
 Tiếng Anh
Tiếng Anh Pháp lý doanh nghiệp
Pháp lý doanh nghiệp Vui lòng đăng nhập tài khoản gói Tiêu chuẩn hoặc Nâng cao để xem Lược đồ.
Nếu chưa có tài khoản Quý khách đăng ký tại đây!
Nếu chưa có tài khoản Quý khách đăng ký tại đây!
Đây là tiện ích dành cho thành viên đăng ký phần mềm.
Quý khách vui lòng Đăng nhập tài khoản LuatVietnam và đăng ký sử dụng Phần mềm tra cứu văn bản.
| Số hiệu: | TCVN IX:2024 | Loại văn bản: | Tiêu chuẩn Việt Nam |
| Cơ quan ban hành: | Bộ Khoa học và Công nghệ | Lĩnh vực: | Y tế-Sức khỏe, Thực phẩm-Dược phẩm |
| Ngày ban hành: | 23/02/2024 | Hiệu lực: | |
| Người ký: | Tình trạng hiệu lực: | Đã biết Vui lòng đăng nhập tài khoản gói Tiêu chuẩn hoặc Nâng cao để xem Tình trạng hiệu lực. Nếu chưa có tài khoản Quý khách đăng ký tại đây! | |
TIÊU CHUẨN QUỐC GIA
TCVN IX:2024
BỘ TIÊU CHUẨN QUỐC GIA VỀ THUỐC
Set of national standards for medicines
Mục lục
Lời nói đầu
Lời giới thiệu
1 Phạm vi áp dụng
2 Tài liệu viện dẫn
3 Chữ viết tắt
Phần 1. Nguyên liệu hóa dược
Alendronat natri
Anastrozol
Betahistin dihydroclorid
Carvedilol
Cefoxitin natri
Citicolin natri
Clobetasol propionat
Clomifen citrat
Desloratadin
Didanosin
Emtricitabin
Flunarizin dihydroclorid
Lamotrigin
Levetiracetam
Levocetirizin dihydroclorid
Lisinopril
Lysin hydroclorid
Mifepriston
Montelukast natri
Nimodipin
Ondansetron hydroclorid
Oxytocin
Rabeprazol natri
Rosuvastatin calci
Sildenafil citrat
Tadalafil
Tenofovir disoproxil fumarat
Thiamazol
Tyrosin
Phần 2. Thành phẩm hóa dược
Bột pha hỗn dịch attapulgit
Bột pha tiêm cefoxitin
Nang emtricitabin
Thuốc tiêm citicolin natri
Thuốc tiêm ondansetron
Thuốc tiêm oxytocin
Thuốc tiêm terbutalin sulfat
Viên nén alendronat natri
Viên nén anastrozol
Viên nén betahistin dihydroclorid
Viên nén bisoprolol fumarat
Viên nén carvedilol
Viên nén citicolin natri
Viên nén clomifen
Viên nén desloratadin
Viên nén didanosin
Viên nén flunarizin
Viên nén lamotrigin
Viên nén levetiracetam
Viên nén levocetirizin dihydroclorid
Viên nén lisinopril
Viên nén mifepriston
Viên nén montelukast
Viên nén nimodipin
Viên nén ondansetron
Viên nén rosuvastatin
Viên nén sertralin
Viên nén sildenafil
Viên nén tadalafil
Viên nén telmisartan và hydroclorothiazid
Viên nén tenofovir disoproxil fumarat
Viên nén terbutalin sulfat
Viên nén thiamazol
Viên nén tramadol hydroclorid và paracetamol
Phần 3. Dược liệu
Hẹ (Hạt)
Táo mèo (Quả)
Thiên hoa phần (Rễ)
Thiên nam tinh (Thân rễ)
Thiên nam tinh đã chế biến
Tổ bọ ngựa
Lời nói đầu
Bộ tiêu chuẩn quốc gia về thuốc TCVN IX:2024 được xây dựng trên nguyên tắc nối tiếp Bộ TCVN I:2017, Bộ TCVN II:2012, Bộ TCVN III:2014, Bộ TCVN IV:2015, Bộ TCVN V:2017 và Bộ TCVN VI:2017, TCVN VII:2021 và TCVN VIII:2022.
Bộ tiêu chuẩn quốc gia về thuốc TCVN IX:2024 do Hội đồng Dược điển Việt Nam biên soạn, Bộ Y tế đề nghị, Tổng cục Tiêu chuẩn Đo lường Chất lượng thẩm định, Bộ Khoa học và Công nghệ công bố.
Lời giới thiệu
Tiêu chuẩn quốc gia về thuốc là văn bản kỹ thuật về tiêu chuẩn hoá và kiểm nghiệm chất lượng thuốc. Bộ tiêu chuẩn quốc gia về thuốc này gồm 69 tiêu chuẩn, chia thành 3 phần như sau:
- Phần 1. Nguyên liệu hóa dược, gồm 29 tiêu chuẩn;
- Phần 2. Thành phẩm hóa dược, gồm 34 tiêu chuẩn;
- Phần 3. Dược liệu, gồm 6 tiêu chuẩn.
Danh pháp, thuật ngữ trong Bộ tiêu chuẩn quốc gia về thuốc được viết theo quy định của Hội đồng Dược điển Việt Nam, Bộ Y tế. Các thuật ngữ dược phẩm được viết dựa trên nguyên tắc việt hoá tên chung quốc tế Latin (DCI Latin) một cách hợp lý nhằm giữ các ký tự cho sát với thuật ngữ quốc tế. Tên hợp chất hữu cơ được viết theo danh pháp do Hiệp hội quốc tế hoá học thuần tuý và ứng dụng (I.U.P.A.C) quy định. Trong một số trường hợp cá biệt, các thuật ngữ tiếng Việt đã quen dùng đối với một số nguyên tố, hoá chất vẫn tiếp tục sử dụng.
BỘ TIÊU CHUẨN QUỐC GIA VỀ THUỐC
Set of national standards for medicines
1 Phạm vi áp dụng
Bộ tiêu chuẩn này quy định các yêu cầu kỹ thuật, phương pháp kiểm nghiệm, bảo quản và các yêu cầu có liên quan đến chất lượng đối với nguyên liệu hóa dược, thành phẩm hóa dược.
2 Tài liệu viện dẫn
Các tài liệu viện dẫn sau rất cần thiết cho việc áp dụng tiêu chuẩn này. Đối với các tài liệu viện dẫn có ghi năm công bố thì áp dụng phiên bản được nêu. Đối với các tài liệu không ghi năm công bố thì áp dụng phiên bản mới nhất, bao gồm cả sửa đổi bổ sung (nếu có).
1. TCVN I-1:2017, Bộ tiêu chuẩn quốc gia về thuốc - Phần 1: Phương pháp kiểm nghiệm thuốc; gồm 18 Phụ lục, cụ thể như sau:
Phụ lục 1: Từ phụ lục 1.1 đến phụ lục 1.24;
Phụ lục 2: Từ phụ lục 2.1 đến phụ lục 2.5;
Phụ lục 3: Từ phụ lục 3.1 đến phụ lục 3.6;
Phụ lục 4: Từ phụ lục 4.1 đến phụ lục 4.4;
Phụ lục 5: Từ phụ lục 5.1 đến phụ lục 5.7;
Phụ lục 6: Từ phụ lục 6.1 đến phụ lục 6.11;
Phụ lục 7: Từ phụ lục 7.1 đến phụ lục 7.11;
Phụ lục 8: Từ phụ lục 8.1 đến phụ lục 8.3;
Phụ lục 9: Từ phụ lục 9.1 đến phụ lục 9.10;
Phụ lục 10: Từ phụ lục 10.1 đến phụ lục 10.19;
Phụ lục 11: Từ phụ lục 11.1 đến phụ lục 11.8;
Phụ lục 12: Từ phụ lục 12.1 đến phụ lục 12.20;
Phụ lục 13: Từ phụ lục 13.1 đến phụ lục 13.10;
Phụ lục 14;
Phụ lục 15: Từ phụ lục 15.1 đến phụ lục 15.41;
Phụ lục 16: Từ phụ lục 16.1 đến phụ lục 16.2;
Phụ lục 17: Từ phụ lục 17.1 đến phụ lục 17.8;
Phụ lục 18.
TCVN I-4:2017, Bộ tiêu chuẩn quốc gia về thuốc - Phần 4: Dược liệu và thuốc từ dược liệu.
TCVN II:2012, Bộ tiêu chuẩn quốc gia về thuốc - Phần 1: Phương pháp kiểm nghiệm thuốc; gồm 5 phụ lục như sau: Phụ lục 1.25; phụ lục 4.5; phụ lục 6.12; phụ lục 12.21 và phụ lục 12.22; Phần 4: Dược liệu.
TCVN III:2014, Bộ tiêu chuẩn quốc gia về thuốc - Phần 1: Phương pháp kiểm nghiệm thuốc, gồm 2 phụ lục như sau: Phụ lục 15.42 và phụ lục 15.43; Phần 4: Dược liệu.
TCVN IV:2015, Bộ tiêu chuẩn quốc gia về thuốc - Phần 1: Phương pháp kiểm nghiệm thuốc, gồm 1 phụ lục 10.20; Phần 5: Dược liệu.
TCVN V:2017, Bộ tiêu chuẩn quốc gia về thuốc - Phần 1: Phương pháp kiểm nghiệm thuốc và chuyên mục, gồm 12 phụ lục như sau: Phụ lục 1.26; phụ lục 6.13; phụ lục 10.21; phụ lục 10.22; phụ lục 11.9; phụ lục 11.10; phụ lục 12.23; phụ lục 13.11; phụ lục 15.44; phụ lục 15.45; phụ lục 15.46 và phụ lục 15.47; Phần 4: Dược liệu.
TCVN VI:2017, Bộ tiêu chuẩn quốc gia về thuốc - Phần 1: Phương pháp kiểm nghiệm thuốc và chuyên mục, gồm 5 phụ lục như sau: Phụ lục 1.27; phụ lục 4.6; phụ lục 4.7; phụ lục 4.8 và phụ lục 12.24; Phần 4: Dược liệu.
TCVN VIII:2022, Bộ tiêu chuẩn quốc gia về thuốc - Phần 1: Phụ lục 17 đồ đựng cấp 1 dùng cho chế phẩm dược; gồm 4 phụ lục như sau: 17.3.4; 17.9.8; 17.9.9; 17.9.10; Phần 2. Dược liệu.
3 Chữ viết tắt
Tên hóa chất, thuốc thử in nghiêng kèm theo các chữ viết tắt sau đây trong ngoặc đơn biểu thị thuốc thử đó phải đạt yêu cầu quy định tại Phụ lục 2.
Chữ viết tắt | Ý nghĩa |
CĐ | Chuẩn độ |
TT | Thuốc thử |
TT1, TT2, TT3... | Thuốc thử 1, thuốc thử 2, thuốc thử 3... |
tt/tt | Thể tích/thể tích |
PHẦN 1
NGUYÊN LIỆU HÓA DƯỢC
ALENDRONAT NATRI

C4H12NNaO7P2.3H2O | P.t.l: 325,1 |
Alendronat natri là natri trihydrogen (4-amino-1-hydroxybutyliden)diphosphonat trihydrat, phải chứa từ 98,0 % đến 102,0 % C4H12NNaO7P2, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu trắng hoặc gần như trắng. Tan trong nước, thực tế không tan trong methanol và methylen clorid.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của alendronat natri chuẩn.
B. Chế phẩm phải cho phản ứng (A) của ion natri (Phụ lục 8.1).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 2,94 g/L natri citrat (TT) và 1,42 g/L dinatri hydrophosphat khan (TT), điều chỉnh đến pH 8,0 bằng acid phosphoric (TT) và lọc qua màng lọc có kích thước lỗ lọc 0,5 μm hoặc nhỏ hơn.
Pha động A: Acetonitril - dung dịch đệm (3 : 17).
Pha động B: Acetonitril - dung dịch đệm (7:3).
Dung dịch C: Dung dịch chứa 4 mg/ml 9-fluorenylmethyl cloroformat (TT) trong acetonitril (TT) (chuẩn bị dung dịch trước khi dùng).
Dung môi pha mẫu: Dung dịch chứa 29,4 g/L natri Citrat (TT).
Dung dịch borat: Dung dịch chứa 19,1 g/L natri borat (TT).
Dung dịch thử: Lấy 5,0 ml dung dịch chứa 0,6 mg/ml chế phẩm trong dung môi pha mẫu vào 1 ống ly tâm bằng Polypropylen có nắp xoáy dung tích 50 ml đã có sẵn 5 ml dung dịch borat. Thêm 5 ml acetonitril (TT) và 5 ml dung dịch C, lắc trong 45 s. Để yên ở nhiệt độ phòng trong 30 min. Thêm 20 ml methylen clorid (TT) và lắc mạnh trong 1 min. Ly tâm 5 min đến 10 min, lấy phần dung dịch trong ở phía trên.
Dung dịch đối chiếu (1): Dung dịch chứa 0,6 mg/ml alendronat natri chuẩn trong dung môi pha mẫu.
Dung dịch đối chiếu (2): Lấy 5,0 ml dung dịch đối chiếu (1) vào 1 ống ly tâm bằng polypropylen có nắp xoáy dung tích 50 ml đã có sẵn 5 ml dung dịch borat. Tiếp tục tiến hành như dung dịch thử bắt đầu từ “Thêm 5 ml acetonitril (TT)...”.
Dung dịch đối chiếu (3): Pha loãng dung dịch đối chiếu (1) thành dung dịch có nồng độ 0,6 pg/ml alendronat natri bằng dung môi pha mẫu. Lấy 5,0 ml dung dịch thu được vào 1 ống ly tâm bằng polypropylen có nắp xoáy dung tích 50 ml đã có sẵn 5 ml dung dịch borat. Tiếp tục tiến hành như dung dịch thử bắt đầu từ “Thêm 5 ml acetonitril (TT) ...”.
Dung dịch mẫu trắng: Lấy 5,0 ml dung môi pha mẫu vào 1 ống ly tâm bằng polypropylen có nắp xoáy dung tích 50 ml đã có sẵn 5 ml dung dịch borat. Tiếp tục tiến hành như dung dịch thử bắt đầu từ “Thêm 5 ml acetonitril (TT)….”.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh styren-divinylbenzen copolymer (3 μm đến 30 μm).
Nhiệt độ cột: 45 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 266 nm.
Tốc độ dòng: 1,8 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 100 | 0 |
15 | 50 | 50 |
25 | 0 | 100 |
27 | 100 | 0 |
32 | 100 | 0 |
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu (2), (3) và dung dịch mẫu trắng
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), hệ số đối xứng của pic chính không quá 2,0; trên sắc ký đồ của dung dịch đối chiếu (3), tỷ số tín hiệu trên nhiễu đối với pic chính ít nhất là 3.
Tính hàm lượng phần trăm của các tạp chất bằng phương pháp chuẩn hóa.
Giới hạn:
Tạp chất bất kỳ: Không được quá 0,1 %.
Tổng tạp: Không được quá 0,5 %.
Bỏ qua các pic tương ứng với pic trên sắc ký đồ của dung dịch mẫu trắng.
Mất khối lượng do làm khô
Từ 16,1 % đến 17,1 % (Phụ lục 9.6).
(1,000 g; áp suất không quá 5 mmHg; 140 °C).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 14,7 g/L natri citrat (TT) và 7,05 g/L dinatri hydrophosphat khan (TT), điều chỉnh đến pH 8,0 bằng acid phosphoric (TT).
Pha động: Acetonitril - methanol - dung dịch đệm (25:5: 70).
Dung dịch A: Dung dịch chứa 0,5 mg/ml 9-fluorenylmethyl cloroformat (TT) trong acetonitril (TT). Chuẩn bị dung dịch trước khi dùng).
Dung môi pha mẫu và dung dịch borat: Như mô tả trong phần Tạp chất liên quan.
Dung dịch thử: Lấy 5,0 ml dung dịch chứa 0,1 mg/ml chế phẩm trong dung môi pha mẫu vào 1 ống ly tâm bằng Polypropylen có nắp xoáy dung tích 50 ml đã có sẵn 5 ml dung dịch borat. Thêm 5 ml dung dịch A, lắc trong 30 s. Để yên ở nhiệt độ phòng trong 25 min. Thêm 25 ml methylen clorid (TT) và lắc mạnh trong 1 min. Ly tâm 5 min đến 10 min, lấy phần dung dịch trong ở phía trên.
Dung dịch chuẩn: Lấy 5,0 ml dung dịch chứa 0,1 mg/ml alendronat natri chuẩn trong dung môi pha mẫu vào 1 ống ly tâm bằng polypropylen có nắp xoáy dung tích 50 ml đã có sẵn 5 ml dung dịch borat.
Tiếp tục tiến hành như dung dịch thử bắt đầu từ “Thêm 5 ml dung dịch A...”.
Dung dịch mẫu trắng: Lấy 5,0 ml dung môi pha mẫu vào 1 ống ly tâm bằng polypropylen có nắp xoáy dung tích 50 ml đã có sẵn 5 ml dung dịch borat. Tiếp tục tiến hành như dung dịch thử bắt đầu từ “Thêm 5 ml dung dịch A...”.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh styren-divinylbenzen copolymer (3 μm đến 30 μm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 266 nm.
Tốc độ dòng: 1,2 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic chính không lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic chính thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của alendronat natri, C4H12NNaO7P2, trong chế phẩm dựa vào diện tích pic của alendronat trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng C4H12NNaO7P2 trong alendronat natri chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Ức chế tiêu xương.
Chế phẩm
Viên nén.
ANASTROZOL
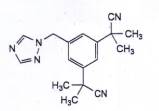
C17H19N5 | P.t.l: 293,4 |
Anastrozol là 2,2’-[5-(1H-1,2,4-triazol-1-ylmethyl)benzen-1,3-dlyl]bis(2-methylpropanenitril), phải chứa từ 98,0 % đến 102,0 % C17H19N5, tính theo chế phẩm khan và không tính đến dung môi.
Tính chất
Bột kết tinh màu trắng hoặc trắng ngà. Rất tan trong acetonitril, dễ tan trong methanol, aceton, ethanol 96 %, và tetrahydroturan.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của anastrozol chuẩn.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Acetonitril - methanol- acid trifluoroacetic - nước (100 : 300 : 0,5 : 600).
Pha động B: Acetonitril - methanol - acid trifluoroacetic - nước (150 : 450 : 0,5 : 400).
Dung dịch thử: Hòa tan 50 mg chế phẩm trong 10 ml acetonitril (TT) và pha loãng thành 25,0 ml bằng pha động A.
Dung dịch đối chiếu: Hòa tan một lượng thích hợp anastrozol chuẩn bằng acetonitril (TT) (thể tích acetonitril bằng 40 % thể tích cuối cùng) và pha loãng bằng pha động A để thu được dung dịch có nồng độ anastrozol 0,2 mg/ml. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động A (nồng độ anastrozol 0,02 mg/ml).
Dung dịch mẫu trắng: Pha loãng 10 ml acetonitril (TT) thành 25 ml bằng pha động A.
Điều kiện sắc ký:
Cột kích thước (10 cm x 3,2 mm) được nhồi pha tĩnh là các hạt silica gel xốp, hình cầu liên kết với nhóm octylsilan và octadecylsilan (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 215 nm.
Tốc độ dòng: 0,75 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động |
(min) | (% tt/tt) | (% tt/tt) |
0 | 100 | 0 |
10 | 100 | 0 |
40 | 0 | 100 |
41 | 100 | 0 |
56 | 100 | 0 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, hệ số đối xứng của pic anastrozol từ 0,9 đến 1,4 và độ lệch chuẩn tương đối của diện tích pic anastrozol thu được từ 6 lần tiêm lặp lại không lớn hơn 5 %.
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu và dung dịch mẫu trắng (hiệu chỉnh diện tích pic nếu trên sắc ký đồ của dung dịch mẫu trắng có xuất hiện pic tại cùng vị trí).
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic anastrozol trên sắc ký đồ của dung dịch đối chiếu và nồng độ anastrozol trong dung dịch đối chiếu.
Thời gian lưu tương đối và giới hạn phần trăm của các tạp chất được trình bày trong Bảng 1. Bỏ qua các tạp chất có hàm lượng nhỏ hơn 0,05 %.
Bảng 1
Tạp chất | Thời gian lưu tương đối | Giới hạn (%) |
Desmethyl anastrozol | 0,6 | 0,2 |
Anastrozol | 1,00 | - |
Anastrozol dimer | 2,0 | 0,2 |
5-Bromomethyl anastrozol | 4,3 | 0,1 |
5-Dibromomethyl anastrozol | 5,4 | 0,1 |
Tạp đơn chưa xác định | - | 0,1 |
Tổng tạp | - | 0,5 |
Ghi chú:
Desmethyl anastrozol: 2-(3-(1-Cyanoethyl)-5-(1H-1,2,4-triazol-1-ylmethyl)phenyl)-2-methylpropionitril.
Anastrozol dimer: 2,3-Bis(3-(1-cyano-1-methylethyl)-5-(1H-1,2,4-triazol-1-ylmethyl)phenyl)-2-methylpropionitril.
5-Bromomethyl anastrozol: 2,2'-(5-(Bromomethyl)-1,3-phenylen)bis(2-methylpropionitril).
5-Dibromomethyl anastrozol: 2,2'-(5-(Dibromomethyl)-1,3-phenylen)bis(2-methylpropionitril).
Nước
Không được quá 0,3 % (Phụ lục 10.3, phương pháp 2).
Tro Sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A, pha động B, chương trình dung môi và điều kiện sắc ký: Như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Hòa tan 25 mg chế phẩm trong 20 ml acetonitril (TT) và pha loãng thành 50,0 ml bằng pha động A.
Dung dịch chuẩn: Hòa tan một lượng thích hợp anastrozol chuẩn bằng acetonitril (TT) (thể tích acetonitril chiếm 40 % thể tích cuối cùng) và pha loãng bằng pha động A để thu được dung dịch có nồng độ anastrozol 0,5 mg/ml.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic anastrozol từ 0,9 đến 1,4 và độ lệch chuẩn tương đối của diện tích pic anastrozol thu được từ 5 lần tiêm lặp lại không lớn hơn 0,73 %.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Tính hàm lượng phần trăm của anastrozol, C17H19N5, trong chế phẩm dựa vào diện tích pic anastrozol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C17H19N5 trong anastrozol chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Ức chế aromatase, điều trị ung thư vú.
Chế phẩm
Viên nén.
BETAHISTIN DIHYDROCLORID

C8H12N2.2HCl | P.t.l: 209,1 |
Betahistin dihydroclorid là N-methyl-2-(pyridin-2-yl)ethanamin dihydroclorid, phải chứa từ 99,0 % đến 101,0 % C8H12N2.2HCl, tính theo chế phẩm đã làm khô.
Tính chất
Bột màu trắng hoặc vàng nhạt, rất hút ẩm. Rất tan trong nước, tan trong ethanol 96 %, thực tế không tan trong 2-propanol.
Định tính
Có thể chọn một trong hai nhóm định tính sau:
Nhóm I: A, D.
Nhóm II: B, C, D.
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của betahistin dihydroclorid chuẩn.
B. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel GF254.
Dung môi khai triển: Amoniac đậm đặc - ethyl acetat - methanol (0,75 : 15 : 30).
Dung dịch thử: Hòa tan 10 mg chế phẩm trong 2 ml ethanol 96 %(TT).
Dung dịch đối chiếu: Hòa tan 10 mg betahistin dihydroclorid chuẩn trong 2 ml ethanol 96 %(TT).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 2 μl mỗi dung dịch trên. Triển khai sắc ký đến khi dung môi đi được khoảng 2/3 chiều dài bản mỏng, sấy bản mỏng ở 110 °C trong 10 min và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm. Vết chính trên sắc ký đồ của dung dịch thử phải giống với vết chính trên sắc ký đồ của dung dịch đối chiếu về vị trí và kích thước.
C. Chế phẩm có nhiệt độ nóng chảy từ 150 °C đến 154 °C (Phụ lục 6.7)
D. Chế phẩm phải cho phản ứng (A) của clorid (Phụ lục 8.1).
Độ trong và màu sắc của dung dịch
Dung dịch S: Hòa tan 5,0 g chế phẩm trong nước không có carbon dioxyd (TT) và pha loãng thành 50 ml với cùng dung môi.
Dung dịch S phải trong (Phụ lục 9.2) và không được có màu đậm hơn màu mẫu N8 (Phụ lục 9.3, phương pháp 2).
pH
pH của dung dịch s phải từ 2,0 đến 3,0 (Phụ lục 6.2).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Hòa tan 2,0 g natri dodecyl sulfat (TT) trong một hỗn hợp chứa 15 ml dung dịch acid sulfuric (TT) 10 % (tt/tt); 35 ml dung dịch tetrabutylamoni hydrosulfat (TT) 1,7 % và 650 ml nước, điều chỉnh đến pH 3,3 bằng dung dịch natri hydroxyd loãng (TT). Sau đó trộn đều với 300 ml acetonitril (TT).
Dung dịch thử: Hòa tan 25 mg chế phẩm trong pha động và pha loãng thành 25,0 ml bằng pha động.
Dung dịch đối chiếu (1): Hòa tan 10 mg betahistin dihydroclorid chuẩn và 10 mg 2-vinylpyrldin (TT) trong pha động và pha loãng thành 50,0 ml bằng pha động. Pha loãng 2,0 ml dung dịch thu được thành 50.0 ml bằng pha động.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng pha động.
Dung dịch đối chiếu (3): Pha loãng 2,0 ml dung dịch đối chiếu (2) thành 10,0 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (15 cm x 3,0 mm) được nhồi pha tĩnh base-deactivated end-capped octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 260 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 4 lần thời gian lưu của betahistin.
Thời gian lưu tương đối so với betahlstin (thời gian lưu khoảng 7 min): Tạp chất B khoảng 0,2; tạp chất A khoảng 0,3; tạp chất C khoảng 3.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), độ phân giải giữa pic 2-vinylpyridin và pic betahistin không nhỏ hơn 3,5.
Để tính hàm lượng, nhân diện tích pic của tạp chất B với hệ số hiệu chỉnh là 0,4.
Giới hạn:
Tạp chất A, B, C: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (3) (0,2 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn 0,5 lần diện tích pic chính trên sắc ký đồ của dung dịch đối chiếu (3) (0,10 %).
Tổng diện tích pic của tất cả các tạp chất không được lớn hơn 0,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,5 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,25 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (3) (0,05 %).
Ghi chú:
Tạp chất A: 2-Ethenylpyridin (2-vinylpyridin).
Tạp chất B: 2-(pyridin-2-yl)ethanol.
Tạp chất C: N-methyl-2-(pyridin-2-yl)-N-[2-(pyridin-2-yl)ethyl]ethanamin.
Mất khối lượng do làm khô
Không được quá 1,0 % (Phụ lục 9.6).
(1,000 g; 105 °C).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Hòa tan khoảng 80,0 mg chế phẩm trong 50 ml ethanol 96 % (TT). Chuẩn độ bằng dung dịch natri hydroxyd 0,1 N (CĐ). Xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2). Đọc thể tích dung dịch natri hydroxyd 0,1 N (CĐ) tiêu thụ tại điểm uốn thứ hai.
1 ml dung dịch natri hydroxyd 0,1 N (CĐ) tương đương với 10,46 mg C8H12N2.2HCl.
Bảo quản
Trong đồ đựng kín
Loại thuốc
Thuốc kháng histamin.
Chế phẩm
Viên nén.
CARVEDILOL

C24H26N2O4 | P.t.l: 406,5 |
Carvedilol là (2RS)-1 -(9H-carbazol-4-yloxy)-3-[[2-(2-methoxyphenoxy)ethyl]amino]propan-2-ol, phải chứa từ 99,0 % đến 101,0 % C24H26N2O4, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu trắng hoặc gần như trắng, đa hình. Thực tế không tan trong nước và các dung dịch acid loãng, hơi tan trong methylen clorid, khó tan trong ethanol 96 %.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của carvedilol chuẩn. Nếu phổ hồng ngoại ở trạng thái rắn của mẫu thử khác với phổ hồng ngoại của carvedilol chuẩn thì hòa tan riêng rẽ chế phẩm và chuẩn trong 2-propanol (TT), bốc hơi tới cắn rồi tiến hành ghi lại phổ của cắn mới.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Hòa tan 1,77 g kali dihydrophosphat (TT) trong nước và pha loãng thành 650 ml với cùng dung môi, điều chỉnh đến pH 2,0 bằng acid phosphoric (TT), sau đó thêm 350 ml acetonitril (TT).
Dung dịch thử: Hòa tan 25 mg chế phẩm trong pha động và pha loãng thành 25,0 ml với cùng dung môi. Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng pha động. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động.
Dung dịch đối chiếu (2): Hòa tan 5 mg tạp chất C chuẩn của carvedilol trong 5,0 ml pha động và pha loãng thành 100,0 ml với cùng dung môi. Pha loãng 4,0 ml dung dịch thu được thành 100,0 ml bằng pha động. Tiếp tục pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động
Dung dịch đối chiếu (3): Hòa tan 5 mg carvedilol chuẩn dùng để kiểm tra tính phù hợp của hệ thống (chứa tạp chất A và D) trong ptia động và pha loãng thành 50,0 ml với cùng dung môi.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh end-capped octylsilyl silica gel dùng cho sắc ký (5 μm).
Nhiệt độ cột: 55 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 6 lần thời gian lưu của carvedilol.
Sử dụng sắc ký đồ cung cấp kèm theo carvedilol chuẩn dùng để kiểm tra tính phù hợp của hệ thống và sắc ký đồ của dung dịch đối chiếu (3) để xác định pic của tạp chất A và D. Sử dụng sắc ký đồ của dung dịch đối chiếu (2) để xác định pic của tạp chất C.
Thời gian lưu tương đối so với carvedilol (thời gian lưu khoảng 4 min): Tạp chất A khoảng 0,5; tạp chất C khoảng 2,9; tạp chất D khoảng 3,8.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic tạp chất A và pic carvedilol ít nhất là 3,5; trên sắc ký đồ của dung dịch đối chiếu (2), tỉ số tín hiệu trên nhiễu ít nhất là 10 đối với pic tạp chất C.
Để tính hàm lượng, nhân diện tích pic của tạp chất A với hệ số hiệu chỉnh là 2,0.
Giới hạn:
Tạp chất A: Diện tích pic tạp chất A không được lớn hơn 2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,2 %).
Tạp chất D: Diện tích pic tạp chất D không được lớn hơn 1,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,15 %).
Tạp chất C: Diện tích pic tạp chất C không được lớn hơn diện tích pic tương ứng thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,02 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic chính trên sắc ký đồ của dung dịch đối chiếu (1) (0,10 %).
Tổng diện tích pic của tất cả các tạp chất (trừ tạp chất C) không được lớn hơn 5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,5 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,05 %).
Ghi chú:
Tạp chất A: 1 -[[9-[2-hydroxy-3-[[2-(2-methoxyphenoxy)ethyl]amino]propyl]-9H-carbazol-4-yl]oxy]-3-[[2-(2-methoxyphenoxy)ethyl]amino]propan-2-ol.
Tạp chất B: 1,1’-[[2-(2-methoxyphenoxy)ethyl]nitrilo]bis[3-(9H-carbazol-4-yloxy)propan-2-ol].
Tạp chất C: (2RS)-1 -[benzyl[2-(2-methoxyphenoxy)ethyl]amino]-3-(9H-carbazol-4-yloxy)propan-2-ol.
Tạp chất D: 1-(9H-carbazol-4-yloxy)-3-[4-[2-hydroxy-3-[[2-(2-methoxyphenoxy)ethyl]amino]propoxy]-9H-carbazol-9-yl]propan-2-ol.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6).
(1,000 g; 105 °C).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Hòa tan 0,350 g chế phẩm trong 60 ml acid acetic khan (TT). Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ). Xác định điểm kết thúc bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2).
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 40,65 mg C24H26N2O4.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc chẹn beta-adrenergic, giãn mạch.
Chế phẩm
Viên nén.
CEFOXITIN NATRI
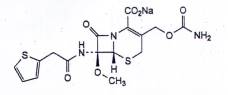
C16H16N3NaO7S2 | P.t.l: 449,4 |
Cefoxitin natri là natri (6R,7S)-3-[(carbamoyloxy)methyl]-7-methoxy-8-oxo-7-[[(thiophen-2-yl) acetyl]amino]-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylat, phải chứa từ 927 μg/mg đến 970 μg/mg cefoxitin (C16H17N3O7S2), tương đương với từ 97,5 % đến 102,0 % cefoxitin natri (C16H16N3NaO7S2), tính theo chế phẩm khan, không tính đến aceton và methanol.
Tính chất
Bột màu trắng hoặc gần như trắng, rất hút ẩm. Rất tan trong nước, hơi tan trong ethanol 96 %.
Định tính
A. Trong phần Định lượng, thời gian lưu của pic chính thu được trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính thu được trên sắc ký đồ của dung dịch chuẩn.
B. Phương pháp quang phổ hấp thụ tử ngoại (Phụ lục 4.1).
Dung dịch đệm: Dung dịch chứa 1,0 g/L kali dihydrophosphat (TT) và 1,8 g/L dinatri hydrophosphat khan (TT) trong nước.
Dung dịch thử: Dung dịch chứa 20 μg/ml chế phẩm trong dung dịch đệm.
Dung dịch đối chiếu: Dung dịch chứa 20 μg/ml cefoxitin chuẩn trong dung dịch đệm.
Phổ hấp thụ tử ngoại trong khoảng bước sóng từ 200 nm đến 350 nm của dung dịch thử phải tương ứng với phổ của dung dịch đối chiếu.
C. Dung dịch chứa 50 mg/ml chế phẩm trong nước phải cho phản ứng (A) của ion natri (Phụ lục 8.1).
pH
Từ 4,2 đến 7,0 (Phụ lục 6.2).
Dùng dung dịch chứa 100 mg/ml chế phẩm trong nước để đo.
Góc quay cực riêng
Từ +206° đến +214°, tính theo chế phẩm khan, không tính đến aceton và methanol (Phụ lục 6.4).
Dùng dung dịch chứa 10 mg/ml chế phẩm trong methanol (TT) để đo. Nhiệt độ đo: 25 °C.
Giới hạn aceton và methanol
Phương pháp sắc ký khí (Phụ lục 5.2).
Dung dịch thử: Cân chính xác 5,0 g chế phẩm vào bình định mức 50 ml, hòa tan và pha loãng bằng nước đến thể tích. Lấy 3,0 ml dung dịch thu được, cho vào ống ly tâm dung tích 15 ml, làm lạnh trong nước đá trong 2 min và thêm 3,0 ml dung dịch acid hydrocloric 0,24 M (TT), vừa thêm vừa lắc xoáy mạnh. Ly tâm để thu được dịch trong.
Dung dịch đối chiếu (1): Dung dịch chứa 0,5 % (tt/tt) aceton (TT) trong nước.
Dung dịch đối chiếu (2): Dung dịch chứa 0,5 % (tt/tt) methanol (TT) trong nước.
Dung dịch đối chiếu (3): Lấy 10,0 ml dung dịch đối chiếu (1) và 1,0 ml dung dịch đối chiếu (2) vào bình định mức 100 ml và thêm nước đến vạch (dung dịch chứa 0,05 % (tt/tt) aceton và 0,005 % (tt/tt) methanol).
Điều kiện sắc ký:
Cột thủy tinh (1,8 m x 6,3 mm) được nhồi styren-divinylbenzen copolymer có diện tích bề mặt nhỏ hơn 50 m2/g và đường kính lỗ xốp trung bình từ 0,3 μm đến 0,4 μm.
Tiền cột được nhồi hạt thủy tinh đã được silan hóa có kích thước từ 60 mesh đến 80 mesh.
Khí mang: Nitrogen dùng cho sắc ký khí.
Tốc độ dòng: 50 ml/min.
Nhiệt độ: Cột 110 °C, buồng tiêm 100 °C, detector 200 °C.
Detector: Ion hóa ngọn lửa.
Thể tích tiêm: 2 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch thử và dung dịch đối chiếu (3).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), số đĩa lý thuyết tính trên pic aceton không nhỏ hơn 160 và tính trên pic methanol không nhỏ hơn 200; hệ số đối xứng của pic aceton không lớn hơn 1,3 và của pic methanol không lớn hơn 2,3; độ lệch chuẩn tương đối của diện tích pic chính thu được từ 6 lần tiêm lặp lại không được lớn hơn 5 %.
Tính hàm lượng μg/mg của aceton và methanol trong chế phẩm dựa vào diện tích pic aceton và methanol thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (3) và nồng độ aceton và methanol (đã đổi từ nồng độ tt/tt sang kl/tt) trong dung dịch đối chiếu (3).
Giới hạn:
Aceton: Không được quá 0,7 %.
Methanol: Không được quá 0,1 %.
Nước
Không được quá 1,0 % (Phụ lục 10.3).
Dùng hỗn hợp dung môi ethylen glycol - pyridin (3 : 1).
Nội độc tố vi khuẩn
Không được quá 0,13 EU/mg cefoxitin (Phụ lục 13.2).
Nếu trên nhãn ghi là chế phẩm vô khuẩn hoặc chế phẩm được dùng để sản xuất các dạng thuốc tiêm thì phải đáp ứng yêu cầu của phép thử này.
Thử vô khuẩn
Nếu trên nhãn ghi là chế phẩm vô khuẩn thì chế phẩm phải đáp ứng yêu của phép thử vô khuẩn, phương pháp màng lọc (Phụ lục 13.7).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 1,0 g kali dihydrophosphat (TT) và 1,8 g dinatri hydrophosphat khan (TT) trong 900 ml nước, điều chỉnh đến pH 7,1 ± 0,1 bằng acid phosphoric (TT) hoặc dung dịch natri hydroxyd 10 M (TT), và pha loãng thành 1 L bằng nước. Lọc qua màng lọc có kích thước lỗ lọc 1 μm hoặc nhỏ hơn.
Pha động: Nước - acetonitril - acid acetic băng (840 : 160 : 10). Lọc qua màng lọc có kích thước lỗ lọc 1 μm hoặc nhỏ hơn.
Dung dịch thử: Dung dịch chứa 0,3 mg/ml chế phẩm trong dung dịch đệm.
Dung dịch chuẩn: Dung dịch chứa 0,3 mg/ml cefoxitin chuẩn trong dung dịch đệm, nếu cần, siêu âm để hòa tan.
Dung dịch thử và dung dịch chuẩn chỉ dùng trong vòng 5 h sau khi pha xong.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4 mm) được nhồi pha tĩnh C (5 μm đến 10 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 254 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic chính không nhỏ hơn 2800; hệ số đối xứng của pic chính không lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic chính thu được từ 5 lần tiêm lặp lại không lớn hơn 1,0 %.
Tính hàm lượng của cefoxitin, C16H17N3O7S2, trong chế phẩm dựa vào diện tích pic cefoxitin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C16H17N3O7S2 trong cefoxitin chuẩn.
Bảo quản
Trong đồ đựng kín, từ 2 °C đến 8 °C.
Loại thuốc
Kháng sinh nhóm cephalosporin.
Chế phẩm
Thuốc tiêm.
CITICOLIN NATRI

C14H25N4NaO11P2 | P.t.l: 510,3 |
Citicolin natri là cytidin-5’-diphosphocholin natri, phải chứa từ 98,0 % đến 102,0 % C14H25N4NaO11P2, tính theo chế phẩm khan.
Tính chất
Tinh thể hoặc bột kết tinh màu trắng, không mùi. Dễ tan trong nước, không tan trong ethanol và aceton.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của citicolin natri chuẩn.
B. Trong phần Định lượng, thời gian lưu của pic chính thu được trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính thu được trên sắc ký đồ của dung dịch chuẩn.
Clorid
Không được quá 150 phần triệu (Phụ lục 9.4.5).
Hòa tan 0,33 g chế phẩm trong 10 ml nước và tiến hành thử.
Sulfat
Không được quá 200 phần triệu (Phụ lục 9.4.14).
Hòa tan 0,75 g chế phẩm trong 15 ml nước và tiến hành thử.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Hòa tan 8 g tetrabutylamoni hydroxyd (TT) và 2,13 g dinatri hydrophosphat khan (TT) trong 1000 ml nước, điều chỉnh đến pH 6,0 bằng dung dịch acid phosphoric (TT) 20 %.
Pha động B: Methanol (TT).
Dung dịch thử: Dung dịch chứa 2,4 mg/ml chế phẩm trong nước.
Dung dịch đối chiếu (1): Dung dịch chứa 2,4 μg/ml citlcolin natri chuẩn trong nước.
Dung dịch đối chiếu (2): Dung dịch chứa 4,8 μg/ml acid 5'-cytidylic chuẩn trong nước.
Dung dịch đối chiếu (3): Dung dịch chứa 24 pg/ml citicolin natri chuẩn và 48 pg/ml acid 5'-cytidylic chuẩn trong nước.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,0 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 262 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 15 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 100 | 0 |
0 | 100 | 0 |
15 | 85 | 15 |
20 | 85 | 15 |
21 | 100 | 0 |
35 | 100 | 0 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic citicolin và pic acíd 5’-cytidylic ít nhất là 12; số đĩa lý thuyết tính theo pic citicolin không nhỏ hơn 5000. Tính hàm lượng phần trăm của acid 5’-cytidyllc trong chế phẩm dựa vào diện tích pic acid 5’-cytidylic thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (2) và nồng độ acid 5’-cytidylic chuẩn trong dung dịch đối chiếu (2).
Tính hàm lượng phần trăm của các tạp chất khác (nếu có) trong chế phẩm dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic citicolin trên sắc ký đồ của dung dịch đối chiếu (1), nồng độ citicolin natri chuẩn trong dung dịch đối chiếu (1); chia hệ số đáp ứng tương đối cho trong Bảng 1.
Bảng 1
Tên | Thời gian lưu tương đối | Hệ số đáp ứng tương đối | Giới hạn (%) |
Citicolin | 1,0 | - | - |
Acid 5'-cytidylic | 2,2 | - | 0,2 |
Ester cytidin-5'- monophosphat methyl | 3,2 | 1,51 | 0,15 |
Tạp chất khác | - | 1,0 | 0,1 |
Tổng các tạp chất | - | - | 0,7 |
Nước
Không được quá 5,0 % (Phụ lục 10.3).
Dùng 0,10 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Dùng pha động A trong phần Tạp chất liên quan.
Dung dịch thử: Dung dịch chứa 24 μg/ml chế phẩm trong nước.
Dung dịch chuẩn: Dung dịch chứa 24 μg/ml citicolin natri chuẩn trong nước.
Điều kiện sắc ký: Như mô tả trong phần Tạp chất liên quan.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính theo pic citicolin không nhỏ hơn 5000; hệ số đối xứng của pic citicolin không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của citicolin natri, C14H25N4NaO11P2, trong chế phẩm dựa vào diện tích pic của citicolin thu được trên sắc ký đồ của dung dịch thử; dung dịch chuẩn và hàm lượng C14H25N4NaO11P2 trong citicolin natri chuẩn.
Bảo quản
Trong đồ đựng kín.
Loại thuốc
Tăng chuyển hóa ở tế bào thần kinh.
Chế phẩm
Viên nén, nang, thuốc tiêm.
CLOBETASOL PROPIONAT
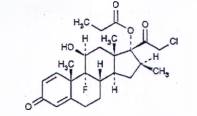
C25H32CIFO5 | P.t.l: 467,0 |
Clobetasol propionat là 21-cloro-9-fluoro-11β,17-dihydroxy-16β-methylpregna-1,4-dien-3,20-dion 17- propionat, phải chứa từ 97,0 % đến 102,0 % C25H32CIFO5, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu trắng đến màu kem. Tan trong aceton, dimethyl sulfoxid, cloroform, methanol và dioxan; hòa tan trong ethanol; khó tan trong benzen và diethyl ether; thực tế không tan trong nước.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của clobetasol propionat chuẩn.
Góc quay cực riêng
Từ +98° đến +104°, tính theo chế phẩm đã làm khô (Phụ lục 6.4).
Dùng dung dịch chứa 10 mg/ml chế phẩm trong dioxan (TT) để đo.
Nhiệt độ nóng chảy
Khoảng 196 °C (Phụ lục 6.7).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch A: Hòa tan 6 g natri dihydrophosphat khan (TT) trong vừa đủ 1000 ml nước, điều chỉnh đến pH 2,5 bằng acid phosphoric (TT).
Pha động: Acetonitril - methanol - dung dịch A (19:4:17).
Dung dịch thử: Dung dịch chứa 0,1 mg/ml chế phẩm trong pha động.
Dung dịch đối chiếu: Dung dịch chứa 0,001 mg/ml tạp chất A của clobetasol propionat chuẩn và 0,1 mg/ml clobetasol propionat chuẩn trong pha động.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Thời gian lưu tương đối so với clobetasol propionat của tạp chất A khoảng 1,1.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, độ phân giải giữa pic clobetasol propionat và pic tạp chất A không nhỏ hơn 1,5; số đĩa lý thuyết tính trên pic clobetasol proplonat không nhỏ hơn 5000.
Tính hàm lượng phần trăm tạp chất (nếu có) trên sắc ký đồ của dung dịch thử bằng phương pháp chuẩn hóa diện tích.
Giới hạn:
Tạp chất bất kỳ: Với mỗi tạp chất, không được quá 1,0 %.
Tổng các tạp chất: Không được quá 2,5 %.
Ghi chú:
Tạp chát A: 9a-Fluoro-11β-hydroxy-16β-methyI 3-oxo-androsta-1,4-dien-17(R)-spiro-2'-[4'-cloro-5'-ethylfuran-3,(2'H)- on].
Tro Sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Mất khối lượng do làm khô
Không được quá 2,0 % (Phụ lục 9.6).
(1,000 g; 105 °C; 3 h)
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch A, pha động, dung dịch đối chiếu và điều kiện sắc ký: Như mô tả ở phần Tạp chất liên quan.
Dung dịch chuẩn nội: Dung dịch chứa 0,2 mg/ml beclomethason dipropionat trong methanol (TT).
Dung dịch chuẩn: Hòa tan một lượng thích hợp clobetasol propionat chuẩn trong methanol (TT) và thêm dung dịch nội chuẩn để thu được dung dịch có chứa 0,04 mg/ml clobetasol propionat và 0,08 mg/ml beclomethason dipropionat.
Dung dịch thử: Hòa tan một lượng thích hợp chế phẩm trong methanol (TT) và thêm dung dịch chuẩn nội để thu được dung dịch có chứa 0,04 mg/ml clobetasol propionat và 0,08 mg/ml beclomethason dipropionat.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Như mô tả ở phần Tạp chất liên quan.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Thời gian lưu tương đối so với clobetasol propionat của beclomethason diproplonat khoảng 1,6.
Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic clobetasol propionat không lớn hơn 2,0 và độ lệch chuẩn tương đối của tỷ số giữa diện tích pic clobetasol propionat và pic beclomethason dipropionat thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của clobetasol propionat, C25H32CIFO5, trong chế phẩm dựa vào tỷ lệ diện tích pic clobetasol propionat và pic beclomethason dipropionat thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C25H32CIFO5 trong clobetasol propionat chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Corticosteroid.
Chế phẩm
Viên nén.
CLOMIFEN CITRAT
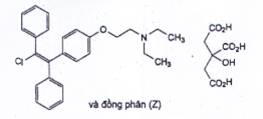
C32H36CINO8 | P.t.l: 598,1 |
Clomifen citrat là 2-[4-[(1EZ)-2-cloro-1,2-diphenylethen-1-yl]phenoxy]-N,N-diethylethan-1-amin dihydro 2-hydroxypropan-1,2,3-tricarboxylat, phải chứa từ 98,0 % đến 101,0 % C32H36CINO8, tính theo chế phẩm khan, trong đó đồng phân Z phải chiếm từ 30,0 % đến 50,0 %.
Tính chất
Bột kết tinh màu trắng hoặc vàng nhạt. Khó tan trong nước, hơi tan trong ethanol 96 %.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của Clomifen Citrat chuẩn.
B. Hòa tan 5 mg chế phẩm trong 5 ml hỗn hợp anhydrid acetic - pyridin (1 : 5) và làm nóng trong cách thủy. Màu đỏ đậm xuất hiện.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Chuẩn bị các dung dịch trong điều kiện tránh ánh sáng, sử dụng các bình thủy tinh màu nâu. Đảm bảo các dung dịch tiếp xúc ít nhất với ánh sáng cho đến khi tiến hành sắc ký.
Pha động: Acetonitril - nước - diethylamin (400 : 600 : 8); điều chỉnh pH hỗn hợp đến 6,2 bằng 1 ml đến 2 ml acid phosphoric (TT).
Dung dịch thử: Hòa tan 25 mg chế phẩm trong pha động và pha loãng thành 20,0 ml bằng pha động.
Dung dịch đối chiếu (1): Hòa tan 12,5 mg Clomifen chuẩn dùng-để kiểm tra tính phù hợp của hệ thống (chứa tạp chất A và C) trong pha động và pha loãng thành 10,0 ml bằng pha động.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng pha động.
Dung dịch đối chiếu (3): Pha loãng 1,0 ml dung dịch đối chiếu (2) thành 10,0 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh end-capped butylsilyl silica gel dùng cho sắc ký (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 233 nm.
Tốc độ dòng: 1,2 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 4 lần thời gian lưu của clomifen.
Sử dụng sắc ký đồ cung cấp kèm theo clomifen chuẩn dùng để kiểm tra tính phù hợp của hệ thống và sắc ký đồ của dung dịch đối chiếu (1) để xác định pic của tạp chất A và C.
Thời gian lưu tương đối so với clomifen (thời gian lưu khoảng 14 min): Citrat khoảng 0,16; tạp chất B khoảng 0,28; tạp chất D khoảng 0,34; Tạp chất C khoảng 0,5; tạp chất A khoảng 0,9; tạp chất G và H có thứ tự rửa giải chưa xác định rõ khoảng 1,44 và 1,49.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), tỷ số đỉnh - hõm (Hp/Hv) ít nhất là 15; trong đó Hp là chiều cao đỉnh pic tạp chất A so với đường nền và Hv là chiều cao tính từ đường nền lên đến đáy hõm giữa pic tạp chất A và pic clomifen.
Tính hàm lượng phần trăm tạp chất A dựa vào nồng độ của clomifen citrat trong dung dịch đối chiếu (2) và tính hàm lượng phần trăm của các tạp chất khác dựa vào nồng độ của clomifen citrat trong dung dịch đối chiếu (3); nhân diện tích tạp chất B với hệ số hiệu chỉnh là 1,9.
Giới hạn:
Tạp chất A: Không được quá 1,0%.
Tạp chất C và D: Mỗi tạp chất không được quá 0,3 %.
Tạp chất B: Không được quá 0,2 %.
Tạp chất G và H: Tổng hai tạp chất không được quá 0,15 %.
Các tạp chất khác: Với mỗi tạp chất, không được quá 0,10 %.
Tổng các tạp chất: Không được quá 2,0 %.
Bỏ qua pic citrat và các tạp chất có hàm lượng dưới 0,05 %.
Ghi chú:
Tạp chất A: 2-[4-[(1EZ)-1,2-diphenylethen-1-yl]phenoxy]-N,N-diethylethan-1-amin.
Tạp chất B: [4-[2-(diethylamino)ethoxy]phenyl]phenylmethanon.
Tạp chất G: (2RS)-2-[4-[2-(diethylamino)ethoxy]phenyl]-1,2-diphenylethan-1-on.
