- Tổng quan
- Nội dung
- VB gốc
- Tiếng Anh
- Hiệu lực
- VB liên quan
- Lược đồ
-
Nội dung hợp nhất
Tính năng này chỉ có tại LuatVietnam.vn. Nội dung hợp nhất tổng hợp lại tất cả các quy định còn hiệu lực của văn bản gốc và các văn bản sửa đổi, bổ sung, đính chính... trên một trang. Việc hợp nhất văn bản gốc và những văn bản, Thông tư, Nghị định hướng dẫn khác không làm thay đổi thứ tự điều khoản, nội dung.
Khách hàng chỉ cần xem Nội dung hợp nhất là có thể nắm bắt toàn bộ quy định hiện hành đang áp dụng, cho dù văn bản gốc đã qua nhiều lần chỉnh sửa, bổ sung.
- Tải về
Văn bản hợp nhất 03/QĐHN-BYT năm 2013 do Bộ Y tế ban hành hợp nhất Quyết định về việc triển khai áp dụng nguyên tắc "Thực hành tốt phòng kiểm nghiệm thuốc"
| Số hiệu: | 03/QĐHN-BYT | Ngày ký xác thực: | 04/10/2013 |
| Loại văn bản: | Văn bản hợp nhất | Cơ quan hợp nhất: | Bộ Y tế |
|
Ngày đăng công báo:
|
Đã biết
|
Người ký:
|
Lê Quang Cường |
|
Số công báo:
Số công báo là mã số ấn phẩm được đăng chính thức trên ấn phẩm thông tin của Nhà nước. Mã số này do Chính phủ thống nhất quản lý.
|
Đã biết
|
Ngày hết hiệu lực:
Ngày hết hiệu lực là ngày, tháng, năm văn bản chính thức không còn hiệu lực (áp dụng).
|
Đang cập nhật |
TÓM TẮT VĂN BẢN HỢP NHẤT 03/QĐHN-BYT
Nội dung tóm tắt đang được cập nhật, Quý khách vui lòng quay lại sau!
Tải văn bản hợp nhất 03/QĐHN-BYT
 Văn bản hợp nhất 03/QĐHN-BYT PDF (Bản có dấu đỏ)
Văn bản hợp nhất 03/QĐHN-BYT PDF (Bản có dấu đỏ) Văn bản hợp nhất 03/QĐHN-BYT DOC (Bản Word)
Văn bản hợp nhất 03/QĐHN-BYT DOC (Bản Word)| BỘ Y TẾ Số: 03/QĐHN-BYT | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM Hà Nội, ngày 04 tháng 10 năm 2013 |
QUYẾT ĐỊNH
VỀ VIỆC TRIỂN KHAI ÁP DỤNG NGUYÊN TẮC “THỰC HÀNH TỐT PHÒNG KIỂM NGHIỆM THUỐC”
------------------
BỘ TRƯỞNG BỘ Y TẾ
Quyết định số 1570/2000/QĐ-BYT ngày 22/5/2000 của Bộ trưởng Bộ Y tế về việc triển khai áp dụng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”, có hiệu lực kể từ ngày 07/6/2000 được sửa đổi, bổ sung bởi:
Thông tư số 45/2011/TT-BYT ngày 21/12/2011 sửa đổi, bổ sung một số điều của Quyết định số 1570/2000/QĐ-BYT ngày 22/5/2000 của Bộ trưởng Bộ Y tế về việc triển khai áp dụng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”; Quyết định số 2701/2001/QĐ-BYT ngày 29/6/2001 của Bộ trưởng Bộ Y tế về việc triển khai áp dụng nguyên tắc “Thực hành tốt bảo quản thuốc”; Thông tư số 06/2004/TT-BYT ngày 28/5/2004 hướng dẫn sản xuất gia công thuốc; Quyết định 3886/2004/QĐ-BYT ngày 03/11/2004 của Bộ Y tế về việc triển khai áp dụng nguyên tắc, tiêu chuẩn “Thực hành tốt sản xuất thuốc” theo khuyến cáo của tổ chức y tế thế giới; Thông tư số 13/2009/TT-BYT ngày 01/9/2009 của Bộ Y tế hướng dẫn hoạt động thông tin quảng cáo thuốc; Thông tư số 22/2009/TT-BYT ngày 24/11/2009 của Bộ Y tế quy định về đăng ký thuốc; thông tư số 47/2010/TT- BYT ngày 29/12/2010 hướng dẫn hoạt động xuất khẩu, nhập khẩu thuốc và bao bì tiếp xúc trực tiếp với thuốc (sau đây gọi tắt là Thông tư số 45/2011/TT-BYT), có hiệu lực kể từ ngày 05 tháng 02 năm 2012.
Căn cứ Luật bảo vệ sức khỏe nhân dân ban hành ngày 11/7/1989 và Điều lệ thuốc phòng bệnh, chữa bệnh ban hành kèm theo Nghị định số 23/HĐBT ngày 24/01/1991 của Hội đồng Bộ trưởng (nay là Chính phủ);
Căn cứ Pháp lệnh chất lượng hàng hóa số 18/1999/PL-UBTVQH10 ngày 24/12/1999 của Ủy ban thường vụ Quốc hội;
Căn cứ Pháp lệnh đo lường số 16/1999/PL-UBTVQH10 ngày 06/10/1999 của Ủy ban thường vụ Quốc hội;
Căn cứ Nghị định số 68/CP ngày 11/10/1993 của Chính phủ quy định chức năng nhiệm vụ, quyền hạn và tổ chức bộ máy Bộ Y tế;
Căn cứ Nghị định số 86/CP ngày 08/12/1995 của Chính phủ quy định phân công trách nhiệm quản lý Nhà nước về chất lượng hàng hóa;
Xét đề nghị của Cục trưởng Cục Quản lý Dược Việt Nam.[1]
QUYẾT ĐỊNH:
Điều 1. Triển khai áp dụng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” ban hành kèm theo Quyết định này ở tất cả các đơn vị kiểm nghiệm thuốc.
Điều 2.[2] Quyết định này có hiệu lực sau 15 ngày kể từ ngày ký ban hành.
Điều 3. Ông Cục trưởng Cục Quản lý dược Việt Nam chịu trách nhiệm hướng dẫn, kiểm tra và theo dõi việc triển khai áp dụng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”.
Điều 4. Các Ông, Bà Chánh văn phòng, Chánh thanh tra Bộ Y tế, Cục trưởng Cục Quản lý dược Việt Nam, Viện trưởng Viện Kiểm nghiệm, Thủ trưởng các đơn vị trực thuộc Bộ, Giám đốc Sở Y tế các tỉnh, thành phố trực thuộc Trung ương, Y tế ngành chịu trách nhiệm thi hành Quyết định này./.
|
| XÁC THỰC VĂN BẢN HỢP NHẤT KT. BỘ TRƯỞNG |
NGUYÊN TẮC
“THỰC HÀNH TỐT PHÒNG KIỂM NGHIỆM THUỐC”
(Ban hành theo Quyết định số 1570/2000/QĐ-BYT ngày 22/5/2000 của Bộ trưởng Bộ Y tế)
Phần 1. PHẦN CHUNG
1. Mục đích
Việc thực hành tốt các nguyên tắc kiểm nghiệm thuốc nhằm nâng cao tính hiệu quả của hệ thống các phòng kiểm nghiệm thuốc trên cả hai mặt quản lý nghiệp vụ và quản lý kỹ thuật, kể cả khu vực quản lý nhà nước và doanh nghiệp, nhằm đảm bảo tính khách quan, trung thực và chính xác trong việc đánh giá chất lượng thuốc.
Chức năng của một phòng kiểm nghiệm thuốc là đánh giá một loại thuốc có đạt tiêu chuẩn chất lượng đã đăng ký hay không. Phòng kiểm nghiệm sẽ là một công cụ đắc lực cho công tác quản lý chất lượng nếu kết quả phân tích mẫu đáng tin cậy và kết luận về chất lượng của thuốc là chính xác. Muốn vậy công tác kiểm nghiệm phải được tiêu chuẩn hóa. Các nguyên tắc thực hành tốt phòng kiểm nghiệm thuốc được soạn thảo với mục đích cung cấp cơ sở cho việc đánh giá các phòng kiểm nghiệm theo yêu cầu nói trên.
Nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” được áp dụng cho các phòng kiểm nghiệm của Nhà nước và của các doanh nghiệp, kể cả các doanh nghiệp có vốn đầu tư nước ngoài, các phòng kiểm nghiệm tư nhân hay phòng kiểm nghiệm độc lập.
2. Giải thích thuật ngữ
Một số thuật ngữ sử dụng trong bản nguyên tắc này được hiểu như sau:
- Đơn vị kiểm nghiệm: là một bộ phận của phòng kiểm nghiệm thuốc được chuyên môn hóa để thực hiện các phân tích theo một kỹ thuật chung, thí dụ phân tích lý, hóa, vi sinh...
- Hệ thống chất lượng: là một hệ thống bao gồm: chính sách, mục tiêu, cơ cấu tổ chức, chức năng nhiệm vụ, quy trình và nguồn lực cần thiết để đảm bảo chất lượng hoạt động của một đơn vị.
Hệ thống chất lượng được xây dựng để đáp ứng yêu cầu quản lý chất lượng nội bộ của một tổ chức, bao gồm luôn cả các mối quan hệ với khách hàng
- Hệ thống phân tích: là một thiết bị hoặc một tập hợp hoàn chỉnh các thiết bị được dùng để tiến hành các phân tích trên một mẫu.
- Lô sản xuất: là một lượng thuốc nhất định được sản xuất ra trong một chu kỳ sản xuất, phải đồng nhất và được nhà sản xuất ghi trên nhãn và/hoặc các bao gói bằng cùng một ký hiệu của lô sản xuất.
- Đơn vị lấy mẫu: là một phần của lô sản xuất như một số đơn vị đóng gói như: một số gói, một số hộp hay một số thùng nguyên vẹn hoặc không nguyên vẹn được chọn ra để lấy mẫu.
- Mẫu ban đầu: là một lượng thuốc được lấy ra từ một đơn vị lấy mẫu.
- Mẫu riêng: là một lượng thuốc được tạo thành bằng cách trộn các mẫu ban đầu với nhau.
- Mẫu chung: là một lượng thuốc được tạo thành bằng cách trộn lẫn các mẫu riêng với nhau. Có thể nghiền trộn với nhau nếu là chất bột rắn, trộn đều với nhau nếu là chất lỏng, hoặc có thể đặt cạnh nhau nếu là các đơn vị thuốc đã phân liều.
- Mẫu cuối cùng: là một phần của mẫu chung đủ để tạo mẫu phân tích và mẫu lưu.
- Mẫu phân tích: là một phần của mẫu cuối cùng dùng để phân tích ở phòng kiểm nghiệm. Lượng thuốc trong mẫu phân tích phải đủ để thực hiện tất cả các phép thử theo yêu cầu của tiêu chuẩn quy định.
- Mẫu lưu: là một phần của mẫu cuối cùng, được giữ lại để kiểm nghiệm lại khi cần thiết. Lượng thuốc trong mẫu lưu cũng phải bằng mẫu phân tích.
- Quy trình thao tác chuẩn: là các hướng dẫn chi tiết có tính chất pháp lý cho việc thực hiện các bước của một thao tác, một công việc hay một quá trình khi kiểm nghiệm thuốc.
- GLP: là từ viết tắt của thuật ngữ tiếng Anh “Good Laboratory Practices”, được dịch là “Thực hành tốt phòng kiểm nghiệm thuốc”.
