thuốc khác nhau về dạng muối, ester, ether, dạng đồng phân, hỗn hợp các đồng phân, phức chất hoặc dẫn chất của cùng một thành phần có hoạt tính hoặc khác nhau về dạng bào chế hoặc hàm lượng.
Các thông số dược động học
Ae(0-t): Lượng thuốc bài tiết tích lũy trong nước tiểu dưới dạng không biến đổi từ khi sử dụng thuốc cho đến thời điểm t;
AUC(0-t): Diện tích dưới đường cong biểu diễn nồng độ thuốc trong huyết tương từ khi sử dụng thuốc cho đến điểm lấy mẫu cuối cùng có thể định lượng tại thời điểm t;
AUC(0-∞): Diện tích dưới đường cong biểu diễn nồng độ thuốc trong huyết tương ngoại suy tới thời điểm ở vô cùng;
AUC(0-זּ): AUC trong một khoảng liều tại trạng thái ổn định;
AUC(0-72h): Diện tích dưới đường cong biểu diễn nồng độ thuốc trong huyết tương từ khi sử dụng thuốc cho đến thời điểm 72 giờ;
Cmax: Nồng độ đỉnh trong huyết tương;
Cmax,ss: Nồng độ đỉnh tại trạng thái ổn định;
Diện tích còn lại Phần diện tích được ngoại suy (AUC(0-∞) - AUC(0-t))/ AUC(0-∞));
 Tốc độ bài tiết tối đa qua nước tiểu;
Tốc độ bài tiết tối đa qua nước tiểu;
tmax: Thời gian đạt được Cmax;
tmax,ss: Thời gian đạt được Cmax,ss;
t1/2: Nửa đời của thuốc trong huyết tương;
λz: Hằng số tốc độ thải trừ pha cuối;
SmPC Tóm tắt đặc tính sản phẩm.
PHỤ LỤC I
Thử độ hòa tan và tính tương đồng của biểu đồ hòa tan
1. Các khía cạnh chung của phép thử độ hòa tan liên quan đến sinh khả dụng
Trong phát triển thuốc, thử nghiệm độ hòa tan được sử dụng như một công cụ để phát hiện các yếu tố về bào chế đang có ảnh hưởng và có thể có tác động lớn đến sinh khả dụng của thuốc. Ngay sau khi công thức và quy trình sản xuất được xác định, thử nghiệm đánh giá độ hòa tan được sử dụng để kiểm tra chất lượng khi nâng cỡ lô sản xuất và để đảm bảo tính đồng nhất giữa các lô cũng như đảm bảo biểu đồ hòa tan của các lô sản xuất sau là tương tự biểu đồ hòa tan của các lô sử dụng trong thử nghiệm lâm sàng then chốt. Ngoài ra, trong một số trường hợp, có thể sử dụng thử nghiệm đánh giá độ hòa tan để làm cơ sở cho việc miễn thử tương đương sinh học. Do đó, nghiên cứu độ hòa tan có thể phục vụ cho một số mục đích sau đây:
i - Đánh giá chất lượng thuốc
● Để thu được thông tin về những lô thuốc thử sử dụng trong các nghiên cứu tương đương sinh học và các nghiên cứu lâm sàng then chốt nhằm cung cấp dữ liệu cho việc xác lập các thông số kỹ thuật trong kiểm tra chất lượng
● Được sử dụng làm công cụ trong kiểm tra chất lượng để chứng minh tính ổn định trong sản xuất
● Để có thông tin về thuốc đối chứng sử dụng trong các nghiên cứu sinh khả dụng/tương đương sinh học và các nghiên cứu lâm sàng then chốt.
ii - Sử dụng thay thế cho nghiên cứu tương đương sinh học
● Để chứng minh tính tương đồng giữa các công thức bào chế khác nhau của một dược chất và thuốc tham chiếu (miễn thử tương đương sinh học, ví dụ đối với các trường hợp thay đổi, thay đổi công thức trong quá trình phát triển và các thuốc generic- xem mục 3.2 và Phụ lục III)
● Để đánh giá tính đồng nhất giữa các lô của các thuốc (thử và đối chứng), được sử dụng làm cơ sở để lựa chọn những lô thích hợp cho nghiên cứu in vivo.
Cần xây dựng phương pháp thử cho chế phẩm liên quan dựa trên những yêu cầu chung và/ hoặc yêu cầu cụ thể được quy định trong dược điển. Trong trường hợp những yêu cầu này được xem là không phù hợp và/ hoặc không phản ánh độ hòa tan in vivo (tương thích sinh học), có thể cân nhắc sử dụng những phương pháp thay thế nếu chứng minh được những phương pháp này có khả năng nhận biết và phân biệt sự khác nhau giữa các lô với kết quả chấp nhận hay không chấp nhận thuốc in vivo. Phải luôn luôn lưu ý đến các thông tin mới nhất bao gồm sự tác động qua lại của các đặc tính suy ra từ hệ thống phân loại sinh dược BCS và dạng bào chế.
Cần lấy đủ số lượng mẫu tại các thời điểm khác nhau để thu được biểu đồ hòa tan có ý nghĩa và cần lấy mẫu tối thiểu mỗi 15 phút một lần. Khuyến cáo lấy mẫu thường xuyên hơn trong giai đoạn có sự thay đổi lớn nhất về biểu đồ hòa tan. Đối với các thuốc có tốc độ hòa tan nhanh (hòa tan hoàn toàn trong 30 phút), có thể phải lấy mẫu 5 hoặc 10 phút một lần để có dữ liệu đầy đủ.
Nếu một dược chất được coi là có khả năng tan tốt, dược chất này thường sẽ không có vấn đề về sinh khả dụng nếu dạng bào chế có khả năng hòa tan nhanh trong khoảng pH sinh lý và các tá dược trong công thức được xác định là không ảnh hưởng đến sinh khả dụng. Trái lại, nếu một dược chất được coi là có độ tan kém hoặc hạn chế, giai đoạn hạn chế tốc độ hấp thu có thể là quá trình hòa tan dược chất từ dạng bào chế. Điều này cũng đúng với trường hợp sử dụng tá dược kiểm soát sự giải phóng và tiếp theo đó là hòa tan dược chất. Trong các trường hợp này, khuyến cáo tiến hành thử ở nhiều điều kiện khác nhau với quy trình lấy mẫu thích hợp.
2. Tính tương đồng của biểu đồ hòa tan
Đánh giá tính tương đồng của biểu đồ hòa tan và bất kì kết luận nào suy ra từ kết quả (ví dụ, làm cơ sở để miễn thử tương đương sinh học) chỉ có thể được coi là có giá trị nếu biểu đồ hòa tan được mô tả đầy đủ với số lượng điểm lấy mẫu đủ lớn.
Đối với các thuốc giải phóng ngay, ngoài những hướng dẫn đã được đề cập tại mục 1 trên đây, cần so sánh tại thời điểm 15 phút để xác định xem thuốc đã đạt được trạng thái hòa tan hoàn toàn trước khi ra khỏi dạ dày hay chưa.
Khi tỷ lệ thuốc hòa tan trong 15 phút vượt qua 85%, biểu đồ hòa tan có thể được coi là tương đồng mà không cần thực hiện thêm những đánh giá về mặt toán học khác.
Trong trường hợp lượng thuốc hòa tan trong vòng 15 phút không đạt trên 85% nhưng đạt được trong vòng 30 phút, cần lấy mẫu tại ít nhất ba thời điểm: thời điểm đầu tiên là trước 15 phút, thời điểm thứ hai tại 15 phút và thời điểm thứ ba khi tỷ lệ thuốc giải phóng được khoảng 85%.
Đối với dạng thuốc giải phóng biến đổi, cần tuân theo hướng dẫn được xây dựng riêng cho dạng bào chế này.
Có thể xác định tính tương đồng của độ hòa tan sử dụng đại lượng thống kế ƒ2 như sau:
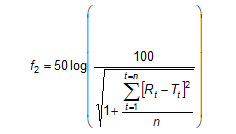
Trong công thức này, ƒ2 là hệ số tương đồng, n là số điểm lấy mẫu, R(t) là trung bình của tỷ lệ phần trăm thuốc đối chứng được hòa tan tại thời điểm t kể từ khi bắt đầu thử nghiệm; T(t) là trung bình của tỷ lệ phần trăm thuốc thử được hòa tan tại thời điểm t kể từ khi bắt đầu thử nghiệm. Đối với cả thuốc thử và thuốc đối chứng, cần xác định tỷ lệ phần trăm thuốc hòa tan.
Đánh giá hệ số tương đồng theo các điều kiện sau đây:
● Tối thiểu 3 thời điểm lấy mẫu (không tính thời điểm bắt đầu)
● Các thời điểm lấy mẫu cần giống nhau giữa hai thuốc so sánh
● Có ít nhất mười hai giá trị riêng biệt cho mỗi thời điểm đối với mỗi thuốc
● Không lấy quá một giá trị trung bình tỷ lệ phần trăm thuốc hòa tan > 85% cho mỗi thuốc.
● Độ lệch chuẩn tương đối hoặc hệ số biến thiên của một chế phẩm cần nhỏ hơn 20% đối với điểm lấy mẫu đầu tiên và dưới 10% đối với điểm thứ hai trở đi.
Giá trị f2 từ 50 đến 100 phản ánh biểu đồ hòa tan của hai thuốc là tương đồng với nhau.
Nếu thống kê giá trị f2 không phù hợp, có thể so sánh tính tương đồng bằng phương pháp phụ thuộc hoặc không phụ thuộc mô hình, ví dụ, so sánh thống kê đa biến giữa các thông số của hàm Weibull hoặc tỷ lệ phần trăm thuốc được hòa tan tại các thời điểm khác nhau.
Các phương pháp thống kê thay thế cho thống kê f2 để đánh giá sự tương đồng của biểu đồ hòa tan cũng được xem xét chấp nhận nếu phương pháp sử dụng có hiệu lực về mặt thống kê và kết quả được giải thích thỏa đáng.
Giới hạn cho phép của tính tương đồng cần được xác định trước và phải được giải thích đầy đủ, không nên khác nhau quá 10%. Ngoài ra, sự biến thiên về độ hòa tan giữa thuốc thử và thuốc đối chứng cũng cần tương tự nhau, tuy nhiên, cũng có thể chấp nhận trường hợp chế phẩm thử có mức độ biến thiên thấp hơn.
Cũng cần cung cấp bằng chứng về việc đã thẩm định phần mềm thống kê. Cần mô tả rõ và giải thích chi tiết về các bước tiến hành khi áp dụng quy trình này kèm theo bảng tóm tắt thích hợp.
PHỤ LỤC II
Yêu cầu về nghiên cứu tương đương sinh học cho các dạng bào chế khác
Mặc dù hướng dẫn này liên quan đến các thuốc giải phóng ngay, Phụ lục II đưa ra một số hướng dẫn chung về những yêu cầu đối với dữ liệu tương đương sinh học của các dạng bào chế khác và kiểu đặc biệt của dạng bào chế giải phóng ngay.
Khi chế phẩm thử khác chế phẩm đối chứng về dạng muối, ester, ether, đồng phân, hỗn hợp đồng phân, phức chất hoặc dẫn chất của hoạt chất, cần chứng minh tương đương sinh học in vivo. Tuy nhiên, khi chế phẩm thử và chế phẩm đối chứng chứa dược chất giống nhau (hoặc chứa các muối có đặc tính tương tự nhau được trình bày tại phụ lục III, mục III), trong một số trường hợp, có thể không cần thực hiện các nghiên cứu tương đương sinh học in vivo như mô tả dưới đây và trong Phụ lục III.
Các dạng bào chế giải phóng ngay dùng đường uống có tác dụng toàn thân
Đối với các dạng bào chế như viên nén, viên nang và hỗn dịch uống, cần thực hiện nghiên cứu tương đương sinh học trừ khi được áp dụng miễn thử sinh học (xem PHỤ LỤC III). Với các viên nén rã trong miệng và các loại dung dịch uống, áp dụng những khuyến cáo riêng được mô tả dưới đây.
Viên nén phân tán trong miệng
Viên nén phân tán trong miệng (ODT) được bào chế để phân tán nhanh trong miệng. Vị trí đặt trong miệng và thời gian tiếp xúc có thể đóng vai trò quan trọng trong trường hợp dược chất được hòa tan trong miệng, đồng thời có thể được hấp thu trực tiếp qua niêm mạc miệng. Tùy thuộc vào từng thuốc, ví dụ, có thể nuốt dược chất đã được bao và quá trình hấp thu từ đường tiêu hóa sẽ xuất hiện sau đó. Nếu có thể chứng minh dược chất không được hấp thu tại khoang miệng mà được nuốt và hấp thu tại đường tiêu hóa, có thể được xem xét miễn thử tương đương sinh học đối với chế phẩm dựa trên BCS (xem Phụ lục III). Nếu không thể chứng minh điều này, cần đánh giá tương đương sinh học trên người.
Nếu chế phẩm thử ODT là sự phát triển tiếp theo của một thuốc dùng đường miệng khác, khuyến cáo thực hiện một nghiên cứu ba giai đoạn để đánh giá viên nén rã trong miệng khi được sử dụng kèm hoặc không kèm chất lỏng. Tuy nhiên, nếu tương đương sinh học được thiết lập giữa thuốc ODT không dùng cùng với nước và thuốc đối chứng được dùng cùng với nước trong một nghiên cứu 2 giai đoạn, có thể ngoại suy tương đương sinh học cho ODT khi dùng cùng với nước.
Nếu ODT là một chế phẩm generic được so sánh với một thuốc đối chứng là ODT đã được phê duyệt, cần lưu ý những khuyến cáo sau đây khi thiết kế nghiên cứu:
● Nếu chế phẩm tham chiếu có thể được sử dụng kèm hoặc không kèm nước, cần chứng minh tương đương sinh học trong điều kiện không dùng cùng với nước do điều kiện này phản ánh tốt hơn mục đích sử dụng của dạng bào chế ODT. Điều này đặc biệt quan trọng khi dược chất có thể được hòa tan và hấp thu một phần tại khoang miệng. Nếu tương đương sinh học được chứng minh khi không dùng cùng với nước, có thể ngoại suy tương đương sinh học trong trường hợp dùng thuốc cùng với nước.
● Nếu chế phẩm tham chiếu chỉ được sử dụng trong một điều kiện (ví dụ, chỉ dùng cùng với nước), cần đánh giá tương đương sinh học trong điều kiện này (sử dụng thiết kế chéo đôi quy ước).
● Nếu chế phẩm tham chiếu chỉ được sử dụng trong một điều kiện (ví dụ, chỉ dùng cùng với nước) và chế phẩm thử được dự kiến có thêm một cách dùng khác (ví dụ, không dùng cùng với nước), cần so sánh chế phẩm thử được dùng theo cách mới và cách thông thường với chế phẩm tham chiếu được dùng theo cách thông thường (thiết kế chéo 6 trình tự, 3 giai đoạn và 3 phương án dùng thuốc).
Trong các nghiên cứu đánh giá thuốc ODT ở điều kiện không dùng cùng với nước, cần làm ướt miệng bằng cách uống 20 ml nước ngay trước khi đặt viên ODT trên lưỡi. Khuyến cáo không dùng chất lỏng trong vòng một giờ kể từ khi dùng thuốc.
Các dạng bào chế dùng đường miệng khác như phim rã trong miệng, viên nén hoặc phim dính trong niêm mạc miệng, viên nén đặt dưới lưỡi và viên nhai có thể được đánh giá tương tự như ODT. Các nghiên cứu tương đương sinh học cần được thực hiện theo khuyến cáo cách sử dụng sản phẩm.
Dung dịch uống
Nếu thuốc thử là một dung dịch uống trong nước tại thời điểm dùng thuốc và chứa dược chất với nồng độ tương tự một dung dịch uống đã được phê duyệt, thuốc thử này có thể được miễn thử tương đương sinh học. Tuy nhiên, nếu sử dụng tá dược có khả năng ảnh hưởng đến nhu động đường tiêu hóa (như sorbitol, mannitol…), khả năng hấp thu (như chất diện hoạt hoặc những tá dược có thể ảnh hưởng đến protein vận chuyển), độ tan in vivo (như các đồng dung môi) hoặc độ ổn định in vivo của dược chất, cần tiến hành đánh giá tương đương sinh học, trừ khi sự khác biệt về lượng của những tá dược này có thể được giải thích đầy đủ bằng cách tham chiếu với các dữ liệu khác. Các yêu cầu về tính tương tự của tá dược áp dụng cho dung dịch uống tương tự các yêu cầu được đề cập trong phần Miễn thử sinh học (xem Phụ lục III, mục IV.2 Tá dược).
Trong trường hợp thuốc thử là một dung dịch uống được dự định tương đương sinh học với một thuốc ở dạng bào chế giải phóng ngay dùng đường miệng khác, cần tiến hành đánh giá tương đương sinh học.
Dạng bào chế phối hợp cố định
Những yêu cầu về tương đương sinh học đối với trường hợp này được trình bày trong “Hướng dẫn phát triển lâm sàng chế phẩm thuốc phối hợp cố định”. Khả năng được miễn thử sinh học đối với Chế phẩm thuốc phối hợp cố định được trình bày tại Phụ lục III, mục V.
Dạng bào chế giải phóng ngay có tác dụng toàn thân không dùng đường miệng
Mục này áp dụng đối với một số dạng thuốc như dạng đặt trực tràng. Nhìn chung, cần tiến hành đánh giá tương đương sinh học. Có thể cân nhắc miễn thử tương đương sinh học trong trường hợp chế phẩm thử là dung dịch chứa dược chất với nồng độ tương tự một dung dịch thuốc đã được phê duyệt và giống với dung dịch tham chiếu về thành phần định tính và định lượng của các tá dược (các điều kiện như đã trình bày với dạng dung dịch uống có thể áp dụng trong trường hợp này).
Dung dịch tiêm
Nhìn chung, không yêu cầu đánh giá tương đương sinh học nếu chế phẩm được sử dụng dưới dạng dung dịch nước để truyền tĩnh mạch chứa cùng dược chất với chế phẩm đã được phê duyệt đang lưu hành. Tuy nhiên, nếu bất kì tá dược nào có tương tác với dược chất (ví dụ, tạo phức) hoặc ảnh hưởng đến sự phân bố của dược chất, cần tiến hành đánh giá tương đương sinh học trừ khi hai chế phẩm chứa các tá dược giống nhau với hàm lượng tương tự nhau và phải chứng minh được bất kì sự khác biệt nào về lượng của các loại tá dược đều không ảnh hưởng đến dược động học của hoạt chất.
Trường hợp dùng thuốc theo những đường tiêm khác, như tiêm bắp hoặc tiêm dưới da, và chế phẩm thử là cùng một loại dung dịch (dung dịch nước hoặc dầu), cùng nồng độ hoạt chất và cùng thành phần tá dược với khối lượng tương đương như chế phẩm đã được phê duyệt đang lưu hành, không yêu cầu đánh giá tương đương sinh học. Ngoài ra, không yêu cầu đánh giá tương đương sinh học đối với dạng thuốc tiêm là dung dịch trong nước chứa các loại tá dược tương tự nhau với khối lượng tương đương, nếu có thể chứng minh tá dược được sử dụng không ảnh hưởng đến độ nhớt của thuốc.
Dạng bào chế liposom, micell và nhũ tương để tiêm truyền tĩnh mạch
● Dạng liposom: Các vấn đề về dược động học liên quan đến dạng bào chế liposom để tiêm truyền tĩnh mạch cần được xem xét đặc biệt và không được đề cập trong hướng dẫn này.
● Nhũ tương: Nhũ tương thường không được miễn thử sinh học. Tuy nhiên, có thể cân nhắc miễn thử sinh học cho dạng bào chế nhũ tương trong các trường hợp sau đây:
(a) chế phẩm thuốc không được thiết kế để kiểm soát sự giải phóng hoặc di chuyển dược chất
(b) cách dùng và tốc độ đưa thuốc vào cơ thể tương tự chế phẩm đã được phê duyệt đang lưu hành
Trong những trường hợp này, thành phần công thức của chế phẩm thử cần tương tự như thành phần công thức của nhũ tương đã được phê duyệt đang lưu hành cả về định tính và định lượng và cần cung cấp dữ liệu thích hợp để chứng minh những đặc tính lý hóa tương đồng bao gồm kích thước phân bố của pha lipid đã phân tán cùng với việc xem xét các đặc tính có liên quan khác của nhũ tương, ví dụ đặc tính bề mặt gồm thể zeta và tính chất lưu biến.
● Lipid dùng để cung cấp chất dinh dưỡng dùng đường tiêm tĩnh mạch có thể được miễn thử sinh học nếu cung cấp đầy đủ dữ liệu để chứng minh tính tương tự về đặc tính lý hóa. Có thể biện giải sự khác biệt về thành phần dựa trên bản chất và mục đích điều trị của dạng bào chế này.
● Dạng bào chế tạo micell: dung dịch micell để dùng theo đường tĩnh mạch có thể được xem là dung dịch ‘phức tạp’ và do đó, thường không được xem xét miễn thử sinh học. Tuy nhiên, có thể cân nhắc miễn thử sinh học với dạng bào chế micell trong những trường hợp sau đây:
(a) Micell được pha loãng nhanh và chế phẩm thuốc không được thiết kế để kiểm soát việc giải phóng hoặc phân bố thuốc.
(b) Cách dùng và tốc độ đưa thuốc vào cơ thể tương tự như chế phẩm đã được phê duyệt đang lưu hành.
(c) Tá dược không ảnh hưởng đến sự phân bố dược chất.
Trong những trường hợp trên, thành phần của dịch truyền micell tại thời điểm ngay trước khi sử dụng cần tương tự với chế phẩm tham chiếu đã được phê duyệt về mặt định tính và định lượng, đồng thời cần cung cấp đầy đủ dữ liệu để chứng minh tính tương tự về đặc tính lý hóa. Ví dụ, nồng độ micell tới hạn, khả năng hòa tan của thuốc (như nồng độ chất tan thêm vào tối đa), dược chất tự do và liên kết, kích thước micell.
Điều này cũng áp dụng đối với những thay đổi nhỏ về thành phần định tính hoặc định lượng trong công thức với điều kiện không có bất cứ thay đổi nào về lượng hoặc loại các chất diện hoạt được sử dụng.
Dạng bào chế giải phóng biến đổi có tác dụng toàn thân
Dạng bào chế giải phóng biến đổi dùng qua da hoặc đường uống
Các yêu cầu thử tương đương sinh học phù hợp với hướng dẫn riêng cho dạng bào chế giải phóng biến đổi dùng qua da hoặc đường uống: Mục II (Đánh giá dược động học và lâm sàng) (CPMP/EWP/280/96).
Dạng bào chế giải phóng biến đổi dùng tiêm bắp hoặc tiêm dưới da
Đối với dạng hỗn dịch hoặc phức chất hoặc bất kỳ phức hợp nào được bào chế với mục đích trì hoãn hoặc kéo dài quá trình giải phóng dược chất để tiêm bắp hoặc tiêm dưới da, việc chứng minh tương đương sinh học cần tuân thủ những quy định đối với dạng bào chế giải phóng biến đổi dùng ngoại mạch, ví dụ, dạng bào chế qua da theo hướng dẫn tương ứng.
Thuốc dùng tại chỗ để có tác dụng tại chỗ
Đối với các thuốc sử dụng tại chỗ (sau khi đưa vào miệng, mũi, phổi, mắt, da, trực tràng, âm đạo…) với mục đích thu được đáp ứng tại vị trí đưa thuốc, các khuyến cáo cụ thể được trình bày trong những hướng dẫn riêng (ví dụ, CPMP/EWP/4151/00 rev 1, CPMP/EWP/239/95).
Có thể miễn cung cấp dữ liệu tương đương nếu chế phẩm thử là dung dịch thuốc, ví dụ thuốc nhỏ mắt, thuốc xịt mũi hoặc dung dịch bôi ngoài da, với điều kiện chế phẩm thử có cùng bản chất dung dịch (nước hoặc dầu), chứa cùng một dược chất với cùng một nồng độ như chế phẩm tham chiếu đã được phê duyệt đang lưu hành. Có thể chấp nhận những khác biệt nhỏ về thành phần tá dược nếu các đặc tính bào chế quan trọng của chế phẩm thử và chế phẩm đối chiếu giống nhau hoặc tương tự nhau. Bất kì khác biệt nào về thành phần định tính và định lượng của các tá dược phải được giải thích thỏa đáng về ảnh hưởng của những khác biệt này đến tương đương điều trị. Cách dùng thuốc và dụng cụ để đưa thuốc vào cơ thể cũng cần tương tự như chế phẩm tham chiếu trừ khi được biện giải thích hợp.
Trong trường hợp sử dụng thuốc tại chỗ dẫn tới phơi nhiễm toàn thân và có nguy cơ gây ra các tác dụng không mong muốn toàn thân, cần xác định mức độ phơi nhiễm toàn thân. Cần chứng minh rằng phơi nhiễm toàn thân của chế phẩm thử không lớn hơn so với chế phẩm đối chứng, tức là giới hạn trên của khoảng tin cậy 90% không được vượt quá giới hạn cho phép trên của tương đương sinh học là 125,00%.
Chất khí
Nếu chế phẩm thuốc ở dạng khí để hít, không yêu cầu thực hiện nghiên cứu tương đương sinh học.
PHỤ LỤC III
Miễn thử sinh học dựa trên BCS
I. Giới thiệu
Miễn thử sinh học dựa trên BCS (Hệ thống phân loại sinh dược) là một hướng tiếp cận nhằm làm giảm các nghiên cứu tương đương sinh học in vivo, tức là có thể sử dụng các dữ liệu khác để thay thế cho dữ liệu tương đương sinh học in vivo. Nghiên cứu tương đương sinh học in vivo có thể được miễn nếu có thể sử dụng các dữ liệu in vitro để dự đoán tương đương sinh học in vivo.
Quy định miễn thử tương đương sinh học chỉ áp dụng cho các dược chất tan tốt, khả năng hấp thu đã được xác định trên người và không thuộc nhóm khoảng điều trị hẹp (xem mục 3.1.9). Quy định này có thể áp dụng với chế phẩm thuốc rắn ở dạng giải phóng ngay dùng đường miệng có tác dụng toàn thân và có cùng dạng bào chế như thuốc đối chứng. Tuy nhiên, điều này không áp dụng đối với dạng bào chế đặt dưới lưỡi, dính vào niêm mạc miệng và dạng bào chế giải phóng biến đổi. Đối với thuốc phân tán trong miệng, chỉ có thể áp dụng hướng tiếp cận miễn thử sinh học dựa trên BCS khi đã chứng minh được thuốc không hấp thu tại khoang miệng.
Cần làm rõ việc áp dụng miễn thử sinh học dựa trên BCS với cơ quan quản lý dược mỗi quốc gia.
II. Tóm tắt các yêu cầu
Miễn thử sinh học dựa trên BCS có thể áp dụng đối với một chế phẩm thuốc giải phóng ngay nếu:
● Dược chất đã được chứng minh là có khả năng tan tốt và hấp thu hoàn toàn (nhóm I theo BCS; xem chi tiết tại mục III.1 và III.2) và
● Đặc tính hòa tan in vitro của thuốc thử và thuốc đối chứng đã được chứng minh là rất nhanh (tan > 85% trong vòng 15 phút) hoặc nhanh và tương tự nhau (tan 85% trong vòng 30 phút), dựa trên các quy định cụ thể (xem tại mục IV.1) và
● Các tá dược có thể ảnh hưởng đến sinh khả dụng phải giống nhau về thành phần định tính và định lượng. Nhìn chung, nên sử dụng cùng loại tá dược với hàm lượng tương tự nhau (xem mục IV.2).
Nhìn chung, cần xem xét kỹ các nguy cơ chế phẩm sẽ không phù hợp để được xem xét miễn thử
sinh học (ví dụ, hấp thu đặc hiệu tại một vị trí, nguy cơ tương tác với protein vận chuyển tại vị trí hấp thu, thành phần tá dược và nguy cơ điều trị).
III. Dược chất
Nhìn chung, có thể sử dụng thông tin từ các tài liệu tổng quan y văn đáng tin cậy đối với các dược chất đã biết để mô tả những đặc tính quan trọng của dược chất khi áp dụng quy định miễn thử sinh học.
Có thể áp dụng quy định miễn thử sinh học khi (các) dược chất trong thuốc thử và thuốc đối chứng giống nhau. Miễn thử sinh học cũng có thể áp dụng nếu thuốc thử và thuốc đối chứng chứa các muối khác nhau nhưng cùng thuốc nhóm I theo phân loại BCS (tan tốt và hấp thu hoàn toàn; xem mục III.1 và III.2). Không áp dụng miễn thử sinh học khi thuốc thử và thuốc đối chứng khác nhau về dạng ester, ether, đồng phân, hỗn hợp đồng phân, phức chất hoặc dẫn chất của dược chất do những khác biệt này có thể dẫn tới sự khác biệt về sinh khả dụng và không thể được ngoại suy bằng cách sử dụng các thử nghiệm theo quy định miễn thử sinh học dựa trên BCS.
Dược chất không được thuộc nhóm các thuốc có ‘khoảng điều trị hẹp’ (xem mục 3.1.9 về các thuốc có khoảng điều trị hẹp).
III.1 Độ tan
Cần xác định và biện giải dữ liệu độ tan của dược chất theo pH. Dược chất được coi là tan tốt nếu liều đơn cao nhất của thuốc ở dạng bào chế giải phóng ngay được hòa tan hoàn toàn trong 250 ml dung dịch đệm có pH 1 - 6, 8 ở 37°C. Thử nghiệm này cần được thực hiện với ít nhất ba dung dịch đệm trong khoảng pH nói trên (ưu tiên pH 1,2, 4,5 và 6,8) và với dung dịch đệm có pH bằng pKa nếu pKa cũng nằm trong khoảng pH này.
Tại mỗi điều kiện pH, cần tiến hành xác định lặp lại vài lần để phân loại độ tan chính xác (ví dụ, phương pháp bình lắc hoặc phương pháp thích hợp khác). pH dung dịch cần được kiểm tra trước và sau khi thêm dược chất vào dung dịch đệm.
III.2 Hấp thu
Chứng minh khả năng hấp thu hoàn toàn trên người là điều kiện ưu tiên để áp dụng miễn thử sinh học dựa trên BCS. Trong trường hợp này, dược chất được coi là được hấp thu hoàn toàn khi mức độ hấp thu ≥ 85 %. Nhìn chung, việc hấp thu hoàn toàn liên quan đến tính thấm cao.
Cần chứng minh khả năng thuốc được hấp thu hoàn toàn dựa vào các nghiên cứu đáng tin cậy trên người. Dữ liệu từ các nghiên cứu
● Sinh khả dụng tuyệt đối hoặc
● Cần bằng khối lượng
có thể được sử dụng làm cơ sở để chứng minh điều này.
Khi sử dụng dữ liệu từ các nghiên cứu cân bằng khối lượng làm cơ sở để chứng minh khả năng hấp thu hoàn toàn, cần đảm bảo rằng chất chuyển hóa đã được tính đến khi xác định phần thuốc được hấp thu sau quá trình hấp thu. Do đó, khi dựa vào tổng hoạt tính phóng xạ được bài tiết qua nước tiểu, cần đảm bảo rằng không xảy ra quá trình giáng hóa hoặc chuyển hóa dược chất không biến đổi trong dịch dạ dày hoặc dịch ruột. Các chất chuyển hóa oxy hóa Pha I và liên hợp Pha II chỉ có thể xuất hiện sau khi hấp thu (tức là không thể xuất hiện trong dịch dạ dày hoặc ruột). Như vậy, dữ liệu từ các nghiên cứu cân bằng khối lượng phản ánh khả năng hấp thu hoàn toàn nếu tổng lượng chất mẹ tìm thấy trong nước tiểu và các chất chuyển hóa oxy hóa Pha I và liên hợp Pha II tìm thấy trong nước tiểu và phân đạt ≥ 85 % liều.
Các quy định chặt chẽ hơn sẽ được áp dụng đối với các chất đề xuất vào nhóm I theo phân loại BCS những khả năng hấp thu hoàn toàn chưa được chứng minh đầy đủ.
Tương đương sinh học đã báo cáo giữa các thuốc ở dạng rắn và dạng nước của một dược chất cụ thể sử dụng theo đường miệng có thể góp phần chỉ ra rằng sự hạn chế khả năng hấp thu gây ra bởi đặc tính của dạng bào chế (giải phóng ngay) có thể là không đáng kể. Các nghiên cứu đánh giá khả năng thấm in vitro được thực hiện tốt với các chất chuẩn đối chiếu cũng có thể được sử dụng để bổ sung cho dữ liệu in vivo.
IV. Chế phẩm thuốc
IV.1 Độ hòa tan in vitro
IV.1.1 Các khía cạnh chung
Các nghiên cứu liên quan đến chế phẩm thuốc cần đảm bảo đặc tính giải phóng ngay và thể hiện tính tương tự giữa các chế phẩm nghiên cứu, tức là, chế phẩm thử và chế phẩm tham chiếu thể hiện độ hòa tan in vitro tương tự nhau ở điều kiện pH thực nghiệm mô phỏng điều kiện sinh lý. Tuy nhiên, điều này chưa đủ để thiết lập mối tương quan in vitro/ in vivo. Độ hòa tan in vitro cần được đánh giá trong khoảng pH 1 - 6,8 (tối thiểu tại 3 giá trị pH 1,2; 4,5 và 6,8). Có thể cần đánh giá thêm tại trị số pH mà tại đó, độ tan của dược chất thấp nhất. Không cho phép sử dụng bất kì chất diện hoạt nào.
Chế phẩm thử và chế phẩm tham chiếu cần đáp ứng các yêu cầu được đưa ra tại mục 3.1.2 trong phần nội dung chính của hướng dẫn này. Theo những yêu cầu đã được đề cập, cần tiến hành nghiên cứu với nhiều hơn một lô của chế phẩm thử và chế phẩm tham chiếu.
Thử nghiệm so sánh độ hòa tan in vitro cần đáp ứng những tiêu chuẩn chính thức hiện hành. Theo đó, cần cung cấp tài liệu mô tả chi tiết cách bố trí thí nghiệm và phương pháp phân tích, bao gồm dữ liệu thẩm định phương pháp phân tích. Khuyến cáo sử dụng 12 đơn vị sản phẩm trong mỗi thí nghiệm để có thể tính toán thống kê. Điều kiện thí nghiệm thông thường gồm:
● Thiết bị: cánh khuấy hoặc giỏ quay
● Thể tích môi trường hòa tan: 900 ml hoặc ít hơn
● Nhiệt độ của môi trường hòa tan: 37±1 °C
● Tốc độ quay: thiết bị cánh khuấy - thường là 50 vòng/phút
thiết bị giỏ quay - thường là 100 vòng/phút
● Kế hoạch lấy mẫu: ví dụ, tại các thời điểm 10, 15, 20, 30 và 45 phút
● Đệm: pH 1,0 – 1,2 (thường dùng HCl 0,1N hoặc dịch dạ dày giả -SGF không có enzym), pH 4,5 và pH 6.8 (hoặc dịch ruột giả -SIF không có enzym); (pH cần được duy trì trong toàn bộ thí nghiệm; khuyến cáo dung dung dịch đệm theoPh.Eur.)
● Những điều kiện khác: không dùng chất diện hoạt; trong trường hợp viên nang gelatin hoặc viên nén bao gelatin, có thể cho phép sử dung enzym.
Hồ sơ hoàn chỉnh về thử nghiệm độ hòa tan in vitro cần bao gồm đề cương nghiên cứu, thông tin về số lô của thuốc thử và thuốc tham chiếu, điều kiện thử nghiệm chi tiết, thẩm định phương pháp thử, kết quả từng mẫu và kết quả trung bình và thống kê tóm tắt tương ứng.
IV.1.2 Đánh giá kết quả thử độ hòa tan in vitro
Chế phẩm thuốc được coi là hòa tan ‘rất nhanh’ khi có trên 85% khối lượng dược chất so với nhãn được hòa tan trong 15 phút. Trong trường hợp cả chế phẩm thử và chế phẩm tham chiếu đều đáp ứng điều kiện này, tính tương đồng của biểu đồ hòa tan có thể được chấp nhận mà không cần thực hiện thêm bất kì tính toán nào khác.
Cần chứng minh tính tương đồng trong trường hợp thời gian cần thiết để chế phẩm hòa tan hoàn toàn (ít nhất 85% so với khối lượng ghi trên nhãn) lớn hơn 15 phút nhưng không vượt quá 30 phút. Cần sử dụng hệ số kiểm định f2 (xem phụ lục I) hoặc những kiểm định thích hợp khác để chứng minh tính tương đồng giữa các biểu đồ hòa tan của thuốc thử và thuốc tham chiếu. Tuy nhiên, việc bàn luận về sự khác nhau của biểu đồ hòa tan và sự liên quan đến hiệu quả lâm sàng/ điều trị của nó được coi là không thích hợp do các nghiên cứu này không phản ánh mối tương quan in vitro/ in vivo.
IV.2 Tá dược
Mặc dù ảnh hưởng của tá dược trong dạng bào chế giải phóng ngay đến sinh khả dụng của các dược chất tan tốt và hấp thu hoàn toàn (tức là thuộc nhóm I theo phân loại BCS) được xem như là không đáng kể, tuy nhiên, không thể loại trừ hoàn toàn. Do đó, ngay cả khi thuốc thuộc nhóm I theo phân loại BCS, thuốc thử cần chứa cùng loại tá dược với cùng lượng như thuốc tham chiếu.
Theo nguyên tắc chung, đối với các thuốc thuộc nhóm I theo phân loại BCS, cần sử dụng những tá dược đã được thiết lập với hàm lượng thường dùng và cần xem xét, bàn luận những tương tác có thể ảnh hưởng đến sinh khả dụng và/ hoặc các đặc tính hòa tan của thuốc. Cần mô tả chức năng của các tá dược kèm theo giải thích khối lượng của mỗi loại tá dược được sử dụng có nằm trong giới hạn thông thường hay không. Những tá dược có thể ảnh hưởng đến sinh khả dụng như sorbitol, mannitol, natri lauryl sulfat hoặc các chất diện hoạt khác cần được xác định và đánh giá tác động của chúng đến:
● Nhu động đường tiêu hóa
● Mức độ nhạy cảm trong tương tác với dược chất (ví dụ, tạo phức)
● Tính thấm của dược chất
● Tương tác với các protein vận chuyển mang tế bào
Thành phần định tính và định lượng của các tá dược có thể ảnh hưởng đến sinh khả dụng của thuốc trong công thức thuốc thử và thuốc đối chứng phải như nhau.
V. Dạng phối hợp cố định (FCs)
Miễn thử sinh học dựa trên BCS có thể được áp dụng với chế phẩm phối hợp cố định ở dạng giải phóng ngay nếu tất cả các dược chất trong chế phẩm phối hợp cố định thuộc nhóm I theo phân loại BCS và các tá dược đáp ứng đầy đủ những yêu cầu được đề cập tại mục IV.2. Trong những trường hợp khác, cần tiến hành đánh giá tương đương sinh học in vivo.
PHỤ LỤC IV
Mẫu báo cáo nghiên cứu tương đương sinh học ASEAN
1. Trang tiêu đề
1.1 Tên nghiên cứu
1.2 Tên và địa chỉ của nhà tài trợ
1.3 Tên, người chịu trách nhiệm và địa chỉ của cơ sở nghiên cứu
1.4 Tên và địa chỉ của Nghiên cứu viên chính
1.5 Tên của Nghiên cứu viên lâm sàng/nhân viên y tế
1.6 Tên, người chịu trách nhiệm và địa chỉ của cơ sở xét nghiệm lâm sàng
1.7 Tên, người chịu trách nhiệm và địa chỉ của cơ sở phân tích
1.8 Tên, người chịu trách nhiệm và địa chỉ cơ sở quản lý dữ liệu, dược động học và phân tích thống kê
1.9 Tên và địa chỉ của các nghiên cứu viên khác và những cá nhân tham gia vào nghiên cứu
1.10 Ngày bắt đầu và ngày kết thúc nghiên cứu phân tích và lâm sàng
1.11 Chữ ký, ngày ký của (các) nghiên cứu viên (kể cả nhân viên hành chính y tế, phụ trách đảm bảo chất lượng - nếu có)
2. Tóm tắt nghiên cứu
3. Mục lục
4. Chữ viết tắt và giải thích thuật ngữ
5. Giới thiệu
5.1 Dược lý
5.2 Dược động học
5.3 Biến cố bất lợi
6. Mục tiêu nghiên cứu
7. Thông tin sản phẩm
7.1 Thông tin thuốc thử
- Tên thương mại
- Dược chất, Hàm lượng và Dạng bào chế
- Số lô, Ngày sản xuất và hạn sử dụng
- Sự phù hợp của cỡ lô (có thể được cung cấp trực tiếp bởi nhà tài trợ)
- Công thức bào chế (có thể được cung cấp trực tiếp bởi nhà tài trợ)
- Tiêu chuẩn chất lượng của thành phẩm (có thể được cung cấp trực tiếp bởi nhà tài trợ)
- Tên và địa chỉ của Nhà sản xuất
7.2 Thông tin chế phẩm đối chứng
- Tên thương mại
- Dược chất, Hàm lượng và Dạng bào chế
- Số lô, Ngày sản xuất và Hạn sử dụng
- Tên và Địa chỉ của Nhà sản xuất
- Tên và địa chỉ của Nhà nhập khẩu hoặc Cơ sở Giữ giấy phép lưu hành
7.3 Dữ liệu tương đương bào chế
- So sánh hàm lượng dược chất / Hoạt lực
- Độ đồng đều đơn vị phân liều
7.4 So sánh biểu đồ hòa tan (có thể được cung cấp trực tiếp bởi nhà tài trợ)
7.5 Bản tuyên bố kèm theo chữ ký của cơ sở đăng ký/ nhà tài trợ xác nhận thuốc thử và thuốc đăng ký lưu hành là một.
8. Kế hoạch nghiên cứu
8.1 Thiết kế nghiên cứu lâm sàng
- Thiết kế nghiên cứu (chéo, song song)
- Trạng thái no, đói
- Tiêu chí chấp nhận, loại trừ, hạn chế
- Tiêu chuẩn hóa điều kiện nghiên cứu
- Cách dùng thuốc
- Loại dữ liệu người tình nguyện không dùng trong đánh giá kết quả
- Sàng lọc sức khỏe
- Thông tin về người tình nguyện, số lượng, các điểm khác so với đề cương
- Quy trình/ thời điểm lấy mẫu, chuẩn bị/ xử lý mẫu, bảo quản, các điểm khác so với đề cương
- Thể tích máu đã lấy
- Giám sát người tình nguyện
- Yếu tố di truyền (nếu áp dụng)
8.2 Dùng thuốc
- Lựa chọn liều thử – đơn liều, đa liều
- Nhận diện các thuốc nghiên cứu, liều dùng
- Phương pháp ngẫu nhiên hóa
- Làm mù (mã hóa)
- Thời kỳ nghỉ giữa các giai đoạn (giai đoạn rửa giải)
- Thể tích nước dùng để uống thuốc
8.3 Dữ liệu lâm sàng và độ an toàn
- Biến cố bất lợi
- Phản ứng bất lợi liên quan đến thuốc
8.4 Các thông số dược động học và cách tính
- Định nghĩa và tính toán
8.5 Phân tích thống kê
- Phân tích dữ liệu đã chuyển log (AUC, Cmax)
- Điều chỉnh theo thời gian lấy mẫu
- t max,
- t ½
- Tiêu chuẩn chấp nhận tương đương sinh học
- Kết quả kiểm định ANOVA
- Hiệu lực (Power)
8.6 Phương pháp định lượng và thẩm định
- Mô tả phương pháp định lượng
- Phương pháp phát hiện
- Quy trình thẩm định và tóm tắt kết quả
● Độ đặc hiệu;
● Độ đúng;
● Độ chính xác;
● Tỷ lệ thu hồi;
● Độ ổn định;
● Giới hạn định lượng (LOQ)
● Độ tuyến tính
8.7 Dữ liệu về đảm bảo chất lượng
9. Kết quả và bàn luận
9.1 Kết quả nghiên cứu lâm sàng
- Đặc điểm nhân khẩu học của người tình nguyện
- Chi tiết hoạt động lâm sàng.
- Các điểm khác so với đề cương, nếu có.
- Dữ liệu về việc hút thuốc/ uống rượu/ sử dụng ma túy, tiền sử bệnh và kiểm tra sức khỏe, những dấu hiệu sinh tồn và kết quả xét nghiệm của người tình nguyện.
- Báo cáo phản ứng/ biến cố bất lợi của thuốc thử và thuốc đối chứng.
9.2 Tóm tắt kết quả phân tích
9.3 Phân tích dược động học
- Nồng độ thuốc tại mỗi thời điểm lấy mẫu, dữ liệu thống kê
- Bằng các thông số dược động học của từng người tình nguyện, dữ liệu thống kê
- Đường biểu diễn nồng độ thuốc trung bình trong huyết tương hoặc nước tiểu theo thời gian
- Đường biểu diễn nồng độ thuốc trong huyết tương hoặc nước tiểu theo thời gian của từng người tình nguyện
9.4 Phân tích thống kê
- Những lưu ý về thống kê
- Các thời điểm được lựa chọn để tính Kel (hằng số tốc độ thải trừ), t1/2
- Thống kê tóm tắt các thông số dược động học: AUCt, % AUC ngoại suy, AUCinf, Cmax, tmax, t1/2
- Tóm tắt mức ý nghĩa thống kê cho AUC và Cmax (dựa trên dữ liệu đã chuyển log được tính theo ước lượng điểm và khoảng tin cậy 90% của trung bình nhân của thuốc thử/ thuốc đối chứng) và cho tmax (dựa trên dữ liệu không chuyển log được biểu thị theo giá trị p)
- Tính toán tương tự cho dữ liệu trong nước tiểu: Ae và dAe/dt (Ae tương ứng với AUC, (dAe/dt)max tương ứng với Cmax).