Tạp chất D: 2,2-bis[4-[2-(diethylamino)ethoxy]phenyl]-1,2-diphenylethan-1-on.
Tạp chất G và H: 2-[2-cloro-4-[(1E)-2-cloro-1,2-diphenylethen-1-yl]phenoxy]-N,N-diethylethan-1-amin (Tạp chất G là đồng phân có nhiệt độ nóng chảy cao hơn, tạp chất H có nhiệt độ nóng chảy thấp hơn).
Nước
Không được quá 1,0 % (Phụ lục 10.3).
Dùng 1,000 g chế phẩm.
Định lượng
Clomifen citrat
Hòa tan khoảng 0,500 g chế phẩm trong 50 ml acid acetic khan (TT). Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ). Xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2).
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 59,81 mg C32H36CINO8.
Đồng phân Z
Phương pháp sắc ký lỏng (Phụ lục 5.3). Chuẩn bị các dung dịch trong điều kiện tránh ánh sáng, sử dụng các bình thủy tinh màu nâu. Đảm bảo các dung dịch tiếp xúc ít nhất với ánh sáng cho đến khi tiến hành sắc ký.
Pha động: Methanol - nước - triethylamin (55 :45 :0,3), điều chỉnh pH hỗn hợp đến 2,5 bằng acid phosphoric (TT).
Dung dịch thử: Hòa tan 25,0 mg chế phẩm trong pha động và pha loãng thành 50,0 ml bằng pha động. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động.
Dung dịch đối chiếu: Hòa tan 25,0 mg clomifen Citrat chuẩn (chứa đồng phân E và Z) trong pha động và pha loãng thành 50,0 ml bằng pha động. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh end-capped butylsilyl silica gel dùng cho sắc ký (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 233 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 50 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 1,5 lần thời gian lưu của đồng phân Z của clomifen.
Sử dụng sắc ký đồ cung cấp kèm theo clomifen citrat chuẩn và sắc ký đồ của dung dịch đối chiếu để xác định pic của từng đồng phân E và Z.
Thời gian lưu tương đối so với đồng phân Z của clomifen (thời gian lưu khoảng 13 min) của đồng phân E khoảng 1,2.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, độ phân giải giữa pic đồng phân E và pic đồng phân Z ít nhất là 1,5.
Tính hàm lượng phần trăm của đồng phân Z trong chế phẩm dựa vào diện tích pic đồng phân Z thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu và hàm lượng đồng phân Z trong clomifen citrat chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc kháng estrogen/thuốc gây phóng noãn.
Chế phẩm
Viên nén.
DESLORATADIN
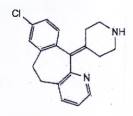
C19H19CIN2 | P.t.l: 310,8 |
Desloratadin là 8-cloro-11-(piperidin-4-yliden)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin phải chứa từ 98,0 % đến 102,0 % C19H19CIN2, tính theo chế phẩm khan.
Tính chất
Bột trắng hoặc gần như trắng, đa hình. Rất khó tan hoặc thực tế không tan trong nước, dễ tan trong ethanol (96 %), khó tan hoặc rất khó tan trong heptan.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của desloratadin chuẩn. Nếu phổ đo được ở trạng thái rắn có sự khác biệt thì hòa tan riêng biệt chế phẩm và chất chuẩn trong methyl isobutyl keton (TT), bay hơi đến khô, ghi phổ của cắn thu được.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Hòa tan 0,865 g natri dodecyl sulfat (TT) trong nước, thêm 0,5 ml acid trifluoroacetic (TT) và pha loãng đến 1000 ml với nước. Trộn 57 thể tích dung dịch thu được với 43 thể tích acetonltril (TT).
Dung dịch thử: Hòa tan 20,0 mg chế phẩm trong pha động và pha loãng thành 25,0 ml bằng pha động. Pha loãng 5,0 ml dung dịch này thành 50,0 ml bằng pha động.
Dung dịch đối chiếu (1): Hòa tan 20,0 mg desloratadin chuẩn trong pha động và pha loãng thành 25,0 ml bằng pha động, Pha loãng 5,0 ml dung dịch này thành 50,0 ml bằng pha động.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng pha động. Pha loãng 1,0 ml dung dịch này thành 10,0 ml bằng pha động.
Dung dịch đối chiếu (3): Hòa tan 4 mg desloratadin chuẩn để kiểm tra tính phù hợp của hệ thống (chứa tạp chất A và B) trong pha động và pha loãng thành 5,0 ml bằng pha động. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh end-capped octadecylsilyl silicagel dùng cho sắc ký (4 μm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 280 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 100 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu (2) và (3).
Thời gian chạy sắc ký bằng 2,5 lần thời gian lưu của desloratadin.
Sử dụng sắc ký đồ được cung cấp kèm desloratadin chuẩn để kiểm tra tính phù hợp của hệ thống và dung dịch đối chiếu (3) để xác định pic của tạp chất A và B.
Thời gian lưu tương đối so với desloratadin (thời gian lưu khoảng 21 min): Tạp chất A khoảng 0,8; tạp chất B khoảng 0,9.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic của tạp chất B và pic desloratadin không nhỏ hơn 2,0.
Tính hàm lượng phần trăm các tạp chất dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic chính trên sắc ký đồ của dung dịch đối chiếu (2) và nồng độ desloratadin trong dung dịch đối chiếu (2), nhân diện tích pic tạp chất A và tạp chất B với hệ số hiệu chỉnh là 1,6.
Giới hạn:
Tạp chất B: Không được quá 0,3 %.
Tạp chất A: Không được quá 0,2 %.
Tạp không xác định: Mỗi tạp chất không được quá 0,10 %.
Tổng các tạp chất: Không được quá 0,4 %,
Bỏ qua các tạp chất có hàm lượng nhỏ hơn 0,05 %.
Ghi chú:
Tạp chất A: (11RS)-8-cloro-11-fluoro-11-(piperidin-4-yl)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2b] pyridin.
Tạp chất B: (11RS)-8-cloro-11-(1,2,3,6-tetrahydropyridin-4-yl)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin.
Nước
Không được quá 0,5 % (Phụ lục 10.3).
Dùng 0,250 g chế phẩm.
Tro sulfat
Không được quá 0,2 % (Phụ lục 9.9, phương pháp 2).
Dùng 0,5 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả trong phần Tạp chất liên quan.
Tiến hành sắc ký với dung dịch thử và dung dịch đối chiếu (1).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), hệ số đối xứng của pic desloratadin phải từ 0,5 đến 1,5.
Tính hàm lượng phần trăm desloratadin, C19H19CIN2, trong chế phẩm dựa vào diện tích pic desloratadin trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (1) và hàm lượng C19H19CIN2 của desloratadin chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc kháng histamin.
Chế phẩm
Viên nén, siro.
DIDANOSIN
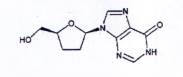
C10H12N4O3 | P.t.I: 236,2 |
Didanosin là 2',3'-dideoxyinosin, phải chứa từ 98,0 % đến 102,0 % C10H12N4O3, tính theo chế phẩm khan.
Tính chất
Bột kết tinh màu trắng hoặc trắng ngà. Rất tan trong dimethyl sultoxid, hơi tan trong nước, khó tan trong aceton và trong methanol,
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của didanosin chuẩn.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Góc quay cực riêng
Từ -28° đến -24°, tính theo chế phẩm khan (Phụ lục 6.4).
Dùng dung dịch chứa 10 mg/ml chế phẩm trong nước, đo ở 25 °C.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 0,77 g/l amoni acetat (TT) trong nước.
Pha động A: Acetonitril - dung dịch đệm (1 : 19).
Pha động B: Acetonitril - dung dịch đệm (1 : 3).
Dung môi pha mẫu: Điều chỉnh pH của dung dịch đệm đến pH 9 bằng dung dịch chứa natri hydroxyd (TT). Chuẩn bị hỗn hợp acetonitril - dung dịch đệm pH 9 (1 : 19).
Dung dịch thử: Dung dịch chứa 0,5 mg/ml chế phẩm trong dung môi pha mẫu.
Dung dịch đối chiếu (1): Dung dịch chứa 0,5 mg/ml didanosin chuẩn dùng để kiểm tra tính phù hợp của hệ thống trong dung môi pha mẫu.
Dung dịch đối chiếu (2): Dung dịch chứa 5 μg/ml tạp chất A chuẩn của didanosin; 1,5 μg/ml didanosin chuẩn và 1,5 μg/ml tạp chất B chuẩn của didanosin trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 254 nm.
Tốc độ dòng: 2 ml/mln.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 100 | 0 |
15 | 100 | 0 |
20 | 0 | 100 |
30 | 0 | 100 |
35 | 100 | 0 |
45 | 100 | 0 |
Thời gian lưu tương đối so với didanosin (thời gian lưu khoảng 6 min đến 7,5 min): Tạp chất A khoảng 0,28; inosin khoảng 0,39; 2'-deoxyinosin khoảng 0,45; 3'-deoxyinosin khoảng 0,51; 2',3’-anhydroinosin khoảng 0,59; dideoxydidehydroinosin khoảng 0,81; tạp chất B khoảng 2,1; 5’-deoxydideoxyadenosin khoảng 3,1.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), độ phân giải giữa pic didanosin và pic dideoxydidehydroinosin không nhỏ hơn 3,0; số đĩa lý thuyết tính trên pic dideoxydidehydroinosin không nhỏ hơn 6000. Trên sắc ký đồ của dung dịch đối chiếu (2), độ lệch chuẩn tương đối của diện tích pic tạp chất A thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của tạp chất A trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất A trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (2) và nồng độ của tạp chất A trong dung dịch đối chiếu (2).
Tính hàm lượng phần trăm của các tạp chất khác trong chế phẩm, nếu có, dựa vào diện tích plc tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic didanosin trên sắc ký đồ của dung dịch đối chiếu (2) và nồng độ của didanosin trong dung dịch đối chiếu (2),
Giới hạn:
Tạp chất A: Không được quá 0,5 %.
2’-Deoxyinosin: Không được quá 0,3 %.
Tạp chất B, inosin, 3'-deoxyinosin; 2',3'-anhydroinosin; dideoxydidehydroinosin; 5’-deoxydldeoxyadenosin:
Với mỗi tạp chất, không được quá 0,2 %.
Tạp chất khác: Với mỗi tạp chất, không được quá 0,1 %.
Tổng tạp chất: Không được quá 1,0 %.
Ghi chú:
Tạp chất A: Hypoxanthin.
Tạp chất B: 2',3'-Dideoxyadenosin.
Nước
Không được quá 2,0 % (Phụ lục 10.3).
Dùng 1,000 g chế phẩm.
Tro sulfat
Không được quá 0,2 % (Phụ lục 9.9, phương pháp 2);
Dùng 1,0 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 0,77 g/l amoni acetat (TT) trong nước.
Pha động: Acetonitril - dung dịch đệm (1 : 21).
Dung dịch thử: Dung dịch chứa 0,1 mg/ml chế phẩm trong nước (lắc kỹ trong 1 h để hòa tan hoàn toàn).
Dung dịch chuẩn: Dung dịch chứa 0,1 mg/ml didanosin chuẩn trong nước.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 254 nm.
Tốc độ dòng: 2 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Thời gian lưu của didanosin khoảng từ 7 min đến 11 min.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic didanosin không nhỏ hơn 6000; hệ số đối xứng của pic didanosin không lớn hơn 2,5; độ lệch chuẩn tương đối của diện tích pic didanosin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của didanosin, C10H12N4O3, trong chế phẩm dựa vào diện tích pic didanosin trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C10H12N4O3 trong didanosin chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc kháng retrovirus nucleosid ức chế enzym phiên mã ngược.
Chế phẩm
Viên nén.
EMTRICITABIN
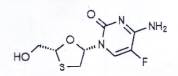
C8H10FN3O3S | P.t.l: 247,2 |
Emtricitabin là 5-fluoro-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosin, phải chứa từ 99,0 % đến 101,0 % C8H10FN3O3S, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu trắng hoặc gần như trắng, đa hình. Dễ tan trong methanol và nước, thực tế không tan trong methylen clorid.
Sản xuất
Quy trình sản xuất dược chất phải đảm bảo chế phẩm đáp ứng chỉ tiêu (2S, 5R)-enantiomer không quá 0,3 %.
Định tính
Có thể chọn một trong hai nhóm định tính sau:
Nhóm I: B, C, D.
Nhóm II: A, C.
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phỗ hấp thụ hồng ngoại của emtricitabin chuẩn hoặc phổ hấp thụ hồng ngoại đối chiếu của emtricitabin. Nếu phổ hồng ngoại ở trạng thái rắn của chế phẩm khác với phổ hồng ngoại của emtricitabin chuẩn thì hòa tan riêng rẽ chế phẩm và emtricitabin chuẩn trong methanol (TT), bốc hơi tới cắn rồi tiến hành ghi lại phổ mới của cắn.
B. Hòa tan 2 mg chế phẩm trong 100 ml nước. Đo phổ hấp thụ tử ngoại (Phụ lục 4.1) trong khoảng bước sóng từ 220 nm đến 350 nm, dung dịch thu được phải cho hai cực đại hấp thụ ở bước sóng 237 nm và 281 nm. Độ hấp thụ riêng tại ở bước sóng 237 nm từ 325 đến 355 và tại ở bước sóng 281 nm từ 340 đến 370.
C. Góc quay cực riêng: Từ -105,0° đến -115,0°, tính theo chế phẩm đã làm khô (Phụ lục 6.4).
Dùng dung dịch chứa 2,5 mg/ml chế phẩm trong nước để đo.
D. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel 60 F254.
Dung môi khai triển: Dicloromethan - methanol - acid acetic băng (90 : 10 : 3).
Dung dịch thử: Dung dịch chứa 5 mg/ml chế phẩm trong methanol (TT).
Dung dịch đối chiếu: Dung dịch chứa 5 mg/ml emtricitabin chuẩn trong methanol (TT).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 5 μl mỗi dung dịch trên. Triển khai sắc ký đến khi dung môi đi được 15 cm. Để khô bản mỏng ngoài không khí và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm. Vết chính trên sắc ký đồ của dung dịch thử phải tương ứng với vết chính trên sắc ký đồ của dung dịch đối chiếu về vị trí, kích thước và độ đậm.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 27,22 g kali dihydrophosphat (TT) trong 1000 ml nước.
Pha động A: Dung dịch đệm - nước (5 : 95).
Pha động B: Acetonitril - dung dịch đệm - nước (70 : 5 : 25).
Dung dịch thử: Dung dịch chứa 0,5 mg/ml chế phẩm trong nước.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng nước. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng nước.
Dung dịch đối chiếu (2): Trộn đều 5 ml dung dịch thử và 2 ml hỗn hợp 115 g phosphoric acid (TT) và 885 ml nước, đặt trong cách thủy sôi trong 15 min.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh base deactivated end-capped octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 280 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 9 | 93 | 7 |
9 - 15 | 93 → 0 | 7 → 100 |
15 - 19 | 0 | 100 |
19 - 19,1 | 0 → 93 | 100 → 7 |
19,1 - 30 | 93 | 7 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic emtricitabin (thời gian lưu khoảng 9 mln) và pic tạp chất có thời gian lưu tương đối so với emtricitabin bằng 1,3 không nhỏ hơn 6.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Các tạp chất rửa giải ra trước pic chính: Với mỗi tạp chất, diện tích pic không được lớn hơn 5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,5 %) và không quá 2 tạp chất có diện tích pic lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,1 %). Các tạp chất rửa giải ra sau pic chính: Với mỗi tạp chất, diện tích pic không được lớn hơn 7 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,7 %) và không quá 2 tạp chất có diện tích pic lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,1 %). Tổng diện tích pic của tất cả các tạp chất (trừ pic chính) không được lớn hơn 10 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (1 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,05 %).
Ghi chú:
Tạp chất A: Acid (2RS,5SR)-5-(4-amino-5-fluoro-2-oxopyrimidin-1(2H)-yl)-1,3-oxathlolan-2-carboxylic.
Tạp chất B: 4-Amlno-5-fluoro-1-[(2RS,5RS)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidln-2(1H)-on (racemic trans-emtricitabin).
Tạp chất C: 4-Amino-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2(1H)-on (lamivudin).
Tạp chất D: 4-Amino-5-fluoro-1-[(2S,5R)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2(1H)-on (emtricitabin enantiomer).
Tạp chất E: 4-Amino-5-fluoropyrimidin-2(1H)-on (5-fluorocytosin).
Tạp chất F: 5-Fluoropyrimidin-2,4(1H,3H)-dion (5-fluorouracil).
Tạp chất G: 4-Amino-5-fluoro-1-[(2R,3S,5S)-2-(hydroxymethyl)-3-oxo-1,3-oxa-λ4-thiolan-5-yl]pyrimidin-2(1H)-on (emtricitabin S-oxid (S)-epimer).
Tạp chất H: 4-Amino-5-fluoro-1-[(2R,3R,5S)-2-(hydroxymethyl)-3-oxo-1,3-oxa-λ4-thiolan-5-yl]pyrimidin-2(1H)-on (emtricitabin S-oxid (R)-epimer).
Tạp chất I: 1,1 '-[Disulfanediylbis(methylen-(2P,5S)-1,3-oxathiolan-2,5-diyl)]bis(4-amino-5-fluoropyrimidin-2(1H)- on).
Tạp chất J: 4-Amino-5-fluoro-1-[(2S,4S)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]pyrimidin-2(1H)-on.
Kim loại nặng
Không được quá 20 phần triệu (Phụ lục 9.4.8).
Hòa tan 1,0 g chế phẩm trong 20 ml nước. Lấy 12,0 ml dung dịch thu được tiến hành thử theo Phương pháp 1. Dùng 10,0 ml dung dịch chì mẫu 1 phần triệu Pb (TT) để chuẩn bị mẫu đối chiếu.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6).
(1,000 g; 105 °C, 3 h)
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Hòa tan 0,150 g chế phẩm trong 40 ml acid acetic khan (TT). Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ). Xác định điểm kết thúc bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2).
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 24,73 mg C8H10FN3O3S tính theo chế phẩm đã làm khô.
Bảo quản
Trong đồ đựng kín.
Loại thuốc
Thuốc kháng retrovirus.
Chế phẩm
Viên nén.
FLUNARIZIN DIHYDROCLORID
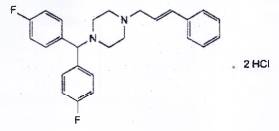
C26H26F2N2.2HCl | P.t.l: 477,4 |
Flunarizin dihydroclorid là 1-[bis(4-fluorophenyl)methyl]-4-[(2E)-3-phenylprop-2-enyl]piperazin dihydroclorid, phải chứa từ 99,0 % đến 101,5 % C26H26F2N2.2HCl, tính theo chế phẩm đã làm khô.
Tính chất
Bột trắng hoặc gần như trắng, hút ẩm. Hơi tan trong methanol, khó tan trong nước, ethanol 96 % và trong methylen clorid.
Điểm chảy ở khoảng 208 °C (Phụ lục 6.7), kèm theo phân hủy.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của flunarizin dihydroclorid chuẩn.
B. Hòa tan 25,0 mg chế phẩm trong 2 ml methanol (TT) và thêm 0,5 ml nước (TT). Dung dịch thu được cho phản ứng (A) của ion clorid (Phụ lục 8.1).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3)
Pha động A: Dung dịch chứa 23,8 g/L tetrabutylamoni hydrosulfat (TT) và 7 g/L amoni acetat (TT).
Pha động B: Acetonitril (TT).
Chuẩn bị các dung dịch sau ngay trước khi dùng và tránh ánh sáng.
Dung dịch thử: Hòa tan 100,0 mg chế phẩm trong methanol (TT) và pha loãng thành 10,0 ml với cùng dung môi.
Dung dịch đối chiếu (1): Hòa tan 10 mg flunarizin dihydroclorid chuẩn dùng để kiểm tra tính phù hợp của hệ thống trong 1,0 ml methanol (TT).
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml với methanol (TT). Pha loãng 5,0 ml dung dịch thu được thành 20,0 ml với methanol (TT).
Điều kiện sắc ký:
Cột kích thước (10 cm x 4,6 mm) được nhồi pha tĩnh base-deactivated octadecylsilyl silica gel dùng cho sắc ký (3 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 230 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 12 | 80 → 40 | 20 → 60 |
12 - 15 | 40 | 60 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), tỷ số đỉnh - hõm (Hp/Hv) ít nhất là 1,5 trong đó Hp là chiều cao đỉnh pic của tạp chất C so với đường nền và Hv là chiều cao tính từ đường nền lên đến đáy hõm giữa pic tạp chất C và pic flunarizin; sắc ký đồ của dung dịch đối chiếu (1) phải tương ứng với sắc ký đồ cung cấp kèm theo flunarizin dihydroclorid chuẩn dùng để kiểm tra tính phù hợp của hệ thống, sử dụng sắc ký đồ này để xác định vị trí các tạp chất A, B, C, D.
Để tính hàm lượng, nhân diện tích pic tạp chất A với hệ số hiệu chỉnh là 1,5.
Giới hạn:
Tạp chất A,D: Với mỗi tạp chất, diện tích pic không được lớn hơn 0,4 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,1 %).
Tạp chất B: Diện tích pic không được lớn hơn 2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,5 %).
Tạp chất C: Diện tích pic không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,25 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn 0,4 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,1 %).
Tổng tất cả các tạp chất không được lớn hơn 4 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (1,0 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,05 %).
Ghi chú:
Tạp chất A: 1-[bis(4-fluorophenyl)methyl]piperazin.
Tạp chất B: 1-[(RS)-(4-fluorophenyl)phenylmethyl]-4-[(2E)-3-phenylprop-2-enyl]piperazln.
Tạp chất C: 1-[(RS)-(2-fluorophenyl)(4-fluorophenyl)methyl]-4-[(2E)-3-phenylprop-2-enyl]piperazin.
Tạp chất D: 1-[bis(4-fluorophenyl)methyl]-4-[(2Z)-3-phenylprop-2-enyl]piperazin.
Mất khối lượng do làm khô
Không được quá 5,0 % (Phụ lục 9.6),
(1,000 g; 105 °C; 4 h).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm, chén platin.
Định lượng
Hòa tan khoảng 0,200 g chế phẩm trong 70 ml ethanol 96% (TT). Tiến hành định lượng bằng phương pháp chuẩn độ đo điện thể (Phụ lục 10.2), dùng dung dịch natri hydroxyd 0,1 N (CĐ). Đọc thể tích dung dịch natri hydroxyd 0,1 N (CĐ) đã tiêu thụ tại điểm uốn thứ hai. Song song tiến hành một mẫu trắng.
1 ml dung dịch natri hydroxyd 0,1 N (CĐ) tương đương với 23,87 mg C26H28CI2F2N2.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Chẹn kênh calci.
Chế phẩm
Viên nén, nang.
LAMOTRIGIN
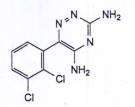
C9H7CI2N5 | P.t.l: 256,1 |
Lamotrigin là 6-(2,3-diclorophenyl)-1,2,4-triazin-3,5-diamin, phải chứa từ 99,0 % đến 101,0 % C9H7CI2N5, tính theo chế phẩm đã làm khô.
Tính chất
Bột màu trắng hoặc gần như trắng. Rất khó tan trong nước, khô tan trong ethanol khan.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của lamotrigin chuẩn.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Trộn đều 1 thể tích triethylamin (TT) và 150 thể tích dung dịch kali dihydrophosphat (TT) 0,27 %, điều chỉnh đến pH 2,0 bằng acid phosphoric (TT).
Pha động B: Acetonitril (TT).
Dung dịch thử: Hòa tan 20 mg chế phẩm trong 5 ml methanol (TT) và pha loãng thành 100,0 ml bằng dung dịch acid hydrocloric 0,1 M (TT).
Dung dịch đối chiếu (1): Hòa tan 5 mg lamotrigin chuẩn dùng để kiểm tra tính phù hợp của hệ thống (chứa tạp chất G) trong 2,5 ml methanol (TT) và pha loãng thành 25,0 ml bằng dung dịch acid hydrocloric 0,1 M (TT).
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng dung dịch acid hydrocloric 0,1 M (TT). Pha loãng 2,0 ml dung dịch thu được thành 10,0 ml bằng dung dịch acid hydrocloric 0,1 M (TT).
Dung dịch đối chiếu (3): Hòa tan 5,0 mg tạp chất E chuẩn của lamotrigin trong hỗn hợp 0,25 ml acid hydrocloric (TT) và 45 ml methanol (TT) và pha loãng thành 50,0 ml bằng methanol (TT). Pha loãng 5,0 ml dung dịch thu được thành 100,0 ml bằng dung dịch acid hydrocloric 0,1 M (TT). Lấy 4,0 ml dung dịch thu được, thêm 5 ml methanol (TT) và pha loãng thành 100,0 ml bằng dung dịch acid hydrocloric 0,1 M (TT).
Dung dịch đối chiếu (4): Hòa tan 10 mg lamotrigin chuẩn dùng để định tính pic (chứa tạp chất A, E và F) trong 2,5 ml methanol (TT) và pha loãng thành 50,0 ml bằng dung dịch acid hydrocloric 0,1 M (TT).
Dung dịch mẫu trắng: Trộn đều 5 thể tích methanol (TT) và 95 thể tích dung dịch acid hydrocloric 0,1 M (TT).
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh base-deactivated end-capped octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 270 nm.
Tốc độ dòng: 1,0 ml/mln.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 4 | 85 | 15 |
4 - 14 | 85 → 20 | 15 → 80 |
Tiến hành sắc ký với dung dịch thử, dung dịch đối chiếu (1), (2), (4) và dung dịch mẫu trắng.
Sử dụng sắc ký đồ cung cấp kèm theo lamotrigin chuẩn dùng để định tính pic và sắc ký đồ của dung dịch đối chiếu (4) để xác định pic của tạp chất A, E và F. Sử dụng sắc ký đồ cung cấp kèm theo lamotrigin chuẩn dùng để kiểm tra tính phù hợp của hệ thống và sắc ký đồ của dung dịch đối chiếu (1) để xác định pic của tạp chất G.
Thời gian lưu tương đối so với lamotrigin (thời gian lưu khoảng 7 min): Tạp chất G khoảng 1,1; tạp chất A khoảng 1,3; tạp chất E khoảng 1,7; tạp chất F khoảng 1,8.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), tỷ số đỉnh - hõm (Hp/Hv) ít nhất là 1,2; trong đó Hp là chiều cao đỉnh pic tạp chất G so với đường nền và Hv là chiều cao tính từ đường nền lên đến đáy hõm giữa pic tạp chất G và pic lamotrigin.
Giới hạn:
Tạp chất F: Diện tích pic tạp chất F sau khi nhân với hệ số hiệu chỉnh là 1,3 không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,2 %).
Tạp chất A, G: Với mỗi tạp chất, diện tích pic không được lớn hơn 0,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,1 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn 0,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,10 %).
Tổng diện tích pic của tất cả các tạp chất không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,2 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,25 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,05 %) và bỏ qua pic của tạp chất E.
Ghi chú:
Tạp chất A: 3-amino-6-(2,3-diclorophenyl)-1,2,4-triazin-5(4H)-on.
Tạp chất F: N-[5-amlno-6-(2,3-diclorophenyl)-1,2,4-triazin-3-yl]-2,3-diclorobenzamid.
Tạp chất G: 6-(2,4-diclorophenyl)-1,2,4-triazin-3,5-diamin.
Tạp chất E
Phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả ở phần Tạp chất liên quan với các thay đổi sau:
Pha động: Acetonitril dùng trong phương pháp sắc ký - pha động A (35 : 65).
Detector quang phổ hấp thụ tử ngoại đặt ở bước sóng 210 nm.
Tiến hành sắc ký với dung dịch thử, dung dịch đối chiếu (3) và (4).
Thời gian chạy sắc ký: 10 min.
Thời gian lưu của tạp chất E khoảng 5,5 min, của tạp chất F khoảng 8,5 min.
Kiểm tra tính phù hợp của hệ thống: sắc ký đồ của dung dịch đối chiếu (4) phải tương tự sắc ký đồ cung cấp kèm theo lamotrigln chuẩn dùng để định tính pic.
Giới hạn:
Tạp chất E: Diện tích pic tạp chất E không được lớn hơn diện tích pic tương ứng thu được trên sắc ký đồ của dung dịch đối chiếu (3) (0,1 %).
Ghi chú:
Tạp chất E: Acid 2,3-diclorobenzoic.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6).
(2,000 g; 105 °C; áp suất không quá 0,7 kPa; 3 h).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 2,0 g chế phẩm.
Định lượng
Hòa tan khoảng 0,200 g chế phẩm trong 60 ml acid acetic khan (TT). Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ). Xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thể (Phụ lục 10.2). Song song tiến hành một mẫu trắng.
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 25,61 mg C9H7CI2N5.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Thuốc chống động kinh.
Chế phẩm
Viên nén.
LEVETIRACETAM

C8H14N2O2 | P.t.l: 170,2 |
Levetiracetam là (2S)-2-(2-oxopyrrolidin-1-yl)butanamid, phải chứa từ 98,0 % đến 102,0 % C8H14N2O2, tính theo chế phẩm khan.
Tính chất
Bột màu trắng hoặc gần như trắng. Rất tan trong nước, tan trong acetonitril, thực tế không tan trong heptan.
Định tính
Có thể chọn một trong hai nhóm định tính sau:
Nhóm I: A, B.
Nhóm II: A, C.
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của levetiracetam chuẩn.
B. Chế phẩm phải đáp ứng phép thử Tạp chất đồng phân đối quang.
C. Góc quay cực riêng: Từ -82° đến -76° (Phụ lục 6.4).
Hòa tan 0,5 g chế phẩm trong nước và pha loãng thành 25,0 ml với cùng dung môi.
Độ trong và màu sắc của dung dịch
Hòa tan 2,0 g chế phẩm trong nước và pha loãng thành 10,0 ml với cùng dung môi.
Dung dịch thu được phải trong (Phụ lục 9.2) và không được có màu đậm hơn màu mẫu VN6 (Phụ lục 9.3, phương pháp 2).
Tạp chất đồng phân đối quang
Không được quá 0,8 %.
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: 2-propanol - heptan (18 : 82).
Dung dịch thử: Hòa tan 0,200 g chế phẩm trong 2-propanol (TT) và pha loãng thành 10,0 ml với cùng dung môi. Pha loãng 1,0 ml dung dịch thu được thành 20,0 ml bằng pha động.
Dung dịch đối chiếu (1): Hòa tan 5 mg chế phẩm và 5 mg tạp chất D chuẩn của levetiracetam trong pha động và pha loãng thành 5,0 ml với cùng dung môi.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng pha động. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh là dẫn xuất celulose của silica gel dùng cho sắc ký phân tách đồng phân đối quang (10 μm),
Detector quang phổ tử ngoại đặt ở bước sóng 205 nm.
Tốc độ dòng: 0,8 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 1,4 lần thời gian lưu của levetiracetam.
Sử dụng sắc ký đồ của dung dịch đối chiếu (1) để xác định pic của tạp chất D.
Thời gian lưu tương đối so với levetiracetam (thời gian lưu khoảng 12 min): Tạp chất D khoảng 0,8.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), độ phân giải giữa pic tạp chất D và pic levetiracetam ít nhất là 1,5 và hệ số đối xứng của pic levetiracetam không lớn hơn 2,4.
Giới hạn:
Tính hàm lượng phần trăm của tạp chất D theo phương pháp chuẩn hóa diện tích pic.
Bỏ qua các tạp chất có diện tích pic nhỏ hơn diện tích pic chính trên sắc ký đồ của dung dịch đối chiếu (2) (0,10%).
Ghi chú:
Tạp chất D: (2R)-2-(2-oxopyrrolidin-1-yl)butanamid [(R)-etiracetam].
Tạp chất G
Không được quá 0,05 %.
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril (TT1) - dung dịch đệm (15 : 85).
Dung dịch đệm: Hòa tan 1,22 g natri decansultonat (TT) trong 850 ml nước dùng cho sắc ký (TT), thêm 1,3 ml acid phosphoric (TT), điều chỉnh đến pH 3,0 bằng dung dịch kali hydroxyd 20 % và pha loãng thành 1000,0 ml bằng nước dùng cho sắc ký (TT).
Dung dịch thử: Hòa tan 20 mg chế phẩm trong pha động và pha loãng thành 10,0 ml bằng pha động.
Dung dịch đối chiếu (1): Hòa tan 2,0 mg tạp chất G chuẩn của levetiracetam trong pha động và pha loãng thành 100,0 ml với cùng dung môi.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch đối chiếu (1) thành 20,0 ml bằng pha động.
Dung dịch đối chiếu (3): Lấy 1,0 ml dung dịch đối chiếu (1), thêm 1,0 ml dung dịch thử và pha loãng thành 20,0 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh base-deactivated octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Nhiệt độ cột: 27 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 200 nm.
Tổc độ dòng: 1,0 ml/min.
Thể tích tiêm: 50 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu (2) và (3).
Tiến hành sắc ký với thời gian gấp 5 lần thời gian lưu của levetiracetam.
Sử dụng sắc ký đồ của dung dịch đối chiếu (2) để xác định pic của tạp chất G.
Thời gian lưu tương đối so với levetiracetam (thời gian lưu khoảng 4 min): Tạp chất G khoảng 3,8.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic levetiracetam và pic tạp chất G ít nhất là 5,0.
Tính hàm lượng phần trăm của tạp chất G dựa vào nồng độ của tạp chất G trong dung dịch đối chiếu (2).
Ghi chú:
Tạp chất G: (2S)-2-aminobutanamid.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Acetonitril (TT1) - dung dịch đệm phosphat pH 5,5 (TT) (5 : 95).
Pha động B: Acetonitril (TT1).
Dung dịch thử (1): Hòa tan 50,0 mg chế phẩm trong pha động A và pha loãng thành 10,0 ml bằng pha động A.
Dung dịch thử (2): Pha loãng 1,0 ml dung dịch thử (1) thành 50,0 ml bằng pha động A.
Dung dịch đối chiếu (1): Hòa tan 5 mg tạp chất A chuẩn của levetiracetam và 5 mg tạp chất E chuẩn của levetiracetam trong pha động A, thêm 1,0 ml dung dịch thử (1) và pha loãng thành 100,0 ml bằng pha động A.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử (1) thành 100,0 ml bằng pha động A. Pha loãng 1,0 ml dung dịch thu được thành 20,0 ml bằng pha động A.
Dung dịch đối chiếu (3): Hòa tan 5,0 mg tạp chất C chuẩn của levetiracetam trong pha động A và pha loãng thành 100,0 ml bằng pha động A. Pha loãng 1,0 ml dung dịch thu được thành 40,0 ml bằng pha động A.
Dung dịch đối chiếu (4): Hòa tan 50,0 mg levetiracetam chuẩn trong pha động A và pha loãng thành 10,0 ml bằng pha động A. Pha loãng 1,0 ml dung dịch thu được thành 50,0 ml bằng pha động A.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh base-deactivated end-capped octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 205 nm.
Tốc độ dòng: 0,9 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0-3 | 100 | 0 |
3-20 | 100 → 71 | 0 → 29 |
20-25 | 71 | 29 |
Tiến hành sắc ký với dung dịch thử (1), dung dịch đối chiếu (1), (2) và (3).
Sử dụng sắc ký đồ của dung dịch đối chiếu (1) để xác định pic của tạp chất A và E; sắc ký đồ của dung dịch đối chiếu (3) để xác định pic của tạp chất C.
Thời gian lưu tương đối so với levetiracetam (thời gian lưu khoảng 11 min): Tạp chất C khoảng 0,5; tạp chất A khoảng 0,7; tạp chất E khoảng 0,9.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), độ phân giải giữa pic tạp chất E và pic levetiracetam ít nhất là 3,5.
Tính hàm lượng phần trăm của tạp chất C dựa vào nồng độ của tạp chất C trong dung dịch đối chiếu (3) và tính hàm lượng phần trăm của các tạp chất khác dựa vào nồng độ của levetiracetam trong dung dịch đối chiếu (2).
Giới hạn:
Tạp chất A: Không được quá 0,3 %.
Tạp chất C: Không được quá 250 phần triệu (0,025 %).
Các tạp chất khác: Với mỗi tạp chất, không được quá 0,05 %.
Tổng các tạp chất: Không được quá 0,4 %.
Bỏ qua các tạp chất có hàm lượng nhỏ hơn 0,03 %, trừ tạp chất C.
Ghi chú:
Tạp chất A: Acid (2RS)-2-(2-oxopyrrolidin-1-yl)butanoic.
Tạp chất B: (2Z)-2-(2-oxopyrrolidin-1-yl)but-2-enamid.
Tạp chất C: Pyridin-2-ol
Tạp chất E: (1R)-1-phenylethan-1-amin.
Nước
Không được quá 0,5 % (Phụ lục 10.3).
Dùng 0,300 g chế phẩm.
Tro Sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả trong phần Tạp chất liên quan.
Tiến hành sắc ký dung dịch thử (2) và dung dịch đối chiếu (4).
Tính hàm lượng phần trăm của levetiracetam, C8H14N2O2, trong chế phẩm dựa vào diện tích pic levetiracetam thu được trên sắc ký đồ của dung dịch thử (2), dung dịch đối chiếu (4) và hàm lượng của C8H14N2O2 trong levetiracetam chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Thuốc chống động kinh.
Chế phẩm
Viên nén.
LEVOCETIRIZIN DIHYDROCLORID
C21H25CIN2O3.2HCl | P.t.l: 461,8 |
Levocetirizin dihydroclorid là acid (2-{4-[(R)-(4-clorophenyl)phenylmethyl]piperazin-1-yl}ethoxy)acetic dihydroclorid, phải chứa từ 98,0 % đến 102,0 % C21H25CIN2O3.2HCl, tính theo chế phẩm đã làm khô.
Tính chất
Bột màu trắng hoặc gần như trắng. Dễ tan trong nước và trong methanol.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của levocetirizin dihydroclorid chuẩn.
B. Trong phần Tạp chất đồng phân đối quang, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic levocetirizin trên sắc ký đồ của dung dịch đối chiếu.
C. Chế phẩm phải cho phản ứng (A) của ion clorid (Phụ lục 8.1).
pH
Từ 1,2 đến 1,8 (Phụ lục 6.2).
Dùng dung dịch chứa 50 mg/ml chế phẩm trong nước để đo.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - nước - dung dịch acid sulfuric 1 M (93: 6,6 : 0,4).
Dung dịch thử: Dung dịch chứa 200 μg/ml chế phẩm trong pha động.
Dung dịch đối chiếu (1): Dung dịch chứa 0,2 μg/ml levocetirizin dihydroclorid chuẩn; 0,2 μg/ml levocetirizin amid chuẩn và 0,2 μg/ml clorobenzhydryl piperazin chuẩn trong pha động.
Dung dịch đối chiếu (2): Dung dịch chứa 0,2 mg/ml levocetirizin dihydroclorid chuẩn; 0,2 μg/ml levocetirizin amid chuẩn và 0,2 μg/ml clorobenzhydryl piperazin chuẩn trong pha động.
Các dung dịch thử, dung dịch đối chiếu (1) và (2): chỉ dùng trong vòng 16 h sau khi pha.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh A (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 230 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian ít nhất bằng 3 lần thời gian lưu của levocetirizin.
Thời gian lưu tương đối so với levocetirizin: của clorobenzhydryl piperazin khoảng 1,3; của levocetirizin amid khoảng 2,5.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic levocetirizin và pic clorobenzhydryl piperazin không nhỏ hơn 3,0 và hệ số đối xứng của pic levocetirizln không lớn hơn 2,0, Trên sắc ký đồ của dung dịch đối chiếu (1), độ lệch chuẩn tương đối của diện tích pic levocetirizin thu được từ 6 lần tiêm lặp lại không lớn hơn 5,0 %.
Tính hàm lượng phần trăm của tạp chất levocetirizin amid hoặc clorobenzhydryl piperazin trong chế phẩm dựa vào diện tích pic levocetirizin amid hoặc clorobenzhydryl piperazin trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (1) và nồng độ của levocetirizin amid chuẩn hoặc clorobenzhydryl piperazin chuẩn trong dung dịch đối chiếu (1).
Tính hàm lượng phần trăm của các tạp chất khác trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic levocetirizin trên sắc ký đồ của dung dịch đối chiếu (1) và nồng độ của levocetlrizin dihydroclorid chuẩn trong dung dịch đối chiếu (1).
Giới hạn:
Clorobenzhydryl piperazin: Không được quá 0,2 %.
Levocetirizin amid: Không được quá 0,2 %.
Các tạp đơn khác: Với mỗi tạp chất, không được quá 0,1 %.
Tổng tạp chất: Không được quá 0,5 %.
Ghi chú:
Clorobenzhydryl piperazin: (R)-1 -[(4-Clorophenyl)phenylmethyl]piperazin.
Levocetirizine amid: (P)-2-(2-{4-[(4-Clorophenyl)phenylmethyl]piperazin-1-yl}ethoxy)acetamid.
Tạp chất đồng phân đối quang
Phương pháp sắc ký lỏng (Phụ lục 5.3). Bảo quản các dung dịch thử, dung dịch đối chiếu tránh ánh sáng.
Dung dịch đệm: Dung dịch chứa 1,5 g/L amoni acetat (TT) trong nước, điều chỉnh đến pH 4,8 bằng acid acetic băng (TT).
Pha động: Acetonitril - dung dịch đệm (30 : 70).
Dung dịch thử: Dung dịch chứa 0,5 mg/ml chế phẩm trong nước.
Dung dịch đối chiếu: Dung dịch chứa 0,5 mg/ml Cetirizin hydroclorid chuẩn trong nước.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh là các hạt silica gel xốp, hình cầu có phủ amylose tris-[(S)-a-methylbenzyl]carbamat (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 230 nm.
Tốc độ dòng: 0,5 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 1,8 lần thời gian lưu của levocetirizin.
Thời gian lưu tương đối của S-enantiomer (đồng phân S của Cetirizin) so với levocetirizin (đồng phân R của Cetirizin) khoảng 0,83.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, độ phân giải giữa pic S- enantiomer và pic levocetirizin không nhỏ hơn 1,4; hệ số đối xứng của pic levocetirizin không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic levocetirizin và pic S-enantiomer thu được từ 5 lần tiêm lặp lại không lớn hơn 1,5 %.
Tính hàm lượng phần trăm của tạp chất S-enantlomer trong chế phẩm dựa vào diện tích pic S- enantiomer trên sắc ký đồ của dung dịch thử và tổng diện tích pic S-enantiomer và pic levocetlrizin trên sắc ký đồ của dung dịch thử.
Giới hạn:
S-enantiomer: Không được quá 2,0 %.
Mất khối lượng do làm khô
Không được quá 1,0 % (Phụ lục 9.6).
(1,000 g; 105 °C).
Tro sulfat
Không được quá 0,2 % (Phụ lục 9,9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - nước - dung dịch acid sulfuric 1 M (93: 6,6 : 0,4).
Dung dịch thử: Dung dịch chứa 0,05 mg/ml chế phẩm trong pha động.
Dung dịch chuẩn: Dung dịch chứa 0,05 mg/ml levocetirizin dihydroclorid chuẩn trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh A (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 230 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic levocetirizin không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic levocetirizin thu được từ 5 lần tiêm lặp lại không lớn hơn 1,0 %.
Tính hàm lượng phần trăm của levocetlrizin dihydroclorid, C21H25CIN2O3.2HCl, trong chế phẩm dựa vào diện tích pic levocetirizin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H25CIN2O3.2HCl trong levocetirizin dihydroclorid chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc kháng histamin.
Chế phẩm
Viên nén.
LISINOPRIL

C21H31N3O5.2H2O | P.t.l: 441,5 |
Lisinopril là 1-[N2-[(S)-1-carboxy-3-phenylpropyl]-L-lysyl]-L-prolin dihydrat, phải chứa từ 98,0 % đến 102,0 % C21H31N3O5, tính theo chế phẩm khan.
Tính chất
Bột kết tinh màu trắng. Nóng chảy ở khoảng 160 °C kèm theo phân hủy. Tan trong nước, hơi tan trong methanol, thực tế không tan trong ethanol, aceton, acetonitril và cloroform.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của lisinopril chuẩn.
B. Trong phần Định lượng, thời gian lưu của pĩc chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Góc quay cực riêng
Từ - 47° đến - 43°, tính theo chế phẩm khan (Phụ lục 6.4).
Dung môi pha mẫu: Thêm 150 ml acid acetic băng (TT) và 54,9 g kẽm acetat (TT) vào 600 ml nước, khuấy đến khi kẽm acetat tan hết. Vừa khuấy vừa thêm 150 ml amoniac (TT), để nguội về nhiệt độ phòng và điều chỉnh đến pH 6,4 bằng amoniac (TT). Chuyển toàn bộ dung dịch thu được vào bình định mức 1000 ml và thêm nước đến vạch.
Dung dịch thử: Dung dịch chứa 10 mg/ml chế phẩm trong dung môi pha mẫu.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 3,53 g/l natri dihydrophosphat (TT) trong nước được điều chỉnh đến pH 4,1 bằng acid phosphoric (TT).
Pha động A: Acetonitril - dung dịch đệm (7 : 193).
Pha động B: Acetonitril - dung dịch đệm (20 : 80).
Dung dịch thử: Dung dịch chứa 2 mg/ml chế phẩm trong pha động A.
Dung dịch đối chiếu (1): Dung dịch chứa 0,006 mg/ml lisinopril chuẩn trong pha động A.
Dung dịch đối chiếu (2): Pha loãng dung dịch đối chiếu (1) bằng pha động A để thu được dung dịch chứa 1,0 μg/ml lisinopril chuẩn.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh B (5 μm).
Nhiệt độ cột: 45 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 210 nm.
Tốc độ dòng: 1,8 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 100 | 0 |
35 | 60 | 40 |
55 | 60 | 40 |
60 | 100 | 0 |
Tiến hành sắc ký dung dịch đối chiếu (1) và (2).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), độ lệch chuẩn tương đối của diện tích pic lisinopril thu được từ 6 lần tiêm lặp lại không lớn hơn 10,0 %. Trên sắc ký đồ của dung dịch đối chiếu (2), tỷ số tín hiệu trên nhiễu của pic lisinopril không nhỏ hơn 10.
Tiến hành sắc ký dung dịch đối chiếu (1) và dung dịch thử.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic lisinopril trên sắc ký đồ dung dịch đối chiếu (1); nồng độ của lisinopril trong dung dịch đối chiếu (1), dung dịch thử, chia diện tích pic tạp chất cho hệ số đáp ứng tương đối F (nếu có).
Thời gian lưu tương đối, hệ số đáp ứng tương đối và giới hạn phần trăm của các tạp chất được trình bày trong Bảng 1.
Bảng 1
Tạp chất | Thời gian lưu tương đối | Hệ số đáp ứng tương đối (F) | Giới hạn |
N-Alkyl-L-lysin | 0,57 | 0,35 | 0,3 |
DL-Homophenylalanin | 0,72 | 1,08 | 0,30 |
Lisinopril | 1,0 | 1,0 | - |
Lisinopril epimer | 1,33 | 0,76 | 0,3 |
Lisinopril cyclohexyl analog | 2,93 | 0,39 | 0,30 |
R, S, S-Diketopiperazin | 3,88 | 0,79 | 0,3 |
S, S, S-Diketopiperazin (tạp chất A) | 4,04 | 0,76 | 0,3 |
N-Alkyl lisinopril | 4,60 | 0,86 | 0,15 |
Tạp đơn chưa xác định | - | 1,00 | 0,1 |
Tổng tạp (không bao gồm tạp chất Lisinopril epimer) | - | - | 0,5 |
Bỏ qua các tạp chất có hàm lượng nhỏ hơn 0,05 %.
Ghi chú:
N-Alkyl-L-lysin: [(S)-1-Carboxy-3-phenylpropyl]-L-lysin.
DL-Homophenylalanin: Acid 2-amino-4-phenylbutanoic.
Lisinopril epimer: [(R)-1-Carboxy-3-phenylpropyl]-L-lysyl-L-prolin.
Lisinopril cyclohexyl analog: [(S)-1-Carboxy-3-cyclohexylpropyl]-L-lysyl-L-prolin.
R,S,S-Diketopiperazin: Acid (S)-2-[(3S,8aR)-3-(4-aminobutyl)-1,4-dioxohexahydropyrrolo[1,2-a]pyrazin-2(1H)-yl]-4-phenylbutanoic.
S,S,S-Diketopiperazin (tạp chất A): Acid (S)-2-[(3S,8aS)-3-(4-Aminobutyl)-1,4-dioxohexahydropyrrolo[1,2- a]pyrazin-2(1H)-yl]-4-phenylbutanoic.
N-Alkyl lisinopril: N2,N6-Bis[(S)-1 -carboxy-3-phenylpropyl]-L-lysyl-L-prolin.
Nước
Từ 8,0 % đến 9,5 % (Phụ lục 10.3).
Dùng 0,200 g chế phẩm.
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 3,12 g natri dihydrophosphat (TT) trong 900 ml nước, điều chỉnh đến pH 5,0 bằng dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml.
Pha động: Acetonitril - dung dịch đệm (4 : 96).
Dung dịch thử: Dung dịch chứa 0,3 mg/ml chế phẩm trong nước.
Dung dịch chuẩn: Dung dịch chứa 0,3 mg/ml lisinopril chuẩn trong nước.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh B (5 μm).
Nhiệt độ cột: 50 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 210 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch chuẩn.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic lisinopril không lớn hơn 1,7; độ lệch chuẩn tương đối của diện tích pic lisinopril thu được từ 5 lần tiêm lặp lại không lớn hơn 0,73 %.
Tiến hành sắc ký dung dịch chuẩn và dung dịch thử.
Tính hàm lượng phần trăm của lisinopril, C21H31N3O5, trong chế phẩm dựa vào diện tích pic lisinopril trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H31N3O5 trong lisinopril chuẩn.
Bảo quản
Trong đồ đựng kín.
Loại thuốc
Thuốc ức chế enzym chuyển angiotensln.
Chế phẩm
Viên nén.
LYSIN HYDROCLORID
C6H14N2O2.HCl | P.t.l: 182,7 |
Lysin hydroclorid là acid (2S)-2,6-diaminohexanoic hydroclorid, phải chứa từ 98,5 % đến 101,5 % C6H14N2O2.HCl, tính theo chế phẩm đã làm khô.
Tính chất
Bột màu trắng, không mùi. Dễ tan trong nước.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của lysln hydroclorid chuẩn.
Góc quay cực riêng
Từ +20,4° đến +21,4°, tính theo chế phẩm đã làm khô (Phụ lục 6.4).
Dùng dung dịch chứa 80 mg/ml chế phẩm trong dung dịch acid hydrocloric 6 M (TT) để đo. Đo ở 25 °C.
Tạp chất liên quan
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel G.
Dung môi khai triển: Isopropanol - amoniac (7 : 3).
Dung dịch thử: Dung dịch chứa 10 mg/ml chế phẩm trong nước.
Dung dịch đối chiếu (1): Dung dịch chứa 0,05 mg/ml lysin hydroclorld chuẩn trong nước.
Dung dịch đối chiếu (2): Dung dịch chứa 0,4 mg/ml lysln hydroclorid chuẩn và 0,4 mg/ml arginin hydroclorid chuẩn trong nước.