Phần 2. NGUYÊN TẮC “THỰC HÀNH TỐT PHÒNG KIỂM NGHIỆM THUỐC”
1. Tổ chức và nhân sự
1.1. Tổ chức
Một phòng kiểm nghiệm thuốc thường được chia làm nhiều đơn vị kiểm nghiệm hoặc các bộ phận được chuyên môn hóa dựa trên kỹ thuật kiểm nghiệm (ví dụ: Hóa lý, Vật lý, Vi sinh vật...) hoặc chia theo các đối tượng là sản phẩm được kiểm nghiệm (ví dụ: kháng sinh, vitamin, dược liệu...). Đôi khi phòng kiểm nghiệm còn có các đơn vị kiểm nghiệm chuyên biệt để phục vụ các yêu cầu như thử độ vô trùng, thử chí nhiệt tố, đo lường vật lý đặc biệt...
Chức năng nhiệm vụ của mỗi đơn vị kiểm nghiệm cần phải được xây dựng và được người có thẩm quyền ban hành chính thức.
Ngoài các đơn vị kiểm nghiệm (hay còn gọi là các phòng chuyên môn), mỗi phòng kiểm nghiệm còn phải có bộ phận đăng ký mẫu và bộ phận lưu trữ hồ sơ tiêu chuẩn chất lượng. Bộ phận đăng ký mẫu có nhiệm vụ nhận mẫu và các tài liệu kèm theo, phân phát mẫu đến các phòng chuyên môn và trả lời kết quả kiểm nghiệm. Bộ phận lưu trữ hồ sơ tiêu chuẩn chất lượng phải đảm bảo cung cấp đầy đủ các tiêu chuẩn chất lượng cập nhật và các tài liệu liên quan cho các đơn vị kiểm nghiệm.
Ngoài ra, phòng kiểm nghiệm cần có một số đơn vị hậu cần để phục vụ cho việc cung ứng trang thiết bị, vật tư, hóa chất, dụng cụ và súc vật thử nghiệm.
1.2. Nhân sự
1.2.1. Phòng kiểm nghiệm phải có đủ nhân viên được đào tạo thích hợp, có chuyên môn và kinh nghiệm để hoàn thành công việc được giao. Các nhân viên cần được quán triệt tầm quan trọng của công tác kiểm nghiệm và không được làm thêm những công việc có mâu thuẫn với công tác kiểm nghiệm.
1.2.2. Yêu cầu trình độ chuyên môn của cán bộ, nhân viên kỹ thuật.
a) Trưởng phòng kiểm nghiệm và trưởng các bộ phận phải có trình độ đại học hoăc sau đại học, có kinh nghiệm trong lĩnh vực phân tích kiểm nghiệm và lĩnh vực kiểm tra, quản lý chất lượng thuốc
b) Kiểm nghiệm viên phải có bằng đại học hoặc sau đại học thuộc một ngành chuyên môn thích hợp như dược, hóa phân tích, dược lý, sinh vật, vi sinh vật...
c) Kỹ thuật viên trung học phải tốt nghiệp các trường trung học chuyên nghiệp thuộc các ngành phù hợp hoặc được đào tạo về công tác kiểm nghiệm.
d) Công nhân kỹ thuật có trình độ sơ học được đào tạo ở một trường dạy nghề hoặc được học tập, kèm cặp tại các đơn vị kiểm nghiệm thuốc ít nhất 01 năm.
1.2.3. Số lượng biên chế phụ thuộc vào quy mô và điều kiện cụ thể của mỗi phòng kiểm nghiệm. Nói chung tỷ lệ giữa kỹ thuật viên (trình độ trung học) và kiểm nghiệm viên (trình độ đại học) nên là 1:3 đối với phòng kiểm nghiệm hóa lý, 2:5 đối với phòng kiểm nghiệm sinh học hoặc vi sinh. Tuy nhiên nếu phòng đảm nhiệm nhiều công việc phức tạp hoặc phải áp dụng nhiều phương pháp phân tích mới, hay kiểm tra nhiều đối tượng phân tích mới thì tỷ lệ kiểm nghiệm viên phải nhiều hơn.
2. Hệ thống chất lượng
2.1. Hệ thống chất lượng được đặt ra nhằm đảm bảo hoạt động của phòng kiểm nghiệm tuân theo các nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”.
2.2. Phòng kiểm nghiệm phải đề ra các quy định, mục tiêu, phương pháp và các hướng dẫn nhằm đảm bảo chất lượng của các kết quả phân tích. Các tài liệu này phải được phân phát tới từng nhân viên có liên quan, để hiểu và thi hành.
2.3. Mỗi phòng kiểm nghiệm phải có một cuốn sổ tay chất lượng gồm có những mục sau:
a) Tổ chức của phòng kiểm nghiệm
b) Các hoạt động chuyên môn và quản lý có liên quan đến chất lượng
c) Các quy trình đảm bảo chất lượng chung
d) Quy định về việc sử dụng chất đối chiếu
e) Thông báo và các biện pháp xử lý khi phát hiện sai lệch trong quá trình thử nghiệm
f) Quy trình giải quyết các khiếu nại
g) Sơ đồ đường đi của mẫu
h) Quy định chất lượng: tiêu chuẩn của công tác kiểm nghiệm, mục đích của hệ thống chất lượng, việc thi hành các quy trình và quy định chất lượng trong công tác kiểm nghiệm.
2.4. Hệ thống chất lượng cần được đánh giá định kỳ và có hệ thống để đảm bảo duy trì tính hiệu quả cũng như áp dụng các biện pháp chấn chỉnh khi cần thiết. Kết quả của việc xem xét đánh giá và các biện pháp chấn chỉnh đã thực hiện phải được ghi chép lại trong hệ thống hồ sơ lưu trữ.
2.5. Một cơ quan kiểm nghiệm hay một phòng kiểm nghiệm cần có một người làm công tác phụ trách chất lượng. Người này, ngoài bổn phận và trách nhiệm riêng của mình, phải có trách nhiệm và quyền hạn xác định nhằm đảm bảo hệ thống chất lượng được thi hành. Người phụ trách chất lượng phải có quyền thảo luận trực tiếp với cấp lãnh đạo cao nhất của phòng kiểm nghiệm về những quyết định có liên quan đến chính sách chất lượng.
3. Cơ sở vật chất
3.1. Phòng kiểm nghiệm phải được thiết kế phù hợp, đảm bảo đủ chỗ cho trang thiết bị dụng cụ chuyên môn, hồ sơ tài liệu và không gian làm việc cho nhân viên.
3.2. Khi bố trí các phòng chuyên môn, phải tạo được sự riêng biệt cho các hoạt động kiểm nghiệm khác nhau. Trong một phòng chuyên môn, phải có đủ các khu vực riêng để đảm bảo sự độc lập của các hệ thống phân tích. Phòng kiểm nghiệm sinh học/vi sinh vật hay chất phóng xạ phải cách biệt với các phòng kiểm nghiệm khác. Riêng đối với phòng kiểm nghiệm vi sinh vật, hệ thống cấp khí sạch phải đảm bảo đúng các tiêu chuẩn kỹ thuật quy định. Phòng kiểm nghiệm dược lý phải có khu chăn nuôi súc vật thử nghiệm thiết kế đúng với các yêu cầu kỹ thuật quy định.
3.3. Nên có những phòng riêng để bảo quản thuốc thử, chất chuẩn, phụ kiện của thiết bị phòng kiểm nghiệm, phòng để lưu mẫu, khu vực chứa dung môi cháy nổ hoặc các hóa chất độc hại. Những khu vực này phải biệt lập với khu vực tiến hành các phân tích và phải được trang bị chống mối mọt, côn trùng, ô nhiễm, cháy nổ... Điều kiện không khí: độ ẩm, nhiệt độ thích hợp.
3.4. Môi trường tiến hành các thử nghiệm phải đảm bảo không làm sai lệch các kết quả hoặc gây ảnh hưởng đến độ chính xác của các phép đo. Phòng kiểm nghiệm phải được bảo vệ tránh các ảnh hưởng quá mức của nhiệt độ, độ ẩm, bụi bặm, tiếng ồn, các rung động và các nhiễu điện từ.
3.5. Việc đi lại của nhân viên và sử dụng phòng kiểm nghiệm phải được kiểm soát và giới hạn theo đúng mục đích và điều kiện quy định. Phải có biện pháp thích hợp giữ cho phòng kiểm nghiệm luôn sạch sẽ, gọn gàng và ngăn nắp.
4. Thiết bị phân tích và hiệu chỉnh thiết bị phân tích
4.1. Phòng kiểm nghiệm phải được trang bị các máy móc, dụng cụ thích hợp cho việc lấy mẫu, phân tích, hiệu chỉnh và xử lý dữ liệu. Các thiết bị phân tích phải phù hợp với phương pháp kiểm nghiệm và phải đáp ứng được yêu cầu kiểm tra chất lượng của đơn vị.
4.2. Thiết bị phân tích phải được thiết kế, cấu tạo, điều chỉnh và bảo trì cho phù hợp với các thao tác được thực hiện trên thiết bị đó. Thiết kế và cấu tạo của thiết bị phải cho phép giảm thiểu tối đa sai số, cho phép việc làm vệ sinh cũng như bảo trì được dễ dàng.
4.3. Các thiết bị phân tích tự động và phần mềm kèm theo phải cho kết quả chính xác như yêu cầu và phải đáp ứng được các tiêu chuẩn kỹ thuật liên quan đến thử nghiệm. Các thiết bị này phải được hiệu chỉnh định kỳ để đảm bảo kết quả phân tích không mắc sai số.
4.4. Tần số hiệu chỉnh: Nếu thiết bị không có quy định riêng, thì phải thực hiện đúng theo quy định chung ở mục B, Phần IV: Phụ lục. Việc hiệu chỉnh thiết bị phải được thực hiện theo một lịch cụ thể và phải được lưu vào sổ lý lịch của máy. Tần số hiệu chỉnh thay đổi tùy loại thiết bị. Ví dụ:
- Máy đo pH được chỉnh ít nhất 1 lần/ngày.
- Máy đo điểm nóng chảy: hiệu chỉnh hàng tháng.
- Máy quang phổ hấp thu tử ngoại: hiệu chỉnh hàng tháng song phải được kiểm tra hàng tuần về độ tin cậy của bước sóng.
- Máy quang phổ hồng ngoại: hiệu chỉnh hàng quý.
- Cân phân tích, máy đo chỉ số khúc xạ và máy quang phổ huỳnh quang: hiệu chỉnh 6 tháng/lần.
4.5. Phải có bản hướng dẫn sử dụng các thiết bị phân tích, trong đó nêu rõ các thao tác căn bản. Bản hướng dẫn này phải được đặt gần thiết bị để người sử dụng tiện tham khảo.
4.6. Mỗi thiết bị phải có một sổ ghi chép gọi là sổ lý lịch máy gồm những thông tin sau:
a) Tên thiết bị.
b) Tên nhà sản xuất, loại thiết bị, số lô sản xuất hoặc mã số khác.
c) Kết quả thẩm định xác nhận thiết bị đạt các tiêu chuẩn kỹ thuật.
d) Vị trí đặt thiết bị.
e) Các hướng dẫn của nhà sản xuất (nếu có) hoặc địa chỉ nhà sản xuất.
f) Ngày, kết quả, bản sao các báo cáo và chứng nhận hiệu chỉnh, giới hạn cho phép và ngày hiệu chỉnh kế tiếp.
g) Các công việc bảo trì cho tới nay và kế hoạch bảo trì.
h) Các báo cáo về hư hỏng, ngừng hoạt động, thay đổi hoặc sửa chữa.