- Sự biến thiên trong cá thể
- Hiệu lực của nghiên cứu
- Đánh giá trình tự, giai đoạn và thuốc/ mô hình thử
- Bảng - Phân tích phương sai, trung bình bình phương tối thiểu hình học của mỗi thông số dược động học.
- Bảng - Tính toán khoảng tin cậy 90% của tỷ lệ các thông số dược động học sử dụng dữ liệu đã chuyển log.
10. Kết luận
11. Phụ lục
11.1 Đề cương và phê duyệt
- Văn bản phê duyệt của cơ quan quản lý dược (nếu áp dụng)
- Đề cương nghiên cứu và những bản sửa đổi bổ sung, văn bản Phê duyệt của Hội đồng đạo đức (IRB/IEC)
- Văn bản đồng thuận tham gia nghiên cứu
- Danh sách những điểm khác so với đề cương
- Danh sách các biến cố bất lợi
- Tiêu chuẩn chất lượng của thuốc thử và CoA (phiếu kiểm nghiệm)
11.2 Báo cáo thẩm định (gồm 20% sắc ký đồ)
11.3 Báo cáo phân tích (gồm 20% sắc ký đồ)
11.4 Chứng nhận cơ sở lâm sàng, cơ sở xét nghiệm lâm sàng và Chứng nhận của cơ sở phân tích (nếu có)
11.5 Biểu đồ so sánh độ hòa tan theo tỷ lệ liều giữa các hàm lượng khác nhau (khi chỉ thực hiện nghiên cứu tương đương sinh học với một hàm lượng nhưng xin đăng ký một số hàm lượng khác nhau).
CÁC THUẬT NGỮ DÙNG TRONG HỒ SƠ KỸ THUẬT ACTD VÀ ACTR
Những định nghĩa sử dụng cho bảng chú giải thuật ngữ này được xây dựng cho hồ sơ kỹ thuật chung ASEAN (ACTD) và những yêu cầu kỹ thuật chung ASEAN (ACTR). Các thuật ngữ này không nhất thiết có ý nghĩa tương tự ngoài phạm vi của những phần cụ thể mà chúng được đề cập đến trong ACTD và ACTR.
Thử Lão hóa cấp tốc [theo Q1AR]/ Tham khảo: ACTD-Q...
Là những nghiên cứu được thiết kế để làm tăng tốc quá trình phân hủy hóa học hay biến đổi vật lý của dược chất hay dược phẩm bằng cách dùng điều kiện bảo quản khắc nghiệt, được coi như là một phần của các nghiên cứu độ ổn định chính thức.
(Dữ liệu nghiên cứu trong điều kiện khắc nghiệt cùng với các nghiên cứu độ ổn định dài hạn, có thể được sử dụng để đánh giá những ảnh hưởng hóa học dài hạn hơn tại điều kiện không lão hóa cấp tốc và ảnh hưởng của tình trạng bảo quản nằm ngoài điều kiện bảo quản ghi trên nhãn trong thời gian ngắn (có thể xảy ra khi vận chuyển). Kết quả thử trong điều kiện lão hóa cấp tốc không phải lúc nào cũng có thể dự báo được những thay đổi vật lý (xem Độ ổn định và những thuật ngữ liên quan)
Tiêu chuẩn chấp nhận [theo Q6B]/ Tham khảo: ACTD-Q...
Là các giới hạn về số, phạm vi các đo lường thích hợp mà một dược chất hoặc dược phẩm hoặc nguyên liệu phải đạt được ở những giai đoạn khác nhau của quy trình sản xuất để kết quả của quy trình phân tích của dược chất hoặc dược phẩm hoặc nguyên liệu đó được chấp nhận.
Độ chính xác [theo Q2A]/ Tham khảo: ACTD-Q...
Độ chính xác của một quy trình phân tích diễn tả độ xác thực giữa giá trị tìm được và một trong các giá trị được chấp nhận (là giá trị thực quy ước đúng hoặc giá trị đối chiếu chấp nhận được).
Hoạt chất (API) [theo WHO]
Một chất hoặc một hợp chất có hoạt tính trị liệu được sử dụng để sản xuất ra một dược phẩm.
Phản ứng có hại của thuốc (ADR) [theo WH]/ Tham khảo: ACDT-E...
Là một phản ứng với thuốc mà phản ứng đó có hại và không mong muốn, xuất hiện ở liều điều trị bình thường đối với con người.
(Trong định nghĩa này, có một lưu ý quan trọng là nó liên quan đến phản ứng của bệnh nhân mà mỗi yếu tố riêng biệt có thể đóng vai trò quan trọng, và hiện tượng là có hại. Chẳng hạn như một phản ứng điều trị không mong muốn có thể là tác dụng phụ (Side Effect) nhưng không phải là phản ứng có hại; xem thêm phần Biến cố ngoại ý (Adverse Event), tác dụng phụ (Side Effect).
Từ web site của Chương Trình Giám Sát Thuốc WHO, www.who-umc.org)
Biến cố ngoại ý (AE) [theo WHO)/Tham khảo: ATCD-E...
Là bất kỳ biến cố y khoa không có lợi nào có thể xảy ra khi điều trị với một thuốc nhưng không nhất thiết có mối quan hệ nhân quả với thuốc điều trị đó. (Điểm cơ bản ở đây là sự trùng hợp cùng lúc mà không nghi ngờ có quan hệ nhân quả; xem thêm phần phản ứng có hại, tác dụng phụ) từ web site của chương trình giám sát thuốc WHO, www.who-umc.org
Quy trình phân tích [theo Q2A]/ Tham khảo: ACTD-Q...
Quy trình phân tích đề cập đến cách tiến hành phân tích; nó phải mô tả chi tiết các bước cần thiết để thực hiện mỗi phép phân tích.
(Nó có thể bao gồm mẫu thử, chất chuẩn, chuẩn bị thuốc thử, sử dụng thiết bị, xây dựng đường chuẩn, sử dụng công thức để tính toán, .v.v.)
Hiện tượng lệch bội lẽ [theo S2A]/Tham khảo: ACTD-S...
Sai số của mẫu các nhiễm sắc thể trong một tế bào hoặc sinh vật.
Cơ sở đăng ký [theo WHO]
Công ty, tập đoàn hay tổ chức pháp nhân hoạt động trong lĩnh vực dược nộp đơn xin cấp giấy phép lưu hành một dược phẩm, nộp tài liệu cập nhật cho một thuốc đã được cấp phép lưu hành hoặc nộp đơn xin phép cho những thay đổi đối với một thuốc đã được cấp phép lưu hành.
Phê duyệt (liên quan đến hội đồng thẩm định) [theo E6] Tham khảo: ACTD-E..
Quyết định có tính khẳng định của hội đồng thẩm định rằng việc thử lâm sàng đã được xem xét và có thể được thực hiện tại một cơ quan được hội đồng thẩm định chỉ định, đáp ứng yêu cầu pháp lý, tiêu chuẩn thực hành lâm sàng tốt (GCP), và các yêu cầu quản lý.
Thanh tra [theo E6]/Tham khảo: ACTD-E...
Việc thẩm tra thử nghiệm có hệ thống và độc lập liên quan đến những hoạt động và tài liệu để xác định xem những hoạt động thử nghiệm đánh giá có được tiến hành hay không, và số liệu có được lưu trữ, phân tích và báo cáo chính xác theo đề cương, quy trình thao tác chuẩn (SOPs) của nhà tài trợ, tiêu chuẩn thực hành lâm sàng tốt (GCP), và những yêu cầu quản lý hay không.
Chứng nhận thanh tra [theo E6]/ Tham khảo: ACTD-E...
Tuyên bố của thanh tra xác nhận rằng việc thanh tra đã được tiến hành.
Báo cáo thanh tra [theo E6]/ Tham khảo: ACTD-E...
Một báo cáo đánh giá bằng văn bản về kết quả thanh tra do thanh tra viên của nhà tài trợ thực hiện.
Bằng chứng thanh tra [theo E6]/ Tham khảo: ACTD-E...
Tài liệu cho phép tái hiện lại diễn tiến sự việc.
Hồ sơ kỹ thuật chung ASEAN (ACTD)
Phần hồ sơ xin cấp giấy phép lưu hành sản phẩm được áp dụng chung cho tất cả các nước thành viên ASEAN.
Yêu cầu kỹ thuật chung ASEAN (ACTR)
Một bộ tài liệu bằng văn bản để hướng dẫn các đơn vị đăng ký chuẩn bị hồ sơ đăng ký một cách đồng bộ theo yêu cầu chung của tất cả cơ quan quản lý thuốc (DRA) ASEAN.
Lô [theo WHO]
Là một lượng xác định nguyên liệu ban đầu, nguyên liệu bao gói hoặc sản phẩm được chế biến đồng nhất từ một công đoạn hoặc một số công đoạn sản xuất.
(Trong sản xuất liên tục, lô phải tương ứng với một phần xác định trong sản xuất, được đặc trưng bởi sự đồng nhất theo dự kiến; đôi khi phải chia lô thành một số mẻ, sau đó tập trung lại để hình thành lô đồng nhất cuối cùng).
Số lô [theo WHO]
Sự kết hợp rõ ràng của các số và/hoặc các chữ cái để nhận dạng cụ thể một lô được ghi trên nhãn, trong hồ sơ lô, và trên phiếu kiểm nghiệm, .v.v.
Hồ sơ lô [theo WHO]
Tất cả các tài liệu liên quan đến việc sản xuất của một lô bán thành phẩm hoặc thành phẩm; chúng thể hiện lịch sử của mỗi lô sản phẩm và tất cả những tình huống liên quan đến chất lượng của sản phẩm cuối cùng.
Thay thế bazơ [theo S2A]/ Tham khảo: ACTD-S…
Sự thay đổi của một hay nhiều bazơ trong chuỗi nucleotide. Điều này có thể tạo ra một protein khác.
Sinh khả dụng [theo WHO]
Tốc độ và mức độ hiện diện của một thành phần hoạt chất thuốc hoặc chất chuyển hóa có hoạt tính được xác định bằng đường cong nồng độ/thời gian của nó trong hệ tuần hoàn hoặc bằng sự thải trừ của nó qua nước tiểu hay thể dịch khác.
Tương đương sinh học [theo WHO]
Hai dược phẩm tương đương sinh học nếu chúng tương đương hoặc thay thế nhau được về mặt dược học, và có sinh khả dụng ở mức độ tương đương nhau để có thể có hiệu quả mong muốn cơ bản là như nhau, sau khi được cho sử dụng cùng 1 liều lượng bằng nhau.
Hoạt tính sinh học [theo Q6B]/ Tham khảo: ACTD-Q...
Năng lực chuyên biệt của một sản phẩm để đạt được một hiệu quả sinh học xác định.
Hoạt lực là đơn vị định lượng của hoạt tính sinh học.
Sản phẩm sinh học [theo WHO]
Bất cứ sản phẩm nào có nguồn gốc sinh học, sản xuất bằng một quá trình sinh học, dẫn xuất từ máu và huyết tương người, hoặc được sản xuất bằng công nghệ sinh học, chứa những chất cao phân tử mà độ tinh khiết, hoạt lực, và cấu tạo của chúng không thể xác định một cách dễ dàng và đáng tin cậy bằng phân tích hóa học hay hóa lý. (Ví dụ của nhóm này là vacxin, những sản phẩm máu, những mô động vật được biến đổi, các hóc- môn phân tử lượng cao, các dị nguyên, và các sản phẩm công nghệ gen hoặc những công nghệ sinh học mới hơn khác. Định nghĩa này không bao gồm các kháng sinh và các chất mặc dù có nguồn gốc sinh học nhưng phân tử lượng thấp và có thể tách được thành những chất tinh khiết, như các steroid và alkaloid tinh khiết. Thông thường sản phẩm sinh học không thể được phê duyệt bởi cơ quan quản lý y tế nếu chỉ dựa vào so sánh in vitro với một sản phẩm so sánh (comparator). Các biệt dược khác nhau có thể có cùng công dụng, ví dụ vắc-xin ho gà, nhưng mỗi sản phẩm phải được chứng minh độc lập về độ an toàn và hiệu quả. Dữ liệu về nồng độ của "thành phần hoạt tính" trong huyết tương cũng thường không giúp ích được bởi vì không thể chắc chắn có thể đo được chính xác cùng lượng hoạt chất đó trong huyết tương. Do đó những sản phẩm này không thể được phê duyệt mà không có số liệu về độ an toàn và hiệu lực; xem Sản Phẩm Công Nghệ Sinh Học)
Sản phẩm công nghệ sinh học [theo WHO]
Bất cứ sản phẩm nào được sản xuất bằng công nghệ gen hoặc công nghệ sinh học tân tiến hơn khác.
(Định nghĩa về tất cả các sản phẩm công nghệ sinh học đều nằm trong định nghĩa về các sản phẩm sinh học; xem Sản Phẩm Sinh Học)
Nghiên cứu mù/ Mặt nạ [theo E6]/ Tham khảo: ACTD- E...
Một quy trình thủ tục trong đó một hoặc nhiều bên tham gia thử nghiệm không biết nội dung điều trị.
(Mù đơn thường là đối tượng/bệnh nhân nghiên cứu không được biết nội dung điều trị và mù đôi thường là cả đối tượng nghiên cứu, các nhà nghiên cứu (nghiên cứu viên; bác sỹ nghiên cứu), người giám sát, và trong một số trường hợp cả những người phân tích số liệu cũng không được biết về nội dung điều trị).
Khoảng quan trắc [theo Q1AR]/ Tham khảo: ACTD-Q...
Một thiết kế về chương trình độ ổn định mà chỉ những mẫu ở các cực điểm của các yếu tố thiết kế nhất định, chẳng hạn hàm lượng, cỡ đóng gói, được thử nghiệm tại tất cả các thời điểm như trong toàn bộ thiết kế.
(Thiết kế thừa nhận rằng độ ổn định của bất cứ mức độ trung gian nào đều được thể hiện qua độ ổn định của các mức cực điểm đã được thử nghiệm. Khi một dãy các hàm lượng được thử nghiệm thì khoảng quan trắc sẽ được áp dụng nếu các hàm lượng là như nhau hoặc liên quan rất mật thiết với nhau trong thành phần công thức [ví dụ đối với những viên nén được tạo ra từ những lực nén khác nhau trên cùng dạng cốm, hay những viên nang được đóng nang ở những khối lượng khác nhau từ cùng một thành phần vào trong các vỏ nang có kích thước khác nhau]. Khoảng quan trắc có thể áp dụng cho các cỡ đóng gói khác nhau hoặc cách đóng gói khác nhau của cùng một hệ bao bì kín).
Bộ dữ liệu bắc cầu [theo E5]/ Tham khảo: ACTD-E...
Thông tin chọn lọc từ Bộ dữ liệu lâm sàng hoàn chỉnh mà cũng phù hợp với dân số của một khu vực mới, bao gồm số liệu về dược động học và mọi dữ liệu sơ bộ về dược lực học và đáp ứng theo liều, và nếu cần, những dữ liệu bổ sung thu được từ nghiên cứu bắc cầu ở khu vực mới mà cho phép ngoại suy các dữ liệu của nước ngoài về tính an toàn và hiệu lực của thuốc cho dân số của khu vực mới.
Nghiên cứu bắc cầu [theo E5]/ Tham khảo: ACTD-E...
Một nghiên cứu bổ sung được thực hiện ở khu vực mới để cung cấp các dữ liệu dược lực học hoặc dữ liệu lâm sàng về hiệu lực, tính an toàn, liều dùng và chế độ liều ở khu vực mới mà cho phép ngoại suy từ dữ liệu lâm sàng ở nước ngoài cho khu vực mới.
(Những nghiên cứu này có thể bao gồm những thông tin bổ sung về dược động học).
Hiệu chuẩn [theo WHO]/ Tham khảo: ACTD-Q...
Là một loạt các thao tác tiến hành trong một điều kiện đặc thù nhằm thiết lập mối quan hệ giữa các giá trị có được do thiết bị/hệ thống đo đạc ghi lại và kiểm soát, hoặc các giá trị đo được bằng phép đo thích hợp, và những giá trị tương ứng đã biết của một chất chuẩn đối chiếu.
(Cần thiết lập giới hạn chấp nhận của các kết quả đo lường)
Mẫu báo cáo dữ liệu (CRF) [theo E6]/ Tham khảo: ACTD-E...
Một tài liệu dạng văn bản in hoặc tài liệu điện tử được thiết kế để ghi lại tất cả thông tin mà đề cương yêu cầu để báo cáo cho nhà tài trợ về mỗi đối tượng thử nghiệm.
Sinh sản tế bào [theo S2A]/ Tham khảo: ACTD-S...
Khả năng tế bào phân chia và hình thành các tế bào con.
Cơ chất tế bào [theo Q5D]/ Tham khảo: ACTD-Q...
Tế bào vi sinh vật hay dòng tế bào lấy từ người hoặc động vật có đầy đủ khả năng tạo ra những sản phẩm công nghệ sinh học/sản phẩm sinh học mong muốn để sử dụng in-vivo hoặc ex-vivo đối với con người.
Giấy chứng nhận dược phẩm (CPP) [theo WHO]
Giấy chứng nhận dược phẩm được quy định theo hệ thống giấy chứng nhận của WHO về chất lượng của dược phẩm được sử dụng trong giao dịch thương mại Quốc tế.
Kiểm soát sự thay đổi [theo WHO]
Một hệ thống chính thức qua đó những đại diện có đủ tiêu chuẩn hoặc các nhà chuyên môn thẩm định những thay đổi dự kiến hoặc thực tế có thể ảnh hưởng đến tình trạng đã được thiết lập.
(Mục đích nhằm xác định nhu cầu tác động để đảm bảo và ghi nhận rằng hệ thống luôn được duy trì ở một tình trạng đã được công nhận.)
Clastogen [theo S2A]/ Tham khảo: ACTD-S...
Một tác nhân gây ra những thay đổi cấu trúc của nhiễm sắc thể, thường có thể phát hiện được bằng kính hiển vi ánh sáng thường.
Vùng khí hậu [theo WHO]
Có 4 vùng được chia theo điều kiện khí hậu hàng năm
Vùng I: ôn đới
Vùng II: cận nhiệt đới, có thể có độ ẩm cao
Vùng III: nóng và khô
Vùng IV: nóng và ẩm
Báo cáo nghiên cứu/thử nghiệm lâm sàng [theo E6]/ Tham khảo: ACTD-E...
Mô tả bằng văn bản về một thử nghiệm/nghiên cứu về bất kỳ loại thuốc nào dùng để chẩn đoán, phòng ngừa hoặc điều trị, được tiến hành trên người, trong đó các mô tả, giới thiệu và phân tích lâm sàng và thống kê được hợp nhất hoàn toàn trong một báo cáo. (Xem hướng dẫn của ICH về cấu trúc và nội dung của các báo cáo nghiên cứu lâm sàng).
Nghiên cứu/ Thử nghiệm lâm sàng [theo E6]/ Tham khảo: ACTD-E...
Bất cứ nghiên cứu nào trên người nhằm phát hiện hoặc xác minh những hiệu quả lâm sàng, dược lý và/ hoặc dược động học của sản phẩm nghiên cứu, và/ hoặc để xác định bất cứ phản ứng có hại nào của sản phẩm nghiên cứu, và/hoặc nghiên cứu về hấp thu, phân bố, chuyển hóa và thải trừ của sản phẩm nghiên cứu với mục đích xác định tính an toàn và/hoặc hiệu quả của sản phẩm nghiên cứu đó.
Hiệu quả tạo dòng vô tính [theo S2A]/ Tham khảo: ACTD-Q ...
Khả năng của một tế bào trong việc tạo dòng vô tính.
(Thường được đo sau khi nuôi cấy số lượng nhỏ các tế bào trong một môi trường thích hợp).
Lô cam kết [theo Q1AR]/ Tham khảo: ACTD-Q...
Lô sản xuất của một dược chất hay một dược phẩm qua đó những nghiên cứu độ ổn định được bắt đầu hoặc hoàn tất sau khi được cấp giấy phép lưu hành theo cam kết trong hồ sơ đăng ký.
Sản phẩm (thuốc) so sánh [theo E6]/ Tham khảo: ACTD-E…
a) Dược phẩm mà sản phẩm mới dự định có thể thay thế lẫn nhau với nó trong thực hành lâm sàng.
(Sản phẩm đối chiếu thường là sản phẩm của nhà phát minh mà hiệu lực, tính an toàn và chất lượng đã được thiết lập. Khi không có sản phẩm của nhà phát minh thì sản phẩm dẫn đầu thị trường được sử dụng là sản phẩm đối chiếu nếu nó đã được cấp phép lưu hành và hiệu lực, tính an toàn và chất lượng đã được thiết lập và ghi thành tài liệu).
b) Một sản phẩm nghiên cứu hoặc đã được đưa ra thị trường (nghĩa là so sánh với thuốc có hoạt tính), hoặc giả dược (placebo) sử dụng làm đối chiếu trong một nghiên cứu lâm sàng. (Xem ‘Sản phẩm đối chiếu’).
Bộ dữ liệu lâm sàng hoàn chỉnh [theo F5]/ Tham khảo: ACTD-E...
Một bộ dữ liệu lâm sàng để đăng ký chứa những dữ liệu lâm sàng đáp ứng đầy đủ những yêu cầu của Cơ quan thẩm quyền của khu vực mới và chứa dữ liệu dược động học liên quan đến dân số ở khu vực mới.
Tuân thủ (liên quan tới các thử nghiệm) [theo F5]/ Tham khảo: ACTD-E...
Làm theo đúng mọi yêu cầu liên quan đến thử nghiệm lâm sàng, yêu cầu của tiêu chuẩn
Thực hành Thử nghiệm Lâm sàng Tốt (GCP), và những yêu cầu theo luật hiện hành.
Thẩm định đồng thời [theo PIC]
Thẩm định tiến hành trong quá trình sản xuất thường qui của sản phẩm dự định bán
Sự bảo mật [theo E6]/ Tham khảo: ACTD-E...
Sự ngăn chặn thất thoát thông tin cá nhân của các đối tượng tham gia nghiên cứu hoặc thông tin độc quyền sở hữu của nhà tài trợ cho những bên không có thẩm quyền.
Hệ bao bì kín[theo Q1AR]/Tham khảo ACTD-Q...
Tất cả các thành phần bao bì tập hợp lại để chứa đựng và bảo vệ dạng bào chế, bao gồm bao bì đóng gói trực tiếp và bao bì đóng gói thứ cấp tạo thành lớp bảo vệ thêm cho sản phẩm thuốc. Hệ đóng gói tương đương với hệ bao bì kín.
Nhãn trên bao bì [theo WHO]
Tất cả các thông tin trên bất kỳ phần nào của bao bì, bao gồm thông tin trên cả bao bì bên ngoài như thùng các-tông.
Chất tạp nhiễm [theo Q6B]/ Tham khảo: ACTD-Q ...
Bất cứ chất nào xuất hiện ngẫu nhiên (chẳng hạn hóa chất, các chủng sinh hóa hay vi khuẩn) không dự tính trong quá trình sản xuất, trong dược chất hay trong thành phẩm thuốc.
Hợp đồng [theo E6]/ Tham khảo: ACTD-E ...
Một thỏa thuận bằng văn bản, ghi ngày tháng và được ký giữa hai hay nhiều bên liên quan nhằm thiết lập bất cứ dàn xếp gì về đại diện hoặc phân công nhiệm vụ và nghĩa vụ, và nếu thích hợp thì cả vấn đề tài chính. Có thể sử dụng một đề cương để làm cơ sở cho một hợp đồng.
Tổ chức nghiên cứu theo hợp đồng (CRO) [theo E6]/ Tham khảo: ACTD-E...
Một pháp nhân hay tổ chức (thương mại, trường, viện,...) do nhà tài trợ ký hợp đồng để thực hiện một hoặc nhiều nhiệm vụ và chức năng liên quan đến thử nghiệm của nhà tài trợ.
Ủy ban điều phối [theo E6]/ Tham khảo: ACTD-E...
Một Ủy ban có thể do nhà tài trợ lập ra để điều phối việc tiến hành thử nghiệm tại nhiều trung tâm.
Nhà điều phối nghiên cứu [theo E6]/ Tham khảo: ACTD-E...
Một nhà nghiên cứu được chỉ định có trách nhiệm điều phối những nhà nghiên cứu tại các trung tâm khác nhau tham gia thử nghiệm đa trung tâm.
Nước xuất xứ
Nước mà dạng bào chế cuối cùng được sản xuất, và/hoặc xuất xưởng lô, hoặc từ đó sản phẩm được vận chuyển đến nước nhập khẩu. Có thể có trường hợp không thể xác định được một nước xuất xứ mà là một số nước xuất xứ. Điều này có thể dẫn đến việc yêu cầu nhiều CPP. (Lưu ý: Định nghĩa này được hiểu liên quan đến thực tế áp dụng hệ thống giấy chứng nhận của WHO áp dụng trong thủ tục đăng ký thuốc).
Quy trình sản xuất quan trọng- [theo ASEAN GMP]
Một quy trình sản xuất có thể dẫn đến biến đổi làm ảnh hưởng đến chất lượng của dược phẩm.
Mật độ tế bào nuôi cấy [theo S2A]/ Tham khảo: ACTD-S...
Là sự xác định mật độ tế bào trong môi trường nuôi cấy (sự sinh sản tế bào thường bị ức chế khi mật độ tế bào tăng cao).
Đánh giá di truyền học tế bào [theo S2B]/ Tham khảo: ACTD-S...
Phân tích cấu trúc nhiễm sắc thể trong gián phân và giảm phân bằng kính hiển vi ánh sáng thường.
Sự phân hủy sản phẩm [theo Q6B]/ Tham khảo: ACTD-Q...
Những biến đổi phân tử trong sản phẩm hoặc trong những chất liên quan đến sản phẩm do những thay đổi gây ra bởi thời gian và/ hoặc tác động của ánh sáng, nhiệt độ, pH, nước, hay do phản ứng với tá dược và/ hoặc hệ bao bì đóng gói trung gian. Những thay đổi này có thể xuất hiện trong quá trình sản xuất và/ hoặc quá trình bảo quản (ví dụ sự thủy phân nhóm amid, oxy hóa, kết tụ, phân giải protein). Sản phẩm phân hủy có thể là các chất liên quan đến sản phẩm hoặc các tạp chất liên quan đến sản phẩm.
Giới hạn phát hiện [theo Q2A]/ Tham khảo: ACTD-Q...…
Giới hạn phát hiện của một quy trình phân tích riêng biệt là lượng nhỏ nhất chất phân tích trong mẫu có thể phát hiện được nhưng không nhất thiết phải định lượng được thành một giá trị cụ thể.
Tiếp cận trực tiếp (liên quan đến thử nghiệm lâm sàng [theo E6]/ Tham khảo: ACTD- E...
Việc cho phép kiểm tra, phân tích, đánh giá và sao chụp lại bất cứ hồ sơ và báo cáo nào quan trọng đối với việc thẩm định thử nghiệm lâm sàng. Bất cứ bên nào (chẳng hạn các cơ quan quản lý trong và ngoài nước, nhà tài trợ, người giám sát và người kiểm tra) được tiếp cận trực tiếp sẽ tiến hành mọi cách phòng ngừa hợp lý trong phạm vi những quy định của cơ quan thẩm quyền để duy trì tính bảo mật cho các thông tin cá nhân của đối tượng nghiên cứu và thông tin thuộc sở hữu của nhà tài trợ.
Tạo dẫn chất với ADN [theo S2B]/ Tham khảo: ACTD-S...
Sự liên kết hóa trị của các chất hóa học với ADN.
Sửa chữa ADN [theo 2B]/ Tham khảo: ACTD-S...
Sự khôi phục chuỗi ADN bị hỏng.
Đứt sợi ADN [theo S2B]/ Tham khảo: ACTD-S...
Sự đứt sợi đơn hoặc sợi kép trong ADN.
Liều [theo E5]
Lượng thuốc đưa vào cơ thể trong 1 lần hay trong một ngày.
Dạng bào chế [theo Q1AR]
Hình thức của dược phẩm (ví dụ viên nén, viên nang, dung dịch, kem) chứa một dược chất, thường phối hợp, nhưng không nhất thiết, với các tá dược.
Chế độ liều [theo E5]/ Tham khảo: ACTD-E...
Đường dùng, tần số dùng và thời hạn dùng theo liều lượng của một thuốc trong một khoảng thời gian.
Thuốc [theo WHO]
Bất cứ một chất hay một dược phẩm dùng cho con người hay thú y với mục đích thay đổi hay thăm dò các hệ sinh lý hay trạng thái bệnh tật vì lợi ích của đối tượng.
Thành phẩm thuốc [theo WHO]
Xem Dược Phẩm.
Dược chất [theo Q1AR]
Là dạng chưa tạo thành công thức của một dược chất nhưng có thể sau đó kết hợp với các tá dược để tạo ra dạng bào chế (Xem Hoạt chất).
Hiệu quả [theo WHO]
Đánh giá hiệu quả của một thuốc được cho rằng sẽ thể hiện trong điều kiện lâm sàng bình thường. Nó phản ánh hiệu quả của việc sử dụng thuốc trong cộng đồng.
Hiệu lực [theo WHO]
Khả năng của một thuốc mang lại hiệu quả có lợi mong muốn cho từng cá nhân trong một dân số xác định có chung một vấn đề y khoa (bệnh lý...), trong các điều kiện sử dụng thuốc lý tưởng.
Yếu tố chủng tộc [theo E5]/ Tham khảo: ACTD-E…
Từ tính chủng tộc được xuất phát từ từ Hy Lạp "ethnos" nghĩa là quốc gia hay dân tộc.
Yếu tố chủng tộc là những yếu tố liên quan đến những nhóm dân cư nhóm lại theo những đặc điểm và tập quán chung. Yếu tố chủng tộc có thể ảnh hưởng đến tính an toàn và hiệu lực của thuốc có thể được phân loại thành yếu tố nội sinh và yếu tố ngoại sinh:
Yếu tố chủng tộc ngoại sinh: là những yếu tố gắn với môi trường và văn hóa nơi một người sinh sống. Yếu tố ngoại sinh có xu hướng ít theo yếu tố di truyền mà theo tính văn hóa và hành vi nhiều hơn. (ví dụ về yếu tố ngoại sinh bao gồm khía cạnh xã hội và văn hóa của vùng như việc thực hành y khoa, chế độ ăn, sử dụng thuốc lá, sử dụng rượu, ảnh hưởng của ô nhiễm và ánh nắng, tình trạng kinh tế-xã hội, tuân thủ dùng thuốc được kê đơn và đặc biệt quan trọng đối với độ tin cậy vào các nghiên cứu từ một vùng khác, các thực hành trong thiết kế và tiến hành thử nghiệm lâm sàng).
● Yếu tố chủng tộc nội sinh: là những yếu tố giúp xác định và nhận dạng một phân nhóm dân số và có thể ảnh hưởng tới khả năng ngoại suy các dữ liệu lâm sàng giữa các khu vực. (Ví dụ của yếu tố nội sinh bao gồm đa hình thái gen, tuổi, giới, chiều cao, cân nặng, độ gầy-béo của cơ thể, cấu tạo cơ thể và những cơ quan bị thiểu năng).
Tá dược [theo Q6B]/ Tham khảo: ACTD-Q...
Một thành phần được chủ định cho vào với dược chất, không có tính chất dược lý ở lượng sử dụng.
Hạn dùng [theo Q1AR]/ Tham khảo: ACTD-Q ...
Thời hạn ghi trên bao bì của sản phẩm nhằm định rõ thời gian mà trước đó lô sản phẩm tiếp tục duy trì được tiêu chuẩn kỹ thuật theo hạn dùng đã được phê duyệt khi được bảo quản ở điều kiện đã được định trước.
(Quá hạn dùng sẽ không đảm bảo là sản phẩm còn đạt các tiêu chuẩn chất lượng được phê duyệt, và do đó sản phẩm có thể không phù hợp để sử dụng và không nên sử dụng).
Kiểm nghiệm sản phẩm mở rộng [theo WHO]
Kiểm nghiệm cuối cùng của sản phẩm trong phạm vi rộng hơn phạm vi được yêu cầu trong kiểm tra chất lượng thường quy.
(Đây là một trong những bước thực tế của quá trình thẩm định, áp dụng chủ yếu đối với những sản phẩm không vô trùng).
Phép ngoại suy từ dữ liệu thử nghiệm lâm sàng nước ngoài [theo E5]/ Tham khảo: ACTD-E.
Sự khái quát hóa và áp dụng dữ liệu về tính an toàn, hiệu lực và đáp ứng liều tiến hành trên dân cư ở khu vực khác cho dân cư ở một khu vực mới.
Thành phẩm [theo WHO]
Một sản phẩm đã trải qua tất cả các công đoạn sản xuất và kiểm tra chất lượng, kể cả đóng gói vào bao bì cuối cùng và dán nhãn.
Nghiên cứu độ ổn định [theo Q1AR]/ Tham khảo: ACTD-Q...
Nghiên cứu dài hạn và nghiên cứu lão hóa cấp tốc (và các nghiên cứu trung gian) được tiến hành trên các lô ban đầu và/hoặc lô cam kết theo một đề cương về độ ổn định để thiết lập hay khẳng định giai đoạn tái kiểm tra của một dược chất hay tuổi thọ của một thuốc.
Công thức [theo WHO]
Thành phần của một dạng bào chế, bao gồm cả đặc tính của các thành phần nguyên liệu thô.
Đột biến gây thay đổi trình tự [theo S2A]/ Tham khảo: ACTD-S...
Là tình trạng đột biến (thay đổi mã di truyền) mà một hay hai bazơ kế cận nhau được thêm vào (chèn vào) hoặc mất đi trong chuỗi nucleotide của gen.
(Điều này có thể dẫn tới thay đổi hoặc cắt ngắn protein).
Đột biến gen [theo S2A]/ Tham khảo: ACTD-S...
Một biến đổi vĩnh viễn phát hiện được trong một gen đơn lẻ hoặc các chuỗi điều khiển của nó. Biến đổi này có thể là đột biến điểm, chèn hoặc xoá.
Sản phẩm Generic (GP) [theo WHO]
Một dược phẩm thường được cho là có thể thay thế sản phẩm phát minh đầu tiên, thường được sản xuất mà không cần giấy phép chấp thuận của công ty phát minh và được bán ra thị trường sau khi bảo hộ bản quyền sáng chế hoặc những độc quyền khác hết hạn.
(Thuật ngữ sản phẩm generic có thể có nghĩa hơi khác nhau tùy vào các pháp chế khác nhau. Do đó nên tránh sử dụng thuật ngữ này càng nhiều càng tốt, thay vào đó nên sử dụng thuật ngữ dược phẩm nhiều nguồn (multisource). Các sản phẩm generic có thể được bán với tên chung [không sở hữu] hoặc với tên biệt dược [tên sở hữu]. Chúng có thể được bán ở những dạng bào chế và/ hoặc hàm lượng khác với những sản phẩm phát minh đầu tiên)
Tiêu chí nghiên cứu về di truyền học [theo S2A]/ Tham khảo: ACTD-S...
Loại hoặc phân loại chính xác thay đổi gen được nghiên cứu (chẳng hạn đột biến gen, bất thường nhiễm sắc thể, sửa chữa ADN, tạo dẫn chất ADN...).
Độc tính gen [theo S2A]/ Tham khảo: ACTD-S...
Một thuật ngữ rộng chỉ bất cứ biến đổi độc hại trong chất liệu gen bất kể là do cơ chế nào gây ra.
Thực hành tốt thử nghiệm lâm sàng (GCP) [theo E6]/ Tham khảo: ACTD-E…
Chuẩn mực để thiết kế, tiến hành, thực hiện, giám sát, kiểm tra, phân tích số liệu và báo cáo các thử nghiệm lâm sàng để đảm bảo số liệu và kết quả báo cáo là tin cậy và chính xác, và đảm bảo các quyền, tính công bằng và bảo mật của các đối tượng tham gia thử nghiệm được bảo vệ.
Khu vực ICH (Thỏa thuận Quốc tế về hòa hợp) [theo E5]/ Tham khảo: ACTD-E ....
Khu vực bao gồm Liên minh Châu Âu, Nhật Bản và Hoa Kỳ.
Dạng bào chế phóng thích nhanh [theo WHO]
Dạng tế bào chế với mục đích giải phóng ngay tất cả các hoạt chất khi đưa vào cơ thể mà không có tác dụng phóng thích chậm hay tác dụng kéo dài.
Nhân chứng khách quan [theo E6]/ Tham khảo: ACTD-E...
Một người độc lập với thử nghiệm, không bị ảnh hưởng bởi những người liên quan đến thử nghiệm, sẽ tham dự vào quá trình ký cam kết của đối tượng tham gia nghiên cứu nếu đối tượng tham gia nghiên cứu hay người đại diện hợp pháp của họ không thể đọc được; là người sẽ đọc các mẫu đơn cam kết và các văn bản thông tin về thử nghiệm được cung cấp cho đối tượng tham gia nghiên cứu.
Bao bì không thấm [theo QIAR/ Tham khảo: ACTD-Q...
Bao bì có rào cản bền vững không cho khí và các dung môi đi qua, chẳng hạn tuýp nhôm kín để chứa đựng chất bán rắn, ống thủy tinh kín để chứa dung dịch.
Tạp chất [theo Q6B]/ Tham khảo: ACTD-Q...
Bất cứ thành phần nào có mặt trong dược chất hay thành phẩm thuốc mà không phải là sản phẩm mong muốn, không phải là chất liên quan đến sản phẩm, không phải là tá dược kể cả tá dược độn. Tạp chất có thể liên quan đến quy trình sản xuất hoặc liên quan đến sản phẩm.
Ủy ban đạo đức độc lập [theo E6]/ Tham khảo: ACTD-E...
Một cơ quan độc lập (một ban hay hội đồng thẩm định, thuộc về một viện, một khu vực, quốc gia và liên quốc gia) được thiết lập gồm các chuyên gia y tế/khoa học và các thành viên không trong ngành y, không phải là chuyên gia khoa học; những người này có trách nhiệm đảm bảo việc bảo vệ các quyền, sự an toàn và sức khỏe của đối tượng nghiên cứu và cung cấp sự đảm bảo cho xã hội đối với việc bảo vệ này bằng cách thẩm định và phê duyệt/cung cấp ý kiến ủng hộ cho đề cương thử nghiệm, sự thích hợp của những nhà nghiên cứu, phương tiện, các phương pháp và tài liệu sử dụng để thu thập và ghi nhận việc thông tin cho đối tượng nghiên cứu và đối tượng ký phiếu đồng ý tham gia thử nghiệm (informed consent). (Tình trạng pháp lý, các thành viên, chức năng, hoạt động và những yêu cầu quản lý của Ủy ban Đạo đức Độc lập có thể khác nhau giữa các quốc gia nhưng phải cho phép Ủy ban Đạo đức Độc lập hoạt động thống nhất với GCP như mô tả trong hướng dẫn liên quan hiện hành).
Ủy ban giám sát dữ liệu độc lập (IDMC) (Ban giám sát dữ liệu và tính an toàn, Ủy ban giám sát, Ủy ban giám sát dữ liệu) [theo E6]/ Tham khảo: ACTD-E..
Một Ủy ban giám sát dữ liệu độc lập có thể do nhà tài trợ thiết lập để đánh giá định kỳ về tiến triển của một thử nghiệm lâm sàng, dữ liệu về tính an toàn, và các tiêu chí chính đánh giá hiệu quả, và để tham vấn cho nhà tài trợ có nên tiếp tục, sửa đổi hay kết thúc một thử nghiệm.
Thông tin cho đối tượng tham gia và ký phiếu đồng ý (Informed Consent) [theo E6]/ Tham khảo: ACTD-E...
Một quá trình mà đối tượng nghiên cứu tự nguyện xác định là có muốn tham gia vào một thử nghiệm cụ thể, sau khi đã được thông tin về tất cả các khía cạnh của thử nghiệm liên quan đến quyết định tham gia của đối tượng. (Việc thông tin và đồng ý này được ghi thành văn bản, ký và ghi ngày tháng vào mẫu).
Vật liệu đối chiếu sơ khởi tự sản xuất [theo Q6B]/ Tham khảo: ACTD-Q...
Vật liệu chuyên biệt do nhà sản xuất pha chế từ một (một số) mẻ đại diện dùng làm chuẩn cho các định lượng sinh học và kiểm nghiệm hóa lý của các mẻ sau, và dựa vào đó để hiệu chỉnh vật liệu đối chiếu vận hành tự sản xuất.
Vật liệu đối chiếu vận hành tự sản xuất [theo Q6B]/ Tham khảo: ACTD-Q...
Vật liệu được pha chế tương tự như vật liệu đối chiếu sơ khởi chỉ với mục đích là đánh giá và kiểm soát những thuộc tính riêng trong tình trạng nghi vấn của những mẻ sau.
(Vật liệu đối chiếu vận hành tự sản xuất luôn được hiệu chỉnh bằng vật liệu đối chiếu sơ khởi tự sản xuất).
Dược phẩm phát minh đầu tiên [theo WHO]
Một dược phẩm phát minh đầu tiên nói chung là sản phẩm lần đầu tiên được cấp giấy phép lưu hành (thường là được bảo hộ bản quyền sáng chế) dựa trên những tài liệu về hiệu quả, độ an toàn và chất lượng (theo những yêu cầu tại thời điểm cấp phép).
(Khi một chất đã xuất hiện trên thị trường trong nhiều năm có thể không xác định được sản phẩm phát minh đầu tiên).
Kiểm tra lắp đặt (IQ) [theo WHO]
Việc tiến hành và văn bản hóa các thử nghiệm để đảm bảo rằng thiết bị (chẳng hạn máy móc, thiết bị đo lường, các công cụ, các khu vực sản xuất) được sử dụng trong quá trình sản xuất được lựa chọn thích hợp, lắp đặt đúng và hoạt động theo đúng các thông số kỹ thuật đã xây dựng.
Đơn vị thử lâm sàng (Institution (medical)) [theo E6]/ Tham khảo: ACTD-E...
Bất cứ cơ sở y/nha khoa hoặc cơ quan y tế công hoặc tư nhân nơi tiến hành thử nghiệm lâm sàng.
Hội đồng xét duyệt (IRB) [theo E6]/ Tham khảo: ACTD-E…
Một cơ quan độc lập bao gồm các thành viên y học, khoa học và không thuộc lĩnh vực khoa học có trách nhiệm đảm bảo bảo vệ các quyền, tính an toàn và sức khỏe của đối tượng nghiên cứu bằng cách thẩm định, phê duyệt và tiếp tục thẩm định đề cương thử nghiệm và các bản sửa đổi, các phương pháp và tài liệu sử dụng để thu thập và ghi nhận việc thông tin cho đối tượng nghiên cứu và đối tượng ký phiếu đồng ý tham gia thử nghiệm (informed consent).
Dược phẩm thay thế [theo WHO]
Sản phẩm tương đương về mặt điều trị với một sản phẩm đối chiếu.
Báo cáo thử nghiệm/ Nghiên cứu Lâm sàng giữa kỳ [theo E6]/ Tham khảo: ACTD-E..
Một báo cáo kết quả giữa kỳ và việc thẩm định dựa trên những phân tích tiến hành trong quá trình thử nghiệm.
Tính chính xác trung gian [theo Q2A]/ Tham khảo: ACTD-EQ...
Tính chính xác Trung gian diễn tả những sai biệt trong cùng phòng thí nghiệm: giữa các ngày khác nhau, giữa những phân tích viên khác nhau, và giữa các thiết bị khác nhau...
Chương trình giám sát Môi trường nội bộ [theo ASEAN]
Một chương trình xác định ghi thành tài liệu mô tả việc giám sát thường quy các tiểu phân và vi sinh của khu vực chế biến và sản xuất, và chương trình này bao gồm cả kế hoạch hiệu chỉnh khi các hoạt động vượt quá mức cho phép.
Sản phẩm nghiên cứu [theo E6]/ Tham khảo: ACTD-E...
Một dạng dược phẩm của một thành phần hoạt chất hay placebo được thử nghiệm hay sử dụng như chất tham chiếu (reference) trong một thử nghiệm lâm sàng, bao gồm cả sản phẩm đã được cấp phép lưu hành khi được sử dụng hoặc bào chế (công thức hay đóng gói) khác với dạng được phê duyệt, hay khi được sử dụng với chỉ định chưa được phê duyệt, hay khi được sử dụng để thu thập thêm những thông tin về công dụng đã được phê duyệt.
Nhà nghiên cứu [theo E6]/ Tham khảo: ACTD-E...
Người có trách nhiệm tiến hành thử nghiệm lâm sàng tại trung tâm thử nghiệm. Nếu thử nghiệm do một nhóm tiến hành tại trung tâm thử nghiệm thì nhà nghiên cứu là người trưởng nhóm chịu trách nhiệm và có thể được gọi là nhà nghiên cứu chính.
Lô quy mô phòng thí nghiệm [theo WHO]
Những lô sản xuất ở giai đoạn thí nghiệm nghiên cứu và bước đầu của phát triển.
(Chúng có thể là những lô có cỡ rất nhỏ [ví dụ chỉ bằng 1/100 hay 1/1000 lần của cỡ lô sản xuất]).
Thư ủy quyền [theo WHO]
Thư của nhà sản xuất hay chủ sở hữu sản phẩm ủy quyền cho một đối tượng trong nước là người giữ giấy phép đăng ký và chịu trách nhiệm đối với tất cả các vấn đề liên quan đến đăng ký sản phẩm.
Tuyến tính [theo Q2A]/ Tham khảo: ACTD-Q...
Tuyến tính của một quy trình phân tích là phạm vi các kết quả thử nghiệm tỉ lệ thuận với nồng độ (lượng) của chất phân tích trong mẫu.
Thử nghiệm dài hạn thời gian thực (liên quan đến độ ổn định) [theo Q1AR]/ Tham khảo: ACTD-Q...