Cách tiến hành: Chấm riêng biệt lên bản mỏng 5 μl mỗi dung dịch trên. Triển khai sắc ký tới khi dung môi đi được 15 cm. Sấy bản mỏng ở 100 °C đến 105 °C để amoniac bay hơi hết. Phun lên bản mỏng dung dịch ninhydrin 0,2 % (TT) và sấy ở 100 °C đến 105 °C trong 15 min. Quan sát dưới ánh sáng thường, bất kỳ vết phụ nào trên sắc ký đồ của dung dịch thử không được đậm hơn vết chính trên sắc ký đồ của dung dịch đối chiếu (1) (0,5 %); tổng độ đậm của các vết phụ trên sắc ký đồ của dung dịch thử không được lớn hơn 2,0 %. Phép thử chỉ có giá trị khi sắc ký đồ của dung dịch đối chiếu (2) cho 2 vết tách rõ rệt.
Hàm lượng clorid
Từ 19,0% đến 19,6%.
Hòa tan khoảng 350 mg chế phẩm trong 140 ml nước, thêm 1 ml dung dịch diclorofluorescein 0,1 % (TT) và chuẩn độ bằng dung dịch bạc nitrat 0,1 N (CĐ) đến khi tủa bạc clorid kết bông và hỗn hợp thu được có màu hồng nhạt. Song song tiến hành mẫu trắng, dùng 140 ml nước.
1 ml dung dịch bạc nitrat 0,1 N (CĐ) tương đương với 3,545 mg Cl.
Sulfat
Không được quá 300 phần triệu (Phụ lục 9.4.14).
Hòa tan 0,5 g chế phẩm trong 15 ml nước cất và tiến hành thử.
Sắt
Không được quá 30 phần triệu (Phụ lục 9.4.13).
Hòa tan 0,33 g chế phẩm trong 10 ml dung dịch acid hydrocloric loãng (TT) trong một bình gạn. Chiết 3 lần, mỗi lần với 10 ml methyl isobutyl keton (TT1) và lắc trong 3 min. Tập trung dịch chiết hữu cơ, thêm 10 ml nước, lắc trong 3 min. Lấy lớp nước và tiến hành thử.
Mất khối lượng do làm khô
Không được quá 0,4 % (Phụ lục 9.6).
(1,000 g; 105 °C; 3 h).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Hòa tan khoảng 90 mg chế phẩm trong hỗn hợp gồm 3 ml acid formic (TT) và 50 ml acid acetic băng (TT). Thêm 10 ml dung dịch thủy ngân acetat 6 % (TT) và chuẩn độ bằng dung dich acid percloric 0,1 N (CĐ). Xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thể (Phụ lục 10.2). Song song tiến hành làm mẫu trắng, dùng hỗn hợp gồm 3 ml acid formic (TT) và 50 ml acid acetic băng (TT).
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 9,133 mg C6H14N2O2.HCl.
Bảo quản
Trong đồ đựng kín.
Loại thuốc
Acid amin.
Chế phẩm
Viên nén, nang.
MIFEPRISTON

C29H35NO2 | P.t.l: 429,6 |
Mifepriston là 17β-hydroxy-11β-(4-dimethylamino)phenyl-17-(1-propynyl)-4,9-estradien-3-on, phải chứa từ 98,0 % đến 102,0 % C29H35NO2, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu vàng nhạt.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của mifepriston chuẩn hoặc phổ đối chiếu của mifepriston.
Góc quay cực riêng
Từ +124° đến +135° (Phụ lục 6.4).
Dùng dung dịch chứa 0,5 % chể phẩm trong dicloromethan (TT).
Độ hấp thụ ánh sáng
Độ hấp thụ ánh sáng (Phụ lục 4.1) của dung dịch chứa 1,0 % chế phẩm trong methanol (TT) ở bước sóng 420 nm không được quá 1,25.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Pha loãng 0,75 ml acid formic (TT) thành 1000 ml bằng nước, điều chỉnh đến pH 5,0 bằng dung dịch amoniac loãng (TT).
Pha động B: Methanol - acetonltril (50 : 50).
Dung môi pha mẫu: Nước - methanol (50 : 50).
Dung dịch thử: Hòa tan 25 mg chế phẩm trong 5 ml methanol (TT) và pha loãng thành 25,0 ml bằng dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4 mm) được nhồi pha tĩnh C (3 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 260 nm.
Tốc độ dòng: 0,8 ml/min.
Thể tích tiêm: 10 μl.
Nhiệt độ autosampler: 5 °C.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 40 | 60 |
30 | 34 | 66 |
50 | 10 | 90 |
60 | 10 | 90 |
65 | 40 | 60 |
70 | 40 | 60 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch thử, độ phân giải giữa pic mifepriston và pic phụ gần nhất không nhỏ hơn 1,5.
Tính hàm lượng phần trăm các tạp chất theo phương pháp chuẩn hóa.
Giới hạn:
Tạp chất bất kỳ: Với mỗi tạp chất, không được quá 0,5 %.
Tổng các tạp chất: Không được quá 1,0 %.
Kim loại nặng
Không được quá 20 phần triệu (Phụ lục 9.4.8).
Dung dịch thử: Thêm 1 lượng vừa đủ acid sulfuric (TT) để làm ướt 1,0 g chế phẩm, sau đó nung cẩn thận ở nhiệt độ thấp đến khi thành than. Thêm 2 ml acid nitric (TT) và 5 giọt acid sulfuric (TT) và đun hỗn hợp cẩn thận đến khi hết khỏi. Sau đó đem hỗn hợp nung ở 500 °C - 600 °C đến khi carbon cháy hoàn toàn. Để nguội, thêm 4 ml acid hydrocloric (TT). Đậy nắp và đun trên cách thủy 15 min, mở nắp để bốc hơi từ từ trên cách thủy đến cắn. Làm ẩm cắn bằng 1 giọt acid hydrocloric (TT), thêm 10 ml nước nóng và đun tiếp 2 min, thêm từng giọt amoniac (TT) đến khi dung dịch chuyển sang kiềm, thử bằng giấy quỳ tím (TT). Pha loãng dung dịch thu được thành 25 ml bằng nước và điều chỉnh về pH 3,0 đến 4,0 bằng acid acetic loãng (TT). Lọc, nếu cần rửa chén nung và phễu lọc bằng 10 ml nước, gộp dịch lọc và dịch rửa vào ống Nessler, và pha loãng thành 35 ml bằng nước.
Dung dịch đối chiếu: Trong một ống Nessler, pha loãng 1,0 ml dung dịch chì mẫu 20 phần triệu Pb (TT) thành 25,0 ml bằng nước, điều chỉnh pH của dung dịch thu được về pH 3,0 đến 4,0 bằng acid acetic loãng (TT) hoặc dung dịch amoniac loãng (TT), và thêm nước vừa đủ 35 ml.
Tiến hành: Thêm 10 ml dung dịch hydrogen sulfid (TT) mới pha vào mỗi ống chứa dung dịch đối chiếu và dung dịch thử, lắc đều và thêm nước vừa đủ 50 ml, để yên 5 min. Quan sát thử và chuẩn trên nền trắng, từ trên xuống.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6).
(1,000 g; 105 °C, 2 h).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 1).
Dùng 1,0 g chế phẩm.
Định lượng
Hòa tan 0,3 g chế phẩm trong 50 ml acid acetic băng (TT). Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ). Xác định điểm kết thúc bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2). Song song tiến hành một mẫu trắng.
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 0,04296 g C29H35NO2.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc tránh thai, thuốc gây sẩy thai.
Chế phẩm
Viên nén.
MONTELUKAST NATRI

C35H35CINNaO3S | P.t.l: 608,2 |
Montelukast natri là natri [1-[[[(1R)-1-[3-[(E)-2-(7-cloroquinolin-2-yl)ethenyl]phenyl]-3-[2-(1-hydroxy-1- methylethyl)phenyl]propyl]sulfanyl]methyl]cyclopropyl]acetat, phải chứa từ 98,0 % đến 102,0 % C35H35CINNaO3S, tính theo chế phẩm khan.
Tính chất
Bột trắng hoặc gần như trắng, hút ẩm. Dễ tan trong nước và trong methylen clorid, dễ tan đến rất tan trong ethanol 96 %.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của montelukast natri chuẩn.
B. Chế phẩm phải đáp ứng phép thử Tạp chất đồng phân đổi quang.
C. Nung 0,1 g đến khi thu được cắn gần như trắng. Hòa tan cắn trong 2 ml nước và lọc, dịch lọc cho phản ứng (A) của ion natri (Phụ lục 8.1).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3). Tiến hành trong điều kiện tránh ánh sáng, sử dụng bình nâu, siêu âm trong nước đá, các dung dịch dùng ngay sau khi pha.
Pha động A: Hòa tan 3,85g amoni acetat (TT) trong 1000 ml nước, thêm 1 ml triethylamin (TT), điều chỉnh đến pH 5,5 bằng acid acetic băng (TT).
Pha động B: Methanol (TT).
Dung môi pha mẫu: Nước - methanol (20 : 80)
Dung dịch thử: Hòa tan chính xác khoảng 100 mg chế phẩm trong 100,0 ml dung môi pha mẫu.
Dung dịch đối chiếu (1): Dung dịch montelukast natri chuẩn 0,1 % trong dung môi pha mẫu.
Dung dịch đối chiếu (2): Pha loãng 10,0 ml dung dịch đối chiếu (1) thành 100,0 ml bằng dung môi pha mẫu. Pha loãng 3,0 ml dung dịch thu được thành 100,0 ml bằng dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm.
Tốc độ dòng: 1,0ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 40 | 60 |
20 | 30 | 70 |
45 | 20 | 80 |
55 | 15 | 85 |
60 | 15 | 85 |
65 | 40 | 60 |
70 | 40 | 60 |
Thời gian lưu tương đối so với montelukast của các đồng phân montelukast sultoxid là khoảng 0,66 và 0,69; của montelukast styren là khoảng 1,38.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ lệch chuẩn tương đối của diện tích pic montelukast thu được từ 6 lần tiêm lặp lại không lớn hơn 5,0 %,
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Tổng diện tích pic của các đồng phân montelukast sulfoxid không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,3 %).
Diện tích pic montelukast styren không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,3 %).
Không được có bất kỳ pic tạp chất nào khác có diện tích lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,3 %).
Tổng diện tích của tất cả các pic tạp chất không được lớn hơn 3,3 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (1,0 %).
Tạp chất đồng phân đối quang
Phương pháp sắc ký lỏng (Phụ lục 5.3). Tiến hành trong điều kiện tránh ánh sáng, sử dụng bình nâu.
Pha động A: Dung dịch chứa 2,3 g/L amoni acetat (TT) được chỉnh pH đến 5,7 bằng acid acetic băng (TT).
Pha động B: Acetonitril - methanol (40 : 60).
Dung môi pha mẫu: Acetonitril - nước (50 : 50).
Dung dịch thử: Hòa tan 50 mg chế phẩm trong dung môi pha mẫu và pha loãng thành 50,0 ml với cùng dung môi.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng dung môi pha mẫu. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (2): Dung dịch chứa 0,1 mg/ml montelukast racemat chuẩn trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,0 mm) được nhồi pha tĩnh là các hạt silica gel hình cầu có gắn α1-acid- glycoprotein (5 μm) (loại cột tương tự L41 của Dược điển Mỹ).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 280 nm.
Tốc độ dòng: 0,9 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 30 | 70 → 60 | 30 → 40 |
30 - 35 | 60 | 40 |
Thời gian lưu tương đối so với montelukast (thời gian lưu khoảng 25 min) của tạp chất A khoảng 0,7.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic tạp chất A và pic montelukast ít nhất là 2,9. Trên sắc ký đồ của dung dịch đối chiếu (1), tỷ số tín hiệu trên nhiễu tính của pic chính ít nhất là 10.
Phần trăm tạp chất A được tính dựa trên tỷ lệ diện tích pic của tạp chất A so với tổng diện tích pic montelukast và tạp chất A thu được trên sắc ký đồ của dung dịch thử.
Giới hạn tạp chất A: Không được quá 0,2 %.
Ghi chú:
Tạp chất A: Acid [1-[[[(1S)-1-[3-[(E)-2-(7-cloroquinolin-2-yl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methyl ethyl)phenyl] propyl]sulfanyl]methyl]cyclopropyl]acetic.
Nước
Không quá 4,0% (Phụ lục 10.3).
Dùng 0,300 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3). Điều kiện tiến hành, pha động A, pha động B, dung môi pha mẫu như mục Tạp chất liên quan.
Pha động: Pha động A - pha động B (22 : 78).
Dung dịch thử: Hòa tan chính xác khoảng 50 mg chế phẩm trong 100,0 ml dung môi pha mẫu. Pha loãng chính xác 5,0 ml dung dịch này thành 50,0 ml bằng dung môi pha mẫu.
Dung dịch chuẩn: Cân chính xác khoảng 50 mg montelukast natri chuẩn hòa tan trong dung môi pha mẫu và thêm dung môi pha mẫu vừa đủ 100,0 ml. Pha loãng 5,0 ml dung dịch thu được thành 50,0 ml bằng dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm,
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic montelukast không nhỏ hơn 2000, hệ số đối xứng của pic montelukast không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic montelukast thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiêm lần lượt dung dịch thử và dung dịch chuẩn.
Tính hàm lượng montelukast natri, C35H35CINNaO3S, trong chế phẩm dựa vào diện tích pic montelukast trên sắc ký đồ của dung dịch thử, dung dịch chuẩn là hàm lượng C35H35CINNaO3S của montelukast natri chuẩn.
Bảo quản
Trong đồ đựng kín và tránh ánh sáng, ở nhiệt độ phòng.
Loại thuốc
Thuốc đối kháng thụ thể leukotrien CysLT1; điều trị hen.
Chế phẩm
Viên nén, viên nhai.
NIMODIPIN
C21H26N2O7 | P.t.l: 418,4 |
Nimodipin là isopropyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-(m-nitrophenyl)-3,5- pyridinedicarboxylat, phải chứa từ 98,0 % đến 102,0 % C21H26N2O7, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu vàng hoặc vàng nhạt, đa hình, nhạy cảm với ánh sáng. Dễ tan trong ethyl acetat, hơi tan trong ethanol 96 %, thực tế không tan trong nước.
Chú ý: Quá trình tiến hành các phép thử cần thực hiện tránh ánh sáng hoặc dưới ánh sáng dịu (có bước sóng lớn hơn 420 nm), các dung dịch chứa nimodipin chỉ pha ngay trước khi dùng và trong các dụng cụ thủy tinh tối màu.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của nimodipin chuẩn.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Góc quay cực riêng
Từ -0,10° đến +0,10°, tính theo chế phẩm đã làm khô (Phụ lục 6.4).
Dùng dung dịch chứa 50 mg/ml chế phẩm trong aceton (TT) để đo.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Methanol - tetrahydroturan - nước (20 : 20 : 60).
Dung dịch thử: Hòa tan 40 mg chế phẩm trong 2,5 ml tetrahydrofuran (TT) và pha loãng thành 25 ml bằng pha động.
Dung dịch đối chiếu (1): Hòa tan một lượng thích hợp nimodipin chuẩn bằng tetrahydrofuran (TT) (thể tích tetrahydroturan đã dùng chiếm 10 % tổng thể tích) và pha loãng bằng pha động để thu được dung dịch có nồng độ nimodipin 1,6 mg/ml. Pha loãng 1,0 ml dung dịch thu được thành 50,0 ml bằng pha động. Tiếp tục pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động (nồng độ nimodipin 3,2 μg/ml).
Dung dịch đối chiếu (2): Hòa tan một lượng thích hợp nimodipin chuẩn và tạp chất A chuẩn của nimodipin bằng tetrahydroturan (TT) (thể tích tetrahydrofuran đã dùng chiếm 10 % tổng thể tích) và pha loãng bằng pha động để thu được một dung dịch có nồng độ nimodipin 0,8 mg/ml và tạp chất A của nimodipin 0,8 mg/ml. Pha loãng 1,0 ml dung dịch thu được thành 50,0 ml bằng pha động. Tiếp tục pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng pha động (nồng độ nimodipin 1,6 pg/ml và tạp chất A của nimodipin 1,6 μg/ml).
Điều kiện sắc ký:
Cột kích thước (12,5 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 235 nm.
Tốc độ dòng: 2 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Thời gian lưu tương đối so với nimodipin của tạp chất A khoảng 0,9.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic tạp chất A và pic nimodipin không nhỏ hơn 1,5 và độ lệch chuẩn tương đối của diện tích pic tạp chất A thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %. Trên sắc ký đồ của dung dịch đối chiếu (1), độ lệch chuẩn tương đối của diện tích pic nimodipin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của tạp chất A trong chế phẩm dựa vào diện tích pic tạp chất A thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (2) và nồng độ tạp chất A chuẩn trong dung dịch đối chiếu (2).
Tính hàm lượng phần trăm của các tạp chất khác trong chế phẩm dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic nimodipln trên sắc ký đồ của dung dịch đối chiếu (1) và nồng độ nimodipin chuẩn trong dung dịch đối chiếu (1).
Giới hạn:
Tạp chất A: Không được quá 0,1 %.
Các tạp chất khác: Với mỗi tạp chất, không được quá 0,2 %.
Tổng các tạp chất: Không được quá 0,5 %,
Ghi chú:
Tạp chất A: 2-Methoxyethyl-1-methylethyl-2,6-dimethyl-4-(3-nitrophenyl)pyridin-3,5-dicarboxylat.
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6).
(1,000 g; 105 °C; 4 h).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động và điều kiện sắc ký: Như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Hòa tan một lượng thích hợp chế phẩm bằng tetrahydrofuran (TT) (thể tích tetrahydroturan đã dùng chiếm 10 % tổng thể tích) và pha loãng bằng pha động để thu được dung dịch có nồng độ nimodipin 0,3 mg/ml.
Dung dịch chuẩn: Hòa tan một lượng thích hợp nimodipin chuẩn bằng tetrahydrofuran (TT) (thể tích tetrahydrofuran đã dùng chiếm 10 % tổng thể tích) và pha loãng bằng pha động để thu được dung dịch có nồng độ nimodipin chuẩn 0,3 mg/ml.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic nimodipin không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic nimodipin thu được từ 5 lần tiêm lặp lại không lớn hơn 0,73 %.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Tính hàm lượng phần trăm của nimodipin, C21H26N2O7, trong chế phẩm dựa vào diện tích pic nimodipin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H26N2O7 trong nimodipin chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ phòng có điều nhiệt
Loại thuốc
Thuốc chẹn kênh calci.
Chế phẩm
Viên nén.
ONDANSETRON HYDROCLORID
C18H19N3O.HCl.2H2O | P.t.l: 365,9 |
Ondansetron hydroclorid là (3RS)-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-1,2,3,9-tetrahydro- 4H-carbazol-4-on hydroclorid dihydrat, phải chứa từ 98,0 % đến 102,0 % C18H19N3O.HCl, tính theo chế phẩm khan.
Tính chất
Bột màu trắng hoặc trắng ngà. Tan trong methanol; hơi tan trong nước và ethanol 96 %; khó tan trong isopropanol và dicloromethan; rất khó tan trong aceton, cloroform và ethyl acetat.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của ondansetron hydroclorid chuẩn.
B. Hòa tan 20 mg chế phẩm trong 2 ml nước, thêm 1 ml dung dịch acid nitric 2 M (TT), lọc. Dịch lọc phải cho phản ứng (A) của ion clorid (Phụ lục 8.1).
Tạp chất D
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 2,72 g kali dihydrophosphat (TT) trong 900 ml nước, điều chỉnh đến pH 5,4 bằng dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml. Lọc và đuổi khí.
Pha động: Dung dịch đệm - acetonitril (80 : 20).
Dung dịch thử: Hòa tan 50 mg chế phẩm trong pha động và pha loãng thành 100,0 ml bằng pha động.
Dung dịch đối chiếu: Dung dịch chứa 0,4 μg/ml tạp chất D chuẩn của ondansetron trong pha động.
Dung dịch phù hợp hệ thống: Dung dịch chứa 0,6 μg/ml tạp chất D chuẩn của ondansetron và 1 μg/ml tạp chất C chuẩn của ondansetron trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh là các hạt silica gel liên kết hóa học với các nhóm nitril (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 328 nm.
Tốc độ dòng: 1,5 ml/mln.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic tạp chất C và pic tạp chất D ít nhất là 1,5; thời gian lưu tương đối của tạp chất C so với tạp chất D khoảng 0,8. Trên sắc ký đồ của dung dịch đối chiếu, số đĩa lý thuyết tính trên pic tạp chất D không nhỏ hơn 400 và độ lệch chuẩn tương đối của diện tích pic tạp chất D thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của tạp chất D trong chế phẩm dựa vào diện tích pic tạp chất D trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu và nồng độ của tạp chất D chuẩn trong dung dịch đối chiếu.
Giới hạn:
Tạp chất D: Không được quá 0,10 %.
Ghi chú:
Tạp chất D: 9-Methyl-3-methylen-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Tạp chất liên quan
Phương pháp 1
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel GF254.
Dung môi khai triển: Cloroform - ethyl acetat - methanol - amoniac (90 : 50 : 40 : 1).
Dung dịch thử: Dung dịch chứa 12,5 mg/ml chế phẩm trong methanol (TT).
Dung dịch đối chiếu gốc: Dung dịch chứa 0,25 mg/ml ondansetron chuẩn trong methanol (TT).
Pha loãng dung dịch đối chiếu gốc bằng methanol (TT) để được các dung dịch sau:
Dung dịch đối chiếu (1): Dung dịch chứa 50 μg/ml ondansetron hydroclorid chuẩn trong methanol (TT).
Dung dịch đối chiếu (2): Dung dịch chứa 25 μg/ml ondansetron chuẩn trong methanol (TT).
Dung dịch đối chiếu (3): Dung dịch chứa 12,5 μg/ml ondansetron chuẩn trong methanol (TT).
Dung dịch phân giải: Dung dịch chứa 12,5 mg/ml hỗn hợp ondansetron chuẩn để thử độ phân giải trong methanol (TT).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 20 μl dung dịch thử, các dung dịch đối chiếu (1), (2), (3) và dung dịch phân giải. Triển khai sắc ký đến khi dung môi đi được 3/4 chiều dài bản mỏng. Lấy bản mỏng ra, để dung môi bay hơi hết và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm.
So sánh độ đậm của bất kỳ vết phụ nào trên sắc ký đồ của dung dịch thử với các vết chính trên sắc ký đồ của các dung dịch đối chiếu: Bất kỳ vết phụ nào trên sắc ký đồ của dung dịch thử có Rf tương ứng với vết phụ cao nhất trên sắc ký đồ của dung dịch phân giải không được lớn hơn hoặc đậm hơn vết chính trên sắc ký đồ của dung dịch đối chiếu (1) (0,4 %) và không có vết phụ nào khác trên sắc ký đồ của dung dịch thử lớn hơn hoặc đậm hơn vết chính trên sắc ký đồ của dung dịch đối chiếu (2) (0,2 %). Phép thử chỉ có giá trị khi sắc ký đồ của dung dịch phân giải cho 3 vết tách rõ rệt.
Ghi chú: Hỗn hợp ondansetron chuẩn để thử độ phân giải là ondansetron hydroclorid chứa 0,4 % tạp chất A và 0,4 % 6,6'-methylen bis-[(1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)-methyl]-4H-carbazol-4-on).
Phương pháp 2
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 3,12 g natri dihydrophosphat (TT) trong 900 ml nước, điều chỉnh đến pH 5,4 bằng dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml. Lọc và đuổi khí.
Pha động: Dung dịch đệm - acetonitril (50 : 50).
Dung dịch thử: Hòa tan 45 mg chế phẩm trong pha động và pha loãng thành 50,0 ml bằng pha động. Pha loãng 5,0 ml dung dịch thu được thành 50 ml bằng pha động.
Dung dịch đối chiếu (1): Dung dịch chứa 90 pg/ml ondansetron hydroclorid chuẩn trong pha động.
Dung dịch đối chiếu (2): Dung dịch chứa 90 μg/ml ondansetron hydroclorid chuẩn và 20 μg/ml tạp chất A chuẩn của ondansetron trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh là các hạt silica gel liên kết hóa học với các nhóm nitril (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 216 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), thời gian lưu tương đối của tạp chất A so với ondansetron khoảng 1,1; độ phân giải giữa pic tạp chất A và pic ondansetron không nhỏ hơn 1,5. Trên sắc ký đồ của dung dịch đối chiếu (1), hệ số đối xứng của pic ondansetron không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic ondansetron thu được từ 5 lần tiêm lặp lại không lớn hơn 1,5 %.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic ondansetron trên sắc ký đồ của dung dịch đối chiếu (1), nồng độ của ondansetron trong dung dịch đối chiếu (1). Chia diện tích pic tạp chất cho hệ số đáp ứng tương đối (F) nếu có.
Thời gian lưu tương đối, hệ số đáp ứng tương đối và giới hạn phần trăm của các tạp chất được trình bày trong Bảng 1.
Bảng 1
Tạp chất | Thời gian lưu tương đối | Hệ số đáp ứng tương đối (F) | Giới hạn (%) |
Tạp chất C | 0,32 | 1,2 | 0,2 |
Tạp chất D* | 0,34 | - | 0,1 |
Imidazol | 0,49 | 0,3 | 0,2 |
2-methylimidazol | 0,54 | 0,4 | 0,2 |
Ondansetron | 1 | - | - |
Tạp chất A | 1,10 | 0,8 | 0,2 |
Tạp chất chưa xác định | - | 1,0 | 0,1 |
Tổng | - | - | 0,5 |
* Xác định trong phép thử tạp chất D
Ghi chú:
Tạp chất A: 3-[(Dimethylamino)methyl] -9-methyl -1,2,3,9-tetrahydro-4H-carbazol-4-on hydroclorid.
Tạp chất C: 9-Methyl-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Nước
Từ 9,0 % đến 10,5 % (Phụ lục 10.3).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động, dung dịch thử và các dung dịch đối chiếu, điều kiện sắc ký và yêu cầu kiểm tra tính phù hợp của hệ thống như mô tả ở phần Tạp chất liên quan (phương pháp 2).
Cách tiến hành:
Tiến hành sắc ký dung dịch thử và dung dịch đối chiếu (1).
Tính hàm lượng phần trăm của ondansetron hydroclorid, C18H19N3O.HCl, trong chế phẩm dựa vào diện tích pic ondansetron thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (1) và hàm lượng của C18H19N3O.HCl trong ondansetron hydroclorld chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở 25 °C.
Loại thuốc
Thuốc chống nôn, đối kháng thụ thể 5-HT3.
Chế phẩm
Viên nén, thuốc tiêm.
OXYTOCIN
![]()
C43H66N12O12S2 | P.t.l: 1007 |
Oxytocin là L-cysteinyl-L-tyrosyl-L-isoleucyl-L-glutamyl-L-asparaglnyl-L-cysteinyl-L-prolyl-L-leucylglycinamid cyclic (1→6)-disulfid, phải chứa từ 93,0 % đến 103,0 % C43H66N12O12S2, tính theo chế phẩm khan và không có acid acetic.
Oxytocin là một noriapeptit vòng tổng hợp có cáu trúc của hormon, được sản xuất bởi thùy sau tuyến yên, có tác dụng kích thích co bóp tử cung và tiết sữa ở động vật có vú. Tồn tại dưới dạng đông khô thành muối acetat.
Theo quy ước, với mục đích ghi nhãn các chể phẩm chứa oxytoxin, 1 mg oxytoxin peptit (CC43H66N12O12S2) tương đương với 600 IU hoạt tính sinh học.
Tính chất
Bột màu trắng hoặc gần như trắng, hút ẩm. Rất tan trong nước, tan trong các dung dịch acid acetic loãng và ethanol.
Sản xuất
Phương pháp sản xuất phải được thẩm định để đảm bảo nếu được tiến hành kiểm tra thì chế phẩm phải đáp ứng yêu cầu của phép thử Các acid amin sau:
Các acid amin
Dùng máy phân tích acid amin thích hợp, đã được hiệu chuẩn. Để chuẩn hóa thiết bị, dùng hỗn hợp cùng nồng độ mol của amonlac, glycin và các acld amin ở dạng đồng phân L sau: Lysin, threonin, alanin, leucin, histidin, serin, valin, tyrosin, arginin, acid glutamic, methlonin, phenylalanin, acid aspartic, prolin, isoleucin và một lượng L-cystin có nồng độ mol bằng 1/2 nồng độ các chất trên. Để thẩm định phương pháp, dùng chất nội chuẩn thích hợp, ví dụ DL-norleucin.
Dung dịch thử: Cân chính xác 1,0 mg chế phẩm vào trong ống thủy tinh cứng dài 100 mm và đường kính trong 6 mm, trước đó đã được làm sạch kỹ lưỡng. Thêm một lượng thích hợp dung dịch acld hydrocloric 18 % (TT). Ngâm ống thủy tinh thu được trong đá ở -5 °C, áp suất dưới 133 Pa và đóng chặt nắp ống. Đun nóng ở 110 °C đến 115 °C trong 16 h.
Để nguội, mở nắp ống và chuyển toàn bộ lượng chất trong ống vào bình dung tích 10 ml, tráng 5 lần,mỗi lần với 0,2 ml nước, làm khô tới cắn bằng kali hydroxyd (TT) trong điều kiện áp suất giảm. Hòa tan cắn vào nước và làm khô tới cắn bằng kali hydroxyd (TT) trong điều kiện áp suất giảm, lặp lại quy trình này thêm một lần nữa. Hòa tan cắn thu được vào dung dịch đệm thích hợp cho máy phân tích acid amin và pha loãng thích hợp với cùng dung môi. Thể tích tiêm phù hợp với máy phân tích.
Biểu thị hàm lượng acid amin theo mol. Tính tỷ lệ tương đối của các acid amin, quy ước rằng 1/6 của tổng số mol của các acid aspartic, acid glutamlc, prolin, glycin, isoleucin và leucln là bằng 1. Giá trị hàm lượng của các acid amin phải nằm trong giới hạn sau: acid aspartic; acid glutamic; prolin; glycin mỗi chất đều từ 0,95 đến 1,05; leucin và isoleucin từ 0,90 đến 1,10; tyrosin từ 0,7 đến 1,05; 1/2 cystin từ 1,4 đến 2,1; các acid amin khác chỉ được có mặt ở dạng vết.
Định tính
Có thể chọn một trong hai nhóm định tính sau:
Nhóm I: A.
Nhóm II: B, C.
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của oxytocin chuẩn hoặc phổ đối chiếu của oxytocin.
B. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel GF254.
Dung môi khai triển: Dicloromethan - methanol - nước - acid acetic băng (70 : 30 : 6 : 1).
Dung dịch thử: Dung dịch chứa 5 mg/ml chế phẩm trong methanol (TT).
Dung dịch đối chiếu: Dung dịch chứa 5 mg/ml oxytocin chuẩn trong methanol (TT).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 10 μl mỗi dung dịch trên. Triển khai sắc ký tới khi dung môi đi được 15 cm. Làm khô bản mỏng dưới luồng không khí lạnh và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm. vết chính trên sắc ký đồ của dung dịch thử phải giống với vết chính trên sắc ký đồ của dung dịch đối chiếu về vị trí, kích thước và độ đậm.
C. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Góc quay cực riêng
Từ -24,0° đến -28,0°, tính theo chế phẩm khan và không có acid acetic (Phụ lục 6.4).
Dùng dung dịch chứa 5,0 mg/ml trong nước.
pH
Từ 3,0 đến 6,0 (Phụ lục 6.2).
Dùng dung dịch chứa 20 mg/ml trong nước không có carbon dioxyd (TT) để đo.
Hàm lượng acid acetic
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Pha loãng 0,7 ml acid phosphoric (TT) trong 900 ml nước, điều chỉnh đến pH 3,0 bằng dung dịch natri hydroxyd 20 % (TT) và thêm nước vừa đủ 1000 ml.
Pha động B: Methanol (TT).
Dung môi pha mẫu: Pha động A - pha động B (95 : 5).
Dung dịch thử: Hòa tan 15,0 mg chế phẩm trong dung môi pha mẫu và pha loãng thành 10,0 ml với cùng dung môi.
Dung dịch đối chiếu: Dung dịch chứa 0,10 g/L acid acetic băng (TT) trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 210 nm.
Tốc độ dòng: 1,2 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0-5 | 95 | 5 |
5-10 | 95 → 50 | 5 → 50 |
10-20 | 50 | 50 |
20-22 | 50 → 95 | 50 → 5 |
22-30 | 95 | 5 |
Thời gian lưu của pic acid acetic khoảng 3 min đến 4 min.
Giới hạn:
Acid acetic: Từ 6,0 % đến 10,0 %.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm phosphat: Hòa tan 31,2 g natri dihydrophosphat (TT) trong 1000 ml nước.
Pha động A: Acetonitril - dung dịch đệm phosphat - nước (15:15:70).
Pha động B: Acetonitril - dung dịch đệm phosphat - nước (70 : 15: 15).
Dung dịch thử: Dung dịch chứa 0,50 mg/ml chế phẩm trong pha động A.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng pha động A.
Dung dịch đối chiếu (2): Lấy 3,0 ml dung dịch thử, thêm 2,0 ml dung dịch acid sulfuric 1 % (TT), đun cẩn thận trong cách thủy sôi trong 20 min.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 220 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 50 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0-5 | 100 | 0 |
5-20 | 100 → 94 | 0 → 6 |
20-50 | 94 → 60 | 6 → 40 |
50-51 | 60 → 100 | 40 → 0 |
51-65 | 100 | 0 |
Tiến hành sắc ký với dung dịch thử, dung dịch đối chiếu (1) và (2).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic oxytocin (thời gian lưu khoảng 25 min) và pic có thời gian lưu tương đối so với oxytocin khoảng 0,9 không nhỏ hơn 1,4.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Diện tích của bất kỳ pic nào, trừ pic chính, không được lớn hơn 1,5 lần diện tích pic chính trên sắc ký đồ của dung dịch đối chiếu (1) (1,5 %).
Tổng diện tích của tất cả các pic, trừ pic chính, không được lớn hơn 5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (5 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,1 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,1 %).
Ghi chú:
Tạp chất A: N-(L-cysteinyl-L-tyrosyl-L-isoleucyl-L-glutaminyl-L-asparaginyl-L-cysteinyl-L-prolyl-L-leucylglycyl)urea cyclic-(1→6)-disulfid (carblmido oxytocin).
Tạp chất B: Acetyl-L-cysteinyl-L-tyrosyl-L-isoleucyl-L-glutaminyl-L-asparaginyl-L-cysteinyl-L-prolyl-L- leucylglycinamid cyclic-(1→6)-disulfid (acetyloxytocin).
Tạp chất C: L-cysteinyl-L-tyrosyl-L-lsoleucyl-L-glutaminyl-L-asparaginyl-L-cysteinyl-L-prolyl-L-leucylglycinamid dimer (1→1’:6→6')-bisdisulfid (α-oxytocin dimer).
Tạp chất D: L-cysteinyl-L-tyrosyl-L-isoleucyl-L-glutaminyl-L-asparaginyl-L-cysteinyl-L-prolyl-L-leucylglycinamid dimer (1→6’:1’→6)-bisdisulfid (β-oxytocin dimer).
Nước
Không được quá 5,0 % (Phụ lục 10.3).
Dùng 0,10 g chế phẩm.
Nội độc tố vi khuẩn
Nếu chế phẩm được dùng để pha chế các dạng thuốc tiêm thì nội độc vi khuẩn không được quá 300 EU/mg (Phụ lục 13.2).
Thử vô khuẩn
Nếu chế phẩm dự định dùng để sản xuất thuốc tiêm mà không tiến hành tiệt khuẩn nữa thì phải đạt yêu cầu Thử vô khuẩn (Phụ lục 13.7).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Điều kiện sắc ký, dung dịch thử và dung dịch đối chiếu (2) được mô tả trong phần Tạp chất liên quan.
Dung dịch chuẩn: Dung dịch chứa 0,50 mg/ml oxytocin chuẩn trong pha động A.
Tiến hành sắc ký với dung dịch thử, dung dịch đối chiếu (2) và dung dịch chuẩn.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic oxytocin (thời gian lưu khoảng 25 min) và pic có thời gian lưu tương đối so với oxytocin khoảng 0,9 không nhỏ hơn 1,4.
Tính hàm lượng phần trăm của oxytocin, C43H66N12O12S2, trong chế phẩm dựa vào diện tích pic thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C43H66N12O12S2 trong oxytocin chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở 2 °C đến 8 °C. Nếu là loại vô khuẩn thì bảo quản trong đồ đựng kín, chống nhiễm khuẩn.
Loại thuốc
Thuốc thúc đẻ - Hormon thùy sau tuyến yên.
Chế phẩm
Thuốc tiêm.
RABEPRAZOL NATRI
C18H20N3NaO3S | P.t.l: 381,4 |
Rabeprazol natri là natri 2-[(RS)-[[4-(3-methoxypropoxy)-3-methylpyridin-2-yl]methyl] sulfinyl] benzimidazol-1-id, phải chứa từ 97,0 % đến 102,0 % C18H20N3NaO3S, tính theo chế phẩm đã làm khô.
Tính chất
Bột màu trắng hoặc trắng ngà, hút ẩm. Rất tan đến dễ tan trong nước, dễ tan trong ethanol khan, thực tế không tan trong heptan.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của rabeprazol natri hydrat chuẩn. Hoà tan riêng biệt chế phẩm và rabeprazol natri hydrat chuẩn trong methanol (TT), bốc hơi đến khô, ghi phổ của các cắn thu được.
B. Chế phẩm phải đáp ứng yêu cầu của phép thử Mất khối lượng do làm khô.
C. Chế phẩm phải cho phản ứng (A) của ion natri (Phụ lục 8.1).
pH
Từ 9,5 đến 11,5 (Phụ lục 6.2).
Hoà tan 0,100 g chế phẩm trong vừa đủ 10 ml nước không có carbon dioxyd (TT).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3). Tiến hành trong điều kiện tránh ánh sáng.
Pha động A: Acetonitril - dung dịch dikali hydrophosphat 4,35 g/L, điều chỉnh tới pH 7,0 bằng acid phosphoric (5 : 95).
Pha động B: Methanol.
Pha động C: Acetonitril.
Dung dịch đệm phosphat 0,1 M pH 11,3: Hòa tan 17,4 g dikali hydrophosphat (TT) trong 950 ml nước, điều chỉnh pH đến 11,3 bằng dung dịch kali hydroxyd (TT) 10 %, thêm nước vừa đủ 1000 ml.
Dung môi pha mẫu: Methanol - dung dịch đệm phosphat 0,1 M pH 11,3 (20 : 80).
Dung dịch thử (1): Cân chính xác khoảng 20 mg chể phẩm vào bình định mức 20 ml, hòa tan bằng dung môi pha mẫu và thêm cùng dung môi đến định mức, lắc đều.
Dung dịch thử (2): Pha loãng 5,0 ml dung dịch thử (1) thành 50,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử (1) thành 100,0 ml bằng dung môi pha mẫu. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (2): Hòa tan 5 mg rabeprazol chuẩn dùng để kiểm tra tính phù hợp của hệ thống (chứa tạp chất A và H) trong 5,0 ml dung môi pha mẫu.
Dung dịch đối chiếu (3): Hòa tan 20,0 mg rabeprazol natri hydrat chuẩn trong dung môi pha mẫu và pha loãng thành 20,0 ml với cùng dung môi. Pha loãng 5,0 ml dung dịch thu được thành 50,0 ml bằng dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh base-deactivated end-capped octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Nhiệt độ cột: 45 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 280 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 5 μl.
Nhiệt độ thiết bị tiêm mẫu tự động: 6 °C.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B | Pha động C |
(min) | (% tt/tt) | (% tt/tt) | (% tt/tt) |
0 - 2 | 100 | 0 | 0 |
2 - 7 | 100 -> 85 | 0 | 0 -> 15 |
7 - 27 | 85 -> 30 | 0 -> 40 | 15 -> 30 |
27 - 32 | 30 -> 15 | 40 -> 55 | 30 |
Tiến hành sắc ký dung dịch thử (1), dung dịch đối chiếu (1) và (2).
Sử dụng sắc ký đồ cung cấp kèm theo rabeprazol chuẩn dùng để kiểm tra tính phù hợp của hệ thống và sắc ký đồ của dung dịch đối chiếu (2) để xác định vị trí pic tạp chất A và H. Thời gian lưu tương đối so với pic rabeprazol (thời gian lưu khoảng 19 min): của tạp chất A là 0,9; tạp chất H là 0,98.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), tỷ số đỉnh - hõm (Hp/Hv) không nhỏ hơn 1,5, trong đó Hp là chiều cao đỉnh pic của tạp chất H so với đường nền, Hv là chiều cao từ đường nền tới đáy hõm phân tách giữa pic tạp chất H và pic rabeprazol.
Tính hàm lượng phần trăm mỗi tạp chất nếu có dựa vào diện tích pic tạp chất thu được trên sắc ký đồ của dung dịch thử (1) và diện tích pic rabeprazol trên sắc ký đồ của dung dịch đối chiếu (1).
Giới hạn:
Tạp chất A: Không được quá 0,8 %.
Tạp chất khác: Mỗi tạp chất không được quá 0,10 %.
Tổng tạp: Không được quá 1,0 %.
Bỏ qua các tạp chất có hàm lượng nhỏ hơn 0,05 %.
Ghi chú:
Tạp chất A: 2-[[[4-(3-methoxypropoxy)-3-methylpyridin-2-yl]methyl]sulfonyl]-1H-benzimidazol.
Tạp chất H: 2-[(RS)-[(4-cloro-3-methylpyridin-2-yl)methyl]sulfinyl]-1H-benzimidazol.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6)
(1,000 g; sấy trong chân không với phosphor pentoxyd, 24 h).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3), điều kiện được mô tả như phần Tạp chất liên quan với sự thay đổi sau:
Tiến hành sắc ký các dung dịch thử (2) và dung dịch đối chiếu (3).
Tính hàm lượng rabeprazol natri C18H20N3NaO3S dựa vào diện tích pic thu được trên sắc ký đồ của dung dịch thử (2), dung dịch đối chiếu (3) và hàm lượng C18H20N3NaO3S của rabeprazol natri hydrat chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ phòng.
Loại thuốc
Thuốc ức chế bơm proton, điều trị viêm loét dạ dày, tá tràng.
Chế phẩm
Viên nén bao tan trong ruột.
ROSUVASTATIN CALCI

C44H54CaF2N6O12S2 | P.t.l: 1001 |
Rosuvastatin calci là calci bis[(3R,5S,6E)-7-[4-(4-fluorophenyl)-2-(N-methylmethanesulfonamido)-6- (propan-2-yl)pyrimidin-5-yl]-3,5-dihydroxyhept-6-enoat], phải chứa từ 97,0 % đến 102,0 % C44H54CaF2N6O12S2, tính theo chế phẩm khan.
Tính chất
Bột màu trắng hoặc gần như trắng, hút ẩm, Khó tan trong nước, dễ tan trong methylen clorid, thực tế không tan trong ethanol khan.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của rosuvastatin calci chuẩn.
B. Chế phẩm phải đáp ứng phép thử Tạp chất đồng phân đối quang.
C. Chế phẩm phải cho phản ứng (A) của ion calci (Phụ lục 8.1)
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3), tiến hành trong điều kiện tránh ánh sáng, chuẩn bị các dung dịch ngay trước khi dùng.
Pha động A: Acetonitril - dung dịch acid trifluoroacetic 1 % (tt/tt) - nước (29 : 1 : 70).
Pha động B: Acetonitril - dung dịch acid trifluoroacetic 1 % (tt/tt) - nước (75 : 1 : 24).
Dung môi pha mẫu: Acetonitril - nước (25 : 75).
Dung dịch thử: Hòa tan 35,0 mg chế phẩm trong 12 ml acetonitril (TT) và pha loãng thành 50,0 ml bằng nước.
Dung dịch đối chiếu (1): Hòa tan 35,0 mg rosuvastatin calci chuẩn trong 12 ml acetonitril (TT) và pha loãng thành 50,0 ml bằng nước.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng dung môi pha mẫu. Pha loãng 2,0 ml dung dịch thu được thành 10,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (3): Hòa tan 7 mg rosuvastatin chuẩn dùng để kiểm tra tính phù hợp của hệ thống (chứa các tạp chất A, B và C) trong 2,5 ml acetonitril (TT) và pha loãng thành 10,0 ml bằng nước.
Điều kiện sắc ký:
Cột kích thước (10 cm x 4,6 mm) được nhồi pha tĩnh là base-deactivated end-capped octadecylsilyl silica gel dùng cho sắc ký (3 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 242 nm.
Tốc độ dòng: 0,75 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 30 | 100 | 0 |
30 - 50 | 100 -> 60 | 0 -> 40 |
50 - 60 | 60 -> 0 | 40 -> 100 |
60 - 70 | 0 | 100 |
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu (2) và (3).
Sử dụng sắc ký đồ cung cấp kèm theo rosuvastatin chuẩn dùng để kiểm tra tính phù hợp của hệ thống và sắc ký đồ của dung dịch đối chiếu (3) để xác định pic của tạp chất A, B và C.
Thời gian lưu tương đối so với rosuvastatin (thời gian lưu khoảng 25 min): Tạp chất A khoảng 0,9; tạp chất B khoảng 1,1; tạp chất C khoảng 1,5.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic rosuvastatin và pic tạp chất B không nhỏ hơn 2,0.
Tính hàm lượng phần trăm mỗi tạp chất nếu có dựa vào diện tích pic tạp chất thu được trên sắc ký đồ của dung dịch thử và diện tích pic rosuvastatin trên sắc ký đồ của dung dịch đối chiếu (2), nhân diện tích pic tạp chất C với hệ số hiệu chỉnh là 1,4.
Giới hạn:
Tạp chất C: Không được quá 0,8 %.
Tạp chất B: Không được quá 0,5 %.
Tạp chất A: Không được quá 0,2 %.
Các tạp chất khác: Với mỗi tạp chất, không được quá 0,10 %.
Tổng các tạp chất: Không được quá 1,2 %.
Bỏ qua các tạp chất có hàm lượng dưới 0,05 %.
Ghi chú:
Tạp chất A: Acid (3R,5S,6E)-7-[4-(4-fluorophenyl)-2-[[(2-hydroxy-2-methylpropyl)sulfonyl](methyI)amino]-6-(1- methylethyl)pyrimidin-5-yl]-3,5-dihydroxyhept-6-enoic.
Tạp chất B: Acid (3RS,5RS,6E)-7-[4-(4-fluorophenyl)-6-(1-methylethyl)-2-[methyl(methylsulfonyl)amino]pyrimidin- 5-yl]-3,5-dihydroxyhept-6-enoic.
Tạp chất C: Acid (3R,6E)-7-[4-(4-fluorophenyl)-6-(1-methylethyl)-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl]-3- hydroxy-5-oxohept-6-enoic.
Tạp chất đồng phân đối quang
Phương pháp sắc ký lỏng (Phụ lục 5.3), tiến hành trong điều kiện tránh ánh sáng.
Pha động: Acetonitril - dung dịch acid trifluoroacetic 0,1 % (tt/tt) (25 : 75).
Dung môi pha mẫu: Acetonitril - nước dùng cho sắc ký (25 : 75).
Dung dịch thử: Hòa tan 25,0 mg chể phẩm trong 6 ml acetonitril (TT) và pha loãng thành 25,0 ml bằng nước.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng dung môi pha mẫu. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (2): Dung dịch chứa tạp chất G của rosuvastatin chuẩn trong dung dịch thử có nồng độ khoảng 0,004 mg/ml.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh là dẫn xuất celulose của silica gel dùng cho sắc ký phân tách đồng phân đối quang (5 μm),
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 242 nm.
Tốc độ dòng: 0,5 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 2,6 lần thời gian lưu của rosuvastatin.
Sử dụng sắc ký đồ của dung dịch đối chiếu (2) để xác định pic của tạp chất G.
Thời gian lưu tương đối so với rosuvastatin (thời gian lưu khoảng 29 min): Tạp chất G khoảng 0,9.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic tạp chất G và pic rosuvastatin không nhỏ hơn 1,5.
Tính hàm lượng phần trăm của tạp chất G dựa vào diện tích pic tạp chất G thu được trên sắc ký đồ của dung dịch thử và diện tích pic rosuvastatin trên sắc ký đồ của dung dịch đối chiếu (1).
Giới hạn:
Tạp chất G: Không được quá 0,15 %.
Ghi chú:
Tạp chất G: Acid (3S,5R,6E)-7-[4-(4-fluorophenyl)-6-(1-methylethyl)-2-[methyl(methylsulfonyl)amino]pyrimidin-5- yl]-3,5-dihydroxyhept-6-enoic.
Nước
Không được quá 6,1 % (Phụ lục 10.3).
Dùng 0,100 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả trong phần Tạp chất liên quan.
Tiến hành sắc ký với dung dịch thử và dung dịch đối chiếu (1).
Tính hàm lượng phần trăm của rosuvastatin calci, C44H54CaF2N6O12S2, trong chế phẩm dựa vào diện tích pic rosuvastatin thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (1) và hàm lượng của C44H54CaF2N6O12S2 trong rosuvastatin calci chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, nhiệt độ từ 2 °C đến 8 °C.
Loại thuốc
Ức chế HMG Co-A reductase; chống tăng lipid máu.
Chế phẩm
Viên nén.
SILDENAFIL CITRAT
C22H30N6O4S.C6H8O7 | P.t.l: 666,7 |
Sildenafil citrat là 1-[[3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxyphenyl]sulfonyl]-4-methylpiperazin citrat, phải chứa từ 98,0 % đến 102,0 % C22H30N6O4S.C6H8O7, tính theo chế phẩm khan.tmai
Tính chất
Bột kết tinh màu trắng hoặc gần như trắng, hơi hút ẩm. Khó tan trong nước và trong methanol, thực tế không tan trong hexan.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của sildenafil citrat chuẩn.
Giới hạn imidazol
Không được quá 0,1 %.
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel F254 (2 - 10 μm).
Dung môi khai triển: Methylen clorid - ethyl acetat - ethanol 96 % - amoniac (50 : 30 : 20 : 1).
Dung môi pha mẫu: Methanol - nước - amoniac (15 : 5 : 1).
Dung dịch thử: Dung dịch chứa 17,5 mg/ml chế phẩm trong dung môi pha mẫu.
Dung dịch đối chiếu (1): Dung dịch chứa 0,035 mg/ml imidazol chuẩn trong dung môi pha mẫu.
Dung dịch đối chiếu (2): Pha loãng dung dịch đối chiếu (1) bằng dung môi pha mẫu để thu được dung dịch chứa 0,0175 mg/ml imidazol chuẩn.
Dung dịch đối chiếu (3): Trộn đồng thể tích dung dịch thử và dung dịch đối chiếu (1).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 10 μl dung dịch thử, dung dịch đối chiếu (2) và (3) (chấm dải khoảng 6 mm). Triển khai sắc ký tới khi dung môi đi được 2/3 chiều dài bản mỏng. Sấy bản mỏng ở 100 °C trong 15 min và để nguội. Để bản mỏng trong bình bão hòa hơi iod đến khi bản mỏng có màu nâu nhạt, quan sát dưới đèn tử ngoại ở bước sóng 254 nm (giá trị Rf của citrat, imidazol, sildenafil lần lượt là 0; 0,25; 0,4). Phép thử chỉ có giá trị khi sắc ký đồ của dung dịch đối chiếu (3) cho 2 vết tách rõ rệt. Vết tương ứng với imidazol trên sắc ký đồ của dung dịch thử không được đậm hơn vết chính trên sắc ký đồ của dung dịch đối chiếu (2).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Pha loãng 7 ml triethylamin (TT) bằng nước thành 1000 ml. Khuấy đều và điều chỉnh đến pH 3,0 ± 0,1 bằng acid phosphoric (TT).