4.7. Tất cả các thiết bị phân tích cần hiệu chỉnh phải được đánh số, dán nhãn, ghi tình trạng hiệu chỉnh và ngày hiệu chỉnh tiếp theo. Nếu vì bất cứ lý do gì thiết bị được đem ra khỏi phòng kiểm nghiệm một thời gian, phải kiểm tra lại hoạt động và tình trạng hiệu chỉnh của thiết bị trước khi đưa vào sử dụng trở lại. Mặc dù thiết bị đã được hiệu chỉnh nhưng mỗi khi phân tích một mẫu không đạt cũng phải xem lại phần kiểm định thiết bị.
4.8. Các thiết bị có dấu hiệu hoạt động quá tải, vận hành không đúng cách, cho kết quả không đáng tin cậy, hỏng hóc hoặc không đạt tiêu chuẩn kỹ thuật phải được đánh dấu và dán nhãn rõ ràng, không được đưa vào sử dụng mà phải cách ly, chờ sửa chữa cho tới khi kiểm tra lại và kết quả hiệu chỉnh đạt yêu cầu mới được phép sử dụng.
5. Thuốc thử và chất đối chiếu
5.1. Thuốc thử
5.1.1. Thuốc thử là những hóa chất, dung môi hay dung dịch chuẩn độ dùng để tiến hành các thử nghiệm định tính và định lượng, thử tinh khiết... vì vậy thuốc thử phải có chất lượng phù hợp để kết quả phân tích được chắc chắn và có độ tin cậy cao.
5.1.2. Thuốc thử phải được mua từ các nhà sản xuất hay các nhà phân phối có uy tín, tốt nhất là ở dạng đóng gói nhỏ, thích hợp để sử dụng trong phòng kiểm nghiệm.
5.1.3. Một số thuốc thử có tính chất độc hại hoặc dễ cháy nổ phải được sử dụng và bảo quản theo một quy chế đặc biệt để đảm bảo an toàn. Các thuốc thử thuộc về chất độc, chất gây nghiện hoặc chất hướng tâm thần phải được dán nhãn rõ ràng, bảo quản trong tủ có khóa và giao cho cán bộ chuyên môn phụ trách dưới sự giám sát của trưởng đơn vị kiểm nghiệm theo các Quy chế tương ứng về Quản lý thuốc độc, thuốc gây nghiện và thuốc hướng tâm thần.
5.1.4. Việc pha chế thuốc thử phải được giao cho người có trình độ thích hợp, và phải theo các quy trình đã được mô tả trong dược điển hoặc các tài liệu chính thức khác.
5.1.5. Thuốc thử sau khi pha phải được dán nhãn đầy đủ với các chi tiết: tên thuốc thử, nồng độ, yếu tố chuẩn hóa (hệ số hiệu chỉnh K), hạn dùng, điều kiện bảo quản, ngày pha chế và tên của người pha chế. Các dung dịch loãng được pha từ các dung dịch gốc đậm đặc cũng cần được dán nhãn ghi tên dung dịch, tên nhà sản xuất, ngày pha chế và tên người pha chế.
5.1.6. Phải có sổ ghi thuốc thử đã pha chế gồm có công thức pha, theo tài liệu nào và tên người pha chế. Một số thuốc thử được sử dụng phổ biến nên tập trung cho một đơn vị pha chế để đảm bảo tính đồng nhất của các thuốc thử này. Không nên di chuyển thuốc thử từ đơn vị này sang đơn vị khác trừ phi thuốc thử còn nguyên. Khi di chuyển thuốc thử phải giữ nguyên bao bì ban đầu nếu được. Khi chia nhỏ thuốc thử phải dùng bao bì sạch và dán nhãn với các chi tiết như nhãn ở chai, lọ gốc.
5.1.7. Phải định kỳ kiểm tra nồng độ của các dung dịch chuẩn độ và dung dịch ion mẫu. Khi kiểm tra thuốc thử, nếu thấy có hiện tượng vẩn đục, kết tủa hay biến màu... thì không được sử dụng.
5.1.8. Nước cất và nước khử khoáng (nước trao đổi ion) phải được coi là một loại thuốc thử. Chúng cần được sản xuất và phân phối sao cho không có sự nhiễm tạp chất cũng như vi khuẩn. Nước phải được kiểm tra ít nhất một lần trong tháng để bảo đảm chất lượng nước đạt tiêu chuẩn dược điển.
5.1.9. Khi nhận thuốc thư, phải kiểm tra để đảm bảo các chai lọ còn nguyên niêm phong. Các thuốc thử bị nghi ngờ là không còn nguyên vẹn, kém phẩm chất hoặc giả mạo phải kiểm tra lại chất lượng. Nếu các kết quả định tính, định lượng và thử độ tinh khiết đạt thì có thể được chấp nhận sử dụng. Nếu không đạt, phải được hủy bỏ.
5.1.10. Tất cả các thuốc thử dự trữ nên được tập trung bảo quản tại kho trung tâm. Kho phải có các chai lọ sạch, phễu, thìa (muỗng), nhãn để tiện cho việc phân phối lẻ. Đối với các dung dịch có tính ăn mòn phải có một dụng cụ đặc biệt để phân phối lẻ. Ngoài kho chính nên có các kho phụ dành riêng cho các chất dễ cháy (ether, benzen, ethanol,...), các chất dễ phát nổ (các kim loại natri, kali,...), các acid đậm đặc dễ bay hơi (acid hydrocloric, acid nitric) và các base, hợp chất amin dễ bay hơi (amoniac, DEA, TEA, bromine...).
Khu vực kho phải đặt ở vị trí có thể ngăn ngừa được hỏa hoạn và được trang bị các phương tiện phòng cháy, chữa cháy thích hợp. Để đảm bảo an toàn và tránh gây ô nhiễm cho phòng kiểm nghiệm, không nên tồn trữ thuốc thử trong phòng nếu không thực sự cần thiết.
5.2. Chất đối chiếu
Các chất đối chiếu dùng trong phòng kiểm nghiệm gồm có chất đối chiếu gốc và các chất đối chiếu thứ cấp được tạo ra trong phòng kiểm nghiệm.
5.2.1. Việc quản lý chất đối chiếu phải do một người chịu trách nhiệm chính và phải mở sổ theo dõi.
Sổ theo dõi chất đối chiếu phải ghi lại những thông tin sau: số thứ tự chất đối chiếu, tên chất đối chiếu, nguồn cung cấp, số lô hay mã nhận dạng (nếu có), công dụng chính (chất đối chiếu cho phổ hồng ngoại, chất đối chiếu tạp chất cho sắc ký lớp mỏng, sắc ký lỏng hiệu năng cao... ), quy cách đóng gói và điều kiện bảo quản.
5.2.2. Ngoài sổ theo dõi chất đối chiếu, tất cả các thông tin về đặc tính, thông số kỹ thuật của chất đối chiếu cũng được lưu lại trong một hồ sơ riêng. Đối với các chất đối chiếu thứ cấp được tạo ra tại phòng kiểm nghiệm, hồ sơ phải lưu lại các phương pháp và kết quả phân tích đánh giá chất đối chiếu, cũng như tên người thực hiện các phân tích này.
5.2.3. Tất cả các chất đối chiếu phải được bảo quản đúng điều kiện quy định và phải được đánh giá định kỳ theo quy trình đánh giá chất đối chiếu của ASEAN để bảo đảm không bị hư hỏng. Kết quả kiểm tra phải được lưu lại trong sổ theo dõi chất đối chiếu cùng với tên người kiểm tra.
5.2.4. Tất cả các loại chất đối chiếu thứ cấp phải được đóng gói theo nguyên tắc đủ để dùng cho một lần kiểm nghiệm nhằm loại trừ yếu tố môi trường ảnh hưởng đến sự ổn định của sản phẩm.
5.3. Súc vật thử nghiệm
5.3.1. Súc vật thử nghiệm phải được nuôi theo đúng những tiêu chuẩn hiện hành về thuần chủng, thức ăn, phương pháp chăm sóc...
5.3.2. Chuồng trại phải đảm bảo đúng tiêu chuẩn hiện hành.
5.3.3. Việc đánh giá chất luợng súc vật thử nghiệm phải được thực hiện định kỳ theo những quy trình thao tác chuẩn được ban hành chính thức.
6. Tiêu chuẩn chất lượng và phương pháp phân tích
6.1. Các tiêu chuẩn chất lượng dùng trong phòng kiểm nghiệm thường dựa vào các chuyên luận của Dược điển Việt Nam hiện hành, Dược điển các nước được Bộ Y tế Việt Nam công nhận và các tiêu chuẩn cơ sở. Các tiêu chuẩn theo Dược điển có thể được thay đổi, bổ sung trong lần xuất bản mới hay được thông báo bởi Hội đồng Dược điển. Riêng các tiêu chuẩn cơ sở muốn sửa đổi phải được sự chấp thuận của cơ quan kiểm nghiệm nhà nước và Cục Quản lý dược - Bộ Y tế.
6.2. Bộ phận lưu trữ tiêu chuẩn của một phòng kiểm nghiệm có trách nhiệm cập nhật và lưu giữ tất cả các tiêu chuẩn chất lượng cần thiết cho công tác kiểm nghiệm, gồm có:
a) Dược điển Việt Nam và các Dược điển nước ngoài, kể cả phụ lục, bản bổ sung và bản hiệu đính.
b) Các tiêu chuẩn chất lượng không có trong Dược điển, đối với những thuốc được kiểm nghiệm dựa trên tiêu chuẩn của nhà sản xuất. Các phương pháp kiểm nghiệm không có trong dược điển do phòng kiểm nghiệm nghiên cứu, ban hành.
6.3. Mỗi tiêu chuẩn cần được đánh số và ghi ngày để dễ dàng nhận ra bản mới nhất. Các bản gốc của tiêu chuẩn phải ghi ngày được duyệt bởi cấp trên hay trưởng đơn vị và có ghi chú về tình trạng của tiêu chuẩn. Tất cả những thay đổi hay hiệu đính phải được ghi vào bản gốc với tên của người hiệu đính và ngày tháng.
6.4. Các bản tiêu chuẩn gốc phải được lưu giữ tại bộ phận lưu trữ tiêu chuẩn. Chỉ dùng các bản sao cho phòng kiểm nghiệm. Các bản sao phải bảo đảm tính chính xác như bản gốc.
6.5. Phương pháp phân tích có thể được rút ra từ các tiêu chuẩn quốc gia hay quốc tế, từ các ấn bản khoa học hay do chính phòng kiểm nghiệm nghiên cứu, ứng dụng.
6.6. Việc chọn phương pháp phân tích phụ thuộc vào đặc thù của mỗi phòng kiểm nghiệm. Ngoại trừ các phương pháp đã được quy định trong các Dược điển chính thức, các phương pháp phân tích khác phải được thẩm định về độ tin cậy và tính chính xác trước khi đưa vào áp dụng chính thức trong phòng kiểm nghiệm. Nếu một phương pháp phân tích được chọn để thay thế một phương pháp có sẵn trong Dược điển, phải chứng minh được phương pháp này là tương đương hoặc ưu việt hơn phương pháp trong Dược điển và phải được Viện Kiểm nghiệm hoặc Phân Viện kiểm nghiệm chấp nhận bằng văn bản.
7. Mẫu thử
7.1. Lấy mẫu
7.1.1. Lấy mẫu là công đoạn nhằm chọn ra một phần của nguyên liệu hay thành phẩm để kiểm nghiệm. Để có kết luận chắc chắn về chất lượng của nguyên liệu hay thành phẩm muốn kiểm tra, việc lấy mẫu phải được tiến hành một cách khoa học và đúng kỹ thuật.