Những nghiên cứu độ ổn định dưới điều kiện bảo quản khuyến cáo được thực hiện trong giai đoạn kiểm nghiệm lại hoặc để nghiên cứu tuổi thọ đề nghị (hoặc tuổi thọ đã được phê duyệt) để đưa vào nhãn.
Thay đổi lớn (MaV) [theo WHO]
Thay đổi đối với một dược phẩm đã được phép lưu hành ảnh hưởng đến một trong những điểm sau:
- đường dùng
- hàm lượng, liều
- chỉ định, hoặc
- những điểm không nằm trong định nghĩa thay đổi nhỏ.
(Hồ sơ nộp xin phép thay đổi lớn thường yêu cầu phải có dữ liệu cần thiết xác định chất lượng, độ an toàn và hiệu quả của công thức mới) (xem Thay Đổi, Thay Đổi Nhỏ)
Sản xuất [theo WHO]
Tất cả các hoạt động mua nguyên liệu và sản phẩm, sản xuất, kiểm tra chất lượng, xuất xưởng, bảo quản, vận chuyển (từ kho đến nhà máy sản xuất) của thành phẩm, và các biện pháp kiểm tra có liên quan.
Nhà sản xuất [theo WHO]
Một công ty thực hiện ít nhất một công đoạn sản xuất và xuất xưởng thành phẩm.
Giấy phép lưu hành
Một tài liệu chính thức do cơ quan quản lý dược có thẩm quyền cấp, cho phép đưa ra thị trường hay phân phối tự do một sản phẩm sau khi đã thẩm định độ an toàn, hiệu quả và chất lượng.
(Bên cạnh những mục khác, phải có tên sản phẩm, dạng bào chế, công thức định lượng (bao gồm cả tá dược) cho một đơn vị liều [sử dụng tên INN hay tên generic quốc gia), tuổi thọ, điều kiện bảo quản, đặc điểm đóng gói. Giấy phép lưu hành cần ghi rõ thông tin làm cơ sở cho việc cấp phép [ví dụ: "sản phẩm phải tuân theo tất cả những chi tiết ghi trong hồ sơ đăng ký và những sửa đổi tương ứng]. Nó cũng chứa những thông tin sản phẩm được phê duyệt dành cho cán bộ y tế và công chúng, phân loại sản phẩm kinh doanh, tên và địa chỉ của người giữ giấy phép và thời hạn hiệu lực của giấy phép).
Đơn vị sở hữu giấy phép lưu hành [theo WHO]
Một công ty hay tập đoàn hay chủ thể hợp pháp trong lĩnh vực dược được cấp giấy phép lưu hành dược phẩm dưới tên của đơn vị đó, chịu trách nhiệm về tất cả vấn đề liên quan đến sản phẩm, bao gồm cả chất lượng và việc tuân thủ những điều kiện của giấy phép lưu hành. Đơn vị sở hữu giấy phép phải chịu trách nhiệm pháp lý tại nước cấp giấy phép lưu hành, tức là thông thường phải có trụ sở tại nước này.
Cân bằng khối lượng [theo QIAR]/ Tham khảo: ACTD-Q...
Tổng các giá trị định lượng và cả độ phân hủy sản phẩm và tính toán xem tổng số này lệch bao nhiêu so với 100% giá trị ban đầu, cân nhắc phù hợp với phạm vi sai số trong phân tích.
Ngân hàng tế bào chủ (MCB) [theo Q5A]/ Tham khảo: ACTD-Q...
Chứa các phần chia nhỏ của 1 nguồn tế bào từ một dòng tế bào chọn lọc dưới những điều kiện xác định- được để vào các vật chứa nhiều ngăn và bảo quản ở điều kiện xác định. (Ngân hàng tế bào chủ được sử dụng để tạo ra các ngân hàng tế bào sử dụng cho thí nghiệm khác) Việc thử nghiệm trên một ngân hàng tế bào chủ mới [từ một dòng tế bào, ngân hàng tế bào chủ và ngân hàng tế bào sử dụng cho thử nghiệm ban đầu] cũng tương tự như cho các ngân hàng tế bào chủ khác, trừ phi có các yêu cầu khác.
Công thức gốc [theo WHO]
Một tài liệu hoặc một bộ tài liệu chỉ rõ những nguyên liệu ban đầu và khối lượng của chúng, nguyên liệu bao gói, cùng với mô tả các quy trình và những điểm cần thận trọng để sản xuất ra một lượng xác định thành phẩm, cũng như các chỉ dẫn về chế biến, kể cả kiểm tra trong quá trình sản xuất.
Ma trận (liên quan đến Độ ổn định) [theo QIAR]/ Tham khảo: ACTD-Q...
Một thiết kế thời gian biểu nghiên cứu độ ổn định mà một tập con được chọn trong tổng số mẫu, sẽ được thử tại một thời điểm cụ thể về tất cả những yếu tố kết hợp.
(Tại một thời điểm sau đó, một tập con khác với tất cả tất cả những yếu tố kết hợp sẽ được thử. Thiết kế này giả định rằng độ ổn định của mỗi tập con của mẫu thử có thể đại diện cho độ ổn định của toàn bộ mẫu tại một thời điểm đưa ra; sự khác biệt giữa các mẫu của cùng một dược phẩm cần được xác định, ví dụ như lô khác nhau, hàm lượng khác nhau, quy cách đóng gói khác nhau của cùng một hệ bao bì kín, và có thể trong vài trường hợp cả hệ bao bì kín khác nhau).
Sản phẩm y học
Xem Dược phẩm
Vi nhân [theo S2A]/ Tham khảo: ACTD-S...
Phần của tế bào chứa ADN nhân có thể phát hiện được bằng kính hiển vi, và có thể chứa (các) nhiễm sắc thể hoàn chỉnh hay (các) phần đứt gãy tâm hoặc không tâm của (các)
nhiễm sắc thể.
(Kích thước của vi nhân thường được xác định là nhỏ hơn 1/5 nhưng lớn hơn 1/20 của nhân chính).
Thay đổi nhỏ (MiV) [theo WHO]
Thay đổi đối với một dược phẩm đã được lưu hành không ảnh hưởng đến một hay nhiều điểm sau:
- đường dùng
- hàm lượng, liều
- chỉ định, và
- hoạt chất
(Hồ sơ nộp xin phép thay đổi nhỏ thường phải có dữ liệu cần thiết chứng minh chất lượng của công thức mới) (Xem Thay đổi, Thay Đổi Lớn).
Chỉ số Phân bào [theo S2A]/ Tham khảo: ACTD-S...
Phần trăm các tế bào trong những giai đoạn khác nhau của sự gián phân trên tổng số các tế bào không phân bào quan sát trên một slide.
Thử nghiệm đa trung tâm [theo E6]/ Tham khảo: ACTD-S…
Một thử nghiệm lâm sàng tiến hành theo một đề cương nhưng tại nhiều hơn một nơi do đó được thực hiện bởi nhiều hơn một nhà nghiên cứu.
Dược phẩm Nhiều nguồn (Generic) [theo WHO]
Dược phẩm nhiều nguồn là những sản phẩm tương đương về dược, có thể tương đương hoặc không tương đương về mặt điều trị.
(Dược phẩm nhiều nguồn tương đương về mặt điều trị thì có thể thay thế nhau).
Cơ quan Quản lý Quốc gia (NRA) / Cơ quan cấp phép [theo WHO]
Một cơ quan quốc gia thực hiện đầy đủ các mặt hoạt động quản lý dược, bao gồm ít nhất các chức năng sau:
● Cấp phép lưu hành các sản phẩm mới và phê duyệt cho những thay đổi của những sản phẩm đã được lưu hành;
● Phòng thí nghiệm kiểm tra chất lượng;
● Theo dõi tác dụng phụ của thuốc;
● Cung cấp thông tin thuốc và thúc đẩy việc sử dụng thuốc hợp lý;
● Thanh tra Thực hành Sản xuất Tốt (GMP) và cấp giấy phép sản xuất, bán buôn và các kênh phân phối;
● Các hoạt động đốc thúc thực thi; và
● Theo dõi sử dụng thuốc
Hoạt chất Mới [theo WHO]
Một hoạt chất hóa học hay sinh học từ trước đến nay chưa được cấp giấy phép lưu hành cho việc sử dụng trong Dược phẩm tại một quốc gia.
(Những chất được cấp phép có điều kiện tại thời điểm thử nghiệm thị trường ban đầu không phải là hoạt chất mới; xem Hóa chất mới, Hoạt chất hóa học hay sinh học mới, Phân tử mới, Thuốc quen dùng, Thuốc phối hợp định liều quen dùng).
Hóa chất mới (NCE) [theo WHO]
Xem Hoạt Chất Mới.
Hoạt chất Hóa học hay Sinh học Mới [theo WHO]
Xem Hoạt chất Mới.
Phân tử Mới [theo Q1AR]/ Tham khảo: ACTD-Q...
Một muối mới, ester mới hay dẫn xuất không có liên kết hóa trị mới của một dược chất đã được lưu hành.
(Nó được cho là phân tử mới để thử nghiệm độ ổn định, xem Hoạt Chất Mới).
Thay đổi Số Nhiễm sắc thể [theo S2B]/ Tham khảo: ACTD-S...
Số nhiễm sắc thể khác với bộ nhiễm sắc thể đơn bội hoặc lưỡng bội gốc ; đối với dòng tế bào, số nhiễm sắc thể khác với bộ nhiễm sắc thể mẫu
Thẩm định vận hành (OQ) [theo WHO] (GMP)
Việc thẩm tra được ghi thành hồ sơ để khẳng định hệ thống hay tiểu hệ thống hoạt động như dự định trong toàn bộ các phạm vi hoạt động dự kiến.
Bệnh án Gốc [theo WHO]
Xem Tài liệu Nguồn
Tờ Hướng dẫn Sử dụng [theo WHO]
Một tài liệu xác định những thông tin do đơn vị sở hữu giấy phép lưu hành cung cấp cùng với dược phẩm.
(Nội dung của tờ hướng dẫn sử dụng thuốc do cơ quan quản lý quốc gia phê duyệt khi cấp giấy phép lưu hành; xem Tờ thông tin cho Bệnh nhân).
Tờ Thông tin cho Bệnh nhân (PIL)
Một tài liệu xác định thông tin cung cấp cho bệnh nhân do đơn vị sở hữu giấy phép lưu hành cung cấp cùng với dược phẩm.
(Nội dung của PIL do cơ quan quản lý quốc gia phê duyệt khi cấp phép lưu hành; xem Tờ hướng dẫn Sử dụng).
Báo cáo trên bố mẹ-con cái/ Thai nhi [theo E2B]/ Tham khảo: ACTD-E ...
Báo cáo về các phản ứng/ hiện tượng ở trẻ con/ bào thai nghi ngờ có liên quan đến việc dùng thuốc của bố/ mẹ.
Thẩm định qui trình thực hiện (PQ) [theo WHO]
Bằng chứng bằng văn bản chứng minh một bước của quy trình, toàn bộ hệ thống quy trình, một phương pháp phân tích thực hiện đúng như dự định, và rằng các vật liệu, sản phẩm hay kết quả phân tích hoàn toàn đáp ứng với những tiêu chuẩn kỹ thuật và những yêu cầu trong đề cương.
(Một điều quan trọng là các tiêu chuẩn chấp nhận cần phải rõ ràng và cụ thể cho mỗi thông số chính).
Tương đương Dược học [theo WHO]
Hai sản phẩm tương đương dược học nếu chúng chứa cùng một lượng hoạt chất trong cùng một dạng bào chế, cùng tiêu chuẩn chất lượng, và dự định có cùng đường sử dụng.
(Tương đương dược học không nhất thiết phải biểu thị tương đương về điều trị vì sự khác nhau về tá dược và/hoặc quy trình sản xuất có thể dẫn đến hiệu quả khác nhau của sản phẩm).
Dược phẩm
Bất cứ chế phẩm nào dùng cho con người với mục đích thay đổi hoặc thăm dò các hệ sinh lý hay trạng thái bệnh tật vì lợi ích của đối tượng.
Nghiên cứu Dược lực học [theo E5]/ Tham khảo: ACTD-E ...
Một nghiên cứu về tác dụng dược lý hay tác dụng lâm sàng của một thuốc trên cá thể để miêu tả mối liên hệ giữa tác dụng của thuốc và liều sử dụng hay nồng độ thuốc. (Tác động dược lực có thể là tác dụng có hại tiềm ẩn [ví dụ: tác động kháng cholinergic của thuốc chống trầm cảm loại 3 vòng (tricyclic)], hay sự đo lường một tác động có liên quan đến lợi ích lâm sàng [như việc đo lường hoạt tính các thuốc chẹn beta, tác động trên các khoảng ECG, hoặc thuốc ức chế ACE hoặc ức chế đáp ứng angiotensin I hay II), một tác động mong muốn ngắn hạn, thường là một tiêu chí thay thế [như huyết áp, cholesterol], hay lợi ích lâm sàng dự kiến cơ bản (tác dụng đối với đau, trầm cảm, đột tử).
Nghiên cứu Dược động học [theo E5]/ Tham khảo: ACTD-E ...
Nghiên cứu về số phận của một dược phẩm trong cơ thể, thường liên quan đến việc đo nồng độ của sản phẩm và chất chuyển hóa của nó trong máu (đôi khi đo nồng độ trong nước tiểu hay trong mô) theo thời gian.
(Nghiên cứu dược động học dùng để mô tả đặc điểm hấp thu, phân phối, chuyển hóa và thải trừ dược phẩm trong máu hoặc ở những vị trí thích hợp khác). Khi phối hợp với các số liệu nghiên cứu về dược lực, số liệu về dược động sẽ đặc trưng hóa sự tương quan giữa nồng độ trong máu với mức độ và thời điểm của các tác động dược lực.
Lô Quy mô Thử nghiệm [theo QIAR]/ Tham khảo: ACTD-Q...
Một lô dược chất hay dược phẩm được sản xuất bằng một quy trình đại diện và mô phỏng để có thể áp dụng cho một lô sản xuất trên quy mô đầy đủ.
(Đối với dạng bào chế rắn dùng đường uống, quy mô thử nghiệm pilot thường tối thiểu bằng 1 phần 10 của quy mô sản xuất đầy đủ hoặc 100.000 viên nén hay viên nang, trừ khi có quy định khác).
Plasmid [theo S2A]/ Tham khảo: ACTD-S...
Thành tố của gen thêm vào hệ gen bình thường của vi khuẩn.
(Một plasmid có thể được chèn vào nhiễm sắc thể của tế bào chủ hay tạo thành một yếu tố ngoại nhiễm sắc thể).
Đột biến Điểm [theo S2A]/ Tham khảo: ACTD-S...
Thay đổi trong mã di truyền thường khu trú trên một cặp bazơ ADN riêng biệt.
Hồng cầu Đa sắc [theo S2A]/ Tham khảo: ACTD-S ...
Một hồng cầu chưa trưởng thành trong một giai đoạn trung gian phát triển chứa ribosome và có thể phân biệt với những hồng cầu màu sắc bình thường trưởng thành (thiếu ribosome) bằng cách nhuộm màu chọn lọc cho ribosome.
Phương pháp Dược động học theo tập hợp [theo E5]/ Tham khảo: ACTD-E ...
Là phương pháp đánh giá việc đo lường nồng độ thuốc trong cơ thể với tối thiểu là 2 phương pháp cho mỗi bệnh nhân ở điều kiện bình thường, từ tất cả hoặc một tập con xác định các bệnh nhân tham gia vào các thử nghiệm lâm sàng.
Hoạt lực [theo Q6B]/ Tham khảo: ACTD-Q ...
Sự đo lường hoạt tính sinh học bằng cách sử dụng một định lượng sinh học phù hợp (cũng còn được gọi là định lượng hoạt lực hay định lượng sinh học) căn cứ trên thuộc tính của sản phẩm có liên hệ với các tính chất sinh học liên quan.
Độ Chính xác [theo Q2A])/ Tham khảo: ACTD-Q...
Độ chính xác của một quy trình phân tích diễn tả mức độ gần đúng (độ phân tán) giữa một loạt các phép đo từ nhiều mẫu khác nhau lấy từ cùng một mẫu đồng nhất dưới những điều kiện mô tả.
(Độ chính xác có thể được chia thành 3 mức: tính lặp lại (repeatability), độ chính xác trung gian và độ lập lại (reproducibility). Độ chính xác nên được nghiên cứu trên các mẫu thực, đồng nhất. Tuy nhiên nếu không thể có được từ một mẫu đồng nhất thì có thể nghiên cứu sử dụng các dung dịch mẫu hoặc mẫu nhân tạo. Độ chính xác của một quy trình phân tích thường được thể hiện là độ biến thiên, độ lệch chuẩn hay hệ số biến thiên của một loạt các phép đo).
Lô Đầu tiên ([theo QIAR]/ Tham khảo: ACTD-Q...
Một lô dược chất hoặc dược phẩm được sử dụng trong nghiên cứu độ ổn định, qua đó dữ liệu về độ ổn định được nộp trong hồ sơ xin đăng ký nhằm thiết lập giai đoạn thử lại của dược chất hoặc tuổi thọ của dược phẩm.
(Lô Đầu tiên của dược chất ít nhất phải là một lô quy mô thử nghiệm. Đối với dược phẩm, tối thiểu hai trong số ba lô phải là lô ở qui mô thử nghiệm, lô thứ ba có thể nhỏ hơn nếu nó đại diện cho các bước sản xuất chính yếu. Tuy nhiên, lô đầu tiên có thể là một lô sản xuất).
Chủ sở hữu sản phẩm
Cá nhân, hoặc công ty hoặc chủ thể là chủ sở hữu hợp pháp/ có đăng ký của công thức sản phẩm và/hoặc qui trình sản xuất sản phẩm và có hợp đồng với đơn vị sở hữu giấy phép lưu hành.
Các tạp chất liên quan đến qui trình [theo Q6B]/ Tham khảo: ACTD-Q...
Những tạp chất xuất hiện trong quá trình sản xuất, chúng có thể từ các chất của tế bào
(ví dụ các protein của tế bào chủ, ADN tế bào chủ), từ môi trường nuôi cấy tế bào (ví dụ chất gây cảm ứng, kháng sinh, các thành phần của môi trường), hay qui trình xử lý qua cột (ví dụ các thuốc thử, chất lọc qua cột).
Thẩm định qui trình [theo FDA]/ Tham khảo ACTD- Q...
Minh chứng bằng văn bản nhằm đảm bảo tốt một qui trình cụ thể sẽ sản xuất sản phẩm đáp ứng những chỉ tiêu kỹ thuật và chất lượng định trước.
Đơn vị sở hữu giấy phép sản phẩm
Xem đơn vị sở hữu giấy phép lưu hành sản phẩm
Các tạp chất liên quan đến sản phẩm [theo Q6B]/ Tham khảo: ACTD-Q...
Những thành phần biến đổi phân tử của sản phẩm (ví dụ các tiền chất, sản phẩm phân hủy của sản phẩm xuất hiện trong quá trình sản xuất và/ hoặc bảo quản) không có các đặc tính mong muốn của sản phẩm về hoạt lực, hiệu lực và độ an toàn.
Các chất liên quan đến sản phẩm [theo Q6B] / Tham khảo: ACTD Q...
Những phân tử biến đổi của sản phẩm hình thành trong quá trình sản xuất và/ hoặc bảo quản có hoạt tính nhưng không gây tác hại đến độ an toàn và hiệu lực của dược phẩm. (Những phần biến đổi này có những đặc tính tương đương với sản phẩm mong muốn và không được coi là tạp chất).
Lô sản xuất [theo QIAR]/ Tham khảo: ACTD-Q...
Một lô dược chất hay dược phẩm được sản xuất ở qui mô sản xuất bằng cách sử dụng những thiết bị sản xuất ghi rõ trong hồ sơ đăng ký.
Tiền thẩm định [theo ASEAN GMP]
Việc thiết lập những bằng chứng bằng văn bản về một quá trình, qui trình, hệ thống, thiết bị hay cơ chế sử dụng trong sản xuất nhằm xác định xem nó có được thực hiện đúng theo mục đích như trong đề cương thẩm định.
Đề cương (liên quan đến thử nghiệm lâm sàng) [theo E6]/ Tham khảo: ACTD-E...
Tài liệu mô tả mục đích, thiết kế, phương pháp luận, phân tích thống kê và tổ chức thử nghiệm lâm sàng.
(Đề cương cũng thường nêu lên cơ sở và tính hợp lý của thử nghiệm lâm sàng, nhưng những phần này cũng có thể được đưa ra ở những tài liệu tham khảo khác của đề cương theo Hướng dẫn của GCP ICH. Thuật ngữ đề cương bao hàm đề cương và phần sửa đổi đề cương).
Đảm bảo chất lượng (QA) [theo E6]/ Tham khảo: ACTD-E ...
Toàn bộ các kế hoạch được xếp đặt (bao gồm cả GMP) với mục đích để đảm bảo các dược phẩm có chất lượng đáp ứng được mục đích sử dụng của nó.
Kiểm tra chất lượng [theo WHO]
Kiểm tra chất lượng xem xét đến việc lấy mẫu, tiêu chuẩn kỹ thuật, kiểm nghiệm và đối với cả việc tổ chức, thiết lập hồ sơ, các qui trình chấp nhận/ lọai trừ nhằm đảm bảo rằng những thử nghiệm liên quan và cần thiết được thực sự tiến hành trên nguyên liệu ban đầu, nguyên liệu trung gian, và thành phẩm; đảm bảo thành phẩm không được chấp nhận đưa vào sử dụng, bán và cung cấp cho đến khi chất lượng của nó được kết luận là đạt yêu cầu.
Giới hạn định lượng [theo Q2]/ Tham khảo: ACTD- Q...
Giới hạn định lượng của một qui trình phân tích đơn lẻ là lượng nhỏ nhất chất phân tích trong một mẫu có thể định lượng được với độ chính xác và độ chuẩn thích hợp. (Giới hạn định lượng là một tham số định lượng cho lượng nhỏ hợp chất trong mẫu ma trận, đặc biệt được sử dụng để xác định tạp chất và/ hoặc sản phẩm phân hủy).
Thử nghiệm Ngẫu nhiên [theo E6]/ Tham khảo: ACTD-E...
Quá trình phân phối đối tượng tham gia thử nghiệm vào nhóm điều trị hay nhóm chứng bằng bốc thăm để hạn chế sự thiên lệch.
Tái tổ hợp [theo S2B]/ Tham khảo: ACTD- S...
Sự cắt và tái kết nối cân xứng hay không cân xứng của ADN.
Nước tham chiếu
Những nước có hệ thống thẩm định thuốc có uy tín được các Cơ Quan Quản Lý Thuốc ASEAN công nhận.
Sản phẩm đối chiếu [theo WHO]
a) Là dược phẩm mà một sản phẩm mới dự định là có thể thay thế nó trong thực tiễn lâm sàng.
(Sản phẩm đối chiếu thường là sản phẩm phát minh đầu tiên vì hiệu quả, độ an toàn và chất lượng đã được xác định. Khi không có sản phẩm phát minh đầu tiên thì sản phẩm đứng đầu thị trường có thể được sử dụng làm sản phẩm đối chiếu nhưng nó phải được cấp giấy phép lưu hành và hiệu lực, độ an toàn và chất lượng của nó phải được xác định rõ và ghi thành văn bản).
b) Là sản phẩm nghiên cứu hay đã được bán trên thị trường (tức là sản phẩm đối chứng có hoạt tính), hoặc có thể là giả dược (placebo) được sử dụng để đối chiếu trong một thử nghiệm lâm sàng.
(Xem Sản Phẩm So Sánh)
Tính lặp lại [theo Q2A]/ Tham khảo: ACTD-Q...
Tính lặp lại diễn tả độ chính xác trong cùng một điều kiện hoạt động trong một khoảng thời gian ngắn.
(Độ lặp lại còn được gọi là độ chính xác trong quá trình định lượng).
Người Báo cáo (liên quan đến thử nghiệm lâm sàng) [theo E2B]/ Tham khảo: ACTD-E
Người cung cấp nguồn thông tin ban đầu, ví dụ người đầu tiên báo cáo lại các dữ kiện.
(Cần phân biệt với người chuyển thông tin, mặc dầu người báo cáo cũng có thể là người chuyển thông tin).
Độ lập lại [theo Q2A]/ Tham khảo: ACTD-Q...
Diễn tả độ chuẩn xác giữa các phòng thí nghiệm. (Những nghiên cứu hợp tác, thường áp dụng để tiêu chuẩn hóa phương pháp).
Ngày thử lại [theo QIAR]/ Tham khảo: ACTD-Q...
Ngày mà sau đó mẫu dược chất được kiểm tra để đảm bảo rằng nguyên liệu vẫn phù hợp với tiêu chuẩn kỹ thuật và do đó thích hợp cho sử dụng để sản xuất một dược phẩm nhất định.
Giai đoạn Thử lại [theo QIAR]/ Tham khảo: ACTD- Q...
Là khoảng thời gian dược chất được cho là vẫn đạt tiêu chuẩn kỹ thuật và do đó có thể sử dụng để sản xuất một dược phẩm nhất định với điều kiện dược chất này phải được bảo quản ở điều kiện xác định.
(Sau giai đoạn này, lô dược chất dự định để sử dụng sản xuất một dược phẩm phải được thử lại để xác định dược chất có còn đạt tiêu chuẩn kỹ thuật và sau đó phải được sử dụng ngay. Một lô dược chất có thể được thử lại nhiều lần và mỗi phần khác nhau của lô vẫn được tiếp tục sử dụng sau mỗi lần thử lại nếu đạt các tiêu chuẩn kỹ thuật. Đối với hầu hết các chất sinh học/ công nghệ sinh học không ổn định, việc xác định tuổi thọ sẽ hợp lý hơn là giai đoạn thử lại. Điều này cũng đúng với một số kháng sinh).
Thẩm định hồi cứu [theo ASEAN GMP]
Thẩm định một quy trình của một sản phẩm đã được bán trên thị trường dựa trên những thông tin tích luỹ được khi sản xuất, kiểm nghiệm và kiểm soát lô.
Thẩm định lại [theo ASEAN GMP]
Lặp lại thẩm định quy trình để khẳng định những thay đổi trong quy trình/ thiết bị sử dụng phù hợp với yêu cầu của quy trình kiểm soát sự thay đổi và không gây ảnh hưởng bất lợi đến đặc tính của quy trình và chất lượng sản phẩm.
Độ ổn định [theo Q2A]/ Tham khảo: ACTD-Q...
Độ ổn định của một qui trình phân tích được đo bằng khả năng không bị ảnh hưởng của qui trình bởi những biến đổi nhỏ nhưng chủ tâm trên các thông số của phương pháp và cho biết mức độ tin cậy của qui trình khi sử dụng bình thường.
Bao bì Bán thấm theo QIAR)/ Tham khảo: ACTD- Q...
Bao bì cho phép dung môi, thường là nước, đi qua nhưng giữ lại chất hòa tan.
(Cơ chế của việc vận chuyển dung môi là hấp thu vào bề mặt bao bì, khuếch tán vào chất liệu bao bì và thoát ra bề mặt kia; Việc vận chuyển là do gradient áp suất từng phần điều khiển; ví dụ về bao bì bán thấm là túi nhựa, túi bán cứng polyethylene mật độ thấp (LDPE) để chứa dịch truyền thể tích lớn (LVP), ống/ chai/ lọ LDPE).
Người gửi (liên quan đến nghiên cứu lâm sàng) [theo E2B]/ Tham khảo: ACTD- E...
Người hay thực thể tạo ra tin nhắn để gửi. (Mặc dù người báo cáo và người gửi có thể là một nhưng chức năng của người gửi không nên bị lẫn với chức năng của người báo cáo).
Biến cố ngoại ý nghiêm trọng (SAE) hay Phản ứng có hại nghiêm trọng [theo E2B]/ Tham khảo: ACTD- E...
Bất cứ một hiện tượng y học bất lợi xuất hiện ở bất cứ liều điều trị nào:
- Gây tử vong;
- Đe dọa tính mạng;
- Đòi hỏi phải nằm viện nội trú hay kéo dài thời gian nằm viện;
- Gây ra tàn tật/mất khả năng lâu dài hoặc nghiêm trọng; hoặc
- Gây ra bất thường hay khuyết tật bẩm sinh
Tuổi thọ (giai đoạn còn hạn dùng của thuốc) [theo QIAR]/ Tham khảo: ACTD-Q...
Khoảng thời gian trong đó một dược phẩm được cho là vẫn đạt các tiêu chuẩn kỹ thuật đã được phê duyệt với điều kiện dược phẩm này được bảo quản trong điều kiện đã ghi trên nhãn bao bì.
Tác dụng phụ (theo Webside Chương trình giám sát thuốc WHO, www.who- umc.org)
Là tác dụng không mong muốn của một dược phẩm xuất hiện ở liều bình thường khi sử dụng trên người, có liên quan đến đặc tính dược lý của thuốc đó.
(Điều thiết yếu ở định nghĩa này là bản chất dược lý của tác dụng, và tác dụng là không mong muốn và không có việc dùng quá liều, xem Biến cố ngoại ý và Phản ứng Có Hại ).
Dữ liệu nguồn (liên quan đến nghiên cứu lâm sàng) [theo E6]/ Tham khảo: ACTD- E...
Tất cả các thông tin trong hồ sơ gốc và bản sao có chứng thực từ hồ sơ gốc về các phát hiện, quan sát lâm sàng hay những hoạt động lâm sàng khác trong một thử nghiệm lâm sàng cần thiết để tái tạo và đánh giá một thử nghiệm.
(Dữ liệu nguồn được giữ trong tài liệu nguồn, các hồ sơ gốc hay các bản sao có chứng thực).
Tài liệu nguồn (liên quan đến nghiên cứu lâm sàng) [theo E6]/ Tham khảo: ACTD-E…
Tài liệu, dữ liệu và các bản ghi gốc (ví dụ các bản ghi tại bệnh viện, các biểu đồ lâm sàng hay văn phòng, các ghi chú kết quả xét nghiệm, các bản ghi nhớ, sổ nhật ký bệnh nhân hay bản đối chiếu đánh giá, bản ghi nhận phát thuốc nghiên cứu từ khoa dược, số liệu được ghi lại từ các thiết bị tự động, bản copy hay sao chép được xác nhận sau khi được xác định là bản sao chính xác, các tấm vi phim, phim âm bản, vi phim hay các phương tiện từ, x- quang, hồ sơ bệnh nhân, và các bản ghi được giữ lại tại khoa dược, tại phòng xét nghiệm và tại phòng y kỹ thuật liên quan đến thử nghiệm lâm sàng).
Tiêu chuẩn kỹ thuật [theo Q6B]/ Tham khảo: ACTD-Q...
Một danh mục các phép thử, các tham chiếu qui trình phân tích và các tiêu chuẩn chấp nhận được thể hiện dưới dạng các giới hạn về số, khoảng số hay các tiêu chuẩn khác của thử nghiệm.
(Nó tạo ra một nhóm các tiêu chuẩn mà một dược chất, dược phẩm hay nguyên liệu tại những thời điểm khác nhau của việc sản xuất phải đáp ứng để được chấp nhận sử dụng. Đáp ứng các tiêu chuẩn kỹ thuật nghĩa là một dược chất hay dược phẩm, khi được thử ở những qui trình phân tích được liệt kê, sẽ đáp ứng với những tiêu chuẩn đề ra. Tiêu chuẩn kỹ thuật là tiêu chuẩn chất lượng then chốt do nhà sản xuất đề nghị và được các cơ quan quản lý thuốc phê duyệt theo những điều kiện phê duyệt).
Tiêu chuẩn kỹ thuật (Xuất xưởng) [theo QIAR]/ Tham khảo: ACTD-Q...
Tiêu chuẩn mà một dược phẩm phải đạt được để xuất xưởng.
Tiêu chuẩn kỹ thuật (tuổi thọ) [theo QIAR]/
Tiêu chuẩn kỹ thuật mà một dược phẩm cần phải đạt được trong suốt giai đoạn kiểm nghiệm lại dược phẩm này, hoặc trong suốt tuổi thọ của nó.
Tính đặc hiệu [theo Q2A]/ Tham khảo: ACTD- Q...
Tính đặc hiệu là khả năng đánh giá chắc chắn một chất phân tích trong sự hiện diện của các thành phần khác được cho là có mặt. (Điển hình bao gồm các tạp chất, các chất phân hủy, thành phần thuốc thử trong định lượng...)
Sự thiếu tính đặc hiệu của một qui trình phân tích đơn lẻ có thể được bù đáp bằng (các) qui trình phân tích hỗ trợ.
Định nghĩa này bao hàm các ý nghĩa sau:
- Định tính: để đảm bảo nhận ra được sự hiện diện của một chất phân tích.
- Thử độ tinh khiết: để đảm bảo tất cả các qui trình phân tích tiến hành đều cho phép đưa ra hàm lượng chính xác tạp chất của một chất phân tích, ví dụ: thử những chất liên quan, kim loại nặng, hàm lượng dung môi còn lại...
- Định lượng (hàm lượng hay hoạt tính): để cung cấp một kết luận chính xác cho phép đưa ra chính xác hàm lượng hay hoạt tính của chất phân tích trong một mẫu.
Độ ổn định [theo WHO]
Khả năng một hoạt chất hay một dược phẩm để duy trì tính chất của nó trong một giới hạn nhất định trong suốt tuổi thọ của nó. (Những khía cạnh hóa học, vật lý, vi sinh và sinh dược học của độ ổn định bắt buộc phải được cân nhắc).
Qui trình thao tác chuẩn (SOP) [theo E6]/ Tham khảo: ACTD- E...
Hướng dẫn bằng văn bản chi tiết để đạt được sự đồng nhất khi thực hiện một chức năng cụ thể.
Nguyên liệu ban đầu [theo WHO]
Bất cứ chất nào có chất lượng theo qui định được sử dụng trong sản xuất một dược phẩm nhưng không bao gồm vật liệu đóng gói.
Tiệt trùng [theo WHO]
Qui trình đã thẩm định được sử dụng làm cho một sản phẩm không có vi sinh vật sống.
Thử độ vô khuẩn [theo WHO]
Phép thử tiến hành để xác định xem vi sinh vật sống có mặt hay không.
Dung sai điều kiện bảo quản (liên quan đến độ ổn định) [theo Q1A]/ Tham khảo: ACTD-Q...
Sư biến thiên nằm trong mức chấp nhận được về nhiệt độ và độ ẩm tương đối của thiết bị bảo quản trong một nghiên cứu độ ổn định chính thức.
(Thiết bị phải có khả năng kiểm soát được điều kiện bảo quản trong giới hạn ghi trong hướng dẫn liên quan. Nhiệt độ và độ ẩm thực tế khi được kiểm soát, nên được theo dõi trong quá trình bảo quản thử độ ổn định. Sự ảnh hưởng trong thời gian ngắn do mở cửa thiết bị là có thể chấp nhận. Sự biến thiên do thiết bị bị hỏng phải được xác định và báo cáo nếu được xem là ảnh hưởng tới kết quả nghiên cứu độ ổn định. Sự biến thiên vượt quá dung sai cho phép trên 24 giờ phải được mô tả trong báo cáo nghiên cứu và phải đánh giá tác động ảnh hưởng của nó).
Thử nghiệm trong điều kiện khắc nghiệt (Dược phẩm) [theo Q1A]/ Tham khảo: ACTD- Q...
Nghiên cứu được thực hiện để đánh giá tác động của các điều kiện khắt nghiệt lên dược phẩm. (bao gồm cả thử nghiệm độ bền với ánh sáng;- xem ICH Q1B- và những thử nghiệm đặc thù trên những sản phẩm nhất định, ví dụ: dạng khí dung phân liều, kem, nhũ dịch, sản phẩm nước lỏng bảo quản lạnh).
Thử nghiệm trong điều kiện khắt nghiệt (Dược chất) [theo Q1A]/ Tham khảo: ACTD-Q...
Nghiên cứu được thực hiện để làm sáng tỏ độ ổn định thực chất của dược chất. Phép thử này là một phần của chiến lược phát triển và nó thường được tiến hành ở những điều kiện khắt khe hơn so với những điều kiện thử lão hóa cấp tốc.
Nghiên cứu viên phụ [theo E6]/ Tham khảo: ACTD-E...
Bất cứ thành viên nào trong nhóm thử lâm sàng được chỉ định và giám sát bởi nghiên cứu viên chính tại một nơi thử nghiệm để tiến hành những qui trình liên quan đến thử nghiệm và/hoặc đưa ra quyết định quan trọng liên quan đến thử nghiệm (ví dụ các cộng sự, sinh viên nội trú, nhà nghiên cứu).
(Xem ‘Người nghiên cứu’).
Mã nhận diện đối tượng tham gia nghiên cứu [theo E6]/ Tham khảo: ACTD- E... Một nhận dạng duy nhất do người nghiên cứu gán cho mỗi đối tượng tham gia thử nghiệm để bảo vệ việc xác định đối tượng và được sử dụng thay cho tên của đối tượng tham gia nghiên cứu khi người nghiên cứu báo cáo những biến cố ngoại ý và/hoặc những số liệu liên quan đến thử nghiệm khác.
Tóm tắt đặc tính sản phẩm (SPC)- [theo EU]
Thông tin sản phẩm được cơ quan quản lý thuốc phê duyệt. (SPC là nguồn thông tin cho các cán bộ y tế và cho người tiêu dùng, thường được ghi trên nhãn và tờ hướng dẫn sử dụng của dược phẩm và để kiểm soát quảng cáo, xem Tờ Hướng dẫn sử dụng, Tờ Thông tin cho bệnh nhân)
Số liệu hỗ trợ (liên quan đến Độ ổn định) [theo E1AR]/ Tham khảo: ACTD-Q...
Số liệu không xuất phát từ những nghiên cứu độ ổn định chính thức, có tác dụng hỗ trợ các qui trình phân tích, thời hạn thử lại dự định hay tuổi thọ và khuyến cáo về bảo quản ghi trên nhãn. (Số liệu này bao gồm (1) số liệu độ ổn định trên những lô dược chất có phương pháp tổng hợp sau đây: những lô nguyên liệu qui mô nhỏ, công thức nghiên cứu không có ý định đưa ra thị trường, các công thức liên quan và sản phẩm được trình bày trong dạng bao bì đóng gói không giống với dạng để đưa ra thị trường; (2) thông tin liên quan đến kết quả thử bao bì; và (3) những cơ sở khoa học khác).
Sự sống sót tế bào (trong ngữ cảnh của thử nghiệm đột biến gen)/ [theo S2A]/ Tham khảo: ACTD-S...
Tỷ lệ tế bào ở một giai đoạn sống trong số những tế bào chết, thường được xác định bằng việc nhuộm màu và phương pháp đếm dòng tế bào sau một thời kỳ điều trị nhất định.
Chuyển gen [theo S2B]/ Tham khảo: ACTD-S...
Một gen ngoại sinh hay gen bên ngoài được chèn vào hệ gen chủ, ở thể tế bào hay thể phôi.
Tương đương trị liệu [theo WHO]
Hai dược phẩm tương đương về mặt trị liệu nếu chúng tương đương về dược học hay có thể sử dụng thay thế cho nhau, và sau khi dùng thuốc ở cùng một liều lượng, tác dụng của chúng trên khía cạnh hiệu lực và độ an toàn căn bản là như nhau, khi được xác định bằng những nghiên cứu tương đương sinh học, dược lực học, lâm sàng hay in vitro thích hợp.
Nơi thử nghiệm [theo WHO]
Vị trí tại đây các hoạt động liên quan đến thử nghiệm thực tế được tiến hành.
Phản ứng có hại không mong muốn của thuốc [theo E6]/ Tham khảo: ACTD-E...
Một phản ứng có hại, bản chất hoặc mức độ trầm trọng của nó không giống với thông tin sản phẩm áp dụng được. (Ví dụ tài liệu của người nghiên cứu về một sản phẩm nghiên cứu chưa được phê duyệt hay tờ hướng dẫn sử dụng/ tóm tắt đặc tính sản phẩm của một thuốc đã được phê duyệt, xem Hướng dẫn ICH về Quản lý dữ liệu an toàn: định nghĩa và tiêu chuẩn báo cáo tiến hành.
Sự tổng hợp ADN lệch pha (UDS) [theo S2A]/ Tham khảo: ACTD-E...
Tổng hợp ADN xuất hiện ở một giai đoạn nào đó trong chu kỳ tế bào nhưng không phải pha S khi có ADN bị hỏng.
(Thường liên quan với quá trình sữa chữa ADN bị cắt).
Thẩm định [theo WHO GMP]
Qui định bằng văn bản chứng minh bất kỳ một qui trình, tiến trình, thiết bị, vật liệu, hoạt động hay hệ thống thực sự mang lại kết quả như mong muốn.
Đề cương thẩm định [theo Q7A]/ Tham khảo: ACTD-Q…
Kế hoạch bằng văn bản xác định cách thức thẩm định sẽ được tiến hành và đưa ra các tiêu chuẩn chấp thuận.
(Ví dụ: đề cương về 1 qui trình sản xuất xác định thiết bị xử lý, các thông số qui trình quan trọng/ phạm vi vận hành quan trọng, đặc tính của sản phẩm, cách lấy mẫu, dữ liệu thử nghiệp thu thập, số lần thẩm định và kết quả thử nghiệm được chấp nhận).
Báo cáo thẩm định [theo ASEAN GMP]
Văn bản đúc kết hồ sơ, kết quả và sự đánh giá một chương trình thẩm định hoàn thiện.
(Có thể bao gồm những đề nghị cải thiện qui trình và/hay thiết bị).
Sự biến thiên [theo WHO]
Sự thay đổi trên bất cứ khía cạnh nào của dược phẩm, kể cả thay đổi thành phần công thức, phương pháp và địa điểm sản xuất, tiêu chuẩn kỹ thuật của thành phẩm và các thành phần khác, bao bì, nhãn bao bì và thông tin sản phẩm.
Làm sạch virus [theo Q5A]/ Tham khảo: ACTD-Q…
Lọai trừ virus đích bằng cách lọai bỏ các tiểu phân virus hay bất hoạt sự lây nhiễm virus.
Các tiểu phân giống virus [theo Q5A]/ Tham khảo: ACTD-Q…
Những cấu trúc có thể quan sát được bằng kính hiển vi điện tử, có hình thái giống với các virus đã biết.
Lọai bỏ virus [theo Q5A]/ Tham khảo: ACTD-Q…
Tách bằng phương pháp vật lý các tiểu phân virus khỏi sản phẩm dự kiến.
Đối tượng tham gia bị lạm dụng (liên quan đến thử nghiệm lâm sàng) [theo E6]/ Tham khảo: ACTD-E…
Các đối tượng mà ý muốn tình nguyện tham gia vào nghiên cứu có thể bị tác động không đúng bởi các lợi ích khi tham gia vào nghiên cứu hay do sự đe dọa nếu từ chối tham gia vào nghiên cứu.
(Ví dụ: các thành viên của những tổ chức phân theo thứ bậc như sinh viên của các khoa y, khoa dược, khoa nha, điều dưỡng, nhân viên phòng thí nghiệm hay bệnh viện tuyến dưới, nhân viên của các công ty dược, thành viên lực lượng vũ trang và tù nhân. Những đối tượng khác bao gồm bệnh nhân bị bệnh không thể chữa lành, đối tượng trong các viện dưỡng lão, người thất nghiệp, nghèo khổ, bệnh nhân trong tình trạng cấp cứu, dân tộc thiểu số, vô gia cư, du cư, di trú, dân nhập cư hay không có khả năng để cam kết).
Dược chất đã được hiểu rõ [theo WHO]
Là những hoạt chất mà:
- Đã lưu hành trên thị trường tối thiểu là 5 năm ở những nước có chính sách theo dõi thuốc tích cực sau khi được cấp phép tiếp thị
- Đã được sử dụng rộng rãi trên một số lượng lớn bệnh nhân, qua đó có thể nắm rõ hiệu quả và tính an toàn của thuốc, và
- Có cùng hàm lượng và đường dùng thuốc, có cùng chỉ định với các hoạt chất đã lưu hành ở các nước
(Xem thêm Sự phối hợp các dược chất đã được hiểu rõ ở liều cố định, và các Dược phẩm đã được hiểu rõ, vì định nghĩa này chỉ đề cập đến hoạt chất chứ không phải sản phẩm, không đề cập đến sự nhạy cảm có thể có của các tá dược hay các yếu tố khác có liên quan đến sự tương đương về mặt trị liệu).
Các phối hợp dược chất đã được hiểu rõ [theo WHO]
Là sự phối hợp các dược chất mà:
- Đã lưu hành trên thị trường tối thiểu là 5 năm ở những nước có chính sách theo dõi thuốc tích cực sau khi được cấp phép tiếp thị
- Đã được sử dụng rộng rãi trên một số lượng lớn bệnh nhân, qua đó có thể nắm rõ hiệu quả và tính an tòan của thuốc, và
- Có cùng hàm lượng và đường dùng thuốc, có cùng chỉ định với các hoạt chất đã lưu hành ở các nước
(Xem thêm Dược chất đã được hiểu rõ và Dược phẩm đã được hiểu rõ, vì định nghĩa này chỉ đề cập đến hoạt chất chứ không phải sản phẩm, không đề cập đến sự nhạy cảm có thể có của các tá dược hay các yếu tố khác có liên quan đến sự tương đương về mặt trị liệu).