Pha động: Dung dịch đệm - methanol - acetonitril (58 : 25 : 17).
Dung dịch thử: Dung dịch chứa 0,7 mg/ml chế phẩm trong pha động.
Dung dịch đối chiếu (1): Pha loãng dung dịch thử bằng pha động để thu được dung dịch chứa 1,4 μg/ml sildenafil citrat.
Dung dịch đối chiếu (2): Pha loãng dung dịch đối chiếu (1) bằng pha động để thu được dung dịch chứa 0,35 μg/ml sildenafil citrat.
Dung dịch đối chiếu (3): Hòa tan 70 mg chế phẩm trong 1 ml hỗn hợp gồm dung dịch hydrogen peroxyd 100 tt - acid formic khan (2 : 1). Để yên ít nhất 10 min để tạo sildenafil N-oxid, và pha loãng thành 250 ml bằng pha động.
Dung dịch đối chiếu (4): Dung dịch chứa 7,5 μg/ml tạp chất A chuẩn của sildenafil trong pha động.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 290 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian bằng 3 lần thời gian lưu của sildenafil.
Thời gian lưu tương đối của sildenafil, sildenafil N-oxid, tạp chất A của sildenafil lần lượt là 1,0; 1,2 và 1,7. Định tính tạp chất A bằng dung dịch đối chiếu (4).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic sildenafil N-oxid và pic sildenafil ít nhất là 2,5; Trên sắc ký đồ của dung dịch đối chiếu (1), hệ số đối xứng của pic sildenafil không lớn hơn 1,5; Trên sắc ký đồ của dung dịch đối chiếu (2), tỉ số tín hiệu trên nhiễu ít nhất là 10 đối với pic sildenafil.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic sildenafil trên sắc ký đồ của dung dịch đối chiếu (1) và nồng độ sildenafil citrat trong dung dịch đối chiếu (1).
Giới hạn:
Tạp chất A: Không được quá 0,3 %.
Các tạp chất chưa xác định khác: Với mỗi tạp chất, không được quá 0,10 %.
Tổng các tạp chất chưa xác định: Không được quá 0,3 %.
Tổng tất cả các tạp chất: Không được quá 0,5 %.
Bỏ qua các tạp chất có hàm lượng dưới 0,05 %.
Ghi chú:
Tạp chất A: 5-[2-Ethoxy-5-[(4-methylpiperazin-1-yl)sulfonyl]phenyl]-1-methyl-3-(2-methylpropyl)-1,6-dihydro-7H- pyrazolo[4,3-d]pyrimidin-7-on.
Nước
Không được quá 2,5 % (Phụ lục 10.3, phương pháp 1).
Dùng 0,200 g chế phẩm.
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 0,5 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm, pha động và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Dung dịch chứa 0,028 mg/ml chế phẩm trong pha động.
Dung dịch chuẩn: Dung dịch chứa 0,028 mg/ml sildenafil citrat chuẩn trong pha động.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic sildenafil không lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic thu được từ 6 lần tiêm lặp lại không lớn hơn 0,85 %.
Tính hàm lượng phần trăm của sildenafil citrat, C22H30N6O4S.C6H8O7, trong chế phẩm dựa vào diện tích pic sildenafil thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng C22H30N6O4S.C6H8O7 trong sildenafil citrat chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Thuốc ức chế men phosphodiesterase tuýp 5 với tác dụng giãn mạch; điều trị rối loạn cương dương.
Chế phẩm
Viên nén.
TADALAFIL

C22H19N3O4 | P.t.l: 389,4 |
Tadalafil là (6R,12aR)-6-(1,3-benzodioxol-5-yl)-2-methyl-2,3,6,7,12,12a-hexahydropyrazino [1',2':1,6]-pyrido[3,4-b]indol-1,4-dion, phải chứa từ 97,5 % đến 102,0 % C22H19N3O4, tính theo chế phẩm đã làm khô.
Tính chất
Bột trắng hoặc gần như trắng. Dễ tan trong dimethyl sulfoxid, khó tan trong methylen clorid, thực tế không tan trong nước.
Định tính
Có thể chọn một trong hai nhóm định tính sau:
Nhóm I: A, B.
Nhóm II: A, C.
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của tadalafil chuẩn.
B. Trong phần Tạp chất A, B và C, pic chính trên sắc ký đồ của dung dịch thử phải có thời gian lưu tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch đối chiếu (1).
C. Góc quay cực riêng (Phụ lục 6.4): Từ +78,0° đến +84,0°, tính theo chế phẩm đã làm khô.
Hòa tan 0,250 g chế phẩm trong dimethyl sulfoxid (TT) và pha loãng thành 25,0 ml với cùng dung môi.
Tạp chất A, B và C
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Hexan - 2-propanol (50 : 50).
Dung môi pha mẫu: Acetonitril - hexan - 2-propanol (20 : 40 : 40)
Dung dịch A: Hòa tan 27 g tetrabutylammonium hydroxyd (TT) trong methanol (TT) và pha loãng thành 100,0 ml với cùng dung môi.
Dung dịch thử: Hòa tan 25,0 mg chế phẩm trong dung môi pha mẫu và pha loãng thành 100,0 ml với cùng dung môi.
Dung dịch đối chiếu (1): Hòa tan 25,0 mg tadalafil chuẩn trong dung môi pha mẫu và pha loãng thành 100,0 ml với cùng dung môi.
Dung dịch đối chiếu (2): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng dung môi pha mẫu. Tiếp tục pha loãng 1,0 ml dung dịch này thành 10,0 ml với cùng dung môi.
Dung dịch đối chiếu (3): Để tạo tạp chất A, hòa tan 25 mg chế phẩm trong 40 ml dung môi pha mẫu, thêm 1 ml dung dịch A, lắc đều và để yên trong 20 min thêm 1 ml acid trifluoroacetic (TT) rồi pha loãng vừa đủ đến 100,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (4): Hút 1,0 ml dung dịch thử và 1,0 ml dung dịch đối chiếu (3) pha loãng thành 50,0 ml bằng dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh là dẫn xuất amylose của silica gel dùng cho sắc ký tách đồng phân đối quang (10 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 222 nm.
Tốc độ dòng: 0,75 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 2,2 lần thời gian lưu của pic tadalafil.
Tiến hành sắc ký dung dịch đối chiếu (4), dung dịch đối chiếu (2) và dung dịch thử
Trên sắc ký đồ của dung dịch đối chiếu (4), pic tạp chất A có thời gian lưu tương đối bằng khoảng 0,8 so với pic chính tadalafil (thời gian lưu khoảng 11 min).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (4) độ phân giải giữa pic tadalafil và pic tạp chất A không nhỏ hơn 2,0.
Giới hạn:
Tạp chất A: Diện tích pic tạp chất A không được lớn hơn 1,5 lần diện tích píc chính trong sắc ký đồ của dung dịch đối chiếu (2) (0,15 %)
Tạp chất khác: Với mỗi tạp chất, có diện tích pic không lớn hơn diện tích pic chính trong sắc ký đồ của dung dịch đối chiếu (2) (0,10 %)
Ghi chú:
Tạp chất A: (6R,12aS)-6-(1,3-benzodioxol-5-yl)-2-methyl-2,3,6,7,12,12a-hexahydropyrazino[1',2,:1,6]pyrido [3,4-b]indol-1,4-dion.
Tạp chất B: (6S,12aS)-6-(1,3-benzodioxol-5-yl)-2-methyl-2,3,6,7,12,12a-hexahydropyrazino[1',2':1,6]pyrido[3,4-b]indol-1,4-dion.
Tạp chất C: (6S,12aR)-6-(1,3-benzodioxol-5-yl)-2-methyl-2,3,6,7,12,12a-hexahydropyrazino[1',2':1,6]pyrido[3,4-b]indol-1,4-dion.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Không siêu âm trong quá trình chuẩn bị các dung dịch.
Pha động A: Pha loãng 1,0 ml acid trifluoroacetic (TT) với 1000 ml nước.
Pha động B: Acetonitril (TT).
Dung môi pha mẫu: Acetonitril - 2-propanol (50 : 50).
Dung dịch A: Hòa tan 27 g tetrabutylammonium hydroxyd (TT) trong methanol (TT) và pha loãng thành 100,0 ml với cùng dung môi.
Dung dịch thử (1): Cân chính xác khoảng 40 mg chế phẩm, hòa tan trong 50 ml acetonitril (TT) và pha loãng thành 100,0 ml bằng pha động A.
Dung dịch thử (2): Cân chính xác khoảng 50 mg chế phẩm, hòa tan trong 50 ml acetonitril (TT) và pha loãng thành 100,0 ml bằng pha động A. Hút 10,0 ml dung dịch này, thêm 25,0 ml acetonitril (TT) và pha loãng thành 50,0 ml bằng pha động A.
Dung dịch đối chiếu (1): Hút 1,0 ml dung dịch thử (1), thêm 50,0 ml acetonitril (TT) và pha loãng thành 100,0 ml bằng pha động A. Hút 1,0 ml dung dịch thu được, thêm 5,0 ml acetonitril (TT) và pha loãng thành 10,0 ml bằng pha động A.
Dung dịch đối chiếu (2): Để tạo tạp chất A, hòa tan khoảng 4 mg chế phẩm trong 50 ml dung môi pha mẫu, thêm 1,0 ml dung dịch A, lắc đều và để yên trong 40 min, thêm 1 ml acid trifluoroacetic (TT) rồi pha loãng thành 100,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (3): Pha loãng 1,0 ml dung dịch đối chiếu (2) thành 50,0 ml bằng dung dịch thử (1).
Dung dịch đối chiếu (4): Cân chính xác khoảng 50,0 mg tadalafil chuẩn, hòa tan trong 50 ml acetonitril (TT) và pha loãng vừa đủ thành 100,0 ml bằng pha động A. Hút 10,0 ml dung dịch này thêm 25 ml acetonitril (TT) rồi pha loãng thành 50,0 ml bằng pha động A.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh B (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 285 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 3 | 85 | 15 |
3 - 30 | 85 -> 5 | 15 -> 95 |
30 - 33 | 5 | 95 |
Tiến hành tiêm lần lượt các dung dịch đối chiếu (3), dung dịch đối chiếu (1) và dung dịch thử (1)
Định tính các tạp: Sử dụng sắc ký đồ của dung dịch đối chiếu (3) để xác định tạp chất A+C.
Thời gian lưu tương đối so với pic tadalafil (thời gian lưu khoảng 16 min) của pic tạp chất A+C khoảng 1,03.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), tỷ số đỉnh - hõm (Hp/Hv) giữa tạp A+C với pic chính tadalafil không nhỏ hơn 3,3, trong đó Hp là chiều cao đỉnh pic tạp chất A+C so với đường nền và Hv là chiều cao tính từ đường nền lên đến đáy hõm phân tách giữa pic tạp chất A+C và pic tadalafil.
Giới hạn:
Với mỗi tạp chất: Diện tích pic của bất kỳ tạp chất nào cũng không được lớn hơn diện tích pic chính trên sắc ký đồ của dung dịch đối chiếu (1) (0,10 %).
Tổng tạp chất: Tổng diện tích các pic tạp chất không được lớn hơn 3 lần diện tích píc chính trên sắc ký đồ của dung dịch đối chiếu (1) (0,30 %)
Bỏ qua tất cả các pic có diện tích nhỏ hơn 0,5 lần diện tích píc chính trên sắc ký đồ của dung dịch đối chiếu (1) (0,05 %); bỏ qua các pic tương ứng với tạp chất A+C.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6)
(1,000 g, chân không, 105 °C, 3 h).
Tro sulphat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3). Tiến hành với các điều kiện như ở mục Tạp chất liên quan với các điều chỉnh như sau:
Pha động: Acetonitril - dung dịch A (45 : 55)
Tốc độ dòng: 1,5 ml/min.
Tiến hành sắc ký với dung dịch thử (2) và dung dịch đối chiếu (4).
Thời gian sắc ký bằng 2 lần thời gian lưu của pic tadalafil (thời gian lưu khoảng 5,2 min).
Tính hàm lượng phần trăm của tadalafil, C22H19N3O4, trong chế phẩm dựa vào diện tích pic tadalafil thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C22H19N3O4 trong tadalafil chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Thuốc ức chế men phosphodiesterase tuýp 5 với tác dụng giãn mạch; điều trị rối loạn cương dương.
Chế phẩm
Viên nén.
TENOFOVIR DISOPROXIL FUMARAT

C19H30N5O10P.C4H4O4 | P.t.l: 635,5 |
Tenofovir disoproxil fumarat là muối của bis({[(1-methylethoxy)carbonyl]oxy}methyl){[(1R)-2-(6-amino- 9H-purin-9-yI)-1-methylethoxy]methyl}phosphonat (ester) và acid fumaric, phải chứa từ 98,5 % đến 101,0 % C19H30N5O10P.C4H4O4, tính theo chế phẩm khan.
Tính chất
Bột kết tinh màu trắng hoặc gần như trắng, có thể ở dạng đa hình. Tan trong methanol, khó tan trong nước và rất khó tan trong dicloromethan.
Sản xuất
Phương pháp sản xuất phải được thẩm định để đảm bảo nếu được tiến hành kiểm tra thì chế phẩm phải đáp ứng yêu cầu sau:
Tạp chất K (9-propenyladenin): Không được quá 5 ppm, dùng phương pháp thích hợp.
Tạp chất G (đồng phân đối quang S của tenofovir disoproxil): Không được quá 1,0 %, dùng phương pháp sắc ký sử dụng cột tách đồng phân đối quang.
Định tính
Có thể chọn một trong hai nhóm định tính sau:
Nhóm I: A.
Nhóm II: B, C, D.
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của tenofovir disoproxil fumarat chuẩn hoặc phổ đối chiếu của tenofovir disoproxil fumarat.
Nếu phổ không phù hợp thì hòa tan riêng biệt chế phẩm và chuẩn trong một lượng nhỏ methanol (TT), bốc hơi đến cắn, dùng cắn thu được để ghi lại phổ mới.
B. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel GF254.
Dung môi khai triển: Dicloromethan - acetonitril - methanol - amoniac (67 : 20 : 10 : 3).
Dung dịch thử: Dung dịch chứa 10 mg/ml chế phẩm trong methanol (TT).
Dung dịch đối chiếu: Dung dịch chứa 10 mg/ml tenofovir disoproxil fumarat chuẩn trong methanol (TT). Cách tiến hành: Chấm riêng biệt lên bản mỏng 5 μl mỗi dung dịch trên. Triển khai sắc ký tới khi dung môi đi được 15 cm. Lấy bản mỏng ra để khô hoàn toàn và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm. Vết chính trên sắc ký đồ của dung dịch thử phải tương ứng với vết chính trên sắc ký đồ của dung dịch đối chiếu về vị trí, hình dạng và độ đậm.
C. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel GF254.
Dung môi khai triển: Heptan - acid acetic băng - dicloromethan (50 : 30 : 20).
Dung dịch thử: Dung dịch chứa 10 mg/ml chế phẩm trong ethanol (TT).
Dung dịch đối chiếu: Dung dịch chứa 2 mg/ml acid fumaric (TT) chuẩn trong ethanol (TT).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 5 μl mỗi dung dịch trên. Triển khai sắc ký trong bình không bão hòa dung môi đến khi dung môi đi được 10 cm. Lấy bản mỏng ra để khô hoàn toàn và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm. Một trong các vết chính thu được trên sắc ký đồ của dung dịch thử phải tương ứng với vết chính trên sắc ký đồ của dung dịch đối chiếu về vị trí, hình dạng và độ đậm.
D. Phổ hấp thụ tử ngoại (Phụ lục 4.1) của dung dịch chứa 25 μg/ml chế phẩm trong nước khi đo ở dải bước sóng từ 220 nm đến 320 nm phải cho một cực đại hấp thụ ở 261 nm và độ hấp thụ riêng tại bước sóng này phải từ 230 đến 250.
Góc quay cực riêng
Từ -15° đến -20°, tính theo chế phẩm khan (Phụ lục 6.4).
Dùng dung dịch chứa 10,0 mg/ml trong dung dịch acid hydrocloric 0,1 M (TT). Tiến hành phép thử ngay khi chuẩn bị dung dịch xong.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm phosphat pH 6,0: Hòa tan 3,50 g kali dihydrophosphat (TT) và 1,70 g tetrabutylamoni hydrosulfat (TT) trong 800 ml nước, điều chỉnh đến pH 6,0 bằng dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml.
Pha động A: Acetonitril - dung dịch đệm phosphat pH 6,0 - nước (2 : 20 : 78).
Pha động B: Acetonitril - dung dịch đệm phosphat pH 6,0 - nước (65 : 20 : 15).
Dung dịch thử: Dung dịch chứa 1,0 mg/ml chế phẩm trong nước.
Dung dịch đối chiếu (1): Pha loãng dung dịch thử trong nước để thu được dung dịch chứa 5 μg/ml tenofovir disoproxil fumarat.
Dung dịch đối chiếu (2): Dung dịch chứa 0,2 mg/ml acid fumaric (TT) trong nước.
Dung dịch đối chiếu (3): Đun nóng cẩn thận dung dịch thử trong cách thủy sôi trong 20 min.
Bảo quản các dung dịch ở 6 °C hoặc sử dụng buồng tiêm có làm lạnh.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh base-deactivated octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 260 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm; 20 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 5 | 81 | 19 |
5 - 40 | 81 -> 49 | 19 -> 51 |
40 - 60 | 49 -> 0 | 51 -> 100 |
60 - 65 | 0 | 100 |
65 - 70 | 0 -> 81 | 100 -> 19 |
70 - 80 | 81 | 19 |
Tiến hành sắc ký dung dịch đối chiếu (3).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic chính (thời gian lưu khoảng 40 min) và pic tạp chất A (có thời gian lưu tương đối so với pic chính khoảng 0,5) không nhỏ hơn 25.
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu (1) và (2).
Thời gian lưu tương đối so với tenofovir disoproxil (thời gian lưu khoảng 40 min) của fumarat khoảng 0,15.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Diện tích của tạp chất A không được lớn hơn 2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (1,0 %).
Diện tích của bất kỳ pic tạp chất khác không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,5 %) và không có quá 2 pic tạp chất có diện tích lớn hơn 0,4 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,2 %).
Tổng diện tích của tất cả các pic tạp chất không được lớn hơn 5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (2,5 %).
Bỏ qua bất kỳ pic nào tương ứng với pic thu được trên sắc ký đồ của dung dịch đối chiếu (2) và bỏ qua những pic có diện tích nhỏ hơn 0,1 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,05 %).
Ghi chú:
Tạp chất A: (1-methylethyl) (8R)-9-(6-amino-9H-purin-9-yl)-5-hydroxy-8-methyl-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoat (tenofovir monosoproxil).
Nước
Không được quá 1,0 % (Phụ lục 10.3).
Dùng 1,0 g chế phẩm.
Tro sulfat
Không được quá 0,2 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Kim loại nặng
Không được quá 20 phần triệu (Phụ lục 9.4.8).
Dùng 1,0 g chế phẩm trong 30 ml methanol (TT) để chuẩn bị mẫu thử và tiến hành thử theo Phương pháp 2. Dùng 10,0 ml dung dịch chì mẫu 2 phần triệu Pb (TT) để chuẩn bị mẫu đối chiếu.
Định lượng
Hòa tan khoảng 0,40 g chế phẩm trong 30 ml acid acetic băng (TT1). Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ). Xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2).
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 63,55 mg tenofovir disoproxil fumarat (C19H30N5O10P.C4H4O4).
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ từ 2 °C đến 8 °C.
Loại thuốc
Thuốc kháng retrovirus.
Chế phẩm
Viên nén.
THIAMAZOL

C4H6N2S | P.t.l: 114,2 |
Thiamazol là 1-methyl-1,3-dihydro-2H-imidazol-2-thion, phải chứa từ 98,0 % đến 101,0 % C4H6N2S, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu trắng hoặc nâu nhạt. Dễ tan trong nước và methylen clorid, dễ tan hoặc tan trong ethanol 96 %.
Định tính
Có thể chọn một trong hai nhóm định tính sau:
Nhóm I: A, C.
Nhóm II: B, C, D.
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của thiamazol chuẩn.
B. Hòa tan 25 mg chế phẩm trong 10 ml dung dịch acid sulfuric (TT) 0,28 % (tt/tt) và pha loãng thành 50,0 ml với cùng dung môi. Pha loãng 1,0 ml dung dịch thu được thành 100,0 ml bằng dung dịch acid sulfuric (TT) 0,28 % (tt/tt).
Đo phổ hấp thụ tử ngoại (Phụ lục 4.1) trong khoảng bước sóng từ 200 nm đến 300 nm, dung dịch thu được phải cho hai cực đại hấp thụ ở bước sóng 211 nm và 251 nm. Tỷ số giữa độ hấp thụ ở bước sóng 251 nm và độ hấp thụ ở bước sóng 211 nm phải từ 2,5 đến 2,7.
C. Nhiệt độ nóng chảy: Từ 143 °C đến 146 °C (Phụ lục 6.7).
D. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel F254.
Dung môi khai triển: Amoniac TT1 - 2-propanol - toluen (1 : 24 : 75).
Dung dịch thử: Hòa tan 5,0 mg chế phẩm trong methanol (TT) và pha loãng thành 5,0 ml với cùng dung môi.
Dung dịch đối chiếu (1): Hòa tan 5,0 mg thiamazol chuẩn trong methanol (TT) và pha loãng thành 5,0 ml với cùng dung môi.
Dung dịch đối chiếu (2): Hòa tan 5,0 mg 2-methylimidazol (TT) trong methanol (TT) và pha loãng thành 5,0 ml với cùng dung môi. Pha loãng 1,0 ml dung dịch thu được thành 2,0 ml bằng dung dịch thử.
Cách tiến hành: Chấm riêng biệt lên bản mỏng 10 μl mỗi dung dịch trên. Triển khai sắc ký đến khi dung môi đi được khoảng 2/3 chiều dài bản mỏng. Để khô bản mỏng ngoài không khí và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm.
Vết chính trên sắc ký đồ của dung dịch thử phải giống với vết chính trên sắc ký đồ của dung dịch đối chiếu (1) về vị trí và kích thước. Phép thử chỉ có giá trị khi sắc ký đồ của dung dịch đối chiếu (2) cho 2 vết tách rõ rệt sau khi cho bản mỏng tiếp xúc với hơi iod trong 30 min.
Độ trong và màu sắc của dung dịch
Dung dịch S: Hòa tan 2,0 g chế phẩm trong nước và pha loãng thành 20,0 ml với cùng dung môi.
Dung dịch thu được phải trong (Phụ lục 9.2) và không được có màu đậm hơn màu mẫu N6 (Phụ lục 9.3, phương pháp 2).
Tạp chất liên quan
Phương pháp sắc ký khí (Phụ lục 5.2).
Dung dịch thử: Hòa tan 0,100 g chế phẩm trong cloroform (TT) và pha loãng thành 10,0 ml với cùng dung môi.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng cloroform (TT). Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng cloroform (TT).
Dung dịch đối chiếu (2): Hòa tan 5,0 mg tạp chất A chuẩn của thiamazol, 5,0 mg 1-methylimidazol (TT1) và 5,0 mg tạp chất C chuẩn của thiamazol trong cloroform (TT) và pha loãng thành 50,0 ml với cùng dung môi. Pha loãng 1,0 ml dung dịch thu được thành 10,0 ml bằng cloroform (TT).
Điều kiện sắc ký:
Cột silica nung chảy kích thước (30 m x 0,25 mm) được phủ pha tĩnh là base-deactivated phenyl(5)methyl(95)polysiloxan (độ dày phim 0,5 μm).
Khí mang: Heli dùng cho sắc ký.
Tỷ lệ chia dòng: 3 : 20.
Tốc độ dòng: 1,5 ml/min.
Nhiệt độ:
| Thời gian | Nhiệt độ |
| (min) | (°C) |
Cột | 0 - 2 | 100 |
2 - 7 | 100 -> 250 | |
7 - 22 | 250 | |
Buồng tiêm |
| 150 |
Detector |
| 250 |
Detector: Ion hóa ngọn lửa.
Thể tích tiêm: 1 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch đối chiếu (2).
Thời gian lưu tương đối so với thiamazol (thời gian lưu khoảng 6,5 min): Tạp chất A khoảng 0,3; tạp chất B khoảng 0,4; tạp chất C khoảng 0,7.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic tạp chất A và pic tạp chất B ít nhất là 1,5.
Tiến hành sắc ký dung dịch thử và dung dịch đối chiếu (1).
Giới hạn:
Tạp chất A, B, C: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic tương ứng thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,1 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,1 %).
Tổng diện tích pic của tất cả các tạp chất không được lớn hơn 5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,5 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,02 %).
Ghi chú:
Tạp chất A: 2,2-dimethoxy-N-methylethanamin.
Tạp chất B: 1-methyl-1H-imidazol.
Tạp chất C: 1-methyl-2-(methylsulfanyl)-1H-imidazol.
Mất khối lượng do làm khô
Không được quá 0,5 % (Phụ lục 9.6).
(1,000 g; 105 °C; 2 h).
Tro sulfat
Không được quá 0,1 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Hòa tan khoảng 0,250 g chế phẩm trong 75 ml nước. Thêm 15,0 ml dung dịch natri hydroxyd 0,1 N (CĐ), trộn đều, sau đó thêm 30 ml dung dịch bạc nitrat 0,1 M (TT), vừa thêm vừa khuấy đều. Chuẩn độ bằng dung dịch natri hydroxyd 0,1 N (CĐ). Xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2).
1 ml dung dịch natri hydroxyd 0,1 N (CĐ) tương đương với 11,42 mg C4H6N2S.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc kháng giáp, dẫn chất thioimidazol.
Chế phẩm
Viên nén.
TYROSIN
L-Tyrosin

C9H11NO3 | P.t.l: 181,2 |
Tyrosin là acid (2S)-2-amino-3-(4-hydroxyphenyl)propanoic, phải chứa từ 98,5 % đến 101,5 % C9H11NO3, tính theo chế phẩm đã làm khô.
Tính chất
Bột kết tinh màu trắng hay gần như trắng, hoặc tinh thể không màu. Rất khó tan trong nước, không tan trong ethanol (96 %), tan trong dung dịch acid vô cơ loãng và trong dung dịch kiềm loãng.
Định tính
Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của chế phẩm phải phù hợp với phổ hấp thụ hồng ngoại của L-tyrosin chuẩn.
Góc quay cực riêng
- 11,2° đến - 9,8°, tính theo chế phẩm đã làm khô (Phụ lục 6.4).
Dung dịch chế phẩm trong dung dịch acid hydrocloric 1 M (TT) nồng độ 50 mg/ml. Đo ở 25 °C.
Tạp chất liên quan
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel G.
Dung môi khai triển: Isopropanol - amoniac (7 : 3).
Dung dịch amoniac loãng: Pha loãng 16 ml amoniac (TT) với nước vừa đủ 100 ml.
Dung dịch thử: Hòa tan 0,1 g chế phẩm trong 6 ml dung dịch amoniac loãng, thêm nước vừa đủ 10 ml.
Dung dịch đối chiếu (1): Dung dịch tyrosin chuẩn 0,05 mg/ml trong nước. Cân một lượng tyrosin chuẩn và chuyển vào bình định mức phù hợp, hòa tan bằng 1 ml dung dịch amoniac loãng, thêm nước và chuyển vào bình định mức phù hợp, thêm nước đến định mức, lắc đều.
Dung dịch đối chiếu (2): Hoà tan hoàn toàn 10 mg tyrosin chuẩn và 10 mg phenylalanin chuẩn trong 1 ml dung dịch amoniac loãng, thêm nước vừa đủ 25 ml.
Cách tiến hành: Chấm riêng biệt lên bản mỏng 5 μl mỗi dung dịch trên, triển khai sắc ký đến khi dung môi đi được khoảng 15 cm, lấy bản mỏng ra, sấy ở 100 °C đến 105 °C để amoniac bay hơi hoàn toàn. Phun lên bản mỏng dung dịch ninhydrin 0,2 % (TT). Sấy bản mỏng ở 100 °C đến 105 °C trong 15 min. Quan sát dưới ánh sáng thường, bất kỳ vết phụ nào trên sắc ký đồ của dung dịch thử không được lớn hơn hoặc đậm hơn vết chính trên sắc ký đồ dung dịch đối chiếu (1) (0,5 %), tổng độ lớn (độ đậm) của các vết phụ trên sắc ký đồ của dung dịch thử không được lớn hơn 2,0 %. Phép thử chỉ có giá trị khi sắc ký đồ của dung dịch đối chiếu (2) cho 2 vết tách rõ rệt.
Clorid
Không được quá 0,04 % (Phụ lục 9.4.5)
0,125 g chế phẩm hòa tan trong 1,5 ml dung dịch acid nitric 2M (TT) và pha loãng thành 15 ml với nước (có thể đun nóng để trợ tan) rồi tiến hành thử.
Sulfat
Không được quá 0,04 % (Phụ lục 9.4.14)
Hòa tan bằng cách đun nóng nhẹ 0,375 g chế phẩm trong 5 ml dung dịch acid hydocloric loãng (TT) và pha loãng thành 15 ml bằng nước rồi tiến hành thử.
Sắt
Không được quá 30 phần triệu (Phụ lục 9.4.13)
Hòa tan 0,333 g chế phẩm trong 10 ml dung dịch acid hydocloric loãng (77). Chiết 3 lần bằng cách mỗi lần lắc với 10 ml 4-methylpentan-2-on (TT1) trong 3 min. Gộp lớp dung môi hữu cơ và thêm 10 ml nước và lắc trong 3 min. Sử dụng lớp nước để thử.
Mất khối lượng do làm khô
Không được quá 0,3 % (Phụ lục 9.6).
(1,000 g; 105 °C, 3h).
Tro sulfat
Không được quá 0,4 % (Phụ lục 9.9, phương pháp 2).
Dùng 1,0 g chế phẩm.
Định lượng
Hoà tan 180 mg chế phẩm trong hỗn hợp gồm 6 ml acid formic khan (TT) và 50 ml acid acetic khan (TT). Chuẩn độ bằng dung dịch acid percloric 0,1 N (CĐ), xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2).
1 ml dung dịch acid percloric 0,1 N (CĐ) tương đương với 18,12 mg C9H11NO3.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Acid amin.
Phần 2
THÀNH PHẨM HÓA DƯỢC
BỘT PHA HỖN DỊCH ATTAPULGIT
Là thuốc bột dùng để pha hỗn dịch uống chứa attapulgit hoạt hóa. Có thể có thêm các tá dược thích hợp tạo mùi vị, tạo màu, chất bảo quản, chất ổn định hỗn dịch....
Hỗn dịch tạo thành sau khi pha theo hướng dẫn trên nhãn thuốc phải đáp ứng các yêu cầu trong chuyên luận “Hỗn dịch thuốc” (Phụ lục 1.5).
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận thuốc bột (Phụ lục 1.7) và các yêu cầu sau đây:
Hàm lượng attapulgit, từ 95,0 % đến 105,0 % so với lượng ghi trên nhãn.
Định tính
A. Nung một lượng chế phẩm chứa khoảng 0,5 g attapulgit với 2 g natri carbonat khan (TT) ở 600 °C trong 60 min, để nguội và chiết bằng 20 ml nước sôi. Để nguội, lọc, rửa cắn bằng nước và gộp nước rửa và dịch lọc. Giữ cắn để dùng cho phép thử định tính B. Acid hóa cẩn thận dịch thu được bằng acid hydrocloric (TT), bay hơi tới khô, làm ẩm cắn bằng 0,2 ml acid hydrocloric (TT), thêm 10 ml nước và khuấy đều. Xuất hiện tủa keo màu trắng.
B. Rửa cắn thu được trong phép thử A bằng nước và hòa tan trong 10 ml dung dịch acid hydrocloric 2 M (TT). Lấy 2 ml dung dịch thu dược, thêm dung dịch amoni thiocyanat (TT) 10 %. Xuất hiện màu đỏ đậm.
C. Lấy 2 ml dung dịch thu được trong phép thử B, thêm 1 ml dung dịch natri hydroxyd 42 % (TT), lọc. Thêm 3 ml dung dịch amoni clorid (TT) 10,7 % vào dịch lọc. Xuất hiện tủa keo màu trắng.
D. Lấy 2 ml dung dịch thu được trong phép thử B, thêm amoni clorid (TT) và thêm một lượng dư amoniac (TT) và lọc. Lấy dịch lọc thu được, thêm 0,15 ml thuốc thử magneson (TT) và một lượng dư dung dịch natri hydroxyd 5 M (TT). Xuất hiện tủa màu xanh dương.
Khả năng hấp phụ
Cân chính xác lượng chế phẩm tương ứng 1,0 g attapulgit đã nghiền mịn, thêm 50,0 ml dung dịch xanh methylen (TT) 0,12 %, lắc trong 5 min để yên và ly tâm. Màu của dung dịch thu được không được đậm hơn màu của dung dịch xanh methylen (TT) 0,0012 %.
Định lượng
Nung chén nung (chọn loại phù hợp) ở 1000 °C ± 50 °C trong ít nhất 30 min. Để nguội trong bình hút ẩm có chứa phosphor pentoxyd (TT) đến nhiệt độ phòng, cân (khối lượng M1).
Cân chính xác một lượng chế phẩm tương ứng với khoảng 3,0 g attapulgit (khối lượng Mt) vào chén nung ở trên. Đốt ở 300 °C trong 30 min, sau đó đốt tiếp ở 600 °C trong 15 min rồi nung ở 1000 °C ± 50 °C trong ít nhất 5h. Để nguội trong bình hút ẩm có chứa phosphor pentoxyd (TT) đến nhiệt độ phòng, cân (khối lượng M2).
Hàm lượng attapulgit trong chế phẩm được tính theo công thức:
![]()
Trong đó:
X(g): Số gam attapulgit có trong 1 đơn vị đóng gói;
M1: Khối lượng chén nung (g);
M2: Khối lượng chén nung và chế phẩm sau khi nung (g);
Mt: Khối lượng chế phẩm đem nung (g);
MTB: Khối lượng trung bình chế phẩm trong 1 đơn vị đóng gói (g);
H: Mất khối lượng do nung (ở 1000 °C) của nguyên liệu dùng để sản xuất chế phẩm (%).
Bảo quản
Trong đồ đựng kín.
Loại thuốc
Chất hấp phụ, bảo vệ niêm mạc dạ dày, ruột.
Hàm lượng thường dùng
1 g; 1,5 g; 2,5 g; 3 g.
BỘT PHA TIÊM CEFOXITIN
Bột pha tiêm cefoxitin là bột vô khuẩn của cefoxitin natri đóng trong lọ thủy tinh nút kín.
Chế phẩm phải đạt các yêu cầu quy định trong chuyên luận chung về “Thuốc tiêm, thuốc tiêm truyền” (Phụ lục 1.19) và các yêu cầu sau đây:
Hàm lượng cefoxitin, C16H17N3O7S2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Trong phần Định lượng, thời gian lưu của pic chính thu được trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính thu được trên sắc ký đồ của dung dịch chuẩn.
B. Phương pháp quang phổ hấp thụ tử ngoại (Phụ lục 4.1).
Dung dịch đệm: Dung dịch chứa 1,0 g/L kali dihydrophosphat (TT) và 1,8 g/L dinatri hydrophosphat khan (TT) trong nước.
Dung dịch thử: Cân một lượng chế phẩm thích hợp, hòa tan trong dung dịch đệm để thu được dung dịch chứa 20 μg/ml cefoxitin.
Dung dịch đối chiếu: Dung dịch chứa 20 μg/ml cefoxitin chuẩn trong dung dịch đệm.
Phổ hấp thụ tử ngoại trong khoảng bước sóng từ 200 nm đến 350 nm của dung dịch thử phải tương ứng với phổ của dung dịch đối chiếu.
C. Dung dịch chế phẩm chứa 50 mg/ml cefoxitin trong nước phải cho phản ứng (A) của ion natri (Phụ lục 8.1).
Độ trong và màu sắc của dung dịch
Dung dịch chế phẩm trong nước không có carbon dioxyd (TT) có chứa 10,0 % cefoxitin phải trong (Phụ lục 9.2) và không được có màu đậm hơn màu mẫu số 5 của dãy dung dịch màu đối chiếu phù hợp nhất (Phụ lục 9.3, phương pháp 2).
pH
pH từ 4,2 đến 7,0 (Phụ lục 6.2).
Dùng dung dịch chế phẩm trong nước chứa 100 mg/ml cefoxitin để đo.
Nội độc tố vi khuẩn
Không được quá 0,13 EU/mg (Phụ lục 13.2).
Nước
Không được quá 1,0 % (Phụ lục 10.3).
Dùng 0,5 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - nước - acid acetic băng (160 : 840 : 10).
Dung dịch thử: Lấy 10 lọ thuốc, cân khối lượng bột thuốc và trộn đều. Cân một lượng bột thuốc tương ứng với 60 mg cefoxitin vào bình định mức 200 ml, hòa tan trong nước. Dùng trong vòng 5 h sau khi pha.
Dung dịch chuẩn: Dung dịch chứa cefoxitin natri chuẩn trong nước có nồng độ 0,3 mg/ml cefoxitin. Dùng trong vòng 5 h sau khi pha.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh C (5 - 10 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 254 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết của cột tính trên pic cefoxitin không nhỏ hơn 2800; hệ số đối xứng của pic cefoxitin không lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic cefoxitin thu được từ 5 lần tiêm lặp lại không lớn hơn 1,0 %.
Tính hàm lượng phần trăm của cefoxitin, C16H17N3O7S2, trong chế phẩm dựa vào diện tích pic cefoxitin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C16H16N3NaO7S2 trong cefoxitin natri chuẩn.
1 mg C16H16N3NaO7S2 tương ứng với 0,9510 mg C16H17N3O7S2.
Bảo quản
Trong bao đồ đựng kín, tránh ánh sáng, ở nhiệt độ không quá 30 °C.
Loại thuốc
Kháng sinh nhóm cephalosporin.
Hàm lượng thường dùng
1 g.
NANG EMTRICITABIN
Là nang cứng chứa emtricitabin.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc nang” (Phụ lục 1.13) và các yêu cầu sau đây:
Hàm lượng emtricitabin, C8H10FN3O3S, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch acid hydrodoric 0,1 M (TT).
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm, bỏ vài mililít dịch lọc đầu. Pha loãng dịch lọc bằng môi trường hòa tan để được dung dịch có nồng độ emtricitabin tương đương nồng độ dung dịch chuẩn.
Dung dịch chuẩn: Dung dịch chứa 10 μg/ml emtricitabin chuẩn trong môi trường hòa tan.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng 280 nm. Mẫu trắng là môi trường hòa tan, dùng cốc đo 1 cm.
Tính lượng emtricitabin, C8H10FN3O3S, đã hòa tan dựa vào độ hấp thụ của dung dịch thử, dung dịch chuẩn và hàm lượng của C8H10FN3O3S trong emtricitabin chuẩn.
Yêu cầu: Không ít hơn 75 % (Q) lượng emtricitabin, C8H10FN3O3S, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 27,2 g kali dihydrophosphat (TT) trong 1000 ml nước.
Pha động A: Dung dịch đệm - nước (5 : 95).
Pha động B: Acetonitril - dung dịch đệm - nước (70 : 5 : 25).
Dung môi pha mẫu: Acetonitril - nước (20 : 80).
Dung dịch thử: Hòa tan một lượng bột thuốc trong nang tương đương với khoảng 50 mg emtricitabin trong 80 ml dung môi pha mẫu và pha loãng thành 100 ml với cùng dung môi, lọc.
Dung dịch đối chiếu: Pha loãng dung dịch thử bằng dung môi pha mẫu để thu được dung dịch chứa 0,50 μg/ml emtricitabin.
Dung dịch phù hợp hệ thống: Trộn đều 5 ml dung dịch thử và 2 ml dung dịch acid phosphoric (TT) 12 %, đặt trong cách thủy sôi trong 15 min.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh base-deactivated octadecylsilyl silica gel dùng cho sắc ký (5 μm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 280 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 9 | 93 | 7 |
9 - 15 | 93 -> 0 | 7 -> 100 |
15 - 19 | 0 | 100 |
19 - 19,1 | 0 -> 93 | 100 -> 7 |
19,1 - 30 | 93 | 7 |
Tiến hành sắc ký với dung dịch thử, dung dịch đối chiếu và dung dịch phù hợp hệ thống.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic emtricitabin (thời gian lưu khoảng 9 min) và pic tạp chất cỏ thời gian lưu tương đối so với emtricitabin bằng 1,3 không nhỏ hơn 6.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Các tạp chất rửa giải ra trước pic chính: Với mỗi tạp chất, diện tích pic không được lớn hơn 5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (0,5 %) và không quá 2 tạp chất có diện tích pic lớn hơn 2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (0,2 %).
Các tạp chất rửa giải ra sau pic chính: Với mỗi tạp chất, diện tích pic không được lớn hơn 8 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (0,8 %) và không quá 2 tạp chất có diện tích pic lớn hơn 2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (0,2 %).
Tổng diện tích pic của tất cả các tạp chất không được lớn hơn 10 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1,0 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (0,05 %).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm, pha động A, pha động B, dung môi pha mẫu, dung dịch phù hợp hệ thống, điều kiện sắc ký, chương trình dung môi và kiểm tra tính phù hợp của hệ thống như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Cân 20 nang, tính khối lượng trung bình của bột thuốc trong nang và nghiền thành bột mịn. Cân chính xác một lượng bột tương ứng khoảng 50 mg emtricitabin vào bình định mức 100 ml, thêm 80 ml dung môi pha mẫu để hòa tan, thêm dung môi pha mẫu đến vạch và lọc.
Dung dịch chuẩn: Dung dịch chứa 0,50 mg/ml emtricitabin chuẩn trong dung môi pha mẫu.
Tính hàm lượng phần trăm của emtricitabin, C8H10FN3O3S, trong chế phẩm dựa vào diện tích pic emtricitabin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C8H10FN3O3S trong emtricitabin chuẩn.
Bảo quản
Trong đồ đựng kín.
Loại thuốc
Thuốc kháng retrovirus.
Hàm lượng thường dùng
200 mg.
THUỐC TIÊM CITICOLIN NATRI
Là dung dịch vô khuẩn có chứa citicolin natri trong nước để pha thuốc tiêm. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc tiêm, thuốc tiêm truyền” (Phụ lục 1.19) và các yêu cầu sau đây:
Hàm lượng citicolin, C14H26N4O11P2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
pH
pH của chế phẩm phải từ 6,0 đến 8,0 (Phụ lục 6.2).
Nội độc tố vi khuẩn
Không được quá 0,30 EU/mg citicolin natri (Phụ lục 13.2).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hỗn hợp đồng thể tích của dung dịch kali dihydrophosphat (TT) 0,1 M và dung dịch tetrabutylamoni hydroxyd (TT) 0,01 M, điều chỉnh đến pH 4,5 bằng acid phosphoric (TT).
Pha động: Dung dịch đệm - methanol (95 : 5).
Dung dịch thử: Pha loãng chế phẩm thích hợp bằng nước để thu được dung dịch chứa 2,5 mg/ml citicolin natri.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 200,0 ml bằng nước.
Dung dịch đối chiếu (2): Dung dịch chứa 7,5 μg/ml acid 5’-cytidylic chuẩn trong nước.
Dung dịch đối chiếu (3): Dung dịch chứa 0,125 mg/ml citicolin natri chuẩn và 0,125 mg/ml acid 5'-cytidylic chuẩn trong nước.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,0 mm) được nhồi pha tĩnh C.
Detector quang phổ hấp thụ tử ngoại đặt ở bước sóng 276 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 μl, riêng dung dịch đối chiếu (3) tiêm 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 2,5 lần thời gian lưu của pic citicolin.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic citicolin và pic acid 5’-cytidylic không nhỏ hơn 1,5.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Acid 5’-cytidylic: Diện tích pic acid 5’-cytidylic không được lớn hơn diện tích pic acid 5’-cytidylic thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,3 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,5 %).
Tổng diện tích pic của tất cả các tạp chất trừ acid 5’-cytidylic không được lớn hơn 1,4 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,7 %).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Trộn đều dung dịch chể phẩm chứa trong 20 đơn vị đóng gói. Pha loãng chế phẩm thích hợp bằng nước để thu được dung dịch chứa 0,25 mg/ml citicolin.
Dung dịch chuẩn: Dung dịch chứa 0,26 mg/ml citicolin natri chuẩn trong nước.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic citicolin không nhỏ hơn 3000, hệ số đối xứng của pic citicolin không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của citicolin, C14H26N4O11P2, trong chế phẩm dựa vào diện tích pic citicolin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C14H26N4O11P2 trong citicolin natri chuẩn. Khối lượng phân tử của citicolin và citicolin natri lần lượt là 488,33 và 510,31.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Tăng chuyển hóa ở tế bào thần kinh.
Hàm lượng thường dùng
100 mg/2 ml, 250 mg/2 ml và 500 mg/2 ml tính theo citicolin.
THUỐC TIÊM ONDANSETRON
Là dung dịch vô khuẩn của ondansetron hydroclorid trong nước để pha thuốc tiêm hoặc của ondansetron trong nước để pha thuốc tiêm có thêm acid hydrocloric để hỗ trợ hòa tan.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc tiêm, thuốc tiêm truyền” (Phụ lục 1.19) và các yêu cầu sau đây:
Hàm lượng ondansetron, C18H19N3O, từ 95,0 % đến 105,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
pH
pH của chế phẩm phải từ 3,3 đến 4,0 (Phụ lục 6.2).
Tạp chất D
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 2,72 g kali dihydrophosphat (TT) trong 900 ml nước, điều chỉnh đến pH 5,4 bằng dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml. Lọc và đuổi khí.
Pha động: Dung dịch đệm - acetonitril (80 : 20), điều chỉnh tỷ lệ nếu cần.
Dung dịch thử: Lấy một lượng thể tích chế phẩm tương ứng với 10 mg ondansetron vào bình định mức 25 ml và thêm pha động đến vạch, trộn đều.
Dung dịch đối chiếu: Dung dịch chứa 0,4 µg/ml tạp chất D chuẩn của ondansetron trong pha động.
Dung dịch phù hợp hệ thống: Dung dịch chứa 0,6 µg/ml tạp chất D chuẩn của ondansetron và 1 µg/ml tạp chất C chuẩn của ondansetron trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh là các hạt silica gel liên kết hóa học với các nhóm nitril (5 µm).
Detector quang phổ tử ngoại đặt ở bước sóng 328 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 20 µl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, thời gian lưu tương đối của tạp chất C so với tạp chất D khoảng 0,8; độ phân giải giữa pic tạp chất C và pic tạp chất D không nhỏ hơn 1,5. Trên sắc ký đồ của dung dịch đối chiếu, số đĩa lý thuyết tính trên pic tạp chất D không nhỏ hơn 400 và độ lệch chuẩn tương đối của diện tích pic tạp chất D thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của tạp chất D trong chế phẩm dựa vào diện tích pic tạp chất D trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu, nồng độ của tạp chất D chuẩn trong dung dịch đối chiếu và nồng độ của ondansetron trong thuốc tiêm (được xác định ở phần Định lượng).
Giới hạn:
Tạp chất D: Không được quá 0,12 %.
Ghi chú:
Tạp chất D: 9-Methyl-3-methylen-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 2,72 g kali dihydrophosphat (TT) trong 900 ml nước, điều chỉnh đến pH 5,4 bằng dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml. Lọc và đuổi khí.
Pha động: Dung dịch đệm - acetonitril (50 : 50), điều chỉnh tỷ lệ nếu cần.
Dung dịch thử: Lấy một lượng thể tích chế phẩm tương ứng với 2 mg ondansetron vào bình định mức 25 ml và thêm pha động đến vạch, trộn đều.
Dung dịch phù hợp hệ thống: Dung dịch chứa 0,6 µg/ml tạp chất D chuẩn của ondansetron và 1 µg/ml tạp chất C chuẩn của ondansetron trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh các hạt silica gel liên kết hóa học với các nhóm nitril (5 µm).
Detector quang phổ tử ngoại đặt ở bước sóng 216 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 20 µl dung dịch phù hợp hệ thống và 10 µl dung dịch thử.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, thời gian lưu tương đối so với ondansetron của pic tạp chất C và tạp chất D lần lượt khoảng 0,35 và 0,37.
Trên sắc ký đồ của dung dịch thử, nếu có bất kỳ tạp chất nào thì hàm lượng phần trăm được tính theo phương pháp chuẩn hóa diện tích pic, bỏ qua pic của tạp chất D.
Giới hạn:
Với mỗi tạp chất, trừ tạp chất D: Không được quá 0,2 %.
Tổng các tạp chất, bao gồm cả tạp chất D (xác định trong phép thử Tạp chất D ở trên), không được quá 0,5 %.
Ghi chú:
Tạp chất A: 3-[(Dimethylamino)methyl]-9-methyl-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Tạp chất C: 9-Methyl-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Nội độc tố vi khuẩn
Không được quá 9,9 EU/mg ondansetron hydroclorid (Phụ lục 13.2).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động, dung dịch thử và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Thể tích tiêm: 10 µl.
Dung dịch chuẩn: Dung dịch chứa 0,1 mg/ml ondansetron hydroclorld chuẩn trong pha động.
Dung dịch phù hợp hệ thống: Dung dịch chứa 0,1 mg/ml ondansetron hydroclorid chuẩn và 50 µg/ml tạp chất A chuẩn của ondansetron trong pha động.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, thời gian lưu tương đối so với ondansetron của tạp chất A khoảng 1,1; độ phân giải giữa pic tạp chất A và ondansetron không nhỏ hơn 1,5. Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic ondansetron không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic ondansetron thu được từ 5 lần tiêm lặp lại không lớn hơn 1,5 %.
Tính hàm lượng phần trăm của ondansetron, C18H19N3O, trong chế phẩm dựa vào diện tích pic ondansetron thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C18H19N3O.HCl trong ondansetron hydroclorid chuẩn. Khối lượng phân tử của ondansetron và ondansetron hydroclorid lần lượt là 293,36 và 329,83.
Bảo quản
Trong đồ đựng kín, tốt nhất là thủy tinh cấp I, tránh ánh sáng, ở 2 °C - 30 °C.
Loại thuốc
Thuốc chống nôn, đối kháng thụ thể 5-HT3.
Hàm lượng thường dùng
2 mg/ml.
THUỐC TIÊM OXYTOCIN
Là dung dịch vô khuẩn có chứa oxytocin trong dung môi phù hợp.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc tiêm, thuốc tiêm truyền” (Phụ lục 1.19) và các yêu cầu sau đây:
Hàm lượng oxytocin, C43H66N12O12S2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Tính chất
Dung dịch trong, không màu
Định tính
Có thể chọn một trong hai phép thử định tính A hoặc B:
A. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel G.
Dung môi khai triển: Dicloromethan - methanol - nước - acid acetic băng (70 : 30 : 6 : 1).