7.1.2. Đối tượng lấy mẫu là các nguyên liệu dùng làm thuốc, các dạng thành phẩm bào chế và các sản phẩm được coi là thuốc dùng trong ngành y tế. Việc lấy mẫu được thực hiện trong các trường hợp tự kiểm tra chất lượng tại các cơ sở sản xuất, trong quá trình bảo quản, lưu thông phân phối thuốc và trong các trường hợp thanh tra, kiểm tra chất lượng thuốc trên thị trường.
7.1.3. Trường hợp tự kiểm tra chất lượng thuốc của các cơ sở sản xuất, bảo quản kinh doanh phân phối thuốc: Việc lấy mẫu do cán bộ chuyên môn của bộ phận kiểm tra chất lượng tiến hành có sự chứng kiến của cán bộ ở bộ phận được lấy mẫu
7.1.4. Trường hợp thanh tra, kiểm tra, giám sát chất lượng thuốc trong sản xuất và lưu thông phân phối:
- Việc lấy mẫu được tiến hành theo kế hoạch kiểm tra, giám sát chất lượng thuốc đã được Bộ Y tế và các Sở Y tế quy định, giao cho các cơ quan kiểm tra chất lượng các cấp thực hiện tại các cơ sở. Tuỳ trường hợp, mỗi loại thuốc cần được lấy mẫu theo một kế hoạch đã định, ưu tiên cho các loại thuốc có tầm quan trọng trong việc điều trị và các thuốc có hiện tượng chất lượng không ổn định.
- Việc lấy mẫu cũng được tiến hành trong trường hợp có những thông tin về thuốc kém chất lượng, không an toàn, ít hiệu lực, có sự khiếu nại của người sử dụng và các trường hợp nghi ngờ có lưu hành thuốc giả. Việc lấy mẫu do thanh tra viên, cán bộ của các cơ quan quản lý, kiểm tra chất lượng nhà nước trực tiếp thực hiện, có sự chứng kiến của cán bộ ở cơ sở được lấy mẫu.
7.1.5. Nơi lấy mẫu là nơi sản xuất, bảo quản hay lưu thông phân phối thuốc. Vị trí lấy mẫu phải sạch, môi trường xung quanh không được gây nhiễm bẩn vào mẫu hoặc tác động làm thay đổi tính chất của mẫu như nhiệt độ, độ ẩm, ánh sáng... đồng thời không để mẫu tác động tới môi trường, nhất là trường hợp mẫu là các sản phẩm bay hơi độc hại.
7.1.6. Người lấy mẫu phải là cán bộ có hiểu biết về phân tích hoặc kiểm nghiệm thuốc, nắm được các văn bản pháp quy về quản lý chất lượng thuốc, các thủ tục pháp lý và các thao tác kỹ thuật lấy mẫu.
7.1.7. Kiểm tra khi lấy mẫu:
Trước khi lấy mẫu phải xem xét bao bì bên ngoài, nếu có hiện tượng hư hỏng thì phân loại để riêng. Quan sát sản phẩm bên trong nếu không đồng nhất hay hư hỏng từng phần, biến đổi màu sắc, ẩm ướt, đóng vón, kết tủa... thì chia ra nhiều phần, phần nào có tính chất giống nhau thì làm một lô riêng. Tất cả các nhận xét về cảm quan của mẫu phải được ghi vào biên bản lấy mẫu.
Phải có đủ dụng cụ lấy mẫu. Dụng cụ lấy mẫu phải có hình dạng và làm bằng vật liệu thích hợp cho việc lấy mẫu, phải sạch, khô, không ảnh hưởng tới tính chất lý hóa của mẫu cũng như không gây nhiễm cho sản phẩm.
Đồ đựng mẫu phải sạch, khô, kín không ảnh hưởng tới tính chất của mẫu. Sau khi lấy mẫu phải đóng gói ngay và phải có nhãn ghi rõ tên cơ sở được lấy mẫu, tên sản phẩm, số lô, cỡ mẫu, ngày và nơi lấy mẫu, tên người lấy mẫu.
7.1.8. Thao tác lấy mẫu phải thận trọng, tỉ mỉ, tránh gây nhiễm. Chú ý quan sát các tình trạng bất thường, các dấu hiệu hư hỏng của sản phẩm. Tất cả các quan sát này đều phải được ghi lại trong biên bản lấy mẫu.
7.1.9. Phương thức lấy mẫu: người lấy mẫu phải tự tay lấy mẫu, ghi nhãn, làm biên bản, đóng gói, niêm phong và bảo quản mẫu.
7.1.10. Số lượng mẫu lấy phải đủ để lặp lại các thử nghiệm (ít nhất 3 lần thử) và để lưu mẫu. Số lượng mẫu lấy của từng dạng thuốc có thể tham khảo mục A: Cơ số mẫu lấy để kiểm tra chất lượng, Phần IV: Phụ lục.
7.1.11. Trình tự chế mẫu
- Từ lô sản xuất, lấy ra một cách ngẫu nhiên các đơn vị đóng gói, mở bao bì để lấy các mẫu ban đầu ở từng đơn vị đóng gói.
- Trộn đều các mẫu ban đầu và gộp lại thành những mẫu riêng của từng đơn vị lấy mẫu.
- Trộn đều các mẫu riêng thành một mẫu chung.
- Từ mẫu chung lấy ra một lượng mẫu cuối cùng để thử nghiệm. Mẫu này chia làm 2 phần bằng nhau, một phần để làm mẫu phân tích và một phần làm mẫu lưu (trường hợp cơ sở tự kiểm tra hoặc cơ quan kiểm tra giám sát chất lượng lấy mẫu bình thường); hoặc chia làm 4 phần bằng nhau: một phần lưu tại cơ sở được lấy mẫu, hai phần do thanh tra viên lấy về để thử và lưu ở bộ phận kiểm nghiệm, một phần do thanh tra viên lấy về để lưu tại cơ quan quản lý (trường hợp lấy mẫu thanh tra khi có vấn đề về chất lượng của thuốc).
7.2. Nhận mẫu
7.2.1. Mẫu và các hồ sơ kèm theo được gửi đến bộ phận đăng ký mẫu. Bộ phận đăng ký mẫu phải kiểm tra mẫu theo các yêu cầu sau:
- Tình trạng niêm phong của mẫu, tình trạng bao bì và nhãn có ghi đầy đủ các thông tin cần thiết như: tên sản phẩm, nơi sản xuất, cỡ mẫu, nồng độ/hàm lượng, số lô, số đăng ký, hạn dùng, điều kiện bảo quản.
- Nếu là mẫu do cơ sở gửi đến phải có phiếu yêu cầu kiểm nghiệm với các chi tiết sau:
• Tên đơn vị gửi mẫu
• Tên cơ sở sản xuất
• Tên sản phẩm, dạng bào chế, nồng độ, hàm lượng
• Số lô và hạn dùng
• Cỡ mẫu
• Lý do kiểm nghiệm
• Yêu cầu kiểm nghiệm
• Tiêu chuẩn áp dụng
• Ngày giao, nhận mẫu
• Tên người giao và nhận mẫu
• Nhận xét tình trạng mẫu gửi
7.2.2. Tất cả mẫu mới nhận và các hồ sơ kèm theo phải được đánh số để tiện theo dõi. Thuốc khác loại, khác dạng bào chế hoặc cùng loại nhưng khác số lô phải có số khác nhau.
7.2.3. Bộ phận đăng ký mẫu phải mở sổ theo dõi, ghi lại tất cả các chi tiết liên quan: số thứ tự của mẫu, ngày nhận mẫu, ngày chuyển mẫu đến các phòng chuyên môn
7.2.4. Mẫu trước khi kiểm nghiệm, mẫu lưu và phần còn lại của mẫu sau khi đã kiểm nghiệm phải được bảo quản phù hợp. Khi chuyển mẫu cho phòng chuyên môn phải kèm theo bản sao tất cả các hồ sơ, tài liệu cần thiết.
7.2.5. Trường hợp mẫu được gửi qua đường bưu điện, cơ quan nhận mẫu phải kiểm tra niêm phong và các thủ tục quy định, sau đó thông báo cho nơi gửi mẫu.
7.3. Lưu mẫu
7.3.1. Mẫu lưu phải có cùng nguồn gốc (lấy từ cùng một lô hàng, lấy cùng một thời điểm) với mẫu thử và được bảo quản trong phòng kiểm nghiệm theo đúng điều kiện quy định, mẫu lưu sẽ được phân tích lại trong trường hợp có tranh chấp về kết quả kiểm nghiệm.
7.3.2. Mẫu lưu phải được lưu trữ đầy đủ bởi bộ phận đăng ký mẫu và có cùng số đăng ký với mẫu thử. Số lượng mẫu lưu tùy thuộc vào số lần lặp lại thử nghiệm, ít nhất phải đủ cho 3 lần kiểm nghiệm tất cả các chỉ tiêu trong tiêu chuẩn chất lượng
7.3.3. Khi kết thúc thử nghiệm, tất cả các chai lọ, hộp, gói đựng mẫu phải được hàn kín ghi ngày bắt đầu lưu và chuyển đến nơi lưu mẫu. Mẫu lưu cần được đặt trong tủ có khóa an toàn và bảo quản đúng điều kiện quy định, chú ý các điều kiện bảo quản đặc biệt (nhiệt độ, độ ẩm, ánh sáng...).
7.3.4. Đối với các cơ sở sản xuất, lưu thông phân phối thuốc, mẫu thuốc được lưu ít nhất 03 tháng sau khi hết hạn dùng của thuốc. Đối với các cơ quan kiểm nghiệm, thời gian lưu mẫu không được dưới 02 năm kể từ ngày lấy mẫu hoặc cơ sở gửi mẫu tới. Khi hết thời gian lưu, cơ quan kiểm nghiệm tổ chức hủy mẫu và lập biên bản hủy mẫu theo đúng quy định.
8. Thử nghiệm và đánh giá kết quả
8.1. Thử nghiệm
8.1.1. Việc kiểm nghiệm mẫu phải được tiến hành càng sớm càng tốt kể từ khi hoàn tất việc ghi chép ban đầu (số đăng ký, tên mẫu thử nghiệm...) để đảm bảo mẫu không bị biến đổi chất lượng so với thời điểm lấy mẫu. Nếu không thể kiểm nghiệm ngay được, cán bộ kiểm nghiệm phải ghi chú trong hồ sơ kiểm nghiệm và tạm cất mẫu trong tủ có khóa, đảm bảo đúng điều kiện bảo quản quy định về nhiệt độ, độ ẩm, ánh sáng...
8.1.2. Thử nghiệm được tiến hành dựa trên các quy trình có sẵn trong các chuyên luận của Dược điển hay tiêu chuẩn cơ sở. Trước khi tiến hành phải tính toán các bước thử thích hợp để tiết kiệm mẫu, tránh tình trạng làm hết mẫu mà không kết luận được.
8.1.3. Khi kết quả thu được rõ ràng, tin cậy:
a) Không cần lặp lại thử nghiệm đối với:
- Các phân tích định tính dựa trên phép so màu, phản ứng kết tủa, phổ hồng ngoại, phổ tử ngoại, sắc ký lớp mỏng.
- Các thử nghiệm về độ tinh khiết, giới hạn tạp chất dựa trên phép so màu hoặc so độ đục, sắc ký lớp mỏng.
b) Luôn luôn phải lặp lại thử nghiệm ít nhất hai lần và lấy giá trị trung bình đối với:
- Các phân tích định lượng cho dù bằng phương pháp nào (chuẩn độ, cân khối lượng, đo quang, quang phổ tử ngoại, sắc ký khí, sắc ký lỏng hiệu năng cao).