Dược phẩm đã được hiểu rõ [theo WHO]
Là những dược phẩm chứa các dược chất đã được hiểu rõ, và:
- Đã lưu hành trên thị trường tối thiểu là 5 năm ở những nước có chính sách theo dõi thuốc tích cực sau khi được cấp phép tiếp thị
- Đã được sử dụng rộng rãi trên một số lượng lớn bệnh nhân, qua đó có thể nắm rõ hiệu quả và tính an tòan của thuốc, và
- Có cùng hàm lượng và đường dùng thuốc, có cùng chỉ định với các dược phẩm đã lưu hành ở các nước
Các phối hợp dược chất đã được hiểu rõ ở liều cố định [theo WHO]
Là sự phối hợp ở liều cố định các dược chất mà:
- Đã lưu hành trên thị trường tối thiểu là 5 năm ở những nước có chính sách theo dõi thuốc tích cực sau khi được cấp phép tiếp thị
- Đã được sử dụng rộng rãi trên một số lượng lớn bệnh nhân, qua đó có thể nắm rõ hiệu quả và tính an tòan của thuốc, và
- Có cùng hàm lượng và đường dùng thuốc, có cùng chỉ định với các hoạt chất đã lưu hành ở các nước
(Xem thêm Dược chất đã được hiểu rõ và Dược phẩm đã được hiểu rõ, vì định nghĩa này chỉ đề cập đến hoạt chất chứ không phải sản phẩm, không đề cập đến sự nhạy cảm có thể có của các tá dược hay các yếu tố khác có liên quan đến sự tương đương về mặt trị liệu).
Ngân hàng tế bào dùng cho thử nghiệm [theo Q5A]/ Tham khảo: ACTD-Q...
Ngân hàng này được tạo ra từ các phân đoạn nhỏ của dịch treo tế bào đồng nhất thu được từ sự nuôi cấy tế bào từ ngân hàng tế bào chủ trong điều kiện nuôi cấy nhất định.
PHỤ LỤC II
CÁC THAY ĐỔI LỚN, THAY ĐỔI NHỎ ÁP DỤNG ĐỐI VỚI THUỐC, NGUYÊN LIỆU LÀM THUỐC ĐÃ ĐƯỢC CẤP SỐ ĐĂNG KÝ LƯU HÀNH
(Ban hành kèm theo Thông tư số /2018 /TT-BYT ngày tháng năm 2018 )
A. THUỐC HÓA DƯỢC
Áp dụng Hướng dẫn của ASEAN về thay đổi, bổ sung đối với thuốc hóa dược đã được cấp số đăng ký lưu hành, gồm các nội dung sau:
MỤC LỤC
1. GIỚI THIỆU
2. PHẠM VI HƯỚNG DẪN
3. KHÁC
4. TỪ VIẾT TẮT
5. THAY ĐỔI LỚN (MaV)
Nội dung thay đổi 1 (MaV- 1) Thay đổi và/hoặc bổ sung chỉ định/liều dùng/đối tượng bệnh nhân/bổ sung thông tin lâm sàng để mở rộng phạm vi sử dụng thuốc
Nội dung thay đổi 2 (MaV-2) Thay đổi/bổ sung nội dung của hướng dẫn sử dụng và/hoặc mẫu nhãn
Nội dung thay đổi 3 (MaV-3) Thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất dược chất (khi không có Giấy chứng nhận tuân thủ Dược điển châu Âu (CEP))
Nội dung thay đổi 4 (MaV-4) Thay đổi địa điểm sản xuất:
Nội dung thay đổi 5 (MaV-5) Thay đổi địa điểm cơ sở đóng gói sơ cấp
Nội dung thay đổi 6 (MaV-6) Thay đổi tiêu chuẩn chất lượng dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] và/ hoặc thuốc thành phẩm
Nội dung thay đổi 7 (MaV-7)
Bổ sung, thay đổi cỡ lô đối với thuốc thành phẩm vô khuẩn
Nội dung thay đổi 8 (MaV-8)
Bổ sung, thay đổi cỡ lô đối với thuốc thành phẩm không vô khuẩn
Nội dung thay đổi 9 (MaV-9)
Thay đổi lớn trong quy trình sản xuất thuốc thành phẩm
Nội dung thay đổi 10 (MaV-10)
Thay đổi loại hoặc lượng tá dược
Nội dung thay đổi 11 (MaV-11)
Thay đổi khối lượng màng bao viên nén hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với thuốc giải phóng biến đổi dùng đường uống
Nội dung thay đổi 12 (MaV-12)
Thay đổi liên quan đến bao bì đóng gói sơ cấp của thuốc thành phẩm vô khuẩn áp dụng đối với các trường hợp sau:
Nội dung thay đổi 13 (MaV-13)
Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và, hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín đối với thuốc vô khuẩn dạng rắn và dạng dung dịch.
Nội dung thay đổi 14 (MaV-14)
Bổ sung hoặc thay thế dung môi, dung dịch hòa tan/ phân tán/ pha loãng thuốc
Nội dung thay đổi 15 (MaV-15) Tăng hạn dùng thuốc thành phẩm
Nội dung thay đổi 16 (MaV-16)
Thay đổi điều kiện bảo quản thuốc thành phẩm (ít khắc nghiệt hơn điều kiện bảo quản đã phê duyệt):
6. THAY ĐỔI NHỎ CẦN PHÊ DUYỆT TRƯỚC KHI THỰC HIỆN
Nội dung thay đổi 1 (MiV- PA1) Thay đổi tên thuốc thành phẩm
Nội dung thay đổi 2 (MiV- PA2)
Thay đổi/bổ sung nội dung của hướng dẫn sử dụng và/hoặc mẫu nhãn
Nội dung thay đổi 3 (MiV- PA3)
Thay đổi cơ sở/địa điểm cơ sở chịu trách nhiệm xuất xưởng lô
Nội dung thay đổi 4 (MiV- PA4)
Thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất của dược chất [khi có giấy chứng nhận tuân thủ dược điển châu Âu (CEP]
Nội dung thay đổi 5 (MiV- PA5)
Thay đổi cỡ lô sản xuất dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)]
Nội dung thay đổi 6 (MiV-PA6)
Thay đổi trong kiểm soát trong quá trình sản xuất đối với dược chất (bao gồm theo hướng chặt chẽ hơn, bổ sung chỉ tiêu kiểm soát mới và khi không có giấy chứng nhận tuân thủ dược điển châu Âu CEP)
Nội dung thay đổi 7 (MiV- PA7)
Thay đổi trong quy trình sản xuất dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)]
Nội dung thay đổi 8 (MiV- PA8)
Thay đổi tiêu chuẩn chất lượng dược chất áp dụng đối với các trường hợp sau:
Nội dung thay đổi 9 (MiV-PA9)
Thay đổi quy trình phân tích trong tiêu chuẩn chất lượng dược chất của các dược chất chưa có trong dược điển
Nội dung thay đổi 10 (MiV- PA10)
Thay đổi hạn dùng hoặc thời hạn phải kiểm tra lại chất lượng dược chất
Nội dung thay đổi 11 (MiV-PA11)
Thay đổi điều kiện bảo quản dược chất
Nội dung thay đổi 12 (MiV-PA12)
Sửa đổi giấy chứng nhận tuân thủ dược điển châu Âu (CEP) của dược chất
Nội dung thay đổi 13 (MiV-PA13)
Bổ sung/Thay đổi cỡ lô đối với thuốc thành phẩm không vô khuẩn
Nội dung thay đổi 14 (MiV-PA14)
Giảm hoặc bỏ lượng đóng dư (overages)
Nội dung thay đổi 15 (MiV–PA15)
Thay đổi loại và/hoặc lượng tá dược
Nội dung thay đổi 16 (MiV–PA16)
Thay đổi khối lượng màng bao viên hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với dạng bào chế rắn giải phóng ngay dùng đường uống
Nội dung thay đổi 17 (MiV- PA17)
Thay đổi chất tạo màu/tạo mùi của thuốc [thêm vào, bỏ bớt hoặc thay thế (các) chất tạo màu/tạo mùi]
Nội dung thay đổi 18 (MiV- PA18)
Bỏ bớt dung môi/dung dịch hòa tan, phân tán, pha loãng thuốc
Nội dung thay đổi 19 (MiV-PA19)
Thay đổi trong kiểm soát trong quy trình sản xuất đối với thuốc thành phẩm (bao gồm theo hướng chặt chẽ hơn và bổ sung phép thử mới)
Nội dung thay đổi 20 (MiV- PA20)
Thay đổi nhỏ trong quy trình sản xuất thuốc thành phẩm không vô khuẩn
Nội dung thay đổi 21 (MiV-PA21)
Thay đổi tiêu chuẩn chất lượng của tá dược áp dụng đối với các trường hợp sau:
Nội dung thay đổi 22 (MiV-PA22)
Thay đổi quy trình phân tích của tá dược, bao gồm thay thế quy trình phân tích đã được duyệt bằng quy trình phân tích mới.
Nội dung thay đổi 23 (MiV-PA23)
Thay đổi nguồn gốc vỏ nang cứng, áp dụng đối với các trường hợp sau:
a) Thay đổi nguồn gốc nguyên liệu sản xuất vỏ nang cứng;
b) Thay đổi nhà sản xuất vỏ nang cứng.
Nội dung thay đổi 24 (MiV-PA24)
Thay đổi trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm, áp dụng đối với các trường hợp sau:
Nội dung thay đổi 25 (MiV-PA25)
Thay đổi vết khắc, hình ảnh hoặc các ký hiệu khác trên viên nén hoặc hình ảnh/ ký hiệu in trên viên nén, viên nang bao gồm thay đổi/ bổ sung mực in dùng để in lên thuốc thành phẩm
Nội dung thay đổi 26 (MiV- PA26)
Thay đổi kích thước và/hoặc hình dạng viên nén, viên nang, viên đạn hoặc viên đặt âm đạo mà không thay đổi loại và lượng của các thành phần trong viên và khối lượng trung bình viên đối với:
Nội dung thay đổi 27 (MiV-PA27)
Thay đổi quy trình phân tích trong tiêu chuẩn chất lượng thuốc thành phẩm (bao gồm thay thế hoặc bổ sung quy trình phân tích)
Nội dung thay đổi 28 (MiV-PA28)
Thay đổi liên quan đến bao bì đóng gói sơ cấp của thuốc thành phẩm không vô khuẩn
Nội dung thay đổi 29 (MiV-PA29)
Thay đổi cơ sở đóng gói thứ cấp
Nội dung thay đổi 30 (MiV-PA30)
Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và/hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín đối với thuốc thành phẩm không vô khuẩn.
Nội dung thay đổi 31 (MiV- PA31)
Thay đổi quy cách đóng gói thứ cấp của thuốc thành phẩm
Nội dung thay đổi 32 (MiV-PA32)
Các thay đổi của bao bì (sơ cấp) không tiếp xúc trực tiếp với thuốc thành phẩm như màu của nắp bật, vạch màu trên ống, thay đổi chụp bảo vệ kim tiêm (sử dụng nhựa khác)
Nội dung thay đổi 33 (MiV-PA33)
Bổ sung hoặc thay thế dụng cụ đo lường của các dạng thuốc lỏng dùng đường uống và các dạng bào chế khác
Nội dung thay đổi 34 (MiV-PA34)
Giảm hạn dùng của thuốc thành phẩm
Nội dung thay đổi 35 (MiV-PA35)
Thay đổi điều kiện bảo quản của thuốc thành phẩm (khắc nghiệt hơn so với điều kiện bảo quản đã được chấp thuận)
Nội dung thay đổi 36 (MiV-PA36)
Thay đổi cơ sở đăng ký
Nội dung thay đổi 37 (MiV-PA37)
Công bố thuốc có chứng minh tương đương sinh học
Nội dung thay đổi 38 (MiV-PA38)
Cập nhật phân loại biệt dược gốc
7. THAY ĐỔI/BỔ SUNG CẦN PHÊ DUYỆT NHƯNG KHÔNG DẪN ĐẾN THAY ĐỔI NHÃN VÀ HƯỚNG DẪN SỬ DỤNG THUỐC
Nội dung thay đổi 39
Bổ sung, cập nhật thông tin để cung cấp thông tin thuốc, quảng cáo thuốc
8. THAY ĐổI NHO CHI YÊU CẦU THÔNG BÁO (NOTIFICATION)
Nội dung thay đổi 1 (MiV-N1)
Thay đổi tên, địa chỉ của cơ sở đăng ký, cập nhật thông tin liên quan đến cơ sở đăng ký
Nội dung thay đổi 2 (MiV-N2) Thay đổi chủ sở hữu sản phẩm
Nội dung thay đổi 3 (MiV-N3)
Thay đổi tên và/hoặc địa chỉ (như mã bưu điện, tên phố) của cơ sở sản xuất dược chất
Nội dung thay đổi 4 (MiV-N4) Bỏ bớt cơ sở sản xuất của dược chất
Nội dung thay đổi 5 (MiV-N5) (Asean:MiV-N8)
Đăng ký lại giấy chứng nhận tuân thủ dược điển châu Âu (CEP)
Nội dung thay đổi 6 (MiV-N6)
Thay đổi tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm và/hoặc dược chất và/hoặc tá dược khi cập nhật dược điển.
Nội dung thay đổi 7 (MiV-N7) Bỏ bớt quy cách đóng gói thuốc thành phẩm
Nội dung thay đổi 8 (MiV-N8)
Thay đổi tên cơ sở sản xuất do thay đổi chủ sở hữu của cơ sở sản xuất
Nội dung thay đổi 9 (MiV-N9)
Thay đổi tên và/hoặc địa chỉ (như mã bưu điện, tên phố) của cơ sở sản xuất thuốc thành phẩm
Nội dung thay đổi 10 (MiV-N10)
Thay đổi tên và/hoặc địa chỉ (như mã bưu điện, tên phố) của cơ sở xuất xưởng lô
Nội dung thay đổi 11 (MiV-N11)
Thay đổi mẫu nhãn, hướng dẫn sử dụng
9. THUẬT NGỮ
10. THAM KHẢO
B. SINH PHẨM VÀ NGUYÊN LIỆU HÓA DƯỢC
C. VẮC XIN, HUYẾT THANH CHỨA KHÁNG THỂ
D. THUỐC DƯỢC LIỆU, NGUYÊN LIỆU LÀM THUỐC DƯỢC LIỆU
HƯỚNG DẪN CỦA ASEAN VỀ THAY ĐỔI, BỔ SUNG ĐỐI VỚI THUỐC HÓA DƯỢC ĐÃ ĐƯỢC CẤP SỐ ĐĂNG KÝ
1. GIỚI THIỆU
Trong suốt quá trình lưu hành của thuốc thành phẩm, cơ sở đăng ký và cơ sở sản xuất chịu trách nhiệm đưa sản phẩm ra thị trường cần xem xét đến tiến bộ khoa học kỹ thuật, cũng như tiến hành bất kỳ thay đổi cần thiết nào để các thuốc được sản xuất và kiểm soát dựa trên các phương pháp khoa học chung được chấp thuận. Các thay đổi này phải được sự chấp thuận của cơ quan quản lý.
Hướng dẫn này nhằm mục đích đưa ra các yêu cầu đối với hồ sơ đề nghị thay đổi/bổ sung của thuốc hóa dược đã cấp số đăng ký. Hồ sơ thay đổi/bổ sung được phân loại thành thay đổi lớn, thay đổi nhỏ (phải được chấp thuận trước khi thực hiện) và thay đổi nhỏ (chỉ yêu cầu thông báo khi thực hiện). Hướng dẫn này sẽ được cập nhật định kỳ theo yêu cầu.
2. PHẠM VI HƯỚNG DẪN
Hướng dẫn về thay đổi, bổ sung của ASEAN áp dụng cho thuốc hóa dược đã được cấp số đăng ký lưu hành.
3. KHÁC
3.1. Trong hướng dẫn này:
3.1.1. Dược điển tham chiếu gồm: Dược điển Việt Nam, Dược điển Anh (BP), Dược điển Mỹ (USP), Dược điển Châu Âu (EP), Dược điển Quốc tế (IP), Dược điển Nhật (JP).
3.1.2. Tiêu chuẩn chất lượng (dược chất/ tá dược/ thành phẩm) được hiểu là bao gồm các chỉ tiêu chất lượng và các mức chất lượng tương ứng kèm theo các quy trình phân tích để thử mỗi chỉ tiêu chất lượng này.
3.1.3. Bán thành phẩm ở dạng bulk product được hiểu là thuốc được sản xuất tới công đoạn cuối cùng trước khi đóng gói vào bao bì sơ cấp.
3.1.4. Các số liệu nghiên cứu độ ổn định, thẩm định quy trình phân tích, thẩm định quy trình sản xuất, nghiên cứu sinh khả dụng/ tương đương sinh học của thuốc...được thực hiện theo các hướng dẫn kỹ thuật ASEAN hiện hành ban hành kèm theo Thông tư quy định việc đăng ký thuốc.
3.1.5. So sánh độ hòa tan được tiến hành và đánh giá kết quả theo quy định tại các hướng dẫn SUPAC-IR và SUPAC-MR của USFDA.
3.2. Bất kỳ thay đổi nào không liệt kê trong hướng dẫn này cần được xem xét và quy định bởi Cục Quản lý Dược. Có thể tham khảo các tài liệu sau:
i. Hướng dẫn phân loại của EMA về thay đổi nhỏ loại IA, thay đổi nhỏ loại IB và thay đổi lớn loại II,
ii. SUPAC-IR: Hướng dẫn của USFDA về nâng quy mô sản xuất và các thay đổi khác đối với các thuốc dạng rắn giải phóng ngay dùng đường uống đã được cấp số đăng ký lưu hành: Các tài liệu cần nộp về hóa học, sản xuất và kiểm soát; thử độ hòa tan in vitro, và nghiên cứu tương đương sinh học.
iii. SUPAC-MR: Hướng dẫn của USFDA về nâng quy mô sản xuất và các thay đổi khác đối với các thuốc dạng rắn giải phóng biến đổi dùng đường uống đã được cấp số đăng ký lưu hành: Các tài liệu cần nộp về hóa học, sản xuất và kiểm soát; thử độ hòa tan in vitro, và nghiên cứu tương đương sinh học in vivo.
iv. Hướng dẫn của WHO về thay đổi đối với hồ sơ thuốc thành phẩm đã được tiền đánh giá chất lượng.
3.3. Các hồ sơ cần nộp quy định tại Hướng dẫn này đều được nộp kèm đơn đăng ký theo mẫu quy định tại Thông tư đăng ký thuốc. Các nội dung của cam kết (nếu có) có thể được đưa vào nội dung của đơn đăng ký và có xác nhận theo đúng yêu cầu.
3.4. Cơ sở đăng ký có các thuốc có cùng một nội dung thay đổi có thể được nộp chung trong cùng 01 hồ sơ kèm theo đơn đăng ký chung cho tất cả sản phẩm theo mẫu quy định tại Thông tư đăng ký thuốc, đối với phần chi tiết sản phẩm: yêu cầu viết riêng cho từng sản phẩm.
3.5. Đối với các thay đổi sau: thay đổi tên, địa chỉ cơ sở đăng ký, thay đổi cơ sở đăng ký, thay đổi tên cơ sở sản xuất, cách ghi địa chỉ cơ sở sản xuất mà địa điểm sản xuất ko thay đổi có thể gộp chung trong cùng một hồ sơ đăng ký thay đổi bao gồm đầy đủ các tài liệu liên quan của từng nội dung theo quy định.
3.6. Ngoài các hồ sơ cần nộp theo Hướng dẫn này, Cục Quản lý Dược có quyền yêu cầu bổ sung thêm thông tin khi thấy cần thiết.
4. TỪ VIẾT TẮT
MaV = Thay đổi lớn
MiV-N = Thay đổi nhỏ (chỉ yêu cầu thông báo)
MiV-PA = Thay đổi nhỏ (cần phê duyệt trước khi thực hiện)
SUPAC = Hướng dẫn của Cơ quan quản lý thuốc và thực phẩm Hoa Kỳ về nâng quy mô sản xuất và các thay đổi sau khi cấp số đăng ký
TSE = Bệnh xốp não có thể lây truyền sang người
BSE = Bệnh xốp não ở bò có thể lây truyền sang người
5. THAY ĐỔI LỚN (MaV)
|
Thay đổi lớn (MaV) |
|
Nội dung thay đổi 1 (MaV- 1) |
Thay đổi và/hoặc bổ sung chỉ định/liều dùng/đối tượng bệnh nhân/bổ sung thông tin lâm sàng để mở rộng phạm vi sử dụng thuốc |
|
Điều kiện cần đáp ứng (C) |
1. Là kết quả của thay đổi Tóm tắt đặc tính sản phẩm (SmPC) hoặc tài liệu tương đương (như USPI) dẫn đến thay đổi hướng dẫn sử dụng và/hoặc mẫu nhãn |
|
Hồ sơ cần nộp (D) |
1. Hướng dẫn sử dụng và/hoặc mẫu nhãn đã được duyệt. 2. Hướng dẫn sử dụng và/hoặc mẫu nhãn mới (02 bản). Bảng so sánh các nội dung thay đổi của hướng dẫn sử dụng (02 bản) và/hoặc mẫu nhãn. 3. Giải trình lý do thay đổi. 4. Tài liệu tham khảo và/hoặc Báo cáo của các chuyên gia lâm sàng (nếu có). 5. Hướng dẫn sử dụng/Tóm tắt đặc tính sản phẩm /Thông tin cho bệnh nhân được chấp thuận bởi cơ quan cấp phép lưu hành thuốc của nước sở tại hoặc nước tham chiếu cho phép thay đổi hoặc công văn của cơ quan cấp phép lưu hành thuốc của nước sở tại hoặc nước tham chiếu cho phép thay đổi/bổ sung (đối với thuốc sản xuất tại nước ngoài có thay đổi dựa trên thay đổi tại nước sở tại hoặc nước tham chiếu) 6. Tài liệu lâm sàng theo Hồ sơ kỹ thuật chung ASEAN (ACTD) phần IV. |
|
Nội dung thay đổi 2 (MaV-2) |
Thay đổi/bổ sung nội dung của hướng dẫn sử dụng và/hoặc mẫu nhãn |
|
Điều kiện cần đáp ứng (C) |
1. Thay đổi này không phải là thay đổi nhỏ và không nằm trong phạm vi MaV-1. 2. Là kết quả của thay đổi Tóm tắt đặc tính sản phẩm (SmPC) hoặc tài liệu tương đương (như USPI). |
|
Hồ sơ cần nộp (D) |
1. Hướng dẫn sử dụng và/hoặc mẫu nhãn đã được duyệt. 2. Hướng dẫn sử dụng và/hoặc mẫu nhãn mới. Bảng so sánh các nội dung thay đổi của hướng dẫn sử dụng và/hoặc mẫu nhãn. 3. Giải trình lý do thay đổi. 4. Tài liệu tham khảo và/hoặc các tài liệu lâm sàng chứng minh. 5. Hướng dẫn sử dụng (PI)/ Tóm tắt đặc tính sản phẩm (SmPC)/Thông tin cho bệnh nhân (PIL) được chấp thuận bởi cơ quan cấp phép lưu hành thuốc của nước sở tại hoặc nước tham chiếu cho phép thay đổi/bổ sung (đối với thuốc sản xuất tại nước ngoài có thay đổi dựa trên thay đổi tại nước sở tại hoặc nước tham chiếu) |
|
Nội dung thay đổi 3 (MaV-3) |
Thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất dược chất (khi không có Giấy chứng nhận tuân thủ Dược điển châu Âu (CEP)) |
|
Điều kiện cần đáp ứng (C) |
1. Tiêu chuẩn chất lượng dược chất không thay đổi. 2. Trường hợp thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất của dược chất khi có Giấy chứng nhận tuân thủ Dược điển châu Âu (CEP) áp dụng MiV-PA4. 3. Trường hợp có thay đổi tiêu chuẩn chất lượng dược chất, áp dụng thêm quy định tại MAV-6 hoặc MiV- PA8 hoặc MiV-N6. |
|
Hồ sơ cần nộp (D) |
1. Có thể nộp một trong những tài liệu sau: a) Toàn bộ các phần từ S1- S7 theo ACTD; b) Hồ sơ tổng thể hoạt chất bao gồm cả phần công khai và phần không công khai (phần không công khai được cơ sở sản xuất dược chất cung cấp trực tiếp cho Cục Quản lý Dược kèm theo thư cho phép tiếp cận tài liệu này); c) Giấy chứng nhận hoặc tài liệu kiểm tra tương đương được cấp từ một trong các nước tham chiếu quy định tại Thông tư đăng ký thuốc. 2. Bảng so sánh sự khác nhau trong quy trình sản xuất dược chất tại địa điểm sản xuất mới và địa điểm sản xuất đã được duyệt (nếu có thay đổi). 3. Số liệu phân tích lô (dưới dạng bảng so sánh) trên ít nhất 02 lô pilot giữa dược chất sản xuất tại địa điểm mới và dược chất sản xuất tại địa điểm đã được duyệt. 4. Thư cam kết của cơ sở sản xuất thuốc thành phẩm về việc tiến hành nghiên cứu độ ổn định của thuốc thành phẩm được sản xuất từ dược chất sản xuất tại địa điểm mới ở điều kiện dài hạn và điều kiện lão hóa cấp tốc và sẽ báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành, đồng thời đề xuất biện pháp xử lý phù hợp. 5. Giấy tờ pháp lý của cơ sở sản xuất dược chất chứng minh cơ sở đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) Trường hợp nguyên liệu đã có Giấy đăng ký lưu hành tại Việt Nam, không yêu cầu tài liệu quy định tại khoản 1, 5 mục này. |
|
Nội dung thay đổi 4 (MaV-4) |
Thay đổi địa điểm sản xuất: a) bán thành phẩm ở dạng bulk product; b) thuốc thành phẩm. |
|
Điều kiện cần đáp ứng (C) |
1. Không áp dụng cho những thay đổi liên quan đến cơ sở chịu trách nhiệm xuất xưởng lô hoặc địa điểm chỉ xuất xưởng lô. 2. Cơ sở sản xuất thuốc thành phẩm/ bán thành phẩm ở dạng bulk product không thay đổi. 3. Địa điểm sản xuất mới phải đạt GMP theo quy định phù hợp với dạng bào chế của thuốc. Đối với thuốc sản xuất tại Việt Nam, địa điểm sản xuất mới phải đã được cấp Giấy chứng nhận đủ điều kiện kinh doanh thuốc. 4. Thay đổi cơ sở/ địa điểm cơ sở chịu trách nhiệm xuất xưởng lô áp dụng MiV-PA3. 5. Nếu có thay đổi về quy trình sản xuất, áp dụng thêm MaV-9. |
|
Hồ sơ cần nộp (D) |
1. Đối với thuốc sản xuất tại nước ngoài: CPP hoặc giấy chứng nhận GMP chứng minh địa điểm sản xuất mới phù hợp để được sản xuất dạng bào chế của thuốc. 2. Bảng so sánh số liệu phân tích lô giữa ít nhất 02 lô sản xuất (hoặc 01 lô sản xuất và 02 lô pilot) của thuốc thành phẩm/ bán thành phẩm ở dạng bulk product sản xuất tại địa điểm mới và 03 lô sản xuất cuối cùng của thuốc thành phẩm/ bán thành phẩm ở dạng bulk product sản xuất tại địa điểm đã được duyệt. Số liệu phân tích lô cho 02 lô sản xuất tiếp theo của thuốc thành phẩm/ bán thành phẩm ở dạng bulk product tại địa điểm mới phải sẵn có để cung cấp khi có yêu cầu. Trong quá trình phân tích lô, nếu có kết quả nào không đạt tiêu chuẩn chất lượng phải có báo cáo đồng thời đề xuất biện pháp xử lý phù hợp. 3. Số liệu độ ổn định của thuốc thành phẩm sau khi thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 4. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất thuốc thành phẩm/ bán thành phẩm ở dạng bulk product tại địa điểm mới. 5. Đối với các thuốc có dạng bào chế rắn dùng đường uống: Số liệu so sánh độ hòa tan giữa các thuốc thành phẩm/ bán thành phẩm ở dạng bulk product trước và sau khi có thay đổi. 6. Công thức bào chế đã được duyệt của thuốc thành phẩm. 7. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 8. Tiêu chuẩn chất lượng đã được duyệt của dược chất. 9. Nghiên cứu ảnh hưởng của thời gian lưu trữ bán thành phẩm ở dạng bulk product trong quá trình bảo quản và vận chuyển từ nơi sản xuất bán thành phẩm đến nơi đóng gói sơ cấp. 10. Đối với cơ sở sản xuất bán thành phẩm ở dạng bulk product theo hợp đồng, thư chỉ định và cho phép sản xuất thuốc bán thành phẩm ở dạng bulk product tại địa điểm mới trong đó nêu rõ các công đoạn nào của quy trình sản xuất được thực hiện tại địa điểm này. |
|
Nội dung thay đổi 5 (MaV-5) |
Thay đổi địa điểm cơ sở đóng gói sơ cấp |
|
Điều kiện cần đáp ứng (C) |
1. Không có sự thay đổi nào ngoại trừ thay đổi địa điểm của cơ sở đóng gói sơ cấp. 2. Địa điểm đóng gói mới phải đạt GMP theo quy định phù hợp với dạng bào chế của thuốc. Đối với thuốc đóng gói tại Việt Nam, địa điểm đóng gói mới phải đã được cấp Giấy chứng nhận đủ điều kiện kinh doanh thuốc. |
|
Hồ sơ cần nộp (D) |
1. Đối với thuốc đóng gói tại nước ngoài: CPP hoặc giấy chứng nhận GMP chứng minh địa điểm đóng gói mới phù hợp để được đóng gói sơ cấp dạng bào chế của thuốc. 2. Đối với cơ sở đóng gói theo hợp đồng, thư chỉ định và cho phép được đóng gói sơ cấp thuốc thành phẩm tại địa điểm mới có nêu cụ thể về công đoạn đóng gói được tiến hành tại địa điểm mới của cơ sở đóng gói. 3. Đối với thuốc vô khuẩn, kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình đóng gói sơ cấp thuốc thành phẩm tại địa điểm mới. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Nghiên cứu ảnh hưởng của thời gian lưu trữ đối với bán thành phẩm ở dạng bulk product trong quá trình bảo quản và vận chuyển từ nơi sản xuất bán thành phẩm đến nơi đóng gói sơ cấp mới. |
|
Nội dung thay đổi 6 (MaV-6) |
Thay đổi tiêu chuẩn chất lượng dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] và/ hoặc thuốc thành phẩm thuộc các trường hợp sau: a) Mở rộng giới hạn của chỉ tiêu chất lượng; b) Bỏ bớt chỉ tiêu chất lượng và mức chất lượng. |
|
Điều kiện cần đáp ứng (C) |
1. Quy trình phân tích không thay đổi hoặc thay đổi rất ít (không cần thiết phải tiến hành thẩm định lại quy trình phân tích). 2. Không áp dụng đối với các trường hợp tiêu chuẩn chất lượng đã được duyệt của dược chất/ thuốc thành phẩm là tiêu chuẩn theo dược điển tham chiếu. 3. Tham khảo MiV-PA12 nếu thay đổi này dẫn đến việc xem xét lại CEP. 4. Thay đổi này không phải là kết quả của những tác động ngoài dự kiến xảy ra trong quá trình sản xuất hoặc liên quan đến độ ổn định. 5. CPP có nội dung đã được thay đổi. |
|
Hồ sơ cần nộp (D) |
(a) Mở rộng giới hạn của chỉ tiêu chất lượng 1. Giải trình về lý do đề nghị thay đổi kèm theo các dữ liệu khoa học để chứng minh. 2. Bảng so sánh sự thay đổi giữa tiêu chuẩn chất lượng mới của dược chất/thuốc thành phẩm và tiêu chuẩn chất lượng đã được duyệt của dược chất/ thuốc thành phẩm có in đậm các nội dung thay đổi. 3. Tiêu chuẩn chất lượng mới của dược chất/ thuốc thành phẩm. 4. Số liệu phân tích lô của dược chất /thuốc thành phẩm đối với tất cả các chỉ tiêu chất lượng trong tiêu chuẩn chất lượng mới tiến hành trên 02 lô (lô pilot hoặc lô sản xuất). 5. Số liệu độ ổn định của dược chất/ thuốc thành phẩm theo tiêu chuẩn chất lượng mới (đối với dược chất) và tiêu chuẩn chất lượng lưu hành mới (đối với thuốc thành phẩm) và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng đồng thời đề xuất biện pháp xử lý phù hợp. (b) Bỏ bớt chỉ tiêu chất lượng và mức chất lượng 1. Cung cấp các tài liệu từ D1- D4. 2. Phiếu kiểm nghiệm dược chất/thuốc thành phẩm theo tiêu chuẩn chất lượng mới (đối với dược chất) và tiêu chuẩn chất lượng lưu hành mới (đối với thuốc thành phẩm). |
|
Nội dung thay đổi 7 (MaV-7) |
Bổ sung, thay đổi cỡ lô đối với thuốc thành phẩm vô khuẩn |
|
Điều kiện cần đáp ứng (C) |
1. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 3. Công thức bào chế thuốc thành phẩm không thay đổi. |
|
Hồ sơ cần nộp (D) |
1. Kế hoạch thẩm định quy trình sản xuất và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô sản xuất theo cỡ lô mới. 2. Bảng so sánh công thức lô sản xuất giữa cỡ lô đã được duyệt và cỡ lô mới. 3. Số liệu phân tích lô (dạng bảng so sánh) của ít nhất 02 lô sản xuất của thuốc thành phẩm trước và sau khi có thay đổi. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành, đồng thời đề xuất biện pháp xử lý phù hợp. |
|
Nội dung thay đổi 8 (MaV-8) |
Bổ sung, thay đổi cỡ lô đối với thuốc thành phẩm không vô khuẩn |
|
Điều kiện cần đáp ứng (C) |
1. Áp dụng đối với thay đổi cỡ lô lớn hơn 10 lần so với cỡ lô đã được duyệt. Đối với thay đổi cỡ lô dưới hoặc bằng 10 lần cỡ lô đã được duyệt, xem MiV-PA13. 2. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. |
|
Hồ sơ cần nộp (D) |
1. Kế hoạch thẩm định quy trình và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô sản xuất theo cỡ lô mới . 2. Bảng so sánh công thức lô sản xuất giữa cỡ lô đã được duyệt và cỡ lô mới. 3. Số liệu phân tích lô (dạng bảng so sánh) giữa ít nhất 01 lô sản xuất thuốc thành phẩm sau khi có thay đổi và 01 lô sản xuất thuốc thành phẩm trước khi có thay đổi kèm theo thư cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký sẽ nộp dữ liệu phân tích lô cho một lô sản xuất đầy đủ kế tiếp. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 6. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh độ hòa tan trên ít nhất 01 lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi. |
|
Nội dung thay đổi 9 (MaV-9) |
Thay đổi lớn trong quy trình sản xuất thuốc thành phẩm |
|
Điều kiện cần đáp ứng (C) |
1. Địa điểm sản xuất không thay đổi. Nếu thay đổi địa điểm sản xuất, áp dụng thêm MaV-4. 2. Thay đổi này không ảnh hưởng tiêu cực đến chất lượng, an toàn và hiệu quả của thuốc. 3. Thay đổi nhỏ trong quy trình sản xuất thuốc thành phẩm không vô khuẩn áp dụng theo MiV-PA20. |
|
Hồ sơ cần nộp (D) |
1. Mô tả quy trình sản xuất mới và giải trình về mặt kỹ thuật các lý do đề nghị thay đổi. 2. Kế hoạch thẩm định quy trình và/hoặc báo cáo thẩm định quy trình sản xuất mới. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Hoặc, có thể thay bằng tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành dự kiến của thuốc thành phẩm để chứng minh thuốc được sản xuất theo quy trình sản xuất mới ít nhất là tương đương hoặc tốt hơn về mặt chất lượng, an toàn và hiệu quả so với thuốc sản xuất theo quy trình sản xuất cũ. 4. Bảng so sánh số liệu phân tích lô trên tối thiểu 01 lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi. 5. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất cứ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 6. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh độ hòa tan giữa các thuốc thành phẩm trước và sau khi có thay đổi. 7. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). |
|
Nội dung thay đổi 10 (MaV-10) |
Thay đổi loại hoặc lượng tá dược a) Đối với dạng bào chế giải phóng ngay dùng đường uống (ở mức 2 và 3, Phần III (Thành phần và công thức)- hướng dẫn SUPAC- IR); b) Đối với dạng bào chế giải phóng biến đổi dùng đường uống; c) Đối với các dạng bào chế đặc biệt khác như các chế phẩm vô khuẩn. |
|
Điều kiện cần đáp ứng (C) |
1. Các chỉ tiêu chất lượng và mức chất lượng tương ứng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc). 2. Số liệu so sánh độ hòa tan giữa các thuốc thành phẩm trước và sau khi thay đổi đạt quy định theo SUPAC-IR hoặc SUPAC-MR. 3. Thay thế một tá dược bằng một tá dược khác tương đương về các đặc tính sử dụng (có cùng chức năng). 4. Đối với những thay đổi khác về loại hoặc lượng tá dược của dạng bào chế giải phóng ngay dùng đường uống và các dạng bào chế không đặc biệt khác, xem MiV-PA15. Trường hợp thay đổi loại tá dược, nhà sản xuất tá dược phải có thêm: 5. CPP của thuốc có thể hiện nội dung đề nghị thay đổi. 6. Giấy tờ pháp lý của cơ sở sản xuất tá dược chứng minh cơ sở đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) (thực hiện theo lộ trình quy định). Không yêu cầu tài liệu nay đối với tá dược đã có Giấy đăng ký lưu hành tại Việt Nam. |
|
Hồ sơ cần nộp (D) |
1. Giải trình về đề nghị thay đổi bằng tài liệu phát triển dược học phù hợp. 2. Số liệu độ ổn định của thuốc thành phẩm sau khi thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành, đồng thời đề xuất biện pháp xử lý phù hợp. 3. Đối với các thuốc có dạng bào chế rắn dùng đương uống: Số liệu so sánh độ hòa tan trên ít nhất 01 lô pilot/ lô sản xuất đại diện giữa các thuốc thành phẩm trước và sau khi có thay đổi. 4. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học) 5. Bảng so sánh công thức bào chế đã được duyệt lần đầu tiên và công thức bào chế đề nghị thay đổi có in đậm các nội dung thay đổi, trong đó lượng từng thành phần được thể hiện dưới dạng phần trăm khối lượng trên tổng khối lượng các thành phần trong công thức 6. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi mô tả hình thức thuốc). 7. Số liệu phân tích lô (dạng bảng so sánh) trên ít nhất 02 lô sản xuất (hoặc 01 lô sản xuất và 02 lô pilot) giữa các thuốc thành phẩm trước và sau khi thay đổi. 8. Tiêu chuẩn chất lượng của tá dược mới (nếu khi thay đổi có sử dụng tá dược chưa có trong công thức bào chế đã được duyệt). 9. Đối với tá dược có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú ý có thẩm quyền cấp. 10. Công thức lô sản xuất mới. 11. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất thuốc thành phẩm sau khi thay đổi trên ít nhất 03 lô sản xuất. 12. Các phần từ P3.1- P3.4 theo ACTD đã cập nhật các thay đổi phù hợp với công thức bào chế mới. 13. Giải trình phù hợp kèm theo các số liệu thực nghiệm chứng minh (nếu cần) về việc các thay đổi trong công thức bào chế này không làm ảnh hưởng đến kết quả của các quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Nếu thay đổi trong công thức bào chế này dẫn đến việc phải thay đổi quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm, yêu cầu tham chiếu MiV-PA27 để bổ sung các tài liệu theo quy định. |
|
Nội dung thay đổi 11 (MaV-11) |
Thay đổi khối lượng màng bao viên nén hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với thuốc giải phóng biến đổi dùng đường uống |
|
Điều kiện cần đáp ứng (C) |
1. Số liệu so sánh độ hòa tan giữa thuốc thành phẩm trước và sau khi thay đổi đạt yêu cầu theo SUPAC-MR. 2. Các chỉ tiêu chất lượng và mức chất lượng tương ứng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc) 3. Thay đổi khối lượng màng bao viên nén hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với dạng thuốc rắn giải phóng ngay dùng đường uống, xem MiV-PA16. |
|
Hồ sơ cần nộp (D) |
1. Số liệu so sánh độ hòa tan trên ít nhất 01 lô pilot/lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi. 2. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi mô tả hình thức thuốc). 4. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc thay đổi này không làm ảnh hưởng đến quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Công thức bào chế cho 01 đơn vị liều và công thức lô sản xuất đã được duyệt và sau khi có thay đổi. 6. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 7. Phần P3.1- P3.4 theo ACTD đã cập nhật các thay đổi phù hợp với công thức bào chế mới. |
|
Nội dung thay đổi 12 (MaV- 12) |
Thay đổi liên quan đến bao bì đóng gói sơ cấp của thuốc thành phẩm vô khuẩn áp dụng đối với các trường hợp sau: a) Thay đổi thành phần và lượng các thành phần trong chất liệu bao bì đóng gói đã được duyệt và/ hoặc b) Thay đổi loại bao bì đóng gói và/ hoặc c) Bổ sung thêm chất liệu bao bì đóng gói. |
|
Điều kiện cần đáp ứng (C) |
1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Thay đổi liên quan đến bao bì đóng gói sơ cấp đối với thuốc thành phẩm không vô khuẩn, xem MiV-PA28. |
|
Hồ sơ cần nộp (D) |
1. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất và quy trình tiệt khuẩn khi sử dụng bao bì đóng gói sơ cấp mới trong sản xuất thuốc thành phẩm. 2. Số liệu độ ổn định của thuốc thành phẩm trong bao bì đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 3. Bằng chứng chứng minh không có sự tương tác giữa thuốc và bao bì đóng gói sơ cấp mới. 4. Bảng so sánh về tiêu chuẩn chất lượng giữa bao bì đóng gói sơ cấp mới và bao bì đóng gói sơ cấp đã được duyệt. 5. Các số liệu khoa học chứng minh sự phù hợp của việc sử dụng bao bì đóng gói sơ cấp mới (số liệu so sánh về tính thấm giữa bao bì đóng gói sơ cấp mới và bao bì đóng gói sơ cấp đã được duyệt đối với các thông số hàm ẩm, O2, CO2) 6. Các phần P3 và P7 theo ACTD đã cập nhật các nội dung thay đổi tương ứng. 7. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). |
|
Nội dung thay đổi 13 (MaV-13) |
Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và, hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín đối với thuốc vô khuẩn dạng rắn và dạng dung dịch. |
|
Điều kiện cần đáp ứng (C) |
1. Không ảnh hưởng đến tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm, ngoại trừ thay đổi trong mô tả quy cách đóng gói của thuốc thành phẩm. 2. Quy cách đóng gói mới phù hợp với liều dùng và thời gian sử dụng đã được duyệt trong hướng dẫn sử dụng. 3. Chất liệu bao bì đóng gói không thay đổi. 4. Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và/hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín của thuốc không vô khuẩn, xem MiV- PA30. |
|
Hồ sơ cần nộp (D) |
1. Chứng minh sự phù hợp giữa quy cách đóng gói mới với liều dùng và thời gian sử dụng đã được duyệt trong hướng dẫn sử dụng. 2. Số liệu thẩm định quy trình sản xuất, hệ thống đóng kín và tiệt khuẩn. 3. Số liệu độ ổn định của thuốc thành phẩm trong các quy cách đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 4. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi/ bổ sung). |
|
Nội dung thay đổi 14 (MaV- 14) |
Bổ sung hoặc thay thế dung môi, dung dịch hòa tan/ phân tán/ pha loãng thuốc |
|
Điều kiện cần đáp ứng (C) |
1. Thay đổi này không gây ra bất kỳ thay đổi nào về dạng bào chế, liều dùng, chỉ định, cách dùng của thuốc. 2. Bỏ bớt dung môi, dung dịch hòa tan/ phân tán/ pha loãng thuốc, xem MiV-PA18. 3. Nếu có thay đổi hạn dùng và/hoặc điều kiện bảo quản thuốc sau khi mở nắp và/hoặc sau khi pha với các dung môi, dung dịch mới, xem MaV-15/MiV-PA34 và/hoặc MaV-16/MiV-PA35 để cung cấp các tài liệu phù hợp với từng loại thay đổi tương ứng. |
|
Hồ sơ cần nộp (D) |
1. Nộp toàn bộ phần P theo ACTD đối với dung dịch hòa tan/ phân tán/ pha loãng mới và nộp phần S theo ACTD đối với các thành phần có tác dụng dược lý trong dung dịch hòa tan/ phân tán/ pha loãng mới nếu đây là các thành phần chưa có trong công thức bào chế của các dung dịch hòa tan/phân tán/ pha loãng đã được duyệt. Nộp tiêu chuẩn chất lượng của dung môi mới (nếu có sử dụng dung môi mới). 2. Tài liệu pháp lý chứng minh cơ sở sản xuất/ địa điểm sản xuất dung môi/ dung dịch hòa tan/ phân tán/ pha loãng mới đạt GMP phù hợp với các dạng bào chế này (nếu các dung môi, dung dịch hòa tan/ phân tán/ pha loãng mới được sản xuất tại cơ sở sản xuất/ địa điểm sản xuất mới). 3. Mẫu nhãn mới và mẫu nhãn đã được duyệt (nếu có thay đổi/ bổ sung). |
|
Nội dung thay đổi 15 (MaV- 15) |
Tăng hạn dùng thuốc thành phẩm: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/ hoặc b) Sau khi mở nắp lần đầu và/ hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. |
|
Điều kiện cần đáp ứng (C) |
1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c)- Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Đối với trường hợp giảm hạn dùng thuốc thành phẩm, xem MiV- PA34. |
|
Hồ sơ cần nộp (D) |
1. Số liệu độ ổn định dài hạn của thuốc thành phẩm phù hợp với hạn dùng mới tương ứng cho ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp trong các trường hợp: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng 2. Thuyết minh về mặt kỹ thuật các lý do cho đề nghị thay đổi này. |
|
Nội dung thay đổi 16 (MaV- 16) |
Thay đổi điều kiện bảo quản thuốc thành phẩm (ít khắc nghiệt hơn điều kiện bảo quản đã phê duyệt): a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. |
|
Điều kiện cần đáp ứng (C) |
1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c)- Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Đối với thay đổi điều kiện bảo quản thuốc thành phẩm (khắc nghiệt hơn so với điều kiện bảo quản đã được chấp thuận), xem MiV-PA35. |
|
Hồ sơ cần nộp (D) |
1. Số liệu độ ổn định dài hạn của thuốc thành phẩm ở điều kiện bảo quản mới phù hợp với hạn dùng đã được duyệt tương ứng của ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp trong các trường hợp: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng 2. Thuyết minh về mặt kỹ thuật các lý do cho đề nghị thay đổi này. |
6. THAY ĐỔI NHỎ CẦN PHÊ DUYỆT TRƯỚC KHI THỰC HIỆN
|
Thay đổi nhỏ (MiV-PA) cần phê duyệt trước khi thực hiện |
|
Nội dung thay đổi 1 (MiV- PA1) |
Thay đổi tên thuốc thành phẩm |
|
Điều kiện cần đáp ứng (C) |
1. Chỉ thay đổi tên thuốc thành phẩm, các phần khác không thay đổi (công thức, tiêu chuẩn chất lượng, nguồn nguyên liệu, quy trình sản xuất…). 2. Tên mới tuân thủ theo quy định về đặt tên thuốc tại Thông tư quy định việc đăng ký thuốc và Thông tư quy định về ghi nhãn thuốc. |
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận dược phẩm (CPP) có tên thuốc mới đối với thuốc sản xuất tại nước ngoài. 2. Giấy đăng ký nhãn hiệu hàng hóa đối với tên thuốc mới. Trường hợp đổi tên thuốc với lý do vi phạm sở hữu trí tuệ đối với nhãn hiệu hàng hoá đã được bảo hộ của một cá nhân, tổ chức khác, cơ sở đăng ký phải cung cấp văn bản của cơ quan có thẩm quyền về sở hữu trí tuệ xác nhận việc vi phạm như lý do đã nêu. |
|
Nội dung thay đổi 2 (MiV- PA2) |
Thay đổi/bổ sung nội dung của hướng dẫn sử dụng và/hoặc mẫu nhãn áp dụng đối với các trường hợp sau: a) Thay đổi thiết kế mà không thay đổi ý nghĩa; b) Bổ sung/bỏ bớt/thay thế hình ảnh, sơ đồ, hoặc nội dung; c) Bổ sung/thay đổi theo hướng chặt chẽ hơn cảnh báo, thận trọng, chống chỉ định và/hoặc tác dụng không mong muốn so với hướng dẫn sử dụng đã được duyệt; d) Thu hẹp nhóm bệnh nhân sử dụng; e) Bỏ bớt chỉ định. |
||
|
Điều kiện cần đáp ứng (C) |
1. Thay đổi không phải là thay đổi lớn và không chứa đựng nội dung quảng cáo, không thuộc quy định tại khoản 2 Điều 35 Thông tư số 01/2018/TT-BYT ngày 18/01/2018 của Bộ Y tế hướng dẫn ghi nhãn thuốc. 2. Đối với thay đổi lớn về mẫu nhãn, xem MaV-1 và MaV-2. |
||
|
Hồ sơ cần nộp (D) |
1. Hướng dẫn sử dụng và/hoặc mẫu nhãn đã được duyệt. 2. Hướng dẫn sử dụng và/hoặc mẫu nhãn mới. Bảng so sánh các nội dung thay đổi của hướng dẫn sử dụng và/hoặc mẫu nhãn.. 3. Giải trình lý do thay đổi. 4. Tài liệu tham khảo cho nội dung thay đổi (nếu có). |
||
|
Nội dung thay đổi 3 (MiV- PA3) |
Thay đổi cơ sở/địa điểm cơ sở chịu trách nhiệm xuất xưởng lô |
||
|
Điều kiện cần đáp ứng (C) |
1. Chỉ áp dụng cho cơ sở xuất xưởng lô. 2. Đã hoàn thành việc chuyển giao quy trình phân tích từ phòng thí nghiệm/ cơ sở xuất xưởng lô cũ sang phòng thí nghiệm/ cơ sở xuất xưởng lô mới. 3. Cơ sở sản xuất thuốc thành phẩm không thay đổi. |
||
|
Hồ sơ cần nộp (D) |
1. Tài liệu pháp lý chứng minh cơ sở/địa điểm xuất xưởng lô mới được cơ quan có thẩm quyền cho phép xuất xưởng lô như: giấy chứng nhận GMP hoặc GLP, hoặc CPP có chứng nhận GMP hoặc GLP cho cơ sở/địa điểm xuất xưởng lô mới. 2. Số liệu thẩm tra quy trình phân tích đã được duyệt tại địa điểm xuất xưởng lô mới hoặc hồ sơ chuyển giao quy trình phân tích đã được duyệt từ địa điểm xuất xưởng lô đã được duyệt sang địa điểm xuất xưởng lô mới. |
||
|
Nội dung thay đổi 4 (MiV- PA4) |
Thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất của dược chất [khi có giấy chứng nhận tuân thủ dược điển châu Âu (CEP] |
||
|
Điều kiện cần đáp ứng (C) |
1. Tiêu chuẩn chất lượng dược chất không thay đổi. 2. Thay đổi và/hoặc bổ sung cơ sở sản xuất/ địa điểm sản xuất dược chất khi không có CEP, xem MaV-3. |
||