Dung dịch thử: Lấy 10,0 ml chế phẩm, đem cô đến cắn ở 30 °C dưới áp suất giảm không quá 0,6 kPa. Hòa tan cắn trong 1 ml methanol (TT).
Dung dịch đối chiếu: Dung dịch chứa 165 µg/ml oxytocin chuẩn trong methanol (TT).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 20 µl mỗi dung dịch trên. Triển khai sắc ký tới khi dung môi đi được 15 cm. Làm khô bản mỏng dưới luồng không khí lạnh và đặt bản mỏng trong bình chứa hơi iod. Quan sát bản mỏng dưới ánh sáng thường, vết chính trên sắc ký đồ của dung dịch thử phải giống với vết chính trên sắc ký đồ của dung dịch đối chiếu về vị trí, kích thước và độ đậm.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của plc chính trên sắc ký đồ của dung dịch đổi chiếu.
pH
pH của chế phẩm phải từ 3,0 đến 5,0 (Phụ lục 6.2).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Acetonitril - dung dịch đệm phosphat - nước (15:15:70).
Pha động B: Acetonitril - dung dịch đệm phosphat - nước (70 : 15 : 15).
Dung dịch đệm phosphat: Hòa tan 31,2 g natri dihydrophosphat (TT) trong 1000 ml nước.
Dung dịch thử: Dung dịch chế phẩm không pha loãng.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 50,0 ml bằng pha động A.
Dung dịch đối chiếu (2): Hòa tan 100 mg clorbutol khan (TT) trong 0,25 ml acid acetic băng (TT) và pha loãng thành 20 ml bằng pha động A.
Dung dịch đối chiếu (3): Lấy 3,0 ml dung dịch thử, thêm 2,0 ml dung dịch acid sulfuric 1 % (TT), đun sôi cẩn thận trong cách thủy trong 20 min.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh C (5 µm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 220 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 50 µl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động |
(min) | (% tt/tt) | (% tt/tt) |
0 - 5 | 100 | 0 |
5 - 20 | 100 → 94 | 0 → 6 |
20 - 50 | 94 → 60 | 6 → 40 |
50 - 51 | 60 → 100 | 40 → 0 |
51 - 65 | 100 | 0 |
Tiến hành sắc ký với dung dịch thử, dung dịch đối chiếu (1), (2) và (3).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic oxytocin (thời gian lưu khoảng 25 min) và pic có thời gian lưu tương đối so với oxytocin khoảng 0,9 không nhỏ hơn 1,4.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Không có quá 1 pic, trừ pic chính, có diện tích lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (2 %).
Không có bất kỳ pic nào, trừ pic chính, có diện tích lớn hơn 2,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (5 %).
Bỏ qua pic tương ứng với pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2).
Nội độc tố vi khuẩn
Không được quá 0,5 EU/IU oxytocin (Phụ lục 13.2).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động, điều kiện sắc ký và kiểm tra tính phù hợp của hệ thống tiến hành như mô tả trong phần Tạp chất liên quan.
Dung dịch thử: Dung dịch chế phẩm.
Dung dịch đối chiếu: Dung dịch chứa 16,7 µg/ml oxytocin chuẩn trong pha động A.
Tính hàm lượng phần trăm của oxytocin, C43H66N12O12S2, trong chế phẩm dựa vào diện tích pic thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu và hàm lượng của C43H66N12O12S2 trong oxytocin chuẩn.
Bảo quản
Tránh ánh sáng, ở nhiệt độ 2 °C đến 8 °C.
Loại thuốc
Thuốc thúc đẻ - Hormon thùy sau tuyến yên.
Hàm lượng thường dùng
10 lU/ml, 5 lU/ml.
THUỐC TIÊM TERBUTALIN SULFAT
Là dung dịch vô khuẩn có chứa terbutalin sulfat trong nước để pha thuốc tiêm. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc tiêm, thuốc tiêm truyền” (Phụ lục 1.19) và các yêu cầu sau đây:
Hàm lượng terbutalin sulfat, (C12H19NO3)2.H2SO4, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel.
Dung môi khai triển: Isopropanol - cyclohexan - acid formic (13 : 5 : 1).
Dung dịch thử: Dung dịch chế phẩm, nếu cần pha loãng với dung dịch natri clorid 0,9 % (TT) để thu được dung dịch có chứa 0,5 mg/ml terbutalin sulfat.
Dung dịch đối chiếu: Dung dịch chứa 0,5 mg/ml terbutalin sulfat chuẩn trong dung dịch natri clorid 0,9 % (TT).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 2 µl mỗi dung dịch trên. Triển khai sắc ký đến khi dung môi đi được 3/4 chiều dài bản mỏng. Lấy bản mỏng ra, để khô bản mỏng ngoài không khí. Phun dung dịch 4-aminoantipyrin (TT) 2 % trong methanol (TT), để khô ngoài không khí. Tiếp tục phun lên bản mỏng dung dịch kali fericyanid (TT) 8 % trong hỗn hợp amoniac - nước (4:1). Vết chính trên sắc ký đồ của dung dịch thử phải có Rf giống với vết chính trên sắc ký đồ của dung dịch đối chiếu.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
pH
pH của chế phẩm phải từ 3,0 đến 5,0 (Phụ lục 6.2).
Nội độc tố vi khuẩn
Không được quá 1250,0 EU/mg terbutalin sulfat (Phụ lục 13.2).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch tạo cặp ion: Hòa tan 3,15 g amoni format (TT) trong 900 ml nước, điều chỉnh đến pH 3,0 bằng acid formic (TT), thêm 5,49 g natri 1-hexansulfonat (TT) và thêm nước vừa đủ 1000 ml, trộn đều. Lọc và đuổi khí.
Pha động: Dung dịch tạo cặp ion - methanol (77 : 23).
Dung dịch thử. Dùng dung dịch chế phẩm. Nếu cần pha loãng với nước để thu được dung dịch có chứa 0,5 mg/ml terbutalin sulfat.
Dung dịch chuẩn: Dung dịch chứa 0,5 mg/ml terbutalin sulfat chuẩn trong pha động.
Dung dịch phân giải: Dung dịch chứa 1,0 mg/ml terbutalin sulfat chuẩn và 0,4 mg/ml tạp chất C chuẩn của terbutalin sulfat trong pha động.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh C (5 µm).
Detector quang phổ tử ngoại đặt ở bước sóng 276 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 µl.
Cách tiến hành:
Tiến hành sắc ký dung dịch phân giải.
Thời gian lưu tương đối so với terbutalin của tạp chất C khoảng 0,9.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phân giải, độ phân giải giữa pic tạp chất C và pic terbutalin không nhỏ hơn 2,0; số đĩa lý thuyết của cột tính trên pic terbutalin không nhỏ hơn 1500; hệ số đối xứng của pic terbutalin không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic terbutalin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Tính hàm lượng phần trăm của terbutalin sulfat, (C12H19NO3)2.H2SO4, trong chế phẩm dựa vào diện tích pic terbutalin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của (C12H19NO3)2.H2SO4 trong terbutalin sulfat chuẩn.
Ghi chú:
Tạp chất C của terbutalin: 1-(3,5-dihydroxyphenyl)-2-[(1,1-dimethylethyl)amino] ethanon.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Kích thích beta2 giao cảm, thuốc giãn phế quản.
Hàm lượng thường dùng
0,5 mg/ml; 1 mg/ml.
VIÊN NÉN ALENDRONAT NATRI
Là viên nén chứa alendronat natri. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng acid alendronic, C4H13NO7P2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml nước.
Tốc độ quay: 50 r/min.
Thời gian: 15 min.
Cách tiến hành:
Xác định hàm lượng acid alendronic đã hòa tan bằng Phương-pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch A, pha động, điều kiện sắc ký và kiểm tra tính phù hợp của hệ thống như mô tả ở phần Định lượng.
Dung dịch đệm citrat 0,6 M: Dung dịch chứa 176,4 g/L natri citrat (TT) trong môi trường hòa tan.
Dung dịch B: Hòa tan 6,2 g acid boric (TT) trong 950 ml nước, điều chỉnh đến pH 9,0 bằng dung dịch natri hydroxyd 1 M (TT) và thêm nước vừa đủ 1000 ml.
Dung dịch C: Dung dịch chứa 0,5 mg/ml 9-fluorenylmethyl cloroformat (TT) trong acetonitril (TT) (Pha ngay trước khi dùng).
Dung dịch thử. Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan, ly tâm ngay. Lấy 5,0 ml phần dịch trong vào ống ly tâm 50 ml có nắp xoáy đã có sẵn 1,0 ml dung dịch đệm citrat 0,6 M, 5,0 ml dung dịch B và trộn đều trong vòng 3 min. Thêm 4,0 ml dung dịch C và lắc mạnh 30 s. Để yên ở nhiệt độ phòng 25 min. Thêm 25,0 ml methylen clorid (TT) và lắc mạnh 40 s. Ly tâm hỗn hợp 5 min. Thu lấy phần chất lỏng trong phía trên.
Dung dịch chuẩn gốc: Dung dịch chứa alendronat natri chuẩn trong môi trường hòa tan có hàm lượng acid alendronic tương đương với hàm lượng 1 viên nén hòa tan trong 900 ml môi trường hòa tan.
Dung dịch chuẩn: Lấy 5,0 ml dung dịch chuẩn gốc và tiến hành tiếp như dung dịch thử từ đoạn “vào ống ly tâm 50 ml...”.
Dung dịch mẫu trắng: Lấy 5,0 ml nước và tiến hành tiếp như dung dịch thử từ đoạn “vào ống ly tâm 50 ml...".
Tính lượng acid alendronic, C4H13NO7P2, đã hòa tan dựa vào diện tích pic acid alendronic thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và nồng độ của acid alendronic trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng acid alendronic, C4H13NO7P2, so với lượng ghi trên nhãn được hòa tan trong 15 min. Với viên nén liều hàng tuần thì không ít hơn 75 % (Q) lượng acid alendronic, C4H13NO7P2, so với lượng ghi trên nhãn được hòa tan trong 15 min.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch A: Hòa tan 14,7 g natri citrat (TT) và 7,05 g dinatri hydrophosphat khan (TT) trong 900 ml nước, điều chỉnh đến pH 8,0 bằng acid phosphoric (TT) và pha loãng thành 1000 ml bằng nước.
Dung dịch B: Dung dịch chứa 38,1 g/L natri borat (TT) trong nước.
Dung dịch C: Dung dịch chứa 1 mg/ml 9-fluorenylmethyl cloroformat (TT) trong acetonitril (TT) (Pha ngay trước khi dùng).
Pha động: Acetonitril - methanol - dung dịch A (20 : 5 : 75).
Dung dịch đệm citrat 0,1 M: Dung dịch chứa 29,4 g/L natri citrat (TT) trong nước.
Dung dịch thử gốc: Cân 10 viên nén, cho vào bình định mức 1000 ml, thêm 500 ml dung dịch đệm citrat 0,1 M, lắc bằng máy lắc cơ học trong 30 min và siêu âm 5 min. Thêm dung dịch đệm citrat 0,1 M đến vạch, và ly tâm dung dịch thu được. Pha loãng phần dịch trong sau khi ly tâm để thu được dung dịch có chứa khoảng 0,03 mg/ml acid alendronic.
Dung dịch thử. Lấy 5,0 ml dung dịch thử gốc vào ống ly tâm 50 ml có nắp xoáy đã có sẵn 5 ml dung dịch B và trộn đều trong vòng 3 min. Thêm 4,0 ml dung dịch C và lắc mạnh 30 s. Để yên ở nhiệt độ phòng 25 min. Thêm 25,0 ml methylen clorid (TT) và lắc mạnh 40 s. Ly tâm hỗn hợp 10 min. Thu lấy phần chất lỏng trong phía trên.
Dung dịch chuẩn gốc: Dung dịch chứa alendronat natri chuẩn có nồng độ acid alendronic khoảng 0,03 mg/ml trong dung dịch đệm citrat 0,1 M.
Dung dịch chuẩn: Lấy 5,0 ml dung dịch chuẩn gốc và tiến hành tiếp như dung dịch thử từ đoạn “vào ống ly tâm 50 ml...."
Dung dịch mẫu trắng: Lấy 5,0 ml nước và tiến hành tiếp như dung dịch thử từ đoạn “vào ống ly tâm 50 ml...."
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,1 mm) được nhồi pha tĩnh styrenedivinylbenzen copolymer (3 - 30 µm) [Cột Hamilton PRP-1,25 cm x 4,1 mm (10 µm), là phù hợp].
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 266 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 50 µl.
Cách tiến hành:
Tiến hành sắc ký dung dịch mẫu trắng, dung dịch chuẩn và dung dịch thử.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ dung dịch chuẩn, hệ số dung lượng không nhỏ hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic acid alendronic thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của acid alendronic, C4H13NO7P2, trong chế phẩm dựa vào diện tích pic acid alendronic thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C4H13NO7P2 trong alendronat natri chuẩn. Khối lượng phân tử của acid alendronic và alendronat natri khan lần lượt là 249,10 và 271,09.
Bảo quản
Trong đồ đựng kín, ở 15 °C đến 30 °C.
Loại thuốc
Ức chế tiêu xương.
Hàm lượng thường dùng
10 mg, 70 mg.
VIÊN NÉN ANASTROZOL
Là viên nén chứa anastrozol.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng anastrozol, C17H19N5, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2).
Lấy một lượng bột viên tương ứng với khoảng 12 mg anastrozol vào bình thích hợp, thêm 10 ml diethyl ether (TT) và siêu âm 5 min. Lọc qua màng lọc nylon 0,45 µm vào cốc có mỏ có chứa sẵn 400 mg kali bromid (TT). Bốc hơi hỗn hợp tới khô bằng dòng khí nitrogen. Sấy khô tiếp trong chân không ở 50 °C trong 1 h. Thêm 400 mg kali bromid (TT) để chuẩn bị đĩa nén.
Chuẩn bị mẫu chuẩn anastrozol tương tự mẫu thử.
Phổ hồng ngoại của mẫu thử cho các dải phổ hấp thụ tại các số sóng ở khoảng 2235 cm-1, 1606 cm-1, 1500 cm-1,1359 cm-1, 1205 cm-1, 1137 cm-1, 1013 cm-1, 875 cm-1 giống với phổ của mẫu chuẩn.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml nước, đuổi khí.
Tốc độ quay: 50 r/min.
Thời gian: 15 min.
Cách tiến hành:
Xác định hàm lượng anastrozol hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động, điều kiện sắc ký và dung môi pha mẫu như mô tả ở mục Định lượng. Thể tích tiêm là 50 µl.
Dung dịch thử. Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 µm. Bỏ vài mililít dịch lọc đầu.
Dung dịch chuẩn gốc: Dung dịch chứa 0,2 mg/ml anastrozol chuẩn trong dung môi pha mẫu.
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc bằng môi trường hòa tan để thu được dung dịch chứa (L/1000) mg/ml anastrozol, L là hàm lượng ghi trên nhãn của viên tính theo mg.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic anastrozol không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic anastrozol thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính lượng anastrozol, C17H19N5, đã hòa tan dựa vào diện tích pic anastrozol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và nồng độ của anastrozol trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng anastrozol, C17H19N5, so với lượng ghi trên nhãn được hòa tan trong 15 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Acetonitril - methanol - acid trifuoroacetic - nước (100 : 200 : 0,7 : 700).
Pha động B: Acetonitril - methanol - acid trifuoroacetic - nước (250 : 500 : 0,7 : 250).
Dung môi pha mẫu: Acetonitril - acid trifuoroacetic - nước (200 : 0,8 : 800).
Dung dịch phù hợp hệ thống gốc: Cân một lượng anastrozol chuẩn và ethyl 4-hydroxybenzoat vào bình định mức thích hợp và thêm dung môi pha mẫu khoảng 50 % thể tích bình. Siêu âm để hòa tan và thêm dung môi pha mẫu đến vạch để thu được dung dịch chứa 0,5 mg/ml anastrozol và 0,3 mg/ml ethyl 4-hydroxybenzoat.
Dung dịch phù hợp hệ thống: Pha loãng dung dịch phù hợp hệ thống gốc bằng dung môi pha mẫu để thu được dung dịch chứa 10 µg/ml anastrozol, 6 µg/ml ethyl 4-hydroxybenzoat.
Dung dịch đối chiếu gốc: Cân một lượng anastrozol chuẩn vào bình định mức thích hợp và thêm dung môi pha mẫu khoảng 50 % thể tích bình. Siêu âm để hòa tan và thêm dung môi pha mẫu đến vạch để thu được dung dịch chứa 0,5 mg/ml anastrozol.
Dung dịch đối chiếu: Pha loãng dung dịch đối chiếu gốc bằng dung môi pha mẫu để thu được dung dịch chứa 10 µg/ml anastrozol.
Dung dịch thử. Cân 25 viên, nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với 10 mg anastrozol vào bình thích hợp, thêm 10,0 ml dung môi pha mẫu. Siêu âm 30 min và để nguội về nhiệt độ phòng. Lọc qua màng lọc 0,45 µm, bỏ vài mililít dịch lọc đầu. Nếu dịch lọc không trong thì lọc lại qua màng lọc 0,2 µm và bỏ vài ml dịch lọc đầu.
Điều kiện sắc ký:
Cột kích thước (10 cm x 4,6 mm) được nhồi pha tĩnh C (5 µm).
Detector quang phổ tử ngoại đặt ở bước sóng 215 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 µl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 100 | 0 |
25 | 100 | 0 |
25,1 | 0 | 100 |
30 | 0 | 100 |
31 | 100 | 0 |
40 | 100 | 0 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, thời gian lưu tương đối của ethyl 4-hydroxybenzoat so với anastrozol khoảng 0,7; độ phân giải giữa pic ethyl 4- hydroxybenzoat và pic anastrozol không nhỏ hơn 4; hệ số đối xứng pic anastrozol từ 0,9 đến 1,3 và độ lệch chuẩn tương đối của diện tích pic anastrozol thu được từ 6 lần tiêm lặp lại không lớn hơn 5 %.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic anastrozol trên sắc ký đồ của dung dịch đối chiếu, nồng độ của anastrozol trong dung dịch đối chiếu và nồng độ lý thuyết của anastrozol trong dung dịch thử. Thời gian lưu tương đối và giới hạn phần trăm của các tạp chất được trình bày trong Bảng 1. Bỏ qua các tạp chất có hàm lượng dưới 0,1 %.
Bảng 1
Tạp chất | Thời gian lưu tương đối | Giới hạn (%) |
Anastrozol diamid | 0,11 | 0,5 |
Anastrozol monoacid monoamid | 0,26 | 0,5 |
Anastrozol monoamid mononitril | 0,30 | 0,5 |
Desmethyl anastrozol | 0,51 | - |
Anastrozol diacid | 0,71 | 0,5 |
Anastrozol monoacid mononitril | 0,87 | 0,5 |
Anastrozol | 1,00 | - |
Tạp đơn chưa xác định | - | 0,5 |
Tổng tạp | - | 1,0 |
Ghi chú:
Anastrozol diamid: 2,2'-{5-[(1H-1,2,4-triazol-1-yl)methyl]-1,3-phenylen}bis(2-methylpropanamid).
Anastrozol monoacid monoamid: Acid 2-{3-[(1H-1,2,4-triazol-1-yl)methyl]-5-(1-amino-2-methyl-1-oxopropan-2- yl)phenyl}-2-methylpropanoic.
Anastrozol monoamid mononitril: 2-{3-[(1H-1,2,4-triazol-1-yl)methyl]-5-(2-cyanopropan-2-yl)phenyl}-2-methylpropanamid.
Desmethyl anastrozol: 2-(3-(1-cyanoethyl)-5-(1H-1,2,4-triazol-1-ylmethyl)phenyl)-2-methylpropannitril (Tạp chất này được kiểm soát ở nguyên liệu, chỉ đưa ra đây để định tính, không tính vào tổng tạp).
Anastrozol diacid: 2,2'-{5-[(1H-1,2,4-triazol-1-yl)methyl]-1,3-phenylene}bis(acid 2-methylpropanoic).
Anastrozol monoacid mononitril: Acid 2-{3-[(1H-1,2,4-triazol-1-yl)methyl]-5-(2-cyanopropan-2-yl)phenyl}-2-methylpropanoic.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9, phương pháp đồng đều hàm lượng.
Phương pháp sắc ký lỏng (Phụ lục 5.3). Pha động, điều kiện sắc ký, dung môi pha mẫu, dung dịch chuẩn và cách tiến hành như mô tả ở mục Định lượng.
Dung dịch thử: Cho 1 viên vào bình định mức có dung tích thích hợp để thu được dung dịch có nồng độ anatrozol 40 µg/ml, thêm nước khoảng 40 % thể tích bình. Lắc trên máy lắc cơ học 10 min cho viên rã hoàn toàn. Thêm tiếp acetonitril (TT) khoảng 40 % thể tích bình, siêu âm 15 mln ở 25 °C, lắc liên tục trong lúc siêu âm. Thêm dung môi pha mẫu đến vạch, ly tâm dung dịch thu được với tốc độ 3500 rpm trong 10 min. Lấy phần dịch trong ở phía trên. Lọc qua màng lọc 0,45 µm nếu cần.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - nước (40 : 60).
Dung môi pha mẫu: Acetonitril - nước (50 : 50).
Dung dịch chuẩn: Dung dịch chứa 40 µg/ml anastrozol chuẩn trong dung môi pha mẫu. Siêu âm để hòa tan nếu cần.
Dung dịch thử: Cân 10 viên. Chuyển 10 viên vào bình định mức có dung tích thích hợp để thu được dung dịch có nồng độ anastrozol 40 µg/ml, thêm nước khoảng 40 % thể tích bình. Lắc trên máy lắc cơ học 10 min cho viên rã hoàn toàn. Thêm tiếp acetonitril (TT) khoảng 40 % thể tích bình, siêu âm 15 min ở 25 °C, lắc liên tục trong lúc siêu âm. Thêm dung môi pha mẫu đến vạch, ly tâm dung dịch thu được với tốc độ 3500 rpm trong 10 min. Lấy phần dịch trong ở phía trên. Lọc qua màng lọc 0,45 µm nếu cần.
Điều kiện sắc ký:
Cột kích thước (10 cm x 4,6 mm) được nhồi pha tĩnh C (5 µm).
Detector quang phổ tử ngoại đặt ở bước sóng 215 nm.
Tốc độ dòng: 1,0 ml/mln.
Thể tích tiêm: 20 µl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic anastrozol không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic anastrozol thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Tính hàm lượng phần trăm của anastrozol, C17H19N5, trong viên dựa vào diện tích pic anastrozol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C17H19N5 trong anastrozol chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Ức chế aromatase, điều trị ung thư vú.
Hàm lượng thường dùng
1 mg.
VIÊN NÉN BETAHISTIN DIHYDROCLORID
Là viên nén chứa betahistin dihydrodorid. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng betahistin dihydroclorid, C8H12N2.2HCl, từ 95,0 % đến 105,0 % so với lượng ghi trên nhãn.
Định tính
A. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
B. Trong phần Định lượng, phổ hấp thụ tử ngoại (Phụ lục 4.1) của pic chính thu được từ dung dịch thử phải tương ứng với phổ hấp thụ tử ngoại của pic betahistin dihydroclorid thu được từ dung dịch chuẩn.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Hòa tan 0,4 g hexylamin (TT) trong 600 ml dung dịch chứa natri dihydrophosphat khan (TT) 0,4% và natri dodecyl sulfat (TT) 0,27 %, thêm 400 ml of acetonitril (TT), trộn đều và điều chỉnh đến pH 3,5 bằng acid phosphoric (TT).
Dung dịch thử: Lắc một lượng bột viên tương ứng với khoảng 32 mg betahistin dihydroclorid với 50 ml pha động trong 10 min, thêm pha động vừa đủ 100,0 ml, trộn đều. Ly tâm và sử dụng dung dịch trong ở phía trên.
Dung dịch đối chiếu (1): Pha loãng 1 thể tích dung dịch thử thành 500 thể tích bằng pha động.
Dung dịch đối chiếu (2): Dung dịch chứa 0,00064 % N-methyl-2-(pyridin-2-yl)-N-[2-(pyridin-2- yl)ethyl]ethanamin trihydroclorid (tạp chất C) trong pha động.
Dung dịch đối chiếu (3): Dung dịch chứa 0,000032 % 2-vinylpyridin (TT) (tạp chất A) trong acetonitril (TT).
Dung dịch đối chiếu (4): Dung dịch chứa 0,00064 % N-methyl-2-(pyridin-2-yl)-N-[2-(pyridin-2- yl)ethyl]ethanamin trihydroclorid và 0,00064 % betahistin dihydroclorid chuẩn trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh C (5 µm) (Cột Zorbax XDB Eclipse là phù hợp).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 254 nm.
Tốc độ dòng: 2 ml/min.
Thể tích tiêm: 20 µl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 4 lần thời gian lưu của betahistin (thời gian lưu của betahistin khoảng 5 min).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (4), độ phân giải giữa 2 pic chính không nhỏ hơn 3,0.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Diện tích của pic tương ứng với pic tạp chất C không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (2 %).
Diện tích của pic tương ứng với pic tạp chất A không được lớn hơn 2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (3) (0,2 %).
Diện tích của bất kỳ pic phụ nào không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,2 %).
Tổng diện tích của các pic tương ứng với tạp chất A, tạp chất C và tất cả các pic phụ khác không được lớn hơn 10 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (2 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,05 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch thử (0,05 %).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đối chiếu (4) và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 32 mg betahistin dihydroclorid, thêm 50 ml pha động và lắc trong 10 min, thêm pha động vừa đủ 100,0 ml, trộn đều. Ly tâm và sử dụng dịch trong phía trên.
Dung dịch chuẩn: Dung dịch chứa 0,032 % betahistin dihydroclorid chuẩn trong pha động.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (4), độ phân giải giữa 2 pic chính không nhỏ hơn 3,0.
Tính hàm lượng phần trăm của betahistln dihydroclorid, C8H12N2.2HCl, trong chế phẩm dựa vào diện tích pic betahistin dihydroclorid thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C8H12N2.2HCl trong betahistin dihydroclorid chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng và ẩm.
Loại thuốc
Thuốc kháng histamin H1.
Hàm lượng thường dùng
8 mg; 16 mg; 24 mg.
VIÊN NÉN BISOPROLOL FUMARAT
Là viên nén chứa bisoprolol fumarat. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng bisoprolol fumarat, (C18H31NO4)2.C4H4O4, từ 90,0 % đến 105,0 % so với lượng ghi trên nhãn.
Định tính
A. Cân chính xác một lượng bột viên tương ứng với khoảng 10 mg bisoprolol fumarat vào bình định mức 100 ml, thêm 60 ml methanol (TT), lắc mạnh 10 min và thêm methanol (TT) đến vạch. Lọc qua màng lọc 0,45 µm. Phổ hấp thụ tử ngoại (Phụ lục 4.1) của dung dịch thu được phải cho cực đại hấp thụ ở bước sóng trong khoảng 271 nm đến 275 nm.
B. Trong mục Định lượng, pic chính thu được trên sắc ký đồ của dung dịch thử phải có thời gian lưu tương ứng với thời gian lưu của pic chính thu được trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml hỗn hợp đệm phosphat chuẩn 0,025 M - nước (1 : 1).
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định hàm lượng bisoprolol fumarat đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3). Pha động và điều kiện sắc ký như mô tả ở phần Định lượng.
Thể tích tiêm: 50 µl.
Dung dịch thử. Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 µm.
Dung dịch chuẩn: Chuẩn bị dung dịch chuẩn bisoprolol fumarat trong môi trường hòa tan có nồng độ tương ứng với nồng độ của dung dịch thử.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic bisoprolol không nhỏ hơn 3000; hệ số đối xứng của pic bisoprolol không quá 2,0; độ lệch chuẩn tương đối của diện tích pic blsoprolol thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính lượng bisoprolol fumarat, (C18H31NO4)2.C4H4O4, đã hòa tan dựa vào diện tích pic bisoprolol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và nồng độ của bisoprolol fumarat trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng bisoprolol tumarat, (C18H31NO4)2.C4H4O4, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3). Pha động, điều kiện sắc ký như mô tả ở phần Định lượng.
Dung dịch thử. Lắc mạnh một lượng bột viên tương ứng khoảng 5 mg bisoprolol fumarat với 20,0 ml hỗn hợp nước - acetonitril (3:1) trong 10 min. Lọc qua màng lọc 0,45 µm.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng hỗn hợp nước - acetonitril (3 : 1).
Dung dịch đối chiếu (2): Pha loãng 2,0 ml dung dịch đối chiếu (1) thành 20,0 ml bằng hỗn hợp nước - acetonitril (3 : 1).
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 5 lần thời gian lưu của bisoprolol.
Trên sắc ký đồ của dung dịch đối chiếu (2) diện tích pic bisoprolol thu được phải bằng 7 % đến 13 % diện tích pic bisoprolol thu được trên sắc ký đồ dung dịch đối chiếu (1).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), số đĩa lý thuyết tính trên pic bisoprolol không nhỏ hơn 5000; hệ số đối xứng của pic bisoprolol không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic bisoprolol thu được từ 6 lần tiêm lặp lại không lớn hơn 1,5 %.
Tính hàm lượng tạp chất bằng phương pháp chuẩn hóa. Nhân diện tích pic tạp chất có thời gian lưu tương đối so với bisoprolol khoảng 1,2 với hệ số hiệu chỉnh là 5. Bỏ qua pic acid fumaric,
Giới hạn:
Tạp chất có thời gian lưu tương đối so với bisoprolol khoảng 1,2 và 3,8: Với mỗi tạp chất, không được quá 1,0 %.
Các tạp chất khác: Với mỗi tạp chất, không được quá 0,2 %.
Tổng tạp chất: Không được quá 2,5 %.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9, phương pháp đồng đều hàm lượng.
Phương pháp sắc ký lỏng (Phụ lục 5.3). Pha động, dung dịch chuẩn và điều kiện sắc ký như mô tả ở phần Định lượng.
Dung dịch thử. Lấy 1 viên cho vào bình định mức phù hợp, thêm một lượng hỗn hợp nước - acetonitril (3:1) bằng khoảng 2/3 thể tích bình, lắc mạnh 10 min, thêm hỗn hợp nước - acetonitril (3:1) đến vạch, thu được dung dịch có nồng độ tương ứng với nồng độ dung dịch chuẩn. Lọc qua màng lọc 0,45 µm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Hòa tan 4,08 g kali dihydrophosphat (TT) trong 1000 ml nước, điều chỉnh đến pH 2,5 bằng acid phosphoric (TT). Trộn đều 800 ml dung dịch thu được với 200 ml acetonitril (TT).
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 20 mg bisoprolol fumarat, thêm 70 ml hỗn hợp nước - acetonitril (3 : 1) lắc mạnh 10 min và pha loãng thành 100,0 ml bằng hỗn hợp nước - acetonitril (3 : 1). Lọc qua màng lọc 0,45 µm.
Dung dịch chuẩn: Cân chính xác khoảng 20 mg bisoprolol fumarat chuẩn vào bình định mức dung tích 100 ml, thêm 70 ml hỗn hợp nước - acetonitril (3 : 1), lắc cho tan và thêm hỗn hợp nước - acetonitril (3 : 1) đến định mức, lắc đều.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh B (5 µm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 225 nm.
Tốc độ dòng: Điều chỉnh tốc độ dòng để thu được thời gian lưu của pic bisoprolol khoảng 8 min.
Thể tích tiêm: 20 µl.
Cách tiến hành:
Thứ tự rửa giải của các pic lần lượt là: acid fumaric, bisoprolol.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, độ lệch chuẩn tương đối của diện tích pic bisoprolol thu được từ 6 lần tiêm lặp lại không lớn hơn 1,0 %.
Tính hàm lượng phần trăm của bisoprolol tumarat, (C18H31NO4)2.C4H4O4, trong chế phẩm dựa vào diện tích pic bisoprolol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của (C18H31NO4)2.C4H4O4 trong bisoprolol fumarat chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc chẹn beta1-adrenergic.
Hàm lượng thường dùng
2,5 mg; 5 mg.
VIÊN NÉN CARVEDILOL
Là viên nén chứa carvedilol.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng carvedilol, C24H26N2O4, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Lắc một lượng bột viên tương ứng với khoảng 12,5 mg carvedilol với 50 ml methanol (TT) trong 15 min, thêm methanol (TT) vừa đủ 100 ml, lọc. Pha loãng 10 ml dịch lọc thành 50 ml bằng methanol (TT).
Đo phổ hấp thụ tử ngoại và khả kiến (Phụ lục 4.1) của dung dịch thu được trong khoảng bước sóng từ 250 nm đến 400 nm, dùng cóng đo 1 cm mẫu trắng là methanol (TT). Song song tiến hành đo dung dịch chuẩn có nồng độ tương đương trong methanol (TT). Phổ hấp thụ tử ngoại của dung dịch thử và dung dịch chuẩn phải cho các cực đại và cực tiểu hấp thụ ở các bước sóng tương tự nhau.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch acid hydrocloric (TT) 0,7 % (tt/tt), điều chỉnh đến pH 1,45 ± 0,2 bằng dung dịch natri hydroxyd (TT) 50 %, đuổi khí,
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 µm.
Dung dịch chuẩn gốc: Cân chính xác khoảng 14 mg carvedilol chuẩn vào bình định mức dung tích 100 ml, thêm 5 ml methanol (TT), siêu âm để hòa tan. Để nguội về nhiệt độ phòng, sau đó thêm môi trường hòa tan đến vạch, trộn đều.
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc bằng môi trường hòa tan để thu được dung dịch có nồng độ 0,028 mg/ml (với viên 25 mg); 0,014 mg/ml (với viên 12,5 mg); 0,007 mg/ml (với viên 6,25 mg); 0,0035 mg/ml (với viên 3,125 mg).
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng 285 nm và 380 nm. Dùng cóng đo 1 cm, mẫu trắng là môi trường hòa tan.
Độ hấp thụ được hiệu chỉnh theo công thức sau:
Ahiệu chỉnh = A285 - A380
Trong đó:
Ahiệu chỉnh là độ hấp thụ đã được hiệu chỉnh của dung dịch chuẩn hoặc dung dịch thử.
A285 là độ hấp thụ của dung dịch chuẩn hoặc dung dịch thử ở bước sóng 285 nm.
A380 là độ hấp thụ của dung dịch chuẩn hoặc dung dịch thử ở bước sóng 380 nm.
Tính lượng carvedilol, C24H26N2O4, đã hòa tan dựa vào độ hấp thụ đã được hiệu chỉnh của dung dịch chuẩn, dung dịch thử và hàm lượng C24H26N2O4 trong carvedilol chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng carvedilol, C24H26N2O4, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều (Phụ lục 11.9)
Dung dịch đệm, pha động, dung môi pha mẫu, dung dịch chuẩn, điều kiện sắc ký và kiểm tra tính phù hợp của hệ thống như mô tả trong phần Định lượng,
Dung dịch thử: Lấy 1 viên cho vào bình định mức dung tích thích hợp để thu được dung dịch có nồng độ carvedilol khoảng 0,25 mg/ml, thêm nước khoảng 10 % thể tích bình, lắc cho viên rã. Thêm dung môi pha mẫu khoảng 75 % thể tích bình và siêu âm 30 min để viên rã hoàn toàn. Lắc bằng máy lắc cơ học trong 30 min, để nguội và thêm dung môi pha mẫu đến định mức, lắc đều. Ly tâm dung dịch trong 10 min ở tốc độ 2400 rpm, lấy 5 ml dịch trong thu được sau ly tâm vào bình định mức 100 ml. Thêm methanol 50 % (tt/tt) đến khoảng 85 % thể tích bình và siêu âm 20 min. Thêm methanol 50 % (tt/tt) đến vạch, lắc đều và lọc qua màng lọc 0,45 µm.
Tính hàm lượng phần trăm của carvedilol, C24H26N2O4, trong viên dựa vào diện tích pic carvedilol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C24H26N2O4 trong carvedilol chuẩn.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 0,7 g kali dihydrophosphat (TT) trong 500 ml nước, thêm 10 ml triethylamin (TT) điều chỉnh đến pH 3,0 ± 0,1 bằng acid phosphoric (TT).
Pha động: Hòa tan 1,04 g natri dodecyl sulfat (TT) trong 150 ml dung dịch đệm trong bình định mức dung tích 2000 ml, siêu âm. Thêm 720 ml acetonitril (TT) và thêm nước đến vạch. Lọc qua màng lọc nylon 66 kích thước lỗ lọc 0,2 µm.
Dung môi pha mẫu: Methanol - dung dịch acid hydrocloric 1 M (9 : 1).
Dung dịch thử gốc: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 25 mg carvedilol cho vào bình định mức 100 ml, thêm 10 ml nước, lắc đều sau đó thêm 70 ml dung môi pha mẫu và siêu âm 30 min. Lắc bằng máy lắc cơ học trong 30 min và thêm dung môi pha mẫu đến vạch. Ly tâm khoảng 50 ml dung dịch thu được trong 10 min ở tốc độ 2000 rpm.
Dung dịch thử. Pha loãng 25 ml dung dịch thử gốc thành 50 ml bằng nước. Lọc qua màng lọc 0,45 µm. Dung dịch đối chiếu (1): Cân chính xác khoảng 25 mg carvedilol chuẩn hòa tan trong hỗn hợp dung môi pha mẫu - nước (9 : 1) và pha loãng thành 100,0 ml với cùng dung môi. Pha loãng 5,0 ml dung dịch thu được thành 100,0 ml bằng methanol 50 % (tt/tt), dung dịch thu được chứa 0,0125 µg/ml carvedilol.
Dung dịch đối chiếu (2): Pha loãng dung dịch đối chiếu (1) bằng hỗn hợp dung môi pha mẫu - nước (1 : 1) để thu được dung dịch chứa 1,25 µg/ml carvedilol.
Điều kiện sắc ký:
Cột kích thước (5,0 cm x 4,6 mm) được nhồi pha tĩnh B (5 µm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 15 µl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ thu được của dung dịch đối chiếu (2), hệ số đối xứng của pic carvedilol không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic carvedilol thu được từ 6 lần tiêm lặp lại không lớn hơn 3,0 %.
Tiến hành sắc ký dung dịch thử với thời gian bằng 4 lần thời gian lưu của carvedilol.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic Carvedilol trên sắc ký đồ của dung dịch đối chiếu (2), nồng độ của carvedilol chuẩn trong dung dịch đối chiếu (2) và nồng độ lý thuyết của carvedilol trong dung dịch thử.
Giới hạn:
Từng tạp chất: Với mỗi tạp chất, không được quá 0,2 %.
Tổng các tạp chất: Không được quá 1,0 %.
Bỏ qua các tạp chất có hàm lượng dưới 0,05 % và các pic tạp chất có thời gian lưu tương đối so với pic carvedilol nhỏ hơn hoặc bằng 0,04.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm, pha động, dung môi pha mẫu, dung dịch thử gốc như mô tả ở phần Tạp chất liên quan.
Dung dịch thử. Pha loãng dung dịch thử gốc bằng methanol 50 % (tt/tt) để thu được dung dịch chứa 0,0125 mg/ml carvediol. Lọc qua màng lọc 0,45 µm, bỏ 5 ml dịch lọc đầu.
Dung dịch chuẩn: Dùng dung dịch đối chiếu (1) trong phần Tạp chất liên quan.
Điều kiện sắc ký.
Cột kích thước (5 cm x 4,6 mm) được nhồi pha tĩnh B (5 µm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 25 µl.
Thời gian chạy sắc ký: 30 min.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ thu được của dung dịch chuẩn, hệ số đối xứng của pic carvedilol không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic carvedilol thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Tính hàm lượng phần trăm của carvedilol, C24H26N2O4, trong chế phẩm dựa vào diện tích pic carvedilol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C24H26N2O4 trong carvedilol chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, tránh ẩm, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc chẹn beta-adrenergic.
Hàm lượng thường dùng
3,125 mg, 6,25 mg, 12,5 mg, 25 mg.
VIÊN NÉN CITICOLIN NATRI
Là viên nén chứa citicolin natri.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng citicolin, C14H26N4O11P2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml nước.
Tốc độ quay: 50 r/min.
Thời gian: 25 min.
Cách tiến hành:
Dung dịch thử. Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan, lọc. Pha loãng dịch lọc nếu cần với môi trường hòa tan để thu được dung dịch chứa 0,11 mg/ml citicolin.
Dung dịch chuẩn: Dung dịch chứa 0,11 mg/ml citicolin natri chuẩn trong môi trường hòa tan.
Tiến hành sắc ký lỏng như mô tả ở phần Định lượng.
Yêu cầu: Không ít hơn 80 % (Q) lượng citicolin, C14H26N4O11P2, so với lượng ghi trên nhãn được hòa tan trong 25 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hỗn hợp đồng thể tích của dung dịch kali dihydrophosphat (TT) 0,1 M và dung dịch tetrabutylamoni hydroxyd (TT) 0,01 M, điều chỉnh đến pH 4,5 bằng acid phosphoric (TT).
Pha động: Dung dịch đệm - methanol (95 : 5).
Dung dịch thử. Hòa tan một lượng bột viên trong nước để thu được dung dịch chứa 2,5 mg/ml citicolin natri và lọc, dùng dịch lọc làm dung dịch thử.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 500,0 ml bằng nước.
Dung dịch đối chiếu (2): Dung dịch chứa 7,5 µg/ml acid 5’-cytidylic chuẩn trong nước.
Dung dịch đối chiếu (3): Dung dịch chứa 0,125 mg/ml citicolin natri chuẩn và 0,125 mg/ml acid 5’- cytidylic chuẩn trong nước.
Điều kiện sắc ký:
Cột kích thước (25 cm x 4,0 mm) được nhồi pha tĩnh C.
Detector quang phổ từ ngoại đặt ở bước sóng 276 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 µl, riêng dung dịch đối chiếu (3) tiêm 20 µl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 2,5 lần thời gian lưu của pic citicolin.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic citlcolin và pic acid 5’-cytidylic không nhỏ hơn 1,5.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Acid 5’-cytidylic: Diện tích pic acid 5’-cytidylic không được lớn hơn diện tích pic acid 5’-cytidylic thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,3 %).
Tạp chất khác: Diện tích của bất kỳ pic tạp chất khác không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,2 %).
Tổng diện tích của tất cả các pic tạp chất, trừ acid 5’-cytidylic, không được lớn hơn 3,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,7 %).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch thử. Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 120 mg citicolin vào bình định mức 100 ml, thêm nước, lắc để hòa tan và thêm nước đến vạch, trộn đều và lọc. Pha loãng 10,0 ml dịch lọc thành 50,0 ml bằng nước.
Dung dịch chuẩn: Dung dịch chứa 0,25 mg/ml citicolin natri chuẩn trong nước.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic citicolin không nhỏ hơn 3000, hệ số đối xứng của pic citicolin không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic citicolin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của citicolin, C14H26N4O11P2, trong chế phẩm dựa vào diện tích pic citicolin natri thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C14H26N4O11P2 trong citlcolin natri chuẩn. Khối lượng phân tử của citicolin và citicolin natri lần lượt là 488,33 và 510,31.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Tăng chuyển hóa ở tế bào thần kinh.
Hàm lượng thường dùng
100 mg, 250 mg, 500 mg tính theo citicolin.
VIÊN NÉN CLOMIFEN
Là viên nén chứa clomifen citrat.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng clomifen citrat, C26H28CINO.C6H8O7, từ 93,0 % đến 107,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, sắc ký đồ của dung dịch thử phải cho 2 pic chính tương ứng với 2 pic đồng phân Z và E trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu giỏ quay.
Môi trường hòa tan: 900 ml nước.
Tốc độ quay: 100 r/min.
Thời gian: 30 min.
Cách tiến hành:
Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc thích hợp, bỏ vài mililít dịch lọc đầu. Pha loãng bằng dung dịch acid hydrocloric 0,1 M (TT) để được dung dịch có nồng độ clomifen citrat khoảng 14 µg/ml. Đo độ hấp thụ ánh sáng của dung dịch thu được ở bước sóng 232 nm (Phụ lục 4.1), cốc đo dày 1 cm. Tiến hành đo song song dung dịch chuẩn clomifen citrat có nồng độ tương đương nồng độ của dung dịch thử pha trong dung dịch acid hydrocloric 0,1 M (TT).
Yêu cầu: Không ít hơn 75 % (Q) clomifen citrat, C26H28CINO.C6H8O7, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Đồng phân Z
Từ 30 % đến 50 % hàm lượng clomifen citrat có trong chế phẩm.
Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động, dung dịch thử, điều kiện sắc ký và cách tiến hành như ở phần Định lượng.
Tính hàm lượng phần trăm của đồng phân Z trong chế phẩm dựa vào diện tích pic đồng phân Z so với tổng diện tích pic đồng phân E và Z trên sắc ký đồ thu được từ dung dịch thử.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3). Chuẩn bị các dung dịch trong các bình thủy tinh màu nâu và tránh ánh sáng.
Pha động: Methanol - nước - triethylamin (55 :45 :0,3), điều chỉnh đến pH 2,5 bằng acid phosphoric (TT).
Dung dịch thử: Cân 20 viên, tính khối lượng trung bình và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với 50 mg clomifen citrat vào bình định mức dung tích 100 ml, thêm 50 ml pha động, lắc siêu âm khoảng 30 min. Để nguội, thêm pha động đến vạch, lắc đều, lọc. Pha loãng 10,0 ml dịch lọc thành 100,0 ml bằng pha động.
Dung dịch chuẩn: Dung dịch clomifen citrat chuẩn (chứa đồng phân E và Z) trong pha động có nồng độ 0,05 mg/ml.
Điều kiện sắc ký.
Cột kích thước (25 cm x 4,6 mm) được nhồi pha tĩnh end-capped butylsilyl silica gel dùng cho sắc ký (5 µm).
Detector quang phổ tử ngoại đặt ở bước sóng 233 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 50 µl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, thời gian lưu tương đối của pic đồng phân E so với pic đồng phân Z khoảng 1,2; độ phân giải giữa pic đồng phân E và pic đồng phân Z không nhỏ hơn 1,5; số đĩa lý thuyết tính trên pic đồng phân E không nhỏ hơn 2000 và hệ số đối xứng của pic đồng phân E không lớn hơn 3,0. Độ lệch chuẩn tương đối của diện tích pic mỗi đồng phân E và Z thu được từ 6 lần tiêm lặp lại dung dịch chuẩn không lớn hơn 2,0 %.
Tính hàm lượng phần trăm clomifen citrat, C26H28CINO.C6H8O7, trong chế phẩm dựa vào tổng diện tích pic đồng phân E và Z thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng C26H28CINO.C6H8O7 của clomifen citrat chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc kháng estrogen/thuốc gây phóng noãn.
Hàm lượng thường dùng
50 mg.
VIÊN NÉN DESLORATADIN
Là viên nén chứa desloratadin. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng desloratadin, C19H19CIN2, từ 93,0 % đến 105,0 % so với lượng ghi trên nhãn.
Định tính
A. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
B. Trong phần Định lượng, phổ hấp thụ tử ngoại xác định trên pic chính của dung dịch thử phải có các cực đại hấp thụ và cực tiểu hấp thụ giống với phổ hấp thụ tử ngoại xác định trên pic chính của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 500 ml dung dịch acid hydrocloric 0,1 M (TT).
Tốc độ quay: 50 r/min.
Thời gian: 45 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan, lọc qua màng lọc 0,45 µm, bỏ 5 ml dịch lọc đầu.
Dung dịch chuẩn: Dung dịch chứa 0,01 mg/ml desloratadin chuẩn trong môi trường hòa tan.
Đo độ hấp thụ (Phụ lục 4.1) của các dung dịch thu được ở bước sóng 282 nm, trong cốc đo dày 1 cm. mẫu trắng là môi trường hòa tan.
Tính lượng desloratadin, C19H19CIN2, hòa tan dựa vào độ hấp thụ của dung dịch thử, dung dịch chuẩn và nồng độ của desloratadin trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng desloratadin so với lượng ghi trên nhãn được hòa tan trong 45 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động, dung môi pha mẫu, dung dịch chuẩn, điều kiện sắc ký và cách tiến hành như mô tả ở phần Định lượng.
Dung dịch thử: Lấy 1 viên cho vào bình định mức 50 ml, thêm 5 ml nước, lắc để viên rã. Thêm methanol (TT) đến khoảng 70 % thể tích bình, lắc siêu âm 10 min, để nguội và thêm methanol (TT) đến vạch. Lắc đều, ly tâm lấy dịch trong. Pha loãng 10,0 ml dung dịch thu được thành 50,0 ml bằng dung môi pha mẫu.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3). Bảo quản các dung dịch chứa desloratadin tránh ánh sáng.
Pha động A: Hòa tan 4,35 g dikali hydrophosphat (TT) trong 1 L nước, điều chỉnh đến pH 3,2 bằng acid phosphoric (TT).
Pha động B: Acetonitril (TT).
Pha động C: Methanol (TT).
Dung môi pha mẫu: Methanol - nước (90 : 10).
Dung dịch thử: Lấy 20 viên nén, cho vào bình định mức dung tích thích hợp. Thêm nước khoáng 10 % thể tích bình, để các viên rã. Thêm methanol (TT) khoảng 50 % thể tích bình, khuấy trong ít nhất 60 min. Để dung dịch về nhiệt độ phòng và thêm methanol (TT) đến vạch. Ly tâm dung dịch thu được, lấy phần dịch trong làm dung dịch thử (dung dịch chứa 0,2 mg/ml desloratadin).
Dung dịch đối chiếu (1): Dung dịch chứa 0,002 mg/ml desloratadin chuẩn và tạp chất F chuẩn của desloratadin trong dung môi pha mẫu.
Dung dịch đối chiếu (2): Dung dịch chứa 0,1 µg/ml desloratadin chuẩn trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh B (3 µm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 241 nm.
Tốc độ dòng: 1,0ml/min.
Thể tích tiêm: 20 µl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B | Pha động C |
(min) | (% tt/tt) | (% tt/tt) | (% tt/tt) |
0 | 70 | 15 | 15 |
12 | 70 | 15 | 15 |
30 | 40 | 30 | 30 |
45 | 40 | 30 | 30 |
47 | 70 | 15 | 15 |
55 | 70 | 15 | 15 |
Thời gian lưu tương đối so với desloratadin: Decloro desloratadin khoảng 0,37; dehydro desloratadin khoảng 1,4; tạp chất F khoảng 1,8; loratadin khoảng 2,7.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), số đĩa lý thuyết tính trên pic desloratadin không nhỏ hơn 1500; độ lệch chuẩn tương đối của diện tích pic desloratadin thu được từ 6 lần tiêm lặp lại không lớn hơn 5,0 %. Trên sắc ký đồ của dung dịch đối chiếu (2), tỷ số tín hiệu trên nhiễu không nhỏ hơn 10 đối với pic desloratadin.
Tính hàm lượng phần trăm của tạp chất F trong chế phẩm dựa vào diện tích pic tạp chất F trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (1) và nồng độ của tạp chất F trong dung dịch đối chiếu (1). Tính hàm lượng phần trăm mỗi tạp chất không xác định trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic desloratadin trên sắc ký đồ của dung dịch đối chiếu (1) và nồng độ của desloratadin trong dung dịch đối chiếu (1).
Giới hạn:
Tạp chất F: Không được quá 0,30 %.
Tạp chất không xác định: Mỗi tạp chất không được quá 0,2 %.