- Các đo lường nhằm xác định tính chất vật lý như pH, năng suất quay cực, chỉ số khúc xạ, điểm nóng chảy...
8.1.4. Khi kết quả thu được không rõ ràng hoặc khi sai lệch giữa những lần lặp lại thử nghiệm vượt ra ngoài giới hạn cho phép:
a) Ít nhất phải lặp lại thử nghiệm 02 lần nữa và do một kiểm nghiệm viên khác tiến hành.
b) Nếu kết quả cho bởi 2 kiểm nghiệm viên không trùng khớp đối với cùng một mẫu thì phải tìm hiểu nguyên nhân, có thể do thao tác của kiểm nghiệm viên chưa thành thạo, thuốc thử hỏng, chất đối chiếu hỏng hoặc do thiết bị gây sai số, độ ẩm cao... Nếu xem xét thấy không phải vì các lý do trên thì kết quả trung bình của mỗi kiểm nghiệm viên được ghi riêng vào phiếu.
8.1.5. Mọi dữ liệu liên quan đến việc kiểm nghiệm mẫu đều phải được ghi vào hoặc kèm với hồ sơ kiểm nghiệm (số liệu cân, kết quả, đồ thị, sắc ký đồ, quang phổ đồ...)
8.2. Đánh giá kết quả phân tích
8.2.1. Khi đã hoàn thành các thử nghiệm, kiểm nghiệm viên phải đối chiếu kết quả thu được với các chỉ tiêu trong tiêu chuẩn quy định. Nếu kết quả kiểm nghiệm phù hợp với các yêu cầu của chỉ tiêu hay mức chất lượng trong tiêu chuẩn thì ghi kết luận đạt. Chỉ khi tất cả các chỉ tiêu đều đạt thì mẫu mới được kết luận là đạt phẩm chất theo tiêu chuẩn quy định.
8.2.2. Nếu có sự khác biệt giữa kết quả thu được và mức chỉ tiêu hay mức chất lượng trong tiêu chuẩn quy định thì mẫu sẽ được làm lại bởi một kiểm nghiệm viên khác hay bởi trưởng đơn vị.
Nếu kết quả kiểm nghiệm lần thứ 2 phù hợp với lần đầu thì kết quả đó được ghi vào phiếu và chuyển đến thủ trưởng đơn vị quyết định và ghi kết luận.
Trường hợp mẫu không đạt hoặc kết quả phân tích không lặp lại, thủ trưởng đơn vị (hay trưởng phòng kiểm nghiệm của doanh nghiệp) là người có thẩm quyền đưa ra kết luận sau cùng.
8.2.3 Trong trường hợp phòng kiểm nghiệm có nhiều đơn vị cùng tham gia kiểm mẫu, nên để đơn vị kiểm nghiệm chính đánh giá kết quả một cách tổng thể..
9. Hồ sơ và tài liệu
9.1. Hồ sơ kiểm nghiệm gồm có:
- Sổ tay kiểm nghiệm viên
- Hồ sơ phân tích
- Phiếu kiểm nghiệm, Phiếu phân tích.
9.1.1. Sổ tay kiểm nghiệm viên ghi lại các kết quả, các tính toán, số liệu và nhận xét có liên quan đến việc phân tích một mẫu. Sổ phải được đánh số trang và không được dùng bút chì để ghi chép, không được tẩy xoá, viết đè.
9.1.2. Hồ sơ phân tích phải có đầy đủ những thông tin về mẫu, phương pháp thử và kết quả phân tích. Hồ sơ phân tích được in sẵn với các chi tiết cần điền vào như sau:
- Số đăng ký của mẫu
- Tên mẫu
- Nơi sản xuất
- Số lô, hạn dùng, số đăng ký
- Người và nơi gửi mẫu
- Yêu cầu phân tích (số, ngày tháng và nội dung)
- Ngày nhận mẫu, người nhận mẫu
- Tiêu chuẩn và phương pháp kiểm nghiệm
- Tình trạng mẫu khi nhận và trước khi phân tích
- Kết quả phân tích (kể cả các phép tính toán)
Hồ sơ phân tích phải được lưu vào hồ sơ kiểm nghiệm cùng với các kết quả in ra từ máy phân tích tự động (phổ hồng ngoại, phổ tử ngoại, sắc ký đồ...). Phiếu phân tích phải có chữ ký của kiểm nghiệm viên và tên người giám sát.
9.1.3. Phiếu kiểm nghiệm hoặc phiếu phân tích xác nhận các kết quả phân tích và đưa ra kết luận cuối cùng về việc kiểm tra một mẫu bao gồm những thông tin sau:
a) Số đăng ký của mẫu
b) Tên mẫu
c) Nơi sản xuất
d) Số lô, hạn dùng, số đăng ký
e) Nơi gửi mẫu hoặc nơi lấy mẫu
f) Ngày nhận mẫu
g) Yêu cầu kiểm nghiệm (số, ngày tháng, nội dung)
h) Tiêu chuẩn kiểm nghiệm
i) Tình trạng mẫu khi nhận và khi kiểm nghiệm
j) Kết quả kiểm nghiệm
k) Kết luận mẫu đạt hay không đạt
l) Ngày kiểm nghiệm
m) Tên cơ quan kiểm nghiệm
n) Chữ ký của người có thẩm quyền
9.1.4. Hồ sơ kiểm nghiệm phải được lưu lại trong suốt thời hạn sử dụng của thuốc và theo các quy định hiện hành về lưu trữ hồ sơ tài liệu. Khi hết thời hạn lưu phải làm thủ tục hủy theo đúng quy định.
9.2. Quy trình thao tác chuẩn:
Phòng kiểm nghiệm phải có các quy trình thao tác chuẩn đã được người có thẩm quyền phê duyệt. Các quy trình này để hướng dẫn nhân viên tiến hành các thao tác chung như:
- Lấy mẫu, nhận mẫu và lưu mẫu
- Kiểm tra mẫu
- Nhận, sử dụng và bảo quản chất đối chiếu
- Vận hành, bảo trì, làm vệ sinh và hiệu chỉnh thiết bị
- Pha chế, dán nhãn và bảo quản thuốc thử
- Xử lý kết quả phân tích, báo cáo kết quả
- Xử lý chất thải
9.3. Các hồ sơ và tài liệu khác:
- Sổ nhận mẫu, lưu mẫu
- Tiêu chuẩn chất lượng
- Sổ theo dõi thuốc thử
- Sổ theo dõi chất đối chiếu
- Hồ sơ hiệu chuẩn thiết bị
10. An toàn trong phòng kiểm nghiệm
10.1. Các quy định chung
- Không được hút thuốc, ăn uống trong phòng kiểm nghiệm.
- Phòng kiểm nghiệm phải được trang bị các thiết bị như: bình cứu hỏa, tủ hút, vòi hoa sen, tủ thuốc cấp cứu,...
- Nhân viên phòng kiểm nghiệm phải biết sử dụng thành thạo tủ hút và các phương tiện phòng cháy, chữa cháy.
- Dây điện, thiết bị điện, tủ lạnh phải được cách điện, nối đất và phòng chống phát sinh tia lửa điện.
- Trong khi làm việc, kiểm nghiệm viên phải mặc áo choàng dùng cho phòng kiểm nghiệm hoặc các trang phục bảo hộ lao động thích hợp khác.
- Tất cả các bình đựng hóa chất phải được dán nhãn và ghi nhãn đặc biệt (ví dụ: “Độc”, “Dễ cháy”, “Ăn mòn”...).
- Kiểm nghiệm viên không được làm việc một mình trong phòng kiểm nghiệm.
- Tất cả nhân viên phòng kiểm nghiệm phải được huấn luyện về cách sơ cứu, cấp cứu và dùng chất giải độc.
10.2. Các phương tiện bảo hộ lao động như kính bảo hộ, khẩu trang, găng tay phải được trang bị đầy đủ. Phải dùng quả bóp cao su khi sử dụng pipet và ống siphon. Nhân viên phòng kiểm nghiệm phải được hướng dẫn cách sử dụng an toàn dụng cụ thủy tinh, hóa chất ăn mòn, các dung môi và các quy định về an toàn lao động trong khi pha chế hoặc tiến hành thử nghiệm. Khi tiến hành các phản ứng hóa học mạnh, nguy hiểm và khó kiểm soát như hòa lẫn nước với acid hay hỗn hợp acetone - chloroform với ammoniac, trộn các chất dễ cháy hay tác nhân oxy hóa... phải đặc biệt thận trọng và tuân theo đúng các hướng dẫn.
10.3. Hóa chất độc hại phải để riêng và dán nhãn cẩn thận. Tránh những tiếp xúc không cần thiết với thuốc thử, đặc biệt là dung môi và hơi dung môi. Hạn chế sử dụng các chất gây ung thư hoặc gây đột biến đã biết, nếu có thể các chất này phải được loại bỏ hoàn toàn. Cố gắng thay thế các thuốc thử và dung môi độc hại bằng các chất ít độc hơn, đặc biệt khi nghiên cứu các phương pháp thử mới.
10.4. Xử lý chất thải
- Cố gắng sử dụng hóa chất ít nhất có thể được nhằm giảm thiểu lượng chất thải.
- Tất cả các hóa chất, dung môi thải có thể gây ảnh hưởng xấu đến môi trường tuyệt đối không được thải trực tiếp vào hệ thống nước thải sinh hoạt mà phải được xử lý bằng những phương tiện, dụng cụ thích hợp, đặc biệt đối với các kim loại độc như thủy ngân, chì, arsen...
- Các hóa chất độc, ăn mòn, cháy nổ, các acid, base mạnh phải được vô hiệu hóa, làm loãng hoặc trung hòa trước khi thải.
Phần 3. HƯỚNG DẪN THỰC HIỆN
1. Quy định:
- Hệ thống kiểm tra chất lượng Nhà nước (Viện Kiểm nghiệm, Phân viện Kiểm nghiệm, Trung tâm Kiểm nghiệm dược phẩm, mỹ phẩm các tỉnh, thành phố trực thuộc Trung ương), hệ thống kiểm tra chất lượng của doanh nghiệp phải xây dựng kế hoạch từng bước đầu tư, nâng cấp Phòng kiểm tra chất lượng thuốc và triển khai áp dụng các nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”. Phấn đấu đến năm 2005, hầu hết các cơ sở kiểm tra chất lượng đạt nguyên tắc, tiêu chuẩn “Thực hành tốt phòng kiểm nghiệm thuốc”
- Các phòng kiểm nghiệm độc lập hay phòng kiểm nghiệm tư nhân phải đạt nguyên tắc tiêu chuẩn “Thực hành tốt phòng kiểm nghiệm thuốc” mới được cấp giấy phép hoạt động trong lĩnh vực kiểm tra chất lượng thuốc và mỹ phẩm ảnh hưởng trực tiếp đến sức khỏe con người.
2. Tổ chức thực hiện:
2.1. Đào tạo:
- Bộ Y tế (Cục Quản lý dược Việt Nam, Viện Kiểm nghiệm, Phân viện Kiểm nghiệm) tổ chức phổ biến, huấn luyện kiến thức về “Thực hành tốt phòng kiểm nghiệm thuốc” cho Trung tâm Kiểm nghiệm dược phẩm, mỹ phẩm các tỉnh, thành phố trực thuộc Trung ương, Y tế các ngành và hệ thống kiểm tra chất lượng của các doanh nghiệp.