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận tuân thủ dược điển châu Âu (CEP) đối với dược chất bản mới nhất còn hiệu lực kèm theo tất cả các phụ lục được ban hành bởi Hội đồng châu Âu về chất lượng thuốc (EDQM). 2. Số liệu phân tích lô (dạng bảng so sánh) của ít nhất 02 lô pilot giữa dược chất sản xuất tại địa điểm mới và dược chất sản xuất tại địa điểm đã được duyệt. 3. Nếu thời hạn phải kiểm tra lại chất lượng dược chất không nêu trong CEP, nộp số liệu nghiên cứu độ ổn định của dược chất sản xuất tại địa điểm mới ở điều kiện dài hạn và điều kiện lão hóa cấp tốc trên 02 lô pilot phù hợp với thời hạn phải kiểm tra lại chất lượng dược chất theo đề xuất của cơ sở sản xuất dược chất. 4. Cam kết của cơ sở sản xuất thuốc thành phẩm về việc tiến hành nghiên cứu độ ổn định của thuốc thành phẩm được sản xuất từ dược chất sản xuất tại địa điểm mới ở điều kiện dài hạn và điều kiện lão hóa cấp tốc, và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. |
||
|
Nội dung thay đổi 5 (MiV- PA5) |
Thay đổi cỡ lô sản xuất dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] |
||
|
Điều kiện cần đáp ứng (C) |
1. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 2. Tiêu chuẩn chất lượng dược chất không thay đổi. |
||
|
Hồ sơ cần nộp (D) |
1. Số liệu phân tích lô (dạng bảng so sánh) theo tiêu chuẩn chất lượng dược chất đã được duyệt của ít nhất 01 lô sản xuất hoặc 01 lô pilot giữa dược chất sản xuất theo cỡ lô mới và dược chất sản xuất theo cỡ lô đã được duyệt. Số liệu phân tích lô cho 02 lô sản xuất đầy đủ kế tiếp phải sẵn có để nộp khi có yêu cầu. Trong quá trình phân tích lô, nếu có kết quả nào không đạt tiêu chuẩn chất lượng phải có báo cáo đồng thời đề xuất biện pháp xử lý phù hợp. 2. Cam kết của cơ sở sản xuất dược chất về việc tiêu chuẩn chất lượng của dược chất không thay đổi và tính ổn định của quy trình sản xuất dược chất không bị ảnh hưởng. 3. Cập nhật mục S theo ACTD. |
||
|
Nội dung thay đổi 6 (MiV-PA6) |
Thay đổi trong kiểm soát trong quá trình sản xuất đối với dược chất (bao gồm theo hướng chặt chẽ hơn, bổ sung chỉ tiêu kiểm soát mới và khi không có giấy chứng nhận tuân thủ dược điển châu Âu CEP) |
||
|
Điều kiện cần đáp ứng (C) |
1. Giới hạn chỉ tiêu kiểm soát trong quy trình sản xuất chặt chẽ hơn hoặc bổ sung chỉ tiêu kiểm soát mới. 2. Thay đổi này không phải là hệ quả của bất kỳ cam kết nào về việc xem xét lại giới hạn trong tiêu chuẩn chất lượng từ lần thẩm định trước. 3. Thay đổi này không phải là kết quả của biến cố ngoại ý phát sinh trong quá trình sản xuất như: tạp chất mới chưa xác định, thay đổi giới hạn tổng tạp chất... 4. Quy trình phân tích của chỉ tiêu kiểm soát mới không liên quan đến một kỹ thuật mới không theo chuẩn hoặc một kỹ thuật cơ bản được sử dụng theo cách mới. |
||
|
Hồ sơ cần nộp (D) |
1. Mô tả quy trình phân tích và tóm tắt dữ liệu thẩm định quy trình phân tích của tất cả các quy trình phân tích mới. 2. Bảng so sánh sự thay đổi về kiểm soát trong quy trình sản xuất có in đậm những nội dung thay đổi liên quan. 3. So sánh số liệu phân tích lô trên 02 lô sản xuất của dược chất đối với tất cả các chỉ tiêu chất lượng trong tiêu chuẩn chất lượng. |
||
|
Nội dung thay đổi 7 (MiV- PA7) |
Thay đổi trong quy trình sản xuất dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] |
||
|
Điều kiện cần đáp ứng (C) |
1. Không dẫn đến các thay đổi bất lợi về loại và/hoặc lượng tạp chất đòi hỏi phải có những nghiên cứu thêm về tính an toàn. 2. Độ ổn định của dược chất không thay đổi. 3. Phương pháp tổng hợp dược chất không thay đổi (ví dụ: chất trung gian không thay đổi). 4. Quy trình sản xuất dược chất không sử dụng bất cứ nguyên liệu nào có nguồn gốc từ người/động vật đòi hỏi phải có đánh giá độ an toàn về nhiễm vi rút. 5. Chỉ tiêu lý hóa và các chỉ tiêu liên quan khác của dược chất không thay đổi. |
||
|
Hồ sơ cần nộp (D) |
1. Hồ sơ tổng thể của dược chất (DMF) hoặc phần hồ sơ cập nhật của dược chất theo ACTD hoặc tài liệu kiểm tra tương đương được cung cấp từ một trong các nước tham chiếu quy định tại Thông tư đăng ký thuốc. 2. Bảng so sánh quy trình sản xuất đã được duyệt và quy trình sản xuất mới có in đậm những nội dung thay đổi. 3. Phiếu kiểm nghiệm của 02 lô dược chất được sản xuất theo quy trình sản xuất mới. 4. Bảng so sánh số liệu phân tích lô của ít nhất 02 lô (lô pilot hoặc lô sản xuất) giữa thuốc thành phẩm sử dụng dược chất sản xuất theo quy trình sản xuất mới và thuốc thành phẩm sử dụng dược chất sản xuất theo quy trình sản xuất đã được duyệt. 5. Thư của cơ sở sản xuất dược chất tuyên bố không xuất hiện tạp chất mới bằng hoặc vượt ngưỡng chấp nhận cũng như không có sự tăng giới hạn tạp chất đòi hỏi phải có nghiên cứu thêm về tính an toàn. 6. Thư của cơ sở sản xuất dược chất tuyên bố không có sự thay đổi trong tiêu chuẩn chất lượng đã được duyệt của dược chất hoặc nếu có bất cứ thay đổi nào trong tiêu chuẩn chất lượng dược chất (ví dụ theo hướng chặt chẽ hơn) thì cần nộp bảng so sánh giữa tiêu chuẩn chất lượng mới và tiêu chuẩn chất lượng đã được duyệt của dược chất. 7. Cam kết của cơ sở sản xuất thuốc thành phẩm về việc đã bắt đầu tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm được sản xuất từ dược chất sản xuất theo quy trình sản xuất mới, và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 8. Đối với dược chất vô khuẩn, nộp báo cáo thẩm định quy trình sản xuất mới của dược chất. 9. Nếu có thay đổi trong tiêu chuẩn chất lượng dược chất, yêu cầu cung cấp các mục S4-S5 theo ACTD đối với dược chất sản xuất theo quy trình sản xuất mới. |
||
|
Nội dung thay đổi 8 (MiV- PA8) |
Thay đổi tiêu chuẩn chất lượng dược chất áp dụng đối với các trường hợp sau: a) Mức giới hạn các chỉ tiêu chất lượng chặt chẽ hơn; b) Bổ sung thêm chỉ tiêu chất lượng và mức chất lượng mới. |
||
|
Điều kiện cần đáp ứng (C) |
1. Chỉ áp dụng cho những trường hợp tiêu chuẩn chất lượng đã được duyệt của dược chất không theo một trong các dược điển tham chiếu và những dược chất generic không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP). 2. Đối với (b) - chỉ áp dụng cho các phương pháp không có trong dược điển. 3. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. 4. Nếu mở rộng giới hạn các chỉ tiêu chất lượng và/ hoặc bỏ bớt chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng dược chất, xem MaV-6. 5. Thay đổi này không phải là kết quả của biến cố ngoại ý xuất hiện trong quá trình sản xuất hoặc do quan ngại về độ ổn định. 6. Quy trình phân tích của các chỉ tiêu chất lượng đã có trong tiêu chuẩn chất lượng đã được duyệt của dược chất không thay đổi hoặc thay đổi không đáng kể (không cần thiết phải tiến hành thẩm định lại quy trình phân tích). |
||
|
Hồ sơ cần nộp (D) |
(a) Mức giới hạn các chỉ tiêu chất lượng chặt chẽ hơn: 1. Bảng so sánh các chỉ tiêu chất lượng và các mức chất lượng tương ứng giữa tiêu chuẩn chất lượng trước và sau khi thay đổi của dược chất, có in đậm các nội dung thay đổi. 2. Bảng so sánh số liệu phân tích lô theo tiêu chuẩn chất lượng mới của dược chất trên 02 lô (lô pilot hoặc lô sản xuất). 3. Giải trình các lý do kỹ thuật dẫn đến đề nghị thay đổi này. (b) Bổ sung thêm chỉ tiêu chất lượng và mức chất lượng mới: Ngoài những tài liệu nêu trên cần bổ sung thêm: Mô tả quy trình phân tích mới và báo cáo số liệu thẩm định quy trình phân tích mới. |
||
|
Nội dung thay đổi 9 (MiV-PA9) |
Thay đổi quy trình phân tích trong tiêu chuẩn chất lượng dược chất của các dược chất chưa có trong dược điển |
||
|
Điều kiện cần đáp ứng (C) |
1. Kết quả thẩm định quy trình phân tích mới cho thấy quy trình phân tích mới ít nhất là tương đương với quy trình phân tích đã được duyệt. 2. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. |
||
|
Hồ sơ cần nộp (D) |
1. Mô tả quy trình phân tích mới, bao cao số liệu thẩm định quy trình phân tích mới và so sánh kết quả phân tích thu được từ quy trình phân tích mới và quy trình phân tích đã được duyệt. 2. Tiêu chuẩn chất lượng mới của dược chất đã cập nhật các thay đổi trong quy trình phân tích. |
||
|
Nội dung thay đổi 10 (MiV- PA10) |
Thay đổi hạn dùng hoặc thời hạn phải kiểm tra lại chất lượng dược chất |
||
|
Điều kiện cần đáp ứng (C) |
1. Các nghiên cứu độ ổn định phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng đã được duyệt của dược chất 2. Không thay đổi điều kiện bảo quản. 3. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. |
||
|
Hồ sơ cần nộp (D) |
1. Số liệu độ ổn định của dược chất trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) phù hợp với hạn dùng mới hoặc thời hạn phải kiểm tra lại chất lượng mới của dược chất. 2. Tiêu chuẩn chất lượng đã được duyệt của dược chất. |
||
|
Nội dung thay đổi 11 (MiV- PA11) |
Thay đổi điều kiện bảo quản dược chất |
||
|
Điều kiện cần đáp ứng (C) |
1. Các nghiên cứu độ ổn định phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng đã được duyệt của dược chất. 2. Không thay đổi hạn dùng/thời hạn phải kiểm tra lại chất lượng của dược chất. 4. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. |
||
|
Hồ sơ cần nộp (D) |
1. Tiêu chuẩn chất lượng đã được duyệt của dược chất. 2. Số liệu độ ổn định của dược chất ở điều kiện bảo quản mới trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) phù hợp với hạn dùng hoặc thời hạn phải kiểm tra lại chất lượng đã được duyệt của dược chất. |
||
|
Nội dung thay đổi 12 (MiV- PA12) |
Sửa đổi giấy chứng nhận tuân thủ dược điển châu Âu (CEP) của dược chất |
||
|
Điều kiện cần đáp ứng (C) |
Không có. |
||
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận tuân thủ dược điển châu Âu (CEP) cho dược chất bản mới nhất còn hiệu lực kèm theo tất cả các phụ lục do EDQM (Hội đồng Châu Âu về chất lượng thuốc) cấp. 2. Kết quả phân tích lô của cơ sở sản xuất dược chất thể hiện chất lượng dược chất đáp ứng chuyên luận tương ứng của dược điển châu Âu đồng thời đáp ứng các chỉ tiêu chất lượng/ mức chất lượng bổ sung thêm được nêu trong CEP (nếu có). 3. Số liệu chứng minh cho bất kỳ thông số nào không được nêu trong CEP do cơ sở sản xuất dược chất tự công bố như dữ liệu về độ ổn định (S7) (trong trường hợp thời hạn phải kiểm tra lại chất lượng dược chất không nêu trong CEP) và các đặc tính lý hóa như kích thước phân tử, hiện tượng đa hình....của dược chất (nếu có). 4. Nếu thay đổi này là do thay đổi tiêu chuẩn chất lượng dược chất, bổ sung cam kết của cơ sở sản xuất thuốc thành phẩm về việc đã tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm sản xuất từ dược chất đạt chất lượng theo tiêu chuẩn chất lượng dược chất mới; và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm đồng thời đề xuất biện pháp xử lý phù hợp. 5. Trong trường hợp tiêu chuẩn chất lượng đã được duyệt của dược chất không phải là tiêu chuẩn theo EP (do nhà sản xuất thành phẩm không áp dụng tiêu chuẩn chất lượng theo EP để kiểm tra chất lượng dược chất trước khi đưa vào sản xuất thành phẩm), nếu có thay đổi trong tiêu chuẩn chất lượng dược chất, yêu cầu nộp lại các phần S4-S5 theo ACTD. |
||
|
Nội dung thay đổi 13 (MiV- PA13) |
Bổ sung/Thay đổi cỡ lô đối với thuốc thành phẩm không vô khuẩn |
||
|
Điều kiện cần đáp ứng (C) |
1. Áp dụng cho thay đổi cỡ lô nhiều nhất đến 10 lần so với cỡ lô hiện tại. 2. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 4. Đối với thay đổi cỡ lô của thuốc thành phẩm vô khuẩn, xem MaV-7. Đối với thay đổi cỡ lô lớn hơn 10 lần so với cỡ lô đã đăng ký của thuốc thành phẩm không vô khuẩn, xem MaV-8. |
||
|
Hồ sơ cần nộp (D) |
1. Kế hoạch thẩm định quy trình và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô sản xuất theo cỡ lô mới. 2. Bảng so sánh công thức lô sản xuất giữa cỡ lô hiện tại và cỡ lô mới. 3. Số liệu phân tích lô (dạng bảng so sánh) trên ít nhất 01 lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi kèm theo bản cam kết của cơ sở sản xuất thuốc thành phẩm sẽ nộp dữ liệu phân tích lô của lô sản xuất tiếp theo. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đã được duyệt đồng thời đề xuất biện pháp xử lý phù hợp. 5. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 6. Các phần từ P1- P3 theo ACTD đã cập nhật phù hợp với cỡ lô sản xuất mới. |
||
|
Nội dung thay đổi 14 (MiV- PA14) |
Giảm hoặc bỏ lượng đóng dư (overages) |
||
|
Điều kiện cần đáp ứng (C) |
1. Chỉ áp dụng cho thay đổi lượng đóng dư của dược chất. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. |
||
|
Hồ sơ cần nộp (D) |
1. Giải trình về các lý do thay đổi. 2. Bảng so sánh công thức lô sản xuất đã được duyệt và công thức lô sản xuất mới. 3. Phiếu kiểm nghiệm của 02 lô thuốc thành phẩm sản xuất theo công thức lô mới. 4. Số liệu độ ổn định của thuốc thành phẩm sản xuất theo công thức lô mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. |
||
|
Nội dung thay đổi 15 (MiV- PA15) |
Thay đổi loại và/hoặc lượng tá dược a) Đối với dạng bào chế giải phóng ngay dùng đường uống (theo mức 1, Phần III (Thành phần và công thức)- hướng dẫn SUPAC-IR); b) Đối với dạng bào chế không đặc biệt khác như dung dịch uống, chế phẩm dùng ngoài. |
||
|
Điều kiện cần đáp ứng (C) |
1. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc). 2. Đối với các thuốc bào chế ở dạng rắn dùng đường uống: Số liệu so sánh độ hòa tan giữa các thuốc thành phẩm trước và sau khi có thay đổi đạt yêu cầu theo SUPAC-IR. 3. Thay thế một tá dược bằng một tá dược khác tương đương về đặc tính sử dụng (có cùng chức năng). 4. Đối với thay đổi về loại hoặc lượng tá dược của dạng bào chế giải phóng ngay dùng đường uống (theo level 2 và 3, phần III (Thành phần và công thức)- hướng dẫn SUPAC-IR), dạng bào chế giải phóng biến đổi dùng đường uống và các dạng bào chế đặc biệt khác, xem MaV-10. |
||
|
Hồ sơ cần nộp (D) |
1. Giải thích về đề nghị thay đổi bằng tài liệu phát triển dược học phù hợp. 2. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 3. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh độ hòa tan trên ít nhất 01 lô (lô pilot hoặc lô sản xuất) đại diện giữa thuốc thành phẩm trước và sau khi có thay đổi. 4. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 5. Bảng so sánh công thức bào chế đã được duyệt và công thức bào chế mới (có in đậm các thay đổi), trong đó lượng mỗi thành phần được thể hiện dưới dạng phần trăm khối lượng trên tổng khối lượng các thành phần trong công thức. 6. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi trong mô tả hình thức thuốc). 7. Số liệu phân tích lô (dạng bảng so sánh) trên ít nhất 02 lô sản xuất (hoặc 01 lô sản xuất và 02 lô pilot) giữa các thuốc thành phẩm trước và sau khi có thay đổi. 8. Tiêu chuẩn chất lượng của tá dược mới (nếu có sử dụng thêm tá dược chưa có trong công thức bào chế đã được duyệt). 9. Đối với tá dược có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú ý có thẩm quyền cấp. 10. Công thức lô sản xuất mới. 11. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô thuốc thành phẩm sản xuất theo công thức bào chế mới. 12. Các phần P3.1 đến P3.4 theo ACTD đã cập nhật phù hợp với công thức bào chế mới. Giải trình phù hợp kèm theo số liệu thực nghiệm chứng minh (nếu cần) về việc các thay đổi này không làm ảnh hưởng đến kết quả của các quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Nếu thay đổi này dẫn đến việc phải thay đổi quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm, yêu cầu tham chiếu MiV-PA27 để bổ sung đầy đủ các tài liệu theo quy định. |
||
|
Nội dung thay đổi 16 (MiV– PA16) |
Thay đổi khối lượng màng bao viên hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với dạng bào chế rắn giải phóng ngay dùng đường uống |
||
|
Điều kiện cần đáp ứng (C) |
1. Đối với các thuốc bào chế ở dạng rắn dùng đường uống: Số liệu so sánh độ hòa tan giữa các thuốc thành phẩm trước và sau khi có thay đổi đạt yêu cầu theo SUPAC-IR. 2. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc). 3. Thay đổi khối lượng màng bao viên hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với dạng bào chế rắn giải phóng biến đổi dùng đường uống, xem MaV-11. |
||
|
Hồ sơ cần nộp (D) |
1. Số liệu so sánh độ hòa tan trên ít nhất 01 lô (lô pilot hoặc lô sản xuất) giữa các thuốc thành phẩm trước và sau khi có thay đổi. 2. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi mô tả hình thức thuốc). 4. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc thay đổi này không ảnh hưởng đến quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Bảng so sánh công thức bào chế cho 01 đơn vị liều và công thức lô sản xuất của các thuốc thành phẩm trước và sau khi có thay đổi. 6. Nếu thay đổi khối lượng màng bao viên: Số liệu nghiên cứu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất cứ kết quả nào không đạt so với tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý. Nếu thay đổi khối lượng và/ hoặc kích cỡ vỏ nang: cam kết của cơ sở sản xuất thuốc thành phẩm về việc đã bắt đầu và sẽ hoàn thành việc nghiên cứu độ ổn định của thuốc thành phẩm sau khi có thay đổi. 7. Phần P3.1 đến P3.4 theo ACTD đã cập nhật các thay đổi tương ứng. |
||
|
Nội dung thay đổi 17 (MiV- PA17) |
Thay đổi chất tạo màu/tạo mùi của thuốc [thêm vào, bỏ bớt hoặc thay thế (các) chất tạo màu/tạo mùi] |
||
|
Điều kiện cần đáp ứng (C) |
1. Các tá dược thay đổi có cùng chức năng. Sự thay đổi này không làm thay đổi đặc tính hòa tan của thuốc thành phẩm đối với các dạng bào chế rắn dùng đường uống. 2. Chất tạo màu/tạo mùi mới không bị cấm dùng trong dược phẩm. 3. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi trừ việc cập nhật mô tả về màu sắc và/ hoặc mùi vị mới của thuốc. |
||
|
Hồ sơ cần nộp (D) |
1. Bảng so sánh các thông tin liên quan đến tính chất và khối lượng sử dụng giữa chất tạo màu/ tạo mùi mới và chất tạo màu/ tạo mùi đang sử dụng . 2. Công thức bào chế cho 01 đơn vị liều và công thức lô sản xuất thuốc thành phẩm sau khi có thay đổi. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi đã cập nhật các mô tả về màu sắc và/ hoặc mùi vị mới của thuốc. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Đối với tá dược có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú ý có thẩm quyền cấp. 6. Giải trình phù hợp kèm theo số liệu thực nghiệm chứng minh (nếu cần) về việc các thay đổi này không làm ảnh hưởng đến kết quả các quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Nếu thay đổi này dẫn đến việc phải thay đổi quy trình phân tích trong tiêu chuẩn chất lượng thành phẩm đã được duyệt, yêu cầu tham chiếu MiV-PA27 để bổ sung các tài liệu theo quy định. 7. Phần P3.1 đến P3.4 theo ACTD đã được cập nhật các thay đổi phù hợp. |
||
|
Nội dung thay đổi 18 (MiV- PA18) |
Bỏ bớt dung môi/dung dịch hòa tan, phân tán, pha loãng thuốc |
||
|
Điều kiện cần đáp ứng (C) |
1. Thay đổi này không dẫn đến bất kỳ thay đổi nào về dạng bào chế, phác đồ điều trị, chỉ định, cách dùng của thuốc. |
||
|
Hồ sơ cần nộp (D) |
1. Giải trình về việc bỏ bớt dung môi, dung dịch hòa tan/ phân tán/ pha loãng của thuốc và trình bày về các biện pháp thay thế khác để có dung môi, dung dịch hòa tan/ phân tán/ pha loãng. |
||
|
Nội dung thay đổi 19 (MiV- PA19) |
Thay đổi trong kiểm soát trong quy trình sản xuất đối với thuốc thành phẩm (bao gồm theo hướng chặt chẽ hơn và bổ sung phép thử mới) |
||
|
Điều kiện cần đáp ứng (C) |
1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Thay đổi này không phải là hệ quả của bất kỳ cam kết nào về việc xem xét lại giới hạn trong tiêu chuẩn chất lượng từ lần thẩm định trước. 3. Thay đổi này không phải là biến cố ngoại ý phát sinh trong quá trình sản xuất như: xuất hiện tạp chất mới chưa xác định, thay đổi giới hạn tổng tạp chất... 4. Bất kỳ quy trình phân tích mới nào đều không liên quan đến kỹ thuật mới không theo chuẩn hoặc một kỹ thuật cơ bản được sử dụng theo cách mới. |
||
|
Hồ sơ cần nộp (D) |
1. Mô tả quy trình phân tích và tóm tắt số liệu thẩm định quy trình phân tích đối với các quy trình phân tích mới (nếu có sử dụng quy trình phân tích mới). 2. Các chỉ tiêu kiểm soát trong quy trình sản xuất sau khi có thay đổi kèm theo các giải trình và số liệu thẩm định quy trình phù hợp. 3. So sánh số liệu phân tích lô trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) của thuốc thành phẩm sau khi có thay đổi. 4. Bảng so sánh kiểm soát trong quy trình sản xuất trước và sau khi có thay đổi |
||
|
Nội dung thay đổi 20 (MiV- PA20) |
Thay đổi nhỏ trong quy trình sản xuất thuốc thành phẩm không vô khuẩn |
||
|
Điều kiện cần đáp ứng (C) |
1. Địa điểm sản xuất không thay đổi. 2. Nguyên tắc sản xuất chung không thay đổi. 3. Thay đổi này không ảnh hưởng tiêu cực đến chất lượng, an toàn và hiệu quả của thuốc thành phẩm. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 5. Đối với các thuốc thành phẩm bào chế ở dạng rắn dùng đường uống: Số liệu so sánh độ hòa tan giữa các thuốc thành phẩm trước và sau khi thay đổi đạt yêu cầu theo quy định tại SUPAC-IR và SUPAC-MR. 6. Đối với thay đổi lớn trong quy trình sản xuất thuốc thành phẩm, áp dụng MaV-9. |
||
|
Hồ sơ cần nộp (D) |
1. Mô tả quy trình sản xuất mới và giải trình về mặt kỹ thuật cho sự thay đổi này. 2. Đối với thuốc dạng bán rắn và dạng hỗn dịch: Kế hoạch thẩm định và/ hoặc báo cáo thẩm định quy trình sản xuất. 3. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh độ hòa tan trên ít nhất 01 lô sản xuất đại diện giữa các thuốc thành phẩm trước và sau khi có thay đổi. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 6. Số liệu phân tích lô thành phẩm (dạng bảng so sánh) trên ít nhất 01 lô sản xuất giữa thuốc thành phẩm trước và sau khi có thay đổi; số liệu phân tích lô cho 02 lô sản xuất đầy đủ kế tiếp phải sẵn có để nộp khi có yêu cầu. 7. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc đã bắt đầu tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm sau khi thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 8. Bảng so sánh quy trình sản xuất mới và quy trình sản xuất đã được duyệt có in đậm những nội dung thay đổi. |
||
|
Nội dung thay đổi 21 (MiV- PA21) |
Thay đổi tiêu chuẩn chất lượng của tá dược áp dụng đối với các trường hợp sau: a) Giới hạn chỉ tiêu chất lượng chặt chẽ hơn; b) Bổ sung chỉ tiêu chất lượng và mức chất lượng mới. |
||
|
Điều kiện cần đáp ứng (C) |
1. Áp dụng cho các trường hợp tiêu chuẩn chất lượng đã được duyệt của tá dược không theo dược điển. Nếu tiêu chuẩn chất lượng đã được duyệt của tá dược theo dược điển, xem MiV-N9. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 3. Thay đổi này không phải là kết quả của biến cố ngoại ý xuất hiện trong quá trình sản xuất hoặc do quan ngại về độ ổn định. |
||
|
Hồ sơ cần nộp (D) |
1. Bảng so sánh tiêu chuẩn chất lượng tá dược trước và sau khi có thay đổi có in đậm những nội dung thay đổi. 2. Mô tả quy trình phân tích mới (trong trường hợp bổ sung chỉ tiêu chất lượng mới). |
||
|
Nội dung thay đổi 22 (MiV- PA22) |
Thay đổi quy trình phân tích của tá dược, bao gồm thay thế quy trình phân tích đã được duyệt bằng quy trình phân tích mới. |
||
|
Điều kiện cần đáp ứng (C) |
1. Đã có số liệu thẩm định quy trình phân tích mới đạt yêu cầu. 2. Kết quả thẩm định quy trình phân tích mới cho thấy quy trình phân tích mới ít nhất là tương đương với quy trình phân tích đã được duyệt. 3. Không có sự thay đổi về giới hạn tổng tạp chất. 4. Chỉ áp dụng đối với các chỉ tiêu chất lượng đã được duyệt trong tiêu chuẩn. 5. Không có sự xuất hiện của các tạp chất mới chưa xác định. 6. Áp dụng cho các trường hợp tiêu chuẩn chất lượng đã được duyệt của tá dược không theo dược điển. |
||
|
Hồ sơ cần nộp (D) |
1. Mô tả quy trình phân tích mới kèm theo bảng so sánh các thay đổi giữa quy trình phân tích mới và quy trình phân tích đã được duyệt. 2. Đối với các quy trình định lượng (bao gồm cả định lượng tạp chất): Số liệu so sánh kết quả thẩm định quy trình phân tích chứng minh quy trình phân tích mới và quy trình phân tích đã được duyệt là tương đương. |
||
|
Nội dung thay đổi 23 (MiV- PA23) |
Thay đổi nguồn gốc vỏ nang cứng, áp dụng đối với các trường hợp sau: a) Thay đổi nguồn gốc nguyên liệu sản xuất vỏ nang cứng; b) Thay đổi nhà sản xuất vỏ nang cứng. |
||
|
Điều kiện cần đáp ứng (C) |
7. Thay đổi từ vỏ nang dùng nguyên liệu có nguồn gốc động vật có nguy cơ gây viêm não dạng bọt (TSE) sang vỏ nang dùng nguyên liệu có nguồn gốc thực vật hoặc vỏ nang tổng hợp và ngược lại. 8. Không thay đổi công thức bào chế và quy trình sản xuất đã được duyệt của thuốc thành phẩm. 9. Không áp dụng khi thay đổi từ vỏ nang cứng sang vỏ nang mềm. 10. Tiêu chuẩn chất lượng đã được duyệt của các tá dược (trừ tiêu chuẩn chất lượng vỏ nang mới), tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. |
||
|
Hồ sơ cần nộp (D) |
1. Số liệu so sánh độ hòa tan trên 01 lô (lô pilot hoặc lô sản xuất) đại diện giữa các thuốc thành phẩm trước và sau khi có thay đổi. 2. Thành phần và tiêu chuẩn chất lượng của vỏ nang cứng mới. 3. Phiếu kiểm nghiệm vỏ nang cứng mới. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Đối với vỏ nang cứng sản xuất từ nguyên liệu có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú ý có thẩm quyền cấp. 6. Cam kết của cơ sở sản xuất thuốc về việc nguyên liệu sản xuất vỏ nang cứng là tinh khiết từ các nguồn tổng hợp hoặc động vật hoặc thực vật. 7. Số liệu chứng minh vỏ nang mới không làm ảnh hưởng đến kết quả thử độ hòa tan theo quy trình phân tích trong tiêu chuẩn chất lượng thuốc thành phẩm đã được duyệt (nếu trong tiêu chuẩn chất lượng thuốc thành phẩm có chỉ tiêu chất lượng này). 11. Giấy tờ pháp lý của cơ sở sản xuất vỏ nang chứng minh cơ sở đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) (thực hiện theo lộ trình quy định). Không yêu cầu tài liệu này đối với vỏ nang đã có Giấy đăng ký lưu hành tại Việt Nam. |
||
|
Nội dung thay đổi 24 (MiV- PA24) |
Thay đổi trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm, áp dụng đối với các trường hợp sau: a) Giới hạn chỉ tiêu chất lượng chặt chẽ hơn; b) Bổ sung chỉ tiêu chất lượng và mức chất lượng. |
||
|
Điều kiện cần đáp ứng (C) |
1. Áp dụng trong trường hợp tiêu chuẩn chất lượng đã được duyệt của thuốc thành phẩm không theo một trong các dược điển tham chiếu. 2. Thay đổi này không phải là kết quả của biến cố ngoại ý phát sinh trong quá trình sản xuất hoặc do quan ngại về độ ổn định. 3. Quy trình phân tích không thay đổi hoặc thay đổi rất ít (không cần thiết phải tiến hành thẩm định lại quy trình). 4. Nếu có thay đổi quy trình phân tích, áp dụng thêm MiV-PA27. 5. Nếu mở rộng giới hạn chỉ tiêu chất lượng, bỏ bớt chỉ tiêu chất lượng và mức chất lượng, xem MaV-6. |
||
|
Hồ sơ cần nộp (D) |
(a) Giới hạn chỉ tiêu chất lượng chặt chẽ hơn: 1. Bảng so sánh tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành trước và sau khi có thay đổi của thuốc thành phẩm có in đậm những nội dung thay đổi. 2. So sánh số liệu phân tích lô của tất cả các chỉ tiêu chất lượng trong tiêu chuẩn chất lượng mới trên ít nhất 02 lô sản xuất của thuốc thành phẩm. 3. Giải trình về mặt kỹ thuật các lý do dẫn đến đề nghị thay đổi này. (b) Bổ sung chỉ tiêu chất lượng và mức chất lượng: Ngoài các tài liệu nêu trên yêu cầu: 4. Mô tả quy trình phân tích các chỉ tiêu chất lượng mới của thuốc thành phẩm và cung cấp số liệu thẩm định các quy trình phân tích của các chỉ tiêu chất lượng mới này (đối với các quy trình phân tích không có trong dược điển, quy trình phân tích trong dược điển mới khác quy trình phân tích dược điển cũ). 5. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành mới đồng thời đề xuất biện pháp xử lý phù hợp. |
||
|
Nội dung thay đổi 25 (MiV- PA25) |
Thay đổi vết khắc, hình ảnh hoặc các ký hiệu khác trên viên nén hoặc hình ảnh/ ký hiệu in trên viên nén, viên nang bao gồm thay đổi/ bổ sung mực in dùng để in lên thuốc thành phẩm |
||
|
Điều kiện cần đáp ứng (C) |
(a) Tất cả các thay đổi tại mục này, trừ đường kẻ/vạch bẻ thuốc: 1. Các ký hiệu mới không gây nhầm lẫn với thuốc thành phẩm khác đã được đăng ký. 2. Loại mực mới dự kiến sử dụng không được nằm trong danh mục các chất cấm dùng và phải đáp ứng các tiêu chuẩn để dùng trong thực phẩm hoặc dược phẩm. 3. Tiêu chuẩn chất lượng xuất xưởng và Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi ngoại trừ thay đổi về mô tả hình thức viên. (b) Thay đổi đường kẻ/vạch bẻ thuốc Ngoài những điều kiện có liên quan nêu trên yêu cầu: 4. Đường kẻ/vạch bẻ thuốc không nhằm mục đích làm đẹp. 5. Áp dụng đối với bổ sung hoặc bỏ đường kẻ/vạch bẻ thuốc. |
||
|
Hồ sơ cần nộp (D) |
(a) Tất cả các thay đổi tại mục này, trừ đường kẻ/vạch bẻ thuốc: 1. Thành phần và tiêu chuẩn chất lượng của mực in mới dự kiến sử dụng. 2. Phiếu kiểm nghiệm của mực in mới/ nguyên liệu dùng để in mới (loại dùng trong thực phẩm, dược phẩm). 3. Mô tả chi tiết (bằng hình ảnh hoặc bản viết) các vết khắc/hình ảnh/ký hiệu trên thuốc thành phẩm trước và sau khi thay đổi. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm đã cập nhật mô tả mới về hình thức viên. (b) Thay đổi đường kẻ/vạch bẻ thuốc: Ngoài những tài liệu có liên quan nêu trên yêu cầu: 5. Giải trình về lý do thay đổi (ví dụ do thay đổi liều dùng). 6. Phiếu kiểm nghiệm cho 02 lô (lô sản xuất hoặc lô pilot) của thuốc thành phẩm sau khi có thay đổi. 7. Số liệu kiểm tra độ đồng đều đơn vị liều giữa các phần của viên thuốc sau khi bẻ tại thời điểm xuất xưởng (trong trường hợp bổ sung đường kẻ/ vạch bẻ thuốc). |
||
|
Nội dung thay đổi 26 (MiV- PA26) |
Thay đổi kích thước và/hoặc hình dạng viên nén, viên nang, viên đạn hoặc viên đặt âm đạo mà không thay đổi loại và lượng của các thành phần trong viên và khối lượng trung bình viên đối với: a) Dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn và viên đặt âm đạo; b) Các trường hợp không phải dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn và viên đặt âm đạo. |
||
|
Điều kiện cần đáp ứng (C) |
1. Có số liệu so sánh độ hòa tan của thuốc thành phẩm trước và sau khi có thay đổi (đối với các trường hợp có áp dụng) đạt quy định theo SUPAC-IR và SUPAC-MR. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi ngoại trừ thay đổi trong mô tả kích thước và/hoặc hình dạng thuốc thành phẩm. |
||
|
Hồ sơ cần nộp (D) |
(a) Dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn, viên đặt âm đạo : 1. Mô tả chi tiết bằng hình vẽ hoặc bản viết hình dạng/ kích thước thuốc thành phẩm trước và sau khi có thay đổi. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm đã cập nhật mô tả mới về hình dạng/ kích thước của thuốc thành phẩm. 3. Số liệu so sánh độ hòa tan trên ít nhất 01 lô (lô pilot hoặc lô sản xuất) giữa các thuốc thành phẩm trước và sau khi có thay đổi . 4. Số liệu kiểm tra độ đồng đều đơn vị liều giữa các phần viên thuốc sau khi bẻ tại thời điểm xuất xưởng đáp ứng các yêu cầu của dược điển (chỉ áp dụng đối với thuốc thành phẩm có đường kẻ/vạch bẻ thuốc). (b) Không phải dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn, viên đặt âm đạo: Ngoài những tài liệu nêu trên yêu cầu: 5. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). |
||
|
Nội dung thay đổi 27 (MiV- PA27) |
Thay đổi quy trình phân tích trong tiêu chuẩn chất lượng thuốc thành phẩm (bao gồm thay thế hoặc bổ sung quy trình phân tích) |
||
|
Điều kiện cần đáp ứng (C) |
1. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng đã được duyệt của thuốc thành phẩm không bị ảnh hưởng ngoại trừ theo hướng chặt chẽ hơn. 2. Kết quả thẩm định quy trình phân tích cho thấy quy trình phân tích mới ít nhất là tương đương với quy trình phân tích đã được duyệt. 3. Thay đổi này không phải là kết quả của biến cố ngoại ý phát sinh trong quá trình sản xuất hoặc do quan ngại về độ ổn định. |
||
|
Hồ sơ cần nộp (D) |
1. Mô tả quy trình phân tích mới. 2. Số liệu thẩm định/ thẩm tra quy trình phân tích mới và bảng so sánh các kết quả phân tích thu được từ quy trình phân tích đã được duyệt và quy trình phân tích mới. 3. Phiếu kiểm nghiệm của 02 lô sản xuất của thuốc thành phẩm được kiểm tra theo tiêu chuẩn chất lượng thuốc thành phẩm đã thay đổi. 4. Giải trình lý do thay đổi. 5. Bảng so sánh tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành trước và sau khi có thay đổi của thuốc thành phẩm. |
||
|
Nội dung thay đổi 28 (MiV- PA28) |
Thay đổi liên quan đến bao bì đóng gói sơ cấp của thuốc thành phẩm không vô khuẩn gồm: a) Thay đổi thành phần và lượng các thành phần của chất liệu bao bì; b) Thay đổi loại bao bì và/ hoặc chất liệu bao bì. |
|
|
|
Điều kiện cần đáp ứng (C) |
1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Đối với thay đổi nguyên liệu bao bì sơ cấp của thuốc thành phẩm vô khuẩn, xem MaV-12. |
||
|
Hồ sơ cần nộp (D) |
1. Giải thích lý do thay đổi chất liệu bao bì đóng gói sơ cấp và các nghiên cứu khoa học phù hợp liên quan đến bao bì đóng gói sơ cấp mới. 2. Đối với dạng bào chế bán rắn và lỏng, cung cấp các bằng chứng chứng minh không có sự tương tác giữa thuốc và chất liệu bao bì đóng gói mới (Thí dụ: các thành phần của chất liệu bao bì không đi vào thuốc và thành phần của thuốc không bị mất vào bao bì). 3. Bảng so sánh tiêu chuẩn chất lượng giữa chất liệu bao bì sơ cấp mới và chất liệu bao bì sơ cấp đã được duyệt. 4. Số liệu độ ổn định của thuốc thành phẩm trong bao bì đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). |
||
|
Nội dung thay đổi 29 (MiV- PA29) |
Thay đổi cơ sở đóng gói thứ cấp |
|
|
|
Điều kiện cần đáp ứng (C) |
Chỉ thay đổi cơ sở đóng gói thứ cấp, tất cả các yếu tố khác không thay đổi |
||
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận GMP hoặc CPP đối với cơ sở đóng gói tại nước ngoài và Giấy chứng nhận đủ điều kiện kinh doanh đối với cơ sở đóng gói trong nước. 2. Thư của chủ sở hữu sản phẩm cho phép cơ sở đóng gói mới thực hiện việc đóng gói thứ cấp (nếu có). |
||
|
Nội dung thay đổi 30 (MiV- PA30) |
Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và/hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín đối với thuốc thành phẩm không vô khuẩn. |
||
|
Điều kiện cần đáp ứng (C) |
1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Quy cách đóng gói mới phù hợp với liều dùng và thời gian sử dụng đã được duyệt trong hướng dẫn sử dụng. 3. Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và/hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín của thuốc vô khuẩn dạng rắn và dạng dung dịch, xem MaV-13. 4. Chất liệu bao bì đóng gói không thay đổi. |
||
|
Hồ sơ cần nộp (D) |
1. Giải thích lý do thay đổi quy cách đóng gói. 2. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). 3. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc đã bắt đầu tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm trong bao bì đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. |
||
|
Nội dung thay đổi 31 (MiV- PA31) |
Thay đổi quy cách đóng gói thứ cấp của thuốc thành phẩm |
||
|
Điều kiện cần đáp ứng (C) |
1. Quy cách đóng gói đề nghị thay đổi phù hợp với liều dùng đã được duyệt trong hướng dẫn sử dụng. |
||
|
Hồ sơ cần nộp (D) |
1. Hướng dẫn sử dụng đã được duyệt. 2. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). 3. Giải trình về việc thay đổi quy cách đóng gói thứ cấp là phù hợp với liều dùng đã được duyệt trong hướng dẫn sử dụng. |
||
|
Nội dung thay đổi 32 (MiV- PA32) |
Các thay đổi của bao bì (sơ cấp) không tiếp xúc trực tiếp với thuốc thành phẩm như màu của nắp bật, vạch màu trên ống, thay đổi chụp bảo vệ kim tiêm (sử dụng nhựa khác) |
||
|
Điều kiện cần đáp ứng (C) |
1. Thay đổi này không liên quan đến phần nguyên liệu bao bì có ảnh hưởng đến sự phân liều, sự sử dụng, tính an toàn hoặc độ ổn định của thuốc. |
||
|
Hồ sơ cần nộp (D) |
1. Các phần hồ sơ liên quan đến thay đổi (trình bày theo quy định của ACTD). 2. Bảng so sánh các nội dung thay đổi. 3. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). |
||
|
Nội dung thay đổi 33 (MiV- PA33) |
Bổ sung hoặc thay thế dụng cụ đo lường của các dạng thuốc lỏng dùng đường uống và các dạng bào chế khác |
||
|
Điều kiện cần đáp ứng (C) |
1. Kích cỡ và nếu cần, mức độ chính xác của dụng cụ đo lường mới phải phù hợp với liều dùng đã được duyệt của thuốc thành phẩm. 2. Dụng cụ mới phải tương thích với thuốc thành phẩm. |
||
|
Hồ sơ cần nộp (D) |
1. Mô tả dụng cụ đo lường mới (bao gồm hình vẽ, nếu có). 2. Thông tin về thành phần nguyên liệu dùng trong sản xuất dụng cụ đo lường. Nguyên liệu dùng trong sản xuất dụng cụ đo lường phải đáp ứng tiêu chuẩn chất lượng của dược điển tham chiếu (nếu có tiêu chuẩn của nguyên liệu này trong dược điển). 3. Thuyết minh về sự phù hợp của kích cỡ và mức độ chính xác của dụng cụ đo lượng mới với liều dùng đã được duyệt của thuốc thành phẩm. |
||
|
Nội dung thay đổi 34 (MiV- PA34) |
Giảm hạn dùng của thuốc thành phẩm a) Đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/ hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng |
||
|
Điều kiện cần đáp ứng (C) |
1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Trường hợp tăng hạn dùng thuốc thành phẩm, xem MaV-15. |
||
|
Hồ sơ cần nộp (D) |
1. Số liệu độ ổn định dài hạn phù hợp với hạn dùng mới trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp của thuốc thành phẩm trong các trường hợp: a) Đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, pha loãng hoặc phân tán 2. Giải trình các lý do đề nghị giảm hạn dùng của thuốc. 3. Báo cáo số lượng thuốc đang lưu hành trên thị trường. |
||
|
Nội dung thay đổi 35 (MiV- PA35) |
Thay đổi điều kiện bảo quản của thuốc thành phẩm (khắc nghiệt hơn so với điều kiện bảo quản đã được chấp thuận) a) Trong bao bì đóng gói sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. |
||
|
Điều kiện cần đáp ứng (C) |
1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Nếu thay đổi điều kiện bảo quản (ít khắc nghiệt hơn so với điều kiện bảo quản đã được duyệt), xem MaV-16. |
||
|
Hồ sơ cần nộp (D) |
1. Số liệu độ ổn định dài hạn của thuốc thành phẩm ở điều kiện bảo quản mới trong khoảng thời gian phù hợp với hạn dùng tương ứng đã được duyệt trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp trong các trường hợp: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/ hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. 2. Giải thích về mặt kỹ thuật các lý do thay đổi điều kiện bảo quản. |
||
|
Nội dung thay đổi 36 (MiV- PA36) |
Thay đổi cơ sở đăng ký |
||
|
Điều kiện cần đáp ứng (C) |
1. Áp dụng đối với thay đổi cơ sở đăng ký |
||
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận đủ điều kiện kinh doanh có tên, địa chỉ mới hoặc của cơ sở đăng ký mới (nếu là cơ sở đăng ký của Việt Nam). 2. Giấy phép sản xuất, kinh doanh thuốc do cơ quan quản lý nhà nước có thẩm quyền nước ngoài cấp và Giấy phép thành lập Văn phòng đại diện tại Việt Nam với tên, địa chỉ mới hoặc của cơ sở đăng ký mới (nếu là cơ sở đăng ký của nước ngoài). |
||
|
Nội dung thay đổi 37 (MiV- PA37) |
Công bố thuốc có chứng minh tương đương sinh học |
||
|
Điều kiện cần đáp ứng (C) |
- Có nghiên cứu chứng minh tương đương sinh học đáp ứng quy định. |
||
|
Hồ sơ cần nộp (D) |
- CPP theo quy định tại điểm h Khoản 4 Điều 23 Thông tư này. - Báo cáo nghiên cứu tương đương sinh học theo hướng dẫn kỹ thuật của ASEAN ban hành theo Thông tư này và quy định tại Thông tư hướng dẫn báo cáo nghiên cứu tương đương sinh học. |
||
|
Nội dung thay đổi 38 (MiV-PA38) |
Cập nhật phân loại biệt dược gốc |
|
|
- Thuốc đã được cấp Giấy phép lưu hành đề nghị phân loại là biệt dược gốc. - Cập nhật thông tin thay đổi của thuốc Biệt dược gốc đã được công bố. |
|
Điều kiện cần đáp ứng (C) |
- Thuốc có đủ dữ liệu an toan hiệu quả. - CPP được cấp bởi một trong các cơ quan quản lý quy định tại khoản 9, 10 Điều 2 Thông tư này hoặc thuốc biệt dược gốc chuyển giao sản xuất tại Việt Nam. |
|
Hồ sơ cần nộp (D) |
1. CPP 2. Tài liệu tiền lâm sàng 3. Tài liệu lâm sàng Tờ hướng dẫn sử dụng đã được phê duyệt, trừ trường hợp thuốc đã được cấp giấy đăng ký lưu hành theo bộ hồ sơ ACTD hoặc ICH-CTD bao gồm hồ sơ lâm sàng của thuốc. 4. Trường hợp Biệt dược gốc đã được công bố, thực hiện các thay đổi, bổ sung theo quy định chung. Thuốc tiếp tục được phân loại là biệt dược gốc nếu sau khi thay đổi thuốc đáp ứng các điều kiện công bố biệt dược gốc. |
Mục MiV-PA 38 tại Mục 6 Thay đổi nhỏ (MiV-PA) cần phê duyệt trước khi thực hiện của Phụ lục II ban hành kèm theo Thông tư này được sửa đổi, bổ sung bởi Phụ lục 01 ban hành kèm theo Thông tư 23/2021/TT-BYT theo quy định tại Điểm g Khoản 3 Điều 1.