Tổng tạp chất: Không được quá 0,50 %.
Bỏ qua các tạp chất có hàm lượng dưới 0,05 % và các tạp chất decloro desloratadin, dehydro desloratadin, loratadin.
Ghi chú:
Decloro desloratadin: 6,11-Dihydro-11-(piperidin-4-yliden)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin.
Dehydro desloratadin: 8-Cloro-11-(piperidin-4-yliden)benzo[5,6]cyclohepta[1,2-b]pyridin.
Loratadin: 8-Cloro-6,11-dihydro-11-(1-ethoxycarbonylpiperidin-4-yliden)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin.
Tạp chất F: 8-Cloro-6,11-dihydro-11-(N-formyl-4-piperidinyliden)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3). Sử dụng bình thủy tinh màu.
Dung dịch đệm: Hòa tan 4,35 g dikali hydrophosphat (TT) trong 1 L nước, điều chỉnh đến pH 3,0 bằng acid phosphoric (TT).
Pha động: Methanol - dung dịch đệm (20 : 80).
Dung môi pha mẫu: Methanol - nước (90 : 10).
Dung dịch thử gốc: Lấy 20 viên nén, cho vào bình định mức dung tích thích hợp. Thêm nước khoảng 10 % thể tích bình, để các viên rã. Thêm methanol (TT) khoảng 50 % thể tích bình, khuấy trong ít nhất 60 min. Để dung dịch về nhiệt độ phòng và thêm methanol (TT) đến vạch. Ly tâm dung dịch thu được, lấy phần dịch trong làm dung dịch thử gốc (dung dịch chứa 0,2 mg/ml desloratadin).
Dung dịch thử: Pha loãng 5,0 ml dung dịch thử gốc thành 50,0 ml bằng dung môi pha mẫu.
Dung dịch chuẩn: Dung dịch chứa 0,02 mg/ml desloratadin chuẩn trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh nitril silica gel dùng cho sắc ký (5 µm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 241 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 µl.
Thời gian chạy sắc ký bằng 4,2 lần thời gian lưu của desloratadin.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic desloratadin không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic desloratadin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của C19H19CIN2 trong chế phẩm dựa vào diện tích pic desloratadin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C19H19CIN2 trong desloratadin chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc kháng histamin.
Hàm lượng thường dùng
5 mg.
VIÊN NÉN DIDANOSIN
Là viên nén chứa didanosin. Viên cần được nhai hoặc phân tán trong nước trước khi uống.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng didanosin, C10H12N4O3, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml nước.
Tốc độ quay: 75 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định lượng didanosin đã hòa tan bằng phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động và điều kiện sắc ký (không cần dùng cột bảo vệ) như mô tả ở phần Định lượng. Thể tích tiêm tương ứng với khoảng 2 µg didanosin.
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 µm. Dung dịch chuẩn gốc: Dung dịch chứa 0,8 mg/ml didanosin chuẩn trong nước (bảo quản ở 5 °C, dùng trong vòng 48 h sau khi pha).
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc bằng nước để thu được dung dịch có chứa (L/900) mg/ml didanosin, L là hàm lượng ghi trên nhãn của viên tính theo mg.
Thời gian chạy sắc ký ít nhất 7 min. Thời gian lưu của didanosin khoảng 4,8 min.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic didanosin không nhỏ hơn 1000; hệ số đối xứng của pic didanosin không lớn hơn 2; độ lệch chuẩn tương đối của diện tích pic didanosin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính lượng didanosin, C10H12N4O3, đã hòa tan dựa vào diện tích pic didanosin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C10H12N4O3 trong didanosin chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng didanosin, C10H12N4O3, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 0,77 g/l amoni acetat (TT) trong nước.
Pha động: Methanol - dung dịch đệm (1 : 99).
Dung dịch thử gốc: Cân 20 viên, tính khối lượng trung bình và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 5 viên nén, cho vào bình định mức 500 ml. Hòa tan trong 250 ml nước, và thêm nước đến vạch, lắc trong 10 min.
Dung dịch thử: Pha loãng dung dịch thử gốc bằng pha động để thu được dung dịch chứa 0,1 mg/ml didanosin.
Dung dịch đối chiếu gốc: Dung dịch chứa 0,125 mg/ml tạp chất A chuẩn của didanosin trong nước (dùng trong vòng 48 h sau khi pha).
Dung dịch đối chiếu: Pha loãng dung dịch đối chiếu gốc bằng nước để thu được dung dịch chứa 1,5 µg/ml tạp chất A chuẩn của didanosin (dùng trong vòng 48 h kể từ khi pha dung dịch đối chiếu gốc).
Điều kiện sắc ký:
Cột kích thước (12,5 cm x 4 mm) được nhồi pha tĩnh B (5 µm). Dùng thêm cột bảo vệ thích hợp. Detector quang phổ tử ngoại đặt ở bước sóng 275 nm.
Tốc độ dòng: 2 ml/min.
Thể tích tiêm: 100 µl.
Cách tiến hành:
Thời gian chạy sắc ký khoảng 30 min.
Thời gian lưu của tạp chất A khoảng 1,5 min đến 2,5 min.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, số đĩa lý thuyết tính trên pic tạp chất A không nhỏ hơn 1000; độ lệch chuẩn tương đối của diện tích pic tạp chất A thu được từ 6 lần tiêm lặp lại không lớn hơn 5,0 %.
Tính hàm lượng phần trăm của tạp chất A trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất A trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu và nồng độ của tạp chất A trong dung dịch đối chiếu.
Tính hàm lượng phần trăm của các tạp chất khác trong chế phẩm, nếu có, dựa theo phương pháp chuẩn hóa.
Giới hạn:
Tạp chất A: Không được quá 0,7 %.
Tạp chất khác: Với mỗi tạp chất, không được quá 0,2 %.
Tổng tạp chất (trừ tạp chất A): Không được quá 1,2 %.
Ghi chú:
Tạp chất A: Hypoxanthin.
Mất khối lượng do làm khô
Không được quá 6 % (Phụ lục 9.6).
(Lấy 4 viên nén; 130 °C; 16 h).
Khả năng trung hòa acid
Không dưới 17 mEq/viên.
Cân ít nhất 20 viên, xác định khối lượng trung bình viên, nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khối lượng trung bình của một viên vào bình nón dung tích 250 ml. Nếu cần có thể làm ẩm bột viên bằng 5 ml ethanol 96 % (TT) (đã được chỉnh đến pH 3,5), trộn đều để làm ẩm bột, thêm 70 ml nước và khuấy từ trong 1 min. Thêm 30,0 ml dung dịch acid hydrocloric 1 N (CĐ) vào mẫu thử, vừa thêm vừa khuấy từ liên tục (nếu khả năng trung hòa acid của mẫu thử lớn hơn 25 mEq, dùng 60,0 ml dung dịch acid hydrocloric 1 N (CĐ) và tiến hành điều chỉnh công thức tính cho phù hợp). Khuấy từ trong chính xác 15 min, sau đó ngay lập tức chuẩn độ acid hydrocloric dư bằng dung dịch natri hydroxyd 0,5 N (CĐ) đến pH 3,5 (ổn định trong 10 s đến 15 s), thời gian chuẩn độ không vượt quá 5 min.
Tính số mEq acid đã dùng theo công thức:
Tổng mEq = (30 x NHCl) - (VNaOH x NNaOH)
Trong đó:
NHCl : Nồng độ thực của dung dịch acid hydrocloric.
VNaOH: Thể tích dung dịch natri hydroxyd đã dùng.
NNaOH: Nồng độ thực của dung dịch natri hydroxyd.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 0,77 g/l amoni acetat (TT) trong nước.
Pha động: Methanol - dung dịch đệm (5 : 95).
Dung dịch thử gốc: Dùng dung dịch thử gốc ở phần Tạp chất liên quan.
Dung dịch thử: Pha loãng dung dịch thử gốc bằng nước để thu được dung dịch chứa 0,1 mg/ml didanosin (dùng trong vòng 72 h sau khi pha).
Dung dịch chuẩn: Dung dịch chứa 0,1 mg/ml didanosin chuẩn trong nước (dùng trong vòng 24 h sau khi pha).
Điều kiện sắc ký:
Cột kích thước (12,5 cm x 4 mm) được nhồi pha tĩnh B (5 µm), Dùng thêm cột bảo vệ thích hợp. Detector quang phổ tử ngoại đặt ở bước sóng 275 nm.
Tốc độ dòng: 2 ml/min.
Thể tích tiêm: 20 µl.
Cách tiến hành:
Thời gian lưu của didanosin lớn hơn 3,0 min.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic didanosin không nhỏ hơn 2000; độ lệch chuẩn tương đối của diện tích pic didanosin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của didanosin, C10H12N4O3, trong chế phẩm dựa vào diện tích pic didanosin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C10H12N4O3 trong didanosin chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng.
Loại thuốc
Thuốc kháng retrovirus nucleosid ức chế enzym phiên mã ngược.
Hàm lượng thường dùng
25 mg; 100 mg.
VIÊN NÉN FLUNARIZIN
Là viên nén chứa flunarizin hydroclorid. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng flunarizin, C26H26F2N2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Lắc một lượng bột viên tương ứng khoảng 50 mg flunarizin hydroclorid với 10 ml ethanol (TT) và lọc. Lấy 2 ml dịch lọc, thêm 2 giọt dung dịch kali hydroxyd (TT) 6,5 %, trộn đều, thêm tiếp 1 đến 2 giọt dung dịch kali permanganat (TT) 0,02 M, màu tím biến mất.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
C. Trong phần Định lượng, phổ hấp thụ tử ngoại (Phụ lục 4.1) của dung dịch thử phải cho cực đại hấp thụ ở bước sóng 226 nm và 253 nm; cực tiểu hấp thụ ở bước sóng 221 nm và 234 nm.
D. Dịch lọc thu được ở mục Định tính A phải cho phản ứng (A) của ion clorid (Phụ lục 8.1).
Độ hòa tan
Thiết bị: Kiểu giỏ quay.
Môi trường hòa tan: 600 ml dung dịch acid hydrocloric được pha như sau: Pha loãng 24 ml dung dịch acid hydrocloric 10 % (TT) thành 1000 ml bằng nước.
Tốc độ quay: 100 r/min.
Thời gian: 30 min.
Cách tiến hành:
Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan, lọc. Đo độ hấp thụ (Phụ lục 4.1) của dịch lọc ở bước sóng 253 nm, trong cốc đo dày 1 cm, mẫu trắng là môi trường hòa tan.
Tính lượng flunarizin, C26H26F2N2, đã hòa tan trong mỗi viên theo A (1 %, 1 cm), lấy 439 là giá trị A (1 %, 1 cm) của flunarizin hydroclorid ở bước sóng 253 nm.
1 mg flunarizin hydroclorid tương đương với 0,8473 mg flunarizin.
Yêu cầu: Không ít hơn 80 % (Q) lượng flunarizin, C26H26F2N2, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 14.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3). Dung dịch acid hydrocloric, dung dịch chuẩn, pha động, điều kiện sắc ký và cách tiến hành như mô tả ở phần Định lượng.
Dung dịch thử: Cho 1 viên vào bình định mức 100 ml, thêm 5 ml ethanol (TT) và 5 ml dung dịch acid hydrocloric, siêu âm để hòa tan, để nguội, thêm dung dịch acid hydrocloric đến vạch, lắc đều và lọc. Pha loãng dịch lọc bằng dung dịch acid hydrocloric để thu được dung dịch có nồng độ flunarizin khoảng 10 µg/ml.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 1,36 g kali dihydrophosphat (TT) trong 1000 ml nước, thêm 4 ml triethylamin (TT), điều chỉnh đến pH 3,5 bằng acid phosphoric (TT).
Pha động: Methanol - dung dịch đệm (75 : 25).
Dung dịch acid hydrocloric: Pha loãng 24 ml dung dịch acid hydrocloric 10 % (TT) thành 1000 ml bằng nước.
Dung dịch thử: Cân 20 viên, tính khối lượng trung bình viên và nghiền thành bột mịn. Cân một lượng bột viên tương ứng khoảng 10 mg flunarizin vào bình định mức 100 ml, thêm 10 ml ethanol (TT) và 5 ml dung dịch acid hydrocloric, siêu âm để hòa tan, để nguội, thêm dung dịch acid hydrocloric đến vạch, lắc đều và lọc. Pha loãng 5,0 ml dịch lọc thành 50,0 ml bằng dung dịch acid hydrocloric.
Dung dịch chuẩn: Cân chính xác một lượng flunarizin chuẩn hòa tan trong 10 ml ethanol (TT) và pha loãng bằng dung dịch acid hydrocloric để thu được dung dịch có nồng độ flunarizin khoảng 10 μg/ml.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,0 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 253 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa ký thuyết tính trên pic flunarizin không nhỏ hơn 3000, độ phân giải giữa pic flunarizin và pic tạp liền kề (nếu có) không nhỏ hơn 1,5.
Tính hàm lượng phần trăm của flunarizin, C26H26F2N2, trong chế phẩm dựa vào diện tích pic flunarizin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C26H26F2N2 trong flunarizin chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc chẹn kênh calci.
Hàm lượng thường dùng
5 mg.
VIÊN NÉN LAMOTRIGIN
Là viên nén chứa lamotrigin.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng lamotrigin, C9H7Cl2N5, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Phổ hấp thụ tử ngoại (Phụ lục 4.1).
Mẫu thử: Hòa tan một lượng bột viên trong dung dịch acid hydrocloric 0,01 M (TT) để thu được dung dịch chứa lamotrigin 0,02 mg/ml.
Mẫu chuẩn: Dung dịch chứa 0,02 mg/ml lamotrigin chuẩn trong dung dịch acid hydrocloric 0,01 M (TT). Phổ hấp thụ tử ngoại của mẫu thử và mẫu chuẩn phải cho các cực đại và cực tiểu hấp thụ tương tự nhau.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch acid hydrocloric 0,1 M (TT).
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định lượng lamotrigin đã hòa tan bằng 1 trong 2 phương pháp sau:
A. Phương pháp quang phổ hấp thụ tử ngoại và khả kiến (Phụ lục 4.1)
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm, bỏ vài mililít dịch lọc đầu. Sử dụng luôn dịch lọc (với viên 25 mg) hoặc pha loãng dịch lọc thu được bằng môi trường hòa tan để thu được dung dịch có chứa nồng độ lamotrigin khoảng 0,028 mg/ml.
Dung dịch chuẩn gốc: Cân chính xác một lượng lamotrigin chuẩn vào bình định mức thích hợp, thêm methanol (TT) khoảng 5 % thể tích bình, lắc để hòa tan sau đó thêm môi trường hòa tan đến vạch để thu được dung dịch có nồng độ lamotrigin 0,14 mg/ml.
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc bằng môi trường hòa tan để thu được dung dịch có nồng độ lamotrigin 0,028 mg/ml.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng 267 nm. Mẫu trắng là môi trường hòa tan.
Tính lượng lamotrigin, C9H7Cl2N5, đã hòa tan dựa vào độ hấp thụ của dung dịch chuẩn, dung dịch thử và hàm lượng C9H7Cl2N5 trong lamotrigin chuẩn.
B. Phương pháp sắc ký lỏng (Phụ lục 5.3)
Dung dịch đệm và pha động như mô tả ở mục Định lượng.
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm.
Dung dịch chuẩn gốc: Cân chính xác một lượng lamotrigin chuẩn vào bình định mức thích hợp, thêm methanol (TT) khoảng 15 % thể tích bình, lắc để hòa tan sau đó thêm môi trường hòa tan đến vạch để thu được dung dịch có nồng độ lamotrigin 0,5 mg/ml.
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc để thu được dung dịch chứa lamotrigin chuẩn có nồng độ khoảng (L/1000) mg/ml trong môi trường hòa tan, trong đó L là hàm lượng trên nhãn của viên tính theo mg.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 310 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 10 μl với viên hàm lượng 100 mg, 150 mg và 200 mg) và 50 μl với viên hàm lượng 25 mg và 50 mg).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic lamotrigin không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic lamotrigin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính lượng lamotrigin, C9H7Cl2N5, đã hòa tan dựa vào diện tích pic lamotrigin thu được trên sắc ký đồ của dụng dịch thử, dung dịch chuẩn và hàm lượng của C9H7Cl2N5 trong lamotrigin chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng lamotrigin, C9H7Cl2N5, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch 0,8 g/l amoni acetat (TT) trong nước, điều chỉnh đến pH 4,5 bằng acid acetic băng (TT).
Pha động: Acetonitril - methanol - dung dịch đệm (10 : 30 : 60).
Dung môi pha mẫu: Methanol - dung dịch đệm (60 : 40).
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 100 mg lamotrigin cho vào bình định mức 250 ml, thêm pha động khoảng 70 % thể tích bình. Siêu âm trong 30 min, thi thoảng lắc đều. Thêm dung môi pha mẫu đến vạch và lọc qua màng lọc 0,45 μm.
Dung dịch đối chiếu: Dung dịch chứa 1,0 μg/ml lamotrigin chuẩn trong dung môi pha mẫu.
Dung dịch phù hợp hệ thống: Dung dịch chứa 1 μg/ml tạp chất E chuẩn của lamotrigin và 0,4 mg/ml lamotrigin chuẩn trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 210 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 5 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic tạp chất E và pic lamotrigin ít nhất là 2,0. Trên sắc ký đồ của dung dịch đối chiếu, hệ số đối xứng của pic lamotrigin không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic lamotrigin thu được từ 6 lần tiêm lặp lại không lớn hơn 10,0 %.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic lamotrigin trên sắc ký đồ của dung dịch đối chiếu và nồng độ lamotrigin trong dung dịch đối chiếu. Chia diện tích pic tạp chất cho hệ số đáp ứng tương đối F nếu có.
Thời gian lưu tương đối, hệ số đáp ứng tương đối và giới hạn phần trăm của các tạp chất được trình bày trong Bảng 1.
Bảng 1
Tạp chất | Thời gian lưu tương đối | Hệ số đáp ứng tương đối (F) | Giới hạn (%) |
Tạp chất E | 0,67 | 0,75 | 0,2 |
Lamotrigin | 1,0 | - | - |
Tạp chất A | 1,5 | 1,0 | 0,5 |
Tạp chất khác | - | 1,0 | 0,2 |
Tổng tạp | - | - | 0,75 |
Ghi chú:
Tạp chất E: Acid 2,3-diclorobenzoic.
Tạp chất A: 3-amino-6-(2,3-diclorophenyl)-1,2,4-triazin-5(4H)-on.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch 0,8 g/l amoni acetat (TT) trong nước, điều chỉnh đến pH 4,5 bằng acid acetic băng (TT).
Pha động: Methanol - dung dịch đệm (60 : 40).
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 100 mg lamotrigin cho vào bình định mức 100 ml, thêm 70 ml pha động, siêu âm trong 20 min và thêm pha động đến vạch. Ly tâm dung dịch thu được. Pha loãng dịch trong thu được sau ly tâm bằng pha động để thu được dung dịch chứa 0,05 mg/ml lamotrigin.
Dung dịch chuẩn: Dung dịch chứa 0,05 mg/ml lamotrigin chuẩn trong pha động.
Điều kiện sắc ký.
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 210 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 10 μl.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic lamotrigin không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic lamotrigin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của lamotrigin, C9H7Cl2N5, trong chế phẩm dựa vào diện tích pic lamotrigin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C9H7Cl2N5 trong lamotrigin chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc chống động kinh.
Hàm lượng thường dùng
25 mg; 50 mg; 100 mg; 150 mg; 200 mg.
VIÊN NÉN LEVETIRACETAM
Là viên nén chứa levetiracetam. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng levetiracetam, C8H14N2O2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2).
Lấy một lượng bột viên tương ứng với khoảng 250 mg levetiracetam vào bình định mức 50 ml, thêm khoảng 35 ml aceton (TT), siêu âm 15 min và thêm aceton (TT) đến vạch, lắc đều. Lấy 10 ml lọc qua màng lọc 0,45 μm và làm bay hơi aceton hoàn toàn tạo thành tinh thể. Cạo lấy tinh thể, chuẩn bị mẫu đo dạng đĩa nén kali bromid bằng cách nghiền 2 mg đến 4 mg tinh thể với 200 mg kali bromid (TT). Đo phổ hấp thụ hồng ngoại trong khoảng từ 4000 cm-1 đến 650 cm-1.
Phổ hấp thụ hồng ngoại của mẫu thử phải phù hợp với phổ hấp thụ hồng ngoại của mẫu chuẩn được chuẩn bị tương tự từ dung dịch chuẩn levetiracetam nồng độ 1mg/ml trong aceton (TT).
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml nước.
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định lượng levetiracetam đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm pH 5,6: Dung dịch chứa 6,8 g/L kali dihydrophosphat (TT) trong nước, điều chỉnh đến pH 5,6 bằng dung dịch kali hydroxyd (TT) 10 %.
Pha động: Acetonitril - dung dịch đệm (15 : 85).
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm.
Dung dịch chuẩn: Dung dịch chứa (L/1000) mg/ml levetiracetam trong môi trường hòa tan, L là hàm lượng ghi trên nhãn của viên tính theo mg.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 220 nm.
Tốc độ dòng: 1,2 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic levetiracetam không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic levetiracetam thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính lượng levetiracetam, C8H14N2O2, đã hòa tan dựa vào diện tích pic levetiracetam thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và nồng độ của levetiracetam trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng levetiracetam, C8H14N2O2, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 6,8 g/L kali dihydrophosphat (TT) và 0,85 g/L natri 1-heptansulfonat (TT) trong nước, điều chỉnh đến pH 2,8 bằng acid phosphoric (TT).
Pha động: Acetonitril - dung dịch đệm (5 : 95).
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên vào bình định mức thích hợp và hòa tan bằng pha động để thu được dung dịch có chứa 1,2 mg/ml levetiracetam (siêu âm để hòa tan nếu cần, ly tâm dung dịch trước khi lọc qua màng lọc).
Dung dịch đối chiếu (1): Dung dịch chứa 3,6 μg/ml levetiracetam chuẩn trong pha động.
Dung dịch đối chiếu (2): Dung dịch chứa 3,6 μg/ml levetiracetam chuẩn và 3,6 μg/ml tạp chất G chuẩn của levetiracetam trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (4 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 200 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải giữa pic tạp chất G và pic levetiracetam không nhỏ hơn 2,0. Trên sắc ký đồ của dung dịch đối chiếu (1), hệ số đối xứng của pic levetiracetam không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic levetiracetam thu được từ 6 lần tiêm lặp lại không lớn hơn 10,0 %.
Thời gian lưu tương đối so với levetiracetam: Tạp chất G khoảng 0,54; tạp chất I khoảng 1,7; levetiracetam acid khoảng 2,1.
Tính hàm lượng phần trăm của từng tạp chất, nếu có, trong chế phẩm dựa vào diện tích pic tạp chất thu được trên sắc ký đồ của dung dịch thử, diện tích pic levetiracetam trên sắc ký đồ của dung dịch đối chiếu (1) và nồng độ của levetiracetam trong dung dịch đối chiếu (1), chia diện tích pic levetiracetam acid cho hệ số đáp ứng tương đối bằng 0,79.
Giới hạn:
Levetiracetam acid: Không được quá 0,3 %.
Tạp chất khác: Mỗi tạp chất, không được quá 0,1 %.
Tổng tạp chất: Không được quá 0,6 %.
Bỏ qua tạp chất I và tạp chất G.
Ghi chú:
Levetiracetam acid: Acid (S)-2-(2-oxopyrrolidin-1-yl)butanoic.
Tạp chất G: (2S)-2-aminobutanamid.
Tạp chất I: (S)-N-(1-amino-1-oxobutan-2-yl)-4-clorobutanamid.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 1,4 g/L kali dihydrophosphat (TT) và 0,6 g/L natri 1-heptansulfonat (TT) trong nước, điều chỉnh đến pH 2,8 bằng acid phosphoric (TT).
Pha động: Acetonitril - dung dịch đệm (8 : 92).
Dung môi pha mẫu: Acetonitril - nước (20 : 80).
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên vào bình định mức thích hợp và hòa tan bằng dung môi pha mẫu để thu được dung dịch có chứa 0,4 mg/ml levetiracetam (siêu âm để hòa tan nếu cần).
Dung dịch chuẩn: Dung dịch chứa 0,4 mg/ml levetiracetam chuẩn trong dung môi pha mẫu (siêu âm để hòa tan nếu cần).
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (4 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 220 nm.
Tốc độ dòng: 2,0 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic levetiracetam không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic levetiracetam thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của levetiracetam, C8H14N2O2, trong chế phẩm dựa vào diện tích pic levetiracetam thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C8H14N2O2 trong levetiracetam chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc chống động kinh.
Hàm lượng thường dùng
250 mg, 500 mg, 750 mg, 1000 mg.
VIÊN NÉN LEVOCETIRIZIN DIHYDROCLORID
Là viên nén chứa levocetirizin dihydroclorid.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng levocetirizin dihydroclorid, C21H25ClN2O3.2HCl, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Lấy 1 viên cho vào bình định mức có dung tích thích hợp để được dung dịch có nồng độ 0,1 mg/ml levocetirizin dihydroclorid, thêm nước khoảng 40 % thể tích bình. Lắc để viên rã hoàn toàn, thêm nước đến vạch. Lọc dung dịch thu được qua màng lọc thích hợp, bỏ vài mililít dịch lọc đầu.
Song song tiến hành chuẩn bị dung dịch chuẩn, thay chế phẩm bằng levocetirizin dihydroclorid chuẩn. Phổ hấp thụ tử ngoại (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn phải cho các cực đại và cực tiểu hấp thụ tương tự nhau.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml nước.
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc thích hợp, bỏ vài mililít dịch lọc đầu.
Dung dịch chuẩn: Dung dịch chứa levocetirizin dihydroclorid chuẩn trong môi trường hòa tan có nồng độ (L/900) mg/ml, L là hàm lượng ghi trên nhãn của viên tính theo mg.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng 230 nm hoặc 231 nm. Mẫu trắng là môi trường hòa tan. Dùng cốc đo 1 cm hoặc 2 cm.
Tính lượng levocetirizin dihydroclorid, C21H25ClN2O3.2HCl, đã hòa tan dựa vào độ hấp thụ của dung dịch chuẩn, dung dịch thử và hàm lượng C21H25ClN2O3.2HCl trong levocetirizin dihydroclorid chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng levocetirizin dihydroclorid, C21H25ClN2O3.2HCl, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động, dung dịch chuẩn, điều kiện sắc ký và cách tiến hành như mô tả ở phần Định lượng.
Dung dịch thử: Lấy 1 viên hòa tan bằng pha động trong bình định mức thích hợp để thu được dung dịch có nồng độ levocetirizin dihydroclorid 0,2 mg/ml. Ly tâm lấy phần dịch trong ở phía trên.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - nước - dung dịch acid sulfuric 1 M (93 : 6,6 : 0,4).
Dung dịch A: Dung dịch acid sulfuric 1 M - nước (5,7 : 94,3).
Dung dịch thử: Cho một lượng viên thích hợp (không ít hơn 10 viên) tương đương với 50 mg levocetirizin dihydroclorid vào bình định mức 250 ml. Thêm 20 ml dung dịch A, lắc bằng máy lắc cơ học 15 min. Thêm 150 ml acetonitril (TT), siêu âm 10 min. Để nguội bình về nhiệt độ phòng và thêm acetonitril (TT) đến vạch, lắc đều. Ly tâm dung dịch thu được trong 5 min, lấy phần dịch trong phía trên.
Dung dịch phù hợp hệ thống: Dung dịch chứa 0,2 mg/ml levocetirizin dihydroclorid chuẩn và 0,2 μg/ml clorobenzhydryl piperazin chuẩn trong pha động.
Dung dịch đối chiếu: Dung dịch chứa 0,002 mg/ml levocetirizin dihydroclorid chuẩn trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh A (5 μm).
Cột bảo vệ (0,3 cm × 4 mm) được nhồi pha tĩnh A (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 230 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 3 lần thời gian lưu của levocetirizin.
Thời gian lưu tương đối so với levocetirizin của clorobenzhydryl piperazin khoảng 1,4; của levocetirizin amid khoảng 2,1.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic levocetirizin và pic clorobenzhydryl piperazin không nhỏ hơn 3,0; hệ số đối xứng của pic levocetirizin không lớn hơn 1,5 và độ lệch chuẩn tương đối của diện tích pic levocetirizin và diện tích pic clorobenzhydryl piperazin thu được từ 6 lần tiêm lặp lại lần lượt không lớn hơn 1,0 % và 5,0 %.
Tính hàm lượng phần trăm của các tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic levocetirizin trên sắc ký đồ của dung dịch đối chiếu và nồng độ của levocetirizin chuẩn trong dung dịch đối chiếu.
Giới hạn:
Mỗi tạp chất: Không được quá 0,30 %.
Tổng tạp chất: Không được quá 1,0 %.
Bỏ qua các pic nhỏ hơn 0,1 % và các pic tạp chất clorobenzhydryl piperazin và levocetirizin amid.
Ghi chú:
Clorobenzhydryl piperazin và levocetirizin amid là các tạp chất đã được kiểm soát ở nguyên liệu, chỉ đưa vào để định tính, không quy định trong thành phẩm.
Levocetirizin amid: (R)-2-(2-{4-[(4-Clorophenyl)phenylmethyl]piperazin-1-yl}ethoxy)acetamid.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch A, pha động, dung dịch phù hợp hệ thống, dung dịch thử và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch chuẩn: Dung dịch chứa 0,2 mg/ml levocetirizin dihydroclorid chuẩn trong pha động.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic levocetirizin và pic clorobenzhydryl piperazin không nhỏ hơn 3,0; hệ số đối xứng của pic levocetirizin không lớn hơn 1,5 và độ lệch chuẩn tương đối của diện tích pic levocetirizin thu được từ 6 lần tiêm lặp lại không lớn hơn 1,0 %.
Tính hàm lượng phần trăm của levocetirizin dihydroclorid, C21H25ClN2O3.2HCl, trong chế phẩm dựa vào diện tích pic levocetirizin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H25ClN2O3.2HCl trong levocetirizin dihydroclorid chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc kháng histamin.
Hàm lượng thường dùng
5 mg.
VIÊN NÉN LISINOPRIL
Là viên nén chứa lisinopril.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng lisinopril, C21H31N3O5, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong mục Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch acid hydrocloric 0,1 M (TT).
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định lượng lisinopril đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3). Pha động và điều kiện sắc ký như mô tả ở mục Định lượng.
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm.
Dung dịch chuẩn: Dung dịch chuẩn có nồng độ lisinopril trong môi trường hòa tan tương tự dung dịch thử.
Tính lượng lisinopril, C21H31N3O5, đã hòa tan dựa vào diện tích pic lisinopril thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H31N3O5 trong lisinopril chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng lisinopril, C21H31N3O5, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3). Dung dịch phosphat, pha động và điều kiện sắc ký như mô tả trong phần Định lượng.
Dung môi pha mẫu: Hòa tan 2,72 g kali dihydrophosphat (TT) trong 800 ml nước, điều chỉnh đến pH 4,0 bằng acid phosphoric (TT) sau đó thêm nước vừa đủ 1000 ml và trộn đều.
Dung dịch thử: Lấy 1 viên cho vào bình định mức dung tích thích hợp để thu được dung dịch có chứa 0,2 mg/ml lisinopril. Thêm một thể tích dung môi pha mẫu bằng 50 % thể tích bình, siêu âm 5 min và lắc bằng máy lắc cơ học 20 min. Thêm dung môi pha mẫu đến vạch, trộn đều và lọc.
Dung dịch chuẩn: Hòa tan một lượng lisinopril chuẩn trong dung môi pha mẫu để thu được dung dịch có chứa 0,2 mg/ml lisinopril.
Tính lượng lisinopril, C21H31N3O5, có trong mỗi viên dựa vào diện tích pic lisinopril trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H31N3O5 trong lisinopril chuẩn.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3). Dung dịch phosphat, pha động, dung môi pha mẫu và điều kiện sắc ký như mô tả trong mục Định lượng.
Dung dịch thử. Dùng dung dịch thử trong phần Định lượng.
Dung dịch đối chiếu: Pha loãng dung dịch chuẩn (trong phần Định lượng) bằng dung môi pha mẫu để thu được dung dịch có chứa 20 μg/ml lisinopril.
Cách tiến hành:
Tiến hành sắc ký với thời gian bằng ít nhất 3 lần thời gian lưu của lisinopril.
Trên sắc ký đồ của dung dịch thử, ghi lại diện tích pic đáp ứng của các pic tạp chất nếu có (bỏ qua các pic của dung môi).
Tính phần trăm tạp chất dựa vào tổng diện tích các pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic lisinopril trên sắc ký đồ của dung dịch đối chiếu và hàm lượng C21H31N3O5 trong lisinopril chuẩn.
Giới hạn:
Tổng tạp chất: Không được quá 2,0 %.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch phosphat: Hòa tan 4,1 g kali dihydrophosphat (TT) trong 900 ml nước, điều chỉnh đến pH 2,0 bằng acid phosphoric (TT) sau đó thêm nước vừa đủ 1000 ml.
Pha động: Hòa tan 1,0 g natri 1-hexansulfonat (TT) trong 820 ml dung dịch phosphat. Thêm 180 ml acetonitril (TT), trộn đều, lọc và đuổi khí.
Dung môi pha mẫu: Nước - methanol (4 : 1).
Dung dịch thử: Lấy 10 viên cho vào bình định mức dung tích thích hợp để thu được dung dịch có chứa 0,2 mg/ml lisinopril. Thêm dung môi pha mẫu, siêu âm 5 min và lắc bằng máy lắc cơ học 20 min. Thêm dung môi pha mẫu đến vạch, trộn đều và lọc.
Dung dịch chuẩn: Hòa tan một lượng lisinopril chuẩn trong dung môi pha mẫu để thu được dung dịch có chứa 0,2 mg/ml lisinopril.
Điều kiện sắc ký:
Cột kích thước (20 cm × 4,6 mm) được nhồi pha tĩnh B (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 215 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic lisinopril không nhỏ hơn 700; hệ số đối xứng của pic lisinopril không lớn hơn 2,0; hệ số phân bố khối lượng k’ của pic lisinopril phải lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic lisinopril thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của lisinopril, C21H31N3O5, trong chế phẩm dựa vào diện tích pic lisinopril trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H31N3O5 trong lisinopril chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt. Tránh ẩm, nóng và tránh bị đông lạnh.
Loại thuốc
Thuốc ức chế enzym chuyển angiotensin.
Hàm lượng thường dùng
5 mg, 10 mg.
VIÊN NÉN MIFEPRISTON
Là viên nén chứa mifepriston.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nên" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng mifepriston, C29H35NO2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Hòa tan một lượng bột viên trong methanol (TT) để thu được dung dịch chứa 0,001 % mifepriston, lọc. Đo phổ hấp thụ tử ngoại (Phụ lục 4.1) của dịch lọc thu được trong khoảng bước sóng từ 200 nm đến 400 nm, dung dịch phải cho hai cực đại hấp thụ ở bước sóng 260 nm và 303 nm.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch đối chiếu.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch chứa 1,0 % natri lauryl sulfat (TT) trong nước.
Tốc độ quay: 75 r/min.
Thời gian: 60 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc, bỏ vài mililít dịch lọc đầu. Pha loãng bằng môi trường hòa tan nếu cần để được dung dịch có nồng độ mifepriston tương đương dung dịch chuẩn.
Dung dịch chuẩn: Cân chính xác một lượng mifepriston chuẩn hòa tan trong một lượng thích hợp methanol (TT), sau đó pha loãng bằng môi trường hòa tan để được dung dịch có nồng độ mifepriston khoảng 0,01 mg/ml.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng 302 nm. Mẫu trắng là môi trường hòa tan.
Tính lượng mifepriston, C29H35NO2, đã hòa tan dựa vào độ hấp thụ của dung dịch thử, dung dịch chuẩn và hàm lượng của C29H35NO2 trong mifepriston chuẩn.
Yêu cầu: Không ít hơn 70 % (Q) lượng mifepriston, C29H35NO2, so với lượng ghi trên nhãn được hòa tan trong 60 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Methanol - acetonitril - nước - triethylamin (21 : 14 : 15 : 0,1), điều chỉnh đến pH 4,5 bằng acid phosphoric (TT).
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 50 mg mifepriston cho vào bình định mức 100 ml, thêm khoảng 20 ml pha động, siêu âm 15 min. Thêm pha động đến vạch và ly tâm. Pha loãng 5,0 ml dịch trong sau khi ly tâm thành 50,0 ml bằng pha động.
Dung dịch đối chiếu: Dung dịch chứa 0,005 % mitepriston chuẩn trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 302 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch thử và dung dịch đối chiếu.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, số đĩa lý thuyết tính theo pic mifepriston không nhỏ hơn 4000; hệ số đối xứng của pic mifepriston không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic mifepriston thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm tạp chất (nếu có) trên sắc ký đồ của dung dịch thử bằng phương pháp chuẩn hóa.
Giới hạn:
Tạp chất bất kỳ: Với mỗi tạp chất, không được quá 1,0 %.
Tổng các tạp chất: Không được quá 2,0 %.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả trong phần Tạp chất liên quan.
Tính hàm lượng phần trăm của mifepriston, C29H35NO2, trong chế phẩm dựa vào diện tích pic thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu và hàm lượng của C29H35NO2 trong mifepriston chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc tránh thai khẩn cấp (hàm lượng thấp).
Thuốc chấm dứt thai kỳ ở giai đoạn sớm (hàm lượng cao).
Hàm lượng thường dùng
10 mg; 25 mg; 200 mg.
VIÊN NÉN MONTELUKAST
Là viên nén chứa montelukast natri. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng montelukast, C35H36ClNO3S, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch 0,5 % natri lauryl sulfat (TT) trong nước.
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định lượng montelukast đã hòa tan bằng phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả ở mục Định lượng.
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan, lọc.
Dung dịch chuẩn: Dung dịch chứa montelukast natri chuẩn trong môi trường hòa tan có hàm lượng montelukast tương đương với dung dịch thử.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic montelukast không nhỏ hơn 2000, hệ số đối xứng của pic montelukast không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic montelukast thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Yêu cầu: Không ít hơn 80 % (Q) lượng montelukast, C35H36ClNO3S, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động, dung môi pha mẫu, dung dịch chuẩn, điều kiện sắc ký và cách tiến hành như mô tả ở phần Định lượng.
Dung dịch thử: Lấy 1 viên chuyển vào bình định mức thích hợp để thu được dung dịch có nồng độ montelukast khoảng 0,1 mg/ml, thêm một lượng dung môi pha mẫu bằng 2/3 thể tích bình, lắc cho viên rã và siêu âm 20 min ở nhiệt độ không quá 10 °C. Thêm dung môi pha mẫu đến vạch, lắc đều và lọc.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Hòa tan 6,0 g amoni acetat (TT) trong 1000 ml nước, điều chỉnh đến pH 5,5 bằng acid acetic (TT).
Pha động B: Methanol (TT).
Dung môi pha mẫu: Dung dịch triethylamin (TT) 0,1 % (tt/tt) trong methanol (TT).
Dung dịch thử: Cân chính xác một lượng bột viên tương ứng với khoảng 100 mg montelukast vào bình định mức 200 ml. Thêm 150 ml dung môi pha mẫu và siêu âm 20 min ở nhiệt độ không quá 10 °C. Thêm dung môi pha mẫu đến vạch và lọc.
Dung dịch đối chiếu (1): Dung dịch chứa 0,0025 % montelukast natri chuẩn trong dung môi pha mẫu.
Dung dịch đối chiếu (2): Pha loãng 5,0 ml dung dịch đối chiếu (1) thành 50,0 ml bằng dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 50 | 50 |
15 | 35 | 65 |
40 | 30 | 70 |
60 | 20 | 80 |
65 | 20 | 80 |
70 | 50 | 50 |
75 | 50 | 50 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ lệch chuẩn tương đối của diện tích pic montelukast thu được từ 6 lần tiêm lặp lại không lớn hơn 5,0 %.
Trên sắc ký đồ của dung dịch thử, tạp chất sulfoxid và tạp chất styren nếu có sẽ xuất hiện ở thời gian lưu tương đối so với montelukast lần lượt là khoảng 0,63 và 1,37.
Giới hạn:
Tạp chất sulfoxid: Diện tích pic tạp chất sulfoxid không được lớn hơn 2 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (1,0 %).
Tạp chất styren: Diện tích pic tạp chất styren không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,5 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,5 %).
Tổng diện tích pic của tất cả các tạp chất không được lớn hơn 4 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (2,0 %).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 3,85 g amoni acetat (TT) trong 1000 ml nước, thêm 1,0 ml triethylamin (TT), điều chỉnh đến pH 5,5 bằng acid acetic (TT).
Pha động: Dung dịch đệm - methanol (20 : 80).
Dung môi pha mẫu: Dung dịch triethylamin (TT) 0,1 % (tt/tt) trong methanol (TT).
Dung dịch thử: Cân 20 viên, tính khối lượng trung bình viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 10 mg montelukast vào bình định mức 100 ml, thêm 70 ml dung môi pha mẫu và siêu âm 20 min ở nhiệt độ không quá 10 °C. Thêm dung môi pha mẫu đến vạch, lắc đều và lọc.
Dung dịch chuẩn: Hòa tan 26 mg montelukast natri chuẩn trong 250,0 ml dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 240 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, số đĩa lý thuyết tính trên pic montelukast không nhỏ hơn 2000; hệ số đối xứng của pic montelukast không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic montelukast thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của montelukast, C35H36ClNO3S, trong chế phẩm dựa vào diện tích pic montelukast thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C35H36ClNO3S trong montelukast natri chuẩn. Khối lượng phân tử của montelukast và montelukast natri lần lượt là 586,2 và 608,2.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng và ẩm.
Loại thuốc
Điều trị hen.
Hàm lượng thường dùng
4 mg, 5 mg, 10 mg.
VIÊN NÉN NIMODIPIN
Là viên nén chứa nimodipin.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng nimodipin, C21H26N2O7, từ 95,0 % đến 105,0 % so với lượng ghi trên nhãn.
Chú ý: Quá trình tiến hành các phép thử cần thực hiện tránh ánh sáng hoặc dưới ánh sáng dịu (có bước sóng lớn hơn 420 nm), các dung dịch chứa nimodipin chỉ pha ngay trước khi dùng và trong các dụng cụ thủy tinh tối màu.
Định tính
A. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel 60 F254.
Dung môi khai triển: Ethyl acetat - cyclohexan (40 : 60).
Dung dịch thử: Lắc một lượng bột viên tương đương với 60 mg nimodipin trong 10 ml ethyl acetat (TT) trong 15 min. Ly tâm và lấy phần dịch trong phía trên.
Dung dịch đối chiếu (1): Dung dịch chứa 0,6 % nimodipin chuẩn trong ethyl acetat (TT).
Dung dịch đối chiếu (2): Trộn đồng thể tích dung dịch thử và dung dịch đối chiếu (1).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 2 μl mỗi dung dịch trên. Triển khai sắc ký đến khi dung môi đi được 15 cm. Để khô bản mỏng ngoài không khí và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm. Phun bản mỏng bằng dung dịch chứa 0,1 % 2,6-dicloroquinonclorimid trong ethanol (TT), sấy ở 110 °C trong 3 min.
Vết chính trên sắc ký đồ của dung dịch thử phải giống với vết chính trên sắc ký đồ của dung dịch đối chiếu (1) về vị trí và màu sắc. Phép thử chỉ có giá trị khi sắc ký đồ của dung dịch đối chiếu (2) chỉ cho một vết nhỏ gọn và duy nhất, vết nimodipin có màu tím xám.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch đệm acetat pH 4,5 có chứa 0,3 % natri lauryl sulfat (TT) (Hòa tan 0,299 g natri acetat (TT) trong 50 ml nước, thêm 0,174 g acid acetic băng (TT) và pha loãng thành 100 ml bằng nước. Hòa tan 0,3 g natri lauryl sulfat (TT) trong 100 ml dung dịch đệm thu được ở trên).
Tốc độ quay: 75 r/min.
Thời gian: 30 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc thích hợp, bỏ vài mililít dịch lọc đầu. Pha loãng bằng môi trường hòa tan nếu cần đề thu được dung dịch có nồng độ khoảng 33 μg/ml.
Dung dịch chuẩn: Dung dịch chứa nimodipin chuẩn trong môi trường hòa tan có nồng độ khoảng 33 μg/ml.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng cực đại khoảng 360 nm. Mẫu trắng là môi trường hòa tan.
Tính lượng nimodipin, C21H26N2O7, đã hòa tan dựa vào độ hấp thụ của dung dịch chuẩn, dung dịch thử và hàm lượng C21H26N2O7 trong nimodipin chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) nimodipin, C21H26N2O7, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - tetrahydrofuran - nước (13 : 26 : 60).
Dung dịch thử: Cân chính xác một lượng bột viên tương ứng với khoảng 60 mg nimodipin, thêm 50 ml methanol (TT), siêu âm 5 min và thêm methanol (TT) vừa đủ 100 ml. Ly tâm và dùng lớp dịch trong phía trên.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 100,0 ml bằng pha động. Pha loãng 2,0 ml dung dịch thu được thành 10,0 ml bằng pha động.
Dung dịch đối chiếu (2): Dung dịch chứa 0,0003 % tạp chất A chuẩn của nimodipin trong pha động.
Dung dịch đối chiếu (3): Dung dịch chứa hỗn hợp 0,0002 % nimodipin chuẩn và 0,0002 % tạp chất A chuẩn của nimodipin trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 235 nm.
Tốc độ dòng: 1,5ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (3), độ phân giải giữa pic nimodipin và pic tạp chất A không nhỏ hơn 1,5 và hệ số đối xứng của pic nimodipin không lớn hơn 2,0.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Tạp chất A: Diện tích pic tương ứng với pic tạp chất A không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,5 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,2 %).
Tổng diện tích pic của tất cả các tạp chất, trừ tạp chất A, không được lớn hơn 2,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,5 %).
Bỏ qua những pic có diện tích nhỏ hơn 0,25 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,05 %).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Cân 20 viên, tính khối lượng trung bình và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 60 mg nimodipin, thêm 50 ml methanol (TT), siêu âm 5 min và thêm methanol (TT) vừa đủ 100 ml. Ly tâm và dùng lớp dịch trong phía trên.
Dung dịch chuẩn: Dung dịch chứa 0,06 % nimodipin chuẩn trong methanol (TT).
Dung dịch phù hợp hệ thống: Dung dịch chứa hỗn hợp 0,0002 % nimodipin chuẩn và 0,0002 % tạp chất A chuẩn của nimodipin trong methanol (TT).
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic nimodipin và pic tạp chất A không nhỏ hơn 1,5 và hệ số đối xứng của pic nimodipin không lớn hơn 2,0.
Tính hàm lượng phần trăm của nimodipin, C21H26N2O7, trong chế phẩm dựa vào diện tích pic nimodipin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C21H26N2O7 trong nimodipin chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc chẹn kênh calci.
Hàm lượng thường dùng
30 mg.
VIÊN NÉN ONDANSETRON
Là viên nén chứa ondansetron hydroclorid.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng ondansetron, C18H19N3O, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn
Định tính
A. Phổ hấp thụ hồng ngoại (Phụ lục 4.2).
Lấy một lượng bột viên tương đương với 100 mg ondansetron hydroclorid vào bình nón thích hợp. Thêm 50 ml ethanol 96 % (TT) và khuấy. Lọc qua màng lọc PTFE 0,45 μm vào cốc có mỏ dung tích 50 ml. Bốc hơi dung môi bằng máy cất quay. Sấy khô cắn ở 105 °C trong 1 h. Dùng cắn thu được để chuẩn bị viên nén với kali bromid và đo phổ hấp thụ hồng ngoại trong khoảng 3800 cm-1 - 650 cm-1. Chuẩn bị mẫu chuẩn tương tự mẫu thử, dùng 50 ml dung dịch chứa 2 mg/ml ondansetron hydroclorid chuẩn trong ethanol 96 % (TT).
Phổ hấp thụ hồng ngoại của mẫu thử cho các dải phổ hấp thụ tại các số sóng ở khoảng 1621 cm-1, 1481 cm-1, 1281 cm-1 và 758 cm-1 tương tự mẫu chuẩn.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 500 ml nước, đuổi khí.
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm, bỏ vài mililít dịch lọc đầu và pha loãng bằng môi trường hòa tan nếu cần để được nồng độ tương đương dung dịch chuẩn.
Dung dịch chuẩn: Dung dịch chứa ondansetron hydroclorid chuẩn trong môi trường hòa tan có nồng độ khoảng 10 μg/ml.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng 310 nm. Mẫu trắng là môi trường hòa tan.
Tính lượng ondansetron, C18H19N3O, đã hòa tan dựa vào độ hấp thụ của dung dịch chuẩn, dung dịch thử và hàm lượng C18H19N3O.HCl trong ondansetron hydroclorid chuẩn. Khối lượng phân tử của ondansetron hydroclorid và ondansetron lần lượt là 329,83 và 293,36.
Yêu cầu: Không ít hơn 70 % (Q) lượng ondansetron, C18H19N3O, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3). Dung dịch đệm, pha động, dung môi pha mẫu và điều kiện sắc ký như mô tả trong phần Định lượng.
Dung dịch thử: Lấy 1 viên cho vào bình định mức dung tích 50 ml, thêm 35 ml dung môi pha mẫu, siêu âm trong 20 min và thêm dung môi pha mẫu đến vạch, lắc đều, ly tâm lấy dịch trong ở phía trên. Pha loãng dung dịch thu được bằng dung môi pha mẫu để thu được dung dịch có nồng độ ondansetron 0,04 mg/ml.
Dung dịch chuẩn: Dung dịch chứa 0,04 mg/ml ondansetron trong dung môi pha mẫu.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Dung dịch chứa 2,7 g/l kali dihydrophosphat (TT) được điều chỉnh đến pH 5,4 bằng dung dịch natri hydroxyd 1 M (TT).
Pha động: Acetonitril - dung dịch đệm (1 : 4).
Dung môi pha mẫu: Acetonitril - dung dịch đệm (1 : 1).
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 50 mg ondansetron vào bình định mức 100 ml, thêm 70 ml pha động, siêu âm trong 20 min và thêm pha động đến vạch. Ly tâm dung dịch thu được. Lọc dịch trong qua màng lọc nylon 0,45 μm.
Dung dịch chuẩn gốc: Dung dịch chứa 0,05 mg/ml ondansetron trong dung môi pha mẫu (dùng ondansetron hydroclorid chuẩn để pha).
Dung dịch đối chiếu: Pha loãng dung dịch chuẩn gốc bằng pha động để thu được dung dịch chứa 1,5 μg/ml ondansetron.
Dung dịch phù hợp hệ thống: Dung dịch chứa 0,05 mg/ml tạp chất A chuẩn của ondansetron và 0,1 mg/ml ondansetron hydroclorid chuẩn trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh là các hạt silica gel liên kết hóa học với các nhóm nitril (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 216 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic tạp chất A và pic ondansetron ít nhất là 2,0. Trên sắc ký đồ của dung dịch đối chiếu, độ lệch chuẩn tương đối của diện tích pic ondansetron thu được từ 6 lần tiêm lặp lại không lớn hơn 5,0 %.