- Các Trung tâm Kiểm nghiệm dược phẩm, mỹ phẩm các tỉnh, thành phố trực thuộc Trung ương, Y tế các ngành và hệ thống kiểm tra chất lượng của các doanh nghiệp có trách nhiệm nghiên cứu, huấn luyện và đào tạo các nguyên tắc, tiêu chuẩn “Thực hành tốt phòng kiểm nghiệm thuốc” cho tất cả cán bộ công nhân viên trong đơn vị.
2.2.[3] Kiểm tra và cấp giấy chứng nhận
2.2.1. Đăng ký kiểm tra:
Các cơ sở sau khi tự kiểm tra, đánh giá đạt nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”, gửi 01 bộ hồ sơ đăng ký kiểm tra đến Cục Quản lý dược - Bộ Y tế. Hồ sơ đăng ký bao gồm:
- Trường hợp đăng ký kiểm tra lần đầu:
(1) Bản đăng ký kiểm tra “Thực hành tốt phòng kiểm nghiệm thuốc” (mẫu 01/GLP Phụ lục số 1);
(2) Bản sao Giấy phép thành lập cơ sở hoặc Giấy đăng ký kinh doanh hoặc Giấy chứng nhận đầu tư có chữ ký của chủ cơ sở và đóng dấu xác nhận của cơ sở;
(3) Sơ đồ tổ chức, biên chế của cơ sở;
(4) Sơ đồ vị trí địa lý và thiết kế của phòng kiểm nghiệm;
(5) Danh mục thiết bị phân tích của cơ sở;
(6) Danh mục các loại phép thử (phương pháp) và/hoặc các loại sản phẩm cơ sở thực hiện kiểm tra chất lượng;
- Trường hợp đăng ký tái kiểm tra:
(1) Bản đăng ký tái kiểm tra “Thực hành tốt phòng kiểm nghiệm thuốc” (mẫu 02/GLP Phụ lục số 1);
(2) Bản sao Giấy phép thành lập cơ sở hoặc Giấy đăng ký kinh doanh hoặc Giấy chứng nhận đầu tư có chữ ký của chủ cơ sở và đóng dấu xác nhận của cơ sở;
(3) Báo cáo khắc phục các tồn tại trong kiểm tra lần trước;
(4) Báo cáo những thay đổi của cơ sở trong 03 năm triển khai “Thực hành tốt phòng kiểm nghiệm thuốc” và hồ sơ liên quan, nếu có.
2.2.2. Kiểm tra và cấp giấy chứng nhận
a) Thẩm quyền thẩm định hồ sơ, kiểm tra, cấp giấy chứng nhận và tiêu chuẩn cán bộ kiểm tra:
* Thẩm quyền:
- Bộ Y tế ra Quyết định thành lập đoàn kiểm tra và cấp Giấy chứng nhận đạt nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” đối với Viện Kiểm nghiệm thuốc Trung ương, Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh và Viện Kiểm định Quốc gia Vắc xin và Sinh phẩm y tế.
- Cục Quản lý dược ra Quyết định thành lập đoàn kiểm tra và cấp Giấy chứng nhận đạt nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” đối với các Trung tâm kiểm nghiệm dược phẩm, mỹ phẩm tỉnh, thành phố trực thuộc Trung ương, các cơ sở kiểm nghiệm của doanh nghiệp, các phòng kiểm nghiệm độc lập hay phòng kiểm nghiệm tư nhân.
* Tiêu chuẩn cán bộ kiểm tra:
- Cán bộ có trình độ đại học trở lên, có kinh nghiệm trong công tác quản lý dược nói chung và công tác kiểm tra chất lượng thuốc nói riêng.
- Có phương pháp thanh tra, kiểm tra khoa học, cương quyết; có khả năng phát hiện nhanh các sai sót của cơ sở đồng thời phải đưa ra được các biện pháp có tính thuyết phục giúp cơ sở khắc phục thiếu sót.
- Trung thực, khách quan và nghiêm chỉnh chấp hành các quy chế, quy định pháp luật trong quá trình kiểm tra.
- Có đủ sức khỏe, không mắc các bệnh truyền nhiễm.
b) Thẩm định hồ sơ và tổ chức kiểm tra
- Sau khi nhận hồ sơ đăng ký kiểm tra của cơ sở theo quy định tại điểm 2.2.1 Phần này, Cục Quản lý dược tiến hành thẩm định hồ sơ (có biên bản thẩm định).
- Trong vòng 05 ngày làm việc kể từ ngày nhận được hồ sơ và phí thẩm định theo quy định, cơ quan quản lý phải thông báo cho cơ sở về tình trạng hồ sơ nếu chưa đạt yêu cầu hoặc kế hoạch kiểm tra.
- Trong vòng 20 ngày làm việc kể từ ngày thông báo kế hoạch kiểm tra, cơ quan quản lý phải tiến hành kiểm tra thực tế tại cơ sở.
c) Kiểm tra, xử lý kết quả kiểm tra và cấp giấy chứng nhận
* Kiểm tra
- Đoàn kiểm tra có trách nhiệm kiểm tra toàn bộ các hoạt động của cơ sở theo các nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” và các quy định chuyên môn hiện hành.
- Các cơ sở đăng ký kiểm tra “Thực hành tốt phòng kiểm nghiệm thuốc” phải tiến hành báo cáo bằng sơ đồ, biểu đồ và các số liệu ngắn gọn về tình hình hoạt động, công tác triển khai áp dụng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”.
- Biên bản kiểm tra phải chỉ rõ các tồn tại trong việc triển khai áp dụng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” tại cơ sở. Trong trường hợp không nhất trí với ý kiến của đoàn kiểm tra, biên bản phải ghi rõ tất cả các ý kiến bảo lưu của cơ sở. Biên bản được phụ trách cơ sở cùng trưởng đoàn kiểm tra ký xác nhận; làm thành 03 bản: 01 bản lưu tại cơ sở, 02 bản lưu tại Cục Quản lý dược.
* Xử lý kết quả kiểm tra và cấp giấy chứng nhận:
- Trường hợp 1: Nếu cơ sở được kiểm tra đáp ứng các nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”, cơ quan quản lý có thẩm quyền theo quy định tại tiết a điểm 2.2.2 Phần này cấp Giấy chứng nhận đạt nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” trong vòng 05 ngày làm việc kể từ ngày kết thúc việc kiểm tra.
- Trường hợp 2: Đối với cơ sở được kiểm tra về cơ bản đáp ứng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” còn một số tồn tại nhưng không ảnh hưởng đến kết quả kiểm nghiệm và có thể khắc phục được trong thời gian ngắn, Đoàn kiểm tra sẽ yêu cầu cơ sở báo cáo khắc phục, sửa chữa.
Cơ sở phải khắc phục, sửa chữa và báo cáo kết quả khắc phục những tồn tại mà đoàn kiểm tra đã nêu ra trong biên bản gửi về Cục Quản lý dược.
Trưởng Đoàn kiểm tra tổng hợp, báo cáo người có thẩm quyền xem xét để cấp Giấy chứng nhận đạt nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” hoặc cơ quan quản lý có thẩm quyền phải có thông báo kết quả chính thức cho cơ sở trong vòng 05 ngày làm việc kể từ ngày nhận được báo cáo khắc phục.
Quá 02 tháng kể từ ngày kết thúc việc kiểm tra, nếu cơ sở không gửi báo cáo khắc phục hợp lệ thì phải tiến hành nộp hồ sơ đăng ký kiểm tra lại từ đầu.
- Trường hợp 3: Đối với cơ sở chưa đáp ứng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”, cơ sở phải tiến hành khắc phục sửa chữa các tồn tại. Sau khi tự kiểm tra đánh giá đạt yêu cầu, cơ sở tiến hành nộp hồ sơ lại từ đầu.
* Đăng ký tái kiểm tra
- Giấy chứng nhận đạt nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” có giá trị 03 năm kể từ ngày ký.
- Định kỳ 03 năm 01 lần, trước khi Giấy chứng nhận đạt nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc” hết hạn 02 tháng, cơ sở phải nộp hồ sơ đăng ký tái kiểm tra, trừ các trường hợp đột xuất do cơ sở hoặc Bộ Y tế (Cục Quản lý dược) yêu cầu.
- Cơ sở sau khi đã nộp hồ sơ đăng ký tái kiểm tra vẫn được phép kiểm nghiệm thuốc theo phạm vi quy định trong Giấy chứng nhận “Thực hành tốt phòng kiểm nghiệm thuốc” và Giấy chứng nhận đủ điều kiện kinh doanh thuốc (đang còn hiệu lực).
- Trong quá trình kiểm tra, nếu phát hiện cơ sở có các tồn tại ở mức độ ảnh hưởng nghiêm trọng tới kết quả kiểm nghiệm hay hoạt động của cơ sở, trưởng đoàn kiểm tra lập biên bản, báo cáo người có thẩm quyền ra quyết định xử lý chính thức.”
Phần 4. PHỤ LỤC
A. Cơ số mẫu lấy để kiểm tra chất lượng
Số lượng mẫu lấy = Mẫu phân tích + Mẫu lưu
1. Trường hợp lấy mẫu ở đầu nguồn (kho xí nghiệp, công ty) và ở cuối nguồn của khâu lưu thông phân phối (nhà thuốc, hiệu thuốc bán lẻ) :
| Số TT | Dạng bào chế | Quy cách | Số lượng lấy | |
| Tại kho công ty, xí nghiệp | Cuối nguồn | |||
| 1 | Thuốc viên - Thuốc thông thường
- Thuốc kháng sinh |
• 1 hoạt chất • ≥ 2 hoạt chất • 1 hoạt chất • ≥ 2 hoạt chất |
100 viên 200 viên 100 viên 200 viên |
50 viên 100 viên 50 viên 100 viên |
| 2 | Thuốc nước - Xiro và các loại thuốc nước đóng chai - ống uống |
• 125 ml • 200 ml trở lên • 5ml – 10ml |
15 chai 10 chai 100 ống |
8 chai 5 chai 50 ống |
| 3 | Hoàn, cốm | Các loại cứng hoặc mềm | 10 hộp | 5 hộp |
| 4 | Bột | Gói chứa hoạt chất tương đương với 01 hoặc 02 viên | 100 gói | 50 gói |
| 5 | Rượu thuốc | • chai 250 ml • chai 650 ml | 10 chai 8 chai | 5 chai 4 chai |
| 6 | Thuốc tiêm - Dịch truyền
- ống tiêm
- Nước cất |
• 250 ml – trở lên • 50 ml – 250 ml • ống 1ml • ống 2 ml trở lên • ống 2 ml • ống 5 ml • ống 10 ml |
10 chai 20 chai 300 ống 200 ống 500 ống 200 ống 150 ống |
5 chai 10 chai 150 ống 100 ống 250 ống 100 ống 80 ống |
| 7 | Thuốc mỡ, thuốc nhỏ mắt, nhỏ mũi, kem ... | Các loại | 50 lọ (tuýp) | 25 lọ (tuýp) |
| 8 | Thuốc bột tiêm | Các loại | 50 lọ | 25 lọ |
| 9 | Dầu xoa | • 1 – 2 ml • 5 ml | 30 lọ 20 lọ | 15 lọ 10 lọ |
| 10 | Cao thuốc | Các loại | 100 g | 50 g |
| 11 | Tinh dầu | Các loại | 150 ml |
|
| 12 | Nguyên liệu | • nguyên liệu quý • nguyên liệu kháng sinh • Nguyên liệu thuốc độc A, B • Nguyên liệu thường • Nhựa hạt | 2 g 30 g 10 g 50 g 200 g |
|
| 13 | Dây truyền dịch | Các loại | 30 bộ |
|
| 14 | ống thủy tinh rỗng | • 2 ml • 5 ml | 500 ống 300 ống |
|
| 15 | Chai đựng dịch truyền | Các loại | 10 chai |
|
Ghi chú
* Việc lấy mẫu thuốc để kiểm tra chất lượng, các cơ sở đi lấy mẫu cần tính toán số lượng mẫu đủ để kiểm tra chất lượng và lưu mẫu .