7.THAY ĐỔI/BỔ SUNG CẦN PHÊ DUYỆT NHƯNG KHÔNG DẪN ĐẾN THAY ĐỔI NHÃN VÀ HƯỚNG DẪN SỬ DỤNG THUỐC
Thay đổi/bổ sung cần phê duyệt nhưng không dẫn đến thay đổi thông tin trên nhãn và hướng dẫn sử dụng thuốc (*)
|
Nội dung thay đổi 39 |
Bổ sung, cập nhật thông tin để cung cấp thông tin thuốc, quảng cáo thuốc |
|
Điều kiện cần đáp ứng (C) |
1. Các thông tin được bổ sung, cập nhật theo quy định tại mục này là các thông tin mới được phát minh, phát hiện qua nghiên cứu khoa học hoặc qua theo dõi sản phẩm trên thị trường nhưng không thuộc trường hợp quy định tại mục từ 1 đến 7 Phụ lục này. 2. Trình bày toàn bộ các thông tin đề nghị theo đúng nội dung sẽ sử dụng để thông tin thuốc. |
|
Hồ sơ cần nộp (D) |
1. Nội dung thông tin đề nghị cập nhật. 2. Tài liệu tham khảo. 3. Bản sao tờ hướng dẫn sử dụng thuốc cập nhật nhất đã được Bộ Y tế phê duyệt có đóng dấu của cơ sở đăng ký thuốc. |
Chú giải:
1. Mục tiêu của việc thay đổi/bổ sung:
Các thông tin đề nghị bổ sung, cập nhật nhằm bổ trợ, làm rõ thêm cho thông tin đã có trong tờ hướng dẫn sử dụng thuốc. Các thông tin sau khi được bổ sung, cập nhật trong hồ sơ đăng ký thuốc là nguồn thông tin để sử dụng trong việc cung cấp thông tin thuốc, quảng cáo thuốc đảm bảo sử dụng thuốc an toàn, hợp lý, hiệu quả.
2. Các loại thông tin đề nghị thay đổi/bổ sung:
- Các thông số dược động học của thuốc trên một quần thể nghiên cứu cụ thể (bao gồm cả việc so sánh thông số dược động học giữa các thuốc).
- Các thông số về dược lực học: sơ đồ cơ chế tác dụng của thuốc; Các nghiên cứu về tính nhạy cảm của kháng sinh đối với các loại vi khuẩn tại một thời điểm cụ thể dựa trên một quần thể nghiên cứu cụ thể...
- Các kết quả cụ thể liên quan đến hiệu quả điều trị: kết quả so sánh hiệu quả điều trị giữa thuốc nghiên cứu (có thể đúng tên Biệt dược hoặc hoạt chất của thuốc đề nghị cập nhật) với giả dược hoặc với một hay nhiều thuốc khác trong cùng nhóm hoặc khác nhóm tác dụng dược lý.
- Các phác đồ hoặc hướng dẫn điều trị liên quan đến thuốc.
- Các kết quả thử tương đương sinh học.
- Các thông tin chi tiết hơn về tác dụng không mong muốn của thuốc đã ghi trong tờ Hướng dẫn sử dụng thuốc;
- Các kết quả nghiên cứu đánh giá mức độ cải thiện chất lượng cuộc sống của người bệnh, sự hài lòng của người bệnh, của bác sĩ đối với sự tiện dụng, dễ tuân thủ điều trị, hiệu quả điều trị...của thuốc.
- Các thông tin khác liên quan đến chất lượng, an toàn, hiệu quả của thuốc.
3. Cách thức trình bày nội dung thông tin:
- Thông tin đề nghị cập nhật phải được trình bày rõ ràng, đầy đủ, chính xác dựa trên bằng chứng, dễ hiểu, phù hợp với đối tượng được cung cấp thông tin;
- Trình bày toàn bộ các thông tin đề nghị chính xác như thông tin sẽ sử dụng để thông tin thuốc.
- Ghi rõ danh mục những Tài liệu tham khảo được sử dụng để xây dựng nội dung thông tin: đánh số thứ tự, ghi rõ tên Tài liệu tham khảo, tên bài nghiên cứu, tác giả, năm xuất bản Tài liệu.
- Trường hợp Tài liệu tham khảo là nghiên cứu lâm sàng thì có phần giới thiệu tóm tắt về nghiên cứu bao gồm các thông tin sau: mục tiêu, đối tượng nghiên cứu, phương pháp nghiên cứu, cỡ mẫu, các bước tiến hành, tiêu chí đánh giá, kết quả nghiên cứu.
- Trường hợp Tài liệu tham khảo là nghiên cứu cho hoạt chất thì thông tin trích dẫn ghi theo đúng dưới dạng tên hoạt chất (không được thay tên hoạt chất bằng tên biệt dược); Trường hợp Tài liệu tham khảo là nghiên cứu cho biệt dược thì thông tin trích dẫn ghi dưới dạng tên biệt dược.
- Ghi rõ số thứ tự Tài liệu tham khảo đằng sau các thông tin trích dẫn.
- Phiên giải đầy đủ kết quả nghiên cứu kèm số liệu, không được chỉ đưa ra các kết luận chung chung.
- Không ghi tên Tài liệu tham khảo, tên tổ chức thực hiện nghiên cứu, tên phương pháp nghiên cứu, tên tổ chức đưa ra phác đồ, hướng dẫn điều trị dưới dạng in thành tít to nổi bật.
4. Yêu cầu đối với Tài liệu tham khảo:
4.1. Các loại tài liệu tham khảo được sử dụng:
- Chuyên luận về thuốc đã được ghi trong phiên bản mới nhất của Dược thư Quốc gia, Martindale, AHFS,BNF, FDA, EMC…;
- Các bài báo nghiên cứu lâm sàng và các nghiên cứu khác liên quan đến thuốc được đăng tải trên các tạp chí thuộc danh mục SCI (Science Citation Index) - Chỉ số trích dẫn khoa học;
- Các phác đồ, hướng dẫn điều trị liên quan đến thuốc;
- Các kết quả nghiên cứu lâm sàng đã được nghiệm thu bởi cơ quan có thẩm quyền.
4.2. Tiêu chí đối với các tài liệu tham khảo:
- Tài liệu tham khảo phải là nguồn tài liệu rõ ràng, đáng tin cậy, là bản ghi đầy đủ, chi tiết, mang tính cập nhật, ghi rõ tên tài liệu, tên tác giả, thời gian xuất bản tài liệu;
- Trường hợp Tài liệu tham khảo là nghiên cứu lâm sàng thì có đầy đủ thông tin của một nghiên cứu lâm sàng (bao gồm: mục tiêu nghiên cứu, phương pháp nghiên cứu, cỡ mẫu, mô tả các bước tiến hành, kết quả, bàn luận, kết luận...).
- Không chấp nhận Tài liệu tham khảo là các nghiên cứu trên động vật, invitro.
- Ngôn ngữ của Tài liệu tham khảo: phải bằng tiếng Việt hoặc tiếng Anh. Nếu tài liệu tham khảo không phải là tiếng Việt hoặc tiếng Anh thì cần nộp kèm bản dịch công chứng bằng tiếng Việt có đóng dấu của đơn vị đăng ký.
- Chú thích rõ ràng các phần thông tin, dữ liệu trong tài liệu tham khảo được trích dẫn ra để cập nhật thông tin.
|
8. THAY ĐỔI NHỎ CHỈ YÊU CẦU THÔNG BÁO (NOTIFICATION) |
|
|
Nội dung thay đổi 1 (MiV-N1) |
Thay đổi tên, địa chỉ của cơ sở đăng ký, cập nhật thông tin liên quan đến cơ sở đăng ký |
|
Điều kiện cần đáp ứng (C) |
Cơ sở đăng ký không thay đổi. |
|
Hồ sơ cần nộp (D) |
1. Đơn đăng ký có nội dung cam kết chịu trách nhiệm về nội dung thay đổi tên, địa chỉ của cơ sở đăng ký. 2. Xác nhận của cơ quan có thẩm quyền về việc thay đổi tên, địa chỉ hoặc các giấy tờ pháp lý của cơ sở đăng ký chứng minh nội dung thay đổi. 3. Giấy tờ pháp lý liên quan . |
|
Nội dung thay đổi 2 (MiV-N2) |
Thay đổi chủ sở hữu sản phẩm |
|
Điều kiện cần đáp ứng (C) |
1. Cơ sở đăng ký không thay đổi. 2. Địa điểm sản xuất không đổi. |
|
Hồ sơ cần nộp (D) |
1. Thư công bố của chủ sở hữu sản phẩm cũ về việc chuyển nhượng quyền sở hữu giữa chủ sở hữu sản phẩm cũ và mới. 2. Công văn của chủ sở hữu mới của sản phẩm công bố sự thay đổi này và cho phép người giữ giấy phép sản phẩm tại mỗi nước chịu trách nhiệm về giấy phép sản phẩm. 3. Văn bản của chủ sở hữu mới cho phép cơ sở sản xuất tiếp tục sản xuất thuốc và văn ban của cơ sở sản xuất về việc chịu trách nhiệm sản xuất và đảm bảo tính hiệu quả, chất lượng và an toàn của thuốc (nếu chủ sở hữu mới của sản phẩm không phải là cơ sở sản xuất thuốc). |
|
Nội dung thay đổi 3 (MiV-N3) |
Thay đổi tên và/hoặc địa chỉ (như mã bưu điện, tên phố) của cơ sở sản xuất dược chất |
|
Điều kiện cần đáp ứng (C) |
1. Địa điểm sản xuất dược chất không đổi. 2. Không có thay đổi nào khác ngoại trừ thay đổi tên và/hoặc địa chỉ cơ sở sản xuất dược chất. |
|
Hồ sơ cần nộp (D) |
1. Cập nhật thông tin cơ sở sản xuất dược chất. 2. Tài liệu chứng minh (nếu có) |
|
Nội dung thay đổi 4 (MiV-N4) |
Bỏ bớt cơ sở sản xuất của dược chất |
|
Điều kiện cần đáp ứng (C) |
1. Cơ sở sản xuất khác đã được đăng ký. |
|
Hồ sơ cần nộp (D) |
1. Công văn nếu rõ lý do rút lại hoặc loại bỏ kèm tài liệu chứng minh (nếu có) |
|
Nội dung thay đổi 5 (MiV-N5) (Asean:Mi V-N8) |
Đăng ký lại giấy chứng nhận tuân thủ dược điển châu Âu (CEP) |
|
Điều kiện cần đáp ứng (C) |
1. Chỉ áp dụng cho đăng ký lại CEP mà không có bất kỳ thay đổi nào. |
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận tuân thủ dược điển châu Âu (CEP) cho dược chất bản mới nhất còn hiệu lực với đầy đủ các phụ lục do EDQM cấp. |
|
Nội dung thay đổi 6 (MiV-N6) |
Thay đổi tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm và/hoặc dược chất và/hoặc tá dược khi cập nhật dược điển. |
|
Điều kiện cần đáp ứng (C) |
1. Chỉ áp dụng trong trường hợp tiêu chuẩn chất lượng đã được duyệt của thuốc thành phẩm/dược chất/ tá dược là theo một trong các dược điển tham chiếu và có ghi cụ thể phiên bản dược điển. 2. Thay đổi nhằm đáp ứng theo chuyên luận tương ứng đã được cập nhật tại phiên bản mới của dược điển. |
|
Hồ sơ cần nộp (D) |
1. Đơn đăng ký thay đổi. 2. Bảng so sánh tiêu chuẩn đã được phê duyệt và tiêu chuẩn đề nghị thay đổi. 3. Dữ liệu phân tích lô. (không yêu cầu đối với trường hợp cập nhật tiêu chuẩn dược chất, tá dược mà không thay đổi tiêu chuẩn thành phẩm) 4. Ban tiêu tiêu chuẩn thay đổi. 5. Đối với thay đổi quy trình phân tích trong tiêu chuẩn thành phẩm, yêu cầu dữ liệu thẩm tra quy trình phân tích mới. Trường hợp cập nhật tiêu chuẩn tá dược theo dược điển, không thay đổi tiêu chuẩn thành phẩm, chỉ yêu cầu 1 đơn, 01 bộ hồ sơ chung cho nhiều thuốc. |
|
Nội dung thay đổi 7 (MiV-N7) |
Bỏ bớt quy cách đóng gói thuốc thành phẩm |
|
Điều kiện cần đáp ứng (C) |
1. Các quy cách đóng gói còn lại phù hợp với liều dùng đã phê duyệt. 2. Đối với thay đổi quy cách đóng gói trong bao bì sơ cấp của thuốc vô khuẩn và không vô khuẩn, xem MaV-13 và MiV-PA30. Đối với thay đổi quy cách đóng gói trong bao bì thứ cấp, xem MiV-PA31. |
|
Hồ sơ cần nộp (D) |
1. Giải trình về lý do bỏ bớt quy cách đóng gói thuốc. 2. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). |
|
Nội dung thay đổi 8 (MiV-N8) |
Thay đổi tên cơ sở sản xuất do thay đổi chủ sở hữu của cơ sở sản xuất |
|
Điều kiện cần đáp ứng (C) |
1. Địa điểm sản xuất không đổi 2. Không có thay đổi nào ngoại trừ thay đổi chủ sở hữu của cơ sở sản xuất. |
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận GMP hoặc CPP với tên mới của cơ sở sản xuất hoặc giấy xác nhận của cơ quan quản lý dược phẩm có thẩm quyền về việc thay đổi chủ sở hữu của cơ sở sản xuất. 2. Thư công bố của cơ sở sở hữu cũ về việc chuyển nhượng quyền sở hữu sang cơ sở sở hữu mới. |
|
Nội dung thay đổi 9 (MiV-N9) |
Thay đổi tên và/hoặc địa chỉ (như mã bưu điện, tên phố) của cơ sở sản xuất thuốc thành phẩm |
|
Điều kiện cần đáp ứng (C) |
1. Địa điểm sản xuất không đổi. 2. Không áp dụng cho trường hợp thay đổi chủ sở hữu của cơ sở sản xuất. Nếu thay đổi chủ sở hữu của cơ sở sản xuất, xem MiV-PA37. 3. Không có thay đổi nào khác ngoại trừ thay đổi tên và/hoặc địa chỉ cơ sở sản xuất thuốc. |
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận GMP hoặc CPP với tên và/hoặc địa chỉ mới của nhà sản xuất. 2. Giấy xác nhận của cơ quan có thẩm quyền về việc đổi tên và/hoặc thay đổi cách ghi địa chỉ mà địa điểm sản xuất không thay đổi đối với thuốc sản xuất tại nước ngoài. 3. Giấy chứng nhận đủ điều kiện kinh doanh thuốc của cơ sở sản xuất với tên và, hoặc địa chỉ mới đối với thuốc sản xuất trong nước. |
|
Nội dung thay đổi 10 (MiV-N10) |
Thay đổi tên và/hoặc địa chỉ (như mã bưu điện, tên phố) của cơ sở xuất xưởng lô |
|
Điều kiện cần đáp ứng (C) |
1. Cơ sở sản xuất thuốc không đổi. 2. Không áp dụng cho trường hợp thay đổi chủ sở hữu của cơ sở sản xuất. Nếu thay đổi chủ sở hữu của cơ sở sản xuất, xem MiV-PA37. 3. Địa điểm cơ sở xuất xưởng lô không đổi. |
|
Hồ sơ cần nộp (D) |
1. Giấy chứng nhận GMP hoặc CPP với tên và/hoặc địa chỉ mới của cơ sở xuất xưởng lô và xác nhận của cơ quan có thẩm quyền về việc đổi tên và/hoặc thay đổi cách ghi địa chỉ mà địa điểm cơ sở xuất xưởng lô không thay đổi đối với thuốc sản xuất tại nước ngoài. 2. Giấy chứng nhận đủ điều kiện kinh doanh thuốc của cơ sở xuất xưởng lô mới đối với thuốc sản xuất trong nước |
|
Nội dung thay đổi 11 |
Thay đổi mẫu nhãn, hướng dẫn sử dụng |
|
Điều kiện cần đáp ứng (C) |
1. Không thuộc các trường hợp thay đổi lớn (MaV-1, MaV-2) và thay đổi nhỏ phải phê duyệt trước khi thực hiện (MiV-PA2). 2. Bổ sung/Thay đổi các nội dung không liên quan đến an toàn, hiệu quả, hướng dẫn sử dụng thuốc và không bắt buộc ghi trên mẫu nhãn, hướng dẫn sử dụng thuốc ngoại trừ các nội dung quy định trong khoản 2 Điều 35 Thông tư số 01/2018/TT-BYT ngày 18/01/2018 của Bộ Y tế hướng dẫn ghi nhãn thuốc. |
|
Hồ sơ cần nộp (D) |
1. Đơn đăng ký có nội dung cam kết chịu trách nhiệm về nội dung thay đổi tên, địa chỉ của cơ sở đăng ký. 2. Giấy tờ pháp lý liên quan (nếu có). |
9. THUẬT NGỮ
Tham khảo thuật ngữ theo ACTD/ACTR
10. THAM KHẢO
1. EMA Variation guidline, 2008
2. Communication from the Commission Guideline on the details of the various categories of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products - Official Journal of the European Union (C 17/1 of 22.01.2010)
3. Commission Regulation (EC) No 1234/2008 Official Journal of the European Union (L334 of 12 December 2008)
4. WHO Guidance on Variations To A Prequalified Product Dossier, 2007
5. SUPAC Guideline Immediate Release Solid Oral Dosage Forms, Scale-up and Post- approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation, November 1995
6. SUPAC-MR: Modified Release Solid, Oral Dosage Forms, Scale-Up and Post - approval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation, September 1997
7. WHO Technical Report Series, No. 953, 2009
8. WHO Quality Assurance of Pharmaceuticals - A Compendium of Guidelines and Related Materials - Volume 1
9. ASEAN Guideline on Stability Study of Drug Product, version 6, May/2013
10. ASEAN Guideline on Submission of Manufacturing Process Validation Data for Drug Registration
11. ASEAN Guideline for Validation of Analytical Procedures
12. ASEAN Guideline for the Conduct of Bioavailability and Bioequivalence Studies, 21 July 2004
B. SINH PHẨM VÀ NGUYÊN LIỆU HÓA DƯỢC
1. Đối với sinh phẩm thực hiện phần hồ sơ hành chính, hồ sơ chất lượng thực hiện theo quy định tại mục A Phụ lục này. Phần dữ liệu lâm sàng theo các hướng dẫn của WHO, US FDA, EMA theo Phụ lục IV ban hành kèm theo Thông tư này.
2. Nguyên liệu làm thuốc là dược chất, tá dược đã có Giấy đăng ký lưu hành thực hiện các thay đổi, bổ sung có liên quan theo quy định tại mục A Phụ lục này.
C. VẮC XIN, HUYẾT THANH CHỨA KHÁNG THỂ
Các thay đổi chỉ liên quan đến hồ sơ hành chính, thực hiện theo quy định tại Mục A Phụ lục này.
I. CÁC THAY ĐỔI LỚN
|
STT |
Nội dung thay đổi/bổ sung |
Điều kiện |
Yêu cầu hồ sơ |
|
1. |
Hàm lượng/nồng độ các thành phần dược chất có tác dụng. |
Áp dụng đối với các dạng bào chế không phân liều |
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc công văn cho phép của cơ quan có thẩm quyền nước sở tại) - yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần III & IV: Phần liên quan |
|
2. |
Đường dùng |
Không thay đổi dạng bào chế |
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc văn bản cho phép của cơ quan có thẩm quyền nước sở tại) - yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần II (Chất lượng): Phần liên quan - Phần III & IV: Phần liên quan |
|
3. |
Thay đổi liều dùng |
|
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc công văn cho phép của cơ quan có thẩm quyền nước sở tại) - yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần III & IV: Phần liên quan |
|
4. |
Chỉ định |
Các nội dung khác không thay đổi |
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc công văn cho phép của cơ quan có thẩm quyền nước sở tại) – yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần III & IV: Phần liên quan |
II- THAY ĐỔI NHỎ
1-CÁC THAY ĐỔI PHẢI ĐƯỢC PHÊ DUYỆT CỦA CƠ QUAN QUẢN LÝ
|
ST T |
Nội dung thay đổi/bổ sung |
Điều kiện |
Yêu cầu hồ sơ |
|
1. |
Thay đổi hoặc bổ sung thành phần tá dược (bao gồm thay đổi tỷ lệ tá dược). |
- Không làm giảm chất lượng của thành phẩm. |
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): phần thay đổi liên quan - Tài liệu pháp lý cua cơ sản xuất tá dược (thực hiện theo lộ trình) |
|
2. |
Thay đổi mô tả đặc tính của thành phẩm |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): phần thay đổi liên quan |
|
3. |
Thay đổi chất chuẩn để kiểm nghiệm thành phẩm |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): phần thay đổi liên quan |
|
4. |
Thay đổi hệ thống đóng kín của bao bì trực tiếp, gián tiếp |
- Chất lượng, độ ổn định ít nhất phải tương đương với chất lượng, độ ổn định của hệ thống đóng gói bao bì cũ |
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): phần thay đổi liên quan |
|
5. |
Thay đổi độ ổn định/hạn dùng của thành phẩm: |
||
|
*Tăng hạn dùng |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
|
* Giảm hạn dùng |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (Chất lượng): Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định - Báo cáo số lượng thuốc đang lưu hành trên thị trường - Cam kết về việc cơ sở đăng ký phải phối hợp với cơ sở sản xuất, cơ sở nhập khẩu tiến hành thu hồi thuốc, nguyên liệu làm thuốc có hạn dùng lớn hơn hạn dùng thay đổi đối với thuốc đã nhập khẩu, lưu hành tại Việt Nam. |
|
|
6. |
Thay đổi điều kiện bảo quản của thành phẩm |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
7. |
Thay đổi qui trình sản xuất của thành phẩm: Sơ đồ, các bước, lô, mẻ, thẩm định qui trình… |
- Theo hướng cải tiến hơn qui trình cũ |
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (Chất lượng): Các tài liệu liên quan. |
|
8. |
Thay đổi tiêu chuẩn và/hoặc phương pháp kiểm nghiệm của thành phẩm (bao gồm cả thẩm định phương pháp phân tích) |
- Theo hướng chặt chẽ hơn |
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (Chất lượng): Các tài liệu liên quan. |
|
9. |
Thay đổi/bổ sung quy cách đóng gói |
|
Phần I (Hành chính): - Đơn - Nhãn cũ đã duyệt + nhãn mới Phần II (Chất lượng): Gồm (1) Tiêu chuẩn bao bì (nếu có thay đổi bao bì, chất lượng bao bì) (2) Hồ sơ theo dõi độ ổn định của quy cách đóng gói mới (nếu có thay đổi bao bì sơ cấp). |
|
10. |
Thay đổi hình thức/ thiết kế bao bì, nhãn |
Theo hướng tốt hơn và nội dung nhãn không thay đổi |
Phần I (Hành chính): - Đơn - Nhãn cũ đã duyệt + nhãn mới Phần II (Chất lượng): Các tài liệu liên quan.. |
|
11. |
Thay đổi hoặc bổ sung nguồn gốc nguyên liệu. |
Không ảnh hưởng đến phần chất lượng (Phải có tài liệu chứng minh tiêu chuẩn nguyên liệu không thay đổi; công thức không thay đổi) |
Phần I (Hành chính): - Đơn - Phần II (Chất lượng): Các tài liệu liên quan. - Tài liệu pháp lý của cơ sở sản xuất nguyên liệu. |
|
12. |
Thay đổi độ ổn định/hạn dùng của nguyên liệu |
Không làm giảm chất lượng của nguyên liệu và thành phẩm. |
Phần I (Hành chính): -Đơn Phần II (Chất lượng): -Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
13. |
Thay đổi/bổ sung các nội dung về an toàn/hiệu quả (Trừ các trường hợp thuộc thay đổi lớn) |
|
Phần I (Hành chính): - Đơn - Nhãn - Thông tin sản phẩm - Tờ hướng dẫn sử dụng, hoặc tóm tắt đặc tính sản phẩm, hoặc thông tin cho bệnh nhân. - Nêu rõ những điểm thay đổi. - Tài liệu chứng minh liên quan đến nội dung thay đổi |
|
14. |
Thay đổi chất chuẩn để kiểm nghiệm nguyên liệu |
|
Phần I (Hành chính): - Đơn Phần II (Chất lượng): - Phần thay đổi liên quan |
|
15. |
Thay đổi điều kiện bảo quản của nguyên liệu |
|
Phần I (Hành chính): - Đơn Phần II (Chất lượng): - Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
16. |
Thay đổi qui trình sản xuất của nguyên liệu: Sơ đồ, các bước, lô, mẻ, thẩm định qui trình |
|
Phần I (Hành chính): - Đơn Phần II (Chất lượng): - Phần thay đổi liên quan. |
|
17. |
Thay đổi tiêu chuẩn và/ hoặc phương pháp kiểm nghiệm của nguyên liệu. |
- Không làm thay đổi và ảnh hưởng đến tiêu chuẩn, chất lượng của thuốc thành phẩm. - Hoặc làm tiêu chuẩn chất lượng của thành phẩm chặt chẽ hơn hoặc tốt hơn. |
Phần I (Hành chính): - Đơn Phần II (Chất lượng): - Phần thay đổi liên quan |
|
18. |
Thay đổi nhà sản xuất nguyên liệu |
|
Phần I (Hành chính): - Đơn - Chứng nhận GMP của nhà sản xuất Phần II (Chất lượng): - Phần thay đổi liên quan |
|
19. |
Cập nhật chủng cúm mùa hàng năm |
|
Phần I (Hành chính): - Đơn - Giấy phép lưu hành của nước sở tại hoặc nước tham chiếu. - Giấy chứng nhận kết quả kiểm định chất lượng, độ an toàn chung của Viện Kiểm định quốc gia vắc xin và sinh phẩm y tế (NICVB) hoặc của cơ quan kiểm định quốc gia nước sản xuất/hoặc nước tham chiếu. - Tài liệu về xuất xứ chủng gốc. - Khuyến cáo của Tổ chức Y tế thế giới của nước sở tại. - Khuyến cáo của Tổ chức Y tế thế giới. - Mẫu nhãn, tờ hướng dẫn sử dụng. - Phần hồ sơ chất lượng liên quan |
2- THAY ĐỔI/ BỔ SUNG CÂN PHÊ DUYỆT NHƯNG KHÔNG DẪN ĐẾN THAY ĐỔI NHÃN VÀ HƯỚNG DẪN SỬ DỤNG THUỐC
|
Thay đổi/bổ sung cần phê duyệt nhưng không dẫn đến thay đổi thông tin trên nhãn và hướng dẫn sử dụng thuốc (*) |
|
|
Nội dung Thay đổi/bổ sung 1 |
Bổ sung, cập nhật thông tin để cung cấp thông tin thuốc, quảng cáo thuốc |
|
Điều kiện cần đáp ứng (C) |
3. Các thông tin được bổ sung, cập nhật theo quy định tại mục này là các thông tin mới được phát minh, phát hiện qua nghiên cứu khoa học hoặc qua theo dõi sản phẩm trên thị trường nhưng không thuộc trường hợp quy định tại mục từ 1 đến 7 Phụ lục này. 4. Trình bày toàn bộ các thông tin đề nghị theo đúng nội dung sẽ sử dụng để thông tin thuốc. |
|
Hồ sơ cần nộp (D) |
1. Nội dung thông tin đề nghị cập nhật. 2. Tài liệu tham khảo. 3. Bản sao tờ hướng dẫn sử dụng thuốc cập nhật nhất đã được Bộ Y tế phê duyệt có đóng dấu của cơ sở đăng ký thuốc. |
Chú giải:
1. Mục tiêu của việc thay đổi/bổ sung:
Các thông tin đề nghị bổ sung, cập nhật nhằm bổ trợ, làm rõ thêm cho thông tin đã có trong tờ hướng dẫn sử dụng thuốc. Các thông tin sau khi được bổ sung, cập nhật trong hồ sơ đăng ký thuốc là nguồn thông tin để sử dụng trong việc cung cấp thông tin thuốc, quảng cáo thuốc đảm bảo sử dụng thuốc an toàn, hợp lý, hiệu quả.
2. Các loại thông tin đề nghị thay đổi/bổ sung:
- Các thông số dược động học của thuốc trên một quần thể nghiên cứu cụ thể (bao gồm cả việc so sánh thông số dược động học giữa các thuốc).
- Các thông số về dược lực học: sơ đồ cơ chế tác dụng của thuốc; Các nghiên cứu về tính nhạy cảm của kháng sinh đối với các loại vi khuẩn tại một thời điểm cụ thể dựa trên một quần thể nghiên cứu cụ thể...
- Các kết quả cụ thể liên quan đến hiệu quả điều trị: kết quả so sánh hiệu quả điều trị giữa thuốc nghiên cứu (có thể đúng tên Biệt dược hoặc hoạt chất của thuốc đề nghị cập nhật) với giả dược hoặc với một hay nhiều thuốc khác trong cùng nhóm hoặc khác nhóm tác dụng dược lý.
- Các phác đồ hoặc hướng dẫn điều trị liên quan đến thuốc.
- Các kết quả thử tương đương sinh học.
- Các thông tin chi tiết hơn về tác dụng không mong muốn của thuốc đã ghi trong tờ Hướng dẫn sử dụng thuốc;
- Các kết quả nghiên cứu đánh giá mức độ cải thiện chất lượng cuộc sống của người bệnh, sự hài lòng của người bệnh, của bác sĩ đối với sự tiện dụng, dễ tuân thủ điều trị, hiệu quả điều trị...của thuốc.
- Các thông tin khác liên quan đến chất lượng, an toàn, hiệu quả của thuốc.
3. Cách thức trình bày nội dung thông tin:
- Thông tin đề nghị cập nhật phải được trình bày rõ ràng, đầy đủ, chính xác dựa trên bằng chứng, dễ hiểu, phù hợp với đối tượng được cung cấp thông tin;
- Trình bày toàn bộ các thông tin đề nghị chính xác như thông tin sẽ sử dụng để thông tin thuốc.
- Ghi rõ danh mục những Tài liệu tham khảo được sử dụng để xây dựng nội dung thông tin: đánh số thứ tự, ghi rõ tên Tài liệu tham khảo, tên bài nghiên cứu, tác giả, năm xuất bản Tài liệu.
- Trường hợp Tài liệu tham khảo là nghiên cứu lâm sàng thì có phần giới thiệu tóm tắt về nghiên cứu bao gồm các thông tin sau: mục tiêu, đối tượng nghiên cứu, phương pháp nghiên cứu, cỡ mẫu, các bước tiến hành, tiêu chí đánh giá, kết quả nghiên cứu.
- Trường hợp Tài liệu tham khảo là nghiên cứu cho hoạt chất thì thông tin trích dẫn ghi theo đúng dưới dạng tên hoạt chất (không được thay tên hoạt chất bằng tên biệt dược); Trường hợp Tài liệu tham khảo là nghiên cứu cho biệt dược thì thông tin trích dẫn ghi dưới dạng tên biệt dược.
- Ghi rõ số thứ tự Tài liệu tham khảo đằng sau các thông tin trích dẫn.
- Phiên giải đầy đủ kết quả nghiên cứu kèm số liệu, không được chỉ đưa ra các kết luận chung chung.
- Không ghi tên Tài liệu tham khảo, tên tổ chức thực hiện nghiên cứu, tên phương pháp nghiên cứu, tên tổ chức đưa ra phác đồ, hướng dẫn điều trị dưới dạng in thành tít to nổi bật.
4. Yêu cầu đối với Tài liệu tham khảo:
4.1. Các loại tài liệu tham khảo được sử dụng:
- Chuyên luận về thuốc đã được ghi trong phiên bản mới nhất của Dược thư Quốc gia, Martindale, AHFS,BNF, FDA, EMC…;
- Các bài báo nghiên cứu lâm sàng và các nghiên cứu khác liên quan đến thuốc được đăng tải trên các tạp chí thuộc danh mục SCI (Science Citation Index) - Chỉ số trích dẫn khoa học;
- Các phác đồ, hướng dẫn điều trị liên quan đến thuốc;
- Các kết quả nghiên cứu lâm sàng đã được nghiệm thu bởi cơ quan có thẩm quyền.
4.2. Tiêu chí đối với các tài liệu tham khảo:
- Tài liệu tham khảo phải là nguồn tài liệu rõ ràng, đáng tin cậy, là bản ghi đầy đủ, chi tiết, mang tính cập nhật, ghi rõ tên tài liệu, tên tác giả, thời gian xuất bản tài liệu;
- Trường hợp Tài liệu tham khảo là nghiên cứu lâm sàng thì có đầy đủ thông tin của một nghiên cứu lâm sàng (bao gồm: mục tiêu nghiên cứu, phương pháp nghiên cứu, cỡ mẫu, mô tả các bước tiến hành, kết quả, bàn luận, kết luận...).
- Không chấp nhận Tài liệu tham khảo là các nghiên cứu trên động vật, invitro.
- Ngôn ngữ của Tài liệu tham khảo: phải bằng tiếng Việt hoặc tiếng Anh. Nếu tài liệu tham khảo không phải là tiếng Việt hoặc tiếng Anh thì cần nộp kèm bản dịch công chứng bằng tiếng Việt có đóng dấu của đơn vị đăng ký.
- Chú thích rõ ràng các phần thông tin, dữ liệu trong tài liệu tham khảo được trích dẫn ra để cập nhật thông tin.
3 - CÁC THAY ĐỔI NHỎ CHỈ YÊU CẦU THÔNG BÁO CHO CƠ QUAN QUẢN LÝ
|
STT |
Nội dung thay đổi/bổ sung |
Điều kiện |
Yêu cầu hồ sơ |
|
1 |
Thay đổi mô tả đặc tính của nguyên liệu |
Không làm thay đổi bản chất của nguyên liệu |
Phần I (Hành chính): -Thông báo Phần II (Chất lượng): - Phần thay đổi liên quan |
|
2 |
Thay đổi nội dung trên mẫu nhãn bao gồm tờ hướng dẫn sử dụng |
- Không thuộc các trường hợp thay đổi lớn và thay đổi nhỏ phải phê duyệt trước khi thực hiện; - Thay đổi thiết kế (màu sắc, hình dáng, kích thước, vị trí thông tin...) trên nhãn và, hoặc các nội dung không liên quan đến thông tin, hướng dẫn sử dụng thuốc. |
Phần I (Hành chính): - Thông báo - Nhãn cũ đã duyệt + nhãn mới - Bảng so sánh nội dung thay đổi - Giấy tờ pháp lý liên quan |
|
3 |
Thay đổi nhà cung cấp bao bì (thay thế, thêm vào hoặc loại bỏ) |
- Không làm thay đổi chất lượng và độ ổn định của thuốc |
Phần I (Hành chính): -Thông báo - Tài liệu liên quan |
|
4 |
Thay thế dụng cụ đo lường thuốc (ví dụ từ muỗng sang cốc) |
|
Phần I (Hành chính): -Thông báo |
D. THUỐC DƯỢC LIỆU, NGUYÊN LIỆU LÀM THUỐC DƯỢC LIỆU
Thực hiện theo quy định tại Mục A Phụ lục này (Thuốc hóa dược) hoặc các thay đổi quy định riêng cho thuốc dược liệu như sau:
I. THAY ĐỔI LỚN
|
STT |
Nội dung thay đổi/bổ sung |
Điều kiện |
Yêu cầu hồ sơ |
|
1 |
Hàm lượng/nồng độ các thành phần dược chất có tác dụng. |
Áp dụng đối với các dạng bào chế không phân liều |
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc công văn cho phép của cơ quan có thẩm quyền nước sở tại) – yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần III & IV: Phần liên quan |
|
2 |
Đường dùng |
Không thay đổi dạng bào chế |
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc văn ban cho phép của cơ quan có thẩm quyền nước sở tại) ) - yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần II (Chất lượng): Phần liên quan - Phần III & IV: Phần liên quan |
|
3 |
Thay đổi liều dùng |
|
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc công văn cho phép của cơ quan có thẩm quyền nước sở tại) - yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần III & IV: Phần liên quan |
|
4 |
Chỉ định |
Các nội dung khác không thay đổi |
- Phần I (hành chính): + Đơn (theo mẫu) + Giấy phép (CPP hoặc công văn cho phép của cơ quan có thẩm quyền nước sở tại) - yêu cầu đối với thuốc nước ngoài + Thông tin sản phẩm + Mẫu nhãn. - Phần III & IV: Phần liên quan |
|
5 |
Thay đổi nguồn dược liệu |
Thay đổi theo hướng nguồn dược liệu được kiểm soát chất lượng ở mức cao hơn |
- Đơn (theo mẫu) - Phần II: tài liệu chất lượng theo quy định tại khoản 1 Điều 29 Thông tư này; Phiếu kiểm nghiệm của thuốc thành phẩm; Nghiên cứu độ ổn định của thuốc thành phẩm theo quy định như đối với thuốc hóa dược thay đổi nguồn nguyên liệu. - Giấy tờ pháp lý của cơ sở sản xuất dược liệu, ban thành phẩm dược liệu đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) (thực hiện theo lộ trình quy định). Không yêu cầu tại liệu này đối với dược liệu, bán thành phẩm dược liệu đã có Giấy đăng ký lưu hành tại Việt Nam. |
II. THAY ĐỔI NHỎ
1- THAY ĐỔI NHỎ PHẢI ĐƯỢC PHÊ DUYỆT CỦA CƠ QUAN QUẢN LÝ
|
STT |
Nội dung thay đổi/bổ sung |
Điều kiện |
Yêu cầu hồ sơ |
|
1 |
Đổi địa điểm sản xuất/cơ sở đóng gói |
-Nhà sản xuất không thay đổi - Địa điểm sản xuất mới trong cùng một quốc gia với địa điểm cũ. |
Phần I (hành chính): - Đơn (theo mẫu) - Giấy phép (CPP hoặc giấy chứng nhận GMP). - Giấy chứng nhận đủ điều kiện sản xuất (đối với thuốc dược liệu sản xuất trong nước). Phần II (Chất lượng) |
|
2 |
Thay đổi hoặc bổ sung thành phần tá dược (bao gồm thay đổi tỷ lệ tá dược). |
- Không làm thay đổi và ảnh hưởng đến tiêu chuẩn, chất lượng của thuốc thành phẩm. |
Phần I (Hành chính): - Đơn (theo mẫu)- Phần II (chất lượng): (Có thể phải chứng minh bằng kết quả tương đương sinh học). - Giấy tờ pháp lý của cơ sở sản xuất tá dược, vỏ nang đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) (thực hiện theo lô trinh quy định. Không yêu cầu tài liệu này đối với tá dược đã có Giấy đăng ký lưu hành tại Việt Nam. |
|
3 |
Thay đổi cơ sở xuất xưởng lô |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (Chất lượng): phần thay đổi liên quan |
|
4 |
Thay đổi mô tả đặc tính của thành phẩm |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): phần thay đổi liên quan |
|
5 |
Thay đổi chất chuẩn để kiểm nghiệm thành phẩm |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): phần thay đổi liên quan |
|
6 |
Thay đổi hệ thống đóng kín của bao bì trực tiếp, gián tiếp |
- Chất lượng tốt hơn - ổn định hơn |
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): Phần thay đổi liên quan |
|
7 |
Thay đổi độ ổn định/hạn dùng của thành phẩm |
||
|
|
*Tăng hạn dùng |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
|
* Giảm hạn dùng |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (Chất lượng): Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định - Báo cáo số lượng thuốc đang lưu hành trên thị trường - Cam kết về việc cơ sở đăng ký phải phối hợp với cơ sở sản xuất, cơ sở nhập khẩu tiến hành thu hồi thuốc, nguyên liệu làm thuốc có hạn dùng lớn hơn hạn dùng thay đổi đối với thuốc đã lưu hành tại Việt Nam. |
|
8 |
Thay đổi điều kiện bảo quản của thành phẩm |
|
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (chất lượng): Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
9 |
Thay đổi qui trình sản xuất của thành phẩm: Sơ đồ, các bước, lô, mẻ, thẩm định qui trình… |
- Theo hướng cải tiến hơn qui trình cũ |
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (Chất lượng): Các tài liệu liên quan. |
|
10 |
Thay đổi tiêu chuẩn và/hoặc phương pháp kiểm nghiệm của thành phẩm (bao gồm cả thẩm định phương pháp phân tích) |
- Theo hướng chặt chẽ hơn |
Phần I (Hành chính): - Đơn (theo mẫu) Phần II (Chất lượng): Các tài liệu liên quan. |
|
11 |
Thay đổi/bổ sung quy cách đóng gói |
|
Phần I (Hành chính): - Đơn - Nhãn cũ đã duyệt + nhãn mới Phần II (Chất lượng): Gồm (1) Tiêu chuẩn bao bì (nếu có thay đổi bao bì, chất lượng bao bì) (2) Hồ sơ theo dõi độ ổn định của quy cách đóng gói mới (nếu có thay đổi bao bì sơ cấp). |
|
12 |
Thay đổi hình thức/ thiết kế bao bì, nhãn |
Theo hướng tốt hơn và nội dung nhãn không thay đổi |
Phần I (Hành chính): - Đơn - Nhãn cũ đã duyệt + nhãn mới Phần II (Chất lượng): Các tài liệu liên quan.. |
2. THAY ĐỔI/BỔ SUNG CẦN PHÊ DUYỆT NHƯNG KHÔNG DẪN ĐẾN THAY ĐỔI NHÃN VÀ HƯỚNG DẪN SỬ DỤNG THUỐC
|
Thay đổi/bổ sung cần phê duyệt nhưng không dẫn đến thay đổi thông tin trên nhãn và hướng dẫn sử dụng thuốc (*) |
|
|
Nội dung Thay đổi/bổ sung 1 |
Bổ sung, cập nhật thông tin để cung cấp thông tin thuốc, quảng cáo thuốc |
|
Điều kiện cần đáp ứng (C) |
1. Các thông tin được bổ sung, cập nhật theo quy định tại mục này là các thông tin mới được phát minh, phát hiện qua nghiên cứu khoa học hoặc qua theo dõi sản phẩm trên thị trường nhưng không thuộc trường hợp quy định tại mục từ 1 đến 7 Mục A Phụ lục này. 2. Trình bày toàn bộ các thông tin đề nghị theo đúng nội dung sẽ sử dụng để thông tin thuốc. |
|
Hồ sơ cần nộp (D) |
1. Nội dung thông tin đề nghị cập nhật. 2. Tài liệu tham khảo. 3. Bản sao tờ hướng dẫn sử dụng thuốc cập nhật nhất đã được Bộ Y tế phê duyệt có đóng dấu của cơ sở đăng ký thuốc. |
Chú giải:
1. Mục tiêu của việc thay đổi/bổ sung:
Các thông tin đề nghị bổ sung, cập nhật nhằm bổ trợ, làm rõ thêm cho thông tin đã có trong tờ hướng dẫn sử dụng thuốc. Các thông tin sau khi được bổ sung, cập nhật trong hồ sơ đăng ký thuốc là nguồn thông tin để sử dụng trong việc cung cấp thông tin thuốc, quảng cáo thuốc đảm bảo sử dụng thuốc an toàn, hợp lý, hiệu quả.