Tiến hành sắc ký dung dịch thử ít nhất 45 min.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic ondansetron trên sắc ký đồ của dung dịch đối chiếu, nồng độ của ondansetron trong dung dịch đối chiếu và nồng độ theo lý thuyết của ondansetron trong dung dịch thử. Chia diện tích pic tạp chất cho hệ số đáp ứng tương đối (F) nếu có.
Thời gian lưu tương đối, hệ số đáp ứng tương đối và giới hạn phần trăm của các tạp chất được trình bày trong Bảng 1.
Bảng 1
Tạp chất | Thời gian lưu tương đối | Hệ số đáp ứng tương đối (F) | Giới hạn (%) |
2-Methyl imidazol* | 0,22 | 0,53 | 0,2 |
Tạp chất C | 0,40 | 1,2 | 0,2 |
Tạp chất D | 0,47 | 1,3 | 0,1 |
Tạp chất A | 0,87 | 0,90 | 0,2 |
Desmethylondansetron* | 0,90 | 0,91 | 0,2 |
Ondansetron | 1,0 | - | .- |
Tạp đơn chưa xác định | - | 1,0 | 0,2 |
Tổng tạp | - | - | 1,0 |
Ghi chú:
* Không tính hai tạp chất này vào tổng tạp.
Tạp chất A: 3[(Dimethylamino)methyl]-9-methyl-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Tạp chất C: 9-Methyl-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Tạp chất D: 9-Methyl-3-methylen-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Desmethylondansetron: 3-[1H-imidazol-1-yl)methyl]-9-methyl-1,2,3,9-tetrahydro-4H-carbazol-4-on.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm, pha động, dung môi pha mẫu, điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch thử gốc: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 50 mg ondansetron cho vào bình định mức 100 ml, thêm 70 ml dung môi pha mẫu, siêu âm trong 20 min và thêm dung môi pha mẫu đến vạch. Ly tâm dung dịch thu được.
Dung dịch thử: Pha loãng dịch trong phía trên của dung dịch thử gốc sau ly tâm bằng dung môi pha mẫu để thu được dung dịch có chứa 0,05 mg/ml ondansetron. Lọc qua màng lọc nylon 0,45 μm.
Dung dịch chuẩn: Dùng dung dịch chuẩn gốc ở phần Tạp chất liên quan.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic ondansetron không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic ondansetron thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của ondansetron, C18H19N3O, trong chế phẩm dựa vào diện tích pic ondansetron thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C18H19N3O.HCl trong ondansetron hydroclorid chuẩn. Khối lượng phân tử của ondansetron hydroclorid và ondansetron lần lượt là 329,83 và 293,36.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc chống nôn, đối kháng thụ thể 5-HT3.
Hàm lượng thường dùng
4 mg, 8 mg, 24 mg.
VIÊN NÉN ROSUVASTATIN
Là viên nén chứa rosuvastatin calci,
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng rosuvastatin, C22H28FN3O6S, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Trong phần Định lượng, phổ hấp thụ tử ngoại của pic chính trên sắc ký đồ của dung dịch thử trong khoảng bước sóng từ 200 nm đến 440 nm phải cho cực đại hấp thụ và cực tiểu hấp thụ ở các bước sóng tương ứng với phổ hấp thụ tử ngoại của pic rosuvastatin trên sắc ký đồ của dung dịch chuẩn.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Tiến hành trong điều kiện tránh ánh sáng.
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch đệm citrat pH 6,6 (dung dịch chứa 14,7 g/l natri citrat (TT) và 0,36 g/l acid citric (TT), điều chỉnh đến pH 6,6 bằng natri citrat hoặc acid citric).
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định lượng rosuvastatin đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - nước - acid phosphoric (400 : 600 : 1).
Dung môi pha mẫu: Acetonitril - nước (25 : 75).
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc thích hợp.
Dung dịch chuẩn gốc: Dung dịch chứa 1 mg/ml rosuvastatin calci chuẩn trong dung môi pha mẫu.
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc bằng môi trường hòa tan để thu được dung dịch có nồng độ rosuvastatin tương đương nồng độ rosuvastatin trong dung dịch thử.
Điều kiện sắc ký:
Cột kích thước (5 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 242 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Tiến hành sắc ký với thời gian gấp ít nhất 2,5 lần thời gian lưu của rosuvastatin.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic rosuvastatin không lớn hơn 1,5.
Tính lượng rosuvastatin, C22H28FN3O6S, đã hòa tan dựa vào diện tích pic rosuvastatin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C44H54CaF2N6O12S2 trong rosuvastatin calci chuẩn. Khối lượng phân tử của rosuvastatin và rosuvastatin calci lần lượt là 481,54 và 1001,14 và 1 mol rosuvastatin calci tương đương 2 mol rosuvastatin.
Yêu cầu: Không ít hơn 75 % (Q) lượng rosuvastatin, C22H28FN3O6S, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động, dung dịch chuẩn, điều kiện sắc ký và cách tiến hành như mô tả ở phần Định lượng.
Dung dịch thử: Lấy 1 viên chuyển vào bình định mức thích hợp để thu được dung dịch có nồng độ 25 μg/ml rosuvastatin. Thêm nước và lắc mạnh để viên rã hoàn toàn, thêm tiếp acetonitril (TT) và lắc mạnh, sau đó thêm nước đến vạch, đảm bảo tỷ lệ acetonitril - nước trong dung dịch thu được là 1 : 3. Lọc dung dịch thu được qua màng lọc thích hợp.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3), tiến hành trong điều kiện tránh ánh sáng.
Pha động: Acetonitril - dung dịch acid trifluoroacetic 1 % (tt/tt) - nước (37 : 1 : 62).
Dung môi pha mẫu: Acetonitril - nước (25 : 75).
Dung dịch thử: Lấy số lượng viên tùy theo hàm lượng rosuvastatin trên nhãn vào bình định mức thích hợp (xem Bảng 1), thêm nước và lắc mạnh để viên rã hoàn toàn. Thêm tiếp acetonitril (TT) và lắc mạnh, sau đó thêm nước đến vạch, đảm bảo tỷ lệ acetonitril - nước trong dung dịch thu được là 1 : 3. Lọc dung dịch thu được qua màng lọc thích hợp.
Bảng 1
Hàm lượng (mg) | Số lượng (viên) | Bình định mức (ml) | Nước (ml) | Acetonitril (ml) |
5 | 20 | 100 | 50 | 25 |
10 | 10 | 100 | 50 | 25 |
20 | 10 | 200 | 100 | 50 |
Dung dịch phù hợp hệ thống gốc: Cân 10 mg rosuvastatin calci chuẩn vào bình định mức 100 ml, thêm khoảng 50 nước. Thêm 10 ml dung dịch acid hydrocloric 1 M (TT). Đun trong cách thủy ở 60 °C trong 2 h và trung hòa bằng cách thêm dung dịch natri hydroxyd 1 M (TT). Để nguội về nhiệt độ phòng, thêm 25 ml acetonitril (TT) rồi thêm nước đến vạch. Dung dịch thu được có chứa khoảng 50 μg/ml rosuvastatin calci và 50 μg/ml rosuvastatin diastereomer (tạp chất B của rosuvastatin).
Dung dịch phù hợp hệ thống: Pha loãng 5,0 ml dung dịch phù hợp hệ thống gốc thành 10,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu: Dung dịch chứa 10 μg/ml rosuvastatin calci chuẩn trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm × 3,2 mm) được nhồi pha tĩnh C (5 μm). Có thể dùng thêm cột bảo vệ.
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 242 nm.
Tốc độ dòng: 0,75 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp ít nhất 2,5 lần thời gian lưu của rosuvastatin.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic rosuvastatin và pic rosuvastatin diastereomer không nhỏ hơn 1,5. Trên sắc ký đồ của dung dịch đối chiếu, hệ số đối xứng của pic rosuvastatin không lớn hơn 1,8 và độ lệch chuẩn tương đối của diện tích pic rosuvastatin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic rosuvastatin trên sắc ký đồ của dung dịch đối chiếu; nồng độ của rosuvastatin calci trong dung dịch đối chiếu và nồng độ lý thuyết của rosuvastatin trong dung dịch thử, chia diện tích pic tạp chất cho hệ số đáp ứng tương đối F (nếu có). Khối lượng phân tử của rosuvastatin và rosuvastatin calci lần lượt là 481,54 và 1001,14 và 1 mol rosuvastatin calci tương đương 2 mol rosuvastatin.
Thời gian lưu tương đối, hệ số đáp ứng tương đối và giới hạn phần trăm của các tạp chất được trình bày trong Bảng 2.
Bảng 2
Tạp chất | Thời gian lưu tương đối | Hệ số đáp ứng tương đối (F) | Giới hạn (%) |
Tạp chất A | 0,9 | - | - |
Rosuvastatin | 1,0 | - | - |
Tạp chất B | 1,1 | - | - |
Rosuvastatin keton | 1,6 | 0,71 | 2,1 |
Rosuvastatin lacton | 2,3 | 1,0 | 1,5 |
Rosuvastatin ethyl ester | 3,8 | 1,0 | 0,5 |
Tạp chất chưa xác định | - | 1,0 | 0,2 |
Tổng tạp | - | - | 3,6 |
Ghi chú:
Tạp chất B: Acid (3RS,5RS,6E)-7-[4-(4-fluorophenyl)-6-(1-methylethyl)-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl]-3,5-dihydroxyhept-6-enoic. Tạp chất B đã được kiểm soát ở nguyên liệu, thông tin cung cấp có ý nghĩa định tính, không tính hàm lượng và không cộng vào tổng tạp.
Rosuvastatin keton: Acid (R,E)-7-[4-(4-fluorophenyl)-6-isopropyl-2-(N-methylmethylsulfonamido)pyrimidin-5-yl]-3-hydroxy-5-oxohept-6-enoic.
Rosuvastatin lacton: N-[4-(4-fluorophenyl)-5-{(E)-2-[(2S,4R)-4-hydroxy-6-oxotetrahydro-2H-pyran-2-yl]vinyl}-6-isopropylpyrimidin-2-yl]-N-methylmethanesulfonamid.
Rosuvastatin ethyl ester: Ethyl (3R,5S,6E)-7-[4-(4-fuorophenyI)-6-isopropyl-2-(N-methylmethylsulfonamido)pyrimidin-5-yl]-3,5-dihydroxyhept-6-enoat.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3), tiến hành trong điều kiện tránh ánh sáng.
Pha động, dung môi pha mẫu và điều kiện sắc ký như mô tả trong phần Tạp chất liên quan, dùng detector diod aray cho Định tính A.
Dung dịch thử: Lấy ít nhất 10 viên vào bình định mức thích hợp, thêm nước và lắc mạnh để viên rã hoàn toàn. Thêm tiếp acetonitril (TT) và lắc mạnh, sau đó thêm nước đến vạch, đảm bảo tỷ lệ acetonitril - nước trong dung dịch thu được là 1 : 3. Lọc dung dịch thu được qua màng lọc thích hợp. Pha loãng dịch lọc thu được bằng dung môi pha mẫu để thu được dung dịch chứa 25 μg/ml rosuvastatin.
Dung dịch chuẩn gốc: Cân chính xác một lượng rosuvastatin calci chuẩn vào bình định mức thích hợp, thêm nước khoảng 50 % thể tích bình. Lắc mạnh hoặc siêu âm để hòa tan hoàn toàn. Thêm acetonitril (TT) khoảng 25 % thể tích bình rồi sau đó thêm nước đến vạch. Dung dịch thu được có chứa 1 mg/ml rosuvastatin calci chuẩn.
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc bằng dung môi pha mẫu để thu được dung dịch chứa 25 μg/ml rosuvastatin calci chuẩn.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp ít nhất 1,3 lần thời gian lưu của rosuvastatin.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic rosuvastatin không lớn hơn 1,8 và độ lệch chuẩn tương đối của diện tích pic rosuvastatin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của rosuvastatin, C22H28FN3O6S, trong chế phẩm dựa vào diện tích pic rosuvastatin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C44H54CaF2N6O12S2 trong rosuvastatin calci chuẩn. Khối lượng phân tử của rosuvastatin và rosuvastatin calci lần lượt là 481,54 và 1001,14 và 1 mol rosuvastatin calci tương đương 2 mol rosuvastatin.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Ức chế HMG Co-A reductase; chống tăng lipid máu.
Hàm lượng thường dùng
5 mg, 10 mg, 20 mg.
VIÊN NÉN SERTRALIN
Là viên nén chứa sertralin hydroclorid. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng sertralin, C17H17Cl2N, từ 95,0 % đến 105,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch đệm natri acetat pH 4,5 (TT).
Tốc độ quay: 75 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định hàm lượng sertralin đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hỗn hợp chứa 0,286 % (tt/tt) acid acetic băng (TT) và 0,348 % (tt/tt) triethylamin (TT) trong nước.
Pha động: Methanol - dung dịch đệm - acetonritril (15 : 40 : 45).
Dung dịch thử: Lấy một phần dung dịch môi trường sau khi hòa tan, lọc. Pha loãng dịch lọc với môi trường hòa tan để thu được dung dịch có nồng độ sertralin tương đương nồng độ của dung dịch đối chiếu (nếu cần).
Dung dịch đối chiếu: Dung dịch sertralin hydroclorid chuẩn trong môi trường hòa tan có nồng độ 0,056 mg/ml.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 265 nm.
Tốc độ dòng: 1,8 ml/min.
Thể tích tiêm: 20 μl.
Tính lượng sertralin, C17H17Cl2N, đã hòa tan dựa vào diện tích pic sertralin thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu và hàm lượng của C17H17Cl2N.HCl trong sertralin hydroclorid chuẩn. 1 mg C17H17Cl2N.HCl tương đương với 0,8936 mg C17H17Cl2N.
Yêu cầu: Không ít hơn 75 % (Q) lượng sertralin, C17H17Cl2N, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Dung dịch chứa 0,272 % kali dihydrophosphat (TT), điều chỉnh đến pH 3,0 bằng acid phosphoric (TT).
Pha động B: Acetonitril (TT1).
Dung môi pha mẫu: Acetonitril - nước (30 : 70).
Dung dịch thử: Lắc một lượng viên tương ứng với 0,45 g sertralin với 150 ml dung môi pha mẫu cho đến khi các viên phân tán hoàn toàn. Siêu âm 5 min và pha loãng thành 200 ml bằng dung môi pha mẫu, trộn đều và lọc.
Dung dịch đối chiếu (1): Pha loãng 1,0 ml dung dịch thử thành 500,0 ml bằng dung môi pha mẫu.
Dung dịch đối chiếu (2): Dung dịch chứa 0,001 % acid (R)-mandelic (TT) trong dung môi pha mẫu.
Dung dịch đối chiếu (3): Dung dịch chứa 0,25 % tạp chất chuẩn của sertralin hydroclorid trong dung môi pha mẫu.
Dung dịch đối chiếu (4): Pha loãng 5,0 ml dung dịch đối chiếu (1) thành 10,0 ml bằng dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 210 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 - 30 | 70 | 30 |
30 - 40 | 70 → 60 | 30 → 40 |
40 - 41 | 60 → 70 | 40 → 30 |
41 - 50 | 70 | 30 |
Kiểm tra tính phù hợp của hệ thống: Sắc ký đồ thu được của dung dịch đối chiếu (3) phải giống với sắc ký đồ cung cấp kèm theo tạp chất chuẩn của sertralin hydroclorid.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Tạp chất C: Diện tích pic tạp chất C không được lớn hơn 4 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,8 %).
Acid (R)-mandelic: Diện tích pic acid (R)-mandelic không được lớn hơn diện tích pic tương ứng thu được trên sắc ký đồ của dung dịch đối chiếu (2) (0,4 %).
Tạp chất khác: Với mỗi tạp chất, diện tích pic không được lớn hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (0,2 %).
Tổng diện tích pic của tất cả các tạp chất, trừ tạp chất C, không được lớn hơn 7,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (1) (1,5 %).
Bỏ qua những pic có diện tích nhỏ hơn diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (4) (0,1 %).
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả ở phần Độ hòa tan.
Dung dịch thử: Cân 20 viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 0,19 g sertralin vào bình định mức 200 ml, thêm 150 ml pha động, siêu âm 45 min. Lắc 30 min, để nguội và thêm nước đến vạch, trộn đều và lọc. Pha loãng 1,0 ml dịch lọc thành 20,0 ml bằng pha động.
Dung dịch chuẩn: Dung dịch sertralin hydroclorid chuẩn trong pha động có nồng độ 0,05 mg/ml.
Tính hàm lượng phần trăm của sertralin, C17H17Cl2N, trong chế phẩm dựa vào diện tích pic sertralin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C17H17Cl2N.HCl trong sertralin hydroclorid chuẩn.
1 mg C17H17Cl2N.HCl tương đương với 0,8936 mg C17H17Cl2N.
Bảo quản
Trong đồ đựng kín, tránh ánh sáng ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc chống trầm cảm (loại ức chế chọn lọc tái hấp thu serotonin).
Hàm lượng thường dùng
50 mg, 100 mg.
VIÊN NÉN SILDENAFIL
Là viên nén chứa sildenafil citrat. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng sildenafil, C22H30N6O4S, từ 95,0 % đến 105,0 % so với lượng ghi trên nhãn.
Định tính
A. Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel F254.
Dung mỗi khai triển: Methanol - ethyl acetat (4 : 8).
Dung dịch thử: Lắc một lượng bột viên tương ứng với khoảng 25 mg sildenafil với 10 ml methanol (TT) và lọc.
Dung dịch đối chiếu (1): Dung dịch chứa 0,35 % sildenafil citrat chuẩn trong methanol (TT).
Dung dịch đối chiếu (2): Trộn đều 1 ml dung dịch thử và 1 ml dung dịch đối chiếu (1).
Cách tiến hành: Chấm riêng biệt lên bản mỏng 10 μl mỗi dung dịch trên. Triển khai sắc ký đến khi dung môi đi được 8 cm. Làm khô bản mỏng dưới luồng không khí ấm và quan sát dưới ánh sáng tử ngoại ở bước sóng 254 nm.
Vết chính trên sắc ký đồ của dung dịch thử phải giống với vết chính trên sắc ký đồ của dung dịch đối chiếu (1) về vị trí và màu sắc. Phép thử chỉ có giá trị khi sắc ký đồ của dung dịch đối chiếu (2) cho 1 vết có cùng Rf và màu sắc với dung dịch đối chiếu (1).
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu giỏ quay.
Môi trường hòa tan: 900 ml dung dịch acid hydrocloric 0,01 M (TT).
Tốc độ quay: 100 r/min.
Thời gian: 45 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan, lọc. Pha loãng dịch lọc thu được với môi trường hòa tan để thu được dung dịch có nồng độ 0,0027 % sildenafil.
Dung dịch chuẩn: Dung dịch chứa 0,0038 % sildenafil citrat chuẩn trong môi trường hòa tan.
Đo độ hấp thụ (Phụ lục 4.1) của các dung dịch thu được ở bước sóng cực đại khoảng 290 nm, cốc đo dày 1 cm, mẫu trắng là môi trường hòa tan.
Tính lượng sildenafil, C22H30N6O4S, đã hòa tan dựa vào độ hấp thụ của dung dịch thử, dung dịch chuẩn và hàm lượng C28H38N6O11S trong sildenafil citrat chuẩn.
1 mg C28H38N6O11S tương đương với 0,7118 mg C22H30N6O4S.
Yêu cầu: Không ít hơn 75 % (Q) lượng sildenafil, C22H30N6O4S, so với lượng ghi trên nhãn được hòa tan trong 45 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Pha loãng 7 ml triethylamin (TT) trong nước thành 1000 ml. Khuấy đều và điều chỉnh đến pH 3,0 bằng acid phosphoric (TT).
Pha động: Dung dịch đệm - methanol - acetonitril (58 : 25 : 17).
Dung môi pha mẫu: Acetonitril - nước (90 : 10).
Dung dịch thử gốc: Chuyển 5 viên nén vào bình định mức dung tích 250 ml, thêm 25 ml dung môi pha mẫu và siêu âm cho rã hoàn toàn. Thêm pha động đến vạch, siêu âm nếu cần. Ly tâm và thu lấy lớp trong phía trên.
Dung dịch thử: Pha loãng dung dịch thử gốc bằng pha động để thu được dung dịch chứa 0,5 mg/ml sildenafil.
Dung dịch đối chiếu: Dung dịch chứa 1,4 μg/ml sildenafil citrat chuẩn trong pha động.
Dung dịch thử độ nhạy: Pha loãng dung dịch đối chiếu bằng pha động để thu được dung dịch chứa 0,35 μg/ml sildenafil citrat.
Dung dịch phân giải: Hòa tan 70 mg sildenafil citrat chuẩn trong 1 ml hỗn hợp gồm dung dịch hydrogen peroxyd 100 tt - acid formic khan (2 : 1). Để yên 10 min để tạo sildenafil N-oxid, và pha loãng thành 250 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 30 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 290 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian bằng 3 lần thời gian lưu của sildenafil.
Thời gian lưu tương đối so với sildenafil của sildenafil N-oxid là 1,2.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, hệ số đối xứng của pic sildenafil không lớn hơn 1,3; độ lệch chuẩn tương đối của diện tích pic sildenafil thu được từ 6 lần tiêm lặp lại không lớn hơn 3,0 %. Trên sắc ký đồ của dung dịch thử độ nhạy, tỷ số tín hiệu trên nhiễu đối với pic sildenafil không nhỏ hơn 10. Trên sắc ký đồ của dung dịch phân giải, độ phân giải giữa pic sildenafil và pic sildenafil N-oxid không nhỏ hơn 2,5.
Tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử, diện tích pic sildenafil trên sắc ký đồ của dung dịch đối chiếu và nồng độ sildenafil citrat trong dung dịch đối chiếu.
1 mg C28H38N6O11S tương đương với 0,7118 mg C22H30N6O4S.
Giới hạn:
Sildenafil N-oxid: Không được quá 0,20 %.
Các tạp chất chưa xác định khác: Với mỗi tạp chất, không được quá 0,20 %.
Tổng tất cả các tạp chất: Không được quá 0,50 %.
Bỏ qua các tạp chất có hàm lượng dưới 0,05 %.
Ghi chú:
Sildenafil N-oxid: 1-{[3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxyphenyl] sulfonyl}-4-methylpiperazin N-oxid.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung môi pha mẫu, dung dịch phân giải và điều kiện sắc ký như mô tả ở phần Tạp chất liên quan.
Dung dịch thử: Chuyển 10 viên nén vào bình định mức dung tích 500 ml, thêm 50 ml dung môi pha mẫu và siêu âm cho rã hoàn toàn. Thêm pha động đến vạch, siêu âm nếu cần. Ly tâm và thu lấy lớp dịch trong phía trên. Pha loãng dịch trong thu được bằng pha động để thu được dung dịch có nồng độ sildenafil khoảng 0,002 %.
Dung dịch chuẩn: Dung dịch chứa 0,0028 % sildenafil citrat chuẩn trong pha động.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic sildenafil không lớn hơn 1,3; độ lệch chuẩn tương đối của diện tích pic sildenafil thu được từ 6 lần tiêm lặp lại không lớn hơn 3,0 %. Trên sắc ký đồ của dung dịch phân giải, độ phân giải giữa pic sildenafil và pic sildenafil N-oxid không nhỏ hơn 2,5.
Tính hàm lượng phần trăm của sildenafil, C22H30N6O4S, trong chế phẩm dựa vào diện tích pic sildenafil thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C28H38N6O11S trong sildenafil citrat chuẩn.
1 mg C28H38N6O11S tương đương với 0,7118 mg C22H30N6O4S.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc ức chế men phosphodiesterase tuýp 5 với tác dụng giãn mạch; điều trị rối loạn cương dương.
Hàm lượng thường dùng
50mg; 100mg.
VIÊN NÉN TADALAFIL
Là viên nén chứa tadalatil.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng tadalafil, C22H19N3O4, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Phương pháp quang phổ hấp thụ hồng ngoại (Phụ lục 4.2).
Mẫu thử: Lấy số lượng viên nén tương đương với 10 - 20 mg tadalafil, thêm 15 ml nước, lắc 10 min hoặc lắc đến khi nào viên nén rã hoàn toàn, ly tâm 10 min và bỏ lớp dịch trong phía trên. Phân tán cắn trong 8 ml ethyl acetat (TT), lắc 5 min, ly tâm 10 min và thu lấy phần dịch trong phía trên. Làm khô dịch trong thu được dưới dòng khí nitrogen, có thể làm nóng tới 70 °C để bốc hơi ethyl acetat nhanh hơn. (Chú ý: ethyl acetat phải được bốc hơi hoàn toàn để tránh ảnh hưởng tới phổ hấp thụ).
Mẫu chuẩn: Thêm 10 mg tadalafil chuẩn vào 15 ml nước, lắc 20 min, ly tâm 10 min và bò lớp dịch trong phía trên. Phân tán cắn trong 8 ml ethyl acetat (TT), lắc 5 min, ly tâm 10 min và thu lấy phần dịch trong phía trên. Làm khô dịch trong thu được dưới dòng khí nitrogen như với mẫu thử.
Chuẩn bị mẫu đo dạng đĩa nén với kali bromid. Phổ hấp thụ hồng ngoại của mẫu thử và mẫu chuẩn trong khoảng số sóng 1700 cm-1 - 400 cm-1 phải tương tự nhau.
B. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch đối chiếu.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 1000 ml dung dịch natri lauryl sulfat (TT) 0,5 %.
Tốc độ quay: 50 r/min, dùng dụng cụ giữ mẫu thử nếu cần.
Thời gian: 10 min và 30 min.
Cách tiến hành:
Xác định lượng tadalafil đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: methanol - nước (50 : 50).
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc thích hợp.
Dung dịch chuẩn gốc: Dung dịch chứa 0,25 mg/ml tadalafil chuẩn trong hỗn hợp acetonitril - nước (1 : 1).
Dung dịch chuẩn: Pha loãng dung dịch chuẩn gốc bằng môi trường hòa tan để thu được dung dịch có nồng độ tadalafil 0,0075 mg/ml.
Điều kiện sắc ký:
Cột kích thước (10 cm × 4,6 mm) được nhồi pha tĩnh B (3,5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 225 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 20 μl.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic tadalafil không lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic tadalafil thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %
Tính lượng tadalafil, C22H19N3O4, đã hòa tan tại thời điểm 10 min (Q10) và 30 min (Q30) theo công thức sau:
Q10 = (rU/rS) × (Cs/L) × V × 100
Q30 = (Q10 × v10/V) + [(rU/rS) × (Cs/L) × (V - v10) × 100]
Trong đó:
rU và rS lần lượt là diện tích pic tadalafil trên sắc ký đồ của dung dịch thử và dung dịch chuẩn.
CS là nồng độ của tadalafil trong dung dịch chuẩn (mg/ml)
L hàm lượng tadalafil trên nhãn của viên (mg)
V là thể tích môi trường hòa tan (1000 ml)
v10 là thể tích mẫu hút ra tại thời điểm 10 min.
Yêu cầu: Không ít hơn 40 % (Q) lượng tadalafil, C22H19N3O4, so với lượng ghi trên nhãn được hòa tan trong 10 min. Không ít hơn 80 % (Q) lượng tadalafil, C22H19N3O4, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều (Phụ lục 11.9)
Dung môi pha mẫu: Acetonitril - nước (1 : 1).
Dung dịch thử: Lấy 1 viên cho vào bình định mức thích hợp để thu được dung dịch có nồng độ 0,1 - 0,2 mg/ml tadalafil. Thêm dung môi pha mẫu khoảng 50 % thể tích bình, lắc cơ học 15 min. Thêm dung môi pha mẫu đến vạch. Lọc qua màng lọc 0,45 μm, bỏ vài ml dịch lọc đầu. Pha loãng 2,0 ml dịch lọc thành 20,0 ml bằng dung môi pha mẫu.
Dung dịch chuẩn: Dung dịch chứa 0,01 - 0,02 mg/ml tadalafil chuẩn trong dung môi pha mẫu.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng 285 nm. Dùng cốc đo 1 cm.
Tính hàm lượng tadalafil, C22H19N3O4, trong 1 viên dựa vào độ hấp thụ của dung dịch chuẩn, dung dịch thử và hàm lượng C22H19N3O4 trong tadalafil chuẩn.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Acetonitril - nước - acid trifluoroacetic (35 : 65 : 0,1).
Dung môi pha mẫu: Acetonitril - nước (1:1).
Dung dịch thử: Lấy 20 viên cho vào bình định mức thích hợp (sao cho nồng độ tadalafil thu được không lớn hơn 6 mg/ml), thêm dung môi pha mẫu khoảng 50 % thể tích bình và lắc 15 min để viên rã (nếu vẫn chưa rã hết, siêu âm 2 min cho rã hoàn toàn). Thêm dung môi pha mẫu đến vạch, trộn đều. Để yên 1 h. Nếu cần, pha loãng dung dịch thu được để được dung dịch có chứa 0,25 mg/ml tadalafil. Ly tâm hoặc lọc dung dịch thu được.
Dung dịch đối chiếu: Dung dịch chứa 0,25 mg/ml tadalafil chuẩn trong dung môi pha mẫu.
Dung dịch phù hợp hệ thống: Chuyển đổi một phần tadalafil thành đồng phân 6R,12aS: Chuyển 25 ml dung dịch đối chiếu vào một bình chứa thích hợp. Thêm 0,25 ml dung dịch natri hydroxyd 5 M (TT), lắc đều và để yên trong 30 min. Trung hòa dung dịch đến pH 7 bằng cách thêm từng giọt acid trifuoroacetic (TT) (dung dịch này ổn định trong vòng 1 tháng khi được bảo quản trong tủ lạnh).
Dung dịch độ nhạy: Pha loãng dung dịch đối chiếu để thu được dung dịch chứa 0,25 μg/ml tadalafil trong dung môi pha mẫu.
Điều kiện sắc ký.
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh B (3,5 μm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 285 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch đối chiếu, dung dịch phù hợp hệ thống và dung dịch độ nhạy.
Thời gian lưu tương đối so với tadalafil của đồng phân 6R,12aS là 1,2.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, hệ số đối xứng của pic tadalafil không lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic tadalafil thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %. Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic tadalafil và pic đồng phân 6R,12aS không nhỏ hơn 3. Trên sắc ký đồ của dung dịch độ nhạy, tỷ số tín hiệu trên nhiễu của pic tadalafil không nhỏ hơn 20.
Tiến hành sắc ký dung dịch thử, tính hàm lượng phần trăm của mỗi tạp chất trong chế phẩm, nếu có, theo phương pháp chuẩn hóa.
Giới hạn:
Từng tạp chất: Với mỗi tạp chất, không được quá 0,2 %.
Tổng các tạp chất: Không được quá 0,3 %.
Bỏ qua các tạp chất có hàm lượng dưới 0,05 %.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3)
Pha động, dung môi pha mẫu, dung dịch thử, dung dịch đối chiếu, dung dịch phù hợp hệ thống và điều kiện sắc ký như mô tả trong phần Tạp chất liên quan.
Tiến hành sắc ký dung dịch đối chiếu và dung dịch phù hợp hệ thống.
Thời gian lưu tương đối so với tadalafil của đồng phân 6R,12aS là 1,2.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, hệ số đối xứng của pic tadalafil không lớn hơn 1,5; độ lệch chuẩn tương đối của diện tích pic tadalafil thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %. Trên sắc ký đồ của dung dịch phù hợp hệ thống, độ phân giải giữa pic tadalafil và pic đồng phân 6R,12aS không nhỏ hơn 3.
Tiến hành sắc ký dung dịch thử và dung dịch đối chiếu.
Tính hàm lượng tadalafil, C22H19N3O4, có trong chế phẩm dựa vào diện tích pic tadalafil thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu và hàm lượng C22H19N3O4 của tadalafil chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc ức chế men phosphodiesterase tuýp 5 với tác dụng giãn mạch; điều trị rối loạn cương dương.
Hàm lượng thường dùng
5 mg, 10 mg, 20 mg.
VIÊN NÉN TELMISARTAN VÀ HYDROCLOROTHIAZID
Là viên nén chứa telmisartan và hydroclorothiazid. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng telmisartan, C33H30N4O2, từ 95,0 % đến 105,0 % so với lượng ghi trên nhãn.
Hàm lượng hydroclorothiazid, C7H8ClN3O4S2, từ 90,0 % đến 107,5 % so với lượng ghi trên nhãn.
Định tính
A. Trong phần Định lượng, phổ hấp thụ tử ngoại xác định trên hai pic chính của dung dịch thử phải giống với phổ hấp thụ tử ngoại xác định trên hai pic tương ứng của dung dịch chuẩn.
B. Trong phần Định lượng, thời gian lưu của hai pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của hai pic chính trên sắc ký đồ của dung dịch đối chiếu (2).
Độ hòa tan
Thiết bị: Kiểu cánh khuấy
Môi trường hòa tan: 900 ml dung dịch đệm pH 7,5.
Tốc độ quay: 75 r/min.
Thời gian: 30 min.
Dung dịch đệm pH 7,5: Hòa tan 6,8 g kali dihydrophosphat (TT) và 1,56 g natri hydroxyd (TT) trong 1000 ml nước, điều chỉnh đến pH 7,5 bằng dung dịch natri hydroxid 10 % (TT).
Cách tiến hành:
Xác định lượng telmisartan và hydroclorothiazid đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch đệm: Hòa tan 2,72 g kali dihydrophosphat (TT) trong 1000 ml nước, thêm 2 ml triethylamin (TT) và điều chỉnh đến pH 2,4 bằng acid phosphoric (TT).
Pha động: Acetonitril - Dung dịch đệm (35 : 65).
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm, bỏ vài ml dịch lọc đầu.
Dung dịch chuẩn gốc telmisartan: Cân chính xác khoảng 45 mg telmisartan chuẩn và chuyển vào bình định mức 100 ml, thêm 50 ml methanol (TT), lắc siêu âm đến khi tan hoàn toàn và thêm môi trường hòa tan đến định mức, lắc đều.
Dung dịch chuẩn gốc hydroclorothiazid: Cân chính xác khoảng 56 mg hydroclorothiazid chuẩn và chuyền vào bình định mức 200 ml, thêm 100 ml methanol (TT), lắc siêu âm đến khi tan hoàn toàn và thêm môi trường hòa tan đến định mức, lắc đều.
Dung dịch chuẩn hỗn hợp: Pha loãng từ dung dịch chuẩn gốc telmisartan và dung dịch chuẩn gốc hydroclorothiazid bằng môi trường hòa tan để thu được dung dịch có nồng độ telmisartan và hydroclorothiazid tương ứng với dung dịch thử.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 270 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 20 μl.
Thời gian chạy sắc ký gấp ít nhất 1,7 lần thời gian lưu của telmisartan.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của hai pic telmisartan và hydroclorothiazid không lớn hơn 2,0; số đĩa lý thuyết của cột không nhỏ hơn 1500 với cả telmisartan và hydroclorothiazid; và độ lệch chuẩn tương đối của diện tích pic telmisartan và pic hydroclorothiazid thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiến hành sắc ký lần lượt với dung dịch chuẩn và dung dịch thử.
Tính hàm lượng telmisartan (C33H30N4O2) và hydroclorothiazid (C7H8ClN3O4S2) đã hòa tan dựa vào diện tích pic telmisartan, hydroclorothiazid thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn tương ứng và nồng độ telmisartan, hydroclorothiazid trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng telmisartan, C33H30N4O2, và hydroclorothiazid, C7H8ClN3O4S2 so với lượng ghi trên nhãn được hòa tan trong 30 min.
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động A: Dung dịch chứa 2,0 g/L amoni dihydrophosphat (TT) trong nước, điều chỉnh đến pH 3,0 bằng acid phosphoric (TT).
Pha động B: Methanol - acetonitril (1 : 1).
Dung môi pha mẫu: Dung dịch natri hydroxyd 0,005 M (TT) trong methanol (TT).
Dung dịch thử gốc: Cân 20 viên, tính khối lượng trung bình, nghiền thành bột mịn. Cân chính xác một lượng bột thuốc tương ứng với 1 viên cho vào bình định mức dung tích phù hợp, thêm dung dịch natri hydroxyd 0,1 M (TT) (khoảng 5 % thể tích bình). Thêm methanol (TT) (khoảng 80 % thể tích bình), siêu âm 10 min và lắc mạnh 30 min, để nguội bình về nhiệt độ phòng sau đó thêm methanol (TT) đến vạch, trộn đều. Nồng độ telmisartan thu được khoảng 1,6 mg/ml (nồng độ hydroclorothiazid phụ thuộc vào hàm lượng trên nhãn). Ly tâm dung dịch thu được ở tốc độ quay 4000 rpm.
(Chú ý, không kéo dài thời gian siêu âm, duy trì nhiệt độ bể siêu âm không quá 22 °C).
Dung dịch thử: Pha loãng 1 ml dung dịch thử gốc thành 5 ml bằng hỗn hợp pha động A - pha động B (1 : 1).
Dung dịch đối chiếu gốc (1): Dung dịch chứa 0,025 mg/ml tạp chất A chuẩn của benzothiadiazin trong dung môi pha mẫu.
Dung dịch đối chiếu (1): Pha loãng 1 ml dung dịch đối chiếu gốc (1) thành 20 ml bằng dung môi pha mẫu (1,25 μg/ml tạp chất A chuẩn của benzothiadiazin). Tiếp tục pha loãng dung dịch thu được bằng hỗn hợp pha động A - pha động B (1 : 1) để thu được dung dịch có nồng độ tạp chất A chuẩn của benzothiadiazin là 0,25 μg/ml (với viên nén hàm lượng 80 mg/12,5 mg) hoặc 0,5 μg/ml (với viên nén hàm lượng 40 mg/12,5 mg và 80 mg/25 mg).
Dung dịch đối chiếu gốc (2): Dung dịch chứa 1,6 mg/ml hoặc 3,2 mg/ml (cho viên nén hàm lượng 80 mg/12,5 mg) telmisartan chuẩn, 0,5 mg/ml hydroclorothiazid chuẩn và 2,5 μg/ml tạp chất A chuẩn của benzothiadiazin [pha từ dung dịch đối chiếu gốc (1)] trong dung môi pha mẫu.
Dung dịch đối chiếu (2): Pha loãng dung dịch đối chiếu gốc (2) bằng hỗn hợp pha động A - pha động B (1 : 1) để thu được dung dịch chứa 0,32 mg/ml telmisartan, 0,1 mg/ml hydroclorothiazid và 0,5 μg/ml tạp chất A chuẩn của benzothiadiazin (với viên nén hàm lượng 80 mg/25 mg và 40 mg/12,5 mg). Hoặc dung dịch chứa 0,32 mg/ml telmisartan, 0,05 mg/ml hydroclorothiazid và 0,25 μg/ml tạp chất A chuẩn của benzothiadiazin (với viên nén hàm lượng 80 mg/12,5 mg).
Dung dịch đối chiếu gốc (3): Dung dịch chứa 1,6 mg/ml telmisartan chuẩn, 0,5 mg/ml hydroclorothiazid chuẩn trong dung môi pha mẫu (với viên nén hàm lượng 40 mg/12,5 mg và 80 mg/25 mg) hoặc dung dịch chứa 1,6 mg/ml telmisartan chuẩn, 0,25 mg/ml hydroclorothiazid chuẩn trong dung môi pha mẫu (với viên nén hàm lượng 80 mg/12,5 mg).
Dung dịch thử độ nhạy: Pha loãng 10 ml dung dịch đối chiếu gốc (3) thành 100 ml bằng dung môi pha mẫu. lấy 1,0 ml dung dịch thu được, thêm 2,0 ml dung dịch đối chiếu gốc (1) và pha loãng thành 100 ml bằng dung môi pha mẫu. Pha loãng 1 ml dung dịch thu được thành 5 ml bằng hỗn hợp pha động A - pha động B (1 : 1).
Điều kiện sắc ký:
Cột kích thước (12,5 cm × 4,0 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 40 °C.
Detector quang phổ hấp thụ tử ngoại đặt ở bước sóng 270 nm với hydroclorothiazid và 298 nm với telmisartan. Detector DAD cho phần Định tính.
Tốc độ dòng: 1,2 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 85 | 15 |
3,50 | 85 | 15 |
3,51 | 45 | 55 |
7,70 | 45 | 55 |
7,71 | 20 | 80 |
12,0 | 20 | 80 |
12,1 | 85 | 15 |
15,5 | 85 | 15 |
Tiến hành sắc ký dung dịch đối chiếu (2) và dung dịch thử độ nhạy.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải của pic hydroclorothiazid và pic tạp chất A của benzothiadiazin không nhỏ hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic telmisartan và pic hydroclorothiazid thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %. Trên sắc ký đồ của dung dịch thử độ nhạy, tỷ số tín hiệu trên nhiễu của các pic telmisartan, pic hydroclorothiazid và pic tạp chất A của benzothiadiazin không nhỏ hơn 3,0.
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu (1) và dung dịch đối chiếu (2).
Tính hàm lượng phần trăm của tạp chất A của benzothiadiazin trong chế phẩm dựa vào diện tích pic tạp chất A thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (1), nồng độ tạp chất A của benzothiadiazin trong dung dịch đối chiếu (1) và nồng độ danh định của hydroclorothiazid trong dung dịch thử.
Tính hàm lượng phần trăm của mỗi tạp chất phân hủy liên quan đến hydroclorothiazid trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử ở bước sóng 270 nm, diện tích pic hydroclorothiazid trên sắc ký đồ của dung dịch đối chiếu (2), nồng độ của hydroclorothiazid trong dung dịch đối chiếu (2) và nồng độ danh định của hydroclorothiazid trong dung dịch thử.
Tính hàm lượng phần trăm của mỗi tạp chất phân hủy liên quan đến telmisartan trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất trên sắc ký đồ của dung dịch thử ở bước sóng 298 nm, diện tích pic telmisartan trên sắc ký đồ của dung dịch đối chiếu (2), nồng độ của telmisartan trong dung dịch đối chiếu (2) và nồng độ danh định của telmisartan trong dung dịch thử.
Giới hạn:
Tạp chất A của benzothiadiazin: Không được quá 1,0 %.
Các tạp chất khác: Với mỗi tạp chất, không được quá 0,2 %.
Tổng các tạp chất: Không được quá 0,2 % với các tạp chất phân hủy liên quan đến telmisartan và không được quá 1,5 % với các tạp chất phân hủy liên quan đến hydroclorothiazid.
Ghi chú:
Tạp chất A của benzothiadiazin: 4-Amino-6-cloro-1,3-benzendisulfonamid.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3) như mô tả trong phần Tạp chất liên quan.
Tiến hành sắc ký dung dịch thử và dung dịch đối chiếu (2).
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (2), độ phân giải của pic hydroclorothiazid và pic tạp chất A của benzothiadiazin không nhỏ hơn 2,0 và độ lệch chuẩn tương đối của diện tích telmisartan và pic hydroclorothiazid thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %. Tính hàm lượng phần trăm của hydroclorothiazid, C7H8ClN3O4S2, và telmisartan, C33H30N4O2, trong chế phẩm dựa vào diện tích pic hydroclorothiazid, telmisartan thu được trên sắc ký đồ của dung dịch thử, dung dịch đối chiếu (2) và hàm lượng của hydroclorothiazid, telmisartan trong dung dịch đối chiếu (2).
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc đối kháng thụ thể angiotensin II phối hợp lợi tiểu nhóm thiazid. Điều trị tăng huyết áp.
Hàm lượng thường dùng
Telmisartan/hydroclorothiazid 40 mg/12,5 mg; 80 mg/12,5 mg, 80 mg/25 mg.
VIÊN NÉN TENOFOVIR DISOPROXIL FUMARAT
Là viên nén chứa tenofovir disoproxil fumarat.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận “Thuốc viên nén” (Phụ lục 1.20) và các yêu cầu sau:
Hàm lượng tenofovir disoproxil fumarat, C19H30N5O10P.C4H4O4 từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
A. Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
B. Lấy một lượng bột viên tương đương 50 mg tenofovir disoproxil fumarat hòa tan trong vừa đủ 100 ml methanol (TT), lọc. Pha loãng 2 ml dịch lọc thành 100 ml bằng methanol (TT). Đo phổ hấp thụ tử ngoại của dung dịch thu được và dung dịch chuẩn có nồng độ tenofovir disoproxil fumarat tương đương trong methanol (TT) ở khoảng bước sóng từ 200 nm đến 400 nm (Phụ lục 4.1). Phổ của dung dịch thử và dung dịch chuẩn phải cho cực đại hấp thụ ở bước sóng tương tự.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch acid hydrocloric 0,1 M (TT).
Tốc độ quay: 50 r/min.
Thời gian: 45 min.
Cách tiến hành:
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc. Pha loãng dịch lọc bằng dung dịch acid hydrocloric 0,1 M (TT) để thu được dung dịch có nồng độ tương đương dung dịch chuẩn.
Dung dịch chuẩn: Cân chính xác một lượng tenofovir disoproxil fumarat chuẩn, hòa tan trong dung dịch acid hydrocloric 0,1 M (TT) để thu được dung dịch có nồng độ tenofovir disoproxil fumarat khoảng 0,025 mg/ml.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử và dung dịch chuẩn ở bước sóng cực đại khoảng 260 nm, mẫu trắng là môi trường hòa tan.
Tính hàm lượng tenofovir disoproxil fumarat, C19H30N5O10P.C4H4O4, hòa tan dựa vào độ hấp thụ của dung dịch chuẩn, dung dịch thử và hàm lượng C19H30N5O10P.C4H4O4 trong tenofovir disoproxil fumarat chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng tenofovir disoproxil fumarat, C19H30N5O10P.C4H4O4, so với lượng ghi trên nhãn được hòa tan trong 45 min
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3). Chuẩn bị các dung dịch ngay trước khi dùng.
Pha động A: Hòa tan 1,9 g amoni acetat (TT) trong 1000 ml nước và điều chỉnh đến pH 3,8 bằng acid acetic băng (TT).
Pha động B: Methanol (TT).
Dung dịch thử: Cân 20 viên và nghiền thành một mịn. Cân một lượng bột viên tương đương với 100 mg tenofovir disoproxil fumarat, phân tán trong 50 ml methanol (TT) và lọc.
Dung dịch đối chiếu (1): Dung dịch có chứa 0,2 % tenofovir disoproxil fumarat chuẩn trong methanol (TT).
Dung dịch đối chiếu (2): Pha loãng 1 ml dung dịch đối chiếu (1) thành 100 ml bằng methanol (TT).
Dung dịch đối chiếu (3): Hòa tan 10 mg acid fumaric (TT) trong vừa đủ 50 ml methanol (TT).
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 260 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian | Pha động A | Pha động B |
(min) | (% tt/tt) | (% tt/tt) |
0 | 95 | 5 |
10 | 50 | 50 |
25 | 50 | 50 |
50 | 20 | 80 |
60 | 95 | 5 |
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu (1), hệ số đối xứng của pic tenofovir disoproxil không lớn hơn 2,0; số đĩa lý thuyết tính trên pic tenofovir disoproxil không nhỏ hơn 2000.
Giới hạn: Trên sắc ký đồ của dung dịch thử:
Tạp chất bất kỳ: Diện tích pic không được lớn hơn 3,5 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (3,5 %).
Tổng diện tích pic của tất cả các tạp chất không được lớn hơn 6 lần diện tích pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (2) (6,0 %).
Bỏ qua pic tương ứng với pic chính thu được trên sắc ký đồ của dung dịch đối chiếu (3).
Nước
Không được quá 5,0 % (Phụ lục 10.3).
Dùng 0,5 g chế phẩm.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3). Chuẩn bị các dung dịch ngay trước khi dùng.
Dung môi pha mẫu: Nước - methanol (1 : 1).
Dung dịch đệm phosphat: Hòa tan 7,8 g natri dihydrophosphat (TT) trong 1000 ml nước, thêm 1 ml triethylamin (TT) và điều chỉnh đến pH 2,3 bằng dung dịch acid phosphoric 10 % (TT).
Pha động: Dung dịch đệm phosphat - acetonitril (60 : 40).
Dung dịch thử: Cân 20 viên và nghiền thành một mịn. Cân một lượng bột viên tương đương với 300 mg tenofovir disoproxil fumarat, phân tán trong 100 ml dung môi pha mẫu và lọc. Pha loãng 5,0 ml dịch lọc thành 50,0 ml bằng pha động.
Dung dịch chuẩn: Hòa tan một lượng tenofovir disoproxil fumarat chuẩn trong dung môi pha mẫu để thu được dung dịch 0,120 %. Pha loãng 5,0 ml dung dịch thu được thành 20,0 ml bằng pha động.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 260 nm.
Tốc độ dòng: 1 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, hệ số đối xứng của pic tenofovir disoproxil không lớn hơn 2,0; số đĩa lý thuyết tính trên pic tenofovir disoproxil không nhỏ hơn 2000; độ lệch chuẩn tương đối của diện tích pic tenofovir disoproxil thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tính hàm lượng phần trăm của tenofovir disoproxil fumarat, C19H30N5O10P.C4H4O4, trong chế phẩm dựa vào diện tích pic tenofovir disoproxil trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của C19H30N5O10P.C4H4O4 trong tenotovir disoproxil tumarat chuẩn.
Bảo quản
Trong đồ đựng kín, tránh ẩm, ở nhiệt độ phòng.
Loại thuốc
Thuốc kháng retrovirus.
Hàm lượng thường dùng
300 mg.
VIÊN NÉN TERBUTALIN SULFAT
Là viên nén chứa terbutalin sulfat. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng terbutalin sulfat, (C12H19NO3)2.H2SO4, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của dung dịch thử phải tương ứng với thời gian lưu của pic chính trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu giỏ quay.
Môi trường hòa tan: 900 ml nước.
Tốc độ quay: 100 r/min.
Thời gian: 45 min.
Cách tiến hành:
Xác định lượng terbutalin sulfat đã hòa tan bằng phương pháp sắc ký lỏng (Phụ lục 5.3) với pha động và điều kiện sắc ký như mô tả ở phần Định lượng, thể tích tiêm là 100 μl.
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan, lọc.
Dung dịch chuẩn: Dung dịch terbutalin sulfat chuẩn trong nước có nồng độ tương đương nồng độ terbutalin sulfat trong dung dịch thử.
Yêu cầu: Không ít hơn 75 % (Q) lượng terbutalin sulfat so với lượng ghi trên nhãn được hòa tan trong 45 min.
Độ đồng đều đơn vị liều
Đáp ứng yêu cầu của Phụ lục 11.9.
Phương pháp sắc ký lỏng (Phụ lục 5.3). Pha động, điều kiện sắc ký, dung dịch chuẩn và cách tiến hành như mô tả ở mục Định lượng.
Dung dịch thử: Cho 1 viên vào bình định mức có dung tích thích hợp, thêm một thể tích dung dịch acid sulfuric 0,05 N (TT) tương đương với 10 % thể tích bình, lắc để viên rã và thêm nước đến vạch, trộn đều, lọc.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dung dịch tạo cặp ion: Hòa tan 3,15 g amoni format (TT) trong 900 ml nước, điều chỉnh đến pH 3,0 bằng acid formic (TT), thêm 5,49 g natri 1-hexansulfonat (TT) và thêm nước vừa đủ 1000 ml, trộn đều. Lọc và đuổi khí.
Pha động: Dung dịch tạo cặp ion - methanol (77 : 23).