* Trường hợp lấy mẫu ở cuối nguồn (nhà thuốc, hiệu thuốc bán lẻ ...) mà không đủ cơ số như quy định ở bảng trên có thể áp dụng cách sau đây đối với thuốc viên (viên nén, bao hay viên nang) :
Khi định lượng hoạt chất trong thuốc viên có thể tiến hành trên một số lượng mẫu ít hơn 20 viên, nhưng không được dưới 05 viên. Khi đó, giới hạn hàm lượng hoạt chất được phép vượt ra ngoài giới hạn quy định một tỷ lệ theo bảng sau :
| Khối lượng hoạt chất trong mỗi viên | Trừ đi % giới hạn dưới nếu mẫu có số viên là | Cộng thêm % giới hạn trên nếu mẫu có số viên là | ||||
| 15 | 10 | 5 | 15 | 10 | 5 | |
| < 0,12g | 0,2 | 0,7 | 1,6 | 0,3 | 0,8 | 1,8 |
| 0,12 – 0,3 g | 0,2 | 0,5 | 1,2 | 0,3 | 0,6 | 1,5 |
| > 0,3 g | 0,1 | 0,2 | 0,8 | 0,2 | 0,4 | 1,0 |
Bảng trên chỉ áp dụng cho thuốc viên có giới hạn hàm lượng hoạt chất trong khoảng từ 90 – 110%. Đối với thuốc viên có giới hạn hàm lượng hoạt chất khác hơn 90 và 110%, một khoảng sai số tỷ lệ thuận với hàm lượng sẽ được áp dụng.
2. Trường hợp lấy mẫu để tự kiểm tra chất lượng tại cơ sở sản xuất.
Cơ số mẫu lấy để kiểm tra chất lượng ở các cơ sở sản xuất cũng phải đảm bảo đủ số lượng để phân tích và lưu mẫu. Dựa trên tiêu chuẩn chất lượng của từng loại thuốc cụ thể, tính toán số lượng mẫu cần lấy để làm toàn bộ các chỉ tiêu trong tiêu chuẩn chất lượng, từ đó tính được số lượng mẫu phải lấy đủ cho 3 lần thử và để lưu mẫu.
B. Tần số hiệu chỉnh thiết bị phân tích dùng trong phòng kiểm nghiệm
| THIẾT BỊ/ HIỆU CHỈNH | PHƯƠNG PHÁP | TẦN SỐ | ||
| Mỗi khi sử dụng | Hàng ngày | Mỗi ... tháng | ||
| Nhiệt kế | Dùng nhiệt kế chuẩn |
|
| 12 tháng |
| Micrometer | Dùng thiết bị đo độ dày chuẩn |
|
| 6 |
| Máy đo độ cứng | Dùng thiết bị thử nghiệm chức năng chuẩn |
|
| 6 |
| Khúc xạ kế | Dùng nước đã khử ion |
|
| 12 |
| Máy đo điểm chảy | Dùng các mẫu điểm chảy chuẩn (Dược điển Mỹ 23 ) |
|
| 12 |
| Nhớt kế | Sử dụng các dầu chuẩn |
|
| 12 |
| Thiết bị Karl-Fischer | Xác định hệ số | 1 |
|
|
| Thiết bị đo độ tan rã | Kiểm tra chu kỳ / phút, độ dài của một nhịp |
|
| 3 |
| Thiết bị đo độ mài mòn | Kiểm tra số vòng quay / phút |
|
| 6 |
| Phân cực kế | Dùng dung dịch chuẩn |
|
| 6 |
| Lò nung | Kiểm tra nhiệt độ |
|
| 12 |
| Tủ ấm / tủ mát / tủ lạnh | Kiểm tra nhiệt độ tối đa, tối thiểu hay theo dõi biến thiên nhiệt độ qua biểu đồ |
| 1 |
|
| Cân phân tích - Cân chính xác vi phân tích - Cân phân tích - Bảo trì và hiệu chỉnh |
Quả cân chuẩn Quả cân chuẩn Bởi các công ty kiểm định |
1 |
|
1 6 |
| Quang phổ tử ngoại khả kiến - Độ chính xác của bước sóng. - Tính tuyến tính của sự đáp ứng |
Kính lọc Holmium oxide Dung dịch chuẩn |
|
|
6 6 |
| Quang phổ hồng ngoại - Độ chính xác của bước sóng/ Độ phân giải |
Màng Polystyrene |
|
|
3 |
| Quang phổ hấp thu nguyên tử - Tính tuyến tính của sự đáp ứng |
Chất chuẩn pha loãng |
1 |
|
|
| pH kế - Bảo trì và hiệu chỉnh | Dung dịch đệm Bởi các công ty kiểm định | 1 |
|
6 |
| Huỳnh quang kế - Tính tuyến tính |
Chất chuẩn pha loãng |
|
|
12 |
| Sắc ký lỏng hiệu năng cao - Pha động - Tốc độ dòng - Bước sóng của detector - Độ lặp lại của hệ thống |
Thể tích Kiểm tra lưu lượng Dung dịch chuẩn Thực hiện nhiều lần tiêm |
1 |
|
3 3 12 |
| Sắc ký khí - Tốc độ dòng khí - Độ lặp lại của hệ thống |
Lưu lượng kế Thực hiện nhiều lần tiêm |
1 |
|
3 |
| Máy thử độ hòa tan - Tốc độ mái chèo - Kiểm soát nhiệt độ - Hiệu chỉnh |
Đo lường Đo lường Dùng chuẩn để hiệu chỉnh (Dược điển Mỹ 23 ) |
1 1 |
|
12 |
C. Hiệu chỉnh dụng cụ thủy tinh chính xác dùng trong phòng kiểm nghiệm
Nguyên tắc chung là cân khối lượng nước cất chứa trong các dụng cụ đó rồi quy ra thể tích chính xác. Đối với một số dụng cụ thủy tinh có kích thước rất nhỏ hoặc có hính dáng lạ thường người ta dùng thủy ngân thay vì nước.
Ở nhiệt độ khoảng 20°C, nước tinh khiết giãn nở xấp xỉ 0,02% khi nhiệt độ tăng 10°C. Điều này có ý nghiã đối với việc hiệu chỉnh các dụng cụ thủy tinh chính xác và đối với các thuốc thử có nồng độ thay đổi theo nhiệt độ.
Tỷ trọng của nước
|
| Thể tích của 1 g nước (cm³) | ||
| Nhiệt độ (°C) | Tỷ trọng (g / cm³) | ở nhiệt độ thử nghiệm | ở 20°C |
| 0 4 5 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 35 37 40 100 | 0,9998425 0,9999750 0,9999668 0,9997026 0,9996081 0,9995004 0,9993801 0,9992474 0,9991026 0,9989460 0,9987779 0,9985986 0,9984082 0,9982071 0,9979955 0,9977735 0,9975415 0,9972995 0,9970479 0,9967867 0,9965162 0,9962365 0,9959478 0,9956502 0,9940349 0,9933316 0,9922187 0,9583665 | - - - 1,0014 1,0015 1,0016 1,0017 1,0018 1,0020 1,0021 1,0023 1,0025 1,0027 1,0029 1,0031 1,0033 1,0035 1,0038 1,0040 1,0043 1,0046 1,0048 1,0051 1,0054 - - - - | - - - 1,0015 1,0016 1,0017 1,0018 1,0019 1,0020 1,0021 1,0023 1,0025 1,0027 1,0029 1,0031 1,0033 1,0035 1,0038 1,0040 1,0042 1,0045 1,0047 1,0050 1,0053 - - - - |
Thủy tinh cũng giãn nở khi bị đun nóng. ở nhiệt độ phòng, thủy tinh pyrex và borosilicate giãn nở khoảng 0,001% khi nhiệt độ tăng 10°C. Có nghĩa là một bình thủy tinh khi bị đun nóng lên 10°C thể tích của nó sẽ tăng khoảng 10 x 0,001% = 0,01%. Trong thực tế sự giãn nở này là không đáng kể. Thủy tinh thô ứng dụng giãn nở nhiều gấp 2 đến 3 lần so với thủy tinh borosilicate.
1- Hiệu chỉnh bình định mức
- Bình định mức phải hoàn toàn sạch và khô trước khi tiến hành hiệu chỉnh. Cân bình định mức sau đó đổ đầy nước khử ion đã loại CO2 đến trên vạch quy định. Phần cổ bình phía trên mực nước phải khô. Lấy bớt nước cho tới đúng vạch sau đó cân lại bình. Khối lượng nước trong bình được quy về khối lượng ở nhiệt độ chuẩn từ đó suy ra thể tích của bình.
- Bình định mức cũng có thể được hiệu chỉnh bằng phương pháp đo quang.
2- Hiệu chỉnh buret
Đổ đầy buret với nước khử ion đã loại CO2 cho tới trên vạch quy định (chú ý tránh bọt khí ). Kiểm tra nước không đọng lại trên thành buret bằng cách mở cho nước chảy ra. Nếu có, rửa sạch buret với nước và xà bông hoặc ngâm buret trong hỗn hợp acid sulfuric – peroxydisulfate. Chỉnh mực nước về tới vạch 0. Lấy đi giọt nước dư ở đầu buret bằng cách chạm đầu buret vào thành một ly bằng thủy tinh.
Cân một bình định mức sạch, khô có nút đậy. Chú ý không chạm tay vào bình vì dấu ngón tay có thể ảnh hưởng tới khối lượng bình. Mở buret cho chảy nước vào bình đã cân bì, đậy chặt nút tránh bay hơi. Chờ 30 giây trước khi đọc mực nước trên buret.
Cân bình định mức để biết khối lượng nước. Tra bảng để đổi khối lượng nước ra thể tích tương ứng.
3- Hiệu chỉnh pipet và micropipet
3.1. Phương pháp cân
Dùng pipet lấy một lượng nước khử ion đã loại CO2 cho tới vạch quy định. Ghi lại nhiệt độ của nước (t)
Cân một lọ sạch, khô có nút đậy (cân chính xác đến 0,1mg). Chú ý không chạm tay vào lọ (Wv).
Chuyển nước từ pipet vào lọ. Đậy nút và cân chính xác đến 0,1mg (Wf)
Tra bảng để tìm yếu tố hiệu chỉnh cho nhiệt độ của nước (Ft)
Tính toán theo công thức sau :
Thể tích thực V (ml) = (Wf – Wv) x Ft
Ví dụ: pipet 10 ml
Wf = 31,9961g
Wv = 22,0391g
t = 240C
Ft = 1,003771 (tra bảng )
Thể tích thực = ( 31,9961 – 22,0391 ) x 1,003771 = 9,9945 ml
Chênh lệch so với thể tích lý thuyết:
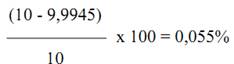
Thông thường chênh lệch ( ≤ 0,1 % là chấp nhận được.
3.2. Phương pháp đo quang
- Tất cả các dụng cụ thủy tinh phải thuộc loại A.
- Thuốc thử
• NaOH 0,01 mol / l
• P – nitrophenol 105 mg / định lượng
Trong một bình định mức 100 ml hòa tan 105 mg p – nitrophenol tinh khiết (NIST SRM 938) trong nước khử ion. Thêm nước tới vạch.