2. Các loại thông tin đề nghị thay đổi/bổ sung:
- Các thông số dược động học của thuốc trên một quần thể nghiên cứu cụ thể (bao gồm cả việc so sánh thông số dược động học giữa các thuốc).
- Các thông số về dược lực học: sơ đồ cơ chế tác dụng của thuốc; Các nghiên cứu về tính nhạy cảm của kháng sinh đối với các loại vi khuẩn tại một thời điểm cụ thể dựa trên một quần thể nghiên cứu cụ thể...
- Các kết quả cụ thể liên quan đến hiệu quả điều trị: kết quả so sánh hiệu quả điều trị giữa thuốc nghiên cứu (có thể đúng tên Biệt dược hoặc hoạt chất của thuốc đề nghị cập nhật) với giả dược hoặc với một hay nhiều thuốc khác trong cùng nhóm hoặc khác nhóm tác dụng dược lý.
- Các phác đồ hoặc hướng dẫn điều trị liên quan đến thuốc.
- Các kết quả thử tương đương sinh học.
- Các thông tin chi tiết hơn về tác dụng không mong muốn của thuốc đã ghi trong tờ Hướng dẫn sử dụng thuốc;
- Các kết quả nghiên cứu đánh giá mức độ cải thiện chất lượng cuộc sống của người bệnh, sự hài lòng của người bệnh, của bác sĩ đối với sự tiện dụng, dễ tuân thủ điều trị, hiệu quả điều trị...của thuốc.
- Các thông tin khác liên quan đến chất lượng, an toàn, hiệu quả của thuốc.
3. Cách thức trình bày nội dung thông tin:
- Thông tin đề nghị cập nhật phải được trình bày rõ ràng, đầy đủ, chính xác dựa trên bằng chứng, dễ hiểu, phù hợp với đối tượng được cung cấp thông tin;
- Trình bày toàn bộ các thông tin đề nghị chính xác như thông tin sẽ sử dụng để thông tin thuốc.
- Ghi rõ danh mục những Tài liệu tham khảo được sử dụng để xây dựng nội dung thông tin: đánh số thứ tự, ghi rõ tên Tài liệu tham khảo, tên bài nghiên cứu, tác giả, năm xuất bản Tài liệu.
- Trường hợp Tài liệu tham khảo là nghiên cứu lâm sàng thì có phần giới thiệu tóm tắt về nghiên cứu bao gồm các thông tin sau: mục tiêu, đối tượng nghiên cứu, phương pháp nghiên cứu, cỡ mẫu, các bước tiến hành, tiêu chí đánh giá, kết quả nghiên cứu.
- Trường hợp Tài liệu tham khảo là nghiên cứu cho hoạt chất thì thông tin trích dẫn ghi theo đúng dưới dạng tên hoạt chất (không được thay tên hoạt chất bằng tên biệt dược); Trường hợp Tài liệu tham khảo là nghiên cứu cho biệt dược thì thông tin trích dẫn ghi dưới dạng tên biệt dược.
- Ghi rõ số thứ tự Tài liệu tham khảo đằng sau các thông tin trích dẫn.
- Phiên giải đầy đủ kết quả nghiên cứu kèm số liệu, không được chỉ đưa ra các kết luận chung chung.
- Không ghi tên Tài liệu tham khảo, tên tổ chức thực hiện nghiên cứu, tên phương pháp nghiên cứu, tên tổ chức đưa ra phác đồ, hướng dẫn điều trị dưới dạng in thành tít to nổi bật.
4. Yêu cầu đối với Tài liệu tham khảo:
4.1. Các loại tài liệu tham khảo được sử dụng:
- Chuyên luận về thuốc đã được ghi trong phiên bản mới nhất của Dược thư Quốc gia, Martindale, AHFS,BNF, FDA, EMC…;
- Các bài báo nghiên cứu lâm sàng và các nghiên cứu khác liên quan đến thuốc được đăng tải trên các tạp chí thuộc danh mục SCI (Science Citation Index) - Chỉ số trích dẫn khoa học;
- Các phác đồ, hướng dẫn điều trị liên quan đến thuốc;
- Các kết quả nghiên cứu lâm sàng đã được nghiệm thu bởi cơ quan có thẩm quyền.
4.2. Tiêu chí đối với các tài liệu tham khảo:
- Tài liệu tham khảo phải là nguồn tài liệu rõ ràng, đáng tin cậy, là bản ghi đầy đủ, chi tiết, mang tính cập nhật, ghi rõ tên tài liệu, tên tác giả, thời gian xuất bản tài liệu;
- Trường hợp Tài liệu tham khảo là nghiên cứu lâm sàng thì có đầy đủ thông tin của một nghiên cứu lâm sàng (bao gồm: mục tiêu nghiên cứu, phương pháp nghiên cứu, cỡ mẫu, mô tả các bước tiến hành, kết quả, bàn luận, kết luận...).
- Không chấp nhận Tài liệu tham khảo là các nghiên cứu trên động vật, invitro.
- Ngôn ngữ của Tài liệu tham khảo: phải bằng tiếng Việt hoặc tiếng Anh. Nếu tài liệu tham khảo không phải là tiếng Việt hoặc tiếng Anh thì cần nộp kèm bản dịch công chứng bằng tiếng Việt có đóng dấu của đơn vị đăng ký.
- Chú thích rõ ràng các phần thông tin, dữ liệu trong tài liệu tham khảo được trích dẫn ra để cập nhật thông tin.
3- THAY ĐỔI NHỎ CHỈ YÊU CẦU THÔNG BÁO (NOTIFICATION)
|
TT |
Nội dung thay đổi/bổ sung |
Điều kiện |
Yêu cầu hồ sơ |
|
1 |
Thay đổi nội dung trên mẫu nhãn bao gồm tờ hướng dẫn sử dụng |
- Không thuộc các trường hợp thay đổi lớn và thay đổi nhỏ phải phê duyệt trước khi thực hiện. - Thay đổi thiết kế (màu sắc, hình dáng, kích thước, vị trí thông tin...) trên nhãn và, hoặc các nội dung không liên quan đến thông tin, hướng dẫn sử dụng thuốc. |
Phần I (Hành chính): - Thông báo - Nhãn cũ đã duyệt + nhãn mới - Bảng so sánh nội dung thay đổi - Giấy tờ pháp lý liên quan (nếu có) |
|
2 |
Thay đổi/bổ sung các nội dung về an toàn/hiệu quả (Trừ các trường hợp thuộc thay đổi lớn) |
Theo hướng sử dụng an toàn, hợp lý và hiệu quả hơn |
Phần I (Hành chính): - Thông báo - Nhãn - Thông tin sản phẩm - Tờ hướng dẫn sử dụng - Nêu rõ những điểm thay đổi. |
|
3 |
Thay đổi mô tả đặc tính của nguyên liệu |
|
Phần I (Hành chính): -Thông báo Phần II (Chất lượng): - Phần thay đổi liên quan |
|
4 |
Thay đổi chất chuẩn để kiểm nghiệm nguyên liệu |
|
Phần I (Hành chính): - Thông báo Phần II (Chất lượng): - Phần thay đổi liên quan |
|
5 |
Thay đổi độ ổn định/hạn dùng của nguyên liệu |
Không làm ảnh hưởng đến chất lượng của thành phẩm. |
Phần I (Hành chính): -Thông báo Phần II (Chất lượng): -Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
6 |
Thay đổi điều kiện bảo quản của nguyên liệu |
|
Phần I (Hành chính): - Thông báo Phần II (Chất lượng): - Phần thay đổi liên quan và theo hướng dẫn về nghiên cứu độ ổn định |
|
7 |
Thay đổi qui trình sản xuất của nguyên liệu: Sơ đồ, các bước, lô, mẻ, thẩm định qui trình…(chỉ áp dụng đối với thuốc thành phẩm hóa dược). |
- Theo hướng cải tiến hơn qui trình cũ. - Không làm thay đổi chất lượng và độ ổn định của nguyên liệu. |
Phần I (Hành chính): - Thông báo Phần II (Chất lượng): - Phần thay đổi liên quan. |
|
8 |
Thay đổi nhà cung cấp bao bì (thay thế, thêm vào hoặc loại bỏ) |
- Không làm thay đổi chất lượng và độ ổn định của thuốc |
Phần I (Hành chính): -Thông báo |
|
9 |
Thay đổi tiêu chuẩn và/ hoặc phương pháp kiểm nghiệm của nguyên liệu |
- Không làm thay đổi và ảnh hưởng đến tiêu chuẩn, chất lượng của thuốc thành phẩm. - Hoặc làm tiêu chuẩn chất lượng của thành phẩm chặt chẽ hơn hoặc tốt hơn. |
Phần I (Hành chính): - Thông báo Phần II (Chất lượng): - Phần thay đổi liên quan |
|
10 |
Thay thế dụng cụ đo lường thuốc (ví dụ từ muỗng sang cốc) |
|
Phần I (Hành chính): -Thông báo |
PHỤ LỤC III
CÁC TRƯỜNG HỢP YÊU CẦU THẨM ĐỊNH LẠI TIÊU CHUẨN CHẤT LƯỢNG VÀ PHƯƠNG PHÁP KIỂM NGHIỆM
1. Các số liệu thẩm định phương pháp chưa chứng minh tính khả thi của phương pháp phân tích.
2. Các số liệu thẩm định phương pháp chưa đủ thuyết phục để chứng minh tính đặc hiệu của phương pháp phân tích (chưa chứng minh được các phương pháp phân tích (định tính, định lượng, độ tinh khiết, tạp chất...) áp dụng đối với một dược chất trong công thức không bị ảnh hưởng bởi các dược chất khác cũng như các thành phần tá dược khác có mặt trong công thức).
3. Các phương pháp phân tích mới hoặc các kỹ thuật phân tích mới được dùng để đánh giá chất lượng của một dạng bào chế chưa được áp dụng tại Việt Nam.
4. Các phương pháp phân tích mới hay các kỹ thuật phân tích mới đối với một hoạt chất chưa được áp dụng tại Việt Nam.
PHỤ LỤC IV
HƯỚNG DẪN VỀ PHÁT TRIỂN LÂM SÀNG THUỐC PHỐI HỢP CỐ ĐỊNH LIỀU, SINH PHẨM TƯƠNG TỰ
Hướng dẫn về phát triển lâm sàng thực hiện theo hướng dẫn của Bộ Y tế Việt Nam, của các tổ chức quốc tế mà Việt Nam là thành viên (WHO, ASEAN), ICH hoặc cơ quan quản lý tham chiếu theo quy định của Thông tư này. Cụ thể theo các nguồn tham khảo sau:
I. Hướng dẫn về phát triển lâm sàng thuốc phối hợp cố định liều:
1. Tổ chức Y tế Thế giới (WHO): Hướng dẫn đăng ký sản phẩm thuốc phối hợp cố định liều (2005):
http://apps.who.int/medicinedocs/en/d/Js19979en/
2. Cơ quan Quản lý Dược phẩm Châu Âu (EMA): Hướng dẫn phát triển lâm sàng của thuốc phối hợp cố định liều (2017):
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/03/WC500224836.pdf
3. Cơ quan Quản lý Dược phẩm, Thực phẩm Mỹ (US-FDA):
- Hướng dẫn về phát triển thuốc Phối hợp cố định liều để điều trị tăng huyết áp (2018):
http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm593825.pdf
- Hướng dẫn về Phối hợp cố định liều, các sản phẩm thuốc đồng đóng gói và các phiên bản đơn lẻ của thuốc kháng vi-rút được cấp phép trước đây để điều trị HIV (2006): http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079742.pdf
II. Hướng dẫn về phát triển lâm sàng sinh phẩm tương tự:
1. Tổ chức Y tế Thế giới (WHO): Hướng dẫn về đánh giá sản phẩm sinh học tương tự (2009):
http://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf
2. Cơ quan Quản lý Dược phẩm Châu Âu (EMA):
Các hướng dẫn chung và chuyên biệt về các sản phẩm thuốc sinh học tương tự:
https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-biosimilar
3. Cơ quan Quản lý Dược phẩm, Thực phẩm Mỹ (US-FDA):
Hướng dẫn về các vấn đề khoa học trong việc chứng minh tương tự với sinh phẩm tham chiếu (2015):
https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
Trường hợp có hướng dẫn cụ thể đối với từng loại thuốc, áp dụng theo các hướng dẫn chung nêu trên và từng hướng dẫn cụ thể. Trường hợp các hướng dẫn nêu trên có bản cập nhật, áp dụng theo bản cập nhật của các hướng dẫn này. Chấp nhận các hướng dẫn được xây dựng trên cơ sở các hướng dẫn nêu trên.
PHỤ LỤC V
HỒ SƠ AN TOÀN, HIỆU QUẢ THUỐC DƯỢC LIỆU
Yêu cầu về cấu trúc của hồ sơ an toàn, hiệu quả thuốc dược liệu bao gồm các phần như sau:
Phần I. Hồ sơ tiền lâm sàng
1. Mục lục của hồ sơ tiền lâm sàng
2. Tóm tắt nghiên cứu tiền lâm sàng
2.1. Mở đầu
2.2. Tóm tắt về dược lý học bằng văn bản và bằng bảng biểu
2.3. Tóm tắt về dược động học (nếu có) bằng văn bản và bằng bảng biểu
2.4. Tóm tắt về độc tính bằng văn bản và bằng bảng biểu
3. Báo cáo nghiên cứu tiền lâm sàng
4. Danh mục tài liệu tham khảo chính
Nếu cần thêm thông tin chi tiết, tham khảo hướng dẫn hồ sơ Tiền lâm sàng theo ASEAN (ACTD) hoặc ICH - CTD.
Phần II. Hồ sơ lâm sàng
1. Mục lục của hồ sơ lâm sàng
2. Tổng quan lâm sàng: Cung cấp thông tin về cơ sở phát triển sản phẩm, chương trình phát triển lâm sàng, các hướng dẫn về phát triển thuốc hoặc các quy định mà chương trình phát triển lâm sàng tuân thủ.
3. Tóm tắt lâm sàng
3.1. Tóm tắt về hiệu quả lâm sàng
3.1.1. Cơ sở nghiên cứu và tổng quan về hiệu quả lâm sàng
3.1.2. Tóm tắt kết quả các nghiên cứu riêng lẻ
3.1.3. So sánh và phân tích các kết quả xuyên suốt các nghiên cứu
3.1.4. Phân tích các thông tin lâm sàng liên quan đến các khuyến cáo về liều dùng
3.1.5. Sự duy trì hiệu quả và/hoặc sự quen thuốc
3.2. Tóm tắt về tính an toàn lâm sàng
3.2.1. Mức độ sử dụng thuốc
3.2.2. Biến cố ngoại ý
3.2.3. Đánh giá kết quả xét nghiệm
3.2.4. Dấu hiệu sinh tồn, triệu chứng thực thể và các ghi nhận khác liên quan đến sự an toàn
3.2.5. Sự an toàn đối với các nhóm dân số đặc biệt và tình huống đặc biệt
3.2.6. Các dữ liệu sau khi đưa thuốc ra thị trường (nếu có)
3.3. Bảng tóm tắt các nghiên cứu riêng lẻ
4. Báo cáo nghiên cứu lâm sàng
Nếu cần thêm thông tin chi tiết, tham khảo hướng dẫn hồ sơ lâm sàng theo ASEAN (ACTD) hoặc ICH - CTD.
MẪU 1/TT
ĐƠN ĐỀ NGHỊ THU HỒI GIẤY ĐĂNG KÝ LƯU HÀNH THUỐC, NGUYÊN LIỆU LÀM THUỐC
|
Kính gửi: |
Cục Quản lý Dược |
|
Tên cơ sở đăng ký: Địa chỉ: Điện thoại: |
Tên cơ sở sản xuất: Địa chỉ: Điện thoại: |
Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài) Địa chỉ: Điện thoại: |
(Cơ sở) đề nghị Cục Quản lý Dược xem xét giải quyết việc rút giấy đăng ký lưu hành đối với thuốc/nguyên liệu làm thuốc sau:
|
Tên thuốc/nguyên liệu làm thuốc: |
Hoạt chất, nồng độ/hàm lượng: |
|
|
Số đăng ký (SĐK): |
Ngày cấp SĐK: |
Ngày hết hạn SĐK: |
Lý do đề nghị rút giấy đăng ký lưu hành: Ghi cụ thể lý do và kèm theo tài liệu (nếu có)
Cơ sở xin rút giấy đăng ký lưu hành cam kết thực hiện đúng các quy định và chịu hoàn toàn trách nhiệm trước pháp luật đối với đề nghị rút giấy đăng ký lưu hành thuốc/nguyên liệu làm thuốc nêu trên.
|
|
Ngày... tháng... năm.....
|
Ghi chú: (*) Người ký đơn theo quy định tại khoản 5 Điều 23 Thông tư này.
MẪU 2A/TT
BÁO CÁO THEO DÕI, ĐÁNH GIÁ AN TOÀN, HIỆU QUẢ CỦA THUỐC
|
Kính gửi: |
|
Thực hiện theo yêu cầu về việc báo cáo theo dõi, đánh giá an toàn, hiệu quả của thuốc trong quá trình lưu hành và khi đăng ký gia hạn đối với những thuốc có yêu cầu báo cáo an toàn, hiệu quả, cơ sở…. báo cáo như sau:
|
1. |
Tên cơ sở đăng ký |
Địa chỉ: |
Điện thoại: |
|
2. |
Tên cơ sở sản xuất: |
Địa chỉ: |
Điện thoại: |
3. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài):
Địa chỉ: Điện thoại:
4. Tên thuốc:
5. Hoạt chất, nồng độ/hàm lượng:
6. Dạng bào chế:
7. Số đăng ký (SĐK): Ngày cấp SĐK: Ngày hết hạn SĐK:
8. Thông tin về an toàn, hiệu quả và sử dụng của thuốc:
8.1. Báo cáo phản ứng có hại của thuốc sau khi đưa thuốc ra lưu hành trên thị trường Việt Nam:
Bảng tổng kết các báo cáo phản ứng có hại của thuốc đã gửi về Trung tâm Quốc gia hoặc Trung tâm khu vực về thông tin thuốc và theo dõi phản ứng có hại của thuốc liên quan đến tác dụng không mong muốn của thuốc sau khi đưa thuốc ra lưu hành trên thị trường Việt Nam (kèm theo bản sao các báo cáo) hoặc thông tin về báo cáo phản ứng có hại của thuốc được ghi nhận từ chương trình giám sát sau khi thuốc được phê duyệt được cơ sở kinh doanh thuốc thực hiện.
8.2. Tình hình sử dụng thuốc tại các cơ sở khám, chữa bệnh trên phạm vi cả nước:
Bảng tổng kết tình hình sử dụng thuốc tại các cơ sở khám, chữa bệnh trên phạm vi cả nước (số lượng thuốc sử dụng tại từng cơ sở khám, chữa bệnh).
8.3. Tổng kết các cập nhật thông tin về an toàn, hiệu quả của thuốc đã thực hiện trong quá trình lưu hành:
Báo cáo tổng hợp các nội dung cập nhật đã được Cục Quản lý Dược phê duyệt, các cập nhật có tính thông báo, các cập nhật theo công văn hướng dẫn của Cục Quản lý Dược (nếu có) liên quan đến thay đổi thông tin trong Tờ hướng dẫn sử dụng thuốc.
8.4. Tóm tắt kết quả của các nghiên cứu lâm sàng được thực hiện tại Việt Nam liên quan đến thuốc (nếu có).
9. Đánh giá lợi ích và nguy cơ liên quan đến thuốc trong quá trình lưu hành; đề xuất liên quan đến việc lưu hành sản phẩm đăng ký:
Cơ sở đăng ký cam kết: những nội dung báo cáo là đúng sự thật, nếu không đúng cơ sở xin hoàn toàn chịu trách nhiệm./.
|
|
Ngày..... tháng..... năm.....
|
(*): Báo cáo theo quy định tại điểm b khoản 2 Điều 5 Thông tư này.
(**): Báo cáo theo quy định tại điểm a khoản 2 Điều 5 Thông tư này.
MẪU 2B/TT
BÁO CÁO THEO DÕI, ĐÁNH GIÁ AN TOÀN, HIỆU QUẢ CỦA VẮC XIN
|
Kính gửi: |
|
Thực hiện theo yêu cầu về việc báo cáo theo dõi, đánh giá an toàn, hiệu quả của vắc xin trong quá trình lưu hành và khi đăng ký gia hạn đối với những vắc xin có yêu cầu báo cáo an toàn, hiệu quả, cơ sở…. báo cáo như sau:
1. Tên cơ sở đăng ký Địa chỉ: Điện thoại:
2. Tên cơ sở sản xuất: Địa chỉ: Điện thoại:
3. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài)
Địa chỉ: Điện thoại:
4. Tên vắc xin:
5. Hoạt chất, nồng độ/hàm lượng:
6. Dạng bào chế:
7. Số đăng ký (SĐK): Ngày cấp SĐK: Ngày hết hạn SĐK:
8. Bảng tổng kết các báo cáo phản ứng có hại của vắc xin ghi nhận được trong quá trình lưu hành vắc xin trên thị trường Việt Nam
Thống kê các trường hợp phản ứng có hại đã được ghi nhận (phản ứng sau tiêm nhẹ, nặng) trong quá trình lưu hành.
9. Bảng tổng kết cập nhật thông tin về an toàn, hiệu quả của vắc xin đã thực hiện trong quá trình lưu hành
Các cập nhật đã được Cục Quản lý Dược phê duyệt; các cập nhật có tính thông báo; các cập nhật theo công văn hướng dẫn của Cục Quản lý Dược (nếu có).
10. Bảng tóm tắt kết quả các nghiên cứu lâm sàng tiến hành tại Việt Nam (nếu có).
11. Đánh giá lợi ích của vắc xin và nguy cơ liên quan đến vắc xin trong quá trình lưu hành; đề xuất liên quan đến việc lưu hành sản phẩm đăng ký:
Cơ sở đăng ký cam kết: những nội dung báo cáo là đúng sự thật, nếu không đúng cơ sở xin hoàn toàn chịu trách nhiệm./.
|
|
Ngày..... tháng..... năm.....
|
(*): Báo cáo theo quy định tại điểm b khoản 2 Điều 5 Thông tư này.
(**): Báo cáo theo quy định tại điểm a khoản 2 Điều 5 Thông tư này.
MẪU 2C/TT
BÁO CÁO TÌNH HÌNH SỬ DỤNG THUỐC
|
Tên cơ sở khám bệnh, |
CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
|
Số………….. |
….., ngày tháng năm |
|
Kính gửi: |
Trung tâm Quốc gia về Thông tin thuốc và |
Thực hiện theo yêu cầu về việc báo cáo theo dõi, đánh giá an toàn, hiệu quả của thuốc trong quá trình lưu hành đối với những thuốc có yêu cầu báo cáo an toàn, hiệu quả, cơ sở…. báo cáo như sau:
1. Tên thuốc:
2. Hoạt chất, nồng độ/hàm lượng:
3. Dạng bào chế:
4. Số đăng ký:
5. Số lượng thuốc đã sử dụng:
6. Số bệnh nhân đã sử dụng thuốc:
7. Thời gian sử dụng:
8. Đánh giá an toàn, hiệu quả của thuốc đã sử dụng (có số liệu kèm theo):
9. Phản ứng có hại của thuốc (ADR): các biểu hiện ADR, số trường hợp, kết quả xử lý ADR (có số liệu kèm theo):
10. Kiến nghị, đề xuất (ghi rõ có tiếp tục sử dụng thuốc tại cơ sở khám bệnh, chữa bệnh hay không?):
(Cơ sở khám bệnh, chữa bệnh) cam kết và chịu trách nhiệm về các nội dung báo cáo nêu trên./.
|
|
Giám đốc/Phó Giám đốc |
(*): Báo cáo theo quy định tại khoản 3 Điều 5 Thông tư này.
MẪU 3/TT
TRANG BÌA
HỒ SƠ ĐĂNG KÝ THUỐC, NGUYÊN LIỆU LÀM THUỐC
1. Tên và địa chỉ cơ sở đăng ký:
2. Tên và địa chỉ cơ sở sản xuất:
3. Tên thuốc, nguyên liệu làm thuốc, nồng độ/hàm lượng, dạng bào chế:
4. Loại thuốc, nguyên liệu làm thuốc đăng ký:
Yêu cầu ghi cụ thể: Thuốc hóa dược/thuốc phóng xạ/vắc xin/sinh phẩm/thuốc dược liệu/nguyên liệu làm thuốc (dược chất/tá dược/vỏ nang/bán thành phẩm dược liệu).
5. Loại hình đăng ký:
Yêu cầu ghi cụ thể: Đăng ký lần đầu/Đăng ký gia hạn/ Đăng ký chuyển giao công nghệ /Đăng ký thay đổi lớn/Đăng ký thay đổi nhỏ cần phê duyệt/Đăng ký thay đổi nhỏ chỉ yêu cầu thông báo/Công bố biệt dược gốc/Công bố tương đương sinh học/Cập nhật thông tin thuốc.
|
|
Mã HS: |
MẪU 4A/TT
THÔNG TIN SẢN PHẨM
(Đăng ký lần đầu)
|
Quy trình thẩm định nhanh |
□ |
Đề nghị công bố biệt dược gốc |
□ |
|
Quy trình thẩm định rút gọn |
□ |
Đề nghị công bố TĐSH |
□ |
|
Chuyển giao công nghệ |
□ |
Đề nghị bảo mật dữ liệu |
□ |
|
Có hồ sơ TĐSH |
□ |
Có hồ sơ lâm sàng |
□ |
|
Tên thuốc/tên nguyên liệu làm thuốc: |
Tên generic: |
||
|
Dạng bào chế: |
Nồng độ/hàm lượng: |
||
|
Tên cơ sở đăng ký: Địa chỉ : Điện thoại : |
Tên cơ sở sản xuất: (Liệt kê đầy đủ các cơ sở tham gia trong quá trình sản xuất và nêu rõ vai trò của từng cơ sở sản xuất) Địa chỉ : Điện thoại : |
||
|
Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài) Địa chỉ: Điện thoại: |
|
||
|
Tên, địa chỉ chủ sở hữu giấy phép lưu hành sản phẩm (Product License Holder) hoặc chủ sở hữu sản phẩm (Product Owner) ghi trên CPP (đối với thuốc nước ngoài):
|
|||
|
Điều kiện bảo quản: |
Hạn dùng: |
|
|
|
Phân loại thuốc (tích vào nội dung phù hợp): - Thuốc kê đơn: - Thuốc không kê đơn: - Thuốc độc: - Thuốc phóng xạ: - Thuốc dược liệu: - Thuốc gây nghiện: - Thuốc thành phẩm dạng phối hợp có chứa hoạt chất gây nghiện: - Thuốc hướng tâm thần: - Thuốc thành phẩm dạng phối hợp có chứa hoạt chất hướng tâm thần: - Tiền chất dùng làm thuốc: - Thuốc thành phẩm dạng phối hợp có chứa tiền chất dùng làm thuốc: - Nguyên liệu làm thuốc: - Mã ATC: |
Đường dùng: |
Tiêu chuẩn (3): |
|
Công thức bào chế (cho một đơn vị liều hoặc đơn vị đóng gói nhỏ nhất)
Thành phần:
|
Hoạt chất (1) |
Hàm lượng (2) |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
|
Tá dược |
Hàm lượng |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
Quy cách đóng gói :
Đề nghị khác (nếu có, đề nghị ghi rõ):
Ghi chú
(1) Ghi chính xác dạng dùng của dược chất (muối ester/các dạng dẫn chất khác).
(2) Nếu liều dùng tính theo gốc có tác dụng dược lý của dược chất (gốc base...), cần bổ sung thêm thông tin về hàm lượng dược chất được quy đổi ra gốc có tác dụng dược lý này.
Nếu dược chất được sử dụng dưới dạng bán thành phẩm đã trộn thêm tá dược, phải ghi đầy đủ cả các thành phần tá dược có trong công thức bào chế các bán thành phẩm có chứa dược chất này.
(3) Nếu là tiêu chuẩn dược điển, đề nghị ghi rõ tên dược điển và phiên bản dược điển hoặc năm phát hành dược điển hoặc ghi theo “dược điển phiên bản hiện hành”.
|
|
Mã HS: |
MẪU 4B /TT
THÔNG TIN SẢN PHẨM
(Đăng ký gia hạn)
|
Tên thuốc/tên nguyên liệu làm thuốc |
Tên generic: |
|
|
Dạng bào chế: |
Nồng độ/hàm lượng: |
|
|
Tên cơ sở đăng ký: Địa chỉ: Điện thoại: |
Tên cơ sở sản xuất: (Liệt kê đầy đủ các cơ sở tham gia trong quá trình sản xuất và nêu rõ vai trò của từng cơ sở sản xuất) Địa chỉ: Điện thoại: |
|
|
Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài) Địa chỉ: Điện thoại: |
|
|
|
Tên, địa chỉ chủ sở hữu giấy phép lưu hành sản phẩm (Product License Holder) hoặc chủ sở hữu sản phẩm (Product Owner) ghi trên CPP (đối với thuốc nước ngoài): |
||
|
Điều kiện bảo quản: |
Hạn dùng: |
|
|
Số đăng ký: |
Ngày cấp SĐK: |
Ngày hết hạn SĐK: |
|
Phân loại thuốc (tích vào nội dung phù hợp): - Thuốc kê đơn: - Thuốc không kê đơn: - Thuốc độc: - Thuốc phóng xạ: - Thuốc dược liệu: - Thuốc gây nghiện: - Thuốc thành phẩm dạng phối hợp có chứa hoạt chất gây nghiện: - Thuốc hướng tâm thần: - Thuốc thành phẩm dạng phối hợp có chứa hoạt chất hướng tâm thần: - Tiền chất dùng làm thuốc: - Thuốc thành phẩm dạng phối hợp có chứa tiền chất dùng làm thuốc: - Nguyên liệu làm thuốc: - Mã ATC: |
Đường dùng: |
Tiêu chuẩn (3): |
Công thức bào chế (cho một đơn vị liều hoặc đơn vị đóng gói nhỏ nhất)
Thành phần:
|
Hoạt chất (1) |
Hàm lượng (2) |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
|
Tá dược |
Hàm lượng |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
Quy cách đóng gói:
Đề nghị khác (nếu có, đề nghị ghi rõ):
Ghi chú
(1) Ghi chính xác dạng dùng của dược chất (muối ester/các dạng dẫn chất khác).
(2) Nếu liều dùng tính theo gốc có tác dụng dược lý của dược chất (gốc base...), cần bổ sung thêm thông tin về hàm lượng dược chất được quy đổi ra gốc có tác dụng dược lý này.
Nếu dược chất được sử dụng dưới dạng bán thành phẩm đã trộn thêm tá dược, phải ghi đầy đủ cả các thành phần tá dược có trong công thức bào chế các bán thành phẩm có chứa dược chất này.
(3) Nếu là tiêu chuẩn dược điển, đề nghị ghi rõ tên dược điển và số phiên bản dược điển hoặc năm phát hành dược điển hoặc ghi theo “dược điển phiên bản hiện hành”.
MẪU 5/TT
MỤC LỤC
1. Tài liệu hành chính
1.1.
1.2.
...
2. Tài liệu chất lượng
2.1.
2.2.
...
3. Tài liệu tiền lâm sàng
3.1.
3.2.
...
4. Tài liệu lâm sàng
4.1.
4.2.
...
5. Tài liệu khác (nếu có)
5.1.
5.2.
...
MẪU 6A/ TT
ĐƠN ĐĂNG KÝ THUỐC, NGUYÊN LIỆU LÀM THUỐC
(Đăng ký lần đầu)
A. Chi tiết về cơ sở đăng ký và cơ sở sản xuất
1. Cơ sở đăng ký
1.1. Tên cơ sở đăng ký:
1.2. Địa chỉ: Website (nếu có):
1.3. Điện thoại: Email:
1.4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài):
Địa chỉ:
Điện thoại:
2. Cơ sở sản xuất (1)
2.1. Tên cơ sở sản xuất:
2.2. Địa chỉ: Website (nếu có):
2.3. Điện thoại: Email:
Các cơ sở sản xuất khác (nếu có) (2):
|
Tên và địa chỉ |
Vai trò (2) |
|
|
|
|
|
|
(1) Cơ sở sản xuất cuối cùng chịu trách nhiệm xuất xưởng lô thuốc
(2) Cơ sở tham gia trong quá trình sản xuất và nêu rõ vai trò của từng cơ sở sản xuất như “sản xuất bán thành phẩm”, “đóng gói sơ cấp”, “đóng gói thứ cấp”, “làm cốm”,…
B. Chi tiết về sản phẩm
1. Tên thuốc/nguyên liệu làm thuốc:
2. Hoạt chất, nồng độ/hàm lượng:
3. Dạng bào chế:
4. Mô tả dạng bào chế:
5. Đường dùng:
6. Tiêu chuẩn chất lượng (3):
7. Hạn dùng:
8. Điều kiện bảo quản:
9. Mô tả quy cách đóng gói:
10. Phân loại (tích vào nội dung phù hợp):
|
Thuốc kê đơn |
|
|
Thuốc không kê đơn |
|
|
Thuốc hướng tâm thần |
|
|
Thuốc thành phẩm dạng phối hợp có chứa hoạt chất hướng tâm thần |
|
|
Thuốc gây nghiện |
|
|
Thuốc thành phẩm dạng phối hợp có chứa hoạt chất gây nghiện |
|
|
Tiền chất dùng làm thuốc |
|
|
Thuốc thành phẩm dạng phối hợp có chứa tiền chất dùng làm thuốc |
|
|
Nguyên liệu làm thuốc |
|
|
Thuốc độc |
|
|
Mã ATC |
|
|
Thuốc phóng xạ |
|
|
|
|
|
Thuốc dược liệu |
|
11. Công thức bào chế (cho một đơn vị liều hoặc đơn vị đóng gói nhỏ nhất)
Thành phần:
|
Hoạt chất (1) |
Nồng độ/ hàm lượng (2) |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
|
|
|
|
|
|
Tá dược |
Nồng độ/ hàm lượng |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
(1) Ghi chính xác dạng dùng của dược chất (muối ester/các dạng dẫn chất khác).
(2) Nếu liều dùng tính theo gốc có tác dụng dược lý của dược chất (gốc base...), cần bổ sung thêm thông tin về hàm lượng dược chất được quy đổi ra gốc có tác dụng dược lý này.
Nếu dược chất được sử dụng dưới dạng bán thành phẩm đã trộn thêm tá dược, phải ghi đầy đủ cả các thành phần tá dược có trong công thức bào chế các bán thành phẩm có chứa dược chất này.
(3) Nếu là tiêu chuẩn dược điển, đề nghị ghi rõ tên dược điển và phiên bản dược điển hoặc năm phát hành dược điển hoặc ghi theo “dược điển phiên bản hiện hành”.
C. Tài liệu kỹ thuật
1. Phần I: Hành chính
2. Phần II: Chất lượng
3. Phần III: Tiền lâm sàng
4. Phần IV: Lâm sàng
Ghi chú: Những tài liệu (Phần I, II, III, IV) phải nộp tùy thuộc vào phân loại sản phẩm/nhóm sản phẩm.
D. Các đề nghị đặc biệt đối với thuốc đăng ký
Thuốc có dữ liệu yêu cầu bảo mật □
Cơ sở đăng ký thuốc đề nghị Cục Quản lý Dược xem xét thực hiện bảo mật đối với các dữ liệu sau đây được nộp kèm theo hồ sơ đăng ký thuốc:
Dữ liệu thử nghiệm độc tính (Tài liệu số .... )
Dữ liệu thử thuốc trên lâm sàng (Tài liệu số .... )
Cơ sở đăng ký thuốc xin cam kết các dữ liệu nêu trên đáp ứng đầy đủ các điều kiện bảo mật dữ liệu theo quy định của pháp luật và cơ sở đăng ký thuốc sẽ thực hiện nghĩa vụ chứng minh khi được cơ quan có thẩm quyền yêu cầu.
Đ. Các nội dung khác (tích vào nội dung phù hợp)
|
Hồ sơ đề nghị theo quy trình thẩm định nhanh |
|
|
Hồ sơ đề nghị theo quy trình thẩm định rút gọn |
|
|
Có hồ sơ tương đương sinh học |
|
|
Có hồ sơ lâm sàng |
|
|
Thuốc đề nghị công bố biệt dược gốc |
|
|
Thuốc đề nghị công bố tương đương sinh học |
|
|
Đề nghị khác (nếu có, đề nghị ghi chi tiết) |
|
E. Tuyên bố của cơ sở đăng ký
Cơ sở đăng ký cam kết:
1. Thực hiện việc cập nhật, bổ sung tờ hướng dẫn sử dụng thuốc theo thuốc biệt dược gốc, sinh phẩm tham chiếu đối với trường hợp quy định tại khoản 3 Điều 38 Thông tư 01/2018/TT-BYT ngày 18/1/2018 của Bộ Y tế quy định ghi nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc (áp dụng đối với thuốc generic, sinh phẩm tương tự).
2. Đã kiểm tra, ký đóng dấu theo quy định và xác nhận là đây là các giấy tờ hợp pháp, nội dung là đúng sự thật. Nếu có sự giả mạo, không đúng sự thật cơ sở đăng ký xin chịu hoàn toàn trách nhiệm và sẽ bị xử phạt theo quy định của pháp luật.
3. Đảm bảo thuốc/nguyên liệu làm thuốc được sản xuất theo đúng hồ sơ đăng ký đã nộp.
4. Thuốc nhập khẩu khi đăng ký lưu hành tại Việt Nam có cùng tiêu chuẩn thành phẩm; tiêu chuẩn dược chất, dược liệu; tên, địa chỉ cơ sở sản xuất dược chất, dược liệu với thuốc lưu hành tại nước sở tại thể hiện trên CPP.
5. Đăng ký thay đổi, bổ sung theo quy định sau khi thuốc/nguyên liệu làm thuốc đã được cấp giấy đăng ký lưu hành.
6. Chịu trách nhiệm hoàn toàn về sở hữu trí tuệ liên quan đến thuốc/nguyên liệu làm thuốc đăng ký.
|
|
Ngày... tháng... năm..... |
MẪU 6B/TT
ĐƠN ĐĂNG KÝ THUỐC, NGUYÊN LIỆU LÀM THUỐC
(Đăng ký gia hạn)
A. Chi tiết về cơ sở đăng ký và cơ sở sản xuất
1. Cơ sở đăng ký
1.1. Tên cơ sở đăng ký:
1.2. Địa chỉ: Website (nếu có):
1.3. Điện thoại: Email:
1.4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài):
Địa chỉ:
Điện thoại:
2. Cơ sở sản xuất (1)
2.1. Tên cơ sở sản xuất:
2.2. Địa chỉ: Website (nếu có):
2.3. Điện thoại: Email:
Các cơ sở sản xuất khác (nếu có) (2):
|
Tên và địa chỉ |
Vai trò (2) |
|
|
|
|
|
|
(1) Cơ sở sản xuất cuối cùng chịu trách nhiệm xuất xưởng lô thuốc
(2) Cơ sở tham gia trong quá trình sản xuất và nêu rõ vai trò của từng cơ sở sản xuất như “sản xuất bán thành phẩm”, “đóng gói sơ cấp”, “đóng gói thứ cấp”, “làm cốm”,…
B. Chi tiết về sản phẩm
1. Tên thuốc/nguyên liệu làm thuốc:
2. Số đăng ký: Ngày cấp: Ngày hết hạn:
3. Hoạt chất, nồng độ/hàm lượng:
4. Dạng bào chế:
5. Mô tả dạng bào chế:
6. Đường dùng:
7. Tiêu chuẩn chất lượng (3):
8. Hạn dùng:
9. Điều kiện bảo quản:
10. Mô tả quy cách đóng gói:
11. Phân loại (tích vào nội dung phù hợp):
|
Thuốc kê đơn |
|
|
Thuốc không kê đơn |
|
|
Thuốc hướng tâm thần |
|
|
Thuốc thành phẩm dạng phối hợp có chứa hoạt chất hướng tâm thần |
|
|
Thuốc gây nghiện |
|
|
Thuốc thành phẩm dạng phối hợp có chứa hoạt chất gây nghiện |
|
|
Tiền chất dùng làm thuốc |
|
|
Thuốc thành phẩm dạng phối hợp có chứa tiền chất dùng làm thuốc |
|
|
Thuốc phóng xạ |
|
|
Thuốc độc |
|
|
Nguyên liệu làm thuốc |
|
|
Thuốc dược liệu |
|
|
Mã ATC |
|
|
|
|
12. Công thức bào chế (cho một đơn vị liều hoặc đơn vị đóng gói nhỏ nhất)
Thành phần:
|
Hoạt chất (1) |
Nồng độ/ hàm lượng (2) |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
|
|
|
|
|
|
Tá dược |
Nồng độ/ hàm lượng |
Cơ sở sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
(1) Ghi chính xác dạng dùng của dược chất (muối ester/các dạng dẫn chất khác).
(2) Nếu liều dùng tính theo gốc có tác dụng dược lý của dược chất (gốc base...), cần bổ sung thêm thông tin về hàm lượng dược chất được quy đổi ra gốc có tác dụng dược lý này.
Nếu dược chất được sử dụng dưới dạng bán thành phẩm đã trộn thêm tá dược, phải ghi đầy đủ cả các thành phần tá dược có trong công thức bào chế các bán thành phẩm có chứa dược chất này.
(3) Nếu là tiêu chuẩn dược điển, đề nghị ghi rõ tên dược điển và phiên bản dược điển hoặc năm phát hành dược điển hoặc ghi theo “dược điển phiên bản hiện hành”.