Dung dịch thử: Cân 20 viên, tính khối lượng trung bình và nghiền thành bột mịn. Cân chính xác một lượng bột thuốc tương ứng với khoảng 10 mg terbutalin sulfat vào bình định mức 100 ml, thêm 10 ml dung dịch acid sulfuric 0,05 N (TT) và 20 ml nước, lắc 15 min sau đó thêm nước đến vạch và trộn đều, lọc.
Dung dịch chuẩn: Hòa tan một lượng thích hợp terbutalin sulfat chuẩn trong pha động để thu được dung dịch chứa 1 mg/ml terbutalin sulfat. Lấy 10,0 ml dung dịch thu được, thêm 10 ml dung dịch acid sulfuric 0,05 N (TT) và pha loãng thành 100,0 ml bằng nước, trộn đều.
Dung dịch phân giải: Dung dịch chứa 1,0 mg/ml terbutalin sulfat chuẩn và 0,4 mg/ml tạp chất C chuẩn của terbutalin sulfat trong pha động.
Điều kiện sắc ký:
Cột kích thước (15 cm x 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 276 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch phân giải.
Thời gian lưu tương đối so với terbutalin của tạp chất C khoảng 0,9.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch phân giải, độ phân giải giữa pic tạp chất C và pic terbutalin không nhỏ hơn 2,0; số đĩa lý thuyết của cột tính trên pic terbutalin không nhỏ hơn 1500; hệ số đối xứng của pic terbutalin không lớn hơn 2,0; độ lệch chuẩn tương đối của diện tích pic terbutalin thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiến hành sắc ký dung dịch thử và dung dịch chuẩn.
Tính hàm lượng phần trăm của terbutalin sulfat, (C12H19NO3)2.H2SO4, trong chế phẩm dựa vào diện tích pic terbutalin thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và hàm lượng của (C12H19NO3)2.H2SO4 trong terbutalin sulfat chuẩn.
Ghi chú:
Tạp chất C của terbutalin: 1-(3,5-dihydroxyphenyl)-2-[(1,1-dimethylethyl)amino]ethanon.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Kích thích beta2 giao cảm, thuốc giãn phế quản.
Hàm lượng thường dùng
2,5 mg, 5 mg.
VIÊN NÉN THIAMAZOL
Viên nén methimazol
Là viên nén chứa thiamazol.
Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng thiamazol, C4H6N2S, từ 94,0 % đến 106,0 % so với lượng ghi trên nhãn.
Định tính
Phân tán đều một lượng bột viên tương ứng với khoảng 10 mg thiamazol với 10 ml cloroform (TT) ấm trong 20 min, lọc và bốc hơi dịch lọc trên cách thủy đến khô. Chuẩn bị mẫu đo dạng đĩa nén kali bromid. Phổ hấp thụ hồng ngoại (Phụ lục 4.2) của mẫu thử phải phù hợp với phổ hấp thụ hồng ngoại của mẫu chuẩn được chuẩn bị trong cùng điều kiện, thay bột viên bằng thiamazol chuẩn.
Độ hòa tan (Phụ lục 11.4)
Thiết bị: Kiểu giỏ quay.
Môi trường hòa tan: 500 ml nước.
Tốc độ quay: 100 r/min.
Thời gian: 30 min.
Cách tiến hành:
Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc. Pha loãng dịch lọc bằng nước để thu được dung dịch có nồng độ thiamazol khoảng 5 μg/ml. Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thu được ở bước sóng 252 nm, cốc đo dày 1 cm, dùng nước làm mẫu trắng. So sánh với dung dịch thiamazol chuẩn có cùng nồng độ pha trong nước.
Tính lượng thiamazol, C4H6N2S, đã hòa tan dựa vào độ hấp thụ của dung dịch thử, dung dịch chuẩn và nồng độ của thiamazol trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng thiamazol, C4H6N2S, so với lượng ghi trên nhãn được hòa tan trong 30 min.
Độ đồng đều đơn vị liều (Phụ lục 11.9)
Dung dịch thử gốc: Chuyển 1 viên (đã được làm vỡ hoặc nghiền thành bột mịn) vào bình định mức 100 ml. Thêm 50 ml nước, lắc bằng máy lắc cơ học trong 30 min. Thêm nước đến vạch, trộn đều và lọc, bỏ 20 ml dịch lọc đầu.
Dung dịch thử: Pha loãng dung dịch thử gốc bằng nước để thu được dung dịch có nồng độ 5 μg/ml thiamazol.
Dung dịch chuẩn: Dung dịch chứa 5 μg/ml thiamazol chuẩn trong nước.
Đo độ hấp thụ (Phụ lục 4.1) của dung dịch thử, dung dịch chuẩn ở bước sóng 252 nm, cốc đo 1 cm, dùng nước làm mẫu trắng.
Tính hàm lượng phần trăm thiamazol, C4H6N2S, trong mỗi viên dựa vào độ hấp thụ của dung dịch thử, dung dịch chuẩn và nồng độ của thiamazol trong dung dịch chuẩn.
Định lượng
Cân 20 viên, tính khối lượng trung bình và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khoảng 120 mg thiamazol vào bình định mức 100 ml. Thêm 80 ml nước, đậy nắp, lắc cơ học trong khoảng 30 min, thêm nước đến vạch, trộn đều và lọc. Thêm 3,5 ml dung dịch natri hydroxyd 0,1 N (CĐ) vào 50,0 ml dịch lọc, trộn đều và thêm 7 ml dung dịch bạc nitrat 0,1 N vừa thêm vừa khuấy đều. Tiếp tục chuẩn độ bằng dung dịch natri hydroxyd 0,1 N (CĐ). Xác định điểm tương đương bằng phương pháp chuẩn độ đo điện thế (Phụ lục 10.2).
1 ml dung dịch natri hydroxyd 0,1 N (CĐ) tương đương với 11,42 mg thiamazol (C4H6N2S).
Bảo quản
Trong đồ đựng kín, tránh ánh sáng.
Loại thuốc
Thuốc kháng giáp dẫn chất thioimidazol.
Hàm lượng thường dùng
5 mg; 10 mg.
VIÊN NÉN TRAMADOL HYDROCLORID VÀ PARACETAMOL
Là viên nén chứa tramadol hydroclorid và paracetamol. Chế phẩm phải đáp ứng các yêu cầu trong chuyên luận "Thuốc viên nén" (Phụ lục 1.20) và các yêu cầu sau đây:
Hàm lượng tramadol hydroclorid, C16H25NO2.HCl, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Hàm lượng paracetamol, C8H9NO2, từ 90,0 % đến 110,0 % so với lượng ghi trên nhãn.
Định tính
Trong phần Định lượng, thời gian lưu của pic chính trên sắc ký đồ của các dung dịch thử tramadol và dung dịch thử paracetamol phải tương ứng với thời gian lưu của pic tramadol và pic paracetamol trên sắc ký đồ của dung dịch chuẩn.
Độ hòa tan
Thiết bị: Kiểu cánh khuấy.
Môi trường hòa tan: 900 ml dung dịch acid hydrodoric 0,1 M (TT).
Tốc độ quay: 50 r/min.
Thời gian: 30 min.
Cách tiến hành:
Xác định hàm lượng tramadol hydroclorid và paracetamol đã hòa tan bằng Phương pháp sắc ký lỏng (Phụ lục 5.3)
Dung dịch đệm: Dung dịch kali dihydrophosphat (TT) 0,68 %, điều chỉnh đến pH 2,5 bằng acid phosphoric (TT).
Pha động: Acetonitril - dung dịch đệm (1 : 4).
Dung dịch thử: Sau thời gian hòa tan quy định, lấy một phần dịch hòa tan và lọc qua màng lọc 0,45 μm.
Dung dịch chuẩn: Dung dịch chứa 0,04 mg/ml tramadol hydroclorid chuẩn và 0,36 mg/ml paracetamol chuẩn trong môi trường hòa tan.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh B (5 μm).
Nhiệt độ cột: 25 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 272 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 25 μl.
Tiến hành sắc ký với thời gian gấp 2 lần thời gian lưu của tramadol.
Tiến hành sắc ký dung dịch chuẩn.
Thời gian lưu tương đối của paracetamol so với tramadol khoảng 0,5.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, độ phân giải giữa pic paracetamol và pic tramadol không nhỏ hơn 5,0 và độ lệch chuẩn tương đối của diện tích pic paracetamol và pic tramadol thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiến hành sắc ký dung dịch thử.
Tính lượng paracetamol, C8H9NO2, và tramadol hydroclorid, C16H25NO2.HCl, đã hòa tan dựa vào diện tích pic paracetamol, pic tramadol thu được trên sắc ký đồ của dung dịch thử, dung dịch chuẩn và nồng độ của paracetamol, tramadol hydroclorid trong dung dịch chuẩn.
Yêu cầu: Không ít hơn 80 % (Q) lượng paracetamol, C8H9NO2 và tramadol hydroclorid, C16H25NO2.HCl so với lượng ghi trên nhãn được hòa tan trong 30 min.
4-Aminophenol
Phương pháp sắc ký lỏng (Phụ lục 5.3). Chuẩn bị các dung dịch ngay trước khi dùng và tránh ánh sáng.
Pha động: Hỗn hợp gồm 250 thể tích methanol (TT) có chứa 4,6 g/l dung dịch tetrabutylamoni hydroxyd 40 %, 375 thể tích dung dịch dinatri hydrophosphat 0,05 M và 375 thể tích dung dịch natri dihydrophosphat 0,05 M.
Dung dịch thử: Cân chính xác một lượng bột viên tương tương với khoảng 0,5 g paracetamol vào bình định mức 50 ml, thêm 35 ml pha động, siêu âm để hòa tan, thêm pha động đến vạch, lắc đều, lọc.
Dung dịch đối chiếu: Dung dịch chứa 0,0015 % 4-aminophenol (TT) và 0,0015 % paracetamol chuẩn trong pha động.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Nhiệt độ cột: 35 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 245 nm.
Tốc độ dòng: 1,5 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 12 lần thời gian lưu của paracetamol.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, độ phân giải giữa 2 pic tương ứng với 4-aminophenol và paracetamol không nhỏ hơn 4,0.
Yêu cầu:
Trên sắc ký đồ thu được của dung dịch thử, pic tương ứng với 4-aminophenol không được có diện tích lớn hơn diện tích pic 4-aminophenol thu được trên sắc ký đồ của dung dịch đối chiếu (0,15 %).
Tạp chất liên quan
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động: Tetrahydrofuran - triethylamin - nước - acid trifluoroacetic (8 : 0,1 : 92 : 0,1), pH của pha động phải từ 2,2 đến 2,4.
Dung môi pha mẫu: Methanol - nước (1 : 9).
Dung dịch thử gốc: Cân 20 viên, tính khối lượng trung bình viên và nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với khối lượng của 1 viên cho vào bình định mức 50 ml, thêm 30 ml dung môi pha mẫu, lắc để phân tán đều bột. Siêu âm 15 min, thỉnh thoảng lắc, sau đó lắc cơ học thêm 30 min, thêm dung môi pha mẫu đến vạch và trộn đều. Ly tâm hỗn dịch thu được và thu lấy phần dịch trong.
Dung dịch thử: Lọc dung dịch thử gốc qua màng lọc nylon kích thước lỗ lọc 0,45 μm, bỏ 4 ml dịch lọc đầu, lấy phần dịch lọc sau.
Dung dịch đối chiếu: Dung dịch chứa 0,75 μg/ml tramadol hydroclorid chuẩn và 0,75 μg/ml tạp chất A chuẩn của tramadol trong dung môi pha mẫu.
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh silica gel liên kết hóa học với nhóm phenyl (1,5 - 10 μm).
Nhiệt độ cột: 50 °C.
Detector quang phổ tử ngoại đặt ở bước sóng 216 nm.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 30 μl.
Cách tiến hành:
Tiến hành sắc ký dung dịch thử, dung dịch đối chiếu và dung môi pha mẫu.
Thời gian lưu tương đối so với tramadol: paracetamol khoảng 0,38; O-desmethyl-tramadol khoảng 0,6; tạp chất A của tramadol khoảng 0,80.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch đối chiếu, độ phân giải giữa pic tạp chất A của tramadol và pic tramadol không nhỏ hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic tramadol hydroclorid thu được từ 6 lần tiêm lặp lại không lớn hơn 6,0 %.
Tính hàm lượng phần trăm của mỗi tạp chất phân hủy trong chế phẩm, nếu có, dựa vào diện tích pic tạp chất thu được trên sắc ký đồ của dung dịch thử, diện tích pic tramadol thu được trên sắc ký đồ của dung dịch đối chiếu và nồng độ của tramadol hydroclorid trong dung dịch đối chiếu.
Giới hạn:
O-desmethyl-tramadol, tạp chất A: Mỗi tạp chất không được quá 0,2 %.
Các tạp chất khác: Với mỗi tạp chất, không được quá 0,2 %.
Tổng các tạp chất phân hủy: Không được quá 0,8 %.
Bỏ qua các pic của dung môi pha mẫu.
Ghi chú:
O-desmethyl-tramadol: 3-{(1RS,2RS)-2- [(dimethylamino)methyl]-1-hydroxycyclohexyl}phenol.
Tạp chất A: RS,SR-1-(3-methoxyphenyl)-2-(dimethylaminomethyl)cyclohexanol hydroclorid.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Pha động, dung môi pha mẫu và dung dịch thử gốc như mô tả ở phần Tạp chất liên quan.
Dung dịch thử tramadol: Pha loãng dung dịch thử gốc trong dung môi pha mẫu để thu được dung dịch có nồng độ tramadol hydroclorid khoảng 75 μg/ml.
Dung dịch thử paracetamol: Pha loãng dung dịch thử gốc trong dung môi pha mẫu để thu được dung dịch có nồng độ paracetamol khoảng 65 μg/ml.
Dung dịch chuẩn: Dung dịch chứa 0,065 mg/ml paracetamol chuẩn và 0,075 mg/ml tramadol hydroclorid chuẩn trong dung môi pha mẫu (siêu âm để hòa tan nếu cần).
Điều kiện sắc ký:
Cột kích thước (15 cm × 4,6 mm) được nhồi pha tĩnh silica gel liên kết hóa học với nhóm phenyl (1,5 - 10 μm).
Nhiệt độ cột: 50 °C.
Detector quang phổ hấp thụ tử ngoại đặt ở bước sóng 216 nm với tramadol hydroclorid và 249 nm với paracetamol.
Tốc độ dòng: 1,0 ml/min.
Thể tích tiêm: 20 μl.
Cách tiến hành:
Tiến hành sắc ký với thời gian gấp 4 lần thời gian lưu của paracetamol.
Kiểm tra tính phù hợp của hệ thống: Trên sắc ký đồ của dung dịch chuẩn, độ phân giải giữa pic paracetamol và pic tramadol không nhỏ hơn 10,0; hệ số đối xứng của pic paracetamol và pic tramadol không lớn hơn 2,0 và độ lệch chuẩn tương đối của diện tích pic paracetamol và pic tramadol thu được từ 6 lần tiêm lặp lại không lớn hơn 2,0 %.
Tiến hành sắc ký dung dịch thử tramadol, dung dịch thử paracetamol và dung dịch chuẩn.
Tính hàm lượng phần trăm của paracetamol, C8H9NO2, và tramadol hydroclorid, C16H25NO2.HCl, trong chế phẩm dựa vào diện tích pic paracetamol diện tích pic tramadol thu được trên sắc ký đồ của dung dịch thử tương ứng, dung dịch chuẩn, hàm lượng C8H9NO2 trong paracetamol chuẩn và C16H25NO2.HCl trong tramadol hydroclorid chuẩn.
Bảo quản
Trong đồ đựng kín, ở nhiệt độ phòng có điều nhiệt.
Loại thuốc
Thuốc giảm đau kết hợp nhóm opioid và nhóm giảm đau, hạ sốt.
Hàm lượng thường dùng
Tramadol 37,5 mg và paracetamol 325 mg.
PHẦN 3
DƯỢC LIỆU
HẸ (Hạt)
Semen Allii Tuberosi
Cửu tử, Phỉ tử, Phỉ thực, Hạt hẹ
Hạt của quả chín đã phơi hay sấy khô của cây Hẹ (Allium tuberosum Rottl. ex Spreng), họ Hành (Liliaceae). Thu hoạch vào mùa thu. Khi thấy chùm quả có quả già đã nứt ra, để lộ các hạt màu đen, cắt lấy cả chùm quả đem phơi khô, đập nhẹ và giũ lấy hạt, loại bỏ tạp chất, phơi hoặc sấy nhẹ đến khô.
Mô tả
Hạt chắc, có dạng nửa hình cầu, hơi dẹt, dài 2 - 4 mm, rộng 1,5 - 3 mm. Bên ngoài màu đen, một mặt hơi nhô lên, thô ráp, có vân dạng lưới, mặt còn lại hơi lõm, có nếp nhăn không rõ. Đỉnh tù, gốc hơi nhọn có núm nhỏ nhô lên. Chất cứng. Mùi thơm nhẹ, vị hơi cay.
Hạt không lép là loại tốt.
Bột
Màu đen xám. Soi kính hiển vi thấy mảnh vỏ hạt tế bào màu nâu hoặc nâu sậm, hình đa giác hoặc hình dạng không xác định, có các vân dạng lưới trên bề mặt. Mảnh nội nhũ tế bào thành dày, đa số bị vỡ thành các mảnh to gần tròn, hơi thuôn dài. Các giọt dầu màu hơi vàng.
Độ ẩm
Không quá 11,0 % (Phụ lục 9.6, 1 g, 100 - 105 °C, 5 h).
Tro toàn phần
Không quá 7,0 % (Phụ lục 9.8).
Tro không tan trong acid
Không quá 1,0 % (Phụ lục 9.7).
Tạp chất
Không quá 1,0 % (Phụ lục 12.11).
Chất chiết được trong dược liệu
Tiến hành theo phương pháp chiết lạnh (Phụ lục 12.10).
Không được ít hơn 7,0 % tính theo dược liệu khô kiệt, dùng ethanol 50 % (TT) làm dung môi.
Không được ít hơn 10,0 % tính theo dược liệu khô kiệt, dùng nước làm dung môi.
Chế biến
Sao muối: Lấy dược liệu sạch, tiến hành như mô tả ở mục Tẩm nước muối sao (Phụ lục 12.20). Dùng dung dịch muối ăn 20 % và sao đến khô và có mùi thơm. Sao muối để tăng cường tính dược đi vào thận.
Bảo quản
Trong đồ đựng kín, tránh mốc mọt. Dược liệu sao muối dùng trong vòng 30 ngày sau khi sao.
Tính vị, quy kinh
Vị cay ngọt, tính ôn. Vào kinh thận, can.
Công năng, chủ trị
Ôn thận, trợ dương, cố tinh. Chủ trị: Thận dương hư suy, dương vật cương cứng kém, xuất tinh sớm, đau lưng, hai chân tê, bàn chân lạnh, đái giắt, đái đục, bạch đới.
Cách dùng, liều lượng
Ngày dùng từ 6 g đến 12 g, dạng thuốc sắc, thường phối hợp trong các bài thuốc.
Kiêng kỵ
Người tỳ vị kém, ăn uống kém khi dùng cần cân nhắc.
TÁO MÈO (Quả)
Fructus Docyniae indicae
Quả già chín, thái lát, phơi hay sấy khô của cây Táo mèo {Docynia indica (Wall.) Decne, Syn. Pyrus indica Wall.} họ Hoa hồng (Rosaceae). Thu hái vào mùa thu đến đầu mùa đông (hoặc tháng 8 - 10), lấy quả già chín, rửa sạch, thái lát ngang quả, phơi hoặc sấy khô.
Mô tả
Lát cắt (phiến) phơi khô có hình tròn hoặc bầu dục, hơi cong queo, đường kính 1,5 - 3,0 cm, dày 0,2 - 0,5 cm, một số phiến mang cuống quả hoặc mang đỉnh quả nhô lên có đài bền ở đỉnh, một số phiến có hạch cứng hoặc không có hạch cứng tùy theo vị trí cắt. Mép phiến nhăn nheo, vỏ mỏng, bóng, đôi khi có các nốt chấm màu nâu sậm. Hạt màu nâu đen (thường có các hạt trong năm khoang dính lại trên các lát cắt giữa quả), hình trứng, vỏ hạt cứng và nhẵn, nhân hạt màu trắng ngà. Thịt quả màu nâu hay nâu nhạt, hơi cứng, có vị chua, chát.
Bột
Bột màu nâu đỏ, khô tơi, có ít xơ, vị chua, chát. Mảnh vỏ ngoài tế bào hình chữ nhật nhỏ, thường tách rời. Mảnh mô mềm thịt quả tế bào hình đa giác, thường dính thành đám, một số tế bào chứa chất màu nâu. Tế bào mô cứng của thịt quả riêng lẻ. Mảnh vỏ hạt tế bào hình chữ nhật xếp khít nhau như mô giậu. Mảnh mô mềm lá mầm chứa giọt dầu béo. Mảnh mạch xoắn.
Định tính
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel GF254.
Dung môi khai triển: Ethyl acetat - methanol - acid formic - nước (25 : 1 : 1 : 1).
Dung dịch thử: Lấy 1 g bột thô dược liệu, thêm 10 ml methanol 50 % (TT), siêu âm trong 30 min. Để nguội. Lọc, thu được dung dịch thử.
Dung dịch dược liệu đối chiếu: Lấy 1 g bột Táo mèo (mẫu chuẩn), chiết như mô tả trong mục Dung dịch thử:
Cách tiến hành: Chấm riêng biệt lên bản mỏng 10 μl mỗi dung dịch trên. Sau khi triển khai sắc ký lấy bản mỏng ra để khô trong không khí ở nhiệt độ phòng, phun dung dịch acid sulfuric trong ethanol (TT). Quan sát dưới ánh sáng tử ngoại bước sóng 365 nm. Trên sắc ký đồ của dung dịch thử phải có các vết giống về màu sắc và vị trí với các vết trên sắc ký đồ của dung dịch dược liệu đối chiếu.
Độ ẩm
Không được quá 13,0 % (Phụ lục 9.6, 1 g, 105 °C, 5 h).
Tạp chất
Tỷ lệ nâu đen (phần thịt quả): Không được quá 1,0 % (Phụ lục 12.11).
Tỷ lệ vụn nát
Qua rây có cỡ mắt rây 4 mm: Không được quá 2 % (Phụ lục 12.12).
Tro toàn phần
Không được quá 8,0 % (Phụ lục 9.8).
Tro không tan trong acid
Không được quá 1,0 % (Phụ lục 9.7).
Chất chiết được trong dược liệu
Không được ít hơn 21,0 % tính theo dược liệu khô kiệt. Tiến hành theo phương pháp chiết lạnh (Phụ lục 12.10), dùng ethanol 70 % (TT) làm dung môi.
Định lượng
Phương pháp sắc ký lỏng (Phụ lục 5.3).
Dãy dung dịch chuẩn: Cân chính xác một lượng acid clorogenic chuẩn và hòa tan trong methanol 50 % (TT) để được dung dịch nồng độ 1 mg/ ml. Pha loãng dung dịch thu được với cùng dung môi để được dãy dung dịch chuẩn có nồng độ 10, 40, 60, 90, 120 μg/ml.
Dung dịch thử: Cân chính xác khoảng 2 g dược liệu (qua rây số 355), thêm 25 ml methanol 50 % (TT), đun hồi lưu trên cách thủy hoặc siêu âm (công suất 390 W, tần số 50 Hz) trong 30 min, để nguội. Lọc và chuyển dịch lọc vào bình định mức 50 ml. Tiếp tục chiết như trên thêm một lần nữa với 15 ml methanol 50 % (TT). Rửa cắn với 5 ml methanol 50 % (TT). Gộp các dịch lọc và dịch rửa vào bình định mức ở trên và thêm methanol 50 % (TT) đến vạch. Trộn đều và lọc qua màng lọc 0,45 μm.
Điều kiện sắc ký:
Cột kích thước (25 cm × 4,6 mm) được nhồi pha tĩnh C (5 μm).
Detector quang phổ tử ngoại đặt ở bước sóng 327 nm.
Tốc độ dòng: 0,7 ml/min.
Thể tích tiêm: 10 μl.
Cách tiến hành:
Tiến hành sắc ký theo chương trình dung môi như sau:
Thời gian (min) | Acetonitril (% tt/tt) | Dung dịch acid phosphoric 0,1 % (% tt/tt) |
0 → 30 | 100 → 95 | 0 → 5 |
Kiểm tra tính phù hợp của hệ thống: Tiến hành sắc ký 5 lần với dung dịch chuẩn 10 μg/ml. Độ lệch chuẩn tương đối của diện tích pic acid clorogenic không lớn hơn 2,0 % và độ lệch chuẩn tương đối thời gian lưu của pic acid clorogenic không lớn hơn 2,0 %. Số đĩa lý thuyết của cột tính theo pic acid clorogenic không nhỏ hơn 6000.
Tiến hành sắc ký với các dung dịch chuẩn ở trên. Vẽ đường hồi qui biểu diễn sự phụ thuộc giữa diện tích pic acid clorogenic và nồng độ các dung dịch chuẩn.
Tiến hành sắc ký dung dịch thử. Xác định pic acid clorogenic trên sắc ký đồ của dung dịch thử bằng cách đối chiếu thời gian lưu của pic này với pic trên sắc ký đồ của dung dịch đối chiếu. Thời gian lưu của acid clorogenic trên hai sắc ký đồ khác nhau không lớn hơn 2,0 %.
Tính hàm lượng acid acid clorogenic trong dược liệu dựa vào diện tích pic acid clorogenic trên sắc ký đồ của dung dịch thử, đường chuẩn đã lập và hàm lượng C16H18O9 trong acid clorogenic chuẩn.
Dược liệu phải chứa không ít hơn 0,1 % acid clorogenic (C16H18O9), tính theo dược liệu khô kiệt.
Chế biến
Sao vàng: Lấy dược liệu đã thái lát sao vàng.
Sao cháy (thán sao): Lấy dược liệu đã thái lát sao cháy.
Cách sao theo hướng dẫn tại Phụ lục 12.20.
Bảo quản
Dược liệu chưa chế biến: Trong đồ đựng kín, tránh ẩm, mốc mọt.
Dược liệu đã chế biến: Trong đồ đựng kín, để nơi khô ráo tránh ẩm mốc, nên dùng trong 60 ngày sau khi chế biến.
Tính vị, quy kinh
Vị chua chát, tính hàn. Vào các kinh can, tỳ, vị.
Công năng, chủ trị
Tán khí hóa ứ, hóa đờm, cầm máu, tiêu thực, giảm đau. Chủ trị: Chứng sán khí, ăn không tiêu đầy hơi chướng bụng. Phụ nữ sau khi sinh đau bụng do ứ huyết. Thán sao để cầm máu, trị chảy máu cam, băng huyết.
Cách dùng, liều lượng
Ngày dùng từ 8 g đến 16 g, có thể dùng đến 20 g tùy theo bệnh chứng của bệnh nhân, dạng thuốc sắc hoặc hoàn tán theo yêu cầu của bài thuốc.
Kiêng kỵ
Tỳ vị hư, ăn kém không có thực tích không được dùng.
THIÊN HOA PHẤN
Radix Trichosanthis
Qua lâu căn
Rễ (rễ củ, còn gọi là củ) đã phơi hay sấy khô của cây Qua lâu (Trichosanthes kirilowii Maxim.) hoặc cây Song biên qua lâu (Trichosanthes rosthornii Harms), họ Bí (Cucurbitaceae). Thu hoạch vào cuối mùa thu đến đầu mùa đông. Đào lấy rễ củ của các cây không có quả (còn gọi là cây đực), rửa sạch, cạo bỏ vỏ thô bên ngoài, cắt khúc dài 10 - 15 cm, ngâm nước vôi loãng trong 7 ngày (để giảm bớt tính đắng), vớt ra, để nguyên hoặc bổ dọc thành 2 hay 4 mảnh với củ to có đường kính trên 3 cm phơi hoặc sấy khô.
Mô tả
Gồm những đoạn rễ củ khô, để nguyên, thẳng hay hơi cong, thiết diện có thể không được tròn, đường kính 1 - 2 cm, dài 9 -15 cm hoặc những đoạn rễ củ đã được bổ dọc, hình của thiết diện tùy thuộc khi bổ dọc. Dược liệu để nguyên hay đã bổ dọc đều có mặt ngoài màu trắng ngà hay nâu vàng nhạt, đôi chỗ còn sót phần vỏ ngoài có màu nâu đen hoặc vết lõm của rễ. Chất thịt, cứng, khó bẻ gẫy, mặt bẻ màu trắng ngà hay ngà vàng, nhiều bột, có các tia (xơ) gỗ xếp hướng tâm. Mùi nhẹ, vị hơi đắng, chua. Loại rễ (củ) khô, chắc nặng, mặt ngoài màu vàng ngà, bên trong màu trắng, nhiều tinh bột là tốt. Nếu củ quá già sẽ nhiều xơ, củ non sẽ xốp, làm thuốc không hiệu quả.
Bột
Màu trắng ngà. Nhiều hạt tinh bột, hạt đơn hình cầu, hơi tròn hoặc hình trứng hoặc hình chuông, đường kính 6 - 48 μm, rốn dạng điểm, vạch ngắn hoặc hình chữ V, có vân hơi rõ; các hạt tinh bột thường tụ thành khối lớn hoặc nhỏ gồm 2 - 14 hạt. Mảnh mạch điểm lớn, hầu hết bị vỡ, một số mạch hình lục giác hoặc hình vuông sít nhau. Tế bào đá màu lục vàng, hình chữ nhật, hình thuôn, gần vuông hoặc hình đa giác hoặc hình thoi, đường kính 27 - 72 μm, thành khá dày, có nhiều lỗ trao đổi nhỏ (ngâm bột trong cloralhydrat sẽ dễ quan sát tế bào đá).
Định tính
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel G.
Dung môi khai triển: n-Butanol - ethanol - acid acetic băng - nước (8 : 2 : 2 : 3).
Dung dịch thử: Cho 2 g dược liệu vào bình nón, thêm 20 ml ethanol 50 % (TT), siêu âm trong 30 min, lọc. Lấy dịch lọc làm dung dịch thử.
Dung dịch chất đối chiếu: Hòa tan L-citrullin chuẩn trong ethanol 50 % (TT) để được dung dịch có nồng độ 1 mg/ml.
Dung dịch dược liệu đối chiếu: Lấy 2 g bột rễ Qua lâu (mẫu chuẩn), chiết như mô tả ở phần dung dịch thử.
Cách tiến hành: Chấm riêng biệt lên bản mỏng 2 μl dung dịch thử và dung dịch dược liệu đối chiếu và 1 μl dung dịch chất đối chiếu. Sau khi triển khai sắc ký lấy bản mỏng ra để khô trong không khí. Phun dung dịch ninhydrin (TT) 2 % trong ethanol 96 % (TT), sấy ở 105 °C đến khi các vết hiện rõ. Quan sát dưới ánh sáng thường. Trên sắc ký đồ của dung dịch thử phải có các vết giống về màu sắc và vị trí với các vết trên sắc ký đồ của dung dịch dược liệu đối chiếu và trong đó có vết giống về màu sắc và vị trí với vết L-citrullin trên sắc ký đồ của dung dịch chất đối chiếu.
Độ ẩm
Không được quá 15,0 % (Phụ lục 9.6, 1 g, 105 °C, 5 h).
Tạp chất
Không được quá 1,0 % (Phụ lục 12.11).
Tro toàn phần
Không được quá 5,0 % đối với dược liệu chưa thái lát và không được quá 4,0 % đối với dược liệu đã thái lát (Phụ lục 9.8).
Tro không tan trong acid
Không được quá 1,0 % (Phụ lục 9.7).
Kim loại nặng
Không được quá 30 phần triệu (Phụ lục 9.4.8, phương pháp 3).
Lưu huỳnh dioxyd
Không được quá 400 phần triệu ( Phụ lục 7.9, phương pháp 2).
Chất chiết được trong dược liệu
Không ít hơn 7,0 % tính theo dược liệu khô kiệt. Tiến hành theo phương pháp chiết lạnh (Phụ lục 12.10), dùng ethanol 70 % (TT) làm dung môi.
Chế biến
Dược liệu thái lát: Lấy dược liệu đã cắt đoạn, tẩm nước, ủ mềm (khoảng 6 h), thái lát mỏng, phơi hoặc sấy khô.
Mô tả: Lát dày 2 - 3 mm, không đều, hình gần tròn hoặc hình bán nguyệt, màu trắng ngà hoặc vàng nâu nhạt, một số lát có viền hơi nâu của phần vỏ thô còn sót lại, mặt cắt nhiều bột. Mùi nhẹ, vị hơi đắng.
Bột, Định tính, Độ ẩm, Tạp chất, Tro toàn phần, Tro không tan trong acid, Kim loại nặng, Lưu huỳnh dioxyd, Chất chiết được trong dược liệu: Yêu cầu và phương pháp thử như dược liệu chưa thái lát.
Ghi chú: Có thể chế biến thành dạng bột bằng cách lấy củ tươi, rửa sạch, cạo bỏ vỏ ngoài, cắt khúc 5 - 10 cm, ngâm nước 4 ngày đêm, giã nhuyễn, lắng lấy bột, phơi khô.
Bảo quản
Dược liệu chưa chế biến: Trong đồ đựng kín, tránh mốc mọt.
Dược liệu đã chế biến nên dùng trong vòng 30 ngày kể từ khi chế.
Tính vị, quy kinh
Vị ngọt hơi đắng, tính hàn. Vào các kinh phế, vị, đại tràng.
Công năng, chủ trị
Thanh nhiệt sinh tân chỉ khát, tiêu thũng bài mủ. Chủ trị: Nhiệt chứng phiền khát, vị nhiệt phản vị (trào ngược), phế nhiệt ho khan, nội nhiệt tiêu khát (tiểu đường), viêm tuyến vú nung mủ, nhọt mạch lươn.
Cách dùng, liều lượng
Ngày dùng từ 8 g đến 12 g, có thể dùng đến 20 g, dạng thuốc sắc hoặc hoàn tán, thường phối hợp trong các bài thuốc.
Kiêng kỵ
Người mắc chứng tỳ hư hàn không dùng. Phản Ô đầu (không dùng chung với thuốc có ô đầu).
THIÊN NAM TINH (Thân rễ)
Rhizoma Arisaematis
Thân rễ (còn gọi là củ) khô đã cạo vỏ ngoài của cây Thiên nam tinh (Arisaema erubescens (Wall.) Schott.), cây Dị diệp thiên nam tinh (Arisaema heterophyllum BI.), hoặc cây Đông bắc thiên nam tinh (Arisaema amurense Maxim.), họ Ráy (Araceae). Thu hoạch vào mùa thu (tiết bạch lộ là tốt nhất), khi thân và lá khô héo, đào lấy thân rễ, gọt bỏ vỏ ngoài, rửa sạch, phơi khô (không nên sấy vì khó chế biến).
Mô tả
Thân rễ dạng củ hình cầu, hơi dẹt, dày 1 - 2 cm, đường kính 1,5 - 6,5 cm. Mặt ngoài màu trắng hơi nhạt hoặc nâu nhạt (loại củ già), tương đối nhẵn, bóng, một số củ lại nhăn nheo. Đỉnh còn vết lõm của gốc thân. Xung quanh có những chấm nhỏ là vết của rễ con. Có khi quanh vết lõm gốc thân có các chồi thân rễ nhỏ hình cầu dẹt. Chất cứng rắn, khó bẻ, mặt bẻ phẳng, màu trắng, có tinh bột, mùi hăng, vị cay tế.
Bột
Màu trắng. Quan sát trên kính hiển vi thấy: Nhiều hạt tinh bột đơn, hình cầu hoặc hình trứng, đường kính 2 - 17 μm, rốn có dạng điểm, dạng kẽ nứt, có thể trông thấy lờ mờ đường vân tăng trưởng ở các hạt lớn. Có ít hạt tinh bột kép 2 - 12. Tinh thể calci oxalat hình kim, dài 63 - 131 μm nằm rải rác hoặc tụ thành bó. Đôi khi thấy tinh thể calci oxalat hình lăng trụ, đường kính 3 - 20 μm.
Định tính
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel G.
Dung môi khai triển: n-Butanol - nước - acid acetic băng (7 : 2 : 1).
Dung dịch thử: Lấy 5 g bột dược liệu cho vào bình nón nút mài 50 ml, thêm 10 ml ethanol (TT), siêu âm 30 min, lọc, được dung dịch chấm sắc ký.
Dung dịch dược liệu đối chiếu: Lấy 1 g bột rễ Thiên nam tinh (mẫu chuẩn), chiết như mô tả ở phần Dung dịch thử:
Cách tiến hành: Chấm riêng biệt lên bản mỏng 6 μl dung dịch thử và 15 μl dung dịch dược liệu đối chiếu. Sau khi triển khai sắc ký, lấy bản mỏng ra để khô trong không khí. Phun dung dịch anisaldehyd trong acid sulfuric (TT), sấy bản mỏng ở 105 °C đến khi hiện rõ vết. Quan sát bản mỏng dưới ánh sáng thường. Trên sắc ký đồ của dung dịch thử phải có các vết giống về màu sắc và vị trí với các vết trên sắc ký đồ của dung dịch dược liệu đối chiếu.
Độ ẩm
Không được quá 14,0 % (Phụ lục 9.6, 1 g, 105 °C, 5 h).
Tro toàn phần
không được quá 5,0 % (Phụ lục 9.8).
Tạp chất
Không được quá 1 % (Phụ lục 12.11).
Chất chiết được trong dược liệu
Không dưới 9,0 %, tính theo dược liệu khô kiệt (Phụ lục 12.10). Tiến hành theo phương pháp chiết nóng, dùng ethanol 50 % (TT) làm dung môi.
Bảo quản
Để nơi khô, thoáng, tránh sâu, mọt.
Cách dùng, liều lượng
Dược liệu chưa chế biến (dược liệu sống) chỉ dùng ngoài trị ung thũng, rắn cắn, côn trùng cắn gây thương tổn. Thực tế ít dùng.
Cách dùng: Tán bột, hoà với giấm hoặc rượu đắp nơi đau, lượng thích hợp.
Làm nguyên liệu để sản xuất Thiên nam tinh đã chế biến.
THIÊN NAM TINH ĐÃ CHẾ BIẾN
Rhizoma Arisaematis praeparata
Thân rễ khô đã chế biến của cây Thiên nam tinh (Arisaema erubescens (Wall.) Schott.), cây Dị diệp thiên nam tinh (Arisaema heterophyllum BI.), hoặc cây Arisaema amurense Maxim., họ Ráy (Araceae).
Chế biến
Lấy Thiên nam tinh sạch, phân loại lớn, nhỏ, ngâm nước sạch, mỗi ngày thay nước 2 - 3 lần. Thời gian ngâm tùy theo củ lớn bé, củ lớn ngâm dài ngày, củ bé ngâm ngắn ngày hơn nhưng phải ngâm đến khi nổi bọt trắng là đạt yêu cầu, vớt ra để ráo nước. Tiếp tục ngâm nước phèn chua (lấy phèn chua hòa tan trong nước sạch vừa đủ để ngâm ngập lượng dược liệu, cứ 10 kg thiên nam tinh dùng 1 - 1,5 kg phèn chua), mỗi ngày thay nước một lần, ngâm cho đến khi bổ ra, nhấm nhẹ không thấy có cảm giác cay, tê, ngứa đầu lưỡi là đạt. Sau đó vớt ra để ráo nước, xếp đều củ vào nồi thành từng lớp, mỗi lớp cho một lớp gừng tươi đã thái lát mỏng, sau đó lấy phèn chua hòa nước vừa đủ cho vào nồi, đun sôi cho đến khi củ mềm, trong suốt không còn lõi trắng là đạt. Vớt ra, bỏ gừng, để se nước nhưng còn độ dẻo không để quá khô, thái lát dày 1 mm, phơi khô (cứ 10 kg thiên nam tinh dùng phèn chua và gừng mỗi loại 1 - 1,5 kg).
Mô tả
Phiến mỏng, hơi tròn, không đều. Chất giòn, dễ bẻ gãy, mặt bẻ phẳng, bóng. Không mùi, vị cay tê nhẹ.
Bột
Màu màu vàng xám hoặc nâu vàng. Mảnh mô mềm mang khối tinh bột bị hồ hóa, mảnh mạch xoắn, mảnh mạch điểm.
Định tính
Phương pháp sắc ký lớp mỏng (Phụ lục 5.4).
Bản mỏng: Silica gel GF254.
Dung môi khai triển: Toluen - ethyl acetat - acid formic - methanol (4 : 4 : 0,5 : 1).
Dung dịch thử: Lấy 2 g bột dược liệu vào bình nón, thêm 25 ml dung dịch acid hydrocloric (TT) 5 %, đun sôi hồi lưu trong 30 min, để nguội, lọc. Chuyển dịch lọc vào bình gạn, lắc kỹ với 30 ml ethyl acetat (TT). Gạn lấy dịch chiết ethyl acetat, cô trên cách thủy đến còn khoảng 5 ml được dung dịch chấm sắc ký.
Dung dịch dược liệu đối chiếu: Lấy 2 g bột rễ Thiên nam tinh (mẫu chuẩn), chiết như mô tả ở phần Dung dịch thử:
Cách tiến hành: Chấm riêng biệt lên bản mỏng 10 μl mỗi dung dịch trên. Sau khi triển khai sắc ký, lấy bản mỏng ra để khô trong không khí ở nhiệt độ phòng. Quan sát bản mỏng dưới ánh sáng tử ngoại ở bước sóng 366 nm. Trên sắc ký đồ của dung dịch thử phải có vết giống về màu sắc và vị trí với các vết trên sắc ký đồ của dung dịch dược liệu đối chiếu.
Độ ẩm
Không được quá 14,0 % (Phụ lục 9.6, 1 g, 105 °C, 5 h).
Tro toàn phần
Không được quá 4,0 % (Phụ lục 9.8).
Tro không tan trong acid
Không được quá 0,5 % (Phụ lục 9.7).
Giới hạn hàm lượng phèn chua
Không được quá 15,0 % phèn chua tính theo kali nhôm sulfat [KAl(SO4)2.12H2O] tính theo dược liệu khô kiệt.
Cân chính xác khoảng 2 g bột dược liệu (qua rây số 250) vào một chén nung bằng sứ hoặc platin, đun nóng để tăng nhiệt độ dần tới 450 °C và nung trong 4 h đến không còn carbon, để nguội, thêm cẩn thận 10 ml dung dịch acid hydrocloric 10 % (TT), đậy chén nung bằng một phiến kính nhỏ để ngăn sự bay hơi của dịch acid, đun trên cách thủy trong 20 min, lấy chén nung ra và rửa phiến kính bằng 5 ml nước nóng. Gộp dịch rửa vào chén nung và lọc, rửa cắn và chén nung bằng 25 ml nước. Gộp dịch lọc và dịch rửa, thêm 1 giọt dung dịch đỏ methyl (TT), trộn đều, thêm từng giọt amomia (TT) cho đến khi dung dịch chuyển từ màu đỏ sang màu vàng. Thêm 25 ml dung dịch dung dịch đệm acetat pH 6,0 (TT), thêm chính xác 25 ml dung dịch natri edetat 0,05 M (CĐ), đun sôi trong 3 - 5 min, để nguội. Thêm 1 ml dung dịch da cam xylenol (TT) và chuẩn độ bằng dung dịch kẽm sulfat 0,05 M (CĐ) đến khi màu của dung dịch chuyển từ màu vàng sang đỏ cam. Song song tiến hành một mẫu trắng.
1 ml dung dịch natri edetat 0,05 M (CĐ) tương đương với 23,72 mg kali nhôm sulfat [KAl(SO4)2.12H2O].
Bảo quản
Trong đồ đựng kín, tránh ẩm, mốc mọt. Nên dùng trong 30 ngày sau khi chế biến.
Tính vị, quy kinh
Vị đắng, cay, tính ôn, có độc, Vào các kinh phế, can, tỳ.
Công năng, chủ trị
Táo thấp, hoá đờm, khu phong, giảm đau, giảm co cứng, tán ứ kết, tiêu thũng, Chủ trị: Thường xuyên có đàm tích khò khè nơi cổ (ngoan đàm) gây ho, bốc hỏa chóng mặt, trúng phong đờm nghẽn, liệt mặt (liệt dây thần kinh số 7 ngoại biên), bán thân bất toại (bại liệt nửa người), động kinh (co giật) cấp kinh phong, mạn kinh phong, uốn ván (phá thương phong).
Cách dùng, liều lượng
Thiên nam tinh đã chế biến: Ngày dùng từ 4 g đến 12 g, dạng thuốc sắc hoặc hoàn tán. Thường phối hợp với các vị thuốc khác.
Kiêng kỵ.
Phụ nữ có thai không nên dùng. Trường hợp bắt buộc dùng phải theo dõi cẩn thận.
TỔ BỌ NGỰA
Mantidis Oötheca
Tang phiêu tiêu, Tổ bọ ngựa trên cây dâu
Toàn tổ chứa trứng của con Bọ ngựa (Tenodera sinensis Saussure, 1871; syn. Tenodera aridifolia sinensis, Saussure 1871), họ Bọ ngựa (Matidae) đã được chế biến rồi phơi hay sấy khô. Tổ bọ ngựa trên cây dâu là tốt nhất. Thu hái tổ bọ ngựa khi trứng còn chưa nở vào khoảng từ mùa thu đến mùa xuân năm sau. Chú ý khi tách lấy tổ cần nhẹ nhàng để tránh làm dập tổ, vỡ trứng.
Mô tả
Toàn tổ bọ ngựa có màu nâu vàng hoặc nâu đen, bên trong có từng nếp nhăn và có vách ngăn thành các ô, mỗi ô có một quả trứng.
Dựa vào hình dạng tổ và màu sắc bên ngoài, có thể phân loại như sau:
Đoàn phiêu tiêu: Hơi hình trụ hoặc hình bán nguyệt, bao gồm nhiều lớp tấm giống như màng, dài 2,5 - 4 cm và rộng 2 - 3 cm. Bên ngoài có màu nâu vàng, phần phình ra ở phía trên như một gờ dài không rõ ràng và phần dưới phẳng hoặc lõm. Chất nhẹ, dai và lỏng lẻo. Mặt cắt ngang có một lớp bên ngoài giống như bọt biển, lớp bên trong bao gồm nhiều khoang nhỏ được sắp xếp theo mô hình xuyên tâm; mỗi khoang có chứa một trứng hình bầu dục nhỏ, màu nâu sẫm và sáng bóng. Vị hơi tanh, hơi mặn.
Trường phiêu tiêu: Hình trụ hơi thon dài, hẹp ở một đầu, dài 2,5 - 5 cm và rộng 1 - 1,5 cm. Bề mặt có màu vàng xám với những chỗ phình hình dải rõ ở trên, có một rãnh nông màu nâu sẫm và vân chéo ở mỗi bên của dải. Chất cứng và giòn.
Hắc phiêu tiêu: Hình bình hành hơi dài, dài 2 - 4 cm, rộng 1,5 - 2 cm. Bề mặt có màu nâu xám hoặc nâu đen, với những chỗ phình hình dải rõ ràng ở trên cùng các vân xiên ở hai bên; ở một đầu có phần nhỏ dài hơi cong nhô lên. Chất cứng và dai.
Bột
Màu nâu vàng nhạt. Soi tiêu bản với glycerin - acid acetic (TT) thấy: Các hạt hình lòng đỏ trứng tụ thành đám (cần phải quan sát ngay lập tức sau khi lên tiêu bản), màu hơi vàng, đường kính 10 - 160 μm, trên bề mặt có các hạt nhỏ hoặc lỗ không đều.
Soi tiêu bản với cloral hydrat (TT) thấy: Mảnh thành tổ không đều, màu hơi vàng đến hơi nâu đỏ, trên bề mặt có các khoang tròn to nhỏ không đều. Một số khối trụ tinh thể acid citric, mỗi khối có đường kính 2 - 10 μm, dài tới 20 μm.
Định tính
Lấy 2,0 g bột dược liệu, thêm 20 ml nước, đun sôi trong 10 min, lọc. Lấy 2 ml dịch lọc, thêm 3 - 4 giọt dung dịch ninhydrin 0,2% (TT), đun sôi nhẹ trong 5 min, xuất hiện màu xanh dương hơi đỏ tía.
Độ ẩm
Không quá 15,0 % (Phụ lục 9.6, 1 g, 100 - 105 °C, 5 h).
Tro toàn phần
Không quá 8,0 % (Phụ lục 9.8).
Tro không tan trong acid
Không quá 3,0 % (Phụ lục 9.7).
Tạp chất
Không quá 1,0 % (Phụ lục 12.11).
Chất chiết được trong dược liệu
Tiến hành theo phương pháp chiết lạnh (Phụ lục 12.10).
Không ít hơn 4,0 % tính theo dược liệu khô kiệt, dùng nước làm dung môi.
Không ít hơn 4,0 % tính theo dược liệu khô kiệt, dùng ethanol 50 % (TT) làm dung môi.
Chế biến
Khi thu hái, nếu cành nhỏ thì cắt cả đoạn cành để lấy tổ, nếu cành to thì bóc lấy cả đoạn vỏ cành nơi tổ đóng, tránh làm vỡ trứng ở trong; nếu dập tổ, vỡ trứng sẽ giảm tác dụng.
Sau khi thu hái, rửa bằng nước phù sa để bảo tồn một phần âm khí trong tổ bọ ngựa. Đồ khoảng 40 - 50 min cho chín trứng, sau đó loại bỏ tạp chất rồi phơi khô. Có thể tẩm nước tương bằng cách đun sôi trong nước tương có cho thêm chút muối để có tác dụng đưa thuốc vào thận, đun cho chín trứng. Phơi hoặc sấy khô để dùng.
Bảo quản
Trong đồ đựng kín, nơi khô, tránh sâu mọt. Nên dùng trong 30 ngày sau khi chế biến.
Tính vị, quy kinh
Vị hơi ngọt, mặn, tính bình. Vào kinh can, thận.
Công năng, chủ trị
Ích thận, cố tinh, bổ hư. Chủ trị: Di tinh, liệt dương, đái giắt, đau eo lưng (đau vùng thắt lưng), bế kinh, đái dầm.
Cách dùng, liều lượng
Ngày dùng từ 4 g đến 12 g, dạng thuốc sắc, trước khi sắc giã dập.
Dạng thuốc hoàn tán: Sao giòn, giã thành bột, liều dùng tùy theo bài thuốc.
Kiêng kỵ
Người hỏa vượng nên dùng liều nhỏ.
Cảm ơn quý khách đã gửi báo lỗi.
Thông tin liên hệ về Tiêu chuẩn Quốc gia - TCVN
Trung tâm Thông tin – Truyền thông (ISMQ), Tổng cục Tiêu chuẩn Đo lường Chất lượng
Số 8 Hoàng Quốc Việt, Quận Cầu Giấy, Hà Nội | ĐT: (024) 37562608 – Email: [email protected]
Hotline (hỗ trợ) của LuatVietnam: 0938361919
Đang tải dữ liệu. Quý khách vui lòng đợi...
 Tiêu chuẩn Việt Nam TCVN IX:2024 PDF
Tiêu chuẩn Việt Nam TCVN IX:2024 PDF Tiêu chuẩn Việt Nam TCVN IX:2024 DOC (Bản Word)
Tiêu chuẩn Việt Nam TCVN IX:2024 DOC (Bản Word)