- Pha dung dịch đối chiếu và dung dịch thử
• Dung dịch đối chiếu: trong 3 bình định mức có chứa 250 ml NaOH 0,01 mol/l, thêm vào mỗi bình 1 ml p – nitrophenol, dùng 3 pipet riêng.
• Dung dịch thử: trong 5 ống nghiệm chứa 2,5 ml NaOH 0,01 mol / l thêm vào mỗi ống 10μl dung dịch p – nitrophenol dùng micropipet muốn hiệu chỉnh. Nếu pipet là loại TD (to-deliver) thì phải tráng pipet với p – nitrophenol.
• Đọc độ hấp thu của dung dịch đối chiếu và dung dịch thử, cốc đo 10 mm, bước sóng 401 nm dùng quang phổ kế dải hẹp.
• Đối với p – nitrophenol NIST SRM 131,48 l x g-1 x cm-1 trong NaOH 0,01 mol / l thì A401 = 0,550.
• Độ hấp thu trung bình của 3 dung dịch đối chiếu A1 phải xấp xỉ 0,550.
• Độ hấp thu trung bình của 5 dung dịch thử là A2.
• Tính toán
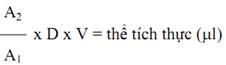
Trong đó:
D = độ pha loãng của dd thử (1/251)
V = thể tích (μl) của dd thử (2510 μl)
Nếu A1 = 0,550; A2 = 0,561
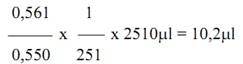
Sai số sẽ là 2%. Thông thường sai số từ 0,5% đến 1,0% là chấp nhận được.
D. Thẩm định phương pháp phân tích
Tất cả các phương pháp phân tích phải được thẩm định kể cả những phương pháp phân tích mới chưa có trong dược điển hoặc những phương pháp sẽ được thay đổi.
Các yếu tố cần được thẩm định đối với một phương pháp phân tích thể hiện trong bảng sau:
| Giới hạn tạp chất | Phân tích định tính | Thử độ tinh khiết | Phân tích định lượng 1. Hàm lượng 2. Độ hòa tan | |
| Định lượng | Giới hạn tạp chất | |||
| Độ đúng | - | + | - | + |
| Độ lặp lại | - | + | - | + |
| Tính đặc hiệu | + | + | + | + |
| Giới hạn phát hiện | - | - | + | - |
| Tính tuyến tính | - | + | - | + |
Các phương pháp phân tích trong Dược điển không cần thẩm định nhưng có thể phải kiểm tra lại.
1. Tính tuyến tính
Tính tuyến tính là sự liên quan giữa diện tích đỉnh hoặc chiều cao đỉnh với nồng độ của một loạt các dung dịch pha loãng của chất đối chiếu. Các nồng độ này nằm trong khoảng từ 40 – 140% so với nồng độ của hoạt chất. Tối thiểu phải xác định trên 06 nồng độ khác nhau. Trong trường hợp sắc ký lỏng cao áp, phải thực hiện tiêm nhiều lần và lấy giá trị trung bình.
2. Độ đúng
Độ đúng được suy ra từ cách tính toán tính tuyến tính ở trên và kết quả không được sai lệch đáng kể ( nghĩa là trong khoảng 95% giới hạn tin cậy được).
3. Tính đặc hiệu (hay còn gọi tính chọn lọc)
Một phương pháp được coi là đặc hiệu nếu tiến hành thử trên mẫu có lẫn tạp chất vẫn cho ra kết quả (đỉnh, thời gian lưu...) giống như khi thử trên mẫu không có tạp chất
4. Độ lặp lại
Độ lặp lại được tính toán từ ít nhất 7 lần phân tích trên cùng một mẫu thử trong cùng một điều kiện. Trường hợp sắc ký lỏng cao áp, thực hiện tiêm nhiều lần và lấy giá trị trung bình.
Phương pháp được coi là chính xác nếu độ lệch tương đối của độ lặp lại ≤ 2%.
5. Giới hạn phát hiện
Đó là kết quả đơn nhỏ nhất có thể đọc được khi so với một mẫu trắng. Trường hợp đối với phương pháp sắc ký lỏng hiệu năng cao giới hạn này tương ứng với 2 lần RSD của nhiễu. Trong phương pháp sắc ký lớp mỏng là chấm nhỏ nhất có thể phát hiện được.
6. Độ thô:
Độ thô là khả năng của qui trình cung cấp các kết quả phân tích có độ chính xác và độ đúng chấp nhận được dưới những điều kiện có sự thay đổi nhỏ về độ ổn định của các dung dịch phân tích, thời gian chiết xuất, trị số pH, nhiệt độ thành phần của hệ dung môi sắc ký, cột sắc ký do các nhà cung cấp khác nhau....Các phương pháp đưa vào Dược diển phải kiểm tra độ thô.
Người ta phân tích các mẫu riêng biệt hầu như giống nhau lấy từ một lô sản phẩm đồng nhất dưới những điều kiện thao tác và môi trường thay đổi, và tính toán độ đúng và độ chính xác. Nếu các kết quả thu được đạt yêu cầu về độ đúng và độ chính xác và vẫn phù hợp với các chỉ tiêu kỹ thuật trong qui trình thì độ thô của qui trình xem như đạt yêu cầu.
E. Hồ sơ đăng ký kiểm tra
"Thực hành tốt phòng kiểm nghiệm thuốc" (GLP)
1[4]. Mẫu số 01/GLP: Mẫu đơn đăng ký kiểm tra “Thực hành tốt phòng kiểm nghiệm thuốc”
| Tên đơn vị chủ quản Tên đơn vị | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
|
| …, ngày.... tháng..... năm..... |
ĐƠN ĐĂNG KÝ KIỂM TRA “THỰC HÀNH TỐT PHÒNG KIỂM NGHIỆM THUỐC”
Kính gửi: Cục Quản lý dược - Bộ Y tế
1. Tên cơ sở:
2. Địa chỉ (trụ sở chính và phòng kiểm nghiệm)
3. Điện thoại: Fax: E-Mail:
4. Quyết định thành lập cơ sở (hoặc Giấy đăng ký kinh doanh hoặc Giấy chứng nhận đầu tư) số............... do................. cấp.
Thi hành Quyết định số 1570/2000/QĐ-BYT ngày 22/5/2000 của Bộ trưởng Bộ Y tế về việc triển khai áp dụng nguyên tắc, tiêu chuẩn “Thực hành tốt phòng kiểm nghiệm thuốc”, sau khi tiến hành tự thanh tra, cơ sở chúng tôi xin đăng ký với Bộ Y tế (Cục Quản lý dược) được kiểm tra “Thực hành tốt phòng kiểm nghiệm thuốc” vào bất kỳ thời gian nào và cam kết khắc phục kịp thời những tồn tại ghi trong biên bản kiểm tra.
Chúng tôi xin gửi kèm bản đăng ký này các tài liệu liên quan sau đây:
(1) Bản sao Giấy phép thành lập cơ sở hoặc Giấy đăng ký kinh doanh hoặc
Giấy chứng nhận đầu tư có xác nhận của cơ sở;
(2) Sơ đồ tổ chức, biên chế của cơ sở;
(3) Sơ đồ vị trí địa lý và thiết kế của phòng kiểm nghiệm;
(4) Danh mục thiết bị phân tích của cơ sở;
(5) Danh mục các loại phép thử (phương pháp) và/hoặc các loại sản phẩm cơ sở thực hiện kiểm tra chất lượng./.
|
| Phụ trách đơn vị |
2.[5] Mẫu số 02/GLP: Mẫu đơn đăng ký tái kiểm tra GLP
| Tên đơn vị chủ quản Tên đơn vị | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
|
| …, ngày.... tháng..... năm..... |
ĐƠN ĐĂNG KÝ TÁI KIỂM TRA “THỰC HÀNH TỐT PHÒNG KIỂM NGHIỆM THUỐC”
Kính gửi: Cục Quản lý dược - Bộ Y tế
1. Tên cơ sở:
2. Địa chỉ (trụ sở chính và phòng kiểm nghiệm)
3. Điện thoại: Fax: E-Mail:
4. Quyết định thành lập cơ sở (hoặc Giấy đăng ký kinh doanh hoặc Giấy chứng nhận đầu tư) số............... do................. cấp.
Thi hành Quyết định số 1570/2000/QĐ-BYT ngày 22/5/2000 của Bộ trưởng Bộ Y tế về việc triển khai áp dụng nguyên tắc, tiêu chuẩn “Thực hành tốt phòng kiểm nghiệm thuốc”, sau khi tiến hành tự thanh tra, cơ sở chúng tôi xin đăng ký với Bộ Y tế (Cục Quản lý dược) được tái kiểm tra “Thực hành tốt phòng kiểm nghiệm thuốc” vào bất kỳ thời gian nào và cam kết khắc phục kịp thời những tồn tại ghi trong biên bản kiểm tra.
Chúng tôi xin gửi kèm bản đăng ký này các tài liệu liên quan sau đây:
(1) Bản sao Giấy phép thành lập cơ sở hoặc Giấy đăng ký kinh doanh hoặc Giấy chứng nhận đầu tư có xác nhận của cơ sở;
(2) Báo cáo khắc phục các tồn tại trong kiểm tra lần trước;
(3) Báo cáo thay đổi của cơ sở trong 03 năm triển khai./.
|
| Phụ trách đơn vị |
[1] Thông tư số 45/2011/TT-BYT có căn cứ ban hành như sau:
"Căn cứ Luật Dược số 34/2005-QH-11 ngày 14 tháng 6 năm 2005;
Căn cứ Nghị định số 188/2007/NĐ-CP ngày 27/12/2007 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và tổ chức bộ máy Bộ Y tế;
Bộ Y tế hướng dẫn việc triển khai áp dụng nguyên tắc, tiêu chuẩn “Thực hành tốt phòng kiểm nghiệm thuốc”, “Thực hành tốt bảo quản thuốc”, “Thực hành tốt sản xuất thuốc”, hướng dẫn sản xuất gia công thuốc, thông tin quảng cáo thuốc, hướng dẫn hoạt động xuất khẩu, nhập khẩu thuốc và bao bì tiếp xúc trực tiếp với thuốc, quy định về đăng ký thuốc như sau:"
[2] Điều 8 Thông tư số 45/2011/TT-BYT, có hiệu lực kể từ ngày 05 tháng 02 năm 2012 quy định như sau:
"Điều 8. Hiệu lực thi hành
Thông tư này có hiệu lực thi hành kể từ ngày 05 tháng 02 năm 2012."
[3] Điểm này được sửa đổi, bổ sung theo quy định tại Khoản 1 Điều 1 của Thông tư số 45/2011/TT-BYT, có hiệu lực kể từ ngày 05 tháng 02 năm 2012.
[4] Mẫu này đã được sửa đổi theo quy định tại Khoản 2 Điều 1 của Thông tư số 45/2011/TT-BYT, có hiệu lực kể từ ngày 05 tháng 02 năm 2012.
[5] Mẫu này được bổ sung theo quy định tại Khoản 2 Điều 1 của Thông tư số 45/2011/TT-BYT, có hiệu lực kể từ ngày 05 tháng 02 năm 2012.
Bạn chưa Đăng nhập thành viên.
Đây là tiện ích dành cho tài khoản thành viên. Vui lòng Đăng nhập để xem chi tiết. Nếu chưa có tài khoản, vui lòng Đăng ký tại đây!
Bạn chưa Đăng nhập thành viên.
Đây là tiện ích dành cho tài khoản thành viên. Vui lòng Đăng nhập để xem chi tiết. Nếu chưa có tài khoản, vui lòng Đăng ký tại đây!