C. Thông tin quá trình lưu hành
1. Liệt kê các nội dung thay đổi, bổ sung đã được phê duyệt trong quá trình lưu hành (kể từ khi cấp SĐK/gia hạn SĐK gần nhất đến thời điểm nộp hồ sơ gia hạn SĐK, kèm bản sao công văn phê duyệt của Cục Quản lý Dược).
2. Liệt kê các nội dung thay đổi, bổ sung đã nộp nhưng chưa được phê duyệt trước thời điểm nộp hồ sơ gia hạn SĐK (nếu có).
3. Liệt kê các nội dung thay đổi, bổ sung về hồ sơ hành chính trong hồ sơ gia hạn SĐK (nếu có).
4. Thuốc đã được công bố biệt dược gốc (nếu có, ghi cụ thể).
5. Thuốc đã được công bố tương đương sinh học (nếu có, ghi cụ thể).
6. Thuốc/nguyên liệu làm thuốc đã cập nhật tiêu chuẩn chất lượng theo quy định (ghi cụ thể).
D. Tài liệu kèm theo quy định
Đ. Tuyên bố của cơ sở đăng ký
Cơ sở đăng ký cam kết:
1. Thực hiện việc cập nhật, bổ sung tờ hướng dẫn sử dụng thuốc theo thuốc biệt dược gốc, sinh phẩm tham chiếu đối với trường hợp quy định tại khoản 3 Điều 38 Thông tư 01/2018/TT-BYT ngày 18/1/2018 của Bộ Y tế quy định ghi nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc (áp dụng đối với thuốc generic, sinh phẩm tương tự).
2. Đã kiểm tra, ký đóng dấu theo quy định và xác nhận là đây là các giấy tờ hợp pháp, nội dung là đúng sự thật. Nếu có sự giả mạo, không đúng sự thật cơ sở đăng ký xin chịu hoàn toàn trách nhiệm và sẽ bị xử phạt theo quy định của pháp luật.
3. Đảm bảo thuốc/nguyên liệu làm thuốc được sản xuất thuốc theo đúng hồ sơ đăng ký đã nộp.
4. Thuốc nhập khẩu khi đăng ký lưu hành tại Việt Nam có cùng tiêu chuẩn thành phẩm; tiêu chuẩn dược chất, dược liệu; tên, địa chỉ cơ sở sản xuất dược chất, dược liệu với thuốc lưu hành tại nước sở tại thể hiện trên CPP.
5. Đăng ký thay đổi, bổ sung theo quy định sau khi thuốc/nguyên liệu làm thuốc đã được gia hạn giấy đăng ký lưu hành.
6. Chịu trách nhiện hoàn toàn về sở hữu trí tuệ liên quan đến thuốc/nguyên liệu làm thuốc đăng ký.
|
|
Ngày... tháng... năm..... |
Đơn đăng ký gia hạn (Mẫu 6B/TT) ban hành kèm theo Thông tư này được sửa đổi, bổ sung bởi Phụ lục 03 ban hành kèm theo Thông tư số 23/2021/TT-BYT theo quy định tại Điểm i Khoản 3 Điều 1.
MẪU 6C/TT
ĐƠN ĐĂNG KÝ THAY ĐỔI, BỔ SUNG (1)
(Thay đổi lớn/Thay đổi nhỏ cần phê duyệt/Chỉ yêu cầu thông báo)
A. Thông tin về sản phẩm (2)
|
1. |
Tên thuốc/nguyên liệu làm thuốc: |
|
|
2. |
Hoạt chất, nồng độ/hàm lượng: |
|
|
3. |
Dạng bào chế: |
|
|
4. |
Số đăng ký: Ngày cấp: |
Ngày hết hạn: |
|
5. |
Tên cơ sở sản xuất: Nước sản xuất: |
|
B. Phân loại thay đổi, bổ sung
|
Loại TĐBS |
|
Mã tham chiếu/Tên |
Loại TĐBS |
|
Ghi chú |
|
Thay đổi lớn |
□ |
|
Công bố biệt dược gốc |
□ |
|
|
Thay đổi nhỏ (có phê duyệt) |
□ |
|
Công bố tương đương sinh học |
□ |
|
|
Thay đổi nhỏ (thông báo) Đề nghị có công văn phê duyệt |
□
□ |
|
Cập nhật cập nhật thông tin thuốc |
□ |
|
C. Chi tiết về cơ sở đăng ký đã được duyệt
1. Tên cơ sở đăng ký:
2. Địa chỉ:
3. Điện thoại:
4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài):
Địa chỉ:
Điện thoại:
D. Mô tả nội dung thay đổi (kèm theo lý do thay đổi) (3)
- Nêu rõ thay đổi, bổ sung thuộc mục nào kèm theo mã thay đổi (nếu có)
dựa trên phân loại các thay đổi, bổ sung:
- Nội dung đã được duyệt (*):
- Nội dung đề nghị thay đổi, bổ sung (*):
(*) có thể nộp dưới dạng bảng so sánh kèm theo đơn đề nghị và có xác nhận của cơ sở đăng ký
Đ. Tài liệu kỹ thuật nộp kèm theo quy định
- Các tài liệu chứng minh, đã được phê duyệt có liên quan.
E. Tuyên bố của cơ sở đăng ký
Cơ sở đăng ký cam kết đã kiểm tra, ký đóng dấu vào những phần liên quan ở tất cả các giấy tờ nộp trong hồ sơ này và xác nhận là đây là các giấy tờ hợp pháp, nội dung là đúng sự thật. Nếu có sự giả mạo, không đúng sự thật cơ sở đăng ký chịu trách nhiệm và sẽ bị xử lý theo quy định của pháp luật.
|
|
Ngày... tháng... năm..... |
(1) Yêu cầu Đơn riêng cho từng thuốc trong các trường hợp: cập nhật công bố biệt dược gốc; thuốc có chứng minh tương đương sinh học; các thay đổi lớn; cập nhật thông tin thuốc.
Chỉ yêu cầu 1 Đơn, 1 bộ hồ sơ đối với các trường hợp: các thuốc có cùng một nội dung thay đổi, gồm: thay đổi tên, địa chỉ cơ sở đăng ký, cơ sở sản xuất thành phẩm, dược chất, tá dược, nội dung thay đổi trên nhãn, tờ hướng dẫn sử dụng; các thay đổi nhỏ của cùng một thuốc;
(2) Trường hợp nhiều thuốc có cùng nội dung thay đổi, bổ sung: Thông tin sản phẩm được liệt kê trong danh mục ứng với từng thuốc.
(3) Các thuốc khác nhau (SĐK khác nhau) của cùng một cơ sở đăng ký và cùng cơ sở sản xuất thành phẩm có cùng các thay đổi nhỏ cần có phê duyệt, thay đổi nhỏ chỉ yêu cầu thông báo (nội dung thay đổi hoàn toàn giống nhau) được phép chung một đơn, một bộ hồ sơ kèm theo danh mục thông tin về sản phẩm quy định tại mục A.
(4) Trường hợp thay đổi cơ sở đăng ký, phải có xác nhận của cơ sở đăng ký cũ và cơ sở đăng ký mới trong đơn đăng ký thay đổi, bổ sung. Trường hợp nhà sản xuất được quyền thay đổi cơ sở đăng ký theo công văn thông báo của Cục Quản lý Dược thì yêu cầu xác nhận của cơ sở sản xuất và cơ sở đăng ký mới.
MẪU 6D/TT
ĐƠN ĐĂNG KÝ THUỐC
(Thuốc chuyển giao công nghệ)
A. Chi tiết về cơ sở đăng ký, cơ sở chuyển giao công nghệ và cơ sở nhận chuyển giao công nghệ
1. Cơ sở đăng ký
1.1. Tên cơ sở đăng ký:
1.2. Địa chỉ: Website (nếu có):
1.3. Điện thoại: Email:
1.4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài):
Địa chỉ:
Điện thoại:
2. Cơ sở chuyển giao công nghệ:
2.1. Tên cơ sở chuyển giao công nghệ:
2.2. Địa chỉ: Website (nếu có):
2.3. Điện thoại: Email:
3. Cơ sở nhận chuyển giao công nghệ (cơ sở sản xuất)
3.1. Tên cơ sở nhận chuyển giao công nghệ :
3.2. Địa chỉ: Website (nếu có):
3.3. Điện thoại: Email:
B. Chi tiết về sản phẩm
1. Tên thuốc:
2. Hoạt chất, nồng độ/hàm lượng:
3. Dạng bào chế:
4. Mô tả dạng bào chế:
5. Đường dùng:
6. Tiêu chuẩn chất lượng (3):
7. Hạn dùng:
8. Điều kiện bảo quản:
9. Mô tả quy cách đóng gói:
10. Số đăng ký (thuốc trước khi chuyển giao công nghệ, nếu có)
Ngày cấp: Ngày hết hạn:
11. Phân loại (tích vào nội dung phù hợp):
|
Thuốc kê đơn |
|
|
Thuốc không kê đơn |
|
|
Thuốc hướng tâm thần |
|
|
Thuốc thành phẩm dạng phối hợp có chứa hoạt chất hướng tâm thần |
|
|
Thuốc gây nghiện |
|
|
Thuốc thành phẩm dạng phối hợp có chứa hoạt chất gây nghiện |
|
|
Tiền chất dùng làm thuốc |
|
|
Thuốc thành phẩm dạng phối hợp có chứa tiền chất dùng làm thuốc |
|
|
Nguyên liệu làm thuốc |
|
|
Thuốc độc |
|
|
Mã ATC |
|
|
Thuốc phóng xạ |
|
|
|
|
|
Thuốc dược liệu |
|
12. Công thức bào chế (cho một đơn vị liều hoặc đơn vị đóng gói nhỏ nhất)
Thành phần:
|
Hoạt chất (1) |
Nồng độ/ hàm lượng (2) |
Nhà sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
|
|
|
|
|
|
Tá dược |
Nồng độ/ hàm lượng |
Nhà sản xuất (tên, địa chỉ chi tiết) |
Tiêu chuẩn (3) |
|
|
|
|
|
C. Tài liệu kỹ thuật
1. Phần I: Hành chính
2. Phần II: Chất lượng
3. Phần III: Tiền lâm sàng
4. Phần IV: Lâm sàng
Ghi chú: Những tài liệu (Phần I, II, III, IV) phải nộp tuỳ thuộc vào phân loại sản phẩm/ nhóm sản phẩm.
D. Các thay đổi giữa thuốc đăng ký và thuốc trước khi chuyển giao công nghệ:
|
TT |
Tên nội dung thay đổi |
Tóm tắt nội dung thuốc trước khi chuyển giao công nghệ |
Tóm tắt nội dung thuốc đăng ký |
Tài liệu kỹ thuật nộp kèm theo |
|
|
|
|
|
|
|
|
|
|
|
|
Đ. Các đề nghị đặc biệt đối với thuốc đăng ký
Thuốc có dữ liệu yêu cầu bảo mật: □
Cơ sở đăng ký thuốc đề nghị Cục Quản lý Dược xem xét thực hiện bảo mật đối với các dữ liệu sau đây được nộp kèm theo hồ sơ đăng ký thuốc:
|
□ □ |
Dữ liệu thử nghiệm độc tính Dữ liệu thử thuốc trên lâm sàng |
(Tài liệu số …) (Tài liệu số …) |
Cơ sở đăng ký thuốc xin cam kết các dữ liệu nêu trên đáp ứng đầy đủ các điều kiện bảo mật dữ liệu theo quy định của pháp luật và cơ sở đăng ký thuốc sẽ thực hiện nghĩa vụ chứng minh khi được cơ quan có thẩm quyền yêu cầu.
|
Hồ sơ đề nghị theo quy trình thẩm định nhanh: Có hồ sơ tương đương sinh học: Có hồ sơ lâm sàng: Thuốc đề nghị phân loại biệt dược gốc: |
□ □ □ □ |
Đề nghị khác (nếu có, đề nghị ghi rõ):
E. Tuyên bố của cơ sở đăng ký
Cơ sở đăng ký thuốc cam kết:
|
1. |
Thực hiện việc cập nhật, bổ sung tờ hướng dẫn sử dụng thuốc theo thuốc biệt dược gốc, sinh phẩm tham chiếu đối với trường hợp quy định tại khoản 3 Điều 38 Thông tư 01/2018/TT-BYT ngày 18/1/2018 của Bộ Y tế quy định ghi nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc (áp dụng đối với thuốc generic, sinh phẩm tương tự). |
|
2. |
Đã kiểm tra, ký đóng dấu theo quy định và xác nhận là đây là các giấy tờ hợp pháp, nội dung là đúng sự thật. Nếu có sự giả mạo, không đúng sự thật cơ sở đăng ký xin chịu hoàn toàn trách nhiệm và sẽ bị xử phạt theo quy định của pháp luật. |
|
3. |
Đảm bảo thuốc/nguyên liệu làm thuốc được sản xuất thuốc theo đúng hồ sơ đăng ký thuốc đã nộp. |
|
4. |
Đăng ký thay đổi, bổ sung theo quy định sau khi thuốc đã được cấp giấy đăng ký lưu hành. |
|
5. |
Chịu trách nhiệm hoàn toàn về sở hữu trí tuệ liên quan đến thuốc đăng ký. |
|
Ngày... tháng... năm..... |
Ngày... tháng... năm..... |
Ngày... tháng... năm..... |
(1) Ghi chính xác dạng dùng của dược chất (muối ester/các dạng dẫn chất khác).
(2) Nếu liều dùng tính theo gốc có tác dụng dược lý của dược chất (gốc base...), cần bổ sung thêm thông tin về hàm lượng dược chất được quy đổi ra gốc có tác dụng dược lý này.
Nếu dược chất được sử dụng dưới dạng bán thành phẩm đã trộn thêm tá dược, phải ghi đầy đủ cả các thành phần tá dược có trong công thức bào chế các bán thành phẩm có chứa dược chất này.
(3)Nếu là tiêu chuẩn dược điển, đề nghị ghi rõ tên dược điển và phiên bản dược điển hoặc năm phát hành dược điển hoặc ghi theo“dược điển phiên bản hiện hành”.
MẪU 6E/TT
ĐƠN ĐỀ NGHỊ
CẤP ĐỔI, CẤP LẠI GIẤY PHÉP LƯU HÀNH SẢN PHẨM
A. Chi tiết về cơ sở đăng ký
1. Tên cơ sở đăng ký:
2. Địa chỉ:
3. Điện thoại:
4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài): Địa chỉ:
Điện thoại:
B. Chi tiết sản phẩm
1. Tên thuốc/nguyên liệu làm thuốc:
2. Số đăng ký: Ngày cấp: Ngày hết hạn:
3. Tên, địa chỉ cơ sở sản xuất:
C. Cơ sở đề nghị cấp đổi, cấp lại giấy phép lưu hành sản phẩm: Ghi rõ cấp đổi hay cấp lại và nêu rõ lý do.
D. Tài liệu nộp kèm theo
- Bản chính giấy phép lưu hành sản phẩm đã cấp trong trường hợp giấy đăng ký lưu hành đã cấp bị hỏng, đính chính hoặc bị ghi sai do lỗi của cơ quan cấp.
- Các tài liệu liên quan khác (nếu có).
Cơ sở đăng ký cam kết và chịu trách nhiệm về tính chính xác về các nội dung nêu trong đơn đề nghị và các tài liệu cung cấp kèm theo.
|
|
Ngày tháng năm |
MẪU 6G/TT
ĐƠN ĐỀ NGHỊ DUY TRÌ HIỆU LỰC GIẤY ĐĂNG KÝ LƯU HÀNH
A. Chi tiết về cơ sở đăng ký
1. Tên cơ sở đăng ký:
2. Địa chỉ:
3. Điện thoại:
4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài): Địa chỉ:
Điện thoại:
B. Chi tiết sản phẩm (hoặc danh mục kèm theo đơn đề nghị và có xác nhận của cơ sở đăng ký)
1. Tên thuốc/nguyên liệu làm thuốc:
|
2. Số đăng ký: |
Ngày cấp: |
Ngày hết hạn: |
|
3. Tên cơ sở sản xuất: |
Nước sản xuất: |
|
C. Cơ sở đề nghị duy trì hiệu lực số đăng ký thuốc tại Mục B nêu trên theo quy định tại Thông tư số ... ngày ... của Bộ Y tế Quy định việc đăng ký thuốc, nguyên liệu làm thuốc.
D. Cơ sở đăng ký cam kết:
1. Đảm bảo duy trì các điều kiện hoạt động của cơ sở đăng ký, cơ sở sản xuất theo quy định.
2. Đảm bảo thuốc/nguyên liệu làm thuốc được sản xuất, lưu hành theo đúng hồ sơ đăng ký đã được phê duyệt.
3. Đã kiểm tra, ký đóng dấu theo quy định và chịu trách nhiệm về tính chính xác, hợp pháp về các nội dung nêu trong đơn đề nghị và các tài liệu cung cấp kèm. Nếu có sự giả mạo, không đúng sự thật cơ sở đăng ký sẽ chịu hoàn toàn trách nhiệm và bị xử lý theo quy định của pháp luật.
|
|
Ngày... tháng... năm..... |
MẪU 6H/TT
ĐƠN ĐỀ NGHỊ CẬP NHẬT THÔNG TIN ĐỂ CUNG CẤP THÔNG TIN THUỐC, QUẢNG CÁO THUỐC
A. Chi tiết về cơ sở đăng ký
1. Tên cơ sở đăng ký:
2. Địa chỉ:
3. Điện thoại:
4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài):
Địa chỉ:
Điện thoại:
|
B. Chi tiết sản phẩm |
|
|
|
1. Tên thuốc: |
||
|
2. Số đăng ký: |
Ngày cấp: |
Ngày hết hạn: |
|
3. Dạng bào chế: |
|
|
4. Hoạt chất, nồng độ/hàm lượng:
5. Tên, địa chỉ cơ sở sản xuất:
C. Cơ sở đề nghị được cập nhật thông tin để cung cấp thông tin thuốc, quảng cáo thuốc theo hướng dẫn tại Phụ lục II ban hành kèm theo Thông tư quy định việc đăng ký thuốc, nguyên liệu làm thuốc.
D. Tài liệu nộp kèm theo quy định
- Nội dung thông tin đề nghị được cập nhật; mục đích và đối tượng tiếp cận thông tin cập nhật.
- Bản sao tờ hướng dẫn sử dụng thuốc đã được phê duyệt.
- Tài liệu chứng minh (ghi rõ tên của các tài liệu chứng minh).
Cơ sở đăng ký cam kết và chịu trách nhiệm về tính chính xác về các nội dung nêu trong đơn đề nghị và các tài liệu cung cấp kèm theo.
|
|
Ngày... tháng... năm..... |
MẪU 6I/TT
BÁO CÁO
TIẾN ĐỘ THỰC HIỆN CHUYỂN GIAO CÔNG NGHỆ SẢN XUẤT THUỐC
A. Chi tiết về cơ sở đăng ký, cơ sở chuyển giao công nghệ và cơ sở nhận chuyển giao công nghệ
1. Cơ sở đăng ký
1.1. Tên cơ sở đăng ký:
1.2. Địa chỉ:
1.3. Điện thoại:
1.4. Tên văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài): Địa chỉ:
Điện thoại:
2. Cơ sở chuyển giao công nghệ:
2.1. Tên cơ sở chuyển giao công nghệ:
2.2. Địa chỉ:
2.3. Điện thoại:
3. Cơ sở nhận chuyển giao công nghệ (cơ sở sản xuất)
3.1. Tên cơ sở nhận chuyển giao công nghệ :
3.2. Địa chỉ:
3.3. Điện thoại:
B. Chi tiết về sản phẩm
1. Tên thuốc:
2. Hoạt chất, nồng độ/hàm lượng:
3. Dạng bào chế:
4. Đường dùng:
5. Tiêu chuẩn chất lượng:
6. Số đăng ký: Ngày cấp: Ngày hết hạn:
C. Các cơ sở báo cáo tiến độ thực hiện chuyển giao công nghệ đối với thuốc đã được cấp giấy đăng ký lưu hành, cụ thể như sau:
1. Nội dung đã triển khai (kèm tài liệu chứng minh):
2. Kế hoạch triển khai trong thời gian tới (nêu rõ nội dung, kế hoạch thời gian hoàn thành):
D. Các kiến nghị, đề xuất (nếu có):
Các cơ sở cam kết và chịu trách nhiệm về tính chính xác về các nội dung nêu trong báo cáo và các tài liệu cung cấp kèm theo.
|
Ngày... tháng... năm..... |
Ngày... tháng... năm..... |
Ngày... tháng... năm..... |
MẪU 7/ TT
GIẤY CHỨNG NHẬN SẢN PHẨM DƯỢC PHẨM (CPP)
Giấy chứng nhận này tuân thủ theo mẫu được Tổ chức Y tế Thế giới khuyến cáo (Hướng dẫn chung và chú giải được đính kèm theo)1
Số giấy chứng nhận:
Nước xuất khẩu (nước chứng nhận):
Nước nhập khẩu (nước yêu cầu chứng nhận):
1. Tên, dạng bào chế và tiêu chuẩn của sản phẩm:
1.1. Hoạt chất2 và hàm lượng cho 1 đơn vị liều lượng3
Thành phần và hàm lượng, bao gồm cả tá dược4
1.2. Sản phẩm này có được cấp phép để lưu hành trên thị trường ở nước xuất khẩu không?5
|
□ |
Có |
□ |
Không |
1.3. Thực tế sản phẩm này có mặt trên thị trường nước xuất khẩu không?
|
□ |
Có |
□ |
Không |
Nếu câu trả lời ở 1.2 là Có, thì tiếp tục với phần 2A, bỏ qua phần 2B
Nếu câu trả lời ở 1.2 là Không, thì bỏ qua phần 2A, tiếp tục với phần 2B6
2A.1 Số đăng ký của sản phẩm7 và ngày cấp:
2A.2 Tên và địa chỉ chủ sở hữu giấy phép lưu hành hoặc chủ sở hữu sản phẩm:
2A.3 Tư cách của chủ sở hữu số đăng ký8 (tích vào mục thích hợp theo định nghĩa ở chú giải 8)
□ a □ b □ c
2A.3.1 Đối với trường hợp b và c, tên và địa chỉ cơ sở sản xuất dạng bào chế này là9:
2A.4 Có bản tóm tắt căn cứ xét duyệt cấp số đăng ký kèm theo không?10
|
□ |
Có |
□ |
Không |
2A.5 Nếu có kèm theo, thì thông tin sản phẩm đã được chính thức phê duyệt có đầy đủ và phù hợp với giấy phép đăng ký đã được cấp không?11
|
□ |
Có |
□ |
Không |
□ |
Không cung cấp |
2A.6 Tên và địa chỉ cơ sở xin giấy chứng nhận, nếu khác với cơ sở sở hữu số đăng ký:12
2B.1 Tên và địa chỉ cơ sở xin cấp giấy chứng nhận:
2B.2 Tư cách của cơ sở xin giấy chứng nhận (tích vào mục thích hợp theo định nghĩa ở chú giải 8)
□ a □ b □ c
2B.2.1 Đối với trường hợp b và c, tên và địa chỉ cơ sở sản xuất dạng bào chế là9:
2B.3 Tại sao không có giấy phép lưu hành?
|
□ □ |
Không có quy định Không được yêu cầu |
□ □ |
Đang được xem xét Bị từ chối |
2B.4 Nhận xét13
3. Cơ quan cấp giấy chứng nhận có tổ chức thanh tra định kỳ đối với nhà máy sản xuất dạng bào chế này không?14
|
□ |
Có |
□ |
Không |
□ |
Không quy định |
Trường hợp Không hoặc Không quy định, thì tiếp tục với câu hỏi 4.
3.1. Định kỳ kiểm tra thường kỳ (năm):
3.2. Việc sản xuất dạng bào chế này đã được kiểm tra chưa?
|
□ |
Đã kiểm tra |
□ |
Chưa kiểm tra |
3.3. Cơ sở vật chất và vận hành của nhà máy có đạt tiêu chuẩn GMP theo khuyến cáo của WHO không?14
|
□ |
Có |
□ |
Không |
□ |
Không quy định15 |
4. Những thông tin mà cơ sở xin giấy chứng nhận nộp có thỏa mãn cơ quan cấp chứng nhận về mọi khía cạnh trong sản xuất sản phẩm này không?16
Nếu Không, giải thích tại sao:
Địa chỉ của cơ quan cấp chứng nhận: Số điện thoại:
Tên, chữ ký của người được ủy quyền ký giấy chứng nhận. Ngày tháng cấp, dấu của cơ quan cấp giấy chứng nhận.
Chú giải:
|
1. |
Giấy chứng nhận này được làm theo mẫu do WHO khuyến cáo, cấu thành nên tình trạng pháp lý của một dược phẩm và của người xin cấp giấy chứng nhận tại nước xuất khẩu. Nó được cấp cho từng sản phẩm riêng lẻ vì thiết kế sản xuất và thông tin được phê duyệt cho các dạng bào chế và hàm lượng khác nhau có thể có sự khác biệt. |
|
2. |
Sử dụng tên chung quốc tế (INN) hoặc tên chung quốc gia bất cứ khi nào có thể. Phải có thông tin về tiêu chuẩn dược chất, dược liệu; tên, địa chỉ cơ sở sản xuất dược chất, dược liệu. Các nội dung này có thể trình bày trong phụ lục đính kèm. |
|
3. |
Công thức (thành phần đầy đủ) của dạng bào chế phải được nêu trên giấy chứng nhận hoặc trong phần phụ lục đính kèm. |
|
4. |
Yêu cầu nêu đầy đủ về thành phần, nồng độ, hàm lượng của từng dược chất, dược liệu và tá dược. Đối với dạng bào chế viên nang mềm, viên nang cứng phải có thêm thông tin về thành phần công thức của vỏ nang. |
|
5. |
Đính kèm chi tiết bất kỳ điều kiện hạn chế nào áp dụng cho việc bán, phân phối hoặc sử dụng sản phẩm này theo đúng như đã nêu trong giấy phép sản phẩm (giấy phép đăng ký), nếu có. |
|
6. |
Mục 2A và 2B miễn trừ lẫn nhau. |
|
7. |
Nếu có, nêu rõ việc giấy phép có kèm theo điều kiện không hoặc sản phẩm vẫn chưa được phê duyệt đăng ký lưu hành. |
|
8. |
Nêu rõ xem người chịu trách nhiệm đưa sản phẩm ra thị trường có: a. sản xuất dạng bào chế đó b.đóng gói và/hoặc dán nhãn dạng bào chế do một công ty độc lập sản xuất; c. không liên quan đến hoạt động nào trên đây |
|
9. |
Thông tin này chỉ có thể được cung cấp nếu được sự đồng ý của chủ sở hữu giấy phép đăng ký sản phẩm hoặc cơ sở xin chứng nhận trong trường hợp sản phẩm chưa đăng ký. Nếu phần này không được điền đầy đủ là dấu hiệu cho thấy bên có liên qua không đồng ý đưa thông tin đó vào. Cần lưu ý là thông tin liên quan đến địa điểm sản xuất là một phần của giấy phép đăng ký sản phẩm. Nếu địa điểm sản xuất thay đổi thì giấy phép đăng ký cũng phải được cập nhật theo nếu không thì sẽ không còn giá trị. |
|
10. |
Là tài liệu mà cơ quan quản lý quốc gia nào đó chuẩn bị trong đó tóm tắt căn cứ chuyên môn kỹ thuật để cấp giấy phép đăng ký sản phẩm. |
|
11. |
Là thông tin sản phẩm đã được cơ quan quản lý quốc gia có thẩm quyền phê duyệt, ví dụ như bản Tóm tắt đặc tính sản phẩm (SmPC). |
|
12. |
Trong trường hợp này, việc cấp giấy chứng nhận cần phải được chủ sở hữu giấy phép đăng ký sản phẩm cho phép. Người xin giấy chứng nhận phải nộp giấy cho phép như vậy cho cơ quan quản lý. |
|
13. |
Xin nêu lý do mà người xin chứng nhận đưa ra để không đề nghị đăng ký sản phẩm: a. sản phẩm đã được phát triển riêng cho việc điều trị những bệnh - đặc biệt là các bệnh nhiệt đới - không có trong mô hình bệnh tật của nước xuất khẩu; b.sản phẩm đã được thay đổi công thức nhằm cải thiện độ ổn định của nó trong điều kiện nhiệt đới; c. sản phẩm đã được thay đổi công thức để loại bỏ các tá dược không được phép sử dụng trong dược phẩm ở nước nhập khẩu; d.sản phẩm đã được thay đổi công thức để đạt một giới hạn liều lượng tối đa khác của hoạt chất; e. nêu bất kỳ nguyên nhân nào khác. |
|
14. |
Quy định về thực hành tốt trong sản xuất và kiểm tra chất lượng thuốc để cập đến trong giấy chứng nhận này là các quy định nêu trong báo cáo lần thứ 32 của Ủy ban chuyên gia về Tiêu chuẩn Dược phẩm (Số báo cáo kỹ thuật WHO 823, 1992, Phụ lục 1). Những khuyến cáo áp dụng chuyên biệt cho sinh phẩm đã được xây dựng với Ủy ban chuyên gia về Tiêu chuẩn hoá sinh phẩm (số báo cáo kỹ thuật WHO 822, 1992, Phụ lục 1). |
|
15. |
Không quy định ở đây nghĩa là việc sản xuất được thực hiện ở một nước khác nước cấp giấy chứng nhận sản phẩm và việc thanh tra do nước sản xuất đảm bảo. |
|
16. |
Phần này sẽ được hoàn thành khi chủ sở hữu giấy phép đăng ký sản phẩm hoặc người xin giấy chứng nhận rơi vào trường hợp (b) hoặc (c) mô tả ở mục 8 trên đây. Vấn đề đặc biệt quan trọng là khi có một bên gia công nước ngoài tham gia vào quá trình sản xuất ra sản phẩm đó. Trong những trường hợp đó, người xin chứng nhận phải nộp cho cơ quan cấp chứng nhận những thông tin về bên gia công chịu trách nhiệm cho từng công đoạn sản xuất của dạng thành phẩm cuối, và phạm vi cũng như tính chất của bất kỳ biện pháp kiểm tra nào đã thực hiện đối với mỗi bên gia công. |
MẪU 8A/TT
ỦY QUYỀN ĐỨNG TÊN CƠ SỞ ĐĂNG KÝ
THƯ UỶ QUYỀN
Chúng tôi, _______________________________________________________________
(Tên, địa chỉ chủ sở hữu giấy phép lưu hành hoặc chủ sở hữu sản phẩm hoặc cơ sở sản xuất)
Bằng văn bản này chỉ định _____________________________________________________
(Tên và địa chỉ cơ sở đăng ký) (*)
Thay mặt chúng tôi đứng tên cơ sở đăng ký sản phẩm sau:
Tên sản phẩm:
Hoạt chất, hàm lượng/nồng độ:
Dạng bào chế:
tại Bộ Y tế (Cục Quản lý Dược) Việt Nam.
Cơ sở (*) sẽ là chủ sở hữu giấy đăng ký lưu hành sản phẩm nêu trên tại Việt Nam và chịu trách nhiệm theo quy định trước Bộ Y tế (Cục Quản lý Dược) và trước pháp luật về những vấn đề có liên quan đến sản phẩm này tại Việt Nam.
|
|
Ngày tháng năm |
(1) Chủ tịch hội đồng thành viên, hội đồng quản trị, tổng giám đốc, giám đốc cơ sở ủy quyền.
MẪU 8B/ TT
ỦY QUYỀN KÝ TÊN TRÊN HỒ SƠ ĐĂNG KÝ
THƯ ỦY QUYỀN
Chúng tôi, __________________________________________________________________
(Tên và địa chỉ cơ sở đăng ký)
Bằng văn bản này ủy quyền cho ông/bà:….....................................................
Chức danh:............................................ thay mặt chúng tôi ký tên và đóng dấu cơ sở đăng ký trên hồ sơ đăng ký sản phẩm:
Tên sản phẩm:
Hoạt chất, hàm lượng/nồng độ:
Dạng bào chế:
tại Bộ Y tế (Cục Quản lý Dược).
Thời hạn hiệu lực của thư ủy quyền:
Người được ủy quyền ký tên trên hồ sơ sẽ chịu trách nhiệm theo quy định trước Bộ Y tế (Cục Quản lý Dược) về những vấn đề có liên quan đến sản phẩm này tại Việt Nam.
|
Ngày tháng năm |
Ngày tháng năm |
(1) Chủ tịch hội đồng thành viên, hội đồng quản trị, tổng giám đốc, giám đốc cơ sở đăng ký.
Trường hợp người được ủy quyền ký tên trên hồ sơ không phải trưởng văn phòng đại diện, trên giấy ủy quyền phải có dấu và chữ ký xác nhận của trưởng văn phòng đại diện tại Việt Nam (đối với cơ sở đăng ký nước ngoài).
MẪU 9/TT
TÓM TẮT ĐẶC TÍNH SẢN PHẨM
1. Thông tin thuốc
1.1. Tên thuốc
1.2. Nồng độ/hàm lượng
1.3. Dạng bào chế
2. Định tính và định lượng
2.1. Công bố về định tính
Cần phải nêu tên hoạt chất bằng tên chung quốc tế (INN), đi kèm với dạng muối hoặc hydrat, nếu có.
2.2. Công bố về định lượng
Phải trình bày lượng hoạt chất trên một đơn vị liều lượng (đối với những sản phẩm xịt phân liều, tính trên một lần xịt), trên một đơn vị thể tích hoặc đơn vị khối lượng.
3. Dạng sản phẩm:
Mô tả hình thức sản phẩm theo quan sát bằng mắt thường (màu sắc, dấu hiệu,...), ví dụ: “viên nén màu trắng, tròn, lồi, cạnh xiên có dập số 100 ở một mặt”.
4. Các đặc tính lâm sàng
4.1. Chỉ định điều trị
4.2. Liều lượng và cách dùng
4.3. Chống chỉ định
4.4. Cảnh báo và thận trọng khi sử dụng
4.5. Tương tác với các thuốc khác và các dạng tương tác khác
4.6. Trường hợp có thai và cho con bú
4.7. Tác dụng đối với khả năng lái xe và vận hành máy móc
4.8. Tác dụng không mong muốn
4.9. Quá liều
5. Các đặc tính dược lý
5.1. Đặc tính dược lực học
5.2. Đặc tính dược động học
5.3. Số liệu an toàn tiền lâm sàng
6. Các đặc tính dược học
6.1. Danh mục tá dược
6.2. Tương kỵ
6.3. Tuổi thọ
Tuổi thọ của thuốc trong bao gói thương phẩm. Tuổi thọ sau khi pha loãng hoặc pha để sử dụng theo hướng dẫn. Tuổi thọ sau khi mở bao bì lần đầu.
6.4. Cảnh báo đặc biệt về bảo quản
6.5. Tính chất và dung lượng của bao bì đóng gói
7. Chủ sở hữu số đăng ký lưu hành sản phẩm
8. Số đăng ký lưu hành sản phẩm
9. Ngày cấp số đăng ký lưu hành lần đầu/gia hạn số đăng ký
10. Ngày xem xét lại bản tóm tắt đặc tính sản phẩm
MẪU 10/TT
KẾ HOẠCH QUẢN LÝ NGUY CƠ ĐỐI VỚI VẮC XIN
I. Thông tin chung về vắc xin
|
Tên vắc xin: |
|
|
Dạng bào chế: |
Thành phần hoạt chất, hàm lượng/nồng độ: |
|
Tên cơ sở đăng ký: Địa chỉ : Điện thoại : |
Tên cơ sở sản xuất: Địa chỉ: Điện thoại: |
|
Tên Văn phòng đại diện tại Việt Nam (nếu có): Địa chỉ: Điện thoại: |
|
|
Điều kiện bảo quản: |
Hạn dùng: |
|
Đường dùng: |
Quy cách đóng gói: |
|
Chỉ định đăng ký tại Việt Nam: |
|
|
Ngày nộp Kế hoạch quản lý nguy cơ lần trước: |
|
|
Quá trình thay đổi/bổ sung Kế hoạch quản lý nguy cơ: |
|
|
Tóm tắt nội dung thay đổi: |
|
|
Lý do thay đổi: |
|
II. Các quan ngại về an toàn vắc xin
Liệt kê các nguy cơ quan trọng đã xác định, nguy cơ quan trọng tiềm ẩn có thể xảy ra và các thông tin quan trọng còn thiếu:
|
Các nguy cơ quan trọng đã xác định (Liệt kê các phản ứng có hại đã được chứng minh có liên quan đến vắc xin) |
|
|
Các nguy cơ quan trọng tiềm ẩn có thể xảy ra (Liệt kê các biến cố bất lợi nghi ngờ liên quan đến vắc xin nhưng ở thời điểm hiện tại chưa có đủ bằng chứng kết luận mối liên quan này) |
|
|
Các thông tin quan trọng còn thiếu |
|
III. Tóm tắt Kế hoạch cảnh giác dược thực hiện tại Việt Nam
Mô tả các hoạt động cảnh giác dược (thường quy và/hoặc bổ sung) được lên kế hoạch để giải quyết các quan ngại về an toàn vắc xin tại Việt Nam:
1. Các hoạt động cảnh giác dược thường quy
|
√ |
Báo cáo các biến cố bất lợi sau tiêm chủng liên quan đến vắc xin theo mẫu quy định gửi về Cục Quản lý Dược, Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc và Cục Y tế dự phòng. |
|
√ |
Kịp thời cập nhật các vấn đề quan trọng liên quan đến an toàn và hiệu quả có thể ảnh hưởng đến tổng quan cân bằng lợi ích - nguy cơ của vắc xin gửi về Cục Quản lý Dược và Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc và Cục Y tế dự phòng. |
|
√ |
Kịp thời cập nhật các thông tin về nguy cơ được công bố hoặc các hoạt động liên quan đến an toàn được thực hiện bởi các cơ quan quản lý khác, đặc biệt là các cơ quan quản lý tham chiếu gửi về Cục Quản lý Dược và Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc và Cục Y tế dự phòng. |
2. Các hoạt động cảnh giác dược bổ sung khác:
- Có thể bao gồm các nghiên cứu phi lâm sàng, lâm sàng, dịch tễ học liên quan đến an toàn của vắc xin.
- Nếu nhận thấy không cần phải có hoạt động cảnh giác dược nào khác, phần này nên được nêu rõ là “Không áp dụng”.
- Nếu áp dụng, nên có kế hoạch về thời gian cụ thể cho các hoạt động này.
Ví dụ: các chương trình giám sát đang tiến hành, các nghiên cứu về an toàn sau khi lưu hành, giám sát các nhóm biến cố,…
IV. Kế hoạch giảm thiểu nguy cơ khi vắc xin lưu hành tại Việt Nam
1. Các hoạt động giảm thiểu nguy cơ thường quy
- Cung cấp đầy đủ thông tin và thường xuyên cập nhật đầy đủ các thông tin về chỉ định, liều dùng, cách dùng, cảnh báo và thận trọng trên nhãn và tờ Hướng dẫn sử dụng của sản phẩm theo quy định hiện hành.
- Cập nhật đầy đủ các công văn hướng dẫn của Cục Quản lý Dược liên quan đến an toàn, hiệu quả của vắc xin.
2. Các hoạt động giảm thiểu nguy cơ bổ sung:
- Nếu nhận thấy không cần thiết tiến hành hoạt động giảm thiểu nguy cơ (RMAs) bổ sung nào, nên nêu rõ trong phần này là “Không áp dụng”.
- Nếu áp dụng cần mô tả rõ hoạt động đề xuất nhằm giảm thiểu nguy cơ khi đưa vắc xin ra lưu hành tại Việt Nam.
Ví dụ: Cung cấp các hướng dẫn, tài liệu đào tạo cho bác sĩ, nhân viên y tế tham gia hoạt động tiêm chủng; hướng dẫn về vắc xin cho bệnh nhân, kiểm soát phân phối, chương trình ngừa thai:
● Các tài liệu đào tạo cho bác sĩ, nhân viên y tế tham gia hoạt động tiêm chủng được xây dựng nhằm nhấn mạnh các quan ngại về an toàn đã xác định, các dấu hiệu và triệu chứng cần theo dõi và nhấn mạnh các nguy cơ tiềm ẩn liên quan đến sai sót trong cấp phát cũng như sai sót y khoa.
● Hướng dẫn về vắc xin cho bệnh nhân được xây dựng nhằm nhấn mạnh các quan ngại về an toàn đã xác định, các dấu hiệu và triệu chứng cần theo dõi và khi nào cần tìm kiếm sự trợ giúp về y tế.
V. Các thông tin khác (nếu có)
Liệt kê các tài liệu quản lý nguy cơ được nộp kèm theo kế hoạch này và có phần thuyết minh, giải trình (nếu có).
Ví dụ: Các tài liệu quản lý nguy cơ sau được nộp kèm theo:
(1) Bản mới nhất của kế hoạch quản lý nguy cơ được phê duyệt ở châu Âu, hoặc chiến lược đánh giá và giảm thiểu nguy cơ (REMS) được FDA Hoa Kỳ phê duyệt;
(2) Bản dự kiến tài liệu đào tạo cho bác sĩ, nhân viên y tế tham gia hoạt động tiêm chủng hoặc hướng dẫn liên quan đến việc sử dụng vắc xin;
Cơ sở đăng ký cam kết và chịu hoàn toàn trách nhiệm về tính chính xác, trung thực của các thông tin cung cấp trong bản kế hoạch này./.
|
|
Ngày..... tháng..... năm..... |
MẪU 11/TT
BÁO CÁO
QUÁ TRÌNH LƯU HÀNH THUỐC, NGUYÊN LIỆU LÀM THUỐC
(Kể từ ngày cấp/gia hạn SĐK gần nhất đến thời điểm nộp hồ sơ gia hạn SĐK)
1. Tên cơ sở đăng ký: Địa chỉ:
2. Tên cơ sở sản xuất: Địa chỉ:
3. Tên thuốc/nguyên liệu làm thuốc:
4. Hoạt chất, nồng độ/hàm lượng:
5. Dạng bào chế:
6. Số đăng ký hiện tại: Ngày cấp SĐK lần đầu:
7. Lưu hành trên thị trường:
|
Có |
□ |
Không |
□ |
8. Vi phạm trong quá trình lưu hành
|
Có |
□ |
Không |
□ |
Nếu có vi phạm thì ghi rõ: số lần vi phạm, loại vi phạm.
9. Thay đổi, bổ sung trong thời gian giấy đăng ký lưu hành còn hiệu lực:
|
Có |
□ |
Không |
□ |
Nếu có thay đổi, bổ sung thì gửi kèm theo bản sao công văn cho phép thay đổi, bổ sung.
10.Thay đổi khi đăng ký gia hạn:
|
Có |
□ |
Không |
□ |
Nếu có thay đổi thì phải ghi rõ nội dung thay đổi so với hồ sơ đã được duyệt cấp số đăng ký.
Cơ sở đăng ký cam kết: ngoài những nội dung xin thay đổi ở mục 10 của báo cáo lưu hành thuốc, không có bất cứ sự thay đổi nào so với hồ sơ đã được duyệt cấp số đăng ký.
|
|
Ngày... tháng... năm..... |
Báo cáo quá trình lưu hành thuốc, nguyên liệu làm thuốc (Mẫu 11/TT) ban hành kèm theo Thông tư này được sửa đổi, bổ sung bởi Phụ lục 03 ban hành kèm theo Thông tư số 23/2021/TT-BYT theo quy định tại Điểm i Khoản 3 Điều 1.
MẪU 12/TT
PHIẾU TIẾP NHẬN HỒ SƠ
Mã hồ sơ:
I. Thông tin thuốc/nguyên liệu làm thuốc
1. Tên và địa chỉ cơ sở đăng ký:
2. Tên và địa chỉ cơ sở sản xuất:
3. Tên thuốc/nguyên liệu làm thuốc:
4. Hoạt chất, nồng độ/hàm lượng:
5. Dạng bào chế:
6. Loại thuốc/nguyên liệu làm thuốc đăng ký:
|
Thuốc hóa dược |
|
|
Vắc xin |
|
|
Sinh phẩm |
|
|
Thuốc dược liệu |
|
|
Thuốc phóng xạ |
|
|
Nguyên liệu làm thuốc (dược chất, tá dược, vỏ nang, bán thành phẩm dược liệu) |
|
7. Loại hình đăng ký:
|
Đăng ký lần đầu |
|
|
Đăng ký gia hạn |
|
|
Đăng ký chuyển giao công nghệ |
|
|
Đăng ký thay đổi lớn |
|
|
Đăng ký thay đổi nhỏ cần phê duyệt |
|
|
Đăng ký thay đổi nhỏ chỉ yêu cầu thông báo |
|
|
Công bố tương đương sinh học |
|
|
Công bố biệt dược gốc |
|
|
Cập nhật thông tin thuốc |
|
II. Các tài liệu nộp trong hồ sơ
|
1. |
|
|
2. |
|
|
|
Ngày tháng năm |
Mẫu 13/TT Bản công bố nguyên tắc, tiêu chuẩn GMP hoặc nguyên tắc, tiêu chuẩn đối với sản xuất tá dược đã được cơ quan quản lý của nước hoặc tổ chức quốc tế khác áp dụng được bổ sung bởi Phụ lục 2 Thông tư số 29/2020/TT-BYT (Theo quy định tại điểm l Khoản 5 Điều 1)
Biểu mẫu 14A/TT, 14B/TT, 14C/TT ban hành kèm theo Thông tư này được bổ sung bởi Phụ lục 02 ban hành kèm theo Thông tư số 23/2021/TT-BYT theo quy định tại Điểm h Khoản 3 Điều 1.
Bạn chưa Đăng nhập thành viên.
Đây là tiện ích dành cho tài khoản thành viên. Vui lòng Đăng nhập để xem chi tiết. Nếu chưa có tài khoản, vui lòng Đăng ký tại đây!
Bạn chưa Đăng nhập thành viên.
Đây là tiện ích dành cho tài khoản thành viên. Vui lòng Đăng nhập để xem chi tiết. Nếu chưa có tài khoản, vui lòng Đăng ký tại đây!
